
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
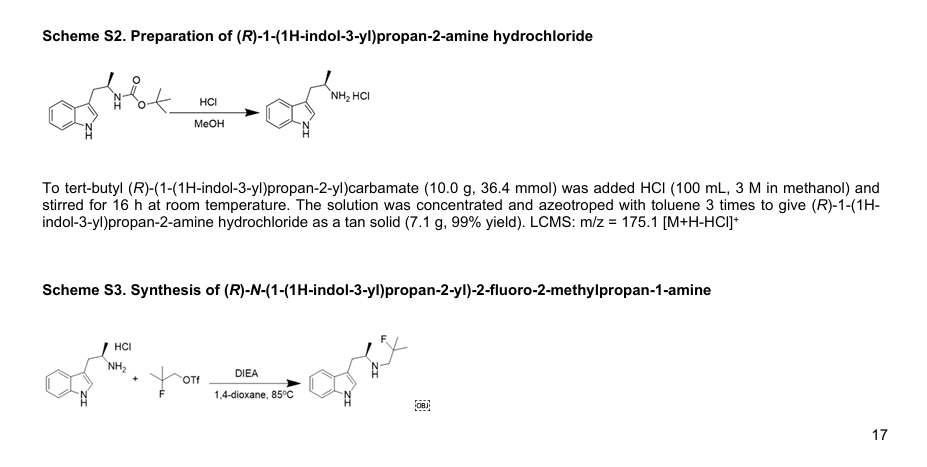
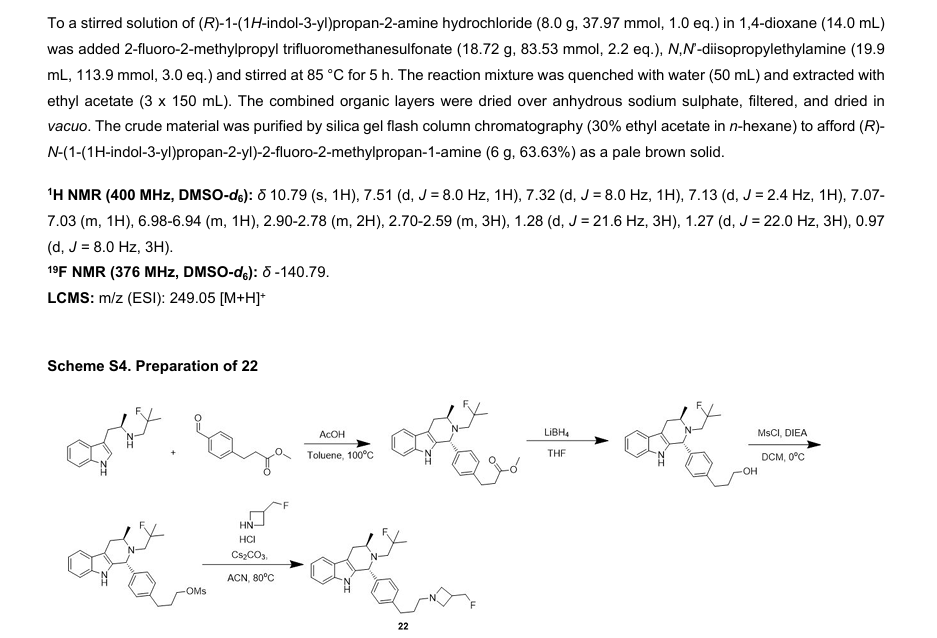
PRITELIVIR MESYLATE
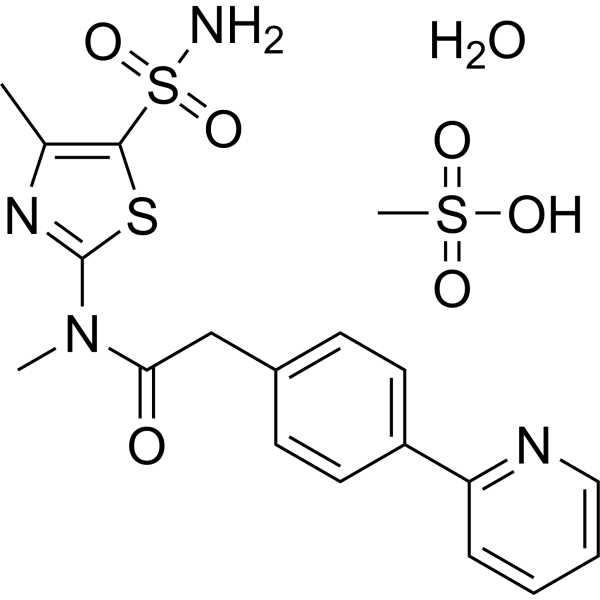
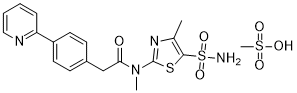
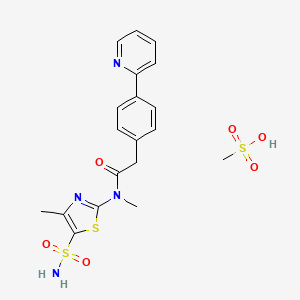
PRITELIVIR MESYLATE
CAS 1428333-96-3
1428321-10-1 HYDRATE
FREE FORM
AIC316 mesylate hydrate; BAY 57-1293 mesylate hydrate
BAY57-1293; BAY 57-1293; BAY-57-1293; BAY571293; BAY 571293; BAY-571293; AIC-316; AIC 316
| Molecular Weight | 516.61 |
|---|---|
| Synonyms | AIC316 mesylate hydrate; BAY 57-1293 mesylate hydrate |
| Formula | C19H24N4O7S3 |
| CAS No. | 1428321-10-1 |
Pritelivir mesylate is an antiviral drug currently under development, specifically targeting herpes simplex virus types 1 and 2 (HSV-1 and HSV-2). It functions by inhibiting the viral helicase-primase enzyme, a crucial component for HSV replication. It is being investigated as a potential treatment for various herpes infections, including those resistant to traditional antivirals like acyclovir.
Key aspects of Pritelivir mesylate:
- Mechanism of Action:Pritelivir is a helicase-primase inhibitor, meaning it blocks the activity of an enzyme essential for the replication of herpes viruses.
- Target Viruses:It is effective against both HSV-1 and HSV-2, the viruses responsible for cold sores and genital herpes, respectively.
- Potential for Resistance:Pritelivir has shown promise in preclinical studies against acyclovir-resistant strains of HSV, making it a potential alternative for patients with drug-resistant infections.
- Clinical Trials:Pritelivir is currently in phase II clinical trials, with ongoing research into its effectiveness and safety.
- Route of Administration:It is being investigated for oral, topical, and vaginal administration.
- Research and Development:Pritelivir is being developed by AiCuris Anti-infective Cures, building upon research from Bayer.
Pritelivir (development codes AIC316 or BAY 57-1293) is a direct-acting antiviral drug in development for the treatment of herpes simplex virus infections (HSV). This is particularly important in immune compromised patients. It is currently in Phase III clinical development by the German biopharmaceutical company AiCuris Anti-infective Cures AG. US FDA granted fast track designation for pritelivir in 2017 and breakthrough therapy designation 2020.
SCHEME
Pritelivir mesylate, an antiviral drug used to treat herpes simplex virus (HSV) infections, is synthesized through a series of chemical reactions, including palladium-catalyzed coupling, ester saponification, and amide coupling reactions. The mesylate salt is then formed by reacting the free base with methanesulfonic acid.
Detailed Synthesis Steps:
- 1. Diaryl Acetic Acid Synthesis:Diaryl acetic acid reagents are synthesized using palladium-catalyzed coupling reactions. These reactions involve the use of organometallic intermediates derived from halo-aryl esters.
- 2. Ester Saponification:The ester group in the synthesized compounds is then converted to a carboxylic acid group through saponification.
- 3. Amide Coupling:The resulting carboxylic acids are coupled with thiazolyl sulfonamides using amide coupling conditions to form the pritelivir molecule.
- 4. Salt Formation:The pritelivir free base is then reacted with methanesulfonic acid to form the mesylate salt, which is the active pharmaceutical ingredient (API).
Key Aspects of Pritelivir Mesylate Synthesis:
- Targeted Mechanism:Pritelivir mesylate inhibits the herpes simplex virus by targeting the viral helicase-primase complex, essential for DNA replication, unlike traditional antivirals that target DNA polymerase.
- Salt and Polymorph Screening:An extensive salt and polymorph screening is performed to optimize the pharmaceutical development of pritelivir, resulting in various salt forms including the mesylate, maleate, and sulfate.
- Solubility and Stability:Pritelivir mesylate is a BCS Class II drug substance with pH-dependent solubility. It exhibits high solubility below pH 3 and poor solubility at neutral pH.
- Formulation Considerations:Due to its limited water solubility, pritelivir mesylate is often formulated with solvents like DMSO, PEG300, Tween-80, and saline or with cyclodextrins like SBE-β-CD.
- Clinical Trials:Pritelivir mesylate is currently under extensive study to evaluate its efficacy and safety profile, with promising results in early clinical trials.
