
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 13)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
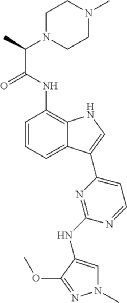
Golidocitinib
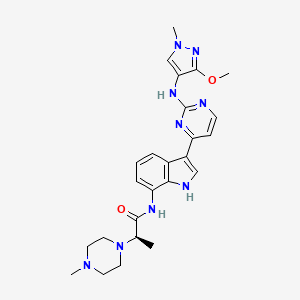
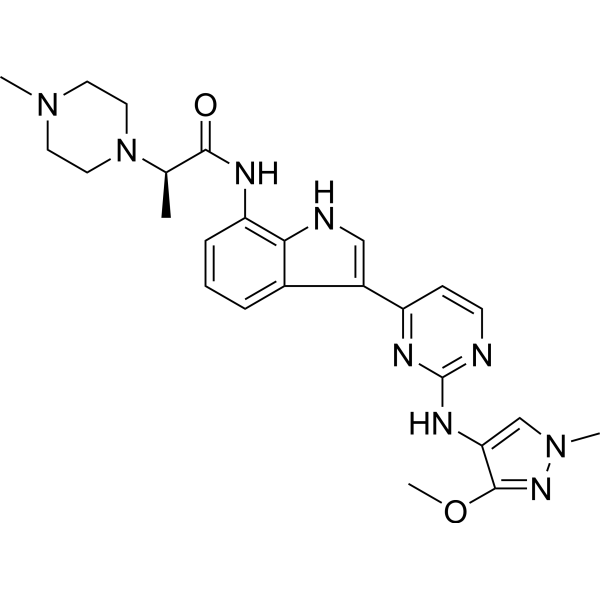
Golidocitinib
CAS 2091134-68-6
- AZD-4205
- AZD4205
- UNII-3BY9Z3M34G
- 3BY9Z3M34G
WeightAverage: 489.584
Monoisotopic: 489.260071274
Chemical FormulaC25H31N9O2
(2R)-N-[3-[2-[(3-methoxy-1-methylpyrazol-4-yl)amino]pyrimidin-4-yl]-1H-indol-7-yl]-2-(4-methylpiperazin-1-yl)propanamide
- (2R)-N-(3-(2-((3-methoxy-1-methylpyrazol-4-yl)amino)pyrimidin-4-yl)-1H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide
- (ALPHAR)-N-(3-(2-((3-METHOXY-1-METHYL-1H-PYRAZOL-4-YL)AMINO)-4-PYRIMIDINYL)-1H-INDOL-7-YL)-ALPHA,4-DIMETHYL-1-PIPERAZINEACETAMIDE
- (2R)-N-[3-[2-[(3-Methoxy-1-methyl-pyrazol-4-yl)amino]pyrimidin-4-yl]-1H-indol-7-yl]-2-(4-methylpiperazin-1-yl)propenamide
- (R)-N-(3-(2-(3-Methoxy-1-methyl-1H-pyrazol-4-ylamino)pyrimidin-4-yl)-1H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide
Approvals 2024, china 2024, DZD 4205, DIZAL, Gao Ruizhe,
Golidocitinib is a pharmaceutical drug for the treatment of cancer. In June 2024, it was given conditional approval in China for the treatment of relapsed or refractory peripheral T-cell lymphoma.[1]
Golidocitinib is classified as a Janus kinase inhibitor.[2][3]
Golidocitinib is an orally available inhibitor of Janus-associated kinase 1 (JAK1), with potential antineoplastic activity. Upon oral administration, golidocitinib inhibits JAK-dependent signaling and may lead to an inhibition of cellular proliferation in JAK1-overexpressing tumor cells. The JAK-STAT (signal transducer and activator of transcription) signaling pathway is a major mediator of cytokine activity and is often dysregulated in a variety of tumor cell types. Additionally, JAK1 may be a primary driver of STAT3 phosphorylation and signaling, which plays a role in neoplastic transformation, resistance to apoptosis, tumor angiogenesis, metastasis, immune evasion, and treatment resistance.
GOLIDOCITINIB is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 4 investigational indications.
PAT
US9714236, https://patentscope.wipo.int/search/en/detail.jsf?docId=US193702885&_cid=P11-MEHX78-54823-1
Example 32: (2R)—N-(3-{2-[(3-Methoxy-1-methyl-1H-pyrazol-4-yl)amino]pyrimidin-4-yl}-1H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide
| The procedure described above for Example 32 was repeated using the indicated Intermediates to give Examples 33-42 described in Table 12: |
[TABLE-US-00012]
| TABLE 12 Starting m/z ExampleIntermediatesNMR δ (400 MHz)[M + H]+Yield % 3325 and 38DMSO-d6 with D2O 1.28 (3H, d), 2.2750413 (3H, s), 2.73 (3H, s), 2.85-3.34 (8H, m), 3.44 (1H, q), 3.63 (3H, s), 374 (3H, s), 7.04 (1H, t), 7.19 (1H, d), 7.55 (1H, s), 7.91 (1H, s), 8.08 (2H, s), 8.26 (1H, s) -two exchangeable protons not observed3425 and 37DMSO-d6 1.26 (3H, d), 2.16 (3H, s),50472 2.33 (3H, s), 2.38 (4H, s), 2.57-2.62 (4H, m), 3.33 (1H, q), 3.67 (3H, s), 3.79 (3H, s), 7.00 (1H, t), 7.41 (1H, d), 7.66 (1H, s), 7.96 (2H, t), 8.14 (1H, s), 8.22 (1H, s), 9.65 (1H, s), 11.28 (1H, s)3530 and 37Methanol-d4 1.34 (3H, t), 1.40 (3H, d),51816 2.32 (3H, s), 2.37 (3H, s), 2.50-2.80 (8H, m), 3.38 (1H, q), 3.69 (3H, s), 4.34 (2H, q), 7.05-7.20 (2H, m), 7.69 (1H, s), 7.85 (1H, s), 8.23 (1H, s), 8.17 (1H, d)-three exchangeable protons not observed3626 and 37DMSO-d6 1.26 (3H, d), 2.27 (3H, s),52448 2.24-2.52 (4H, m), 2.53-2.70 (4H, m), 3.30-3.36 (1H, m), 3.69 (3H, s), 3.78 (3H, s), 7.02 (1H, s), 7.40 (1H, d), 7.65 (1H, s), 8.32 (1H, s), 8.48 (1H, s), 9.69 (1H, s), 11.42 (1H, s)3727 and 37DMSO-d6 1.26 (3H, d), 2.17 (3H, s),56849 2.23-2.45 (4H, m), 2.46-2.71 (4H, m), 3.30-3.32 (1H, m), 3.68 (3H, s), 3.78 (3H, s), 7.01 (1H, s), 7.37 (1H, d), 7.64 (1H, s), 8.42 (1H, s), 8.45-8.56 (2H, m), 9.70 (1H, s), 11.36 (1H, s)3825 and 39Chloroform-d 1.19 (3H, d), 1.35 (3H, d),51819 2.10 (1H, m), 2.26 (1H, m), 2.38 (6H, m), 2.69 (2H, t), 2.89 (3H, m), 3.72 (3H, s), 3.91 (1H, q), 4.00 (3H, s), 6.57 (1H, s), 6.80 (1H, d), 7.15 (1H, t), 7.68 (1H, d), 7.84 (1H, s), 8.06-8.36 (2H, m), 9.88 (1H, s), 11.15 (1H, s)3929 and 37Methanol-d4 1.34 (3H, t), 1.43 (3H, d),52225 2.35 (3H, s), 2.50-2.85 (8H, m), 3.41 (1H, q), 3.79 (3H, s), 4.24 (2H, q), 7.10- 7.22 (2H, m), 7.68 (1H, s), 8.13 (1H, d), 8.16 (1H, d), 8.43 (1H, s)-three exchangeable protons not observed4031 and 37Methanol-d4 1.33 (3H, t), 1.42 (3H, d),53822 2.35 (3H, s), 2.63-2.71 (4H, m), 2.77- 2.81 (4H, m), 3.42 (1H, q), 3.76 (3H, s), 4.26 (2H, q), 7.10-7.20 (2H, m), 7.70 (1H, s), 8.28 (2H, m), 8.48 (1H, m)-three exchangeable protons not observed4128 and 37Chloroform-d 1.41 (3H, d), 2.29 (3H, s),48836 2.36 (3H, s), 2.42 (3H, s), 2.67-2.80 (8H, m), 3.38 (1H, q), 3.80 (3H, s), 6.42 (1H, s), 6.82 (1H, d), 7.12 (1H, t), 7.69 (1H, d), 7.88 (1H, s), 8.21 (2H, m), 9.74 (1H, s), 11.18 (1H, s)4228 and 38DMSO-d6 1.27 (3H, d), 2.12 (3H, s),4884 2.17 (3H, s), 2.35 (3H, s), 2.40 (4H, s), 2.57-2.63 (4H, m), 3.72 (3H, s), 7.03 (1H, t), 7.43 (1H, d), 7.81 (1H, s), 7.97 (1H, d), 8.19 (2H, m), 8.37 (1H, s), 9.68 (1H, s), 11.33 (1H, s) |
SYN
CN108368091
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN225024309&_cid=P11-MEHXD5-59000-1
| Example 32: (2R)-N-(3-{2-[(3-methoxy-1-methyl-1H-pyrazol-4-yl)amino]pyrimidin-4-yl}-1H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide |
| |
| 3-{2-[(3-methoxy-1-methyl-1H-pyrazol-4-yl)amino]pyrimidin-4-yl}-1H-indol-7-amine (180 mg, 0.54 mmol, Intermediate 23), (R)-2-(4-methylpiperazin-1-yl)propanoic acid dihydrochloride (158 mg, 0.64 mmol, Intermediate 37) and HATU (408 mg, 1.1 mmol) were stirred together in THF (5 mL) to give an orange solution. Diisopropylethylamine (0.38 mL, 2.2 mmol) was added at 25°C. The resulting suspension was stirred at 25°C for 3 hours. The reaction mixture was diluted with ethyl acetate (100 mL) and washed with saturated NaCl. 2 CO 3 The mixture was stirred for 2 hours at 4 ℃ for 10 minutes.Then the mixture was stirred for 2 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 10 minutes.Then the mixture was stirred for 2 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 10 minutes.Then the mixture was stirred for 2 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 4 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 4 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 3 hours.Then the mixture was stirred for 4 hours.Then the mixture was stirred for 3 hours . δ (DMSO, 400 MHz) 1.26 (3H, d), 2.16 (3H, s), 2.25-2.45 (4H, m), 2.51-2.70 (4H, m), 3.71 (3H, s), 3.80 (3H, s), 7.05 (1H, t), 7.13 (1H, d), 7.38 (1H, d), 7.70 (1H, s), 8.16-8.31 (4H, m), 9.62 (1H, s), 11.35 (1H, s) – the α-proton of the amide is obscured by the residual water peak; m/z (ES+), [M+H]+=490. |
| The above procedure for Example 32 was repeated using the indicated intermediates to obtain Examples 33-42 described in Table 12: |
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Golidocitinib, also known as DZD4205, is an oral, selective Janus kinase 1 (JAK1) inhibitor developed by Dizal Pharmaceutical. It is designed to target aberrant JAK/STAT signaling pathways implicated in
various malignancies, particularly peripheral T-cell lymphoma (PTCL) [31]. In 2024, Golidocitinib was granted conditional approval by the NMPA under the brand name Gao Ruizhe, for the treatment of adult patients with relapsed or refractory PTCL who have received at least one line of systemic therapy. This agent exerts its therapeutic effects through selective inhibition of JAK1, thereby disrupting the JAK/STAT signaling pathway [32]. This inhibition leads to reduced proliferation and increased apoptosis of malignant T-cells in PTCL [33]. The clinical efficacy of Golidocitinib was demonstrated in the Phase II JACKPOT8 Part B study (NCT04105010), a multinational, single-arm trial evaluating its use in patients with r/r PTCL [34]. The investigation demonstrated an ORR of 44.3 % in patients with PTCL, with sustained efficacy noted across diverse PTCL subtypes. In terms of safety profile, Golidocitinib exhibited favorable tolerability. Hematologic adverse events such as anemia, neutropenia, and thrombocytopenia were the predominant treatment-related toxicities, yet they were effectively controlled through dose modifications and supportive interventions.
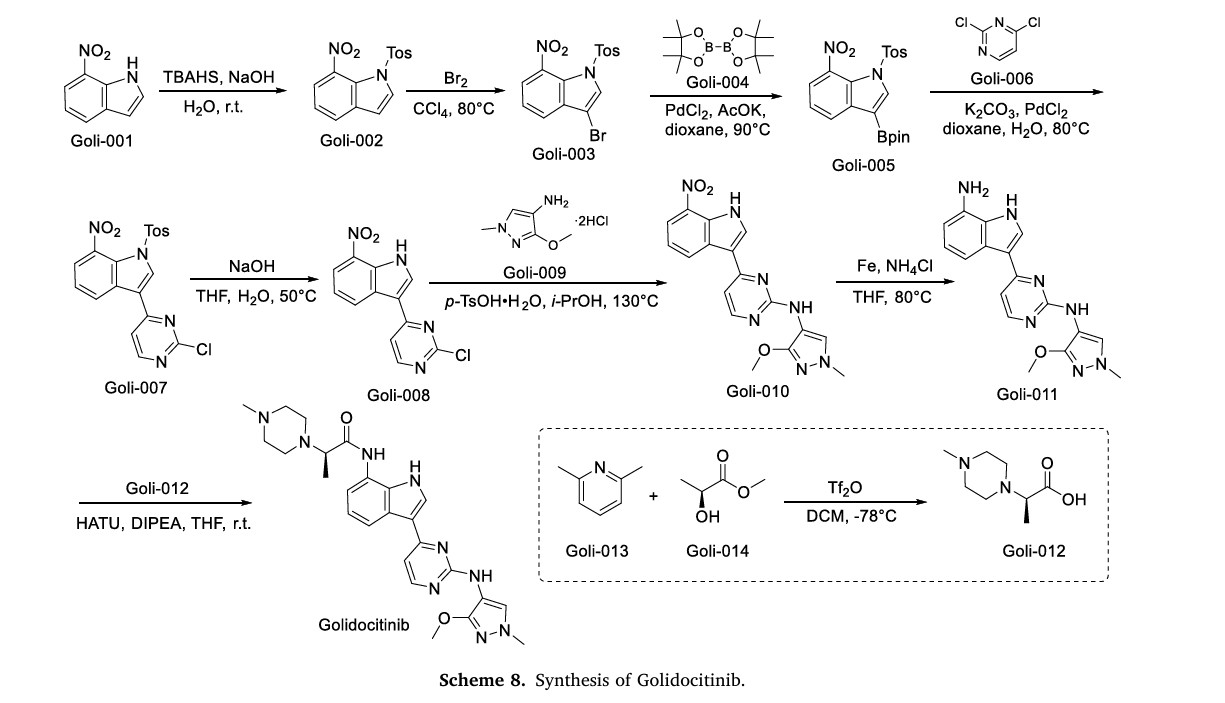
The synthetic route of Golidocitinib, shown in Scheme 8, initiates with amino protection of Goli-001 to afford Goli-002 [35]. Bromination of Goli-002 with Br2 yields Goli-003, which undergoes Miyaura bor
ylation with Goli-004 to form Goli-005. Suzuki-Miyaura coupling of Goli-005 with Goli-006 generates Goli-007. Deprotection of Goli-007 produces Goli-008, which undergoes p-TsOH-mediated nucleophilic
substitution with Goli-009 to yield Goli-010. Reduction of Goli-010 affords Goli-011, followed by amidation with Goli-012 to deliver Golidocitinib. Concurrently, Goli-012 is prepared via Tf2 0- Mediated
nucleophilic substitution between Goli-013 and Goli-014.
[31] S.J. Keam, Golidocitinib: first approval, Drugs 84 (2024) 1319–1324.
[32] K. Chen, X. Guan, Z. Yang, Y. Zhou, Z. Liu, X. Deng, D. Liu, P. Hu, R. Chen,
Pharmacokinetic characteristics of golidocitinib, a highly selective JAK1 inhibitor,
in healthy adult participants, Front. Immunol. 14 (2023) 1127935.
[33] M.B. Nierengarten, Golidocitinib favorable for relapsed/refractory T-cell
lymphoma, Cancer 130 (2024) 1191–1192.
[34] Y. Song, L. Malpica, Q. Cai, W. Zhao, K. Zhou, J. Wu, H. Zhang, N. Mehta-Shah,
K. Ding, Y. Liu, Z. Li, L. Zhang, M. Zheng, J. Jin, H. Yang, Y. Shuang, D.H. Yoon,
S. Gao, W. Li, Z. Zhai, L. Zou, Y. Xi, Y. Koh, F. Li, M. Prince, H. Zhou, L. Lin, H. Liu,
P. Allen, F. Roncolato, Z. Yang, W.S. Kim, J. Zhu, Golidocitinib, a selective JAK1
tyrosine-kinase inhibitor, in patients with refractory or relapsed peripheral T-cell
lymphoma (JACKPOT8 part B): a single-arm, multinational, phase 2 study, Lancet
Oncol. 25 (2024) 117–125.
[35] A.B.M. Aastrand, N.P. Grimster, S. Kawatkar, J.G. Kettle, M.K. Nilsson, L.L. Ruston,
Q. Su, M.M. Vasbinder, J.J. Winter-Holt, D. Wu, W. Yang, T. Grecu, J. McCabe, R.
D. Woessner, C.E. Chuaqui, Preparation of Substituted 2-(piperazin-1-yl)-N-[3-[2-
[(1H-pyrazol-4-yl)amino]pyrimidin-4-yl]-1H-indol-7-yl] Propanamide as Selective
JAK1 Inhibitors for Treating Cancers and Immune Disorders, 2017
CN108368091A.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Keam SJ (October 2024). “Golidocitinib: First Approval”. Drugs. 84 (10): 1319–1324. doi:10.1007/s40265-024-02089-2. PMID 39298087.
- Song Y, Malpica L, Cai Q, Zhao W, Zhou K, Wu J, et al. (January 2024). “Golidocitinib, a selective JAK1 tyrosine-kinase inhibitor, in patients with refractory or relapsed peripheral T-cell lymphoma (JACKPOT8 Part B): a single-arm, multinational, phase 2 study”. The Lancet. Oncology. 25 (1): 117–125. doi:10.1016/S1470-2045(23)00589-2. PMID 38092009.
- Jin J, Zhang L, Zou L, Li Z, Wu H, Zhou K, et al. (2024). “Maintenance Therapy of Golidocitinib, a JAK1 Selective Inhibitor, in Patients with Peripheral T Cell Lymphomas after First-Line Systemic Therapy: Updates of the Phase 2 Study (JACKPOT26)”. Blood. 144: 6368. doi:10.1182/blood-2024-211891.
| Clinical data | |
|---|---|
| Trade names | 高瑞哲 (Gao Ruizhe) |
| Other names | AZD-4205, AZD4205, JAK1-IN-3 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2091134-68-6 |
| PubChem CID | 126715380 |
| DrugBank | DB18057 |
| ChemSpider | 71117616 |
| UNII | 3BY9Z3M34G |
| KEGG | D12502 |
| ChEMBL | ChEMBL4577523 |
| Chemical and physical data | |
| Formula | C25H31N9O2 |
| Molar mass | 489.584 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Golidocitinib: First ApprovalPublication Name: DrugsPublication Date: 2024-09-19PMID: 39298087DOI: 10.1007/s40265-024-02089-2
- Recent Developments in the Use of Kinase Inhibitors for Management of Viral InfectionsPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-02-04PMID: 33539089DOI: 10.1021/acs.jmedchem.0c01467
- Discovery of (2R)-N-[3-[2-[(3-Methoxy-1-methyl-pyrazol-4-yl)amino]pyrimidin-4-yl]-1H-indol-7-yl]-2-(4-methylpiperazin-1-yl)propenamide (AZD4205) as a Potent and Selective Janus Kinase 1 InhibitorPublication Name: Journal of Medicinal ChemistryPublication Date: 2020-04-16PMID: 32297743DOI: 10.1021/acs.jmedchem.9b01392
- Sexuality in a healthcare settingPublication Name: Modern healthcare. [Short-term care ed.]Publication Date: 1976-05PMID: 5656
//////////Golidocitinib, approvals 2024, china 2024, DZD 4205, DIZAL, Gao Ruizhe, AZD-4205, AZD4205, UNII-3BY9Z3M34G, 3BY9Z3M34G
Oritinib
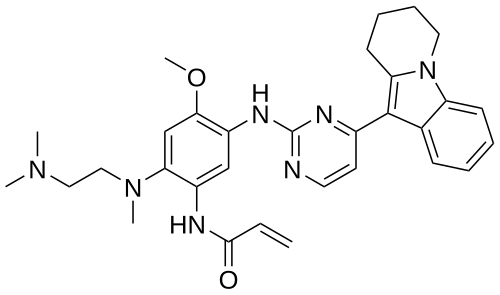
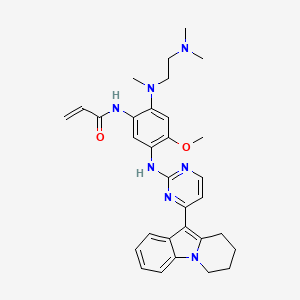
Oritinib
- CAS 2035089-28-0
- MESYLATE CAS 2180164-79-6
- SH-1028
- SK593H37SC
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
- 539.7 g/mol, C31H37N7O2
- rilertinib
CHINA 2024, Nanjing Sanhome Pharmaceutical.
N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
Oritinib is an investigational new drug currently under investigation for its potential use in cancer treatment.[1][2] As a epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, oritinib targets specific enzymes involved in the signaling pathways that regulate cell division and survival, which are often dysregulated in cancer cells.[1]
Oritinib (SH-1028), an irreversible third-generation EGFR TKI, overcomes T790M-mediated resistance in non-small cell lung cancer. Oritinib (SH-1028), a mutant-selective inhibitor of EGFR kinase activity, inhibits EGFRWT, EGFRL858R, EGFRL861Q, EGFRL858R/T790M, EGFRd746-750 and EGFRd746-750/T790M kinases, with IC50s of 18, 0.7, 4, 0.1, 1.4 and 0.89 nM, respectively.
PAT
https://patents.google.com/patent/CN115974845B/en
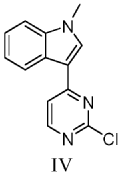
Reaction condition optimization experiment:
The experimental group numbered 1 referred to in table 1 below is the preparation of 1-methyl-3- (2-chloro-4-pyrimidinyl) indole, which was prepared as follows:
To a 10mL reaction tube, 2, 4-dichloropyrimidine (74.5 mg,0.05 mol), zinc triflate (67.3 mg,0.37 equiv), scandium triflate (7.4 mg,0.03 equiv) and 1-methylindole (78.6 mg,1.2 equiv) were added under inert gas atmosphere, and acetonitrile (2.5 mL) were heated to 80℃to react for 24 hours. The reaction was quenched with 30ml of ethyl acetate, the above mixture was added to a separating funnel, 50ml of saturated aqueous sodium carbonate and 50ml of saturated aqueous ammonium chloride were added thereto, and the mixture was shaken for 2 minutes, and the organic phase was taken after the liquid in the separating funnel had settled and separated. The aqueous phase was rinsed with 30ml of ethyl acetate under shaking for 2 times, the whole organic phase was collected, silica gel powder and anhydrous sodium sulfate were added thereto, and the mixture was dried under reduced pressure and packed into a silica gel column. Sequential gradient elution was performed using 250ml (PE: EA: triethylamine 16:4:1), 250ml (PE: EA: triethylamine 15:5:1), 250ml (PE: EA: triethylamine 40:20:3) as developing reagent. The eluent is collected and dried under reduced pressure to obtain pale yellow solid with the yield of 90 percent.
The nuclear magnetic resonance spectrum of 1-methyl-3- (2-chloro-4-pyrimidinyl) indole is as follows:
1H NMR(400MHz,DMSO-d6)δ8.51(d,J=5.9Hz,2H),8.40(dd,1H),7.82(d,J=5.4Hz,1H),7.56(dd,1H),7.28(pd,J=7.1,1.4Hz,2H),3.88(s,3H).
13C NMR(101MHz,DMSO)δ164.55,160.32,158.75,137.84,134.83,125.30,122.81,121.74,121.64,114.43,110.90,110.76,33.31.
PAT
CN109705118
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN242181067&_cid=P20-MEGI3F-20821-1
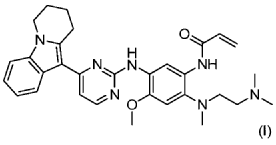
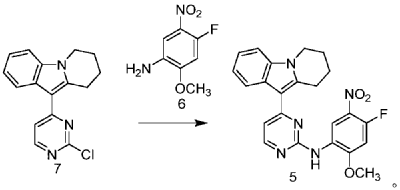
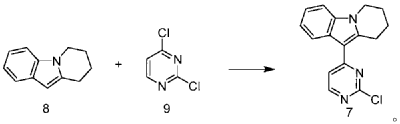
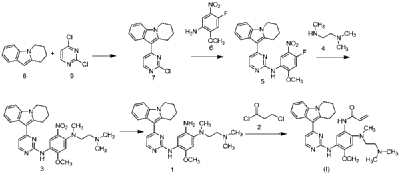
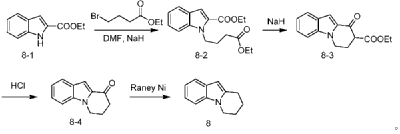
| Step 1: Synthesis of 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole |
| |
| In a 100L vertical jacketed glass reactor, add ethylene glycol dimethyl ether (39.15kg) and 2,4-dichloropyrimidine (3.915kg). Cool the solid-liquid mixture to below 10°C, then add anhydrous aluminum chloride (3.855kg) in batches, controlling the addition rate to keep the temperature below 30°C. After the addition is complete, stir at 25±5°C for 30 minutes, then add 6,7,8,9-tetrahydropyrido[1,2-a]indole (4.500kg). Raise the temperature to 60±5°C and react for 3 hours. Monitor by HPLC until the 6,7,8,9-tetrahydropyrido[1,2-a]indole content does not exceed 1.0%, confirming the reaction is complete. The reaction solution was cooled to below 25° C., purified water (90.0 kg) was added, stirred, and filtered. The filter cake was added to acetonitrile (17.8 kg), slurried, filtered, and dried to obtain a yellow powdery solid, a total of 6.652 kg, with a yield of 89.2%. |
