

Ono Pharmaceutical Co has become the first company in the world to get an approval for a PD-1 checkpoint inhibitor, as regulators in Japan gave the green light to nivolumab, developed with Bristol-Myers Squibb, as a treatment for melanoma.
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 254)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HSD-621 is a potent and selective 11β-HSD1 inhibitor


(R)-3,3,3-Trifluoro-2-(5-(((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-1-yl)sulfonyl)thiophen-2-yl)-2-hydroxypropanamide
2-Thiopheneacetamide, 5-[[(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-1-piperazinyl]sulfonyl]-α-hydroxy-α-(trifluoromethyl)-, (αR)-
1257229-37-0
C19 H18 F7 N3 O4 S2
…………………
The glucocorticoid receptor (GR) signaling pathway has been linked to the pathophysiology of diabetes and metabolic syndrome. We developed a series of potent and selective 11ï¢-HSD1 inhibitors. These compounds showed excellent potency against both human and mouse 11ï¢-HSD1 enzymes and displayed good pharmacokinetics and ex vivo inhibition of the target in mice.Compounds HSD-016 and HSD-621 were ultimately selected as clinical development candidates. Both compounds have attractive overall pharmaceutical profiles and demonstrated good oral bioavailability in mouse, rat and dog. When orally dosed in C57/BL6 dietï€-induced-ï€obesity (DIO) mice, HSD-016 and HSDï€621 were efficacious and showed a significant reduction in both fed and fasting glucose and insulin levels. Furthermore, both compoundswere well tolerated in drug safety assessment studies.
| Discovery of HSD-621 as a Potential Agent for the Treatment of Type 2 Diabetes (ACS Medicinal Chemistry Letters) Wednesday November 28th 2012 Author(s): Zhao-Kui Wan, Eva Chenail, Huan-Qiu Li, Manus Ipek, Jason Xiang, Vipin Suri, Seung Hahm, Joel Bard, Kristine Svenson, Xin Xu, Xianbin Tian, Mengmeng Wang, Xiangping Li, Christian E. Johnson, Ariful Qadri, Darrell Panza, Mylene Perreault, Tarek S. Mansour, James F. Tobin, Eddine Saiah, DOI:10.1021/ml300352x GO TO: [Article] http://pubs.acs.org/doi/full/10.1021/ml300352xandhttp://pubs.acs.org/doi/suppl/10.1021/ml300352x/suppl_file/ml300352x_si_001.pdf nmr data as 18b |
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) catalyzes the conversion of inactive glucocorticoid cortisone to its active form, cortisol. The glucocorticoid receptor (GR) signaling pathway has been linked to the pathophysiology of diabetes and metabolic syndrome. Herein, the structure–activity relationship of a series of piperazine sulfonamide-based 11β-HSD1 inhibitors is described. (R)-3,3,3-Trifluoro-2-(5-(((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-1-yl)sulfonyl)thiophen-2-yl)-2-hydroxypropanamide 18a (HSD-621) was identified as a potent and selective 11β-HSD1 inhibitor and was ultimately selected as a clinical development candidate. HSD-621 has an attractive overall pharmaceutical profile and demonstrates good oral bioavailability in mouse, rat, and dog. When orally dosed in C57/BL6 diet-induced obesity (DIO) mice, HSD-621 was efficacious and showed a significant reduction in both fed and fasting glucose and insulin levels. Furthermore, HSD-621 was well tolerated in drug safety assessment studies.
WO 2010141550
http://www.google.com/patents/WO2010141550A2?cl=en
EXAMPLES The title compounds of Examples 1.1, 1.2, and 1.3 were prepared as shown in
Scheme 1 below. Detailed synthesis procedures are provided below.
Scheme 1

