
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 418)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bausch + Lomb Receives FDA Approval for Prolensa, Bromfenac

bromfenac
april 2013
Bausch + Lomb, the global eye health company, today announced that the U.S. Food and Drug Administration (FDA) has approved the company’s New Drug Application (NDA) for Prolensa (bromfenac ophthalmic solution) 0.07 percent prescription eye drop, an innovative once-daily nonsteroidal anti-inflammatory drug (NSAID) for the treatment of postoperative inflammation and reduction of ocular pain in patients who have undergone cataract surgery. Prolensa will be available in 1.6ml and 3ml bottle sizes.
Prolensa provides powerful and rapid resolution of inflammation and pain by leveraging the unique potency of the bromfenac molecule in a formulation designed to facilitate ocular penetration. The advanced formulation allows for a lower concentration of bromfenac in a once daily dosing regimen. Prolensa is a solution that does not require shaking to deliver a consistent dose in each drop.
“The data show that once-daily dosing with Prolensa provides powerful and rapid control of inflammation and pain following cataract surgery, confirming the potency of this NSAID and the benefits of the new formulation,” said Steven M. Silverstein, M.D., FACS, founder of Silverstein Eye Centers in Kansas City, MO. “Prolensa reduces the amount of medication placed on the healing eye while maintaining a high degree of efficacy and ocular comfort.”
The efficacy of Prolensa was evaluated in two randomized, double-masked, vehicle-controlled studies of patients undergoing cataract surgery. Each randomized patient received Prolensa or vehicle starting with one drop into the surgical eye on the day prior to and the day of surgery, and for 14 days following surgery. The primary efficacy endpoint was complete clearing of ocular inflammation (assessed by the summed ocular inflammation score, SOIS, which includes cells and flare) by day 15. The secondary efficacy endpoint was the number of subjects that were pain free on day one after surgery.
About Bausch + Lomb
Bausch + Lomb is a leading global eye health company that is solely focused on protecting, enhancing, and restoring people’s eyesight. Our core businesses include ophthalmic pharmaceuticals, contact lenses and lens care products, and ophthalmic surgical devices and instruments. We globally develop, manufacture and market one of the most comprehensive product portfolios in our industry, which are available in more than 100 countries. Founded in 1853, our company is headquartered in Rochester, NY, and employs more than 11,000 people worldwide.
Prolensa™ is a trademark of Bausch & Lomb Incorporated or its affiliates.
Bromfenac is a non-steroidal anti-inflammatory drug (NSAID) marketed in the US as an ophthalmic solution (current brand name Bromday, prior formulation brand name Xibrom, which has since been discontinued.) by ISTA Pharmaceuticals for short-term, local use. Bromday® is the once-daily formulation of bromfenac, while Xibrom®, which has since been discontinued, was the twice-daily formulation. Bromfenac is indicated for the treatment of ocular inflammation and pain after cataract surgery, though it may be prescribed in an off-label manner by the physician.
For ophthalmic use, bromfenac has been prescribed more than 20,000,000 times across the world. As an eye drop, it has been available since 2000, starting in Japan where it was sold as Bronuck®. It was first FDA approved for use in the United States in 2005 and it was marketed as Xibrom®, twice-daily. October 2010 was the FDA approval of the new once-daily formulation of bromfenac called Bromday®. The bromfenac molecule will be marketed in Europe and other worldwide markets with agreements from Bausch & Lomb, Croma Pharma, and other companies.
Bromfenac was formerly marketed in the United States by Wyeth-Ayerst in an oral formulation called Duract® for short-term relief of pain (less than 10 days at a time). It was brought to market in July, 1997, and was withdrawn June 22, 1998 following numerous reports of hepatotoxicity in patients who had taken the medication for longer than the recommended 10-day period. The dose was one 25 mg capsule every 6 to 8 hours, or two capsules if taken with a high-fat meal, up to a maximum of 150 mg per day.

GSK licenses out Tykerb in China, signs Singapore pact with A*Star

LAPATINIB
Lapatinib (INN), used in the form of lapatinib ditosylate, (USAN) (Tykerb/Tyverb, GSK) is an orally active drug for breast cancer and other solid tumours.It is a dual tyrosine kinase inhibitor which interrupts the HER2/neu and epidermal growth factor receptor (EGFR) pathways. It is used in combination therapy for HER2-positive breast cancer. It is used for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress HER2 (ErbB2)
Expanding its Asia focus, GlaxoSmithKline has signed up Hong Kong-based Eddingpharm to sell its breast cancer drug Tykerb in China, having just entered into an alliance with Singapore’s A*Star to develop new medicines for emerging markets.
First up, Eddingpharm is to acquire exclusive rights in mainland China to import, market and promote Tykerb (lapatinib), which has recently been given the green light by the country’s state Food and Drug Administration. Specifically the drug is approved for use in combination with Roche’s Xeloda (capecitabine) for the treatment of patients with metastatic breast cancer whose tumours overexpress HER2 and who have received prior therapy with an anthracycline, a taxane and another Roche drug, Herceptin (trastuzumab).
Xin Ni, chief executive at Eddingpharm, said the deal, the financials for which have not been disclosed, is an important milestone, not least because “this is the first time a Chinese pharmaceutical company will participate in the launch of a proprietary global oncology drug”. He added that the partnership with GSK “helps us fully leverage our rich market experience and mature marketing platforms to open a fast track for Tykerb’s China launch”.
Tykerb has been approved in the aforementioned indication in more than 100 countries and Eddingpharm noted that in China there are 170,000 new cases of breast cancer diagnosed each year.
The deal came a day after GSK linked up with the Institute of Chemical and Engineering Sciences (ICES), owned by the Agency for Science, Technology and Research (A*Star) in Singapore.
A five-year strategic agreement has been signed to develop new evidence-based formulations (EBFs) specifically for emerging markets. The latter are medicines which are reformulated to provide additional patient benefit.
Keith Carpenter, executive director at ICES, which has been working with GSK since 2003, said that this latest deal “provides an opportunity for us to further our research and deepen our capabilities in formulation science with skilled scientists and technical expertise”. He added that the venture “will enable us to develop future scientists and laboratory analysts with the right skills to grow this industry in Singapore”.
Duncan McKay, head of emerging markets and Asia Pacific R&D at GSK, said that within the firm’s portfolio of off-patent products, “EBFs are an important part of our growth strategy”. He went on to say that “our hope is that together with ICES, we will create a sustainable, scalable model to meet both specific market conditions and patient requirements”.
Tapping into Singapore scientific talent
Commenting on the agreement, Kevin Lai, director of biomedical sciences at the Singapore Economic Development Board, said the deal is a strong endorsement of the country’s “scientific talent and capabilities”. He added that it also “further reinforces Singapore’s offering as the base for companies to generate insights and develop new solutions and market access strategies for the fast-growing emerging markets”.
GSK is a major player in Singapore, which is home to the drug giant’s headquarters for the emerging markets and Asia Pacific. It also has an R&D facility in Biopolis, two global manufacturing and supply sites (Jurong and Quality Road), a manufacturing facility for its Stiefel unit and a state-of-the-art vaccines plant in Tuas.
A*Star oversees 14 biomedical sciences and physical sciences and engineering research institutes, plus six consortia and centres and says it supports Singapore’s “key economic clusters by providing intellectual, human and industrial capital to its partners in industry”.
Links
AYURVEDA- BITTER MELON (Momordica charantia)

BITTER MELON (Momordica charantia): This edible gourd should be every physician’s “go-to” plant for the 16 million or more Americans with high-normal glucose readings or ‘boderline diabetic/metabolic syndrome patients.
Preliminary evidence suggests bitter melon’s hypoglycemic action can be explained through several independent mechanisms: for one, it has been shown to increase peripheral glucose oxidation as well as glucose tolerance and insulin signaling in induced insulin resistance models (Sridhar MG, et al: Br J Nutr. 2008;99(4):806-12. Basch E, et al. Am J Health Syst Pharm. 2003;60:356-9). It also decreases hepatic gluconeogenesis, while increasing glycogen synthesis.
Bitter Melon increases insulin output from the pancreas, and it also provides a unique compound called polypeptide-P, which is an insulin mimetic with a similar structure to bovine insulin (Krawinkel MB, Keding GB. Nutr Rev. 2006;64(7 Pt 1):331-7).
 Bitter Melon slices.
Bitter Melon slices.Compounds produced by this intriguing gourd have been shown to reduce triglyceride levels in a dose-dependent manner in animal trials (Jayasooriya AP, et al. J Ethnopharmacol. 2000;72:331-6). Though we don’t yet have human data corroborating this effect, the animal studies suggest that bitter melon may have a role in reducing cardiovascular risk, particularly in people with diabetes or metabolic syndrome.
Bitter melon products are typically standardized to their constituents, momordicosides and charantin, and usually dispensed in 500-600 mg doses, twice daily, following meals. As it does have an insulin mimetic action, it may be necessary to adjust the dose of concurrently prescribed hypoglycemic drugs.
DRUG SPOTLIGHT-Ambrisentan