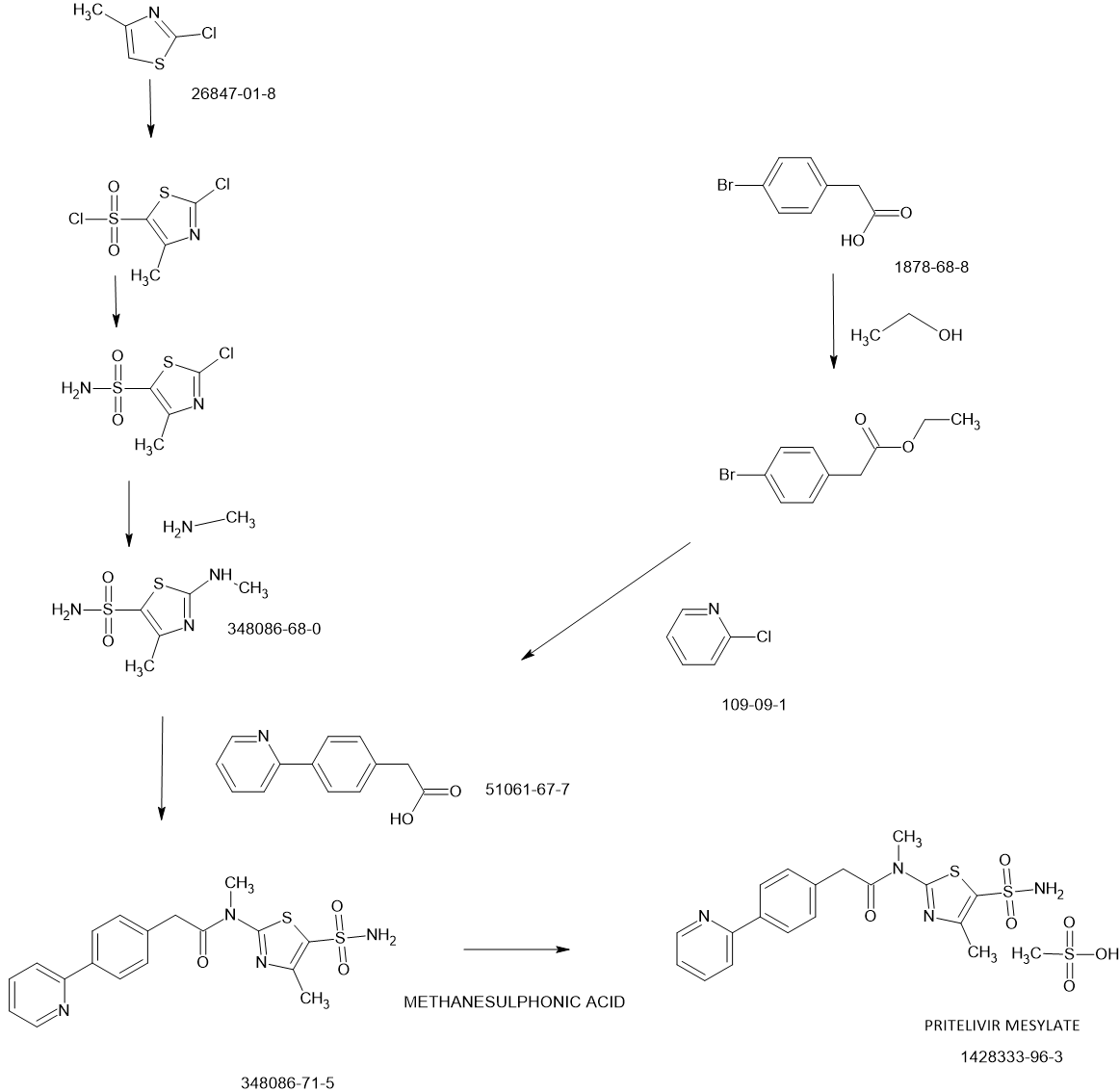
PATENT
WO2018096170
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018096170&_cid=P20-MCVCP5-34284-1
PATENT
WO2018096177
PATENT
https://patents.google.com/patent/WO2018096177A1/en
Likewise, EP 2 598 502 Al describes the crystalline mono mesylate monohydrate salt of N- [5-(aminosulfonyl)-4-methyl- 1 ,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]-acetar^ in a definite particle size distribution and a specific surface area range, which has demonstrated increased long term stability and release kinetics from pharmaceutical compositions, as well as to pharmaceutical compositions containing said N-[5- (aminosulfonyl)-4-methyl- 1 ,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]acetaniide mono mesylate monohydrate having the afore-mentioned particle size distribution and specific surface area range.
WO 2013/045479 Al describes an improved and shortened synthesis process of N-[5- (ammosulfonyl)-4-methyl-l,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]acetaniide and the mesylate salt thereof by using boronic acid derivatives or borolane reagents while avoiding toxic organic tin compounds. Moreover, also the crystalline mesylate monohydrate salt of N- [5 -(aminosulfonyl)-4-methyl- 1 ,3 -thiazol-2-yl] -N-methyl-2- [4-(2-pyridinyl)-phenyl] – acetamide is described therein with increased long-term stability and release kinetics from pharmaceutical compositions thereof.
Said pritelivir is an innovative, highly active and specific inhibitor of herpes simplex virus (HSV) infections. As a compound derived from the chemical class of thiazolylamides, pritelivir is active against both types of herpes simplex virus causing labial and genital herpes, respectively, and retains activity against viruses which have become resistant to marketed drugs. Pritelivir has a mode of action that is distinct from other antiviral agents currently in use for treatment of HSV infections (i.e., the nucleoside analogues acyclovir and its prodrug valacyclovir as well as famciclovir, the prodrug of penciclovix). Whereas nucleoside analogs terminate ongoing DNA chain elongation through inhibition of viral DNA polymerase, pritelivir prevents de novo synthesis of virus DNA through inhibition of the helicase-primase complex. In addition, it does not require activation within an HSV infected cell by viral thymidine kinase and therefore, is also protective to uninfected cells.
With the context of the invention, similar expressions which all would denote the compound pritelivir are “BAY 57-1293”, “AIC090096” and “AIC316”.
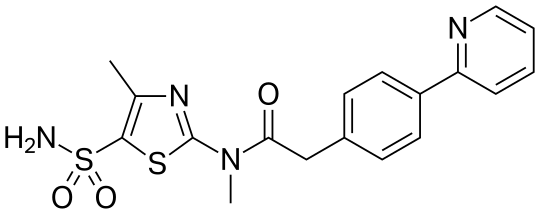
Likewise, the terms ”pritelivir”, “BAY 57-1293”, “AIC090096” and “AIC316″ or the compound *’N-[5-(ammosulfonyl)-4-methyl-l,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridm phenyl] -acetamide” would reflect throughout the text a compound having the structural formula:

Synthetic route – Manufacture of N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-Yl]-N-methyl- 2-[4-(2-pyridinyl)-phenyl]-acetamide free base hemihydrate
The starting materials (4-pyridine-2-yl-phenyl)-acetic acid (PP-acetic acid; C-023930) and aminothiazole sulfonic acid amide (C-023936) are coupled using standard reaction conditions
(N-Ethyl-N’-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC x HCl), tetrahydrofuran (THF)/N-methylpyrrolidone (NMP) to deliver N-[5-(aminosulfonyl)-4- methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide free base hemihydrate (C-023931). To obtain the hemihydrate, N-[5-(aminosulfonyl)-4-methyl-1,3- thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide hemihydrate free base is recrystallized from THF/water. A flowchart showing the synthesis of N-[5-(aminosulfonyl)- 4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide is provided in below in the reaction scheme below 1.
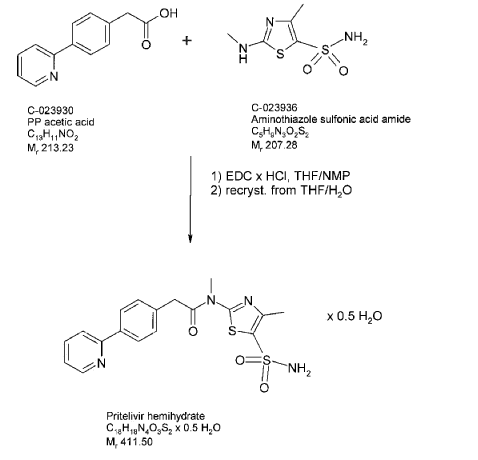
Description of the manufacturing process of N-r5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide free base hemihydrate
PP-acetic acid and aminothiazole sulfonic acid amide are mixed in THF/NMP, the mixture is cooled and then EDC x HCl is added in portions. The reaction mixture is stirred for several hours, and then added slowly to purified water. The suspension is stirred and filtered; the product cake is washed with purified water and dried at room temperature in a nitrogen stream and then under vacuum. Purified water is added slowly at elevated temperature, the suspension is stirred for several hours. The suspension is cooled to 5°C and stirred further for several hours. The product is isolated by filtration and washed with purified water. The product is dried at 65°C under vacuum until the criterion for water content is reached. A major advantage of the synthesis of free base of N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide hemihydrate is the absence impurities related to the presence of mesylate ester that might be present in the N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide mesylate.
PAPER
By: Carta, Fabrizio ; et al. Journal of Medicinal Chemistry (2017), 60(7), 3154-3164
SULPHURIC ACID FOR SULPHATE A SIMILAR REACTION BUT NOT SAME
PAPER
https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00668
Chemistry of Pritelivir
Synthesis of pritelivir and its analogues is based on the reported methods in the literature (18−20) and presented in Figure 4. A simple retrosynthetic disconnection of the target compound suggests a coupling of the thiazolyl sulfonamide and diaryl acetic acid (Figure 4a). During the course of development an optimized route applying the principles of green chemistry was developed and will be used for the commercial phase.
