
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 218)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Epelsiban being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.

Epelsiban
557296
GSK-557296
GSK-557296-B
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione
(3R, 6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[(1R)- 1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1-methylpropyl]-2,5- piperazinedione
Glaxo Group Limited INNOVATOR
Epelsiban (GSK-557,296-B)[1][2] is an oral drug which acts as a selective, sub-nanomolar (Ki=0.13 nM) oxytocin receptor antagonist with >31000-fold selectivity over the related vasopressin receptors and is being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.[3][4]


benzenesulfonic acid;(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione,CAS 1159097-48-9
UNII-H629P9T4UN, GSK557296B, Epelsiban besylate (USAN), Epelsiban besylate [USAN], 1159097-48-9, H629P9T4UN
GSK-557296 is being developed in early clinical studies at GlaxoSmithKline for enhancement of embryo and or blastocyst implantation in women undergoing IVF treatment. The product has been in phase II clinical development for the treatment of premature ejaculation.
Preterm labor is a major clinical problem leading to death and disability in newborns and accounts for 10% of all births and causes 70% of all infant mortality and morbidity.
Oxytocin (OT) is a potent stimulant of uterine contractions and is responsible for the initiation of labor via the interaction with the OT receptors in the mammalian uterus. OT antagonists have been shown to inhibit uterine contractions and delay preterm delivery. So there is increasing interest in OT antagonists because of their potential application in the prevention of preterm labor. Although several tocolytics have already been approved in clinical practice, they have harmful maternal or fetal side effects.
The first clinically tested OT antagonist atosiban has a much more tolerable side effect profile and has recently been approved for use in Europe. However, atosiban is a peptide and a mixed OT/vasopressin V1a receptor antagonist that has to be given by iv infusion and is not suitable for long-term maintenance treatment, as it is not orally bioavailable.
Hence there has been considerable interest in overcoming the shortcomings of the peptide OT antagonists by identifying orally active nonpeptide OT antagonists with a higher degree of selectivity toward the vasopressin receptors (V1a, V1b, V2) with good oral bioavailability. Although several templates have been investigated as potential selective OT antagonists, few have achieved the required selectivity for the OT receptor vs the vasopressin receptors combined with the bioavailability and physical chemical properties required for an efficacious oral drug.
Therefore our objective was to design a potent, orally active OT antagonist with high levels of selectivity over the vasopressin receptor with good oral bioavailability in humans that would delay labor safely by greater than seven days and with improved infant outcome, as shown by a reduced combined morbidity score.
| Patent | Submitted | Granted |
|---|---|---|
| Compounds [US7919492] | 2010-12-02 | 2011-04-05 |
| Piperazinediones as Oxytocin Receptor Antagonists [US7550462] | 2007-11-01 | 2009-06-23 |
| Compounds [US8202864] | 2011-06-23 | 2012-06-19 |
| Novel compounds [US2009247541] | 2009-10-01 |
………………………………………
PATENT
https://www.google.com/patents/US7919492
Example 3
Method A

(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
as a white lyophilisate (88 mg, 23%) after freeze-drying from 1,4-dioxane
HPLC Rt=2.70 minutes (gradient 2); m/z [M+H]+=519
1H NMR (CDCl3) δ 7.49 (d, 1H), 7.27-7.15 (m, 4H), 7.10 (d, 1H), 6.68 (s, 1H), 6.40 (d, 1H), 4.10 (dd, 1H), 4.01 (d, 1H), 3.74-3.52 (m, 5H), 3.28-3.07 (m, 5H), 2.97-2.84 (m, 2H), 2.79-2.71 (m, 1H), 2.62 (s, 3H), 2.59 (s, 3H), 1.65-1.53 (m, 1H), 0.98-0.80 (m, 2H), 0.70 (t, 3H), 0.45 (d, 3H).
Example 3
Method B

(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
A suspension of {(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-6-[(1S)-1-methylpropyl]-2,5-dioxo-1-piperazinyl}(2,6-dimethyl-3-pyridinyl)acetic acid hydrochloride (5.0 g, 10.3 mmol) (intermediate 5) in dry dichloromethane (50 ml) was treated with 1,1-carbonyldiimidazole (2.6 g, 16 mmol) and the reaction mixture was stirred under nitrogen for 18 hours. Morpholine (4.8 ml, 55 mmol) was added and the resultant solution was left to stand under nitrogen for 18 hours. The solvent was removed in vacuo and the residue was separated between ethyl acetate and water. The organic phase was washed with brine and dried over anhydrous magnesium sulphate. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. This was applied to a basic alumina cartridge (240 g) and eluted using a gradient of 0-7.5% methanol in diethyl ether (9CV), 7.5-10% methanol in diethyl ether (1CV) and 10% methanol in diethyl ether (1CV). The required fractions were combined and evaporated in vacuo to give (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione as a white solid (2.4 g, 45%).
HPLC Rt=2.72 minutes (gradient 2); m/z [M+H]+=519
………………………………………
WO 2011051814
http://www.google.com/patents/WO2011051814A1?cl=en
This invention relates to novel crystalline forms of (3R, 6R)-3-(2,3-dihydro-1 H- inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1 – methylpropyl]-2,5-piperazinedione benzenesulfonate salt, processes for their preparation, pharmaceutical compositions containing them and to their use in medicine. The benzenesulfonate salt of Compound A is represented by the following structure:

In one aspect, the present invention provides a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form provides an X-ray powder diffraction pattern substantially in accordance with Figure 1 .
In another aspect, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

In an additional aspect, the invention includes a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2.
In certain aspects, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2 In one aspect, the invention also provides a crystalline form of {3R, 6R)-3-(2,3- dihydro-1 H-inden-2-yl)-1-[(1 R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]- 6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

Experimental
Process Scheme

Stage 4
Acetone / Water Recrystallisation
Compound A-form I Ste8e 5 Besylate salt
MW 676.83 Acetone / Water
Recrystallisation MW 676.83 Process description for isolation of Compound A-Form 1
Stage 0
methyl d-alloisoleucinate hydrochloride (Compound 2) was charged to ethyl acetate. A solution of potassium carbonate in water was then added. The mixture was then stirred vigorously at room temperature for 1 hour. The two layers were separated and the aqueous layer further extracted with ethyl acetate. The organic layers were combined and washed with brine. The organic layers were then concentrated in vacuo and filtered to yield methyl D-alloisoleucinate (Compound 3) as a pale yellow oil.
Stage 1
2,6-dimethyl-3-pyridinecarbaldehyde (Compound 4) in methanol at ambient temperature was treated with D-alloisoleucinate (Compound 3) in methanol followed by 2,2,2- trifluoroethanol and the reaction mixture was warmed to 40°C. When formation of the intermediate imine (methyl A/-[(2,6-dimethyl-3-pyridinyl)methylidene]-D-alloisoleucine) was complete Compound 5 was added followed by 1-isocyano-2- [(phenylmethyl)oxy]benzene (Compound 6) and the reaction mixture was stirred at 40°C until formation of Compound 7 was deemed complete.
Stage 2
Palladium on carbon catalyst was treated with a solution of Compound 7 in methanol and 2,2,2-trifluoroethanol and diluted with acetic acid. The vessel was purged with nitrogen and the reaction mixture warmed to 50°C and hydrogenated at 4.0-4.5 barg. When the reaction was deemed complete it was cooled to ambient temperature and the catalyst removed by filtration and washed through with methanol. The organic solution of 2- {(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1-piperazinyl}- 2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) was concentrated at reduced pressure and then diluted with /‘so-propyl acetate and concentrated at reduced pressure.
The residue was diluted with /‘so-propyl acetate and washed with aqueous ammonia. The aqueous phase was separated and extracted into another portion of /‘so-propyl acetate. The combined organic phases were washed with water, concentrated by distillation at reduced pressure, diluted with /‘so-propyl acetate and concentrated by distillation at reduced pressure, to leave a concentrated solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8). The product was finally dissolved in 1 ,4-dioxane for the next stage and stored into drums.
Stage 3 Solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) in 1 ,4-dioxane was treated with 1 ,1 ‘-carbonyl diimidazole at ambient temperature to form a solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1 -[1-(2,6-dimethyl-3-pyridinyl)- 2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1 -methylpropyl]-2,5- piperazinedione (Compound 9).
In a separate vessel morpholine in 1 ,4-dioxane was heated to 80-85°C. The solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[1 – (2,6-dimethyl-3-pyridinyl)-2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1- methylpropyl]-2,5-piperazinedione (Compound 9) was slowly added to the morpholine in 1 ,4-dioxane. The reaction mixture was stirred for one hour at 80-85°C and cooled before concentration by distillation at reduced pressure.
The concentrated solution of Compound A was diluted with /‘so-propyl acetate and washed with aqueous sodium hydroxide followed by water. The /so-propyl acetate solution of COMPOUND A was then concentrated by distillation at reduced pressure and cooled to ambient temperature. The concentrated solution of Compound A was then diluted with acetone and treated with benzenesulfonic acid and seed crystals were added and the reaction mixture stirred until crystallisation occurred. The slurry of Compound A besylate was heated to 50°C, a temperature cycle was performed, and finally the slurry was cooled to -10°C and isolated by filtration. The filter cake was washed with cold acetone (-10°C) to give Compound A besylate (intermediate grade) as a wet cake.
Yield: 44% from Compound 5
39% from Compound 5
Stage 4
Compound A besylate (intermediate grade wet cake, Compound A besylate ) was suspended in acetone (17.4 vol including acetone content of wet cake) and heated to 55- 60°C. Water (0.66 vol) was added until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (3.2 vol). The temperature of the reaction mixture was adjusted to 45-50°C before the addition of seed crystals (0.00025wt). When crystallisation was complete the reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins.
The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to -3-2°C over 4.5 h and stirred for at least 1 h before the product was isolated by filtration. The wet cake was washed with acetone at 0°C (3 x 3.1 vol) and blown dry before being unloaded. COMPOUND A besylate was dried at 50°C under vacuum for 3 days. Compound A besylate was then milled. Yield: 66% Stage 5
Compound A besylate (OBU-D-02) was suspended in acetone (8 vol) and water (1 .1 vol) and heated to 48-52°C until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (2 vol). The reaction mixture was cooled to 20-25°C before the addition of Form 1 seed crystals (0.0025wt). When crystallisation was complete the reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins. The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins.
The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to -12— 8°C over 3.5 h and stirred for 15 h before the product was isolated by filtration. The wet cake was washed with acetone at -10°C (2 x 3 vol) and blown dry before being unloaded. Compound A besylate was dried at ambient temperature under vacuum for 6 days with a wet nitrogen bleed to afford Form 1 . Compound A besylate was then milled. Yield: 67%
Recrystallisation of Compound A besylate anhydrate (Form 2)