Ambrisentan
(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy- 3,3-diphenylpropanoic acid
177036-94-1 cas no
Ambrisentan (U.S. trade name Letairis; E.U. trade name Volibris; India trade namepulmonext by MSN labs ) is a drug indicated for use in the treatment of pulmonary hypertension.
It functions as an endothelin receptor antagonist, and is selective for the type A endothelin receptor (ETA).[1] Once daily oral ambrisentan 2.5 to 10 mg/day significantly improved exercise capacity (6-minute walk distance) compared with placebo in two double-blind, multicenter trials (ARIES-1 & ARIES-2).[2]
Ambrisentan was approved for sale by the U.S. Food and Drug Administration (FDA) on June 15, 2007 for the once-daily treatment of pulmonary arterial hypertension.[3][4][5] It was later approved by the European Medicines Agency for use in the EU on April 2008.[6]Ambrisentan had previously been designated an orphan drug by both the FDA and the European Commission, in August 2004 and May 2005 respectively.[7]
Ambrisentan is indicated for the treatment of pulmonary arterial hypertension (WHO Group 1) in patients with WHO class II or III symptoms to improve exercise capacity and delay clinical worsening.
The LETAIRIS Education and Access Program (LEAP) is a program to help physicians and patients learn about the risks of LETAIRIS, including the serious risks of liver injury and birth defects.
LEAP works by:
- Providing information to prescribers on the risks of LETAIRIS
- Providing comprehensive education to patients and assistance with obtaining LETAIRIS
- Requiring enrollment of both prescriber and patient in LEAP
- Controlling dispensing through a specialized distribution network (specialty pharmacies)
- Letairis website run by Gilead Sciences
- Prescribing information
- Information on the LETAIRIS Education and Access Program (LEAP)
- Vatter H, Seifert V (2006). “Ambrisentan, a non-peptide endothelin receptor antagonist”. Cardiovasc Drug Rev 24 (1): 63–76.doi:10.1111/j.1527-3466.2006.00063.x. PMID 16939634.
- Frampton JE. Ambrisentan. American Journal of Cardiovascular Drugs August 1, 2011; 11 (4): 215-226.Link text
- Pollack, Andrew (2007-06-16). “Gilead’s Drug Is Approved to Treat a Rare Disease”. New York Times. Archived from the original on 20 June 2007. Retrieved 2007-05-25.
- “U.S. Food and Drug Administration Approves Gilead’s Letairis Treatment of Pulmonary Arterial Hypertension” (Press release).Gilead Sciences. 2007-06-15. Retrieved 2007-06-16.
- “FDA Approves New Orphan Drug for Treatment of Pulmonary Arterial Hypertension” (Press release). Food and Drug Administration. 2007-06-15. Archived from the original on 23 June 2007. Retrieved 2007-06-22.
- “GlaxoSmithKline’s Volibris (ambrisentan) receives authorisation from the European Commission for the treatment of Functional Class II and III Pulmonary Arterial Hypertension” (Press release). GlaxoSmithKline. 2008-04-25. Archived from the original on 30 April 2008. Retrieved 2008-04-29.
- Waknine, Yael (2005-05-09). “International Approvals: Ambrisentan, Oral-lyn, Risperdal”. Medscape. Retrieved 2007-06-16.


Patent EP2547663A1


READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
DRUG SPOTLIGHT–Treprostinil

Treprostinil (marketed under the trade names Remodulin for infusion and Tyvaso for inhalation) is a synthetic analog of prostacyclin (PGI2).
Treprostinil sodium, Uniprost, LRX-15, U-62840, UT-15, BW-15AU, 15AU81, Remodulin
289480-64-4, 81846-19-7 (free acid

During the 1960s a U.K. research team, headed by Professor John Vane began to explore the role of prostaglandins in anaphylaxis and respiratory diseases. Working with a team from the Royal College of Surgeons, Vane discovered that aspirin and other oral anti-inflammatory drugs worked by inhibiting the synthesis of prostaglandins. This finding opened the door to a broader understanding of the role of prostaglandins in the body.

Vane and a team from the Wellcome Foundation had identified a lipid mediator they called “PG-X,” which inhibited platelet aggregation. PG-X, which later would become known as prostacyclin, was 30 times more potent than any other known anti-aggregatory agent.
By 1976, Vane and fellow researcher Salvador Moncada published the first paper on prostacyclin, in the scientific journal Nature. The collaboration produced a synthetic molecule which was given the name epoprostenol. But like native prostacyclin, the structure of the epoprostenol molecule proved to be unstable in solution, prone to rapid degradation. This presented a challenge for both in vitro experiments and clinical applications. To overcome this challenge, the research team that discovered prostacyclin was determined to continue the research in an attempt to build upon the success they had seen with the prototype molecule. The research team synthesized nearly 1,000 analogs.
Treprostinil has demonstrated a unique effect on PPAR gamma, a transcription factor important in vascular pathogenesis as a mediator of proliferation, inflammation and apoptosis. Through a complementary, yet cyclic AMP-independent pathway, treprostinil activates PPARs, another mechanism that contributes to the anti-growth benefits of the prostacyclin class.
Treprostinil is indicated for the treatment of pulmonary arterial hypertension in patients with NYHA Class II-IV symptoms to diminish symptoms associated with exercise.[1] It may be administered as a continuous subcutaneous infusion or continuous intravenous infusion; however, because of the risks associated with chronic indwelling central venous catheters, including serious blood stream infections, continuous intravenous infusion should be reserved for patients who are intolerant of the subcutaneous route, or in whom these risks are considered warranted.
In patients with pulmonary arterial hypertension requiring transition from epoprostenol sodium (Flolan), treprostinil is indicated to diminish the rate of clinical deterioration. The risks and benefits of each drug should be carefully considered prior to transition.
The pharmacokinetics of continuous subcutaneous treprostinil are linear over the dose range of 1.25 to 125 ng/kg/min (corresponding to plasma concentrations of about 15 pg/mL to 18,250 pg/m) and can be described by a two-compartment model. Dose proportionality at infusion rates greater than 125 ng/kg/min has not been studied.
The major effects of treprostinil are vasodilation of arteries in the pulmonary (lung) and body. Treprostinil also inhibits platelet aggregation.
Treprostinil may be administered as a continuous subcutaneous infusion or continuous intravenous infusion via a small infusion pumpthat the patient must wear at all times. Treprostinil can be given subcutaneously by continuous infusion using an infusion set connected to an infusion pump, but also may be given intravenously via a central venous catheter if the patient is unable to tolerate subcutaneous administration because of severe site pain or reaction.
1. Remodulin Full Prescribing Information US Patent No. 5,153,222
2. UT_OpinVEvid_FEB09v.1
3. Cost-minimization analysis of treprostinil vs. epoprostenol as an alternate to oral therapy nonresponders for the treatment of pulmonary arterial hypertension L. Narine, L. K. Hague, J. H. Walker, C. Vicente, R. Schilz, O. Desjardins, T. R. Einarson and M. Iskedjian

…..
(+)-Treprostinil (also known as UT-15) is the active ingredient in Remodulin®, a commercial drug approved by FDA for the treatment of pulmonary arterial hypertension (PAH). It was first described in U.S. Pat. No. 4,306,075. Treprostinil is a stable analog of prostacyclin (PGI2) belonging to a class of compounds known as benzindene prostacyclins, which are useful pharmaceutical compounds possessing activities such as platelet aggregation inhibition, gastric secretion reduction, lesion inhibition, and bronchodilation.

