
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 14)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Asandeutertinib
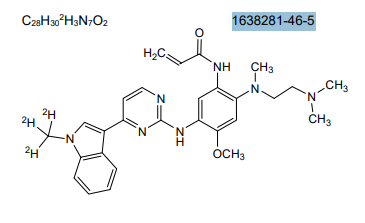
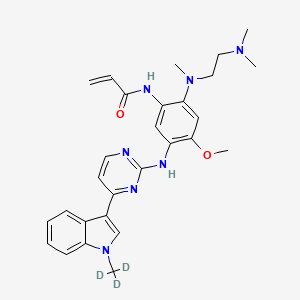
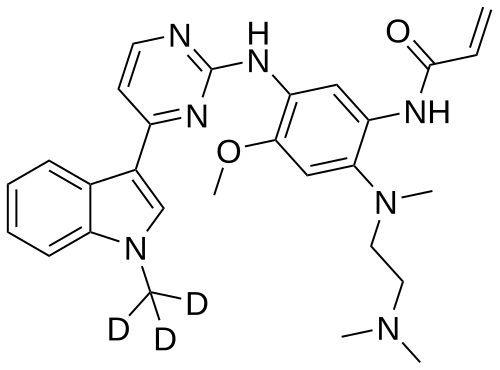
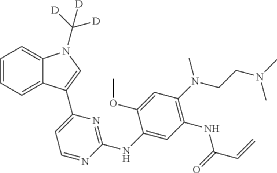
Asandeutertinib, Osimertinib-d3
CAS 1638281-46-5
- 9EKD2E8BM5
- N-(2-(2-(dimethylamino)ethyl-methylamino)-4-methoxy-5-((4-(1-(trideuteriomethyl)indol-3-yl)pyrimidin-2-yl)amino)phenyl)prop-2-enamide
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(trideuteriomethyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide
N-[2-{2-(dimethylamino)ethylamino}-4-methoxy-5-({4-[1-(2H3)methyl-1H-indol-3-yl]pyrimidin-2-
yl}amino)phenyl]prop-2-enamide
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, antineoplastic
MF C28H30. 2H3. N7O2, C28H30D3N7O2 MW 502.6 g/mol
Asandeutertinib is an investigational new drug that is being evaluated for the treatment of cancer. It is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) with antineoplastic properties.[1][2] Developed by TYK Medicines, this small molecule drug is currently being investigated for the treatment of non-small cell lung cancer (NSCLC), particularly in patients with EGFR mutations.[1][3]
PAT
- 2-(2,4,5-substituted aniline) pyrimidine derivative, pharmaceutical composition and use thereofPublication Number: US-10414756-B2Priority Date: 2014-08-15Grant Date: 2019-09-17
- 2-(2,4,5-substituted aniline) pyrimidine derivative, pharmaceutical composition and use thereofPublication Number: US-2018016258-A1Priority Date: 2014-08-15
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US210080627&_cid=P21-MFT3HT-86141-1
Embodiment 3A
N-(2-{2-dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-(1-(D3-methyl)indol-3-yl)pyrimidin-2-yl]amino}phenyl)-2-acrylamide
| Under ice bath condition, to N 1-(2-dimethylaminoethyl)-5-methoxy-N 1-methyl-N 4-[4-(1-[D 3-methylindol]-3-yl)pyrimidin-2-yl]phenyl-1,2,4-triamine (intermediate 3, 20 g) in THF (200 mL) and water (20 mL), was added 6.9 g NaOH. Acryloyl chloride 4.05 g was added while stirring, the reaction mixture was stirred for 30 min at room temperature, then stirred for 1 h at room temperature. After the result of TLC showed that the reaction was complete, 200 mL water and 20 mL aqueous ammonia were added into the reaction mixture, the solid was precipitated and filtered out. The solid was collected and washed with water, dried for 8 h at 50° C. to deliver the title compound (yield 87%). |



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Asandeutertinib”. PatSnap.
- “Asandeutertinib”. IUPHAR/BPS Guide to PHARMACOLOGY.
- Han B, Zhang W, Wu L, Chen B, Zhao Y, Liu J, et al. (October 2024). “P1. 12A. 07 A Phase 1 Study of TY-9591 in Advanced Non-Small Cell Lung Cancer (NSCLC) Patients with EGFR Positive Mutation”. Journal of Thoracic Oncology. 19 (10): S195. doi:10.1016/j.jtho.2024.09.353.
| Clinical data | |
|---|---|
| Other names | Runnor-9591, TY 9591 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1638281-46-5 |
| PubChem CID | 87056175 |
| IUPHAR/BPS | 13201 |
| ChemSpider | 129431787 |
| UNII | 9EKD2E8BM5 |
| Chemical and physical data | |
| Formula | C28H30D3N7O2 |
| Molar mass | 502.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////////Asandeutertinib, antineoplastic, 9EKD2E8BM5, Osimertinib-d3
Admilparant
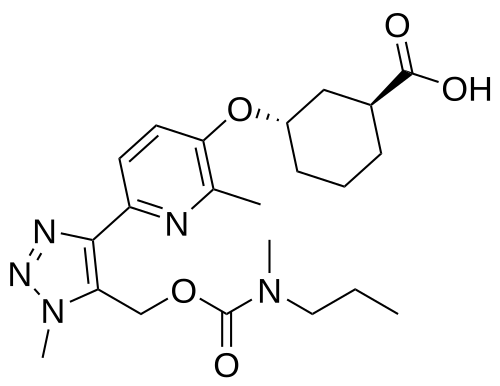
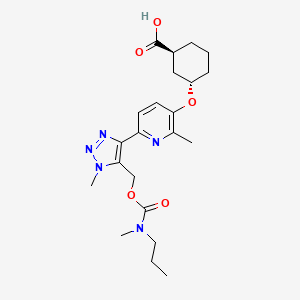
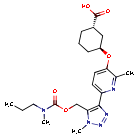
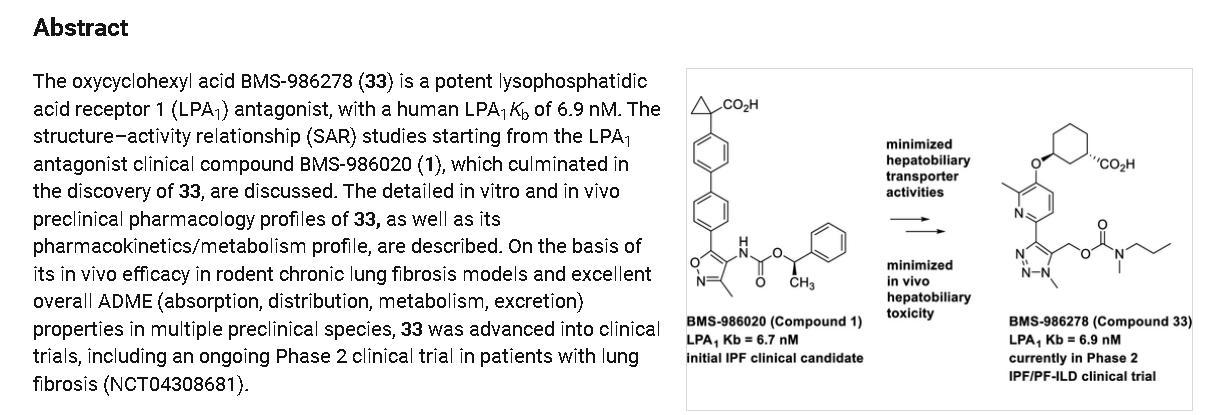
Admilparant, (BMS-986278)
CAS 2170126-74-4
MF C22H31N5O5 MW 445.5 g/mol
(1S,3S)-3-({2-methyl-6-[1-methyl-5-({[methyl(propyl)carbamoyl]oxy}methyl)-1H-1,2,3-triazol-4-l]pyridin-3-yl}oxy)cyclohexane-1-carboxylic acid
lysophosphatidic acid receptor 1 (LPA1) antagonist
- 4UN9AOU6G8
- BMS986278
- (1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic acid
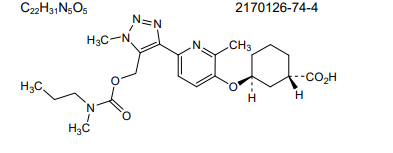
Admilparant is an investigational new drug being developed by Bristol-Myers Squibb for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a first-in-class lysophosphatidic acid receptor 1 (LPA1) antagonist.[1][2]
As of 2024, admilparant is in Phase III clinical trials for both IPF and PPF.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry, Publication Date: 2021-10-28, PMID: 34709814
DOI: 10.1021/acs.jmedchem.1c01256
(1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)-oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic Acid (33). Compound 33 was prepared using the same
synthetic sequence as 25, except that intermediate 42 was reacted with
N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine. 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 11.99−11.46 (m,1H), 7.82 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 5.65 (s, 2H),
4.89−4.62 (m, 1H), 4.10 (s, 3H), 3.12 (br t, J = 7.2 Hz, 2H), 2.79 (s,3H), 2.69 (tt, J = 9.4, 4.4 Hz, 1H), 2.44 (s, 3H), 2.03 (dt, J = 13.8, 4.5Hz, 1H), 1.92−1.86 (m, 1H), 1.86−1.79 (m, 2H), 1.74−1.68 (m, 1H),
1.68−1.58 (m, 2H), 1.58−1.51 (m, 1H), 1.43 (dq, J = 14.4, 7.1 Hz,2H), 0.76 (br t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, 100°C) δ 175.4, 154.7, 150.1, 147.7, 143.9, 141.4, 129.6, 120.0, 118.6, 71.8,
54.5, 49.5, 37.4, 34.4, 33.4, 31.6, 28.7, 27.2, 19.8, 19.4, 18.6, 10.1. m/z446 [M + H]+
. HPLC/UV purity: 99.9% using the following reverse phase chromatographic conditions: Agilent HPLC; Phenomenex Kinetex-C-18; 100 (L) × 4.6 mm2 (i.d.) column; 2.6 μm particle size; wavelength, 220−380 nm; flow rate, 1.0 mL/min; temperature, 35°C; injection volume, 4 μL of 0.25 mg/mL in 1:1 MeCN:H2O; mobilephase A, H2O−0.05% TFA; mobile phase B, MeCN−0.05% TFA; gradient elution, starting at 10−80% B over 10 min and ending at 95% Bafter an additional 4 min; retention time = 8.28 min. Stereoisomeric purity was >99.5% using the following chiral chromatographic conditions: UPC2 Analytical SFC, ChromegaChiral CC4; 250 (L) ×4.6 mm2 (i.d.); 5 μm column; flow rate, 3 mL/min; temperature, 40 °C;injection volume, 10 μL of 0.25 mg/mL in MeCN:MeOH (1:1);mobile phase, 30% MeOH and 70% CO2 at 120 bar retention time =6.05 min. Accurate mass, [M + H]+ at m/z = 446.2398 (−2.03 ppmfrom theoretical for C22H32N5O5). [α]20D = +28.24° (MeOH, c = 0.51).
Elem. Anal. (theoretical): C, 59.31; H, 7.01; N, 15.72. Found: C, 59.35;H, 6.78; N, 15.69. UV (MeOH) at 254 nm (ε = 17,856), 290 nm (ε =7,519), and 296 nm (ε = 8,288). Concentration: adjusted for purity,
0.05154840 g/L or 0.0001157047 mol/L. Melting point = 152−154°C. Accurate mass, [M + H]+ at m/z 466.2398 (−2.03 ppm fromtheoretical for C22H32N5O5).
synthetic sequence as 25, except that intermediate 42 was reacted with N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine
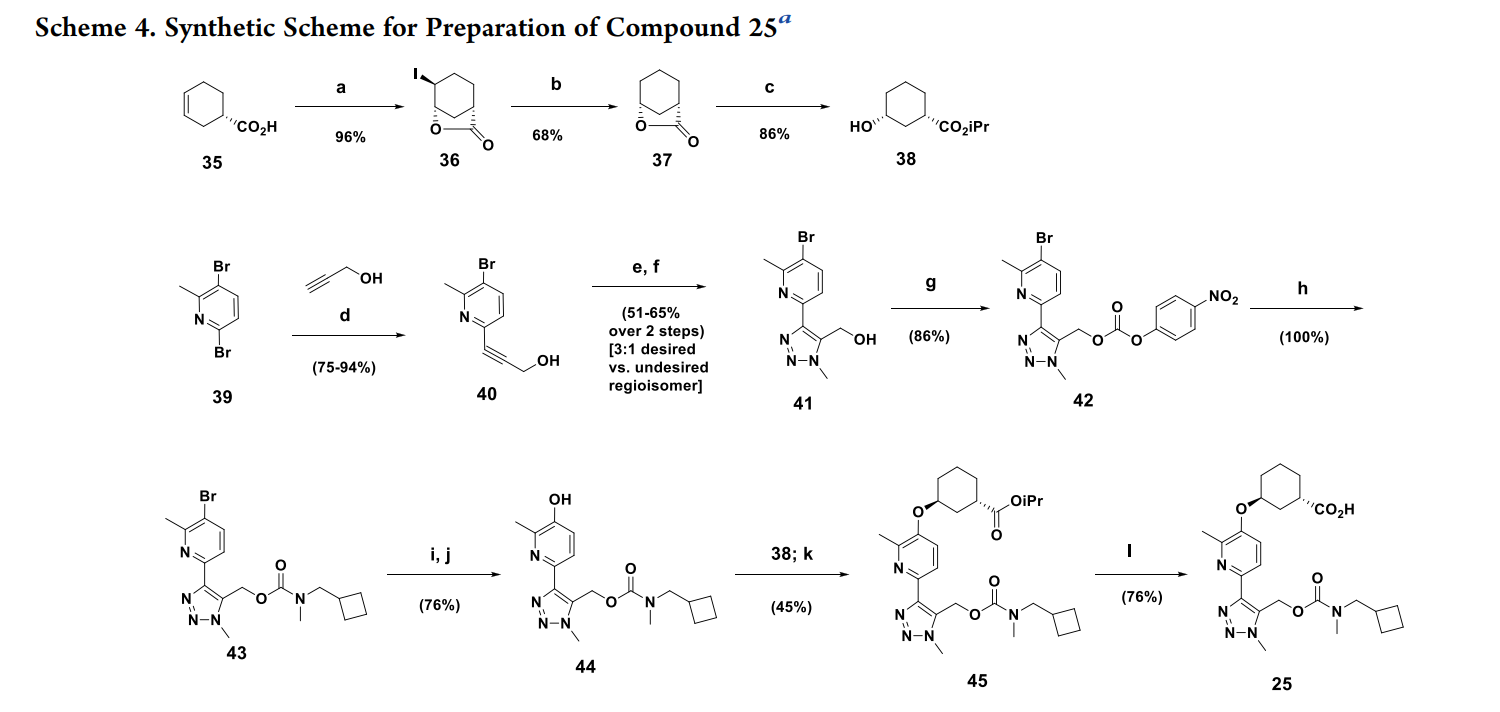
a
Reagents and conditions: (a) I2 (1.1 equiv)/KI (2.5 equiv)/NaHCO3 (3 equiv)/water (96%); (b) H2 (50 psi)/ Pd/C (cat)/Et3N (2 equiv)/EtOAc (68%); (c) CH3COCl (2.5 equiv)/iPrOH (87−95%); d) (Ph3P)2PdCl2 (5%)/ Et3N/CuI (5%)/RT (75−94%); (e) Ru(II)-(Ph3P)2(Me5Cyp)Cl (5%)/TMSCH2N3/dioxane 50 °C/15 h; (f) Bu4NF/0 °C to RT (51−65% over 2 steps; 3:1 desired:undesired regioisomer); (g) 4-nitrophenyl chloroformate/pyridine/CH2Cl2 (86%); (h) N-cyclobutyl N-methylamine/iPr2NEt/CH2Cl2 (100%); (i) B2(pin)2/KOAc/PdCl2(dppf)/THF/80 °C; (j) NaH2BO4/H2O/RT (76% over 2 steps); (k) 38; 1,1′-(azodicarbonyl)dipiperidine/Bu3P/toluene/50 °C (45%); (l)LiOH/H2O/MeOH (76%).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US208146892&_cid=P20-MFS2PF-83792-1
PATENT
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2022249443-A1Priority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: US-RE49352-EPriority Date: 2016-06-21Grant Date: 2023-01-03
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: AU-2021209334-B2Priority Date: 2016-06-21Grant Date: 2023-06-01
- Carbamoyloxymethyltriazole cyclohexylates as LPA antagonistsPublication Number: JP-7312295-B2Priority Date: 2016-06-21Grant Date: 2023-07-20
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2023390249-A1Priority Date: 2016-06-21
- Carbamoyloxymethyltriazolylcyclohexanes as LPA antagonistsPublication Number: CN-109963843-BPriority Date: 2016-06-21Grant Date: 2022-03-11
- Carbamoyloxymethyltriazole cyclohexyl acid as LPA antagonistPublication Number: CN-114601830-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acid as an LPA antagonistPublication Number: KR-102377340-B1Priority Date: 2016-06-21Grant Date: 2022-03-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-20220038537-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-102463621-B1Priority Date: 2016-06-21Grant Date: 2022-11-03
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Admilparant (BMS-986278): Idiopathic Pulmonary Fibrosis Likelihood of Approval”. Pharmaceutical Technology. 25 December 2023. Retrieved 2024-11-23.
- Corte TJ, Behr J, Cottin V, Glassberg MK, Kreuter M, Martinez FJ, et al. (October 2024). “Efficacy and Safety of Admilparant, an LPA1 Antagonist in Pulmonary Fibrosis: A Phase 2 Randomized Clinical Trial”. American Journal of Respiratory and Critical Care Medicine. 211 (2): 230–238. doi:10.1164/rccm.202405-0977OC. PMID 39393084.
