
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
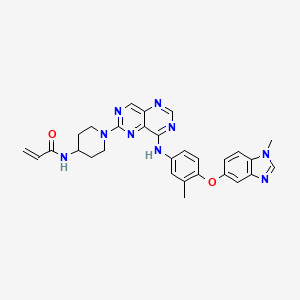
Iruplinalkib
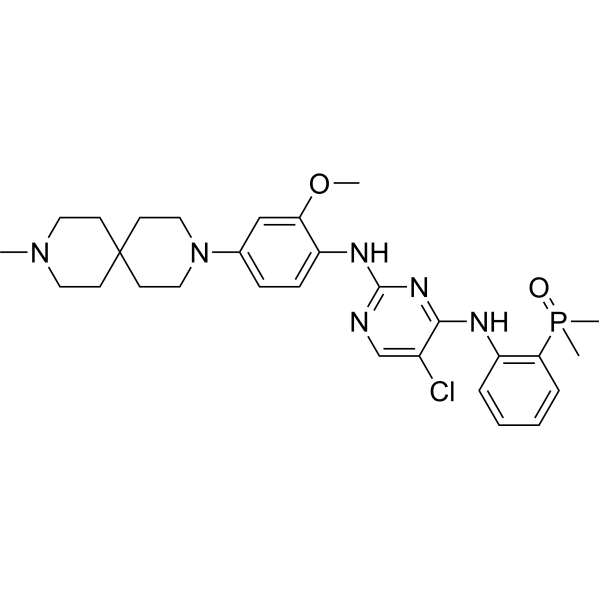
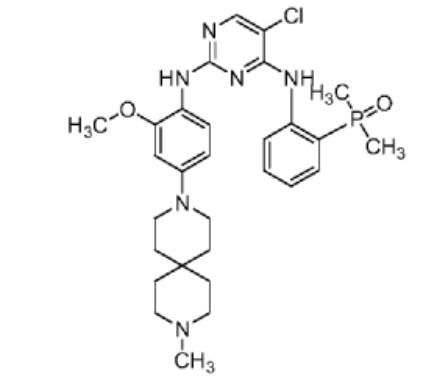
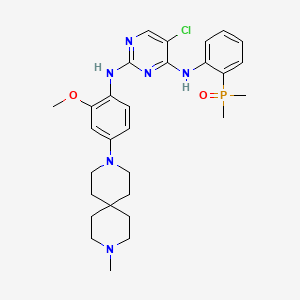
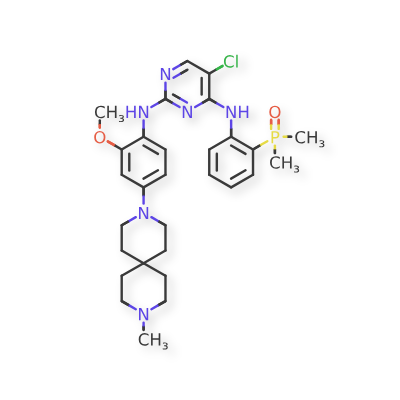
Iruplinalkib
CAS No. : 1854943-32-0
| Molecular Weight | 569.08 |
|---|---|
| Formula | C29H38ClN6O2P |
5-chloro-4-N-(2-dimethylphosphorylphenyl)-2-N-[2-methoxy-4-(9-methyl-3,9-diazaspiro[5.5]undecan-3-yl)phenyl]pyrimidine-2,4-diamine
Iruplinalkib (WX-0593) is an orally active and selective ALK/ROS1 inhibitor. Iruplinalkib can effectively inhibit tyrosine autophosphorylation of ALK and mutant ALK, EGFR, with the IC50 between 5.38 and 16.74 nM. Iruplinalkib is also a suppressive agent of the transporter MATE1, MATE2K, P-gp and BCRP. Iruplinalkib can be used in the study of non-small cell lung cancer.
Iruplinalkib is an orally available, small molecule inhibitor of the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK), with potential antineoplastic activity. Upon oral administration, iruplinalkib binds to and inhibits ALK tyrosine kinase, ALK fusion proteins, ALK point mutation variants ALK L1196M, ALK C1156Y, and EGFR L858R/T790M. Inhibition of ALK leads to the disruption of ALK-mediated signaling and the inhibition of cell growth in ALK-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a series of tumors. Additionally, ALK mutations are associated with acquired resistance to small molecule tyrosine kinase inhibitors.
SYN
Bioorg. Chem. 2023, 140, No. 106807
Bioorg. Med. Chem. Lett. 2022, 66, No. 128730.
CN106928275, 2017.
EP 3165530 A1, 2017
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US196228122&_cid=P22-ME5G3V-88608-1
Example 9
(2-((5-Chloro-2-((2-methoxy-4-(9-methyl-3,9-diazaspiro[5.5]undecan-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide
This Example was prepared according to the process as described in Example 7, take the place of (2-((5-chloro-2-((2-methoxy-4-(2,6-diazaspiro[3.4]octan-6-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide was replaced with (2-((5-chloro-2-((2-methoxy-4-(3,9-diazaspiro[5.5]undecan-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethyl phosphine oxide to give the title compound as yellow solid, yield 57%. 1H NMR (400 MHz, CD 3OD): δ, 8.28 (s, 1H), 8.12 (br. s., 1H), 7.81-7.68 (m, 3H), 7.65 (d, J=2.0 Hz, 1H), 7.56-7.49 (m, 1H), 7.37 (d, J=8.8 Hz, 1H), 4.02 (s, 3H), 3.74 (br. s., 4H), 3.47 (d, J=12.8 Hz, 2H), 3.24 (t, J=12.8 Hz, 2H), 2.93 (s, 3H), 2.42-1.97 (m, 5H), 1.93-1.75 (m, 9H). LCMS (ESI) (0-60AB): m/z: 569.2 [M+1].
PATENT
CN 110407877, 2019
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN276468167&_cid=P22-ME5FTE-83156-1
| Experimental Example 11 |
| (2-((5-chloro-2-((2-methoxy-4-(9-methyl-3,9-diaza-spiro[5.5]undec-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethylphosphine oxide (Compound I) |
| NMP (N-methylpyrrolidone, 102.6 g) was added to the reaction bottle (room temperature), and 20 g of compound (e) was added to the reaction bottle under stirring. 21.85 g of compound (f) was added to the reaction bottle, and 19.93 g (3 eq) of MeSO 3 H was added dropwise to the reaction flask (temperature was controlled at <40°C) 2 Bubble for 15-20 minutes. Heat the reaction to 85-90°C and react at 85-90°C for 12 hours before sampling and testing (HPLC). Sample testing (test method: dissolve 0.1 ml of the reaction solution in 2 ml of MeOH). Stop the reaction when compound (f) is <2.5%. Add NaOH solution (1M, 287 g) to the reaction solution to adjust the pH to 13. A large amount of solid precipitates and continue stirring for 2 hours. Filter the solid and wash the filter cake with water (40 g) until the filtrate is colorless. Collect the filter cake, filter and dry (55-60°C) to obtain an off-white crude product. Add methanol (268 g) to the reaction flask, then add the obtained solid, heat to 60°C to dissolve, add activated carbon (5.2 g) to the reaction flask, then stir at 60°C for 2.5 hours, cool to 30°C, filter, and concentrate the mother liquor under reduced pressure to obtain a gray-green solid. MeOH (155.5 g) was added to the reaction flask, followed by the solid obtained above. The mixture was heated under reflux until clear. Purified water (388 g) was added to the reaction flask and the temperature was naturally lowered to 15-20°C. A white solid precipitated. The solid was filtered and washed once with purified water (194 g). The filter cake was collected and dried to yield Compound I (32.5 g). 1 H NMR (400MHz, CD3OD): 8.36 (dd, J=8.0, 4.4Hz, 1H), 8.03 (s, 1H), 7.69 (d, J=8.8Hz, 1H), 7.65-7.55 (m, 1H), 7.51 (dd, J=8.0 8.0Hz,1H),7.32-7.20(m,1H),6.66(d,J=2.4Hz,1H),6.45(dd,J=8.8,2.4Hz,1H),3.85(s,3H) ,3.18-3.06(m,4H),2.54-2.38(m,4H),2.30(s,3H),1.85(d,J=13.6Hz,6H),1.74-1.54(m,8H). |
| Example 1: Preparation of Form A of Compound I: |
| 10 g of (2-((5-chloro-2-((2-methoxy-4-(9-methyl-3,9-diaza-spiro[5.5]undec-3-yl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)dimethylphosphine oxide (Compound I) was heated to reflux for complete dissolution with 45 ml of ethanol and 20 ml of purified water. The mixture was then cooled to 10-20° C., 50 ml of purified water was added, and the mixture was stirred at this temperature for 2-3 hours. The mixture was filtered and dried in vacuo at 50-60° C. to obtain 9.0 g of an off-white solid with a yield of 90%. X-ray powder diffraction analysis showed an XRPD pattern as shown in FIG1 . DSC-TGA analysis yielded pattern 2. |
Syn
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
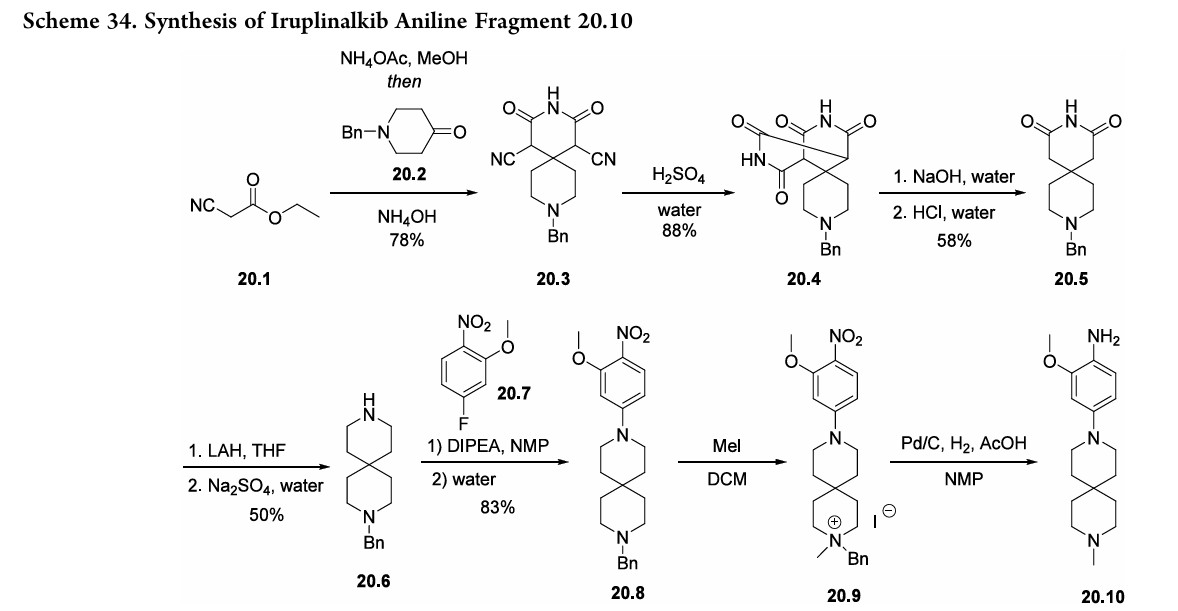
Iruplinalkib. The final NSCLC treatment approved in 2023 was iruplinalkib (20), developed by Qilu Pharmaceutical Co., Ltd. and approved for use in China in June 2023.156,157 Iruplinalkib is a highly selective oral anaplastic kinase (ALK) and ROS1TKI,andis, therefore, an oral treatment for ALK-positive (ALK+) or ROS1-positive (ROS1+) NSCLC.156,157 This treatment is specifically meant for use by patients with locally advanced or metastatic ALK-positive NSCLC whose disease has progressed after crizotinib therapy, often due to the develop ment of crizotinib resistance. Although early syntheses were published 158 further optimized routes were disclosed by Qilu Pharmaceutical Co., Ltd.(Scheme 34 and Scheme 35).First, ethyl 2-cyanoacetate 20.1) and cyclic ketone20.2 were combined to generate spirocyclic imide 20.3 (Scheme 34). The cyano substituents were then removed by treating 20.3 with sulfuric acid followed
by sodiumhydroxide to form 20.5. Subsequent amide reduction via lithium aluminum hydride treatment revealed secondary amine 20.6, which underwent a substitution reaction with fluorophenyl 20.7 to produce intermediate 20.8. Next, addition of methyl iodide generated a quaternary amine (20.9). The
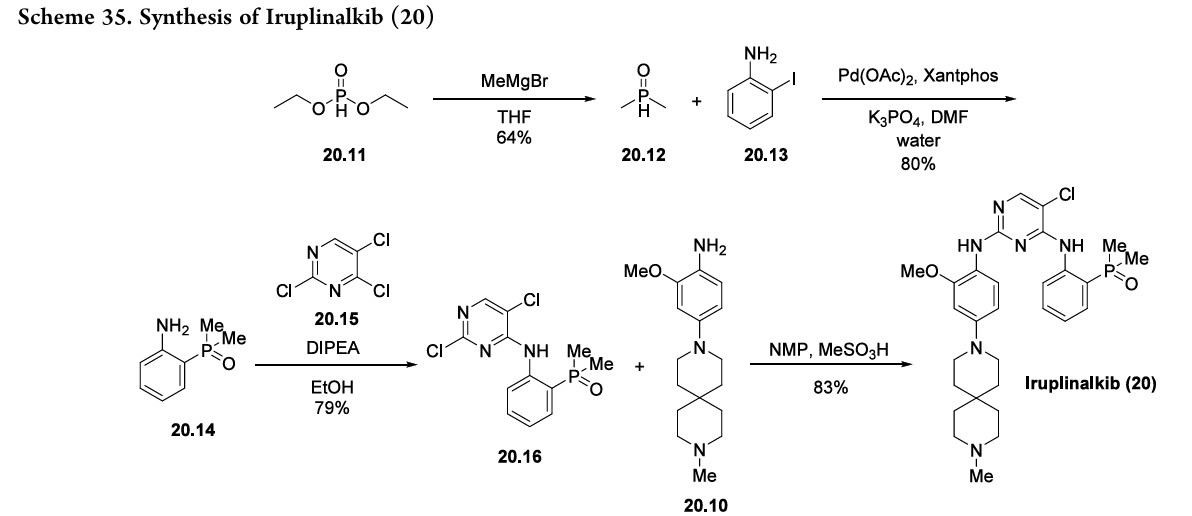
following hydrogenolysis step reduced the nitro group and removed the benzyl substituent using palladium on carbon to produce the final aniline fragment, 20.10. To generate the other fragment (20.16) to construct iruplinalkib, diethylphosphite (20.11) and methyl magnesium bromide were combinedtoproducedimethylphosphineoxide 20.12(Scheme35).Couplingthisproductwith2-iodoaniline
20.13andsubsequent selectiveSNArwithtrichloropyrimidine20.15 generated the resultingdichloropyrimidine20.16.Finally, a selective SNAr reaction yielded the desired product,
iruplinalkib(20).
(156) Yang, Y.; Zheng, Q.; Wang, X.; Zhao, S.; Huang, W.; Jia, L.; Ma,
C.; Liu, S.; Zhang, Y.; Xin, Q.; et al. Iruplinalkib (WX-0593), a novel
ALK/ROS1 inhibitor, overcomes crizotinib resistance in preclinical
modelsfornon-smallcell lung cancer. Invest. New Drugs 2023, 41,254−
266.
(157) Keam, S. J. Iruplinalkib: first approval. Drugs 2023, 83, 1717−
1721.
(158) Liu, X.; Zhang, L.; Wan, H.; Zhu, Z.; Jin, J.; Qin, Y.; Mao, W.;
Yan, K.; Fang, D.; Jiang, W.; et al. Discovery and preclinical evaluations
of WX-0593, a novel ALK inhibitor targeting crizotinib-resistant
mutations. Bioorg. Med. Chem. Lett. 2022, 66, No. 128730.
(159) Ding, Z.; Chen, S.; Liu, X.; Wan, H.; Zhang, L. Preparation,
intermediate and crystal form of spiroamine type arylphosphine oxide.
CN106928275, 2017.
(160) Ding, Z.; Zhang, M.; Chen, S.; Liu, X.; Zhu, Y.; Fan, C.;
Baoping, Z.; Chang, L.; Yang, Y.; Zheng, Q.; et al. Preparation,
intermediate and crystal form of spiroamine type arylphosphine oxide.
EP 3165530 A1, 2017.
(161) Lin, D.; Zhou, G.; Li, S.; Wang, X.; Zhang, Z.; Liu, Z.; Wang, X.
Polymorph of spirocycloaryl phosphorus oxide. CN 110407877, 2019.
(162) Gao, H.; Zhang, J.-Y.; Zhao, L.-J.; Guo, Y.-Y. Synthesis and
clinical application of small-molecule inhibitors and PROTACs of
anaplastic lymphoma kinase. Bioorg. Chem. 2023, 140, No. 106807.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- [1]. Yang Y, et al. Iruplinalkib (WX 0593), a novel ALK/ROS1 inhibitor, overcomes crizotinib resistance in preclinical models for non-small cell lung cancer. Invest New Drugs. 2023 Apr;41(2):254-266. [Content Brief][2]. Shi Y, et al. Safety and activity of WX-0593 (Iruplinalkib) in patients with ALK- or ROS1-rearranged advanced non-small cell lung cancer: a phase 1 dose-escalation and dose-expansion trial. Signal Transduct Target Ther. 2022;7(1):25. [Content Brief]
/////////Iruplinalkib, china 2023, approvals 2023, Qilu Pharmaceutical, Z5F65W1YAZ, Qixinke, WX0593, WX 0593, FL006, FL 006, FL-006
Zongertinib
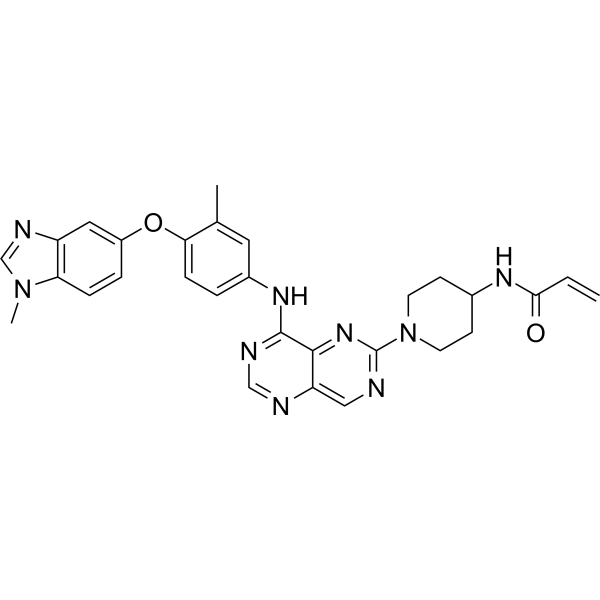
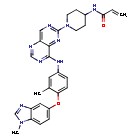
Zongertinib
CAS No. : 2728667-27-2,
BI-1810631, BI1810631
| Molecular Weight | 535.60 |
|---|---|
| Formula | C29H29N9O2 |
FDA 8/8/2025, Hernexeos, To treat adults with unresectable or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 tyrosine kinase domain activating mutations, as detected by an FDA-approved test, and who have received prior systemic therapy
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo(d)imidazol-5-yl)oxy)phenyl)amino)pyrimido(5,4-d)pyrimidin-2-yl)piperidin-4-yl)acrylamide
- 884-819-6
Zongertinib is an orally bioavailable inhibitor of the receptor tyrosine kinase human epidermal growth factor receptor 2 (HER2; ErbB2; HER-2), with potential antineoplastic activity. Upon oral administration, zongertinib covalently binds to and inhibits the activity of both wild-type and HER2 mutants, including HER2 mutants with exon 20 insertion (ex20ins) mutations. This prevents HER2-mediated signaling and may lead to cell death in HER2-expressing tumor cells. HER2, a receptor tyrosine kinase overexpressed on a variety of tumor cell types, plays an important role in tumor cell proliferation and tumor vascularization.
REF
Synthesis of zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-
548 yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
Methods
Synthesis of Zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
An overview of the synthetic routes to zongertinib and BI-3999 is shown in Supplementary Fig. S1, and graphical NMR spectra are shown in Supplementary Fig. S2.
