
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 255)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Edible flowers may inhibit chronic diseases
A new study in the Journal of Food Science, published by the Institute of Food Technologists (IFT), found that common edible flowers in China are rich in phenolics and have excellent antioxidant capacity.
Edible flowers, which have been used in the culinary arts in China for centuries, are receiving renewed interest. Flowers can be used as an essential ingredient in a recipe, provide seasoning to a dish, or simply be used as a garnish. Some of these flowers contain phenolics that have been correlated with anti-inflammatory activity and a reduced risk of cardiovascular disease and certain cancers.
The findings of this study show that common edible flowers have the potential to be used as an additive in food to prevent chronic disease, help health promotion and prevent food oxidization. However, the antioxidant mechanisms, the anti-tumor, anti-inflammation and anti-aging activity of the edible flower extracts should be further studied to…
View original post 13 more words
Cebranopadol GRT 6005 セブラノパドール a Potent Analgesic NOP and Opioid Receptor Agonist

- C24H27FN2O
- Average mass: 378.482391 Da

Neuropathic pain
Neuropathic pain is caused when peripheral nerves are damaged by mechanical, metabolic or inflammatory way. The pain occurring images are mainly due to the occurrence of spontaneous pain, hyperalgesia and allodynia (pain is already triggered by non-noxious stimuli) in. As a result, the lesions to increased expression of Na + channels and thus to spontaneous activity in the damaged axons and their Nachbaraxonen (England et al., Neurology, 1996, 47, 272-276).The excitability of the neurons is increased and they react to incoming stimuli with an increased discharge frequency. This results in an increased sensitivity to pain, which contributes to the development of hyperalgesia and spontaneous pain (Baron, Clin J Pain 2000;. 16 (2 Suppl), 12-20). The causes and manifestations, and therefore the treatment needs of neuropathischerm pain are varied. They arise as a result of injury or disease of the brain, spinal cord or peripheral nerves.Causes may be operations, such as phantom pain after amputation, stroke, multiple sclerosis, spinal cord injury, alcohol or drug abuse or other toxins, cancers but also
Metabolic diseases such as diabetes, gout, kidney failure or liver cirrhosis, or infectious diseases such as mononucleosis, ehrlichiosis, typhoid, diphtheria, HIV, syphilis or Lyme disease. The pain experience is very different signs and symptoms that can change over time in number and intensity. Paradoxically, patients with neuropathic pain outline a slowdown or failure of acute pain perception and the simultaneous increase of neuropathic pain. The typical symptoms of neuropathic pain as tingling, burning, shooting or described, or radiating electrifying. Pharmacological basis for treatment of neuropathic pain include tricyclic antidepressants and anticonvulsants, which are used as monotherapy or in combination with opioids. These drugs usually provide only a certain pain relief during a pain-free but is often not achieved. The often-adjusting side effects are dose increases while the drug to achieve adequate pain relief often in the way. In fact, a higher dosage of a μ-opioid is often required as the treatment of acute pain, thereby reducing the side effects get even more important for satisfactory treatment of neuropathic pain. By the occurrence of typical μ-opioid tolerance development and the concomitant need for dose escalation of this problem is exacerbated. In summary it can be stated that neuropathic pain is difficult to treat and today is alleviated by high doses of μ-opioids only partially (Saudi Pharm J. 2002, 10 (3), 73-85). There is therefore an urgent need for medicines for the treatment of chronic pain, the dose should not be increased until the occurrence of intolerable side effects to ensure a satisfactory pain treatment.
……………
http://www.google.com/patents/US7547707
Example 24 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Non-polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (651 mg, 3 mmoles) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 537 mg, 3 mmoles) were initially introduced into abs. MC (20 ml) under argon. Trifluoromethanesulfonic acid trimethylsilyl ester (0.6 ml, 3.1 mmoles) was then added very rapidly. The mixture was stirred at RT for 20 h. For working up, 1 M NaOH (30 ml) was added to the reaction mixture and the mixture was stirred for 30 min. The organic phase was separated, and the aqueous phase which remained was extracted with MC (3×60 ml). The combined organic phases were washed with water (2×30 ml) and dried over sodium sulfate. Methanol (30 ml) was added to the solid residue obtained after the solvent had been distilled off, and the mixture was heated, and stirred for 15 hours. The solid contained in the suspension was filtered off with suction and dried. 955 mg of the more non-polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole were obtained (m.p. 284-292° C.). 850 mg of this were dissolved in hot ethanol (900 ml), and a similarly hot solution of citric acid (1 g, 5.2 mmoles) in ethanol (20 ml) was added. After approx. 15 minutes, crystals precipitated out at the boiling point. After cooling to approx. 5° C., the mixture was left to stand for 2 h. The solid formed was filtered off with suction. 640 mg of the hemicitrate were obtained as a white solid (m.p. 258-282° C.).
Example 25 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (217 mg, 1 mmole) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 179 mg, 1 mmole) were dissolved in conc. acetic acid (4 ml). Phosphoric acid (1 ml, 85 wt. %) was slowly added dropwise to this mixture. The mixture was stirred at RT for 16 h. For working up, the mixture was diluted with water (20 ml), brought to pH 11 with 5 M NaOH and extracted with MC (3×20 ml). The combined organic phases were dried with sodium sulfate and evaporated. The residue (364 mg of white solid) was suspended in hot ethanol (20 ml), and a similarly hot solution of citric acid (185 mg, 0.96 mmole) in ethanol (5 ml) was added. The residue thereby dissolved completely and no longer precipitated out even on cooling to approx. 5° C. Ethanol was removed on a rotary evaporator and the hemicitrate of the more polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole was obtained in this way in a yield of 548 mg as a white solid (m.p. 148-155° C.).
| 24 |
 |
hemicitrate | more non-polar diastereomer |
| 25 |
 |
hemicitrate | more polar diastereomer |
(1 r,4r)-6′-fluoro-N,N- dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1 ,1 ‘-pyrano[3,4-b]indol]-4-amine (free base), has the following structural formula (I):
http://www.google.com/patents/WO2013113690A1?cl=en


One particular drug that is of great interest for use in treating cancer pain (and other acute, visceral, neuropathic and chronic pain pain disorders) is (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine. This drug is depicted below as the compound of formula (I).

The solid forms of (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine that are known so far are not satisfactory in every respect and there is a demand for advantageous solid forms

In a previous communication, our efforts leading from 1 to the identification of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amine 2a as analgesic NOP and opioid receptor agonist were disclosed and their favorable in vitro and in vivo pharmacological properties revealed. We herein report our efforts to further optimize lead 2a, toward trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (cebranopadol, 3a), which is currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol
http://pubs.acs.org/doi/full/10.1021/ml500117c
b]indol]-4-amine, trans-, 2-hydroxy-1,2,3-propanetricarboxylate (2:1)
2.76 (m,6 H); 3.88 (t, 2 H); 6.86 (dt, 1 H); 7.10 (dd, 1 H); 7.30-7.43 (m, 6 H); 10.91 (br
s, 1 H).
overlap); 71.5; 72.2; 102.3 (2JC,F = 23 Hz); 105.6 (3JC,F = 4 Hz); 108.3 (2JC,F = 26 Hz);
156,7 (1JC,F = 231 Hz); 171.3 (2 C), 175.3.HPLC-MS: m/z 378.9 [M + H]+
| US20120034297 * | Aug 4, 2011 | Feb 9, 2012 | Gruenenthal Gmbh | Pharmaceutical dosage forms comprising 6′-fluoro-(N-methyl- or N,N-dimethyl-)-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3’h-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| WO2004043967A1 | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2008040481A1 | Sep 26, 2007 | Apr 10, 2008 | Gruenenthal Gmbh | MIXED ORL 1/μ AGONISTS FOR TREATING PAIN |
-
CORAL – Cebranopadol Versus Morphine Prolonged-release in Patients With Chronic Moderate to Severe Pain Related to Cancer
Efficacy, Safety, and Tolerability of Oral Cebranopadol Versus Morphine Sulfate PR in Subjects With Chronic Moderate to Severe Pain Related to Cancer.Average amount of daily rescue medication at the end of the maintenance period.
UK Clinical Trials Gateway, 07 October 2013
-
CORAL XT – Open-label Extension Trial of the CORAL Trial
An Open-label, Multi-site Trial to Describe the Safety and Tolerability of Oral Cebranopadol Administered for 26 Weeks in Subjects With Cancer-related Pain Who Have Completed Treatment in the KF6005/07 Trial.Absolute…
UK Clinical Trials Gateway, 12 December 2013
| WO2004043967A1 * | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2005066183A1 * | Dec 21, 2004 | Jul 21, 2005 | Gruenenthal Gmbh | Spirocyclic cyclohexane derivatives with affinity for the orl1-receptor |
| US20050153998 * | Aug 19, 2004 | Jul 14, 2005 | Fumitaka Ito | Tetrahydroisoquinoline or isochroman compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7799931 * | Feb 17, 2009 | Sep 21, 2010 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7951948 * | Apr 19, 2010 | May 31, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7960404 | Aug 21, 2009 | Jun 14, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8034936 | Nov 4, 2010 | Oct 11, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds useful to treat substance dependency |
| US8053576 | Feb 17, 2009 | Nov 8, 2011 | Gruenenthal Gmbh | Treating conditions associated with the nociceptin/ORL1 receptor system, e.g. pain, drug withdrawal, anxiety, muscle relaxants, anxiolytic agents; e.g. 1,1-[3-dimethylamino-3-(pyridin-2-yl)pentamethylene]-3,4-dihydro-1H-2,9-diazafluorene |
| US8288406 | Sep 22, 2010 | Oct 16, 2012 | Gruenenthal Gmbh | Hydroxymethylcyclohexylamines |
| US8288430 | Mar 25, 2009 | Oct 16, 2012 | Grunenthal Gmbh | Spiro(5.5)undecane derivatives |
| US8293758 * | Mar 25, 2009 | Oct 23, 2012 | Grunenthal Gmbh | Substituted spirocyclic cyclohexane derivatives |
| US8357705 | Mar 25, 2009 | Jan 22, 2013 | Gruenenthal Gmbh | Substituted cyclohexyldiamines |
| US8404740 | Aug 21, 2009 | Mar 26, 2013 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8614245 * | Jan 8, 2013 | Dec 24, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US8618156 * | Jul 6, 2012 | Dec 31, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3’h-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
http://www.stumbleupon.com/stumbler/amcrasto
VIETNAM
ICELAND
RUSSIA