- Splete H (16 September 2024). “Admilparant Affects Biomarkers in Pulmonary Fibrosis”. Medscape. Retrieved 2024-11-23.
| Clinical data | |
|---|---|
| Other names | BMS-986278 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2170126-74-4 |
| PubChem CID | 132232205 |
| DrugBank | DB18011 |
| ChemSpider | 115009679 |
| UNII | 4UN9AOU6G8 |
| KEGG | D12657 |
| ChEMBL | ChEMBL5087506 |
| Chemical and physical data | |
| Formula | C22H31N5O5 |
| Molar mass | 445.520 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
/////////Admilparant, BMS 986278, PHASE 3, Bristol-Myers Squibb, idiopathic pulmonary fibrosis, 4UN9AOU6G8
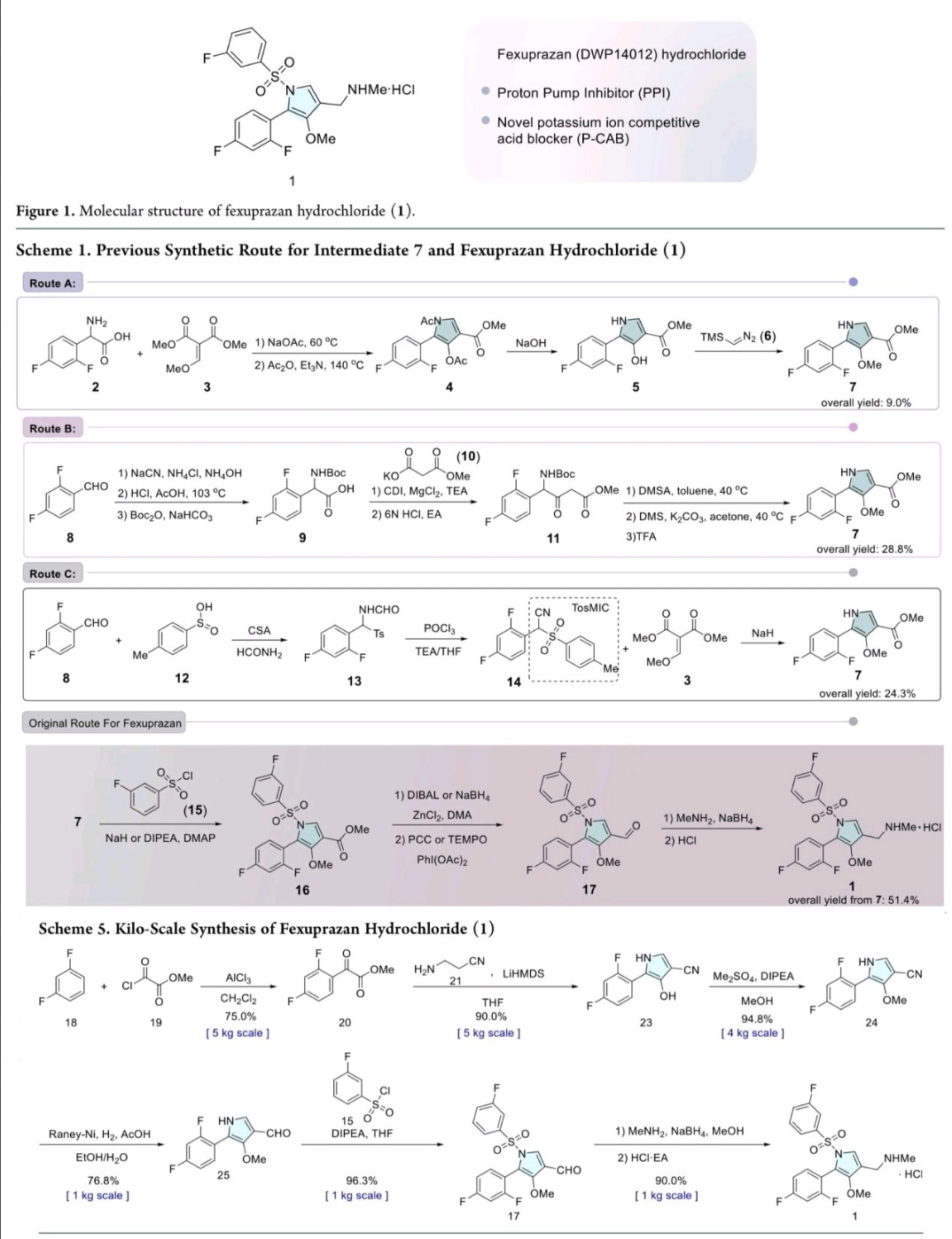
Fexuprazan, Abeprazan
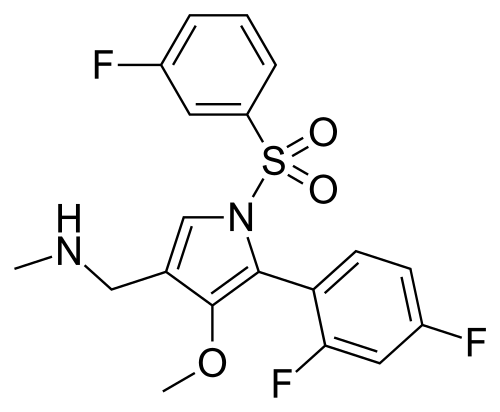
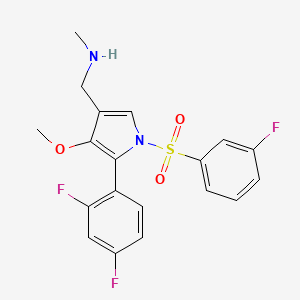
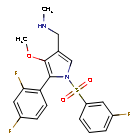
Fexuprazan, Abeprazan; DWP14012; DWP-14012
- CAS 1902954-60-2
- BE52S2C1QT
1-[5-(2,4-difluorophenyl)-1-(3-fluorophenyl)sulfonyl-4-methoxypyrrol-3-yl]-N-methylmethanamine
WeightAverage: 410.41
Monoisotopic: 410.091198078
Chemical FormulaC19H17F3N2O3S
Fexuprazan (trade name Fexuclue) is a drug for the treatment of gastroesophageal reflux disease (GERD).[1] It is a potassium-competitive acid blocker,[2] which is a class of drugs suppressing gastric acids.[3][4]
Fexuprazan is approved for clinical use in South Korea,[4][5] Mexico,[6] Philippines,[7] Chile,[8] and Ecuador.[9]
Abeprazan is under investigation in clinical trial NCT04341454 (Study to Evaluate the Efficacy and Safety of DWP14012 in Patients With Acute or Chronic Gastritis).
Proton pump inhibitors (PPIs) typified by omeprazole, which inhibit gastric acid secretion, are widely used in clinical settings. However, existing PPIs are accompanied by problems in terms of effectiveness and side effects. Specifically, since existing PPIs are unstable under acidic conditions, they are often formulated as enteric agents. in need. In addition, the existing PPI exhibits variation in therapeutic effect due to metabolic enzyme polymorphism and drug interaction with drugs such as diazepam, so improvement is desired.
In addition, since PPI is a prodrug activated by gastric acid and acts only on the active proton pump, the maximum drug expression time is delayed, the effect of suppressing acid secretion at night is poor, and it has disadvantages such as having to take it before meals. exist. In addition, PPI is mainly metabolized through the CYP2C19 enzyme, and there is a large difference in efficacy between individuals due to the genetic polymorphism of the CYP2C19 enzyme.
In order to improve the disadvantages of PPI as described above, a potassium-competitive gastric acid secretion inhibitor (Potassium-Competitive Acid Blocker, P-CAB) is attracting attention. Potassium competitive gastric acid secretion inhibitor strongly and rapidly inhibits gastric acid secretion by reversibly and competitively binding with K + ions to proton pump (H + /K + -ATPase), an enzyme involved in the final stage of gastric acid secretion in parietal cells. These P-CAB formulations show strong inhibition of the normal acidity (pH 1-3) in the stomach compared to the PPI formulations. However, pharmacological activity, which decreases as the pH increases, is required for gastric P-CAB preparations, and some P-CAB preparations show pharmacological activity that maintains pharmacological activity even when the pH increases, and some related side effects have been reported. In addition, since P-CAB preparations are mainly metabolized through the CYP3A4 enzyme, the difference in efficacy between individuals is relatively small, and concerns about interactions with drugs metabolized by the CYP2C19 enzyme are relatively low.
International Patent Publication No. WO2019/013310 A1 discloses bonoprazan as a potassium-competitive acid secretion inhibitor.
However, it was confirmed that vonoprazan induces severe hypergastrinemia compared to the existing PPI drug lansoprazole. Such hypergastrinemia can include enterochromaffin-like (ECL)-cell hyperplasia; parietal cell hyperplasia; fundic gland polyp; It can cause problems such as bone loss, damaged bone quality, and fractures. In fact, it has been reported that vonoprazan is associated with the development of gastric neuroendocrine tumors in carcinogenicity studies in mice and rats. However, discontinuation of administration of P-CAB or PPI-based drugs such as vonoprazan restores excess gastric acid and causes indigestion, so despite the above problems, drug administration cannot be easily stopped.
On the other hand, PPI is used for the prevention of gastric and duodenal ulcers by administration of nonsteroidal anti-inflammatory drugs (NSAIDs). However, it has been reported that bonoprazan aggravates the damage to the small intestine caused by various types of NSAIDs. For example, NSAID-induced gastrointestinal damage includes edema, erythema, submucosal hemorrhage, erosion, and ulceration. From this point of view, clinically, in the case of vonoprazan, there may be significant limitations in combination with NSAID drugs.
There are two major mechanisms by which drugs such as NSAIDs or alcohol cause damage to the gastrointestinal mucosa: a local irritant effect and a systemic effect. The local irritant effect occurs due to ion-trap and mitochondrial damage, and systemically due to the decrease in prostaglandin and NO (nitric oxide). In addition to mitochondrial damage caused by oxidative stress, damage to vascular endothelial cells causes microcirculation disorders, making the gastrointestinal mucosa very vulnerable to damage and interfering with the mucosal damage recovery mechanism. Due to the combined action of these mechanisms, damage to the mucous membrane of the gastrointestinal tract, ie, gastric ulcer, enteropathy, etc. may occur or be severe.
Accordingly, even considering the effect of bonoprazan in terms of suppressing gastric acid secretion, its use is bound to be very limited due to the above potential problems.
Separately, Helicobacter pylori ( H. pylori ) is known as one of the main causes of gastrointestinal diseases such as chronic gastritis and peptic ulcer and gastric cancer. Although the prevalence of Helicobacter pylori in our country is gradually decreasing, a prevalence of more than 50% is still being reported. In particular, Helicobacter pylori is related to digestive diseases, and the importance of antibacterial treatment agents is increasing day by day. In particular, as reported in several studies, antibacterial treatment of Helicobacter pylori reduces the occurrence of bleeding in peptic ulcer. For this antibacterial therapy, in general, patients take clarithromycin and amoxicillin along with gastric acid inhibitors such as PPI as the first-line treatment. For multi-drug use of PPIs and antibiotics, the risk of drug-drug interactions must be low, and the risk of such interactions can be predicted through in vitro CYP inhibition, CYP/UGT phenotyping, and CYP induction tests.
However, additional or repeated administration of various antibiotics is required up to the second and third treatment, and side effects and resistance have been reported. Therefore, by reducing gastric acidity, the antibacterial effect of antibiotics on Helicobacter pylori (H. pylori ) is enhanced, and long-term dose reduction of gastric acidity, for example, proton-potassium pump inhibitory ability, etc. The need to develop a visible drug is emerging.
In addition, in the case of an oral drug, the bioavailability, which is the rate at which the administered drug enters the systemic circulation and is used in the body, is measured. High bioavailability is one of the essential elements of oral drugs because the higher the bioavailability, the higher the rate and extent to which the active ingredient or part of the drug is absorbed and utilized at the site of action. In general, such bioavailability increases as absorption through the gastrointestinal tract is higher and the degree of first-pass effect is lower. , is affected by the size and shape of the particles, and the surface area of the particles.
It is also important that the concentration of the drug in the target organ, in this case the gastric tissue, is maintained as well as the bioavailability in the circulatory system. Therefore, drug distribution and maintenance to the target organ, gastric tissue, is judged to be an important pharmacokinetic characteristic in P-CAB drug development.
On the other hand, somatostatin, also known as growth hormone-inhibiting hormone (GHIH), is a cyclic peptide expressed in the gastrointestinal tract, pancreas, hypothalamus and central nervous system. It is secreted by D cells of the stomach and pancreas and acts as a paracrine regulator of gastric acid secretion, and suppresses gastric acid secretion by inhibiting gastric G cell gastrin secretion and parietal cell acid secretion. Activation of somatostatin receptors by somatostatin analogs and somatostatin receptor agonists inhibit gastrin secretion, thereby regulating histamine release from ECL cells and inhibiting acid secretion. In actual animal models and hypergastrinemia patients, it has been reported that the somatostatin analogue decreased the total gastric acid secretion by decreasing gastrin secretion and gastric acid response.
Gastric acid suppression by taking drugs such as PPI suppresses somatostatin secretion by D cells and promotes gastrin secretion by G cells by a feedback mechanism to induce hypergastrinemia. Gastrin promotes epithelial cell growth to induce oxyntic cell hyperplasia in the gastric body and increase parietal cell mass. This leads to proliferation of adenoma cells and hyperplasia of ECL cells, which may increase the risk of neuroendocrine tumors. In addition, the frequency of neuroendocrine tumors among tumors occurring in the duodenum is relatively high, and it is known that gastrin secretion is the most common form in neuroendocrine tumors occurring in the duodenum, accounting for approximately 65% of the total. It has been confirmed that the group taking bonoprazan tends to have a higher blood gastrin level than the group taking the existing PPI formulation due to the feedback mechanism of excessive gastric acid suppression. Because hypergastrinemia stimulates intestinal endocrine cells and may increase the risk of neuroendocrine tumors, studies are ongoing regarding the safety of long-term use.
Inhibition of gastrin secretion through somatostatin receptor activation has been reported to inhibit ECL cell hyperproliferation. In fact, synthetic peptide analogues of somatostatin with indications for endocrine diseases such as acromegaly, neuroendocrine tumors (NETs), and digestive system diseases such as upper gastrointestinal bleeding Sandostatin® (octreotide acetate) and Somatuline® Depot (lanreotide) are gastric neuroendocrine It has been reported to inhibit the overgrowth of ECL cells by inhibiting gastrin secretion in tumors.
In addition, there have been reports of anti-inflammatory responses through somatostatin receptor activation. Somatostatin is a type of neuropeptide that suppresses neurogenic inflammation and regulates the secretion of hormones and neurotransmitters. It is known to inhibit neurogenic inflammation and to be involved in nociception. Somatostatin is known to control the secretion of hormones and neurotransmitters to suppress neuronal inflammation and to be involved in nociception. Inflammatory somatostatin inhibits the proliferation of T lymphocytes and granulocytes in addition to controlling the neuroendocrine system. Somatostatin analogs are known to increase the expression of the anti-inflammatory factor IL-10 and inhibit the expression of the pro-inflammatory factors IFN-γ and TNF-α. As a result, the anti-inflammatory role of somatostatin has been mainly reported in studies related to inflammatory bowel disease (IBD). It is known that the level of intestinal somatostatin is reduced in patients with IBD, and it is known that the higher the level of inflammation in the intestine, the lower the level of somatostatin. In fact, it has been reported that the somatostatin analogue octreotide improved the symptoms of IBD in patients and animal models.