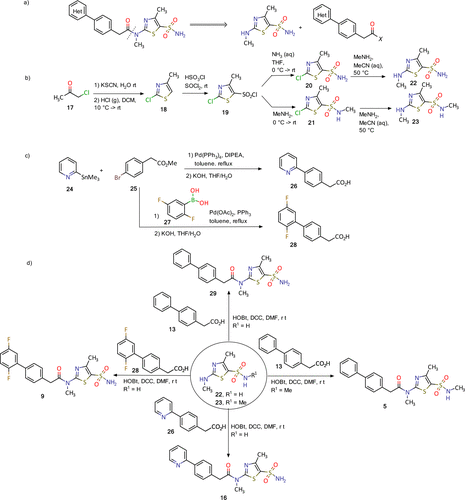
Figure 4. Synthesis of pritelivir (16) and analogues: (a) disconnection approach to target molecules; (b) synthesis of thiazolyl sulfonamide reagents; (c) synthesis of diaryl acetic acids; (d) synthesis of some representative examples of pritelivir and analogues. (18−20)
The synthesis of the thiazolyl sulfonamide reagents begins with a reaction of chloroacetone (17) and potassium thiocyanide to give an intermediate ketone which was cyclized to the thiazole 18 by treatment with gaseous hydrochloride (Figure 4b). Chlorosulfonylation with chlorosulfonic acid and thionyl chloride resulted in the sulfonyl chloride 19 that was converted to the corresponding sulfonamides 20 and 21 after treatment with ammonia or methylamine, respectively. The 2-chloro substituent in 20 and 21 was converted to the methyl amine in an SNAr reaction to deliver the building blocks 22–23. Diaryl acetic acid reagents were synthesized using palladium catalyzed coupling reactions with organometallic intermediates formed from the corresponding halo-aryl esters (Figure 4c). Ester saponification then delivered the corresponding carboxylic acids (e.g., 26 and 28, Figure 4c). Finally, the target molecules, for instance, 5, 11, 16, and 9, were obtained using amide coupling reaction conditions with corresponding diaryl acetic acids and the thiazolyl sulfonamides (Figure 4d). (18−20)
Medical use
Pritelivir is currently being developed for the treatment of immunocompromised patients with mucocutaneous HSV lesions that are resistant to acyclovir.
HSV in immunocompromised patients
Although HSV infection is very common in the general population, it rarely causes serious disease and is effectively contained by the immune system. In those with a weakened immune system such as transplant recipients, people receiving chemo- or radiotherapy, or HIV patients, an active HSV infection can cause disease in 35-68% of patients and may become severe or even life-threatening.[1]
Standard of care treatments
HSV treatment revolves around the use of nucleoside analogues (NA) which act via the viral DNA polymerase, causing DNA chain termination and prevention of viral replication. First-line treatment is generally acyclovir or its prodrug valacyclovir. Resistance to acyclovir is more common in HSV patients with weakened or suppressed immune systems, affecting between 4 and 25% of cases.[2][3][4][5][6]
Resistance to standard treatments
If HSV drug resistance is mediated by mutation(s) of the viral UL23 gene, which encodes the viral thymidine kinase (TK), then the pyrophosphate analogue foscarnet may be effective as a rescue treatment, since it does not require activation by TK. The use of foscarnet is commonly accompanied by restrictive toxicity, particularly nephrotoxicity.[7] If the virus also acquires resistance to foscarnet, then there is currently no FDA approved treatment.
Clinical research
Completed phase II clinical trials in otherwise healthy patients with genital herpes
- A Double-blind Randomized Placebo Controlled Dose-finding Trial to Investigate Different Doses of a New Antiviral Drug in Subjects With Genital HSV Type 2 Infection.[8][9]
- A Double-blind, Double Dummy, Randomized Crossover Trial to Compare the Effect of “AIC316 (Pritelivir)” 100 mg Once Daily Versus Valacyclovir 500 mg Once Daily on Genital HSV Shedding in HSV-2 Seropositive Adults.[10][11]
Ongoing phase II / phase III clinical trials with pritelivir
A phase II / III multinational, comparator-controlled, clinical trial in immunocompromised patients with acyclovir-resistant mucocutaneous lesions is listed on ClinicalTrials.gov[12] and EudraCT.[13]
Pharmacology
Mechanism of action
Pritelivir is a member of the helicase-primase inhibitors (HPI), a novel class of direct-acting antiviral drugs acting specifically against HSV-1 and HSV-2.[14][15] As the name suggests, the drugs act through inhibition of the viral helicase primase complex, encoded by the UL5 (helicase), UL8 (scaffold protein) and UL52 (primase) genes, which is essential for HSV replication.[16] The helicase primase complex is encoded separately from the viral DNA polymerase (encoded by the UL30 gene). Because HPIs i) do not target the viral DNA polymerase and ii) do not require activation by the viral thymidine kinase enzyme (encoded by the UL23 gene), mutations causing resistance to NAs are not protective against HPIs. Similarly, resistance to HPIs does not confer resistance to NAs.
References
- ^ Wilck, M.B.; Zuckerman, R.A.; A. S. T. Infectious Diseases Community of Practice (2013). “Herpes simplex virus in solid organ transplantation”. Am J Transplant. 13 (Suppl 4): 121–7. doi:10.1111/ajt.12105. PMID 23465005. S2CID 44969727.
- ^ Zuckerman, R.; Wald, A.; A. S. T. Infectious Diseases Community of Practice (2009). “Herpes simplex virus infections in solid organ transplant recipients”. Am J Transplant. 9 (Suppl 4): S104-7. doi:10.1111/j.1600-6143.2009.02900.x. PMID 20070669. S2CID 205846431.
- ^ Frobert, E.; Burrel, S.; Ducastelle-Lepretre, S.; Billaud, G.; Ader, F.; Casalegno, J.S. (2014). “Resistance of herpes simplex viruses to acyclovir: an update from a ten-year survey in France”. Antiviral Res. 111: 36–41. doi:10.1016/j.antiviral.2014.08.013. PMID 25218782.
- ^ Patel, D.; Marchaim, D.; Marcus, G.; Gayathri, R.; Lephart, P.R.; Lazarovitch, T.; Zaidenstein, R.; Chandrasekar, P. (2014). “Predictors and outcomes of acyclovir-resistant herpes simplex virus infection among hematopoietic cell transplant recipients: case-case-control investigation”. Clin Transplant. 28 (1): 1–5. doi:10.1111/ctr.12227. PMID 24033498. S2CID 37729458.
- ^ Danve-Szatanek, C.; Aymard, M.; Thouvenot, D.; Morfin, F.; Agius, G.; Bertin, I. (2004). “Surveillance network for herpes simplex virus resistance to antiviral drugs: 3-year follow-up”. J Clin Microbiol. 42 (1): 242–9. doi:10.1128/JCM.42.1.242-249.2004. PMC 321677. PMID 14715760.
- ^ Chakrabarti, R.; Pillay, D.; Ratcliffe, D.; Cane, P.A.; Collingham, K.E.; Milligan, D.W. (2000). “Resistance to antiviral drugs in herpes simplex virus infections among allogeneic stem cell transplant recipients: risk factors and prognostic significance”. J Infect Dis. 181 (6): 2055–8. doi:10.1086/315524. PMID 10837192.
- ^ SmPC
- ^ NCT01047540
- ^ Wald, A.; Timmler, B.; Magaret, A.; Warren, T.; Trying, S. (2014). “Helicase-primase inhibitor pritelivir for HSV-2 infection”. N Engl J Med. 370 (3): 201–10. doi:10.1056/NEJMoa1301150. PMID 24428466.
- ^ NCT01658826
- ^ Wald, A.; Timmler, B.; Warren, T.; Trying, S.; Johnston, C. (2016). “Effect of Pritelivir Compared With Valacyclovir on Genital HSV-2 Shedding in Patients With Frequent Recurrences: A Randomized Clinical Trial”. JAMA. 316 (23): 2495–2503. doi:10.1001/jama.2016.18189. hdl:1805/14200. PMID 27997653.
- ^ NCT03073967
- ^ 2020-004940-27
- ^ Biswas, S.; Jennens, L.; Field, H.J. (2007). “Single amino acid substitutions in the HSV-1 helicase protein that confer resistance to the helicase-primase inhibitor BAY 57-1293 are associated with increased or decreased virus growth characteristics in tissue culture”. Arch Virol. 152 (8): 1489–500. doi:10.1007/s00705-007-0964-7. PMID 17404685. S2CID 23688945.
- ^ Field, H.J.; Biswas, S. (2011). “Antiviral drug resistance and helicase-primase inhibitors of herpes simplex virus”. Drug Resist Updat. 14 (1): 45–51. doi:10.1016/j.drup.2010.11.002. PMID 21183396.
- ^ Crute, J.J.; Tsurumi, T.; Zhu, L.A.; Weller, S.K.; Olivo, P.D.; Challberg, M.D. (1989). “Herpes simplex virus 1 helicase-primase: a complex of three herpes-encoded gene products”. Proc. Natl. Acad. Sci. U.S.A. 86 (7): 2186–2189. Bibcode:1989PNAS…86.2186C. doi:10.1073/pnas.86.7.2186. PMC 286876. PMID 2538835.