Example 1.1

3,3,3-trifluoro-2-r5-({(2R)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2-methylpiperazin- l-yl}sulfonyl)-2-thienyll-2-hvdroxypropanamide Step IA: A mixture of (R)-2-methyl-piperazine (25.0 g, 250 mmol), 2-bromo 5- fluoro benzotrifluoride (55.1 g, 227 mmol), tris(dibenzylidineacetone)dipalldium (0) (2.08g, 2.27 mmol), rac-2,2′-bis(diphenylphosphino)-l,r-binaphthyl (4.24 g, 6.81 mmol) and sodium tert-butoxide (27.3 g, 280 mmol) was mixed and purged with N2. Anhydrous toluene (500 mL) was added and purged with N2 again. The resulting mixture was heated in an oil bath at 105 0C under N2 for 3.5 hours. After cooling, the reaction mixture was concentrated and then filtered through a pad of Celite, washed with Et2O. The organic layer was concentrated, diluted with Et2O (500 mL), filtered through a pad of Celite again, and washed with IN aq. HCl (2 x 150 mL). The aqueous layer was basified with NaOH at 0 0C (pH = -10) and then was extracted with Et2O (3 x 200 mL). The combined organic layer was dried over MgSO4 and concentrated under vacuum to give (3i?)-l-[4- fluoro-2-(trifluoromethyl)phenyl]-3-methylpiperazine as a brown oil (58.5 g, 98%), which was used without further purification.
Step IB: To a solution of 5-bromothiophene-2-sulfonyl chloride (26.2 g, 100 mmol) and (3R)-l-(4-fluoro-2-(trifluoromethyl)phenyl)-3-methylpiperazine (27.6 g, lOOmmol) in DCM (200 ml) was added Et3N (41.8 ml, 300 mmol) at room temp. The reaction mixture was stirred at room temperature until completion of the reaction (about 6 hours) and then washed with aq. NaHCO3. The basic washes were back extracted with dichloromethane (DCM). The combined organic layers were washed with brine and dried over Na2SO4. The crude product was purified on a SiO2 column using hexanes/DCM as the eluent to give (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine as a white solid (38 g, 78 mmol, 78 % yield).
Step 1C: To a solution of (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine (28.1 g, 57.7 mmol) in anhydrous THF (200 ml) was added Butyllithium (28.8 ml, 57.7 mmol) at -780C. The reaction mixture was Stirred under N2 for 15 min. and then a solution of methyl 3,3,3-trifluoropyruvate (6.07 ml, 57.7 mmol) in THF (20 mL) was added via a cannula. The reaction mixture was stirred at -780C for 2 h. and then quenched with a 10 mL of 10% aq. HCl. The reaction mixture was dried over MgSO4 and CombiFlashed with DCM/hexane (15 – 100%) to provide methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanoate as a sticky, light yellow solid (22 g, 39.0 mmol, 67.6 % yield).
Step ID, Method 1: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro- 2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (21.5 g, 38.1 mmol) in MeOH (200 ml) was added aq. NH3 (-28-
30%, 50 mL). The reaction mixture was stirred at room temperature o/n and then diluted with ice water (700 mL). The resultant white ppt was collected by filtration, washed with water, and dried in an oven at 60 0C to give the desired product 3,3,3-trifluoro-2-(5-((R)- 4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide (15 g, 27.3 mmol, 71.7 % yield). The aqueous layer was extracted with DCM (4 x 100 mL), and the combined organic layers were concentrated. Purification of the concentrate by column chromatography with EA/DCM (0-40%) gave an additional 1.5 g of product.
Method 2: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-
(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (200 mg) in MeOH (20 ml) at -780C was bubbled NH3 gas. The resultant mixture was stirred at room temperature overnight, concentrated, and dissolved in fresh DCM. The organic layer was washed with aq. NaHCO3 and dried to give 3,3,3- trifluoro-2-(5 -((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin- 1 – ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide as a white solid (150 mg). It was found that competing hydrolysis of the ester group to the corresponding acid occurred to a greater extent when using Method 1. Thus, in some instances, it may be preferable to use Method 2 when performing step D.
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI-FTMS,
[M+H]1+), 550.07165. Example 1.2
 desired
desiredαR)-3,3,3-trifluoro-2-r5-ααR)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)thiophen-2-yll-2-hvdroxypropanamide
13.5 grams of 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide (prepared according to a procedure similar to that described in Example 1.1) was separated was separated with a chiral column (Chiralpak ADH) in SFC Analytical Instrument; Mobile Phase was 90% CO2 /10%Methanol at flow rate 5mL/min. Early fraction (Retention 4.4min) was collected to give the title compound (5.7g); late fraction was collected to give the diastereomer described in Example 1.3 (6g, retention time 6. lmin).
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0697. Example 1.3
 undesired
undesiredαS)-3,3,3-trifluoro-2-r5-ααR)-4-r4-fluoro-2-qrifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)thiophen-2-yll-2-hvdroxypropanamide The title compound was obtained as the late fraction using the separation method described in Example 1.2.
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0701.
| US8524894 | Jun 4, 2010 | Sep 3, 2013 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2005063247A1 * | Dec 20, 2004 | Jul 14, 2005 | Amgen Sf Llc | Aryl sulfonamide compounds and uses related thereto |
| WO2007092435A2 * | Feb 7, 2007 | Aug 16, 2007 | Wyeth Corp | 11-beta hsd1 inhibitors |
BMS-582949 in phase 2 for Treatment of Antipsoriatics , Rheumatoid arthritis
BMS 582949, PS-540446
UNII-CR743OME9E
CAS 623152-17-0
4-[5-(N-Cyclopropylcarbamoyl)-2-methylphenylamino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide
4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide
Bristol-Myers Squibb Company
M.Wt: 406.48
Cas : 623152-17-0 Formula: C22H26N6O2
BMS-582949 had been in phase II clinical trials at Bristol-Myers Squibb for the oral treatment of moderate to severe psoriasis and for the treatment of rheumatoid arthritis (RA) in combination with methotrexate and for the treatment of inflammation in atherosclerotic plaque. However, no recent development has been reported for this research.
…………………..
http://www.google.com/patents/WO2012031057A1?cl=en
The present invention generally relates to a method of treating resistant rheumatic disease, such as refractory rheumatoid arthritis, with a therapeutically effective amount of a dual action p38 inhibitor that is safe and well-tolerated. A dual action p38 kinase inhibitor is a compound that inhibits both activation of p38 kinase and p38 kinase activity in cells.
A large number of cytokines participate in the inflammatory response, including IL- 1 , IL-6, IL-8 and TNF-a. Overproduction of cytokines such as IL-1 and TNF-a are implicated in a wide variety of diseases, including inflammatory bowel disease, rheumatoid arthritis, psoriasis, multiple sclerosis, endotoxin shock, osteoporosis, Alzheimer’s disease, and congestive heart failure, among others. See e.g., Henry et al., Drugs Fut. , 24: 1345- 1354 ( 1999); Salituro et al., Curr. Med. Ckem., 6:807-823 (1999)]. Important mediators of proinflammatory cytokines such as TNFct and IL-1 β,. as well as cellular responses to such cytokines production, are the mitogen-activated protein (MAP) kinases, and in particular, p38 kinase. See e.g., Schieven, G.L., “The biology of p38 kinase: a central role in inflammation”, Current Topics in Medicinal Chemistry, 5 :921 – 928 (2005). Accordingly, modulation of p38 kinase may be useful in the treatment of inflammatory disease including rheumatic diseases such as rheumatoid arthritis (RA).
Compounds that reportedly inhibit p38 kinase and cytokines such as IL-1 and TNF-a for use in treating inflammatory diseases are disclosed in U.S. Patent Nos.
6,277,989 and 6, 130,235 to Scios, Inc; U.S. Patent. Nos. 6, 147,080 and 5,945,41 8 to Vertex Pharmaceuticals Inc; U.S. Patent Nos. 6,251 ,914, 5,977, 103 and 5,658,903 to Smith-Kline Beecham Corp.; U.S. Patent Nos. 5,932,576 and 6,087,496 to G.D. Searle & Co.; WO 00/56738 and WO 01 /27089 to Astra Zeneca; WO 01/34605 to Johnson & Johnson; WO 00/12497 (quinazoHne derivatives as p38 kinase inhibitors); WO 00/56738 (pyridine and pyrimidine derivatives for the same purpose); WO 00/12497 (discusses the relationship between p38 kinase inhibitors); and WO 00/12074 (piperazine and piperidine compounds useful as p38 inhibitors). Other compounds that inhibit p38 kinase are pyrrolotriazine aniline compounds, information on these compounds is disclosed in U.S. Patent Nos. 6,670,357; 6,867,300; 7,034, 151 ; 7, 160,883; 7,21 1,666; 7,253, 167; and U.S. Publication Nos. 2003/023283 1 (published Dec. 18, 2003); 2004/0229877 (published Nov. 1 8, 2004); 2005/0043306 (published Feb. 24, 2005; 2006/0003967 (published Jan. 5, 2006); 2006/0030708 (published Feb. 9, 2006); 2006/0041 124 (published Feb. 23, 2006); 2006/0229449 (published Oct. 12, 2006); 2006/0235020 (published Oct. 19, 2006); and 2007/0213300 (published Sept 13, 2007).
In particular, WO 2003/090912 (U.S. Patent Nos. 7, 160,883, 7,388,009, p38 inhibitor, BMS-582949 (Example 7,

including processes of making and uses thereof.
……………………
http://www.google.com/patents/WO2003090912A9?cl=en
Examples 4-22

Compounds having the formula (Id), above, wherein R4 has the values listed in the following Table, were prepared following the same procedure described for Example 3, using the appropriate amine in place of ra-butylamine.



…………………………
WO 2006020904
http://www.google.com.br/patents/WO2006020904A1?cl=en
EXAMPLE IA St


Part a.
A solution of Example 1 (0.86 g, 2.20 mmol, 1.0 eq.) in THF (4.0 mL) and 1 N aqueous NaOH (9.0 mL, 4.1 eq.) was stirred at 6O0C overnight. After cooling to RT, the reaction mixture was concentrated in vacuo but not to dryness. To the solution at O0C was added 1 N aqueous hydrochloric acid until it was acidic and the precipitate was collected and dried to afford crude Example IA acid (0.51 g, 64.0 % yield). HPLC Ret. t. = 2.400 min.; LC/MS (M+H) + = 366.06+. The filtrate was then extracted with EtOAc (3x) and the organic layers were combined, dried over sodium sulfate, and concentrated in vacuo to give Example IA acid (0.035 g, 4.4 % yield). Part b.

A solution of Part a. acid (0.026 g, 0.071 mmol, 1.0 eq.), EDC (0.021 g, 0.11 mmol, 1.5 eq.), HOBt (0.015 g, 0.11 mmol, 1.5 eq), ^-propylamine (0.015 mL, 0.15 mmol, 2.1 eq.) and DIPEA (0.040 mL, 0.23 mmol, 3.2 eq.) in DMF (0.20 mL) was shaken at RT overnight. Water (1 mL) was added and the precipitate collected by filtration, washed with water, and dried to give Example IA amide (0.021 g, 70% yield); HPLC Ret. t. = 2.883 min.; LC/MS (M+H)+ = 421.18 +.
EJiAMPLE 2 Direct Aminolysis Procedure

