
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 330)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CHINESE MEDICINE-Xuezhikang , A blood lipid regulator

Xuezhikang
Xuezhikang, the extract of red yeast rice, has been widely used as a Chinese traditional medicine for the therapy of patients with cardiovascular diseases. It contains natural Lovastatin and its homologues, as well as unsaturated fatty acids, flavonoids, plant sterols and other biologically active substances
The product is a world-recognized blood lipid regulator, which is made by extracting from “specially-made red yeast rice”. It combines modern high-tech biotechnology with traditional Chinese medicine, which can safely and effectively regulate blood lipids in a comprehensive way with proven curative effects and reliable safety.
Pharmacological Effects: the product can reduce blood cholesterol, triglycerides, low density lipoprotein cholesterol, improve high density lipoprotein cholesterol, inhibit atherosclerotic plaque formation, and protect vascular endothelial cells; and inhibit lipid deposition in the liver. The large-scale evidence-based research has proven that long-term use of XUEZHIKANG can greatly reduce the risk of CHD occurrence and decrease the mortality. XUEZHIKANG is the only Chinese medicine with blood lipids regulating function which is listed into the National Basic Medicine List.
Beijing Peking University WBL Biotech (WPU) has developed and launched Xuezhikang, a capsule formulation of Monascus purpureus-fermented rice, for the oral treatment of hyperlipidemia and cardiovascular disease
CLINICAL TRIAL, NCT01327014 PHASE 2
The data had shown that Xuezhikang significantly reduced the level of low density lipoprotein cholesterin (LDL-C) in patients in a similar manner to statins and increased the level of the beneficial high density lipoprotein cholesterin (HDL-C). It had a good safety profile with no significant liver enzyme abnormal events observed. Besides regulation of dyslipidemia, the drug also signifcantly reduced cardiovascular events and general mortality rate of patients
NCT01686451 PHASE 4
Both XueZhiKang and Statins are cholesterol-lowering medications that are often prescribed for individuals with high cholesterol and who are at risk for cardiovascular disease (CVD). Several studies, including one randomized, double-blind, placebo-controlled clinical trial, have suggested that the use of statins is more frequently associated with fatigue. And XueZhiKang may be not. The purpose of this study is to compare the effect of these two medications on fatigue in persons who are at moderate to low CVD risk based on the risk estimation system in ESC(European Society of Cardiology)/ESA(European Atherosclerosis Society) guidelines (2011) for the management of dyslipidemias.

Those of you with high cholesterol will be happy to learn that there are some legitimate options to your statin pills. Many people cannot tolerate the extremely popular statin pills, especially from side effects of muscle aches. But there’s now some very strong evidence that herbal medicines, including red yeast rice, can be at least as effective as a statin, and without the side effects. Too good to be true? Maybe not…
![]() Red yeast rice is a bright reddish purple fermented rice, which acquires its colour from being cultivated with the mold Monascus purpureus. Red yeast rice is known as Zhi Tai when in powdered form but is called Xue Zhi Kang in alcohol extract form. This has been used in China for many centuries for many reasons, but researchers have been very interested in its effectiveness in lowering cholesterol and preventing heart disease (similar benefits from statins). It seems that the main active ingredient is indeed the natural form of a common statin, lovastatin — but researchers feel that other ingredients inside may add more protective effects. There is an official patented Chinese TCM formulation, called Xue Zhi Kang (xue2 zhi1 kang2 jiao nang 血脂康 胶囊), which has the equivalent of 10mg of lovastatin. The ScienceDaily website has a nice 2008 review of a well-designed study, printed in American Journal of Cardiology, which followed 5,000 persons after their first heart attack, and divided them into two groups taking either xuezhikang or placebo. After 5 years:
Red yeast rice is a bright reddish purple fermented rice, which acquires its colour from being cultivated with the mold Monascus purpureus. Red yeast rice is known as Zhi Tai when in powdered form but is called Xue Zhi Kang in alcohol extract form. This has been used in China for many centuries for many reasons, but researchers have been very interested in its effectiveness in lowering cholesterol and preventing heart disease (similar benefits from statins). It seems that the main active ingredient is indeed the natural form of a common statin, lovastatin — but researchers feel that other ingredients inside may add more protective effects. There is an official patented Chinese TCM formulation, called Xue Zhi Kang (xue2 zhi1 kang2 jiao nang 血脂康 胶囊), which has the equivalent of 10mg of lovastatin. The ScienceDaily website has a nice 2008 review of a well-designed study, printed in American Journal of Cardiology, which followed 5,000 persons after their first heart attack, and divided them into two groups taking either xuezhikang or placebo. After 5 years:
Frequencies of the primary end point were 10.4% in the placebo group and 5.7% in the XZK-treated group, with absolute and relative decreases of 4.7% and 45%, respectively. Treatment with XZK also significantly decreased CV and total mortality by 30% and 33%, the need for coronary revascularization by 1/3, and lowered total and low-density lipoprotein cholesterol and triglycerides, but raised high-density lipoprotein cholesterol levels. In conclusion, long-term therapy with XZK significantly decreased the recurrence of coronary events and the occurrence of new CV events and deaths, improved lipoprotein regulation, and was safe and well tolerated.
This is impressive data, and the study design is very well done, which means the evidence is quite strong. One co-author, Dr Capuzzi, has a nice summary:
“It’s very exciting because this is a natural product and had very few adverse side effects including no abnormal blood changes,” said Capuzzi. “People in the Far East have been taking Chinese red yeast rice as food for thousands of years, but no one has ever studied it clinically in a double-blind manner with a purified product against a placebo group until now and we are pleased with the results. However, people in the United States should know that the commercially available over-the-counter supplement found in your average health food store is not what was studied here. Those over-the-counter supplements are not regulated, so exact amounts of active ingredient are unknown and their efficacy has not been studied yet.”
- XueZhiKang
In another randomized trial study, printed last year in the Annals of Internal Medicine, patients who had previously failed treatment of statins due to side effects were given 1800mg of red yeast rice twice a day versus placebo. The red yeast rice group had a significant improvement in cholesterol numbers — with no major reports of severe muscle aches they previously had on the statins.
There are other studies that also show similar benefits. In fact, the evidence is so strong that it is classified as Grade A evidence: “Strong scientific evidence for use”. This is the highest grade that any therapy can get. There are a number of good reviews of red yeast rice in Western literature, including from Medscape; the Mayo Clinic; WebMD; MedlinePlus; and NCCAM. There’s also more informal information from the TCM blog Qi Spot. You can find more scholarly information in the 2008 review from Chinese Medical Journal.
http://www.hindawi.com/journals/ecam/2012/636547/
(U.S. patent #6,046,022), ethanol extract of red yeast rice, with a total monacolins content of approx. 0.8%.
1 Heber D et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. American Journal of Clinical Nutrition 1999;69(2): 231-236
2) SoRelle R. Appeals court says Food and Drug Administration can regulate cholestin. Circulation 200;102 (7): E9012?E9013.
3) Li, C et al. Monascus purpureus-fermented rice (red yeast rice): a natural food product that lowers blood cholesterol in animal models of hypercholesterolemia. Nutrition Research 1998;18 (1): 71-81
4) Becker DJ et al. Red yeast rice for dyslipidemia in statin-intolerant patients: a randomized trial.Ann Intern Med. 2009 Jun 16;150(12):830-9, W147-9
5) Lu Z et al.Effect of Xuezhikang, an extract from red yeast Chinese rice, on coronary events in a Chinese population with previous myocardial infarction. Am J Cardiol. 2008 Jun 15;101(12):1689-93.
Hypochol is the same product. Xuezhikang is the brand name marketed in China. Hypochol, is manufactured by a Singapore comapany who have a joint venture agreement with Peking University who perfected the processing and quality control of the Red Yeast Rice Extract Product. You can order directly from: http://www.hypocol.com/wbm.html
or thru their New Zealand distributor (very good service) at: http://www.hypocol.co.nz/