3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)aniline (500 mg, 1.97 mmol) and 8-chloro-2-(methylthio)pyrimido[5,4-d]pyrimidine hydrochloride (492 mg, 1.97 mmol) were suspended in isopropanol, and the resulting reaction mixture stirred at 50°C for 3 hours, at which time high-performance liquid chromatography–mass spectrometry (HPLC-MS) indicated full conversion. The reaction mixture was concentrated under reduced pressure, and the crude product was redissolved in dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–15% methanol in dichloromethane) to afford the product (840 mg).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylthio)pyrimido[5,4-d]pyrimidin-4-amine (860 mg, 90%, 1.80 mmol) was suspended in dichloromethane (30 mL), and the resulting mixture was cooled to 0°C to 5°C. mCPBA (3-chloroperbenzoic acid, 444 mg, 77%, 1.98 mmol) was added portionwise over 1 hour, and the resulting reaction mixture was stirred at room temperature overnight, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product which was used directly in the next step (767 mg, crude).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylsulfinyl)pyrimido[5,4-d]pyrimidin-4-amine (5.42 g, 80%, 9.73 mmol) was dissolved in N,N-dimethyl formamide (DMF, 50 mL) and diisopropylethylamine (2.8 mL, 16 mmol). 4-Boc-amino-1-piperidine (2.39 g, 11.9 mmol) was added, and the reaction was stirred at 60°C overnight. Then, the reaction mixture was concentrated, and the crude product was used directly in the next step (5.66 g, crude).
Tert-butyl (1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)carbamate (5.66 g, 9.73 mmol) was dissolved in dichloromethane (100 mL) and methanol (30 mL). Four mol/L HCl in dioxane (11 mL, 44 mmol) was added, and the resulting reaction mixture was heated to 45°C for 7 hours. HPLC-MS indicated some remaining starting material; therefore, the reaction mixture was stirred at room temperature overnight. Four mol/L HCl in dioxane (1 mL, 0.40 mmol) was added, and the reaction mixture was reheated to 45°C for 4 hours, at which time HPLC-MS indicated full conversion. The reaction mixture was concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (4.5 g, 70% purity).
1-[8-({3-methyl-4-[(1-methyl-1H-1,3-benzodiazol-5-yl)oxy]phenyl}amino)-[1,3]diazino[5,4-d]pyrimidin-2-yl]piperidin-4-amine (4.5 g, 70%, 6.9 mmol) was suspended in dichloromethane (150 mL) and triethyl amine (4 mL, 28 mmol), and dimethylaminopyridine (115 mg, 0.941 mmol) was added. Then, acroyloyl anhydride (1.36 g, 95%, 10.3 mmol) was added, and the resulting reaction mixture was stirred at room temperature for 1 hour, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane (50 mL) and washed with aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (2.49 g).
1H NMR (DMSO-d6, 500 MHz) δ 9.58 (s, 1H), 9.08 (s, 1H), 8.39 (s, 1H), 8.19 (s, 1H), 8.10 (d, 1H, J = 7.6 Hz), 7.84 (d, 1H, J = 2.2 Hz), 7.77 (dd, 1H, J = 8.8 Hz, J = 2.2 Hz), 7.57 (d, 1H, J = 8.8 Hz), 7.09 (d, 1H, J = 2.2 Hz), 7.00 (dd, 1H, J = 2.2, 8.5 Hz), 6.89 (d, 1H, J = 8.8 Hz), 6.20 (dd, 1H, J = 10.1, 17.0 Hz), 6.10 (dd, 1H, J = 2.2, 17.0 Hz), 5.6 (dd, 1H, J = 2.2, 9.8 Hz), 4.86 (m, 2H), 3.99 (m, 1H), 3.84 (s, 3H), 3.25 (m, 2H), 2.26 (s, 3H), 1.92 (m, 2H), and 1.43 (m, 2H).
Synthesis of BI-3999 (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acetamide)
6-(4-aminopiperidin-1-yl)-N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)pyrimido[5,4-d]pyrimidin-4-amine (100 mg, 208 mmol) and 4-dimethylaminopyridine (2.5 mg, 0.02 mmol) were suspended in 5 mL dichloromethane. Acetic anhydride (25 μL, 0.23 mmol) was added, and the resulting reaction mixture was stirred at room temperature for one hour. Then, the reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3 and brine. Then, the layers were separated, and the organic layer was dried over MgSO4 and concentrated. The crude product was purified by column chromatography (SiO2, gradient of 0%–10% methanol in dichloromethane) to afford the product (75 mg).
1H NMR (DMSO-d6, 400 MHz) δ 9.58 (s, 1H), 9.07 (s, 1H), 8.39 (s, 1H), 8.17 (s, 1H), 7.88 (d, 1H, J = 7.9 Hz), 7.84 (d, 1H, J = 2.5 Hz), 7.77 (dd, 1H, J = 2.7, 8.7 Hz), 7.57 (d, 1H, J = 8.9 Hz), 7.09 (d, 1H, J = 2.3 Hz), 7.00 (dd, 1H, J = 2.3, 8.6 Hz), 6.89 (d, 1H, J = 8.6 Hz), 4.85 (m, 2H), 3.90 (m, 1H), 3.84 (s, 3H), 3.23 (m, 2H), 2.26 (s, 3H), 1.88 (m, 2H), 1.82 (s, 3H), and 1.38 (m, 2H).
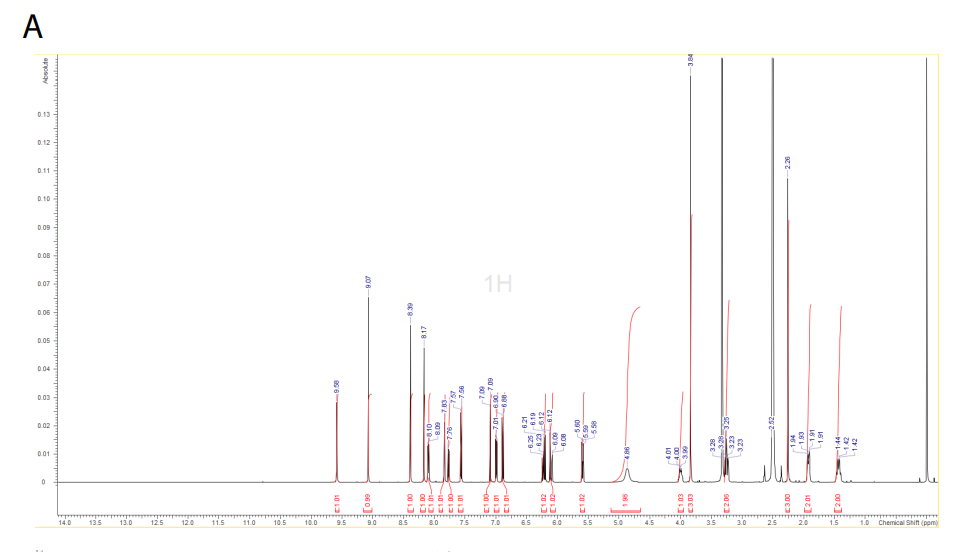
A) 1H NMR spectrum of zongertinib
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021213800&_cid=P10-ME52KD-62836-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. WHO Drug Informat ion – World Health Organization (WHO).[2]. Wilding Birgit, et al. Synthesis of diazino-pyrimidines as anticancer agents: World Intellectual Property Organization, WO2021213800. 2021-10-28.[3]. Li S, et al. Emerging Targeted Therapies in Advanced Non-Small-Cell Lung Cancer. Cancers (Basel). 2023 May 24;15(11):2899. [Content Brief]
////////////Zongertinib, Hernexeos, APPROVALS 2025, FDA 2025, lung cancer, BI-1810631, BI1810631, DRH7R67UVL
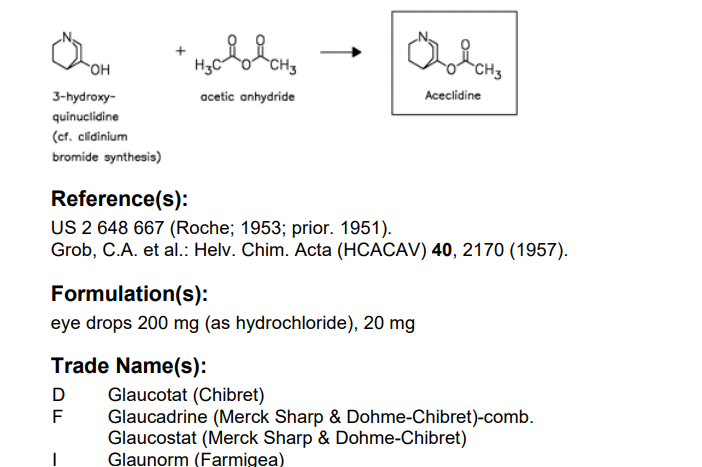
Aceclidine
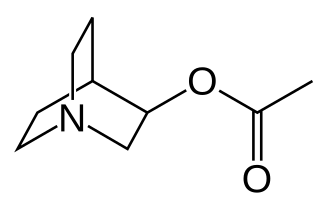
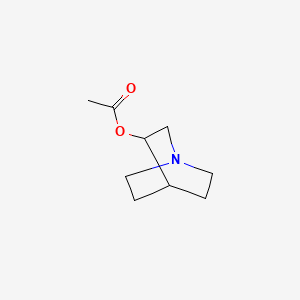
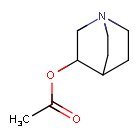
Aceclidine
WeightAverage: 169.224
Monoisotopic: 169.110278727
Chemical FormulaC9H15NO2
CAS 827-61-2, 3-Acetoxyquinuclidine, 3-Quinuclidinol acetate (ester), Aceclidina, 0578K3ELIO
APROVAL 7/31/2025, Vizz. To treat presbyopia
1-azabicyclo[2.2.2]octan-3-yl acetate
Acetic acid 1-aza-bicyclo[2.2.2]oct-3-yl ester(aceclidine)
MW: 169.22 MF: C9H15NO2
LD50: 78 mg/kg (M, i.p.); 36 mg/kg (M, i.v.); 165 mg/kg (M, p.o.); 102 mg/kg (M, s.c.);
45 mg/kg (R, i.v.); 225 mg/kg (R, s.c.)
CN: 1-azabicyclo[2.2.2]octan-3-ol acetate (ester)
WeightAverage: 205.68
Monoisotopic: 205.0869565
Chemical FormulaC9H16ClNO2
LD50: 27 mg/kg (M, i.v.); 165 mg/kg (M, p.o.);
45 mg/kg (R, i.v.)
Aceclidine (Glaucostat, Glaunorm, Glaudin, Vizz) is a parasympathomimetic miotic agent used in the treatment of narrow angle glaucoma.
Aceclidine was approved for medical use in the United States in July 2025.[2]
Medicinal properties
Aceclidine decreases intraocular pressure. It acts as a muscarinic acetylcholine receptor agonist.[3]
Chemistry
Aceclidine is an organic compound that is structurally related to quinuclidine. As such its alternative name is 3-acetoxyquinuclidine. Its protonated derivative has a pKa of 9.3.[4]
SYN
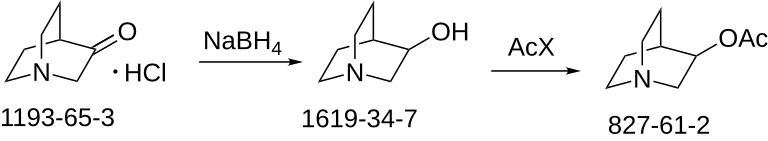
E. E. Mikhlina and M. V. Rubtsov, Zhur. Obschei
Khim, 30, 163 (1960). L. H. Sternbach and S. Kaiser, J. Am. Chem. Soc., 74, 2215 (1952). C. A. Grob, A. Kaiser and E. Renk, Helv. Chim.Acta, 40, 2170 (1957).
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/218585s000lbl.pdf
- “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 4 August 2025. Retrieved 5 August 2025.
- Shannon HE, Hart JC, Bymaster FP, Calligaro DO, DeLapp NW, Mitch CH, et al. (August 1999). “Muscarinic receptor agonists, like dopamine receptor antagonist antipsychotics, inhibit conditioned avoidance response in rats”. The Journal of Pharmacology and Experimental Therapeutics. 290 (2): 901–907. doi:10.1016/S0022-3565(24)34979-1. PMID 10411607.
- Aggarwal VK, Emme I, Fulford SY (February 2003). “Correlation between pK(a) and reactivity of quinuclidine-based catalysts in the Baylis-Hillman reaction: discovery of quinuclidine as optimum catalyst leading to substantial enhancement of scope”. The Journal of Organic Chemistry. 68 (3): 692–700. doi:10.1021/jo026671s. PMID 12558387.
External links
- Clinical trial number (NCT05656027 for “Phase 3 Evaluation of the Safety and Efficacy of LNZ101 for the Treatment of Presbyopia (CLARITY)” at ClinicalTrials.gov
- Clinical trial number (NCT05728944 for “Phase 3 Efficacy Study of LNZ101 for the Treatment of Presbyopia (CLARITY)” at ClinicalTrials.gov
- Clinical trial number (NCT05753189 for “Phase 3 Safety Study for the Treatment of Presbyopia Subjects” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Other names | LNZ101 |
| AHFS/Drugs.com | Vizz |
| License data | US DailyMed: Aceclidine |
| Routes of administration | Topical (ophthalmic solution) |
| ATC code | S01EB08 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]In general: ℞ (Prescription only) |
| Identifiers | |
| IUPAC name | |
| CAS Number | 827-61-2 6109-70-2 |
| PubChem CID | 1979 |
| ChemSpider | 1902 |
| UNII | 0578K3ELIO |
| KEGG | D02750 |
| ChEMBL | ChEMBL20835 |
| CompTox Dashboard (EPA) | DTXSID2045658 |
| ECHA InfoCard | 100.011.431 |
| Chemical and physical data | |
| Formula | C9H15NO2 |
| Molar mass | 169.224 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
///////////Aceclidine, APPROVALS 2025, FDA 2025, Vizz. To treat presbyopia, 827-61-2, 3-Acetoxyquinuclidine, 3-Quinuclidinol acetate (ester), Aceclidina, 0578K3ELIO, Glaucostat
Ropotrectinib
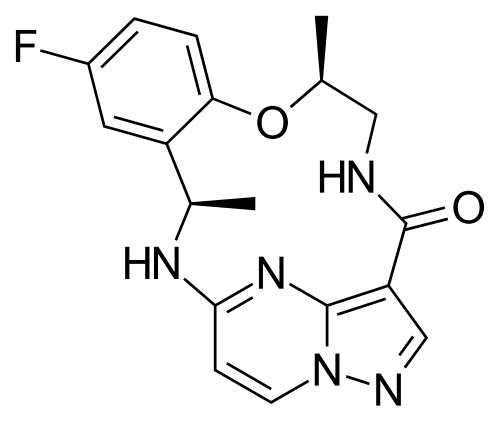
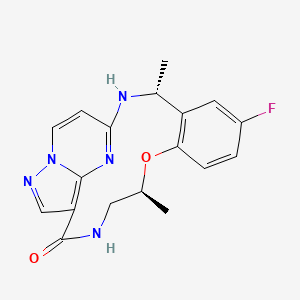
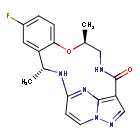
Ropotrectinib
- CAS 1802220-02-5
- TPX-0005
- Augtyro
- 08O3FQ4UNP
WeightAverage: 355.373
Monoisotopic: 355.144453003
Chemical FormulaC18H18FN5O2
- repotrectinibum
- (3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
- 1,15-Etheno-1H-pyrazolo(4,3-F)(1,4,8,10)benzoxatriazacyclotridecin-4(5H)-one, 11-fluoro-6,7,13,14-tetrahydro-7,13-dimethyl-, (7S,13R)-
- 1,15-Etheno-1H-pyrazolo(4,3-f)(1,4,8,10)benzoxatriazacyclotridecin-4(5H)-one, 11-fluoro-2,6,7,13-tetrahydro-7,13-dimethyl-, (14Z)-
- (1Z)-6-Fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo(13.5.2.04,9.018,22)docosa-1,4,6,8,15,19,21-heptaen-14-one
- (3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo[13.5.2.0,.0,]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
- (7S,13R)-11-fluoro-7,13-dimethyl-6,7,13,14- tetrahydro-1,15-ethenopyrazolo[4,3- f][1,4,8,10]benzoxatriazacyclotridecin-4(5H)- one
- (3R,6S,)-45-FLUORO-3,6-DIMETHYL-5-OXA-2,8-DIAZA-1(5,3)-PYRAZOLO(1,5-A)PYRIMIDINA-4(1,2)-BENZENANONAPHAN-9-ONE
- (7S,13R)-11-Fluoro-7,13-Dimethyl-6,7,13,14-Tetrahydro-1,15-Ethenopyrazolo[4,3-F][1,4,8,10]Benzoxatriazacyclotridecin-4(5H)-One
- 1,15-ETHENO-1H-PYRAZOLO(4,3-F)(1,4,8,10)BENZOXATRIAZACYCLOTRIDECIN-4(5H)-ONE, 11-FLUORO-6,7,13,14-TETRAHYDRO-7,13-DIMETHYL-, (7S,13R)-
Repotrectinib, sold under the brand name Augtyro, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[2][5] It is taken by mouth.[2] Repotrectinib is an inhibitor of proto-oncogene tyrosine-protein kinase ROS1 (ROS1) and of the tropomyosin receptor tyrosine kinases (TRKs) TRKA, TRKB, and TRKC.[2]
The most common adverse reactions include dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, ataxia, fatigue, cognitive disorders, and muscular weakness.[5]
Repotrectinib was approved for medical use in the United States in November 2023,[5][6] and in the European Union in January 2025.[3][4] CHINA 2024
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.202405153
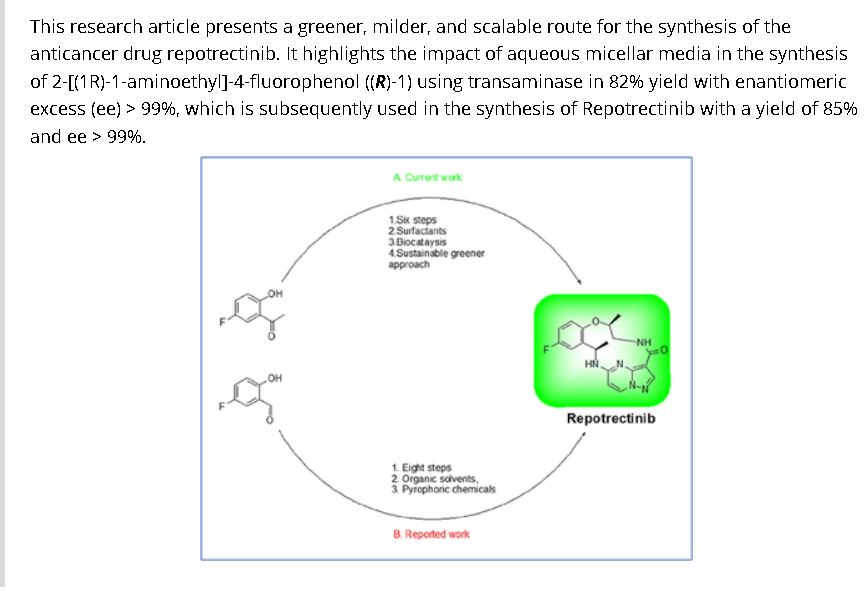
Synthesis of Repotrectinib
To a stirred solution of 5-{[(1R)-1-(2-{[(2S)-1-aminopropan-2-yl]oxy}-5-fluorophenyl)ethyl]amino}pyrazolo[1,5-a]pyrimidine-3-carboxylic acid 15 (0.25 g, 0.000611 mol, 1.0 eq.) in DMF (4.0 mL, 16V) was slowly added to solution of DIPEA (0.6 mL, 0.00488 mol, 8.0 eq.) in DCM (1.8 mL, 7V) at 0-5 °C. Then FDPP (0.25 g, 0.000672 mol, 1.1 eq.) was added at 0-5 °C. The reaction mixture was allowed to stirr for 1-2h at 25-30 °C. The reaction was monitored by TLC for disappearance of starting material. Then the resulting reaction mixture was diluted with ethyl acetate (50 mL), washed with water (20 mL) and brine solution (20 mL). The separated organic layer was dried over sodium sulphate and concentrated under reduced pressure at 45 °C. The obtained crude product was purified by silica gel (60-120 mesh) column chromatography to get repotrectinib asawhite solid (0.18 g, 85%).
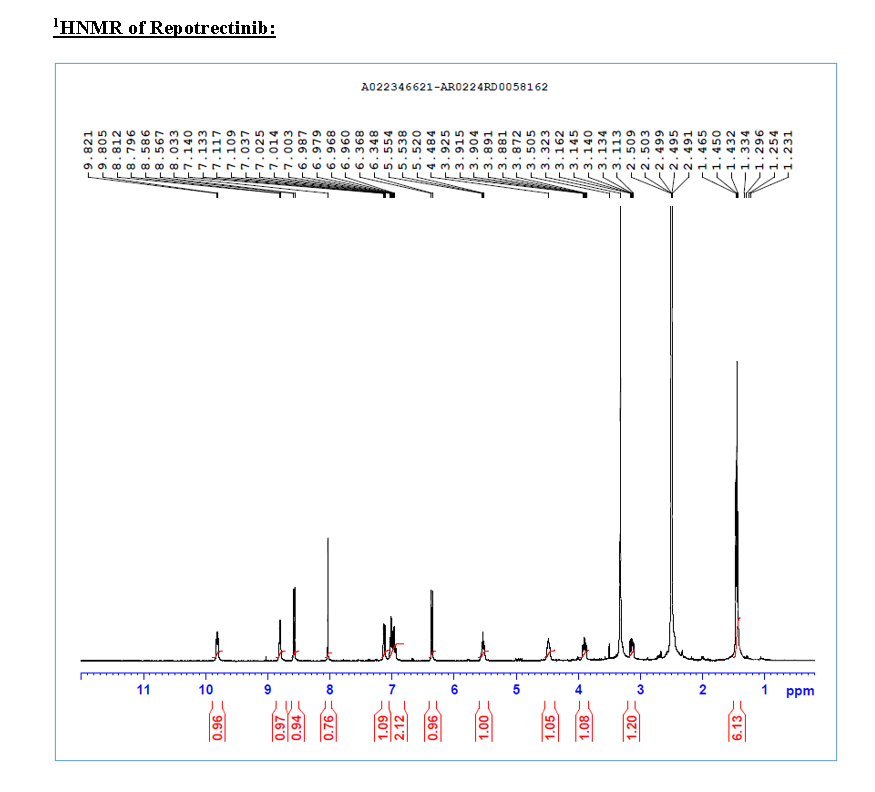

