
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 8)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sevabertinib
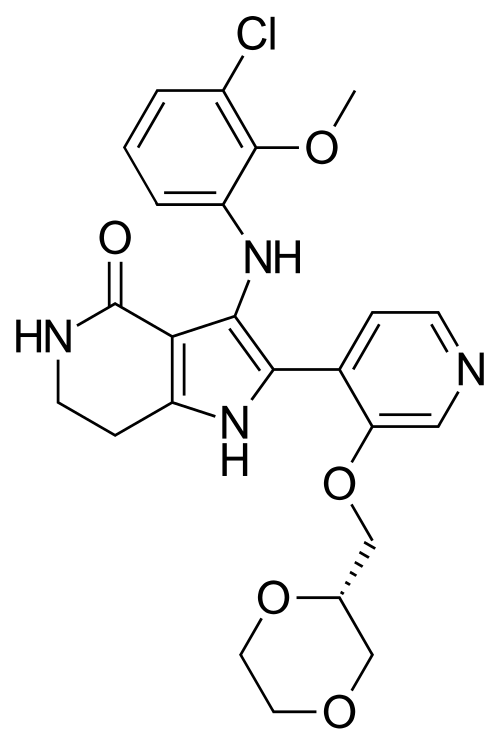
Sevabertinib
CAS 2521285-05-0
MF C24H25ClN4O5, 484.9 g/mol
3-(3-chloro-2-methoxyanilino)-2-[3-[[(2S)-1,4-dioxan-2-yl]methoxy]-4-pyridinyl]-1,5,6,7-tetrahydropyrrolo[3,2-c]pyridin-4-one
11/19/2025, FDA 2025, APPROVALS 2025, Hyrnuo, 2A7VPM5RWH, BAY-2927088, BAY 2927088
To treat locally advanced or metastatic non-squamous non-small cell lung cancer with tumors that have activating HER2 tyrosine kinase domain activating mutations in patients who received a systemic therapy
Sevabertinib, sold under the brand name Hyrnuo, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1] Sevabertinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Sevabertinib was approved for medical use in the United States in November 2025.[2]
SYN
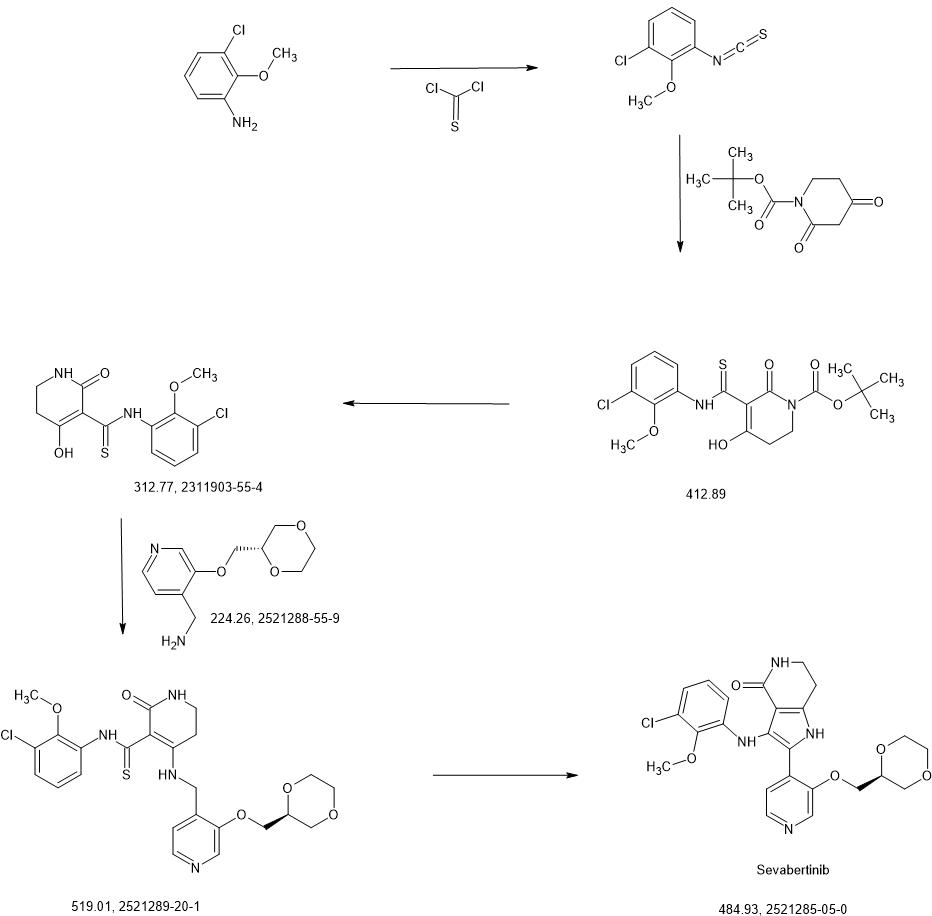
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020216781&_cid=P22-MICIVF-33261-1
Intermediate 3-1
1-chloro-3-isothiocyanato-2-methoxybenzene
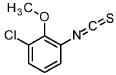
3-chloro-2-methoxyaniline (CAS 511 14-68-2, 8.4 ml, 63 mmol) was solved in DCM (100 ml) and sat. sodium bicarbonate solution (100 ml) was added. To the ice cooled mixture was slowly added thiophosgene (5.4 ml, 70 mmol). The reaction was stirred at 0°C for 2 h. At RT the DCM layer was separated and washed with sat. sodium bicarbonate solution, filtered through a hydrophobic filter and concentrated under reduced pressure to give the title compound (12.97 g, 100 % yield) which was used directly in the next step.
1H-NMR (400MHz, DMSO-de): d [ppm]= 7.51 (dd, 1 H), 7.35 (dd, 1 H), 7.20 (t, 1 H), 3.85 -3.91 (m, 3H).
Intermediate 4-1
tert- butyl 5-[(3-chloro-2-methoxyphenyl)carbamothioyl]-4-hydroxy-6-oxo-3,6-dihydropyridine-1(2/-/)-carboxylate
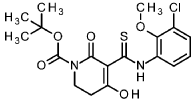
To an ice-cooled solution of 1-chloro-3-isothiocyanato-2-methoxybenzene (intermediate 3-1 , 4.00 g, 20.0 mmol) and tert- butyl 2,4-dioxopiperidine-1-carboxylate (CAS 845267-78-9, 4.27 g, 20.0 mmol) in acetonitrile (92 ml) was added dropwise DBU (4.5 ml, 30 mmol). The reaction was stirred at RT overnight. To the reaction mixture was added ice-water (200 ml_) and cone. HCI (2 ml_). The mixture was stirred for 20 min. and extracted with DCM. The organic phase was filtered over a water-repellent filter, conentrated under reduced pressure and purified by flash chromatography (silica, hexane / EtOAc gradient 0-50 %) to give 6.54 g of the title compound (71 % yield).
1H-NMR (400MHz, DMSO-de): d [ppm]= 13.36 (br s, 1 H), 7.73 (d, 1 H), 7.47 (dd, 1 H), 7.22 (t, 1 H), 3.76 – 3.82 (m, 5H), 2.88 (t, 2H), 1.48 (s, 9H).
LC-MS (method 1): Rt = 1.49 min; MS (ESIpos): m/z = 413.1 [M+H]+
Intermediate 5-1
A/-(3-chloro-2-methoxyphenyl)-4-hydroxy-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide
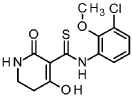
To a solution of tert- butyl 5-[(3-chloro-2-methoxyphenyl)carbamothioyl]-4-hydroxy-6-oxo-3,6-dihydropyridine-1 (2/-/)-carboxylate (intermediate 4-1 , 6.54 g, 15.8 mmol) in dichloromethane (94 ml) was added TFA (12 ml, 160 mmol) and the mixture was stirred 1.5 h at RT. The reaction mixture was concentrated under reduced pressure and the residue was solved in EtOAc and washed with sat. sodium bicarbonate solution and brine. The organic layer was filtered through a hydrophobic filter and the filtrate was dried to dryness. The residue was purified by flash chromatography (silica, hexane / EtOAc gradient 20-100 %) to give 4.06 g of the title compound (78 % yield).
1H-NMR (400 MHz, DMSO-de): d [ppm]= 16.45 (d, 1 H), 14.69 (s, 1 H), 14.33 (s, 1 H), 9.37 (br s, 1 H), 8.18 (br s, 1 H), 7.76 – 7.87 (m, 1 H), 7.37 – 7.45 (m, 1 H), 7.15 – 7.23 (m, 1 H), 3.73 – 3.76 (m, 3H), 3.43 (td, 1 H), 3.27 – 3.32 (m, 1 H), 2.79 (t, 1 H), 2.59 – 2.69 (m, 1 H).
LC-MS (method 1): Rt = 1.19 min; MS (ESIpos): m/z = 313 [M+H]+
ntermediate 6-2
A/-(3-chloro-2-methoxyphenyl)-4-{[(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methyl]amino}-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide
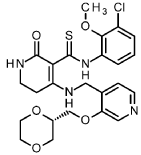
A mixture of A/-(3-chloro-2-methoxyphenyl)-4-hydroxy-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioa ide (intermediate 5-1 , 866 mg, 2.77 mmol) and 1-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methanamine (intermediate 2-8, 776 mg, 80% purity, 2.77 mmol) in ACN (22 ml) was treated with A/,0-bis(trimethylsilyl)acetamide (2.05 ml, 8.6 mmol, CAS 10416-59-8) and stirred at 80°C for 4 h. The reaction mixture was concentrated under reduced pressure and purified by flash chromatography (silica, DCM / EtOH gradient 0-20%) to give 1.23 g (95% purity, 81 % yield) of the title compound.
1H-NMR (400MHz, DMSO-d6): d [ppm]= 2.78 (t, 2H), 3.16 (td, 2H), 3.40 – 3.54 (m, 3H), 3.59 – 3.69 (m, 2H), 3.71 (s, 3H), 3.73 – 3.79 (m, 1 H), 3.83 – 3.95 (m, 2H), 4.16 (t, 2H), 4.67 (d, 2H), 7.11 (t, 1 H), 7.27 – 7.33 (m, 2H), 7.73 (br s, 1 H), 7.81 (dd, 1 H), 8.24 (d, 1 H), 8.39 (s, 1 H), 13.69 (s, 1 H), 14.79 (s, 1 H).
LC-MS (method 2): Rt = 1.09 min; MS (ESIpos): m/z = 519 [M+H]+
Example 2
3-(3-chloro-2-methoxyanilino)-2-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)-1 ,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridin-4-one (Stereoisomer 1)

The title compound from example 1 (140 mg) was separated into enantiomers by preparative chiral HPLC to give title compound (enantiomer 1 , 27 mg at Rt = 14.0 – 17.0 min) and enantiomer 2 (25 mg at Rt = 20.0 – 24.8 min, see example 3).
Preparative chiral HPLC method:
Instrument: Labomatic HD5000, Labocord-5000; Gilson GX-241 , Labcol Vario 4000; column: Cellulose SB 5m, 250×30 mm; eluent A: hexane + 0.1 vol. % diethylamine (99 %); eluent B: 2-propanol; isocratic: 50 % A + 50 % B; flow 50 ml/min; UV: 254 nm.
Analytical chiral HPLC method:
Instrument: Agilent HPLC 1260; column: Cellulose SB 3m, 100×4.6 mm; eluent A: hexane + 0.1 vol. % diethylamine (99 %); eluent B: 2-propanol; isocratic: 50 % A + 50 % B, flow 1.4 ml/min; temperature: 25°C; UV: 254 nm
Analytical chiral HPLC: Rt = 4.49 min.
Optical rotation:[a]D = 1.7° +/- 0.98° (c = 3.6 mg/2 ml, methanol)
Enantioselective synthesis confirmed the title compound as 3-(3-chloro-2-methoxyanilino)-2-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)-1 ,5,6,7-tetrahydro-4/-/-pyrrolo[3,2-c]pyridin-4-one. 872 mg (95% purity, 72% yield) of the title compound were prepared in analogy to example 1 using A/-(3-chloro-2-methoxyphenyl)-4-{[(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methyl]amino}-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide (intermediate 6-2, 1.23 g, 2.36 mmol) as starting material, followed by purification with preparative HPLC (method 10, gradient: 0.00-0.50 min 15% B, 0.50-6.00 min 15-55% B).
1H-NMR (400MHz, DMSO-d6): d [ppm]= 2.86 (t, 2H), 3.38 – 3.47 (m, 3H), 3.53 (td, 1 H), 3.69
– 3.78 (m, 2H), 3.83 (dd, 1 H), 3.88 (s, 3H), 3.90 (m, 1 H), 3.98 – 4.08 (m, 1 H), 4.12 – 4.18 (m, 1 H), 4.28 (dd, 1 H), 6.12 – 6.17 (quin, 1 H), 6.66 – 6.71 (m, 2H), 7.16 (s, 1 H), 7.28 (d, 1 H),
7.52 (s, 1 H), 8.04 (d, 1 H), 8.39 (s, 1 H), 11.07 (s, 1 H).
Analytical chiral HPLC: Rt = 4.46 min.
Optical rotation:[a]D = -12.5° +/- 0.52° (c = 5.6 mg/ l, chloroform)
PAT
- 4H-pyrrolo[3,2-c]pyridin-4-one compoundPublication Number: CN-114127064-BPriority Date: 2019-04-24Grant Date: 2023-12-26
- 4H-Pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: CN-117946100-APriority Date: 2019-04-24
- 4H-pyrrolo [3,2-c ] pyridin-4-one compoundsPublication Number: CN-117986251-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: TW-I849114-BPriority Date: 2019-04-24Grant Date: 2024-07-21
- 4H-pyrrolo[3,2-c]pyridin-4-one compoundPublication Number: KR-20220004103-APriority Date: 2019-04-24
- 4H-PYRROLO[3,2-C]PYRIDIN-4-ONE COMPOUNDSPublication Number: PE-20220254-A1Priority Date: 2019-04-24
- 4H-Pyrrolo [3,2-C] Pyridine-4-one CompoundPublication Number: JP-2022532850-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: US-2022298157-A1Priority Date: 2019-04-24
- 4H-PYRROL[3,2-C]PYRIDIN-4-ONE, ITS USES, PHARMACEUTICAL COMPOSITION, AND KIT OF PARTSPublication Number: BR-112021019998-B1Priority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: WO-2020216781-A1Priority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: TW-202106683-APriority Date: 2019-04-24
- 4H-pyrrolo(3,2-c)pyridin-4-one compoundsPublication Number: AU-2020262221-A1Priority Date: 2019-04-24
- 4H-pyrrolo [3,2-c ] pyridin-4-one compoundsPublication Number: CN-114127064-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: EP-3959211-A1Priority Date: 2019-04-24