Dried grain red yeast rice
Red yeast rice (simplified Chinese: 红曲米; traditional Chinese: 紅麴米); pinyin: hóng qū mǐ; literally “red yeast rice”), red rice koji (べにこうじ, lit. ‘red koji‘) or akakoji (あかこぎ, also meaning ‘red koji‘), red fermented rice, red kojic rice, red koji rice, anka, or ang-kak, is a bright reddish purple fermented rice, which acquires its colour from being cultivated with the mold Monascus purpureus.
Red yeast rice is what is referred to, in Japanese, as a koji, meaning ‘grain or bean overgrown with a mold culture’, a food preparation tradition going back to ca. 300 BC.[1] In both the scientific and popular literature in English that draws principally on Japanese, it is most often known as “red rice koji“. English works favoring Chinese sources may prefer the translation “red yeast rice”.
In addition to its culinary use, red yeast rice is also used in Chinese herbology and traditional Chinese medicine. Its use has been documented as far back as the Tang Dynasty in China in 800 AD. It is taken internally to invigorate the body, aid in digestion, and revitalize the blood. A more complete description is in the traditional Chinese pharmacopoeia, Ben Cao Gang Mu-Dan Shi Bu Yi, from the Ming Dynasty (1378–1644).
What other names is Red Yeast known by?
Arroz de Levadura Roja, Cholestin, Hong Qu, Koji Rouge, Levure de Riz Rouge, Monascus, Monascus purpureus, Monascus Purpureus Went, Red Rice, Red Rice Yeast, Red Yeast Rice, Red Yeast Rice Extract, Riz Rouge, Xue Zhi Kang, XueZhiKang, XZK, Zhibituo, Zhi Tai.
What is Red Yeast?
Red yeast is the product of rice fermented with Monascus purpureus yeast. Red yeast supplements are different from red yeast rice sold in Chinese grocery stores. People use red yeast as medicine.
Possibly Effective for…
- High cholesterol.
- High cholesterol and triglyceride levels caused by human immunodeficiency virus (HIV) disease (AIDS).
Insufficient Evidence to Rate Effectiveness for…
- Indigestion, diarrhea, improving blood circulation, spleen and stomach problems, and other conditions.
In the late 1970s, researchers in the United States and Japan were isolating lovastatin from Aspergillus and monacolins fromMonascus, respectively, the latter being the same fungus used to make red yeast rice but cultured under carefully controlled conditions. Chemical analysis soon showed that lovastatin and monacolin K are identical. The article “The origin of statins” summarizes how the two isolations, documentations and patent applications were just months apart.[5] Lovastatin became the patented, prescription drug Mevacor for Merck & Co. Red yeast rice went on to become a contentious non-prescription dietary supplement in the United States and other countries.
Lovastatin and other prescription “statin” drugs inhibit cholesterol synthesis by blocking action of the enzyme HMG-CoA reductase. As a consequence, circulating total cholesterol and LDL-cholesterol are lowered. In a meta-analysis of 91 randomized clinical trial of ≥12 weeks duration, totaling 68,485 participants, LDL-cholesterol was lowered by 24-49% depending on the statin. Different strains ofMonascus fungus will produce different amounts of monacolins. The ‘Went’ strain of Monascus purpureus (purpureus = dark red in Latin), when properly fermented and processed, will yield a dried red yeast rice powder that is approximately 0.4% monacolins, of which roughly half will be monacolin K (identical to lovastatin). Monacolin content of a red yeast rice product is described in a 2008 clinical trial report.
- Website about medicinal use of Monascus purpureus
- Medicinal use of Red yeast rice
- Fermentiertes Rotes Reismehl (in German)







Europace Publishes Data Supporting Use Of BRINAVESS™ (Vernakalant) As A First Line Agent For Pharmacological Cardioversion Of Atrial Fibrillation

Vernakalant, MK-6621, RSD 1235
(3R)-1-{(1R,2R)-2-[2-(3,4-dimethoxyphenyl)
ethoxy]cyclohexyl}pyrrolidin-3-ol
C20H31NO4 , 349.47, Brinavess , Kynapid
cas no 794466-70-9
748810-28-8 (HCl)
EMA:Link click here
PATENT WO 2004099137
VANCOUVER, Nov. 21, 2013 /PRNewswire/ – Cardiome Pharma Corp. (NASDAQ: CRME / TSX: COM) today announced that a publication titled, Pharmacological Cardioversion of Atrial Fibrillation with Vernakalant: Evidence in Support of the ESC Guidelines, was published in Europace, the official Journal of the European Heart Rhythm Association, and was made available in the advanced online article access section. The authors conclude that BRINAVESS is an efficacious and rapid acting pharmacological cardioversion agent, for recent-onset atrial fibrillation (AF,) that can be used first line in patients with little or no underlying cardiovascular disease and in patients with moderate disease, such as stable coronary and hypertensive heart disease.
Vernakalant (INN; codenamed RSD1235, proposed tradenames Kynapid and Brinavess) is an investigational drug under regulatory review for the acute conversion of atrial fibrillation. It was initially developed by Cardiome Pharma, and the intravenous formulation has been bought for further development by Merck in April 2009.[1] In September 2012, Merck terminated its agreements with Cardiom and has consequently returned all rights of the drug back to Cardiom.
On 11 December 2007, the Cardiovascular and Renal Drugs Advisory Committee of the USFood and Drug Administration (FDA) voted to recommend the approval of vernakalant,[2]but in August 2008 the FDA judged that additional information was necessary for approval.[1] The drug was approved in Europe on 1 September 2010.[3]
An oral formulation underwent Phase II clinical trials between 2005 and 2008.[4][5]
Like other class III antiarrhythmics, vernakalant blocks atrial potassium channels, thereby prolonging repolarization. It differs from typical class III agents by blocking a certain type of potassium channel, the cardiac transient outward potassium current, with increased potency as the heart rate increases. This means that it is more effective at high heart rates, while other class III agents tend to lose effectiveness under these circumstances. It also slightly blocks the hERG potassium channel, leading to a prolonged QT interval. This may theoretically increase the risk of ventricular tachycardia, though this does not seem to be clinically relevant.[6]
The drug also blocks atrial sodium channels.[6]
- “Merck and Cardiome Pharma Sign License Agreement for Vernakalant, an Investigational Drug for Treatment of Atrial Fibrillation”. FierceBiotech. 9 April 2009. Retrieved 12 October 2010.
- “FDA Advisory Committee Recommends Approval of Kynapid for Acute Atrial Fibrillation”. Drugs.com. Retrieved 2008-03-15.
- “BRINAVESS (vernakalant) for Infusion Approved in the European Union for Rapid Conversion of Recent Onset Atrial Fibrillation” (Press release). Merck & Co., Inc. 1 September 2010. Retrieved 28 September 2010.
- ClinicalTrials.gov NCT00267930 Study of RSD1235-SR for the Prevention of Atrial Fibrillation/Atrial Flutter Recurrence
- ClinicalTrials.gov NCT00526136 Vernakalant (Oral) Prevention of Atrial Fibrillation Recurrence Post-Conversion Study
- Miki Finnin, Vernakalant: A Novel Agent for the Termination of Atrial Fibrillation: Pharmacology, Medscape Today, retrieved 12 October 2010
- Arzneimittel-Fachinformation (EMA)
- Cheng J.W. Vernakalant in the management of atrial fibrillation. Ann Pharmacother, 2008, 42(4), 533-42Pubmed

- Dobrev D., Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet, 2010, 375(9721), 1212-23 Pubmed
- Finnin M. Vernakalant: A novel agent for the termination of atrial fibrillation. Am J Health Syst Pharm, 2010, 67(14), 1157-64 Pubmed
- Mason P.K., DiMarco J.P. New pharmacological agents for arrhythmias. Circ Arrhythm Electrophysiol, 2009, 2(5), 588-97 Pubmed
- Naccarelli G.V., Wolbrette D.L., Samii S., Banchs J.E., Penny-Peterson E., Stevenson R., Gonzalez M.D. Vernakalant – a promising therapy for conversion of recent-onset atrial fibrillation. Expert Opin Investig Drugs, 2008, 17(5), 805-10 Pubmed
- European Patent No. 1,560,812
- WO 2006138673
 , WO 200653037
, WO 200653037 - WO 200597203, WO 200688525
- Vernakalant HydrochlorideDrugs Fut 2007, 32(3): 234
//////////////////////////////////////////////////////

![]()
Nitrogen: dark blue, oxygen: red, hydrogen: light blue
![]()