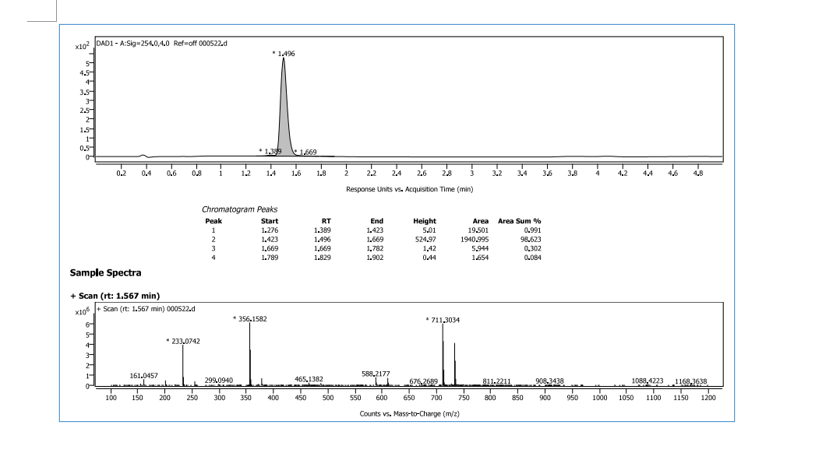
HRMS
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.3c00152
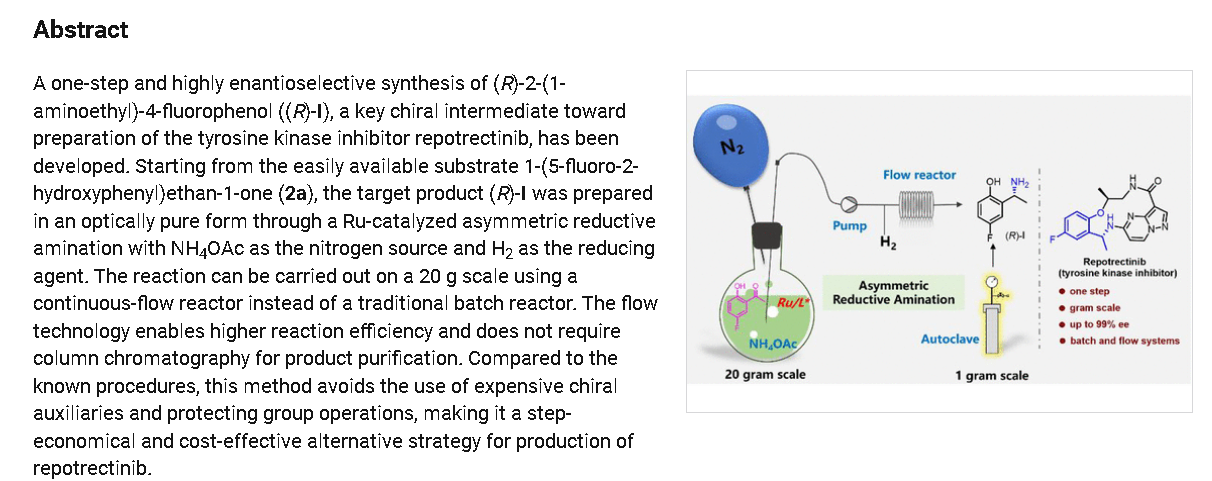
REF
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00061
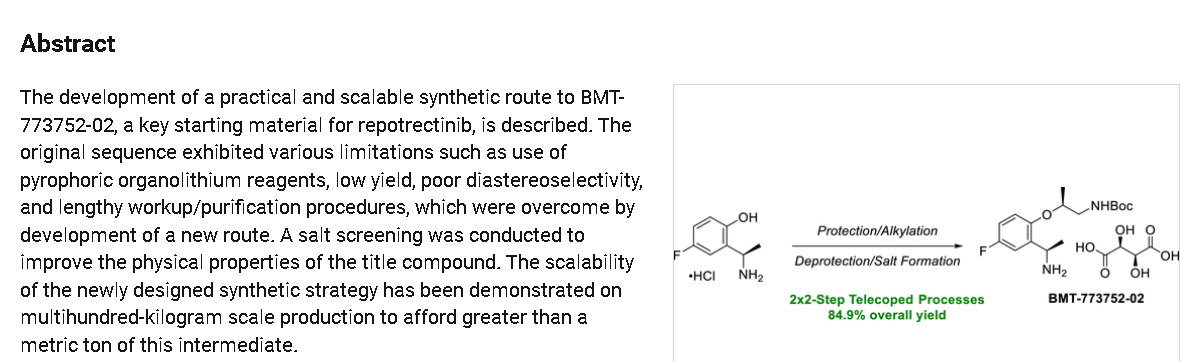
REF
US20180194777
https://patentscope.wipo.int/search/en/detail.jsf?docId=US222923082&_cid=P11-ME283N-03701-1
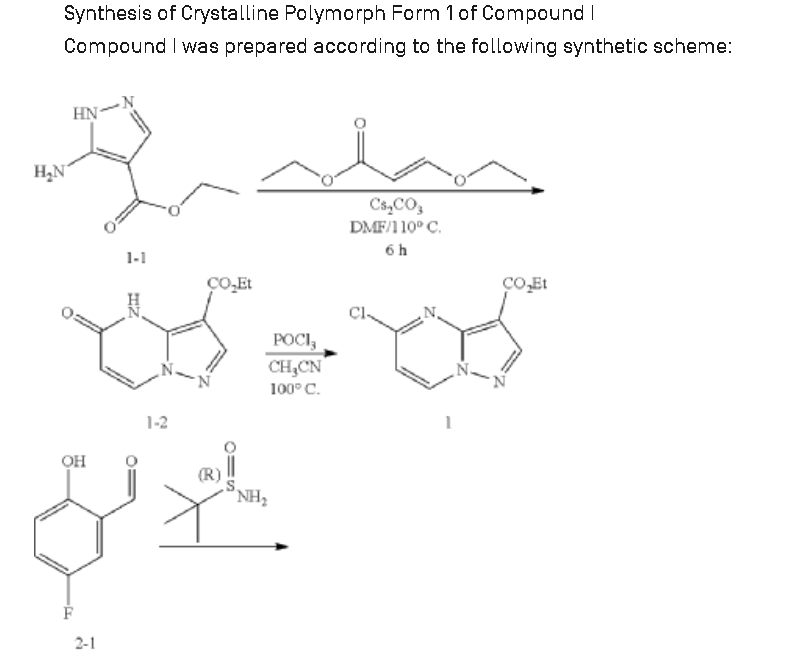
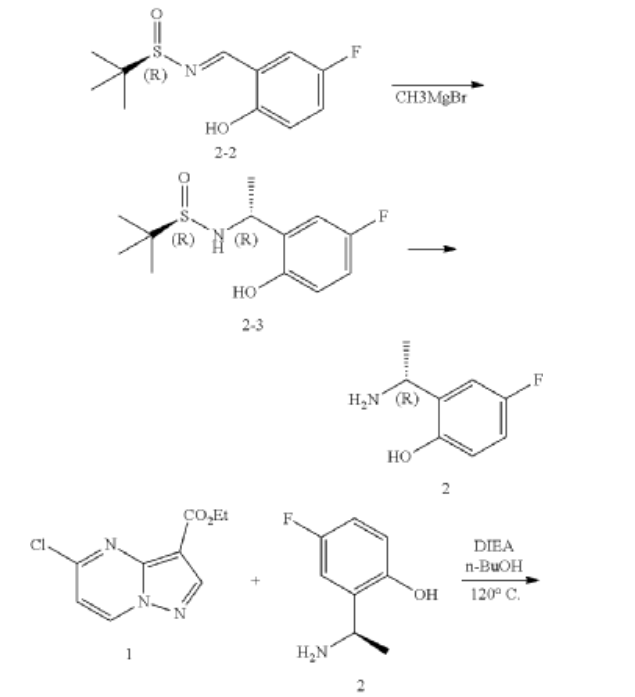
Example 1: Preparation of 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (1)
Step 1: Preparation of ethyl 5-oxo-4H-pyrazolo[1,5-a]pyrimidine-3-carboxylate (1-2)
Step 2: Preparation of 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (1)
PATENT
https://patents.google.com/patent/US10246466B2/en
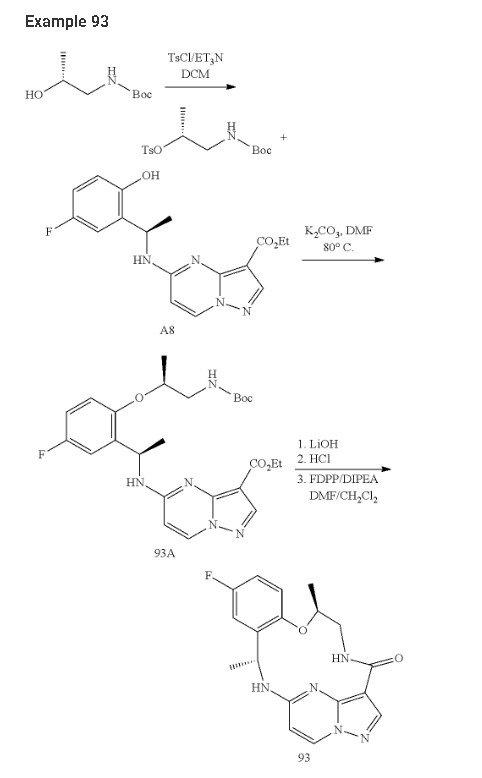
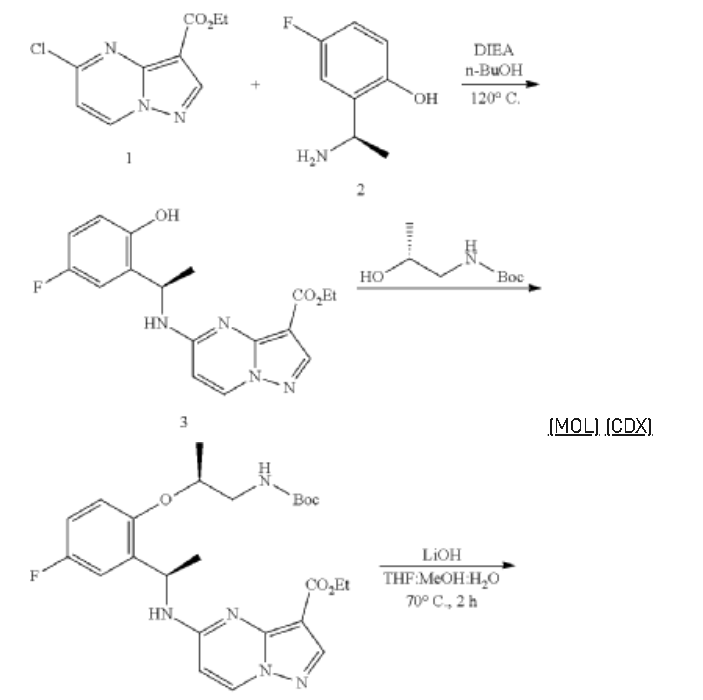
Step 1. To a solution of tert-butyl (R)-(2-hydroxypropyl)carbamate (1.00 g, 5.71 mmol) and tosyl chloride (1.14 g, 6.00 mmol) in DCM (29 mL) was added triethylamine (1.44 g, 14.28 mmol and the mixture was stirred at room temp for 48 hour. The reaction solution was concentrated under reduced pressure and the residue was purified with flash chromatography (ISCO system, silica (40 g), 0-20% ethyl acetate in hexane) to provide (R)-1-((tert-butoxycarbonyl)amino)propan-2-yl 4-methylbenzenesulfonate (1.12 g, 3.40 mmol, 59.54% yield).
Step 2. To a solution of A8 (100.00 mg, 0.290 mmol) and (R)-1-((tert-butoxycarbonyl)amino)propan-2-yl 4-methylbenzenesulfonate (143.50 mg, 0.436 mmol) in DMF (1.45 mL) was added K2CO3 (200.7 mg, 1.45 mmol) and heated at 80° C. with stirring for 16 hour. The reaction was cooled to ambient temperature and diluted with DCM (3 mL), filtered through a syringe filter, and concentrated under reduced pressure. Flash chromatography (ISCO system, silica (12 g), 0-60% ethyl acetate in hexane) provided 93A (32.90 mg, 0.0656 mmol, 22.59% yield).
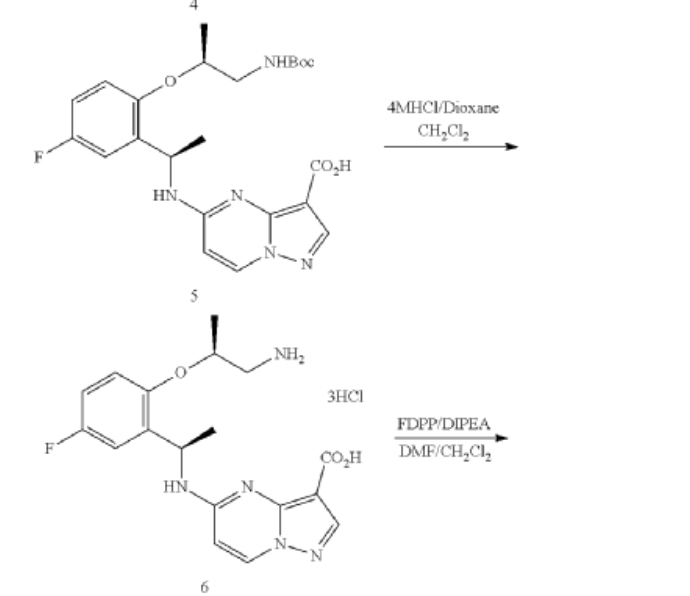
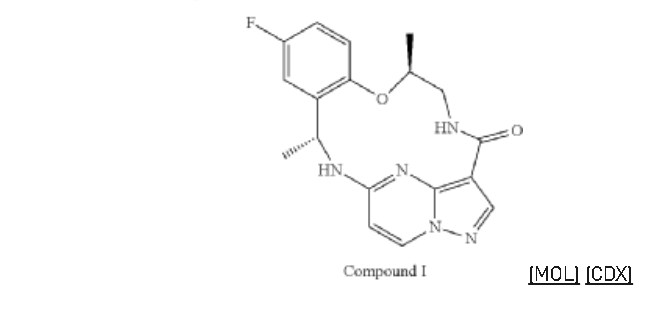
Step 3. To a solution of 93A (32.90 mg, 0.0656 mmol) in MeOH (3 mL) and THF (2 mL) was added LiOH aqueous solution (2M, 2 mL) at ambient temperature. The reaction solution was heated at 70° C. for 2 hours The reaction flask was cooled to ambient temperature, diluted with water and methanol, and then quenched with HCl aqueous solution (2 M, 2 mL) to pH<5. The mixture was extracted with DCM (3×5 mL), dried with Na2SO4, concentrated under reduced and dried on high vacuum overnight. To a solution of the acid product in DCM (4 mL) was added 4 M HCl in 1,4-dioxane (2.0 mL). The mixture was stirred at room temperature for 3 hours, and then concentrated under reduced pressure and dried on high vacuum. To a solution of the de-Boc product and FDPP (27.62 mg, 0.0719 mmol) in DMF (1.6 mL) was added Hunig’s base (42.23 mg, 0.327 mmol) at room temperature. The mixture was stirred for 2.5 hours, and then quenched the reaction with 2 M Na2CO3 solution (2 mL). The mixture was stirred for 15 min then extracted with DCM (4×10 mL). The combined extracts were dried with Na2SO4 and concentrated under reduced pressure. The residue was purified with flash chromatography (ISCO system, silica (12 g), 0-10% methanol in dichloromethane) to provide 93 (10.1 mg, 0.0284 mmol, 43.49% yield for three steps).
PATENT
SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Repotrectinib (Augtyro) Repotrectinib, developed by Turning Point Therapeutics, Inc., was granted FDA approval on November 15, 2023. It is indicated to treat locally advanced or metastatic ROS proto-oncogene 1, receptor tyrosine kinase (ROS1)-positive non-small cell lung cancer (NSCLC). Repotrectinib is a highly effective inhibitor of ROS1 (ICtyrosine receptor kinase (TRK) (IC5050= 0.07 nM) and
=0.83/0.05/0.1 nM for TRKA/B/C) [87]. After undergoing currently approved targeted therapies, patients with tumors containing ROS1 and neurotrophic tyrosine kinase receptor (NTRK) gene fusions frequently acquire resistance mutations [88,89]. These mutations restrict the ability of drugs to bind to their
targets, ultimately resulting in the advancement of tumors. Repotrectinib, a novel tyrosine kinase inhibitor (TKI), is the pioneering drug developed to specifically target ROS1 or NTRK-positive metastatic
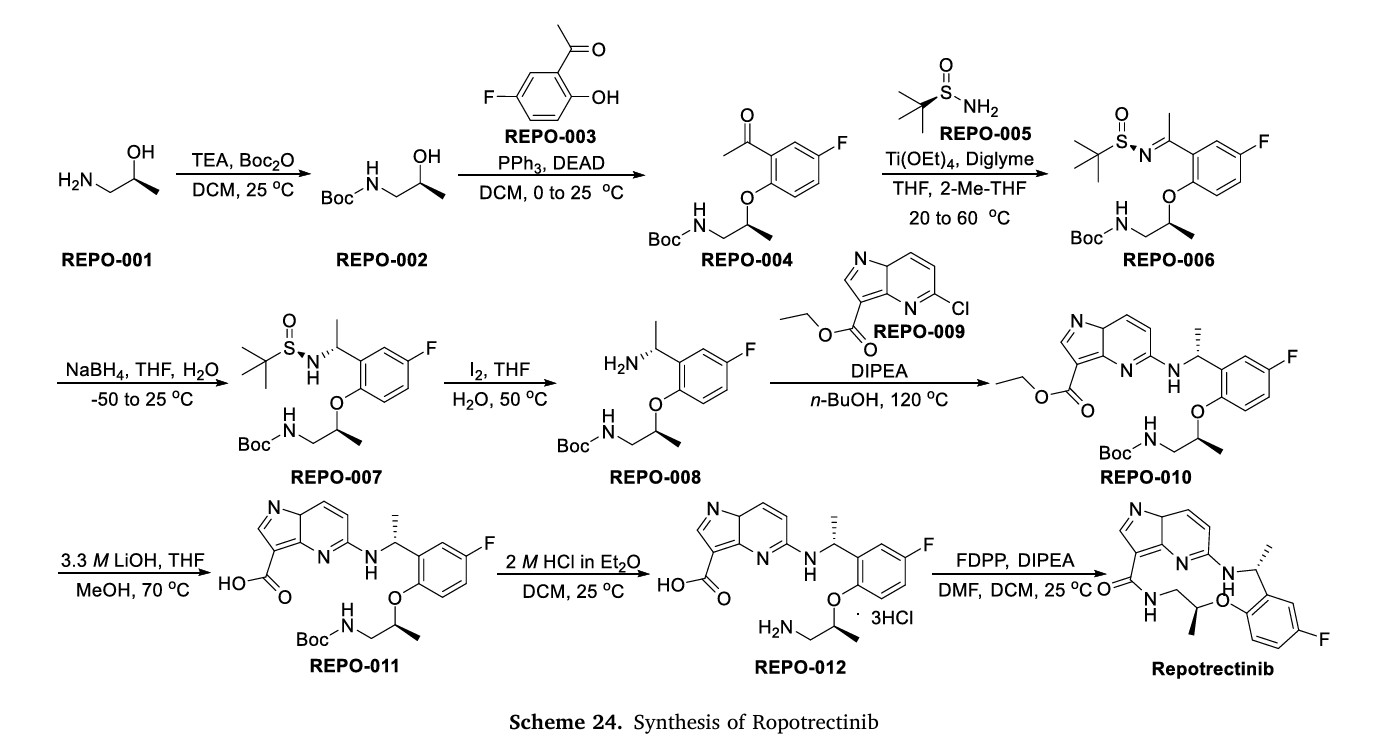
NSCLC and effectively combat the primary factors contributing to disease advancement [90].Preparation of Repotrectinib is described as Scheme 24 [91].Protecting the amino group of REPO-001 with Boc group in the presence of Kgave REPO-002, followed by intermolecular dehydration with
1-(5-fluoro-2-hydroxyphenyl)ethan-1-one (REPO-003) to give the ester REPO-004. REPO-004 was reacted with chiral auxiliary REPO-005 to give REPO-006, which was reduced by NaBH4
to obtain REPO-007. Then REPO-008 was obtained by removing the chiral auxiliary under iodine conditions. Substitution of REPO-008 with REPO-009 gave REPO-010, which was further hydrolyzed under alkaline conditions to obtain REPO-011. Salt formation of REPO-011 with hydrochloric acid
yielded REPO-012, which underwent intramolecular condensation to obtain the product Repotrectinib.
[87] D. Zhai, W. Deng, Z. Huang, E. Rogers, J.J. Cui, The novel, rationally-designed,
ALK/SRC inhibitor TPX-0005 overcomes multiple acquired resistance
mechanisms to current ALK inhibitors, Cancer Res. 76 (2016) 2132.
[88] C. Keddy, P. Shinde, K. Jones, S. Kaech, R. Somwar, U. Shinde, M.A. Davare,
Resistance profile and structural modeling of next-generation ROS1 tyrosine
kinase inhibitors, Mol. Cancer Therapeut. 21 (2022) 336–346.
[89] E. Cocco, M. Scaltriti, A. Drilon, NTRK fusion-positive cancers and TRK inhibitor
therapy, Nat. Rev. Clin. Oncol. 15 (2018) 731–747.
[90] A. Drilon, S.I. Ou, B.C. Cho, D.W. Kim, J. Lee, J.J. Lin, V.W. Zhu, M.J. Ahn, D.
R. Camidge, J. Nguyen, D. Zhai, W. Deng, Z. Huang, E. Rogers, J. Liu, J. Whitten,
J.K. Lim, S. Stopatschinskaja, D.M. Hyman, R.C. Doebele, J.J. Cui, A.T. Shaw,
Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that
potently inhibits ROS1/TRK/ALK solvent-front mutations, Cancer Discov. 8
(2018) 1227–1236.
[91] J.J. Cui, E.W. Rogers, Gialir Macrocyclic Polymorph, 2018. US20180194777A1.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Repotrectinib, developed by Bristol-Myers Squibb and marketed under the brand name Augtyro, is an oral tyrosine kinase inhibitor (TKI) targeting ROS1 and TRK oncogenic drivers. In 2024, NMPA condition
ally approved Repotrectinib for adult patients with ROS1-positive locally advanced or metastatic NSCLC [15]. Repotrectinib exerts its antitumor activity by inhibiting ROS1 and TRK kinases, thereby disrupting the downstream signaling pathways that facilitate tumor cell proliferation and survival [16]. This argeted mechanism is particularly effective against tumors that harbor ROS1 or NTRK gene fusions. The clinical efficacy of Repotrectinib has been through validated the Phase 1/2 TRIDENT-1 trial (NCT03093116) [17]. In the study cohort, treat ment-naïve patients harboring ROS1-positive NSCLC exhibited an overall response rate (ORR) of 79 %, characterized by a median duration of response (DOR) reaching 34.1 months. Conversely, among those who had previously received ROS1 TKI therapy, the ORR was documented at 38 %, accompanied by a median DOR of 14.8 months. With respect to safety profiles, the adverse event spectrum commonly encompassed dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea
[18,19]. These side effects are generally manageable, but patients should be monitored for potential severe adverse events.
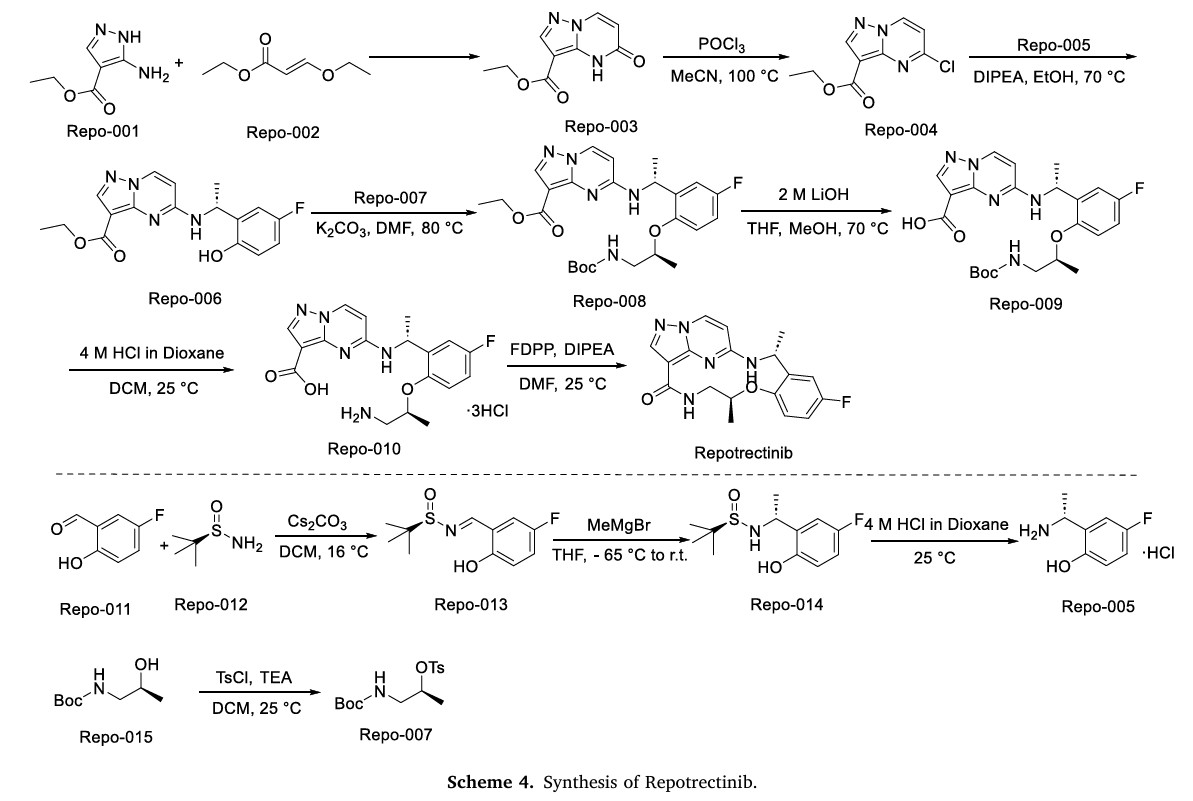
The synthetic route of Repotrectinib, shown in Scheme 4, begins with condensation reaction between Repo-001 and Repo-002 to afford Repo-003, which is chlorinated to yield Repo-004 [20]. This intermediate undergoes nucleophilic substitution with Repo-005 to form Repo-006,
followed by second nucleophilic substitution with Repo-007 to produce Repo-008. Ester hydrolysis of Repo-008 affords Repo-009, which undergoes acid-mediated deprotection to generate Repo-010. Final
intramolecular amidation of Repo-010 delivers Repotrectinib. In parallel, Repo-011 and Repo-012 undergo condensation to form imine Repo-013, which undergoes Grignard addition to afford Repo-014.
Acidification of Repo-014 then yields Repo-005. Concurrently, Repo-015 undergoes nucleophilic substitution to generate Repo-007.
[15] S. Dhillon, Repotrectinib: first approval, Drugs 84 (2024) 239–246.
[16] T. Rais, A. Shakeel, L. Naseem, N. Nasser, M. Aamir, Repotrectinib: a promising
new therapy for advanced nonsmall cell lung cancer, Ann Med Surg (Lond) 86
(2024) 7265–7269.
[17] A. Drilon, S.I. Ou, B.C. Cho, D.W. Kim, J. Lee, J.J. Lin, V.W. Zhu, M.J. Ahn, D.
R. Camidge, J. Nguyen, D. Zhai, W. Deng, Z. Huang, E. Rogers, J. Liu, J. Whitten, J.
K. Lim, S. Stopatschinskaja, D.M. Hyman, R.C. Doebele, J.J. Cui, A.T. Shaw,
Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that
potently inhibits ROS1/TRK/ALK solvent-front mutations, Cancer Discov. 8 (2018)
1227–1236.
[18] Repotrectinib, Drugs and Lactation Database (Lactmed®), National Institute of
Child Health and Human Development, Bethesda (MD), 2006.
[19] H. Zhong, J. Lu, M. Wang, B. Han, Real-world studies of crizotinib in patients with
ROS1-positive non-small-cell lung cancer: experience from China, J Comp Eff Res
14 (2024) e240043.
[20] J.J. Cui, E.W. Rogers, Preparation of
Fluorodimethyltetrahydroethenopyrazolobenzoxatriazacyclotridecinone
Derivatives for Use as Antitumor Agents, 2017. US20180194777A1.
Repotrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[2][5]
In June 2024, the US Food and Drug Administration (FDA) expanded the indication to include the treatment of people twelve years of age and older with solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion, are locally advanced or metastatic or where surgical resection is likely to result in severe morbidity, and that have progressed following treatment or have no satisfactory alternative therapy.[7][8]
References
- “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 23 May 2025.
- “Augtyro- repotrectinib capsule”. DailyMed. 15 November 2023. Archived from the original on 12 December 2023. Retrieved 12 December 2023.
- “Augtyro EPAR”. European Medicines Agency (EMA). 14 November 2024. Retrieved 16 November 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Augtyro PI”. Union Register of medicinal products. 14 January 2025. Retrieved 16 January 2025.
- “FDA approves repotrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 15 November 2023. Archived from the original on 16 November 2023. Retrieved 17 November 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “U.S. Food and Drug Administration Approves Augtyro (repotrectinib), a Next-Generation Tyrosine Kinase Inhibitor (TKI), for the Treatment of Locally Advanced or Metastatic ROS1-Positive Non-Small Cell Lung Cancer (NSCLC)” (Press release). Bristol Myers Squibb. 16 November 2023. Archived from the original on 16 November 2023. Retrieved 17 November 2023 – via Business Wire.
- “FDA grants accelerated approval to repotrectinib for adult and pediatric participants with neurotrophic tyrosine receptor kinase gene fusion-positive solid tumors”. U.S. Food and Drug Administration. 13 June 2024. Archived from the original on 13 June 2024. Retrieved 13 June 2024. This article incorporates text from this source, which is in the public domain.