U.S. Pat. No. 5,153,222 describes use of treprostinil for treatment of pulmonary hypertension. Treprostinil is approved for the intravenous as well as subcutaneous route, the latter avoiding potential septic events associated with continuous intravenous catheters. U.S. Pat. Nos. 6,521,212 and 6,756,033 describe administration of treprostinil by inhalation for treatment of pulmonary hypertension, peripheral vascular disease and other diseases and conditions. U.S. Pat. No. 6,803,386 discloses administration of treprostinil for treating cancer such lung, liver, brain, pancreatic, kidney, prostate, breast, colon and head-neck cancer. U.S. patent application publication No. 2005/0165111 discloses treprostinil treatment of ischemic lesions. U.S. Pat. No. 7,199,157 discloses that treprostinil treatment improves kidney functions. U.S. Pat. No. 7,879,909 discloses treprostinil treatment of neuropathic foot ulcers. U.S. publication No. 2008/0280986 discloses treprostinil treatment of pulmonary fibrosis, interstitial lung disease with treprostinil and asthma. U.S. Pat. No. 6,054,486 discloses treatment of peripheral vascular disease with treprostinil. U.S. patent application publication No. 2009/0036465 discloses combination therapies comprising treprostinil. U.S. publication No. 2008/0200449 discloses delivery of treprostinil using a metered dose inhaler. U.S. Pat. Nos. 7,417,070, 7,384,978 and 7,544,713 as well as U.S. publications Nos. 2007/0078095, 2005/0282901, and 2008/0249167 describe oral formulations of treprostinil and other prostacyclin analogs as well as their use for treatment of a variety of conditions. U.S. provisional application No. 61/354,949 filed Jun. 15, 2010 discloses the use of orally administered treprostinil for treatment of Raynaud’s phenomenon, systemic sclerosis and digital ischemic lesions.
Treprostinil and other prostacyclin derivatives have been prepared as described in Moriarty, et al in J. Org. Chem. 2004, 69, 1890-1902, Drug of the Future, 2001, 26(4), 364-374, U.S. Pat. Nos. 4,306,075, 6,441,245, 6,528,688, 6,700,025, 6,765,117, 6,809,223 and US Publication No. 2009/0163738. The entire teaching of these documents are incorporated herein by reference in their entirety. The methods described in these patent documents, however, do not describe a feasible production method for producing stereochemically pure treprostinil because, for example, the methods require the use of expensive reagents and tedious chromatographic purification techniques. Therefore, there is a need in the art for an economical, efficient and simplified method for preparing treprostinil and its synthetic intermediates.

NMR
The 1HNMR and HPLC of the samples were compared with reference UT-15 and were identical; 1H NMR (CDCl3, 300 MHz) δ 0.90 (t, 3H, 6 Hz), 1.05-1.78 (m, 13H), 2.85-2.85-2.98 (m, 1H), 2.03 2.12 (m, 1H), 2.21-2.32 (m, 1H), 2.45-2.53 (m, 1H), 2.61-2.81 (m, 3H), 3.52 (br s, 1H), 3.58-3.69 (m, 1H), 4.62 (s, 2H), 6.69 (d, 1H, J=8 Hz), 6.78 (d, 1H, J=8 Hz), 7.04 (dd, 1H, J=8 Hz).
J. Org. Chem. 2004, 69, 1890-1902
mp 126−127 °C;
[α]25D +52.6 (c 0.453, MeOH), [α]25D + 34.0° (c 0.457, EtOH).
IR 3385, 2928, 2856, 1739, 1713, 1585, and 779 cm–1;
1H NMR (CDCl3, 300 MHz) δ 0.87 (t, 3 H, J = 6 Hz), 1.21−1.86 (m, 13H), 2.02−2.44 (m, 4H), 3.42−3.76 (m, 3H), 3.81 (s, 2H), 3.82−3.94 (m, 1H), 4.63−4.68 (m, 1H), 4.88−4.92 (m, 1H), 4.94−4.98 (m, 1H), 4.99−5.02 (m, 1H), 5.60 (s, 1H), 5.92−6.06 (m, 1H), 6.85 (d, 1H, J = 6 Hz), 7.20−7.27 (m, 1H), 7.31−7.37 (m, 1H);
13C NMR (MeOH, 75 MHz) δ 13.1, 22.4, 25.1, 25.3, 28.3, 31.8, 32.7, 33.2, 34.7, 36.9, 40.7, 41.0, 51.3, 65.2, 71.6, 76.3, 109.5, 121.1, 125.8, 127.4, 140.8, 155.2, 171.5; UV, λmax MeOH, 217 nm;
HPLC, Hypersil ODS column (4.6 × 250 mm2), 5 μm; flow rate 2.0 mL/min; mobile phase A, water (60%):acetonitrile (40%):trifluoroacetic acid (0.1%), and mobile phase B, water (22%):acetonitrile (78%):trifluoroacetic acid (0.1%); retention time, 15 min (purity 99.7%). Anal. Calcd for C23H34O5: C, 70.74; H, 8.78. Found: C, 70.41; H, 8.83.
Peramivir, a Flu Treatment

PERAMIVIR
CAS 330600-85-6
- Molecular FormulaC15H28N4O4
- Average mass328.407 Da
(1S,2S,3R,4R)-4-carbamimidamido-3-[(1S)-1-acetamido-2-ethylbutyl]-2-hydroxycyclopentane-1-carboxylic acid
- RWJ 270201
- RWJ-270201
- RWJ270201
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Peramivir hydrate | QW7Y7ZR15U | 1041434-82-5 | RFUCJKFZFXNIGB-ZBBHRWOZSA-N |
| Molecular Weight | 382.45 |
| Formula | C15H28N4O4 • 3H2O |
| CAS No. | 330600-85-6 (Peramivir); 1041434-82-5 (Peramivir Trihydrate); |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Rapivab | Solution | 600 mg/60mL | Intravenous | BioCryst Pharmaceuticals, Inc. | 2014-12-20 | Not applicable |  |
|
| Rapivab | Solution | 600 mg/60mL | Intravenous | Seqirus USA Inc. | 2014-12-20 | Not applicable | |
|
| Rapivab | Solution | 10 mg | Intravenous | Biocryst Pharmaceuticals Inc | Not applicable | Not applicable |  |
- Drug Name:
- Peramivir Hydrate
- Research Code:
- S-021812
- Trade Name:
- Rapiacta® / Peramiflu®
- MOA:
- Neuraminidase inhibitor
- Indication:
- Influenza infection
- Status:
- Approved
- Company:
- BioCryst (Originator) , Shionogi
- Sales:
- $21.6 Million (Y2015);

$23.7 Million (Y2014);
$20 Million (Y2013);
$24.1 Million (Y2012);
$17.7 Million (Y2011); - ATC Code:
Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-12-19 | Marketing approval | Rapivab | Influenza infection | Solution | 200 mg/20 ml | BioCryst | Standard |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-01-13 | Marketing approval | Rapiacta | Influenza infection | Injection, Solution | Eq. 150 mg/300 mg Peramivir | BioCryst, Shionogi |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-04-05 | Marketing approval | 力纬 | Influenza infection | Injection, Solution | 100ml | 广州南新制药 | 1.1 |
Shionogi , GC Pharma and Seqirus , under license from BioCryst Pharmaceuticals (which licensed the program from the University of Alabama ), have developed and launched an iv formulation of the influenza neuraminidase inhibitor peramivir.
Peramivir hydrate was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on January 13, 2010, and approved by the U.S. Food and Drug Administration (FDA) on December 19, 2014. It was co-developed and co-marketed as Rapiacta® by BioCryst and Shionogi in Japan.
Peramivir hydrate is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells. It is indicated for the treatment of influenza A and B viral infections.
Rapiacta® is available as injection solution for intravenous, containing 150 mg/300 mg Peramivir hydrate. The recommended dose is once a day by an intravenous drip infusion over a period of 15 minutes or longer.
Peramivir (trade name Rapivab) is an antiviral drug developed by BioCryst Pharmaceuticals for the treatment of influenza. Peramivir is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells. It is approved for intravenous administration.[1]
In October 2009, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for the use of peramivir based on safety data from phase I, phase II, and limited phase III trial data. The emergency use authorization for peramivir expired in June 2010.[2][3] On 19 December 2014, the FDA approved peramivir to treat influenza infection in adults.[1]
Peramivir has also been approved in Japan and South Korea and is available in Japan as Rapiacta and in South Korea as Peramiflu.[ As of 2015, it is the only intravenous option for treating swine flu.
Peramivir is an antiviral agent developed by Biocryst Pharmaceuticals to treat influenza A/B. The development of peramivir has been supported by the US Department of Health and Human Services as part of the government’s effort to prepare for a flu pandemic. Being an influenza virus neuraminidase inhibitor, peramivir works by preventing new viruses from emerging from infected cells. Due to the poor oral bioavailability, the oral formulation of the drug was previously abandoned by Johnson and Johnson Company. The injectable intravenous formulation of peramivir was approved by the FDA in September 2017 for the treatment of acute uncomplicated influenza to pediatric patients 2 years and older who have been symptomatic for no more than two days.
History
An intramuscular (IM) peramivir phase II study for seasonal influenza in 2008–2009 found no effect for the primary endpoint of improvement in the median time to alleviation of symptoms in subjects with confirmed, acute, uncomplicated influenza infection versus placebo.
In October 2009, it was reported that the experimental antiviral drug peramivir had been “life-saving” effective in intravenous treating 8 serious cases of swine flu.[4] On October 23, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization for peramivir, allowing the use of the drug in intravenous form for hospitalized patients only in cases where the other available methods of treatment are ineffective or unavailable;[5] for instance, if oseltamivir resistance develops and a person is unable to take zanamivir via the inhaled route. The U.S. government (department of Health and Human Services) gave BioCryst Pharmaceuticals more than $77 million to finish the Phase III clinical development of peramivir. In 2009 the department of Health and Human Services had already given about $180 million to the program.[6] Biocryst also donated 1200 courses of treatment to the US department of Health and Human Services.[7] The Emergency Use Authorization expired on June 23, 2010. In 2011 a phase III trial found the median durations of influenza symptoms were the same with 1 intravenous injection of peramivir against 5 days of oral oseltamivir for people with seasonal influenza virus infection.[8]
In 2012 BioCryst reported that it should halt enrollment on its study for intravenous peramivir in potentially life-threatened people after an interim analysis led trial monitors to conclude that it would be futile to continue and the trial should be terminated. The difference between peramivir and control group (oral oseltamivir) for the primary endpoint, clinical or virologic, was small.[9] In 2013 the Biomedical Advanced Research and Development Authority (BARDA/HHS) released new funding under the current $234.8 million contract to enable completion of a New Drug Application filing for intravenous (IV) peramivir.[10]
According to a research report published in June 2011, a new variant of swine flu had emerged in Asia with a genetic adaptation (a S247N neuraminidase mutation) giving some resistance to oseltamivir and zanamivir, but no significant reduction in sensitivity to peramivir.[11][12] But a H274Y virus mutation showed resistance to oseltamivir and peramivir, but not to zanamivir, and only in N1 neuraminidases.[13] Ultimately 3.2% (19/599) of A(H1N1)pdm09 viruses collected between 2009 and 2012 had highly reduced peramivir inhibition due to the H275Y NA mutation.[14]
BioCryst Pharmaceuticals submitted a new drug application (NDA) to the U.S. Food and Drug Administration (FDA) for intravenous peramivir in December 2013.[15] Peramivir (Rapivab) was approved for intravenous administration in December 2014.[1][16]