REF
- The efficacy and safety of fexuprazan in treating erosive esophagitis: a phase III, randomized, double‐blind, multicenter studyPublication Name: Journal of Gastroenterology and HepatologyPublication Date: 2024-01-22PMID: 38251791DOI: 10.1111/jgh.16471
- Review of the clinical development of fexuprazan for gastroesophageal reflux–related diseasePublication Name: European Journal of Clinical PharmacologyPublication Date: 2023-06-22PMID: 37344679DOI: 10.1007/s00228-023-03521-4
- Efficacy and Safety of Fexuprazan in Patients with Acute or Chronic GastritisPublication Name: Gut and LiverPublication Date: 2023-02-15PMCID: PMC10651377PMID: 36789577DOI: 10.5009/gnl220457
- Randomized controlled trial to evaluate the efficacy and safety of fexuprazan compared with esomeprazole in erosive esophagitisPublication Name: World Journal of GastroenterologyPublication Date: 2022-11-28PMCID: PMC9730436PMID: 36504556DOI: 10.3748/wjg.v28.i44.6294
- Editorial: acid suppression with potassium‐competitive acid blockers—dismissing genotype concerns. Authors’ replyPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2020-12-17PMID: 33333601DOI: 10.1111/apt.16158
- Editorial: acid suppression with potassium‐competitive acid blockers dismissing genotype concernsPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2020-12-17PMID: 33333607DOI: 10.1111/apt.16139
- Pharmacodynamics and pharmacokinetics of DWP14012 (fexuprazan) in healthy subjects with different ethnicitiesPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2020-10-27PMID: 33111337DOI: 10.1111/apt.16131
- Safety, tolerability, pharmacodynamics and pharmacokinetics of <scp>DWP</scp>14012, a novel potassium‐competitive acid blocker, in healthy male subjectsPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2018-06-04PMID: 29863280DOI: 10.1111/apt.14818
PATENTS
SearchSubmit searchSort byPublication Number – A to ZPublication Number – Z to APriority Date – OldestPriority Date – Most RecentGrant Date – OldestGrant Date – Most Recent
- Novel 4-methoxypyrrole derivative or salt thereof and pharmaceutical composition containing the samePublication Number: JP-6244498-B1Priority Date: 2015-04-27Grant Date: 2017-12-06
- Novel 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the samePublication Number: KR-20160127646-APriority Date: 2015-04-27
- Novel 4-methexipirols derivatives or salts thereof and pharmaceutical compositions thereofPublication Number: RU-2663895-C1Priority Date: 2015-04-27Grant Date: 2018-08-13
- 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the samePublication Number: US-10100010-B1Priority Date: 2015-04-27Grant Date: 2018-10-16
- Novel 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the samePublication Number: WO-2016175555-A2Priority Date: 2015-04-27
SYN
https://pubs.acs.org/doi/10.1021/acsomega.4c04507
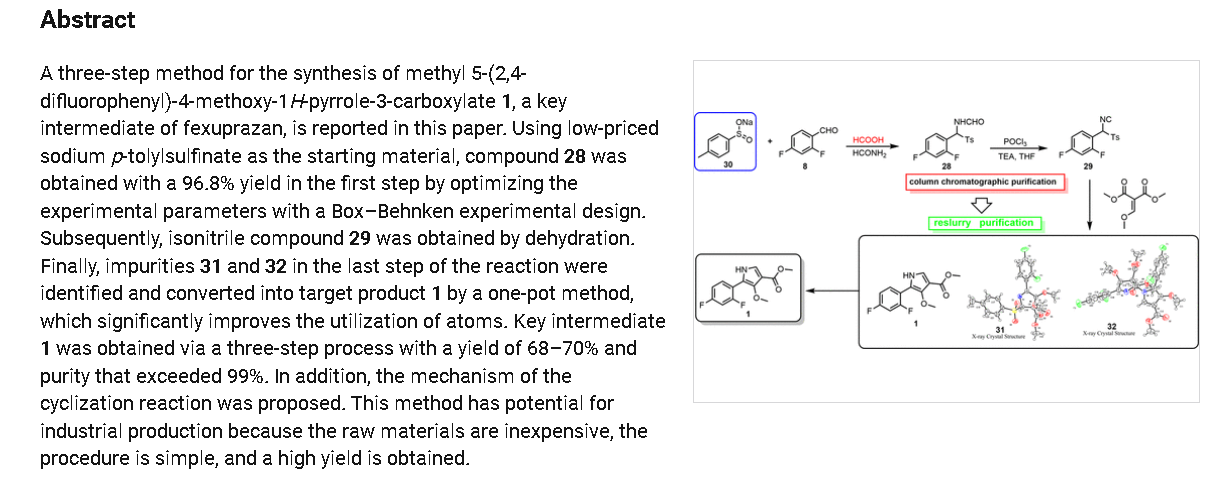
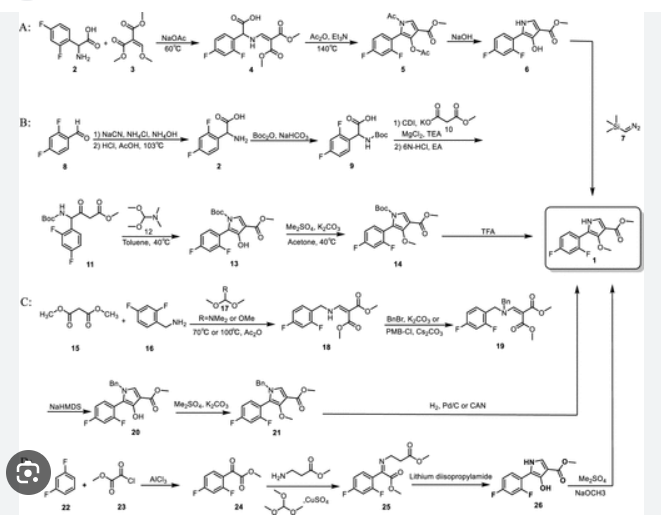
PAT
https://patents.google.com/patent/WO2023211843A1/en
Patent Citations (4)
Publication numberPriority datePublication dateAssigneeTitle
WO2016175555A2 *2015-04-272016-11-03Daewoong Pharmaceutical Co., Ltd.Novel 4-methoxy pyrrole derivatives or salts thereof and pharmaceutical composition comprising the same
US20190031609A1 *2016-03-252019-01-31Daewoong Pharmaceutical Co., Ltd.Novel acid addition salt of 1-(5-(2,4-difluorophenyl)-1-((3-fluorophenyl)sulfonyl)-4-methoxy-1h-pyrrol-3-yl)-n-methylmethanamine
US20200146974A1 *2017-07-072020-05-14Cj Healthcare CorporationComposition for injection
WO2021256861A1 *2020-06-172021-12-23일동제약(주)Novel acid secretion inhibitor and use thereof
PAT
https://patents.google.com/patent/WO2016175555A2/en
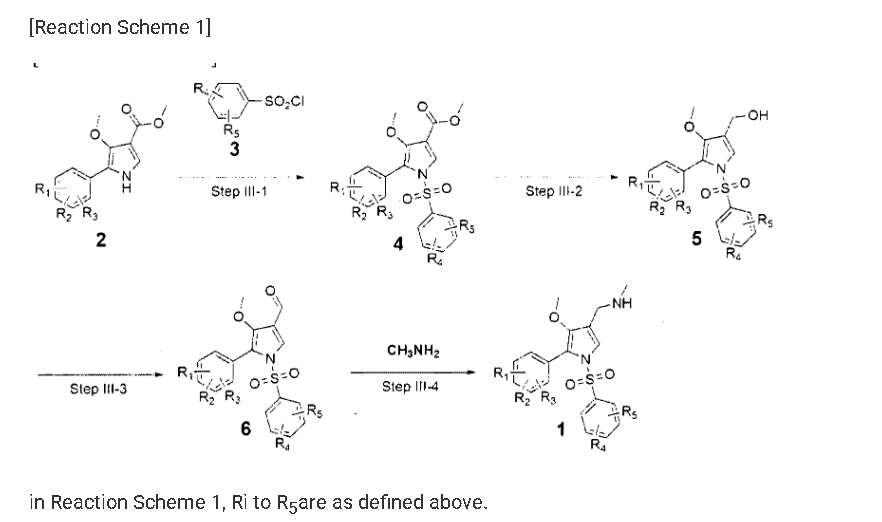
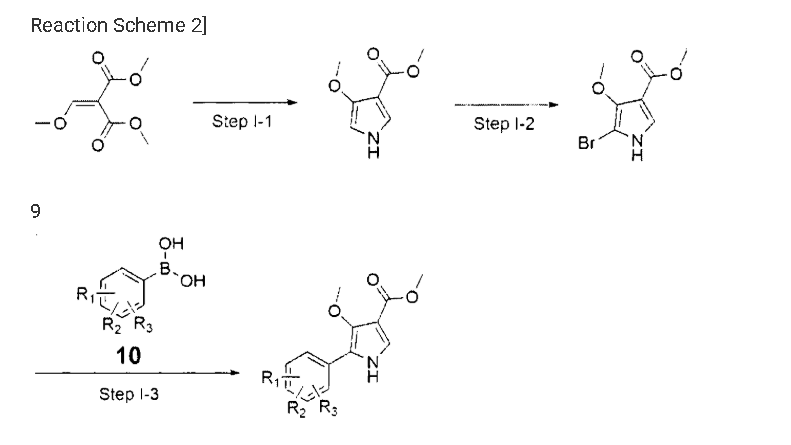
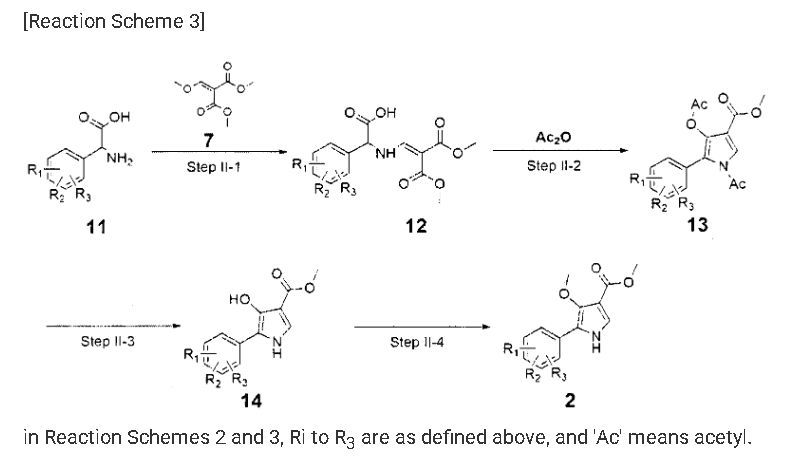
Example 8: Preparation of l-(5-(2,4-difluorophenyI)-l-((3-fluorophenyl)sulfonyI)-
4- metho\ -lH-pyrrol-3-yl)-N-methylmethanamine hydrochloride

(Step 8-1) Preparation of 2-(2,4-difluorophenyl)-2-((3-methoxy-2- (methoxycarbonyl)-3-oxoprop-l-en-l-yl)amino)acetic acid
2,4- Di fluorophenyl glycine (150.0 g, 801.5 mmol), dimethyl 2- (methoxymethylene)malonate (126.9 g, 728.6 mmol), and sodium acetate (65.8 g, 801 .5 mmol) were added to methanol (800.0 ml), and then refJuxed at 60°C for 4 hours. The reaction mixture was cooled to room temperature, and concentrated under reduced pressure to remove about 70% of methanol, and then filtered. The resulting solid was dried reduced pressure to give 190.0 g of the title compound. (Yield: 79.2%) Ή-NMR (500 MHz, CDC13): 8.02-7.99 (m, 1H), 7.45-7.40 (m, lH), 7.00-6.95 (m, 2H), 5.16 (s, lH), 3.74 (s, 3H), 3.76 (s, 3H)
PAT
https://patents.google.com/patent/WO2021256861A1/en
PAT
https://patents.google.com/patent/CN112094219A/en

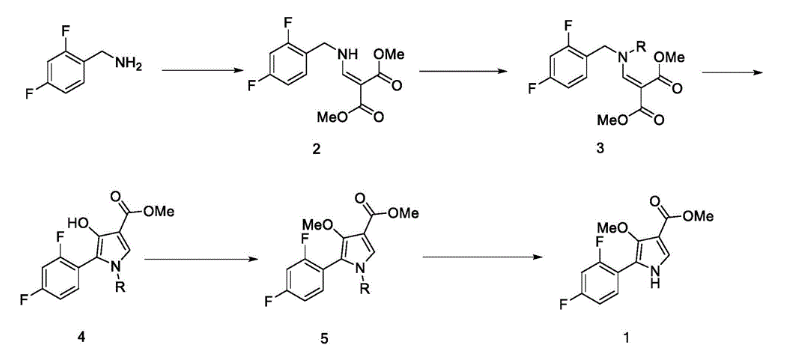
Synthesis of Compound 1
In a 500ml reaction flask were charged 10 g of compound 5B, 100 ml of acetonitrile, 50 ml of water, 56 g of ceric ammonium nitrate, and reacted at room temperature for 12 hours. 100 ml of water and 100 ml of ethyl acetate are added. The mixture was allowed to stand for separation, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with water and saturated brine in this order, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a crude product of Compound 1. The crude product was crystallized from ethyl acetate and n-heptane to give 6.1 g of compound 1 in 85.6% yield as a pale yellow solid.
Syn
https://pubs.acs.org/doi/10.1021/acs.oprd.5c00255
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ramani A, Merchant A, Cash BD (August 2023). “Review of the clinical development of fexuprazan for gastroesophageal reflux-related disease”. European Journal of Clinical Pharmacology. 79 (8): 1023–1029. doi:10.1007/s00228-023-03521-4. PMID 37344679. S2CID 259222741.
- Kim GH, Choi MG, Kim JI, Lee ST, Chun HJ, Lee KL, et al. (November 2023). “Efficacy and Safety of Fexuprazan in Patients with Acute or Chronic Gastritis”. Gut and Liver. 17 (6): 884–893. doi:10.5009/gnl220457. PMC 10651377. PMID 36789577.
- Jeong YS, Kim MS, Lee N, Lee A, Chae YJ, Chung SJ, et al. (May 2021). “Development of Physiologically Based Pharmacokinetic Model for Orally Administered Fexuprazan in Humans”. Pharmaceutics. 13 (6): 813. doi:10.3390/pharmaceutics13060813. PMC 8229463. PMID 34072547.
- Kim MS, Lee N, Lee A, Chae YJ, Chung SJ, Lee KR (June 2022). “Model-Based Prediction of Acid Suppression and Proposal of a New Dosing Regimen of Fexuprazan in Humans”. Pharmaceuticals. 15 (6): 709. doi:10.3390/ph15060709. PMC 9230547. PMID 35745628.
- “펙수클루정40밀리그램(펙수프라잔염산염)” [Fexuclue tablets 40 mg (fexuprazan hydrochloride)]. nedrug.mfds.go.kr (in Korean).
- “Daewoong Pharma’s GER drug gets product OK from Mexico”. Korea Economic Daily. 19 October 2023.
- Park IH. “Daewoong launches GERD treatment Fexuclu in Philippines”. KED Global. Retrieved 4 April 2025.
- Lee JH (14 March 2023). “Daewoong wins approval for GERD treatment Fexuclu in Chile”. KED Global. Retrieved 4 April 2025.
- Kim JE. “Daewoong receives approval for its GERD drug Fexuclue in Ecuador”. KED Global. Retrieved 4 April 2025.
| Clinical data | |
|---|---|
| Trade names | Fexuclue |
| Other names | Abeprazan; DWP14012; DWP-14012 |
| ATC code | A02BC10 (WHO) |
| Legal status | |
| Legal status | Rx in South Korea, Mexico |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1902954-60-2 |
| PubChem CID | 122662112 |
| DrugBank | DB16078 |
| ChemSpider | 68006985 |
| UNII | BE52S2C1QT |
| KEGG | D13012 |
| ChEMBL | ChEMBL4594445 |
| Chemical and physical data | |
| Formula | C19H17F3N2O3S |
| Molar mass | 410.41 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Fexuprazan, Abeprazan, DWP14012, DWP-14012
Atigliflozin
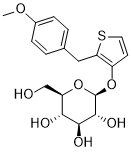
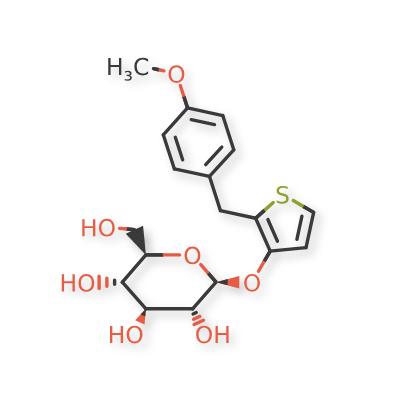
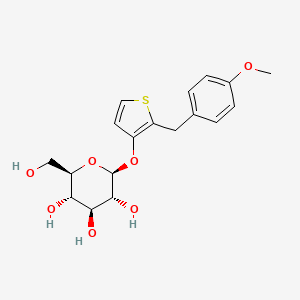
Atigliflozin
CAS 647834-15-9
Chemical Formula: C18H22O7S
Exact Mass: 382.1086
Molecular Weight: 382.43
AVE 2268; AVE-2268; AVE2268; Y0H7UPE4WJ
(2R,3S,4S,5R,6S)-2-(hydroxymethyl)-6-((2-(4-methoxybenzyl)thiophen-3-yl)oxy)tetrahydro-2H-pyran-3,4,5-triol
Atigliflozin (AVE-2268) is an orally active and selective SGLT-2 inhibitor, with IC50s of 10 nM and 8.2 μM for hSGLT-2 and hSGLT-1) respectively. Atigliflozin can lower the blood glucose and improve the impaired oral glucose tolerance. Atigliflozin can be used for research of type II diabetes mellitus.
Patent
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: KR-20240160678-APriority Date: 2008-08-06
- Use of thiophene glycoside derivatives for producing medicaments for treatment of hypertensionPublication Number: WO-2009138195-A2Priority Date: 2008-05-16
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: KR-20200118243-APriority Date: 2008-08-06
- Treatment of diabetes in patients who are inadequate for metformin treatmentPublication Number: JP-2021035998-APriority Date: 2008-08-06
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: US-2021093633-A1Priority Date: 2008-08-06
- Treatment of diabetes in patients unsuitable for metformin treatmentPublication Number: JP-2023011007-APriority Date: 2008-08-06
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: US-2022323434-A1Priority Date: 2008-08-06
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: US-2018271859-A1Priority Date: 2008-08-06
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: US-2019105321-A1Priority Date: 2008-08-06
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: US-8853156-B2Priority Date: 2008-08-06Grant Date: 2014-10-07
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: US-9486526-B2Priority Date: 2008-08-06Grant Date: 2016-11-08
- Treatment for diabetes in patients inappropriate for metformin therapyPublication Number: WO-2010015664-A1Priority Date: 2008-08-06
SYN
https://www.sciencedirect.com/science/article/abs/pii/S022352342400223X
Atigliflozin is developed by Sanofi and is currently in phase II clinical development. It is used for the treatment of T2DM (IC50= 13 nmol/L)[74]. In mice, Atigliflozin led to a rise in urinary glucose excretion that was dependent on the dosage administered (ID3030=79±8.1 mg/kg p.o.). Similarly, in rats, Atigliflozin caused a dose-dependent increase in UGE(ID= 39.8±4.0 mg/kg p.o.). When glucose was administered intraperitoneally, Atigliflozin was found to be more effective in reducing blood glucose levels in mice (IDorally administered glucose (ID5050= 13.2±3.9 mg/kg) compared to =26.1±3.9 mg/kg). This suggests that Atigliflozin does not have an impact on SGLT 1 in the gut in vivo, which
aligns with its very low affinity to SGLT1 in vitro Additionally, studies have demonstrated that the combined use of metformin and Atigliflozin can effectively lower glucose levels by inhibiting the body’s natural glucose production. This coapplication may offer a sustainable solution for improving glycemic control in in dividuals with T2DM [75].