========================

Trifarotene
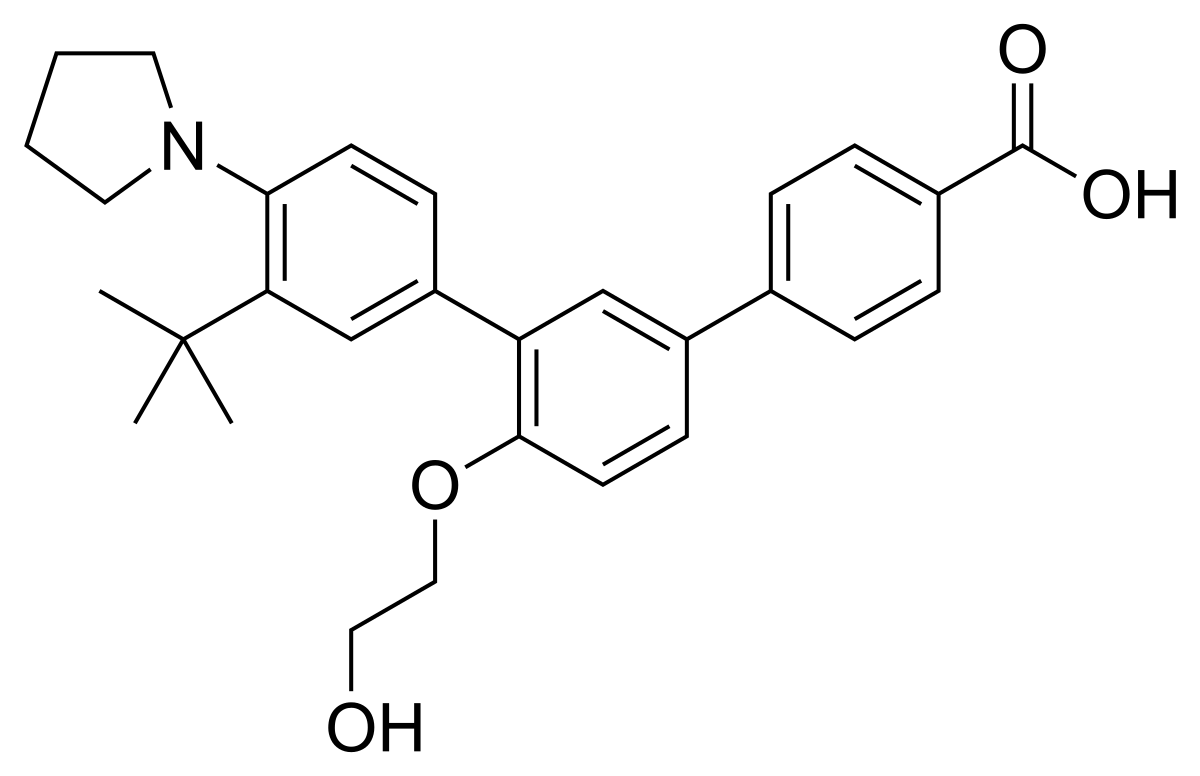
Trifarotene
CAS 895542-09-3
3”-Tert-butyl-4′-(2-hydroxyethoxy)-4”-(pyrrolidin-1-yl)(1,1′:3′,1”)terphenyl-4-carboxylic acid
3′-[3-tert-butyl-4-(pyrrolidin-1-yl)phenyl]-4′-(2-hydroxyethoxy)-[1,1′-biphenyl]-4-carboxylic acid
UNII-0J8RN2W0HK,
Galderma Research & Development
459.5766
C29 H33 N O4
- CD-5789
- CD5789
Trifarotene, sold under the brand name Aklief, is a medication for the topical treatment of acne vulgaris in those nine years of age and older.[1] It is a retinoid;[2] more specifically, it is a fourth generation selective retinoic acid receptor (RAR)-γ agonist.[3]
It was approved for medical use in the United States in 2019,[1][4][5] but is not approved in the European Union as of January 2021.[6] Trifarotene was granted orphan drug designation for the treatment of congenital ichthyosis by both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).[7][8]
State Solid
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 245C | FDA Label |
| pKa | 5.69 (pKa1) | FDA Label |
USFDA
The drug substance, trifarotene, a terphenyl acid derivative, is a retinoic
acid receptor (RAR) aQonist and is classified as a rotenoid. Trifarotene
intended as a drug for the treatment of acne vulgaris. Since trifarotene
has not been previously approved as an active ingredient in any drug
product in the United States, it is classified as a new molecular entity
(NME).
Trifarotene is produced as a white to off-white to slightly yellow crystalline
powder. It is slightly soluble in acetone, ethanol, and toluene, very slight
soluble in isopropanol, and practically insoluble in water (tiJT4
1
Cb><“JTrifarotene is nonhygroscopic and has pKa1 of 5.69 and pKa2 of 4.55. The chemical name
for trifarotene is 4-{3-[3-tert-butyl-4-(pyrrolidin-1-yl) phenyl]-4-(2-
hydroxyethoxy) phenyl} benzoic acid. It has the chemical formula of
C29H33NQ4, the molecular weiQht of 459.59, …………https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211527Orig1s000ChemR.pdf
Prescription Products
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Aklief | Cream | 50 mcg | Topical | Galderma | 2019-11-28 | Not applicable |  |
|
| Aklief | Cream | 50 ug/1g | Topical | Galderma Laboratories, L.P. | 2019-10-04 | Not applicable |  |
For treatment of congenital ichthyosis, PRECLINICAL, Galderma Res & Dev,
Galderma announced that the U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation status for the company’s trifarotene molecule for the treatment of congenital ichthyosis. Based on this decision, Galderma plans to implement a clinical development plan, reinforcing its commitment to exploring new treatment options for rare diseases, as well as meeting the needs of all patients with skin diseases over the course of their lives.
Galderma治療先天性魚鱗癬的Trifarotene分子取得FDA的孤兒藥資格認定
http://news.msn.com.tw/market3773054.aspx

The company’s molecule trifarotene is a selective agonist of the gamma retinoic acid receptor (RAR-gamma), which is currently in clinical development for use in other more common dermatological conditions. It is the drug’s retinoid functionality and potent keratolytic properties that make it a potentially viable treatment of the lamellar ichthyosis pathology. Galderma has already initiated the program for investigating the treatment of lamellar ichthyosis with trifarotene and is currently working in collaboration with regulatory authorities to implement an innovative and expedient clinical development plan.
Ichthyoses comprise a large group of skin scaling disorders with diverse etiologies. The stereotypic pathophysiology is epidermal hyperplasia and abnormal desquamation, leading to visible accumulation of squames (scales) on the skin’s surface. Congenital ichthyosis is a term used to refer to a specific group of rare inherited forms of ichthyoses that are generally more severe than non-inherited forms of the disease. Lamellar ichthyosis is one such disorder that falls within the congenital ichthyosis category. Lamellar ichthyosis is recognized as a severe disease which persists throughout life. After birth, during the first post-natal weeks, the hyperkeratotic (colloidion) membrane patients are typically born with, is gradually shed and is replaced by scaling and lichenification that involves the entire body, including face, scalp, palms and soles. While usually not life threatening, lamellar ichthyosis can result in disability, partial deafness, poor adaptation to environmental conditions (due to hypohydrosis), severe discomfort (pruritus, fissuring of the skin), and significant psycho-social impact. The estimated prevalence of LI in the US is in the range of 1 per 100,000 to 1 per 200,000 persons.
Synthesis Reference
Thoreau, E. et. al. Structure-based design of Trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of acne. Bioorganic & Medicinal Chemistry Letters, Volume 28, Issue 10. 2018. Pages 1736-1741
https://www.sciencedirect.com/science/article/abs/pii/S0960894X18303482
Trifarotene – Synthetic Route 1

Synthetic Description
Reference: Biadatti, Thibaud; Dumais, Laurence; Soulet, Catherine; Talano, Sandrine; Daver, Sebastien. Preparation of [1,1′:3′,1”]terphenyl-4-carboxylic acid and esters a novel ligands modulating retinoic acid receptors (RAR), and use thereof in human medicine and in cosmetics. Assignee Galderma Research & Development, S.N.C., Fr. WO 2006066978. (2016).
PATENT
WO 2006066978
http://www.google.com/patents/WO2006066978A1?cl=en
Example 25 – 3″-ter.-Butyl-4′-(2-hvdroxyethoxy)-4″-pyrrolidin-1-ylM,1′:3′,1″1- terphenyl-4-carboxylic acid

In a manner similar to that of Example 6b, by reacting 500 mg (0.9 mmol) of ethyl 4′-(2- acetoxyethoxy)-3″-terf-butyl-4″-pyrrolidin-1 -yl[1 , 1 ‘;3’, 1 “]terphenyl-4-carboxylate with
300 mg (8 mmol) of sodium hydroxide, 242 mg of 3″-tert-butyl-4′-(2-hydroxyethoxy)-4″- pyrrolidin-1-yl[1l1′;3′,1″]terphenyl-4-carboxylic acid are obtained (yield = 55 %) in the form of a white solid (m.p. = 2230C).
1H NMR (DMSO. 400 MHz): 1.43 (s, 9H); 1.90 (m, 4H); 3.0 (m, 4H); 3.73 (d, J=4.7Hz, 2H); 4.1 (m, 2H); 4.7 (s, 1H); 7.2 (d, 1H, J=8.6Hz); 7.48 (m, 2H); 7.59 (d, J=1.6Hz, 1H); 7.64 (d, J=UHz, 1H); 7.68 (dd, J=2Hz, 7.8Hz, 1H); 7.82 (d, J=8.3Hz, 2H); 7.99 (d, J=8.4Hz, 2H).
PATENT
WO 2013178759
http://www.google.com/patents/WO2013178759A1?cl=en
PATENT
WO 2013178758
http://www.google.com/patents/WO2013178758A1?cl=en
PATENT
WO 2013178760
http://www.google.com/patents/WO2013178760A1?cl=en
The details of skin application are given in the table below.

SYN
New Drug Approvals for 2019: Synthesis and Clinical Applications
New Drug Approvals for 2019: Synthesis and Clinical Applications
Shuo Yuan, Bin Yu, Hong-Min Liu
PII: S0223-5234(20)30639-5
DOI: https://doi.org/10.1016/j.ejmech.2020.112667
Reference: EJMECH 112667
To appear in: European Journal of Medicinal Chemistry
Trifarotene (Aklief). In October 2019, trifarotene, a topical retinoid that
selectively targets retinoic acid receptor gamma (RAR-γ), was approved by the FDA
for the treatment of acne vulgaris [142]. The drug was developed and marketed by
Galderma Pharmaceutical in Switzerland. Trifarotene is considered as the first of the
‘fourth-generation’ retinoids due to its uniquely selective agonism at RAR-γ. The
selective agonism leads to downstream alterations, confering improved efficacy and
reduced side effects [143]. In two phase 3 clinical trials of 2420 patients with
moderate acne on the face and trunk, trifarotene was well tolerated and significantly
reduced inflammatory lesions as early as two weeks on the face and four weeks on the
back, shoulders and chest compared to vehicle (p<0.05) [144].
The synthetic approach of this drug was disclosed by Galderma Research &
Development (Scheme 25) [145]. Bromination of commercially available
2-(tert-butyl)aniline 171 gave 4-bromo-2-(tert-butyl)aniline 172 in quantitative yield,
which then reacted with 1-dibromobutane 173 to give phenylpyrrolidine 174 in 52%
yield. Miyaura reaction of 174 was realized by employing n-BuLi and triisopropyl
borate (TIPB) followed by washed with aqueous HCl, resulting in arylboronic acid
adduct 175 in 66% yield. Treatment of 175 with aromatic bromide 176 in the presence
of Pd(PPh3)4 gave the coupling product 177 in 47% yield, which then underwent
hydrolysis delivering trifarotene (XIX) in 55% yield.
The preparation of coupling partner 176 is depicted in Scheme 26. Esterification of
4-hydroxy-4-biphenylcarboxylic acid 178 gave ethyl benzoate derivative 179 upon
treatment with catalytic H2SO4 in the refluxing EtOH [145]. The resulting ester was
subjected to treatment with tetrabutylammonium bromide (TBAB) in THF, resulting
in bromide 180 in good yields, further NaH-mediated Williamson ether synthesis with
2-bromoethyl acetate 181 gave 176 in 95% yield.