Reference:1. WO9933781A1 / US6562861B1.

Reference:1. CN102372657A.

Reference:1. CN102633686A.

Reference:1. J. Med. Chem. 2001, 44, 4379-4392.



PATENT
peramivir’s product case, WO9933781 ,
hold SPC protection in the EU states until December 2023, and contain one of the Orange Book (OB) listed patents, US6562861 , that was due to expire in the US in December 2023. In January 2021, the US FDA’s OB was seen to list patents describing peramivir that expire in 2027.
PATENT
WO-2021002689
Process for preparing an inhibitor of neuraminidase infection ie peramivir trihydrate crystal from the free form of the drug ie peramivir. Discloses the use of peramivir trihydrate in treating influenza infection. Represents the first PCT filing from CKD Bio Corp that focuses on peramivir; however, this case was first seen as a Korean national filing that is published in February 2020.
PATENT
https://patents.google.com/patent/WO2012145932A1/en
Peramivir has the chemical name of
(1^,2^,3^, 4R)-3-[(llS)-l-acetamido-2-ethyl-butyl] -4-(diaminomethylideneamino)- 2-hydroxy-cyclopentane-l-carboxylicacid, and has the following structure:

(I)
[0003] Peramivir is currently being developed as an antiviral drug, and in particular, for treatment of influenza. Acting as a neuraminidase inhibitor, peramivir can efficiently inhibit the replication of all type of influenza viruses. Peramivir can be administered via injection, and is known to be well-tolerated and cause only mild adverse effect.
[0004] Several processes relating to the preparation of peramivir are disclosed in CN1227466, CN1282316, and WO01/00577A1.
[0005] As shown in Scheme 1, CN 1227466 discloses a process comprising ring-opening of chiral 2-azabicyclo [2.2.1] hept-5-en-3-one, followed by
amino-protecting reaction, Diel- Alder conjugate cycloaddition, reduction, acetylation, guanidylation and finally hydrolyzation to yield peramivir. The major drawback of this process is the use of highly expensive starting material 1. In addition, this process is not suitable for scale-up.
Scheme 1

[0006] WO2009021404 discloses a method comprising reacting
N-Boc-protected chiral 2-azabicyclo [2.2.1] hept-5-en-3-one and
2-ethylbutylaldehyde as starting material to prepare peramivir as illustrated in Scheme 2.
Scheme 2

eram v r
EXAMPLES
Example 1
1 (liS’,4J/?)-methyl-4-(2,3-bis(ieri-butoxycarbonyl)guanidino)cyclopent-2-ene- carboxylate (13)

11 12 13 [0063] To a mixture of (lS^R^methyl 4-aminocyclopent-2-enecarboxylate tartaric acid salt 11 ( 7.29 g, 25 mmol) in dichloromethane (150 mL), was added Et3N (9 mL, 65 mmol) at 0 °C, and the resulting mixture was stirred for 15 min. To this, tert-butyl (lH-pyrazol-l-yl)methylenedicarbamate 12 was added. After addition, the reaction was monitored for completion by TLC ( PE: EtOAc=5 : 1 ) . The organic layers were washed with water and brine and dried over anhydrous Na2S04. The mixture was filtered and concentrated to give 13 as a white solid, which was used in the next step without further purification.
[0064] MS (M+l ) : 384.
[0065] ‘H NMR (400 MHz, CDC13) δ 11.49 (s, 1H), 8.53 (d, J= 8.4 Hz, 1H), 5.94 – 5.83 (m, 2H), 5.38 – 5.31 (m, 1H), 3.73 (s, 3H), 3.56 – 3.44 (m, 1H), 2.60 (dt, J= 14.0, 8.5 Hz, 1H), 1.94 (dt, J= 13.9, 4.7 Hz, 1H), 1.50 (d, J= 7.4 Hz, 18H) (See attached Chart 1)
Example 2
2 (3aJ/?,4J/?,6iS’,6aiS -methyl-4-(2,3-bis(ieri-butoxycarbonyl)guanidino)-3-(pentan -3-yl)-4,5,6,6a-tetrahydro-3aH-cyclopenta[d]isoxazole-6-carboxylate (5)

14 85% a) Preparation of 2-Ethyl-N-hydroxybutanimidoyl chloride (14):
[0066] Hydroxylamine hydrochloride (7.2g, 0.1 mol) was dissolved in water (7 mL). Toluene (27 mL) was added, followed by addition of 2-ethylbutylraldehyde (lOg, 0.1 mol). The two-phase mixture was stirred vigorously while cooling.
Sodium hydroxide solution (ca.30%, 14.6g, O. l lmol) was added slowly (addition is very exothermic) to maintain a temperature between 15-25 °C. The mixture was stirred for 60 min, then allowed to stand to separate the layers. The organic extract was washed with water and brine, dried over Na2S04, and directly used in the next step.
[0067] N-Chlorosuccinimide (NCS) (13.3g, 0.1 mol) was suspended in dimethylformamide (DMF) (17ml) and cooled to about 10 °C. The toluene solution prepared above (3 .15 mol) was added dropwise with sufficient cooling to maintain the reaction temperature betweenlO-25°C. After addition, the reaction was monitored by TLC until completion of the reaction. Water (100ml) was added slowly (slightly exothermic) while maintaining the temperature at 15-25 °C. The two-phase mixture was stirred for 15 min at 15-25 °C and the layers were separated. The water layer was extracted with toluene (10ml) and the organic layer washed with water (3 X 20ml) and brine, dried over Na2S04, and directly used in the next step. b)
Preparation of (3aJ/?,4J/?,6iS’,6aiS -methyl-4-(2,3-bis(ieri-butoxycarbonyl)-guan idine)-3 -(pentan-3 -yl)-4,5,6,6a-tetrahy dro-3 aH-cy clopenta [d] is oxazole-6- carboxylate (15)
[0068] 13 (from example 1, 9.2g, 0.024 mol) was dissolved in toluene (100 mL) and triethylamine (10. Og, 0.099 mol) and the reaction mixture was heated to 60-70 °C. 2- Ethyl-N-hydroxylbutanimidoyl chloride 14 (from example 2a, 14.8 g, 0.099 mol) in toluene (40 mL) was added to the above solution. A white solid
(triethylammonium chloride) was formed. After addition, the reaction was
monitored for completion by TLC ( PE: EtOAc=3 : 1. The reaction mixture was cooled to 20-25 °C, the precipitate was removed by filtration and the filter cake was washed with toluene (50 g). The organic filtrate was washed with water, brine, and dried over anhydrous Na2S04. The mixture was filtered and concentrated by rotary evaporation. The resulting residue was purified by silica gel flash column
chromatography using PE/EtOAc (30 : 1-4: 1, v/v) to give 15 as a white solid.
[0069] Yield : 10.0 g (85%).
[0070] MS (M+l ) :497.
[0071] ‘H NMR (400 MHz, CDC13) δ 11.29 (s, 1H), 8.55 (d, J= 6.4 Hz, 1H), 5.30 (dd, J= 9.1, 1.5 Hz, 1H), 4.53 (d, J= 4.8 Hz, 1H), 3.78 (s, 3H), 3.70 (d, J = 9.1 Hz, 1H), 3.25 (t, J= 5.4 Hz, 1H), 2.93 – 2.84 (m, 1H), 2.20 (dd, J= 7.6, 3.7 Hz, 2H), 1.87 – 1.60 (m, 4H), 1.49 (d, J= 5.0 Hz, 18H), 0.95 (t, J= 7.4 Hz, 3H), 0.87 (t, J= 7.5 Hz, 3H). (See attached Chart 2)
Example 3 3. (1^,2^,3^,4^)-ιη6ίΗγ1-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4-(2,3- Ϊ8(ί^ί- butoxycarb -onyl) guanidino)-2-hydroxycyclopentanecarboxylate (16):