n-Buli/THF
Ester Compound I or Hexyllithium/THF
-^
,NH9

1. Aminolysis with hexyllithium
To a dried 100 ml flask was added THF (10 ml) under nitrogen, which was then cooled to -100C. Hexyllithium (2.3 M in hexane, 6.5 ml, 15.0 mmol) was added slowly (exothermic, temperature was up to 5°C), followed by dropwise addition of propylamine (1.01 g, 1.4 ml, 17.1 mmol) at such a rate to maintain the temperature below 5°C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (exothermic, T<5°C). After being stirred at 00C for 20 minutes, the mixture was allowed to warm to room temperature and stirred for 5 hours. Ester compound I was <0.1 AP at this point by HPLC analysis. The mixture was cooled to -50C. Acetic acid (2 ml) was added slowly to maintain the temperature <10°C. The resulting thick slurry was stirred at room temperature for 20 minutes, and then solvents were exchanged with DMF (15 ml) on a rotavapor. To the resulting yellow slurry, water (15 ml) was added slowly to keep T<25°C. During the addition of water, the slurry became a clear solution, and a new slurry was formed. The slurry was stirred at room temperature for overnight. In the morning the slurry was filtered and the solid was washed with DMF/water (1:1, 5 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for 24 hours to afford 0.90 g of amide product II (yield: 87.2%) as a white solid. HPLC: 99.70 AP.
2. Aminolysis with n-butyllithium
To a dried 100 ml of flask was added THF (10 ml) under nitrogen and then cooled to -100C. n-Butyllithium (2.5 M in hexane, 6.0 ml, 15.0 mmol) was added slowly, followed by dropwise addition of propylamine (0.98 g, 16.5 mmol) at such a rate to keep the temperature below 00C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (T<5°C). After being stirred at O0C for 30 minutes, the mixture was allowed to warm to room temperature and stirred for overnight (~22h, Note 1). Compound I was not detected at this point by HPLC analysis. The mixture was cooled to -7°C. Acetic acid (2 ml) was added dropwise to maintain the temperature <10°C. The resulting thick slurry was stirred at 50C for 2 hours and at room temperature for 20 minutes, followed by evaporation on a rotavapor to give a wet yellow solid. To this solid was added acetone (10 ml) and water (20 ml). The slurry was stirred at room temperature for one and half hours. Filtration gave a white solid. This solid was washed with 35% acetone in water (10 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for the weekend to afford 0.94g of amide product II (yield: 91.0%) as a white solid. HPLC: 99.76 AP. Note 1: Compound I was -0.056 AP at 2.5 hours.
……………………
WO 2003090912
http://www.google.com/patents/WO2003090912A1?cl=en
……………………..
Discovery of 4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38a MAP kinase inhibitor for the treatment of inflammatory diseases
J Med Chem 2010, 53(18): 6629
http://pubs.acs.org/doi/abs/10.1021/jm100540x
The discovery and characterization of 7k (BMS-582949), a highly selective p38α MAP kinase inhibitor that is currently in phase II clinical trials for the treatment of rheumatoid arthritis, is described. A key to the discovery was the rational substitution of N-cyclopropyl for N-methoxy in 1a, a previously reported clinical candidate p38α inhibitor. Unlike alkyl and other cycloalkyls, the sp2 character of the cyclopropyl group can confer improved H-bonding characteristics to the directly substituted amide NH. Inhibitor 7k is slightly less active than 1a in the p38α enzymatic assay but displays a superior pharmacokinetic profile and, as such, was more effective in both the acute murine model of inflammation and pseudoestablished rat AA model. The binding mode of 7k with p38α was confirmed by X-ray crystallographic analysis.

4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (7k)
A mixture of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methylpyrrolo[1,2-f][1,2,4]triazine-6-carboxylic acid (6b) (2.16 g, 5.91 mmol), n-propylamine (1.0 mL, 12.2 mmol), BOP (3.40 g, 7.69 mmol), and N-methylmorpholine (2.5 mL, 22.7 mmol) in DMF (10 mL) was stirred at 50 °C for 3 h. The mixture was poured into a mixture prepared from saturated NaHCO3 solution (60 mL) and water (60 mL). The precipitating product was collected by suction filtration was washed with water. This crude product was suspended into ethyl acetate (100 mL) and stirred at 70 °C for 1 h. Upon cooling to rt, the title compound (2.07 g, 86% yield) was collected as a white solid by suction filtration; 98% purity by HPLC. LCMS (EI)
m/z Calcd for C22H26N6O2 (M + H)+ = 407.21. Found: 407.22.
1H NMR (500 MHz, DMSO-d6) δ 8.49 (d, J = 3.6 Hz, 1H), 8.23 (s, 1H), 8.21 (s, 1H), 7.86 (s, 1H), 7.80 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 3.20 (m, 2H), 2.87 (m, 1H), 2.82 (s, 3H), 2.25 (s, 3H), 1.54 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H), 0.68 (m, 2H), 0.59 (m, 2H).
13C NMR (125 MHz, DMSO-d6) δ 167.3, 164.45, 155.3, 148.7, 138.8, 137.1, 133.0, 130.6, 127.2, 125.8, 119.6, 118.8, 114.4, 113.3, 41.0, 23.6, 23.1, 18.5, 12.1, 12.0, 6.2.
EXAMPLE 3