[142] L.J. Scott, Trifarotene: first approval, Drugs 79 (2019) 1905-1909.
[143] E. Thoreau, J.M. Arlabosse, C. Bouix-Peter, S. Chambon, L. Chantalat, S.
Daver, L. Dumais, G. Duvert, A. Feret, G. Ouvry, J. Pascau, C. Raffin, N.
Rodeville, C. Soulet, S. Tabet, S. Talano, T. Portal, Structure-based design of
trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of
acne, Bioorg. Med. Chem. Lett. 28 (2018) 1736-1741.
[144] J. Tan, D. Thiboutot, G. Popp, M. Gooderham, C. Lynde, J.D. Rosso, J. Weiss,
U. Blume-Peytavi, J. Weglovska, S. Johnson, L. Parish, D. Witkowska, N.S.
Colon, A.A. Saenz, F. Ahmad, M. Graeber, L.S. Gold, Randomized phase 3
evaluation of trifarotene 50 µg/g cream treatment of moderate facial and truncal
acne, J. Am. Acad. Dermatol. 80 (2019) 1691-1699.
[145] T. Biadatti, L. Dumais, C. Soulet, S. Talano, S. Daver, Novel ligands that
modulate rar receptors, and use thereof in human medicine and in cosmetics,
2006. WO2006066978.
| WO2006066978A1 * | Dec 21, 2005 | Jun 29, 2006 | Galderma Res & Dev | Novel ligands that modulate rar receptors, and use thereof in human medicine and in cosmetics |
| EP0826366A2 | Aug 1, 1997 | Mar 4, 1998 | Unilever N.V. | Cosmetic compositions containing hydroxy acid or retinoid |
| EP0989846A2 | Sep 22, 1998 | Apr 5, 2000 | E-L Management Corp. | Non-irritating cosmetic and pharmaceutical compositions |
| EP1831149A1 | Dec 21, 2005 | Sep 12, 2007 | Galderma Research & Development | Novel ligands that modulate rar receptors and use thereof in human medicine and in cosmetics |
| FR2915682A1 * | Title not available | |||
| US5851538 | Dec 29, 1995 | Dec 22, 1998 | Advanced Polymer Systems, Inc. | Retinoid formulations in porous microspheres for reduced irritation and enhanced stability |
| WO1999010308A1 * | Aug 21, 1998 | Mar 4, 1999 | Bernardon Jean Michel | Biphenyl derivatives substituted by an aromatic or heteroaromatic radical and pharmaceutical and cosmetic compositions containing same |
| US6150413 * | May 26, 1998 | Nov 21, 2000 | Centre International De Recherches Dermatologiques | Treatment of dermatological, rheumatic, respiratory, cardiovascular, bone and ophthalmological disorders, as well as mammalian skin and hair conditions; 4-(4-(biphenyl-2-yl)but-3-en-1-ynyl)benzoic acid, for example |
 |
|
| Clinical data | |
|---|---|
| Trade names | Aklief |
| Other names | CD5789 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620004 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Topical |
| Drug class | Skin and mucous membrane agents |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.278.901 |
| Chemical and physical data | |
| Formula | C29H33NO4 |
| Molar mass | 459.586 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Jump up to:a b “Drug Trials Snapshots: Aklief”. U.S. Food and Drug Administration (FDA). 11 October 2019. Archived from the original on 19 November 2019. Retrieved 18 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Trifarotene Monograph
- ^ Scott LJ (November 2019). “Trifarotene: First Approval”. Drugs. 79 (17): 1905–1909. doi:10.1007/s40265-019-01218-6. PMID 31713811.
- ^ “Aklief (trifarotene) FDA Approval History”. Drugs.com. 7 October 2019. Retrieved 19 November 2019.
- ^ “Drug Approval Package: Aklief”. U.S. Food and Drug Administration (FDA). 21 October 2019. Archived from the original on 19 November 2019. Retrieved 18 November 2019.
- ^ “Trifarotene”. European Medicines Agency. Retrieved 17 June 2020.
- ^ “Trifarotene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 19 August 2020.
- ^ “EU/3/20/2264”. European Medicines Agency (EMA). 12 August 2020. Retrieved 19 August 2020.
External links
- “Trifarotene”. Drug Information Portal. U.S. National Library of Medicine (NLM).
- Aubert J, Piwnica D, Bertino B, Blanchet-Rethore S, Carlavan I, Deret S, Dreno B, Gamboa B, Jomard A, Luzy AP, Mauvais P, Mounier C, Pascau J, Pelisson I, Portal T, Rivier M, Rossio P, Thoreau E, Vial E, Voegel JJ: Nonclinical and human pharmacology of the potent and selective topical retinoic acid receptor-gamma agonist trifarotene. Br J Dermatol. 2018 Aug;179(2):442-456. doi: 10.1111/bjd.16719. Epub 2018 Jul 4. [PubMed:29974453]
- Balak DMW: Topical trifarotene: a new retinoid. Br J Dermatol. 2018 Aug;179(2):231-232. doi: 10.1111/bjd.16733. [PubMed:30141539]
- Blume-Peytavi U, Fowler J, Kemeny L, Draelos Z, Cook-Bolden F, Dirschka T, Eichenfield L, Graeber M, Ahmad F, Alio Saenz A, Rich P, Tanghetti E: Long-term safety and efficacy of trifarotene 50 mug/g cream, a first-in-class RAR-gamma selective topical retinoid, in patients with moderate facial and truncal acne. J Eur Acad Dermatol Venereol. 2019 Jul 15. doi: 10.1111/jdv.15794. [PubMed:31306527]
- Tan J, Thiboutot D, Popp G, Gooderham M, Lynde C, Del Rosso J, Weiss J, Blume-Peytavi U, Weglovska J, Johnson S, Parish L, Witkowska D, Sanchez Colon N, Alio Saenz A, Ahmad F, Graeber M, Stein Gold L: Randomized phase 3 evaluation of trifarotene 50 mug/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019 Jun;80(6):1691-1699. doi: 10.1016/j.jaad.2019.02.044. Epub 2019 Feb 22. [PubMed:30802558]
- Chien A: Retinoids in Acne Management: Review of Current Understanding, Future Considerations, and Focus on Topical Treatments J Drugs Dermatol. 2018 Dec 1;17(12):s51-55. [PubMed:30586483]
- FDA Approved Drugs: Aklief® [Link]
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Cream | Topical | 50 mcg |
| Cream | Topical | 50 ug/1g |
| Cream | Topical | 50 MICROGRAMMI/G |
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 4 | Enrolling by Invitation | Treatment | Acne Vulgaris | 1 |
| 3 | Completed | Treatment | Acne Vulgaris | 4 |
| 2 | Completed | Treatment | Acne Vulgaris | 1 |
| 2 | Recruiting | Treatment | Lamellar Ichthyosis | 1 |
| 1 | Completed | Treatment | Malignant Lymphomas | 1 |
FDA Approves Beleodaq (belinostat) for Peripheral T-Cell Lymphoma

Belinostat (PXD101)
FAST TRACK FDA , ORPHAN STATUS
July 3, 2014 — The U.S. Food and Drug Administration today approved Beleodaq (belinostat) for the treatment of patients with peripheral T-cell lymphoma (PTCL), a rare and fast-growing type of non-Hodgkin lymphoma (NHL). The action was taken under the agency’s accelerated approval program.
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101

| Identifiers | |
|---|---|
| CAS | 414864-00-9 |
| PubChem | 6918638 |
| ChemSpider | 5293831 |
| UNII | F4H96P17NZ |
| ChEBI | CHEBI:61076 |
| ChEMBL | CHEMBL408513 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C15H14N2O4S |
| Molar mass | 318.35 g mol−1 |
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
PTCL comprises a diverse group of rare diseases in which lymph nodes become cancerous. In 2014, the National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die. PTCL represents about 10 to 15 percent of NHLs in North America.
Beleodaq works by stopping enzymes that contribute to T-cells, a type of immune cell, becoming cancerous. It is intended for patients whose disease returned after treatment (relapsed) or did not respond to previous treatment (refractory).
“This is the third drug that has been approved since 2009 for the treatment of peripheral T-cell lymphoma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval expands the number of treatment options available to patients with serious and life-threatening diseases.”
The FDA granted accelerated approval to Folotyn (pralatrexate) in 2009 for use in patients with relapsed or refractory PTCL and Istodax (romidepsin) in 2011 for the treatment of PTCL in patients who received at least one prior therapy.
The safety and effectiveness of Beleodaq was evaluated in a clinical study involving 129 participants with relapsed or refractory PTCL. All participants were treated with Beleodaq until their disease progressed or side effects became unacceptable. Results showed 25.8 percent of participants had their cancer disappear (complete response) or shrink (partial response) after treatment.
The most common side effects seen in Beleodaq-treated participants were nausea, fatigue, fever (pyrexia), low red blood cells (anemia), and vomiting.
The FDA’s accelerated approval program allows for approval of a drug based on surrogate or intermediate endpoints reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs. Drugs receiving accelerated approval are subject to confirmatory trials verifying clinical benefit. Beleodaq also received orphan product designation by the FDA because it is intended to treat a rare disease or condition.
Beleodaq and Folotyn are marketed by Spectrum Pharmaceuticals, Inc., based in Henderson, Nevada. Istodax is marketed by Celgene Corporation based in Summit, New Jersey.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
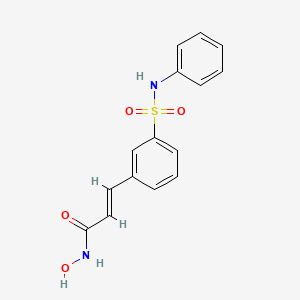
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..