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Medical uses
Sevabertinib is indicated for the treatment of adults with locally advanced or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 (ERBB2) tyrosine kinase domain activating mutations.[1][2]
Adverse effects
The US prescribing information includes warnings and precautions for diarrhea, hepatotoxicity, interstitial lung disease/pneumonitis, ocular toxicity, pancreatic enzyme elevation, and embryo-fetal toxicity.[2]
History
Efficacy was evaluated in people with unresectable or metastatic, non-squamous non-small cell lung cancer with HER2 (ERBB2) tyrosine kinase domain activating mutations who had received prior systemic therapy and received sevabertinib in SOHO-01 (NCT05099172), an open-label, single-arm, multi-center, multi-cohort clinical trial.[2] HER2 (ERBB2) activating mutations were determined in tumor tissue or plasma by local laboratories prior to enrollment.[2]
The US Food and Drug Administration granted the application for sevabertinib priority review, breakthrough therapy, and orphan drug designations.[2]
Society and culture
Legal status
Sevabertinib was approved for medical use in the United States in November 2025.[3][4]
Names
Sevabertinib is the international nonproprietary name.[5]
Sevabertinib is sold under the brand name Hyrnuo.[1][3]
References
- “HYRNUO (sevabertinib) tablets, for oral use” (PDF). Bayer HealthCare Pharmaceuticals Inc. U.S. Food and Drug Administration.
- “FDA grants accelerated approval to sevabertinib for non-squamous non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 19 November 2025. Retrieved 21 November 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “U.S. FDA Approves Hyrnuo (sevabertinib) for Previously Treated Patients with HER2-Mutated Locally Advanced or Metastatic Non-Squamous Non-Small Cell Lung Cancer” (Press release). Bayer. 20 November 2025. Retrieved 21 November 2025 – via Business Wire.
- “U.S. FDA grants accelerated approval to Bayer’s Hyrnuo (sevabertinib) for patients with previously treated advanced HER2-mutant non-small cell lung cancer”. Bayer (Press release). 20 November 2025. Retrieved 21 November 2025.
- World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 93”. WHO Drug Information. 39 (1). hdl:10665/381075.
Further reading
- Le X, Kim TM, Loong HH, Prelaj A, Goh BC, Li L, et al. (November 2025). “Sevabertinib in Advanced HER2-Mutant Non-Small-Cell Lung Cancer”. The New England Journal of Medicine. 393 (18): 1819–1832. doi:10.1056/NEJMoa2511065. PMID 41104928.
- Siegel F, Siegel S, Kotýnková K, Karsli Uzunbas G, Korr D, Tomono H, et al. (October 2025). “Sevabertinib, a Reversible HER2 Inhibitor with Activity in Lung Cancer”. Cancer Discovery: OF1 – OF14. doi:10.1158/2159-8290.CD-25-0605. PMID 41090369.
External links
- “Sevabertinib”. NCI Drug Dictionary.
- “Sevabertinib ( Code – C185187 )”. EVS Explore.
- Clinical trial number NCT05099172 for “First in Human Study of BAY2927088 in Participants Who Have Advanced Non-small Cell Lung Cancer (NSCLC) With Mutations in the Genes of Epidermal Growth Factor Receptor (EGFR) and/or Human Epidermal Growth Factor Receptor 2 (HER2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Hyrnuo |
| Other names | BAY2927088, sevabertinib hydrate (JAN JP) |
| License data | US DailyMed: Sevabertinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2521285-05-0 |
| PubChem CID | 155234713 |
| DrugBank | DB21667 |
| ChemSpider | 129786615 |
| UNII | 2A7VPM5RWH |
| KEGG | D13098 |
| Chemical and physical data | |
| Formula | C24H25ClN4O5 |
| Molar mass | 484.94 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////sevabertinib, FDA 2025, APPROVALS 2025, Hyrnuo, 2A7VPM5RWH, BAY-2927088, BAY 2927088
Plozasiran
Plozasiran
CAS 2379776-40-4
2379776-41-5 SODIUM SALT
RNA, ([1′-de(6-amino-9H-purin-9-yl)]dA-(5′→5′)-sp-Am-Cm-Gm-Gm-Gm-Am-Cm-Am-(2′-deoxy-2′-fluoro)G-(2′-deoxy-2′-fluoro)U-(2′-deoxy-2′-fluoro)A-Um-Um-Cm-Um-Cm-Am-Gm-Um-Im-Am-(3′→3′)-sp-[1′-de(6-amino-9H-purin-9-yl)]dA), 3′-[O-[cis-4-[(3S,8S)-17-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-3,8-bis[[[2-[2-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]ethoxy]ethyl]amino]carbonyl]-1,6,11-trioxo-15-oxa-2,7,12-triazaheptadec-1-yl]cyclohexyl] hydrogen phosphorothioate], complex with RNA (Um-sp-(2′-deoxy-2′-fluoro)C-sp-Am-sp-(2′-deoxy-2′-fluoro)C-Um-(2′-deoxy-2′-fluoro)G-Am-Gm-Am-Am-Um-(2′-deoxy-2′-fluoro)A-Cm-(2′-deoxy-2′-fluoro)U-Gm-(2′-deoxy-2′-fluoro)U-Cm-(2′-deoxy-2′-fluoro)C-Cm-(2′-deoxy-2′-fluoro)G-sp-Um) (1:1
FDA 2025, 11/18/2025, APPROVALS 2025, Redemplo, ARO-APOC3, VSA001, ARO-APOC3, VSA 001, ADS 005, XG9ARL6P25
To reduce triglycerides in adults with familial chylomicronemia syndrome
Plozasiran, sold under the brand name Redemplo, is a medication usd for the treatment of familial chylomicronemia syndrome.[1] Plozasiran is an apolipoprotein C-III (apoC-III)-directed small interfering ribonucleic acid (siRNA).[1] It is given by injection under the skin (subcutaneously).[1]
Plozasiran was approved for medical use in the United States in November 2025.[2]
Plozasiran is under investigation in clinical trial NCT05089084 (Study of ARO-APOC3 (Plozasiran) in Adults With Familial Chylomicronemia Syndrome (FCS)).
Plozasiran (ARO-APOC3) is an investigational RNAi therapeutic targeting apolipoprotein C-III (APOC3). It received an Orphan Drug designation by the FDA for the treatment of familial chylomicronemia syndrome.1
Plozasiran, a novel therapeutic agent, is a small interfering RNA (siRNA) developed by Silence Therapeutics. This innovative medication targets proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein involved in cholesterol metabolism, and is specifically indicated for the treatment of hypercholesterolemia, a condition characterized by elevated levels of low-density lipoprotein cholesterol (LDL-C) in the blood. Hypercholesterolemia is a significant risk factor for cardiovascular diseases, making effective treatments crucial for patient health.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Plozasiran is indicated as an adjunct to diet to reduce triglycerides in adults with familial chylomicronemia syndrome.[1]
Familial chylomicronemia syndrome is a rare genetic disorder that affects the body’s ability to break down fats (triglycerides) in the bloodstream.[2] This leads to abnormally high levels of chylomicrons, which are particles that carry triglycerides.[2] Normal triglyceride levels are less than 150 mg/dL; levels above 500 mg/dL are considered severely high (severe hypertriglyceridemia).[2] People with familial chylomicronemia syndrome can have triglyceride levels in the thousands.[2] These high triglyceride levels can cause severe abdominal pain, inflammation of the pancreas (acute pancreatitis), and fatty deposits in the skin (xanthomas).[2] Some of these symptoms, specifically acute pancreatitis, can be life-threatening.[2]
Side effects
The most common side effects include hyperglycemia (high blood sugar), headache, nausea, and injection site reaction.[2]
History
The efficacy of plozasiran was demonstrated in a randomized, placebo-controlled, double-blind trial (NCT05089084) in adults with genetically confirmed or clinically diagnosed familial chylomicronemia syndrome maintained on a low-fat diet (≤20 grams fat per day).[2] Participants were randomly assigned to receive four total doses of plozasiran 25 mg or matching placebo, injected subcutaneously (under the skin) once every three months over a twelve-month treatment period.[2] The primary endpoint was percent change in fasting triglycerides from baseline to month ten.[2] The median percent change in triglycerides from baseline to month ten in the plozasiran treatment group was -59% compared to the placebo group.[2]
The US Food and Drug Administration granted the application for plozasiran breakthrough therapy, orphan drug, and fast track designations.[2]
Society and culture
Legal status
Plozasiran was approved for medical use in the United States in November 2025.[3]
Names
Plozasiran is the international nonproprietary name.[4]
Plozasiran is sold under the brand name Redemplo.[2][3]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219947s000lbl.pdf
- “FDA approves drug to reduce triglycerides in adults with familial chylomicronemia syndrome”. U.S. Food and Drug Administration. 18 November 2025. Retrieved 21 November 2025. This article incorporates text from this source, which is in the public domain.
- “Arrowhead Pharmaceuticals Announces FDA Approval of Redemplo (plozasiran) to Reduce Triglycerides in Adults with Familial Chylomicronemia Syndrome (FCS)” (Press release). Arrowhead Pharmaceuticals. 18 November 2025. Retrieved 21 November 2025 – via Business Wire.
- World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 92”. WHO Drug Information. 38 (3). hdl:10665/379650.
Further reading
- Alla SS, Shah DJ, Meyur S, Agarwal P, Alla D, Moraboina SL, et al. (2025). “Small Interfering RNA (siRNA) in Dyslipidemia: A Systematic Review on Safety and Efficacy of siRNA”. Journal of Experimental Pharmacology. 17: 249–267. doi:10.2147/JEP.S521579. PMC 12126973. PMID 40453040.
- Olatunji G, Ogieuhi IJ, Kokori E, Oluwatomiwa AV, Ajimotokan OI, Odukudu GO, et al. (November 2024). “Olezarsen and Plozasiran in Dyslipidemia Management: A Narrative Review of Clinical Trials”. High Blood Pressure & Cardiovascular Prevention. 31 (6): 567–576. doi:10.1007/s40292-024-00677-7. PMID 39352667.
- Pan Z, Zaman MA, Kalsoom S, Zhang Y (November 2024). “Messenger interference RNA therapies targeting apolipoprotein C-III and angiopoietin-like protein 3 for mixed hyperlipidemia: the future of plozasiran and zodasiran”. Expert Review of Clinical Pharmacology. 17 (11): 1017–1023. doi:10.1080/17512433.2024.2423724. PMID 39469883.
- Sydhom P, Al-Quraishi B, Gohar A, El-Shawaf M, Shehata N, Ataya M, et al. (November 2025). “The Efficacy and Safety of Plozasiran on Lipid Profile in Dyslipidemic Disorders: A Systematic Review and Meta-Analysis”. Cardiovascular Drugs and Therapy. doi:10.1007/s10557-025-07798-8. PMID 41251855.
External links
- Clinical trial number NCT05089084 for “Study of ARO-APOC3 (Plozasiran) in Adults With Familial Chylomicronemia Syndrome (FCS) (PALISADE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Redemplo |
| Other names | ARO-APOC3 |
| AHFS/Drugs.com | Redemplo |
| License data | US DailyMed: Plozasiran |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2379776-40-4 |
| DrugBank | DB18997 |
| UNII | |
//////////Plozasiran, FDA 2025, APPROVALS 2025, Redemplo, ARO-APOC3, VSA001, ARO-APOC3, VSA 001, ADS 005, XG9ARL6P25
Ziftomenib

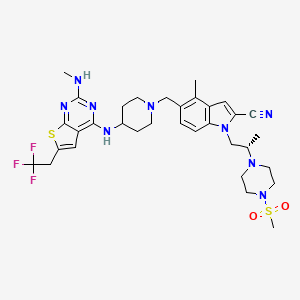
Ziftomenib
CAS 2134675-36-6
4MOD1F4ENC, KO 539
717.9 g/mol, C33H42F3N9O2S2
APPROVALS 2025, FDA 2025, 11/13/2025, Komzifti
4-methyl-5-[[4-[[2-(methylamino)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl]amino]piperidin-1-yl]methyl]-1-[(2S)-2-(4-methylsulfonylpiperazin-1-yl)propyl]indole-2-carbonitrile
To treat adults with relapsed or refractory acute myeloid leukemia with a susceptible nucleophosmin 1 mutation who have no satisfactory alternative treatment options
Ziftomenib, sold under the brand name Komzifti, is an anti-cancer medication used for the treatment of acute myeloid leukemia.[1] Ziftomenib is a menin inhibitor.[1] It is taken by mouth.[1]
Ziftomenib blocks the interaction between two proteins, menin (MEN1) and KMT2A (also known as mixed lineage leukemia protein, MLL).[2][3]
Ziftomenib was approved for medical use in the United States in November 2025.[4][5]
Ziftomenib, also known as KO539, is an orally bioavailable inhibitor of the menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) fusion protein, with potential antineoplastic activity. Upon oral administration, ziftomenib prevents the interaction between the two proteins menin and MLL, and thus the formation of the menin-MLL complex. This reduces the expression of downstream target genes and results in an inhibition of the proliferation of MLL-rearranged leukemic cells. The menin-MLL complex plays a key role in the survival, growth and proliferation of certain kinds of leukemia cells
SYN

syn
WO2022086986
above similar not same
pat
WO2020069027
WO2018175746
WO2017161028
WO2018106820
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US239825810&_cid=P20-MI88RV-91969-1

