
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 422)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Europe grants conditional OK to Pfizer’s Bosulif
Good News For Pfizer’s Orphan Drug Bosulif (Bosutinib) in Europe

mar 28,2013
Regulators in Europe have given a partial green light to Pfizer ‘s leukaemia drug Bosulif.
The European Commission has granted conditional marketing authorisation for Bosulif (bosutinib) for the treatment of adults with chronic, accelerated or blast phase Philadelphia chromosome-positive (Ph+) chronic myelogenous leukaemia (CML). The drug can be given to patients previously treated with one or more tyrosine kinase inhibitors ie Novartis’ Gleevec (imatinib) and Tasigna (nilotinib) or Bristol-Myers Squibb’s Sprycel (dasatinib).
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on January 17, 2013, adopts a positive opinion, recommending a conditional marketing authhorization for Pfizer’s orphan drug Bosulif (Bosutinib) for Chronic Leukemia (CML). Bosutinib receives orphan designation from the European Commission (EC) on August 4, 2010, for CML.
Pfizer receives FDA approval on September 4, 2012, for orphan drug Bosulif (Bosutinib) for CML. Pfizer receives on February 24, 2009, FDA Orphan Drug Designation (ODD) for Bosutinib for CML.

Bosutinib (rINN/USAN; codenamed SKI-606, marketed under the trade name Bosulif) is atyrosine kinase inhibitor undergoing research for use in the treatment of cancer. [1] [2]Originally synthesized by Wyeth, it is being developed by Pfizer.
Some commercial stocks of bosutinib (from sources other than the Pfizer material used for clinical trials) have recently been found to have the incorrect chemical structure, calling the biological results obtained with them into doubt.[3]
Bosutinib received US FDA approval on September 5, 2012 for the treatment of adult patients with chronic, accelerated, or blast phase Philadelphia chromosome-positive (Ph+)chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.[4][5][6]
- Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006 Dec 1;66(23):11314-22. Epub 2006 Nov 17.
- Vultur A, Buettner R, Kowolik C, et al. (May 2008). “SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells”.Mol. Cancer Ther. 7 (5): 1185–94. doi:10.1158/1535-7163.MCT-08-0126.PMC 2794837. PMID 18483306.
- Derek Lowe, In The Pipeline (blog), “Bosutinib: Don’t Believe the Label!”
- Cortes JE, Kantarjian HM, Brümmendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, Besson N, Abbas R, Wang J, Leip E, Gambacorti-Passerini C. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011 Oct 27;118(17):4567-76. Epub 2011 Aug 24.
- Cortes JE, Kim DW, Kantarjian HM, Brümmendorf TH, Dyagil I, Griskevicus L, Malhotra H, Powell C, Gogat K, Countouriotis AM, Gambacorti-Passerini C. Bosutinib Versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia: Results From the BELA Trial. J Clin Oncol. 2012 Sep 4. [Epub ahead of print]
- “Bosulif Approved for Previously Treated Philadelphia Chromosome-Positive Chronic Myelogenous Leukemia”. 05 Sep 2012.
Ospemifene ….EMA accepts MAA submission of Shionogi’s ospemifene for the treatment of VVA

Ospemifene
CAS Number: 128607-22-7
OSPHENA is indicated for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause
- CCRIS 9205
- Deamino-hydroxytoremifene
- Fc-1271
- FC-1271a
- Ospemifene
- Osphena
- UNII-B0P231ILBK
Molecular Formula: C24H23ClO2
Molecular Weight: 378.89 g.mol-1