- “Cancer Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 6 December 2024.
- Turning Point Therapeutics, Inc. (5 February 2024). A Phase 1/2, Open-Label, Multi-Center, First-in-Human Study of the Safety, Tolerability, Pharmacokinetics, and Anti-Tumor Activity of TPX-0005 in Patients With Advanced Solid Tumors Harboring ALK, ROS1, or NTRK1-3 Rearrangements (TRIDENT-1) (Report). clinicaltrials.gov. Archived from the original on 18 June 2024. Retrieved 18 June 2024.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 11-14 November 2024”. European Medicines Agency (EMA). 15 November 2024. Retrieved 16 November 2024.
Further reading
- Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. (October 2018). “Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations”. Cancer Discovery. 8 (10): 1227–1236. doi:10.1158/2159-8290.CD-18-0484. PMID 30093503.
External links
- “Repotrectinib (Code C133821)”. NCI Thesaurus. 25 September 2023. Retrieved 17 November 2023.
| Clinical data | |
|---|---|
| Trade names | Augtyro |
| Other names | TPX-0005 |
| AHFS/Drugs.com | Augtyro |
| License data | US DailyMed: Repotrectinib |
| Routes of administration | By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code | L01EX28 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| CAS Number | 1802220-02-5 |
| PubChem CID | 135565923 |
| DrugBank | DB16826 |
| ChemSpider | 64853849 |
| UNII | 08O3FQ4UNP |
| KEGG | D11454 |
| ChEBI | CHEBI:229220 |
| ChEMBL | ChEMBL4298138 |
| PDB ligand | 7GI (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H18FN5O2 |
| Molar mass | 355.373 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- (3R,6S,)-45-FLUORO-3,6-DIMETHYL-5-OXA-2,8-DIAZA-1(5,3)-PYRAZOLO(1,5-A)PYRIMIDINA-4(1,2)-BENZENANONAPHAN-9-ONE
- (7S,13R)-11-Fluoro-7,13-Dimethyl-6,7,13,14-Tetrahydro-1,15-Ethenopyrazolo[4,3-F][1,4,8,10]Benzoxatriazacyclotridecin-4(5H)-One
- 1,15-ETHENO-1H-PYRAZOLO(4,3-F)(1,4,8,10)BENZOXATRIAZACYCLOTRIDECIN-4(5H)-ONE, 11-FLUORO-6,7,13,14-TETRAHYDRO-7,13-DIMETHYL-, (7S,13R)-
////////Ropotrectinib, FDA 2023, APPROVALS 2023, Turning Point , EU 2025, APPROVALS 2025, EMA 2025, Augtyro, TPX 0005, CHINA 2024, APPROVALS 2024
Donafenib
Donafenib
CAS 1130115-44-4, CM-4307, Zepsun, 41XGO0VS1U
4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-(trideuteriomethyl)pyridine-2-carboxamide
- 4-(4-(((4-chloro-3-(trifluoromethyl)phenyl)carbamoyl)amino)phenoxy)-N-(2H3)methylpyridine-2-carboxamide
- Sorafenib-d3
- Sorafenib D3
- Donafenib (Sorafenib D3)
- Sorafenib-methyl-d3
- d3-sorafenib
CM-4307 is under investigation in clinical trial NCT03602495 (Donafenib in 131I-Refractory Differentiated Thyroid Cancer).
Donafenib, sold under the brand name Zepsun, is a pharmaceutical drug for the treatment of cancer.
In China, donafenib is approved for the treatment of unresectable hepatocellular carcinoma in patients who have not previously received systemic treatment.[1][2]
Donafenib is a kinase inhibitor that targets Raf kinase and various receptor tyrosine kinases.[3] It is a deuterated derivative of sorafenib with improved pharmacokinetic properties.[4][5]
Donafenib is an orally available multikinase inhibitor that targets Raf kinase and various receptor tyrosine kinases (RTKs), with potential antineoplastic activity. Upon oral administration, donafenib binds to and blocks the activity of Raf kinase, and inhibits Raf-mediated signal transduction pathways. This inhibits cell proliferation in Raf-expressing tumor cells. In addition, this agent may inhibit unidentified RTKs, and thus may further block tumor cell proliferation in susceptible tumor cells. Raf, a serine/threonine protein kinase, plays a key role in the Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway. Deregulation of this pathway often results in tumor cell proliferation and survival.
SYN
ACS Omega 2021, 6, 5532−5547.
https://pubs.acs.org/doi/10.1021/acsomega.0c05908
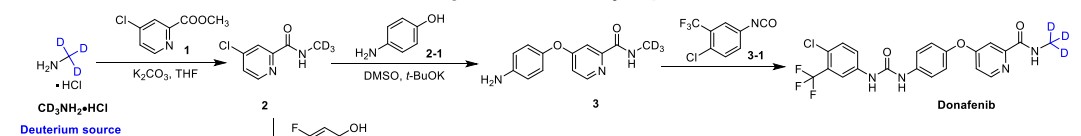
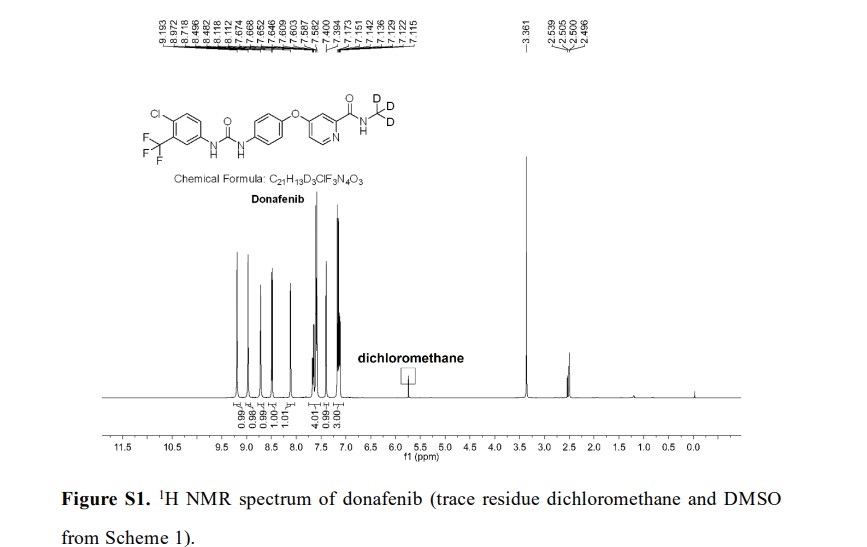
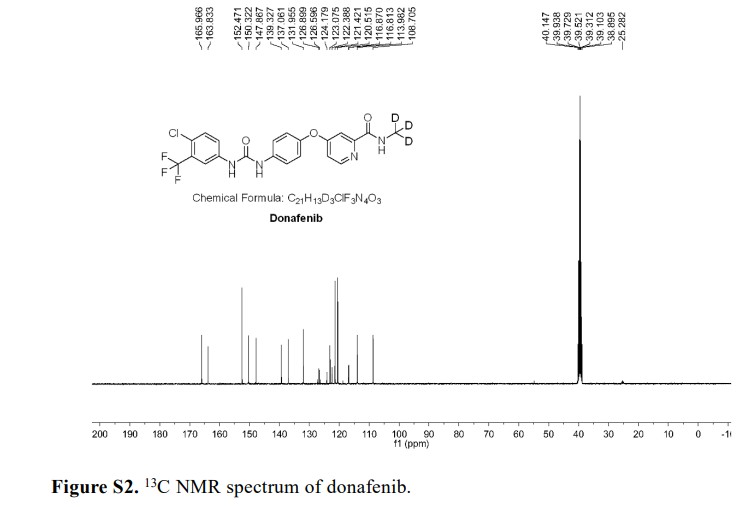
Syn
Donafenib (Zepsun). Donafenib (31), developed by Suzhou Zelgen Biopharmaceuticals, is a deuterated derivative of sorafenib, a multikinase inhibitor for the treatment of advanced hepatocellular carcinoma (HCC). 222 HCC is the most common type of primary liver cancer in adults and the third leading cause of cancer-related deaths worldwide.223,224 Donafenib inhibits Raf kinase and VEGFR tyrosine kinases,
thereby preventing the proliferation of tumor cells. 225 The presence of the deuterated methyl group in donafenib improves metabolic stability with prolonged half-life, lower systemic clearance, and higher systemic exposure.226 Donafenib has been shown to significantly improve the overall survival
of patients with HCC when compared against sorafenib, with favorable safety and tolerability.227 228
In June 2021, donafenib was first approved in China for treating unresectable HCC in patients who have not previously received systemic treatment.
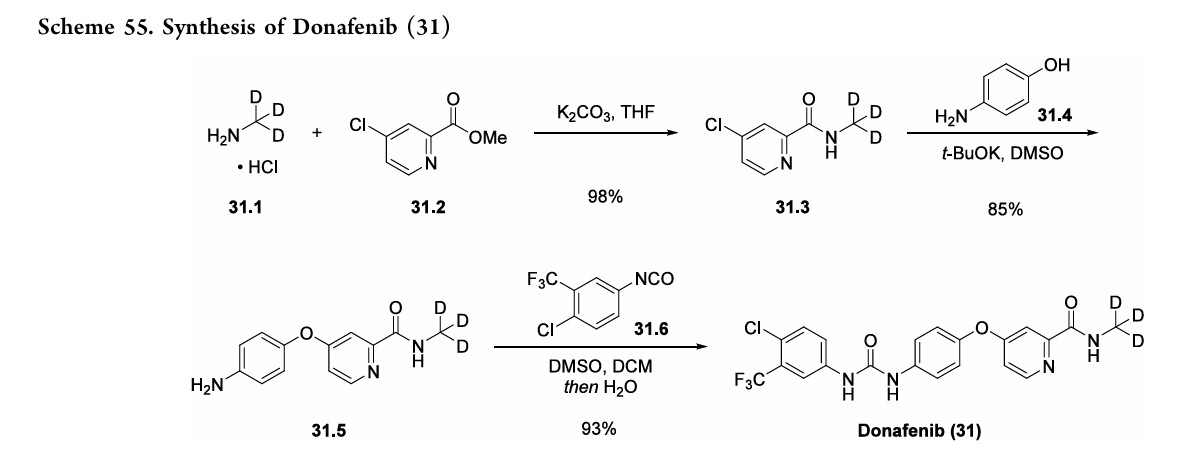
A gram-scale synthesis of donafenib was recently disclosed by Luo and co-workers (Scheme 55).229
The synthetic sequence commenced with amidation of methyl ester 31.2 using methan-d3-amine hydrochloride (31.1) as the deuterium source, affording CD 3-amide 31.3 in high yield (98%). SNAr
displacement with aminophenol 31.4 in DMSO provided diaryl ether 31.5. Finally, reaction of the aniline moiety with isocyanate 31.6 delivered donafenib (31) in 79% yield from 31.3
(222) Mousa, A. B. Sorafenib in the treatment of advanced
hepatocellular carcinoma. Saudi J. Gastroenterol 2008, 14, 40−42.
(223) Forner, A.; Llovet, J. M.; Bruix, J. Hepatocellular carcinoma.
Lancet 2012, 379, 1245−1255.
(224) Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski,
A. Hepatocellular carcinoma. Lancet 2022, 400, 1345−1362.
(225) Gong, X.; Qin, S. Study progression of anti-angiogenetic
therapy and its combination with other agents for the treatment of
advanced hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2018, 7,
466−474.
(226) Zhong, L.; Hou, C.; Zhang, L.; Zhao, J.; Li, F.; Li, W.
Synthesis of deuterium-enriched sorafenib derivatives and evaluation
of their biological activities. Mol. Divers. 2019, 23, 341−350.
(227) Qin, S.; Bi, F.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu,
Y.; Meng, Z.; Pan, H.; et al. Donafenib versus sorafenib in first-line
treatment of unresectable or metastatic hepatocellular carcinoma: A
randomized, open-label, parallel-controlled phase II-III trial. J. Clin.
Oncol. 2021, 39, 3002−3011.
(228) Keam, S. J.; Duggan, S. Donafenib: First approval. Drugs 2021,
81, 1915−1920.
(229) Li, C.; Zhong, J.; Liu, B.; Yang, T.; Lv, B.; Luo, Y. Study on
typical diarylurea drugs or derivatives in cocrystallizing with strong H
bond acceptor DMSO. ACS Omega 2021, 6, 5532−5547.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Keam SJ, Duggan S (November 2021). “Donafenib: First Approval”. Drugs. 81 (16): 1915–1920. doi:10.1007/s40265-021-01603-0. PMID 34591285.
- Chen R, Ielasi L, di Carlo A, Tovoli F (February 2023). “Donafenib in hepatocellular carcinoma”. Drugs of Today. 59 (2): 83–90. doi:10.1358/dot.2023.59.2.3507751. hdl:11585/955557. PMID 36811408.
- “Donafenib”. NCI Cancer Dictionary. National Cancer Institute, National Institutes of Health.
- Qin S, Bi F, Gu S, Bai Y, Chen Z, Wang Z, et al. (September 2021). “Donafenib Versus Sorafenib in First-Line Treatment of Unresectable or Metastatic Hepatocellular Carcinoma: A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial”. Journal of Clinical Oncology. 39 (27): 3002–3011. doi:10.1200/JCO.21.00163. PMC 8445562. PMID 34185551.
- Qin S, Bi F, Xu J, Du C, Fan Q, Zhang L, et al. (2020). “P-86 Comparison of the pharmacokinetics of donafenib and sorafenib in patients with advanced hepatocellular carcinoma: An open-label, randomized, parallel-controlled, multicentre phase II/III trial”. Annals of Oncology. 31: S117 – S118. doi:10.1016/j.annonc.2020.04.168.
| Clinical data | |
|---|---|
| Trade names | Zepsun |
| Other names | CM-4307 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1130115-44-4 |
| PubChem CID | 25191001 |
| DrugBank | DB15414 |
| ChemSpider | 23937167 |
| UNII | 41XGO0VS1U |
| ChEMBL | ChEMBL4297490 |
| CompTox Dashboard (EPA) | DTXSID90648995 |
| Chemical and physical data | |
| Formula | C21H16ClD3F3N4O3 |
| Molar mass | 470.87 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////Donafenib, ZEPSUN, CHINA 2021, APPROVALS 2021, Suzhou Zelgen, 1130115-44-4, CM 4307, 41XGO0VS1U, Sorafenib D3
Pamiparib
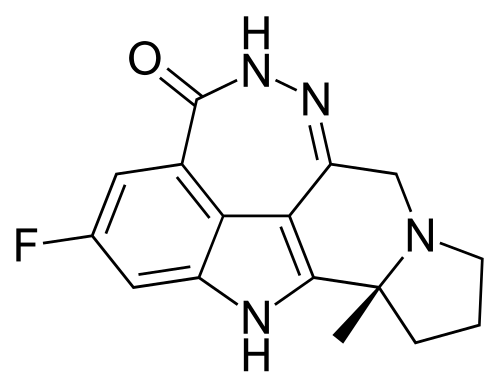
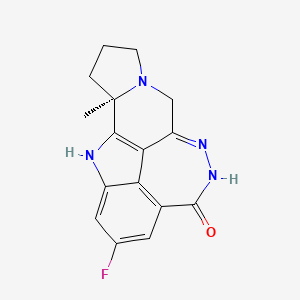
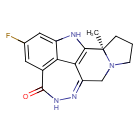
Pamiparib
BGB-290 APPROVED CHINA 2022, BEIGENE
(2R)-14-fluoro-2-methyl-6,9,10,19-tetrazapentacyclo[14.2.1.02,6.08,18.012,17]nonadeca-1(18),8,12(17),13,15-pentaen-11-one
- 1446261-44-4
- 8375F9S90C
- 5,6,7a,11-Tetraazacyclohepta(def)cyclopenta(a)fluoren-4(7H)-one, 2-fluoro-5,8,9,10,10a,11-hexahydro-10a-methyl-, (10aR)-
- 5,6,7a,11-Tetraazacyclohepta[def]cyclopenta[a]fluoren-4(7H)-one, 2-fluoro-5,8,9,10,10a,11-hexahydro-10a-methyl-, (10aR)-
- 298.31 g/mol, C16H15FN4O
Pamiparib, sold under the brand name Partruvix, is a pharmaceutical drug used for the treatment of various types of cancer. Pamiparib is a member of the PARP inhibitor drug class.[1]
In China, it is approved for the treatment of germline BRCA mutation-associated recurrent advanced ovarian, fallopian tube, and primary peritoneal cancers previously treated with two or more lines of chemotherapy.[2]
It is currently under investigation for the treatment of other forms of cancer.[3][1]
Pamiparib is under investigation in clinical trial NCT03933761 (Pamiparib in Fusion Positive, Reversion Negative High Grade Serous Ovarian Cancer or Carcinosarcoma With BRCA1/2 Gene Mutations If Progression on Substrate Poly ADP Ribose Polymerase Inhibitbor (PARPI) or Chemotherapy).
Pamiparib is an orally bioavailable inhibitor of the nuclear enzyme poly(ADP-ribose) polymerase (PARP), with potential antineoplastic activity. Upon administration, pamiparib selectively binds to PARP and prevents PARP-mediated repair of single-strand DNA breaks via the base-excision repair (BER) pathway. This enhances the accumulation of DNA strand breaks, promotes genomic instability, and eventually leads to apoptosis. PARP is activated by single-strand DNA breaks and, subsequently, catalyzes post-translational ADP-ribosylation of nuclear proteins which then transduce signals to recruit other proteins to repair damaged DNA. Pamiparib may both potentiate the cytotoxicity of DNA-damaging agents and reverse tumor cell chemo- and radioresistance.
REF
https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-0040-1719372
REF
J. Med. Chem. 2020, 63, 15541−15563.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.0c01346
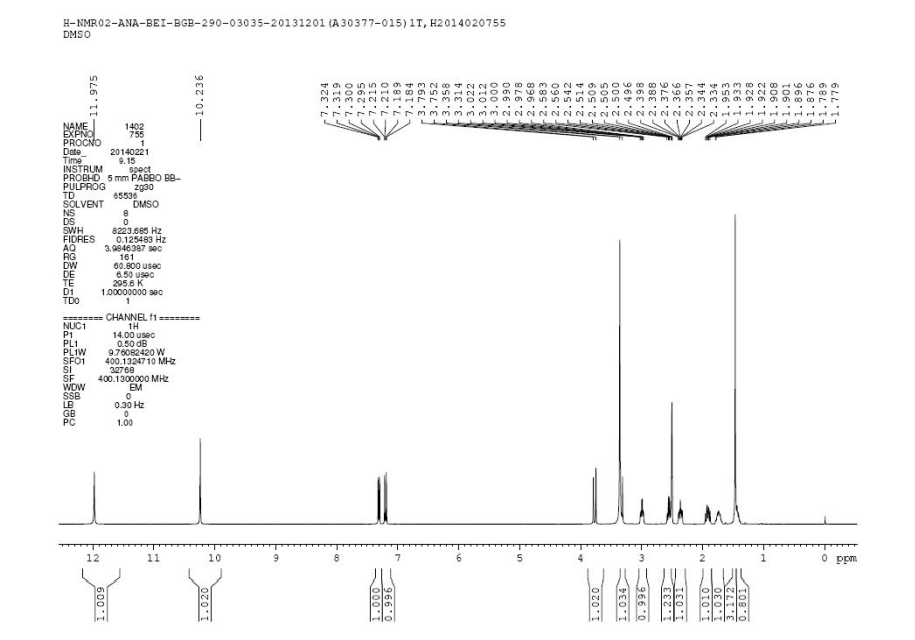
1HNMR 400, DMSO D6
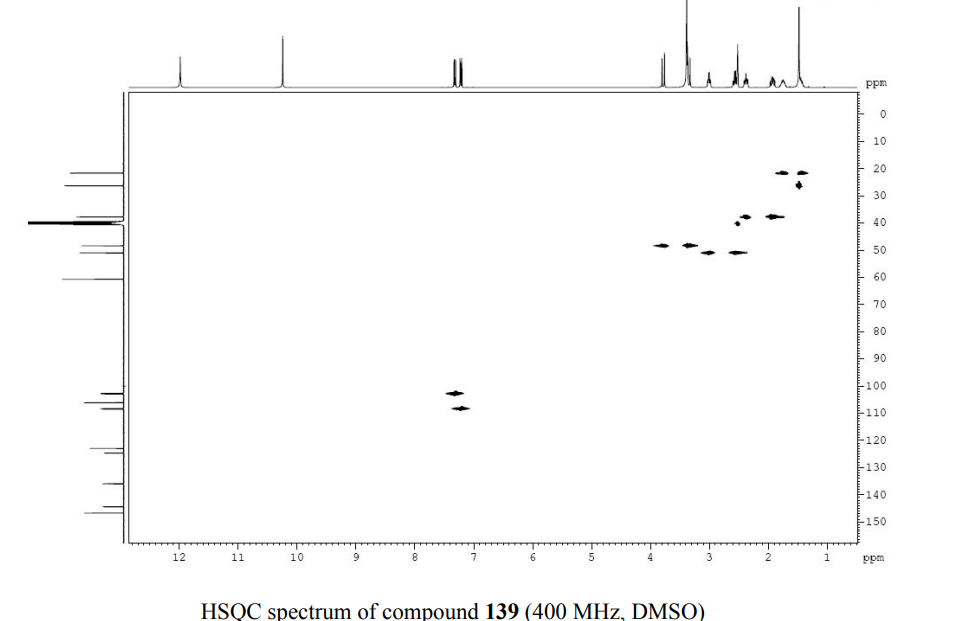
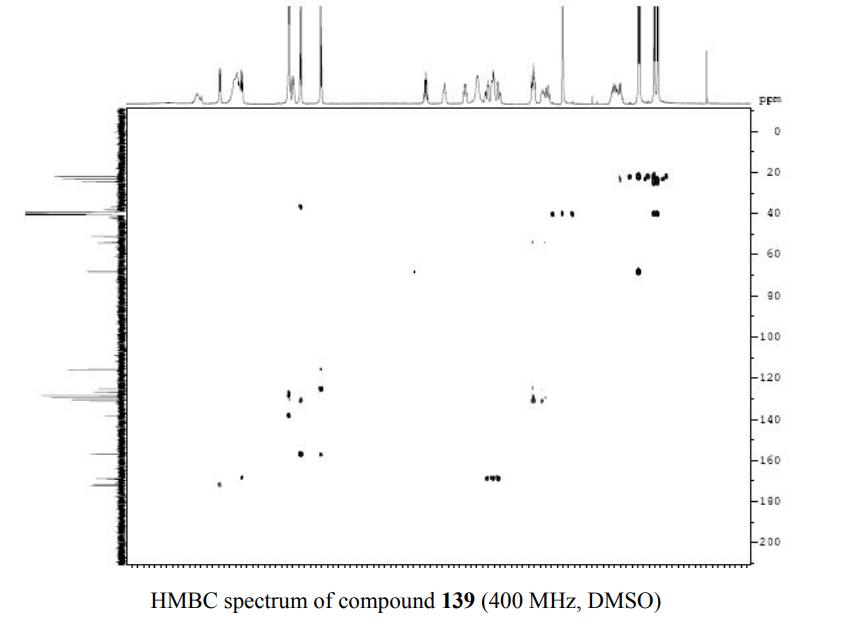
PATENT
WO 2018157794
https://patents.google.com/patent/WO2018157794A1/en
(R) -2-fluoro-10a-methyl-7, 8, 9, 10, 10a, 11-hexahydro-5, 6, 7a, 11-tetraazacyclohepta [def] cyclopenta – [a] fluoren-4 (5H) -one (hereafter Compound 1) , has been disclosed as a highly selective and potent Parp1/2 inhibitor, See WO 2013/097225 A1, which is incorporated herein by reference.