PAT
- Inhibitors of the myst family of lysine acetyl transferasesPublication Number: US-2025051343-A1
- Small molecule inhibitors of dyrk/clk and uses thereofPublication Number: US-2025051325-A1
- VCP/p97 INHIBITOR FOR THE TREATMENT OF CANCERPublication Number: US-2025049800-A1
- RNAi agents for inhibiting expression of HIF-2 alpha (EPAS1), compositions thereof, and methods of usePublication Number: US-12221610-B2Grant Date: 2025-02-11
- Inhibitors of the myst family of lysine acetyl transferasesPublication Number: US-2025051343-A1
- Small molecule inhibitors of dyrk/clk and uses thereofPublication Number: US-2025051325-A1
- VCP/p97 INHIBITOR FOR THE TREATMENT OF CANCERPublication Number: US-2025049800-A1
- RNAi agents for inhibiting expression of HIF-2 alpha (EPAS1), compositions thereof, and methods of usePublication Number: US-12221610-B2Grant Date: 2025-02-11
- N-(3-hydroxy-4-piperidinyl)benzamide derivatives and pharmaceutical compositionsPublication Number: NZ-201856-APriority Date: 1981-10-01
- Novel n-(3-hydroxy-4-piperidinyl)benzamide derivativesPublication Number: EP-0076530-B1Priority Date: 1981-10-01Grant Date: 1985-12-11
- Novel N-(3-hydroxy-4-piperidinyl)benzamide derivativesPublication Number: EP-0076530-A2Priority Date: 1981-10-01
- Boron-containing polyphosphonates for the treatment of calcogenic tumorsPublication Number: JP-S5817120-APriority Date: 1981-06-30
- Novel n-aryl-piperazinealkanamidesPublication Number: IE-53465-B1Priority Date: 1981-06-23
- Novel((bis(aryl)methylene)-1-piperidinyl)-alkyl-pyrimidinonesPublication Number: IE-56180-B1Priority Date: 1982-11-01
- Novel ((bis(aryl)methylene)-1-piperidinyl)alkyl-pyrimidinonesPublication Number: EP-0110435-B1Priority Date: 1982-11-01Grant Date: 1989-01-04
- Novel ((bis(aryl)methylene)-1-piperidinyl)alkyl-pyrimidinonesPublication Number: EP-0110435-A1Priority Date: 1982-11-01
- NEW // BIS (ARYL) METHYLENE / -1-PIPERIDINYL / -ALKYL-PyrimidinonesPublication Number: BG-60538-B2Priority Date: 1982-11-01
- Process for preparing n-(3-hydroxy-4-piperidinyl)benzamide derivativesPublication Number: KR-860001584-B1Priority Date: 1982-07-30Grant Date: 1986-10-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Ziftomenib is indicated for the treatment of adults with relapsed or refractory acute myeloid leukemia with a susceptible nucleophosmin 1 mutation who have no satisfactory alternative treatment options.[1]
Adverse effects
The US prescribing information includes warnings and precautions for differentiation syndrome, QTc interval prolongation, and embryo-fetal toxicity.[4]
History
Efficacy was evaluated in KO-MEN-001 (NCT04067336), an open-label, single, arm, multi-center trial in 112 adults with relapsed or refractory acute myeloid leukemia with an nucleophosmin 1 mutation identified using next-generation sequencing or polymerase chain reaction.[4] Participants with nucleophosmin 1 mutations, including type A, B, and D mutations and other nucleophosmin 1 mutations likely to result in cytoplasmic localization of the nucleophosmin 1 protein, were enrolled.[4]
The US Food and Drug Administration granted the application for ziftomenib priority review, breakthrough therapy, and orphan drug designations.[4]
Society and culture
Legal status
Ziftomenib was approved for medical use in the United States in November 2025.[6]
Names
Ziftomenib is the international nonproprietary name.[7][8]
Ziftomenib is sold under the brand name Komzifti.[6]
References
- https://kuraoncology.com/wp-content/uploads/prescribinginformation.pdf
- “Ziftomenib”. NCI Cancer Dictionary. National Cancer Institute.
- Rausch J, Dzama MM, Dolgikh N, Stiller HL, Bohl SR, Lahrmann C, et al. (October 2023). “Menin inhibitor ziftomenib (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax”. Haematologica. 108 (10): 2837–2843. doi:10.3324/haematol.2022.282160. PMC 10543165. PMID 37102614.
- “FDA approves ziftomenib for relapsed or refractory acute myeloid leukemia with a NPM1 mutation”. U.S. Food and Drug Administration (FDA). 13 November 2025. Retrieved 14 November 2025. This article incorporates text from this source, which is in the public domain.
- “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 13 November 2025. Retrieved 14 November 2025.
- “Kura Oncology and Kyowa Kirin Announce FDA Approval of Komzifti (ziftomenib), the First and Only Once-Daily Targeted Therapy for Adults with Relapsed or Refractory NPM1-Mutated Acute Myeloid Leukemia” (Press release). Kura Oncology. 13 November 2025. Retrieved 14 November 2025 – via GlobeNewswire News Room.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Wang ES, Issa GC, Erba HP, Altman JK, Montesinos P, DeBotton S, et al. (October 2024). “Ziftomenib in relapsed or refractory acute myeloid leukaemia (KOMET-001): a multicentre, open-label, multi-cohort, phase 1 trial”. The Lancet. Oncology. 25 (10): 1310–1324. doi:10.1016/S1470-2045(24)00386-3. PMID 39362248.
External links
- Clinical trial number NCT04067336 for “First in Human Study of Ziftomenib in Relapsed or Refractory Acute Myeloid Leukemia” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Komzifti |
| Other names | KO-539; KO539 |
| AHFS/Drugs.com | Komzifti |
| License data | US DailyMed: Ziftomenib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2134675-36-6 |
| PubChem CID | 138497449 |
| IUPHAR/BPS | 11680 |
| DrugBank | DB17171 |
| ChemSpider | 115009296 |
| UNII | 4MOD1F4ENC |
| KEGG | D12419 |
| ChEMBL | ChEMBL5095038 |
| PDB ligand | K5O (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C33H42F3N9O2S2 |
| Molar mass | 717.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Ziftomenib, APPROVALS 2025, FDA 2025, 4MOD1F4ENC, Komzifti
Potrasertib

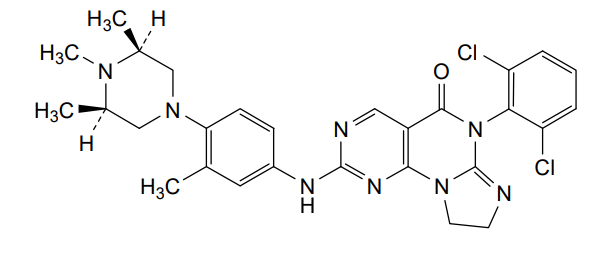
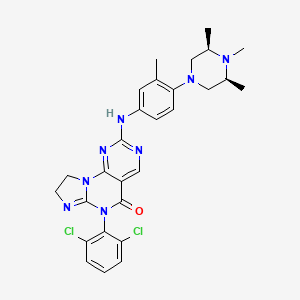
Potrasertib
CAS 2226938-19-6
MFC28H30Cl2N8O MW 565.5 g/mol
6-(2,6-dichlorophenyl)-2-{3-methyl-4-[(3R,5S)-3,4,5-trimethylpiperazin-1-yl]anilino}-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one
7-(2,6-dichlorophenyl)-12-[3-methyl-4-[(3S,5R)-3,4,5-trimethylpiperazin-1-yl]anilino]-2,5,7,11,13-pentazatricyclo[7.4.0.02,6]trideca-1(13),5,9,11-tetraen-8-one
serine/ threonine kinase inhibitor, antineoplastic, IMP 7068, WEE1-IN-10, orb2664172, 621K13UG4B, Phase 1, Solid tumours
- OriginatorIMPACT Therapeutics
- ClassAntineoplastics; Small molecules
- Mechanism of ActionWEE1 protein inhibitors
- Phase ISolid tumours
- 28 Mar 2024No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy) in Taiwan (PO)
- 28 Mar 2024No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy) in USA (PO)
- 20 Oct 2023Efficacy, adverse events, pharmacodynamics and pharmacokinetics data from the phase I WEE1 trial in Solid tumours presented at the 48th European Society for Medical Oncology Congress (ESMO-2023)
Potrasertib is an investigational drug that is a selective inhibitor of WEE1 kinase, a protein crucial for the cell cycle. It is being studied for the treatment of various advanced solid tumors, including small cell lung cancer, ovarian, and colorectal cancers. By blocking the WEE1 kinase, potrasertib causes cancer cells with DNA damage to undergo premature, error-prone mitosis, which leads to cell death.
How it works
- Potrasertib is a serine/threonine kinase inhibitor.
- It works by targeting WEE1 kinase, which regulates the cell’s response to DNA damage.
- By inhibiting WEE1, it prevents cancer cells from repairing DNA damage before dividing, forcing them into a state that leads to cell death.
- This mechanism is particularly effective in tumors with a defective p53 gene, as these tumors rely more heavily on the WEE1 checkpoint for survival.
Potential uses
- Combination therapy: It is being explored in combination with chemotherapy (like gemcitabine and cisplatin) or radiotherapy to enhance their effectiveness against cancer.
- Monotherapy: It is also being studied as a standalone treatment for certain cancers, including ovarian, colorectal, and non-small cell lung cancer, especially those with high replication stress or WEE1 dependency.
Current status
- Potrasertib is still an investigational drug and is not yet approved for widespread clinical use.
- It is undergoing clinical trials to evaluate its safety and effectiveness in treating advanced cancers.
Potrasertib is an investigational new drug that is being evaluated by IMPACT Therapeutics for the treatment of advanced solid tumors. It is oral inhibitor of WEE1 kinase, a key regulator of cell cycle checkpoints.[1][2]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018090939&_cid=P21-MI6TEY-70275-1