Marja Sodervall, Maire Eloranta, Arja Kalapudas, Brian Kearton, Michael McKenzie, “METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE.” U.S. Patent US20080214860, issued September 04, 2008.
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 6245819 | 2013-02-26 | 2020-07-21 |
| United States | 8236861 | 2013-02-26 | 2026-08-11 |
Data
| Patent No
US |
PatentExpireyDate | patent use code |
|---|---|---|
| 6245819 | Jul 21, 2020 | U-1369 |
| 8236861 | Aug 11, 2026 | U-1369 |
| 8236861 | Aug 11, 2026 | U-1370 |
| Exclusivity Code | ExclusivityDate |
|---|---|
| NCE | Feb 26, 2018 |
UPDATE……….ON OCT 2015
| Date of issue ofmarketing authorisation valid throughout the European Union | 15/01/2015 |
|---|
NMR……http://file.selleckchem.com/downloads/nmr/S428501-Ospemifene-HNMR-Selleck.pdf
HPLC….http://file.selleckchem.com/downloads/hplc/S428501-Ospemifene-HPLC-Selleck.pdf
Ospemifene appears as a white to almost white, non-hygroscopic crystalline powder. It is insoluble in water, soluble in ethanol and propanol, very slightly soluble in isopropanol. The partition coefficient was found 4.43 and the pKa was calculated 14.26. The molecule has two geometrical isomeric forms. The active substance ospemifene is the Z-isomer. Polymorphism was not observed.
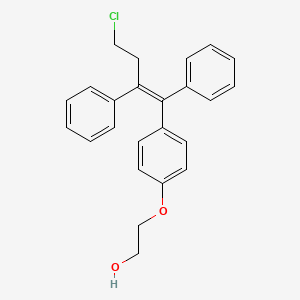
The chemical name of the active substance ospemifene is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl) phenoxy]ethanol, corresponding to the molecular formula C24H23O2Cl and has a relative molecular mass of 378.9.
Ospemifene is a new selective non-hormonal estrogen receptor modulator (SERM) that is used for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause. FDA approved on February 26, 2013.
Article
 OSPEMIFINE
OSPEMIFINEArticle 27 March 2013
Shionogi Limited, the London-based European subsidiary of Shionogi & Co., Ltd announced today that on 26th March 2013 the European Medicines Agency (EMA) accepted its Marketing Authorisation Application (MAA) submission for ospemifene for the treatment of vulvar and vaginal atrophy (VVA) in post-menopausal women.
this is already approved by FDA
“We are pleased to announce the MAA submission for ospemifene to the EMA following the US Food and Drug Administration (FDA) approval last month. The acceptance of the MAA submission for ospemifene not only represents an important step forward in expanding the treatment options for women living in Europe with this condition, but it is also an important milestone for Shionogi as it continues to build its business in Europe” said Takashi Takenoshita, CEO of Shionogi Limited.
Osphena (ospemifene) to treat women experiencing moderate to severe dyspareunia (pain during sexual intercourse), a symptom of vulvar and vaginal atrophy due to menopause.
Dyspareunia is a condition associated with declining levels of estrogen hormones during menopause. Less estrogen can make vaginal tissues thinner, drier and more fragile, resulting in pain during sexual intercourse.
Osphena, a pill taken with food once daily, acts like estrogen on vaginal tissues to make them thicker and less fragile, resulting in a reduction in the amount of pain women experience with sexual intercourse.
“Dyspareunia is among the problems most frequently reported by postmenopausal women,” said Victoria Kusiak, M.D., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Osphena provides an additional treatment option for women seeking relief.”
Osphena’s safety and effectiveness were established in three clinical studies of 1,889 postmenopausal women with symptoms of vulvar and vaginal atrophy. Women were randomly assigned to receive Osphena or a placebo. After 12 weeks of treatment, results from the first two trials showed a statistically significant improvement of dyspareunia in Osphena-treated women compared with women receiving placebo. Results from the third study support Osphena’s long-term safety in treating dyspareunia.
Common side effects reported during clinical trials included hot flush/flashes, vaginal discharge, muscle spasms, genital discharge and excessive sweating.
Osphena is marketed by Florham Park, N.J.-based Shionogi, Inc.
- Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy – May 9, 2012
- END OF ARTICLE
OSPHENA is an estrogen agonist/antagonist. The chemical structure of ospemifene is shown in Figure 1.
Figure 1: Chemical structure
 |
The chemical designation is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanol, and has the empirical formula C24H23ClO2, which corresponds to a molecular weight of 378.9. Ospemifene is a white to off-white crystalline powder that is insoluble in water and soluble in ethanol.
Each OSPHENA tablet contains 60 mg of ospemifene. Inactive ingredients include colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized starch, sodium starch glycolate, titanium dioxide, and triacetin.
INTRODUCTION
Ospemifene (commercial name Osphena produced by Shionogi) is an oral medication indicated for the treatment of dyspareunia – pain during sexual intercourse – encountered by some women, more often in those who are post-menopausal. Ospemifene is aselective estrogen receptor modulator (SERM)[1] acting similarly to an estrogen on thevaginalepithelium, building vaginal wall thickness which in turn reduces the pain associated with dyspareunia. Dyspareunia is most commonly caused by “vulval and vaginal atrophy.”[2]
The medication was approved by the FDA in February 2013.[3]
Ospemifene is used to treat dyspareunia. It is available as a 60 mg tablet that is taken by mouth once a day. The fact that ospemifene can be taken orally is an advantage over other products that are used to treat dyspareunia, because these are generally in a topical dosage form and have to be applied locally.[2] The oral dosage form is much easier and more convenient for patients to administer.
It is “an agonist/antagonist that makes vaginal tissue thicker and less fragile resulting in a reduction in the amount of pain women experience with sexual intercourse.”[2] This drug should be used for the shortest amount of time possible due to associated adverse effects.[2]
Approval process
Hormos Medical Ltd., which is a part of QuatRx Pharmaceuticals, filed a patent on January 19, 2005 for a solid dosage form of ospemifene.[5] In March of 2010, QuatRX Pharmaceuticals licensed ospemifene to Shionogi & Co., Ltd. for them to develop it into a product and put it on the market.[6] A New Drug Application (NDA) was submitted to the FDA on April 26, 2012.[7] Amendments to the NDA were submitted in June, July, August, October, and November 2012, and January and February of 2013.[7] It was ultimately approved by the FDA on February 26, 2013.[6]
Preclinial and clinical trials
Preclinical trials were performed in ovariectomized rats to model menopause.[8] Oral ospemifene was compared with raloxifene (another SERM), its metabolites 4-hydroxy ospemifene and 4′-hydroxy ospemifene, estradiol, and ospemifene administered as an intravaginal suppository.[8] Estradiol was used as a positive control and raloxifene was used because it is in the same drug class as ospemifene.[8]Multiple doses of oral ospemifene were tested.[8] 10 mg/kg/day of Ospemifene was found to cause a greater increase in vaginal weight and vaginal epithelial height than 10 mg/kg/day of raloxifene.[8] Vaginal weight had a 1.46x increase after a two week treatment of 10mg/kg/day of ospemifene.[8] The number of progesterone receptors was increased in the vaginal stroma and epithelium, which indicates that ospemifene has “estrogenic activity.”[8]
A binding assay was also performed to measure the affinity of ospemifene for the estrogen receptor (ERα and ERβ).[8] The study showed that ospemifene bound ERα and ERβ with similar affinity.[8] Ospemifene bound the estrogen receptors with a lower affinity than estradiol.[8] Ospemifene was shown to be an antagonist of “ERE-mediated transactivation on MCF-7 cells,” which the authors concluded indicates “anti-estrogenic activity in breast cancer cells.”[8]
Two 12 week phase 3 clinical trials were performed for ospemifene.[9] To evaluate the efficacy of the drug, 4 signs and symptoms of dyspareunia were measured. These included the “change in percent parabasal cells,” “change in percent superficial cells,” “change in vaginal pH,” and “change in most bothersome symptom (vaginal dryness and vaginal pain associated with sexual activity.”[9]Ospemifene was more effective than placebo in all four of these categories.[9] A dose-response was also seen in the trial; ospemifene 60 mg had greater efficacy than ospemifene 30 mg.[9] Safety was also evaluated in these phase 3 trials. There was a 5.2% increase in the incidence of hot flushes, 1.6% increase in urinary tract infections, and 0.5% increase in the incidence of headache with ospemifene over placebo.[9] One of the phase 3 trials was double-blinded and randomized and involved 826 women who were post-menopausal.[10]The women in the study were required to have one or more vulvovaginal atrophy (VVA) symptom that was moderate or severe in nature, no more than 5% of cells that were superficial when given a vaginal smear, and have a vaginal pH of at least 5.0.[10] Another phase 3 trial involved 605 women who were between the ages of 40 and 80, were diagnosed with VVA, and whose worst symptom was dyspareunia.[11]
 OSPEMIFENE
OSPEMIFENE
In the first half of the 2013 fiscal year, Osphena® generated 0.1 B yen in revenue, which is probably roughly equivalent to $974, 944 U.S. dollars.[12] When Osphena® was put onto the market, it was predicted to earn $495 million in 2017.[13]
OSPHENA is an estrogen agonist/antagonist. The chemical structure of ospemifene is shown in Figure 1.
Figure 1: Chemical structure
 |
The chemical designation is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanol, and has the empirical formula C24H23ClO2, which corresponds to a molecular weight of 378.9. Ospemifene is a white to off-white crystalline powder that is insoluble in water and soluble in ethanol.
Each OSPHENA tablet contains 60 mg of ospemifene. Inactive ingredients include colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized starch, sodium starch glycolate, titanium dioxide, and triacetin.
“SERM”s (selective estrogen receptor modulators) have both estrogen-like and antiestrogenic properties (Kauffman & Bryant, 1995). The effects may be tissue-specific as in the case of tamoxifen and toremifene which have estrogen-like effects in the bone, partial estrogen-like effect in the uterus and liver, and pure antiestrogenic effect in breast cancer.
Raloxifene and droloxifen are similar to tamoxifen and toremifene, except that their antiestrogenic properties dominate. Based on the published information, many SERMs are more likely to cause menopausal symptoms than to prevent them. They have, however, other important benefits in elderly women: they decrease total and LDL cholesterol, thus deminishing the risk of cardiovascular diseases, and they may prevent osteoporosis and inhibit breast cancer growth in postmenopausal women.
Ospemifene is the Z-isomer of the compound of formula (I)