Besylate salt ………………………………………………………………Besylate salt
C30H38 4O4■ C6H603S C30H38 4O4■
MW 676.83 MW 676.83
COMPOUND A besylate is charged to the vessel and treated with methyl ethyl ketone (MEK) (8vol) and water (0.35vol) and the solution heated until dissolution is observed (ca. 55-60°C). The solution is then filtered and recharged to the vessel. Pressure is then reduced to 650mbar and the reaction mixture heated further to distil out solvent. MEK is added at the same rate as solvent is removed by distillation keeping the reaction mixture volume constant. After 4 volumes of MEK have been added the reaction mixture is treated with Form 2 seed crystals (2%wt) and the distillation continued in the same manner until another 7 volumes of MEK has been added. The vacuum is then released to an atmospheric pressure of nitrogen and the temperature of the reaction mixture adjusted to 65°C. The reaction mixture is then filtered and washed with pre heated MEK (2vol at 65°C). The purified COMPOUND A besylate anhydrate is then sucked dry and dried further in a vacuum oven at 65°C at l OOmbar with a nitrogen bleed. Yield 89%
NMR data is the same for Forms 1 and 2.
1 H NMR (500MHz, DMSO-d6) 5ppm 0.71-0.80(m, 6H) 0.87-0.98(m, 1 H) 1 .31 (br. S, 1 H) 1.69(br. S, 1 H) 2.68(s, 3H) 2.69(s, 3H) 2.72-2.79(m, 1 H) 2.80-2.87(m, 1 H) 2.88-3.01 (m, 3H) 3.18-3.25(m, 1 H) 3.27-3.33(m, 1 H) 3.38-3.46(m, 1 H) 3.47-3.52(m, 1 H)3.53-3.57(m, 1 H) 3.60-3.71 (m, 3H) 3.83(dd, J=9.46,3.15 Hz, 1 H) 3.89 (br. S, 1 H)6.10(br. S, 1 H) 7.1 1 – 7.14(m, 2H) 7.19-7.23(m, 2H) 7.30-7.35(m, 3H)7.59-7.63(m, 2H) 7.67(d, J=7.25Hz, 1 H) 8.12(br. S, 1 H) 8.50(d, J=3.78Hz, 1 H)
………………………………………..
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm201287w