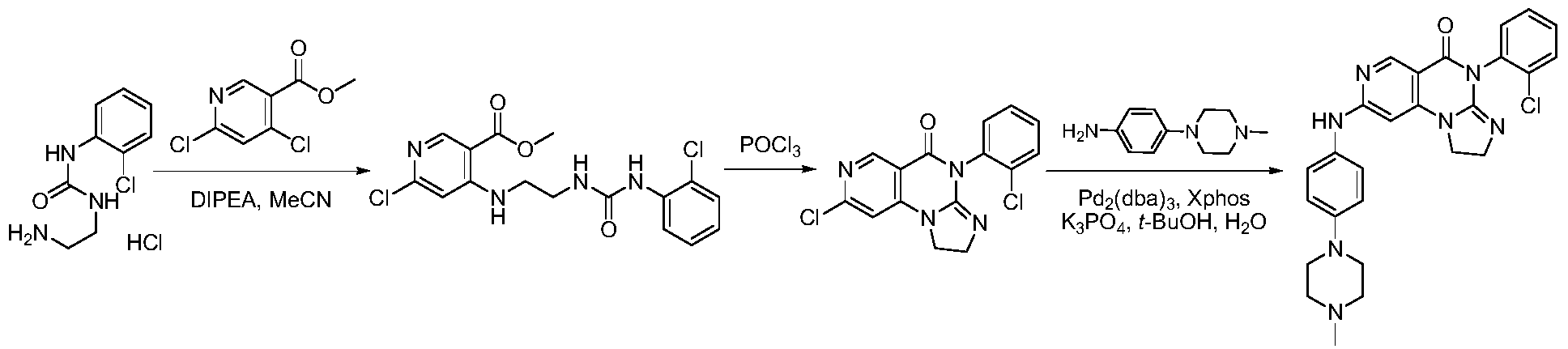
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021073491&_cid=P21-MI6TF3-70349-1
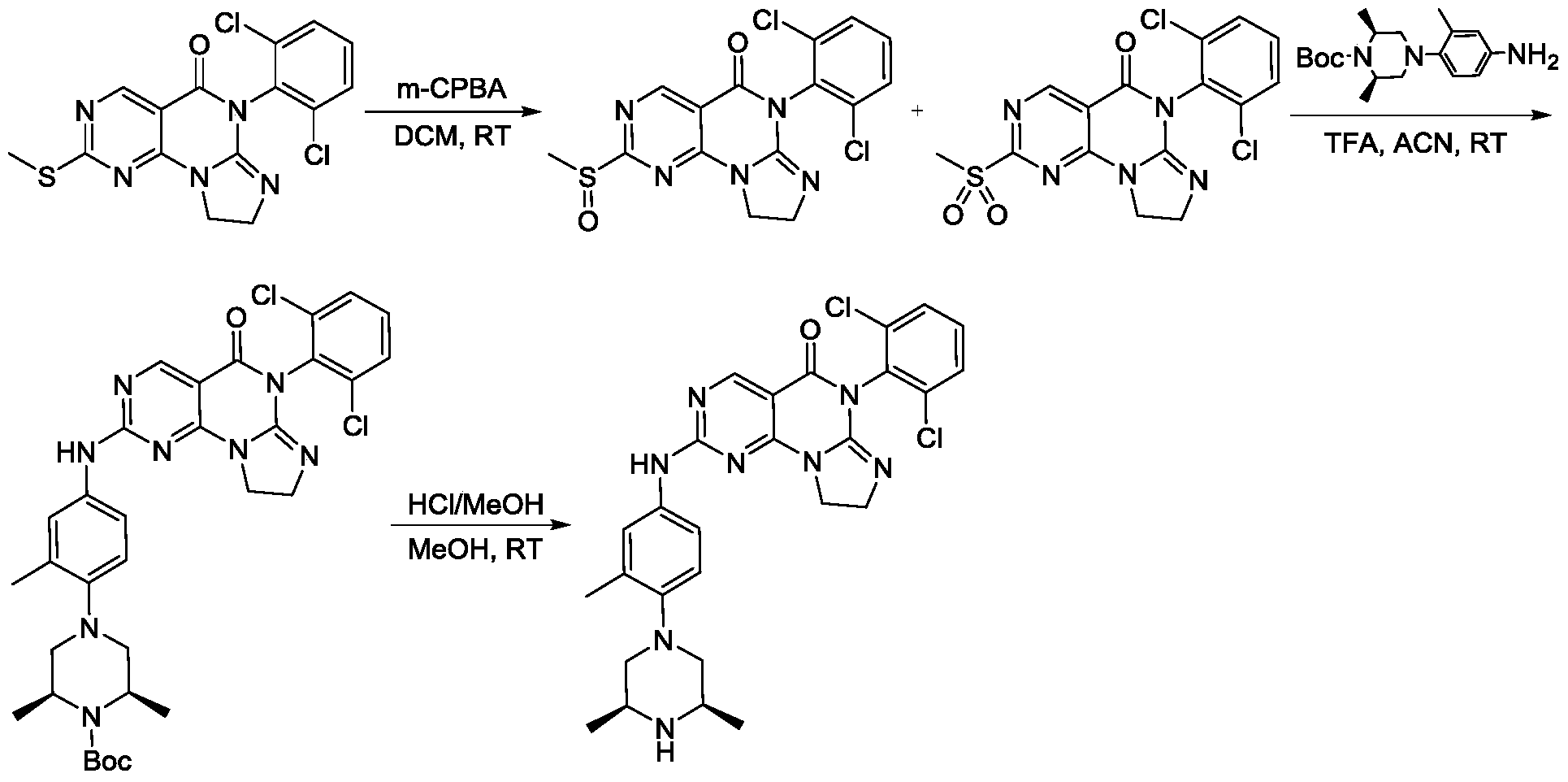
Example 1
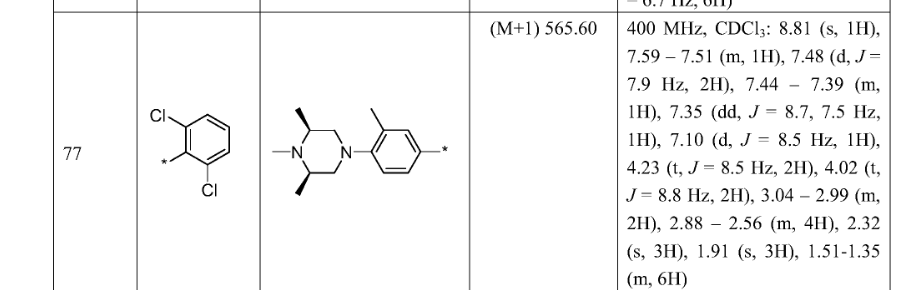
SIMILAR NOT SAME
[0117]6-(2,6-dichlorophenyl)-2-((4-((3S,5R)-3,5-dimethylpiperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one
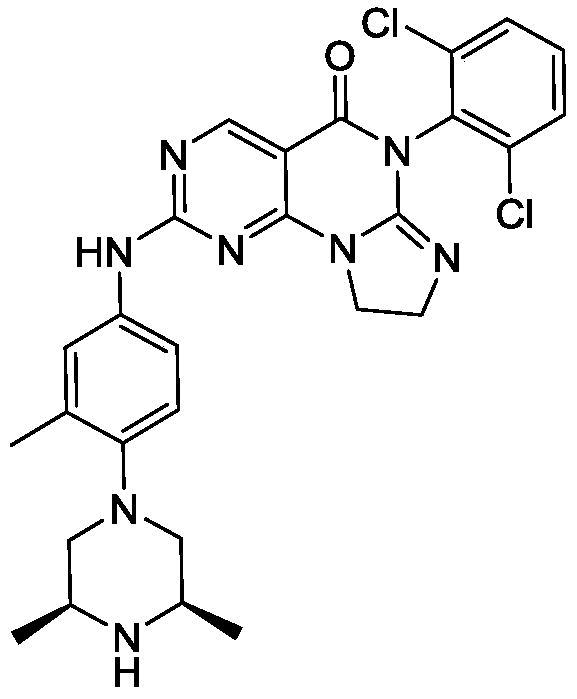
SIMILAR NOT SAME
[0128]6-(2,6-dichlorophenyl)-2-((4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one
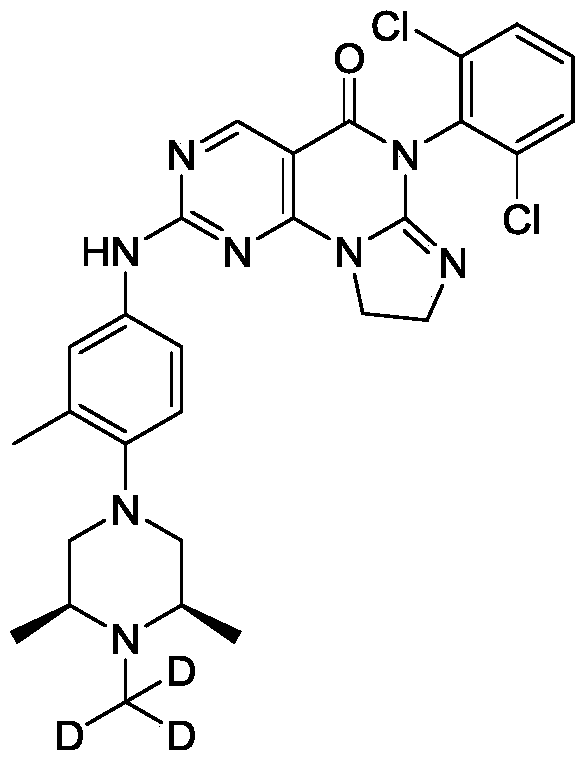
[0130]a) Preparation of (2S,6R)-2,6-dimethyl-1-(methyl-d3)-4-(2-methyl-4-nitro)piperazine: Sodium hydride (385.03 mg, 9.63 mmol, 60% purity) was added to a solution of (3S,5R)-3,5-dimethyl-1-(2-methyl-4-nitro)piperazine (2 g, 8.02 mmol) in N,N-dimethylformamide (15 mL). The mixture was stirred at 0 °C for 25 hours, then trideuterated iodomethane (1.16 g, 8.02 mmol, 499.09 μL) was added, and the mixture was stirred at 0 °C for 2 hours. The reaction was quenched by adding an aqueous sodium bicarbonate solution (30 mL) at 0 °C, extracted with ethyl acetate (50 mL × 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the target crude product (1.5 g, yellow-green solid). LC-MS(ESI): m/z(M+1) + 267.1. 1 H NMR (400MHz, CDCl
3 ): δ8.04-8.01 (m, 2H), 6.96 (d, J = 12.0Hz, 1H), 3.10 (d, J = 12Hz, 2H), 2.65 (t , J=12Hz, 2H), 2.45-2.43 (m, 2H), 2.36 (s, 3H), 1.16-1.15 (d, J=4.0Hz, 6H).
[0131]b) Preparation of 4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylaniline: Under nitrogen protection, palladium on carbon (281.58 μmol, 10% purity) was added to a methanol (5 mL) solution of (2S,6R)-2,6-dimethyl-1-(methyl-d3)-4-(2-methyl-4-nitro)piperazine (1.5 g, 5.63 mmol). The resulting suspension was purified multiple times under vacuum with hydrogen. The mixture was stirred at 25 °C for 12 hours under a hydrogen atmosphere (15 psi). The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to give the target crude product (1.3 g, black solid). LC-MS (ESI): m/z (M+1) + 237.1.
[0132]c) Preparation of 6-(2,6-dichlorophenyl)-2-((4-(((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one: 4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylaniline (459.32 mg, 1.94 mmol) and the prepared 6-(2,6-dichlorophenyl)-2- A mixture (700 mg, crude) of crude (methanesulfonyl)-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one and 6-(2,6-dichlorophenyl)-2-(methanesulfonyl)-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one was dissolved in acetonitrile (5 mL) and trifluoroacetic acid (20.14 mg, 0.177 mmol, 13.08 μL) was added. The mixture was stirred at 20–25 °C for 2 hours, filtered, and the filtrate was concentrated under reduced pressure to give the crude product. The crude product was purified by reversed-phase HPLC to give the target compound (56.89 mg, 100.00 μmol, yellow solid, 5.66% yield). LC-MS (ESI): m/z (M+1) + 568.0.
1 H NMR (400MHz, CDCl 3 ): δ8.81 (s, 1H), 7.49 (d, J=3.8Hz, 3H), 7.41-7.34 (m, 3H), 7.02 (d, J=4.2Hz, 1H), 4.25-4.21 (m, 2H), 4.02 (t, J=8.0Hz, 2H), 2.95 (d, J=6.0Hz 2H), 2.62 (t, J=6.0Hz, 2H), 2.46-2.41 (m, 2H), 2.34 (s, 6H), 1.15 (d, J=6.4Hz, 6H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022188802&_cid=P21-MI6TVM-79837-1
PAT
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: US-11345711-B2Priority Date: 2016-11-16Grant Date: 2022-05-31
- 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidine-5(6h)-ketone compoundPublication Number: EP-3543242-B1Priority Date: 2016-11-16Grant Date: 2024-01-03
- Compound 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-ketonePublication Number: ES-2968252-T3Priority Date: 2016-11-16Grant Date: 2024-05-08
- 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidine-5(6h)-ketone compoundPublication Number: EP-3543242-A1Priority Date: 2016-11-16
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: US-10703759-B2Priority Date: 2016-11-16Grant Date: 2020-07-07
- 8,9-DIHYDROIMIDAZO[1,2-a]PYRIMIDO[5,4-e]PYRIMIDIN-5(6H)-ONESPublication Number: US-2019308984-A1Priority Date: 2016-11-16
- 8,9-DIHYDROIMIDAZO[1,2-a]PYRIMIDO[5,4-e]PYRIMIDIN-5(6H)-ONESPublication Number: US-2020385394-A1Priority Date: 2016-11-16
- 8,9-Dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: CN-109906227-BPriority Date: 2016-11-16Grant Date: 2022-03-11
- Dihydroimidazo pyrimido pyrimidinone compoundPublication Number: WO-2021073491-A1Priority Date: 2019-10-16
- DihydroimidazopyrimidopyrimidinonesPublication Number: CN-114502559-APriority Date: 2019-10-16
- Dihydroimidazopyrimidopyrimidinone compoundsPublication Number: CN-114502559-BPriority Date: 2019-10-16Grant Date: 2024-02-02
- Dihydroimidazo pyrimido pyrimidinone compoundPublication Number: US-2024010655-A1Priority Date: 2019-10-16
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6h)-onesPublication Number: CA-3043945-A1Priority Date: 2016-11-16
- Use of Wee1 kinase inhibitors in the treatment of cancerPublication Number: CN-118338905-APriority Date: 2021-11-26
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: WO-2023093840-A1Priority Date: 2021-11-26
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: WO-2022188802-A1Priority Date: 2021-03-10
- The use of Wee1 kinase inhibitors in the treatment of cancer diseasesPublication Number: CN-117202908-APriority Date: 2021-03-10
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: US-2024091233-A1Priority Date: 2021-03-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | IMP7068 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2226938-19-6 |
| PubChem CID | 139503236 |
| UNII | 621K13UG4B |
| Chemical and physical data | |
| Formula | C28H30Cl2N8O |
| Molar mass | 565.50 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “IMP 7068”. AdisInsight. Springer Nature Switzerland AG.
- Wang Z, Li W, Li F, Xiao R (January 2024). “An update of predictive biomarkers related to WEE1 inhibition in cancer therapy”. Journal of Cancer Research and Clinical Oncology. 150 (1): 13. doi:10.1007/s00432-023-05527-y. PMC 10794259. PMID 38231277.
///////potrasertib, antineoplastic, IMP 7068, WEE1-IN-10, orb2664172, 621K13UG4B, Phase 1, Solid tumours
Plosaracetam
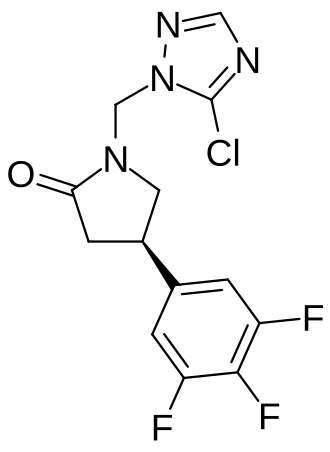
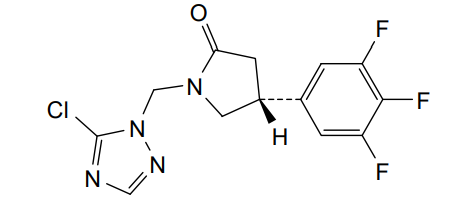

Plosaracetam
CAS 1651179-19-9
MF C13H10ClF3N4O MW330.69 g/mol
(4R)-1-[(5-chloro-1H-1,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one
(4R)-1-[(5-chloro-1,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one
(4R)-1-[(5-Chloro-1H-1,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)-2-pyrrolidinone
(4R)-1-[(5-chloro-1H-1,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one
2-Pyrrolidinone, 1-[(5-chloro-1H-1,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)-, (4R)-
synaptic vesicle glycoprotein 2A (SV2A) positive modulator, ABBV-552, ABBV552, SDI-118, SDI118, ABBV 552, ABBV552, SDI 118, SDI118, W3LYF2KQ6F
Plosaracetam (INNTooltip International Nonproprietary Name; developmental code names ABBV-552, SDI-118) is a synaptic vesicle glycoprotein 2A (SV2A) ligand which is under development for the treatment of Alzheimer’s disease and other cognition disorders.[1][3][4][2] In contrast to earlier SV2A ligands like levetiracetam and brivaracetam, polsaracetam does not have anticonvulsant activity and instead shows pro-cognitive effects.[2] The drug is being developed by UCB Biopharma and AbbVie.[1][3] As of October 2024, it is in phase 2 clinical trials for Alzheimer’s disease and phase 1 trials for cognition disorders.[1][3]
Plosaracetam is a small molecule drug. The usage of the INN stem ‘-racetam’ in the name indicates that Plosaracetam is a piracetam type amide type nootrope agent. Plosaracetam is under investigation in clinical trial NCT05199142 (A Study to Evaluate the Safety, Tolerability, and Pharmacodynamics of SDI-118 in Elderly Male and Female Study Participants With Cognitive Decline). Plosaracetam has a monoisotopic molecular weight of 330.05 Da.
PAT
- Compounds for Enhancing the Cognitive FunctionPublication Number: US-2016185761-A1Priority Date: 2013-08-02
- Compounds for enhancing the cognitive functionPublication Number: US-9630948-B2Priority Date: 2013-08-02Grant Date: 2017-04-25
- Compounds for enhancing the cognitive functionPublication Number: WO-2015014785-A1Priority Date: 2013-08-02
- Connections to improve cognitive functionPublication Number: DK-3027606-T3Priority Date: 2013-08-02Grant Date: 2018-04-30
- Compounds for enhancing the cognitive functionPublication Number: EP-3027606-A1Priority Date: 2013-08-02
- Compounds for enhancing the cognitive functionPublication Number: EP-3027606-B1Priority Date: 2013-08-02Grant Date: 2018-02-28
- Compounds to enhance cognitive functionPublication Number: ES-2666134-T3Priority Date: 2013-08-02Grant Date: 2018-05-03
- Compounds for enhancing the cognitive functionPublication Number: SI-3027606-T1Priority Date: 2013-08-02
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015014785&_cid=P11-MI5R7J-79014-1
Example 1 : Synthesis of (4R)-1 -[(5-chloro-1H-1,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 7.