BI launches COPD drug Striverdi, olodaterol in UK and Ireland

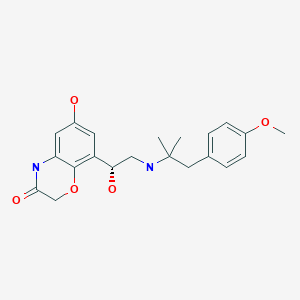
Olodaterol
BI-1744
BI-1744-CL (hydrochloride) marketed as drug
Boehringer Ingelheim Pharma innovator
synthesis…..http://wendang.baidu.com/view/d4f95541e518964bcf847c22.html
Olodaterol (trade name Striverdi) is a long acting beta-adrenoceptor agonist used as an inhalation for treating patients with chronic obstructive pulmonary disease (COPD), manufactured by Boehringer-Ingelheim.[1]
see……….https://www.thieme-connect.de/DOI/DOI?10.1055/s-0029-1219649 ……… synfacts
Olodaterol is a potent agonist of the human β2-adrenoceptor with a high β1/β2 selectivity. Its crystalline hydrochloride salt is suitable for inhalation and is currently undergoing clinical trials in man for the treatment of asthma. Olodaterol has a duration of action that exceeds 24 hours in two preclinical animal models of bronchoprotection and it has a better safety margin compared with formoterol.
Olodaterol hydrochloride [USAN]
Bi 1744 cl
Bi-1744-cl
Olodaterol hydrochloride
Olodaterol hydrochloride [usan]
UNII-65R445W3V9
868049-49-4 [RN] FREE FORM
CAS 869477-96-3 HCL SALT
R ENANTIOMER
2H-1,4-Benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
2H-1,4-benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
6-Hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)-1,1-dimethylethyl)amino)ethyl)- 2H-1,4-benzoxazin-3(4H)-one hydrochloride
clinical trialshttp://clinicaltrials.gov/search/intervention=Olodaterol+OR+BI+1744
Boehringer Ingelheim has launched a new chronic obstructive pulmonary disease drug, Striverdi in the UK and Ireland.
Striverdi (olodaterol) is the second molecule to be licenced for delivery via the company’s Respimat Soft Mist inhaler, following the COPD blockbuster Spiriva (tiotropium). The drug was approved in Europe in November based on results from a Phase III programme that included more than 3,000 patients with moderate to very severe disease.http://www.pharmatimes.com/Article/14-07-01/BI_launches_COPD_drug_Striverdi_in_UK_and_Ireland.aspx
Olodaterol hydrochloride is a drug candidate originated by Boehringer Ingelheim. The product, delivered once-daily by the Respimat Soft Mist Inhaler, was first launched in Denmark and the Netherlands in March 2014 for the use as maintenance treatment of chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. In 2013, approval was obtained in Russia and Canada for the same indication, and in the U.S, the product was recommended for approval. Phase III clinical trials for the treatment of COPD are ongoing in Japan.
| Systematic (IUPAC) name | |
|---|---|
| 6-hydroxy-8-{(1R)-1-hydroxy-2-{[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino}ethyl}-4H-1,4-benzoxazin-3-one | |
| Clinical data | |
| Trade names | Striverdi |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy cat. | No experience |
| Legal status | POM (UK) |
| Routes | Inhalation |
| Identifiers | |
| CAS number | 868049-49-4; 869477-96-3 (hydrochloride) |
| ATC code | R03AC19 |
| PubChem | CID 11504295 |
| ChemSpider | 9679097 |
| UNII | VD2YSN1AFD |
| ChEMBL | CHEMBL605846 |
| Synonyms | BI 1744 CL |
| Chemical data | |
| Formula | C21H26N2O5 free form C21 H26 N2 O5 . Cl H; of hcl salt |
| Mol. mass | 386.44 g/mol free form; 422.902 as hyd salt |
Medical uses
Olodaterol is a once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD including chronic bronchitis and/or emphysema, and is administered in an inhaler called Respimat Soft Mist Inhaler.[2][3][4][5][6][7]
As of December 2013, olodaterol is not approved for the treatment of asthma. Olodaterol monotherapy was previously evaluated in four Phase 2 studies in asthma patients. However, currently there are no Phase 3 studies planned for olodaterol monotherapy in patients with asthma.
In late January 2013, Olodaterol CAS# 868049-49-4 was the focus of an FDA committee reviewing data for the drug’s approval as a once-daily maintenance bronchodilator to treat chronic obstructive pulmonary disease (COPD), as well as chronic bronchitis and emphysema. The FDA Pulmonary-Allergy Drugs Advisory Committee recommended that the clinical data from the Boehringer Ingelheim Phase III studies be included in their NDA.
Also known as the trade name Striverdi Respimat, Olodaterol is efficacious as a long-acting beta-agonist, which patients self-administer via an easy to use metered dose inhaler. While early statistics from clinical trials of Olodaterol were encouraging, a new set of data was released earlier this week, which only further solidified the effectual and tolerable benefits of this COPD drug.
On September 10, 2013 results from two Phase 3 studies of Olodaterol revealed additional positive results from this formidable COPD treatment. The conclusion from these two 48 week studies, which included over 3,000 patients, showed sizable and significant improvements in the lung function of patients who were dosed with Olodaterol. Patients in the aforementioned studies were administered either a once a day dosage of Olodaterol via the appropriate metered-dose inhaler or “usual care”. The “usual care” included a variety of treatment options, such as inhaled corticosteroids (not Olodaterol), short and long acting anticholinergics, xanthines and beta agonists, which were short acting. The clinical trial participants who were dosed with Olodaterol displayed a rapid onset of action from this drug, oftentimes within the first five minutes after taking this medication. Additionally, patients dispensed the Olodaterol inhaler were successfully able to maintain optimum lung function for longer than a full 24 hour period. The participants who were given Olodaterol experienced such an obvious clinical improvement in their COPD symptoms, and it quickly became apparent that the “usual care” protocol was lacking in efficacy and reliability.
A staggering 24 million patients in the United States suffer from chronic obstructive pulmonary disease, and this patient population is in need of an effectual, safe and tolerable solution. Olodaterol is shaping up to be that much needed solution. Not only have the results from studies of Olodaterol been encouraging, the studies themselves have actually been forward thinking and wellness centered. Boehringer Ingelheim is the first company to included studies to evaluate exercise tolerance in patients with COPD, and compare the data to those patients who were dosed with Olodaterol. By including exercise tolerance as an important benchmark in pertinent data for Olodaterol, Boehringer Ingelheim has created a standard for COPD treatment expectations. The impaired lung function for patients with COPD contributes greatly to their inability to exercise and stay healthy. Patients who find treatments and management techniques to combat the lung hyperinflation that develops during exercise have a distinct advantage to attaining overall good health.
– See more at: http://www.lgmpharma.com/blog/olodaterol-offers-encouraging-results-patients-copd/#sthash.DOjcrGxc.dpuf
Data has demonstrated that Striverdi, a once-daily long-acting beta2 agonist, significantly improved lung function versus placebo and is comparable to improvements shown with the older LABA formoterol. The NHS price for the drug is £26.35 for a 30-day supply.
Boehringer cited Richard Russell at Wexham Park Hospital as saying that the licensing of Stirverdi will be welcomed by clinicians as it provides another option. He added that the trial results showing improvements in lung function “are particularly impressive considering the study design, which allowed participants to continue their usual treatment regimen. This reflects more closely the real-world patient population”.
Significantly, the company is also developing olodaterol in combination with Spiriva, a long-acting muscarinic antagonist. LAMA/LABA combinations provide the convenience of delivering the two major bronchodilator classes.
 Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.Adverse effects
Adverse effects generally were rare and mild in clinical studies. Most common, but still affecting no more than 1% of patients, were nasopharyngitis (running nose), dizziness and rash. To judge from the drug’s mechanism of action and from experiences with related drugs, hypertension (high blood pressure), tachycardia (fast heartbeat), hypokalaemia (low blood levels of potassium), shaking, etc., might occur in some patients, but these effects have rarely, if at all, been observed in studies.[1]
Interactions
Based on theoretical considerations, co-application of other beta-adrenoceptor agonists, potassium lowering drugs (e. g. corticoids, many diuretics, and theophylline), tricyclic antidepressants, and monoamine oxidase inhibitors could increase the likelihood of adverse effects to occur. Beta blockers, a group of drugs for the treatment of hypertension (high blood pressure) and various conditions of the heart, could reduce the efficacy of olodaterol.[1] Clinical data on the relevance of such interactions are very limited.
Pharmacology
Mechanism of action
Like all beta-adrenoceptor agonists, olodaterol mimics the effect of epinephrine at beta-2 receptors (β₂-receptors) in the lung, which causes the bronchi to relax and reduces their resistance to airflow.[3]
Olodaterol is a nearly full β₂-agonist, having 88% intrinsic activity compared to the gold standard isoprenaline. Its half maximal effective concentration (EC50) is 0.1 nM. It has a higher in vitro selectivity for β₂-receptors than the related drugs formoterol and salmeterol: 241-fold versus β₁- and 2299-fold versus β₃-receptors.[2] The high β₂/β₁ selectivity may account for the apparent lack of tachycardia in clinical trials, which is mediated by β₁-receptors on the heart.
Pharmacokinetics
Once bound to a β₂-receptor, an olodaterol molecule stays there for hours – its dissociation half-life is 17.8 hours –, which allows for once-a-day application of the drug[3] like with indacaterol. Other related compounds generally have a shorter duration of action and have to be applied twice daily (e.g. formoterol, salmeterol). Still others (e. g. salbutamol, fenoterol) have to be applied three or four times a day for continuous action, which can also be an advantage for patients who need to apply β₂-agonists only occasionally, for example in an asthma attack.[8]
History
On 29 January 2013 the U.S. Food and Drug Administration (FDA) Pulmonary-Allergy Drugs Advisory Committee (PADAC) recommended that the clinical data included in the new drug application (NDA) for olodaterol provide substantial evidence of safety and efficacy to support the approval of olodaterol as a once-daily maintenance bronchodilator treatment for airflow obstruction in patients with COPD.[9]
On 18 October 2013 approval of olodaterol in the first three European countries – the United Kingdom, Denmark and Iceland – was announced by the manufacturer.[10]

Figure Chemical structures of salmeterol, formoterol, inda- caterol, and emerging once-daily long-acting β2-agonists
CLIP
Synthetic approaches to the 2013 new drugs – ScienceDirect

Olodaterol hydrochloride was approved for long-term, once-daily maintenance treatment of chronic
obstructive pulmonary disease (COPD) in 2013 in the following countries: Canada, Russia, United
Kingdom, Denmark, and Iceland.142, 143 The drug has been recommended by a federal advisory panel for
approval by the FDA.142, 143 Developed and marketed by Boehringer Ingelheim, olodaterol is a longacting
β2-adrenergic receptor agonist with high selectivity over the β1- and β3-receptors (219- and 1622-fold, respectively).144 Upon binding to and activating the β2-adrenergic receptor in the airway, olodaterol
stimulates adenyl cyclase to synthesize cAMP, leading to the relaxation of smooth muscle cells in the
airway. Administered by inhalation using the Respimat®
Soft Mist inhaler, it delivers significant
bronchodilator effects within five minutes of the first dose and provides sustained improvement in
forced expiratory volume (FEV1) for over 24 hours.143 While several routes have been reported in the
patent and published literature,144-146 the manufacturing route for olodaterol hydrochloride disclosed in
2011 is summarized in Scheme 19 below.147
Commercial 2’,5’-dihydroxyacetophenone (122) was treated with one equivalent of benzyl bromide
and potassium carbonate in methylisobutylketone (MIBK) to give the 5’-monobenzylated product in
76% yield. Subsequent nitration occurred at the 4’-position to provide nitrophenol 123 in 87% yield.
Reduction of the nitro group followed by subjection to chloroacetyl chloride resulted in the construction
of benzoxazine 124 in 82% yield. Next, monobromination through the use of tetrabutylammonium
tribromide occurred at the acetophenone carbon to provide bromoketone 125, and this was followed by
asymmetric reduction of the ketone employing (−)-DIP chloride to afford an intermediate bromohydrin,
which underwent conversion to the corresponding epoxide 126 in situ upon treatment with aqueous
NaOH. This epoxide was efficiently formed in 85% yield and 98.3% enantiomeric excess. Epoxide
126 underwent ring-opening upon subjection to amine 127 to provide amino-alcohol 128 in in 84-90%
yield and 89.5-99.5% enantiomeric purity following salt formation with HCl. Tertiary amine 127 was
itself prepared in three steps by reaction of ketone 129 with methylmagnesium chloride, Ritter reaction
of the tertiary alcohol with acetonitrile, and hydrolysis of the resultant acetamide with ethanolic
potassium hydroxide. Hydrogenative removal of the benzyl ether within 128 followed by
recrystallization with methanolic isopropanol furnished olodaterol hydrochloride (XVI) in 63-70%
yield. Overall, the synthesis of olodaterol hydrochloride required 10 total steps (7 linear) from
commercially available acetophenone 122.
142. Gibb, A.; Yang, L. P. H. Drugs 2013, 73, 1841.
143. http://www.boehringeringelheim.com/news/news_releases/press_releases/2013/18_october_2013_olodaterol.html.
144. Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine,
C.; Buettner, F. H.; Schnapp, A.; Konetzki, I. Bioorg. Med. Chem. Lett. 2010, 20, 1410.
145. Trunk, M. J. F.; Schiewe, J. US Patent 20050255050A1, 2005.
146. Lustenberger, P.; Konetzki, I.; Sieger, P. US Patent 20090137578A1, 2009.
147. Krueger, T.; Ries, U.; Schnaubelt, J.; Rall, W.; Leuter, Z. A.; Duran, A.; Soyka, R. US Patent
20110124859A1, 2011.
PATENT
WO 2004045618 or
http://www.google.com/patents/EP1562603B1?cl=en
Example

a)
To a solution of 3.6 g 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine in 100 mL of ethanol at 70 ° C. 7.5 g of (6-benzyloxy-4H-benzo [1,4] oxazin-3-one )-glyoxal added and allowed to stir for 15 minutes. Then within 30 minutes at 10 to 20 ° C. 1 g of sodium borohydride added. It is stirred for one hour, with 10 mL of acetone and stirred for another 30 minutes. The reaction mixture is diluted with 150 mL ethyl acetate, washed with water, dried with sodium sulfate and concentrated. The residue is dissolved in 50 mL of methanol and 100 mL ethyl acetate and acidified with conc. Hydrochloric acid. After addition of 100 mL of diethyl ether, the product precipitates. The crystals are filtered, washed and recrystallized from 50 mL of ethanol. Yield: 7 g (68%; hydrochloride), mp = 232-234 ° C.
b)
6.8 g of the above obtained benzyl compound in 125 mL of methanol with the addition of 1 g of palladium on carbon (5%) was hydrogenated at room temperature and normal pressure. The catalyst is filtered and the filtrate was freed from solvent. Recrystallization of the residue in 50 mL of acetone and a little water, a solid is obtained, which is filtered and washed.
Yield: 5.0 g (89%; hydrochloride), mp = 155-160 ° C.
The (R) – and (S)-enantiomers of Example 3 can be obtained from the racemate, for example, by chiral HPLC (for example, column: Chirobiotic T, 250 x 1.22 mm from the company Astec). As the mobile phase, methanol with 0.05% triethylamine and 0.05% acetic acid. Silica gel with a grain size of 5 microns, to which is covalently bound the glycoprotein teicoplanin can reach as column material used. Retention time (R enantiomer) = 40.1 min, retention time (S-enantiomer) = 45.9 min. The two enantiomers can be obtained by this method in the form of free bases. According to the invention of paramount importance is the R enantiomer of Example 3
PATENT
WO 2005111005
http://www.google.fm/patents/WO2005111005A1?cl=en
Scheme 1.