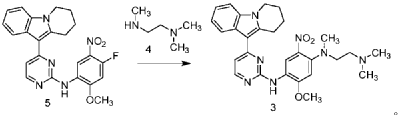
| Step 2: Synthesis of N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-amine |
| |
| To a 500L glass-lined reactor, sec-butyl alcohol (80.82kg), 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole (6.652kg), 4-fluoro-2-methoxy-5-nitroaniline (4.363kg), and p-toluenesulfonic acid monohydrate (4.816kg) were added to obtain a solid-liquid mixture. The reaction mixture was heated to reflux, and the solid gradually dissolved. As the reaction proceeded, a yellow solid precipitated. After reflux for 7.5 hours, the reaction was monitored by HPLC to confirm completion. Heating was stopped, the reaction mixture was cooled to below 15°C, stirred for 1 hour, and the solid was centrifuged and filtered. Acetonitrile (31.5kg) was added to the filter cake, and the mixture was slurried at 25±5°C for 1.5 hours. The mixture was centrifuged and dried to obtain the title compound, a total of 9.548kg, with a yield of 94.0%. |
| Step 3: Synthesis of N 1 -(2-dimethylaminoethyl)-5-methoxy-N 1 -methyl-2-nitro-N 4 -(4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)phenyl-1,4-diamine |
| |
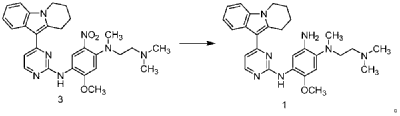
| To a 100 L vertical jacketed glass reactor, add N,N-dimethylacetamide (44.7 kg), N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-amine (9.548 kg), N,N,N’-trimethylethylenediamine (3.380 kg), and N,N-diisopropylethylamine (4.841 kg). Under nitrogen, the reaction mixture was reacted at 85±5°C for 2 hours and monitored by HPLC until the reaction was complete. The reaction solution was cooled to below 70°C, purified water (95.5 kg) was added, filtered, and dried to obtain the title compound, a total of 8.206 kg, with a yield of 72.2%. |
| Step 4: Synthesis of N 1 -(2-(dimethylamino)ethyl)-5-methoxy-N 1 -methyl-N 4 -(4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)benzene-1,2,4-triamine |
| |
| A 100 L vertical jacketed reactor was charged with anhydrous ethanol (32.39 kg), purified water (14.32 kg), N 1 -(2-dimethylaminoethyl)-5-methoxy-N 1 -methyl-2-nitro-N 4 -(4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)phenyl-1,4-diamine (4.103 kg), reduced iron powder (2.224 kg), and ammonium chloride (2.129 kg). The reaction mixture was refluxed for 1.5 hours and monitored by HPLC until the reaction was complete. The reaction mixture was cooled to below 50°C and filtered through diatomaceous earth to remove the solid. The filtrate was concentrated, and tetrahydrofuran (3.45 kg) and purified water (34.71 kg) were added to the residue. The mixture was slurried, filtered, and dried to obtain 3.244 kg of the title compound in an 84.0% yield. |
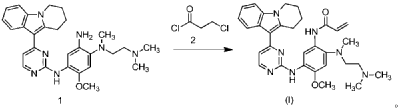
| Step 5: Synthesis of N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide |
| |
| Add N,N-dimethylacetamide (48.6 kg) to a 100 L vertical jacketed glass reactor. Raise the temperature to 40°C, then add N₁- ( 2-(dimethylamino)ethyl)-5-methoxy- N₁ -methyl- N₄- (4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)benzene-1,2,4-triamine (6.487 kg). Then, begin the dropwise addition of 3-chloropropionyl chloride (1.777 kg). Control the addition rate to no more than 60°C. After the addition is complete, cool the reaction mixture and stir at 40±5°C for 1 hour. Sample the mixture and monitor the reaction by HPLC until complete. Add purified water (0.253 kg) and stir for 30 minutes. |
| The reaction mixture was heated at 80±5°C, triethylamine (13.52 kg) was added, and the temperature was raised to 95±5°C. After reacting for 2 hours, the reaction was complete as determined by HPLC. The temperature was then lowered, and methanol (83.0 kg) was added. The mixture was then cooled and crystallized, filtered, and dried to obtain 4.953 kg of the title compound, with a yield of 68.6% and a purity of 97.37%. |
| Step 6: Purification of N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide |
| Anhydrous ethanol (31.25 kg) was added to a 100 L reactor and heated to above 70°C. The crude N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide prepared in step 5 was added. The reaction mixture was heated and stirred under nitrogen until dissolved. The reaction mixture was cooled to below 10°C, the precipitated solid was centrifuged and dried under vacuum at 60±5°C for more than 12 hours to obtain 4.559 kg of the title compound with a yield of 92.1% and a purity of 98.73%. 1 H NMR (300 MHz, DMSO-d 6 )δ10.20(s,1H),8.65(s,1H),8.34(d,1H),8.11(s,1H),8.06(d,1H),7.43(d, 1H),7.19-7.03(m,3H),6.98(s,1H),6.57-6.41(m,1H),6.28-6.15(m,1H),5.8 2-5.71(m,1H),4.09(t,2H),3.84(s,3H),3.18(t,2H),3.06-2.92(m,2H),2.66 (s,3H),2.47-2.40(m,2H),2.27(s,6H),2.08-1.96(m,2H),1.87-1.74(m,2H). ESI-Ms m/z: 540.3 [M+H] + . |
| Example 2: Synthesis of N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide |
| |
| The preparation method is the same as that in step 5 of Example 1, except that N,N-dimethylacetamide is replaced by N,N-dimethylformamide. The purity of the obtained title compound is 69%. |
| The N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide of the present invention prepared according to the above method has a high yield and purity, mild reaction conditions, easy purification, stable process, easy operation, environmental friendliness, and can meet the requirements of industrial-scale production and application. |
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Oritinib represents a third-generation EGFR TKI engineered by Nanjing Sanhome Pharmaceutical. This agent specifically targets both EGFR-sensitizing mutations and the T790 M resistance mutation,
thereby addressing resistance mechanisms linked to prior-generation EGFR-TKIs. In 2024, the NMPA granted approval for Oritinib to treat adult patients with locally advanced or metastatic NSCLC who have experienced disease progression during or following EGFR-TKI therapy and possess confirmed EGFR T790 M mutation-positive status. The mechanism of action of Oritinib involves irreversible binding to mutant EGFR, including the T790 M variant, which in turn suppresses down stream signaling pathways responsible for tumor cell proliferation and survival [28]. The mechanism of Oritinib effectively inhibits tumor growth in patients harboring T790M-mediated resistance to first- and second-generation EGFR-TKIs. Clinical efficacy was established in a Phase II trial (NCT03823807) enrolling patients with EGFR T790 Mmutation-positive NSCLC who had experienced disease progression following prior EGFR-TKI therapy. This study documented an ORR of 60.5 % and a median PFS of 9.6 months, highlighting substantial anti
tumor efficacy in this specific patient cohort. In terms of safety, Oritinib exhibited favorable tolerability. The predominant treatment-related adverse events were rash, diarrhea, and elevated liver enzymes, pri
marily of mild (Grade 1) or moderate (Grade 2) severity. No dose-limiting toxicities were encountered, and the overall safety profile aligned with those observed for other third-generation EGFR-TKIs [29].
The synthetic route of Oritinib Mesylate, shown in Scheme 7, begins with nucleophilic substitution reaction between Orit-001 and Orit-002 to yield Orit-003, which further reacts with Orit-004 via nucleophilic substitution to produce Orit-005 [30]. Orit-005 subsequently undergoes another nucleophilic substitution with Orit-006 to generate Orit-007. Following this, Orit-007 is reduced to form Orit-008. Finally, an amidation reaction between Orit-008 and Orit-009 affords Oritinib.
[28] C. Zhou, A. Xiong, L. Miao, J. Chen, K. Li, H. Liu, Z. Ma, H. Wang, Z. Lu, J. Shen,
P51.03 oritinib (SH-1028), a third-generation EGFR-TKI in advanced NSCLC
patients with positive EGFR T790M: results of a single-arm phase Ib trial,
J. Thorac. Oncol. 16 (2021) S1119–S1120.
[29] C. Zhou, A. Xiong, J. Zhao, W. Li, M. Bi, J. Chen, K. Li, L. Miao, Y. Mao, D. Wang,
7MO oritinib (SH-1028) a third-generation EGFR tyrosine kinase inhibitor in
locally advanced or metastatic NSCLC patients with positive EGFR T790M: results
of a single-arm phase II trial, Ann. Oncol. 33 (2022) S31.
[30] L. Zhao, W. Fu, W. Wu, J. Liu, J. Jin, Method for Preparing Tricyclic Compound as
EGFR Kinase Inhibitor, 2019. CN109705118A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Xiong A, Ren S, Liu H, Miao L, Wang L, Chen J, et al. (October 2022). “Efficacy and Safety of SH-1028 in Patients With EGFR T790M-Positive NSCLC: A Multicenter, Single-Arm, Open-Label, Phase 2 Trial”. Journal of Thoracic Oncology. 17 (10): 1216–1226. doi:10.1016/j.jtho.2022.06.013. PMID 35798241.
- “Rilertinib – Nanjing Sanhome Pharmaceutical”. AdisInsight. Springer Nature Switzerland AG.
| Clinical data | |
|---|---|
| Other names | SH-1028 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2035089-28-0 |
| PubChem CID | 122666966 |
| ChemSpider | 115007246 |
| UNII | SK593H37SC |
| Chemical and physical data | |
| Formula | C31H37N7O2 |
| Molar mass | 539.684 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Safety, efficacy, and pharmacokinetics of SH‐1028 in EGFR T790M‐positive advanced non–small cell lung cancer patients: A dose‐escalation phase 1 studyPublication Name: CancerPublication Date: 2023-02-22PMID: 36813747DOI: 10.1002/cncr.34697
- SH-1028, An Irreversible Third-Generation EGFR TKI, Overcomes T790M-Mediated Resistance in Non-Small Cell Lung CancerPublication Name: Frontiers in PharmacologyPublication Date: 2021-04-27PMCID: PMC8111447PMID: 33986687DOI: 10.3389/fphar.2021.665253
- [1]. Luwei Han, et al. SH-1028, An Irreversible Third-Generation EGFR TKI, Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Front Pharmacol. 2021 Apr 27;12:665253. [Content Brief]
/////////Oritinib, CHINA 2024, APPROVALS 2024, 2035089-28-0, SH 1028, SK593H37SC, rilertinib, Oritinib mesylate, Nanjing Sanhome Pharmaceutical,
Envonalkib
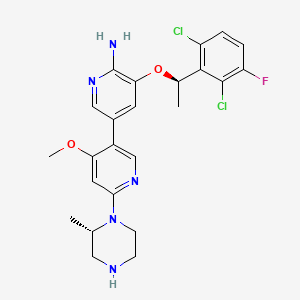
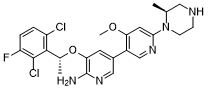
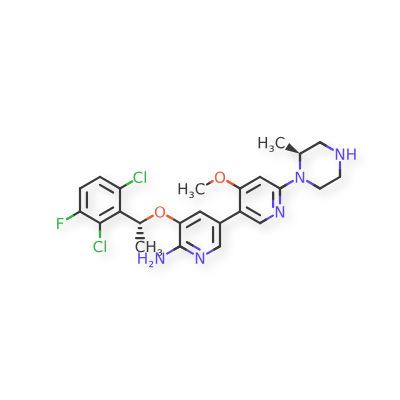
Envonalkib
- CAS 1621519-26-3
- QB7KTQ7VW9
- 5-((1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((2S)-2-methyl-1-piperazinyl)(3,3′-bipyridin)-6-amine
- 506.4 g/mol, C24H26Cl2FN5O2
TQ-B3139, Chia Tai Tianqing, Anluoqing, cancer
ENVONALKIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
SYN
https://patentscope.wipo.int/search/en/WO2014117718
Example 27: 5-[(2,6-dichloro-3-fluorophenyl)ethoxy-4′-methoxy-6′ …
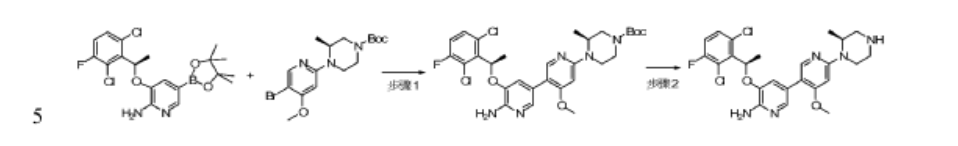
Step 1: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methyl-4-tert-butoxycarbonylpiperazin-1-yl)-3,3′-bipyridin-6-amine
To dioxane (10 mL) and water (1.5 mL) were added tert-butyl (S)-4-(5-bromo-4-methoxypyridin-2-yl)-3-methylpiperidin-1-carboxylate (106 mg, 0.275 mmol), (R)-3-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-aminopyridine (140 mg, 0.33 mmol), tetrakis(triphenylphosphine)palladium (32 mg, 0.0275 mmol) and cesium carbonate (179 mg, 0.55 mmol), the atmosphere was replaced with nitrogen, and the reaction was carried out at 100 ° C. overnight. After cooling, the mixture was separated by silica gel column chromatography to give 5-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6-(5-(2-methyl-4-tert-butoxycarbonylpiperidin-1-yl)-3,3′-bipyridin-6-amine) (70 mg) in a yield of 42%. MS m/z [ESI]: 606.2 [M+1].
Step 2: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-3,3′-bipyridin-6-amine
To a stirred dichloromethane solution (10 mL) of 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methyl-4-tert-butoxycarbonylpiperidin-1-yl)-3,3′-bipyridin-6-amine (67 mg, 0.11 mmol) was added trifluoroacetic acid (1 mL) and stirred for 1 hour. The pH was adjusted to greater than 13 with sodium hydroxide solution, and the mixture was extracted with dichloromethane. The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated. The product was separated and purified by column chromatography (with dichloromethane:methanol = 8:1 as eluent) to give 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperidin-1-yl)-3,3′-bipyridin-6-amine (30 mg). Yield: 55%, MS m/z [ESI]: 506.1[M+1]. 1H-NM (400 MHz, CDC1 3 ):5= 7.94(1H, s), 7.71(1H, s), 7.28-7.32(lH, m), 7.07(1H, t, J=8.4Hz), 6.97(1H, s), 6.04-6.13(2H, m), 4.86 (2H : s), 4.57-4.59(lH, m), 4.03 (1H, d, J=14Hz), 3.76(3H, s), 3.07-3.33(4H, m), 2.88-3.00(lH, m), 1.84(3H, d, J=6.8Hz), 1.34 (3H, d, J=6.8Hz).
SYN
CN107949560
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US154015806&_cid=P11-MEF9W1-27198-1
Example 27: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-[3,3′-bipyridin]-6-amine
General Synthetic Methods:
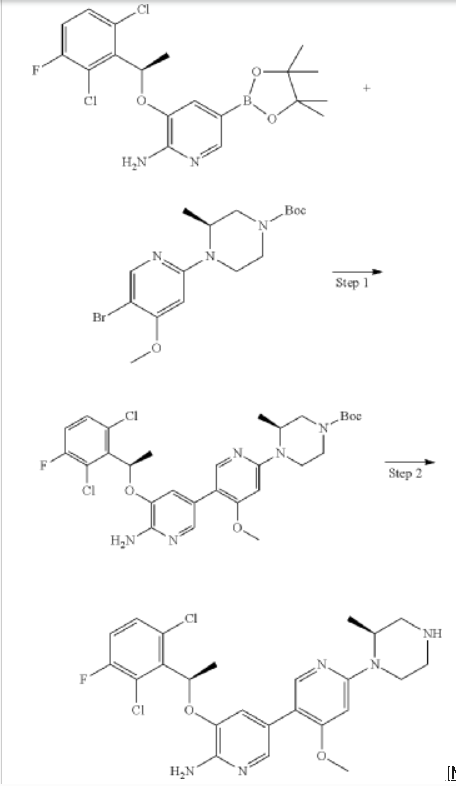
Step 1: (S)-tert-butyl 4-(6′-amino-5′-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4-methoxy-[3,3′-bipyridin]-6-yl)-3-methylpiperazine-1-carboxylate
Step 2: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-[3,3′-bipyridin]-6-amine
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Envonalkib, also known as TQ-B3139, is a novel small-molecule TKI, developed by Chia Tai Tianqing Pharmaceutical Group. It targets ALK, ROS1, and c-Met kinases, exhibiting potent antitumor activity against cancers harboring these genetic alterations. In 2024, the NMPA approved Envonalkib under the brand name Anluoqing for the treatment of adult patients with ALK-positive locally advanced or metastatic NSCLC who have not received prior ALK inhibitor therapy [24]. Envonalkib exerts its therapeutic effects through selective inhibition of the kinase activities of ALK, ROS1, and c-Met, thereby interrupting the downstream signaling pathways that are crucial for tumor cell proliferation and survival [25]. The inhibition of these targets results in cell cycle arrest and apoptosis in cancer cells。The clinical efficacy of Envonalkib was evidenced in a Phase III randomized, open-label, multicenter clinical trial (NCT04009317), which compared Envonalkib with crizotinib in treatment-naïve patients with ALK-positive advanced NSCLC [25,26]. In the reported study, Envonalkib demonstrated a me dian PFS of 24.87 months, which was markedly superior to the 11.60 months achieved with crizotinib (hazard ratio [HR] = 0.47, p < 0.0001). Notably, in patients harboring brain metastases, Envonalkib exhibited a
central nervous system objective response rate (CNS-ORR) of 78.95 %, a substantial improvement over the 23.81 % observed with crizotinib. In terms of safety profile, Envonalkib was generally well-tolerated. Treat ment-related adverse events (TRAEs) of Grade ≥3 were noted in 55.73 % of patients receiving Envonalkib, contrasting with the 42.86 % incidence in the crizotinib cohort. The predominant TRAEs encompassed elevated liver enzymes, neutropenia, and gastrointestinal symptoms, all of which
were amenable to effective management through appropriate support ive care measures. The regulatory approval of Envonalkib thus in troduces a novel therapeutic modality for patients with ALK-positive NSCLC, effectively addressing a significant unmet medical need within this patient population [25].
The synthesis of Envonalkib, illustrated in Scheme 6, initiates with Mitsunobu coupling of Envo-001 and Envo-002, affording Envo-003 [27]. Sequential reduction and NBS-bromination converts Envo-003 to
Envo-005 via Envo-004. Miyaura borylation of Envo-005 constructs Envo-006, which undergoes Suzuki-Miyaura cross-coupling with Envo-007 followed by deprotection to deliver Envonalkib. In parallel,
Envo-009 reacts with Envo-010 through Buchwald-Hartwig cross coupling to form Envo-011. This intermediate is brominated to produce Envo-007, which is used in the Suzuki-Miyaura coupling with Envo-006
[24] X. Li, Y. Xia, C. Wang, S. Huang, Q. Chu, Efficacy of ALK inhibitors in Asian
patients with ALK inhibitor-naïve advanced ALK-Positive non-small cell lung
cancer: a systematic review and network meta-analysis, Transl. Lung Cancer Res.
13 (2024) 2015–2022.
[25] Y. Yang, J. Min, N. Yang, Q. Yu, Y. Cheng, Y. Zhao, M. Li, H. Chen, S. Ren, J. Zhou,
W. Zhuang, X. Qin, L. Cao, Y. Yu, J. Zhang, J. He, J. Feng, H. Yu, L. Zhang, W. Fang,
Envonalkib versus crizotinib for treatment-naive ALK-Positive non-small cell lung
cancer: a randomized, multicenter, open-label, phase III trial, Signal Transduct
Target Ther 8 (2023) 301.
[26] R. Garcia-Carbonero, A. Carnero, L. Paz-Ares, Inhibition of HSP90 molecular
chaperones: moving into the clinic, Lancet Oncol. 14 (2013) e358–e369.
[27] F. Gong, X. Li, R. Zhao, X. Zhang, X. Xu, X. Liu, D. Xiao, Y. Han, Process for
Preparation of Pyridine Substituted 2-aminopyridine Protein Kinase Inhibitor
Crystal, 2017. CN107949560B.