Direct Aminolysis
Ester Compound I

Amide Product II
Method A:
A solution of n-propylamine (6.5 eq) in THF (20 ml/g of ester compound I) was cooled to — 5°C and was slowly treated with 2.5 M solution of n-butyllithium (6.1 eq). The mixture was stirred for 10 minutes. At the end of the period, a slurry of ester compound I (1 eq) in THF (14 ml/g of ester compound I) was cannulated into the performed Li-NHPr solution. The reaction mixture was warmed to 25°C and stirred till all of ester compound I was consumed (~ 3 hours). After the reaction was judged to be completed by HPLC, the reaction mixture was cooled to ~0°C and was slowly treated with acetic acid (5 ml/g of ester compound I). The slurry was then warmed to -2O0C and was stirred for 1 hour. At the end of the period, the solvent was distilled under vacuum to the minimum volume and the concentrated slurry was diluted with a solution of acetone (10 ml/g of ester compound I) and water (20 ml/g of ester compound I). The slurry was stirred for 1 hour and was cooled to ~5°C. The slurry was filtered and the cake was washed with acetone (5 ml/g of ester compound I). The cake was dried to give the amide product II (typically in 85% yield and 99 AP).
Method B:
A solution of n-propylamine (20 eq) in 2,2,2-trifmoroethanol (10 ml/g of ester compound I) was slowly treated with 2.5 M solution of n-butyllithium (1.5 eq). The mixture was stirred for 5 minutes. At the end of the period, the starting material, ester compound I, was added and the reaction mixture was warmed to 900C. The reaction mixture was held at 900C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then analyzed by HPLC. Typically, analysis indicated there was only 1.57 AP of starting material left.
Method C:
A solution of n-propylamine (2 eq) in methylene chloride (10 ml/g of ester compound I) at 200C was slowly treated with 2.0 M solution of trimethylaluminum (4 eq) in hexanes. The mixture was stirred for 15 minutes. At the end of the period, the starting material, ester compound 1 (1 eq), was added and the reaction mixture was warmed to 600C. The reaction mixture was held at 600C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then slowly quenched with aqueous HCl solution and analyzed by HPLC. Typically, analysis indicated there was 96.8AP of amide compound II product with 0.03 AP of the dipropylamide impurity.
…………………………………….
| WO2003090912A1 * | 15 abr. 2003 | 6 nov. 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors |
Liu C, Lin J, Everlof G, Gesenberg C, Zhang H, Marathe PH, Malley M, Galella MA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
Bioorg Med Chem Lett. 2013 May 15;23(10):3028-33. doi: 10.1016/j.bmcl.2013.03.022. Epub 2013 Mar 15.
Freebern WJ, Bigwarfe TJ, Price KD, Haggerty HG.
J Immunotoxicol. 2013 Jan-Mar;10(1):106-17. doi: 10.3109/1547691X.2012.736427. Epub 2012 Nov 23.
Liu C, Lin J, Wrobleski ST, Lin S, Hynes J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, Pitt S, Shen DR, Zhang RF, McIntyre KW, Salter-Cid L, Shuster DJ, Zhang H, Marathe PH, Doweyko AM, Sack JS, Kiefer SE, Kish KF, Newitt JA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
J Med Chem. 2010 Sep 23;53(18):6629-39. doi: 10.1021/jm100540x.
BMS-582949: crystalline form of a p38alpha inhibitor? WO2008079857.
Norman P.
Expert Opin Ther Pat. 2009 Aug;19(8):1165-8. doi: 10.1517/13543770902816160.
| WO2000012074A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Use of piperidines and/or piperazines as inhibitors of p38-alpha kinase | |
| WO2000012497A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Quinazoline derivatives as medicaments | |
| WO2000056738A1 | Mar 17, 2000 | Sep 28, 2000 | Astrazeneca Ab | Pyridine and pyrimidine derivatives and their use as inhibitors of cytokine mediated disease | |
| WO2001027089A1 | Oct 10, 2000 | Apr 19, 2001 | Astrazeneca Ab | Pyrimidine derivatives | |
| WO2001034605A1 | Oct 27, 2000 | May 17, 2001 | Ortho Mcneil Pharm Inc | SUBSTITUTED 2-ARYL-3-(HETEROARYL)-IMIDAZO[1,2-a]PYRIMIDINES, AND RELATED PHARMACEUTICAL COMPOSITIONS AND METHODS | |
| WO2003090912A1 | Apr 15, 2003 | Nov 6, 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US4200750 | Dec 8, 1977 | Apr 29, 1980 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines | |
| US5658903 | Jun 3, 1996 | Aug 19, 1997 | Smithkline Beecham Corporation | Cytokine inhibitors | |
| US5932576 | May 22, 1998 | Aug 3, 1999 | G. D. Searle & Company | 3(5)-heteroaryl substituted pyrazoles as p38 kinase inhibitors | |
| US5945418 | Mar 20, 1997 | Aug 31, 1999 | Vertex Pharmaceuticals Incorporated | Administering to the mammal to inhibit a mammalian protein kinase p38 which causes cell proliferation, cell death and response to extracellular stimuli | |
| US5977103 | Jan 10, 1997 | Nov 2, 1999 | Smithkline Beecham Corporation | Substituted imidazole compounds | |
| US6087496 | Apr 1, 1999 | Jul 11, 2000 | G. D. Searle & Co. | Enzyme inhibitors | |
| US6130235 | Aug 3, 1998 | Oct 10, 2000 | Scios Inc. | Piperidine moieties coupled to indole, benzimidazole or benzotriazole. | |
| US6147080 | Jun 10, 1997 | Nov 14, 2000 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 | |
| US6251914 | Jul 1, 1998 | Jun 26, 2001 | Smithkline Beecham Corporation | Treating cytokine mediated diseases | |
| US6277989 | Mar 14, 2000 | Aug 21, 2001 | Scios, Inc. | Quinazoline derivatives as medicaments | |
| US6670357 | Nov 7, 2001 | Dec 30, 2003 | Bristol-Myers Squibb Company | Antiinflammatory agents | |
| US6867300 | Nov 6, 2002 | Mar 15, 2005 | Bristol-Myers Squibb Company | Methods for the preparation of pyrrolotriazine compounds useful as kinase inhibitors | |
| US7034151 | Feb 5, 2004 | Apr 25, 2006 | Bristol-Myers Squibb Company | 1,4-dihydro-4-oxo-pyrrolo[2,1-f][1,2,4]triazine-6-carboxylates; novel approach to the formation of the bicyclic heterocyclic ring system | |
| US7041501 | Oct 31, 2002 | May 9, 2006 | Bristol-Myers Squibb Company | Methods of screening for toxicity of test compounds | |
| US7160883 | Apr 22, 2003 | Jan 9, 2007 | Bristol-Myers-Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7211666 | Dec 22, 2004 | May 1, 2007 | Bristol-Myers Squibb Company | N-Cyclopropyl-4-[[5-[(methoxyamino)carbonyl]-2-methylphenyl]amino]-5-methylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide; aminating with chloramine to produce a pyrrole with a Nitrogen nitrogen bond; reacting with formamide, cyclizing to form the pyrrolotriazine core; kinase inhibitors | |
| US7253167 | Jun 29, 2005 | Aug 7, 2007 | Bristol-Myers Squibb Company | Tricyclic-heteroaryl compounds useful as kinase inhibitors | |
| US7388009 | Oct 3, 2003 | Jun 17, 2008 | Bristol-Myers Squibb Company | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US7462616 | Oct 24, 2006 | Dec 9, 2008 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7759343 | Oct 28, 2008 | Jul 20, 2010 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US61379001 | Title not available | ||||
| US20030232831 | Apr 22, 2003 | Dec 18, 2003 | Alaric Dyckman | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors | |
| US20040229877 | Oct 29, 2003 | Nov 18, 2004 | Katerina Leftheris | Administering pyrrolotriazine carboxamide and benzamide compounds for therapy of p38 kinase-associated conditions | |
| US20050043306 | Oct 3, 2003 | Feb 24, 2005 | Katerina Leftheris | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US20060003967 | Jun 28, 2005 | Jan 5, 2006 | Zhongping Shi | Method for preparing pyrrolotriazine compounds | |
| US20060030708 | Aug 5, 2005 | Feb 9, 2006 | Lobben Paul C | Methods for the preparation of pyrrolotriazine compounds | |
| US20060041124 | Oct 14, 2005 | Feb 23, 2006 | Bang-Chi Chen | Process for preparing pyrrolotriazine kinase inhibitors | |
| US20060229449 | Apr 3, 2006 | Oct 12, 2006 | Apurba Bhattacharya | Reacting with chloramine in presence of aqueous base, phase transfer catalyst; anti-cancer agents, kinase inhibitors | |
| US20060235020 | Apr 4, 2006 | Oct 19, 2006 | Soojin Kim | Process for preparing salts of 4-[[5-[(cyclopropylamino)carbonyl]-2-methylphenyl]amino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide and novel stable forms produced therein | |
| US20070213300 | Mar 6, 2007 | Sep 13, 2007 | Bristol-Myers Squibb Company | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
Japan First to Approve Alectinib アレクチニブ 塩酸塩 (AF 802) for ALK+ NSCLC
Alectinib (AF802, CH5424802, RG7853, RO5424802)
CAS 1256580-46-7 FREE
1256589-74-8 (Alectinib Hydrochloride)
9-Ethyl-6,11-dihydro-6,6-dimethyl-8-[4-(4-morpholinyl)-1-piperidinyl]-11-oxo-5H-benzo[b]carbazole-3-carbonitrile
| Formula: | C30H34N4O2 |
| M.Wt: | 482.62 |
Mechanism of Action:ALK inhibitor
Indication:Non-small cell lung cancer (NSCLC)
Current Status:Phase II (US,EU,UK), NDA(Japan)
Company:中外製薬株式会社 (Chugai), Roche
Japan First to Approve Alectinib for ALK+ NSCLC
Roche announced that the Japanese Ministry of Health, Labor and Welfare (MHLW) has approved alectinib for the treatment of people living with non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase fusion gene-positive (ALK+). The approval was based on results from a Japanese Phase 1/2 clinical study (AF-001JP) for people whose tumors were advanced, recurrent or could not be removed completely through surgery (unresectable).

| Company | Chugai Pharmaceutical Co. Ltd. |
| Description | Anaplastic lymphoma kinase (ALK) inhibitor |
| Molecular Target | Anaplastic lymphoma kinase (ALK) |
| Mechanism of Action | Anaplastic lymphoma kinase (Ki-1) (ALK) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Registration |
| Standard Indication | Non-small cell lung cancer (NSCLC) |
| Indication Details | Treat advanced ALK-positive non-small cell lung cancer (NSCLC); Treat non-small cell lung cancer (NSCLC); Treat unresectable progressive or recurrent ALK-positive non-small cell lung cancer (NSCLC) |
| Regulatory Designation |
U.S. – Breakthrough Therapy (Treat advanced ALK-positive non-small cell lung cancer (NSCLC)); |
| Partner |