A six-stage stereoselective synthesis of indanyl-7-(3′-pyridyl)-(3R,6R,7R)-2,5-diketopiperazines oxytocin antagonists from indene is described. SAR studies involving mono- and disubstitution in the 3′-pyridyl ring and variation of the 3-isobutyl group gave potent compounds (pKi > 9.0) with good aqueous solubility. Evaluation of the pharmacokinetic profile in the rat, dog, and cynomolgus monkey of those derivatives with low cynomolgus monkey and human intrinsic clearance gave 2′,6′-dimethyl-3′-pyridyl R–sec-butyl morpholine amide Epelsiban (69), a highly potent oxytocin antagonist (pKi = 9.9) with >31000-fold selectivity over all three human vasopressin receptors hV1aR, hV2R, and hV1bR, with no significant P450 inhibition. Epelsiban has low levels of intrinsic clearance against the microsomes of four species, good bioavailability (55%) and comparable potency to atosiban in the rat, but is 100-fold more potent than the latter in vitro and was negative in the genotoxicity screens with a satisfactory oral safety profile in female rats.
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione (69 EPELSIBAN)
References
- Borthwick AD, Liddle J, Davies DE, Exall AM, Hamlett C, Hickey DM, Mason AM, Smith IE, Nerozzi F, Peace S, Pollard D, Sollis SL, Allen MJ, Woollard PM, Pullen MA, Westfall TD, Stanislaus DJ (January 2012). “Pyridyl-2,5-diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists: synthesis, pharmacokinetics, and in vivo potency”. Journal of Medicinal Chemistry 55 (2): 783–96. doi:10.1021/jm201287w. PMID 205501.
- 2 Borthwick, A. D.; Liddle, J. (January 2013). “Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists”. In Domling, A. Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. ISBN 978-3-527-33107-9.
- 3 World Health Organization (2011). “International Nonproprietary Names for Pharmaceutical Substances (INN): Proposed INN: List 105”. WHO Drug Information 25 (2): 179.
- 4 USAN Council (2011). “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). Retrieved 2011-10-28.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-(morpholin-4-yl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]piperazine-2,5-dione | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 872599-83-2 1159097-48-9 (besylate) |
| ATC code | None |
| PubChem | CID 11634973 |
| ChemSpider | 9809717 |
| KEGG | D10117 |
| Chemical data | |
| Formula | C30H38N4O4 |
| Molecular mass | 518.6 g/mol |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| WO2003053443A1 | Dec 20, 2002 | Jul 3, 2003 | Glaxo Group Ltd | Substituted diketopiperazines as oxytocin antagonists | |
| WO2006000399A1 | Jun 21, 2005 | Jan 5, 2006 | Glaxo Group Ltd | Novel compounds | |
| EP2005006760W | Title not available | ||||
| US6914160 | Jul 31, 2003 | Jul 5, 2005 | Pfizer Inc | Oxytocin inhibitors | |
| US20070254888 | Jun 21, 2005 | Nov 1, 2007 | Glaxo Group Limited | Piperazinediones as Oxytocin Receptor Antagonists |
| US8202864 * | Feb 25, 2011 | Jun 19, 2012 | Glaxo Group Limited | Compounds |
| US8716286 | Oct 28, 2010 | May 6, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US8742099 | May 20, 2013 | Jun 3, 2014 | Glaxo Group Limited | Compounds |
| US8815856 | Mar 18, 2014 | Aug 26, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US20120202811 * | Apr 19, 2012 | Aug 9, 2012 | Glaxo Group Limited | Novel compounds |
2014 in review……Dr ANTHONY’S “NEW DRUG APROVALS” BLOG



The WordPress.com stats helper monkeys prepared a 2014 annual report for this blog.

Here’s an excerpt:
The Louvre Museum has 8.5 million visitors per year. This blog was viewed about 350,000 times in 2014. If it were an exhibit at the Louvre Museum, it would take about 15 days for that many people to see it.
 Click here to see the complete report.
Click here to see the complete report.
/////////////////////






Important Industrial Procedures Revisited in Flow: Very Efficient Oxidation and N-Alkylation Reactions with High Atom-Economy

http://www.akademiai.com/content/u87p126856085276/?p=2f48c96a10a64882aeb5c47c657a10b7&pi=4
| Journal | Journal of Flow Chemistry |
| Publisher | Akadémiai Kiadó |
| ISSN | 2062-249X (Print) 2063-0212 (Online) |
| Subject | Flow Chemistry |
| Issue | Volume 3, Number 2/June 2013 |
| Pages | 51-58 |
| DOI | 10.1556/JFC-D-12-00025 |

Authors
1ThalesNano Zahony u. 7 1031 Budapest Hungary
 László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.
László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.
![]()
Abstract
The atom economy concept is one of the earliest recognition for green and sustainable aspects of organic synthesis. Over the years, novel technologies emerged that made this important feature of reactions into practice. Continuous-flow devices increased the efficiency of the chemical transformations with novel process windows (high T, high p and heterogeneous packed catalysts etc.) and increased safety which turned the attention to reexamine old, industrial processes. Oxidation can be performed under flow catalytic conditions with molecular oxygen; alcohols can be oxidized to carbonyl compounds with high atom economy (AE = 87 %). Using O2 and 1 % Au/TiO2, alcohol oxidation in flow was achieved with complete conversion and >90 % yield. N-alkylation is another good example for achieving high atom economy. Under flow catalytic conditions (Raney Ni), amines were successfully reacted with alcohols directly (AE = 91 %) with >90 % conversion and selectivity. In both examples, the effective residence time was less than 1 min. These two examples demonstrate the significant contribution of flow technology to the realization of key principles in green and sustainable chemistry.
ThalesNano Nanotechnology Inc, GraphisoftPark. Záhony u. 7. H-1031 Budapest HUNGARY