and it is one of the main metabolites of toremifene, is known to be an estrogen agonist and antagonist (Kangas, 1990; International patent publications WO 96/07402 and WO 97/32574 ). The compound is also called (deaminohydroxy)toremifene and it is also known under the code FC-1271a. Ospemifene has relatively weak estrogenic and antiestrogenic effects in the classical hormonal tests (Kangas, 1990). It has anti-osteoporosis actions and it decreases total and LDL cholesterol levels in both experimental models and in human volunteers (International patent publications WO 96/07402 and WO 97/32574 ). It also has antitumor activity in an early stage of breast cancer development in an animal breast cancer model.
Ospemifene is also the first SERM which has been shown to have beneficial effects in climacteric syndromes in healthy women. The use of ospemifene for the treatment of certain climacteric disorders in postmenopausal women, namely vaginal dryness and sexual dysfunction, is disclosed in WO 02/07718 . The published patent application WO 03/103649 describes the use of ospemifene for inhibition of atrophy and for the treatment or prevention of atrophyrelated diseases or disorders in women, especially in women during or after the menopause.
SYNTHESIS

SYNTHESIS

SYNTHESIS
Ospemifene simple structure, its point is to control the synthesis of the product cis-trans isomerization of the double bond. Chloride 1 and benzene ( 2 ) occurs pay – acylation reaction 3 . Ester4 aluminum trichloride under the action of Fries rearrangement of 5 , 5 on the propylene oxide under alkaline conditions to obtain 6 , 6 and 3 McMurry coupling occurs directly generated Ospemifene.
……………………………………………
SYNTHESIS
https://www.google.com/patents/EP2121553B1
-
Ospemifene is the Z-isomer of the compound of formula (Ib)

-
The common starting material in the syntheses of (Ib), namely compound (II), is previously known (Toivola, 1990; EP 0095875 ). According to a method disclosed in EP 095875 , this compound was prepared by dealkylation of a corresponding ether to give (II). The method may be used to produce a mixture of isomers of compounds (Ib), but most preferably is used to prepare the pure E- and Z-isomers of this compound.
-
Particularly in case the Z-isomer of the compound (Ib) is desired, a preferable method for the synthesis of compound (II) is a McMurry reaction of commercially available starting materials, 4-hydroxybenzophenone with 3-chloropropiophenone. The McMurry reaction is a well-known reductive coupling of ketones involving two steps: (1) a single electron transfer to the carbonyl groups from an alkali metal, followed by (2) deoxygenation of the 1,2-diol with low-valent titanium to yield the alkene. This reaction produces mainly the Z-isomer of compound (II)