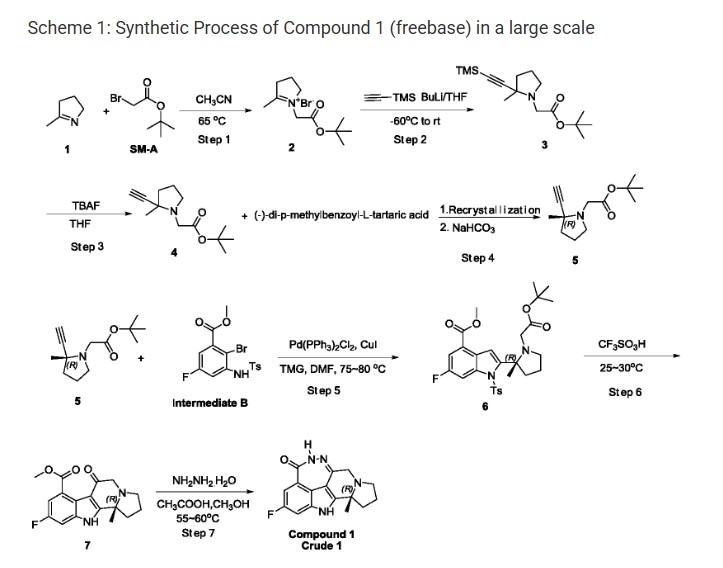
Step 1: Synthesis of Compound-2

t-Butyl bromoacetate (51.7 Kg) was dissolved in anhydrous acetonitrile (72 Kg) . The temperature was raised to 65-75 ℃, then methyl pyrroline (22 Kg) was added. The reaction mixture was condensed after the reaction was completed, the residual acetonitrile was removed by adding THF and then condensing. After GC showed a complete removal of acetonitrile, more THF was added and stirred. The resulting solid was filtered and collected. 44.1 Kg of off white solid Compound-2 was obtained. 1H NMR (400 MHz, DMSO-d6) δ 4.91 (s, 2H) , 4.15 (m, 2H) , 3.29 (m, 2H) , 2.46 (s, 3H) , ) , 2.14 (m, 2H) , 1.46 (s, 9H) ppm.
Step 2: Synthesis of Compound-3

To a cool (-60 ℃) solution of trimethylsilyl acetyne (12.4 Kg) in THF was added a solution of n-butyl lithium in hexane (43.4 Kg) . After complete addition of n-butyl lithium solution, the resulting mixture was stirred for additional 1-2 h and then the entire solution was transferred into a suspension of Compound-2 (31 Kg) in THF cooled at -60 ℃. After transfer completion, the resulting mixture was warmed to room temperature and stirred for 1 h. The reaction was quenched with water, extracted with petroleum. The organic phase was washed with brine, dried over sodium sulfate, condensed to give 25.1 Kg of Compound-3. 1H NMR (400 MHz, DMSO-d6) δ 3.34 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.78 (d, J = 16.0 Hz, 1H) , 2.27 (m, 1H) , 1.93 (m, 1H) , 1.68 (m, 3H) , 1.41 (s, 9H) , 1.24 (s, 3H) , 0.13 (s, 9 H) ppm.
Step 3: Synthesis of Compound-4

To a cool (0-5 ℃) solution of 70.1 Kg of Compound-3 in THF was added tetrabutylammonium fluoride (13.3 Kg) in THF. After de-silylation was completed, the reaction was quenched with water, extracted with petroleum (290 Kg) and the organic phase was condensed and passed through a pad of silica gel. The filtrate was condensed to give 48 Kg of Compound-4. 1H NMR (400 MHz, DMSO-d6) δ 3.36 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.82 (d, J = 16.0 Hz, 1H) , 2.28 (m, 1H) , 1.97 (m, 1H) , 1.70 (m, 3H) , 1.41 (s, 9H) , 1.26 (s, 3H) ppm.
Step 4: Syntheses of Compound-5

A solution of Compound-4 (48 Kg) in THF was warmed to 50-60 ℃. To the above solution was added a solution of (-) -di-p-methylbenzoyl-L-tartaric acid (69.6 Kg) in THF. The resulting mixture was stirred at 50-60 ℃ 1-2 h and then gradually cooled to 0-10 ℃. The resulting salt solid was filtered and re-suspended in methyl tert-butyl ether and heated at 50-60 ℃ for 1 h. The mixture was gradually cooled to 0-5 ℃. The resulting solid was filtered to give 13.1 Kg of off-white solid. The solid was treated with aqueous sodium hydroxide, extracted with petroleum, condensed to give 13.1 Kg of Compound-5 (ee≥96%) . 1H NMR (400 MHz, DMSO-d6) δ 3.36 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.82 (d, J = 16.0 Hz, 1H) , 2.29 (m, 1H) , 1.97 (m, 1H) , 1.70 (m, 3H) , 1.41 (s, 9H) , 1.26 (s, 3H) ppm.
Step 5: Syntheses of Compound-6