- [1]. Ligat G, et al. Identification of Amino Acids Essential for Viral Replication in the HCMV Helicase-PrimaseComplex. Front Microbiol. 2018 Oct 23;9:2483. [Content Brief][2]. Wald A, et al. Helicase-primase inhibitor Pritelivir for HSV-2 infection. N Engl J Med. 2014 Jan 16;370(3):201-10. [Content Brief][3]. Quenelle DC, et al. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antiviral Res. 2018 Jan;149:1-6. [Content Brief]
| Names | |
|---|---|
| Systematic IUPAC nameN-Methyl-N-(4-methyl-5-sulfamoyl-1,3-thiazol-2-yl)-2-[4-(pyridin-2-yl)phenyl]acetamide | |
| Identifiers | |
| CAS Number | 348086-71-5 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 430613 |
| KEGG | D12811 |
| PubChem CID | 491941 |
| UNII | 07HQ1TJ4JE |
| CompTox Dashboard (EPA) | DTXSID70188344 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C18H18N4O3S2 |
| Molar mass | 402.49 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).Infobox references | |
///////////PRITELIVIR MESYLATE, AIC316 mesylate hydrate, BAY 57-1293 mesylate hydrate, AIC 316 mesylate hydrate, BAY 57-1293 mesylate hydrate, BAY57-1293, BAY 57-1293, BAY-57-1293, BAY571293, BAY 571293, BAY-571293, AIC-316, AIC 316



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
PIMICOTINIB
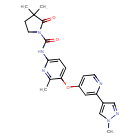
PIMICOTINIB
CAS 2253123-16-7
ABSK021
WeightAverage: 420.473
Monoisotopic: 420.190988657
Chemical FormulaC22H24N6O3
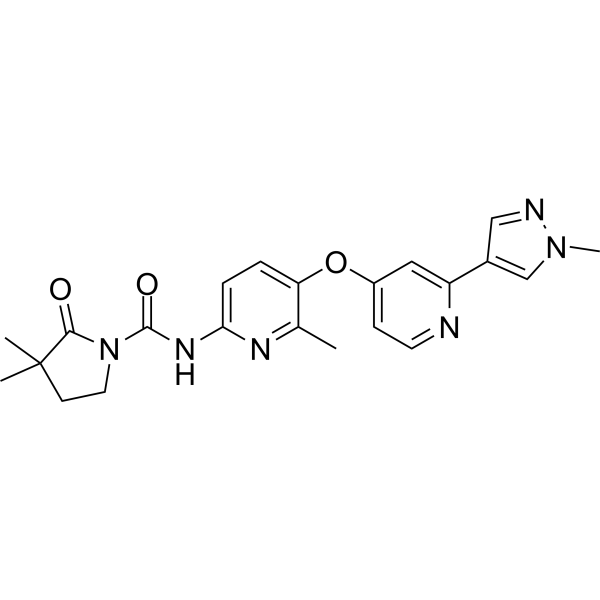
3,3-dimethyl-N-[6-methyl-5-[2-(1-methylpyrazol-4-yl)pyridin-4-yl]oxypyridin-2-yl]-2-oxopyrrolidine-1-carboxamide
CSF-1R inhibitor Pimicotinib (ABSK021) of Abbisko Therapeutics, HV1XI8HST2
Pimicotinib (ABSK021), an oral, highly potent and selective small molecule blocker of the colony-stimulating factor 1 receptor (CSF-1R) independently discovered by Abbisko Therapeutics. A number of studies have shown that blocking the CSF-1R signaling pathway could effectively modulate and change macrophage functions, and potentially treat many macrophage-dependent human diseases.[1]
Pimicotinib is under investigation in clinical trial NCT05804045 (Study of Pimicotinib (ABSK021) for Tenosynovial Giant Cell Tumor (MANEUVER)).
History
In December 2023, Abbisko Therapeutics entered into a licensing agreement for pimicotinib in all indications for China rights with Merck KGaA.[2] [3][4]
In April 2023, a global phase III, randomized, double-blind, placebo-controlled, multicenter clinical trial designed to evaluate the safety and efficacy of pimicotinib in patients with tenosynovial giant cell tumor was started (NCT05804045).[5]
Following with pimicotinib for tenosynovial giant cell tumor treatment in phase III, pimicotinib has also entered into a phase II trial in June 2023 for cGVHD treatment in China.[6]
The U.S. Food and Drug Administration (FDA) and the Center for Drug Evaluation (CDE) of NMPA granted pimicotinib breakthrough therapy designation (BTD) for the treatment of tenosynovial giant cell tumor patients that are not amenable to surgery in January 2023 and July 2022, respectively.[7]
Research
Pimicotinib is being investigated as a treatment for tenosynovial giant cell tumor,[8][9] chronic graft-versus-host-disease (cGVHD), and pancreatic cancer.
PATENTS
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018214867&_cid=P10-MCTXKZ-70170-1
Example 41: Preparation of 3-hydroxy-3-methyl-N-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)-2-carbonylpyrrolidine-1-carboxamide
[0463]
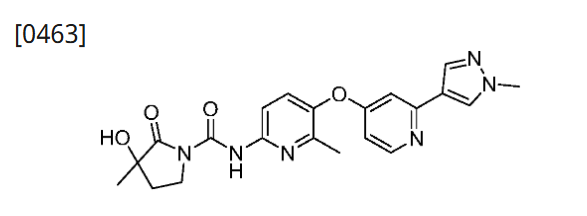
[0464]Palladium carbon (50 mg) was added to a solution of 3-(benzyloxy)-3-methyl-N-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)-2-carbonylpyrrolidine-1-carboxamide (80 mg, 0.15 mmol) in methanol (10 mL). The reaction was stirred at 50° C. for 2 hours in the presence of hydrogen. Filtered and concentrated. Plate chromatography (dichloromethane/methanol=18:1) gave 3-hydroxy-3-methyl-N-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)-2-carbonylpyrrolidine-1-carboxamide (10 mg, yield 15%). MS m/z(ESI):423[M+1]
[0465]
1H NMR(400MHz,DMSO-d 6)δ10.93(s,1H),8.37(d,J=5.7Hz,1H),8.27(s,1H),8.01–7.85(m,2H),7.67(d,J=8.8Hz,1H),7.19(d,J=2.4Hz,1H),6.62(dd,J=5.7,2.4Hz,1H),5.89(s,1H),3.86(s,3H),3.83–3.76(m,1H),3.67–3.61(m,1H),2.29(s,3H),2.09–1.98(m,2H),1.34(s,3H)。
PATENTS
EP3643715
WO2018233527
US20200140431
US11180495 WO2018233527 US20200140431
References
- ^ Vaynrub A, Healey JH, Tap W, Vaynrub M (2022). “Pexidartinib in the Management of Advanced Tenosynovial Giant Cell Tumor: Focus on Patient Selection and Special Considerations”. OncoTargets and Therapy. 15: 53–66. doi:10.2147/OTT.S345878. PMC 8763255. PMID 35046667.
- ^ Merck Strengthens Oncology Portfolio Through Commercialization Agreement With Abbisko for Phase III Asset, Pimicotinib. (2023) https://www.merckgroup.com/en/news/abbisko-pimicotinib-agreement-04-12-2023.html
- ^ Merck KGaA buys into Abbisko’s late-stage joint tumor med for $70M upfront. Fierce Biotech. (2023) https://www.fiercebiotech.com/biotech/merck-kgaa-buys-abbiskos-late-stage-joint-tumor-med-70m-upfront
- ^ “Abbisko Therapeutics Announced the Entry into a Licensing Agreement for Pimicotinib (ABSK021) with Merck”. http://www.prnewswire.com (Press release). Retrieved 18 April 2024.
- ^ Study of Pimicotinib (ABSK021) for Tenosynovial Giant Cell Tumor (MANEUVER). U. S. National Institutes of Health, National Cancer Institute. https://classic.clinicaltrials.gov/ct2/show/NCT05804045
- ^ A Phase II Study Evaluating the Efficacy and Safety of ABSK021 (Pimicotinib)) in the Treatment of cGvHD Chronic Graft Versus Host Disease (cGvHD)U. S. National Institutes of Health, National Cancer Institute.https://classic.clinicaltrials.gov/ct2/show/NCT06186804
- ^ “FDA Grants Breakthrough Therapy Designation to Abbisko’s Pimicotinib”. Global genes. Retrieved 18 April 2024.
- ^ “Pimicotinib”. TGCT Support. Retrieved 18 April 2024.
- ^ “A Phase 3, Randomized, Double-blind, Placebo-Controlled, Multicenter Study of ABSK021 to Assess the Efficacy and Safety in Patients With Tenosynovial Giant Cell Tumor”. clinicaltrials. clinicaltrials.gov. 10 April 2024. Retrieved 18 April 2024.
External links
- “Pimicotinib”. NCI Drug Dictionary. National Cancer Institute.
- Clinical trial number NCT06186804 for “A Phase II Study Evaluating the Efficacy and Safety of ABSK021 (Pimicotinib)) in the Treatment of cGvHD Chronic Graft Versus Host Disease (cGvHD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Other names | ABSK021 |
| Routes of administration | Oral |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2253123-16-7 |
| PubChem CID | 139549388 |
| ChemSpider | 128942304 |
| UNII | HV1XI8HST2 |
| KEGG | D12938 |
| Chemical and physical data | |
| Formula | C22H24N6O3 |
| Molar mass | 420.473 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Zhao BW, et al. N-(azaaryl)cyclolactam-1-carboxamide derivative, preparation method and application. World Intellectual Property Organization, WO2018214867 A1. 2018-11-29.[2]. Yang S, et al. Abstract LB-288: A highly selective small molecule CSF-1R inhibitor demonstrates strong immunomodulatory activity in syngeneic models. Cancer Research, 2018, 78(13_Supplement): LB-288-LB-288.[3]. Zhang N, et al. Abstract LB077: CSF-1R inhibition with Pimicotinib (ABSK021) enhanced anti-tumor efficacy of KRASG12C inhibitors in preclinical non-small cell lung cancer mouse models. Cancer Research, 2024, 84(7_Supplement): LB077-LB077.