NMR
1H NMR (300 MHz, CDCI3) 5 6.75 (m, 3H), 4.22 (m, 1H), 3.87 (s, 3H), 3.85 (m, 3H), 3.74 (m, 1H), 3.57 (m, 1H), 3.32 (td, J =
7.7, 3.5, 1H), 2.96-2.75 (m, 5H), 2.64 (dd, J= 10.0, 5.0, 1H), 2.49-2.37 (m, 2H), 2.05-1.98 (m, 2H), 1.84 (m, 1H), 1.69-1.62 (m, 3H), 1.35-1.19 (m, 4H).
IN
WO 201240846
Arrhythmias are abnormal rhythms of the heart. The term “arrhythmia” refers to a deviation from the normal sequence of initiation and conduction of electrical impulses that cause the heart to beat. Arrhythmias may occur in the atria or the ventricles. Atrial arrhythmias are widespread and relatively benign, although they place the subject at a higher risk of stroke and heart failure. Ventricular arrhythmias are typically less common, but very often fatal.
Arrhythmia is a variation from the normal rhythm of the heart beat and generally represents the end product of abnormal ion-channel structure, number or function. Both atrial arrhythmias and ventricular arrhythmias are known. The major cause of fatalities due to cardiac arrhythmias is the subtype of ventricular arrhythmias known as ventricular fibrillation (VF). Conservative estimates indicate that, in the U.S. alone, each year over one million Americans will have a new or recurrent coronary attack (defined as myocardial infarction or fatal coronary heart disease). About 650,000 of these will be first heart attacks and 450,000 will be recurrent attacks. About one-third of the people experiencing these attacks will die of them. At least 250,000 people a year die of coronary heart disease within 1 hour of the onset of symptoms and before they reach a hospital. These are sudden deaths caused by cardiac arrest, usually resulting from ventricular fibrillation.
Atrial fibrillation (AF) is the most common arrhythmia seen in clinical practice and is a cause of morbidity in many individuals (Pritchett E.L., N. Engl. J. Med. 327(14):1031 Oct. 1, 1992, discussion 1031-2; Kannel and Wolf, Am. Heart J. 123(l):264-7 Jan. 1992). Its prevalence is likely to increase as the population ages and it is estimated that 3-5% of patients over the age of 60 years have AF (Kannel W.B., Abbot R.D., Savage D.D., McNamara P.M., N. Engl. J. Med. 306(17): 1018-22, 1982; Wolf P.A., Abbot R.D., Kannel W.B. Stroke. 22(8):983-8, 1991). While AF is rarely fatal, it can impair cardiac function and is a major cause of stroke (Hinton R.C., Kistler J.P., Fallon J.T., Friedlich A.L., Fisher CM., American Journal of Cardiology 40(4):509-13, 1977; Wolf P.A., Abbot R.D., Kannel W.B., Archives of Internal Medicine 147(9): 1561 -4, 1987; Wolf P. A., Abbot R.D., Kannel W.B. Stroke. 22(8):983-8, 1991; Cabin H.S., Clubb K.S., Hall C, Perlmutter R.A., Feinstein A.R., American Journal of Cardiology 65(16): 1112-6, 1990).
WO95/08544 discloses a class of aminocyclohexylester compounds as useful in the treatment of arrhythmias.
WO93/ 19056 discloses a class of aminocyclohexylamides as useful in the treatment of arrhythmia and in the inducement of local anaesthesia.
WO99/50225 discloses a class of aminocyclohexylether compounds as useful in the treatment of arrhythmias.
Antiarrhythmic agents have been developed to prevent or alleviate cardiac arrhythmia. For example, Class I antiarrhythmic compounds have been used to treat supraventricular arrhythmias and ventricular arrhythmias. Treatment of ventricular arrhythmia is very important since such an arrhythmia can be fatal. Serious ventricular arrhythmias (ventricular tachycardia and ventricular fibrillation) occur most often in the presence of myocardial ischemia and/or infarction. Ventricular fibrillation often occurs in the setting of acute myocardial ischemia, before infarction fully develops. At present, there is no satisfactory pharmacotherapy for the treatment and/or prevention of ventricular fibrillation during acute ischemia. In fact, many Class I antiarrhythmic compounds may actually increase mortality in patients who have had a myocardial infarction.
Class la, Ic and HI antiarrhythmic drugs have been used to convert recent onset AF to sinus rhythm and prevent recurrence of the arrhythmia (Fuch and Podrid, 1992; Nattel S., Hadjis T., Talajic M., Drugs 48(3):345-7l, 1994). However, drug therapy is often limited by adverse effects, including the possibility of increased mortality, and inadequate efficacy (Feld G.K., Circulation. <°3(<5):2248-50, 1990; Coplen S.E., Antman E.M., Berlin J.A., Hewitt P., Chalmers T.C., Circulation 1991; S3(2):714 and Circulation 82(4):1106-16, 1990; Flaker G.C., Blackshear J.L., McBride R., Kronmal R.A., Halperin J.L., Hart R.G., Journal of the American College of Cardiology 20(3):527-32, 1992; CAST, N. Engl. J. Med. 321:406, 1989; Nattel S., Cardiovascular Research. 37(3):567 -77, 1998). Conversion rates for Class I antiarrhythmics range between 50-90% (Nattel S., Hadjis T., Talajic M., Drugs 48(3)345-71, 1994; Steinbeck G., Remp T., Hoffmann E., Journal of Cardiovascular Electrophysiology. 9(8 Suppl):S 104-8, 1998). Class ILT antiarrhythmics appear to be more effective for terminating atrial flutter than for AF and are generally regarded as less effective than Class I drugs for terminating of AF (Nattel S., Hadjis T., Talajic M., Drugs. 48(3):345-71, 1994; Capucci A., Aschieri D., Villani G.Q., Drugs & Aging 13(l):5l- 70, 1998). Examples of such drugs include ibutilide, dofetilide and sotalol. Conversion rates for these drugs range between 30-50% for recent onset AF (Capucci A., Aschieri D., Nillani G.Q., Drugs & Aging J3(l):5l-70, 1998), and they are also associated with a risk of the induction of Torsades de Pointes ventricular tachyarrhythmias. For ibutilide, the risk of ventricular proarrhythmia is estimated at ~4.4%, with ~1.7% of patients requiring cardioversion for refractory ventricular arrhythmias (Kowey P.R., NanderLugt J.T., Luderer J.R., American Journal of Cardiology 78(8A):46-52, 1996). Such events are particularly tragic in the case of AF as this arrhythmia is rarely a fatal in and of itself.
Atrial fibrillation is the most common arrhythmia encountered in clinical practice. It has been estimated that 2.2 million individuals in the United States have paroxysmal or persistent atrial fibrillation. The prevalence of atrial fibrillation is estimated at 0.4% of the general population, and increases with age. Atrial fibrillation is usually associated with age and general physical condition, rather than with a specific cardiac event, as is often the case with ventricular arrhythmia. While not directly life threatening, atrial arrhythmias can cause discomfort and can lead to stroke or congestive heart failure, and increase overall morbidity.
There are two general therapeutic strategies used in treating subjects with atrial fibrillation. One strategy is to allow the atrial fibrillation to continue and to control the ventricular response rate by slowing the conduction through the atrioventricular (AV) node with digoxin, calcium channel blockers or beta-blockers; this is referred to as rate control. The other strategy, known as rhythm control, seeks to convert the atrial fibrillation and then maintain normal sinus rhythm, thus attempting to avoid the morbidity associated with chronic atrial fibrillation. The main disadvantage of the rhythm control strategy is related to the toxicities and proarrhythmic potential of the anti-arrhythmic drugs used in this strategy. Most drugs currently used to prevent atrial or ventricular arrhythmias have effects on the entire heart muscle, including both healthy and damaged tissue. These drugs, which globally block ion channels in the heart, have long been associated with life-threatening ventricular arrhythmia, leading to increased, rather than decreased, mortality in broad subject populations. There is therefore a long recognized need for antiarrhythmic drugs that are more selective for the tissue responsible for the arrhythmia, leaving the rest of the heart to function normally, less likely to cause ventricular arrhythmias.
One specific class of ion channel modulating compounds selective for the tissue responsible for arrhythmia has been described in U.S. Pat. No. 7,057,053, including the ion channel modulating compound known as vernakalant hydrochloride. Vernakalant hydrochloride is the non-proprietary name adopted by the United States Adopted Name (USAN) council for the ion channel modulating compound (1R,2R)-2-[(3R)-hydroxypyrrolidinyl]-1-(3,4-dimethoxyphenethoxy)-cyclohexane monohydrochloride, which compound has the following formula:

Vernakalant hydrochloride may also be referred to as “vernakalant” herein.
Vernakalant hydrochloride modifies atrial electrical activity through a combination of concentration-, voltage- and frequency-dependent blockade of sodium channels and blockade of potassium channels, including, e.g., the ultra-rapidly activating (lKur) and transient outward (lto) channels. These combined effects prolong atrial refractoriness and rate-dependently slow atrial conduction. This unique profile provides an effective anti-fibrillatory approach suitable for conversion of atrial fibrillation and the prevention of atrial fibrillation.

C20H32ClNO4, Mr = 385.9 g/mol
Pfizer’s Xalkori Granted Regular FDA Approval
Pfizer’s XALKORI® Granted Regular FDA Approval
Standard of Care for Patients With Metastatic ALK-Positive Non-Small Cell Lung Cancer
NEW YORK, November 21, 2013–(BUSINESS WIRE)–Pfizer Inc. announced today that the U.S. Food and Drug Administration (FDA) has granted Pfizer’s XALKORI® (crizotinib) regular approval for the treatment of patients with metastatic ALK-positive non-small cell lung cancer (NSCLC) as detected by an FDA-approved test. XALKORI was previously granted accelerated approval in August 2011 due to the critical need for new agents for people living with ALK-positive NSCLC
read all at
http://www.pharmalive.com/pfizer%E2%80%99s-xalkori-granted-regular-fda-approval
Daiichi Sankyo anticoagulant edoxaban succeeds in Phase III

Edoxaban, DU-176b
Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery
for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
NOV20, 2013
Daiichi Sankyo will file edoxaban on both sides of the Atlantic shortly after the bloodthinner proved as effective and safer than warfarin in a Phase III trial of patients with atrial fibrillation.
The company has presented data on edoxaban, a once-daily oral factor Xa inhibitor, at the American Heart Association meeting in Dallas, from a study involving 21,105 patients across 46 countries. The drug, evaluated in 60mg and 30mg doses, met its primary endpoint of non-inferiority compared to warfarin for the prevention of stroke or systemic embolic events in patients with non-valvular AF.http://www.pharmatimes.com/Article/13-11-20/Daiichi_Sankyo_anticoagulant_edoxaban_succeeds_in_Phase_III.aspx
Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]
In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]
- “First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost. 6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID 18624979.
- Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis 104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID 20589317. edit
- “Phase III Trial Finds Edoxaban Outclasses Enoxaparin in Preventing Venous Thromboembolic Events”. 8 Dec 2010.
- Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost. 104 (3): 633–41. doi:10.1160/TH10-01-0066.
- Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
- “Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID 23991658.
- WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
- J Am Chem Soc 1978, 100(16): 5199
Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274
-
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :
-


-
is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
-
For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):
-

-
with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
Citation ListPatent Literature
-
- Patent Literature 1: International Publication No. WO 03/000657
- Patent Literature 2: International Publication No. WO 03/000680
- Patent Literature 3: International Publication No. WO 03/016302
- Patent Literature 4: International Publication No. WO 04/058715
- Patent Literature 5: International Publication No. WO 05/047296
- Patent Literature 6: International Publication No. WO 07/032498
- Patent Literature 7: International Publication No. WO 08/129846
- Patent Literature 8: International Publication No. WO 08/156159
SIMILAR

OTHER SALTS

Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:

formula ( 1 ) While Edoxaban is reported to be soluble in strongly acidic aqueous solutions, its solubility is considered to be very low in neutral or alkaline aqueous media. EP 2 140 867 A 1 claims an edoxaban-containing pharmaceutical composition comprising a water-swelling additive and/or a sugar alcohol. Further, it is alleged that compositions comprising lactose or cornstarch do not have good dissolution properties. The claimed pharmaceutical compositions in EP 2 140 867 Al are considered to show good dissolution properties in a neutral aqueous medium as well. Tablets comprising said composition were produced by wet granulation. However, it turned out that prior art pharmaceutical formulations comprising edoxaban being suitable for oral administration are still improvable with regards to dissolution rate and bioavailability. Further, stability and content uniformity of the known formulations could be improved. Further, due to the intolerance of many people to sugar alcohol(s), such as sorbitol, the use of sugar alcohol(s) should be avoided.
New anticoagulant Edoxaban is non inferior to Warfarin, and cuts bleeding risks
November 21,2013 | By Márcio Barra