-
The alkylation in step a) is carried out in an organic solvent, preferably carried out in tetrahydrofuran. It is also preferable to add a base to the solvent, most preferably sodium hydride
-
Zinc (15.0 g, 0.23 mol) and tetrahydrofuran (THF) (180 ml) was added to the reaction vessel and cooled to -10 °C. Titan tetrachloride was added dropwise to the mixture (21.6 g, 0.114 mol) at about -10 °C. After the addition was completed the mixture was refluxed for two hours. Then the mixture was cooled to 40 °C and 4-hydroxybenzophenone (7.68 g, 0.039 mol) and 3-chloropropiophenone (6.48 g, 0.039 mol) dissolved in THF (75 ml) were added to the mixture. Refluxing was continued for additional 3.5 hours. The cooled reaction mixture was poured in aqueous potassium carbonate solution (21 g K2CO3 + 210 ml water) and allowed to stand overnight at the ambient temperature. The mixture was filtered and the precipitate was washed with THF. The filtrate was evaporated to dryness. The residue was dissolved in ethyl acetate and washed with water. Ethyl acetate phase was evaporated to dryness and the residue was crystallized first from methanol-water (8:2) and then from methanol-water (9:1). Yield 5.4 g.
-
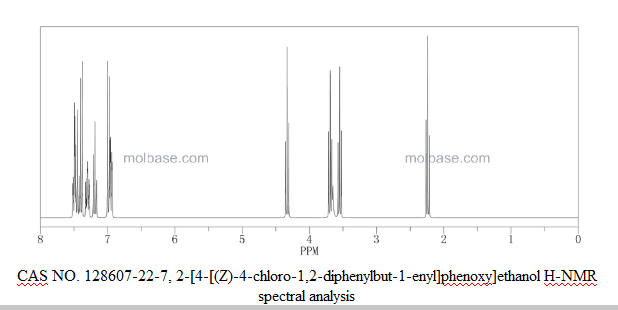
Z-isomer:1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 6.48 (d, 2H, aromatic proton ortho to hydroxy), 6.75 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.4 (m, 10H, aromatic protons)
- EXAMPLE 14-(4-Chloro-1,2-diphenyl-but-1-enyl)phenol (Compound II)
EXAMPLE 2
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenol (0.23 g, 0.689 mmol) was dissolved in tetrahydrofuran (3 ml) under nitrogen atmosphere. Sodium hydride (0.025 g, 1.03 mmol) was added to the solution and the mixture was stirred at room temperature for an hour. 2-(2-iodo-ethoxy)-tetrahydropyran (0.3 g, 1.17 mmol) was added and the mixture was refluxed for 2 hours. Additional portions of 2-(2-iodo-ethoxy)-tetrahydro-pyran (0.5 g, 2 mmol) were added to the mixture during seven hours. After cooling and adding water, THF was evaporated and the mixture was extracted three times with ethyl acetate. The organic phase was washed with 2 N aqueous sodium hydroxide and water, dried with sodium sulphate and evaporated to dryness. The residue (which is Compound (IV) where Pr is tetrahydropyranyl) was dissolved in ethanol and acidified with 2 N aqueous hydrogen chloride solution. The mixture was stirred at room temperature over night, evaporated and extracted with dichloromethane. After washing with water the organic phase was dried (Na2SO4) and evaporated. The residue was purified by flash chromatography with dichloromethane/methanol 9.5/0.5 as eluent. Yield 0.17 g, 59 %.
-
Z-isomer, 1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 3.85-3.89 (m, 4H, OCH2CH2), 6.56 (d, 2H, aromatic proton ortho to hydroxy), 6.80 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.43 (m, 10H, aromatic protons).
EXAMPLE 3
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
The compound was prepared by the same method as described in Example 2 using 2-(2-iodo-ethoxymethyl)-benzene as a reagent and removing the benzylic protecting group using the method described in Example (e) ofUS Patent No. 6,891,070 B2 . Briefly, the removal is carried out under a nitrogen atmosphere, in the presence of Zn powder and acetyl chloride.
- EXAMPLE 5
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-acetic acid ethyl ester (Example 4) was dissolved in tetrahydrofuran at room temperature under nitrogen atmosphere. Lithium aluminium hydride was added to the solution in small portions until the reaction was complete. The reaction was quenched by adding saturated ammonium chloride solution to the mixture. The product was extracted into toluene, which was dried and evaporated in vacuo. The yield 100 mg, 43 %.
-
1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 3.85-3.89 (m, 4H, OCH2CH2), 6.56 (d, 2H, aromatic proton ortho to hydroxy), 6.80 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.43 (m, 10H, aromatic protons).
PATENT
https://www.google.com/patents/US6891070
e) 2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethoxy}ethanol:
Z-1-{4-[2-(2-Benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene (3.8 g, 7.4 mmol) is dissolved in ethyl acetate under nitrogen atmosphere, Zn powder (0.12 g, 1.85 mmol) and acetyl chloride (1.27 g, 16.3 mmol) are added and the mixture is stirred at 50° C. for 3 h (Bhar, 1995). The reaction mixture is cooled to room temperature, water (10 ml) is added and stirring is continued for additional 10 min. The aqueous layer is separated and the organic phase is washed with 1 M aqueous hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in methanol (16 ml) and water (4 ml). The acetate ester of the product is hydrolysed by making the mixture alkaline with sodium hydroxide (1 g) and stirring the mixture at room temperature for 1 h. Methanol is evaporated, water is added and the residue is extracted in ethyl acetate and washed with 1 M hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in toluene (25 ml), silica gel (0.25 g) is added and mixture is stirred for 15 min. Toluene is filtered and evaporated to dryness.
The residue is crystallised from heptane-ethyl acetate (2:1). The yield is 71%.
Z-isomer: 1H NMR (CDCl3): 2.92 (t, 2H), 3.41 (t, 2H), 3.58-3.63 (m, 2H), 3.69-3.80 (m, 4H), 3.96-4.01 (m, 2H), 6.56 (d, 2H), 6.78 (d, 2H), 7.10-7.40 (m, 10H).
E-2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethoxy}ethanol is prepared analogously starting from E-1-{4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene. The product is purified by flash chromatography with toluene-methanol (10:0.5) as eluent.
E-isomer: 1H NMR (CDCl3): 2.97 (t, 2H), 3.43 (t, 2H), 3.65-3.79 (m, 4H), 3.85-3.90 (m, 2H), 4.13-4.17 (m, 2H), 6.85-7.25 (m, 2H).
Debenzylation of 1-{4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene is also carried out by hydrogenation with Pd on carbon as a catalyst in ethyl acetate-ethanol solution at room temperature.
PATENT
http://www.google.com/patents/WO2014060639A1?cl=en
EXAMPLE 5. Preparation of (Z)-2-[4-(4-chloro-l,2-diphenyl-but-l-enyl)- phenoxy]ethanol (ospemifene) by base hydrolysis of pivaloyl-groiip
; . (Z)-2-(4-(4-Chloro- l ,2-diphenylbut-l-en- l-yl)phenqxy)ethyl pivalate ( 1 g, 2.16 mmol) was dissolved in THF (8 ml) followed by addition of MeOH (1 ml) and water (1 ml). Sodium hydroxide (0.1 g, 2.5 mmol) was added in orie portion and the reaction was stirred at room temperature for 12 h. After completion of the reaction the mixture was partitioned between water (20 ml) and EtOAc (20 ml). Organic phase was washed with water (20 ml) and brine (20 ml); dried (Na2S04), filtered, and concentrated: The residue was crystallized from -PrOH yielding ospernifene (0:29 g, 35 %) as a white solid.
1H-NMR (400 MHz, CDC13) δ (ppm): 7.37 (2H, t, 7=8Hz, ArH), 7.29 (3Η, t, J=7.2Hz, ArH), 7.20 (2Η, t,7=7.6Hz, ArH), 7.16-7.13 (3Η, m, ArH), 6.80 (2Η, d, J=8.8Hz, ArH), 6.57 (2Η, d, 7=8.8Hz, ArH), 3.94 (2Η, t, y=4.4Hz, ArOCH2CH2OH), 3.87 (2H, m, ArOCH2CH OH), 3.42 (2H, t, J=7.2Hz, C1CH2CH2), 2.92 (2H, t, 7=7.2Hz, C1CH2CH2), 1.95 (1Η, t, 7=6.4Hz, OH).
13C- NMR (100 MHz, CDC13) δ (ppm): 157.2, 143.2, 142.1 , 141.3, 2 x 135.7, 132.2, 130.0, 129.8, 128.8, 128.7, 127.4, 127.0, 113.9, 69.3, 61.8, 43.3, 39.0.
EXAMPLE 6. Preparation of (Z)-2-[4-(4-chloro-l,2-diphenyl-but-l-enyl)- phenoxy]ethanol (ospernifene) by reductive cleavage of pivaloyl-grou
(Z)-2-(4-(4-Chloro- 1 ,2-diphenylbut- 1 -en- 1 -yl)phenoxy)ethyl pivalate (3.5 g, 7.56 mmol) was dissolved in toluene (35 ml) and stirred under nitrogen for 5 min at room temperature. Lithium aluminium hydride solution (1 M in THE) (7.56 ml, 7.56 n mbi) was added dropwise to the reaction and the mixture was stirred at room temperature for 30 min. After HPLC indicated completion, the reaction was quenched by addition of saturated NH4Cl-sblution (75 ml). Additional amount of toluene (30 ml) was added and the phases were separated. The organic phase was washed with water (50 ml), brine (50 ml), dried (Na2S04), filtered and concentrated in vacuo. The residue was crystallized from 90 % MeOH yielding ospernifene (1 ,75 g, 61 9c) as a white solid.
1H NMR PREDICT