[0072] Compound 15 ( from example 2, 5.0 g, 10.08 mmol) and nickel chloride hexahydrate (2.5g, 10.5 mmol) were dissolved in methanol (40 mL). The green solution was cooled to -15 °C, while a suspension formed. Sodium borohydride
( 0.456 g, 12 mmol) was added to the reaction mixture at -10 to -5 °C (reaction is highly exothermic). A black suspension was formed along with gas evolution.
After complete addition of the sodium borohydride solution, the reaction mixture was stirred until TLC showed 15 was fully consumed. A solution of acetic
anhydride ( 15g, 0.13 mol) was added slowly and maintained the reaction temperature at 0-5 °C, the reaction mixture was stirred for 2-12 h at 0 °C (The black solution change into green solution ), The pH of the mixture was adjusted to ~9 by addition of 25% aq. ammonium hydroxide. The mixture was concentrated by rotary evaporator. The resulting residue was diluted with water (30 mL) and extracted with EtOAc (50 mL><3). The combined organic extracts were washed with water and brine and dried over anhydrous Na2S04. The mixture was filtered and concentrated by rotary evaporation. The residue was purified by flash chromatography using DCM/Methanol (100:0 to 100:2, v/v) to give 16 as a white solid. [0073] Yield: 3.8 g (71%).
[0074] MS (M+l ) : 543.
[0075] ‘H NMR (400 MHz, CDC13) δ 11.39 (s, 1H), 8.72 (d, J= 9.9 Hz, 1H), 8.59 (d, J= 8.5 Hz, 1H), 4.53 – 4.39 (m, 1H), 4.26 (d, J= 16.4 Hz, 2H), 3.96 (t, J = 10.2 Hz, 1H), 3.71 (s, 3H), 2.90 – 2.75 (m, 1H), 2.53 (dt, J= 13.6, 8.8 Hz, 1H), 2.10 (s, 3H), 2.03 (d, J= 6.3 Hz, 1H), 1.90 – 1.76 (m, 1H), 1.38 (dd, J= 73.9, 7.9 Hz, 18H), 1.25 (ddd, J= 15.2, 13.1, 7.3 Hz, 4H), 0.79 (t, J= 7.3 Hz, 3H), 0.75 (dd, J = 14.1, 6.9 Hz, 3H). (See attached Chart 3)
Example 4
4. (1^,2^,3^,4^)-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4-(2,3- Ϊ8(ί^ί- butoxycarbonyl)gu -anidino)-2-hydroxycyclopentanecarboxylic acid (17)

[0076] To a mixture of compound 16 ( from example 3, 2.0 g, 3.69 mmol) in MeOH/THF (1 : 1, v/v, 12 mL), was added aq. NaOH (IN, 7 mL) at room
temperature. After completion of the reaction (monitored by TLC,
DCM:MeOH=10: l, the mixture was concentrated by rotary evaporation. The resulting solution was neutralized to pH 7 using ice-cold 1 N HCl aq. solution and quickly extracted with EtOAc twice. The combined organic extracts were washed with water, brine, and dried over anhydrous Na2S04. The mixture was filtered and the filtrate was concentrated by rotary evaporation. The resulting white foam was washed triturated by hexane, filtered, dried to give 17 as a white solid
[0077] Yield: 1.6 g (84%).
[0078] MS (M+l ) : 529 o
[0079] ‘H NMR (400 MHz, CDC13) δ 11.41 (s, 1H), 8.80 (d, J= 9.8 Hz, 1H), 8.62 (d, J= 8.3 Hz, 1H), 4.43 (dd, J= 23.3, 14.3 Hz, 2H), 4.00 (t, J= 9.8 Hz, 1H), 2.83 (s, 1H), 2.53 (dt, J= 16.9, 8.4 Hz, 1H), 2.14 (s, 3H), 1.91 (dd, J= 12.5, 6.0 Hz, 1H), 1.46 (dd, J = 30.1, 9.5 Hz, 18H), 1.47 – 1.14 (m, 6H), 0.97 – 0.69 (m, 6H). (See attached Chart 4)
Example 5
5. (1^,2^,3^,4^)-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4^υαηΐ(1ΐηο-2- hydroxycyclopent -anecarboxylic acid (Peramivir I)

[0080] Compound 17 ( from example 4, 1.1 g, 2 mmol) was dissolved in aq. HC1 ( 6N, 6 mL, 36 mmol) at 0 °C. The mixture was stirred at room temperature overnight. The resulting solution was neutralized to pH 6-7 using ice-cold 1 N NaOH aq. solution. The mixture was concentrated to 1.5 ml by rotary evaporation. To this, methanol (20 mL) was added. The precipitate was filtered, and the filtrate was concentrated. The resulting white solid was recrystallized from
methanol/water (1 : 1, v/v) to give Peramivir I as a white solid.
[0081] Yield: 500 mg (73%).
[0082] MS (M+l ) : 329.
[0083] H NMR (400 MHz, D20) δ 4.21 (d, J= 10.6 Hz, 2H), 3.70 (dd, J= 14.6, 9.0 Hz, 1H), 2.57 (d, J= 4.8 Hz, 1H), 2.40 (dt, J= 17.7, 8.9 Hz, 1H), 2.14 – 2.01 (m, 1H), 1.81 (s, 3H), 1.75 – 1.58 (m, 1H), 1.31 (s, 3H), 0.78 (ddd, J= 21.6, 18.6, 6.8 Hz, 8H). (See attached Chart 5)
SYN