Scheme 1:
Example 1 6-Hydroxy-8-{(1-hydroxy-2-r2-(4-methoxy-phenyl) – 1, 1-dimethyl-ethylamino]-ethyl)-4H-benzor 41oxazin-3-one – Hvdrochlorid

a) l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone
To a solution of 81.5 g (0.34 mol) l-(5-benzyloxy-2-hydroxy-phenyl)-ethanone in 700 ml of acetic acid are added dropwise under cooling with ice bath, 18 mL of fuming nitric acid, the temperature does not exceed 20 ° C. increases. The reaction mixture is stirred for two hours at room temperature, poured onto ice water and filtered. The product is recrystallized from isopropanol, filtered off and washed with isopropanol and diisopropyl ether. Yield: 69.6 g (72%), mass spectroscopy [M + H] + = 288
b) l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone
69.5 g (242 mmol) of l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone are dissolved in 1.4 L of methanol and in the presence of 14 g of rhodium on carbon (10%) as catalyst at 3 bar room temperature and hydrogenated. Then the catalyst is filtered off and the filtrate concentrated. The residue is reacted further without additional purification. Yield: 60.0 g (96%), R f value = 0.45 (silica gel, dichloromethane).
c) 8-acetyl-6-benzyloxy-4H-benzoπ .4] oxazin-3-one
To 60.0 g (233 mmol) of l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone and 70.0 g (506 mmol) of potassium carbonate while cooling with ice bath, 21.0 ml (258 mmol) of chloroacetyl chloride added dropwise. Then stirred overnight at room temperature and then for 6 hours under reflux. The hot reaction mixture is filtered and then concentrated to about 400 mL and treated with ice water. The precipitate is filtered off, dried and purified by chromatography on a short silica gel column (dichloromethane: methanol = 99:1). The product-containing fractions are concentrated, suspended in isopropanol, diisopropyl ether, and extracted with
Diisopropyl ether. Yield: 34.6 g (50%), mass spectroscopy [M + H] + = 298
d) 6-Benzyloxy-8-(2-chloro-acetyl)-4H-benzoFl, 4] oxazin-3-one 13.8 g (46.0 mmol) of 8-benzyloxy-6-Acetyl-4H-benzo [l, 4] oxazin -3-one and 35.3 g (101.5 mmol) of benzyltrimethylammonium dichloriodat are stirred in 250 mL dichloroethane, 84 mL glacial acetic acid and 14 mL water for 5 hours at 65 ° C. After cooling to room temperature, treated with 5% aqueous sodium hydrogen sulfite solution and stirred for 30 minutes. The precipitated solid is filtered off, washed with water and diethyl ether and dried. Yield: 13.2 g (86%), mass spectroscopy [M + H] + = 330/32.
e) 6-Benzyloxy-8-((R-2-chloro-l-hydroxy-ethyl)-4H-benzori ,41-oxazin-3-one The procedure is analogous to a procedure described in the literature (Org. Lett ., 2002, 4, 4373-4376).
To 13:15 g (39.6 mmol) of 6-benzyloxy-8-(2-chloro-acetyl)-4H-benzo [l, 4] oxazin-3-one and 25.5 mg (0:04 mmol) Cρ * RhCl [(S, S) -TsDPEN] (Cp * = pentamethylcyclopentadienyl and TsDPEN = (lS, 2S)-Np-toluenesulfonyl-l ,2-diphenylethylenediamine) in 40 mL of dimethylformamide at -15 ° C and 8 mL of a mixture of formic acid and triethylamine (molar ratio = 5: 2) dropwise. It is allowed for 5 hours at this temperature, stirring, then 25 mg of catalyst and stirred overnight at -15 ° C. The reaction mixture is mixed with ice water and filtered. The filter residue is dissolved in dichloromethane, dried with sodium sulfate and the solvent evaporated. The residue is recrystallized gel (dichloromethane / methanol gradient) and the product in diethyl ether / diisopropyl ether. Yield: 10.08 g (76%), R f value = 00:28 (on silica gel, dichloromethane ethanol = 50:1).
f) 6-Benzyloxy-8-(R-oxiranyl-4H-benzo [“L4] oxazin-3-one 6.10 g (30.1 mmol) of 6-benzyloxy-8-((R)-2-chloro-l-hydroxy- ethyl)-4H-benzo [l, 4] oxazin-3-one are dissolved in 200 mL of dimethylformamide. added to the solution at 0 ° C with 40 mL of a 2 molar sodium hydroxide solution and stirred at this temperature for 4 hours. the reaction mixture is poured onto ice water, stirred for 15 minutes, and then filtered The solid is washed with water and dried to give 8.60 g (96%), mass spectroscopy [M + H] + = 298..
g) 6-Benyloxy-8-{(R-l-hydroxy-2-r2-(4-methoxy-phenyl)-dimethyl-ll-ethvIaminol-ethyl)-4H-benzo-3-Tl A1oxazin
5.25 g (17.7 mmol) of 6-benzyloxy-8-(R)-oxiranyl-4H-benzo [l, 4] oxazin-3-one and 6.30 g (35.1 mmol) of 2 – (4-methoxy-phenyl 1, 1 – dimethyl-ethyl to be with 21 mL
Of isopropanol and stirred at 135 ° C for 30 minutes under microwave irradiation in a sealed reaction vessel. The solvent is distilled off and the residue chromatographed (alumina, ethyl acetate / methanol gradient). The product thus obtained is purified by recrystallization from a mixture further Diethylether/Diisopropylether-. Yield: 5:33 g (63%), mass spectroscopy [M + H] + = 477 h) 6-Hydroxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino] – ethyl}-4H-benzo [1, 4, 1 oxazin-3-one hydrochloride
A suspension of 5:33 g (11.2 mmol) of 6-Benyloxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino]-ethyl}-4H -benzo [l, 4] oxazin-3-one in 120 mL of methanol with 0.8 g of palladium on carbon (10%), heated to 50 ° C and hydrogenated at 3 bar hydrogen pressure. Then the catalyst is filtered off and the filtrate concentrated. The residue is dissolved in 20 mL of isopropanol, and 2.5 mL of 5 molar hydrochloric acid in isopropanol. The product is precipitated with 200 mL of diethyl ether, filtered off and dried. Yield: 4.50 g (95%, hydrochloride), mass spectroscopy [M + H] + = 387
PATENT
WO 2007020227
http://www.google.com.ar/patents/WO2007020227A1?cl=en
PATENT
WO 2008090193
or
http://www.google.com/patents/EP2125759B1?cl=en
PAPER
Discovery of olodaterol, a novel inhaled beta(2)-adrenoceptor agonist with a 24h bronchodilatory efficacy
Bioorg Med Chem Lett 2010, 20(4): 1410
http://www.sciencedirect.com/science/article/pii/S0960894X09018101
The discovery of the β2-adrenoceptor agonist (R)-4p designated olodaterol is described. The preclinical profile of the compound suggests a bronchoprotective effect over 24 h in humans.

CLIP
Australia
http://www.tga.gov.au/pdf/auspar/auspar-olodaterol-140327-pi.pdf
CLIP
DUTCH
http://mri.medagencies.org/download/NL_H_2498_001_PAR.pdf
FDA
Click to access 203108Orig1s000ChemR.pdf
NDA 203108
Striverdi® Respimat® (olodaterol) Inhalation Spray
Boehringer Ingelheim Pharmaceuticals, Inc.
References
- Striverdi UK Drug Information
- Bouyssou, T.; Casarosa, P.; Naline, E.; Pestel, S.; Konetzki, I.; Devillier, P.; Schnapp, A. (2010). “Pharmacological Characterization of Olodaterol, a Novel Inhaled 2-Adrenoceptor Agonist Exerting a 24-Hour-Long Duration of Action in Preclinical Models”. Journal of Pharmacology and Experimental Therapeutics 334 (1): 53–62. doi:10.1124/jpet.110.167007. PMID 20371707.
- Casarosa, P.; Kollak, I.; Kiechle, T.; Ostermann, A.; Schnapp, A.; Kiesling, R.; Pieper, M.; Sieger, P.; Gantner, F. (2011). “Functional and Biochemical Rationales for the 24-Hour-Long Duration of Action of Olodaterol”. Journal of Pharmacology and Experimental Therapeutics 337 (3): 600–609. doi:10.1124/jpet.111.179259. PMID 21357659.
- Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine, C.; Büttner, F. H.; Schnapp, A.; Konetzki, I. (2010). “Discovery of olodaterol, a novel inhaled β2-adrenoceptor agonist with a 24h bronchodilatory efficacy”. Bioorganic & Medicinal Chemistry Letters 20 (4): 1410–1414. doi:10.1016/j.bmcl.2009.12.087. PMID 20096576.
- Joos G, Aumann JL, Coeck C, et al. ATS 2012 Abstract: Comparison of 24-Hour FEV1 Profile for Once-Daily versus Twice-Daily Treatment with Olodaterol, A Novel Long-Acting ß2-Agonist, in Patients with COPD[dead link]
- Van Noord, J. A.; Smeets, J. J.; Drenth, B. M.; Rascher, J.; Pivovarova, A.; Hamilton, A. L.; Cornelissen, P. J. G. (2011). “24-hour Bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD”. Pulmonary Pharmacology & Therapeutics 24 (6): 666–672. doi:10.1016/j.pupt.2011.07.006. PMID 21839850.
- van Noord JA, Korducki L, Hamilton AL and Koker P. Four Weeks Once Daily Treatment with BI 1744 CL, a Novel Long-Acting ß2-Agonist, is Effective in COPD Patients. Am. J. Respir. Crit. Care Med. 2009; 179: A6183[dead link]
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Hollis A (31 January 2013). “Panel Overwhelmingly Supports Boehringer COPD Drug Striverdi”. FDA News/Drug Industry Daily.
- “New once-daily Striverdi (olodaterol) Respimat gains approval in first EU countries”. Boehringer-Ingelheim. 18 October 2013.
External links
The active moiety olodaterol is a selective beta2-adrenergic bronchodilator. The drug substance, olodaterol hydrochloride, is chemically described as 2H-1,4- Benzoxazin-3H(4H)-one, 6-hydroxy-8-[(1R)-1-hydroxy-2-[[2-(4-methoxyphenyl)-1,1-dimethylethyl]-amino]ethyl]-, monohydrochloride. Olodaterol hydrochloride is a white to off-white powder that is sparingly-slightly soluble in water and slightly soluble in ethanol. The molecular weight is 422.9 g/mole (salt): 386.5 g/mole (base), and the molecular formula is C21H26N2O5 x HCl as a hydrochloride. The conversion factor from salt to free base is 1.094.
The structural formula is:
 |
The drug product, STRIVERDI RESPIMAT, is composed of a sterile, aqueous solution of olodaterol hydrochloride filled into a 4.5 mL plastic container crimped into an aluminum cylinder (STRIVERDI RESPIMAT cartridge) for use with the STRIVERDI RESPIMAT inhaler.
Excipients include water for injection, benzalkonium chloride, edetate disodium, and anhydrous citric acid. The STRIVERDI RESPIMAT cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler. The STRIVERDI RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow-moving aerosol cloud of medication from a metered volume of the drug solution. The STRIVERDI RESPIMAT inhaler has a yellow-colored cap.
When used with the STRIVERDI RESPIMAT inhaler, each cartridge containing a minimum of 4 grams of a sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (1 dose equals 2 actuations) from the STRIVERDI RESPIMAT inhaler delivers 5 mcg olodaterol in 22.1 mcL of solution from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
| WO2002030928A1 | 28 Sep 2001 | 11 Apr 2003 | Boehringer Ingelheim Pharma | Crystalline monohydrate, method for producing the same and the use thereof in the production of a medicament |
| WO2003000265A1 | 8 Jun 2002 | 3 Jan 2003 | Boehringer Ingelheim Pharma | Crystalline anticholinergic, method for its production, and use thereof in the production of a drug |
| WO2004045618A2 * | 11 Nov 2003 | 3 Jun 2004 | Boehringer Ingelheim Pharma | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| EP0073505A1 * | 28 Aug 1982 | 9 Mar 1983 | Boehringer Ingelheim Kg | Benzo-heterocycles |
| EP0321864A2 * | 15 Dec 1988 | 28 Jun 1989 | Boehringer Ingelheim Kg | Ammonium compounds, their preparation and use |
| US4460581 | 12 Oct 1982 | 17 Jul 1984 | Boehringer Ingelheim Kg | Antispasmodic agents, antiallergens |
| US4656168 * | 13 Oct 1983 | 7 Apr 1987 | Merck & Co., Inc. | Vision defects; adrenergic blocking and hypotensive agents |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com
BMS 587101…….The LFA-1 receptor antagonist in preclinical for the treatment of a variety of autoimmune and inflammatory diseases such as rheumatoid arthritis and psoriasis.