Alectinib (also known as CH5424802,RO5424802), a second generation oral inhibitor of anaplastic lymphoma kinase (ALK), is being developed by Chugai and Roche for the treatment of patients with ALK-positive non-small cell lung cancer (NSCLC) that has progressed on Xalkori (Crizotinib).
Alectinib was discovered by Chugai Pharmaceutical Co. Ltd. Chugai became a subsidiary of Roche in 2002 and the Swiss group currently owns 59.9 percent of the company.
On October 8, 2013, Chugai Pharmaceutical announced that it has filed a new drug application to Japan’s Ministry of Health, Labour and Welfare (MHLW) for alectinib hydrochloride for the treatment of ALK fusion gene positive non-small cell lung cancer (NSCLC).
IT is a potent and selective ALK inhibitor with IC50 of 1.9 nM.Alterations in the anaplastic lymphoma kinase (ALK) gene have been implicated in human cancers. Among these findings, the fusion gene comprising EML4 and ALK has been identified in non-small cell lung cancer (NSCLC) and fusion of ALK to NPM1 has been observed in anaplastic large cell lymphoma (ALCL). The possibility of targeting ALK in human cancer was advanced with the launch of crizotinib for NSCLC in the U.S. in 2011. The development of resistance to crizotinib in tumors, however, has led to the need for second-generation ALK inhibitors. One of these, alectinib hydrochloride, has been found to be an orally active, potent and highly selective ALK inhibitor with activity in ALK-driven tumor models. Alectinib has shown preclinical activity against cancers with ALK gene alterations, including NSCLC cells expressing the EML4-ALK fusion and ALCL cells expressing the NPM-ALK fusion. Alectinib was well tolerated and active in a phase I/II study conducted in Japan in patients with ALK-rearranged advanced NSCLC and in patients with ALK-positive NSCLC who had progressed on crizotinib. Alectinib has been submitted for approval in Japan for the treatment of ALK fusion gene-positive NSCLC and is in phase I/II development for ALK-rearranged NSCLC in the U.S.

……………..

………………….
WO2012023597
http://www.google.fm/patents/WO2012023597A1?cl=en
(Preparation 30)
Compound F6-20
9 – ethyl-6, 6 – dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) -11 – oxo-6 ,11 – dihydro-5H-benzo [b] carbazol-3 – carbonitrile

Under the same conditions as the synthesis of the compound B3-13-1, and the title compound was synthesized from compound F5-49.
1 H-NMR (400MHz, DMSO-D 6) δ: 12.70 (1H, s), 8.32 (1H, d, J = 7.9 Hz), 8.04 (1H, s), 8.00 (1H, s), 7.61 (1H , d, J = 8.5 Hz), 7.34 (1H, s), 3.64-3.57 (4H, m), 3.27-3.18 (2H, m), 2.82-2.66 (4H, m), 2.39-2.28 (1H, m ), 1.96-1.87 (2H, m), 1.76 (6H, s), 1.69-1.53 (2H, m), 1.29 (3H, t, J = 7.3 Hz)
LCMS: m / z 483 [M + H] +
HPLC retention time: 1.98 minutes (analysis conditions U)
Hydrochloride 9 of compound F6-20 – ethyl-6, 6 – dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) -11 – oxo-6 ,11 – dihydro-5H-benzo [b I was dissolved at 60 ℃ in a mixture of 10 volumes of methyl ethyl ketone, 3 volumes of water and acetic acid volume 4-carbonitrile -] carbazol-3. I was dropped hydrochloric acid (2N) 1 volume of solution. After stirring for 30 minutes at 60 ℃, and the precipitated solid was filtered and added dropwise to 25 volume ethanol, 9 – Dry ethyl -6,6 – dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) I got a one-carbonitrile hydrochloride – 11 – oxo-6 ,11 – dihydro-5H-benzo [b] carbazol-3. Ethyl-6, 6 – 9 – obtained dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) -11 – oxo-6 ,11 – dihydro-5H-benzo [b] carbazol-3 – I was pulverized with a jet mill carbonitrile monohydrochloride.
1 H-NMR (400MHz, DMSO-D 6) δ: 12.78 (1H, s), 10.57 (1H, br.s), 8.30 (1H, J = 8.4 Hz), 8.05 (1H, s), 7.99 (1H , s), 7.59 (1H, d, J = 7.9 Hz), 7.36 (1H, s) ,4.02-3 .99 (2H, m) ,3.84-3 .78 (2H, m) ,3.51-3 .48 (2H, m), 3.15-3.13 (1H, s) ,2.83-2 .73 (2H, s) ,2.71-2 .67 (2H, s) ,2.23-2 .20 (2H, m) ,1.94-1 .83 (2H, m), 1.75 (6H, s ), 1.27 (3H, t, J = 7.5 Hz)
FABMS: m / z 483 [M + H] +
I was dissolved at 90 ℃ to 33 volume dimethylacetamide F6-20 F6-20 mesylate. Was added to 168 volumes mesylate solution (2 N) 1.2 volume, ethyl acetate solution was stirred for 4 hours. The filtered crystals were precipitated, and dried to obtain a F6-20 one mesylate. I was milled in a jet mill F6-20 one mesylate salt was obtained.
……………………
Journal of Medicinal Chemistry, 54(18), 6286-6294; 2011
http://pubs.acs.org/doi/abs/10.1021/jm200652u
| WO2002043704A1 * | 30 Nov 2001 | 6 Jun 2002 | Yasuki Kato | Composition improved in solubility or oral absorbability |
| WO2008051547A1 * | 23 Oct 2007 | 2 May 2008 | Cephalon Inc | Fused bicyclic derivatives of 2,4-diaminopyrimidine as alk and c-met inhibitors |
| WO2009073620A2 * | 1 Dec 2008 | 11 Jun 2009 | Newlink Genetics | Ido inhibitors |
| WO2010143664A1 * | 9 Jun 2010 | 16 Dec 2010 | Chugai Seiyaku Kabushiki Kaisha | Tetracyclic compound |
| JP2008280352A | Title not available | |||
| JP2009100783A | Title not available | |||
| JPH0892090A * | Title not available |
|
References |
1: Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, Casey C, He J, Ali SM, Klempner SJ, Miller VA. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014 Apr;9(4):549-53. doi: 10.1097/JTO.0000000000000094. PubMed PMID: 24736079.
2: Gouji T, Takashi S, Mitsuhiro T, Yukito I. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. 2014 Mar;9(3):e27-8. doi: 10.1097/JTO.0000000000000113. PubMed PMID: 24518097.
3: Latif M, Saeed A, Kim SH. Journey of the ALK-inhibitor CH5424802 to phase II clinical trial. Arch Pharm Res. 2013 Sep;36(9):1051-4. doi: 10.1007/s12272-013-0157-8. Epub 2013 May 23. Review. PubMed PMID: 23700294.
4: Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, Ohe Y, Nogami N, Takeuchi K, Shimada T, Tanaka T, Tamura T. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013 Jun;14(7):590-8. doi: 10.1016/S1470-2045(13)70142-6. Epub 2013 Apr 30. PubMed PMID: 23639470.
5: Kinoshita K, Asoh K, Furuichi N, Ito T, Kawada H, Hara S, Ohwada J, Miyagi T, Kobayashi T, Takanashi K, Tsukaguchi T, Sakamoto H, Tsukuda T, Oikawa N. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem. 2012 Feb 1;20(3):1271-80. doi: 10.1016/j.bmc.2011.12.021. Epub 2011 Dec 22. PubMed PMID: 22225917.
6: Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011 May 17;19(5):679-90. doi: 10.1016/j.ccr.2011.04.004. PubMed PMID: 21575866.
Gadgeel S, Ou SH, Chiappori A, et al: A phase I dose escalation study of a new ALK inhibitor, CH542480202, in ALK+ non-small cell lung cancer patients who have failed crizotinib. Abstract O16.06. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia, October 29, 2013.
Ou SH, Gadgeel S, Chiappori AA, et al: Consistent therapeutic efficacy of CH5424802/RO5424802 in brain metastases among crizotinib-refractory ALK-positive non-small cell lung cancer patients in an ongoing phase I/II study. Abstract O16.07. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia, October 29, 2013.
Kinoshita, Kazuhiro et al,Preparation of tetracyclic compounds such as 11-oxo-5,6-dihydrobenzo[b]carbazole-3-carbonitrile derivatives as anaplastic lymphoma kinase (ALK) inhibitors,Jpn. Kokai Tokkyo Koho, 2012126711, 05 Jul 2012
Furumoto, Kentaro et al, Composition containing tetracyclic compound and dissolution aid (4環性化合物を含む組成物), PCT Int. Appl., WO2012023597, 23 Feb 2012, Also published as CA2808210A1, CN103052386A, EP2606886A1, EP2606886A4, US20130143877
Kinoshita, Kazutomo et al,Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802), Bioorganic & Medicinal Chemistry, 20(3), 1271-1280; 2012
Kinoshita, Kazutomo et al,9-Substituted 6,6-Dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazoles as Highly Selective and Potent Anaplastic Lymphoma Kinase Inhibitors, Journal of Medicinal Chemistry, 54(18), 6286-6294; 2011
Kinoshita, Kazuhiro et al, Preparation of tetracyclic compounds such as 11-oxo-5,6-dihydrobenzo[b]carbazole-3-carbonitrile derivatives as anaplastic lymphoma kinase (ALK) inhibitors,Jpn. Tokkyo Koho, 4588121, 24 Nov 2010