Method of synthesis
i. Lactan is dissolved I alcoholic solution, esterification by ring opening under HCl and acid catalysis.
ii. Compound formed is reacted with hydrochloride and tert-butyl dicarbonate.
iii. Reaction with oxammonium hydrochloride reactant salt in alcohol-carbonate system.
iv. Cycloaddition reaction takes place under base catalysis.
v. Hydrogenation in alcoholic solution to generate intermediate compound.
vi. Taking off BOC protect hydrochloride
vii. Two guanidine radicals are put in the reaction vessel, system made of alcohol, organic solvent, aqueous sodium hydroxide solution.
viii. Taking off BOC, product is obtained under organic acid.
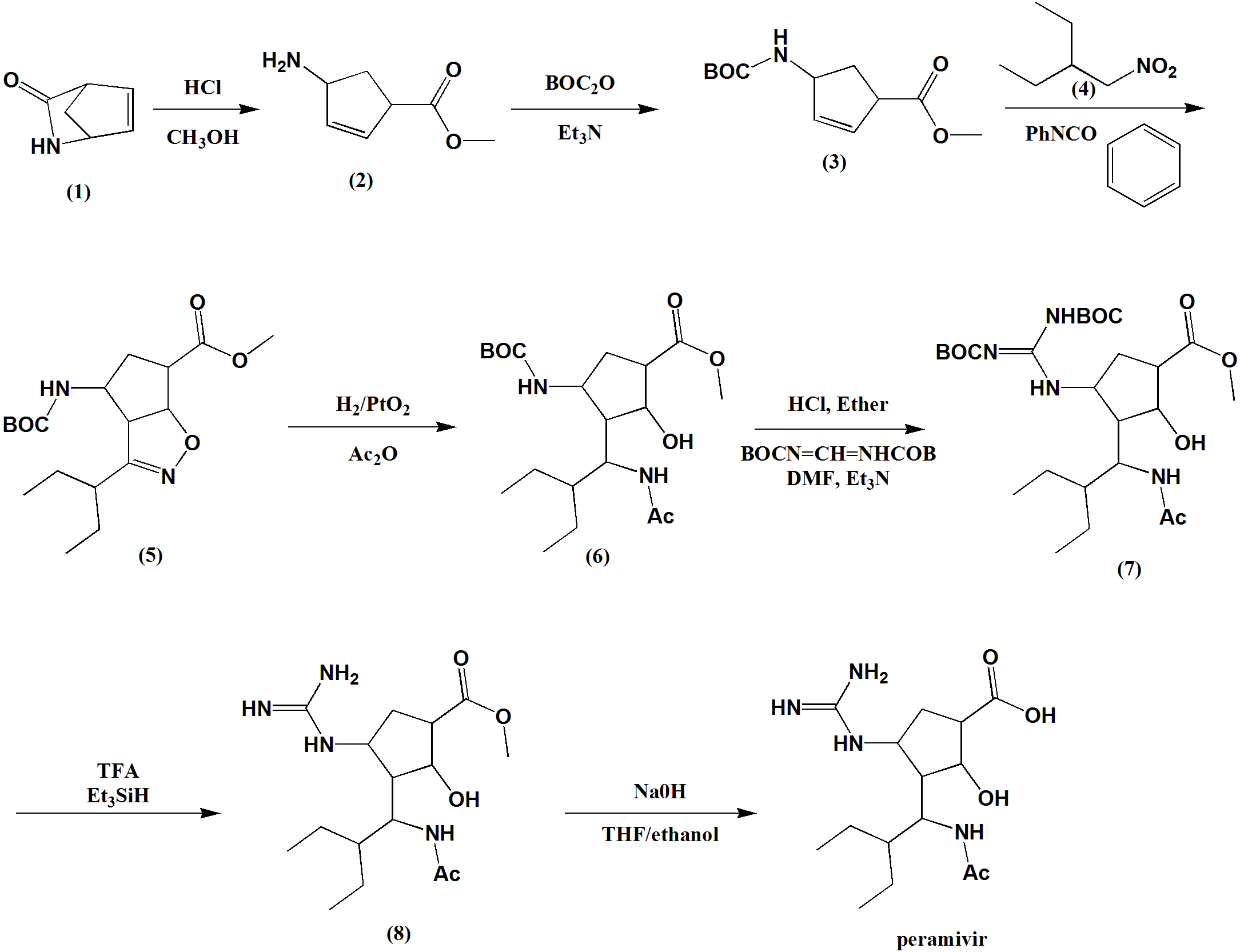
SYN
https://www.tandfonline.com/doi/abs/10.1080/00397911.2012.729279?journalCode=lsyc20
Abstract
An improved and convenient synthetic route for the synthesis of peramivir has been developed with a total 34% yield. The process was improved from previous methods in three key reaction steps including 1,3-dipolar cycloaddition, reductive ring cleavage of the isoxazoline, and incorporation of the peripheral guanidino group. First, an activated sodium hypochlorite (Cl% = 10%) was employed for the catalytic 1,3-dipolar cycloaddition, and 61–68% yields were obtained. Second, the NaBH4-NiCl2 was used as a new reducing reagent instead of the expensive catalyst PtO2. Most important, an innovative and environmentally friendly method of guanylation in the final step was developed using chloroformamidine hydrochloride as the amidino reagent, which avoided the use of the highlytoxic reagent of HgCl2 and made the process greener.
Supplemental materials are available for this article. Go to the publisher’s online edition of Synthetic Communications® to view the free supplemental file.
GRAPHICAL ABSTRACT


Peramivir – Synthetic Route 1

Synthetic Reference
Chand, Pooran; Kotian, Pravin L.; Dehghani, Ali; El-Kattan, Yahya; Lin, Tsu-Hsing; Hutchison, Tracy L.; Babu, Y. Sudhakar; Bantia, Shanta; Elliott, Arthur J.; Montgomery, John A. Systematic Structure-Based Design and Stereoselective Synthesis of Novel Multi-Substituted Cyclopentane Derivatives with Potent Anti-influenza Activity. Journal of Medicinal Chemistry. Volume 44. Issue 25. Pages 4379-4392. Journal. (2001).
Synthetic Reference
Jia, Fei; Hong, Juan; Sun, Ping-Hua; Chen, Jian-Xin; Chen, Wei-Min. Facile synthesis of the neuraminidase inhibitor peramivir. Synthetic Communications. Volume 43. Issue 19. Pages 2641-2647. Journal; Online Computer File. (2013).
Peramivir – Synthetic Route 3

Peramivir – Synthetic Route 4

Synthetic Reference
Li, Mingjie; Zhu, Quanming; Wang, Chunyan. A process for preparing peramivir. Assignee Shandong Luoxin Pharmacy Stock Co., Ltd., Peop. Rep. China. CN 103524383. (2014).
Synthetic Reference
Ge, Min; Li, Liang; Hu, Chunchen; Wang, Huaiqiu; Fu, Mingwei; Li, Lingchao. Synthesis of Peramivir via 3+2 ring closure. Assignee Acesys Pharmatech Ltd., Peop. Rep. China. CN 108997171. (2018).
SYN 1
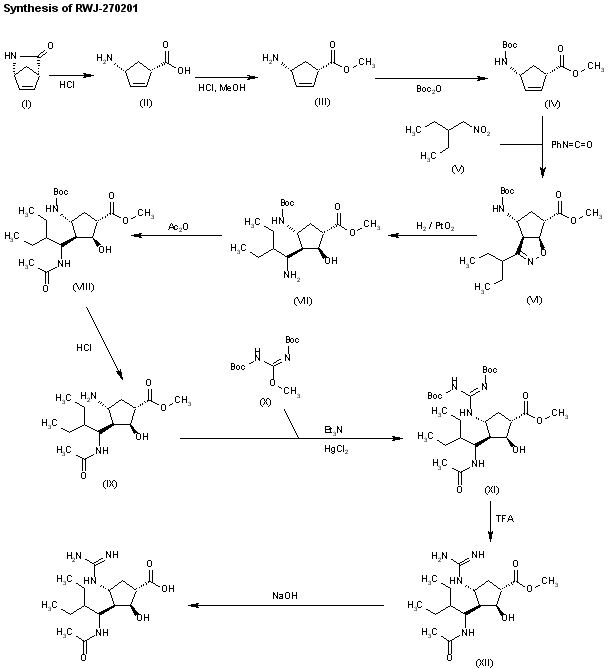
WO 9933781
Ring opening of cis-2-azabicyclo[2.2.1]hept-5-en-3-one (I) with HCl in refluxing methanol gives cis-4-amino-2-cyclopentene-1-carboxylic acid (II), which is esterified with HCl/methanol, yielding the methyl ester (III). The protection of (III) with tert-butoxycarbonyl anhydride and triethylamine in dichloromethane affords carbamate (IV), which is cyclized with 2-ethyl-1-nitrobutane (V) by means of phenyl isocyanate in refluxing benzene to give the cyclopenta-oxazoline (VI). Ring opening of (VI) by hydrogenation with H2 over PtO2 in HCl/methanol yields amine (VII), which is acylated with acetic anhydride and triethylamine in dichloromethane to afford acetamide (VIII). The deprotection of (VIII) with HCl in ethyl ether gives the cyclopentylamine (IX), which is condensed with N,N’-bis(tert-butoxycarbonyl)-O-methylisourea (X) by means of HgCl2 in DMF to yield the protected guanidino compound (XI). Deprotection of the guanidino group of (XI) with trifluoroacetic acid in dichloromethane affords the methyl ester (XII), which is finally hydrolyzed with NaOH in THF/methanol.
SYN 2
J Med Chem 2000,43(19),3482
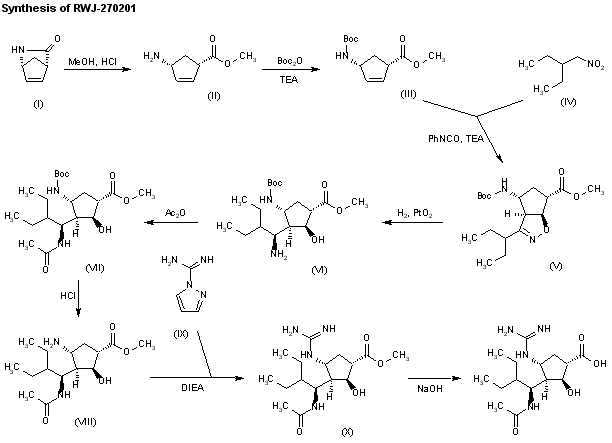
The stereospecific synthesis of BCX-1812(RWJ-270201) has been reported: Ring opening of (-)-2-azabicyclo[2.2.1]hept-5-en-3-one (I) with methanolic HCl gives the methyl ester (II), which is N-protected with Boc2O and TEA yielding the carbamate (III). Cyclization of (III) with 2-ethyl-1-nitrobutane (IV) by means of phenyl isocyanate and TEA affords the bicyclic compound (V) and other isomers. Compound (V) is isolated from the mixture and then hydrogenated with H2 over PtO2 in MeOH and a catalytic amount of HCl to provide amine (VI). Reaction of (VI) with acetic anhydride gives the acetamide (VII), which is Boc-deprotected with ethereal HCl yielding amine (VIII). The guanylation of (VIII) with pyrazolecarboxamidine hydrochloride (IX) and DIEA affords the guanidino derivative (X), which is finally hydrolyzed with NaOH to the desired (1S,2S,3R,4R,1’S)-diastereomer.
SYN 3
J Med Chem 2001,44(25),4379
The ring opening of (-)-2-azabicyclo [2,2,1]hept-5-en-3-one (I) with methanolic HCl gives the methyl ester (II), which is N-protected with Boc2O and TEA, yielding the carbamate (III). The cyclization of (III) with 2-ethyl-1-nitrobutane (IV) by means of phenyl isocyanate and TEA affords the bicyclic compound (V) along with other isomers that are separated by column chromatography. Compound (V) is hydrogenated with H2 over PtO2 in methanol to provide amine (VI), which is acylated with Ac2O, giving the acetamide (VII). N-Boc deprotection of (VII) by means of HCl in ethyl ether yields the amine (VIII), which is condensed with protected isothiourea (IX) by means of HgCl2 to afford the guanidine derivative (X). The hydrolysis of (X) with NaOH provides the carboxylic acid (XI), which is finally deprotected with trifluoroacetic acid to yield the target cyclopentanecarboxylic acid derivative.