//////////PIMICOTINIB, ABSK 021, Abbisko Therapeutics, HV1XI8HST2
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
PALTUSOTINE



PALTUSOTINE
CAS 2172870-89-0
- CRN00808
- F2IBD1GMD3
WeightAverage: 456.497
Monoisotopic: 456.17616767
Chemical FormulaC27H22F2N4O
3-[4-(4-Amino-1-piperidinyl)-3-(3,5-difluorophenyl)-6-quinolinyl]-2-hydroxybenzonitrile
fda 2025, approvals 2025, To treat acromegaly in adults who had an inadequate response to surgery and/or for whom surgery is not an option
- OriginatorCrinetics Pharmaceuticals
- ClassAmines; Antineoplastics; Antisecretories; Fluorobenzenes; Nitriles; Piperidines; Quinolines; Small molecules
- Mechanism of ActionSomatostatin receptor 2 agonists
- Orphan Drug Status – Acromegaly
- PreregistrationAcromegaly
- Phase IIMalignant carcinoid syndrome
- 08 May 2025Crinetics Pharmaceuticals expects potential EMA decision for paltusotine in Acromegaly, in the first half of 2026
- 08 May 2025FDA assigns PDUFA action date of 25/09/2025 for paltusotine for acromegaly
- 08 May 2025Crinetics Pharamceuticals plans the phase III CAREFNDR trial for Malignant carcinoid syndrome (PO), in the second quarter of 2025
Paltusotine is a selective somatostatin receptor type 2 (SST2) agonist in development by Crinetics Pharmaceuticals for the treatment of acromegaly and certain neuroendocrine tumors. It is a small molecule delivered orally.[1][2][3][4]
SCHEME
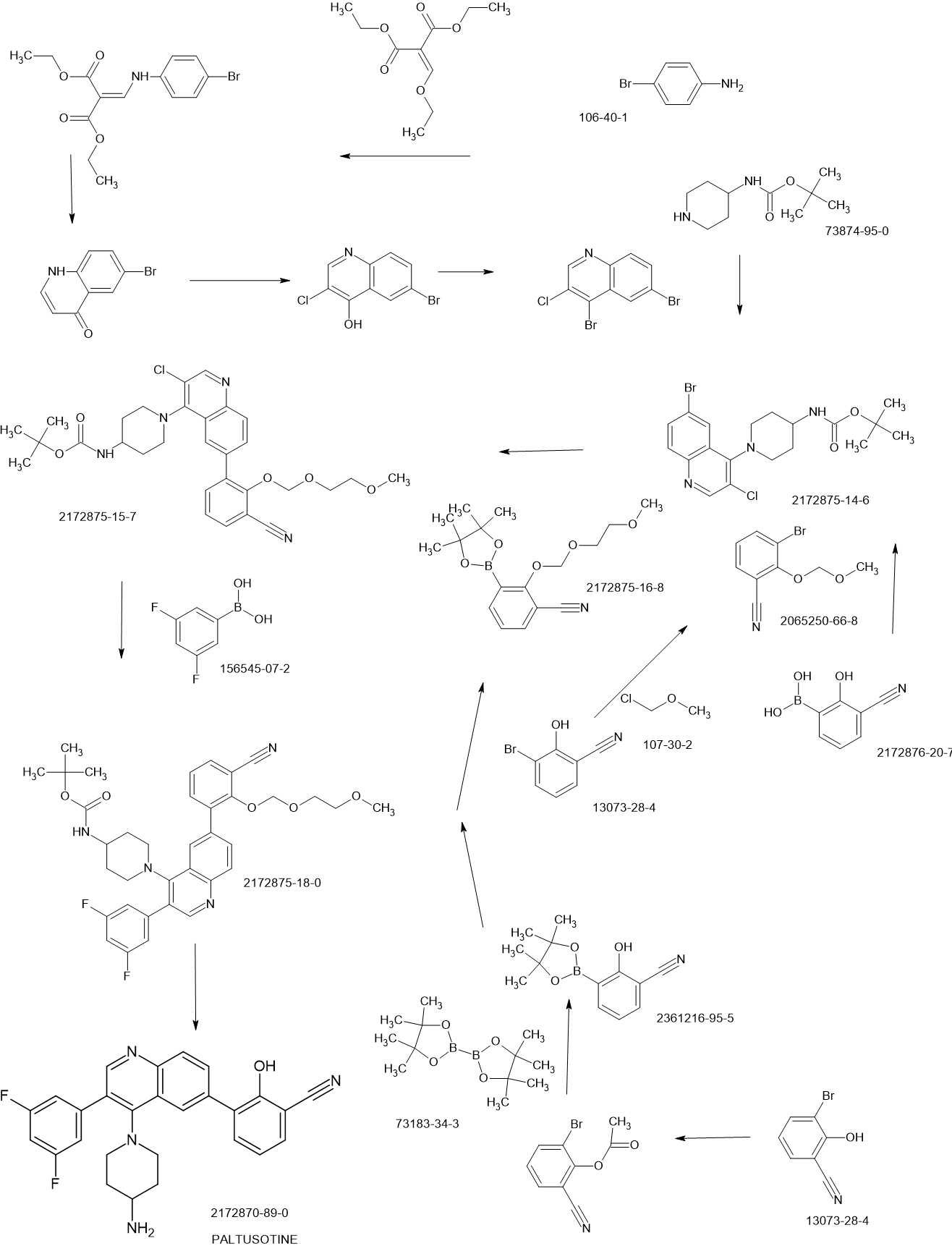

PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00431
Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist
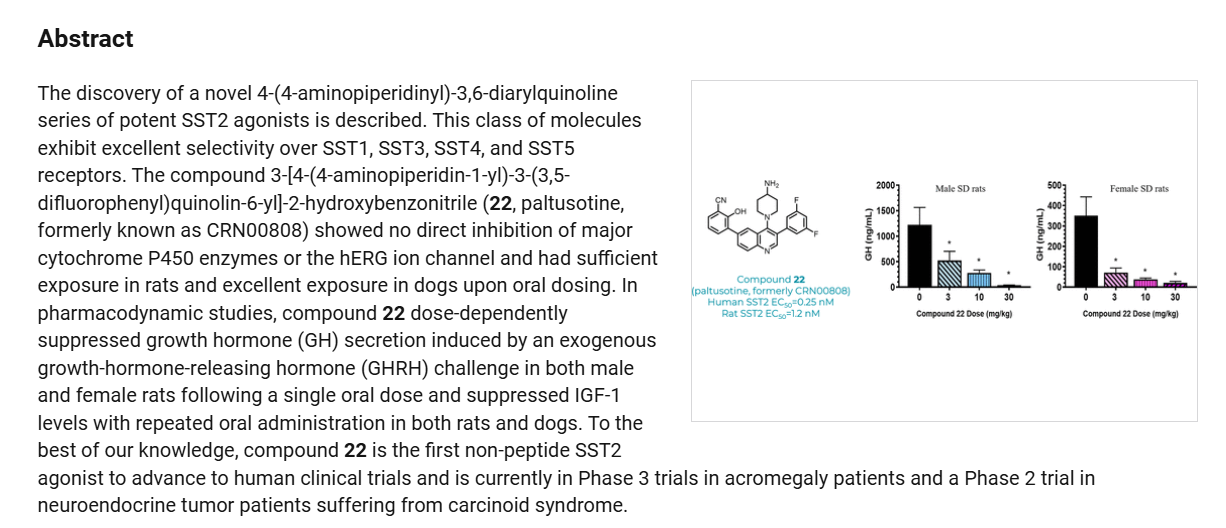

Step 2-1, preparation of [1-(6-bromo-3-chloro-quinolin-4-yl)-piperidin-4-yl]-carbamic acid tertbutyl ester: To a DMSO solution of 6-bromo-3,4-dichloroquinoline (950 mg, 1 Eq, 3.43 mmol)
was added tert-butyl piperidin-4-ylcarbamate (841 mg, 98% Wt, 1.2 Eq, 4.12 mmol) and DIPEA
(1.19 g, 1.60 mL, 3 Eq, 10.3 mmol). The resulting mixture was heated at 60 °C for overnight.
The reaction crude was quenched with water, extracted with EtOAc, washed with brine,
concentrated and purified by silica gel chromatography to afford tert-butyl (1-(6-bromo-3-
chloroquinolin-4-yl)piperidin-4-yl)carbamate (0.95 g, 2.2 mmol, 63 %) as an off-white solid. 1H
NMR (500 MHz, CDCl3) δ 8.66 (s, 1H), 8.25 (d, J=5 Hz, 1H), 7.94 (d, J=10 Hz, 1H), 7.74 (d,
J=10 Hz, 1H), 4.61 (s, 1H), 3.76 (s, 1H), 3.51 (m, 2H), 3.37 (m, 2H), 2.13-2.15 (m, 2H), 1.73-
1.65 (m, 2H), 1.48 (s, 9H). MS [M+H]
+= 442.0.
Step 4-2, preparation of 1-{3-chloro-6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-
quinolin-4-yl}-piperidin-4-yl)-carbamic acid tert-butyl ester: To a THF (5.0 mL) solution of [1-
(6-bromo-3-chloro-quinolin-4-yl)-piperidin-4-yl]-carbamic acid tert-butyl ester (1.0 mmol, 440
mg) and 2-(2-methoxy-ethoxymethoxy)-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
benzonitrile (1.4 eq., 1.4 mmol, 460 mg) was added PdCl2dppf (0.1 eq., 0.1 mmol, 75 mg) and
KOAc (3.0 eq., 3.0 mmol, 300 mg). N2 was bubbled through the reaction solution for 5 min and
0.5 mL water was added. The resulting mixture was heated at 80 °C for 1 h. LCMS analysis
showed about 50% of the starting material has been converted to the desired product. Additional
2-(2-methoxy-ethoxymethoxy)-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzonitrile
(1.4 eq., 1.4 mmol, 460 mg), PdCl2dppf (0.1 eq., 0.1 mmol, 75 mg) and KOAc (3.0 eq., 3.0
mmol, 300 mg) were added and the resulting solution was heated at 80 °C for another 2 h. The
reaction solution was combined with silica gel and concentrated. The residue obtained was
purified by silica gel chromatography eluting with ethyl acetate/hexane (0~50%) to give 0.512 g
of the desired product as white solid. MS [M+H]
+= 567.6.