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Efficacy of ALK inhibitors in Asian patients with ALK inhibitor-naïve advanced ALK-positive non-small cell lung cancer: a systematic review and network meta-analysisPublication Name: Translational Lung Cancer ResearchPublication Date: 2024-08-31PMCID: PMC11384493PMID: 39263024DOI: 10.21037/tlcr-24-604
- Envonalkib versus crizotinib for treatment-naive ALK-positive non-small cell lung cancer: a randomized, multicenter, open-label, phase III trialPublication Name: Signal Transduction and Targeted TherapyPublication Date: 2023-08-14PMCID: PMC10423717PMID: 37574511DOI: 10.1038/s41392-023-01538-w
- Pharmacokinetic, pharmacodynamic, and behavioural studies of deschloroketamine in Wistar ratsPublication Name: British Journal of PharmacologyPublication Date: 2021-10-31PMID: 34519023DOI: 10.1111/bph.15680
//////////Envonalkib, china 2024, approvals 2024, TQ-B3139, TQ B3139, Chia Tai Tianqing, Anluoqing, cancer, QB7KTQ7VW9
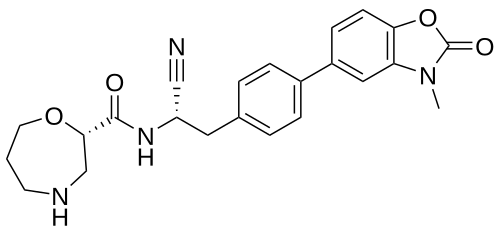
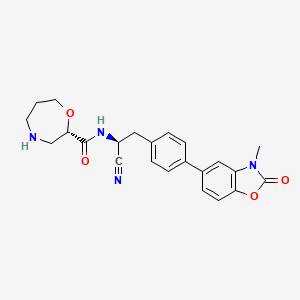
Brensocatib