A Method to Identify Best Available Technologies (BAT) for Hydrogenation Reactors in the Pharmaceutical Industry




J. Flow Chem. 2012, 2(3), 77–82
http://www.akademiai.com/content/8652651g3378x686/?p=ab7c1bc4cd7740e1855623297649f542&pi=3
http://www.akademiai.com/content/8652651g3378x686/fulltext.pdf
| Journal of Flow Chemistry | |
| Publisher | Akadémiai Kiadó |
| ISSN | 2062-249X (Print) 2063-0212 (Online) |
| Subject | Flow Chemistry |
| Issue | Volume 2, Number 3/September 2012 |
| Pages | 77-82 |
| DOI | 10.1556/JFC-D-12-00014 |
Authors
1CNRS, CPE Lyon University of Lyon Villeurbanne France
Abstract
A methodology that may be applied to help in the choice of a continuous reactor is proposed. In this methodology, the chemistry is first described through the use of eight simple criteria (rate, thermicity, deactivation, solubility, conversion, selectivity, viscosity, and catalyst). Then, each reactor type is also analyzed from their capability to answer each of these criteria. A final score is presented using “spider diagrams.” Lower surfaces indicate the best reactor choice. The methodology is exemplified with a model substrate nitrobenzene and a target pharmaceutical intermediate, N-methyl-4-nitrobenzenemethanesulphonamide, and for three different continuous reactors, i.e., stirred tank, fixed bed, and an advanced microstructured reactor. Comparison with the traditional batch reactor is also provided.



Fanetizole

Fanetizole
Fanetizole shows immunoregulating activity.
RN: 79069-95-7
Fanetizole mesylate [USAN]
CP-48,810-27
Fanetizole mesylate
UNII-D3OG7B0G4M
Synthesis
Thioureas serve as a convenient starting material for 2-aminothiazoles.

Fanetizole synthesis.
Reaction of β-phenethylamine with ammonium isothiocyanate gives the corresponding thiourea. Treatment of that product with phenacyl bromide thus affords the thiazole product.[1]
- Lombardino, J. G.; 1981, U.S. Patent 4,307,106

| Systematic (IUPAC) name | |
|---|---|
| 4-Phenyl-N-(2-phenylethyl)-1,3-thiazol-2-amine | |
| Clinical data | |
| Legal status |
?
|
| Pharmacokinetic data | |
| Protein binding | % |
| Identifiers | |
| CAS number | 79069-94-6 |
| ATC code | ? |
| PubChem | CID 54339 |
| ChemSpider | 49083 |
| UNII | BH48F620JA |
| Chemical data | |
| Formula | C17H16N2S |
| Mol. mass | 280.39 g/mol |
………………………………………….
Journal of the Chinese Chemical Society, 2009, 56, 455-458
http://proj3.sinica.edu.tw/~chem/servxx6/files/paper_10990_1246593848.pdf
Fanetizole (3j)
mp 114-115 C (Lit.,30 116-117 C). IR (KBr) :3192, 2957, 1562, 1481, 1445, 1332, 698 cm-1;
1H NMR(CDCl3) : 2.81 (t, J = 7.4 Hz, 2H), 3.42 (dd, J = 6.8, 10.8
Hz, 2H), 6.32 (s, 1H), 6.64 (s, 1H), 7.08 (d, J = 6.8 Hz, 2H),
7.15-7.28 (m, 4H), 7.34-7.37 (m, 2H), 7.77-7.80 (m, 2H).
30=. Potewar, T. M.; Ingale, S. A.; Srinivasan, K. V. Tetrahedron
2008, 64, 5019-5022.
…………………………………………
A remarkably high-speed solution-phase combinatorial synthesis of 2-substituted-amino-4-aryl thiazoles in polar solvents in the absence of a catalyst under ambient conditions and study of their antimicrobial activities
ISRN Organic Chemistry (2011), 434613, 6 pp. Publisher: (Hindawi Publishing Corp., )
http://www.hindawi.com/journals/isrn/2011/434613/
……………………………………………
Fanetizole
Ley et al had previously developed a tube-in-tube reactor based on a semipermeable polymer membrane to enable the transfer of gases into liquid flow streams. and here, we demonstrate the scalability and throughput of this reactor when used with ammonia gas. This was made possible by a the inclusion of a titration method to assess parameters including the liquid and gas configuration, reactor temperatures, flow rates, and solvent polarity. These data were then employed in a scaling-up process affording alkyl thioureas which were ultimately used in a telescoped procedure for the preparation of anti-inflammatory agent fanetizole on a multigram scale.