1.1 Synthesis of tert-butyl 2-oxo-4-(3,4,5-trifluorophenyl)pyrrolidine-1 -carboxylate 3 and enantiomers.
To a solution of tert-butyl 2-oxo-2,5-dihydro-1 H-pyrrole-1-carboxylate 1 (10 g, 1 eq., 54.6 mmol) in dioxane/water (100 ml/30 ml) are added at room temperature (3,4,5-trifluorophenyl)boronic acid 2 (19.2 g, 2 eq., 109.2 mmol), cesium fluoride (24.9 g, 3 eq., 163.8 mmol), (±)-2,2′-bis(diphenyl-phosphino)-1 , 1′-binaphthyl (1.5 g, 4.5%, 2.5 mmol), potassium carbonate (22.6 g, 3 eq., 163.8 mmol) and chloro(1 ,5-cyclooctadiene)rhodium(l)dimer (0.82 g, 1.5%, 8.2 mmol). The mixture is heated at 1 10°C for 2 h. Solvent are removed under reduced pressure and the residue is purified by chromatography over silicagel (eluent: CI-^C^/MeOH/NI-^OH 96/3.5/0.5 v/v/v) to afford tert-butyl 2-oxo-4-(3,4,5-trifluorophenyl)pyrrolidine-1-carboxylate 3. The enantiomers are
resolved by chiral chromatography (chiralpak IC, 150*4.6 mm, eluent: heptane/AcOEt/diethylamine 80/20/0.1 v/v/v) to afford tert-butyl (4R)-2-oxo-4-(3,4,5-trifluorophenyl)pyrrolidine-1-carboxylate 3A (second eluted, 5.1 g), and its enantiomer tert-butyl (4S)-2-oxo-4-(3,4,5-trifluorophenyl)pyrrolidine-1-carboxylate 3B (first eluted, 5.2 g) as white solids.
Compound 3A:
Yield: 30%.
LC-MS (MH+): 316.
alphaD (MeOH, 25°C): -19.9.
1.2 Synthesis of (4R)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 4.
At 0°C, TFA (20 ml, 261 mmol) is added to a solution of tert-butyl (4R)-2-oxo-4-(3,4,5-trifluorophenyl)pyrrolidine-1-carboxylate 3A (8 g, 1 eq., 25.4 mmol) in dichloromethane (100 ml). The mixture is stirred at room temperature for 2 h. Then, TFA and solvent are removed under reduced pressure. The crude mixture is poured in an aqueous saturated solution of NaHCC>3 (100 ml) and extracted with AcOEt (3*200 ml). The combined organic extracts are dried over MgS04 and concentrated under reduced pressure. The conversion is total and the evaporation affords 5.5 g of (4R)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 4, which is used in the next step without any further purification.
LC-MS (MH+): 216; LC-MS (MKT): 214.
alphaD (MeOH, 22°C): -20.1.
1.3 Synthesis of (4R)-1 -(hydroxymethyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 5.
To a solution of (4R)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 4 (5.5 g, 1 eq., 25.6 mmol) in THF (20 ml) are added potassium tert-butoxide (0.049 g, 0.02 eq., 0.44 mmol) and paraformaldehyde (0.95 g, 1.2 eq., 31.1 mmol) at room temperature. After overnight stirring at 60°C, the mixture is quenched with brine (100 ml) and the aqueous phase is extracted with AcOEt (2*100 ml). The combined organic extracts are dried over MgS04 and concentrated under reduced pressure yielding 4.7 g of (4R)-1-(hydroxymethyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 5, which is used in the next step without any further purification.
LC-MS (MH+): 246.
H NMR (DMSO) δ 7.34 (dd, J-| =9.2 Hz, J2=6.8 Hz, 2 H), 5.87 (t, J=6.8 Hz, 1 H), 4.70 (m, 2 H), 3.78 (m, 1 H), 3.62 (m, 1 H), 3.40 (m, 1 H), 2.68 (m, 1 H), 2.43 (dd, J<l =16.6 Hz, J2=8.6 Hz, 1 H).
1.4 Synthesis of (4R)-1 -[(5-chloro-1 H-1 ,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluoro- phenyl)pyrrolidin-2-one 7.
1 ) To a cold solution (0°C) of (4R)-1-(hydroxymethyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 5 (4.7 g, 1 eq., 19.4 mmol) in CH2CI2 (200 mL) is added oxalyl chloride (3.7 ml, 2 eq., 38 mmol). After stirring for 30 minutes at 0°C, the reaction mixture is evaporated in vacuum yielding (4R)-1-(chloromethyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 6 which is dissolved in THF (100 ml) to afford Solution A.
2) To a cold solution (0°C) of 5-chloro-1 H-1 ,2,4-triazole (3.0 g, 1.5 eq., 29.1 mmol) in THF (100 ml) is added NaH 95% in mineral oil (0.9 g, 2 eq., 38.7 mmol). The reaction mixture is stirred during 30 minutes at 0°C to afford Solution B.
3) Solution A is added to solution B at 0°C and the reaction mixture is maintained under stirring overnight at room temperature. The mixture is quenched with water (100 ml) and extracted with AcOEt (2*100 mL). The combined organic extracts are washed with brine (100 ml), dried over MgS04 then concentrated under reduced pressure yielding 7 g of compound 7 as crude material. The crude residue is purified by chromatography on silicagel (eluent: CH2Cl2/MeOH/NH4OH 95/5/0.5 v/v/v) and recrystallized from iPr20/EtOH affording 1.6 g of (4R)-1-[(5-chloro-1 H-1 ,2,4-triazol-1-yl)methyl]-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one 7 as a white solid.
Yield: 25%.
LC-MS (MH+): 331/333.
H NMR (DMSO) δ 8.12 (s, 1 H), 7.32 (dd, J-| =9.2 Hz, J2=6.9 Hz, 2 H), 5.63 (d, J=1.5 Hz, 2 H), 3.81 (t, J=8.6 Hz, 1 H), 3.62 (t, J=8.4 Hz, 1 H), 3.39 (m, 1 H), 2.71 (dd, J<l =16.7 Hz, J2=8.8 Hz, 1 H), 2.54 (d, J=9.1 Hz, 1 H).
alphaD (MeOH, 25°C): + 9.2.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | ABBV-552; ABBV552; SDI-118; SDI118 |
| Routes of administration | Oral[1] |
| Drug class | Synaptic vesicle glycoprotein 2A (SV2A) ligand[2] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1651179-19-9 |
| PubChem CID | 90467376 |
| ChemSpider | 129532952 |
| UNII | W3LYF2KQ6F |
| KEGG | D13077 |
| ChEMBL | ChEMBL5314929 |
| Chemical and physical data | |
| Formula | C13H10ClF3N4O |
| Molar mass | 330.70 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “ABBV 552”. AdisInsight. 28 October 2024. Retrieved 26 February 2025.
- Botermans W, Koole M, Van Laere K, Savidge JR, Kemp JA, Sunaert S, et al. (2022). “SDI-118, a novel procognitive SV2A modulator: First-in-human randomized controlled trial including PET/fMRI assessment of target engagement”. Frontiers in Pharmacology. 13 1066447. doi:10.3389/fphar.2022.1066447. PMC 9887116. PMID 36733374.
- “Delving into the Latest Updates on Plosaracetam with Synapse”. Synapse. 22 February 2025. Retrieved 26 February 2025.
- “ABBV-552”. ALZFORUM. 28 February 2023. Retrieved 26 February 2025.
/////////Plosaracetam, ABBV-552, ABBV552, SDI-118, SDI118, ABBV 552, ABBV552, SDI 118, SDI118, W3LYF2KQ6F
Pilavapadin
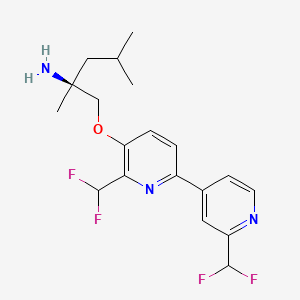

Pilavapadin
CAS1815613-42-3
MFC19H23F4N3O MW 385.4 g/mol
(2S)-1-{[2′,6-bis(difluoromethyl)[2,4′-bipyridin]-5-yl]oxy}-2,4-dimethylpentan-2-amine
(2S)-1-[[2-(difluoromethyl)-6-[2-(difluoromethyl)-4-pyridinyl]-3-pyridinyl]oxy]-2,4-dimethylpentan-2-amine
adaptor protein 2-associated kinase 1 (AAK1) inhibitor, LX9211, BMS-986176, LX 9211, BMS 986176, Phase 2, Neuropathic pain, Postherpetic neuralgia, AAK1-IN-1, 9G4RLM5X6Z
Pilavapadin (also known as LX9211 or BMS-986176) is an investigational, orally available small molecule developed by Lexicon Pharmaceuticals for the treatment of neuropathic pain, primarily diabetic peripheral neuropathic pain (DPNP).
Key Information
- Mechanism of Action: Pilavapadin is a selective inhibitor of AAK1 (AP2 associated kinase 1), a novel target identified through Lexicon’s gene science research. It is designed to inhibit the reuptake and recycling of neurotransmitters involved in pain signaling in the central nervous system without affecting opiate pathways.
- Indication: It is being investigated for the management of chronic and debilitating conditions such as diabetic peripheral neuropathic pain (DPNP), chemotherapy-induced peripheral neuropathy (CIPN), and multiple sclerosis (MS) pain.
- Development Stage: Pilavapadin has completed Phase 2 clinical trials for DPNP and is expected to advance to a Phase 3 trial.
- Status/Designation: The U.S. Food and Drug Administration (FDA) has granted Fast Track designation for the development of pilavapadin in DPNP.
Clinical Trial Results
Phase 2 studies (RELIEF-DPN-1 and PROGRESS) demonstrated that pilavapadin can provide meaningful pain reduction in adults with DPNP.
- In a post-hoc analysis of the PROGRESS study, the 10 mg dose was found to be effective, achieving a clinically meaningful, two-point reduction in average daily pain scores from baseline, with an acceptable safety and tolerability profile.
- The data has been presented at several medical meetings, including the European Association for the Study of Diabetes (EASD).
- OriginatorBristol-Myers Squibb; Lexicon Pharmaceuticals
- DeveloperLexicon Pharmaceuticals
- ClassAnalgesics; Small molecules
- Mechanism of ActionAdaptor-associated kinase 1 inhibitors
- Phase IINeuropathic pain; Postherpetic neuralgia
- 20 Jun 2025Updated efficacy data from the phase II PROGRESS trial in Neuropathic pain presented at 85th Annual Scientific Sessions of the American Diabetes Association (ADA-2025)
- 13 May 2025Lexicon Pharmaceuticals plans an End of Phase 2 meeting with FDA for Pilavapadin
- 13 May 2025Updated efficacy data from the phase II PROGRESS trial in Neuropathic pain released by Lexicon Pharmaceuticals
- A Dose-ranging Study in Patients With Diabetic Peripheral Neuropathic Pain (DPNP)CTID: NCT06203002Phase: Phase 2Status: CompletedDate: 2025-08-29
- Efficacy, Safety, and PK of LX9211 in Participants With Diabetic Peripheral Neuropathic PainCTID: NCT04455633Phase: Phase 2Status: CompletedDate: 2025-06-25
- Efficacy and Safety of LX9211 in Participants With Postherpetic NeuralgiaCTID: NCT04662281Phase: Phase 2Status: CompletedDate: 2023-11-18
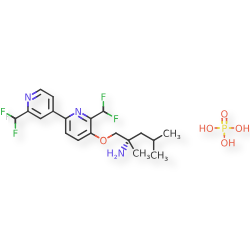
Molecular FormulaC19H23F4N3O.H3O4P
Molecular Weight483.4
CAS 2977251-24-2
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US215884039&_cid=P11-MI4ESM-19570-1
Example 123
(S)-1-((2′,6-bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine

Part A: (2-(difluoromethyl)pyridin-4-yl)boronic acid

Part B: (S)-1-((2′,6-bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021216441&_cid=P11-MI4EP8-16561-2
REF
- Discovery and Optimization of Biaryl Alkyl Ethers as a Novel Class of Highly Selective, CNS-Penetrable, and Orally Active Adaptor Protein-2-Associated Kinase 1 (AAK1) Inhibitors for the Potential Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-09PMID: 35261239DOI: 10.1021/acs.jmedchem.1c02132
- Discovery of (S)-1-((2′,6-Bis(difluoromethyl)-[2,4′-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine (BMS-986176/LX-9211): A Highly Selective, CNS Penetrable, and Orally Active Adaptor Protein-2 Associated Kinase 1 Inhibitor in Clinical Trials for the Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-08PMID: 35257579DOI: 10.1021/acs.jmedchem.1c02131
- Discovery, Structure–Activity Relationships, and In Vivo Evaluation of Novel Aryl Amides as Brain Penetrant Adaptor Protein 2-Associated Kinase 1 (AAK1) Inhibitors for the Treatment of Neuropathic PainPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-07-16PMID: 34270254DOI: 10.1021/acs.jmedchem.1c00472
PAT
- Biaryl kinase inhibitorsPublication Number: US-2021277001-A1Priority Date: 2014-04-02
- Biaryl kinase inhibitorsPublication Number: KR-102379518-B1Priority Date: 2014-04-02Grant Date: 2022-03-25
- Biaryl kinase inhibitorsPublication Number: US-12065437-B2Priority Date: 2014-04-02Grant Date: 2024-08-20
- Biaryl kinase inhibitorsPublication Number: US-2024360131-A1Priority Date: 2014-04-02
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////////Pilavapadin, LX9211, BMS-986176, LX 9211, BMS 986176, Phase 2, Neuropathic pain, Postherpetic neuralgia, AAK1-IN-1, 9G4RLM5X6Z
Paluratide
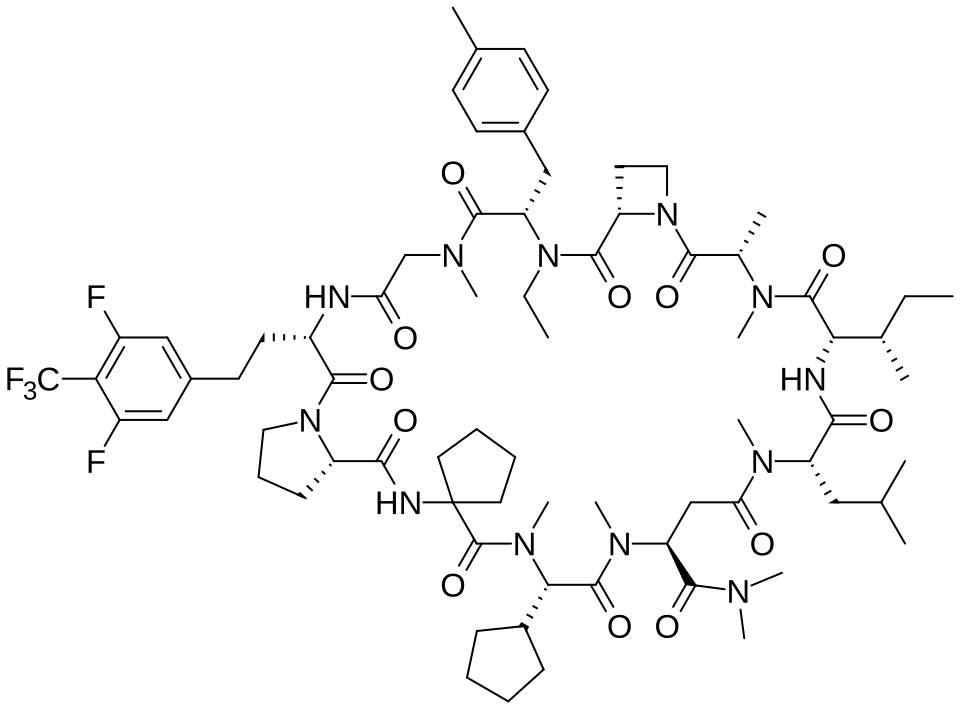