Brensocatib
WeightAverage: 420.469
Monoisotopic: 420.179755269
Chemical FormulaC23H24N4O4
- AZD7986
- CAS 1802148-05-5
- INS1007
- AZD 7986
- WHO 11097
(2S)-N-[(1S)-1-cyano-2-[4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
- 1,4-Oxazepine-2-carboxamide, N-((1S)-1-cyano-2-(4-(2,3-dihydro-3-methyl-2-oxo-5-benzoxazolyl)phenyl)ethyl)hexahydro-, (2S)-
- (2S)-N-((1S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide
- (2S)-N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
- Brensocatib [USAN]
- (S)-N-((S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide
- UNII-25CG88L0BB
- (2S)-N-[(1S)-1-cyano-2-[4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
FDA 8/12/2025. Brinsupri, To treat non-cystic fibrosis bronchiectasis
Brensocatib is an investigational new drug that is being evaluated to treat bronchiectasis.[1] It is a dipeptidyl-peptidase I (also known as cathepsin C) inhibitor.[2]
A phase 3 clinical trial, known as the ASPEN trial, was conducted to evaluate the safety and efficacy of brensocatib in patients with non-cystic fibrosis bronchiectasis.[3] Brensocatib tablets (Brinsupri) by Insmed Inc. was approved by the FDA in August 2025 after it received breakthrough therapy designation and was reviewed on a priority timeline.
Brensocatib is an orally bioavailable, small molecule, reversible inhibitor of dipeptidyl peptidase 1 (DPP1), with potential anti-inflammatory activity. Upon oral administration, brensocatib reversibly binds to and inhibits the activity of DPP1, thereby inhibiting the activation of neutrophil serine proteases (NSPs), including neutrophil elastase (NE), during neutrophil maturation. This inhibits the activity of NSPs, and may prevent lung inflammation and injury and improve lung function associated with NSPs-induced respiratory diseases. NSPs, serine proteases released by neutrophils during inflammation, is upregulated in a number of respiratory diseases.
SYN
J. Med. Chem. 2016, 59, 9457–9472, DOI: 10.1021/acs.jmedchem.6b01127
https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0040-1719365.pdf
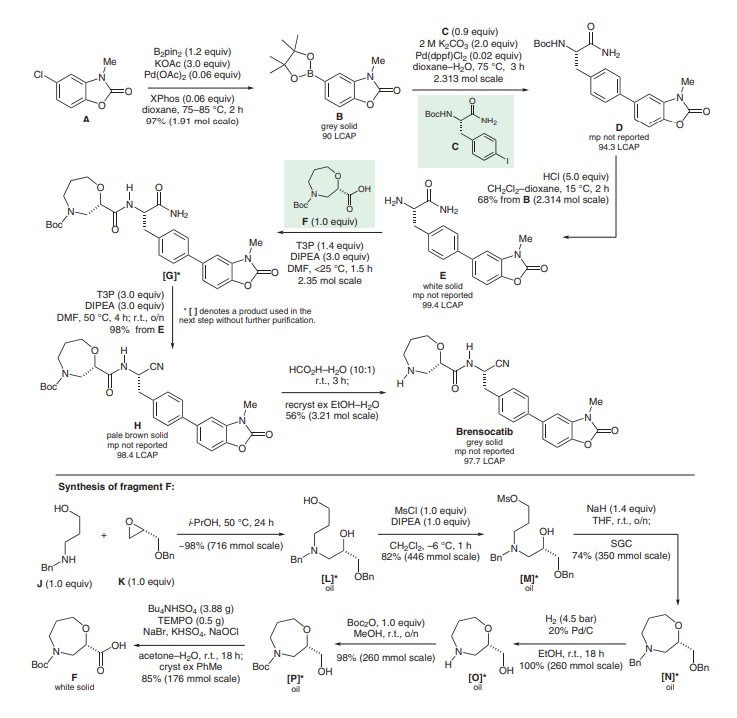
SYN
Brensocatib is now a clinical candidate to impair proteasedriven tissue degradation in COVID-19 (B. Korkmaz,
A. Lesner, S. Marchand-Adam, C. Moss, D. E. Jenne
J. Med. Chem. 2020, 63, 13258).
PAT
https://patents.google.com/patent/US9522894B2/en
A compound according to claim 1
which is (2S)—N-{(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide

EXAMPLESExample 1(2S)—N-[(1S)-1-Cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]-1,4-oxazepane-2-carboxamide

i) tert-Butyl(2S)-2-{[(1S)-1-cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]carbamoyl}-1,4-oxazepane-4-carboxylate
2-Pyridinol-1-oxide (0.155 g, 1.4 mmol), TEA (0.36 g, 3.6 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.268 g, 1.4 mmol) were added to a solution of (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (Intermediate 3, 0.294 g, 1.2 mmol) in DCM (15 mL). After 20 min
4′-[(2S)-2-amino-2-cyanoethyl]biphenyl-4-carbonitrile (Intermediate 1, 0.296 g, 1.2 mmol) was added and the mixture was stirred for 3 h and allowed to stand at rt for 18 h. The mixture was heated at 40° C. for 4 h before water (15 mL) was added. After 10 min the DCM was dried (phase separating cartridge) and evaporated under reduced pressure. The resultant yellow oil was purified by silica gel column chromatography to give the subtitled compound (0.29 g, 52%). Used without further purification in the next step.ii) (2S)—N-[(1S)-1-Cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]-1,4-oxazepane-2-carboxamide
Prepared according to procedure in Method A step ii) using tert-butyl(2S)-2-{[(1S)-1-cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]carbamoyl}-1,4-oxazepane-4-carboxylate to afford the title compound as a white solid (60 mg, 28%).
1H NMR (400 MHz, CDCl3): δ 7.77-7.65 (m, 4H), 7.62-7.57 (m, 2H), 7.40 (d, 2H), 7.11 (d, 1H), 5.18-5.11 (m, 1H), 4.19-4.14 (m, 1H), 4.06-3.96 (m, 2H), 3.75-3.69 (m, 1H), 3.56-3.48 (m, 2H), 3.18-3.05 (m, 3H), 2.95-2.90 (m, 1H), 2.70 (ddd, 1H) (1 exchangeable proton not observed).
LCMS (10 cm_ESCI_Formic_MeCN) tR 2.57 (min) m/z 375 (MH+).Example 2(2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide

i) tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate

N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (468 mg, 2.44 mmol) and 2-pyridinol 1-oxide (271 mg, 2.44 mmol) were added to a solution of (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (Intermediate 3, 490 mg, 2.0 mmol) in DCM (15 mL). The reaction was stirred at rt for 30 min before the addition of (2S)-2-amino-3-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]propanenitrile (Intermediate 2, 586 mg, 2.0 mmol) and DiPEA (1.79 mL, 10 mmol). The reaction was stirred at rt for 18 h before transferring to a separating funnel. The mixture was washed with 2 M hydrochloric acid, saturated sodium hydrogen carbonate solution and brine. The organic extract was run through a hydrophobic frit/phase separator and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography eluting with 0-60% EtOAc in iso-hexane to afford the subtitled compound as an oil (457 mg, 44%). 1H NMR (400 MHz, CDCl3): δ 7.63-7.52 (m, 2H), 7.38 (d, 2H), 7.36-7.24 (m, 2H), 7.35-6.98 (m, 2H), 5.18 (t, 1H), 4.22-3.97 (m, 2H), 3.76-3.67 (m, 0.5H), 4.10-2.94 (m, 4.5H), 3.35-3.26 (m, 1H), 3.24-3.04 (m, 3H), 2.06-1.82 (m, 2H), 1.47 (s, 10H).ii) (2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate (457 mg, 0.85 mmol) was dissolved in formic acid (3 mL) and heated at 50° C. for 10 min on a pre-heated stirrer hotplate. After this time the reaction was concentrated under reduced pressure, dissolved in DCM and washed with saturated sodium hydrogen carbonate solution. The organic extract was run through a hydrophobic frit/phase separator and concentrated under reduced pressure. The resultant foam was purified by silica gel column chromatography eluting with 0-5% methanolic ammonia (7 N) in DCM to afford the title compound as solid material (230 mg, 64%).
1H NMR (400 MHz, CDCl3): δ 7.59-7.51 (m, 2H), 7.39 (dd, 2H), 7.33-7.23 (m, 3H), 7.14 (d, 1H), 5.23-5.12 (m, 1H), 4.12-4.06 (m, 1H), 4.05-3.95 (m, 1H), 3.81-3.71 (m, 1H), 3.46 (s, 3H), 3.34-3.26 (m, 1H), 3.19-3.00 (m, 3H), 2.99-2.82 (m, 2H), 1.92-1.77 (m, 2H) (one exchangeable proton not observed).
LCMS (10 cm_ESCI_Formic_MeCN) tR 2.48 (min) m/z 375 (MH+).Example 2Alternative Synthesis(2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamidei) 5-Chloro-1,3-benzoxazol-2(3H)-one