Researchers at Cambridge have shown how it is possible to calibrate a ‘tube-in-tube’ reactor containing ammonia gas using a simple in-line colourimetric titration technique.
This information was then used to deliver an ammonia solution of stoichiometrically to effect the telescoped 2 stage synthesis of the anti-inflammatory agent Fanetizole.
The automated continuous flow synthesiser was able to produce drug substance at a rate of approximately 10 g per hour, isolating the product by direct precipitation from the outflow reaction stream.
Fanetizole: Scaling-up of continuous flow processes with gases using a tube-in-tube reactor: in-line titrations and fanetizole synthesis with ammonia J. Pastre, D.L. Browne, M. O’Brien and S.V. Ley, Org. Proc. Res. Dev. 2013, 17, 1183-1191.
http://pubs.acs.org/doi/full/10.1021/op400152r
………………………..
A Hantzsch synthesis of 2-aminothiazoles performed in a heated microreactor system
DOI: 10.1039/B109360F…….http://pubs.rsc.org/en/content/articlelanding/2002/lc/b109360f/unauth#!divAbstract
………………………………
Bioorganic and Medicinal Chemistry Letters, 1996 , vol. 6, 12 pg. 1409 – 1414
http://www.sciencedirect.com/science/article/pii/0960894X96002417

………………………………………
ref
Heterocycles, 2010 , vol. 81, 12 pg. 2849 – 2854
Journal of the Chinese Chemical Society, 2009 , vol. 56, 3 pg. 455 – 458
Bioorganic and Medicinal Chemistry Letters, 1996 , vol. 6, 12 pg. 1409 – 1414
Pfizer Patent: DD144055DE2922523 , 1979 ;Chem.Abstr., vol. 92, 111001
Organic Process Research and Development, 2013 , vol. 17, 9 pg. 1183 – 1191
Tetrahedron, 2007 , vol. 63, 45 pg. 11066 – 11069
Tetrahedron, 2008 , vol. 64, 22 pg. 5019 – 5022
The application of flow microreactors to the preparation of a family of casein kinase I inhibitors

|
The Application of Flow Microreactors to the Preparation of a Family of Casein Kinase I Inhibitors.
|
||
|
Org. Biomol. Chem. 2010, 8, 1798-1806.
Link: 10.1039/b925327k
|
In this article we demonstrate how a combination of enabling technologies such as flow synthesis, solid-supported reagents and scavenging resins utilised under fully automated software control can assist in typical medicinal chemistry programmes. In particular automated continuous flow methods have greatly assisted in the optimisation of reaction conditions and facilitated scale up operations involving hazardous chemical materials. Overall a collection of twenty diverse analogues of a casein kinase I inhibitor has been synthesised by changing three principle binding vectors.
DOI: 10.1039/B925327K

Meclinertant (SR48692)

2-[[1-(7-chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)pyrazole-3-carbonyl]amino]adamantane-2-carboxylic acid
Meclinertant (SR-48692) is a drug which acts as a selective, non-peptide antagonist at the neurotensin receptor NTS1, and was the first non-peptide antagonist developed for this receptor.[1][2] It is used in scientific research to explore the interaction between neurotensin and other neurotransmitters in the brain,[3][4][5][6][7][8] and produces anxiolytic, anti-addictive and memory-impairing effects in animal studies.[9][10][11][12]
PatentSubmittedGranted1-(7-chloroquinolin-4-yl)pyrazole-3-carboxamide N-oxide derivatives, method of preparing them, and their pharmaceutical compositions [US5561234]1996-10-01
Substituted 1-naphthyl-3-pyrazolecarboxamides which are active on neurotensin [US5585497]1996-12-17
3-amidopyrazole derivatives, process for preparing these and pharmaceutical composites containing them [US5420141]1995-05-30
Substituted 1-naphthyl-3-pyrazolecarboxamides which are active on neurotensin, their preparation and pharmaceutical compositions containing them [US5523455]1996-06-04
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them [US5607958]1997-03-04
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them [US5616592]1997-04-01
3-amidopyrazole derivatives, process for preparing these and pharmaceutical compositions containing them [US5635526]1997-06-03
Substituted 1-phenyl-3-pyrazolecarboxamides active on neurotensin receptors, their preparation and pharmaceutical compositions containing them [US5965579]1999-10-12
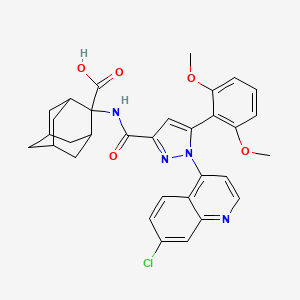
| Systematic (IUPAC) name | |
|---|---|
| 2-([1-(7-Chloro-4-quinolinyl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carbonyl]amino)admantane-2-carboxylic acid | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 146362-70-1 |
| ATC code | ? |
| PubChem | CID 119192 |
| IUPHAR ligand | 1582 |
| UNII | 5JBP4SI96H |
| Chemical data | |
| Formula | C32H31ClN4O5 |
| Mol. mass | 587.064 |