13C NMR PREDICT


References
- Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O (2003). “Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial”. Menopause10 (5): 433–9.doi:10.1097/01.GME.0000063609.62485.27.PMID14501605.
- Tanzi MG (April 2013). “Ospemifene: New treatment for postmenopausal women.”. Pharmacy Today. American Pharmacists Association.
- “FDA approves Osphena for postmenopausal women experiencing pain during sex”. FDA News Release (U.S. Food and Drug Administration). 2013-02-26.
- “Ospemifene: Indications, Side Effects, Warnings”. Drugs.com.
- EP application 2286806, Lehtola V-M, Halonen K, “Solid formulations of ospemifene”, published 2011-02-23, assigned to Hormos Medical Ltd.
- “Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy”. Drugs.com.
- Kusiak V (2013-02-13). “NDA Approval” (PDF). U.S. Food and Drug Administration.
- Unkila M, Kari S, Yatkin E, Lammintausta R (November 2013). “Vaginal effects of ospemifene in the ovariectomized rat preclinical model of menopause”. J. Steroid Biochem. Mol. Biol.138: 107–15.doi:10.1016/j.jsbmb.2013.04.004. PMID23665515.
- Center for Drug Evaluation and Research (2013-02-26). “Clinical Pharmacology and Biopharmaceutics Review Application Number 203505Orig1s000” (PDF). Office of Clinical Pharmacology Review. U.S. Food and Drug Administration.
- Bachmann GA, Komi JO (2010). “Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study”. Menopause17 (3): 480–6.doi:10.1097/gme.0b013e3181c1ac01. PMID20032798.
- Portman DJ, Bachmann GA, Simon JA (June 2013). “Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy”. Menopause20 (6): 623–30.doi:10.1097/gme.0b013e318279ba64. PMID23361170.
- http://www.shionogi.co.jp/en/ir/pdf/e_p131101.pdf. First Half of Fiscal 2013 Financial Results. Nov. 1, 2013.
- http://www.thepharmaletter.com/article/fda-approves-shionogi-s-osphena-for-postmenopausal-women-experiencing-pain-during-sex. ThePharmaLetter
PATENTS
|
8-8-2012
|
Method for enhancing the bioavailablity of ospemifene
|
|
|
1-21-2011
|
METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES
|
|
|
11-5-2010
|
METHODS FOR THE INHIBITION OF ATROPHY OR FOR TREATMENT OR PREVENTION OF ATROPHY-RELATED SYMPTOMS IN WOMEN
|
|
|
10-13-2010
|
METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES
|
|
|
3-18-2009
|
METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE
|
|
|
5-11-2007
|
Novel oral formulations of ospemifene
|
|
|
5-11-2007
|
Formulations of fispemifene
|
|
|
1-11-2006
|
Methods for the inhibition of atrophy or for treatment or prevention of atrophy-related symptoms in women
|
|
|
12-9-2005
|
Methods for the inhibition of atrophy or for treatment or prevention of atrophy-related symptoms in women
|
|
|
8-26-2005
|
Solid formulations of ospemifene
|
|
8-26-2005
|
Method for treatment or prevention of osteoporosis in individuals with high bone turnover
|
|
|
4-6-2005
|
Triphenylalkene derivatives and their use as selective estrogen receptor modulators
|
|
|
6-11-2003
|
Triphenylalkene derivatives and their use as selective estrogen receptor modulators
|
|
|
6-13-2001
|
Method for the treatment of vaginal dryness and sexual dysfunction in women during or after the menopause
|
Phase 2 , Sarepta, shows eteplirsen for Duchenne muscular dystrophy
ETEPLIRSEN
27 MAR 2013
The clock is ticking for Sarepta Therapeutics , a multitude of biotech investors and the boys who suffer from Duchenne muscular dystrophy.
Armed with promising Phase IIb data from a small study of eteplirsen involving only 12 patients with the lethal disease, Sarepta CEO Chris Garabedian is completing one of the most closely-watched high wire acts in the industry. At a time most companies would be focused solely on organizing a pivotal late-stage study, there’s intense speculation that the biotech will shoot for an accelerated approval with the data in hand. And it all comes down to their sit-down with the FDA to review mid-stage data.
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
The drug is a Morpholino antisense oligomer which triggers excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.

Lundbeck, Otsuka reach agreement to develop and commercialize Lu AE58054

Lu AE58054
467459-31-0
C20H19F5N2O, 398.37
2-(6-fluoro-1H-indol-3-yl)-N-(3-(2,2,3,3,3-pentafluoropropoxy)benzyl)ethanamine
1H-Indole-3-ethanamine, 6-fluoro-N-[[3-(2,2,3,3-tetrafluoropropoxy)phenyl]methyl]-
N-[2-(6-Fluoro-1H-indol-3-yl)ethyl]-3-(2,2,3,3-tetrafluoropropoxy)benzylamine
CAS No: 467458-02-2 hydrochloride, M.Wt: 434.83
Lu AE58054 Hydrochloride Formula: C20H20ClF5N2O

MAR26.2013
H. Lundbeck A/S (Lundbeck) and Otsuka Pharmaceutical Co., Ltd. (Otsuka) today announced a license and development agreement for Lu AE58054, a selective 5HT6receptor antagonist currently in development for the treatment of Alzheimer’s disease. Under the terms of the agreement, Lundbeck will grant Otsuka co-development and co-commercialization rights to Lu AE58054 in the U.S., Canada, East Asia including Japan, major European countries and Nordic countries.
Lu AE58054 is a potent and selective 5-HT6 receptor antagonist under development byLundbeck as an augmentation therapy for the treatment of cognitive deficits associated with Alzheimer’s disease and schizophrenia.[1][2]
Phase III trials recently began for Lu-AE58054, a novel 5-HT6 antagonist for Alzheimer’s disease (AD) from H Lundbeck and Otsuka. Lu-AE58054 already demonstrated significant improvement of cognitive function in an earlier phase II trial when administered with Aricept.
This orally available drug is an antagonist of the serotonin 6 (5-HT6) receptor. This receptor subtype is expressed primarily in the brain, particularly in the cerebral cortex and hippocampus, where it has been proposed to play a role in cognitive impairments associated with schizophrenia and Alzheimer’s disease. The 5-HT6 receptor antagonists are thought to enhance cholinergic, glutamatergic, noradrenergic, and dopaminergic neurotransmission. Apart from some affinity for adrenergic receptors, Lu AE58054 has been reported to be highly selective over other G-protein coupled receptors. The compound enters the brain and dose-dependently reversed deficits in a rat model of cognitive impairment (Upton et al., 2008; Arnt et al., 2010).
Lu AE58054 is being developed as a symptomatic adjunct to cholinesterase inhibitor treatment in Alzheimer’s disease. Lu AE58054 was originally discovered by Lilly, which licensed it to the biotechnology company Saegis for the development of cognitive impairment in thinking disorders such as schizophrenia. In 2006, Saegis was acquired by Lundbeck, which in October 2013 launched a global Phase 3 program in AD. This program consists of four trials planned to enroll a total of about 3,000 patients (see company press release).
No Phase 1 trials on this drug are listed in publicly available databases. In 2005, Saegis conducted a Phase 2a trial in 20 schizophrenia patients in the United States, evaluating the safety, tolerability, pharmacokinetics, and pharmacodynamics of giving this drug as an add-on to Risperidone (see company press release). In 2009 and 2010, Lundbeck conducted a Phase 2 trial in Europe and Asia to evaluate the compound as an adjuct to Risperidone for its effect on cognitive deficits in 124 patients with schizophrenia. Results were not published in the peer-reviewed literature, but development of Lu AE58054 for cognitive deficits in schizophrenia appears to have ended.
In 2010 and 2011, Lundbeck evaluated Lu AE58054 in a Phase 2 study in 278 patients with probable Alzheimer’s disease. Conducted in Australia, Canada, and Europe, the trial compared the effects of a six-month course of an undisclosed dose of Lu AE58054 with 10 mg/day of Donepezil to the same dose of placebo. In June 2012, the company announced that the trial had met its primary cognition endpoint as measured by the ADAS-cog. On secondary endpoints, such as measures of global status and activities of daily living, Lu AE58054 treatment showed trends for a benefit but fell short of achieving statistical significance (see Jun 2012 news story).
In July 2013, a Phase 1 study evaluating pharmacokinetics of single and multiple ascending doses in 42 healthy volunteers was added, and in October 2013, the first of four planned Phase 3 studies began. This study is set to enroll 930 patients with mild to moderate Alzheimer’s disease who are already taking a stable dose of 10 mg/day of Donepezil. The trial compares a six-month course of once-daily 30 or 60 mg capsules of drug to placebo for added benefit on cognition as measured by the ADAS-cog. Secondary outcomes will assess various aspects of global clinical function and behavior. No biomarkers are embedded in this trial, which is expected to last until September 2015. For all listed trials on Lu AE58054, seeclinicaltrials.gov.