Daiichi Sankyo’s edoxaban proved as effective and safer than warfarin in a Phase III trial of patients with atrial fibrillation, according to the results presented at the American Heart Association Scientific Sessions yesterday and published simultaneously online in The New England Journal of Medicine.
The trial, entitled ENGAGE AF-TIMI 48, compared two daily dosages of Edoxaban, 60 mg and 30 mg, against warfarin, dose-adjusted to achieve an international normalized ratio (INR) of 2.0 to 3.0, in the largest – 21,105 patients across 46 countries – and longest – 2.8 years average follow-up – clinical trial in the company’s history.
View original post 299 more words
Temsirolimus

TEMSIROLIMUS
Proline CCI-779
Torisel, NCGC00167518-01
LAUNCHED 2007
PFIZER
- CCI 779
- CCI-779
- HSDB 7931
- Temsirolimus
- Torisel
- UNII-624KN6GM2T
- WAY-CCI 779
Inhibits mTOR protein
For the treatment of renal cell carcinoma (RCC). Also investigated for use/treatment in breast cancer, lymphoma (unspecified), rheumatoid arthritis, and multiple myeloma.
An ester analog of rapamycin. Temsirolimus binds to and inhibits the mammalian target of rapamycin (mTOR), resulting in decreased expression of mRNAs necessary for cell cycle progression and arresting cells in the G1 phase of the cell cycle. mTOR is a serine/threonine kinase which plays a role in the PI3K/AKT pathway that is upregulated in some tumors
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo-1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,31,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate
cas 162635-04-3
Temsirolimus is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by Wyeth Pharmaceuticals and approved by the FDA in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Molecular Formula: C56H87NO16
Molecular Weight: 1030.28708
Temsirolimus (CCI-779) is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by WyethPharmaceuticals and approved by the U.S. Food and Drug Administration (FDA) in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
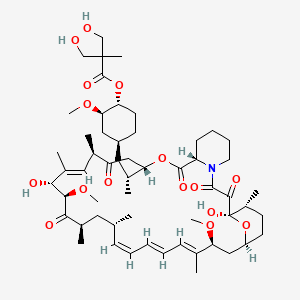
Temsirolimus is a specific inhibitor of mTOR and interferes with the synthesis of proteins that regulate proliferation, growth, and survival of tumor cells. Treatment with temsirolimus leads to cell cycle arrest in the G1 phase, and also inhibits tumor angiogenesis by reducing synthesis of VEGF.
The product had been under development by Wyeth Pharmaceutical for the treatment of pancreas cancer and metastatic breast cancer, multiple sclerosis (MS) and rheumatoid arthritis (RA); however, no recent development for these indications has been reported. Pfizer had been developing the compound for the treatment of sarcoma.
Temsirolimus holds orphan drug designation in both the U.S. and the E.U. for the treatment of renal cell carcinoma. Orphan drug designation was received in the U.S. in 2006 for the treatment of mantle-cell lymphoma.
mTOR (mammalian target of rapamycin) is a kinase enzyme inside the cell that collects and interprets the numerous and varied growth and survival signals received by tumor cells. When the kinase activity of mTOR is activated, its downstream effectors, the synthesis of cell cycle proteins such as cyclin D and hypoxia-inducible factor-1a (HIF-1a) are increased. HIF-1a then stimulates VEGF. Whether or not mTOR kinase is activated, determines whether the tumor cell produces key proteins needed for proliferation, growth, survival, and angiogenesis.
mTOR is activated in tumor cells by various mechanisms including growth factor surface receptor tyrosine kinases, oncogenes, and loss of tumor suppressor genes. These activating factors are known to be important for malignant transformation and progression.mTOR is particularly important in the biology of renal cancer (RCC) owing to its function in regulating HIF-1a levels. Mutation or loss of the von Hippel Lindau tumor-suppressor gene is common in RCC and is manifested by reduced degradation of HIF-1a. In RCC tumors, activated mTOR further exacerbates accumulation of HIF-1a by increasing synthesis of this transcription factor and its angiogenic target gene products.
Rapamycin 42-ester with 3-hydroxy-2-(hydroxymethyl)-2-methylpropionic acid (CCl-779) is an ester of rapamycin which has demonstrated significant inhibitory effects on tumor growth in both in vitro and in vivo models.
CCl-779 may delay the time to progression of tumors or time to tumor recurrence which is more typical of cytostatic rather than cytotoxic agents. CCl-779 is considered to have a mechanism of action that is similar to that of sirolimus. CCl-779 binds to and forms a complex with the cytoplasmic protein FKBP, which inhibits an enzyme, mTOR (mammalian target of rapamycin, also known as FKBP12-rapamycin associated protein [FRAP]). Inhibition of mTOR’s kinase activity inhibits a variety of signal transduction pathways, including cytokine-stimulated cell proliferation, translation of mRNAs for several key proteins that regulate the G1 phase of the cell cycle, and IL-2-induced transcription, leading to inhibition of progression of the cell cycle from G1 to S. The mechanism of action of CCl-779 that results in the G1-S phase block is novel for an anticancer drug.
The preparation and use of hydroxyesters of rapamycin, including CCl-779, are disclosed in U.S. Pat. No. 5,362,718. A regiospecific synthesis of CCl-779 is described in U.S. Pat. No. 6,277,983.
CCl-779 can be synthesized by the non-regioselective acylation of rapamycin, as described in U.S. Pat. No. 5,362,718. The synthesis, however, is complicated by mixtures of the desired 42-ester, with 31-esterified rapamycin, as well as 31, 42-diesterified rapamycin and unreacted rapamycin.
CCl-779 can also be prepared by the acylation of the 31-silyl ether of rapamycin with a ketal of bis-(hydroxymethyl)propionic acid, followed by removal of the 31-silyl ether and ketal protecting group from the bis-(hydroxymethyl) propionic acid, as described in U.S. Pat. No. 6,277,983. However, the crude 42-monoester produced from this regioselective synthesis requires further purification by column chromatography to remove residual amounts of diester by-products and unreacted rapamycin starting material.
Temsirolimus (CCI-779), an mTOR kinase Inhibitor of formula (I) is an antineoplastic agent indicated for the treatment of advanced renal cell carcinoma.Temsirolimus is a Rapamycin 42 ester with [3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid and was first disclosed by Skotnicki et al in US Patent No. 5,362,718.

Several processes for the preparation of Temsirolimus have been reported in the literature such as those described in US 5,362,718; US 6,277,983 and US 7, 153,957.
US Patent No 5,362,718 discloses a process for the preparation of different rapamycin 42 esters including Temsirolimus as per the scheme given below (Scheme-I).

Scheme-I: Synthesis of Temsirolimus as disclosed in US Patent No. 5,362,718
The process is non-regioselective and hence results in 31-estehfied rapamycin, 31 , 42 diesterified rapamycin and unreacted rapamycin along with the desired rapamycin-42 ester.
US Patent No. 6,277,983 reports a process for the preparation of Temsirolimus by using 31 , 42 bis silyl intermediates as per the scheme shown below (Scheme-ll).

Scheme-ll: Synthesis of Temsirolimus as disclosed in US Patent No. 6,277,983 US Patent No. 7, 153,957 reports a process for the preparation of Temsirolimusby using boronate intermediate as per the scheme shown below (Scheme-Ill).

Scheme-Ill: Synthesis of Temsirolimus as disclosed in US Patent No. 7, 153,957
Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS “Amano” (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
………………………………………………..
SYNTHESIS
https://www.google.co.in/patents/EP0763039A1
Example 11
Rapamycin 42-ester with 2.2-bis-(hydroxymethyl)propionic acid
A solution of the product of Example 10 (2.8 g, 2.65 mmol) in 50 mL THF and
25 mL IN HCl was stirred at room temperature for 4 h. The mixture was diluted with water and extracted three times with EtOAc. The combined organic phases were washed with saturated NaHCO3 solution, saturated NaCl solution, dried over MgSO4, filtered and evaporated to a yellow oily solid. Purification by flash chromatography (3X with EtOAc) afforded the title compound (1.6 g, 59 %).
(-)FAB-MS mlz 1029.6 (M-), 590.4 (southern fragment), 437.3 (northern fragment). !H NMR (400 MHz, d-6 DMSO) δ 4.5 (m, 1 H, C(42)H), 3.45 (s, 4 H), 1.04 (s, 3 H).
*3C NMR (100.6 MHz, d-6 DMSO) δ 174.2, 63.7, 63.6, 49.9, 16.8.
Example 10 Rapamycin 42-ester with 2.2.5-trimethyl.1.3_dioxane-5-carboxyric acid
To a solution of the 2,2-bis(hydroxymethyl)propionic acid isopropylidene ketal (1.041 g, 5.98 mmol) (prepared according to the procedure of Bruice, J. Am. Chem. Soc. 89: 3568 (1967)) and triethylamine (0.83 mL, 5.98 mmol) in 20 mL anhydrous THF at 0 °C under nitrogen was added 2, 4, 6-trichlorobenzoyl chloride (0.93 mL, 5.98 mmol) and the resultant white suspension was stirred 5 h at room temperature. The precipitate was removed by vacuum filtration, rinsing the flask and filter cake with an additional 10 mL dry THF. The filtrate was concentrated by rotary evaporation to a white solid. The residue was dissolved in 20 mL dry benzene, then rapamycin (5.47 g, 5.98 mmol) and DMAP (0.731 g, 5.98 mmol) were added. After stirring overnight at room temperature, the mixture was diluted with EtOAc, washed with H2O and saturated NaCl (aq), dried over MgSO4, filtered and evaporated to a yellow oil. Flash chromatography (5X with 60% EtOAc-hexane) afforded the title compound (2.2 g, 34 %) as a white solid.
(-)FAB-MS mlz 1069.5 (M-), 590.3 (southern fragment), 477.2 (northern fragment). –■H NMR (400 MHz, d-6 DMSO) δ 4.57 (m, 1 H, C(42)H), 4.02 (d, 2 H), 3.60 (d, 2 H), 1.34 (s, 3 H), 1.24 (s, 3 H), 1.06 (s, 3 H). 1 C NMR (100.6 MHz, d-6 DMSO) δ 173.2, 99.0, 65.0, 22.2, 18.1.
…………………………………………..
SYNTHESIS
https://www.google.co.in/patents/US7153957
This scheme