Intermediate B (14 Kg) , bis (triphenyl) palladium dichloride (0.7 Kg) , CuI (0.42 Kg) and tetramethyl guanidine (11.5 Kg) were dissolved in DMF (48.1 Kg) . The resulting solution was stirred and de-gassed and then heated under nitrogen. A solution of Compound-5 (9.24 Kg) in DMF (16 Kg) was added dropwise. After coupling, the organic phase was condensed, the resiue was stirred with water (145 Kg) and methyl t-butyl ether (104 Kg) , the entire mixture passed trough a pad of celite, separated. The organic phase was washed with a solution of thiourea (14 Kg) in water (165 kg) and brine (100 Kg) , condensed. The residue was dissolved in a mixture of n-heptane (120 Kg) and ethyl acetate (28 Kg) . The solution was mixed with charcoal (1.4 kg) , heated at 40-50 ℃ for 1-2 h, fltered though a pad of silica gel. The filtrate was condensed to give Compound-6 solid (14.89 Kg) and the liquid filtrate (13 Kg heptane solution, contains 1.24 Kg of Compound-6) . 1H NMR (400 MHz, DMSO-d6) δ 7.85 (d, J = 9.6 Hz, 1H) , 7.55 (m, 3H) , 7.32 (m, 2H) , 3.87 (s, 3H) , 3.37 (d, J = 16.0 Hz, 1H) , 3.22 (m , 1H) , 2.94 (d, J = 16.0, Hz, 1H) , 2.60 (m, 1H) , 2.48 (m, 1H) , 2.29 (s, 3h) , 2.26 (m, 1 H) , 1.82 (m, 2H) , 1.49 (s, 3H) , 1.43 (s, 9H) ppm.
Step 6: Syntheses of Compound-7

The above heptane solution of Compound-6 was added into a cold trifluoromethane sulfonic acid (66.1 Kg) while maintaining the internal temperature below 25 ℃. Then solid Compound-6 (14.87 Kg) was added batchwise. After complete addition of Compound-6, the reaction mixture was warmed to 25-30℃ and stiired until the reaction was completed. The entire mixture was poured into a solution of sodium acetate (123.5 Kg) in water (240 Kg) . pH of the solution was then adjusted to 7-8 by adding solid potassium carbonate (46.1 Kg) . The mixture was extracted wuth dichloromethane (509 Kg) , condensed. The residue was mixed with n-heptane (41 Kg) , condensed again to give the precipitate which was filtered and washed by n-heptane (8 Kg) and dried. 8.78 Kg of Compound-7 was obtained. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H) , 7.35 (dd, J = 9.2, 1.6 Hz, 1H) , 7.08 (dd, J = 9.2, 1.6 Hz, 1H) , 3.79 (s, 3H) , 3.68 (d, J = 17.2 Hz, 1H) , 3.21 (d, J = 17.2 Hz, 1H) , 3.06 (m, 1H) , 2.68 (m, 1H) , 1.96 (m, 1H) , 1.74 (m, 1H) , 1.49 (s, 3H) ppm.
Step 7: Syntheses of Compound 1 –Crude 1

Compound-7 (8.76 Kg) was dissolved in methanol (69 Kg) and internally cooled below 25 ℃. Acetic acid (9.3 Kg) and hydrazine hydrate (7.4 Kg, 85%) were added while maintaining internal temperature below 25 ℃. After de-gassed and re-filled with nitrogen (repeated three times) , the reaction mixture was stirred at 55-60 ℃ for 4 h. After a complete reaction, the mixture was mixed with water (29 Kg) . The organic phase was condensed and potassium carbonate (12.5 Kg) in water (40 Kg) was added. The resulting solid was filtered, washed with water (18.3 Kg) . The solid was slurred with water (110 Kg) , centrifuged, dried and slurred with ethanol (9.4 Kg) , centrifuged, filtered, washed with ethanol, dried in vacuum to give Compound 1-Crude 1 (7.91 Kg) . 1H-NMR (600 MHz, DMSO-d 6) δ 12.0 (s, 1H) , 10.2 (s, 1H) , 7.31 (dd, 1H, J=9.6, 2.0 Hz) , 7.19 (dd, 1H, J=9.6, 2.0 Hz) , 3.77 (d, 1H, J=16.4 Hz) , 3.34 (d, 1H, J=16.4 Hz) , 2.97-3.02 (m, 1H) , 2.54-2.58 (m, 1H) , 2.35-2.40 (m, 1H) , 1.90-1.94 (m, 1H) , 1.73-1.75 (m, 1H) , 1.47 (s, 3H) , 1.43-1.45 (m, 1H) ppm. MS (ESI) m/e [M+1] + 299.
Step 8: Synthesis of Compound 1-Crude 2

Under nitrogen protection, Compound 1 (Crude 1) (7.88 Kg) was stirred with isopropanol (422 Kg) and heated at 70-80 ℃ for 1-2 h until the solid disappeared completely. A solution of (+) -di-p-methylbenzoyl-D-tartaric acid (10.25 Kg) in isopropanol (84.4 Kg) was added. The mixture was stirred for 14-16 h, filtered and washed with isopropanol (16 Kg) , dried. The resulting salt was added into a stirred solution of potassium carbonate (6.15 Kg) in water (118 Kg) . The precipitate was centrifuged, filtered, washed with water (18 Kg) . The solid was slurred with water (110 Kg) , centrifuged, dried. The solid was dissolved in THF (75 Kg) , active carbon (0.8 Kg) was added. The mixture was degassed and re-protected by nitrogen, stirred and heated at 40-45 ℃ for 1-2 h, cooled, filtered through celite, condensed to give the solid which was further slurred with ethanol (6.5 Kg) , filtered to give 5.6 Kg of Compound
1 crude
2. 1H NMR (400 MHz, DMSO-d6) δ 12.0 (s, 1H) , 10.2 (s, 1H) , 7.31 (dd, 1H, J=9.6, 2.0 Hz) , 7.19 (dd, 1H, J=9.6, 2.0 Hz) , 3.77 (d, 1H, J=16.4 Hz) , 3.34 (d, 1H, J=16.4 Hz) , 2.97-3.02 (m, 1H) , 2.54-2.58 (m, 1H) , 2.35-2.40 (m, 1H) , 1.90-1.94 (m, 1H) , 1.73-1.75 (m, 1H) , 1.47 (s, 3H) , 1.43-1.45 (m, 1H) ppm. MS (ESI) m/e [M+1] + 299.
PATENT
WO 2017032289
https://patents.google.com/patent/WO2017032289A1/en
Scheme 1: Synthetic Process of Compound A in a large scale

PATENT
WO 2013097225
https://patents.google.com/patent/WO2013097225A1/en
Example 36: Synthesis of Compound 69 Compound 69: (RV2-fluoro-10a-methyl-7,8,9 JO .10a.l l-hexahydro-5,6,7a,l 1- tetraazacvcloheptardeflcyclopentara1fluoren-4(5H)-one

Step 1 : Methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate

To a solution of methyl 3-amino-2-bromo-5-fluorobenzoate (25. Og, 100 mmol) and K2CO3 (42.0g, 302 mmol) in DCM (250mL) were added 2,2,2-trifluoroacetic anhydride (249.0g, 1.197mol) at 5 -10°C under nitrogen atmosphere. The mixture was stirred for overnight at 25°C. The reaction mixture was diluted with DCM, washed with H20 (200mLx2) and saturared
NaHCC”3 aq (200mLx2), dried over anhydrousNa2S04, and concentrated to give 34.0 g (98%) of methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate as white solid. 1H NMR (CDCI3– dl) δ 8.87 (s, 1H), 8.36 (d, 1H,J=6.4 Hz), 7.43 (d, 1H,J=5.2 Hz), 3.98 (s, 3H).
Step 2: (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6- (2,2,2trifluoroacetamido)phenyl)ethvnyl)-2-methylpyrrolidine-l-carboxylate

A mixture of methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate (27.52g, 80 mmol), (PPh3)2PdCl2 (2.8 g, 4 mmol), (R)-benzyl 2-ethynyl-2-methylpyrrolidine-l-carboxylate (19.44 g, 80 mmol),copper(I) iodide (764 mg, 4 mmol) and tetramethylguanidine (27.6 g, 240 mmol) in DMF (200 mL) was heated at 80 °C with nitrogen protection system for 16 hours. The cooled reaction mixture was diluted with EA (3×200 mL) and water (800 mL). The organic layer was separated, washed with water (2×200 mL), dried (Na2S04), and concentrated. The remaining residue was chromatographed on silica gel, eluted with gradient 0-30% EtOAc in hexane to give the product (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6-
(2,2,2trifluoroacetamido)phenyl)ethynyl)-2-methylpyrrolidine-l-carboxylate (21 g, 53%) as white solid. 1H NMR (DMSO-dl) δ 11.01 (s, 1H), 7.64-7.77 (m, 1H), 7.36 (m, 5H),7.19-7.31 (m, 1H), 5.04-5.12 (m, 2H), 3.85(s, 3H ), 3.44-3.47 (m, 2H), 2.0-2.29 (m, 2H), 1.90-1.97 (m, 2H), and 1.69 (s, 3H).MS (ESI) m/e [M+l]+ 507.0.
Step 3: (R)-methyl 6-fluoro-2-(2 -methyl- l-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl)-lH-indole-4- carboxylate

To a solution of (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6- (2,2,2trifiuoroacetamido)phenyl)ethynyl)-2-methylpyrrolidine- 1 -carboxylate(5.0g, 1 Ommol) in toluene was added zinc(II) bromide(l 1.25g, 50 mmol) at room temperture. The reaction mixture was heated at 80 °C with nitrogen protection system for 15 hours. The solvent was removed under reduced pressure, and the residue was treated with DCM (500 mL) and water (800 mL). The organic layer was separated, washed with water (2×200 mL), dried (Na2S04), and
concentrated. The remaining residue was chromatographed on silica gel ,eluted with gradient 0- 50% EtOAc in hexane to give the product(R)-methyl 6-fluoro-2-(2 -methyl- 1 -(2,2,2- trifluoroacetyl)pyrrolidin-2-yl)-lH-indole-4-carboxylate (1.9 g, 51%) as yellow solid. 1H NMR (CDCls-dl) δ 9.97 (s, 1H), 7.62 (d,lH, J=10.2 Hz), 7.27 (d,lH, J=9.6 Hz), 7.05 (d,lH, J=1.2 Hz), 3.98 (s, 3H), 3.86-3.88 (m,2H),2.91-2.96 (m,lH), 2.25-2.28 (m,lH), 2.12-2.16 (m, 2H), and 1.99 (s, 3H). MS (ESI) m/e [M+l]+ 507.0.
Step 4: (R)-methyl 6-fluoro-2-(2-methylpyrrolidin-2-yl)-lH-indole-4-carboxylate

To a solution of (R)-methyl 6-fluoro-2-(2 -methyl- l-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl)- lH-indole-4-carboxylate (1.0 g, 1.9 mmol) in MeOH was added NaBH4 (706 mg, 11.4 mmol) at room temperature. The reaction mixture was refluxed for 4 hours with nitrogen protection system. The solvent was removed under reduced pressure. The residue was dissolved in DCM (200 mL), which was washed with water (200 mL)and brine (200 mL), dried over Na2S04, and concentrated to give the desire product as yellow oil. (R)-methyl 6-fluoro-2-(2-methylpyrrolidin- 2-yl)-lH-indole-4-carboxylate (727 mg, 98%). 1H NMR (CD3OD-dl) δ 7.50(dd,lH, J=10.2, 2.4 Hz), 7.32 (d,lH, J=9.0, 2.4 Hz), 6.93 (s, 1H),3.97 (s, 3H), 3.03-3.12 (m, 2H), 2.27-2.32 (m, 1H),1.88-1.98 (m, 3H), and 1.60 (s, 3H). MS (ESI) m/e [M+l]+ 276.0.
Step 5: (R)-Methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate

To a stirred mixture of (R)-methyl 6-fluoro-2-(2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate (1.0, 1.27 mol), CH3CN (50 ml) and methylbromoacetate (0.58 g, 3.82mmol) was added DIPEA(0.82 g, 6.35 mmol). The reaction mixture was stirred at room temperature for about 20 hours. The reaction mixture was then diluted with CH2CI2 (15 ml) and washed with water three times. The organic layer was dried with MgS04 and concentrated to give 0.85 g of (R)-methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate. 1H NMR (CD3OD-d4) δ 7.47 (dd, 1H, J=2.4, 12.0 Hz), 7.27 (dd, 1H, J=2.4, 9.0 Hz), 6.89 (s,lH), 3.95 (s, 3H), 3.66-3.68 (m, 1H), 3.64 (s, 3H), 3.16-3.17 (m, 2H), 2.72-2.75 (m, 1H), 1.88-2.02 (m, 4H), and 1.44 (s, 3H).MS (ESI) m/e [M+l]+ 349.0.
Step 6: (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3, 5,6, 11,1 lb-hexahydro-lH-indolizinor8,7- blindole-7-carboxylate

In a 25 -mL flask, (R)-methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2- yl)-lH-indole-4-carboxylate (100 mg) was treated with anhydrous MeS03H (6 mL). The flask was fitted with a reflux condenser and heated at 60 °C for 1 h. Then, the reaction mixture was cooled in an ice-bath and diluted with distilled water (6.0 mL). The pH of the solution was increased to pH~10 by the addition of saturated aq. NaHC03. The reaction mixture was then extracted with EtOAc (3×5 mL). Theorganic extracts were combined and washed with brine (lx5mL), dried over Na2S04, filtered, and concentrated. The residue was purified by Pre-TLC to give (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3, 5,6,11,1 lb-hexahydro-lH-indolizino[8,7- b]indole-7-carboxylate(30 mg). 1H NMR (CDCl3-d) δ 7.14-7.224 (m, 2H), 4.03 (s, 3H), 3.81- 3.84 (m, 1H), 3.57-3.59 (m, 1H), 3.22-3.24 (m, 1H), 2.92-2.94 (m, 1H), 2.39-2.40 (m,lH), 2.16- 2.17 (m,lH),1.93-1.94 (m, 1H), 1.63 (s, 3H), and 1.56-1.57 (m, 1H).MS (ESI) m e [M+l]+ 317.0. Step 7: (RV2-fluoro-10a-methyl-7,8,9 JO JOa.l l-hexahvdro-5.6.7a.l 1- tetraazacyclohepta[def|cyclopenta[alfluoren-4(5H)-one