- C26H20Cl2N4O4S
- mass: 555.432373 Da
read poster
http://www.cerep.fr/cerep/users/pages/news/Publications/123.pdf
5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-ylmethyl]-thiophene-3-carboxylic Acid
3-Thiophenecarboxylic acid, 5-[[(5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]methyl]- [ACD/Index Name]
5-{[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]methyl}-3-thiophenecarboxylic acid [ACD/IUPAC Name]
5-{[(5S,9R)-9-(4-Cyanphenyl)-3-(3,5-dichlorphenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]methyl}-3-thiophencarbonsäure [German] [ACD/IUPAC Name]
Acide 5-{[(5S,9R)-9-(4-cyanophényl)-3-(3,5-dichlorophényl)-1-méthyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]méthyl}-3-thiophènecarboxylique [French] [ACD/IUPAC Name]
2IC
BMS-587101
BMS-688521
data
Interaction between leukocyte function-associated antigen-1 (LFA-1), expressed on the surface of cytokine-stimulated cells, and intercellular adhesion molecule (I-CAM), found on the surface of both leukocytes and endothelium, plays a key function in the intercellular immune response, causing T-cell adhesion and subsequent migration through the blood vessel wall to the inflamed area.(1)
Small molecules which inhibit the LFA-1/I-CAM interaction are targeted as potential drugs for the treatment of a variety of autoimmune and inflammatory diseases such as rheumatoid arthritis and psoriasis.(2, 3) The LFA-1 receptor antagonist, BMS-587101, 1,(4, 5) was selected for clinical development, and we required a synthesis that would reliably generate kilogram quantities of API. This paper details the identification and development of a synthesis which enabled the realization of this goal.
BMS-587101 inhibits the interaction between leukocyte function-associated antigen-1 (LFA-1) and the intercellular adhesion molecule (ICAM), thereby offering a potential treatment for various autoimmune and inflammatory diseases, such as rheumatoid arthritis and psoriasis. A four-step multikilogram route to BMS-587101 (22% overall yield ) from the commercial hydantoin B features an efficient dipolar cycloaddition of an azomethine ylide generated by reaction of glycine with hexamethylenetetramine (HMTA).

………….
paper

http://pubs.acs.org/doi/abs/10.1021/op9003168
The process development and the kilogram-scale synthesis of BMS-587101 (1) are described. The synthesis features a [3 + 2] azomethine ylide cycloaddition to efficiently build the spirocyclic core in a diastereoselective fashion followed by a classical resolution which affords the desired enantiomer in >98% enantiomeric excess. The target was prepared in four steps in an overall yield of 22%.
 22.9 kg) was added until a pH of 6.5 was attained. After agitating for 15 min and holding for 30 min, the aqueous layer was discarded, and the organic layer was washed with H2O (470 kg). The solution was then polish filtered, and isopropylacetate (52.2 kg) was used to rinse the polish filter assembly. The solution was concentrated under reduced pressure (240 Torr) to a volume of 718 L at <45 °C. Seeds (500 g) were charged, and the distillation was continued until a volume of 207 L was attained. Heptane (117.8 kg) was charged, the slurry was cooled to 20 °C over 1.5 h and was subsequently wet milled until d90 < 60 μm. The slurry was held for >2 h and filtered. The cake was washed with a 1:1 isopropyl acetate/heptane solution (109.7 kg) isopropyl acetate and dried in vacuum at 35−40 °C to a constant weight. Acid 1 (39.6 kg, 91.5% yield and 99.33 HPLC area % purity) was obtained as a white and sandy crystalline solid.
22.9 kg) was added until a pH of 6.5 was attained. After agitating for 15 min and holding for 30 min, the aqueous layer was discarded, and the organic layer was washed with H2O (470 kg). The solution was then polish filtered, and isopropylacetate (52.2 kg) was used to rinse the polish filter assembly. The solution was concentrated under reduced pressure (240 Torr) to a volume of 718 L at <45 °C. Seeds (500 g) were charged, and the distillation was continued until a volume of 207 L was attained. Heptane (117.8 kg) was charged, the slurry was cooled to 20 °C over 1.5 h and was subsequently wet milled until d90 < 60 μm. The slurry was held for >2 h and filtered. The cake was washed with a 1:1 isopropyl acetate/heptane solution (109.7 kg) isopropyl acetate and dried in vacuum at 35−40 °C to a constant weight. Acid 1 (39.6 kg, 91.5% yield and 99.33 HPLC area % purity) was obtained as a white and sandy crystalline solid.
…………………………
U.S. Patent 7,381,737 B2
http://www.google.com/patents/US7381737
IIIn:

Also provided are crystalline forms of solvates and salts of the substituted spiro-hydantoin compound (IIIn).
5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid.
EXAMPLES
The following examples illustrate embodiments of the inventive process, and are not intended to limit the scope of the claims. For ease of reference, the following abbreviations are used herein:
ABBREVIATIONS
- DMSO=dimethyl sulfoxide
- DTTA=(+)-Di-p-toluoyl-D-tartaric acid
Preparation 13-(3,5-dichlorophenyl)-1-methylimidazolidine-2,4-dione

Triethylamine (0.78 kg, 7.75 mol) was added in 15-30 minutes with stirring to a thin suspension of sarcosine ethylene hydrochloride (1.00 kg, 6.51 mol) in dichloromethane (6.00 L). After stirring at room temperature for 1.5-2.0 hours, the mixture was filtered to remove the resulting triethylamine hydrochloride salt. The salt cake was washed with dichloromethane (2.00 L). The filtrate was cooled to 0-5° C.
A solution of 3,5-dichlorophenyl isocyanate (1.47 kg, 7.81 mol) in dichloromethane was prepared at 20-25° C. The solution was added to the above cooled filtrate slowly in 30-60 minutes. The temperature was maintained below 10° C. during the addition. After the addition, the mixture was stirred at 20-25° C. for 12-14 hours. The completeness of the reaction was followed by HPLC. Upon reaction completion, TBME (16.00 L) was added in one portion. The resulting suspension was stirred at 20-25° C. for 2-3 hours and was then filtered. The filter cake was washed with TBME (4.50 L) and dried at maximum 40° C. to a constant weight. A suspension of the above filter cake in water (17.0 L, 10 L/kg input) was prepared and stirred at 20-25° C. for at least 16 hours. The suspension was filtered and the filter cake was washed with water (3×1.36 L) and dried at maximum 40° C. to a constant weight to a constant weight. 3-(3,5-dichlorophenyl)-1-methylimidazolidine-2,4-dione (1.52 kg, 90%) was obtained as a white crystalline solid. mp=202-204° C. 1H NMR (DMSO-d6): 7.66 (1H, m), 7.51 (2H, m), 4.10 (2H, s), 3.35 (3H, s). 13C NMR (DMSO-d6): 8 Carbons (169.30, 155.00, 134.98, 134.15, 127.59, 125.30, 51.75, 29.79). Anal. Calcd for C10H8Cl2N2O2: C, 46.35; H, 3.11; N, 10.81; Cl, 27.36. Found: C, 46.43; H, 2.9; N, 10.73; Cl, 27.33.
Preparation 2(E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile

A mixture of 3-(3,5-dichlorophenyl)-1-methylimidazolidine-2,4-dione (1.00 kg, 3.86 mol), 4-cyanobenzaldehyde (0.70 kg, 5.79 mol) and pyrrolidone (0.27 kg, 3.86 mmol) was refluxed in EtOH (13.00 L) for 20-24 hours at a temperature of 78° C. The completeness of the reaction was followed by HPLC. Upon reaction completion, the suspension was cooled to 65° C. and THF (4.33 L) was added in 5-10 minutes. The suspension was cooled to 20-25° C. in 3-4 hours and was then filtered. The filter cake was washed with EtOH (4×2.00 L) and dried at maximum 40° C. to a constant weight. (E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (1.24 kg, 86%) was obtained as a fluffy, yellowish crystalline solid. mp=239-241° C. 1H NMR (DMSO-d6): 8.07 (2H, d, J=8.3 Hz), 7.86 (2H, d, J=8.4 Hz), 7.72 (1H, m), 7.59 (2H, m), 6.72 (1H, s), 3.35 (3H, s). 13C NMR (DMSO-d6): 14 Carbons (159.80, 151.48, 137.64, 133.83, 133.70, 131.80, 130.80, 130.68, 127.71, 125.51, 118.83, 114.48, 110.32, 26.72). Anal. Calcd for C18H11Cl2N3O2: C, 58.08; H, 2.97; N, 11.29; Cl, 19.05. Found: C, 58.14; H, 2.72; N, 11.14; Cl, 19.15.
Example 14-[(5S*,9R*)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile hydrochloride salt

A mixture of (E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (1.00 kg, 2.69 mol), glycine (0.50 kg, 6.72 mol) and hexamethylenetetramine (0.28 kg, 2.02 mol) in 1-methyl-2-pyrrolidinone (5.00 L) and toluene (2.50 L) was heated at 140° C. for 7-8 hours. The completeness of the reaction was followed by HPLC. Upon reaction completion, the mixture was cooled to 40-50° C. and filtered. The filtered solid was washed with toluene (0.67 L). To the filtrate was added HCl (1M, 13.33 L, 13.33 mol). The resulting biphasic mixture was heated to 50-60° C. and was stirred for 10-15 minutes. The aqueous phase was separated and the organic phase was washed with HCl (1M, 1.67 L, 1.67 mol) at 60-80° C. The aqueous phases were combined and were stirred at 80° C. for 2 hours. The solution was cooled slowly in 3-4 hours to 20-25° C. with gentle stirring and seeding. Crystallization occurred and the resulting suspension was put aside at 20-25° C. for at least 16 hours with occasional stirring, cooled to 0-5° C. in 2 hours, stirred gently at 0-5° C. for 2 hours and then filtered. The filter cake was washed with ice water (2×2.50 L) and dried at maximum 40° C. to a constant weight. 4-[(5S*,9R*)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile hydrochloride salt (1.09 kg, 90%) was obtained as beige crystalline solid. mp=183-185° C. 1H NMR (DMSO-d6): 7.87(2H, d, J=8.1 Hz), 7.61 (1H, m), 7.40 (2H, d, J=8.1 Hz), 6.68 (2H, m), 4.17 (1H, m), 3.85 (2H, m), 3.76 (2H, m), 3.43 (3H, s), 3.24(2H, s). 13C NMR (DMSO-d6): 14 Carbons (170.84, 152.92, 137.35, 133.94, 132.87, 132.35, 128.01, 124.50, 118.12, 111.30, 71.42, 46.57, 45.11, 25.51). Anal. Calcd for C20H17Cl3N4O2+1.3 H2O: C, 50.51; H, 3.91; N, 11.79; Cl, 22.39. Found: C, 50.56; H, 3.86; N, 11.58; Cl, 21.98; KF, 5.12.
Example 2a4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt

To a suspension of 4-[(5S*,9R*)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile hydrochloric acid salt (1.00 kg, 2.21 mol) in dichloromethane (10.67 L) was added diispopropylethylamine (0.29 kg, 2.21 mol). The mixture was stirred to a clear solution, to which (+)-Di-p-toluoyl-D-tartaric acid (0.21 kg, 0.55 mol) was added. The resulting solution was warmed to 34-36° C. and seeded immediately. It was cooled to 20-25° C. in 1.5-2.0 hours. Crystallization occurred during cooling. TBME (2.75 L) was added in 0.5 hours. The suspension was stirred at 20-25° C. for 16 hours and then filtered. The filter cake was washed with dichloromethane/TBME (2/1, 1.00 L), TBME (1 L) and dried at maximum 35° C. to a constant weight. 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt (0.47 kg, 35%) was obtained as a white crystalline solid. mp=175-177° C. 1H NMR (DMSO-d6): 7.86 (2H, d, J=8.1 Hz), 7.81 (2H, d, J=8.3 Hz), 7.61 (1H, m), 7.28 (2H, d, J=8.1 Hz), 7.22 (2H, 8.5 Hz), 6.68 (2H, m), 5.71 (1H, s), 3.81(1H, m), 3.50 (4H, m), 3.06 (3H, s), 2.34 (3H, s). 13C NMR (DMSO-d6): 24 Carbons (171.45, 169.40, 165.04, 152.88, 143.61, 138.99, 133.88, 133.08, 132.16, 129.26, 129.20, 128.76, 127.84, 126.99, 124.51, 118.25, 110.78, 72.81, 73.38, 48.15, 47.51, 46.30, 24.90, 21.14). Anal. Calcd for C30H25Cl2N4O6+0.5 H2O: C, 58.40; H, 4.17; N, 9.08; Cl, 11.49. Found C, 58.58; H, 4.06; N, 8.94; Cl, 11.38; KF, 1.59.
Example 2b4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt

A mixture of (E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (10.0 g, 26.9 mmol), glycine (5.06 g, 67.4 mmol), hexamethylenetetramine (2.82 g, 20.1 mmol) in 50 mL N-methylpyrrolidinone and 25 mL of toluene under nitrogen was heated to 138° C. for approximately 12 h. Next, 25 mL toluene and 25 mL H2O were added. The aqueous and nonaqueous layers were split, and the aqueous layer was washed with 25 mL of toluene, and the nonaqueous layers were combined to form a nonaqueous mixture. The nonaqueous mixture was heated to 45-50° C. and ethylene diamine (7.0 mL) was added. The nonaqueous mixture was stirred for 3 hours and then cooled to room temperature. Next, 50 mL H2O was added, followed by the addition of 10 mL brine. The next addition was 25 mL toluene, which was followed by the addition of 125 mL CH2Cl2. The bottom layer of the mixture was removed through a filter. Next, (+)-Di-p-toluoyl-D-tartaric acid (2.59 g, 6.7 mmol) was added and the mixture was stirred for 18 h to form a slurry. Slowly 40 mL of MTBE was added to the slurry. A wash solution containing 7 mL of MTBE and 11 mL of CH2Cl2 was prepared. Filter paper was wetted with 1 mL of the wash solution. The slurry was filtered and then the filtered to form a cake. The filter, the wash reaction flask, and the cake were washed with the remaining 16 mL of the wash solution. Next, the cake was washed with 10 mL MTBE. 4-[(5S, 9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt (4.0 g, 20% yield) was obtained as a white solid (98.7% HPLC AP and 98.3% ee).
Example 2c4-[(5S,9R)-3-3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4,4]non-9-yl]-benzonitrile semi (+)-DTTA salt
A mixture of (E)-4-((1-(3,5)-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (40.0 g, 107.5 mmol), glycine (19.76 g, 263.2 mmol), hexamethylenetetramine (9.07 g, 64.7 mmol) in 200 mL N-methyl-2-pyrrolidinone and 100 mL of toluene was heated under nitrogen to 143° C. for approximately 5.5 h. Next, the mixture was cooled to 50° C. and a solution of 25 mL of ethylenediamine in 200 mL of tetrahydrofuran was added. The mixture was maintained at a temperature of 50° C. for 30 minutes and then was cooled to room temperature. Next, 520 mL of 20 wt % NaCl aqueous solution was added. The aqueous and nonaqueous layers were separated. The nonaqueous layer was transferred to a vacuum distillation apparatus and solvent was distilled off until the temperature of the residue in the flask reached 58° C. at a pressure of 60 torr. Next, 360 mL of methylene chloride was added, followed by the additions of 20 mL of methanol and 2 mL of water. The next addition was (+)-Di-p-toluoyl-D-tartaric acid (10.38 g, 26.9 mmol), followed by 120 mL of methylene chloride and 0.200 g of seeds of 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4,4]non-9-yl]-benzonitrile semi (+)-DTTA salt. A-slurry was formed and was stirred at room temperature for 24 hours. The slurry was filtered and the cake of crystals was washed with 200 mL of methylene chloride in two portions. The washed cake was then dried at 50° C. under vacuum for 24 hours. A total amount of 20.11 g (yield 31%) of 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4,4]non-9-yl]-benzonitrile semi (+)-DTTA salt, which was of greater than 99.5% area percent purity, 98.4% potency and 99.2% ee was obtained after drying.
Example 35-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid methyl ester hydrochloride salt

To a suspension of 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt (7.50 kg, 12.30 mmol) and methyl 5-formylthiophene-3-carboxylate (2.2 kg, 13.10 mol) was added triethylamine (2.08 kg, 20.60 mol) at 20-25° C. The mixture was stirred to a clear solution, to which acetic acid (1.24 kg, 20.60 mol) was added. The resulting mixture was stirred at 20-25° C. for 1 hour and then cooled to 15° C. Solid sodium triacetoxyborohydride (1.31 kg, 6.17 mol) was added and the reaction mixture was stirred for 0.5 hours. The addition of sodium triacetoxyborohydride was repeated three more times. At the end, a total of 5.22 kg (24.7 mol) sodium triacetoxyborohydride was added in 2 hours. The reaction mixture was stirred at 20-25° C. for 16 hours. The completeness of the reaction was followed by HPLC. Upon reaction completion, TBME (48.1 L) was added to the resulting jelly reaction mixture. The mixture was washed with saturated sodium hydrogen carbonate solution (60.0 L×3). The combined aqueous phase was extracted with TBME (48.1 L). All organic layers were combined, washed with brine (48.1 L) and concentrated in vacuum to a volume of 10.6 L. Isopropanol (192.3 L) was added to the residue and the resulting oil precipitates were dissolved upon warming up to 70-75° C. The solvent volume was reduced to 160.0 L by distillation at 70-75° C. Concentrated HCl (1.5 L) was added at 75° C. in 10 minutes followed by the addition of seed crystals. Crystallization occurred upon cooling to 20-25° C. in 16 hours. The mixture was filtered. The cake was washed with isopropanol (9.6 L×2) and dried at maximum 40° C. to a constant weight. 5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid methyl ester hydrochloride salt (6.57 kg, 88.0%) was obtained as white crystalline solid. mp=204-207° C. 1H NMR (CDCl3): 14.22 (1H, b), 8.18 (1H, d, J=0.9 Hz), 7.86 (1H, m), 7.67 (2H, d, J=8.1 Hz), 7.24 (1H, m), 7.23 (2H, d, J=8.1 Hz), 6.67 (2H, m), 4.76 (2H, m), 4.46 (1H, m), 4.16 (1H, m), 4.02 (2H, m), 3.86 (3H, s), 3.75 (1H, m), 3.38 (3H, s). 13C NMR (CDCl3): 18 Carbons (171.24, 162.32, 152.98, 136.05, 135.27, 134.03, 132.83, 131.94, 130.46, 128.85, 128.56, 123.92, 117.52, 113.43, 71.13, 52.43, 52.22, 46.73). Anal. Calcd for C27H23Cl3N4O4S: C, 53.52; H, 3.83; N, 9.25; S, 5.29; Cl, 17.55. Found: C, 53.07; H, 3.69; N, 9.08; S, 5.23; Cl, 17.20.
Example 45-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid

To a solution of 5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid methyl ester hydrochloride salt (20.00 g, 33.00 mmol) and 1,2-propanediol (5.0 g) in tetrahydrofuran (200 mL) and water (100 mL) was added slowly potassium hydroxide solution (0.85M, 116 mL) at 8-12° C. in 0.5 hours. The resulting biphasic mixture was stirred at 8-12° C. for 20-27 hours until the reaction was complete. The reaction mixture was washed with n-heptane (200 mL). The pH was adjusted to 6.5 with addition of water (100 mL) and acetic acid (2.5 mL). Tetrahydrofuran was removed under reduced pressure at internal temperature <40° C. The pH was adjusted to 4.5 with addition of isopropyl acetate (400 mL) and acetic acid (11 mL). After 10 minutes of stirring, the aqueous layer was separated and was extracted with isopropylacetate (200 mL). The organic layers were combined, washed with water (100 mL) and concentrated under reduced pressure to a volume of 190 mL at bath temperature <40° C. Crystallization occurred during concentration. The crystal slurry was stirred at 20-25° C. for 16 hours and was then filtered. The cake was washed with cold isopropylacetate (15 mL×3) and dried in vacuum at 35-40° C. to a constant weight.
5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid (14.35 g, 78.3%) was obtained as white and sandy crystalline solid.
mp=209-230° C. 1H NMR (Acetone-d6): 8.19 (1H, d, J=1.3 Hz), 7.76 (2H, d, J=8.4 Hz), 7.49 (2H, d, J=8.2 Hz), 7.43 (1H, d, J=1.0 Hz), 7.41 (1H, t, J=1.9 Hz), 6.87 (2H, d, J=1.9 Hz), 4.16 (1H, dd, J1=13.9 Hz J2=0.8 Hz), 4.10 (1H, dd, J1=11.7 Hz, J2=6.2 Hz), 3.99 (1H, d, J=14.0 Hz), 3.48(1H, d, J=10.6 Hz), 3.47 (1H, dd, J1=9.6 Hz, J2=6.2 Hz), 3.25 (3H, s), 3.24 (1H, dd, J1=9.6 Hz, J2=11.7 Hz), 3.01 (1H, d, J=11.3 Hz).
13C NMR (Acetone-d6): 22 Carbons (172.69, 163.7, 153.98, 144.55, 142.23, 135.26, 135.09, 134.41, 133.89, 132.96, 130.33, 128.27, 126.98, 125.18, 119.07, 112.44, 74.28, 59.09, 56.45, 54.33, 50.73, 25.75).
Anal. Calcd for C26H20Cl2N4O4S: C, 56.22; H, 3.62; N, 10.08; S, 5.77; Cl, 12.76. Found: C, 56.27; H, 3.20; N, 9.97; S, 5.65; Cl, 12.68.
…………………………..
paper
J. Med. Chem. 2006, 49, 6946
http://pubs.acs.org/doi/abs/10.1021/jm0610806