Japan approves world’s first PD-1 drug, nivolumab
http://www.pharmatimes.com/Article/14-07-07/Japan_approves_world_s_first_PD-1_drug_nivolumab.aspx
old article cut paste

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Rheumatoid arthritis & Ginger
As ageing populations grow, diseases such as Rheumatoid arthritis are becoming more prevelant. With advancing years this disease can lead to massive bone destruction with inflammation and pain. The researchers (Al-Nahain et al) in this recent paper study and review ginger (Zingiber official). This spice has traditionally been used for treatment of Rheumatoid arthritis in many countries like India where Ayurvedic doctore have been using it for many hundreds of years.
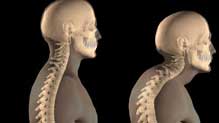
This review attempts to list the constituents and mechanisms of action.
The study concludes that phytochemicals from Ginger can form the basis of discovery of new drugs, which not only can provide symptomatic relief but also may provide total relief from diseases like Rheumatoid arthritis inhibiting bone destruction.

PARP Inhibitor.. Veliparib (ABT-888) 维利帕尼

Veliparib
2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide
CAS number: 912444-00-9 (Veliparib),
912445-05-7 (Veliparib dihydrochloride)
Mechanism of Action:poly (adenosine diphosphate [ADP]–ribose) polymerase (PARP) inhibitor
Indiction:cancer treatment
Development Status:Phase III
Drug Company: AbbVie
PARP Inhibitor Veliparib (ABT-888)
Also known as: ABT-888, 912444-00-9, ABT 888, carboxamide, CHEBI:62880, 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, ABT888, Veliparib
Molecular Formula: C13H16N4O Molecular Weight: 244.29234
| Systematic (IUPAC) name | |
|---|---|
| 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide | |
| Clinical data | |
| Legal status | experimental |
| Identifiers | |
| ATC code | None |
| PubChem | CID 11960529 |
| DrugBank | DB07232 |
| ChemSpider | 10134775 |
| UNII | 01O4K0631N |
| ChEMBL | CHEMBL506871 |
| Chemical data | |
| Formula | C13H16N4O |
| Mol. mass | 244.29 g/mol |
|
2-10-2012
|
PARP1 TARGETED THERAPY
|
|
|
4-17-2009
|
2-{(R)-2-METHYLPYRROLIDIN-2-YL)-1H-BENZIMIDAZOLE-4-CARBOXAMIDE CRYSTALLINE FORM 1
|
Veliparib (ABT-888)[1] is a potential anti-cancer drug acting as a PARP inhibitor. It kills cancer cells by blocking a protein called PARP, thereby preventing the repair of DNA or genetic damage in cancer cells and possibly making them more susceptible to anticancer treatments. Veliparib may make whole brain radiation treatment work more effectively against brain metastases from NSCLC.
It inhibits both PARP1 and PARP2.[2][3]
AbbVie’s Veliparib (ABT-888,), an inhibitor of poly ADP-ribose polymerase 1 and 2 (PARP 1 and PARP 2), is being investigated in multiple tumor types, including 3 phase III studies, all initiated this year, in neoadjuvant treatment of triple-negative breast cancer (clinical trial number:NCT02032277), non-small cell lung cancer (NSCLC, clinical trial number:NCT02106546) and HER2-negative, BRCA1 and/or BRCA2-positive breast cancer (clinical trial number:NCT02163694).
AbbVie, which was spun off from Abbott Laboratories in early 2013, is currently looking to buy Irish drug maker Shire for $46 billion. The proposed deal follows Pfizer’s failed $120 billion attempt to buy AstraZeneca. Humira, AbbVie’s rheumatoid arthritis drug and the world’s top-selling drug last year, accounts for 60% of company revenue and is going off-patent in at the end of 2016. The threat of growing competition for Humira may be a major motivation for AbbVie.


Clinical trials
Numerous phase I clinical trials are in progress.[4]
A phase I/II clinical trial for use with/out doxorubicin (for Metastatic or Unresectable Solid Tumors or Non-Hodgkin Lymphoma) started in 2008 and is due to complete in 2010.[5] Results (inc MTD) with topotecan.[6]
A phase II clinical trial for metastatic melanoma has started recruiting.[7] Due to end Dec 2011.
A phase II clinical trial for metastatic breast cancer has started recruiting.[8] Due to end Nov 2011.
A phase II clinical trial for add-on to Radiation Therapy for Patients with Brain Metastases from Non-Small Cell Lung Cancer.
It was included in the I-SPY2 breast cancer trial,[9] and there are encouraging data from that study [10]
A phase I clinical trial for prostate cancer in men who carry the BRCA mutation is underway and is now recruiting (as of May 2013).[11]