Step 4-3, preparation of {1-[6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-3-(3,5-difluorophenyl)-quinolin-4-yl]-piperidin-4-yl}-carbamic acid tert-butyl ester: To a dioxane (5 mL)
solution of (1-{3-chloro-6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-quinolin-4-yl}-
piperidin-4-yl)-carbamic acid tert-butyl ester (0.5 mmol, 283 mg) was added Pd(amphos)Cl2 (0.1
eq., 0.05 mmol, 37 mg), 3, 5-difluorophenyl boronic acid (3.0 eq., 1.5 mmol, 250 mg) and
K2CO3 (4.0 eq., 2.0 mmol, 276 mg). N2 was bubbled through the reaction solution for 5 min and
0.5 mL water was added. The resulting mixture was heated at 95 °C for 0.5 h and LCMS analysis
showed that starting material was completely consumed. The reaction solution was concentrated
with silica gel and purified by silica gel chromatography eluting with ethyl acetate/hexane
(0~50%) to give 0.170 g of the desired product as white solid. MS (M+H)+= 645.6.
Step 4-4, preparation of 3-[4-(4-amino-piperidin-1-yl)-3-(3,5-difluoro-phenyl)-quinolin-6-yl]-2-hydroxybenzonitrile: to the dichloromethane (5.0 mL) solution of {1-[6-[3-cyano-2-(2-methoxyethoxymethoxy)-phenyl]-3-(3,5-difluoro-phenyl)-quinolin-4-yl]-piperidin-4-yl}-carbamic acid
tert-butyl ester (0.264 mmol, 170 mg) was added trifluroroacetic acid (2.0 mL) and the resulting
mixture was stirred at ambient temperature for 2 h. The reaction solution was concentrated and
purified by C18 reversed phase chromatography eluting with MeCN/water (0~40%). Pure
fractions were combined, neutralized with saturated NaHCO3, extracted with ethyl acetate and
dried with MgSO4. The organic solution was concentrated with HCl in ether (2.0 M) to give the
final compound as HCl salt (68 mg, 0.138 mmol, 52%).
1H NMR (500 MHz, DMSO-d6) δ 10.77
(br s, 1H), 8.78 (s, 1H), 8.29-8.15 (m, 5H), 7.79 (dd, J=20 Hz, 5 Hz, 2H), 7.41 (m, 1H), 7.26-
7.19 (m, 3H), 3.59 (t, J=12 Hz, 2H), 3.31 (m, 1H), 3.00 (t, J=12 Hz, 2H), 2.05-1.99 (m, 2H),
1.76-1.74 (m, 2H). MS [M+H]
+= 457.5. 13C NMR (DMSO-d6) δ 30.2, 47.4, 50.8, 102.4, 103.2,
113.4, 117.2, 121.4, 124.6, 130.7, 133.1, 134.6, 136.0, 141.7, 156.6, 161.2, 163.2. LCMS purity
98% (254&220 nM). HRMS m/z [M+H]+ Calcd for C27H23F2N4O 457.1834; found 457.1833.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US235548187&_cid=P20-MCSHXW-73235-1
PATENTS
WO2021011641
WO2018013676
References
- ^ Madan, Ajay; Markison, Stacy; Betz, Stephen F.; Krasner, Alan; Luo, Rosa; Jochelson, Theresa; Lickliter, Jason; Struthers, R. Scott (April 2022). “Paltusotine, a novel oral once-daily nonpeptide SST2 receptor agonist, suppresses GH and IGF-1 in healthy volunteers”. Pituitary. 25 (2): 328–339. doi:10.1007/s11102-021-01201-z. PMC 8894159. PMID 35000098.
- ^ Zhao, Jian; Wang, Shimiao; Markison, Stacy; Kim, Sun Hee; Han, Sangdon; Chen, Mi; Kusnetzow, Ana Karin; Rico-Bautista, Elizabeth; Johns, Michael; Luo, Rosa; Struthers, R. Scott; Madan, Ajay; Zhu, Yunfei; Betz, Stephen F. (12 January 2023). “Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist”. ACS Medicinal Chemistry Letters. 14 (1): 66–74. doi:10.1021/acsmedchemlett.2c00431. PMC 9841592. PMID 36655128.
- ^ Gadelha, Monica R; Gordon, Murray B; Doknic, Mirjana; Mezősi, Emese; Tóth, Miklós; Randeva, Harpal; Marmon, Tonya; Jochelson, Theresa; Luo, Rosa; Monahan, Michael; Madan, Ajay; Ferrara-Cook, Christine; Struthers, R Scott; Krasner, Alan (13 April 2023). “ACROBAT Edge: Safety and Efficacy of Switching Injected SRLs to Oral Paltusotine in Patients With Acromegaly”. The Journal of Clinical Endocrinology & Metabolism. 108 (5): e148 – e159. doi:10.1210/clinem/dgac643. PMC 10099171. PMID 36353760. S2CID 253445337.
- ^ Zhao, Jie; Fu, Hong; Yu, Jingjing; Hong, Weiqi; Tian, Xiaowen; Qi, Jieyu; Sun, Suyue; Zhao, Chang; Wu, Chao; Xu, Zheng; Cheng, Lin; Chai, Renjie; Yan, Wei; Wei, Xiawei; Shao, Zhenhua (21 February 2023). “Prospect of acromegaly therapy: molecular mechanism of clinical drugs octreotide and paltusotine”. Nature Communications. 14 (1): 962. Bibcode:2023NatCo..14..962Z. doi:10.1038/s41467-023-36673-z. ISSN 2041-1723. PMC 9944328. PMID 36810324.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2172870-89-0 |
| PubChem CID | 134168328 |
| ChemSpider | 81367268 |
| UNII | F2IBD1GMD3 |
| Chemical and physical data | |
| Formula | C27H22F2N4O |
| Molar mass | 456.497 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////PALTUSOTINE, ORPHAN DRUG, Acromegaly, CRN 00808, F2IBD1GMD3, fda 2025, approvals 2025
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
PALAZESTRANT
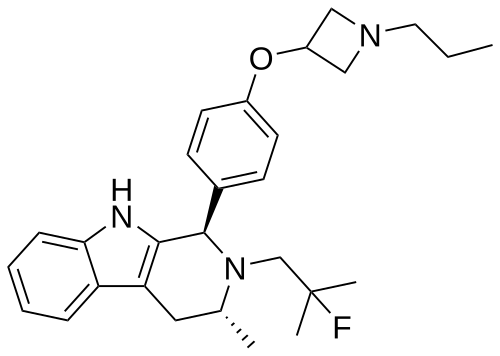
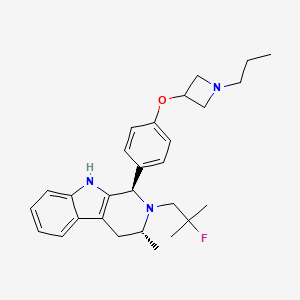
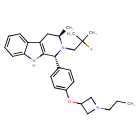
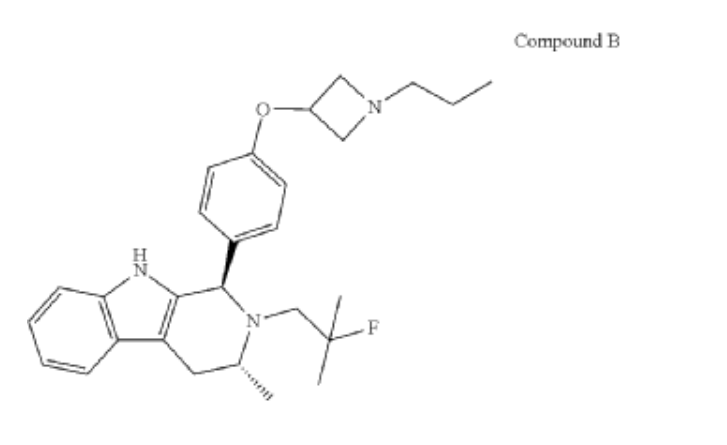
PALAZESTRANT
CAS 2092925-89-6
OP-1250, VU35KM56Q4
449.6 g/mol, C28H36FN3O
(1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-1-[4-(1-propylazetidin-3-yl)oxyphenyl]-1,3,4,9-tetrahydropyrido[3,4-b]indole
- (1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-1-[4-(1-propylazetidin-3-yl)oxyphenyl]-1,3,4,9-tetrahydropyrido[3,4-b]indole
- (1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-1-{4-[(1-propylazetidin-3- yl)oxy]phenyl}-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole
Palazestrant (OP-1250) is an investigational drug being developed for estrogen receptor-positive (ER+) breast cancer. It is a small molecule with a dual mechanism of action, acting as both a complete estrogen receptor antagonist and a selective estrogen receptor degrader (SERD). This means it can block estrogen receptor activity and also degrade the receptor itself, potentially offering a more effective treatment approach.
Here’s a more detailed breakdown:
- Dual Mechanism:Palazestrant is a complete ER antagonist, meaning it blocks all estrogen receptor activity. It is also a SERD, which means it degrades the estrogen receptor, preventing it from functioning.
- Oral Administration:Palazestrant is an orally available drug.