References
- ^ Jump up to:a b c “Drug Approval Package: Rapivab (peramivir) Injection NDA #206426”. U.S. Food and Drug Administration (FDA). 16 January 2015. Retrieved 11 February 2020.
- ^ Thorlund K, Awad T, Boivin G, Thabane L (May 2011). “Systematic review of influenza resistance to the neuraminidase inhibitors”. BMC Infectious Diseases. 11 (1): 134. doi:10.1186/1471-2334-11-134. PMC 3123567. PMID 21592407.
- ^ “Peramivir authorized for Emergency use”. LifeHugger. 2009-12-04. Archived from the original on 2011-07-13. Retrieved 2009-12-04.
- ^ “Life-Saving H1N1 Drug Unavailable to Most”. CBS Evening News. Atlanta, GA, USA: CBS Interactive. 2009-10-19. Retrieved 2009-10-20.
- ^ “Emergency Use Authorization Granted For BioCryst’s Peramivir”. Reuters. 2009-10-24.
- ^ “Feds hand BioCryst $77M for anti-viral trial”. Fierce biotech. September 21, 2009.
- ^ “FDA Authorizes Emergency Use of Intravenous Antiviral Peramivir for 2009 H1N1 Influenza for Certain Patients, Settings”. Reuters. 2009-10-24.
- ^ Kohno S, Yen MY, Cheong HJ, Hirotsu N, Ishida T, Kadota J, et al. (November 2011). “Phase III randomized, double-blind study comparing single-dose intravenous peramivir with oral oseltamivir in patients with seasonal influenza virus infection”. Antimicrobial Agents and Chemotherapy. 55 (11): 5267–76. doi:10.1128/AAC.00360-11. PMC 3195028. PMID 21825298.
- ^ “BioCryst scraps $235M late-stage flu drug program backed by feds”. Fierce Biotech. November 8, 2012.
- ^ “BioCryst to File Peramivir NDA Supported by BARDA/HHS Funding”. Fierce Biotech. July 11, 2013.
- ^ Hurt, A.C. (9 June 2011). “Increased detection in Australia and Singapore of a novel influenza A(H1N1)2009 variant with reduced oseltamivir and zanamivir sensitivity due to a S247N neuraminidase mutation”. Eurosurveillance.
- ^ Hirschler, Ben (2011-06-10). “Swine flu starting to show resistance to drugs”. Reuters.
- ^ McKimm-Breschkin JL (January 2013). “Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance”. Influenza and Other Respiratory Viruses. 7 Suppl 1: 25–36. doi:10.1111/irv.12047. PMC 4942987. PMID 23279894.
- ^ Leang SK, Kwok S, Sullivan SG, Maurer-Stroh S, Kelso A, Barr IG, Hurt AC (March 2014). “Peramivir and laninamivir susceptibility of circulating influenza A and B viruses”. Influenza and Other Respiratory Viruses. 8 (2): 135–9. doi:10.1111/irv.12187. PMC 4186459. PMID 24734292.
- ^ “BioCryst Files Peramivir NDA for the Treatment of Influenza”(Press release). BioCryst Pharmaceuticals. 2013-12-20.
- ^ “Rapivab: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 February 2020.
External links
- “Peramivir”. Drug Information Portal. U.S. National Library of Medicine.
- “Peramivir: Requirements for Administration under EUA”. Lifehugger. Archived from the original on 2011-07-13.
- “CDC H1N1 Flu – Termination of the Emergency Use Authorization (EUA) of Medical Products and Devices”. U.S. Centers for Disease Control and Prevention (CDC).
- Clinical trial number NCT00957996 for “Safety Study of IV Peramivir in Hospitalized Subjects With Confirmed or Suspected Influenza” at ClinicalTrials.gov
- “A Novel Anti-viral Drug for Influenza, RAPIACTA Launch” (PDF). Shionogi & Co., Ltd. January 26, 2010. Archived from the original (PDF) on 2013-05-22.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rapivab |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Elimination half-life | 7.7 to 20.8 hours (in patients with normal renal function) |
| Excretion | Kidney |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C15H28N4O4 |
| Molar mass | 328.413 g·mol−1 |
| 3D model (JSmol) | |
| |
|
| Influenza (Flu) |
|---|
 |
////////////////PERAMIVIR, RWJ 270201, RWJ-270201, RWJ270201M, BCX 1812, RWJ 270201
/////////////////

PERAMIVIR
(1S,2S,3S,4R)-3-[(1S)-1-acetamido-2-ethyl-butyl]-4- (diaminomethylideneamino)-2-hydroxy-cyclopentane- 1-carboxylic acid
8th, APRIL 2013
In the wake of the growing bird flu outbreak, China’s SFDA will fast-track approvals of a flu treatment – not a vaccine – known as peramivir (peramivir sodium chloride injection). The drug is a neuraminidase inhibitor, which works by preventing the virus from moving from an infected cell to infect other cells. Guangzhou Nanxin Pharma has been approved to produce the medicine, and it expects to begin distributing the drug in 30 days. Other China pharmas have also filed for approval to make the drug or conduct human clinical trials
Peramivir is an experimental antiviral drug developed by BioCryst Pharmaceuticals for the treatment of influenza. It has been authorized for the emergency use of treatment of certain hospitalized patients with known or suspected 2009 H1N1 influenza.[1]
Peramivir is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells.
The development of peramivir is supported by the US Department of Health and Human Services as part of the US government’s effort to prepare against the threat of an influenza pandemic.[2]
Peramivir is already available in Japan as RAPIACTA (R) and also available in South Korea as PERAMIFLU. Peramivir is currently the only intravenous option for treating swine flu. The drug is in Phase III studies in US.[3][4]
Use in treating Influenza A (H1N1) “Swine Flu”
In October 2009, it was reported that the experimental antiviral drug peramivir had been effective in treating serious cases of swine flu.[5] On October 23, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization for Peramivir, allowing the use of the drug in intravenous form for hospitalized patients only in cases where the other available methods of treatment are ineffective or unavailable;[6] for instance, if oseltamivir (Tamiflu) resistance develops and a person is unable to take Relenza via the inhaled route. The Emergency Use Authorization expired on June 23, 2010.
Biocryst also donated 1200 courses of treatment to the US department of Health and Human Services.[7]
According to a research report published in June 2011, a novel variant of swine flu has emerged in Asia with a genetic adaptation giving some resistance to Roche’s (ROG.VX) Tamiflu and GlaxoSmithKline’s (GSK.L) Relenza, the two mainstay drugs used to tackle the disease. There was no significant reduction in sensitivity to Peramivir. [8]
Initial treatment courses are for 5 to 10 days duration. Treatment beyond 10 days is permitted depending on clinical presentation such as critical illness (e.g., respiratory failure or intensive care unit admission), continued viral shedding or unresolved clinical influenza illness.[9]
- “Peramivir authorized for Emergency use”. LifeHugger. 2009-12-04. Retrieved 2009-12-04.
- “HHS Pursues Advance Development of New Influenza Antiviral Drug” (Press release). USDepartment of Health and Human Services. 2007-01-04. Retrieved 2007-05-25.
- “Evaluation of the Efficacy and Safety of Peramivir in Subjects With Uncomplicated Acute Influenza”.National Institutes of Health. 2007-03-16. Retrieved 2007-05-25.
- “Evaluation of the Efficacy and Safety of Peramivir in Adults With Acute Serious or Potentially Life-Threatening Influenza”. National Institutes of Health. 2007-03-28. Retrieved 2007-05-25.
- “Life-Saving H1N1 Drug Unavailable to Most”. CBS Evening News (Atlanta, GA, USA: CBS Interactive). 2009-10-19. Retrieved 2009-10-20.
- “Emergency Use Authorization Granted For BioCryst’s Peramivir”. Reuters. 2009-10-24.
- “FDA Authorizes Emergency Use of Intravenous Antiviral Peramivir for 2009 H1N1 Influenza for Certain Patients, Settings”. Reuters. 2009-10-24.
- Hirschler, Ben (2011-06-10). “Swine flu starting to show resistance to drugs”. Reuters.
- “Peramivir: Dosage and Administration”. LifeHugger. 2009-12-04. Retrieved 2009-12-04.
Gilead Submits NDA to FDA for Sofosbuvir for the Treatment of Hepatitis C