Paluratide
CAS 2676177-63-0
MFC73H105F5N12O12 MW 1437.7 g/mol
1,11-anhydro[N-methyl-L-alanyl-(2S)-azetidine-2-carbonyl-N-ethyl-4-methyl-L-phenylalanyl-N-methylglycyl-3-{[3,5-difluoro-4-(trifluoromethyl)phenyl]methyl}-L-alanyl-L-prolyl-2-
aminocyclopentane-1-carbonyl-(2S)-N-methyl-3-cyclopentylglycyl-1-
(dimethylamino)-N-methyl-L-aspart-4-yl-N-methyl-L-leucyl-Lisoleucine]
(3S,9S,12S,17S,20S,23S,27S,30S,36S)-20-[(2S)-butan-2-yl]-30-cyclopentyl-3-[2-[3,5-difluoro-4-(trifluoromethyl)phenyl]ethyl]-10-ethyl-N,N,7,17,18,24,28,31-octamethyl-9-[(4-methylphenyl)methyl]-23-(2-methylpropyl)-2,5,8,11,16,19,22,25,29,32,35-undecaoxospiro[1,4,7,10,15,18,21,24,28,31,34-undecazatricyclo[34.3.0.012,15]nonatriacontane-33,1′-cyclopentane]-27-carboxamide
G-protein Ras (rat sarcoma virus) inhibitor, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Paluratide (development code LUNA18) was an investigational cyclic peptide KRAS inhibitor developed by Chugai Pharmaceutical, a member of the Roche Group, for the treatment of cancers with KRAS mutations.[1] The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular protein-protein interactions, a class of targets traditionally considered “undruggable.”[2]
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.[3]
Ras Inhibitor LUNA18 is an orally bioavailable cyclic peptide and Ras inhibitor, with potential antineoplastic activity. Upon oral administration, Ras inhibitor LUNA18 selectively targets, binds to and inhibits Ras, thereby inhibiting Ras-dependent signaling and inhibits proliferation of tumor cells in which Ras is overexpressed and/or mutated. Ras serves an important role in cell signaling, division and differentiation. Mutations of Ras may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
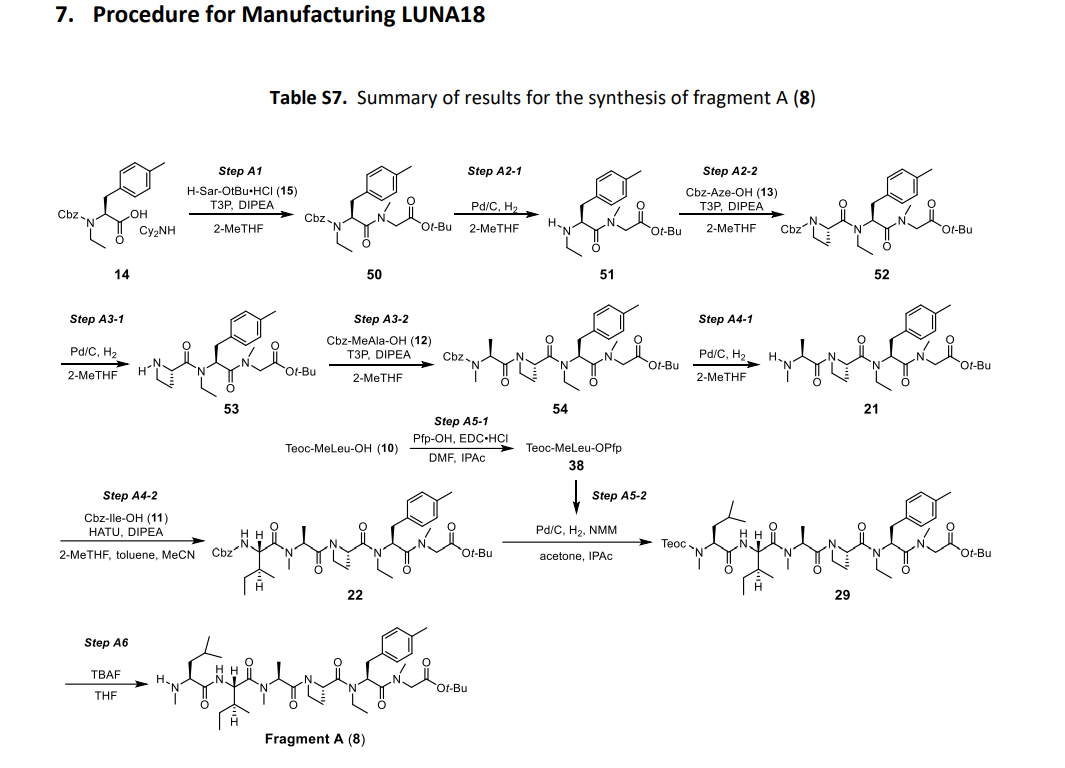
Paluratide (LUNA18 is synthesized using a novel liquid-phase peptide synthesis (LPPS) method, not traditional solid-phase methods, to overcome challenges with N-alkylated cyclic peptides. This process involves a convergent route of 24 telescoped chemical transformations, a final crystallization step, and a focus on specific strategies to manage side reactions like diketopiperazine formation and low reactivity of sterically hindered amino acids.
Key aspects of the synthesis
- Liquid-phase synthesis: A novel, high-yielding LPPS process was developed to enable the large-scale production of paluratide. This is a departure from traditional solid-phase methods, which have limitations with solubility and waste.
- Convergent synthetic route: The synthesis uses a convergent approach, meaning smaller fragments of the peptide are synthesized separately and then joined together. The overall process includes 24 telescoped chemical transformations followed by a final crystallization step.
- Addressing synthesis challenges: Specific strategies were employed to overcome key difficulties:
- Low reactivity: Amino acids with N-alkylation are sterically hindered, so more reactive and stable protecting groups were used to ensure efficient coupling.
- Side reactions: The method was designed to prevent side reactions like diketopiperazine formation in intermediates and incomplete hydrolysis of active esters.
- Instability: The peptide backbone is sensitive to acidic conditions, so a mildly acidic aqueous medium was chosen for workup and purification to maintain stability.
- Protecting group selection: Cbz-protected amino acid active esters were preferred over Boc-protected ones because they are less prone to forming N-carboxyanhydrides (NCA) under activating conditions, which can reduce yield and purity.
- Purification: A final crystallization step is used for purification.
PAT
- Method for producing eutectic of cyclic peptidePublication Number: WO-2024195801-A1Priority Date: 2023-03-20
- Method for producing cyclic peptide crystalsPublication Number: WO-2024085235-A1Priority Date: 2022-10-20
- Composition containing peptide, surfactant, and polymerPublication Number: WO-2024080308-A1Priority Date: 2022-10-12
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: EP-4086272-A1Priority Date: 2021-05-07
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: US-2022411462-A1Priority Date: 2021-05-07
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.5c00260?ref=PDF