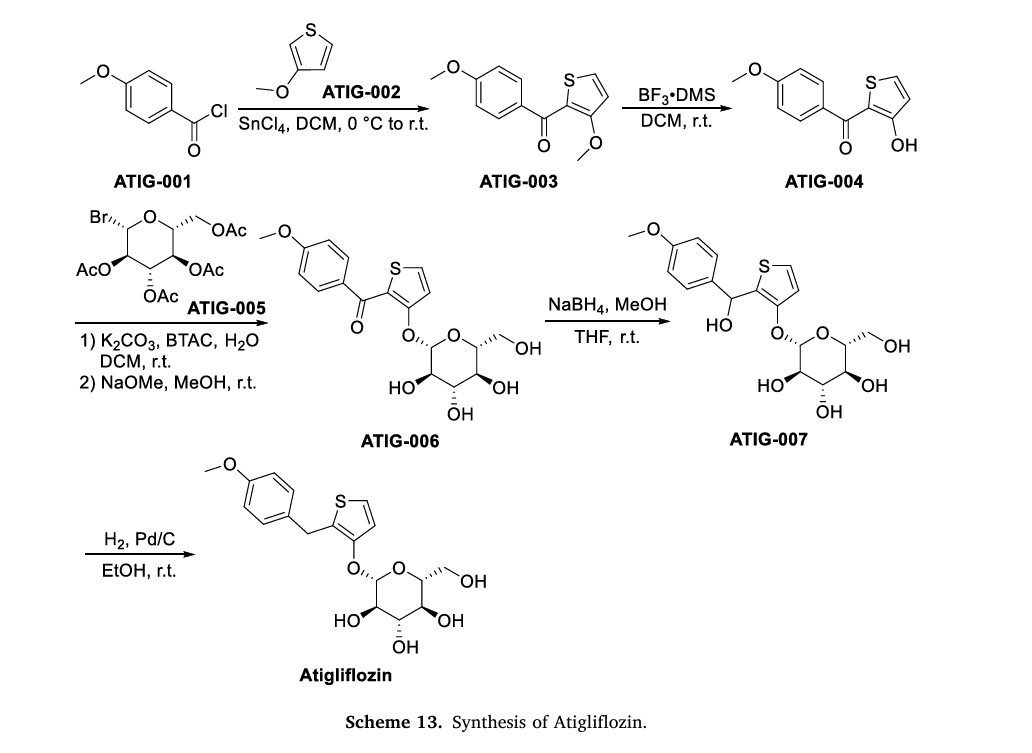
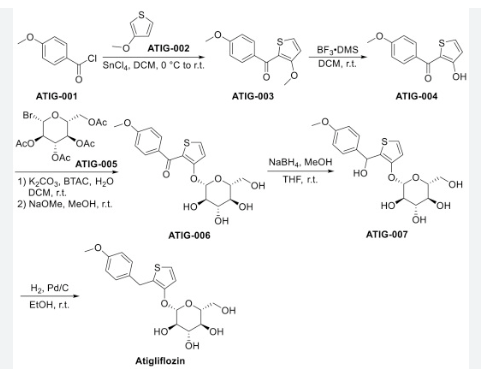
The original synthesis route of Atigliflozin is showed in Scheme 13 [76,77]. Friedel-Crafts acylation of 4-methoxybenzoyl chloride (ATIG-001) with 3-methoxythiophene (ATIG-002) catalyzed by SnCl114to give the ketone ATIG-003. In the presence of borane-methyl sulfide (DMS) complex, ATIG-003 is demethylated to give the thiophenol ATIG-004. Next, nucleophilic substitution of ATIG-004 with 2,3,4,
6-tetra-O-acetyl αD-glucopyranosyl bromide (ATIG-005), followed by hydrolysis in the presence of sodium methanolate give ether ATIG-006. ATIG-006 is reduced by sodium borohydride to give the alcohol ATIG-007. Finally, further reduction of ATIG-007 catalyzed by Pd/C with H2 provides Atigliflozin.
[74] M. Bickel, H. Brummerhop, W. Frick, H. Glombik, A.W. Herling, H.O. Heuer,
O. Plettenburg, S. Theis, U. Werner, W. Kramer, Effects of AVE2268, a substituted
glycopyranoside, on urinary glucose excretion and blood glucose in mice and rats,
Arzneimittelforschung 58 (2008) 574–580.
[75] S. Neschen, M. Scheerer, A. Seelig, P. Huypens, J. Schultheiss, M. Wu, W. Wurst,
B. Rathkolb, K. Suhre, E. Wolf, J. Beckers, M. Hrab´e de Angelis, Metformin
supports the antidiabetic effect of a sodium glucose cotransporter 2 inhibitor by
suppressing endogenous glucose production in diabetic mice, Diabetes 64 (2015)
284–290.
[76] G. Heiner, F. Wendelin, H. Hubert, K. Werner, Novel Thiophenylglycoside
Derivatives, Methods for Production Thereof, Medicaments Comprising Said
Compounds and Use Thereof, 2014 WO2004007517A1.
[77] H. Glombik, W. Frick, H. Heuer, W. Kramer, Thiophene Glycoside Derivatives,
Processes for the Preparation, Medicaments Comprising These Compounds, and the
Use Thereof, 2010 US7666848B2.
- The magic of small structure differences in a sodium‐glucose cotransporter drug discovery project—14C‐labelled drug candidates in a key‐differentiating studyPublication Name: Journal of Labelled Compounds and RadiopharmaceuticalsPublication Date: 2020-07-14PMID: 32633850DOI: 10.1002/jlcr.3869
- Metformin Supports the Antidiabetic Effect of a Sodium Glucose Cotransporter 2 Inhibitor by Suppressing Endogenous Glucose Production in Diabetic MicePublication Name: DiabetesPublication Date: 2014-07-28PMID: 25071027DOI: 10.2337/db14-0393
- Energy loss via urine and faeces – a combustive analysis in diabetic rats and the impact of antidiabetic treatment on body weightPublication Name: Diabetes, Obesity and MetabolismPublication Date: 2012-11-22PMID: 23121319DOI: 10.1111/dom.12030
- Effects of AVE2268, a Substituted Glycopyranoside, on Urinary Glucose Excretion and Blood Glucose in Mice and RatsPublication Name: Arzneimittel-ForschungPublication Date: 2011-12-19PMID: 19137908DOI: 10.1055/s-0031-1296559
- [1]. Schudok M, et al. The magic of small structure differences in a sodium-glucose cotransporter drug discovery project-14 C-labelled drug candidates in a key-differentiating study. J Labelled Comp Radiopharm. 2021 Feb;64(2):73-76. [Content Brief][2]. Bickel M, et al. Effects of AVE2268, a substituted glycopyranoside, on urinary glucose excretion and blood glucose in mice and rats. Arzneimittelforschung. 2008;58(11):574-80. [Content Brief]
////////// Atigliflozin, AVE 2268, AVE-2268, AVE2268, Y0H7UPE4WJ
Abarelix
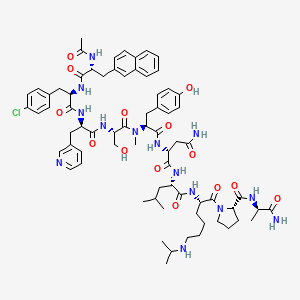
Abarelix
CAS 183552-38-7
785804-17-3 (acetate) 183552-38-7 (free base)
PPI149, PPI-149, PPI 149, R3827, R-3827, R 3827, Abarelix, Abarelix acetate, Plenaxis,
W486SJ5824
Chemical Formula: C72H95ClN14O14
Exact Mass: 1414.6841
Molecular Weight: 1416.06
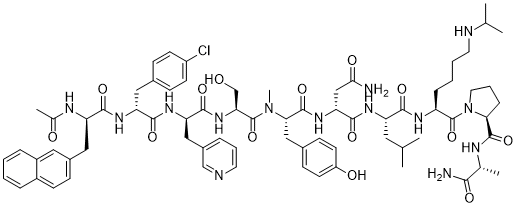
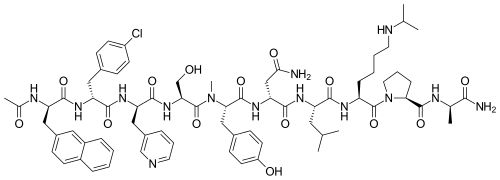
Ac-D-Nal-[D-(pCl)Phe]-D-Pal-Ser-[Nalpha-Me-Tyr]-D-Asn-Leu-ILys-Pro-DAla-NH2

(2R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]-methylamino]-3-(4-hydroxyphenyl)propanoyl]amino]-N-[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]butanediamide
(2R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]-methylamino]-3-(4-hydroxyphenyl)propanoyl]amino]-N-[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]butanediamide
Abarelix is a synthetic decapeptide and antagonist of naturally occurring gonadotropin-releasing hormone (GnRH). Abarelix directly and competitively binds to and blocks the gonadotropin releasing hormone receptor in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with prostate hypertrophy or prostate cancer, since testosterone is required to sustain prostate growth.
Abarelix, sold under the brand name Plenaxis, is an injectable gonadotropin-releasing hormone antagonist (GnRH antagonist) which is marketed in Germany and the Netherlands. It is primarily used in oncology to reduce the amount of testosterone made in patients with advanced symptomatic prostate cancer for which no other treatment options are available.[2][3]
It was originally marketed by Praecis Pharmaceuticals as Plenaxis,[2] and is now marketed by Speciality European Pharma in Germany[4] after receiving a marketing authorization in 2005. The drug was introduced in the United States in 2003, but was discontinued in this country in May 2005 due to poor sales and a higher-than-expected incidence of severe allergic reactions.[5] It remains marketed in Germany and the Netherlands however.[6]
Pat
https://patents.google.com/patent/CN107778354B/en
Example 1: synthesis of peptide resin 1
Dissolving 0.15mol of Fmoc-D-Ala and 0.15mol of HOBt by using a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Taking 0.05mol of MOBHA resin (the substitution value is about 0.6mmol/g), swelling with DMF for 25 minutes, washing and filtering, adding the activated solution, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3 times, wherein the washing time is 3min each time, obtaining Fmoc-D-Ala-MOBHA resin, namely the peptide resin 1, removing Fmoc protection with 20% PIP/DMF solution for 25 minutes before carrying out the next coupling reaction, washing and filtering to obtain the D-Ala-MOBHA resin.
Example 2: synthesis of peptide resin 1
Dissolving 0.15mol of Boc-D-Ala and 0.15mol of HOBt with a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Taking 0.05mol of MOBHA resin (the substitution value is about 0.6mmol/g), swelling with DMF for 25 minutes, washing and filtering, adding an activated Fmoc-D-Ala solution, stirring at room temperature for 3 hours, pumping out the reaction solution, washing 3 times with DMF, washing 3 times with DCM, wherein each washing time is 3min, obtaining Boc-D-Ala-MOBHA resin, namely peptide resin 1, deprotecting with 30% TFA/DCM solution for 30 minutes, neutralizing with DIEA/DCM solution, washing and filtering with DMF and DCM, and obtaining D-Ala-MOBHA resin.
Example 3: synthesis of Abarelix peptide resin
Dissolving 0.15mol of Fmoc-Pro and 0.15mol of HOBt in a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Adding the activated Fmoc-Pro solution into the peptide resin 1 obtained in example 1, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3 minutes each time, removing Fmoc protection with 20% PIP/DMF solution for 25 minutes, washing and filtering to obtain Pro-D-Ala-MOBHA resin.
Boc-Lys (iPr, Z), Fmoc-Leu, Fmoc-D-Asn (Trt), Fmoc-N-Me-Tyr (tBu), Fmoc-Ser (tBu), Fmoc-D-Pal, Fmoc-D-Cpa and Ac-D-Nal are sequentially added in the same method, and the Abarelix peptide resin, Ac-D-Nal-D-Cpa-D-Pal-Ser (tBu) -N-Me-Tyr (tBu) -D-Asn (Trt) -Leu-Lys (iPr, Z) -Pro-D-Ala-MOBHA resin are obtained by washing and filtering.
Example 4: synthesis of Abarelix peptide resin
Dissolving 0.15mol of Boc-Pro and 0.15mol of HOBt by using a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Adding the activated Boc-Pro solution into the peptide resin 1 obtained in example 1, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3min each time, deprotecting with 30% TFA/DCM solution for 30 minutes, neutralizing with DIEA/DCM solution, washing with DMF and DCM, and filtering to obtain Pro-D-Ala-MBHA resin.
Boc-Lys (iPr, Z), Fmoc-Leu, Fmoc-D-Asn (Trt), Fmoc-N-Me-Tyr (tBu), Fmoc-Ser (tBu), Fmoc-D-Pal, Fmoc-D-Cpa and Ac-D-Nal are sequentially added in the same method, and the Abarelix peptide resin, Ac-D-Nal-D-Cpa-D-Pal-Ser (tBu) -N-Me-Tyr (tBu) -D-Asn (Trt) -Leu-Lys (iPr, Z) -Pro-D-Ala-MOBHA resin are obtained by washing and filtering.
Example 5: preparation of crude Abarelix
Taking the abarelix peptide resin prepared in the example 3, adding 8% HBr/TFA solution (acidolysis solution 10mL/g abarelix resin), stirring and reacting for 6 hours, filtering and collecting filtrate, washing the resin with a small amount of TFA for 3 times, combining the filtrates, concentrating under reduced pressure, adding anhydrous ether for precipitation, washing the precipitate with anhydrous ether for 3 times, and draining to obtain white-like powder, namely a crude product of abarelix, wherein the purity of the crude product is 79.3%.
Example 6: preparation of crude Abarelix
Taking the abarelix peptide resin prepared in the example 4, adding 8% HBr/TFA solution (acidolysis solution 10mL/g abarelix resin), stirring and reacting for 6 hours, filtering and collecting filtrate, washing the resin with a small amount of TFA for 3 times, combining the filtrates, concentrating under reduced pressure, adding anhydrous ether for precipitation, washing the precipitate with anhydrous ether for 3 times, and draining to obtain white-like powder, namely a crude product of abarelix, wherein the purity of the crude product is 77.4%.
Example 7: purification and trans-salt conversion of crude Abarelix
Taking the crude Abarelix product obtained in the example 5, dissolving the Abarelix product in 20 percent acetic acid solution, filtering the solution by using a 0.45 mu m microporous membrane, and purifying for later use;
purifying by high performance liquid chromatography, wherein a chromatographic filler is 10 mu m reverse phase C18, a mobile phase system is 0.1% TFA/water solution-0.1% TFA/acetonitrile solution, a chromatographic column with the flow rate of 77mm x 250mm is 90mL/min, eluting by a gradient system, circularly sampling and purifying, sampling a crude product solution in the chromatographic column, starting the mobile phase for elution, collecting a main peak, and evaporating acetonitrile to obtain an abarelix purified intermediate concentrated solution;
taking the Abarelix purified intermediate concentrated solution, and filtering with a 0.45-micrometer filter membrane for later use;
performing salt exchange by high performance liquid chromatography, wherein the mobile phase system is 1% acetic acid/water solution-acetonitrile, the purification is performed by reversed phase C18 with chromatographic packing of 10 μm, the flow rate of a chromatographic column of 77mm × 250mm is 90mL/min, gradient elution and circular sample loading method are adopted, the sample is loaded in the chromatographic column, the mobile phase elution is started, the chromatogram is collected, the change of the absorbance is observed, the main peak of salt exchange is collected and the purity is detected by analyzing the liquid phase, the main peak solutions of salt exchange are combined, the concentration is performed under reduced pressure to obtain the aqueous solution of abarelix acetic acid, and freeze drying is performed to obtain 39.4g abarelix pure product
The total yield was 55.6%, molecular weight: 1417.2, purity: 99.6%, maximum single impurity of 0.13%, no toxic hydantoin degradation products were detected.
Example 8: purification and trans-salt conversion of crude Abarelix
Taking the crude Abarelix product obtained in the example 6, dissolving the Abarelix product by using a purification mobile phase A, and filtering the solution by using a 0.45 mu m microporous filter membrane to purify the Abarelix product for later use;
purifying by high performance liquid chromatography, wherein a chromatographic filler is 10 mu m reverse phase C18, a mobile phase system is 0.1% TFA/water solution-0.1% TFA/acetonitrile solution, a chromatographic column with the flow rate of 77mm x 250mm is 90mL/min, eluting by a gradient system, circularly sampling and purifying, sampling a crude product solution in the chromatographic column, starting the mobile phase for elution, collecting a main peak, and evaporating acetonitrile to obtain an abarelix purified intermediate concentrated solution;
taking the Abarelix purified intermediate concentrated solution, and filtering with a 0.45-micrometer filter membrane for later use;
performing salt exchange by adopting a high performance liquid chromatography, wherein a mobile phase system is 1% acetic acid/water solution-acetonitrile, a chromatographic filler for purification is reversed phase C18 with the diameter of 10 mu m, the flow rate of a chromatographic column with the diameter of 77mm × 250mm is 90mL/min, a gradient elution method and a circular sample loading method are adopted, loading the chromatographic column, starting the mobile phase elution, collecting a spectrum, observing the change of the absorbance, collecting a main salt exchange peak, detecting the purity by using an analysis liquid phase, combining main salt exchange peak solutions, concentrating under reduced pressure to obtain an abarelix acetic acid water solution, and performing freeze drying to obtain 41.7g of an abarelix pure product.
The total yield is 58.9%, molecular weight: 1417.0, purity: 99.5%, maximum single impurity 0.09%, no toxic hydantoin degradation products were detected.
SYN
Ma, Zhonggang; Guo, Dewen; Zeng, Dezhi; Wen, Yongjun. Method for synthesizing abarelix. Assignee Chengdu Shengnuo Biopharm Co., Ltd.. 2018.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
1: Tombal B. New treatment paradigm for prostate cancer: abarelix initiation therapy for immediate testosterone suppression followed by a luteinizing hormone-releasing hormone agonist. BJU Int. 2012 Mar;109(6):E16; author reply E16-7. doi: 10.1111/j.1464-410X.2012.10983.x. PubMed PMID: 22360806.