Preparation of 5-Methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [A]
To a suspension of 2,2-bis(hydroxymethyl)propionic acid (131 g, 0.98 mole) in tetrahydrofuran (500 ml) was added a solution of phenylboronic acid (122 g, 1.0 mole) in tetrahydrofuran (500 ml). The mixture was stirred for 3 h and toluene (1.0 L) was added. Water was removed by azeotropic distillation with toluene. Heptanes (500 ml) was added to the precipitated product, heated to reflux and cooled. The mixture was filtered and washed with heptanes (2×300 ml). The solids were dried under vacuum at 70–75° C. until constant weight to give 94% yield. 1H NMR: δ (DMSO-d6) 7.65 (d, 2H, Ar), 7.40 (m, 3H, Ar), 4.35 (d, 2H, CH2), 3.92 (d, 2H, CH2), 1.17 (s, 3H, CH3)
Preparation of Rapamycin 42-ester with 5-methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [B]
As described in U.S. Pat. No. 6,277,983 (2001) a 3 L flask was charged with rapamycin (100 g, 0.104 mole) and dissolved in ethyl acetate (1.50 L). The solution was cooled to 5–10° C. Imidazole (30 g, 0.44 moles, 4.23 eq.) was added and dissolved. Under nitrogen protection, trimethylsilyl chloride (44 g, 0.405 mole, 4.0 eq.) was added over 30–40 min while maintaining the temperature at 0–5° C. during the addition. The mixture was held for a minimum of 0.5 h. The reaction was monitored by TLC (30:70 acetone:heptane eluent). The reaction was complete when all of the rapamycin was consumed.
Two to three drops of the reaction mixture were removed and retained as a 31,42-bis(trimethylsilyl) rapamycin reference standard. 0.5 N Sulfuric acid (300 mL) was added to the 3 L flask over 0.5 h maintaining the temperature 0–5° C. The mixture was stirred vigorously and held for 5 h. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone:heptane eluent). The reaction was complete when essentially no 31,42-bis-(trimethylsilyl) rapamycin was present. The layers were separated and the lower aqueous layer was back extracted with ethyl acetate (500 mL). The combined organic layers were washed with saturated brine (500 mL) and saturated sodium bicarbonate (2×200 mL) until pH 8 was obtained. The organic layer was washed with water (2×500 mL) and brine (500 ml) until pH 6 to 7 was obtained. The solution was dried over magnesium sulfate (100 g) for 30 min, filtered into a 2 L flask and concentrated to a volume of 135 ml. Ethyl acetate (500 ml) was added and concentrated to a volume of 135 ml. The water chase was repeated once more with ethyl acetate (500 ml). Methylene chloride (300 ml) was added and the solution held until needed in the next step.
A 3 L flask equipped with mechanical stirrer was charged with compound [A] (75 g, 0.341 mole) in methylene chloride (400 mL). Diisopropylethylamine (66.1 g, 0.51 mole) was added dropwise over 20 mins and rinsed with methylene chloride (25 mL). 2,4,6-Trichlorobenzoyl chloride (80 g, 0.328 mole) was added and rinsed with methylene chloride (25 mL). The mixture was held at 0–5° C. for 4 h, and cooled to −10±5° C.
The solution of 31-trimethylsilyl rapamycin was added to the 3 L flask containing the mixed anhydride, and rinsed with methylene chloride (25 mL). A solution of dimethylamino pyridine (48.5 g, 0.397 mole) in methylene chloride (150 mL) was prepared, added over 1.5 h, maintaining the temperature <−8° C., and rinsed with methylene chloride (25 mL). The mixture was held for 12 h at −11 to −5° C. The reaction mixture was quenched with 1 N sulfuric acid (600 ml) keeping the temperature <10° C. The mixture was stirred and held for 30 mins. The pH of the upper aqueous layer was ≦2. The layers were separated, and the lower organic layers washed with brine (450 ml), saturated sodium bicarbonate (500 mL) until pH ≧8. The organic layer was washed with water (450 ml) until pH 6–7 was obtained. The solution was concentrated, acetone (250 ml) added and concentrated. This was repeated with another portion of acetone (250 ml) and concentrated.
The solution was diluted with acetone. 0.5 N Sulfuric acid (500 ml) was added dropwise over 30 mins keeping the pot temperature 0–5° C. The mixture was held for a minimum of 5 h, during which time, the product precipitated out of solution. Aqueous sodium bicarbonate (30 g in 375 ml water) was added dropwise over 30 minutes keeping the pot temperature 0 to 5° C.; the mixture was held for a minimum of 30 minutes. Acetic acid (25 ml) was added until pH was 5–6 keeping the pot temperature <10° C. The mixture was warmed to room temperature and held for 16 h. The solid product was filtered and washed with water (2×100 ml) followed by 1:1 acetone:water (2×100 ml). The cake was purified in acetone (375 ml) to give 65 g (58% overall from rapamycin) of product [B]. LC/MS: using an electrospray interface in the positive ion mode afforded the molecular ion [M+Na]=1138.5 atomic mass units (amu).
Preparation of Rapamycin 42-ester with 2,2-bis(hydroxymethyl)-propionic acid, [C]
Compound [B] (200 g, 0.179 mole), was dissolved in tetrahydrofuran (600 ml), 2-methyl-2,4-pentanediol (42.3 g, 0.358 mole, 2.0 eq.) was added and the mixture stirred for a minimum of 3 h. The reaction mixture was concentrated to a foam. Diethyl ether (1.0 L) was added and the mixture stirred for 2 h. Heptanes (1.0 L) was added dropwise over 1 h and the mixture stirred for 2 h. The mixture was filtered and the solid product washed with heptanes (500 ml). The solids were re-dissolved in acetone (400 ml), re-treated with 2-methyl-2,4-pentanediol (21.1 g, 0.179 mole, 1 eq.) in acetone (200 ml), clarified through a 0.2 micron cartridge filter, and rinsed with acetone (200 ml). The solution was concentrated to a foam, diethyl ether (1.0 L), pre-filtered through a 0.2 micron cartridge filter, was added and the mixture stirred for 2 h. The mixture was co-precipitated by adding pre-filtered heptanes (1.0 L). The precipitated solids were filtered and washed with ether:heptane (2×500 ml). The solids were dried (55 to 60° C., 10 mm Hg, minimum 24 h) to give 159 g (86%) of product [C]. LC/MS: using APCl in the positive ion mode afforded the molecular ion [M+NH4]=1047.0 amu. The 1H NMR of the product (CCl-779) was identical to the product described in example 11 of U.S. Pat. No. 5,362,718 (1994).
…………………………………
Synthesis
http://www.google.com/patents/WO2005100366A1
Example 1 – Synthesis of Proline CCI-779