A Machine-Assisted Flow Synthesis of SR48692: A Probe for the Investigation of Neurotensin Receptor-1 (pages 7917–7930)
Dr. Claudio Battilocchio, Benjamin J. Deadman, Dr. Nikzad Nikbin, Dr. Matthew O. Kitching, Prof. Ian R. Baxendale and Prof. Steven V. Ley
Article first published online: 16 APR 2013 | DOI: 10.1002/chem.201300696
Flow and pharmaceuticals? An investigation into whether machine-assisted technologies can be of true help in the multistep synthesis of a potent neurotensin receptor-1 probe, Meclinertant (SR48692; see structure), is reported.
Meclinertant (SR 48692)
We developed an improved synthesis of the neurotensin antagonist biological probe SR 48692. The preparation includes an number of chemical conversions and strategies involving the use of flow chemistry platforms which helped overcome some of the limiting synthetic transformations in the original chemical route .

Meclinertant (SR 48692): The synthesis of neurotensin antagonist SR 48692 for prostate cancer research I.R. Baxendale, S. Cheung, M.O. Kitching, S.V. Ley, J.W. Shearman Bio. Org. Med. Chem. 2013, 21, 4378-4387.

A synthesis of the neurotensin 1 receptor probe Merclinertant (SR48692) has been reported using a range of continuous flow through synthesis, in-line reaction monioring and purification techniques. This strategy has been contrasted with a more conventional batch synthesis approach.
Notably the safe use of phosgene gas (generated in situ), the superheating of solvents to accelerate reaction rates, the processing of a reagent suspension under continuous flow-through conditions and the application of semi-permeable membrane technology to facilitate work-up and purification were all techniques that could be beneficially applied in the synthetic scheme.
Abstract:


Meclinertant, Reminertant, SR-48692
The condensation of 2′,6′-dimethoxyacetophenone (I) with diethyl oxalate (II) by means of sodium methoxide in refluxing methanol gives the dioxobutyrate (III), which is cyclized with 7-chloroquinoline-4-hydrazine (IV) in refluxing acetic acid yielding the pyrazole derivative (V). The hydrolysis of the ester group of (V) with KOH in refluxing methanol/water affords the corresponding carboxylic acid (VI), which is finally treated with SOCl2 in refluxing toluene and condensed with 2-aminoadamantane-2-carboxylic acid.
EP 0477049; FR 2665898; JP 1992244065; US 5420141; US 5607958; US 5616592; US 5635526; US 5744491; US 5744493
…………………………….