LFA-1 (leukocyte function-associated antigen-1), is a member of the β2-integrin family and is expressed on all leukocytes. This letter describes the discovery and preliminary SAR of spirocyclic hydantoin based LFA-1 antagonists that culminated in the identification of analog 8 as a clinical candidate. We also report the first example of the efficacy of a small molecule LFA-1 antagonist in combination with CTLA-4Ig in an animal model of transplant rejection.
http://pubs.acs.org/doi/suppl/10.1021/jm0610806/suppl_file/jm0610806si20060913_101747.pdf synthesis as compd 8
says
a white solid: Anal.RP-HPLCtR= 3.09min (method D, purity 99%);
………………….
U.S. Patent 7,199,125 B2
http://www.google.com/patents/US7199125
………………………..
.U.S. Patent 6,710,064 B2
http://www.google.com/patents/US6710064
………….
REFERENCES
-
For a discussion on the inhibition of LFA-1/ICAM-1as an approach to treating autoimmune diseases see:
Yusuf-Makagiansar, H.; Anderson, M. E.; Yakovleva, T. V.; Murray, J. S.; Siahaan, T. J. Medicinal Research Reviews 2002, 22, 146 -
2.
For a discussion of therapeutic options for treatment of psoriasis, see:
Gottlieb, A. B. J. Acad. Dermatol 2005, 53, S3Larson, R. S.; Davis, T.; Bologa, C.; Semenuk, G.; Vijayan, S.; Li, Y.; Oprea, T.; Chigaev, A.; Buranda, T.; Wagner, C. R.; Sklar, L. A. -
3.
For other small molecule LFA-1/ICAM-1 antagonists as potential drugs please see:
(a) Pei, Z.; Xin, Z.; Liu, G.; Li, Y.; Reilly, E. B.; Lubbers, N. L.; Huth, J. R.; Link, J. T.; von Geldern, T. W.; Cox, B. F.; Leitza, S.; Gao, Y.; Marsh, K. C.; DeVries, P.; Okasinski, G. F. J. Med. Chem. 2001, 44, 2913(b) Liu, G.; Huth, J. R.; Olejniczak, E. T.; Mendoza, R.; DeVries, P.; Leitza, S.; Reilly, E. B.; Olasinski, G. F.; Fesik, S. W.; von Geldern, T. W. J. Med. Chem. 2001, 44, 1202(c) Wu, J.-P.; Emeigh, J.; Gao, D. A.; Goldberg, D. R.; Kuzmich, D.; Miao, C.; Potocki, I.; Qian, K. C.; Sorcek, R. J.; Jeanfavre, D. D.; Kishimoto, K.; Mainolfi, E. A.; Nabozny, G.; Peng, C.; Reilly, P.; Rothlein, R.; Sellati, R. H.; Woska, J. R.; Chen, S.; Gunn, J. A.; O’Brien, D.; Norris, S. H.; Kelly, T. A. J. Med. Chem. 2004, 47, 5356(d) Last-Barney, K.; Davidson, W.; Cardozo, M.; Frye, L. L.; Grygon, C. a.; Hopkins, J. L.; Jeanfavre, D. D.; Pav, S.; Qian, C.; Stevenson, J. M.; Tong, L.; Zindell, R.; Kelly, T. A. J. Am. Chem. Soc. 2001, 123, 5643(e) Wang, G. T.; Wang, S.; Gentles, R.; Sowin, T.; Leitza, S.; Reilly, E. B.; von Geldern, T. W. Bioorg. Med. Chem. Lett. 2005, 15, 195(f) Wattanasin, S.; Albert, R.; Ehrhardt, C.; Roche, D.; Savio, M.; Hommel, U.; Welzenbach, K.; Weitz-Schmidt, G. Bioorg. Med. Chem. Lett. 2003, 12, 499 -
4.
The Discovery work towards this target compound BMS-587101 is described in:
Potin, D.; Launay, M.; Monatlik, F.; Malabre, P.; Fabreguettes, M.; Fouquet, A.; Maillet, M.; Nicolai, E.; Dorgeret, L.; Chevallier, F.; Besse, D.; Dufort, M.; Caussade, F.; Ahmad, S. Z.; Stetsko, D. K.; Skala, S.; Davis, P. M.; Balimane, P.; Patel, K.; Yang, Z.; Marathe, P.; Postelneck, J.; Townsend, R. M.; Goldfarb, V.; Sheriff, S.; Einspahr, H.; Kish, K.; Malley, M. F.; DiMarco, J. D.; Gougoutas, J. Z.; Kadiyala, P.; Cheney, D. L.; Tejwani, R. W.; Murphy, D. K.; Mcintyre, K. W.; Yang, X.; Chao, S.; Leith, L.; Xiao, Z.; Mathur, A.; Chen, B.-C.; Wu, D.-R.; Traeger, S. C.; McKinnon, M.; Barrish, J. C.; Robl, J. A.; Iwanowicz, E. J.; Suchard, S. J.; Dhar, M. T. G. J. Med. Chem. 2006, 49, 6946 -
5.
For additional information related to this compound see:
(a) Chen, B.-C.; DelMonte, A. J.; Dhar, T. G. M.; Fan, Y.; Gougoutas, J. Z.; Malley, M. F.; McLeod, D. D.; Waltermire, R.; Wei, C. Crystalline Forms and Process for Preparing Spiro-Hydantoin Compounds. (Bristol-Myers Squibb). U.S. Patent 7,381,737 B2 .(b) Dhar, T. G. M.; Potin, D.; Maillet, M.; Launay, M.; Nicolai, E.; Iwanowicz, E. Spiro-cyclic compounds useful as anti-inflammatory agents. Bristol-Myers Squibb and Cerep). U.S. Patent 7,199,125 B2.(c) Launay, M.; Potin, D.; Maillet, M.; Nicolai, E.; Dhar, T. G. M.; Iwanowicz, E. Hydantoin compounds useful as anti-inflammatory agents. (Bristol-Myers Squibb).U.S. Patent 6,710,064 B2.For the radiolabelled synthesis of BMS-587101 see:
Tran, S. B.; Maxwell, B. D.; Chen, S.-Y.; Bonacorsi, S. J.; Leith, L.; Ogan, M.; Rinehart, J. K.; Balasubramanian, B. J. Labelled Compd. Radiopharm. 2009, 52, 236
|
10-31-2008
|
CRYSTALLINE FORMS AND PROCESS FOR PREPARING SPIRO-HYDANTOIN COMPOUNDS
|
|
|
6-4-2008
|
Crystalline forms and process for preparing spiro-hydantoin compounds
|
|
|
3-7-2007
|
Pyridyl-substituted spiro-hydantoin compounds and use thereof
|
|
|
7-19-2006
|
Spiro-hydantoin compounds useful as anti-inflammatory agents
|
|
|
6-30-2006
|
Pyridyl-substituted spiro-hydantoin crystalline forms and process
|
|
|
12-21-2005
|
Spiro-hydantoin compounds useful as anti-inflammatory agents
|
| US8710058 * | Dec 4, 2009 | Apr 29, 2014 | Merck Patent Gmbh | Polymorphic forms of 3-(1-{3-[5-(1-methyl-piperidin-4-ylmethoxy)-pyrimidin-2-yl]-benzyl}-6-oxo-1,6-dihydro-pyridazin-3-yl)-benzonitrile hydrochloride salt and processes of manufacturing thereof |
| US20110269767 * | Dec 4, 2009 | Nov 3, 2011 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Novel Polymorphic Forms of 3-(1–6-oxo-1,6-dihydro-pyridazin-3-yl)-benzonitrile Hydrochloride Salt and Processes of Manufacturing Thereof |
USPTO Guidance On Patentable Subject Matter: Impediment to Biotech Innovation?
USPTO Guidance On Patentable Subject Matter: Impediment to Biotech Innovation?
http://commercialbiotechnology.com/index.php/jcb/article/view/664
Abstract
In June 2013, the U.S. Supreme Court issued a unanimous decision upending more than three decades worth of established patent practice when it ruled that isolated gene sequences are no longer patentable subject matter under 35 U.S.C. Section 101.While many practitioners in the field believed that the USPTO would interpret the decision narrowly, the USPTO actually expanded the scope of the decision when it issued its guidelines for determining whether an invention satisfies Section 101. The guidelines were met with intense backlash with many arguing that they unnecessarily expanded the scope of the Supreme Court cases in a way that could unduly restrict the scope of patentable subject matter, weaken the U.S. patent system, and create a disincentive to innovation. By undermining patentable subject matter in this way, the guidelines may end up harming not only the companies that patent medical innovations, but also the patients who need medical care. This article examines the guidelines and their impact on various technologies.
FDA Guidance for Industry: Electronic Source Data in Clinical Investigations
FDA Guidance for Industry: Electronic Source Data in Clinical Investigations
The FDA published its new Guidance for Industry (GfI) – “Electronic Source Data in Clinical Investigations” in September 2013. The Guidance defines the expectations of the FDA concerning electronic source data generated in the context of clinical trials. Find out more about this Guidance.
|
FDA Guidance for Industry: Electronic Source Data in Clinical Investigations |
|
After more than 5 years and two draft versions, the final version of the Guidance for Industry (GfI) – “Electronic Source Data in Clinical Investigations” was published in September 2013. This new FDA Guidance defines the FDA’s expectations for sponsors, CROs, investigators and other persons involved in the capture, review and retention of electronic source data generated in the context of FDA-regulated clinical trials. In an effort to encourage the modernization and increased efficiency of processes in clinical trials, the FDA clearly supports the capture of electronic source data and emphasizes the agency’s intention to support activities aimed at ensuring the reliability, quality, integrity and traceability of this source data, from its electronic source to the electronic submission of the data in the context of an authorization procedure. The Guidance addresses aspects as data capture, data review and record retention. When the computerized systems used in clinical trials are described, the FDA recommends that the description not only focus on the intended use of the system, but also on data protection measures and the flow of data across system components and interfaces. In practice, the pharmaceutical industry needs to meet significant requirements regarding organisation, planning, specification and verification of computerized systems in the field of clinical trials. The FDA also mentions in the Guidance that it does not intend to apply 21 CFR Part 11 to electronic health records (EHR). Author: Source: |
Low levels of omega-3 fatty acids may cause memory problems
09 Mar 2012
ST. PAUL, Minn. – A diet lacking in omega-3 fatty acids, nutrients commonly found in fish, may cause your brain to age faster and lose some of its memory and thinking abilities, according to a study published in the February 28, 2012, print issue of Neurology®, the medical journal of the American Academy of Neurology. Omega-3 fatty acids include the nutrients called docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA).
View original post 225 more words
Biomarker predicts effectiveness of brain cancer treatment
Researchers at the University of California, San Diego School of Medicine have identified a new biomarker that predicts whether glioblastoma – the most common form of primary brain cancer – will respond to chemotherapy. The findings are published in the July print issue of Oncotarget.
“Every patient diagnosed with glioblastoma is treated with a chemotherapy called temozolomide. About 15 percent of these patients derive long-lasting benefit,” said Clark C. Chen, MD, PhD, vice-chairman of Academic Affairs, Division of Neurosurgery, UC San Diego School of Medicine and the study’s principal investigator. “We need to identify which patients benefit from temozolomide and which another type of treatment. All therapies involve risk and the possibility of side-effects. Patients should not undergo therapies if there’s no likelihood of benefit.”
To pinpoint which patients were most likely respond to temozolomide, the researchers studied microRNAs that control the expression of a protein called methyl-guanine-methyl-transferase…
View original post 163 more words