- Clinical Trials:Palazestrant is currently in clinical trials, including Phase 1/2 and Phase 3 studies, for the treatment of ER+, HER2- metastatic breast cancer.
- Combination Therapy:Palazestrant is being evaluated in combination with other drugs like CDK4/6 inhibitors (e.g., ribociclib).
- Promising Results:Preliminary results from clinical trials have shown promising antitumor efficacy and favorable pharmacokinetic properties for palazestrant.
- FDA Fast Track Designation:The FDA has granted Fast Track designation for the treatment of ER+/HER2- metastatic breast cancer that has progressed following endocrine therapy with a CDK4/6 inhibitor.
- Brain Metastasis:Palazestrant has shown activity in brain metastasis animal models.
- ESR1 Mutation Status:Palazestrant has demonstrated activity against both wild-type and mutant ER (ESR1) breast cancer models.
Palazestrant is an investigational new drug which is being evaluated for the treatment of estrogen receptor-positive (ER+) breast cancer, with a dual mechanism of action as both a complete estrogen receptor antagonist (CERAN) and a selective estrogen receptor degrader (SERD). This orally bioavailable small molecule has demonstrated potent activity against both wild-type and mutant forms of the estrogen receptor.[1]
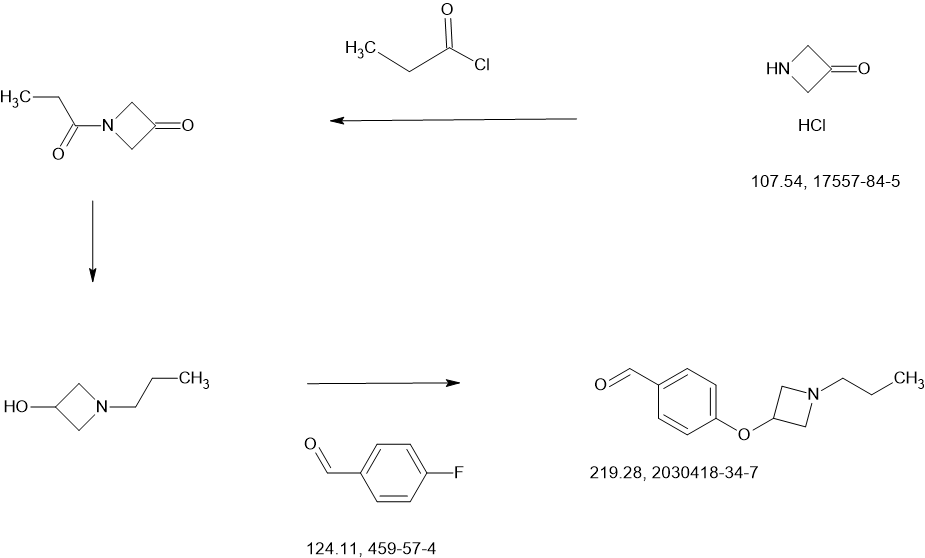
SCHEME
MAIN
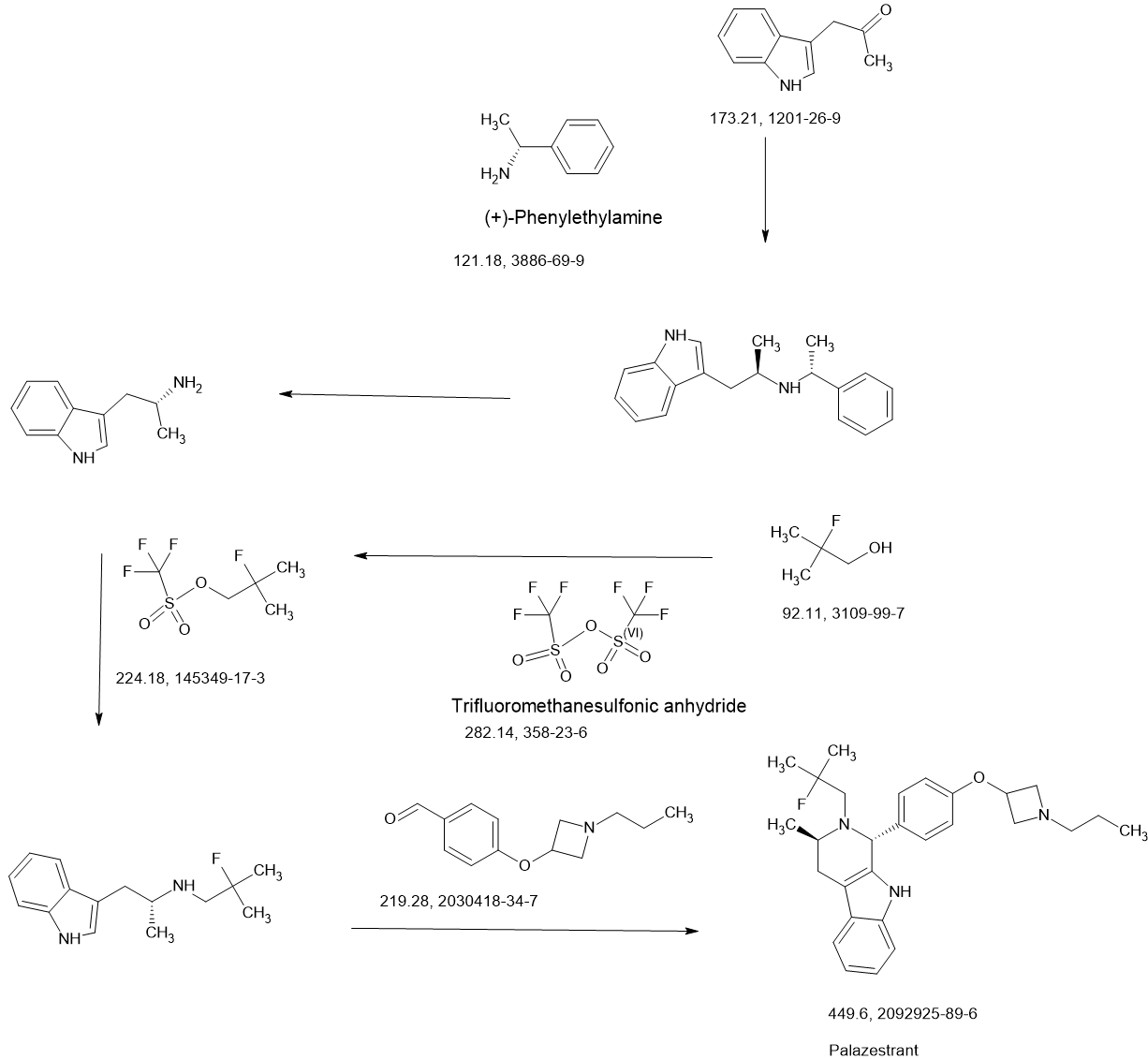
PAPER
https://pubs.acs.org/doi/10.1021/acsomega.4c11023



PATENTS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US379744130&_cid=P22-MCPZ5L-11621-1
PATENTS’
WO2017059139
WO2023225354
WO2023091550
WO2023283329
WO2021178846
References
- ^ Parisian AD, Barratt SA, Hodges-Gallagher L, Ortega FE, Peña G, Sapugay J, et al. (March 2024). “Palazestrant (OP-1250), A Complete Estrogen Receptor Antagonist, Inhibits Wild-type and Mutant ER-positive Breast Cancer Models as Monotherapy and in Combination”. Molecular Cancer Therapeutics. 23 (3): 285–300. doi:10.1158/1535-7163.MCT-23-0351. PMC 10911704. PMID 38102750.