- Gully D, Canton M, Boigegrain R, Jeanjean F, Molimard JC, Poncelet M, Gueudet C, Heaulme M, Leyris R, Brouard A (January 1993).“Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor”. Proceedings of the National Academy of Sciences of the United States of America 90 (1): 65–9. doi:10.1073/pnas.90.1.65. PMC 45600. PMID 8380498.
- Gully D, Jeanjean F, Poncelet M, Steinberg R, Soubrié P, Le Fur G, Maffrand JP (1995). “Neuropharmacological profile of non-peptide neurotensin antagonists”. Fundamental & Clinical Pharmacology 9 (6): 513–21. doi:10.1111/j.1472-8206.1995.tb00528.x.PMID 8808171.
- Rostene W, Azzi M, Boudin H, Lepee I, Souaze F, Mendez-Ubach M, Betancur C, Gully D (April 1997). “Use of nonpeptide antagonists to explore the physiological roles of neurotensin. Focus on brain neurotensin/dopamine interactions”. Annals of the New York Academy of Sciences 814: 125–41. doi:10.1111/j.1749-6632.1997.tb46151.x. PMID 9160965.
- Jump up^ Jolas T, Aghajanian GK (August 1997). “Neurotensin and the serotonergic system”. Progress in Neurobiology 52 (6): 455–68.doi:10.1016/S0301-0082(97)00025-7. PMID 9316156.
- Jump up^ Dobner PR, Deutch AY, Fadel J (June 2003). “Neurotensin: dual roles in psychostimulant and antipsychotic drug responses”. Life Sciences73 (6): 801–11. doi:10.1016/S0024-3205(03)00411-9. PMID 12801600.
- Jump up^ Chen L, Yung KK, Yung WH (September 2006). “Neurotensin selectively facilitates glutamatergic transmission in globus pallidus”.Neuroscience 141 (4): 1871–8. doi:10.1016/j.neuroscience.2006.05.049. PMID 16814931.
- Petkova-Kirova P, Rakovska A, Della Corte L, Zaekova G, Radomirov R, Mayer A (September 2008). “Neurotensin modulation of acetylcholine, GABA, and aspartate release from rat prefrontal cortex studied in vivo with microdialysis”. Brain Research Bulletin 77 (2–3): 129–35. doi:10.1016/j.brainresbull.2008.04.003. PMID 18721670.
- Petkova-Kirova P, Rakovska A, Zaekova G, Ballini C, Corte LD, Radomirov R, Vágvölgyi A (December 2008). “Stimulation by neurotensin of dopamine and 5-hydroxytryptamine (5-HT) release from rat prefrontal cortex: possible role of NTR1 receptors in neuropsychiatric disorders”.Neurochemistry International 53 (6–8): 355–61. doi:10.1016/j.neuint.2008.08.010. PMID 18835308.
- Griebel G, Moindrot N, Aliaga C, Simiand J, Soubrié P (December 2001). “Characterization of the profile of neurokinin-2 and neurotensin receptor antagonists in the mouse defense test battery”. Neuroscience and Biobehavioral Reviews 25 (7–8): 619–26. doi:10.1016/S0149-7634(01)00045-8. PMID 11801287.
- Tirado-Santiago G, Lázaro-Muñoz G, Rodríguez-González V, Maldonado-Vlaar CS (October 2006). “Microinfusions of neurotensin antagonist SR 48692 within the nucleus accumbens core impair spatial learning in rats”. Behavioral Neuroscience 120 (5): 1093–102. doi:10.1037/0735-7044.120.5.1093. PMID 17014260.
- Felszeghy K, Espinosa JM, Scarna H, Bérod A, Rostène W, Pélaprat D (December 2007). “Neurotensin receptor antagonist administered during cocaine withdrawal decreases locomotor sensitization and conditioned place preference”. Neuropsychopharmacology 32 (12): 2601–10. doi:10.1038/sj.npp.1301382. PMC 2992550. PMID 17356568.
- Lévesque K, Lamarche C, Rompré PP (October 2008). “Evidence for a role of endogenous neurotensin in the development of sensitization to the locomotor stimulant effect of morphine”.European Journal of Pharmacology 594 (1–3): 132–8. doi:10.1016/j.ejphar.2008.07.048. PMID 18706409.

Continuous Flow Synthesis of alpha-Halo Ketones: Building Blocks for Anti-retroviral Agents

Chiral alpha-halo ketones derived from N-protected amino acids are key building blocks for the synthesis of HIV protease inhibitors such as atazanavir used in HAART combination therapy.
Kappe and De Souza have reported a continuous flow through route to these intermediates which utilises a tube-in-tube reactor to introduce diazomethane generated on demand into the reaction stream containing mixed anhydride derivatives of N-protected amino acids. The resulting alpha-diazo ketones are then decomposed with HCl or HBr to afford the corresponding alpha-halo ketones.
This process allows the safe generation, separation and use of diazomethane in a continuous integrated multi-step synthesis of important API intermediates.

The development of a continuous flow process for the multistep synthesis of α-halo ketones starting from N-protected amino acids is described. The obtained α-halo ketones are chiral building blocks for the synthesis of HIV protease inhibitors, such as atazanavir and darunavir. The synthesis starts with the formation of a mixed anhydride in a first tubular reactor.
The anhydride is subsequently combined with anhydrous diazomethane in a tube-in-tube reactor. The tube-in-tube reactor consists of an inner tube, made from a gas-permeable, hydrophobic material, enclosed in a thick-walled, impermeable outer tube. Diazomethane is generated in the inner tube in an aqueous medium, and anhydrous diazomethane subsequently diffuses through the permeable membrane into the outer chamber.
The α-diazo ketone is produced from the mixed anhydride and diazomethane in the outer chamber, and the resulting diazo ketone is finally converted to the halo ketone with anhydrous ethereal hydrogen halide.
This method eliminates the need to store, transport, or handle diazomethane and produces α-halo ketone building blocks in a multistep system without racemization in excellent yields. A fully continuous process allowed the synthesis of 1.84 g of α-chloro ketone from the respective N-protected amino acid within ∼4.5 h (87% yield).

Arteflene