2: Garnick MB, Mottet N. New treatment paradigm for prostate cancer: abarelix initiation therapy for immediate testosterone suppression followed by a luteinizing hormone-releasing hormone agonist. BJU Int. 2012 Aug;110(4):499-504. doi: 10.1111/j.1464-410X.2011.10708.x. Epub 2011 Nov 16. PubMed PMID: 22093775.
3: Koechling W, Hjortkjaer R, Tankó LB. Degarelix, a novel GnRH antagonist, causes minimal histamine release compared with cetrorelix, abarelix and ganirelix in an ex vivo model of human skin samples. Br J Clin Pharmacol. 2010 Oct;70(4):580-7. doi: 10.1111/j.1365-2125.2010.03730.x. PubMed PMID: 20840449; PubMed Central PMCID: PMC2950992.
4: Retraction statement: Reconstitution of Plenaxis® (Abarelix) 100 mg for injection is more effective with a vortex-like mixer than when performed manually. J Pharm Pract. 2010 Feb;23(1):78. doi: 10.1177/0897190009360369. PubMed PMID: 21507797.
5: Kirby RS, Fitzpatrick JM, Clarke N. Abarelix and other gonadotrophin-releasing hormone antagonists in prostate cancer. BJU Int. 2009 Dec;104(11):1580-4. doi: 10.1111/j.1464-410X.2009.08924.x. Review. PubMed PMID: 20053189.
6: Debruyne F, Bhat G, Garnick MB. Abarelix for injectable suspension: first-in-class gonadotropin-releasing hormone antagonist for prostate cancer. Future Oncol. 2006 Dec;2(6):677-96. Review. PubMed PMID: 17155895.
7: Beer TM, Ryan C, Bhat G, Garnick M; Abarelix Study Group. Dose-escalated abarelix in androgen-independent prostate cancer: a phase I study. Anticancer Drugs. 2006 Oct;17(9):1075-9. PubMed PMID: 17001181.
8: Hogle WP. Abarelix (plenaxis). Clin J Oncol Nurs. 2004 Dec;8(6):663-5. PubMed PMID: 15637961.
9: Mongiat-Artus P, Teillac P. Abarelix: the first gonadotrophin-releasing hormone antagonist for the treatment of prostate cancer. Expert Opin Pharmacother. 2004 Oct;5(10):2171-9. Review. PubMed PMID: 15461552.
10: Wong SL, Lau DT, Baughman SA, Fotheringham N, Menchaca D, Garnick MB. Pharmacokinetics and pharmacodynamics of a novel depot formulation of abarelix, a gonadotropin-releasing hormone (GnRH) antagonist, in healthy men ages 50 to 75. J Clin Pharmacol. 2004 May;44(5):495-502. PubMed PMID: 15102870.
References
- “Abarelix”. PubChem. 2017-07-29.
- “Abarelix”. Drugs.com. Archived from the original on 2018-02-10. Retrieved 2018-01-23.
- Boccon-Gibod L, van der Meulen E, Persson BE (June 2011). “An update on the use of gonadotropin-releasing hormone antagonists in prostate cancer”. Therapeutic Advances in Urology. 3 (3): 127–40. doi:10.1177/1756287211414457. PMC 3159401. PMID 21904569.
- Pharmazeutische Zeitung online: Abarelix (in German)
- Minev B (13 January 2011). Cancer Management in Man: Chemotherapy, Biological Therapy, Hyperthermia and Supporting Measures. Springer Science & Business Media. pp. 182–. ISBN 978-90-481-9704-0.
- “Abarelix”. Drugs.com. Archived from the original on 2019-08-29. Retrieved 2018-08-27.
| Clinical data | |
|---|---|
| Trade names | Plenaxis |
| AHFS/Drugs.com | Monograph |
| Routes of administration | Intramuscular injection |
| Drug class | GnRH analogue; GnRH antagonist; Antigonadotropin |
| ATC code | L02BX01 (WHO) |
| Pharmacokinetic data | |
| Protein binding | 96–99% |
| Identifiers | |
| IUPAC name | |
| CAS Number | 183552-38-7 |
| PubChem CID | 16131215 |
| IUPHAR/BPS | 1188 |
| DrugBank | DB00106 |
| ChemSpider | 10482301 |
| UNII | W486SJ5824 |
| KEGG | D02738 |
| ChEBI | CHEBI:337298 |
| ChEMBL | ChEMBL1252 |
| CompTox Dashboard (EPA) | DTXSID20171443 |
| Chemical and physical data | |
| Formula | C72H95ClN14O14 |
| Molar mass | 1416.09 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Abarelix, PPI149, PPI-149, PPI 149, R3827, R-3827, R 3827, Abarelix, Abarelix acetate, Plenaxis,
W486SJ5824
O=C(N[C@@H](CC(C)C)C(N[C@@H](CCCCNC(C)C)C(N1[C@H](C(N[C@H](C)C(N)=O)=O)CCC1)=O)=O)[C@H](NC([C@@H](N(C([C@@H](NC([C@H](NC([C@H](NC([C@H](NC(C)=O)CC2=CC=C3C=CC=CC3=C2)=O)CC4=CC=C(Cl)C=C4)=O)CC5=CC=CN=C5)=O)CO)=O)C)CC6=CC=C(O)C=C6)=O)CC(N)=O
Rongliflozin, Olorigliflozin
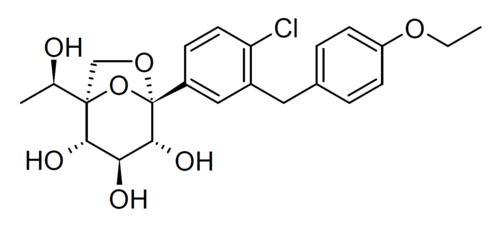
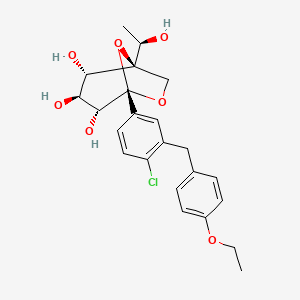
Rongliflozin
Olorigliflozin, 6FP3NST6ZQ, DJT1116PG
Cas 2035989-50-3
450.9 g/mol, C23H27ClO7
(1R,2S,3S,4R,5S)-5-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-[(1R)-1-hydroxyethyl]-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- (1R,2S,3S,4R,5S)-5-(4-Chloro-3-(4-ethoxybenzyl)phenyl)-1-((R)-1-hydroxyethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- 1,6-Anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-beta-L-idopyranose
- beta-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-
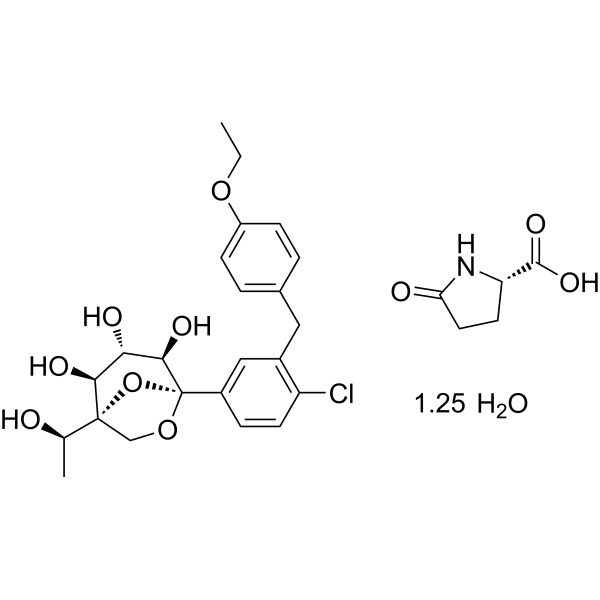
Rongliflozin 화학구조
CAS No. : 2648020-91-9
| MW | 602.55 |
|---|---|
| MF | C23H27ClO7.C5H7NO3.5/4H2O |
- OriginatorHEC Pharm
- DeveloperSunshine Lake Pharma
- ClassAntihyperglycaemics; Small molecules
- Mechanism of ActionSodium-glucose transporter 2 inhibitors
- PreregistrationType 2 diabetes mellitus
- 04 Sep 2025Chemical structure information added.
- 31 Dec 2023Preregistration for Type 2 diabetes mellitus in China (PO), in December 2023
- 31 Dec 2023Efficacy and adverse events data from a phase IIIa trial in Type 2 diabetes mellitus released by Sunshine Lake Pharma, before December 2023
Rongliflozin is an SGLT2 inhibitor developed as a potential treatment for diabetes.[1][2]
Rongliflozin (DJT1116PG) is a selective and orally active inhibitor of sodium-glucose co-transporter-2 (SGLT-2). Rongliflozin can be used for the research of type 2 diabetes mellitus (T2DM).
PAT
- (1R,2S,3S,4R,5S)-5-(4-Chloro-3-(4-ethoxybenzyl)phenyl)-1-((R)-1-hydroxyethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- 1,6-Anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-beta-L-idopyranose
- beta-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-
- Complexes of glucopyranosyl derivatives and methods for their preparation and usePublication Number: JP-2018535237-APriority Date: 2015-11-27
- Complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: US-10555930-B2Priority Date: 2015-11-27Grant Date: 2020-02-11
- Complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: US-2018344689-A1Priority Date: 2015-11-27
- A complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: WO-2017088839-A1Priority Date: 2015-11-27
- Glucopyranosyl derivative complex and its preparation method and usePublication Number: JP-6916180-B2Priority Date: 2015-11-27Grant Date: 2021-08-11
- Preparation method and intermediate of glucopyranosyl derivativesPublication Number: CN-113195510-BPriority Date: 2019-01-08Grant Date: 2022-12-23
- Crystalline forms of glucopyranosyl derivativesPublication Number: CN-107778336-BPriority Date: 2016-08-24Grant Date: 2022-09-27
- Glucopyranosyl derivative compound, preparation method and applicationPublication Number: CN-106810582-APriority Date: 2015-11-27
- Glucopyranosyl derivative compound, preparation method and applicationPublication Number: CN-106810582-BPriority Date: 2015-11-27Grant Date: 2019-12-31
- A complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: EP-3371199-A1Priority Date: 2015-11-27
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: WO-2022007838-A1Priority Date: 2020-07-08
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: EP-4178970-A1Priority Date: 2020-07-08
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: US-2023250121-A1Priority Date: 2020-07-08
- Preparation methods of glucopyranosyl derivatives and intermediates thereofPublication Number: CN-113912567-BPriority Date: 2020-07-08Grant Date: 2024-01-16
- Preparation method for glucopyranosyl derivative and intermediate thereofPublication Number: WO-2020143653-A1Priority Date: 2019-01-08
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: WO-2022036506-A1Priority Date: 2020-08-17
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: EP-4197543-A1Priority Date: 2020-08-17
- Compositions of SGLT-2 inhibitors and angiotensin receptor antagonists and uses thereofPublication Number: KR-20230057388-APriority Date: 2020-08-17
- Composition and application of SGLT-2 inhibitor and angiotensin receptor blockerPublication Number: CN-116490178-APriority Date: 2020-08-17
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: US-2023346817-A1Priority Date: 2020-08-17
- Nintedanib targeted combinationPublication Number: CN-118021812-APriority Date: 2023-12-30
- Preparation method of L-pyroglutamic acid co-crystal of glucopyranosyl derivativesPublication Number: CN-115141235-APriority Date: 2021-03-30
- Preparation method of L-pyroglutamic acid cocrystal of pyranose glucopyranose derivativePublication Number: CN-115141235-BPriority Date: 2021-03-30Grant Date: 2024-08-09
- Fixed-dose combination of sglt-2 inhibitor and angiotensin converting enzyme inhibitor, and use thereofPublication Number: WO-2022104621-A1Priority Date: 2020-11-19
- Compositions and uses of fixed-dose SGLT-2 inhibitors and angiotensin-converting enzyme inhibitorsPublication Number: CN-116234545-APriority Date: 2020-11-19
SYN
https://pubs.rsc.org/en/content/articlelanding/2021/ce/d1ce01305j/unauth
Rongliflozin L-pyroglutamic acid, a highly active SGLT-2 inhibitor cocrystal discovered and developed by our group, is currently undergoing clinical trials for the treatment of diabetes. Here, we report and design a simple and robust process to obtain a single and pure crystalline form I (1) of the cocrystal, containing Rongliflozin (2) with L-pyroglutamic acid (L-PA), based on coformer-induced purification (CoIP). Extensive experiments showed that the addition of L-pyroglutamic acid in the eluent was key to suppression of the dissociation equilibrium of the cocrystal during lessivation, with high efficiency. Importantly, based in this profile, this process exhibited strong robustness and margin of safety at multigram and multikilogram scales
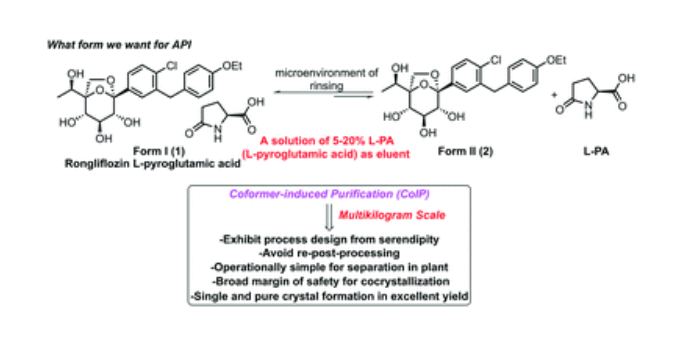
Kilogram scale Process of 1
A mixture of (1R,2S,3S,4R,5S)-5-(4-chloro-3-(4-ethoxybenzyl) phenyl)-1-((R)-1-
hydroxyethyl)-6,8-dioxabicyclo [3.2.1] octane-2,3,4-triol ethanolate form III (3) (23.45 kg, 47.3
mol), L-pyroglutamic acid (24.31 kg, 4.0 equiv.), EtOH (35.9 L) and H2O (70 L) was added into a
300 L reactor at room temperature. The slurry was heated to 65 °C and stirred until it is clear. The
clear solution was cooled to 35±5 °C typically. Seed crystal form I (1) (0.70 kg, 3% g/g) was added
when the solution was cooled to 34 °C and maintained for 1.5 h. Gradually, the slurry was cool to
30 °C and 25 °C in 3 hours, and finally stirred at 25 °C for 24 h. The slurry was collected on a
centrifuge filter. The filter cake was washed with a mixed solution of EtOH (31.3 L)/H2O (62.7 L)
with L-pyroglutamic acid (1.64 kg, 7% g/g) pre-cooled to -15°C. The wet cake was dried under
vacuum at 45 °C for 8 h. Pure cocrystal form I (1) was obtained as a white solid (24.91 kg, yield
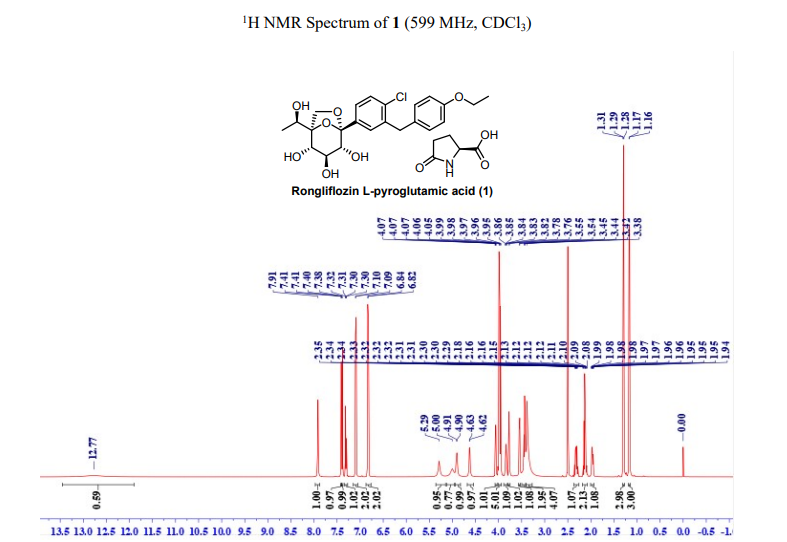
91%). MP (DSC onset) = 96.91 ℃. 1H NMR (599 MHz, DMSO-d6) δ 12.77 (br, 1H), 7.91 (s, 1H),
7.41 (d, J = 2.0 Hz, 1H), 7.39 (d, J = 12.0 Hz, 1H), 7.31 (dd, J = 12.0, 2.0 Hz, 1H), 7.10 (d, J = 2.0
Hz , 2H), 6.83 (d, J = 2.0 Hz, 2H), 5.29 (s, 1H), 5.00 (s, 1H), 4.91 (d, J = 6.7 Hz, 1H), 4.63 (d, J =
6.1 Hz, 1H), 4.06 (dd, J = 12.0, 6.0 Hz, 1H), 3.99– 3.95 (m, 5H), 3.84 (p, J = 6.0 Hz, 1H), 3.77 (d,
J = 12.0 Hz, 1H), 3.55 (d, J = 6.0 Hz, 1H), 3.44 (t, J = 12.0 Hz, 2H), 3.38 (s, 4H), 2.35-2.29 (m,
1H), 2.18-2.08 (m, 2), 1.99-1.94 (m, 1H), 1.29 (t, J = 12.0 Hz, 3H), 1.17 (d, J = 6.0 Hz, 3H). 13C
NMR (151 MHz, DMSO-d6) δ 177.06, 174.48, 156.96, 138.17, 137.69, 131.16, 129.64, 129.42,
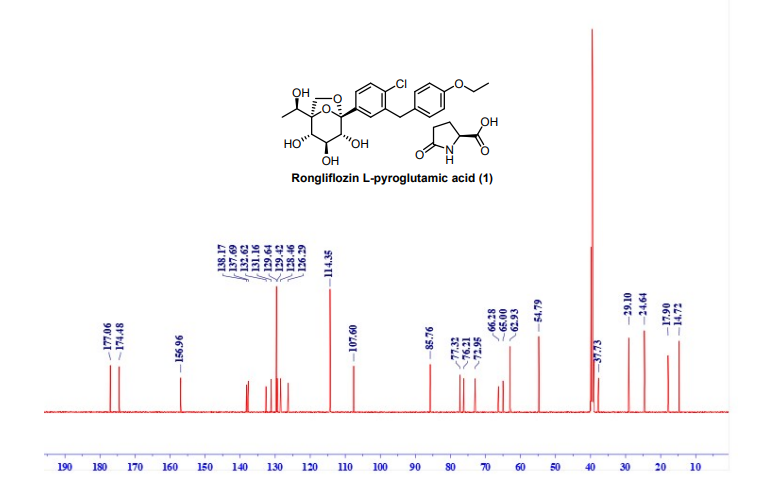
128.46, 126.29, 114.35, 107.60, 85.76, 77.32, 76.21, 72.95, 66.28, 65.00, 62.93, 54.79, 37.73, 29.10,
24.64, 17.90, 14.72. HRMS: (ESI) Calcd for C23H27ClO7 [M+NH4]+: 468.1784, C5H7NO3 [M+H]+
:130.0499; Found: 468.1774, 130.0490 respectively. IR (KBr, cm-1): 3257, 2986, 2927, 1750, 1648,
1513, 1476, 1371, 1264, 1239, 1223, 1206, 1088, 1061, 821
13C NMR
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Zhang H, Liu J, Zhu X, Li X, Chen H, Wu M, et al. (May 2020). “A Phase I Study on the Pharmacokinetics and Pharmacodynamics of DJT1116PG, a Novel Selective Inhibitor of Sodium-glucose Cotransporter Type 2, in Healthy Individuals at Steady State”. Clinical Therapeutics. 42 (5): 892–905.e3. doi:10.1016/j.clinthera.2020.03.007. PMID 32265061.