Sofosbuvir
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate

9 APRIL 2013
Gilead Sciences today announced that the company has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of sofosbuvir, a once-daily oral nucleotide analogue for the treatment of chronic hepatitis C virus (HCV) infection. The data submitted in this NDA support the use of sofosbuvir and ribavirin (RBV) as an all-oral therapy for patients with genotype 2 and 3 HCV infection, and for sofosbuvir in combination with RBV and pegylated interferon (peg-IFN) for treatment-naïve patients with genotype 1, 4, 5 and 6 HCV infection.
Chronic HCV infection affects up to four million Americans, particularly individuals born between 1946 and 1964. The disease is the leading cause of liver cancer and liver transplantation in the United States. Treatment for HCV currently includes 24-48 weeks of therapy with peg-IFN, which has to be injected and is associated with significant side effects, leaving some patients unable to complete therapy. If approved, sofosbuvir would shorten HCV therapy to 12 to 16 weeks, and depending on the genotype, would either eliminate or reduce the duration of peg-IFN injections.
“Current therapies are not suitable for large numbers of patients with HCV infection, and are challenging to take and tolerate,” said John C. Martin, PhD, Chairman and Chief Executive Officer of Gilead Sciences. “Sofosbuvir’s antiviral potency, safety profile and once-daily administration have the potential to improve cure rates by simplifying and shortening therapy for patients with this disease.”
The sofosbuvir NDA is supported primarily by data from four phase 3 studies, NEUTRINO, FISSION, POSITRON and FUSION, in which 12 or 16 weeks of sofosbuvir-based therapy was found to be superior or non-inferior to currently available treatment options or historical controls, based on the proportion of patients who had a sustained virologic response (HCV undetectable) 12 weeks after completing therapy (SVR12). Patients who achieve SVR12 are considered cured of HCV.
Gilead plans to file for regulatory approval of sofosbuvir in other geographies, including the European Union, in the second quarter of 2013. The European Medicines Agency (EMA) has accepted Gilead’s request for accelerated assessment for sofosbuvir, a designation that is granted to new medicines of major public health interest. Accelerated assessment could shorten the EMA’s review time of sofosbuvir by two months. Granting of accelerated assessment does not guarantee a positive opinion from the CHMP or approval by the European Commission.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]
- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S. et al. (2010). “Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus”. Journal of Medicinal Chemistry 53 (19): 7202–7218. doi:10.1021/jm100863x. PMID 20845908. edit
- “PSI-7977″. Gilead Sciences.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M. M.; Bao, D.; Chang, W.; Espiritu, C. et al. (2010). “Mechanism of Activation of PSI-7851 and Its Diastereoisomer PSI-7977″. Journal of Biological Chemistry 285 (45): 34337–34347. doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890. edit
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- Tom Murphy (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”. Bloomberg Businessweek.
- http://www.gilead.com/pr_1757156
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. Gane, E et al. New England Journal of Medicine 368:3444. January 3, 2013.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
Lundbeck has presented promising data on Brintellix, its recently-filed investigational antidepressant co-developed with Takeda.

vortioxetine
9 APRIL 2013
Lundbeck has presented promising data on Brintellix, its recently-filed investigational antidepressant co-developed with Takeda.
Vortioxetine (code name Lu AA21004) is an experimental drug currently under development by Lundbeck and Takeda for the treatment of major depressive disorder(MDD) and generalized anxiety disorder (GAD).Commercial name chosen is Brintellix.
Regulatory approval for the treatment of MDD for the European market has been filed in September 2012, for the United States in October 2012, and filing for Canada should follow. Filing for the Japanese market is expected in 2013
The Danish drugmaker announced results for the REVIVE study which compared Brintellix (vortioxetine) with Servier’s Valdoxan (agomelatine), Servier’s in adults with major depression (MDD) who changed antidepressant after an inadequate response to commonly-prescribed selective serotonin reuptake inhibitors (SSRIs) or serotonin–norepinephrine reuptake inhibitors (SNRIs). Lundbeck noted that as one of the newest antidepressants, agomelatine was chosen as a comparator because of its different mode of action from conventional SSRI/SNRI therapies.
Lundbeck noted that few randomised, double-blind trials looking at MDD patients who were unresponsive to first-line antidepressants have been conducted and “this is one of these few studies which also shows a significant difference between treatments.” On the primary efficacy endpoint for REVIVE, Brintellix was statistically significantly superior to agomelatine by 2.2 points on the Montgomery–Asberg Depression Rating Scale (MADRS), a ten-item questionnaire used to measure severity of the disorder.
Brintellix is under review on both sides of the Atlantic and is one of three new products, Lundbeck hopes to launch this year. The other two, which are already approved in some territories, are its once-monthly version of Abilify (aripiprazole) for schizophrenia and the alcohol dependence treatment Selincro (nalmefene); indeed, Lundbeck also presented new data on the later from three Phase III studies that “consistently show a significant reduction in alcohol consumption” in patients with high-risk drinking levels.
Bayer PAH drug Riociguat gets priority review at FDA

RIOCIQUAT
CAS NO 625115-55-1
Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
9 APRIL2013
Bayer has been boosted by the news that regulators in the USA are fast-tracking the German group’s investigational pulmonary arterial hypertension riociguat.
The US Food and Drug Administration has granted priority review to the New Drug Application for riociguat, which Bayer filed in February on both sides of the Atlantic for PAH and a related condition, inoperable chronic thromboembolic pulmonary hypertension (CTEPH). The FDA bestows a priority review on medicines that offer major advances in care or that provide a treatment where no adequate therapy exists. The agency aims to complete its assessment within eight months from the submission of the NDA, rather than the standard 12 months.
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators
The submissions are based on two Phase III studies and riociguat, the first member of a novel class of compounds called stimulators of soluble guanylate cyclase (sGC), met its primary endpoint in both trials, a change in exercise capacity after 12- or 16 weeks respectively. The drug was generally well tolerated, with a good safety profile.

If approved, riociguat would be going up against Actelion’s Tracleer (bosentan) and Gilead Sciences/GlaxoSmithKline’s Letairis/Volibris (ambrisentan). Actelion, which has dominated the PAH market, has already filed its follow-up to Tracleer, Opsumit (macitentan).
Links
FDA Approves Diclegis for Pregnant Women Experiencing Nausea and Vomiting

doxylamine

pyridoxine
8 april 2013
The U.S. Food and Drug Administration today approved Diclegis (doxylamine succinate and pyridoxine hydrochloride) to treat pregnant women experiencing nausea and vomiting. Diclegis is a delayed-release tablet intended for women who have not adequately responded to conservative management of nausea and vomiting during pregnancy, such as dietary and lifestyle modifications.
These modifications include eating several small meals instead of three large meals, eating bland foods that are low in fat and easy to digest and avoiding smells that can trigger nausea. “Many women experience nausea and vomiting during pregnancy, and sometimes these symptoms are not adequately managed through recommended changes in diet and lifestyle,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Division of Reproductive and Urologic Products in the FDA’s Center for Drug Evaluation and Research.
“Diclegis is now the only FDA-approved treatment for nausea and vomiting due to pregnancy, providing a therapeutic option for pregnant women seeking relief from these symptoms.” Diclegis was studied in 261 women experiencing nausea and vomiting due to pregnancy. Study participants in the clinical trial were at least 18 years old and had been pregnant for at least 7 weeks and up to 14 weeks. Women were randomly assigned to receive two weeks of treatment with Diclegis or a placebo.
The study results showed that women taking Diclegis experienced greater improvement in nausea and vomiting than those taking placebo. Additionally, observational (epidemiological) studies have shown that the combination of active ingredients in Diclegis does not pose an increased risk of harm to the fetus. Diclegis is taken daily. Tablets must be taken whole on an empty stomach. The recommended starting dose is two tablets taken at bedtime.
If symptoms are not adequately controlled, the dose can be increased to a maximum recommended dose of four tablets daily (one in the morning, one mid-afternoon and two at bedtime). Nausea and vomiting due to pregnancy usually improve after the first trimester. Health care professionals should reassess their patients for continued need for Diclegis as pregnancy progresses. Drowsiness or sleepiness, which can be severe, is the most common side effect reported by women taking Diclegis.
Women should avoid using Diclegis when engaging in activities requiring mental alertness, such as driving or operating heavy machinery, until cleared to do so by their health care provider.
Diclegis is marketed by Duchesnay Inc., based in Blainville, Québec, Canada.