To a solution of 2-amino-4-chlorophenol (400 g, 2.79 mol) in 2-MeTHF (6 L) was added CDI (497 g, 3.07 mol) under N2 (exotherm 11.0° C.-22.0° C.). The reaction mixture was heated at reflux for 1 h. The mixture was cooled to rt, washed with 2 M HCl(aq) (6 L), 8% NaHCO3(aq) (6 L) and brine (3 L). The organic layer was dried over MgSO4, filtered and evaporated. This gave the product as a pale brown solid (456.1 g, 97% yield, LC purity >99%).
1H NMR (270 MHz, DMSO-d6): δ 12.0-11.5 (br s, 1H), 7.31 (d, 1H), 7.12 (m, 2H).
LCMS (5 cm_ESCI, aq. formic acid_methanol) tR 3.87 (min) m/z 169.8 (MH+).ii) 5-Chloro-3-methyl-1,3-benzoxazol-2(3H)-one

To a solution of 5-chloro-1,3-benzoxazol-2(3H)-one (stage i) (1111.8 g, 6.56 mol) in DMF (4.12 L) was added Cs2CO3 (2136.4 g, 6.56 mol) maintaining the temperature between 0-5° C. MeI (450 ml, 7.21 mol) was then added slowly maintaining the temperature between 0-5° C. The reaction mixture was allowed to warm-up to rt and stirred overnight. The mixture was cooled to 0-5° C. and H2O (4.12 L) was added slowly. The reaction mixture was then warmed to rt and stirred for 15 min. The solids were filtered off and washed with water (4×980 ml). The filter cake was dried under vacuum at 55° C. overnight (1149.9 g, 96% yield, LC purity >99%, H2O: (Karl Fischer) 0.1%).
1H NMR (270 MHz, DMSO-d6): δ 7.45 (d, 1H), 7.35 (d, 1H), 7.15 (dd, 1H), 3.35 (s, 3H). LCMS (5 cm_ESCI_aq. formic acid_methanol) tR 4.13 (min) m/z 183.8 (M+).iii) 3-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2(3H)-one

A solution of 5-chloro-3-methyl-1,3-benzoxazol-2(3H)-one (stage ii)) (350 g, 1.91 mol), B2pin2 (581.0 g, 2.29 mol) and KOAc (561.3 g, 5.72 mol) was vacuum degassed and purged with N2 (×3). Pd(OAc)2 (12.9 g, 57.2 mmol) and XPhos (54.6 g, 114 mmol) were added and the mixture was vacuum degassed and purged with N2 (×3). The mixture was heated to 75° C. A large exotherm was observed at ˜70° C. which warmed-up the mixture to reflux (100° C.). The reaction mixture was stirred for 1 h with no heating. HPLC analysis indicated 2.5% of the starting material remaining therefore the mixture was heated at 85° C. for 1 h. At this stage, no further change was observed. Additional portions of B2pin2 (14.6 g, 57.2 mmol), KOAc (5.7 g, 57.2 mmol), Pd(OAc)2 (12.9 g, 57.2 mmol) and XPhos (27.3 g, 57.2 mmol) were added and the mixture was stirred for 1 h at 75° C. HPLC analysis showed no starting material remaining. The mixture was cooled to rt, filtered through a pad of Celite (501 g) and the cake was washed with EtOAc (2240 ml). The filtrate was combined with two other batches prepared in the same way (2×350 g) and evaporated. This gave 1865.1 g of the product as a grey solid (97% yield, 90.0% pure by LC, 82±2% pure by 1H NMR (DMSO-d6) assay vs TCNB).
1H NMR (270 MHz, DMSO-d6): δ 7.40-7.50 (m, 2H), 7.30 (d, 1H), 3.40 (s, 3H), 1.30 (s, 12H).
LCMS (5 cm_ESCI_aq. formic acid_methanol_) tR 4.91 (min) m/z 276.1 (MH+).iv) Nα-(tert-Butoxycarbonyl)-4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide

To a suspension of 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2(3H)-one (stage iii)) (859 g, 700 g active, 2.544 mol) and tert-butyl (S)-1-carbamoyl-2-(4-iodophenypethylcarbamate (prepared according to the procedure in WO 2009/074829 p. 47), (903 g, 2.313 mol) in dioxane (4.1 L) was added 2 M K2CO3 (2.3 L). The suspension was vacuum degassed and purged with N2 (×3). Pd(dppf)Cl2.DCM (28.33 g, 0.0347 mol) was added and the reaction mixture was heated at 75° C. for 3 h. The mixture was cooled to rt and diluted with water (6.4 L). The suspension was stirred at rt overnight; the solid was filtered off and washed with water (3×1 L). The product was dried at 45° C. for 3 days (1269.1 g, yield 133%—by 1H NMR contains pinacol related impurity and dioxane, LC 94.3% pure, H2O: (Karl Fischer) 3.35%).
1H NMR (270 MHz, DMSO-d6): δ 7.62-7.34 (m, 7H), 7.04 (brs, 2H), 6.86 (d, 1H) 4.12 (m, 1H), 3.40 (s, 3H), 3.00 (dd, 1H), 2.78 (dd, 1H), 1.30 (s, 9H).
LCMS (5 cm_ESI_Water_MeCN) tR 4.51 (min) m/z 312 (MH+).v) 4-(3-Methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide

To a very thick suspension of Nα-(tert-butoxycarbonyl)-4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide (stage iv)) (1269 g, active 952 g assumed 100% conversion at stage iv), 2.3138 mol) in DCM (2.1 L) under N2 was added dropwise 4.1 M HCl in dioxane (2.7 L, 11.06 mol) over 1 h maintaining the temperature at 15° C. (suspension became more mobile after addition of approx. 0.5 L of 4.1 M HCl dioxane). After 2 h, the mixture was diluted with water (5.6 L) and stirred for 30 min at rt. The mixture was then filtered through a pad of Celite (500 g) to remove undissolved material—very slow filtration; the Celite was checked for product by LC. The pad was washed with water (400 ml). The layers DCM/dioxane-water were separated. The aqueous layer was cooled to ˜5° C. and 35% NH3 (aq) (700 ml) was added slowly to achieve pH=9-10. The suspension was stirred overnight then the product was filtered off and washed with water (3×400 ml). The product was dried at 45° C. in vacuo for 2 days (off white solid, 489.4 g, 68% yield over two stages, 99.4% pure by LC, >99% EP, 98±2% pure by 1H NMR assay vs TCNB in DMSO, H2O: (Karl Fischer) 0.92%).
1H NMR (270 MHz, DMSO-d6): δ 7.59-7.30 (m, 7H), 6.98 (brs, 1H), 3.36 (m, 4H), 2.95 (dd, 1H), 2.67 (dd, 1H) 1.86 (brs, 2H).
LCMS (5 cm_ESI_Water_MeCN) tR 2.76 (min) m/z 312 (MH+).vi) tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate

To a solution of 4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide (stage v)) (756 g, active 733 g, 2.354 mol) and (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (577 g, 2.354) (Intermediate 3) in DMF (3 L) was added DiPEA (1230 ml, 7.062 mol) under N2. T3P in DMF (50% w/w, 1924 ml, 3.296 mol) was added dropwise over 1.5 h maintaining the temperature<25° C. After 30 min, LC completion check indicated completion of the coupling reaction. DiPEA (1230 ml, 7.062 mol) was then added and the reaction mixture was heated to 50° C. T3P in DMF (50% w/w, 3986 ml, 6.827 mol) was added portionwise over 1 h (no exotherm observed). The reaction mixture was stirred at 50° C. for 4 h and then at rt overnight. The mixture was cooled to 10° C., diluted with 2-MeTHF (4 L) and water (5.6 L, exothermic). The layers were separated and the aqueous layer was extracted with 2-MeTHF (2×4 L). The combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. This delivered the product as a pale brown solid in 98% yield (1242 g (active 1205 g), corrected yield 98%, LC purity 98.4%, 1H NMR assay vs TCNB 97±2%, main impurities by 1H NMR: 2-MeTHF 1.9%, DMF 0.6%).vii) (2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
A solution of tert-butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate (stage vi)) (1776 g, active 1671 g, 3.210 mol) in formic acid/water (4.2 L/440 ml) was stirred on a buchi at 35-37° C. under reduced pressure (300-500 mbar). After 3 h, LCMS completion check indicated 93.95% of the product and 0.5% of the starting material. The mixture was concentrated (4 h) to give an oily residue. The residue was dissolved in water (4.4 L) and washed with TBME (2.2 L). The aqueous layer was vigorously stirred and treated with NH3(aq) (1.8 L) at <25° C. to achieve pH=9-10. The mixture was stirred at rt for 3 h. The solid was filtered off and washed with water (3×1 L). The filter cake was dried at 45° C. overnight. This gave the product as a pale brown solid (1498 g, active 1333 g, LC 91.5%, 1H NMR assay vs TCNB 89±2%, H2O: (Karl Fischer) 4.63%).
The crude product was re-crystallised from EtOH/H2O in two batches (2×747 g).
Batch A: The crude product (747 g) was dissolved in EtOH (8 L) at reflux under N2. Water (1.6 L) was added slowly. The mixture was hot filtered (65° C.) to remove black particles (filtrate temperature
50° C.) and then stirred at 40° C. overnight. The suspension was cooled to 10° C. over 4 h and held at that temperature for 3 h. The product was filtered off and washed with EtOH/H2O (8:2, 3×500 ml) then water (3×500 ml). The filter cake was dried at 45° C. overnight (473 g, 97.7% pure by LC, Pd level 71.4 ppm).
Batch B gave 436 g of the product (95.8% pure by LC, Pd level 65.8 ppm).
The liquors from both batches were combined and concentrated to ˜8 L. The liquors were left overnight at rt. The solids were filtered off and washed with EtOH/H2O (8:2, 3×400 ml) then water (3×400 ml). The product was dried at 45° C. overnight. This gave additional 88 g of the product (LC purity 95.0%).
The products (LC purity of the blend 95.69%) were re-crystallised from EtOH/H2O in two batches (Batch C: 520 g, Batch D: 520 g).
Batch C: The crude product (520 g) was dissolved in EtOH (6.24 L) at reflux under N2. Water (1248 ml) was added slowly. The mixture was allowed to cool down to 40° C. (3 h), seeded with 0.5 g of the title compound and stirred at 40° C. for 10 h. The mixture was then cooled to 26° C. over 7 h. The resulting suspension was cooled to 10° C. and stirred at that temperature for 6 h. The product was filtered off, washed with EtOH/water (8:2, 3×500 ml) and water (3×500 ml). The filter cake was dried at 45° C. for 2 d. The product was obtained as a grey solid (418 g, yield ˜56%, LCMS purity 97.5%, chiral LC
100%, 1H NMR (DMSO-d6) assay vs TCNB
100±2%).
Batch D: 418 g, yield 56%, LCMS purity 97.5%, chiral LC
100%, 1H NMR (DMSO-d6) assay vs TCNB
100±2%
The product was blended with the material from an intermediate scale reaction performed in the same way and re-analysed (968 g, LC purity 98.04%, chiral LC
100%, 1H NMR assay vs TCNB 99±2%, 0.35% EtOH by 1H NMR, H2O: (Karl Fischer) 4.58%, Pd 57.6 ppm, XRPD (X-ray powder diffraction) Form A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Brensocatib – Insmed”. AdisInsight. Springer Nature Switzerland AG.
- Chalmers JD, Usansky H, Rubino CM, Teper A, Fernandez C, Zou J, et al. (October 2022). “Pharmacokinetic/Pharmacodynamic Evaluation of the Dipeptidyl Peptidase 1 Inhibitor Brensocatib for Non-cystic Fibrosis Bronchiectasis”. Clinical Pharmacokinetics. 61 (10): 1457–1469. doi:10.1007/s40262-022-01147-w. PMC 9553789. PMID 35976570.
- Chalmers JD, Burgel PR, Daley CL, De Soyza A, Haworth CS, Mauger D, et al. (April 2025). “Phase 3 Trial of the DPP-1 Inhibitor Brensocatib in Bronchiectasis”. The New England Journal of Medicine. 392 (16): 1569–1581. doi:10.1056/NEJMoa2411664. PMID 40267423.
| Clinical data | |
|---|---|
| Other names | AZD7986; INS1007 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1802148-05-5 |
| PubChem CID | 118253852 |
| IUPHAR/BPS | 9412 |
| DrugBank | DB15638 |
| ChemSpider | 67896269 |
| UNII | 25CG88L0BB |
| KEGG | D12120 |
| ChEMBL | ChEMBL3900409 |
| Chemical and physical data | |
| Formula | C23H24N4O4 |
| Molar mass | 420.469 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Brensocatib, APPROVALS 2025, FDA 2025, Brinsupri, non-cystic fibrosis, AZD7986, 1802148-05-5, INS1007, AZD 7986, WHO 11097
Unecritinib
Unecritinib
- CAS 1418026-92-2
- 4T3Z98RR86
- TQ-B3101
492.4 g/mol, C23H24Cl2FN5O2
N-[3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-yl]acetamide
- Acetamide, N-[3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-[1-(4-piperidinyl)-1H-pyrazol-4-yl]-2-pyridinyl]-
- N-{3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-[1- (piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-yl}acetamide
Chia Tai Tianqing Pharmaceutical Group
Unecritinib is an orally available, small molecule inhibitor of the receptor tyrosine kinases anaplastic lymphoma kinase (ALK), C-ros oncogene 1 (ROS1) and Met (hepatocyte growth factor receptor; HGFR; c-Met), with potential antineoplastic activity. Upon oral administration,unecritinib targets, binds to and inhibits the activity of ALK, ROS1 and c-Met, which leads to the disruption of ALK-, ROS1- and c-Met-mediated signaling and the inhibition of cell growth in ALK-, ROS1- and c-Met-expressing tumor cells. ALK, ROS1 and c-Met, overexpressed or mutated in many tumor cell types, play key roles in tumor cell proliferation, survival, invasion and metastasis.
UNECRITINIB is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 3 investigational indications.
- OriginatorChia Tai Tianqing Pharmaceutical Group
- ClassAcetamides; Antineoplastics; Benzofurans; Chlorobenzenes; Esters; Ethers; Fluorobenzenes; Ketones; Morpholines; Piperidines; Pyrazoles; Pyridines; Small molecules
- Mechanism of ActionAnaplastic lymphoma kinase inhibitors; Proto-oncogene protein c-met inhibitors; ROS1 protein inhibitors
- RegisteredNon-small cell lung cancer
- No development reportedAnaplastic large cell lymphoma
- 07 Sep 2024Efficacy and adverse events data from a phase II trial in Non-small cell lung cancer presented at the 25th World Conference on Lung Cancer (WCLC-2024)
- 17 May 2024Chemical structure information added
- 17 May 2024No development reported – Phase-II for Anaplastic large cell lymphoma (In adolescents, In children, Late-stage disease, Refractory metastatic disease, Second-line therapy or greater, In adults) in China (PO)
PATENT
https://patentscope.wipo.int/search/en/WO2013041038
Example 11: Synthesis of
(R)-N-(3-(l-(2,6-dichloro-3-fluorophenyl)ethoxy)- 5-(l -(piperidin-4-yl)-lH-pyrazol-4-yl)pyridin-2-yl)acetamide (Compound 18)
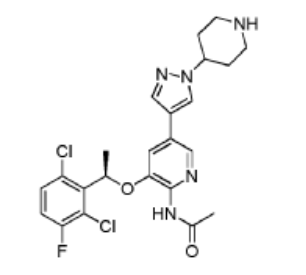
Step 1. To a solution of (R)-tert-butyl 4-(4-(6-amino-5-(l-(2,6-dichloro-3-fluorophenyl)ethoxy)pyridin-3 -yl)- 1 H-pyrazol- 1 -yl)piperidine- 1 -carboxylate ( 4g, 7.27 mmol, 1.0 eq) and pyridine ( 2.3g, 29.1 mmol, 4.0 eq) in 50 ml DCM was added acetyl chloride (0.86g, 10.9 mmol, 1.5 eq) in an ice bath. The reaction mixture was stirred at room temperature for overnight. The resulting mixture was washed with H20 (3×20 mL). The organic layer was dried and concentrated. The crude product was purified on silica gel column to give (R)-tert-butyl 4-(4-(6-acetamido-5-(l-(2,6-dichloro-3-fluorophenyl)ethoxy)pyridin-3-yl)-lH-pyrazol-l-yl)piperidine-l-carboxylatel .66g (38.6% yield).
Step 2. To a solution of (R)-tert-butyl 4-(4-(6-acetamido-5-(l-(2,6-dichloro-3 -fluorophenyl)ethoxy)pyridin-3 -yl)- 1 H-pyrazol- 1 -yl)piperidine- 1 -carboxylate (500 mg, 0.84 mmol, 1.0 eq) in DCM (5 mL) was added trifluoroacetic acid (2 ml) in an ice bath. The reaction mixture was stirred at room temperature for 2 hours. The pH of the reaction mixture was adjusted to 9 by saturated bicarbonate sodium in an ice bath. The aqueous solution was extracted with ethyl acetate (3×20 mL), the combined organic layers were washed with brine, dried over (MgSC^), filtered, and concentrated. The crude product was purified by silica gel column to give (R)-N-(3 -( 1 -(2,6-dichloro-3 -fluorophenyl)ethoxy)-5-( 1 -(piperidin-4-yl)- 1 H-pyrazol-4-yl)pyridin-2-yl)acetamide 250 mg (60.2% yield).
^-NMR^DC , 400Hz): 51.88(d, J=6.4Hz, 3H), 51.90-1.94(m, 2H), 52.16-2.20(m, 2H), 52.48(s, 3H), 52.76-2.824(m, 2H), 53.25-3.28(m, 2H), 53.69-3.74(m, 1H), 54.22-4.26 (m, 1H), 56.10-6.15(m, 1H), 57.05-7.07 (m, 1H), 57.09(s, 1H), 57.30-7.33 (m, 1H), 57.59(s, 1H), 57.62(s, 1H), 58.06(s, 1H),
58.12(s, 1H). MS m/z 493 [M+l]
PATENT
CN102850328
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN85774618&_cid=P12-MECPSG-91316-1
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Unecritinib, developed by Chia Tai Tianqing Pharmaceutical Group, is a novel small-molecule tyrosine kinase inhibitor. It targets c-rosoncogene 1 (ROS1), anaplastic lymphoma kinase (ALK), and c-mesen
chymal-epithelial transition factor (c-MET) kinases, exhibiting potent antitumor activity against cancers harboring these genetic alterations. In 2024, the NMPA approved Unecritinib under the brand name Anbaini for the treatment of adult patients with ROS1-positive locally advanced or metastatic non-small cell lung cancer (NSCLC). Unecritinib exerts its therapeutic effects through selective inhibition of the kinase activities of ROS1, ALK, and c-MET, which effectively disrupts the downstream signaling pathways that are crucial for the proliferation and survival of tumor cells. Consequently, this inhibition induces cell cycle arrest and apoptosis in cancer cells that express these specific targets [13]. The clinical efficacy of Unecritinib was established in a Phase II single-arm, multicenter clinical trial (NCT03750739) enrolling patients with ROS1-positive advanced NSCLC. Among 111 evaluable patients, an ORR of 80.2 % was achieved, along with a median PFS of 16.5 months. These findings underscore the robust antitumor activity of Unecritinib in this specific patient cohort. In terms of safety, Unecritinib exhibited a
favorable tolerability profile. The most frequently reported treatment-related adverse events were neutropenia, leukopenia, vomit ing, and nausea, which were predominantly of mild (Grade 1) or mod
erate (Grade 2) severity. Importantly, no dose-limiting toxicities were observed, and the maximum tolerated dose was not established, further supporting its favorable safety profile. The approval of Unecritinib represents a novel therapeutic strategy for patients with ROS1-positive NSCLC, effectively addressing a significant unmet medical need within this population [13].
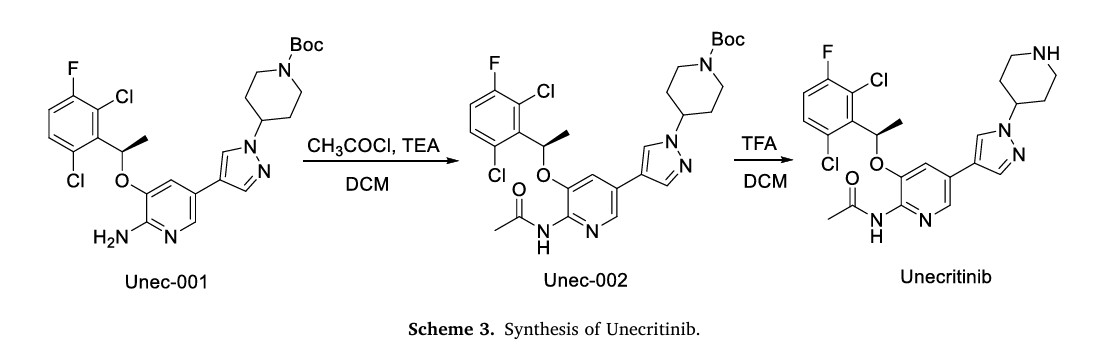
The synthesis of Unecritinib, depicted in Scheme 3, initiates with acetylation of Unec-001 to yield Unec-002, which undergoes deprotection to afford Unecritinib [14]
[13] S. Lu, H. Pan, L. Wu, Y. Yao, J. He, Y. Wang, X. Wang, Y. Fang, Z. Zhou, X. Wang,
X. Cai, Y. Yu, Z. Ma, X. Min, Z. Yang, L. Cao, H. Yang, Y. Shu, W. Zhuang, S. Cang,
J. Fang, K. Li, Z. Yu, J. Cui, Y. Zhang, M. Li, X. Wen, J. Zhang, W. Li, J. Shi, X. Xu,
D. Zhong, T. Wang, J. Zhu, Efficacy, safety and pharmacokinetics of unecritinib
(TQ-B3101) for patients with ROS1 positive advanced non-small cell lung cancer: a
phase I/II trial, Signal Transduct Target Ther 8 (2023) 249.
[14] A. Zhang, M. Geng, Y. Wang, J. Ai, X. Peng, Preparation of Pyridine Compounds as
Inhibitors of c-Met And/Or ALK Kinases, Shanghai Institute of Materia Medica,
2013 CN102850328A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Unecritinib, Chia Tai Tianqing Pharmaceutical Group, 1418026-92-2, 4T3Z98RR86, TQ B3101, APPROVALS 2024, CHINA 2024
Dordaviprone
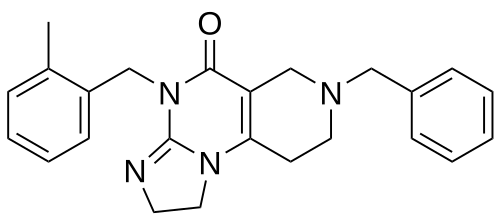
Dordaviprone
WeightAverage: 386.499
Monoisotopic: 386.210661473
Chemical FormulaC24H26N4O
- TIC10
- CAS 1616632-77-9
- Dordaviprone
- ONC201
- ONC 201
- 9U35A31JAI
- NSC-350625
11-benzyl-7-[(2-methylphenyl)methyl]-2,5,7,11-tetrazatricyclo[7.4.0.02,6]trideca-1(9),5-dien-8-one
Product Ingredients
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Dordaviprone dihydrochloride | 53VG71J90J | 1638178-82-1 | Not applicable |
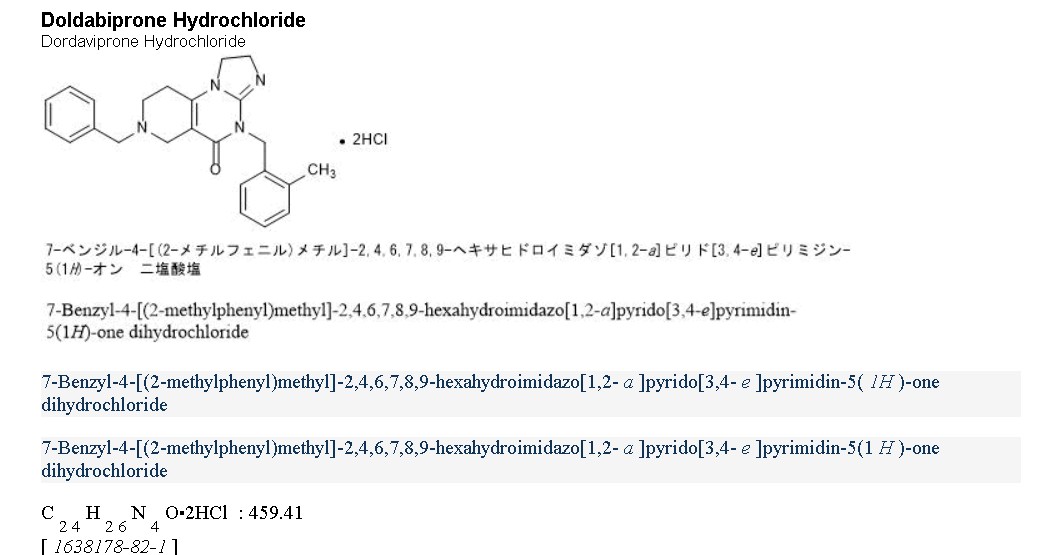
- 11-benzyl-7-(o-tolylmethyl)-2,5,7,11-tetrazatricyclo[7.4.0.02,6]trideca-1(9),5-dien-8-one
- 11-benzyl-7-[(2-methylphenyl)methyl]-2,5,7,11-tetrazatricyclo[7.4.0.0(2,6)]trideca-1(9),5-dien-8-one
- 7-Benzyl-4-(2-methylbenzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one
- 7-benzyl-4-[(2-methylphenyl)methyl]-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one
FDA 8/6/2025, Modeyso, To treat diffuse midline glioma harboring an H3 K27M mutation with progressive disease following prior therapy
Dordaviprone, sold under the brand name Modeyso is an anti-cancer medication used for the treatment of diffuse midline glioma (a type of brain tumor).[1][2] Dordaviprone is a protease activator of the mitochondrial caseinolytic protease P.[1] It is dopamine receptor D2 antagonist and an allosteric activator of the mitochondrial caseinolytic protease P.[3]
Dordaviprone was approved for medical use in the United States in August 2025.[2] It is the first approval of a systemic therapy for H3 K27M-mutant diffuse midline glioma by the US Food and Drug Administration.[2]
Dordaviprone is an organic heterotricyclic compound that is 2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one substituted by 2-methylbenzyl and benzyl groups at positions 4 and 7, respectively. It is a selective antagonist of the dopamine receptor D2 and an allosteric agonist of mitochondrial protease caseinolytic protease P. It has a role as an antineoplastic agent, a dopamine receptor D2 antagonist and an apoptosis inducer. It is a member of toluenes, a member of benzenes and an organic heterotricyclic compound.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- https://pp.jazzpharma.com/pi/modeyso.en.USPI.pdf [bare URL PDF]
- “FDA grants accelerated approval to dordaviprone for diffuse midline glioma”. U.S. Food and Drug Administration (FDA). 6 August 2025. Retrieved 7 August 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J, et al. (December 2020). “ONC201 and imipridones: Anti-cancer compounds with clinical efficacy”. Neoplasia. 22 (12). New York, N.Y.: 725–744. doi:10.1016/j.neo.2020.09.005. PMC 7588802. PMID 33142238.
- “Jazz Pharmaceuticals Announces U.S. FDA Approval of Modeyso (dordaviprone) as the First and Only Treatment for Recurrent H3 K27M-mutant Diffuse Midline Glioma” (Press release). Jazz Pharmaceuticals. 6 August 2025. Retrieved 10 August 2025 – via PR Newswire.
- World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
External links
- Clinical trial number NCT02525692 for “Oral ONC201 in Adult Recurrent Glioblastoma” at ClinicalTrials.gov
- Clinical trial number NCT03295396 for “ONC201 in Adults With Recurrent H3 K27M-mutant Glioma” at ClinicalTrials.gov
- Clinical trial number NCT03416530 for “ONC201 in Pediatric H3 K27M Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT05392374 for “Expanded Access Use of ONC201 in a Patient With Diffuse Intrinsic Pontine Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT03134131 for “Expanded Access to ONC201 for Patients With H3 K27M-mutant and/or Midline High Grade Gliomas” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Modeyso |
| Other names | ONC201, ONC-201 |
| AHFS/Drugs.com | Modeyso |
| License data | US DailyMed: Dordaviprone |
| Routes of administration | By mouth |
| Drug class | Protease activator |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1616632-77-9as HCl: 1638178-82-1 |
| PubChem CID | 73777259 |
| DrugBank | DB14844as HCl: DBSALT003291 |
| ChemSpider | 30904994 |
| UNII | 9U35A31JAIas HCl: 53VG71J90J |
| KEGG | D12733as HCl: D12734 |
| ChEBI | CHEBI:232328 |
| ChEMBL | ChEMBL4297310 |
| Chemical and physical data | |
| Formula | C24H26N4O |
| Molar mass | 386.499 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Dordaviprone, Modeyso, FDA 2025, APPROVALS 2025, TIC10, 1616632-77-9, Dordaviprone, ONC201, ONC 201, 9U35A31JAI, NSC 350625
Tunlametinib
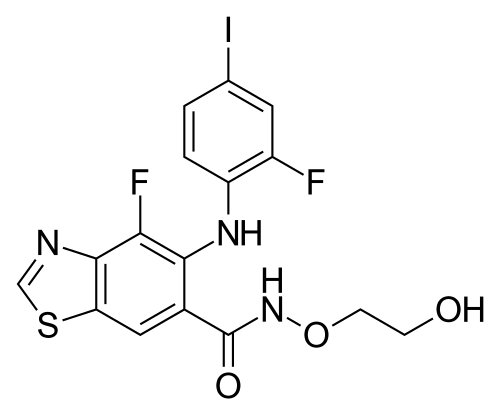
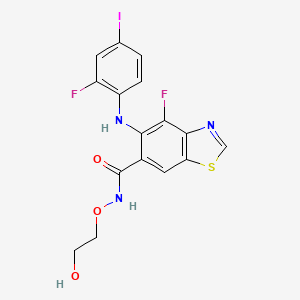
Tunlametinib
- CAS 1801756-06-8
- IF25NR1PV3
- HL085
- C16H12F2IN3O3S
491.3 g/mol
4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-6-benzothiazolecarboxamide
- 4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 6-Benzothiazolecarboxamide, 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping,
Tunlametinib is a pharmaceutical drug for the treatment of cancer. It is an inhbitor of mitogen-activated protein kinase kinase.[1]
In China, tunlametinib was approved in 2024 for the treatment of patients with NRAS-mutated advanced melanoma who were previously treated with a PD-1/PD-L1 targeting agent.[2][3]
It is also being studied for use in combination with vemurafenib in patients with advanced BRAF V600-mutant solid tumors.[4]
PAT
US9937158
PAT
https://patents.google.com/patent/WO2013107283A1/en
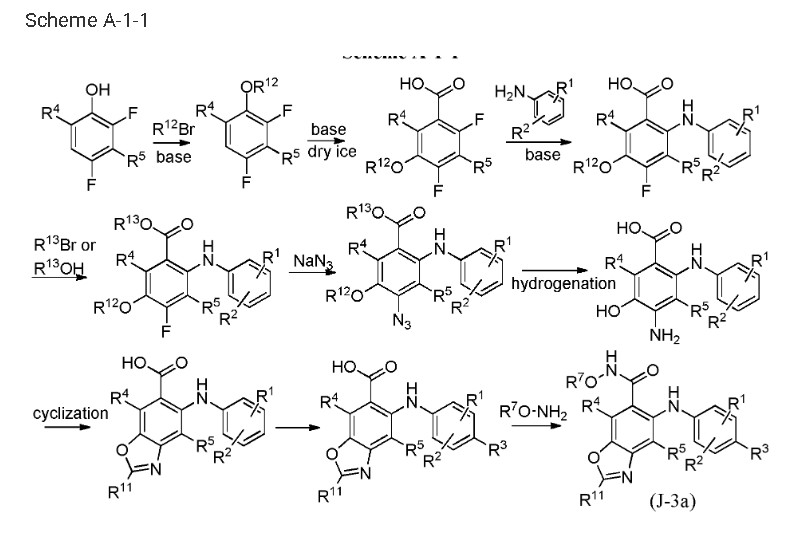
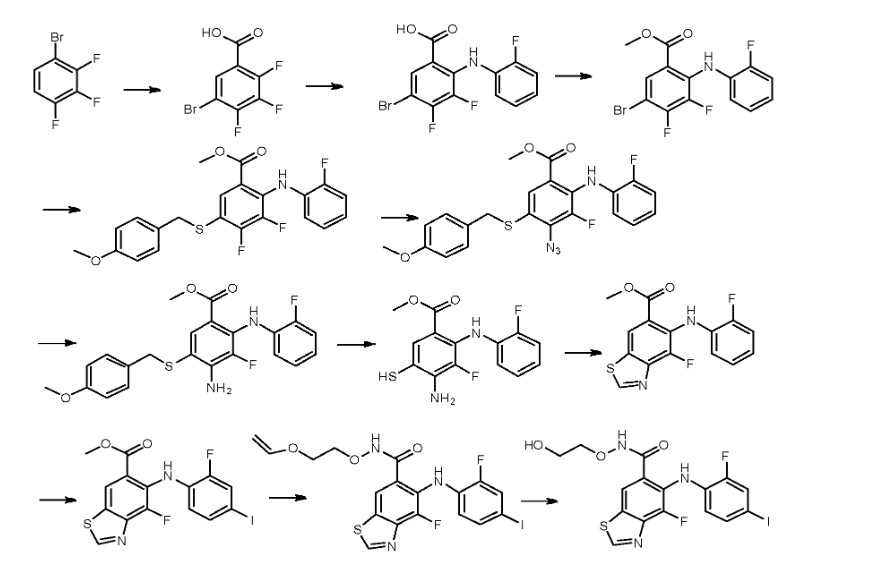
Step 1:

[0435] To a solution of 2,3,4-trifluorobromobenzene in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) was added strong base (such as LDA, nBuLi,
LiHDMS) at low temperature (-50 °C 80 °C, prefer -78 °C) under nitrogen atmosphere. The reaction is kept stirring for some time (0.5-12 h, prefer 0.5-2 h) and is added dry ice. After several hours (3-12 h, prefer 5-10 h), 5-bromo-2,3,4-trifluorobenzoic acid is obtained after conventional workup.
Step 2:

[0436] 5-Bromo-2,3,4-trifluorobenzoic acid can be reacted with halogenated aniline (such as o-fluoroaniline, o-chloroaniline, o-bromoaniline, o-iodoaniline) in the presence of base (such as LDA, n-BuLi, LiHDMS) in appropriate solvent (include aliphatic and aromatic
hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2- methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) at low temperature (-50 °C— -80 °C, prefer -78 °C) for some time (such as 3-12 h, prefer 5-10 h). 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid is obtained after conventional workup.
Step 3:

[0437] 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid can be reacted with MeOH in the presence of SOCl2 in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol and ethanol). The reaction proceeds for several hours (3-12 h, prefer 5-10 h). Methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate is obtained after conventional workup.
Step 4:

[0438] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dioxane) was added base (such as aliphatic and aromatic amine(such as, but not limited to, N-ethyl-N-isopropylpropan-2-amine, triethylamine, diethylamine, DBU, t-butylamine, cyclopropanamine, dibutylamine, diisopropylamine, 1,2- dimethylpropanamine), inorganic base(such as Na2C03, K2C03, NaHC03, KHC03, t-BuONa, t- BuOK), prefer N-ethyl-N-isopropylpropan-2-amine) at ambient temperature under nitrogen atmosphere, followed by Pd catalyst (such as tris(dibenzylideneacetone)dipalladium,
bis(dibenzylideneacetone) palladium, bis(triphenylphosphine)palladium(II) chloride, palladium diacetate, tetrakis(triphenylphosphine)palladium, bis(triphenylphosphinepalladium)acetate, prefer tris(dibenzylideneacetone) dipalladium) and phosphine ligand (such as
dimethylbisdiphenylphosphinoxanthene, tri-tert-butylphosphine, tri-p-tolylphosphine, tris(4- chlorophenyl)phosphine, triisopropylphosphine, tris(2,6-dimethoxyphenyl)phosphine, 1, 1 ‘- bis(diphenylphosphino)ferrocene, prefer dimethylbisdiphenylphosphinoxanthene). The reaction is kept stirring at high temperature (80-130 °C, prefer 90-110 °C) for some time (8-24 h, prefer 12-18 h). Methyl 3,4-difluoro-2- ((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 5:

[0439] Methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy benzyl)thio)benzoate can be reacted with azide (such as NaN3, KN3) at high temperature (60-120 °C, prefer 80-100 °C) in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1-12 h, prefer 3-10 h). Methyl 4-azido-3-fluoro-2-((2-fluorophenyl) amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup.
Step 6:

[0440] Methyl 4-azido-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy
benzyl)thio)benzoate can be hydrogenated catalyzed by appropriate catalyst (such as Pd/C, Pt, Ni) in the solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), amide (such as N,N-dimethylformamide, N,N- dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol, ethanol, propan-l-ol and water) for some time (1-12 h, prefer 3-10 h). Methyl 4- amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 7:

[0441] 4-Amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate can be deprotected in the presence of acid (such as CF3COOH, HCOOH, CH3COOH and n- C5H11COOH, prefer CF3COOH) at certain temperature (20-75 °C, prefer 25-75 °C) in
appropriate aromatic aliphatic ether (such as anisole and phenetole, prefer anisole) for some time (1-12 h, prefer 3-10 h). Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate is obtained after conventional workup.
Step 8:

[0442] Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercapto benzoate can be cyclized in the presence of acid (such as ^-toluenesulfonic acid, pyridinium toluene-4- sulphonate, formic acid, acetic acid, sulfuric acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as
dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methyl acetate, ethyl acetate and trimethoxymethane) for some time (0.2-12 h, prefer 0.5-10 h). Methyl 4-fluoro-5-((2- fluorophenyl)amino) benzo[d]thiazole-6-carboxylate is obtained after conventional workup. Step 9:

[0443] Methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate can be reacted with halogenations reagent (such as NIS) in the presence of acid (such as trifluoroacetic acid, trifluoromethanesulfonic acid, methanesulfonic acid, formic acid, acetic acid) at ambient temperature in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N- dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1- 12 h, prefer 3-10 h). Methyl 4-fluoro-5-((2-fluoro-4-iodophenyl) amino)benzo[d]thiazole-6- carboxylate is obtained after conventional workup.
Step 10:

[0444] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid can be reacted with O-(2-(vinyloxy)ethyl)hydroxylamine in the presence of coupling reagent(such as HOBt, EDCI, HATU, TBTU) at ambient temperature in appropriate solvent(include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane, 1,2- dichloroethane and N,N-dimethylformamide) for some time (1-12 h, prefer 3-10 h). 4-Fluoro-5- ((2-fluoro-4-iodophenyl) amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide is obtained after conventional workup. Step 11:

[0445] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole- 6-carboxamide can be reacted in the presence of acid (such as HCl, H2S04, trifluoroacetic acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane and 1,2-dichloroethane) for some time (1-12 h, prefer 3-10 h). 4-Fluoro- 5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxy ethoxy)benzo[d]oxazole-6-carboxamide is obtained after conventional workup.
Example 9: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyDamino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic acid
[0510] To a solution of diisopropylamine (10.14 g, 100.20 mmol) in THF (100 mL) was added «-BuLi (40.08 mL, 2.5 M in hexane, 100.20 mmol) at -78 °C under nitrogen atmosphere. The stirring was maintained at this temperature for 1 h. Then a solution of l-bromo-2,3,4- trifluorobenzene (17.62 g, 83.50 mmol) in THF (120 mL) was added. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HCl and pH was adjusted to 1- 2. The mixture was extracted with ethyl acetate (100 mL x 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (20.12 g, 94.5% yield). 1H NMR (400 MHz, DMSO-d6): δ 13.95 (s, 1H), 7.97 (m, 1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid
[0511] To a solution of 2-fluoroaniline (17.54 g, 157.80 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (20.12 g, 78.90 mmol) in THF (120 mL) was added LiHMDS (236.7 mL, 1 M in THF, 236.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with water (100 mL) and acidified to pH 2-3 with 10% HCl (aq.). The mixture was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 24.24 g, 88.8% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.22 (s, 1H), 8.01 (dd, J= 7.4, 2.1 Hz, 1H), 7.25 (m, 1H), 7.10 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate
[0512] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino) benzoic acid (24.24 g, 70.04 mmol) in MeOH (300 mL) was added thionyl chloride (20 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. After purification by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v), the corresponding product was obtained as a white solid (22.33 g, 88.5% yield). 1H NMR (400 MHz, CDC13): δ 9.06 (s, 1H), 8.01 (dd, J= 7.1, 2.3 Hz, 1H), 7.04 (m, 4H), 3.92 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate
[0513] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate (22.33 g, 62.01 mmol) in anhydrous 1,4-dioxane (200 mL) was added N,N- diisopropylethylamine (16.03 g, 124.04 mmol). Then Pd2(dba)3 (2.84 g, 3.10 mmol) followed by Xantphos (3.59 g, 6.20 mmol) and 4-methoxy-a-toluenethiol (10.27 g, 65.11 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to warm to ambient temperature. The insoluble matter was filtered off and the filter cake was washed ethyl acetate. The filtrate was diluted with water (300 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 24.35 g, 90.6% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.25 (m, 6H), 6.85 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H). Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate
[0514] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2- ((2- fluorophenyl)amino)benzoate (24.35 g, 56.18 mmol) in DMF (200 mL) was added NaN3 (4.38 g, 67.41 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (200 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 21.04 g, 82.1% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.10 (m, 6H), 6.84 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H). Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (21.04 g, 46.09 mmol) in MeOH (500 mL) was added and 10% palladium on carbon (3.40 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred for 2 h at ambient temperature. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (19.46 g, 98.1% yield). 1H NMR (400 MHz, CDC13): δ 9.07 (s, 1H), 7.77 (s, 1H), 7.06 (m, 4H), 6.95 (m, 2H), 6.81 (d, J = 8.3 Hz, 2H), 4.68 (s, 2H), 3.85 (s, 5H), 3.81 (s, 3H).
Step 7: dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2-fluorophenyl)amino)benzoate)
[0515] To a solution of methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (19.46 g, 45.21 mmol) in CH2C12 (180 mL) was added DDQ (11.29 g, 49.73 mmol) followed by water (20 mL). After stirring at ambient temperature for 10 h, the reaction was quenched by saturated sodium bicarbonate (aq., 100 mL). The aqueous layer was extracted by CH2C12 (100 mL χ 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 5: 1, v/v) to give the desired product (pale yellow solid, 9.81 g, 35.1% yield). 1H NMR (400 MHz, CDC13): δ 9.34 (s, 2H), 7.46 (s, 2H), 7.06 (m, 8H), 4.89 (br, 4H), 3.75 (s, 6H). Step 8: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate
[0516] To a solution of dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2- fluorophenyl)amino)benzoate) (9.81 g, 15.86 mmol) in THF/MeOH (100 mL, 10: 1, v/v) was added NaBH4 (3.00 g, 79.29 mmol) portion-wise in 1 h. After stirring at ambient temperature for 1 h, the reaction was quenched with 10% HCl (aq.) and pH was adjusted to 1-2. The aqueous layer was extracted with CH2C12 (50 mL χ 3). The combined organic phase was washed with water (50 mL) and brine (50 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was used directly in the next step without further purification.
Step 9: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate
[0517] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate in trimethyl orthoformate (50 mL) was added p-TsOU (0.61 g, 3.17 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (pale yellow solid, 8.64 g, 85.1% yield for two steps). 1H MR (400 MHz, CDC13): δ 9.13 (s, 1H), 8.68 (s, 1H), 8.46 (s, 1H), 7.10 (m, 1H), 7.01 (m, 1H), 6.92 (s, 2H), 3.97 (s, 3H).
Step 10: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate
[0518] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (8.64 g, 26.97 mmol) in DMF (100 mL) was added NIS (6.68 g, 29.67 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 5 h at ambient temperature, the reaction was treated by water (150 mL). The precipitate was filtered off and the filter cake was washed with water. The desired product was obtained as a yellow solid (10.34 g, 86.0% yield). 1H NMR (400 MHz, CDC13): δ 9.14 (s, 1H), 8.66 (s, 1H), 8.46 (s, 1H), 7.42 (d, J= 10.4 Hz, 1H), 7.31 (d, J= 8.8 Hz, 1H), 6.63 (dd, J= 15.0, 8.7 Hz, 1H), 3.97 (s, 3H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid
[0519] To a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino) benzo[d]thiazole-6- carboxylate (10.34 g, 23.17 mmol) in THF and MeOH (20 mL, 4: 1, v/v) was added 5.0 M LiOH (aq., 2 mL, 10 mmol). After stirring at ambient temperature for 2 h, the reaction was treated with 1.0 M HCl (aq.) till the solution was acidic. The aqueous layer was extracted with ethyl acetate (50 mL x 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (9.51 g, 95.0% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.10 (s, 1H), 9.18 (s, 1H), 8.68 (s, 1H), 8.45 (s, 1H), 7.41 (m, 1H), 7.30 (m, 1H), 6.65 (m, 1H). Step 12: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)etho
carboxamide
[0520] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6- carboxylic acid (519 mg, 1.20 mmol) in CH2C12 (10 mL) was added HOBt (254 mg, 1.63 mmol) and EDCI (314 mg, 1.63 mmol). The mixture was stirred for 1 h and O-(2-
(vinyloxy)ethyl)hydroxyl -amine (172 mg, 1.62 mmol) was added. After stirring for 4 h at ambient temperature, the reaction was treated with saturated H4C1 (aq.). The resultant mixture was extracted with CH2C12 (30 mL χ 3). The combined organic extracts were washed with water (30 mL) and brine (30 mL), dried over Na2S04 filtered, and concentrated in vacuo. The crude product (492 mg) was used directly in the next step without further purification.
Step 13: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzo[d]thiazole-6- carboxamide
[0521] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (492 mg, 1.00 mmol) in CH2C12 (10 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol). After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The aqueous layer was washed with CH2C12 (30 mL). The combined organic layer was washed with water (30 mL x 2) and brine (30 mL), dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 50: 1, v/v) and gave the desired product as a white solid (446 mg, 75.9% yield for the two steps). 1H MR (400 MHz, DMSO-d6): δ 11.80 (s, 1H), 9.55 (s, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.55 (d, J= 11.0 Hz, 1H), 7.31 (d, J= 8.5 Hz, 1H), 6.48 (d, J= 9.2 Hz, 1H), 4.72 (s, 1H), 3.84 (m, 2H), 3.57 (m, 2H). MS APCI(+)m/z: 491.8, [M+H].
Example 9A: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic aci

[0522] To a solution of l-bromo-2,3,4-trifluorobenzene (13.64 g, 64.6 mmol) in THF (120 mL) was added lithium diisopropylamide (2.0 M in THF, 33.9 mL, 67.8 mmol) at -78 °C under nitrogen atmosphere. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HC1 (300 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with 5% sodium hydroxide (300 mL). The aqueous layer was acidized to pH 1 and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (white solid, 13.51 g, 82% yield). 1H MR (400 MHz, CDC13): δ 13.94 (s, 1H), 7.95 (m,
1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic

[0523] To a solution of 2-fluoroaniline (10.2 mL, 105.8 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (13.51 g, 52.9 mmol) in THF (120 mL) was added LiHMDS (158.7 mL, 1 M in THF, 158.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with 10% HC1 (aq., 100 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 13.73 g, 75% yield). 1H MR (400 MHz, DMSO-d6): δ 9.21 (s, 1H), 8.01 (d, 1H), 7.26 (m, 1H), 7.01-7.16 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2- -fluorophenyl)amino)benzoate

[0524] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid (13.73 g, 39.6 mmol) in MeOH (300 mL) was added SOCl2 (60 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (300 mL χ 3). The combined organic extract was washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the corresponding product (gray solid, 12.58 g, 90% yield). 1H MR (400 MHz, CDC13): δ 9.09 (s, 1H), 8.05 (d, 1H), 7.00-7.14 (m, 4H), 3.94 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate

[0525] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate (12.85 g, 35.6 mmol) in anhydrous 1,4-dioxane (30 mL) was added N,N-diisopropylethylamine (9.21 g, 71.2 mmol). Then Pd2(dba)3 (1.63 g, 1.78 mmol) followed by Xantphos (2.06 g, 3.56 mmol) and 4-methoxy-a-toluenethiol (5.48 g, 35.6 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to cool to ambient temperature. The reaction was quenched with water (150 mL) and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was washed with water (200 mL χ 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 12.64 g, 82% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.06-7.44 (m, 6H), 6.82-6.88 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H).
Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0526] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2-((2- fluorophenyl)amino)benzoate (12.64 g, 29.2 mmol) in DMF (30 mL) was added NaN3 (2.28 g, 35.0 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (150 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL χ 3) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 10.38 g, 78% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.02-7.28 (m, 6H), 6.83- 6.85 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H).
Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0527] To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (10.38 g, 22.7 mmol) in MeOH (100 mL) was added and 10% palladium on carbon (1.55 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred at ambient temperature for 6 h. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (9.79 g, 100% yield).1H MR (400 MHz, CDC13): δ 9.08 (s, 1H), 7.78 (s, 1H), 6.93-7.28 (m, 8H), 4.65 (s, 2H), 4.00 (s, 2H), 3.89 (s, 3H), 3.75 (s, 3H).
Step 7: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate

[0528] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4- methoxybenzyl)thio)benzoate (9.79 g, 22.7 mmol) in anisole (12 mL) was added CF3COOH (20 mL). After stirring at ambient temperature for 23 h, the solvent was removed in vacuo. To the residue was added water (30 mL). The mixture was neutralized with 25% aqueous ammonia and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (white solid, 5.28 g, 75% yield). The product was used directly in the next step without further purification.
Step 8: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate

[0529] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate (2.07 g, 6.67 mmol) in trimethyl orthoformate (20 mL) was added p-TsOU (166 mg, 0.65 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (white solid, 1.963 g, 92% yield for two steps). 1H NMR (400 MHz, DMSO-d6): δ 9.01 (s, 1H), 8.08 (s, 1H), 7.90 (s, 1H), 7.15-6.78 (m, 4H), 3.91 (s, 3H).
Step 9: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate

[0530] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (1.963 g, 6.14 mmol) in DMF (10 mL) was added NIS (1.5 g, 6.5 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 4 h at ambient temperature, the reaction was treated by saturated H4C1 (aq.). The aqueous layer was extracted with ethyl acetate (150 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. After purification by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained as white solid (1.889 g, 69% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.10 (s, 1H), 7.93 (s, 1H), 7.18-6.72 (m, 3H), 3.91 (s, 3H).
Step 10: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy
carboxamide

[0531] To a solution of O-(2-(vinyloxy)ethyl)hydroxyl-amine (172 mg, 1.62 mmol) in THF (6 mL) was added LiHMDS (2.5 mL, 1 M in THF, 2.5 mmol) at -78 °C. After stirring at this temperature for 10 min, a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d] thiazole-6-carboxylate (360 mg, 0.81 mmol) in THF was syringed dropwisely. Then the mixture was allowed to warm to ambient temperature, quenched with saturated NH4C1 (aq., 20 mL) and extracted with ethyl acetate (15 mL χ 3). The combined organic extract was washed with water (10 mL x 3) and brine (10 mL), dried over Na2S04, filtered and concentrated in vacuo. After purification by flash chromatography (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained (410 mg, 98% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.85 (s, 1H),
8.98 (s, 1H), 8.04 (s, 1H), 7.89 (s, 1H), 7.55 (d, J= 10.8 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 6.53 (dd, J= 13.9, 6.6 Hz, 1H), 6.42 (d, J= 6.0 Hz, 1H), 4.21 (d, J= 14.5 Hz, 1H), 4.01 (m, 3H), 3.83 (m, 2H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzofdJthiazole-6- carboxamide