- Zhang H, Zhu X, Li X, Chen H, Wu M, Li C, et al. (February 2020). “Pharmacokinetics and pharmacodynamics of rongliflozin, a novel selective inhibitor of sodium-glucose co-transporter-2, in people with type 2 diabetes mellitus”. Diabetes, Obesity & Metabolism. 22 (2): 191–202. doi:10.1111/dom.13887. PMID 31588657.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2035989-50-3 |
| PubChem CID | 122660464 |
| UNII | 6FP3NST6ZQ |
| ChEMBL | ChEMBL5314927 |
| Chemical and physical data | |
| Formula | C23H27ClO7 |
| Molar mass | 450.91 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Rongliflozin, diabetes, Olorigliflozin, 6FP3NST6ZQ, 2035989-50-3, DJT1116PG, DJT 1116PG,
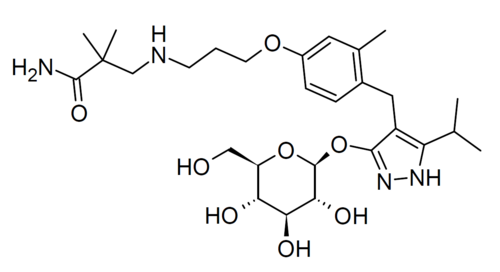
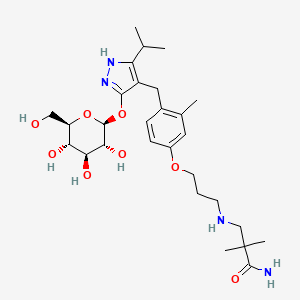
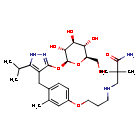
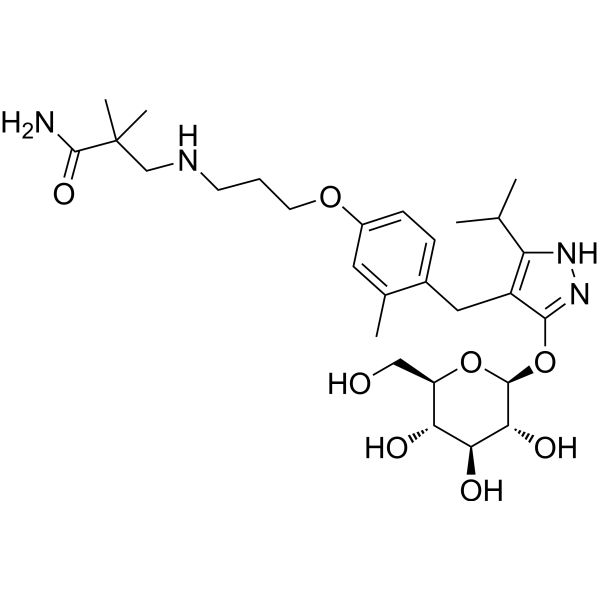
Mizagliflozin
Mizagliflozin
- CAS 666843-10-3
- 1X96A704XV
- DSP-3235
- KGA-3235
WeightAverage: 564.68
Monoisotopic: 564.315914393
Chemical FormulaC28H44N4O8
- Dsp-3235 free base
- GSK-1614235 free base
- Kga-3235 free base
2,2-dimethyl-3-[3-[3-methyl-4-[[5-propan-2-yl-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-1H-pyrazol-4-yl]methyl]phenoxy]propylamino]propanamide
- 3-((3-(4-((3-(beta-D-Glucopyranosyloxy)-5-(propan-2-yl)-1H-pyrazol-4-yl)methyl)-3-methylphenoxy)propyl)amino)-2,2-dimethylpropanamide
- Propanamide, 3-((3-(4-((3-(beta-D-glucopyranosyloxy)-5-(1-methylethyl)-1H-pyrazol-4-yl)methyl)-3-methylphenoxy)propyl)amino)-2,2-dimethyl-
Mizagliflozin is an SGLT1 inhibitor developed as a potential treatment for chronic constipation.[1][2] It progressed as far as Phase II trials in humans but was not approved for medical use, however it has since been investigated for other applications.[3][4]
MIZAGLIFLOZIN is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Mizagliflozin is under investigation in clinical trial NCT05721729 (Effect of Mizagliflozin Repeat Dosing on Adverse Events and Postprandial Glucose Excursions).
an SGLT1 inhibitor; structure in first source
- OriginatorKissei Pharmaceutical
- DeveloperKissei Pharmaceutical; Vogenx
- ClassAmides; Antihypoglycaemics; Laxatives; Pyrazoles; Small molecules
- Mechanism of ActionSodium-glucose transporter 1 inhibitors
- Phase IIHypoglycaemia
- Phase IGastroparesis
- PreclinicalUnspecified
- DiscontinuedConstipation
- 18 Jun 2025Phase-I clinical trials in Gastroparesis in USA (PO) (Vogenx pipeline, June 2025)
- 18 Jun 2025Preclinical trials in Undisclosed rare disease in USA (PO) (Vogenx pipeline, June 2025)
- 01 Oct 2019Chemical structure information added
LIT
- Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereofPublication Number: US-7635684-B2Priority Date: 2002-08-23Grant Date: 2009-12-22
- Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereofPublication Number: US-8324176-B2Priority Date: 2002-08-23Grant Date: 2012-12-04
- Monosebacate of pyrazole derivativePublication Number: US-2010279962-A1Priority Date: 2007-12-27
- Monosebacate of pyrazole derivativePublication Number: US-8399418-B2Priority Date: 2007-12-27Grant Date: 2013-03-19
- Monosebacate of pyrazole derivativePublication Number: WO-2009084531-A1Priority Date: 2007-12-27
- Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereofPublication Number: US-2005272669-A1Priority Date: 2002-08-23
- Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereofPublication Number: US-2009203633-A1Priority Date: 2002-08-23
- Hemifumarate of a pyrazole derivativePublication Number: WO-2009128421-A1Priority Date: 2008-04-16
- Monosebacate of pyrazole derivativePublication Number: AU-2008344436-B2Priority Date: 2007-12-27Grant Date: 2013-08-29
- Monosebacate of pyrazole derivativePublication Number: EP-2228378-A1Priority Date: 2007-12-27
- Monosebacate salt of pyrazole derivativePublication Number: JP-5144683-B2Priority Date: 2007-12-27Grant Date: 2013-02-13
- Monosebacate salt of pyrazole derivativePublication Number: JP-WO2009084531-A1Priority Date: 2007-12-27
- Hemifumarate of a pyrazole derivativePublication Number: EP-2275430-B1Priority Date: 2008-04-16Grant Date: 2012-05-16
- 1/2 fumarate salt of pyrazole derivativePublication Number: JP-5467040-B2Priority Date: 2008-04-16Grant Date: 2014-04-09
- 1/2 fumarate salt of pyrazole derivativePublication Number: JP-WO2009128421-A1Priority Date: 2008-04-16
- Hemifumarate of a pyrazole derivativePublication Number: US-2011034679-A1Priority Date: 2008-04-16
- Hemifumarate of a pyrazole derivativePublication Number: US-8354382-B2Priority Date: 2008-04-16Grant Date: 2013-01-15
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 666843-10-3 |
| PubChem CID | 10460535 |
| ChemSpider | 8635948 |
| UNII | 1X96A704XV |
| ChEMBL | ChEMBL5314923 |
| Chemical and physical data | |
| Formula | C28H44N4O8 |
| Molar mass | 564.680 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Inoue T, Takemura M, Fushimi N, Fujimori Y, Onozato T, Kurooka T, et al. (July 2017). “Mizagliflozin, a novel selective SGLT1 inhibitor, exhibits potential in the amelioration of chronic constipation”. European Journal of Pharmacology. 806: 25–31. doi:10.1016/j.ejphar.2017.04.010. PMID 28410751.
- Fukudo S, Endo Y, Hongo M, Nakajima A, Abe T, Kobayashi H, et al. (September 2018). “Safety and efficacy of the sodium-glucose cotransporter 1 inhibitor mizagliflozin for functional constipation: a randomised, placebo-controlled, double-blind phase 2 trial”. The Lancet. Gastroenterology & Hepatology. 3 (9): 603–613. doi:10.1016/S2468-1253(18)30165-1. PMID 30056028.
- Ishida N, Saito M, Sato S, Tezuka Y, Sanbe A, Taira E, et al. (October 2021). “Mizagliflozin, a selective SGLT1 inhibitor, improves vascular cognitive impairment in a mouse model of small vessel disease”. Pharmacology Research & Perspectives. 9 (5): e00869. doi:10.1002/prp2.869. PMC 8480397. PMID 34586752.
- Tsunokake S, Iwabuchi E, Miki Y, Kanai A, Onodera Y, Sasano H, et al. (October 2023). “SGLT1 as an adverse prognostic factor in invasive ductal carcinoma of the breast”. Breast Cancer Research and Treatment. 201 (3): 499–513. doi:10.1007/s10549-023-07024-9. PMID 37439959.
- [1]. Inoue T, et al. Mizagliflozin, a novel selective SGLT1 inhibitor, exhibits potential in the amelioration of chronic constipation. Eur J Pharmacol. 2017 Jul 5;806:25-31. [Content Brief][2]. Ohno H, et al. Absorption, disposition, metabolism and excretion of [14C]mizagliflozin, a novel selective SGLT1 inhibitor, in rats. Xenobiotica. 2019 Apr;49(4):463-473. [Content Brief]
/////////666843-10-3, 1X96A704XV, DSP 3235, KGA 3235, Mizagliflozin, Dsp-3235 free base, GSK-1614235 free base, Kga-3235 free base
Ebselen
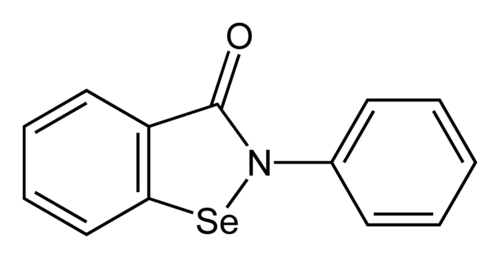
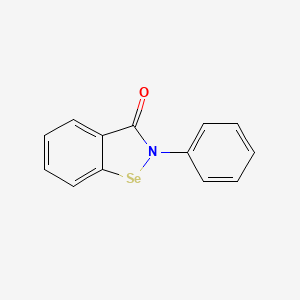
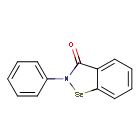
Ebselen
274.19 g/mol,
C13H9NOSe
2-phenyl-1,2-benzoselenazol-3-one
- CAS 60940-34-3
- 2-phenyl-1,2-benzoselenazol-3-one
- 2-Phenyl-1,2-benzisoselenazol-3(2H)-one
- Ebselene
- PZ 51, DR3305, and SPI-1005
- 40X2P7DPGH
Ebselen is a benzoselenazole that is 1,2-benzoselenazol-3-one carrying an additional phenyl substituent at position 2. Acts as a mimic of glutathione peroxidase. It has a role as a neuroprotective agent, an apoptosis inducer, an anti-inflammatory drug, an antioxidant, a hepatoprotective agent, a genotoxin, a radical scavenger, an enzyme mimic, an EC 1.3.1.8 [acyl-CoA dehydrogenase (NADP(+))] inhibitor, an EC 1.8.1.12 (trypanothione-disulfide reductase) inhibitor, an EC 1.13.11.33 (arachidonate 15-lipoxygenase) inhibitor, an EC 1.13.11.34 (arachidonate 5-lipoxygenase) inhibitor, an EC 2.5.1.7 (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) inhibitor, an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor, an EC 3.5.4.1 (cytosine deaminase) inhibitor, an EC 5.1.3.2 (UDP-glucose 4-epimerase) inhibitor, a ferroptosis inhibitor, an antifungal agent, an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor, an anticoronaviral agent, an antibacterial agent, an antineoplastic agent and an EC 3.1.3.25 (inositol–phosphate phosphatase) inhibitor.
Ebselen (also called PZ 51, DR3305, and SPI-1005), is a synthetic organoselenium molecule under preliminary investigation as a drug candidate.[1] It belongs to the class of compounds related to benzene and its derivatives.[1] It is being developed by the Seattle biotechnology company, Sound Pharmaceuticals, Inc.[1] It has also been reported to target tubulin, blocking its polymerization.[2]
Ebselen has been investigated for the treatment and basic science of Meniere’s Disease, Type 2 Diabetes Mellitus, and Type 1 Diabetes Mellitus.
Ebselen has been entered into clinical trials as a lead compound intended for the potential treatment of various diseases.[3] Its most advanced clinical trial is a Phase III study in people with Meniere’s disease, completed in July 2024.[4]
In vitro, ebselen is a mimic of glutathione peroxidase and reacts with peroxynitrite.[5] It is purported to have antioxidant and anti-inflammatory properties.[1][5]
Synthesis
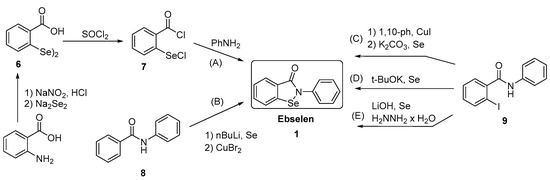
Generally, synthesis of the characteristic scaffold of ebselen, the benzoisoselenazolone ring system, can be achieved either through reaction of primary amines (RNH2) with 2-(chloroseleno)benzoyl chloride (Route I),[6] by ortho-lithiation of benzanilides followed by oxidative cyclization (Route II) mediated by cupric bromide (CuBr2),[7] or through the efficient Cu-catalyzed selenation / heterocyclization of o-halobenzamides, a methodology developed by Kumar et al.[8] (Route III).

SYN
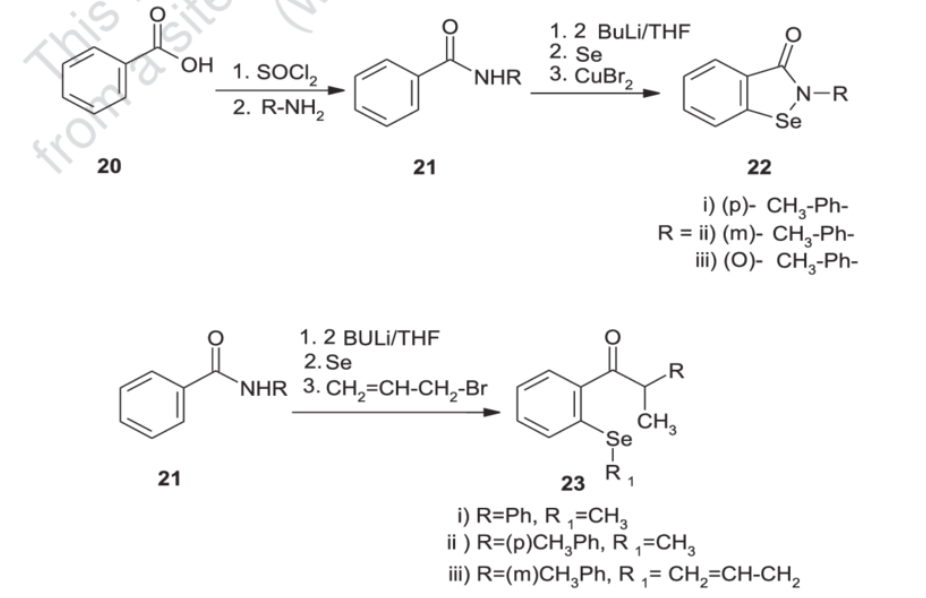
Synthesis of ebselen from benzoic acid by ortholithiation of benzanilide SOCl 2 =Thionyl chloride, R-NH 2 =Substituted aryl mine, BuLi/THF=n-butyllithium/ tetrahydrofuran, CuBr 2 =Cupper bromide, CH 2 =CH- CH 2 -Br = Allyl bromide.