……………….
http://www.google.com/patents/US20060229289
EXAMPLE 1
2-(2-methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide EXAMPLE 1A 1-benzyl 2-methyl 2-methylpyrrolidine-1,2-dicarboxylate
A solution of 1-benzyl 2-methyl pyrrolidine-1,2-dicarboxylate (15.0 g, 57 mmol) and iodomethane (7.11 ml, 114 mmol) in THF (100 mL) was treated with NaN(TMS)2 (1.0 M solution in THF, 114 mL, 114 mmol) at −75° C. under nitrogen. The temperature of the cooling bath was then slowly raised to −20° C. within 1 h and the mixture was stirred at the same temperature for another 3 h. After quenching with water, the mixture was acidified with 2 N HCl (˜100 mL) and was partitioned between water (400 mL) and EtOAc (400 mL). The organic phase was washed with brine and concentrated. The residue was purified by flash column chromatography (silica gel, EtOAc/hexane) to give Example 1A (15.15 g, Yield: 96%). MS (DCI/NH3) m/z 278 (M+H)+.
EXAMPLE 1B
1-[(benzyloxy)carbonyl]-2-methylpyrrolidine-2-carboxylic acid
A solution of Example 1A (15.15 g, 54.63 mmol) in a mixture of THF (100 mL) and water (50 mL) was treated with LiOH.H2O (4.58 g, 109.26 mmol) in water (50 mL). Methanol was added until a transparent solution formed (60 mL). This solution was heated at 60° C. for overnight and the organic solvents were removed under vacuum. The residual aqueous solution was acidified with 2 N HCl to pH 2 and was partitioned between ethyl acetate and water. The organic phase was washed with water, dried (MgSO4), filtered and concentrated to give Example 1B as a white solid (13.72 g, 95.4% yield). MS (DCI/NH3) m/z 264 (M+H)+.
EXAMPLE 1C
benzyl 2-({[2-amino-3-(aminocarbonyl)phenyl]amino}carbonyl)-2-methylpyrrolidine-1-carboxylate
A solution of Example 1B (13.7 g, 52 mmol) in a mixture of pyridine (60 mL) and DMF (60 mL) was treated with 1,1′-carbonyldiimidazole (9.27 g, 57.2 mmol) at 45° C. for 2 h. 2,3-Diamino-benzamide dihydrochloride (11.66 g, 52 mmol), which was synthesized as described in previous patent application WO0026192, was added and the mixture was stirred at rt overnight. After concentration under vacuum, the residue was partitioned between ethyl acetate and diluted sodium bicarbonate aqueous solution. The slightly yellow solid material was collected by filtration, washed with water and ethyl acetate, and dried to give Example 1C (16.26 g). Extraction of the aqueous phase with ethyl acetate followed by concentration, filtration and water-EtOAc wash, provided additional 1.03 g of Example 1C. Combined yield: 84%. MS (APCI) m/z 397 (M+H)+.
EXAMPLE 1D
benzyl 2-[4-(aminocarbonyl)-1H-benzimidazol-2-yl]-2-methylpyrrolidine-1-carboxylate
A suspension of Example 1C (17.28 g, 43.6 mmol) in acetic acid (180 mL) was heated under reflux for 2 h. After cooling, the solution was concentrated and the residual oil was partitioned between ethyl acetate and sodium bicarbonate aqueous solution. The organic phase was washed with water and concentrated. The residue was purified by flash column chromatography (silica gel, 3-15% CH3OH in 2:1 EtOAc/hexane) to provide Example 1D (16.42 g, Yield: 99%).
MS (APCI) m/z 379 (M+H)+.
EXAMPLE 1E 2-(2-methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide
A solution of Example 1D (15.0 g, 40 mmol) in methanol (250 ml) was treated with 10% Pd/C (2.8 g) under 60 psi of hydrogen for overnight. Solid material was filtered off and the filtrate was concentrated. The residual solid was recrystallized in methanol to give 7.768 g of Example 1E as free base. The bis-HCl salt was prepared by dissolving the free base in warm methanol and treating with 2 equivalents of HCl in ether (10.09 g). MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): δ 1.92 (s, 3 H), 2.00-2.09 (m, 1 H), 2.21-2.29 (m, 1 H), 2.35-2.41 (m, 1 H), 2.52-2.57 (m, 1 H), 3.54-3.65 (m, 2 H), 7.31 (t, J=7.93 Hz, 1 H), 7.68 (dd, J=8.24, 0.92 Hz, 1 H), 7.72 (dd, J=7.63, 0.92 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72N, 17.66. Found: C, 49.30; H, 5.60; N, 17.39.
EXAMPLE 3 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide EXAMPLE 3A benzyl(2R)-2-[4-(aminocarbonyl)-1H-benzimidazol-2-yl]-2-methylpyrrolidine-1-carboxylate
Example 1D (1.05 g, 2.8 mmol) was resolved on chiral HPLC (Chiralcel OD, 80/10/10 hexane/EtOH/MeOH). The faster eluting peak was collected and concentrated to provide Example 3A (99.4% e.e., 500 mg). MS (APCI) m/z 379 (M+H)+.
EXAMPLE 3B 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide
A solution of Example 3A (500 mg, 1.32 mmol) in methanol (10 ml) was treated with 10% Pd/C (150 mg) under hydrogen for overnight (balloon). Solid material was filtered off and the filtrate was concentrated. The residual solid was further purified by HPLC (Zorbax C-18, CH3CN/H2O/0.1%TFA) and was converted to bis-HCl salt to provide Example 4 as white solid (254 mg). Co-crystallization of the free base with 1 equivalent of L-tartaric acid in methanol gave a single crystal that was suitable for X-ray study. The X-ray structure with L-tartaric acid was assigned the R-configuration. MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): δ 2.00 (s, 3 H), 2.10-2.19 (m, 1 H), 2.30-2.39 (m, 1 H), 2.45-2.51 (m, 1 H), 2.61-2.66 (m, 1 H), 3.64-3.73 (m, 2 H), 7.40 (t, J=7.95 Hz, 1 H), 7.77 (d, J=8.11 Hz, 1 H), 7.80 (d, J=7.49 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72; N, 17.66. Found: C, 49.10; H, 5.52; N, 17.61.
……………….
WO2009049111
http://www.google.com/patents/WO2009049111A1?cl=en
EXAMPLE 1 Preparation of ABT-888 Crystalline Form 1 A mixture of ABT-888 dihydrochloride (10 g) was stirred in saturated potassium bicarbonate (50 mL) and n-butanol (50 mL) until the ABT-888 dihydrochloride completely dissolved. The aqueous layer was extracted with a second portion of n-butanol then discarded. The extracts were combined, washed with 15% sodium chloride solution (50 mL) and concentrated. The concentrate was chase distilled three times with heptane (50 mL),dissolved in refluxing 2-propanol (45 mL) and filtered hot. The filtrate was cooled to ambient temperature with stirring over 18 hours, cooled to 0-50C, stirred for 1 hour, and filtered. The filtrant was washed with 2-propanol and dried in a vacuum oven at 45-500C with a slight nitrogen purge.
EXAMPLE 2
Preparation of ABT-888 Crystalline Form 2
A mixture of ABT-888 in methanol, in which the ABT-888 was completely dissolved, was concentrated at about 35 0C, and the concentrate was dried to a constant weight.
EXAMPLE 3 Preparation of ABT-888 Crystalline Form 1

15 16
Step 1 : 2-(2-methyl-2-pyrrolidino)-benzimidazole-4-carboxamide 2 HCl (15) is dissolved in water (3.5 kg / kg 15) at 20 + 5 0C. Dissolution of 15 in water results in a solution of pH 0 – 1.
Step 2: The reaction is run at 20 – 25 0C. One equivalent of sodium hydroxide is added, raising the pH to 2 – 3 with only a mild exotherm (100C observed with rapid addition of 1.0 equiv.). This generates a solution that remains clear for several days even when seeded with free base crystals. 3N NaOH (1.0 equiv., 1.25 kg / kg 15) is charged and the solution polish filtered into the crystallizer/ reactor.
Step 3: 5% Na2CO3 (1.5 equiv., 10.08 kg / kg 15) is then filtered into the crystallizer over 2 hours. Nucleation occurs after approximately l/6th of the Na2CO3 solution is added (-0.25 equiv.)
Step 4: The slurry is mixed for NLT 15 min before sampling (typically 1 to 4 hours (2.5 mg/mL product in the supernatant)). The slurry is filtered at 200C and washed with 6 portions of water (1.0 kg / kg 15 each). Each wash was applied to the top of the cake and then pressured through. No mixing of the wetcake was done.
Step 5 : The solids are then dried. Drying was performed at 500C keeping the Cogeim under vacuum while applying a slight nitrogen bleed. The agitator blade was left in the cake to improve heat transfer to the cake. It was rotated and lifted out of the cake once per hour of drying to speed the drying process while minimizing potential crystal attrition that occurs with continuous agitator use. In one embodiment of Step 1, the volume of water for dissolution of the Dihydrochloride (15) is about 1.3 g water/g 15. In another embodiment of Step 1,, the volume of water for dissolution is about 1.3 g to about 4 g water/g 15. In another embodiment of Step 1, the volume of water for dissolution is 1.3 g to 3.5 g water/g 15. In another embodiment of Step 1, the volume of water for dissolution is 3.5 g water/g 15.
……………………
J. Med. Chem., 2009, 52 (2), pp 514–523
DOI: 10.1021/jm801171j