This example describes a method for the synthesis of the proline analog of CCI- 779, which is illustrated in the scheme provided above.
A.
Preparation of 31, 42-Bis (trimethylsilyl) proline rapamycin (Compound B)
A 3 -neck 50 mL flask was charged with proline rapamycin (compound A in the scheme) (1.47 g, 1.63 mmol), imidazole (0.45 g, 6.6 mmol, 4 eq.) and ethyl acetate (22.5 mL). The magnetically stirred mixture became cloudy. The mixture was cooled to 0-5°C. Under nitrogen protection, trimethylsilyl chloride (0.62 g, 5.7 mmol, 3.5 eq.) was added over 0.5 h via syringe while maintaining the temperature at 0-5°C during the addition. The syringe was rinsed with 2.5 ml ethyl acetate and the mixture held for 0.75 hours (0.75 h), whereupon a white precipitate was formed. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone :heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The mixture was shaken and allowed to settle. The upper organic layer was spotted against the starting material (proline rapamycin). The reaction was complete when no more starting material was present.
B.
Preparation of 31 -trimethylsilyl proline rapamycin, Compound E
When the above reaction was complete, 2-3 drops of the reaction mixture was removed and retained for the following step as the 31,42-bis(trimethylsilyl) proline rapamycin reference standard. To the 50 ml flask was added 0.5 N sulfuric acid (4.5 mL) over 0.5 h maintaining the temperature at 0-5 °C. The mixture became less cloudy. The mixture was held for 2.5 h and was monitored by thin layer chromatography (TLC, 30:70 acetone:heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The reaction aliquot was shaken and allowed to settle. The upper organic layer was spotted against the 31 ,42-bis(trimethylsilyl) proline rapamycin reference. The reaction was complete when essentially no 31,42-bis(trimethylsilyl) proline rapamycin was present. Ethyl acetate (5 mL) was added and the layers separated. The lower aqueous layer is extracted with ethyl acetate (7.5 mL). The combined organic layers were washed with brine (7.5 mL), by washing with saturated sodium bicarbonate (6 mL) followed by washing water (3 x 7.5 mL), in that order. The pH of the last water wash was 6-7. The organic layer was washed again with brine (7.5 mL) and dried over sodium sulfate (4 g) for 20 min. The mixture was filtered into a 250 mL flask and concentrated to dryness.
The solid was dried at room temperature under high vacuum (10 mmHg or less) for 20 h.
Weight = 1.51 g of an off-white foam.
C.
Preparation of Intermediate, Compound F:
A 3 -neck 100 mL flask equipped with mechanical stirrer was charged with
2,2,5-trimethyl[l,3-dioxane]-5-carboxylic acid, Compound C (0.63 g, 3.6 mmol) in methylene chloride (7.5 mL). Dusopropylethylamine (0.77 g, 5.9 mmol) was added, followed by a rinse with methylene chloride (1 mL). 2,4,6-Trichlorobenzoyl chloride (0.85 g, 3.5 mmol) was added, followed by a rinse with methylene chloride (1.5 mL).
The mixture was held at room temperature for 4.5 h. The solution was cooled to -12 ±
2°C. 31 -Trimethylsilyl proline rapamycin, compound E, (1.51 g) in methylene chloride (8 mL) was dissolved and added to the 100 mL flask. Methylene chloride (2 mL) was added as a rinse. A solution of dimethylamino pyridine (DMAP) (0.77 g, 6.8 mmol) in methylene chloride (3 mL) was prepared and added to the 100 mL flask over
2.5 h maintaining the temperature -12 ± 2 °C. Methylene chloride (1 mL) was added as a rinse. The mixture was held for 16 h and was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL water and 0.2 mL ethyl acetate. The HPLC sample was prepared by withdrawing 2 drops of the upper organic layer, blowdrying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04
Wavelength : λ = 280 nm Flow rate : 1 mL / min
Time : 60 min
Retention times : Compound E ~14.0-14.5 min Compound F -33.4-33.8 min
The reaction was complete when < 0.5% of starting material was present. The reaction mixture was quenched with water (6 mL). Methylene chloride (10 mL) was added and the layers separated. The aqueous layer was extracted with methylene chloride (10 mL). The combined organic layers were washed with 0.5 N sulfuric acid (12 mL), brine (10 mL), saturated sodium bicarbonate (6 mL), and water (3 x 10 mL) in that order. The pH of the last water wash was 6-7. The clear yellow solution was concentrated to a foam. The solid was dried at room temperature under high vacuum (10 mmHg or less) for 24 h. Weight = 1.88 g of a yellow foam.
D.
Preparation of crude proline CCI-779
A 1-neck 50 mL flask equipped with mechanical stirrer was charged with Compound F in THF (18.8 mL, 10 vols) and then cooled to 0 – 5 °C (or about -2.5°C). 2 N sulfuric acid (9.4 mL, 5 vols) was added over 2.5 h. After complete addition, the mixture was warmed to 2.5 °C and then held for 45 h. The reaction was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 0.25 mL ethyl acetate. The HPLC sample was prepared by withdrawing 5 drops of the upper organic layer, blow drying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04 Wavelength : λ= 280 nm Flow rate : 1 mL / min Time : 60 min Retention times Compound F ~33.4-33.8 min Desilylated Compound F ~10.5-11.5 min (intermediate) Proline CCI-779 -5.0-5.5 min The desilylated intermediate of compound F was formed first. The reaction was complete when < 0.5% of the silylated analog remained. Ethyl acetate (27 mL) and brine (7.5 mL) was added and the layers separated. The aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layers were washed with brine (10 mL), saturated sodium bicarbonate (7.5 mL), and water (3 x 7.5 mL) in that order. The pH of the last water wash was 6-7. The mixture was dried over sodium sulfate (5 g) for 30 min, filtered into a 250 L flask and concentrated to dryness. Weight = 1.58 g of a yellow foam.
E.
Chromatographic purification of crude proline CCI-779
A silica gel column (31.6 g, 60 A, 200-400 mesh) (22 cm length x 2.5 cm diameter) was prepared and conditioned with 15:85 acetone:HPLC grade hexane (1 L). The yellow crude proline CCI-779 (1.58 g) in acetone (1.58 mL) was prepared and chromatographed. The column was eluted with the remaining 15:85 acetone :hexane mixture followed by 25:75 acetone:hexane (4 L). The positive fractions were combined and concentrated to dryness. The resulting foam was dried at 35 °C, high vacuum (i.e., 10 mmHg or less) for 24 h. Weight = 1.12 g of a light yellow foam.
F.
Ether treatment of proline CCI-779
A 1 -neck 50 mL flask was charged with proline CCI-779 ( 1.12 g) and dissolved in ether (1.5 mL). The mixture was held for 2 h. The ether was stripped to give a foam. The foam was dried at 35 °C, under high vacuum (10 mmHg or less) for 12 h then at room temperature overnight (12 h). Weight = 1.09 g.
*H NMR (500 and 600 MHz, DMSO-d6) δ 5.45 (H-l), 6.12 (H-2), 6.27 (H-3), 6.41 (H-4), 6.20 (H-5), 3.66 (H-7), 1.14 and 1.86 (H-8), 4.02 (H-9), 1.19 and 1.81 (H-10), 1.52 (H-11), 2.03 (H-12), 3.23 and 3.54 (H-18), 1.76 (H-19), 2.20 and 1.89 (H-21), 4.22 (H-22), 4.87 (H-25), 2.28 and 2.70 (H-26), 3.22 (H-28), 5.11 (H-29), 4.04 (H-31), 4.17 (H-32), 2.25 (H-34), 0.985 and 1.38 (H-35), 2.22 (H-36), 1.76 (H-37), 0.961 and 1.11 (H-38), 1.31 (H-39), 0.726 and 1.90 (H- 40), 3.14 (H-41), 4.46 (H-42), 1.22 and 1.81 (H-43), 0.888 and 1.60 (H-44), 1.60 (H-45), 3.05 (H-46, OCH3), 0.697 (H-47), 6.48 (H-48), 0.821 (H-49), 1.76 (H-50), approx. 5.1- 5.3 (H-51), 3.17 (H-52, OCH3), 0.755 (H-53), 0.966 (H-54), 0.805 (H-55), 3.29 (H-56, OCH3), 3.46 (H-59), 1.01 (H-60), approx. 4.3-4.7 (0-61)
13C NMR (75 MHz, DMSO- d6) δ 139.12 (C-1), 130.53 (C-2), 132.49 (C-3), 127.08 (C-4), 127.21 (C-5), 137.12 (C-6), 81.93 (C-7), 40.40 (C-8), 65.83 (C-9), 29.45 (C-10), 25.87 (C-l l), 34.21 (C-12), 99.25 (C-13), 198.17 (C-15), 165.55 (C-16), 47.01 (C-18), 24.04 (C-19), 28.93 (C-21), 58.50 (C-22), 170.44 (C-23), 73.24 (C-25), 39.96 (C-26), 207.67 (C-27), 44.51 (C-28), 123.92 (C-29), 136.56 (C-30), 75.84 (C-31), 84.86 (C-32), 209.49 (C-33), 40.76 (C-34), 39.20 (C-35), 35.05 (C-36), 32.73 (C-37), 38.42 (C-38), 32.06 (C-39), 36.01 (C-40), 80.12 (C- 41), 75.92 (C-42), 29.25 (C-43), 30.24 (C-44), 10.27 (C-45), 55.48 (C-46, OCH3), 15.46 (C-47), 15.59 (C-49), 14.41 (C-50), 56.56 (C-52, OCH3), 12.67 (C-53), 21.50 (C-54), 14.89 (C-55), 57.27 (C-56, OCH3), 174.22 (C-57), 49.90 (C-58), 63.59 and 63.98 (C-59), 16.82 (C-60). MS [M+NH ] 1033.5, [ESI(+), M+Na+] 1038.7.
Example 3 – Synthesis of CCI-779:

A. Synthesis of CCI-779 via intermediate A Method 1 : A mixture of rapamycin (6 g), vinyl ester I (2 g), lipase PS-C “Amano” II (6 g) in anhydrous TBME (36 mL) was heated at 45 °C under Ar2 for 2 days. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, the oily residue was added to heptane while stirring. The batch was then cooled to -15 °C for 2 h, collect the solid on the Buchner funnel and washed with cold heptane, A was obtained as off-white solid, crude yield : 98%.MS (El): 1070 Above crude A (6g), dissolved in n-PrOH (24 mL) cooled to 0 °C with an ice-water bath, to this solution was added aqueous H2S04 (12 mL, 1.2N). The mixture was stirred for 24 h at 0°C and was then added to cold phosphate buffer (300 ml, pH=7.8), collect the solid on a Buchner funnel and washed with DI water and dry under vacuum, silica gel column purification eluting with hexane-acetone furnished CCI-779 as a white solid (5.2 g, 90%). MS (El): 1030 Method 2: A mixture of rapamycin (30.0 g, 32.8 mmol), vinyl ester I (10.0 g, 50 mmol), lipase PS-C “Amano” II (30 g) and molecular sieves (5 A) (10.0 g) in anhydrous TBME (150 mL) was heated at 42-43 °C under Ar2 for 48 hours. THF (100 mL) was added to dissolve the precipitation and the mixture was cooled to room temperature. Enzyme was removed by filtration and washed with THF (200 mL), the filtrate was concentrated to about 60 mL and diluted with THF (320 mL). The solution was then cooled to 0-5 °C, H2S04 (180 mL, 2N) was added dropwise over lh. The mixture was stirred for 48 h at 0-5 °C or until the disappearance of A as monitored by TLC. The mixture was diluted with brine (300 mL) and extracted with EtOAc (three times). The combined organic layer was washed with H20, 5% NaHC03, then brine and dried
(MgS04). Evaporation of solvent gave a light yellowish semi solid which was purified by flash chromatography (hexane/acetone, 2:1) to give CCI-779 as a white solid (30.77 g, 91% for two steps). B. Synthesis of CCI-779 via intermediate B: A mixture of rapamycin (3 g), vinyl ester II (1.2 g), lipase PS-C “Amano” II (5 g) in anhydrous TBME (45 mL) was heated at 45 °C under Ar2 for 60 h. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, MeOH (20 mL) was added to the residue and concentrated to dryness. Silica gel column purification of crude eluting with hexane-acetone furnished CCI-779 as a white solid (2.3 g), and recovered rapamycin (0.81 g). The yield is 93% based on the recovered rapamycin.
proline analog of CCI-779 (proline-rapamycin42-ester with 2,2-bis(hydroxymethyl)propionic acid or proline-CCI-779) and methods of synthesizing same. Proline-CCI-779 is an active drug substance useful in oncology and other associated indications (immunosuppression, anti-inflammatory, anti-proliferation and anti-tumor). In one aspect, the synthesis of proline-CCI-779 is accomplished through bis- silylation of proline rapamycin, mono-de-protecting 31 ,42-bis-trimethylsilyl proline rapamycin, and acylating the mono-silyl proline rapamycin followed by hydrolysis. In another aspect, the invention provides a two-step enzymatic process involving a regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to give CCI-779.
Example 4 – Synthesis of Proline-CCI-779 The enzymatic procedure of the invention can also be applied to the synthesis of proline CCI-779 from proline-rapamycin under essentially the same conditions as described in Example 2, procedure A for the synthesis of CCI-779 from rapamycin.

proline-rapamycin proline-CCI-779
………………….
more info added for readers
synthesis of CCI-779 or Proline CCI-779 (Temsirolimus) which is useful as an antineoplastic agent having the structure

It is stated to be effective in multiple applications, including inhibition of tumor growth, the treatment for multiple sclerosis and rheumatoid arthritis.
2. The Prior Arts

U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.


A selective synthesis of 42-monoacylated product was previously conducted by reacting rapamycin 31,42-bis-silyl ether, and then the 42-sily ether protection group is selectively removed to provide rapamycin-OH-31-sily ether (U.S. Pat. No. 5,563,145). In addition, a regioselective process for the preparation of CCI-779 is also described in U.S. Pat. No. 6,277,983 (Scheme2). First, rapamycin (compound 4b) is treated with excess chlorotrimethylsilane to form rapamycin31,42-bis-trimethylsilyl ether (compound 5), and then 42-trimethylsilyl ether protection group is selectively removed in mild acid to provide rapamycin 42-OH-31-trimethylsilyl ether (compound 6). This free 42-OH was then acylated with 2,4,6-trichlorobenzyl mixed anhydride of 2,2,5-trimethyl[1,3-dioxane]-5-carboxylic acid (compound 7) at −15° C. for 16 h to give rapamycin 31-trimethylsilyl ether 42-ester (compound 8). Following treatment with mild acid for a certain period, CCI-779 can be isolated. 2,4,6-trichlorobenzyl chloride is irritant, moisture sensitive and costly.

Further, as below-depicted in Scheme 3, U.S. Pat. No. 7,153,957 disclose another method for the CCI-779. It can be prepared by the acylation of 31-silyl ether of rapamycin with the anhydride derived from the 2-phenylboronate acid to give rapamycin 31-silyl ether, 42-boronate. Thereafter, it is hydrolyzed under mild acid condition to form rapamycin 42-ester boronate. After being treated with a suitable diol, CCI-779 was obtained (Scheme 3). Mixed anhydride is not satisfactory for commercial scale synthesis because it can be kept stable only for 48 hr at −5˜0° C., not durable for longer time.
synthesis ofTemsirolimus in a more economic way.

…………..
PAPERS
CCI-779
Drugs Fut 2002, 27(1): 7
Drugs Fut 2002, 27(1): 7
Organic Letters, 2005 , vol. 7, 18 pg. 3945 – 3948 seenmr
PATENTS
| United States | 5362718 | APPROVED 1994-04-18 | EXPIRY 2014-04-18 |
| Canada | 2429020 | 2009-05-26 | 2021-11-13 |
| Canada | 2187024 | 2004-08-10 | 2015-04-14 |
|
6-13-2012
|
N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF
|
|
|
11-18-2011
|
N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF
|
|
|
8-17-2011
|
N-Hydroxyamide Derivatives and Use Thereof
|
|
|
7-6-2011
|
Sulfonyl Amino Cyclic Derivatives and Use Thereof
|
|
|
11-24-2010
|
Benzothiazole Formulations and Use Thereof
|
|
|
11-19-2010
|
Indazole Compounds for Treating Inflammatory Disorders, Demyelinating Disorders and Cancers
|
|
|
9-31-2010
|
Process for preparation of temsirolimus
|
|
|
4-23-2010
|
COMBINATION OF BENZIMIDAZOLE ANTI-CANCER AGENT AND A SECOND ANTI-CANCER AGENT
|
|
|
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
|
|
|
6-12-2009
|
Administration of an Inhibitor of HDAC and an mTOR Inhibitor
|
|
6-8-2007
|
Methods for preparing crystalline rapamycin and for measuring crystallinity of rapamycin compounds using differential scanning calorimetry
|
|
|
4-11-2007
|
Proline CCI-779, production of and uses therefor, and two-step enzymatic synthesis of proline CCI-779 and CCI-779
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
7-12-2006
|
CCI-779 Isomer C
|
| US5362718 | 18 Apr 1994 | 8 Nov 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US6197967 | 13 Dec 1999 | 6 Mar 2001 | Clariant Gmbh | Process for the preparation of paraoxadiazolyphenylboronic acids |
| US6277983 | 27 Sep 2000 | 21 Aug 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO1995028406A1 | 14 Apr 1995 | 26 Oct 1995 | American Home Prod | Rapamycin hydroxyesters, process for their preparation and pharmaceutical compositions containing them |
| US7553843 | 6 Dec 2006 | 30 Jun 2009 | Wyeth | Process for the preparation of purified crystalline CCI-779 |
| US7605258 | 16 Oct 2007 | 20 Oct 2009 | Wyeth | Processes for the synthesis of individual isomers of mono-peg CCI-779 |
| US7622578 | 6 Dec 2006 | 24 Nov 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US7625726 | 29 Sep 2008 | 1 Dec 2009 | Wyeth | Process for preparing rapamycin 42-esters and FK-506 32-esters with dicarboxylic acid, precursors for rapamycin conjugates and antibodies |
| US7875612 | 24 Apr 2002 | 25 Jan 2011 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US7910594 | 13 May 2003 | 22 Mar 2011 | Endocyte, Inc. | Vitamin-mitomycin conjugates |
| US8026276 | 25 Jul 2003 | 27 Sep 2011 | Wyeth Llc | Parenteral CCI-779 formulations containing cosolvents, an antioxidant, and a surfactant |
| US8044200 | 14 Mar 2006 | 25 Oct 2011 | Endocyte, Inc. | Synthesis and purification of pteroic acid and conjugates thereof |
| US8105568 | 10 Jul 2009 | 31 Jan 2012 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| US8288557 | 22 Jul 2005 | 16 Oct 2012 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| US8299116 | 10 Aug 2011 | 30 Oct 2012 | Wyeth Llc | CCI-779 concentrate formulations |
| US8455539 | 15 Oct 2012 | 4 Jun 2013 | Wyeth Llc | CCI-779 concentrate formulations |
| US8465724 | 18 Aug 2006 | 18 Jun 2013 | Endocyte, Inc. | Multi-drug ligand conjugates |
| US8470822 | 7 May 2010 | 25 Jun 2013 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US8524893 | 28 Jan 2011 | 3 Sep 2013 | Fresenius Kabi Oncology Limited | Process for the preparation of temsirolimus and its intermediates |
| WO2011092564A2 | 20 Jan 2011 | 4 Aug 2011 | Fresenius Kabi Oncology Ltd | Process for the preparation of temsirolimus and its intermediates |
Orphan Drug Designation Granted for Epidiolex in Dravet syndrome by the FDA