SYN
New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential
Molecules, March 2017, 22(3):492
SYN
https://pubs.acs.org/doi/10.1021/ol102027j
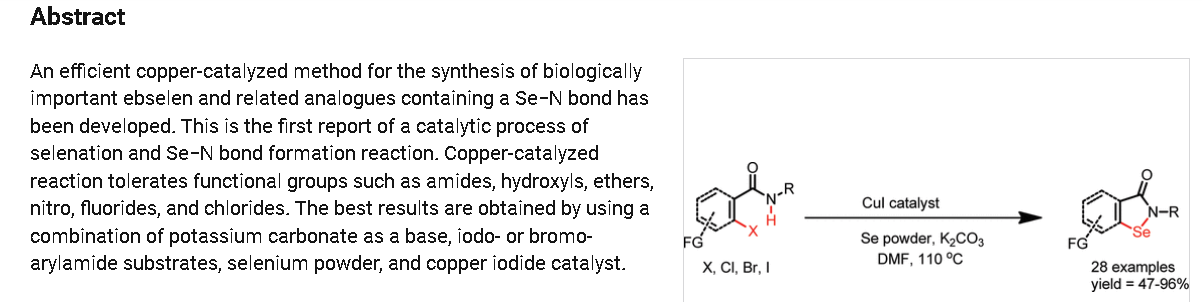

2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide (Typical
Procedure): Copper iodide (114 mg, 0.6 mmol) and 1,10-phenanthroline (108 mg, 0.6 mmol)
were added into DMF (3 mL) in a single neck flask. Resulted brownish solution was stirred for
15 min and then 2-iodo-N-phenylbenzamide1 (0.97 g, 3.0 mmol), selenium powder (0.29 g, 3.6
mmol), and potassium carbonate powder (0.65 g, 4.7 mmol) were added sequentially to same reaction flask. Brown colored reaction mixture was refluxed at 110oC using refluxing condenser
under nitrogen atmosphere. Progress of reaction was monitored by TLC. Reaction mixture was
refluxed for 8h. After this, reaction mixture poured over brine solution (60 mL) and stirred for 3
h. Product was precipitated as white solid which was collected by filtration over Buchner funnel,
product was washed with water (15 mL x 2), dried in air, dissolved in ethyl acetate, concentrated
over rotary evaporator, resulted brown solid which was purified by column chromatography
using hexane/ ethyl acetate (8:2) over silica gel. Yield 0.69 g (84%), mp 182-183 °C (180-181
°C).14,15 1H NMR (400 MHz, DMSO-d6) 8.09 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H),
7.71-7.62 (m, 3H), 7.51-7.43 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H). 1H NMR (400 MHz, CDCl3)
8.12 (d, 7.6 Hz, 1H), 7.68-7.62 (m, 4H), 7.52-7.41 (m, 3H), 7.29 (m, 1H). IR (plate): 3057, 2921,
1598, 1443, 1346, 1263, 1028 cm-1; ESMS m/z: 276 (M+H+).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide at 74 mmol
scale: Reaction was carried out at 74 mmol scale using 2-iodo-N-phenylbenzamide (24.00 g,
74.3 mmol), selenium powder (7.04 g, 89.1 mmol), CuI (2.83 g, 14.9 mmol), 1,10
phenanthroline (2.69 g, 14.9 mmol), and anhydrous potassium carbonate powder (15.40 g, 111.4
mmol) in DMF (50 mL) and procedure and workup followed are similar to 3.6 mmol scale
reaction. Yield 16.28 g (80%), Figure S1.
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Bromo-N-phenylbenzamide: Ebselen 1
was prepared from 2-bromo-N-phenylbenzamide2 (1.00 g, 3.6 mmol), selenium powder (0.34 g,
4.3 mmol), K2CO3 powder (0.74 g, 5.4 mmol), CuI (137 mg, 0.7 mmol), and 1,10-phenanthroline
(130 mg, 0.7 mmol) in DMF (3 mL). Reaction mixture was refluxed for 16 h at 110oC. Progress of reaction was monitored by TLC. After completion of reaction, mixture was poured into brine
solution (60 mL) and the resulted white precipitate was washed with water (20 mL x 2), and
dried in air. Purification by column chromatography on silica gel using CH2Cl2 provided white
crystalline solid (0.77 g, 78%).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Chloro-N-phenylbenzamide: Reaction
was carried out at 4 mmol scale using 2-chloro-N-phenylbenzamide3 (1.00 g, 4.3 mmol), CuI
(172 mg, 0.9 mmol), 1,10-phenanthroline (162 mg, 0.9 mmol), selenium powder (0.41 g, 5.2
mmol), K2CO3 (0.89 g, 6.4 mmol) in DMF (4 mL). Reaction mixture was refluxed for 24 h at
110oC. Workup procedure is similar as followed for bromo substrate. Yield 0.55 g (47%).
History
The first patent for 2-phenyl-1,2-benzoselenazol-3(2H)-one was filed in 1980 and granted in 1982.[9]
Research
Ebselen is in preliminary clinical development for the potential treatment of hearing loss and depression, among other medical indications.[3][10]
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Ebselen”. DrugBank. 29 January 2025. Retrieved 4 February 2025.
- Baksheeva VE, La Rocca R, Allegro D, Derviaux C, Pasquier E, Roche P, Morelli X, Devred F, Golovin AV, Tsvetkov PO (2025). “NanoDSF Screening for Anti-tubulin Agents Uncovers New Structure–Activity Insights”. Journal of Medicinal Chemistry. doi:10.1021/acs.jmedchem.5c01008.
- “Ebselen pipeline”. Sound Pharmaceuticals, Inc. 2025. Retrieved 4 February 2025.
- “SPI-1005 for the Treatment of Meniere’s Disease (STOPMD-3)”. ClinicalTrials.gov, US National Library of Medicine. 1 August 2024. Retrieved 4 February 2025.
- Schewe T (October 1995). “Molecular actions of ebselen – an antiinflammatory antioxidant”. General Pharmacology. 26 (6): 1153–69. doi:10.1016/0306-3623(95)00003-J. PMID 7590103.
- Kamigata N, Iizuka H, Izuoka A, Kobayashi M (July 1986). “Photochemical Reaction of 2-Aryl-1, 2-benzisoselenazol-3 (2 H)-ones”. Bulletin of the Chemical Society of Japan. 59 (7): 2179–83. doi:10.1246/bcsj.59.2179.
- Engman L, Hallberg A (1989-06-01). “Expedient synthesis of ebselen and related compounds”. The Journal of Organic Chemistry. 54 (12): 2964–2966. doi:10.1021/jo00273a035. ISSN 0022-3263.
- Balkrishna SJ, Bhakuni BS, Chopra D, Kumar S (December 2010). “Cu-catalyzed efficient synthetic methodology for ebselen and related Se-N heterocycles”. Organic Letters. 12 (23): 5394–7. doi:10.1021/ol102027j. PMID 21053969.
- DE3027073A1, Etschenberg, Eugen Dr; Renson, Marcel Prof Dipl-Chem Jupille & Winkelmann, Johannes Dr 5000 Köln, “2-phenyl-1,2-benzisoselenazol-3(2h)-on enthaltende pharmazeutische praeparate und ihre verwendung”, issued 1982-02-18
- “Ebselen search: list of clinical trials sponsored by Sound Pharmaceuticals”. ClinicalTrials.gov, US National Library of Medicine. 2025. Retrieved 4 February 2025.
External links
| Names | |
|---|---|
| Preferred IUPAC name2-Phenyl-1,2-benzoselenazol-3(2H)-one | |
| Identifiers | |
| CAS Number | 60940-34-3 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:77543 |
| ChEMBL | ChEMBL51085 |
| ChemSpider | 3082 |
| ECHA InfoCard | 100.132.190 |
| PubChem CID | 3194 |
| UNII | 40X2P7DPGH |
| CompTox Dashboard (EPA) | DTXSID7045150 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C13H9NOSe |
| Molar mass | 274.17666 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
////////Ebselen, Ebselene, PZ 51, DR 3305, SPI 1005, PHASE 3, 40X2P7DPGH, Meniere’s Disease, Type 2 Diabetes Mellitus, Type 1 Diabetes Mellitus
Myself, NIPER-G and NDTL Collaborate to Synthesize ‘Methandienone LTM’ for Global Anti-Doping Efforts
A proud moment for me [ ANTHONY MELVIN CRASTO ] as Scientific Advisor at Niper-G Dept Pharma Ministry of Chemicals and Fertilizers Govt of India 
Congrats team Niper-G and team National Dope Testing Lab Govt of India Prof. (Dr.) P. L. Sahu and Myself being a part of it as Scientific Advisor Niper-G for few years in Medicinal chem dept. Launched by Union minister Mansukh Mandaviya on 4th sept 2025 in Delhi
will be distributed across the world via World Anti-Doping agency WADA
Methandienone LTM is high purity rare reference material
Hope my interactions and guidance has given fruitful results. Thanks to Dr Murty for appointing me as advisor. Methandienone LTM will make India proud across the World 
A great achievement for India as nation and advanced capability demonstration
https://www.pib.gov.in/PressReleasePage.aspx?PRID=2163812
India Develops Rare Reference Material for Enhanced Anti-Doping Testing in Sports
NIPER Guwahati and NDTL Collaborate to Synthesize ‘Methandienone Long-Term Metabolite’ for Global Anti-Doping Efforts

//////////NIPER-G, NDTL, Methandienone LTM, Global Anti-Doping, INDIA
Sergliflozin Etabonate

Sergliflozin Etabonate
408504-26-7 Cas no
Ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2……….TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
- etabonate de sergliflozine
- etabonato de sergliflozina
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.

Sergliflozin
[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of …onlinelibrary.wiley.com974 × 740Search by imageDesign, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.

sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
PATENT
US6872706B2
https://patentscope.wipo.int/search/en/detail.jsf?docId=US40677423&_cid=P20-MF4ZUQ-42384-1
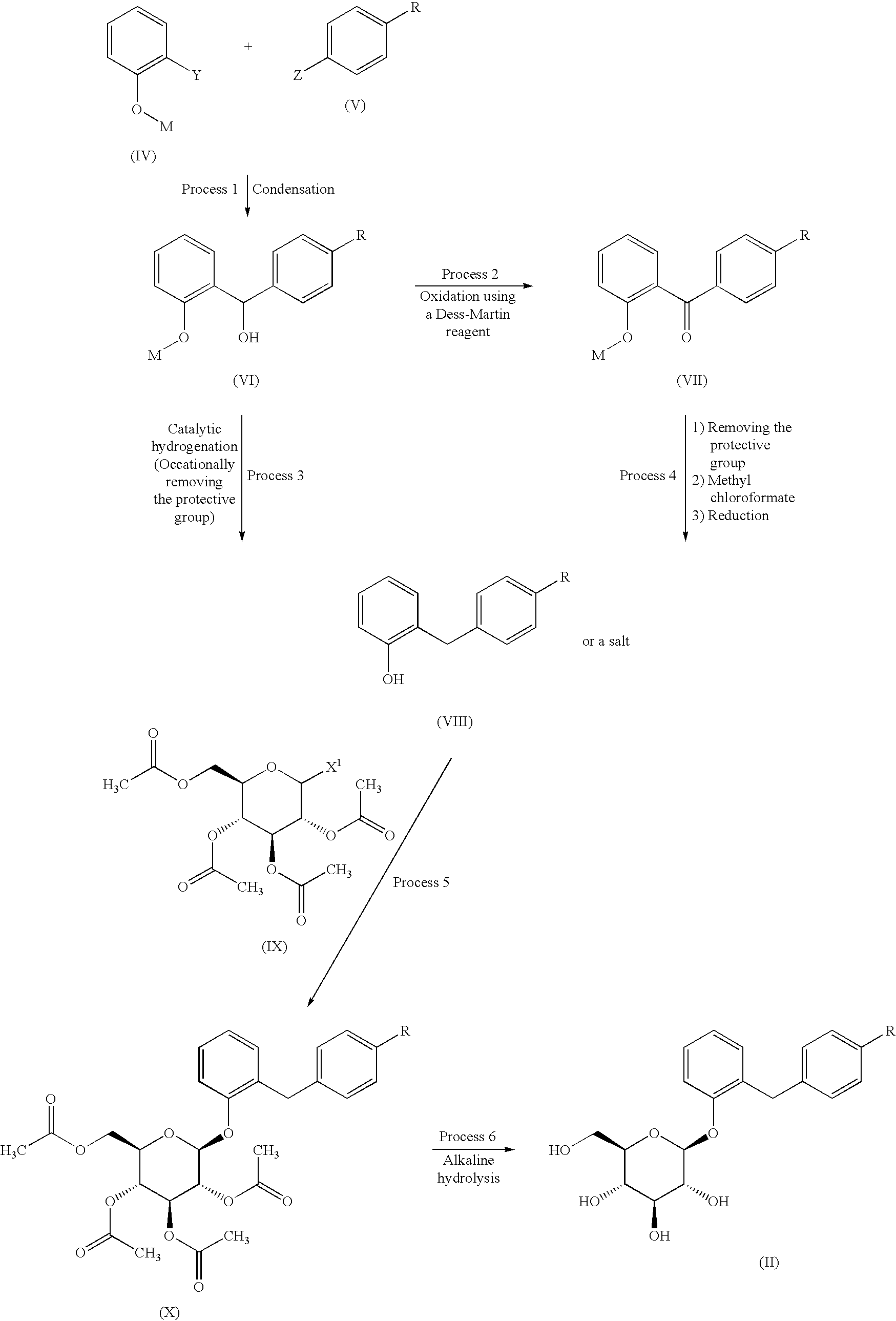
PATENT
WO2001068660A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001068660&_cid=P20-MF4ZXC-45172-1
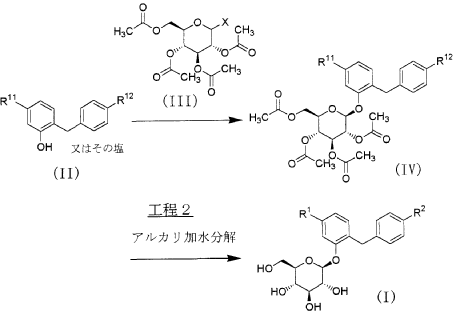
SYN
Heterocycles 2016, 92, 1599
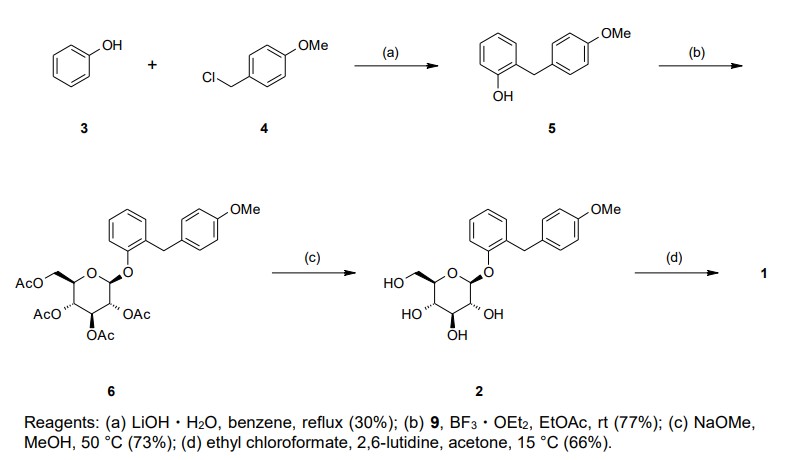
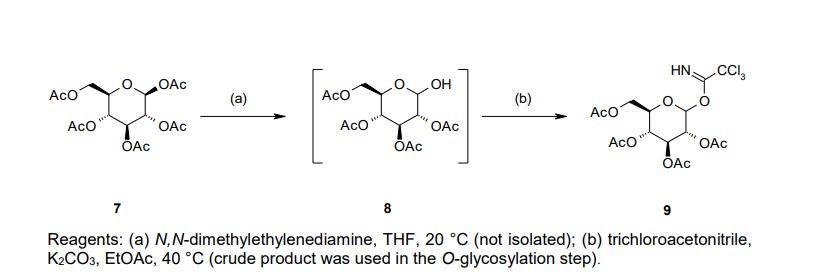
Our initial synthetic route of Serglifrozin etabonate (1) in early development consisted of six steps,
including synthesis of tetra-O-acetyl-D-glucopyranosyl trichloroacetimidate (9), as shown in Scheme 1
and Scheme 2 The first step is the coupling reaction of phenol (3) and 4-methoxybenzyl chloride (4) in the presence of
lithium hydroxide monohydrate (LiOH·H2O) to provide the aglycon 5 in a 30% yield following
chromatographic purification (Scheme 1). We prepared 9 separately by mono-deacetylation of
penta-O-acetyl-β-D-glucopyranose (7) with N,N-dimethylethylenediamine in THF followed by reaction of
the crude product of 8 with trichloroacetonitrile in the presence of potassium carbonate (K2CO3) in ethyl
acetate (EtOAc) (Scheme 2). Next, we carried out glycosylation of 5 with 9 in the presence of boron
trifluoride diethyl etherate (BF3·OEt2) in EtOAc to produce 6 in a 77% yield. The obtained 6 was
deacetylated with sodium methoxide (NaOMe) in MeOH to produce Serglifrozin (2) in a 73% yield, and
reaction of the isolated 2 with ethyl chloroformate in the presence of 2,6-lutidine in acetone provided 1 in
a 66% yield. The overall yield from 3 was 11%. While this route was capable of supplying small
amounts of 1, it suffered from several disadvantages.
The coupling reaction between 3 and 4 provided the aglycon 5 in low yield (30%); thus, chromatographic
purification was required to obtain highly pure 5. The trichloroimidation reaction of 8 is too hazardous
for large-scale manufacturing, because an excess amount of trichloroacetonitrile, a volatile and highly
toxic reagent, is required to obtain the trichloroacetimidate 9. Furthermore, 9 is too unstable to use
conveniently in large-scale manufacturing. Trichloroacetamide, a sublimation compound, is formed as a
by-product from the glycosylation of 5 with 9. Thus, the vacuum line and the vacuum pump of the
manufacturing equipment would be polluted by trichloroacetamide.