Description: IC50 Value: 0.83 nm [1] Lu AE58054 is an in-vitro potency and selectivity, in-vivo binding affinity and effect of the 5-HT (6) R antagonist in vitro:. Lu AE58054 displayed high affinity to the human 5-HT (6) receptor (5-HT (6) R) with a Ki of 0.83 nm. In a 5-HT (6) GTPgammaS efficacy assay Lu AE58054 showed no agonist activity, but demonstrated potent inhibition of 5-HT- . mediated activation Besides medium affinity to adrenergic alpha (1A) – and alpha (1B)-adrenoreceptors, Lu AE58054 demonstrated> 50-fold selectivity for more than 70 targets examined [1] in vivo: Orally administered Lu AE58054 potently inhibited striatal in. -vivo binding of the 5-HT (6) antagonist radioligand [(3) H] Lu AE60157, with an ED (50) of 2.7 mg / kg. Steady-state modelling of an acute pharmacokinetic/5-HT (6) R occupancy time-course experiment indicated a plasma EC (50) value of 20 ng / ml. Administration of Lu AE58054 in a dose range (5-20 mg / kg po) leading to above 65% striatal 5-HT (6) R binding occupancy in vivo, reversed cognitive impairment in a rat novel object recognition task induced after subchronic treatment for 7 d with phencyclidine (PCP 2 mg / kg bid, ip for 7 d, followed by 7 d drug free). The results indicate that Lu AE58054 is a selective antagonist of 5-HT (6) Rs with good oral bioavailability and robust efficacy in a rat model of cognitive impairment in schizophrenia [1] Clinical trial:. Lu-AE58054 Added to Donepezil for the Treatment for Moderate Alzheimer’s Disease Phage2.
Dementia is a clinical syndrome characterized by deficits in multiple areas of cognition that cannot be explained by normal aging, a noticeable decline in function, and an absence of delirium. In addition, neuropsychiatric symptoms and focal neurological findings are usually present, Dementia is further classified based on etiology. Alzheimer’s disease (AD) is the most common cause of dementia, followed by mixed AD and vascular dementia, vascular dementia, Lewy body dementia (DLB), and fronto- temporal dementia.
The incidence of Alzheimer’s disease is expected to increase through the year 2050 with an estimated prevalence of 1 1 to 16 million cases. Currently, two classes of medications are FDA approved for managing symptoms of AD – acetylcholinesterase inhibitors (AChEIs) and an N-methyl-D-aspartase (NMDA) receptor antagonist. AChEIs are commonly used as initial treatment on diagnosis. The AChEIs – donepezil, rivastigmine, galantamine, and tacrine – are indicated for mild-to-moderate AD; only donepezil is approved for the severe stage.
Despite the available therapies, there are no treatments to cure AD or to prevent or stop disease progression. Acetylcholinesterase inhibitors do not help everyone who has AhJieimers disease and in fact are not efficacious in many patients. Considering that AChEIs and memantine have only a modest symptomatic effect, and cannot prevent AD decline and slow disease progression, mere is a high unmet need for more effective symptomatic treatments and for a disease modifying/slowing therapies. The use of selective 5-HTe receptor antagonists to treat cognitive dysfunction has been suggested and is based on several lines of reasoning. For example, selective 5-HT« receptor antagonists have been shown to modulate cholinergic and glutamatergic neuronal function. The activity of selective 5-HT6 receptor antagonists has been demonstrated in animal models of cognitive function. Since the disclosure of the first selective 5-HT4 receptor antagonists, there have been several reports on the activity of these selective compounds in in-vivo models of cognitive function, N*(2-(6-fluoro-lH-indol-3-yI)ethyI)-3- (2,2,3,3-tetrafluoropropoxy)benzylamine (herein referred to as “Compound Γ) is a potent and selective1
RECTIFIED SHEET RULE 91 ISA/EP 5-HT6 receptor antagonist which has been in clinical development for treating cognition impairment associated with schizophrenia and as a treatment for AD.

In November 2008, a multi-centre, randomised, double-blind, fixed-dose study (120 mg/day BID) was initiated to explore the efficacy and safety of Compound I as adjunctive treatment to risperidone in patients with schizophrenia. Overall improvement in schizophrenia symptoms was assessed by using the Positive and Negative Syndrome Scale (PANSS) total score. Compound I did not offer any treatment advantage over placebo as measured by the PANSS total score. In 2010, it was announced that there did not appear to be any treatment advantage over placebo in improving patients’ overall neurocognitive performance as assessed using the BACS composite Z-score and the PANSS cognitive subscale scores.
In 2012, it was reported that a randomized, double blind, placebo controlled trial conducted in Europe, Canada and Australia met its primary endpoint in the treatment of AD. Data demonstrated that Compound I plus 10 mg/day of donepezil significantly improved cognitive function in 278 patients with Alzheimer’s disease compared to placebo plus donepezil, when measured by Alzheimer’s Disease Assessment Scale-cognitive sub-scale (ADAS-cog). Compound I showed positive results in secondary endpoints including measures of global impression and daily living activities compared to donepezil treated patients.
The daily dose of 90 mg of Compound I in the AD study was administered three times daily (3 x 30 mg) to overcome the relative short half-life observed in subjects in previous clinical studies. An issue for that dose selection was to ensure that the maximum exposure level fell under the maximum exposure limit which had been established from non-clinical toxicology studies. Accordingly, a fixed dose of three times in the study was introduced.
As the 5-HT6 receptor is a novel target predominately localized in the brain, a key problem in the development is to determine the amount of receptor occupancy and the correlation with plasma exposure. With CNS targets, further challenges exist that revolve around whether a drug will pass the blood brain barrier and whether it will reach the target at a suitable concentration and for a sufficient length of receptor occupancy. Direct brain measures of 5 -HT6 receptor occupancy may be valuable to many decision-making processes during the development of centrally acting drugs targeted at 5-HT6 to ensure adequate proof-of-concept testing and to optimize dosing regimens. In humans, tools such as positron emission tomography (PET) with specific radiolabeled ligands have been used to quantitatively assess in-vivo occupancy of a number of neurotransmitter receptors, including those for dopamine, serotonin, and benzodiazepines (Talbot, et al., European Neuropsychopharmacology, 2002, 12, 503-511).
An effective PET ligand, [nC]-LuPET was developed and has since been successfully evaluated for human use. The ligand was subsequently used to determine the 5-HT6 receptor occupancy following multiple dose ranges of Compound I. In the assessment for receptor occupancy, human subjects were administered the compound for at least three days at several dosage regimens.
The inventors discovered that high receptor occupancies were observed after multiple dosages of Compound I and that receptor occupancy was maintained 24 hrs post dose. Data generated from a separate Phase I P study in the elderly and data generated from the above AD study have shown that the elimination half life of Compound I in the elderly population was longer (about 19 hours) compared to young healthy subjects (about 12 hours).
With these convergent discoveries, the inventors have identified improved methods of treating AD by introducing a new and improved dosage regime comprising once daily administration in a novel dosage range. Based on the findings described herein, the dose range contemplated is expected to be efficacious while providing exposure levels below the NOAEL, thus improving the safety ratio.
………………………
WO 2009073118
http://www.google.st/patents/WO2009073118A1?cl=en
General Synthetic Scheme