A solution of compound (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3,5,6,l 1,1 lb-hexahydro- lH-indolizino[8,7-b]indole-7-carboxylate (90 mg), acetic acid (0.54 g), and hydrazine hydrate (0.28g) in methanol (30 mL) was heated at reflux. After 5 h, the reaction was cooled and water (5 mL) was added.The mixture was extracted with EtOAc (3×5 mL). The combined organic layers were washed with brine (10 mL) and driedover MgSC^. The mixture was filtered, and the filtrate was evaporated to dryness, and the residue was purified by Pre-TLC using CH2CI2 as eluent to give 80 mg of (R)-2-fluoro-10a-methyl-7,8,9,10,10a,l l-hexahydro-5,6,7a,l l- tetraazacyclohepta[defJcyclopenta[a]fluoren-4(5H)-one. 1H NMR (DMSO-d6) δ 11.9 (s, 1H), 10.2 (s, 1H), 7.30 (d, 1H, J=9.6 Hz), 7.20 (d, 1H, J=10.2 Hz), 3.76 (d, 1H, J=16.4 Hz), 3.34 (d, 1H, J=16.4 Hz), 2.99-3.02 (m, 1H), 2.54-2.58 (m, 1H), 2.35-2.40 (m, 1H), 1.90-1.94 (m, 1H), 1.73-1.75 (m, 1H), 1.48 (s, 3H), and 1.43-1.45(m, 1H). MS (ESI) m/e [M+l]+ 299.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Pamiparib (Partruvix). Pamiparib (27) is an orally active, potent, and highly selective PARP1 and PARP2
inhibitor being developed by BeiGene Limited.190 The drug was approved in China in 2022 for the treatment of germline BRCA-mutated recurrent advanced ovarian, fallopian tube, or primary peritoneal cancer.190BRCA1 and BRCA2 are critical tumor suppressors that help DNA double-strand break (DSB)
repair by functional homologous recombination (HR). 191,192 192 It was claimed that pamiparib showed good brain penetration ability for the treatment of cancer patients with brain metastasis. A small-scale synthesis of pamiparib (27) was first disclosed by BeiGene Limited in 2013.193 Later, they reported a
modified route for industrial scale preparation of the API which is described below. 194,195
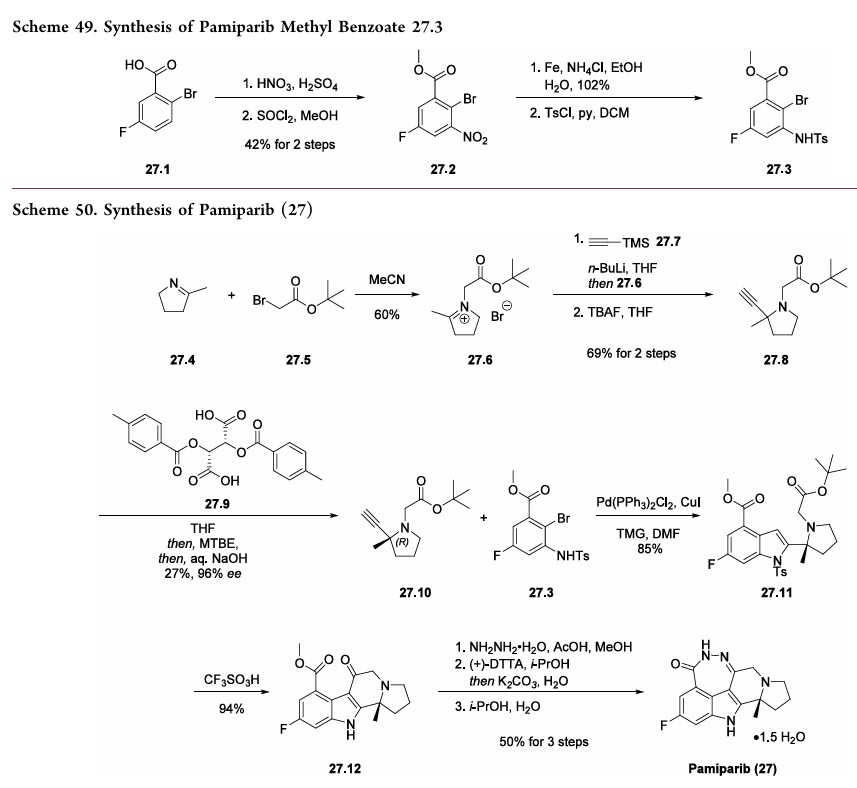
The synthesis commenced with 2-bromo-5-fluorobenzoic acid (27.1) which was subjected to nitration followed by esterification to deliver methyl benzoate 27.2 in 42% overall yield (Scheme 49). The nitro
derivative 27.2 was reduced to an aniline and subsequently protected as a tosylate to obtain the key aryl bromide fragment 27.3. It should be noted that a yield for the tosylation step was not provided by the inventors.
Preparation of the other key fragment 27.10 and endgame of the pamiparib synthesis are described in Scheme 50. First,pyrroline 27.4 was treated with t-butyl bromoacetate 27.5 to generate iminium bromide salt 27.6. An acetylide derived from trimethylsilyl acetylene 27.7 was then added to the iminium to
install the tertiary center. A TBAF-mediated removal of the silyl moiety delivered racemic alkyne 27.8 in 69% yield over two steps. The enantiomers were separated via a classical salt resolution with (−)-di-p-methylbenzolyl-L-tartaric acid (27.9). The desired (R)-enantiomer 27.10 was obtained in 96%
enantiomeric excess (ee) after isolation as the free-base amine. The authors explored several routes to access 27.10; however,the salt resolution approach was selected due to its scalability and reproducibility.
192With the alkyne subunit 27.10 and bromide subunit 27.3 in hand, the next objective was combining them in a convergent manner. This was achieved via an efficient Larock heteroannulation reaction, affording indole 27.11 in 85% yield. Treatment of diester 27.11 with triflic acid triggered removal of both t-butyl ester and N-tosyl protecting groups, as well as cyclization to generate tetracycle 27.12 in 94% yield. The ketoester 27.12 was subjected to hydrazine hydrate in the presence of acetic acid to deliver the
crude cyclized material which was purified via salt formation with (+)-DTTA. Finally, treatment of the amine precursor with water in hot isopropanol delivered pamiparib (27) as a sesquihydrate crystalline solid in 50% over 3 steps.
(190) Markham, A. Pamiparib: First approval. Drugs 2021, 81,1343−1348.
(191) Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.;Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent andselective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431−440.
(192) Wang, H.; Ren, B.; Liu, Y.; Jiang, B.; Guo, Y.; Wei, M.; Luo,L.; Kuang, X.; Qiu, M.; Lv, L.; et al. Discovery of pamiparib (BGB290), a potent and selective poly (ADP-ribose) polymerase (PARP)inhibitor in clinical development. J. Med. Chem. 2020, 63, 15541−15563.
(193) Zhou, C.; Ren, B.; Wang, H. Fused tetracyclic and pentacyclic
dihydrodiazepinocarbazolones as PARP inhibitors and their prepara
tion. WO 2013097225 A1, 2013.
(194) Wang, H.; Zhou, C.; Ren, B.; Kuang, X. Process for preparing
(R)-2-fluoro-10a-methyl-7,8,9,10,10a,11-hexahydro-5,6,7a,11
tetraazacyclohepta[def]cyclopenta[a]fluoren-4(5H)-one as PARP in
hibitor, crystalline forms, and uses thereof. WO 2017032289 A1,
2017.
(195) Wang, H.; Kuang, X.; Zhou, C. Crystalline forms of salts of
fused tetra or penta-cyclic dihydrodiazepinocarazolones, and uses
thereof. WO 2018157794 A1, 2018.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Xiong Y, Guo Y, Liu Y, Wang H, Gong W, Liu Y, et al. (September 2020). “Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor”. Neoplasia. 22 (9): 431–440. doi:10.1016/j.neo.2020.06.009. PMC 7350150. PMID 32652442.
- Markham A (July 2021). “Pamiparib: First Approval”. Drugs. 81 (11): 1343–1348. doi:10.1007/s40265-021-01552-8. PMID 34287805.
- Friedlander M, Mileshkin L, Lombard J, Frentzas S, Gao B, Wilson M, et al. (September 2023). “Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-expansion stage of a multicentre, open-label, phase I trial”. British Journal of Cancer. 129 (5): 797–810. doi:10.1038/s41416-023-02349-0. PMC 10449784. PMID 37474720.
| Clinical data | |
|---|---|
| Trade names | Partruvix |
| Other names | BGB-290 |
| ATC code | L01XK06 (WHO) |
| Legal status | |
| Legal status | US: Investigational New DrugRx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1446261-44-4 |
| PubChem CID | 135565554 |
| DrugBank | DB14769 |
| ChemSpider | 58805610 |
| UNII | 8375F9S90C |
| KEGG | D11426 |
| ChEMBL | ChEMBL4112930 |
| Chemical and physical data | |
| Formula | C16H15FN4O |
| Molar mass | 298.321 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Pamiparib, APPROVALS 2022, CHINA 2022, BeiGene, BGB 290
Sepiapterin
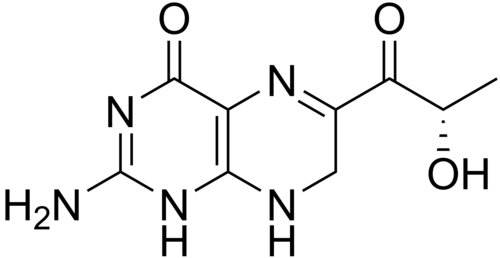
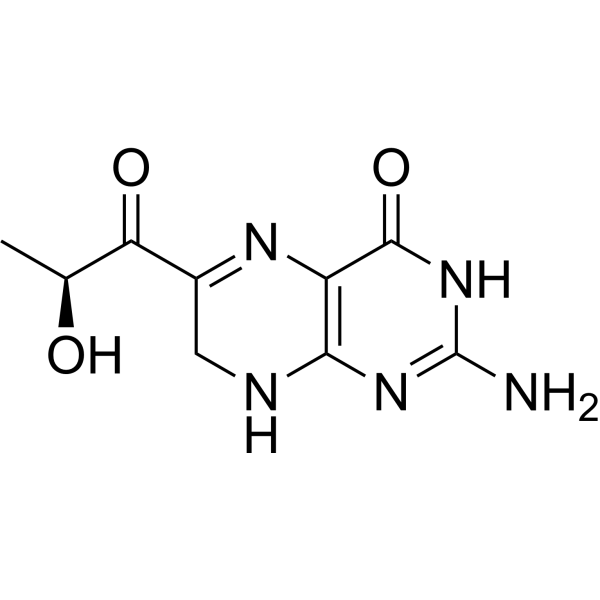
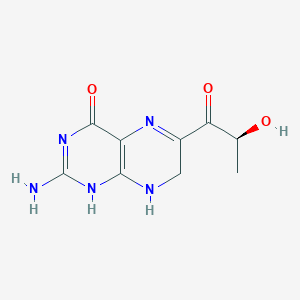
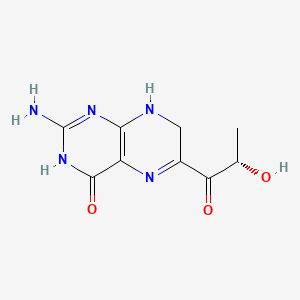
Sepiapterin
- Sepiapterine
- CNSA-001
- CJQ26KO7HP
| Molecular Weight | 237.22 |
|---|---|
| Formula | C9H11N5O3 |
2-amino-6-[(2S)-2-hydroxypropanoyl]-7,8-dihydro-3H-pteridin-4-one
(S)-2-Amino-6-(2-hydroxypropanoyl)-7,8-dihydropteridin-4(3H)-one
- 1-(2-amino-7,8-dihydro-4-hydroxy-6-pteridinyl)-2-hydroxy-1-Propanone
- 2-amino-6-[(2S)-2-hydroxypropanoyl]-7,8-dihydropteridin-4(1H)-one
- 2-amino-7,8-dihydro-6-[(2S)-2-hydroxy-1-oxopropyl]-4(1H)Pteridinone
- S(-)-2-Amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-4(1H)-pteridione
- S-(-)-2-Amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-4(1H)-pteridinone
- 2-AMINO-7,8-DIHYDRO-6-((2S)-2-HYDROXY-1-OXOPROPYL)-4(3H)-PTERIDINONE
- 4(1H)-Pteridinone, 2-amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-, (S)-
7/28/2025 fda approved, Sephience, To treat hyperphenylalaninemia in patients with sepiapterin-responsive phenylketonuria, in conjunction with a phenylalanine-restricted diet
Sepiapterin, sold under the brand name Sephience, is a medication used for the treatment of hyperphenylalaninemia.[2][3] Sepiapterin is a phenylalanine hydroxylase activator.[1]
The most common side effects are upper respiratory tract infection, headache, diarrhea, abdominal pain, hyperphenylalaninemia and discoloration of feces.[2]
Syn
https://patents.google.com/patent/WO2013168693A1/en
Sepiapterin is synthesized by a method of reacting 7,8-dihydropterin and α-keto-β-hydroxybutyric acid in the presence of zinc chloride (Non-patent Document 1), and a method of oxidizing BH4 in air for 6 days. (Non-Patent Document 2) is known.
As a method for synthesizing lactoylpterin, it is known that it can be obtained by oxidizing sepiapterin (Non-patent Documents 3 and 4).International Publication No. 2011/132435
However, the method described in Non-Patent Document 1 produces only a trace amount of sepiapterin and cannot be a stable supply method. Further, in the method of Non-Patent Document 2, very expensive BH4 is used as a raw material, and this cannot be a method that can be industrially stably supplied. Further, the method of Non-Patent Document 2 has a problem that the reaction time is long and many by-products such as biopterin in which BH4 is oxidized and deoxysepiapterin from which the β-position hydroxyl group of the side chain is eliminated are also generated. . In addition, the methods for synthesizing lactoylpterin of Non-Patent Documents 3 and 4 use sepiapterin, which is difficult to obtain industrially, as a raw material, and the yield is low, which cannot be a stable supply method.
Accordingly, an object of the present invention is to provide a novel production method capable of stably supplying sepiapterin, lactoylpterin and tetrahydrolactoylpterin, which have recently been found to be useful as pharmaceuticals.
Therefore, the present inventor has studied a method for synthesizing sepiapterin, lactoylpterin, and tetrahydrolactoylpterin using available raw materials. As a starting material, the compound of the following formula (1) or the compound of formula (7) is used. As a result, it was found that sepiapterin, lactoylpterin and tetrahydrolactoylpterin can be obtained in good yield, and these compounds can be stably supplied as a medicine for the first time, thereby completing the present invention.
Example 1
Synthesis of S-lactoylpterin (2)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxymethoxypropan-1-one (compound (1)) 24.7 g (68.2 mmol) in methanol 50 mL, 3 mol / L hydrochloric acid 250 mL And stirred at 50 ° C. for 3 hours. The reaction solution was adjusted to pH = 7 with an aqueous sodium hydroxide solution, collected by filtration, and dried under reduced pressure to obtain 15.1 g (64.2 mmol, 94% yield) of S-lactoylpterin.
(S-lactoylpterin: (2))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.32 (d, 3H, J = 6.8Hz), 5.16 (br, 1H), 5.32 (q, 1H, J = 6.8Hz), 9.09 (s, 1H )
Example 2 (Synthesis of 1- (2-amino-4-cyclohexyloxypteridin-6-yl) -2S-hydroxypropan-1-one)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2St-butyldimethylsilanoxypropan-1-one (compound (1)) (4.0 g, 9.27 mmol) was added to THF 40 mL, 70% 6.92 g (18.5 mmol) of tetrabutylammonium fluoride was added and stirred at 10 ° C. or lower for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dehydrated and concentrated under reduced pressure. The crude product was purified by flash chromatography to give 2.09 g (6.59 mmol, 71% yield) of 1- (2-amino-4-cyclohexyloxypteridin-6-yl) -2S-hydroxypropan-1-one. Got.
1 H NMR (DMSO-d 6 ): δ / ppm = 1.38 (d, 3H, J = 6.6Hz), 1.37-1.79 (m, 8H), 1.98-1.99 (m, 2H), 5.20 (d, 1H, J = 6.3Hz), 5.34 (dq, 1H, J = 6.6Hz), 5.29-5.37 (m, 1H), 7.68 (br, 1H), 7.82 (br, 1H), 9.22 (s, 1H)
Example 3
Synthesis of S-lactoylpterin hydrochloride

To 500 mg (2.13 mmol) of S-lactoylpterin were added 1.25 mL of 6 mol / L hydrochloric acid and 10 mL of ethanol, and the mixture was stirred for 30 minutes. The crystals were collected by filtration and dried under reduced pressure, and 465 mg of S-lactoylpterin hydrochloride (1. 71 mmol, yield 80%).
(S-lactoylpterin hydrochloride)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.34 (d, 3H, J = 6.9Hz), 3.91 (br, 3H), 5.34 (q, 1H, J = 6.9Hz), 9.12 (s, 1H )
Example 4 Synthesis of 2-amino-6- (2S-hydroxypropionyl) -7,8-dihydro-3H-pteridin-4-one (S-sepiapterin)

To 500 mg (2.13 mol) of S-lactoylpterin were added 125 mL of methanol, 2.08 mL (14.9 mmol) of triethylamine, 250 mg of 8.4% Pd / C (Ph 2 S) (containing 50% water), and an external temperature of 40 ° C. The hydrogenation reaction was carried out for 3 hours. After completion of the reaction, the reaction solution was stirred in air at room temperature for 1 hour, and then the catalyst was filtered off from the reaction solution and concentrated under reduced pressure. The crude product was separated and purified by flash chromatography and 296 mg (1.25 mmol) of S-sepiapterin. Yield 59%).
(S-sepiapterin: (3))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.21 (d, 3H, J = 6.6Hz), 4.11 (s, 2H), 4.89 (d, 1H, J = 6.6Hz), 5, 10 (quin ., 1H, J = 6.6Hz), 6.81 (br-s, 2H), 7.51 (s, 1H), 10.26 (s, 1H)
Example 5
To 20 mg (0.085 mmol) of S-lactoylpterin were added 2 mL of saturated aqueous sodium hydrogen carbonate and 76 mg (0.44 mmol) of sodium dithionite, and the mixture was stirred at room temperature for 2 hours to give S-sepiapterin as a mixture.
Example 6
A reaction was carried out in the same manner as in Example 5 except that 20% (0.085 mmol) of S-lactoylpterin was used and the saturated aqueous sodium bicarbonate solution was changed to an aqueous sodium borate solution to give S-sepiapterin as a mixture.
Example 7
Synthesis of S-sepiapterin hydrochloride

To 620 mg (2.61 mmol) of S-sepiapterin were added 2.5 mL of 6 mol / L hydrochloric acid and 5.0 mL of ethanol, and the mixture was stirred at 0 ° C. for 30 minutes. The crystals were collected by filtration and dried under reduced pressure to obtain 650 mg (2.38 mmol, yield 91%) of S-sepiapterin hydrochloride.
(S-sepiapterin hydrochloride)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.22 (d, 3H, J = 6.9Hz), 4.14 (s, 2H), 4.89 (d, 1H, J = 6.6Hz), 5.11 (q, 1H , J = 6.9Hz), 7.40 (br-s, 4H), 7.80 (br-s, 1H)
Example 8 (Synthesis of 2-amino-6- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (S-tetrahydrolactoylpterin) dihydrochloride)

Methanol 50 mL, 6 mol / L hydrochloric acid 5 mL, and borane pyridine complex 593 mg (6.38 mmol) were added to 1.00 g (4.25 mmol) of S-lactoylpterin, and the mixture was stirred at an external temperature of 0 ° C. for 1 hour. After completion of the reaction, 5 mL of acetone was added, concentrated under reduced pressure, azeotropically dehydrated with ethanol, ethanol was added, the crystals were filtered and dried under reduced pressure, and a mixture 1 of S-tetrahydrolactoylpterin dihydrochloride (4a) and (4b) 1 .12 g (3.59 mmol, 85% yield) was obtained.
(6S—S-tetrahydrolactoylpterin dihydrochloride: (4a))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.24 (d, 3H, J = 6.9Hz), 3.45 (dd, 1H, J = 7.2, 13.5Hz), 3.87 (dd, 1H, J = 3.3, 13.5Hz), 4.34 (q, 1H, J = 6.9Hz), 4.53 (dd, 1H, J = 3.3, 7.2Hz), 7.03 (br-s, 4H), 7.67 (br-s, 1H)
(6R-S-tetrahydrolactoylpterin dihydrochloride: (4b))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.24 (d, 3H, J = 6.9Hz), 3.45 (dd, 1H, J = 6.9, 13.5Hz), 3.91 (dd, 1H, J = 3.3, 13.5Hz), 4.31 (q, 1H, J = 6.6Hz), 4.55 (dd, 1H, J = 3.3, 6.9Hz), 7.12 (br-s, 3H), 7.71 (br-s, 2H)
Example 9
To 3.00 g (12.8 mmol) of S-lactoylpterin was added 150 mL of methanol, 15 mL of 6 mol / L hydrochloric acid, and 1.78 g (19.1 mmol) of borane pyridine complex, and the mixture was stirred at an external temperature of 0 ° C. for 1 hour. After completion of the reaction, 45 mL of concentrated hydrochloric acid was added, and the mixture was stirred overnight at the same temperature. The crystals were collected by filtration and dried under reduced pressure, and 1.63 g (5.2 mmol, yield) of 6S-S-tetrahydrolactoylpterin dihydrochloride (4a) 41%). The filtrate was concentrated under reduced pressure, azeotropically dehydrated with ethanol, ethanol was added, the crystals were collected by filtration and dried under reduced pressure, and 1.38 g (4.4 mmol) of 6R-S-tetrahydrolactoylpterin dihydrochloride (4b) was collected. Yield 35%). It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 10
Methanol 5 mL, 6 mol / L hydrochloric acid 0.5 mL, and borane pyridine complex 59 mg (0.64 mmol) were added to 100 mg (0.43 mmol) of S-lactoylpterin, and the mixture was stirred overnight at an external temperature of 0 ° C. The precipitated crystals were collected by filtration and dried under reduced pressure to obtain 46 mg (0.15 mmol, yield 35%) of 6S—S-tetrahydrolactoylpterin dihydrochloride (4a). It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 11
To 200 mg (0.85 mol) of S-lactoylpterin, 50 mL of methanol, 0.62 mL (5.95 mmol) of diethylamine and 100 mg of 8.4% Pd / C (Ph 2 S) (containing 50% water) were added, and the external temperature was 40 ° C. The hydrogenation reaction was carried out for 2.5 hours. After completion of the reaction, concentrated hydrochloric acid is added, the catalyst is filtered off, concentrated under reduced pressure, azeotropically dehydrated with ethanol, ethanol is added, the crystals are filtered and dried under reduced pressure, and S-tetrahydrolactoylpterin dihydrochloride (4a) and 122 mg (0.39 mmol, 46% yield) of a mixture of (4b) was obtained. It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 12 (Synthesis of 2-amino-6- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (S-tetrahydrolactoylpterin) ditoluenesulfonate)

To 100 mg (0.43 mmol) of S-lactoylpterin was added 5 mL of methanol, 0.5 mL of water, 566 mg (2.98 mmol) of p-toluenesulfonic acid monohydrate, and 59 mg (0.64 mmol) of borane pyridine complex. Stir at 0 ° C. for 1 hour. After completion of the reaction, 0.5 mL of acetone was added and concentrated under reduced pressure. After azeotropic dehydration with ethanol, acetone was added, the crystals were collected by filtration and dried under reduced pressure, and S-tetrahydrolactoylpterin ditoluenesulfonate 158 mg (0.27 mmol, Yield 63%) was obtained.
(S-tetrahydrolactoylpterin ditoluenesulfonate)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.25 (d, 3H, J = 7.2Hz), 2.29 (S, 6H), 3.35 (dd, 1H, J = 7.5, 13.5Hz), 3.84 (dd , 1H, J = 3.0, 13.5Hz), 4.35 (q, 1H, J = 6.9Hz), 4.49 (dd, 1H, J = 3.0, 7.5Hz), 6.72 (br-s, 2H), 7.13 (d, 4H, J = 8.1Hz), 7.49 (d, 4H, J = 8.1Hz), 7.62 (br-s, 1H), 10.66 (br-s, 1H)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.25 (d, 3H, J = 7.2Hz), 2.29 (S, 6H), 3.33 (dd, 1H, J = 7.5, 13.5Hz), 3.84 (dd , 1H, J = 3.0, 13.5Hz), 4.32 (q, 1H, J = 6.9Hz), 4.49 (dd, 1H, J = 3.0, 7.5Hz), 6.72 (br-s, 2H), 7.13 (d, 4H, J = 8.1Hz), 7.49 (d, 4H, J = 8.1Hz), 7.62 (br-s, 1H), 10.66 (br-s, 1H)
Example 13 Synthesis of 2-amino-6- (2S-hydroxypropionyl) -7,8-dihydro-3H-pteridin-4-one (S-sepiapterin)