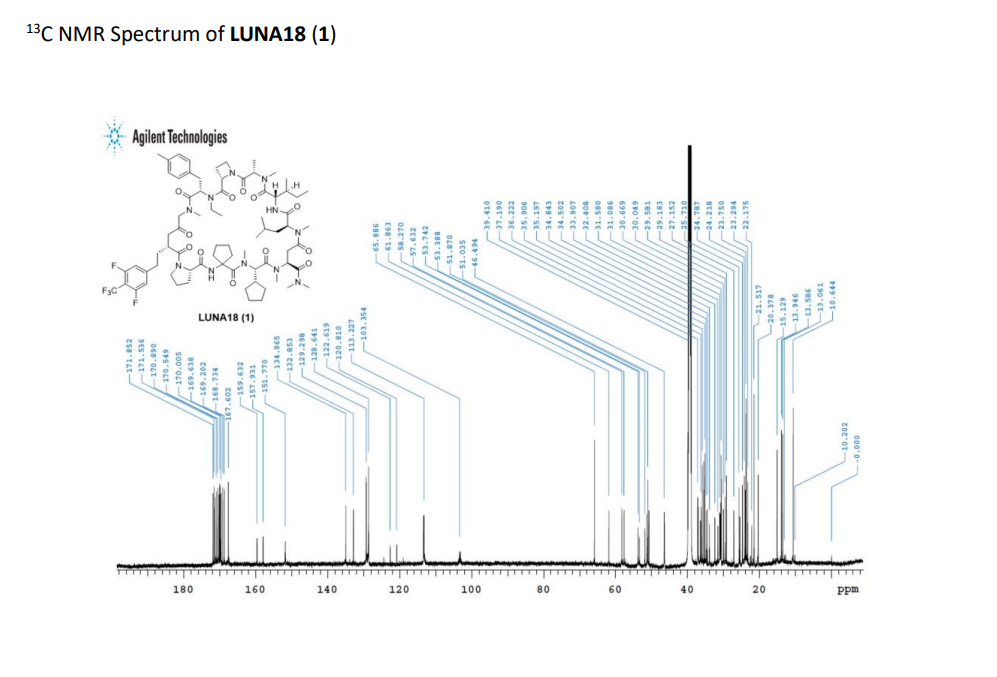
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US383248369&_cid=P20-MI3YXS-80609-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Mechanism of action
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including KRAS, NRAS, and HRAS.[1] The compound binds with high affinity to KRASG12D, with a dissociation constant (Kd) of 0.043 nM, and blocks the interaction between KRASG12D and the guanine nucleotide exchange factor SOS1 with an IC50 of less than 2.2 nM.[4]
Unlike covalent KRAS inhibitors that target specific mutations (such as sotorasib for KRASG12C), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with guanine nucleotide exchange factors (GEFs).[1] This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting ERK and AKT phosphorylation.[4]
Medical uses
Paluratide was being developed for the treatment of locally advanced or metastatic solid tumors harbouring RAS gene alterations.[5] The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including colorectal cancer, gastric cancer, non-small cell lung cancer, and pancreatic cancer.[1]
Chemistry
Paluratide is an 11-member (11-mer) cyclic peptide with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.[1] The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.[2] Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.[1]
Discovery
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an mRNA display library screening approach.[1] The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).[1] The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.[1]
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.[6] This method overcame three major technical challenges: formation of diketopiperazine, insufficient reactivity of amidation due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.[6]
Clinical trials
A Phase 1 dose-escalation and cohort expansion study (NCT05012618) was initiated in August 2021 to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary activity of paluratide administered as a single agent or in combination with other anti-cancer drugs.[5] The study, in the United States and Japan, was designed to enrol approximately 195 patients with locally advanced or metastatic solid tumors positive for documented RAS alterations.[5]
Paluratide was administered orally as capsules.[5] The study also evaluated combination therapy with cetuximab, an EGFR inhibitor.[5]
References
- Tanada M, Tamiya M, Matsuo A, Chiyoda A, Takano K, Ito T, et al. (August 2023). “Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor”. Journal of the American Chemical Society. 145 (30): 16610–16620. Bibcode:2023JAChS.14516610T. doi:10.1021/jacs.3c03886. PMID 37463267.
- Ohta A, Tanada M, Shinohara S, Morita Y, Nakano K, Yamagishi Y, et al. (November 2023). “Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets”. Journal of the American Chemical Society. 145 (44): 24035–24051. Bibcode:2023JAChS.14524035O. doi:10.1021/jacs.3c07145. PMID 37874670.
- Taylor NP (24 October 2025). “Roche axes 4 Chugai solid tumor assets in early-phase clear-out”. Fierce Biotech.
- “LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor”. MedChemExpress.
- “A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion)”. ClinicalTrials.gov. 29 July 2025. NCT05012618.
- Nomura K, Hashimoto S, Takeyama R, Tamiya M, Kato T, Muraoka T, et al. (October 2022). “Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides”. Journal of Medicinal Chemistry. 65 (19): 13401–13412. doi:10.1021/acs.jmedchem.2c01296. PMID 36109865.
- “Chugai Announces 2025 2nd Quarter Results” (Press release). Chugai Pharmaceutical. 24 July 2025.
External links
- Phase 1 Clinical Trial Information at ClinicalTrials.gov
- Development of LUNA18 at Journal of the American Chemical Society
| Clinical data | |
|---|---|
| Other names | LUNA18 |
| Routes of administration | Oral administration |
| Legal status | |
| Legal status | Development discontinued |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2676177-63-0 |
| PubChem CID | 166509683 |
| ChemSpider | 129321315 |
| UNII | AW3YP3CD9X |
| Chemical and physical data | |
| Formula | C73H105F5N12O12 |
| Molar mass | 1437.707 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Paluratide, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Padoprazan
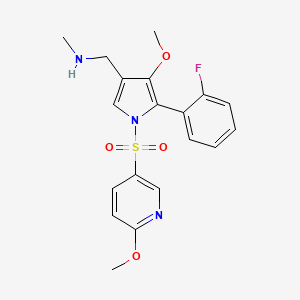
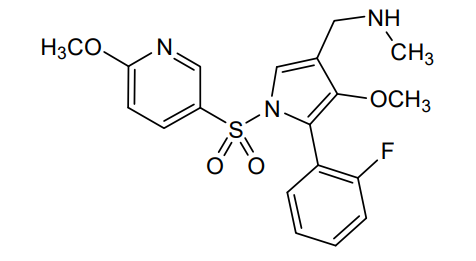
Padoprazan
CAS 2756367-23-2
MF C19H20FN3O4S MW 405.4 g/mol
1-[5-(2-fluorophenyl)-4-methoxy-1-(6-methoxypyridine-3-sulfonyl)-1Hpyrrol-3-yl]-N-methyl methanamine
1-[5-(2-fluorophenyl)-4-methoxy-1-[(6-methoxy-3-pyridinyl)sulfonyl]pyrrol-3-yl]-N-methylmethanamine
1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)- N -methylmethanamine
proton pump inhibitor, 95BJ28E2RP, ID-120040002, ID 120040002
Padoprazan is a new-generation potassium-competitive acid blocker (P-CAB) used to treat acid-related disorders like gastroesophageal reflux, according to MedchemExpress.com and Patsnap Synapse. It works by inhibiting the proton pump in the stomach and is different from traditional proton pump inhibitors (PPIs) because it is not dependent on acid activation. Padoprazan is currently undergoing Phase 3 clinical trials in Korea, notes THE BIO (더바이오).
Key facts about padoprazan
- Drug class: Potassium-competitive acid blocker (P-CAB), a type of proton pump inhibitor, according to DrugBank and GlpBio.
- Mechanism: It inhibits the proton pump in the stomach to reduce acid production and is not acid-activated like older PPIs, per DrugBank.
- Indications: Used for acid-related conditions like gastroesophageal reflux, reports Patsnap Synapse.
- Status: Currently undergoing Phase 3 clinical trials in Korea, says THE BIO (더바이오).
- Development: It is a new-generation drug being developed by companies like Daewon Pharmaceutical.
Padoprazan is a small molecule drug. The usage of the INN stem ‘-prazan’ in the name indicates that Padoprazan is a proton pump inhibitor, not dependent on acid activation. Padoprazan has a monoisotopic molecular weight of 405.12 Da.
PAT
- NEW INHIBITOR OF ACID SECRETION AND USE OF THE SAMEPublication Number: PE-20231652-A1Priority Date: 2020-06-17
- Novel acid secretion inhibitors and use thereofPublication Number: TW-I839161-BPriority Date: 2020-06-17Grant Date: 2024-04-11
- Novel acid secretion inhibitor and use thereofPublication Number: US-2023373954-A1Priority Date: 2020-06-17
- Novel acid secretion inhibitor and use thereofPublication Number: EP-4148050-B1Priority Date: 2020-06-17Grant Date: 2024-12-18
- Novel acid secretion inhibitors and use thereofPublication Number: TW-I797645-BPriority Date: 2020-06-17Grant Date: 2023-04-01
- Acid secretion inhibitor and use thereofPublication Number: US-11767311-B2Priority Date: 2020-06-17Grant Date: 2023-09-26
- Novel Acid Secretion Inhibitor and use thereofPublication Number: AU-2021293694-B2Priority Date: 2020-06-17Grant Date: 2023-12-21
- Novel acid secretion inhibitor and use thereofPublication Number: CN-115884968-BPriority Date: 2020-06-17Grant Date: 2024-06-21
- Novel acid secretion inhibitors and their usesPublication Number: JP-7404561-B2Priority Date: 2020-06-17Grant Date: 2023-12-25
- Novel acid secretion inhibitors and use thereofPublication Number: KR-102432523-B1Priority Date: 2020-06-17Grant Date: 2022-08-16
- Novel acid secretion inhibitor and use thereofPublication Number: CN-115884968-APriority Date: 2020-06-17
- Novel acid secretion inhibitor and use thereofPublication Number: JP-2023524172-APriority Date: 2020-06-17
- Novel acid secretion inhibitor and use thereofPublication Number: US-2023192650-A1Priority Date: 2020-06-17
- Novel acid secretion inhibitors and use thereofPublication Number: TW-202325702-APriority Date: 2020-06-17
- Novel acid secretion inhibitor and use thereofPublication Number: WO-2021256861-A1Priority Date: 2020-06-17
- Novel acid secretion inhibitors and use thereofPublication Number: TW-202214588-APriority Date: 2020-06-17
- Novel Acid Secretion Inhibitor and use thereofPublication Number: AU-2021293694-A1Priority Date: 2020-06-17
- Novel acid secretion inhibitor and use thereofPublication Number: CA-3182882-A1Priority Date: 2020-06-17
- Novel acid secretion inhibitor and use thereofPublication Number: EP-4148050-A1Priority Date: 2020-06-17
- Novel salt of 1-sulfonyl pyrrole derivative, preparation method thereof and pharmaceutical composition comprising thereofPublication Number: TW-I828476-BPriority Date: 2021-12-15Grant Date: 2024-01-01
- Novel salt of 1-sulfonyl pyrrole derivative, preparation method thereof and pharmaceutical composition comprising thereofPublication Number: WO-2023113458-A1Priority Date: 2021-12-15
- Novel salt of 1-sulfonyl pyrrole derivative, method for preparing same, and pharmaceutical composition including samePublication Number: WO-2023113474-A1Priority Date: 2021-12-15
- Novel salt of 1-sulfonyl pyrrole derivative, method for preparing same, and pharmaceutical composition including samePublication Number: US-2025042872-A1Priority Date: 2021-12-15
- Novel acid secretion inhibitors and use thereofPublication Number: KR-20210156234-APriority Date: 2020-06-17
- Novel salt of 1-sulfonylpyrrole derivative, preparation method thereof and pharmaceutical composition comprising the samePublication Number: CN-118541361-APriority Date: 2021-12-15
- Novel salt of 1-sulfonyl pyrrole derivative, preparation method thereof and pharmaceutical composition comprising thereofPublication Number: KR-20230091056-APriority Date: 2021-12-15
- Novel salts of 1-sulfonyl pyrrole derivatives, methods for producing the same, and pharmaceutical compositions containing the samePublication Number: KR-20240119083-APriority Date: 2021-12-15
- Novel salt of 1-sulfonyl pyrrole derivative, preparation method thereof and pharmaceutical composition comprising thereofPublication Number: TW-202334114-APriority Date: 2021-12-15
- Novel formulation comprising acid secretion inhibitorsPublication Number: KR-20240161598-APriority Date: 2023-05-04
- Method for preparation of 6-methoxypyridine-3-yl derivativesPublication Number: TW-202411216-APriority Date: 2022-05-23
- Method for preparing 6-methoxypyridin-3-yl derivativesPublication Number: WO-2023229322-A1Priority Date: 2022-05-23
- Method for preparation of 6-methoxypyridine-3-yl derivativesPublication Number: KR-20230163283-APriority Date: 2022-05-23
- NOVEL SALT OF A DERIVATIVE OF 1-SULFONYLPYRROL, METHOD OF PREPARATION THEREOF AND PHARMACEUTICAL COMPOSITION THAT INCLUDES THE SAMEPublication Number: AR-127964-A1Priority Date: 2021-12-15
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021256861&_cid=P22-MI13VU-05837-1
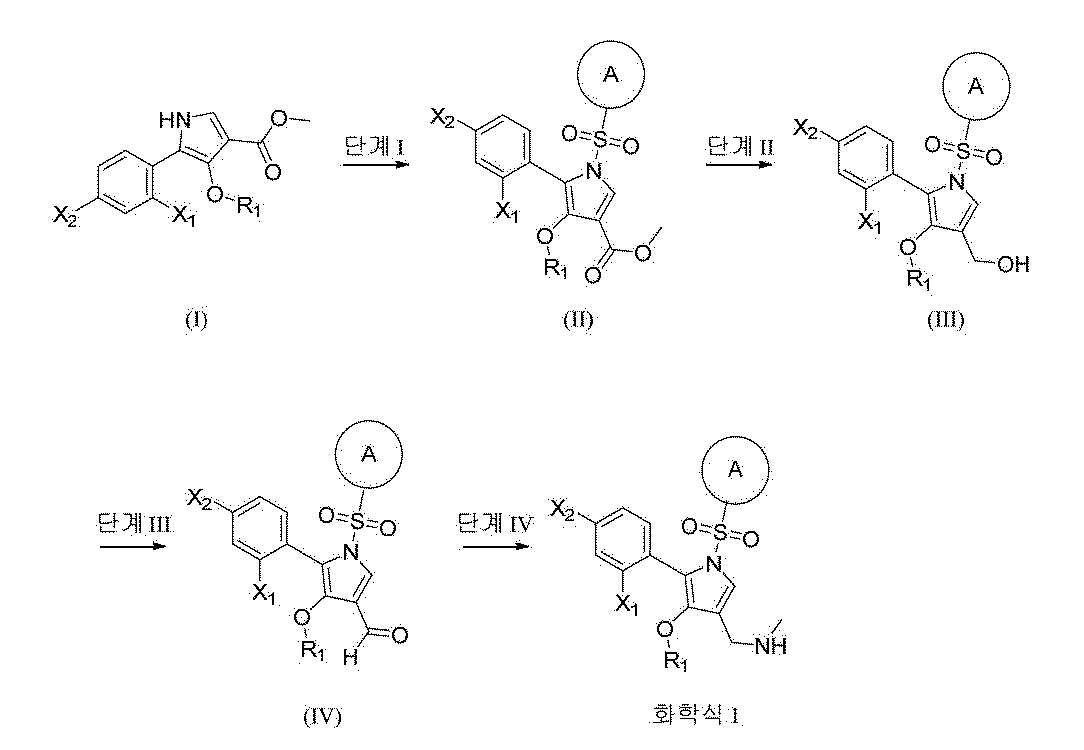
Synthesis Example 1. Synthesis of Example 1
[267]
[Example 1] 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)- N -methylmethanamine
[268]
(1) Synthesis of step methyl 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrole-3-carboxylate
[269]Methyl 5-(2-fluorophenyl)-4-methoxy-1
H -pyrrole-3-carboxylate (intermediate 1, 1.0 eq., 1.2 g, 4.8 mmol) was dissolved in THF (20.0 mL), and NaH (2.0 eq., 384.8 mg, 9.6 mmol) was added dropwise at 0 °C and stirred at room temperature for 10 min. 6-Methoxypyridine-3-sulfonyl chloride (1.5 eq., 1.6 g, 7.2 mmol) was added and stirred at room temperature for 1 h. Water was added to the reaction solution, and the mixture was extracted with EA. The organic layer was dried over anhydrous magnesium sulfate, filtered, concentrated, and purified by column chromatography to obtain methyl 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carboxylate as a light brown solid. (1.85 g, 91.6%)
[270]
(2) Synthesis of step 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)methanol
[271]Methyl 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carboxylate (1.0 eq., 1.0 g, 2.38 mmol) was dissolved in THF (5.0 mL), and 1.0 M DIBAL in
n -hexane solution (5.0 eq., 11.9 mL, 11.9 mmol) was added dropwise at 0 °C, followed by stirring at room temperature for 1 h. The reaction solution was cooled to 0 °C, quenched with an aqueous Rochelle salt solution, and extracted with EA. The organic layer was dried over anhydrous magnesium sulfate, filtered, concentrated, and purified by column chromatography to obtain 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrol-3-yl)methanol as a yellow oil. (654.8 mg, 70.2%)
[272]
(3) Synthesis of step 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrole-3-carbaldehyde
[273]5-(2-Fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrol-3-yl)methanol (1.0 eq., 500.0 mg, 1.3 mmol) and Dess-Martin periodinane (1.0 eq., 540.4 mg, 1.3 mmol) were dissolved in DCM (10.0 mL) and stirred at room temperature for 1 h. The reaction mixture was concentrated and purified by column chromatography to give 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carbaldehyde as a pale brown solid. (388.2 mg, 78.1%)
[274]
(4) Step 1 Synthesis of (5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)- N -methylmethanamine
[275]5-(2-Fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carbaldehyde (1.0 eq., 385.0 mg, 0.99 mmol) was dissolved in THF (5.0 mL), and 2.0 M methylamine in THF (10 eq., 4.9 mL, 9.9 mmol) was added. After stirring at room temperature for 1 h, the reaction mixture was cooled to 0 °C, and NaBH
4 (10 eq., 373.4 mg, 9.9 mmol) was added, followed by stirring at room temperature for 1 h. 6.0
N aqueous hydrogen chloride solution was slowly added dropwise to the reaction solution, and the resulting solid was filtered. The filtered solid was dissolved in water, 1
N aqueous sodium hydroxide solution was added, and extraction was performed with EA. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated to obtain 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)-
N -methylmethanamine as a white solid. (125.8 mg, 28.3%) [M+H] + : 405
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023113474&_cid=P22-MI1405-08231-1
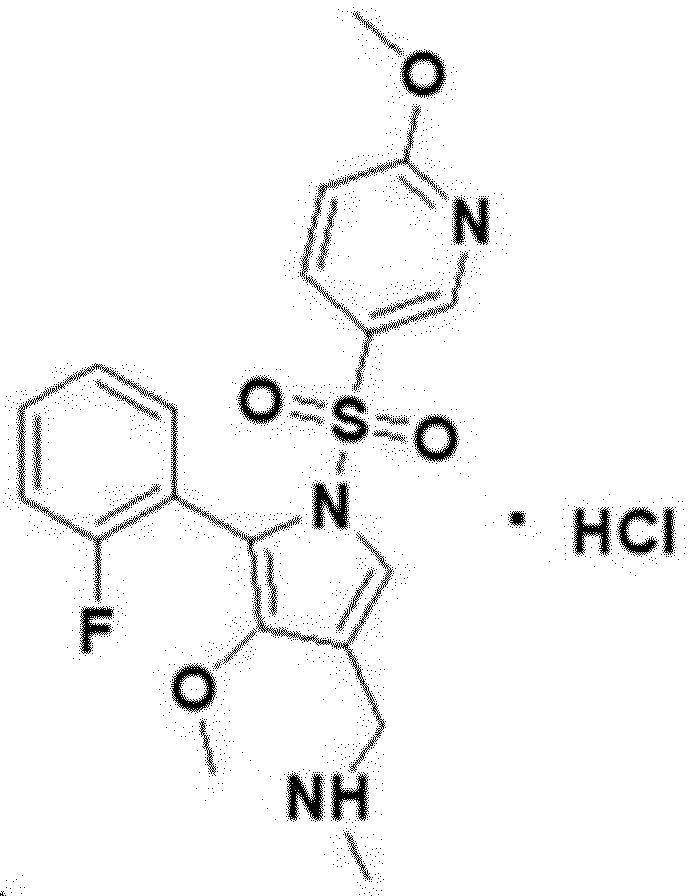
7) Preparation of 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine free base[211]
(1) Step: Synthesis of methyl 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrole-3-carboxylate[212]Methyl 5-(2-fluorophenyl)-4-methoxy-1
H -pyrrole-3-carboxylate (intermediate 1, 1.0 eq., 920 g, 3.69 mol) was dissolved in DMF (9.2 L), and t-BuOK (2.0 eq., 828 g, 7.38 mmol) was added dropwise at 0 °C and stirred for 30 min. 6-Methoxypyridine-3-sulfonyl chloride (1.5 eq., 1.15 kg, 5.54 mol) was added and stirred at 0 °C for 1 h. Water was added to the reaction solution, which was then extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, concentrated, and purified by column chromatography to obtain methyl 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carboxylate as a white solid. (1.20 kg, 77.4%) [213]
(2) Step: Synthesis of 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)methanol[214]Methyl 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carboxylate (1.0 eq., 1.1 kg, 2.62 mol) was dissolved in THF (11.0 L), and DIBAL 2.0 M in THF solution (3.0 eq., 3.93 L, 7.86 mol) was added dropwise at 0 °C, followed by stirring for 30 min. The reaction solution was quenched with 5% aqueous Rochelle’s salt solution and extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated to obtain 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrol-3-yl)methanol as a light yellow oil. (870 g, 84.8%) [215]
(3) Step: Synthesis of 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrole-3-carbaldehyde[216]5-(2-Fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrol-3-yl)methanol (1.0 eq., 830 g, 2.12 mol) and TEA (4.0 eq., 1.59 kg, 15.7 mol) were dissolved in DMSO (4.15 L), and SO
3 -pyridine (4.0 eq., 1.35 kg, 8.48 mol) was added dropwise, and the mixture was stirred at room temperature for 1.5 h. Water was added to the reaction mixture at 0 °C, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated to obtain 5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carbaldehyde as a yellow solid. (722 g, 87.6%) [217]
(4) Step: Synthesis of 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)- N -methylmethanamine[218]5-(2-Fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1
H -pyrrole-3-carbaldehyde (1.0 eq., 715 g, 1.83 mol) was dissolved in methanol (7.2 L), and methylamine in methanol (5.0 eq., 916 g, 9.16 mol) was added. After stirring at room temperature for 1 h, the reaction mixture was concentrated, dissolved in ethanol (7.2 L), cooled to 0 °C, and NaBH
4 (2.0 eq., 139 g, 3.66 mol) was added, and stirred at 0 °C for 1 h. Water was added to the reaction solution, and extracted with ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, concentrated, and purified by column chromatography to obtain 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1 H -pyrrol-3-yl)-
N -methylmethanamine as a brown oil. (347 g, 46.7%)
<Example 1> Preparation of hydrochloric acid salt of 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine About 500 mg of the free base of 1-(5-(2-fluorophenyl)-4-methoxy-1-((6-methoxypyridin-3-yl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine was weighed and placed in a glass vial, and then dissolved in 2 mL of ethanol while heating at 25°C. Then, 647.44 μL (2 M) hydrochloric acid was added to the vial. The sample was continuously stirred on a magnetic stirrer at room temperature for 24 hours, and after stirring for 24 hours, the solid precipitate was separated by centrifugation. Subsequently, the wet solid was dried at 40°C for 20 hours to obtain a grayish white dried powder.
SYN
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Padoprazan, proton pump inhibitor, 95BJ28E2RP, ID-120040002, ID 120040002
Ofirnoflast
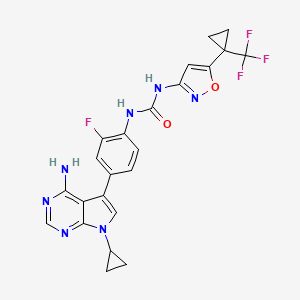
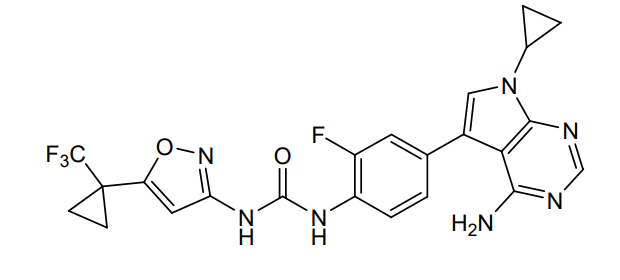
Ofirnoflast
CAS 2731294-23-6
MFC23H19F4N7O2 MW501.4 g/mol
N-[4-(4-amino-7-cyclopropyl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)-2-fluorophenyl]-N’-{5-[1-
(trifluoromethyl)cyclopropyl]-1,2-oxazol-3-yl}urea
N-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO(2,3-D)PYRIMIDIN-5-YL)-2-FLUOROPHENYL)-N’-(5-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)-3-ISOXAZOLYL)UREA
N-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO(2,3-D)PYRIMIDIN-5-YL)-2-FLUOROPHENYL)-N’-(5-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)-1,2-OXAZOL-3-YL)UREA
OFIRNOLAST [USAN]
OFIRNOFLAST
UREA, N-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO(2,3-D)PYRIMIDIN-5-YL)-2-FLUOROPHENYL)-N’-(5-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)-3-ISOXAZOLYL)-
OFIRNOFLAST [INN]
serine/ threonine-protein kinase Nek7 inhibitor, antiinflammatory, HT-6184, HT 6184, 54PY2PBN7S
Ofirnoflast is an investigational drug, a NEK7 inhibitor, that targets and disrupts the formation of the NLRP3 inflammasome, a key driver of chronic inflammation. Developed by Halia Therapeutics, it is being explored for conditions like myelodysplastic syndromes (MDS), obesity, and Alzheimer’s disease. The drug’s unique mechanism aims to address inflammation at a root cause level, potentially offering a new approach to treating these diseases.
How it works
- Ofirnoflast is a “first-in-class” molecule that selectively inhibits the NEK7 protein.
- NEK7 is essential for the assembly of the NLRP3 inflammasome, a molecular complex that causes chronic inflammation.
- By inhibiting NEK7, ofirnoflast prevents the inflammasome from forming and promotes its disassembly.
- This approach aims to reduce inflammation without causing broad immunosuppression.
Therapeutic applications
- Myelodysplastic Syndromes (MDS): Ofirnoflast has completed a Phase 2 study for this condition and received Orphan Drug Designation from the FDA. It is being investigated for its potential to improve blood cell production by targeting the underlying inflammation.
- Obesity: An ongoing Phase 2 study is exploring ofirnoflast in combination with semaglutide to target inflammation and metabolic issues.
- Alzheimer’s Disease: Ofirnoflast is part of an early-stage program looking into its potential for this disease.
Ofirnoflast is a first-in-class, orally bioavailable NEK7 inhibitor currently undergoing Phase 2 clinical evaluation. It disrupts NLRP3 inflammasome assembly by targeting NEK7’s scaffolding function—blocking complex formation independently of NLRP3 activation status, upstream of caspase activation, pyroptosis, and inflammatory cytokine release. This mechanism offers a novel therapeutic approach for chronic inflammation. Unlike NSAIDs, corticosteroids, cytokine-neutralising biologics, and NLRP3-directed small molecules—which are frequently limited by off-target effects, immunosuppression, or incomplete efficacy—ofirnoflast provides a targeted approach with fewer anticipated liabilities
- A Ph2 Study to Evaluate the Safety, Efficacy and Tolerability of HT-6184 and Semaglutide in Obese Participants With T2DMCTID: NCT07172867Phase: Phase 2Status: Not yet recruitingDate: 2025-09-15
- HT-6184 in Subjects With MDSCTID: NCT07052006Phase: Phase 2Status: Active, not recruitingDate: 2025-07-14
- Evaluating Ability of HT-6184 to Reduce Inflammation and Pain After Third Molar ExtractionCTID: NCT06241742Phase: Phase 2Status: CompletedDate: 2025-03-30
- Study to Evaluate HT-6184 in Healthy SubjectsCTID: NCT05447546Phase: Phase 1Status: CompletedDate: 2023-08-28
SYN
https://www.tandfonline.com/doi/full/10.1080/1061186X.2025.2542856
SYN
COMPD 10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021242505&_cid=P11-MHZPDU-32878-1
INTERMEDIATE D1
5-(4-AMINO-3-FLUOROPHENYL)-7-CYCLOPROPYL-7H-PYRROLO[2,3-D]PYRIMIDIN-4- AMINE
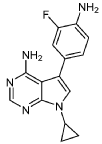
A mixture of 7-cyclopropyl-5-iodo-7H-pyrrolo[2,3-d]pyrimidin-4-amine (C1, 0.160 g, 0.533 mmol), 2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (0.190 g, 0.800 mmol), and K2CO3 (0.221 g, 1.599 mmol) in 1,4-dioxane (1 mL) and water (0.3 mL) was purged with N2 for 10 min. Pd(PPh3)4 (0.062 g, 0.053 mmol) was then added and the reaction mixture was stirred at 100 °C for 12 h. Following completion of the reaction (as indicated by TLC), the mixture was filtered through a pad celite which was then rinsed with EtOAc (2 x 10 mL). The combined filtrates were concentrated under reduced pressure to yield crude material which was purified by flash chromatography (silica gel 230-400 mesh, eluting with 3% MeOH in DCM), affording
the title compound as a yellow solid (0.110 g, 73% yield).1H NMR (400 MHz, DMSO-d6) δ = 8.14 (s, 1H), 7.13 (s, 1H), 7.05-7.09 (m, 1H), 6.95-6.98 (m, 1H), 6.82-6.86 (m, 1H), 6.10 (bs, 2H), 5.22 (bs, 2H), 3.52-3.58 (m, 1H), 1.00-1.04 (m, 4H). LCMS: 284.1 [M+H].
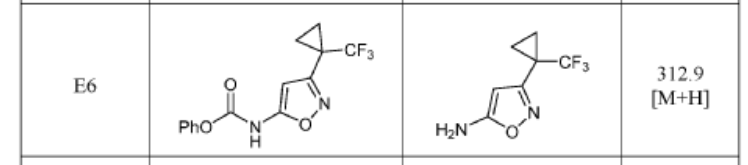
3-(1-(Trifluoromethyl)cyclopropyl)isoxazol-5-amine (precursor to E6) and 5-(1-(trifluoromethyl)cyclopropyl)isoxazol-3-amine (precursor to E7) were synthesized as reported in Synthesis 2013, 45, 171–173
EXAMPLE 5
1-(4-(4-AMINO-7-CYCLOPROPYL-7H-PYRROLO[2,3-D]PYRIMIDIN-5-YL)-2- FLUOROPHENYL)-3-(3-(1-(TRIFLUOROMETHYL)CYCLOPROPYL)ISOXAZOL-5-YL)UREA
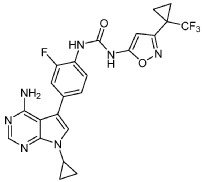
The title compound was prepared following the general procedure for urea formation (Method A), starting from 5-(4-amino-3-fluorophenyl)-7-cyclopropyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (D1, 0.080 g, 0.282 mmol) and phenyl (3-(1-(trifluoromethyl)cyclopropyl)isoxazol-5-yl)carbamate (E6, 0.088 g, 0.282 mmol), and was obtained as a white solid (0.031 g, 22% yield).1H NMR (400 MHz, DMSO-d6) δ = 10.59 (bs, 1H), 8.84 (bs, 1H), 8.11-8.17 (m, 2H), 7.26-7.37 (m, 3H), 6.20 (s, 1H), 6.16 (bs, 2H), 3.55-3.61 (m, 1H), 1.45-1.49 (m, 2H), 1.38-1.43 (m, 2H), 1.03-1.08 (m, 4H). LCMS: 502.1 [M+H].
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024249257&_cid=P11-MHZP9H-30149-1
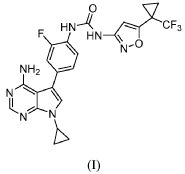
PAT
- Targeted nek7 inhibition for modulation of the nlrp3 inflammasomePublication Number: US-2023210853-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: US-11713321-B2Priority Date: 2020-05-08Grant Date: 2023-08-01
- Inhibitors of nek7 kinasePublication Number: EP-4146348-B1Priority Date: 2020-05-08Grant Date: 2024-07-03
- Inhibitors of nek7 kinasePublication Number: US-2023416259-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: US-12091413-B2Priority Date: 2020-05-08Grant Date: 2024-09-17
- Inhibitors of nek7 kinasePublication Number: TW-202208356-APriority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: AU-2021280893-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: CN-115843272-APriority Date: 2020-05-08
- Inhibitors of nek7 kinasePublication Number: EP-4146348-A1Priority Date: 2020-05-08
- Inhibitors of NEK7 kinasePublication Number: KR-20230008763-APriority Date: 2020-05-08
- Polymorphs of nek 7 inhibitorsPublication Number: WO-2024249257-A1Priority Date: 2023-05-26
- Inhibitors of NEK7 kinasePublication Number: US-11161852-B1Priority Date: 2020-05-08Grant Date: 2021-11-02
- Inhibitors of nek7 kinasePublication Number: US-2021355130-A1Priority Date: 2020-05-08
- Inhibitors of nek7 kinasePublication Number: US-2022064173-A1Priority Date: 2020-05-08
- Inhibitors of nek7 kinasePublication Number: WO-2021242505-A1Priority Date: 2020-05-08
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////ofirnoflast, serine/ threonine-protein kinase Nek7 inhibitor, antiinflammatory, HT-6184, HT 6184, 54PY2PBN7S
Odentegravir