(2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide
excellent PARP enzyme potency as well as single-digit nanomolar cellular potency. These efforts led to the identification of 3a (2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, ABT-888), currently in human phase I clinical trials. Compound 3a displayed excellent potency against both the PARP-1 and PARP-2 enzymes with a Ki of 5 nM and in a C41 whole cell assay with an EC50 of 2 nM. In addition, 3a is aqueous soluble, orally bioavailable across multiple species, and demonstrated good in vivo efficacy in a B16F10 subcutaneous murine melanoma model in combination with temozolomide (TMZ) and in an MX-1 breast cancer xenograft model in combination with either carboplatin or cyclophosphamide.
References
- “ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models” May 2007
- http://www.cancer.gov/drugdictionary/?CdrID=496464
- “ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models”, 2007
- http://clinicaltrialsfeeds.org/clinical-trials/results/term=Drug:+ABT-888
- “ABT-888 and Cyclophosphamide With Versus Without Doxorubicin in Treating Patients With Metastatic or Unresectable Solid Tumors or Non-Hodgkin Lymphoma”
- Phase I Study of ABT-888, a PARP Inhibitor, in Combination with Topotecan Hydrochloride in Adults with Refractory Solid Tumors and Lymphomas.. July 2011. doi:10.1158/0008-5472.CAN-11-1227.
- “A Study Evaluating Efficacy of ABT-888 in Combination With Temozolomide in Metastatic Melanoma”
- “ABT-888 and Temozolomide for Metastatic Breast Cancer”
- “Breast cancer study aims to speed drugs, cooperation”, March 2010
- http://www.centerwatch.com/news-online/article/5737/new-presurgery-combination-therapy-for-triple-negative-breast-cancer
- “Veliparib in Treating Patients With Malignant Solid Tumors That Did Not Respond to Previous Therapy. Clinical Trial NCT00892736”
|
4-1-2013
|
Design, synthesis and biological evaluation of novel imidazo[4,5-c]pyridinecarboxamide derivatives as PARP-1 inhibitors.
|
Bioorganic & medicinal chemistry letters
|
|
8-15-2013
|
Discovery of novel benzo[b][1,4]oxazin-3(4H)-ones as poly(ADP-ribose)polymerase inhibitors.
|
Bioorganic & medicinal chemistry letters
|
|
|
8-1-2013
|
Identification of potent Yes1 kinase inhibitors using a library screening approach.
|
Bioorganic & medicinal chemistry letters
|
|
5-1-2010
|
A rapid and sensitive method for determination of veliparib (ABT-888), in human plasma, bone marrow cells and supernatant by using LC/MS/MS.
|
Journal of pharmaceutical and biomedical analysis
|
|
1-22-2009
|
Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer.
|
Journal of medicinal chemistry
|
External links
http://kdwn.com/2013/12/16/new-drug-study-method-show-breast-cancer-promise/
| US8013168 | Oct 10, 2008 | Sep 6, 2011 | Abbott Laboratories | Veliparib crystal structure; an anticancer PARP inhibitor |
| US8372987 | Oct 10, 2008 | Feb 12, 2013 | Abbvie Inc. | Title compound is Veliparib, a Poly(ADP-ribose) polymerase i.e. PARP inhibitor; anticancer agent |
| US20060229289 * | Apr 11, 2006 | Oct 12, 2006 | Gui-Dong Zhu | 2-(2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide, aka veliparib, for example; poly(ADP-ribose)polymerase inhibitors; antiinflammatory, antitumor agents; Parkinson’s disease |
Penning, Thomas D. et al. Discovery of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the Treatment of Cancer. Journal of Medicinal Chemistry, 52(2), 514-523; 2009
Zhu, Guidong. 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide crystalline form 2 compositions and preparation for treating cancer. PCT Int. Appl. (2009), WO2009049109 A1 20090416
Kolaczkowski, Lawrence . 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide (ABT-888) crystalline form I and its pharmaceutical composition for cancer treatment. PCT Int. Appl. (2009), WO2009049111 A1 20090416.
Zhu, Gui-Dong; Gong, Jianchun; Gandhi, Virajkumar B.; Penning, Thomas D.; Giranda, Vincent L. Preparation of 1H-benzimidazole-4-carboxamides as poly(ADP-ribose)polymerase (PARP) inhibitors. U.S. Pat. Appl. Publ. (2006), US20060229289 A1 20061012.

Your Own Saliva Better For Wound Healing Than Yunnan Baiyao Alone
There are a few herbal formulas within Chinese Medicine that are worth their weight in gold. Yin qiao and/or Gan Mao Ling for colds/flus, Bao Ji Wan for food poisioning/acute digestive disturbances, and Yunnan Baiyao for acute bleeding, among others… In our clinic many of our patients, particularly those prone to getting cuts and scrapes such as construction workers, landscapers, etc. are aware of Yunnan Baiyao. We usually tell them to first rinse the wound if possible, then pour some of the Yunnan Baiyao powder on the wound and then rub in some saliva, then cover lightly. The bleeding stops quickly and the wound heals easily time and time again. Yunnan Baiyao is a top level Chinese military secret, originally developed for healing gun shot wounds in battle, and there is only one manufacturer.
Researchers from the Department of Pathology within the College of Medicine at Xi’an Jiaotong University in…
View original post 186 more words
‘Master switch’ for myelination in human brain stem cells is identified
Scientists at the University at Buffalo have identified the single transcription factor or “master switch” that initiates the critical myelination process in the brain. The research will be published online in Proceedings of the National Academy of Sciences (PNAS) on June 30.
The identification of this factor, SOX10, in human brain cells, brings researchers closer to the goal of treating multiple sclerosis (MS) by transplanting into patients the brain cells that make myelin.
“Now that we have identified SOX10 as an initiator of myelination, we can work on developing a viral or pharmaceutical approach to inducing it in MS patients,” says Fraser Sim, PhD, senior author on the paper and assistant professor in the UB Department of Pharmacology and Toxicology in the School of Medicine and Biomedical Sciences.
“If we could create a small molecule drug that would switch on SOX10, that would be therapeutically important,” he adds.
View original post 514 more words
Eating flavonoids protects men against Parkinson’s disease
07 Apr 2012
Men who eat flavonoid-rich foods such as berries, tea, apples and red wine significantly reduce their risk of developing Parkinson’s disease, according to new research by Harvard University and the University of East Anglia (UEA).
Published today in the journal Neurology ®, the findings add to the growing body of evidence that regular consumption of some flavonoids can have a marked effect on human health. Recent studies have shown that these compounds can offer protection against a wide range of diseases including heart disease, hypertension, some cancers and dementia.
This latest study is the first study in humans to show that flavonoids can protect neurons against diseases of the brain such as Parkinson’s.
Around 130,000 men and women took part in the research. More than 800 had developed Parkinson’s disease within 20 years of follow-up. After a detailed analysis of their diets and adjusting for age and…
View original post 472 more words
Japan scientists find ageing cure – for flowers
Japanese scientists say they have found a way to slow down the ageing process in flowers by up to a half, meaning bouquets could remain fresh for much longer.
Researchers at the National Agriculture and Food Research Organisation in Tsukuba, east of Tokyo, said they had found the gene believed to be responsible for the short shelf-life of flowers in one Japanese variety of morning glory.
“Morning glory” is the popular name for a hundreds of species of flowering plants whose short-lived blooms usually unfold early in the day and are gone by nightfall.
By suppressing the gene—named “EPHEMERAL1″—the lifespan of each flower was almost doubled, said Kenichi Shibuya, one of the lead researchers in a study carried out jointly with Kagoshima University in southern Japan.
“Unmodified flowers started withering 13 hours after they opened, but flowers that had been genetically modified stayed open for 24 hours,” he said.
This…
View original post 166 more words