MeOH (-H2O); NaBH4 BoC2O



…………………………
WO 2002078693
http://www.google.com/patents/WO2002078693A2?cl=en
Example 402 N-(2-(6-Fluoro-lH-indol-3-yl)ethyl)-3-(2.2,3,3-tetrafluoropropoxy)benzylamine

Combine 6-fluorotryptamine hydrochloride (90 g, 0.419 mol) and water (900 ml). Add an aqueous solution of NaOH (2N, 230 ml) and dichloromethane (900 ml). After 1 hour, separate the organic layer, extract the aqueous layer with dichloromethane, combine the organic layers, wash water, dry over MgSO , and evaporate to a residue. Combine the residue and toluene (200 ml) and evaporate to give 78.45 g of a brown oil. Combine the above 78.45 g with another 41.4 g batch to provide 6-fluorotryptamine. Combine 6- fluorotryptamine (119.85) and ethanol (3.325 L), add 2,2,3,3- tetrafluoropropylbenzaldehyde (176 g, 0.745 moles, 1.2 equiv.) and 150 g of molecular sieve 3A. Heat to reflux. After 2 hours, cool to RT room temperature and add NaBH4 (35.2 g, 0.93 mol, 1.5 equiv.). After 1 hour, filter through celite and wash with 500 ml of ethanol. Evaporate the filtrate under reduced pressure to give an oily residue. Partition the residue between water and dichloromethane. Separate the layers, extract the aqueous later with dichloromethane, combine organic layers, wash with brine and dry over MgSO4. Filter and evaporate under reduced pressure to give the title compound. The HCI salt is formed as follows: Combine N-(2-(6-fluror-lH-indol-3-yl)ethyl)-
3-(2,2,3,3-tetrafluoropropyl)benzylamine (387 g, 0.97 moles) and diethyl ether (3.95 L) of at room temperature. Add dropwise a solution of HCl/Et2O (298 ml) over 15 minutes until the pH is about 3 to give a solid. Stir for 1 hour and collect the solid, wash with ether, and dry under reduced pressure for at 40°C to give the title compound as the hydrochloride.
……………………..
http://www.google.com/patents/US20130210829
Compound 6, also known as 2-(6-fluoro-1H-indol-3-yl)-N-(3-(2,2,3,3-tetrafluoropropoxy)benzyl)ethanamine,


Combine 6-fluorotryptamine hydrochloride (90 g, 0.419 mol) and water (900 ml). Add an aqueous solution of NaOH (2N, 230 ml) and dichloromethane (900 ml). After 1 hour, separate the organic layer, extract the aqueous layer with dichloromethane, combine the organic layers, wash water, dry over MgSO4, and evaporate to a residue. Combine the residue and toluene (200 ml) and evaporate to give 78.45 g of a brown oil. Combine the above 78.45 g with another 41.4 g hatch to provide 6-fluorotryptamine. Combine 6-fluorotryptamine (119.85) and ethanol (3.325 L), add 2,2,3,3-tetrafluoropropoxybenzaldehyde (176 g, 0.745 moles, 1.2 equiv.) and 150 g of molecular sieve 3A. Heat to reflux. After 2 hours, cool to RT room temperature and acid NaBH4 (35.2 g, 0.93 mol, 1.5 equiv.). After 1 hour, filter through celite and wash with 500 nil of ethanol. Evaporate the filtrate under reduced pressure to give an oily residue. Partition the residue between water and dichloromethane. Separate the layers, extract the aqueous later with dichloromethane, combine organic layers, wash with brine and dry over MgSO4. Filter and evaporate under reduced pressure to give the title compound.
The HCl salt is formed as follows: Combine N-(2-(6-fluoror-1H-indol-3-yl)ethyl)-3-(2,2,3,3-tetrafluoropropyl)benzylamine (387 g, 0.97 moles) and diethyl ether (3.95 L) of at room temperature. Add dropwise a solution of HCl/Et2O (298 ml) over 15 minutes until the pH is about 3 to give a solid. Stir for 1 hour and collect the solid, wash with ether, and dry under reduced pressure for at 40° C. to give the title compound as the hydrochloride.
- “U.S. Development Programs. – Lundbeck”.
- “Search of: Lu AE58054 – List Results – ClinicalTrials.gov”.
- WO 2002078693
- WO 2009073118
- Novel and Potent 5-Piperazinyl Methyl-N1-aryl Sulfonyl Indole Derivatives as 5-HT6 Receptor Ligands
ACS Medicinal Chemistry Letters (2010), 1(7), 340-344, http://pubs.acs.org/doi/full/10.1021/ml100101u - WO 2012002583
- WO 2013055386
- US 20130210829
- WO 2014037532
- Lundbeck expands its pipeline – initiating phase II clinical trials with a new compound for the treatment of schizophrenia
- Lundbeck initiates clinical phase II trials with Lu AE58054 as augmentation treatment in Alzheimer’s disease
References on Lu AE58054 Hydrochloride:
NDA PA32540, Pozen Submits NDA for Aspirin Alternatives
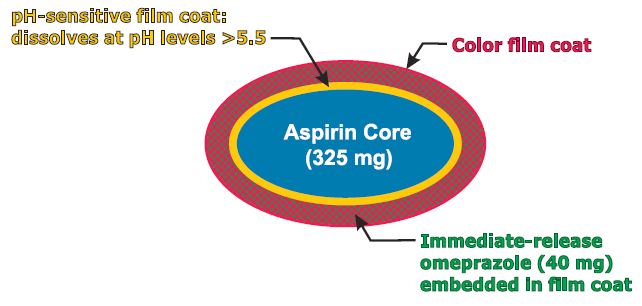
PA32540
PA-325/40 (EC-ASA 325 mg + IR omeprazole 40 mg)
enteric-coated aspirin (EC-ASA) 325 mg
MAR 27,2013
Pozen Inc. announced the submission of a new drug application to the U.S. Food and Drug Administration for the marketing approval of two potential cardiovascular drugs.
The move is a major step toward commercializing the drugs and means that the company thinks it has proven that the drugs are safe and effective.
Pozen’s PA32540 and PA8140 are both intended as alternatives to plain aspirin for the prevention of cardiovascular disease. Many people take aspirin to prevent heart problems, but long-term use of aspirin can cause ulcers.
Pozen’s drugs contain aspirin and the omeprazole, the active ingredient in heartburn drugs like Prilosec. The omeprazole is released as soon as the drug is taken and the aspirin is released over time.
Pozen is seeking approval for use in patients at risk of aspirin-induced ulcers.
Pozen CEO John Plachetka called the move “an important milestone for the drug” and said the company looks forward to completing a commercial deal with a partner “in the upcoming months.”
Pozen’s PA-325/40 is a combination pill containing enteric-coated aspirin 325 mg surrounded by a pH-sensitive film surrounded by an immediate-release omeprazole 40 mg.
SPOTLIGHT-Ganetespib Shows Potency Against Lung Cancer
Ganetespib
http://www.ama-assn.org/resources/doc/usan/ganetespib.pdf
A drug that indirectly impairs the function of several cancer-driving proteins, including anaplastic lymphoma kinase, may be an effective new treatment for patients with anaplastic lymphoma kinase-positive non-small cell lung cancer.
The drug, ganetespib, may also be effective for treating patients who have become resistant to the only FDA-approved targeted therapy for this disease, crizotinib, according to data published in Cancer Discovery, a journal of the American Association for Cancer Research.
“Lung cancer, a leading cause of death, is no longer thought of as a single disease, but rather as a group of diseases, each with a distinct genetic profile,” according to David Proia, Ph.D., associate director of cancer biology at Synta Pharmaceuticals Corporation, the company that funded the research. “This realization has paved the way for the design of new treatments tailored to the specific biological characteristics of a patient’s tumor.
“For example, patients with lung cancer caused by alterations in the anaplastic lymphoma kinase (ALK) protein typically respond well to crizotinib, which blocks that activity of the modified ALK and consequently kills off the cancer cells,” said Proia. “However, as is the case for many cancer drugs, most patients treated with crizotinib eventually become resistant to the drug.”
Proia and colleagues investigated ganetespib as an alternative treatment for ALK-positive non-small cell lung cancer (NSCLC). Ganetespib targets heat shock protein 90 (Hsp90), a chaperone for many different proteins, including ALK, to ensure proper functioning. When Hsp90 is blocked, ALK can no longer work properly and is destroyed by the cell. Once ALK is lost, the cancer cells die and the tumors shrink.
Ganetespib had 30 times greater potency than crizotinib against a cultured ALK-positive NSCLC cell line, resulting in the complete loss of ALK protein expression. In addition, the drug was active against ALK-positive lung cancer cell lines that had become resistant to the effects of crizotinib.
The researchers then compared ganetespib and crizotinib in mice xenografted with human ALK-positive NSCLC cancer cells. Ganetespib displayed greater antitumor activity and prolonged animal survival as compared to crizotinib. It was also shown that ganetespib had meaningful activity in a patient with ALK-driven NSCLC who had responded to, and then progressed, following crizotinib therapy.
“Ganetespib therapy represents a new option for treating ALK-dependent lung cancer in sequence with direct ALK inhibitors and/or for treating patients who relapse following direct ALK inhibitor therapy,” said Proia.
The U.S. Food and Drug Administration today approved Tecfidera (dimethyl fumarate) capsules to treat adults with relapsing forms of multiple sclerosis (MS).