Odentegravir
CAS 2495436-99-0
MF C20H18F3N3O4 MW421.4 g/mol
(7S)-12-hydroxy-1,11-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-1,4,5,6,7,11-hexahydro-3H-2,7-
methanopyrido [1,2-a][1,4]diazonine-10-carboxamide
(7S)-1,4,5,6,7,11-HEXAHYDRO-12-HYDROXY-1,11-DIOXO-N-((2,4,6-TRIFLUOROPHENYL)METHYL)-3H-2,7-METHANOPYRIDO(1,2-A)(1,4)DIAZONINE-10-CARBOXAMIDE
(7S)-12-HYDROXY-1,11-DIOXO-N-((2,4,6-TRIFLUOROPHENYL)METHYL)-1,4,5,6,7,11-HEXAHYDRO-3H-2,7-METHANOPYRIDO(1,2-A)(1,4)DIAZONINE-10-CARBOXAMIDE
3H-2,7-METHANOPYRIDO(1,2-A)(1,4)DIAZONINE-10-CARBOXAMIDE, 1,4,5,6,7,11-HEXAHYDRO-12-HYDROXY-1,11-DIOXO-N-((2,4,6-TRIFLUOROPHENYL)METHYL)-, (7S)-
antiviral, H8B26JZ4A4, orb2664247
Odentegravir is a small molecule drug classified as a
HIV integrase inhibitor, indicated by the “-tegravir” stem in its name. It is a chemical compound with the molecular formula
has been used in research for its antiviral properties.
- Drug Class: HIV integrase inhibitor
- Chemical Formula:
C20H18F3N3O4cap C sub 20 cap H sub 18 cap F sub 3 cap N sub 3 cap O sub 4𝐶20𝐻18𝐹3𝑁3𝑂4
- Molecular Weight:
421.12421.12421.12 Da (monoisotopic)
- Classification: Small molecule drug
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020197991&_cid=P12-MHY8KB-06018-1
Example 23: Preparation of racemic-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26), (7R)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-
methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-1) and (7S)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-2):

Synthesis of 12-Hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26):
[0335] 12-(Benzyloxy)-1,11-dioxo-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxylic acid (57 mg, 0.155 mmol) was dissolved in DCM (2 mL) with (2,4,6-trifluorophenyl)methanamine (27 mg, 0.17 mmol) and triethylamine (60 mg, 0.464 mmol). HATU (60 mg, 0.186 mmol) was added and the mixture was stirred at room
temperature. After overnight reaction, the reaction was concentrated to dryness, purified by silicon gel chromatography to obtain compound 12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a) MS (m/z) 512.06 [M+H]+.
[0336] Compound 12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a) (7 mg, 0.014 mmol) was dissloved in Tolune (1 mL), then followed by the addition of TFA (1 mL). The resulting mixture was stirred at rt for overnight. The solvent was removed under vacuo an the residue was purifed by HPLC to obtain the title compound (26). MS (m/z) 422.091 [M+H]+.1H NMR (400 MHz, DMSO-d6) d 10.39 (t, J = 5.8 Hz, 1H), 8.45 (s, 1H), 7.24 – 7.11 (m, 2H), 4.72 (dd, J = 5.9, 2.9 Hz, 1H), 4.54 (dd, J = 6.0, 2.4 Hz, 2H), 4.11 (d, J = 13.3 Hz, 1H), 3.88 – 3.79 (m, 1H), 3.64 (dd, J = 14.7, 1.9 Hz, 1H), 3.05 (dq, J = 9.5, 3.4 Hz, 1H), 2.06 – 1.91 (m, 1H), 1.89 – 1.74 (m, 3H), 1.61 (d, J = 7.7 Hz, 1H), 1.11 (d, J = 12.7 Hz, 1H).
Synthesis of (7S)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-2) and (7R)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-1):
[0337] Racemic 12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a) was separated by chiral HPLC separation (SFC chromatography on an IB 4.6X100mm 5mic column using MeOH(20) as co-solvent) to obtain compounds (7R)-12-(Benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-1) and (7S)-12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-2)
[0338] Compound (7S)-12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-2) (20 mg, 0.039 mmol) was dissloved in Tolune (1 mL), then followed by the addition of TFA (1 mL). The resulting mixture was stireed at rt for overnight. The solvent was removed under vacuo an the residue was purifed by HPLC to obtain the title compound (26-2). (MS (m/z) 422.123 [M+H]+. 1H NMR (400 MHz, DMSO-d6) d 10.59 (s, 1H), 10.39 (d, J = 5.9 Hz, 1H), 8.45 (s, 1H), 7.18 (t, J = 8.6 Hz, 2H), 4.72 (s, 1H), 4.59 – 4.48 (m, 2H), 4.11 (d, J = 13.2 Hz, 1H), 3.85 (d, J = 14.6 Hz, 1H), 3.69 – 3.59 (m, 1H), 3.05 (ddd, J = 11.3, 6.7, 3.6 Hz, 1H), 1.97 (m, 1H), 1.87 – 1.71 (m, 3H), 1.67 – 1.55 (m, 1H), 1.10 (m, 1H).
[0339] Compound (7R)-12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-1) ((20 mg, 0.039 mmol) was dissloved in Tolune (1 mL), then followed by the addition of TFA (1 mL). The resulting mixture was stireed at rt for overnight. The solvent was removed under vacuo an the residue was purifed by HPLC to obtain the title compound (26-1). MS (m/z) 422.116 [M+H]+. 1H NMR (400 MHz, DMSO-d6) d 10.58 (s, 1H), 10.39 (t, J = 5.8 Hz, 1H), 8.45 (s, 1H), 7.18 (dd, J = 9.2, 8.0 Hz, 2H), 4.73 (s, 1H), 4.58 – 4.49 (m, 2H), 4.11 (d, J = 13.3 Hz, 1H), 3.85 (d, J = 14.6 Hz, 1H), 3.65 (d, J = 14.2 Hz, 1H), 3.10 – 3.00 (m, 1H), 1.96 (m, 1H), 1.82 (d, J = 12.2 Hz, 3H), 1.61 (d, J = 7.4 Hz, 1H), 1.18 – 1.05 (m, 1H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023196875&_cid=P12-MHY8FJ-02517-1
PAT
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usesPublication Number: JP-2025013503-APriority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usesPublication Number: KR-102714084-B1Priority Date: 2019-03-22Grant Date: 2024-10-08
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: KR-20240151256-APriority Date: 2019-03-22
- Bridged Tricyclic Carbamoylpyridone Compounds and Their Pharmaceutical UsePublication Number: ES-2927041-T3Priority Date: 2019-03-22Grant Date: 2022-11-03
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-11548902-B1Priority Date: 2019-03-22Grant Date: 2023-01-10
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-2023027019-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: AU-2020245350-B2Priority Date: 2019-03-22Grant Date: 2023-04-20
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-2023203061-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-11084832-B2Priority Date: 2019-03-22Grant Date: 2021-08-10
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: AU-2020245350-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: EP-3938047-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: EP-3938047-B1Priority Date: 2019-03-22Grant Date: 2022-06-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: EP-4122537-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and uses thereofPublication Number: US-2023339971-A1Priority Date: 2022-04-06
- Bridged tricyclic carbamoylpyridone compounds and uses thereofPublication Number: US-2023339972-A1Priority Date: 2022-04-06
- Bridged tricyclic carbamoylpyridone compounds and uses thereofPublication Number: WO-2023196875-A1Priority Date: 2022-04-06
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-2020317689-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: WO-2020197991-A1Priority Date: 2019-03-22
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////Odentegravir, antiviral, H8B26JZ4A4, orb2664247

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}