6 mL of water and 6 mL of ethanol were added to 1.00 g (3.20 mmol) of S-tetrahydrolactoylpterin dihydrochloride, and 363 mg (3.20 mmol) of 30% aqueous hydrogen peroxide was added at an external temperature of −10 ° C. at the same temperature. Stir for 2 hours. A sodium sulfite aqueous solution was added to the reaction solution, and the crystals were collected by filtration and dried under reduced pressure to obtain 676 mg (2.85 mmol, yield 89%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 14
46 mg of S-sepiapterin was prepared in the same manner as in Example 13 except that 100 mg (0.32 mmol) of S-tetrahydrolactoylpterin dihydrochloride was changed to 68 mg (0.32 mmol) of 36% peracetic acid with 30% hydrogen peroxide. 0.19 mmol, 61% yield). It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 15
S-Sepia was prepared in the same manner as in Example 13 except that 100 mg (0.32 mmol) of S-tetrahydrolactoylpterin dihydrochloride was changed to 85 mg of m-CPBA (content 65%, 0.32 mmol) with 30% hydrogen peroxide. 35 mg (0.15 mmol, 46% yield) of pterin was obtained. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 16
20 mL of methanol and 0.89 mL (6.40 mmol) of triethylamine were added to 200 mg (0.64 mmol) of S-tetrahydrolactoylpterin dihydrochloride, and the mixture was stirred at room temperature for 1 hour in air. The reaction mixture was concentrated under reduced pressure, water was added, the crystals were collected by filtration and dried under reduced pressure to obtain 105 mg (0.44 mmol, yield 69%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 17
Add 20 mL of methanol to 200 mg (0.64 mmol) of S-tetrahydrolactoylpterin dihydrochloride, neutralize with 0.16 mL (1.28 mmol) of 8 mol / L aqueous sodium hydroxide solution, and stir in air at room temperature for 1 hour did. The reaction mixture was concentrated under reduced pressure, water was added, the crystals were collected by filtration and dried under reduced pressure to obtain 87 mg (0.37 mmol, yield 58%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 18 (Synthesis of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxymethoxypropan-1-one)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxymethoxypropan-1-one (1.00 g, 2.77 mmol), ethyl acetate 60 mL, 10% Pd—C 500 mg, potassium carbonate 3 .82 g (27.6 mmol) was added, and the hydrogenation reaction was performed at an external temperature of 50 ° C. for 3 hours. After the catalyst was filtered off, the reaction solution was concentrated under reduced pressure. The crude product was separated and purified by flash chromatography to obtain 257 mg (0.71 mmol) of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxymethoxypropan-1-one. Yield 26%).
1 H NMR (CDCl 3 ): δ / ppm = 1.33-1.47 (m, 3H), 1.44 (d, 3H, J = 6.9Hz), 1.54-1.63 (m, 3H), 1.79 (m, 2H), 1.91 (m, 2H), 3.37 (s, 3H), 4.36 (d, 1H, J = 15.6), 4.43 (d, 1H, J = 15.6), 4.71 (d, 1H, J = 6.6Hz), 4.74 (d , 1H, J = 6.6Hz), 4.90 (br-s, 2H), 5.00 (br-s, 1H), 5.05-5.11 (m, 1H), 5.34 (q, 1H, J = 6.9Hz)
Example 19 (Synthesis of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxyethoxymethoxypropan-1-one)

100 mg (0.56 mmol) of ascorbic acid was weighed and 2 mL of water was added. 1- (2-amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxyethoxymethoxypropane- dissolved in 2 mL of methanol after neutralizing the pH of the solution with 1 mol / L aqueous sodium hydroxide solution 20 mg (0.054 mmol) of 1-one was added. To this, 80 mg (0.46 mmol) of Na 2 S 2 O 4 was added and stirred at room temperature for 1 hour. Water was added to the reaction solution, and the mixture was extracted with ethyl acetate. After dehydrating the organic phase, the solvent was concentrated under reduced pressure. Separation and purification by silica gel column chromatography gave 4.4 mg (0.011 mmol) of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxyethoxymethoxypropan-1-one. Yield 20%).
1 H NMR (CDCl 3 ): δ / ppm = 1.13 (m, 1H), 1.44 (d, 3H, J = 6.8 Hz), 1.63 (m, 1H), 1.80 (m, 2H), 1.93 (m, 2H ), 2.06 (m, 2H), 3.37 (s, 3H), 3.52 (m, 2H), 3.70 (t, J = 4.6 Hz, 2H), 4.40 (m, 2H), 4.81 (m, 2H), 5.11 (tt, J = 3.9, 8.5 Hz, 1H), 5.35 (q, J = 6.8 Hz, 1H)
Example 20
Synthesis of S-sepiapterin (3)

To 10 mg of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxymethoxypropan-1-one (compound (5-2)) was added 0.1 mL of concentrated hydrochloric acid. , Warmed up. The reaction solution was diluted with water, neutralized to pH 6-7 with an aqueous sodium hydroxide solution, and the precipitated crystals were filtered off. The filtrate was concentrated under reduced pressure to obtain S-sepiapterin as a mixture. The resulting compound was consistent with the spectral data described in Example 4.
Example 21
Synthesis of S-sepiapterin (3)

4.0 mg (9.8 μmol) of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxyethoxymethoxypropan-1-one in 2 mL of methanol, ascorbic acid 3 0.02 mg was added, 2 mL of 3 mol / L hydrochloric acid was added thereto, and the mixture was stirred at 50 ° C. for 6 hours while shielding light. The solution was adjusted to pH 7 with 28% aqueous ammonia, washed with ethyl acetate, and purified by Florisil column chromatography to obtain 2.0 mg (8.4 μmol, yield 86%) of S-sepiapterin. As a result of HPLC measurement, the retention time and the UV waveform of the peak coincided with the standard S-sepiapterin.
Example 22 (Synthesis of 2-amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl] -3H-pteridin-4-one (6))

To 3.00 g (12.8 mmol) of S-lactoylpterin were added 30 mL of DMF, 2.61 g (38.3 mmol) of imidazole and 3.84 g (25.5 mmol) of TBSCl, and the mixture was stirred for 1 hour under ice cooling. Water was added to the reaction mixture, and the crystals were collected by filtration and dried under reduced pressure to give 3.91 g of 2-amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl] -3H-pteridin-4-one (6). (11.2 mmol, 88% yield) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.01 (s, 3H), 0.06 (s, 3H), 0.83 (s, 9H), 1.36 (d, 3H, J = 6.9Hz), 5.55 (q , 1H, J = 6.9Hz), 9.10 (s, 1H), 11.73 (br-s, 1H)
Example 23 (Synthesis of 2-amino-6- [2S- (triisopropylsilanyl) -propionyl] -3H-pteridin-4-one (6))
2-amino-6- [2S- (triisopropylsilanyl) -propionyl] -3H- was prepared in the same manner as in Example 22 except that TBSCl was changed to TIPSCl from 300 mg (1.28 mmol) of S-lactoylpterin. Pteridin-4-one (6) (339 mg, 0.87 mmol, yield 68%) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.89-1.15 (m, 21H), 1.40 (d, 3H, J = 6.9Hz), 5.71 (q, 1H, J = 6.9Hz), 9.13 (s , 1H), 11.74 (br-s, 1H)
Example 24 (Synthesis of 2-amino-6- [2S- (tert-butyldiphenylsilanyl) -propionyl] -3H-pteridin-4-one (6))

2-amino-6- [2S- (triisopropylsilanyl) -propionyl] -3H- was prepared in the same manner as in Example 22 except that TBSCl was changed to TBDPSCl from 300 mg (1.28 mmol) of S-lactoylpterin. 498 mg (1.05 mmol, yield 82%) of pteridin-4-one (6) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 1.03 (s, 9H), 1.38 (d, 3H, J = 6.9Hz), 5.71 (q, 1H, J = 6.9Hz), 7.23-7.33 (m , 3H), 7.37-7.45 (m, 3H), 7.50-7.59 (m, 2H), 7.61-7.71 (m, 2H), 8.96 (s, 1H), 11.67 (br-s, 1H)
Example 25 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one Synthesis of (7))

2-amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl] -3H-pteridin-4-one 1.50 g (4.29 mmol) and 75 mL of ethyl acetate, di-tert-butyl dicarbonate 4 .68 g (21.4 mmol) and N, N-dimethylaminopyridine 52 mg (0.43 mmol) were added, and the mixture was heated to reflux for 1 hour. The reaction solution is washed with water, and the organic layer is dehydrated and concentrated under reduced pressure to give 2- (N, N-di-tert-butylcarbonyl) -amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl]- 2.18 g (3.35 mmol, yield 78%) of 3H-pteridin-4-one (7) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.01 (s, 3H), 0.08 (s, 3H), 0.75 (s, 9H), 1.40 (d, 3H, J = 6.6Hz), 1.48 (s , 18H), 1.71 (s, 9H), 5.59 (q, 1H, J = 6.6Hz), 9.53 (s, 1H)
Example 26 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butoxycarbonyloxypropan-1-one (7 )

To 1.00 g (4.25 mmol) of S-lactoylpterin was added 50 mL of THF, 4.64 g (21.3 mmol) of di-tert-butyl dicarbonate, and 30 mg (0.25 mmol) of N, N-dimethylaminopyridine. Heated to reflux for hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by flash chromatography, and 1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] There was obtained 0.30 g (0.47 mmol, yield 11%) of -2S-tert-butoxycarbonyloxypropan-1-one (7).
1 H NMR (CDCl 3 ): δ / ppm = 1.26 (s, 9H), 1.27 (d, 3H, J = 7.2Hz), 1.45 (s, 18H), 1.71 (s, 9H), 6.11 (q, 1H , J = 7.2Hz), 6.73 (s, 1H)
Example 27 (Synthesis of 1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-methoxymethoxypropan-1-one)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxymethoxypropan-1-one (1.00 g, 2.76 mmol) was added with 20 mL of THF and 1.27 g of di-tert-butyl dicarbonate ( 5.82 mmol) and 3.4 mg (0.03 mmol) of N, N-dimethylaminopyridine were added, and the mixture was heated to reflux for 1 hour. The reaction solution was concentrated under reduced pressure to give 1- [4-cyclohexyloxy-2- (N, N-di-tert-butylcarbonyl) aminopteridin-6-yl] -2S-methoxymethoxypropan-1-one (7) 1 Obtained .55 g (2.76 mmol, 100% yield).
1 H NMR (DMSO-d 6 ): δ / ppm = 1.45-1.88 (m, 8H), 1.53 (s, 18H), 1.58 (d, 3H, J = 6.9Hz), 2.10-2.14 (m, 2H) , 3.38 (s, 3H), 4.78 (d, 1H, J = 6.9Hz), 4.84 (d, 1H, J = 6.9Hz), 5.36-5.45 (m, 1H), 5.55 (q, 1H, J = 6.9 Hz), 9.65 (s, 1H)
Example 28 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-7,8-dihydropteridin-6-yl] -2S-tert-butyldimethylsilanyl Synthesis of oxypropan-1-one)

1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 1.31 g ( 2.02 mmol) was added with 130 mL of ethyl acetate, 655 mg of 10% Pd—C and 2.78 g (20.1 mmol) of potassium carbonate, and the hydrogenation reaction was carried out for 1 hour at an external temperature of 50 ° C. under normal pressure (H 2 balloon). . After the catalyst was filtered off, the reaction solution was stirred in air at room temperature overnight, and the reaction solution was concentrated under reduced pressure. The crude product was separated and purified by flash chromatography, and 1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-7,8-dihydropteridin-6-yl]- 684 mg (1.05 mmol, 66% yield) of 2S-tert-butyldimethylsilanyloxypropan-1-one was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.01 (s, 3H), 0.07 (s, 3H), 0.82 (s, 9H), 1.24 (d, 3H, J = 6.6Hz), 1.42 (s , 18H), 1.53 (s, 9H), 4.23 (d, 1H, J = 16.5Hz), 4.32 (d, 1H, J = 16.5Hz), 5.39 (q, 1H, J = 6.6Hz), 7.92 (s , 1H)
Example 29 (Synthesis of 2-amino-6S- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (6S-S-tetrahydrolactoylpterin) dihydrochloride)

1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 4.92 g ( 7.57 mmol) was added 250 mL of ethyl acetate, 2.46 g of 10% Pd—C, and 10.5 g (76.0 mmol) of K 2 CO 3 , and the hydrogenation reaction was performed at an external temperature of 50 ° C. under normal pressure (H 2 balloon). It went for 1 hour. After the catalyst was filtered off, the reaction solution was concentrated under reduced pressure, 49 mL of concentrated hydrochloric acid was added, and the mixture was concentrated under reduced pressure. Ethanol was added to the concentrate, and the crystals were collected by filtration and dried under reduced pressure to obtain 1.79 g (5.73 mmol, yield 76%) of 6S—S-tetrahydrolactoylpterin dihydrochloride (4a). The compound obtained agreed with the spectral data described in Example 8.
Example 30
1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 500 mg (0. 164 mg (0.53 mmol, 68% yield) of 6S—S-tetrahydrolactoylpterin dihydrochloride (4a) was obtained in the same manner as in Example 29 except that the amount of 10% Pd / C was changed to 100 mg from 77 mmol). Obtained. It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 31 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6-yl] -2S-tert- Synthesis of butyldimethylsilanyloxypropan-1-one)

1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 100 mg (0. 15 mmol), 10 mL of ethyl acetate, 20 mg of 10% Pd—C and 156 mg (1.54 mmol) of triethylamine were added, and the hydrogenation reaction was carried out for 1 hour at an external temperature of 50 ° C. under normal pressure (H 2 balloon). After the catalyst was filtered off, the reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by flash chromatography to give 1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino. -5,6,7,8-tetrahydropteridin-6S-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one (8a) 30 mg (0.045 mmol, 30% yield) and 1- [4 -Tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6R-yl] -2S-tert-butyldimethylsilanyloxypropane-1 -30 mg (0.045 mmol, 30% yield) of ONE (8b) was obtained.
(8a)
1 H NMR (DMSO-d 6 ): δ / ppm = 0.08 (s, 3H), 0.09 (s, 3H), 0.89 (s, 9H), 1.21 (d, 3H, J = 6.6Hz), 1.37 (s , 18H), 1.49 (s, 9H), 3.56-3.67 (m, 2H), 4.39 (m, 1H), 4.42 (q, 1H, J = 6.6Hz), 4.79 (s, 1H), 7.00 (s, 1H)
(8b)
1 H NMR (DMSO-d 6 ): δ / ppm = 0.08 (s, 3H), 0.09 (s, 3H), 0.89 (s, 9H), 1.23 (d, 3H, J = 6.6Hz), 1.37 (s , 18H), 1.49 (s, 9H), 3.40-3.53 (m, 2H), 4.35 (m, 1H), 4.44 (q, 1H, J = 6.6Hz), 4.93 (s, 1H), 7.09 (s, 1H)
Example 32 (1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6-yl] -2S-methoxymethoxypropane- Synthesis of 1-one)

From Example 1 from 200 mg (0.36 mmol) of 1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-methoxymethoxypropan-1-one In a similar manner, 1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6-yl] -2S-methoxymethoxypropane- 76 mg (0.13 mmol, yield = 38%) of 1-one was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 1.21 (d, 3H, J = 6.9Hz), 1.32-1.37 (m, 3H), 1.38 (s, 18H), 1.43-1.51 (m, 3H) , 1.73 (m, 2H), 1.89-1.91 (m, 2H), 3.27 (s, 3H), 3.51-3.56 (m, 2H), 4.33-4.35 (m, 1H), 4.41 (q, 1H, J = 6.9Hz), 4.59 (d, 1H, J = 6.9Hz), 4.67 (d, 1H, J = 6.9Hz), 4.86-4.89 (m, 1H), 4.95 (d, 1H, J = 2.7Hz), 7.08 (s, 1H)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.24 (d, 3H, J = 6.9Hz), 1.32-1.37 (m, 3H), 1.38 (s, 18H), 1.43-1.51 (m, 3H) , 1.73 (m, 2H), 1.89-1.91 (m, 2H), 3.32 (s, 3H), 3.51-3.56 (m, 2H), 4.33-4.35 (m, 1H), 4.39 (q, 1H, J = 6.9Hz), 4.59 (d, 1H, J = 6.9Hz), 4.67 (d, 1H, J = 6.9Hz), 4.86-4.89 (m, 1H), 5.01 (d, 1H, J = 2.4Hz), 7.08 (s, 1H)
Example 33 Synthesis of 2-amino-6- (2S-hydroxypropionyl) -7,8-dihydro-3H-pteridin-4-one (S-sepiapterin)

1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-7,8-dihydropteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropane-1 -To 300 mg (0.46 mmol) of ON was added 3 mL of acetonitrile and 6 mL of 2 mol / L hydrochloric acid, and the mixture was stirred at an external temperature of 40 ° C for 3 hours. The reaction solution was adjusted to pH = 7 with an aqueous sodium hydroxide solution, and the crystals were collected by filtration and dried under reduced pressure to obtain 96 mg (0.40 mmol, yield 88%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 34 (Synthesis of 2-amino-6R- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (6R-S-tetrahydrolactoylpterin) dihydrochloride)

1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6R-yl] -2S-tert-butyldimethylsilanyl To 393 mg (0.60 mmol) of oxypropan-1-one (8b) was added 10 mL of concentrated hydrochloric acid, and the mixture was concentrated under reduced pressure. Ethanol was added to the concentrate, and the crystals were collected by filtration and dried under reduced pressure to obtain 106 mg (0.34 mmol, yield 56%) of 6R—S-tetrahydrolactoylpterin dihydrochloride (4b). The compound obtained agreed with the spectral data described in Example 8.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Sepiapterin is indicated for the treatment of hyperphenylalaninemia in people with phenylketonuria.[1][2]
Side effects
The most common side effects are upper respiratory tract infection, headache, diarrhea, abdominal pain, hyperphenylalaninemia and discoloration of feces.[2]
Society and culture
Legal status
In April 2025, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Sephience, intended for the treatment of hyperphenylalaninemia in adults and children with phenylketonuria.[2] The applicant for this medicinal product is PTC Therapeutics International Limited.[2] Sepiapterin was authorized for medical use in the European Union in June 2025.[2][3]
Sepiapterin was approved for medical use in the United States in July 2025.[1]
Research
Deficiency of tetrahydrobiopterin can cause toxic buildup of phenylalanine (phenylketonuria) as well as deficiencies of dopamine, norepinephrine, and epinephrine, leading to dystonia and other neurological illnesses. This has led to clinical study of sepiapterin in humans to treat tetrahydrobiopterin deficiency.[4]
Since atherosclerosis and other circulatory diseases associated with diabetes are also associated with tetrahydrobiopterin deficiency, animal studies of the value of sepiaterin in these vascular diseases have been done. These studies show that relaxation of the blood vessels studied was impaired after animals were given sepiapterin, even though their levels of tetrahydrobiopterin were replenished.[5]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219666s000lbl.pdf
- “Sephience EPAR”. European Medicines Agency (EMA). 25 April 2025. Retrieved 2 May 2025. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Sephience Product information”. Union Register of medicinal products. 25 June 2025. Retrieved 27 June 2025.
- Smith N, Longo N, Levert K, Hyland K, Blau N (April 2019). “Phase I clinical evaluation of CNSA-001 (sepiapterin), a novel pharmacological treatment for phenylketonuria and tetrahydrobiopterin deficiencies, in healthy volunteers”. Molecular Genetics and Metabolism. 126 (4): 406–412. doi:10.1016/j.ymgme.2019.02.001. ISSN 1096-7192. PMID 30922814. S2CID 85564348.
- Vasquez-Vivar J, Duquiane D, Whitsett J, Kalyanaraman B, Rajagopalan S (October 2002). “Altered Tetrahydrobiopterin Metabolism in Atherosclerosis”. Arteriosclerosis, Thrombosis, and Vascular Biology. 22 (10): 1655–1661. doi:10.1161/01.ATV.0000029122.79665.D9. PMID 12377745.
| Names | |
|---|---|
| IUPAC name2-amino-6-[(2S)-2-hydroxypropanoyl]-7,8-dihydro-1H-pteridin-4-one | |
| Other namesSephience | |
| Identifiers | |
| CAS Number | 17094-01-8 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL1255653 |
| ChemSpider | 58746 |
| KEGG | C00835 |
| PubChem CID | 65253 |
| UNII | CJQ26KO7HP |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C9H11N5O3 |
| Molar mass | 237.22 g/mol |
| Pharmacology | |
| ATC code | None |
| Routes of administration | By mouth |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
- [1]. Pannirselvam M, et al. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br J Pharmacol. 2003;140(4):701‐706. [Content Brief][2]. Cho YR, et al. Sepiapterin inhibits cell proliferation and migration of ovarian cancer cells via down-regulation of p70S6K-dependent VEGFR-2 expression. Oncol Rep. 2011;26(4):861‐867. [Content Brief]
//////////Sepiapterin, approvals 2025, fda 2025, Sephience, Sepiapterine, CNSA 001, CJQ26KO7HP, PTC 923, WHO 11848,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}