Because of these issues, this synthetic method is unsuitable for large-scale manufacturing. Therefore,
we investigated alternative processes for the preparation of 1, suitable for large-scale manufacturing. An improved synthetic method for 1 was achieved in a five-step procedure without purification of 6
(intermediate), as shown in Scheme 3.
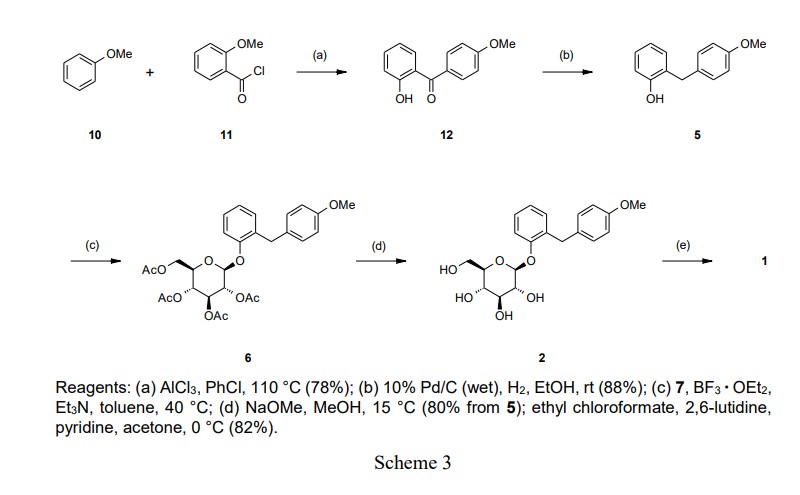
The Friedel-Crafts acylation of anisole (10) with 2-methoxybenzoyl chloride (11) in the presence of
aluminum chloride (AlCl3) at 110 °C provided benzophenone (12), which was selectively demethylated
on the methoxy group at the 2-position. The crude product of 12 was crystallized from MeOH to
provide highly pure 12 in a 78% yield. Hydrogenation of 12 in EtOH with 0.3–0.4 MPa H2 at room
temperature in the presence of 10% Pd/C provided 5. The crude product of 5 was crystallized from
toluene/n-heptane to provide highly pure 5 in an 88% yield.
The key step of the synthesis was the formation of the O-glycosylated product 6. In the initial synthesis,
it was necessary to isolate 6 to remove trichloroacetamide. Consequently, 2 was provided in a 56%
yield from 5. To obtain 6 efficiently without using the trichloroacetimidate (9), we evaluated several
conditions for the direct O-glycosylation of 5 with 7. The results are summarized in Table 1. The
O-glycosylation of 5 with 7 (200 mol%) in the presence of boron trifluoride diethyl etherate (BF3·OEt2;
100 mol%) in dichloromethane (DCM) at room temperature provided the crude product of 6 with a good
yield (80%) and β-selectivity (94/6), and then the deacetylation of the crude product of 6 in the presence
of sodium methoxide (NaOMe) in MeOH proceeded almost quantitatively to provide 2 in a 71% isolated
yield from 5 (run 1). Using this method, it was not necessary to isolate 6 because the excess amount of
7 was converted to glucose and removed to the aqueous layers in the deacetylation step. Use of DCM is
undesirable for large-scale manufacturing because quenching of O-glycosylation with water is highly
exothermic and washing of the DCM layer with water is a complicated procedure. Additionally, it is
strongly desirable to avoid using DCM in a manufacturing process due to environmental issues. For the reasons mentioned above, we attempted to use toluene as an alternative solvent. The O-glycosylation in
the presence of BF3·OEt2 (100 mol%) in toluene at 30 °C did not proceed completely, and the yield of 6
was lower than run 1 (run 2). We concluded that the lower solubility of 7 in toluene, compared with
DCM, caused the low yield. Because it was difficult to increase the amount of toluene from the
perspective of manufacturing efficiency, we tried to improve its solubility by optimizing the reagent
equivalent. Fortunately, we found that an excess amount of BF3·OEt2 enhanced the solubility of 7 in
toluene, and using 300 mol% of BF3·OEt2 in toluene provided 6 in a good yield (80%), similar to that
when using DCM (run 3). In contrast, reducing the amount of 7 provided 6 in an insufficient yield, and
2 was consequently provided in a lower yield (60%) (run 4). To achieve higher β-selectivity and an
increased yield, triethylamine (Et3N) was added to the O-glycosylation of 5 with 7 in the presence of
BF3·OEt2, according to the method of Lee et al.
9 Addition of Et3N (30 mol%) at 30 °C resulted in both
higher yield (89%) and higher β-selectivity (97/3) to provide 2 with a 79% isolated yield (run 5).
Increasing the amount of Et3N to 60 mol% at 30 °C resulted in a lower yield (85%) of 6 compared with
run 5, and the yield of 2 decreased (74%) (run 6). Increasing the reaction temperature to 40 °C in the
presence of 60 mol% of Et3N achieved the best results for both high yield (90%) and high β-selectivity
(99/1) to provide 2 in an 80% yield (run 7).
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (407 mg, 3.75 mol) was added drop-wise to the mixture of 2 (1.13 g, 3.0 mmol) and
2,6-lutidine (563 mg, 5.25 mmol) in acetone (4 mL) while maintaining the temperature between 12 and
18 °C. The reaction mixture was stirred at 15 °C for 23 h. Water (5 mL) was added drop-wise while
maintaining the temperature below 30 °C, and EtOAc (10 mL) was then added to the mixture. The
biphasic solution was transferred to a separating funnel for phase separation. The aqueous layer was
extracted with EtOAc (5 mL). The EtOAc layers were combined, washed successively with an aqueous
solution of 10% citric acid (5 mL × 2), an aqueous solution of 10% NaCl (5 mL), an aqueous solution of
5% NaHCO3 (5 mL × 2), and an aqueous solution of 10% NaCl (5 mL). They were then dried over
Na2SO4 and the filtrate was concentrated under reduced pressure. EtOH was added to the residue, and
the weight was adjusted to 7.2 g. The mixture was heated to 65 °C to dissolve solids. The solution was
cooled to 55 °C and seeded with 1. The solution was aged for 1 h at 50 °C, during which time the
product began to crystallize. After the slurry was cooled to 25 °C, n-heptane (11 mL) was added
drop-wise to the slurry at 25 °C followed by stirring for 1 h at 25 °C. The slurry was cooled to 3 °C and
then stirred for 2 h at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent
of EtOH (1.5 mL) and n-heptane (3 mL). The precipitate was dried in vacuo at 70 °C to give 888 mg
(66% yield) of 1 as a white solid. [α]
20
D -43.5 (c 1.0, DMSO). IR (KBr) cm-1
: 3495, 1744, 1514, 1488,
1454, 1467, 1411, 1372, 1340, 1266. 1H-NMR (CDCl3) δ: 1.27 (3H, t, J=7.0 Hz), 2.00 (1H, d, J=1.6
Hz), 3.46–3.54 (3H, m), 3.56–3.61 (2H, m), 3.72 (1H, d, J=2.1 Hz), 3.75 (3H, s), 3.82 (1H, d, J=15.5 Hz),
4.03 (1H, d, J=15.5 Hz), 4.11–4.22 (2H, m), 4.42 (2H, d, J=3.8 Hz), 4.69 (1H, d, J=7.4 Hz), 6.79–6.83
(2H, m), 6.97–7.02 (2H, m), 7.04–7.07 (2H, m), 7.15–7.22 (2H, m). 13C-NMR (CDCl3) δ: 14.2 (q), 36.1
(t), 55.4 (q), 64.4 (t), 66.4 (t), 69.6 (d), 73.4 (d), 73.8 (d), 75.7 (d), 100.8 (d), 114.1 (d×2), 114.4 (d), 122.7
(d), 128.0 (d), 129.2 (d×2), 130.0 (s), 131.1 (d), 133.4 (s), 155.2 (s), 155.4 (s), 157.8 (s). HRMS (ESI)
m/z: 466.2070 [M+NH4]
+
(Calcd for C23H32NO9: 466.2072)
6-O-Ethoxycarbonyl-2-[(4-methoxyphenyl)methyl]phenyl-β-D-glucopyranoside (1). Ethyl
chloroformate (21.6 g, 0.199 mol) was added drop-wise to the mixture of 2 (65.0 g, 0.173 mol),
2,6-lutidine (27.8 g, 0.259 mol) and pyridine (0.33 g, 4.2 mmol) in acetone (210 mL), maintaining the
temperature between -1 and 5 °C. The reaction mixture was stirred at 0 °C for 2 h. The reaction was
monitored by HPLC.15 Water (200 mL) was added drop-wise, maintaining the temperature below 30 °C,
and then EtOAc (220 mL) was added to the mixture. The biphasic solution was transferred to a
separating funnel for phase separation. The aqueous layer was extracted with EtOAc (140 mL). The
EtOAc layers were combined, washed successively with an aqueous solution of 10% citric acid (180 mL
× 2), an aqueous solution of 10% NaCl (66 g), an aqueous solution of 5% NaHCO3 (65 g × 2), and an aqueous solution of 10% NaCl (100 g), and then dried over Na2SO4 (65 g). After acetic acid (10 g,
0.167 mol) was added to the filtrate, the mixture was concentrated under reduced pressure. The residue
was dissolved in EtOH (660 mL) at 65 °C. The solution was concentrated under reduced pressure until
more than 330 mL distillate had been collected. EtOH was added to the residue, and the weight was
adjusted to 370 g. n-Heptane (120 mL) was added, and the resulting slurry was heated to 65 °C to
dissolve solids. The solution was cooled to 55 °C and seeded with 1. The solution was aged for 1 h at
50 °C, during which time the product began to crystallize. n-Heptane (480 mL) was added drop-wise to
the slurry, maintaining the temperature between 50 and 60 °C, and the slurry was stirred for 0.5 h at 55 °C.
The slurry was allowed to cool slowly over 2.5 h to 30 °C, then cooled to 3 °C, and then stirred for 1.5 h
at 3 °C. The slurry was filtered, and the wet cake was washed with a mixed solvent of EtOH (80 mL)
and n-heptane (180 mL). The precipitate was dried in vacuo at 70 °C to give 63.6 g (82% yield) of 1 as
a white solid.
REFERENCES (AND NOTES)
- W. N. Washburn, Expert Opin. Ther. Patents, 2009, 19, 1485.
- A. M. Pajor and E. M. Wright, J. Biol. Chem., 1992, 267, 3557.
- E. M. Wright, Am. J. Physiol. Renal Physiol., 2001, 280, F10.
- Y. Kanai, W. S. Lee, G. You, D. Brown, and M. A. Hediger, J. Clin. Invest., 1994, 93, 397.
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, and M. Isaji, PCT, WO 02/28872 (2002).
- H. Fujikura, N. Fushimi, T. Nishimura, K. Tatani, K. Katsuno, M. Hiratochi, Y. Tokutake, and M.
Isaji, PCT, WO 01/688660 (2001). - K. Katsuno, Y. Fujimori, Y. Takemura, M. Hiratochi, F. Itoh, Y. Komatsu, H. Fujikura, and M. Isaji,
J. Pharmacol. Exp. Ther., 2007, 320, 323. - M. Isaji, Curr. Opin. Investig. Drugs, 2007, 8, 285.
- S. Y. Lee, S. E. Rho, K. Y. Min, T. B. Kim, and H. K. Kim, J. Carbohydr. Chem., 2001, 20, 503.
- M. Yamaguchi, A. Horiguchi, A. Fukuda, and T. Minami, J. Chem. Soc., Perkin Trans. 1, 1990,
1079. - K. Ishihara, H. Kurihara, and H. Yamamoto, J. Org. Chem., 1993, 58, 3791.
- I. T. Akimova, A. V. Kaminsky, and V. I. Svistunova, Chem. Heterocycl. Compd., 2005, 41, 1374.
- B. N. Cook, S. Bhakta, T. Biegel, K. G. Bowman, J. I. Armstrong, S. Hemmerich, and C. R. Bertozzi,
J. Am. Chem. Soc., 2000, 122, 8612. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, isocratic elution with acetonitrile / 0.02 M KH2PO4, pH 3 = 6/4; flow rate, 1.0 mL/min;
column oven temperature, 40 °C; wave length, 225 nm; retention times, 5 = 16 min, α-anomer of 5 =18 min. - HPLC conditions: column, Inertsil ODS-3 (5 µm) 4.6 mm × 250 mm (GL Science Inc.); mobile
phase, gradient elution with 5 min 4/6 → 15 min 6/4 → 30 min 6/4 of acetonitrile/0.02 M KH2PO4,
pH 3; flow rate, 1.0 mL/min; column oven temperature, 40 °C; wavelength, 225 nm; retention times,
1 = 17 min, 2,6- and 4,6-bis-O-ethoxycarbonyl derivatives = 24 min, 3,6-bis-O-ethoxycarbonyl
derivative = 25 min.
SYN
Synthesis 2024, 56, 906–943
Sergliflozin etabonate (16), also known as GW869682X, was developed collaboratively by GlaxoSmithKline and Kissei Pharmaceutical (Japan). Unfortunately, it did not pass phase III trials. It belongs to the class of sodium–glucose linked transporter 2 (SGLT2) inhibitors and acts as a prodrug of sergliflozin, with the ethyl carbonate group referred to as etabonate. When compared to phlorizin, sergliflozin etabonate demonstrated significantly higher activity against SGLT2 than SGLT1. The initial synthetic route for the preparation of sergliflozin was described and patented by Kissei Pharmaceutical Co., Ltd. This particular route for Oaryl-glycoside-type derivatives was registered in the United States under patent application number US6872706B2.73 The first reported synthesis of sergliflozin etabonate
(16), which involves six steps, can be found in the patents US6872706B2 73a and WO2001068660A1 (Scheme 48).73b Compound 271 was prepared in a high yield of 96% follow ing a literature procedure. The selective monodeacetylationof penta-O-acetyl-b-D-glucopyranose, compound 269, was
achieved using N,N-dimethylethylenediamine in THF, resulting in the formation of compound 270. Subsequently, a reaction with trichloroacetonitrile and potassium carbonate led to the synthesis of intermediate 271 in excellent yield. To prepare the aglycone intermediate 268, phenol (235) was condensed with 4-methoxybenzyl chloride (267) using LiOH under reflux conditions. Further,O-glycosyla
tion of compound 268 with 271 was accomplished using boron trifluoride–diethyl etherate (BF3·OEt2), yielding intermediate 272. Removal of the acetyl groups from intermediate 272 was carried out using NaOMe in methanol to obtain sergliflozin (16a) in a yield of 73%. Finally, sergliflozin etabonate (16) was obtained by reacting compound 16a with ethyl chloroformate and 2,6-lutidine, resulting in a yield of
66%. The overall yield of sergliflozin etabonate (16a) was calculated to be 11%. It is important to note that the trichloroimidation reaction used in the synthesis of trichloroacetimidate 271 is considered hazardous and is not recommended for commercial use due to the highly toxic reagent, trichloroaceto
nitrile. Additionally, the process poses challenges in effectively removing the unwanted by-product, trichloroacetamide, formed during the preparation.A recently published approach presents an alternative synthesis of sergliflozin etabonate (16) that avoids the use of a trichloroacetimidate intermediate (Scheme 49).74a The five-step synthesis of compound 16a commenced from
readily available anisole (273a). An efficient Friedel–Crafts reaction was performed on anisole (273a) using the acid chloride 273 in the presence of aluminum chloride in chlorobenzene, leading to formation of benzophenone 274. Notably, demethylation of 274 was also observed under these
conditions. Next, ketone group reduction was achieved us ing 10% Pd/C and ethanol under 0.3–0.4 MPa of H2, providing compound 268 in 88% yield and high purity. Subsequently, O-glycosylation of 268 with penta-acetylated com pound 269 was carried out using BF3·Et2O and triethylamine, resulting in the formation of 272 in 90% yield with a high b-selectivity (99:1).74b Deacetylation of compound 272 was performed using NaOMe in methanol, affording sergliflozin (16a) in 80% yield. Further reaction with
ethyl chloroformate in the presence of 2,6-lutidine resulted in sergliflozin etabonate (16). The overall yield of compound 16 was calculated to be 41%. This novel synthetic route offers a promising alternative to the traditional method and demonstrates improved efficiency in the preparation of sergliflozin etabonate (16)
(73) (a) Fujikura, H.; Fushimi, N. US6872706B2, 2005. (b) Fujikura, H.; Fushimi, N.; Nishimura, T.; Tatani, K.; Katsuno, K.; Hiratochi, M.; Tokutake, Y.; Isaji, M. WO2001068660A1, 2001.
(74) (a) Kobayashi, M.; Isawa, H.; Sonehara, J.; Kubota, M. Heterocycles 2016, 92, 1599. (b) Lee, Y. S.; Rho, S. E.; Min, K. Y.; Kim, T. B.; Kim, H. K. J. Carbohydr. Chem. 2001, 20, 503.
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
| Systematic (IUPAC) name | |
|---|---|
| 2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside | |
| Clinical data | |
| Routes of administration | Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
References
- World Health Organization (2008). “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). WHO Drug Information. 22 (1): 66. Archived from the original (PDF) on February 19, 2009.
- “Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate” (PDF). American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”. J Pharmacol Exp Ther. 320 (1): 323–30. doi:10.1124/jpet.106.110296. PMID 17050778. S2CID 8306408.
- “Prous Science: Molecule of the Month November 2007”. Archived from the original on 2007-11-05. Retrieved 2008-10-28.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////// etabonate, Sergliflozin etabonate, Sergliflozin, PHASE 3, GW869682X, GSK, KISSEI, GW-869682; GW-869682X; KGT-1251
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}