[0532] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (410 mg, 0.8 mmol) in CH2C12 (5 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol) dropwise. After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The organic layer was separated, washed with water (30 mL x 2) and brine (30 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 15: 1, v/v) and the desired product was obtained as a white solid (290 mg, 52 % yield). 1H MR (400 MHz, DMSO-de): δ 11.83 (s, 1H), 8.92 (s, 1H), 8.03 (s, 1H), 7.90 (s, 1H), 7.56 (d, J= 9.4 Hz, 1H), 7.30 (d, J= 8.7 Hz, 1H), 6.41 (m, 1H), 4.72 (m, 1H), 3.85 (m, 2H), 3.59 (m, 2H). MS (ES+): m/z 492.35 [MH+].
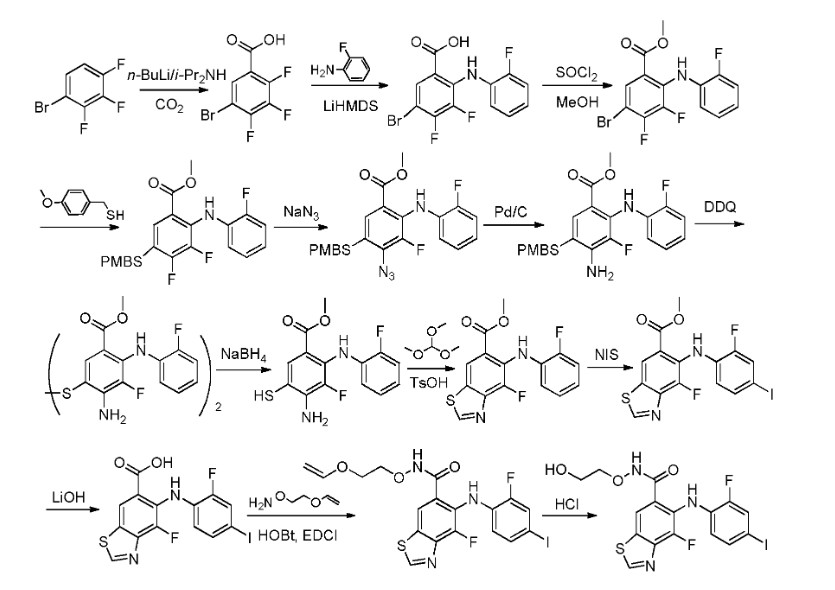
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping, it received conditional approval from the NMP in 2024 for the treatment of patients with advanced melanoma harboring NRAS mutations, particularly those who have not responded to anti-PD-1/PD-L1therapies [1]. Tunlametinib exerts its antitumor effects by targeting the MEK1/2 kinases within the RAS-RAF-MEK-ERK signaling pathway, thereby disrupting downstream signaling cascades and inhibiting tumor cell growth and proliferation [2]. Its clinical efficacy was demonstrated in a Phase II pivotal registration study (NCT05217303) involving patients with advanced NRAS-mutant melanoma [3]. The study reported a confirmed objective response rate (ORR) of 34.8 % and a median progression-free survival (mPFS) of 4.2 months. These findings suggest that Tunlametinib holds promise as a treatment option for NRAS-mutant melanoma, including in patients who have failed immunotherapy. In terms of safety, Tunlametinib has been generally well-tolerated [4]. Adverse events frequently encountered during treatment primarily consist of increased blood creatine phosphokinase (CPK) levels, diarrhea, and edema. Additionally, warnings and precautions pertinent to Tunlametinib therapy encompass decreased left ventricular ejection fraction (LVEF), skin toxicity, ocular toxicity, interstitial lung disease,
gastrointestinal reactions, and elevated CPK levels [5].
The synthetic pathway of Tunlametinib, illustrated in Scheme 1, begins with carboxylation of Tunl-001 to yield Tunl-002 [6]. Nucleophilic substitution of Tunl-002 with Tunl-003 then produces Tunl-004,
which undergoes esterification to form Tunl-005. Subsequent nucleophilic substitution between Tunl-05 and Tunl-006 generates Tunl-007. This intermediate undergoes azidation to afford Tunl-008, followed by
reduction to Tunl-009. Treatment of Tunl-009 with DDQ converts it to Tunl-010, which is deprotected to yield Tunl-011. Cycloaddition of Tunl-011 with Tunl-012 forms Tunl-013. Iodination of Tunl-013 gives
Tunl-014, which is hydrolyzed to produce Tunl-015. Amidation of Tunl-015 with Tunl-016 yields Tunl-017, and its subsequent acidolysis completes the synthesis of Tunlametinib.
[1] Y. Liu, Y. Cheng, G. Huang, X. Xia, X. Wang, H. Tian, Preclinical characterization of
tunlametinib, a novel, potent, and selective MEK inhibitor, Front. Pharmacol. 14
(2023) 1271268.
[2] S.J. Keam, Tunlametinib: first approval, Drugs 84 (2024) 1005–1010.
[3] X. Wei, Z. Zou, W. Zhang, M. Fang, X. Zhang, Z. Luo, J. Chen, G. Huang, P. Zhang,
Y. Cheng, J. Liu, J. Liu, J. Zhang, D. Wu, Y. Chen, X. Ma, H. Pan, R. Jiang, X. Liu,
X. Ren, H. Tian, Z. Jia, J. Guo, L. Si, A phase II study of efficacy and safety of the MEK inhibitor tunlametinib in patients with advanced NRAS-Mutant melanoma,
Eur. J. Cancer 202 (2024) 114008.
[4] Q. Zhao, T. Wang, H. Wang, C. Cui, W. Zhong, D. Fu, W. Xi, L. Si, J. Guo, Y. Cheng,
H. Tian, P. Hu, Phase I pharmacokinetic study of an oral, small-molecule MEK
inhibitor tunlametinib in patients with advanced NRAS mutant melanoma, Front.
Pharmacol. 13 (2022) 1039416.
[5] Y. Shi, X. Han, Q. Zhao, Y. Zheng, J. Chen, X. Yu, J. Fang, Y. Liu, D. Huang, T. Liu,
H. Shen, S. Luo, H. Yu, Y. Cao, X. Zhang, P. Hu, Tunlametinib (HL-085) plus
vemurafenib in patients with advanced BRAF V600-mutant solid tumors: an open-
label, single-arm, multicenter, phase I study, Exp. Hematol. Oncol. 13 (2024) 60.
[6] H. Tian, C. Ji, C. Liu, L. Kong, Y. Cheng, G. Huang, Benzoheterocyclic Compounds
and Use Thereof, 2014. US9937158B2.
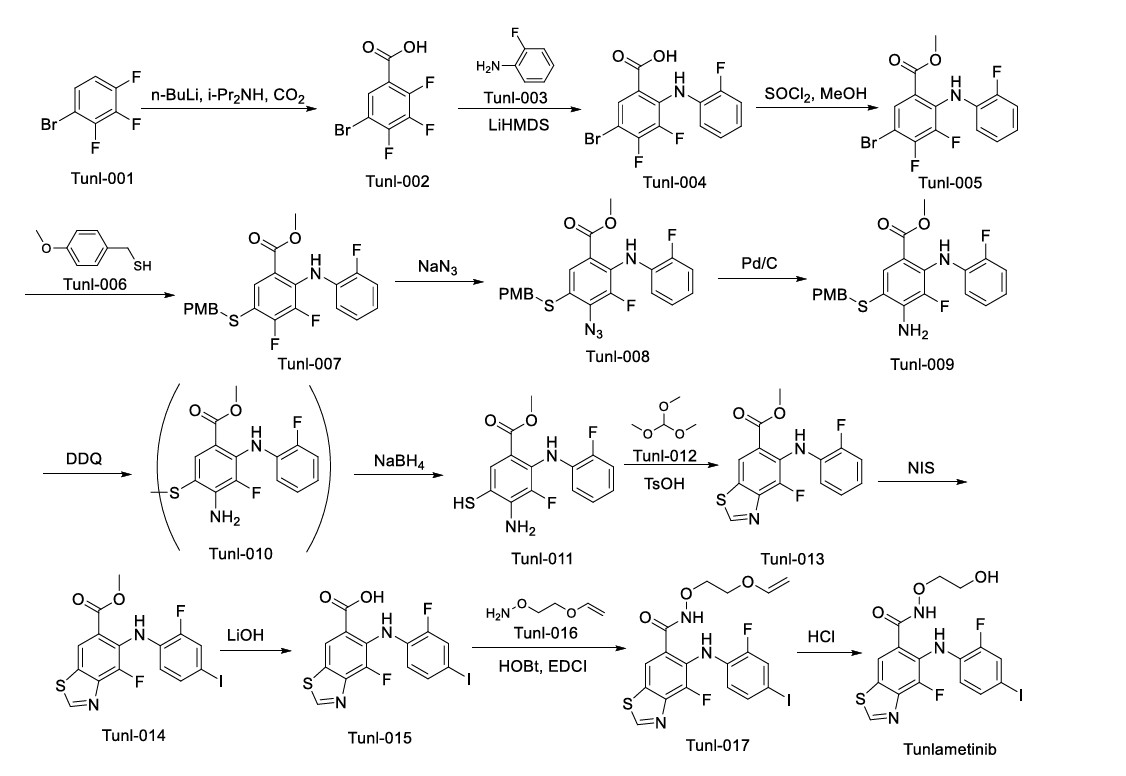
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Tunlametinib”. NCI Drug Dictionary. National Cancer Institute.
- “Tunlametinib Wins Approval in China for NRAS+ Advanced Melanoma After PD-1/PD-L1 Therapy”. 18 March 2024.
- Keam SJ (2024). “Tunlametinib: First Approval”. Drugs. 84 (8): 1005–1010. doi:10.1007/s40265-024-02072-x. PMID 39034326.
- Shi Y, Han X, Zhao Q, Zheng Y, Chen J, Yu X, et al. (2024). “Tunlametinib (HL-085) plus vemurafenib in patients with advanced BRAF V600-mutant solid tumors: An open-label, single-arm, multicenter, phase I study”. Experimental Hematology & Oncology. 13 (1): 60. doi:10.1186/s40164-024-00528-0. PMC 11167782. PMID 38867257.
| Clinical data | |
|---|---|
| Other names | HL-085 |
| ATC code | None |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1801756-06-8 |
| PubChem CID | 71621329 |
| ChemSpider | 115006753 |
| UNII | IF25NR1PV3 |
| ChEMBL | ChEMBL5095241 |
| Chemical and physical data | |
| Formula | C16H12F2IN3O3S |
| Molar mass | 491.25 g·mol−1 |
/////////Tunlametinib, CHINA 2024, APPROVALS 2024, Shanghai KeChow, Keluping,1801756-06-8, IF25NR1PV3, HL 085
Befotertinib
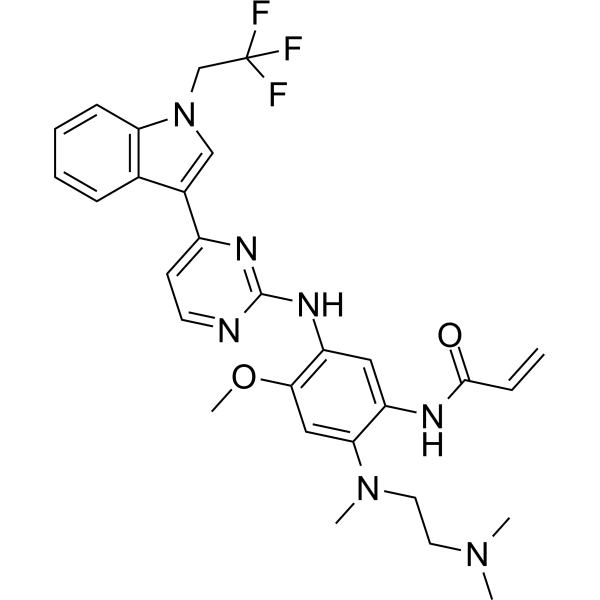
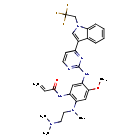
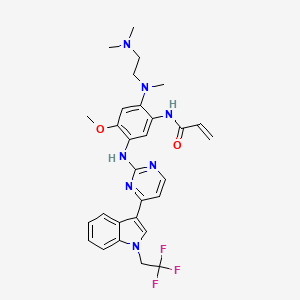
Befotertinib
D-0316, 0XT2CPR891
CAS No. : 1835667-63-4, MESYLATE CAS No. 2226167-02-6
- 2-propenamide, n-(2-((2-(dimethylamino)ethyl)methylamino)-4-methoxy-5-((4-(1-(2,2,2-trifluoroethyl)-1h-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-
- N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-(2,2,2-trifluoroethyl)-1h-indol-3-yl)pyrimidin-2-yl)amino)phenyl)prop-2-enamide
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(2,2,2-trifluoroethyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide
| Molecular Weight | 567.61 |
|---|---|
| Formula | C29H32F3N7O2 |
Befotertinib (D-0316) is an orally active EGFR tyrosine kinase inhibitor. Befotertinib can inhibit the proliferation of tumor cells. Befotertinib can be used in the research of EGFR T790M-positive non-small cell lung cancer (NSCLC).
Befotertinib is an orally available inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, befotertinib specifically binds to and inhibits EGFR T790M, a secondarily acquired resistance mutation, which prevents EGFR-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. Compared to some other EGFR inhibitors, befotertinib may have therapeutic benefits in tumors with T790M-mediated drug resistance. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization.
PAPER
J. Med. Chem. 2017, 60, 6480−6515.
PATENT
WO 2019218987
https://patentscope.wipo.int/search/en/WO2019218987
[0054]
U.S. Publication No. 2017/0355696 A1 describes a method of preparing Compound 4 and various pharmaceutically acceptable salts thereof. The exemplified synthetic process in U. S. Publication No. 2017/0355696 A1 includes a two-step conversion from the aniline compound, corresponding to Compound 1 of this disclosure, into the bismesylate of Compound 4, which has a low yield.
[0055]
As shown herein, representative methods of preparation of Compound 4, or a pharmaceutically acceptable salt, (or alternatively referred to as synthetic methods) , can provide the desired Compound 4, or a pharmaceutically acceptable salt, in improved yield and high purity and can be adapted for large-scale manufacture.
[0056]
In various embodiments, the present invention provides a novel method of preparing Compound 4, or a pharmaceutically acceptable salt thereof. The method typically includes converting a compound of Formula III, or a salt thereof, into compound 4, typically under an elimination reaction condition:
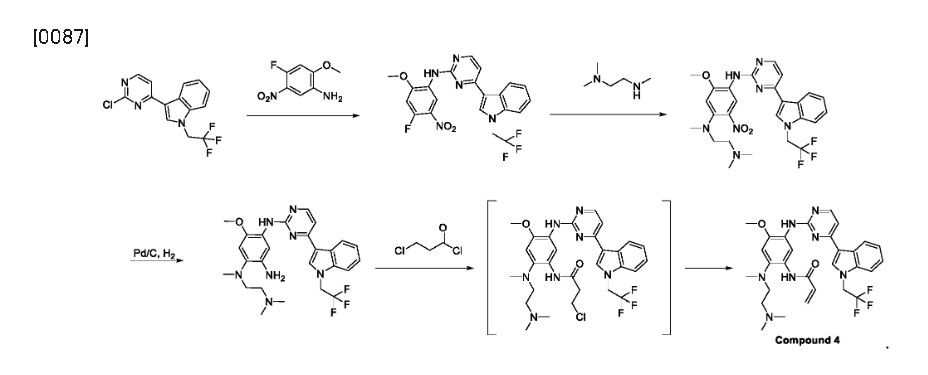
Syn
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Befotertinib (Surmana). Befotertinib (17), an oral, highly selective, third generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) developed by Betta Pharmaceuticals and InventisBio, was approved in China in May 2023 for the second-line treatment of patients
with locally advanced or metastatic nonsmall cell lung cancer (NSCLC) with positive EGFR T790 M mutation who have disease progression on previous EGFR TKI therapy. 140 139 NSCLC
has a high incidence and disease burden in China, which has spurred the development of multiple EGFR TKIs by Chinese companies.
Achromatography-free process route to befotertinib (17) has been reported in the patent literature by researchers at InventisBio (Scheme 29), although details about scale and yields were not provided.
141 142 The reaction sequence closely follows that of osimertinib, a third generation EGFR inhibitor
that was first approved in 2015 and was covered in our previous review.
Osimertinib and befotertinib share a common backbone, differing only in N-substitution on the indole ring.
Friedel−Crafts arylation of 1H-indole with 2,4-dichloropyrimidine (17.1) gave the 3-pyrimidinyl indole 17.2. The trifluoroethyl moiety in indole 17.4 was introduced via Nalkylation of 17.2 with triflate 17.3. This was followed by an SAr reaction with nitroaniline 17.5 to provide amino pyrimidine 17.6. Next, N,N,N′-trimethylethylenediamine (17.7) displaced the electrophilic aryl fluoride in an SNArreaction to generate intermediate 17.8. The acrylamide moiety was installed using a three-step sequence: hydrogenolytic
reduction of the nitro group to the corresponding aniline, acylation with 3-chloropropanoyl chloride, and immediate elimination to the acrylamide. Mesylate salt formation and crystallization furnished befotertinib mesylate (17) in eight steps from 17.1.
(139) Blair, H. A. Befotertinib: first approval. Drugs 2023, 83, 1433−
1437.
(140) Lau, S. C. M.; Ou, S.-H. I. And still they come over troubled
waters: can Asia’s third-generation EGFR tyrosine kinase inhibitors
(Furmonertinib, Aumolertinib, Rezivertinib, Limertinib, Befotertinib,
SH-1028, and Lazertinib) affect global treatment of EGFR+ NSCLC. J.
Thorac. Oncol. 2022, 17, 1144−1154.
(141) Dai, X.; Jiang, Y. Preparation of pyrimidine derivative and its
pharmaceutical salt as EGFR inhibitors for the treatment of cancer and
other diseases. WO 2019218987, 2019.
(142) Flick, A. C.; Ding, H. X.; Leverett, C. A.; Kyne, R. E.; Liu, K. K.
C.; Fink, S. J.; O’Donnell, C. J. Synthetic approaches to the new drugs
approved during 2015. J. Med. Chem. 2017, 60, 6480−6515.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Nagasaka M, et, al. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors For Advanced EGFR+ NSCLC. J Thorac Oncol. 2021 May;16(5):740-763. [Content Brief][2]. Blair HA. Befotertinib: First Approval. Drugs. 2023 Oct;83(15):1433-1437. [Content Brief]
/////////Befotertinib, APPROVALS 2023, CHINA 2023, Betta Pharmaceuticals, InventisBio, CANCER, D-0316, D 0316, 0XT2CPR891
Iruplinalkib
Iruplinalkib
CAS No. : 1854943-32-0
| Molecular Weight | 569.08 |
|---|---|
| Formula | C29H38ClN6O2P |
5-chloro-4-N-(2-dimethylphosphorylphenyl)-2-N-[2-methoxy-4-(9-methyl-3,9-diazaspiro[5.5]undecan-3-yl)phenyl]pyrimidine-2,4-diamine
Iruplinalkib (WX-0593) is an orally active and selective ALK/ROS1 inhibitor. Iruplinalkib can effectively inhibit tyrosine autophosphorylation of ALK and mutant ALK, EGFR, with the IC50 between 5.38 and 16.74 nM. Iruplinalkib is also a suppressive agent of the transporter MATE1, MATE2K, P-gp and BCRP. Iruplinalkib can be used in the study of non-small cell lung cancer.
Iruplinalkib is an orally available, small molecule inhibitor of the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK), with potential antineoplastic activity. Upon oral administration, iruplinalkib binds to and inhibits ALK tyrosine kinase, ALK fusion proteins, ALK point mutation variants ALK L1196M, ALK C1156Y, and EGFR L858R/T790M. Inhibition of ALK leads to the disruption of ALK-mediated signaling and the inhibition of cell growth in ALK-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a series of tumors. Additionally, ALK mutations are associated with acquired resistance to small molecule tyrosine kinase inhibitors.
SYN
Bioorg. Chem. 2023, 140, No. 106807
Bioorg. Med. Chem. Lett. 2022, 66, No. 128730.
CN106928275, 2017.
EP 3165530 A1, 2017
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US196228122&_cid=P22-ME5G3V-88608-1
Example 9
(2-((5-Chloro-2-((2-methoxy-4-(9-methyl-3,9-diazaspiro[5.5]undecan-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide
This Example was prepared according to the process as described in Example 7, take the place of (2-((5-chloro-2-((2-methoxy-4-(2,6-diazaspiro[3.4]octan-6-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide was replaced with (2-((5-chloro-2-((2-methoxy-4-(3,9-diazaspiro[5.5]undecan-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide to give the title compound as yellow solid, yield 57%. 1H NMR (400 MHz, CD 3OD): δ, 8.28 (s, 1H), 8.12 (br. s., 1H), 7.81-7.68 (m, 3H), 7.65 (d, J=2.0 Hz, 1H), 7.56-7.49 (m, 1H), 7.37 (d, J=8.8 Hz, 1H), 4.02 (s, 3H), 3.74 (br. s., 4H), 3.47 (d, J=12.8 Hz, 2H), 3.24 (t, J=12.8 Hz, 2H), 2.93 (s, 3H), 2.42-1.97 (m, 5H), 1.93-1.75 (m, 9H). LCMS (ESI) (0-60AB): m/z: 569.2 [M+1].
PATENT
CN 110407877, 2019
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN276468167&_cid=P22-ME5FTE-83156-1
| Experimental Example 11 |
| (2-((5-chloro-2-((2-methoxy-4-(9-methyl-3,9-diaza-spiro[5.5]undec-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethylphosphine oxide (Compound I) |
| NMP (N-methylpyrrolidone, 102.6 g) was added to the reaction bottle (room temperature), and 20 g of compound (e) was added to the reaction bottle under stirring. 21.85 g of compound (f) was added to the reaction bottle, and 19.93 g (3 eq) of MeSO 3 H was added dropwise to the reaction flask (temperature was controlled at <40°C) 2 Bubble for 15-20 minutes. Heat the reaction to 85-90°C and react at 85-90°C for 12 hours before sampling and testing (HPLC). Sample testing (test method: dissolve 0.1 ml of the reaction solution in 2 ml of MeOH). Stop the reaction when compound (f) is <2.5%. Add NaOH solution (1M, 287 g) to the reaction solution to adjust the pH to 13. A large amount of solid precipitates and continue stirring for 2 hours. Filter the solid and wash the filter cake with water (40 g) until the filtrate is colorless. Collect the filter cake, filter and dry (55-60°C) to obtain an off-white crude product. Add methanol (268 g) to the reaction flask, then add the obtained solid, heat to 60°C to dissolve, add activated carbon (5.2 g) to the reaction flask, then stir at 60°C for 2.5 hours, cool to 30°C, filter, and concentrate the mother liquor under reduced pressure to obtain a gray-green solid. MeOH (155.5 g) was added to the reaction flask, followed by the solid obtained above. The mixture was heated under reflux until clear. Purified water (388 g) was added to the reaction flask and the temperature was naturally lowered to 15-20°C. A white solid precipitated. The solid was filtered and washed once with purified water (194 g). The filter cake was collected and dried to yield Compound I (32.5 g). 1 H NMR (400MHz, CD3OD): 8.36 (dd, J=8.0, 4.4Hz, 1H), 8.03 (s, 1H), 7.69 (d, J=8.8Hz, 1H), 7.65-7.55 (m, 1H), 7.51 (dd, J=8.0 8.0Hz,1H),7.32-7.20(m,1H),6.66(d,J=2.4Hz,1H),6.45(dd,J=8.8,2.4Hz,1H),3.85(s,3H) ,3.18-3.06(m,4H),2.54-2.38(m,4H),2.30(s,3H),1.85(d,J=13.6Hz,6H),1.74-1.54(m,8H). |
| Example 1: Preparation of Form A of Compound I: |
| 10 g of (2-((5-chloro-2-((2-methoxy-4-(9-methyl-3,9-diaza-spiro[5.5]undec-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethylphosphine oxide (Compound I) was heated to reflux for complete dissolution with 45 ml of ethanol and 20 ml of purified water. The mixture was then cooled to 10-20° C., 50 ml of purified water was added, and the mixture was stirred at this temperature for 2-3 hours. The mixture was filtered and dried in vacuo at 50-60° C. to obtain 9.0 g of an off-white solid with a yield of 90%. X-ray powder diffraction analysis showed an XRPD pattern as shown in FIG1 . DSC-TGA analysis yielded pattern 2. |
Syn
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Iruplinalkib. The final NSCLC treatment approved in 2023 was iruplinalkib (20), developed by Qilu Pharmaceutical Co., Ltd. and approved for use in China in June 2023.156,157 Iruplinalkib is a highly selective oral anaplastic kinase (ALK) and ROS1TKI,andis, therefore, an oral treatment for ALK-positive (ALK+) or ROS1-positive (ROS1+) NSCLC.156,157 This treatment is specifically meant for use by patients with locally advanced or metastatic ALK-positive NSCLC whose disease has progressed after crizotinib therapy, often due to the develop ment of crizotinib resistance. Although early syntheses were published 158 further optimized routes were disclosed by Qilu Pharmaceutical Co., Ltd.(Scheme 34 and Scheme 35).First, ethyl 2-cyanoacetate 20.1) and cyclic ketone20.2 were combined to generate spirocyclic imide 20.3 (Scheme 34). The cyano substituents were then removed by treating 20.3 with sulfuric acid followed
by sodiumhydroxide to form 20.5. Subsequent amide reduction via lithium aluminum hydride treatment revealed secondary amine 20.6, which underwent a substitution reaction with fluorophenyl 20.7 to produce intermediate 20.8. Next, addition of methyl iodide generated a quaternary amine (20.9). The
following hydrogenolysis step reduced the nitro group and removed the benzyl substituent using palladium on carbon to produce the final aniline fragment, 20.10. To generate the other fragment (20.16) to construct iruplinalkib, diethylphosphite (20.11) and methyl magnesium bromide were combinedtoproducedimethylphosphineoxide 20.12(Scheme35).Couplingthisproductwith2-iodoaniline
20.13andsubsequent selectiveSNArwithtrichloropyrimidine20.15 generated the resultingdichloropyrimidine20.16.Finally, a selective SNAr reaction yielded the desired product,
iruplinalkib(20).
(156) Yang, Y.; Zheng, Q.; Wang, X.; Zhao, S.; Huang, W.; Jia, L.; Ma,
C.; Liu, S.; Zhang, Y.; Xin, Q.; et al. Iruplinalkib (WX-0593), a novel
ALK/ROS1 inhibitor, overcomes crizotinib resistance in preclinical
modelsfornon-smallcell lung cancer. Invest. New Drugs 2023, 41,254−
266.
(157) Keam, S. J. Iruplinalkib: first approval. Drugs 2023, 83, 1717−
1721.
(158) Liu, X.; Zhang, L.; Wan, H.; Zhu, Z.; Jin, J.; Qin, Y.; Mao, W.;
Yan, K.; Fang, D.; Jiang, W.; et al. Discovery and preclinical evaluations
of WX-0593, a novel ALK inhibitor targeting crizotinib-resistant
mutations. Bioorg. Med. Chem. Lett. 2022, 66, No. 128730.
(159) Ding, Z.; Chen, S.; Liu, X.; Wan, H.; Zhang, L. Preparation,
intermediate and crystal form of spiroamine type arylphosphine oxide.
CN106928275, 2017.
(160) Ding, Z.; Zhang, M.; Chen, S.; Liu, X.; Zhu, Y.; Fan, C.;
Baoping, Z.; Chang, L.; Yang, Y.; Zheng, Q.; et al. Preparation,
intermediate and crystal form of spiroamine type arylphosphine oxide.
EP 3165530 A1, 2017.
(161) Lin, D.; Zhou, G.; Li, S.; Wang, X.; Zhang, Z.; Liu, Z.; Wang, X.
Polymorph of spirocycloaryl phosphorus oxide. CN 110407877, 2019.
(162) Gao, H.; Zhang, J.-Y.; Zhao, L.-J.; Guo, Y.-Y. Synthesis and
clinical application of small-molecule inhibitors and PROTACs of
anaplastic lymphoma kinase. Bioorg. Chem. 2023, 140, No. 106807.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Yang Y, et al. Iruplinalkib (WX 0593), a novel ALK/ROS1 inhibitor, overcomes crizotinib resistance in preclinical models for non-small cell lung cancer. Invest New Drugs. 2023 Apr;41(2):254-266. [Content Brief][2]. Shi Y, et al. Safety and activity of WX-0593 (Iruplinalkib) in patients with ALK- or ROS1-rearranged advanced non-small cell lung cancer: a phase 1 dose-escalation and dose-expansion trial. Signal Transduct Target Ther. 2022;7(1):25. [Content Brief]
/////////Iruplinalkib, china 2023, approvals 2023, Qilu Pharmaceutical, Z5F65W1YAZ, Qixinke, WX0593, WX 0593, FL006, FL 006, FL-006
Zongertinib
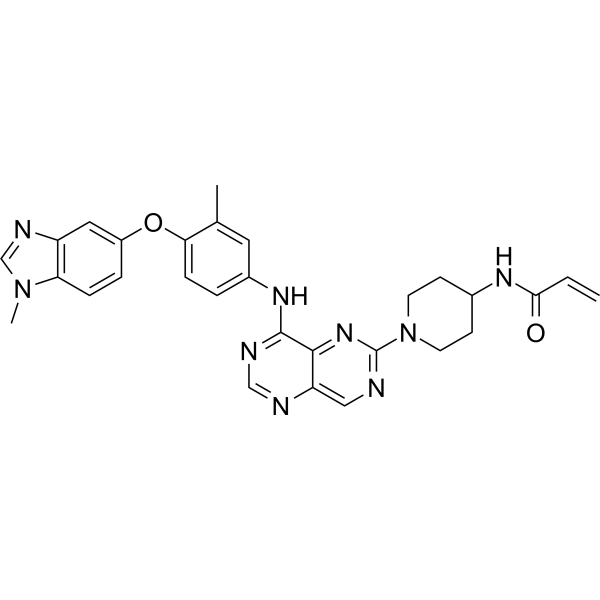
Zongertinib
CAS No. : 2728667-27-2,
BI-1810631, BI1810631
| Molecular Weight | 535.60 |
|---|---|
| Formula | C29H29N9O2 |
FDA 8/8/2025, Hernexeos, To treat adults with unresectable or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 tyrosine kinase domain activating mutations, as detected by an FDA-approved test, and who have received prior systemic therapy
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo(d)imidazol-5-yl)oxy)phenyl)amino)pyrimido(5,4-d)pyrimidin-2-yl)piperidin-4-yl)acrylamide
- 884-819-6
Zongertinib is an orally bioavailable inhibitor of the receptor tyrosine kinase human epidermal growth factor receptor 2 (HER2; ErbB2; HER-2), with potential antineoplastic activity. Upon oral administration, zongertinib covalently binds to and inhibits the activity of both wild-type and HER2 mutants, including HER2 mutants with exon 20 insertion (ex20ins) mutations. This prevents HER2-mediated signaling and may lead to cell death in HER2-expressing tumor cells. HER2, a receptor tyrosine kinase overexpressed on a variety of tumor cell types, plays an important role in tumor cell proliferation and tumor vascularization.
REF
Synthesis of zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-
548 yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
Methods
Synthesis of Zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
An overview of the synthetic routes to zongertinib and BI-3999 is shown in Supplementary Fig. S1, and graphical NMR spectra are shown in Supplementary Fig. S2.
3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)aniline (500 mg, 1.97 mmol) and 8-chloro-2-(methylthio)pyrimido[5,4-d]pyrimidine hydrochloride (492 mg, 1.97 mmol) were suspended in isopropanol, and the resulting reaction mixture stirred at 50°C for 3 hours, at which time high-performance liquid chromatography–mass spectrometry (HPLC-MS) indicated full conversion. The reaction mixture was concentrated under reduced pressure, and the crude product was redissolved in dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–15% methanol in dichloromethane) to afford the product (840 mg).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylthio)pyrimido[5,4-d]pyrimidin-4-amine (860 mg, 90%, 1.80 mmol) was suspended in dichloromethane (30 mL), and the resulting mixture was cooled to 0°C to 5°C. mCPBA (3-chloroperbenzoic acid, 444 mg, 77%, 1.98 mmol) was added portionwise over 1 hour, and the resulting reaction mixture was stirred at room temperature overnight, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product which was used directly in the next step (767 mg, crude).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylsulfinyl)pyrimido[5,4-d]pyrimidin-4-amine (5.42 g, 80%, 9.73 mmol) was dissolved in N,N-dimethyl formamide (DMF, 50 mL) and diisopropylethylamine (2.8 mL, 16 mmol). 4-Boc-amino-1-piperidine (2.39 g, 11.9 mmol) was added, and the reaction was stirred at 60°C overnight. Then, the reaction mixture was concentrated, and the crude product was used directly in the next step (5.66 g, crude).
Tert-butyl (1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)carbamate (5.66 g, 9.73 mmol) was dissolved in dichloromethane (100 mL) and methanol (30 mL). Four mol/L HCl in dioxane (11 mL, 44 mmol) was added, and the resulting reaction mixture was heated to 45°C for 7 hours. HPLC-MS indicated some remaining starting material; therefore, the reaction mixture was stirred at room temperature overnight. Four mol/L HCl in dioxane (1 mL, 0.40 mmol) was added, and the reaction mixture was reheated to 45°C for 4 hours, at which time HPLC-MS indicated full conversion. The reaction mixture was concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (4.5 g, 70% purity).
1-[8-({3-methyl-4-[(1-methyl-1H-1,3-benzodiazol-5-yl)oxy]phenyl}amino)-[1,3]diazino[5,4-d]pyrimidin-2-yl]piperidin-4-amine (4.5 g, 70%, 6.9 mmol) was suspended in dichloromethane (150 mL) and triethyl amine (4 mL, 28 mmol), and dimethylaminopyridine (115 mg, 0.941 mmol) was added. Then, acroyloyl anhydride (1.36 g, 95%, 10.3 mmol) was added, and the resulting reaction mixture was stirred at room temperature for 1 hour, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane (50 mL) and washed with aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (2.49 g).
1H NMR (DMSO-d6, 500 MHz) δ 9.58 (s, 1H), 9.08 (s, 1H), 8.39 (s, 1H), 8.19 (s, 1H), 8.10 (d, 1H, J = 7.6 Hz), 7.84 (d, 1H, J = 2.2 Hz), 7.77 (dd, 1H, J = 8.8 Hz, J = 2.2 Hz), 7.57 (d, 1H, J = 8.8 Hz), 7.09 (d, 1H, J = 2.2 Hz), 7.00 (dd, 1H, J = 2.2, 8.5 Hz), 6.89 (d, 1H, J = 8.8 Hz), 6.20 (dd, 1H, J = 10.1, 17.0 Hz), 6.10 (dd, 1H, J = 2.2, 17.0 Hz), 5.6 (dd, 1H, J = 2.2, 9.8 Hz), 4.86 (m, 2H), 3.99 (m, 1H), 3.84 (s, 3H), 3.25 (m, 2H), 2.26 (s, 3H), 1.92 (m, 2H), and 1.43 (m, 2H).
Synthesis of BI-3999 (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acetamide)
6-(4-aminopiperidin-1-yl)-N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)pyrimido[5,4-d]pyrimidin-4-amine (100 mg, 208 mmol) and 4-dimethylaminopyridine (2.5 mg, 0.02 mmol) were suspended in 5 mL dichloromethane. Acetic anhydride (25 μL, 0.23 mmol) was added, and the resulting reaction mixture was stirred at room temperature for one hour. Then, the reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3 and brine. Then, the layers were separated, and the organic layer was dried over MgSO4 and concentrated. The crude product was purified by column chromatography (SiO2, gradient of 0%–10% methanol in dichloromethane) to afford the product (75 mg).
1H NMR (DMSO-d6, 400 MHz) δ 9.58 (s, 1H), 9.07 (s, 1H), 8.39 (s, 1H), 8.17 (s, 1H), 7.88 (d, 1H, J = 7.9 Hz), 7.84 (d, 1H, J = 2.5 Hz), 7.77 (dd, 1H, J = 2.7, 8.7 Hz), 7.57 (d, 1H, J = 8.9 Hz), 7.09 (d, 1H, J = 2.3 Hz), 7.00 (dd, 1H, J = 2.3, 8.6 Hz), 6.89 (d, 1H, J = 8.6 Hz), 4.85 (m, 2H), 3.90 (m, 1H), 3.84 (s, 3H), 3.23 (m, 2H), 2.26 (s, 3H), 1.88 (m, 2H), 1.82 (s, 3H), and 1.38 (m, 2H).
A) 1H NMR spectrum of zongertinib
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021213800&_cid=P10-ME52KD-62836-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. WHO Drug Informat ion – World Health Organization (WHO).[2]. Wilding Birgit, et al. Synthesis of diazino-pyrimidines as anticancer agents: World Intellectual Property Organization, WO2021213800. 2021-10-28.[3]. Li S, et al. Emerging Targeted Therapies in Advanced Non-Small-Cell Lung Cancer. Cancers (Basel). 2023 May 24;15(11):2899. [Content Brief]
////////////Zongertinib, Hernexeos, APPROVALS 2025, FDA 2025, lung cancer, BI-1810631, BI1810631, DRH7R67UVL

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}