Dimethyl fumarate
March 27, 2013
The U.S. Food and Drug Administration today approved Tecfidera (dimethyl fumarate) capsules to treat adults with relapsing forms of multiple sclerosis (MS).
MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function (relapses) are initially followed by recovery periods (remissions). Over time, recovery periods may be incomplete, leading to progressive decline in function and increased disability. MS patients often experience muscle weakness and difficulty with coordination and balance. Most people experience their first symptoms of MS between the ages of 20 and 40.
Dimethyl fumarate (DMF) is the methyl ester of fumaric acid. DMF was initially recognized as a very effective hypoxic cell radiosensitizer. Later, DMF combined with three other fumaric acid esters (FAE) was licensed in Germany as oral therapy for psoriasis (Fumaderm). Other diseases, such as necrobiosis lipoidica, granuloma annulare, and sarcoidosis were also found to respond to treatment with DMF in case reports or small patient series. Recently, phase III clinical trials found that DMF (BG-12) successfully reduced relapse rate and time to progression of disability in multiple sclerosis. DMF is thought to have immunomodulatory properties without significant immunosuppression.
In a non-medical use, DMF was applied as a biocide in furniture or shoes to prevent growths of mold during storage or transport in a humid climate. However, due to incidences of allergic reactions after skin contact the European Union banned DMF in consumer products since 1998, and since January 2009 the import of products containing DMF was also banned.
MHLW Japan approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome

Rufinamide
mar26, 2013
The agency approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome
Rufinamide is an anticonvulsant medication. It is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders. Rufinamide, a triazole derivative, was developed in 2004 by Novartis Pharma, AG, and is manufactured by Eisai.
Rufinamide was approved by the US Food and Drug Administration on November 14, 2008 as adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in children 4 years and older and adults. Its official FDA-approved labeling does not mention use in the treatment of partial seizures inasmuch as clinical trials submitted to the FDA were marginal. However, several recent clinical trials suggest that the drug has efficacy for partial seizures It is marketed under the brand name Banzel. It is also marketed in the European Union under the brand name Inovelon.
The mechanism of action of rufinamide is unknown. However, it is presumed to involve stabilization of the sodium channel inactive state, effectively keeping these ion channels closed. Although the direct mechanism of action may be different, several other antiepileptic agents also stabilize a sodium channel inactive state including phenytoin, carbamazepine, and lacosamide (stabilizes the slow inactive state).
MHLW Japan, GlaxoSmithKline and Genmab announced the approval of Arzerra (ofatumumab) by the MHLW for use in patients with relapsed/refractory CD20-positive chronic lymphocytic leukaemia.
mar 26, 2013
GlaxoSmithKline and Genmab announced the approval of Arzerra (ofatumumab) by the MHLW for use in patients with relapsed/refractory CD20-positive chronic lymphocytic leukaemia.
The thumbs-up triggers a milestone payment of 20 million Danish kroner to Genmab.
Ofatumumab(trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.
MHLW JAPAN has approved Nippon Shinyaku Co’s Regtect (acamprosate) for support maintenance of abstinence in patients with alcohol dependence

ACAMPROSATE
MAR 26 2013
MHLW JAPAN has approved Nippon Shinyaku Co’s Regtect (acamprosate) for support maintenance of abstinence in patients with alcohol dependence.
Acamprosate, also known as N-acetyl homotaurine and by the brand name Campral, is a drug used for treating alcohol dependence.
Acamprosate is thought to stabilize the chemical balance in the brain that would otherwise be disrupted by alcoholism, possibly by antagonizing glutamatergic N-methyl-D-aspartate receptors and agonizing gamma-aminobutyric acid (GABA) type A receptors. Reports indicate that acamprosate only works with a combination of attending support groups and abstinence from alcohol.Certain serious side effects include diarrhea, allergic reactions, irregular heartbeats, and low or high blood pressure, while less serious side effects include headaches, insomnia, and impotence. Acamprosate should not be taken by people with kidney problems or allergies to the drug.
Campral is manufactured and marketed in the United States by Forest Laboratories, while Merck KGaA markets it outside the US. It is sold as 333 mg white and odorless tablets of acamprosate calcium, which is the equivalent of 300 mg of acamprosate.