Cannabidiol Seven Expanded Access INDs granted by FDA to U.S. physicians to treat with Epidiolex 125 children suffering from intractable epilepsy syndromes -
LONDON, Nov. 15, 2013
GW Pharmaceuticals plc (AIM: GWP, Nasdaq: GWPH, “GW”) announced today that the U.S. Food and Drug Administration (FDA) has granted orphan drug designation for Epidiolex(R), our product candidate that contains plant-derived Cannabidiol (CBD) as its active ingredient, for use in treating children with Dravet syndrome, a rare and severe form of infantile-onset, genetic, drug-resistant epilepsy syndrome. Epidiolex is an oral liquid formulation of a highly purified extract of CBD, a non-psychoactive molecule from the cannabis plant. Following receipt of this orphan designation, GW anticipates holding a pre-IND meeting with the FDA in the near future to discuss a development plan for Epidiolex in Dravet syndrome.
Dravet syndrome is a rare pediatric epilepsy syndrome with a distinctive but complex electroclinical presentation. Onset of Dravet syndrome occurs during the first year of life with clonic and tonic-clonic seizures in previously healthy and developmentally normal infants. Prognosis is poor and patients typically develop intellectual disability and life-long ongoing seizures. There are approximately 5,440 patients with Dravet in the United States and an estimated 6,710 Dravet patients in Europe. These figures may be an underestimate as this syndrome is reportedly underdiagnosed.
In addition to GW’s clinical development program for Epidiolex in Dravet syndrome, which is expected to commence in 2014, GW has also made arrangements to enable independent U.S. pediatric epilepsy specialists to treat high need pediatric epilepsy cases with Epidiolex immediately. To date in 2013, a total of seven “expanded access” INDs have been granted by the FDA to U.S. clinicians to allow treatment with Epidiolex of approximately 125 children with epilepsy. These children suffer from Dravet syndrome, Lennox-Gastaut syndrome, and other pediatric epilepsy syndromes. GW is aware of further interest from additional U.S. and ex-U.S. physicians to host similar INDs for Epidiolex. GW expects data generated under these INDs to provide useful observational data during 2014 on the effect of Epidiolex in the treatment of a range of pediatric epilepsy syndromes.
“I, together with many colleagues in the U.S. who specialize in the treatment of childhood epilepsy, very much welcome the opportunity to investigate Epidiolex in the treatment of Dravet syndrome. The FDA’s timely approval of the orphan drug designation for Epidiolex in Dravet syndrome is a key milestone that comes after many years of reported clinical cases that suggest encouraging evidence of efficacy for CBD in this intractable condition,” stated Dr. Orrin Devinsky, Professor of Neurology, Neurosurgery and Psychiatry in New York City. “With GW now making plans to advance Epidiolex through an FDA development program, we have the prospect for the first time of fully understanding the science of CBD in epilepsy with a view to making an appropriately tested and approved prescription medicine available in the future for children who suffer from this debilitating disease.”
“GW is proud to be at the forefront of this important new program to treat children with Dravet Syndrome and potentially other forms of intractable childhood epilepsy. For families in these circumstances, their lives are significantly impacted by constant and often times very severe seizures in children where all options to control these seizures have been exhausted,” stated Dr. Stephen Wright, GW’s R&D Director. “GW intends to advance a full clinical development program for Epidiolex in Dravet syndrome as quickly as possible, whilst at the same time helping families in the short term through supporting physician-led INDs to treat intractable cases. Through its efforts, GW aims to provide the necessary evidence to confirm the promise of CBD in epilepsy and ultimately enabling children to have access to an FDA-approved prescription CBD medicine.”
“This orphan program for Epidiolex in childhood epilepsy is an important corporate strategic priority for GW. Following receipt of today’s orphan designation, GW now intends to commence discussions with the FDA regarding the U.S. regulatory pathway for Epidiolex,” stated Justin Gover, GW’s Chief Executive Officer. “GW intends to pursue this development in-house and retains full commercial rights to Epidiolex.”
About Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition — generally a disease or condition that affects fewer than 200,000 individuals in the U.S. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication.
About GW Pharmaceuticals plc
Founded in 1998, GW is a biopharmaceutical company focused on discovering, developing and commercializing novel therapeutics from its proprietary cannabinoid product platform in a broad range of disease areas. GW commercialized the world’s first plant-derived cannabinoid prescription drug, Sativex(R), which is approved for the treatment of spasticity due to multiple sclerosis in 22 countries. Sativex is also in Phase 3 clinical development as a potential treatment of pain in people with advanced cancer. This Phase 3 program is intended to support the submission of a New Drug Application for Sativex in cancer pain with the U.S. Food and Drug Administration and in other markets around the world. GW has established a world leading position in the development of plant-derived cannabinoid therapeutics and has a deep pipeline of additional clinical-stage cannabinoid product candidates targeting epilepsy (including an orphan pediatric epilepsy program), Type 2 diabetes, ulcerative colitis, glioma and schizophrenia. For further information, please visit http://www.gwpharm.com.
Cannabidiol (CBD) is one of at least 85 cannabinoids found in cannabis.It is a major constituent of the plant, second to tetrahydrocannabinol (THC), and represents up to 40% in its extracts. Compared with THC, cannabidiol is not psychoactive in healthy individuals, and is considered to have a wider scope of medical applications than THC, including to epilepsy, multiple sclerosis spasms, anxiety disorders, bipolar disorder,schizophrenia,nausea, convulsion and inflammation, as well as inhibiting cancer cell growth. There is some preclinical evidence from studies in animals that suggests CBD may modestly reduce the clearance of THC from the body by interfering with its metabolism.Cannabidiol has displayed sedative effects in animal tests. Other research indicates that CBD increases alertness. CBD has been shown to reduce growth of aggressive human breast cancer cells in vitro, and to reduce their invasiveness.

Amgen: Phase III Melanoma treatment, Talimogene Laherparepvec Improves Survival
 |
|
|---|---|
| Transmission electron micrograph of an unmodified herpes simplex virus |
Talimogene Laherparepvec
Amgen Presents Interim Overall Survival Data From Phase 3 Study Of Talimogene Laherparepvec In Patients With Metastatic Melanoma
T-VEC was engineered from herpes simplex 1 (HSV-1), a relatively innocuous virus that normally causes cold sores. A number of genetic modifications were made to the virus in order to:
- Attenuate the virus (so it can no longer cause herpes)
- Increase selectivity for cancer cells (so it destroys cancer cells while leaving healthy cells unharmed)
- Secrete the cytokine GM-CSF (a protein naturally secreted in the body to initiate an immune response)
T-VEC has a dual mechanism of action, destroying cancer both by directly attacking cancer cells and also by helping the immune system to recognize and destroy cancer cells. T-VEC is injected directly into a number of a patient’s tumors. The virus invades both cancerous and healthy cells, but it is unable to replicate in healthy cells and thus they remain unharmed. Inside a cancer cell, the virus is able to replicate, secreting GM-CSF in the process. Eventually overwhelmed, the cancer cell lyses (ruptures), destroying the cell and releasing new viruses, GM-CSF, and an array of tumor-specific antigens (pieces of the cancer cell that are small enough to be recognized by the immune system).
The GM-CSF attracts dendritic cells to the site. Dendritic cells are immune cells that process and present antigens to the immune system so that the immune system can then identify and destroy whatever produced the antigen. The dendritic cells pick up the tumor antigens, process them, and then present them on their surface to cytotoxic (killer) T cells. Now the T cells are essentially “programmed” to recognize the cancer as a threat. These T cells lead an immune response that seeks and destroys cancer cells throughout the body (eg, tumors and cancer cells that were not directly injected with T-VEC).

In this way, T-VEC has both a direct effect on injected tumors and a systemic effect throughout the entire body. Because the adaptive immune system “remembers” a target once it has been identified, there is high likelihood that the effect of an oncolytic virus like T-VEC will be durable (eg, prevent relapse). And it is for this reason that T-VEC does not need to be injected into every tumor, just a few in order to start the immune process.
Clinical efficacy in unresectable melanoma has been demonstrated in Phase II and Phase III clinical trials.
The Phase II clinical trial was published in the Journal of Clinical Oncology in 2009. 50 patients with advanced melanoma (most of whom had failed previous treatment) were treated with T-VEC. The overall response rate (patients with a complete or partial response per RECIST criteria) was 26% (16% complete responses, 10% partial responses). Another 4% of patients had a surgical complete response, and another 20% had stable disease for at least 3 months. On an extension protocol, 3 more patients achieved complete responses, and overall survival was 54% at 1 year and 52% at 2 years—demonstrating that responses to T-VEC are quite durable.
Consistent with other immunotherapies, some patients exhibited initial disease progression before responding to therapy because of the time it takes to generate the full immune response. Responses were seen in both injected and uninjected tumors (including those in visceral organs), demonstrating the systemic immunotherapeutic effect of T-VEC. Treatment was extremely well tolerated, with only Grade 1 or 2 drug-related side effects, the most common being mild flu-like symptoms.

|

|
Amgen announced the initial results of the Phase III OPTiM trial on Mar. 19, 2013. This global, randomized, open-label trial compared T-VEC with subcutaneously administered GM-CSF (2:1 randomization) in 430 patients with unresectable stage IIIB, IIIC or IV melanoma. The primary endpoint was durable response rate (DRR), defined as a complete or partial tumor response lasting at least 6 months and starting within 12 months of treatment.

T-VEC was proven to offer superior benefits in metastatic melanoma. DRR was achieved in 16% of patients receiving T-VEC compared with only 2% in the GM-CSF control group (P<.0001). The greatest benefit was seen in patient with stage IIIB or IIIC melanoma, with a 33% DRR vs 0% with GM-CSF. The objective response rate (any response) with T-VEC was 26%, with an impressive 11% of patients experiencing a complete response (complete disappearance of melanoma throughout the body). This demonstrated once again that T-VEC has a systemic immune effect that destroys distant, uninjected tumors. According to Financial Times one of the investigators involved questioned the ethics of the trial design, as the control arm received subcutaneous GM-CSF instead of standard care

A trend toward improved survival with T-VEC was observed in a pre-specified interim analysis of this endpoint, with the final survival data (event-driven) expected in late 2013. At the interim analysis, T-VEC was associated with a 21% reduced risk of death. The most common side effects with T-VEC were fatigue, chills, and fever. No serious side effect occurred in more than 3% of patients in either arm of the study.
The investigators concluded that “T-VEC represents a novel potential [treatment] option for melanoma with regional or distant metastases.” The success of T-VEC in the OPTiM trial represents the first Phase III proof of efficacy for a virus-based oncolytic immunotherapy.