| Clinical data | |
|---|---|
| Other names | OP-1250 |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2092925-89-6 |
| PubChem CID | 135351887 |
| DrugBank | DB18971 |
| ChemSpider | 128922074 |
| UNII | VU35KM56Q4 |
| KEGG | D12827 |
| ChEMBL | ChEMBL5314475 |
| Chemical and physical data | |
| Formula | C28H36FN3O |
| Molar mass | 449.614 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////////PALAZESTRANT, OP 1250, A1AEA, VU35KM56Q4
Orforglipron’
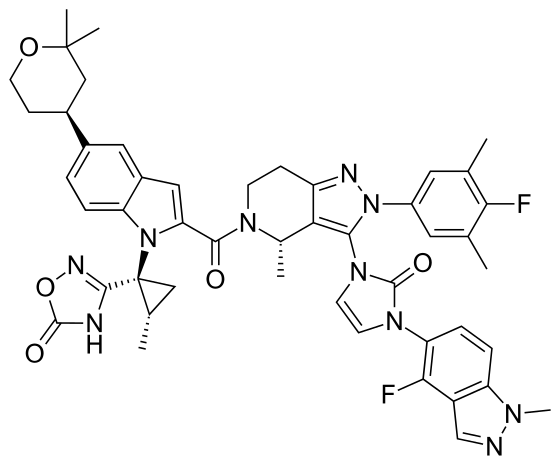
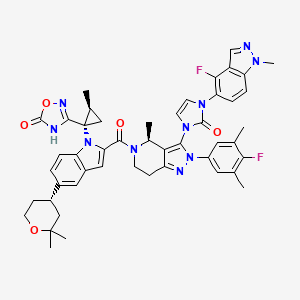
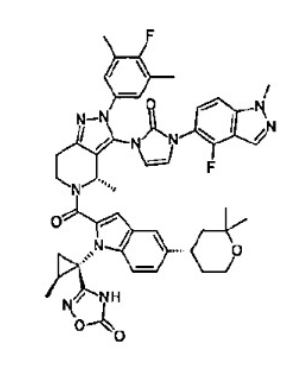
Orforglipron’
CAS 2212020-52-3
C48H48F2N10O5
883.0 g/mol MW
LY-3502970
- OWL833
- 3-[(1S,2S)-1-[5-[(4S)-2,2-dimethyloxan-4-yl]-2-[(4S)-2-(4-fluoro-3,5-dimethylphenyl)-3-[3-(4-fluoro-1-methylindazol-5-yl)-2-oxoimidazol-1-yl]-4-methyl-6,7-dihydro-4H-pyrazolo[4,3-c]pyridine-5-carbonyl]indol-1-yl]-2-methylcyclopropyl]-4H-1,2,4-oxadiazol-5-one
- 3-[(1S,2S)-1-[5-[(4S)-2,2-dimethyloxan-4-yl]-2-[(4S)-2-(4-fluoro-3,5-dimethylphenyl)-3-[3-(4-fluoro-1-methylindazol-5-yl)-2-oxoimidazol-1-yl]-4-methyl-6,7-dihydro-4H-pyrazolo[4,3-c]pyridine-5-carbonyl]indol-1-yl]-2-methylcyclopropyl]-4H-1,2,4-oxadiazol-5-one
SCHEME
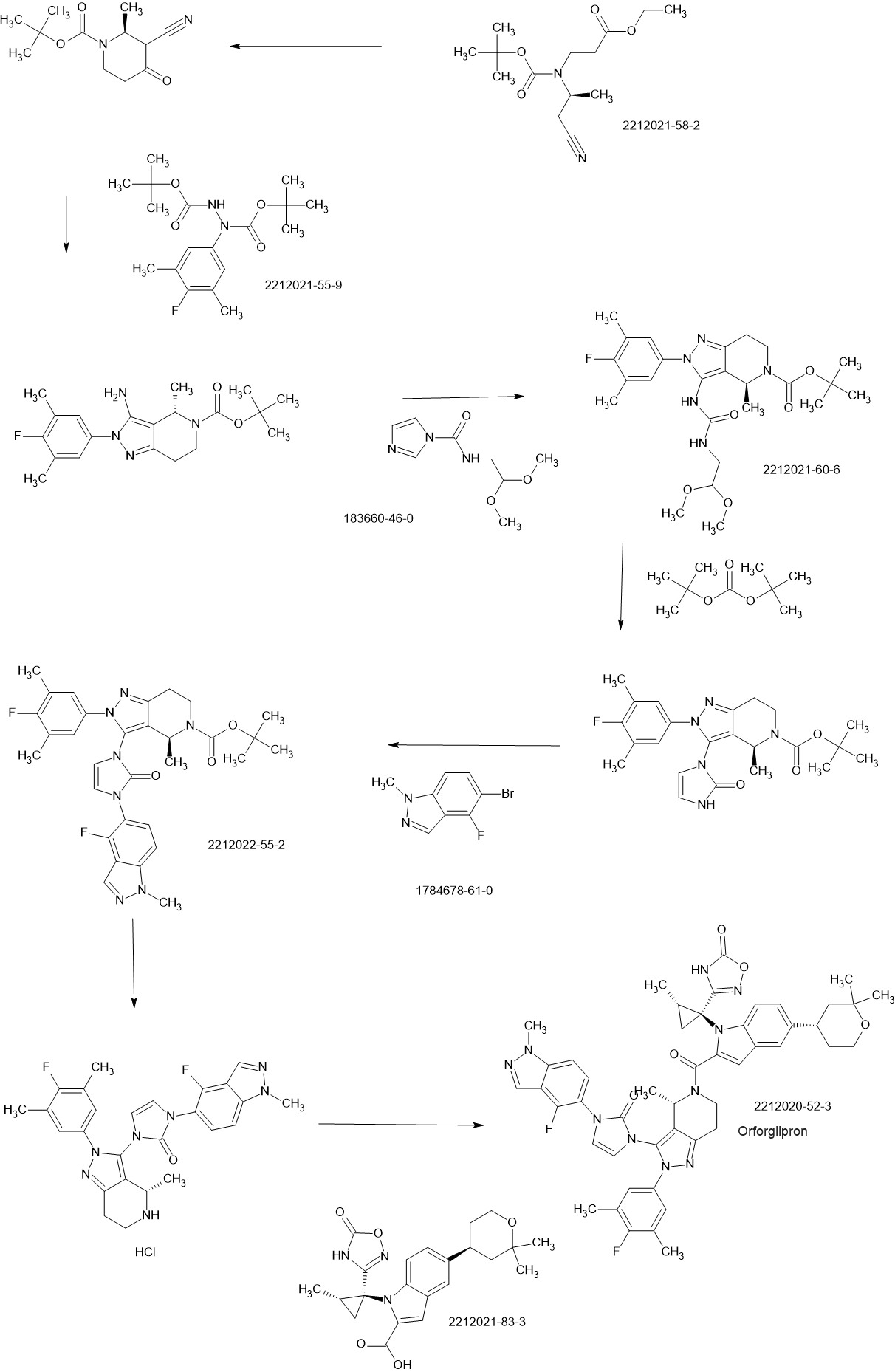
PATENT
JP2019099571
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018056453&_cid=P22-MCLODW-73083-1
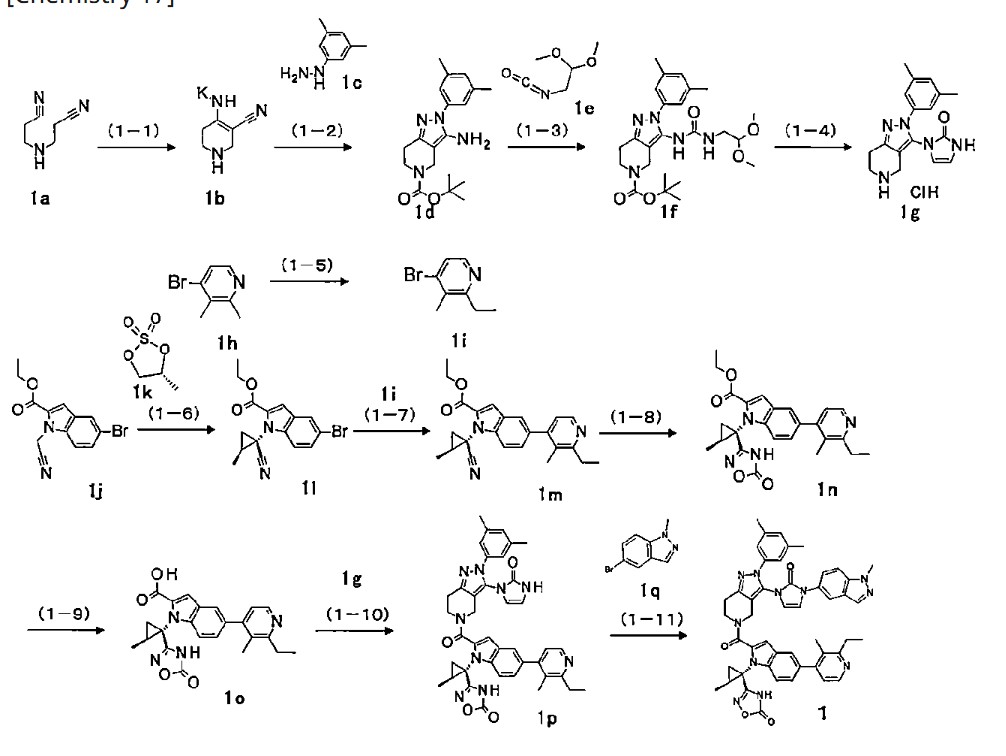
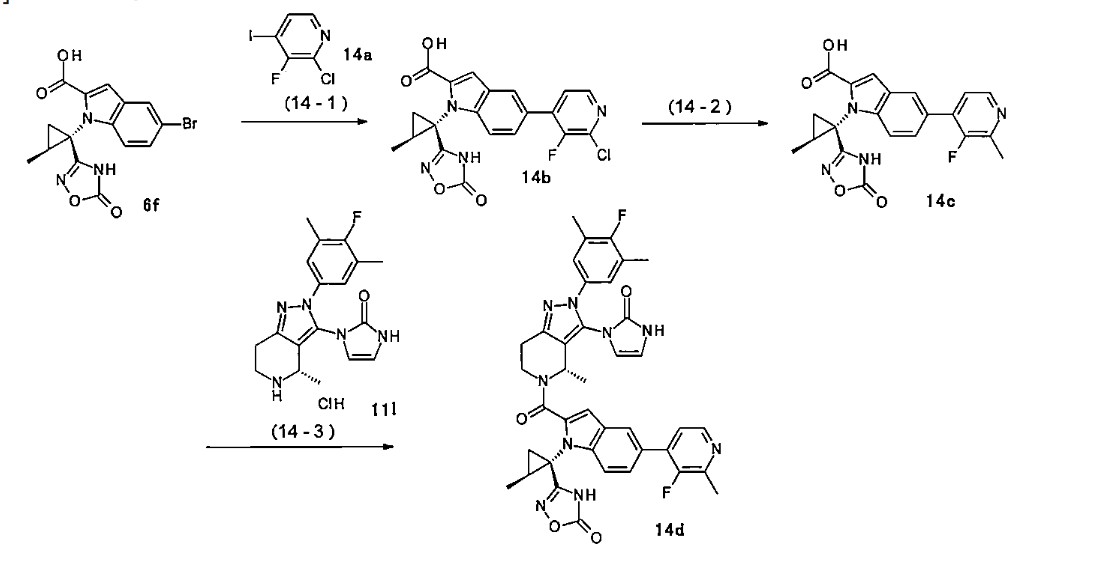
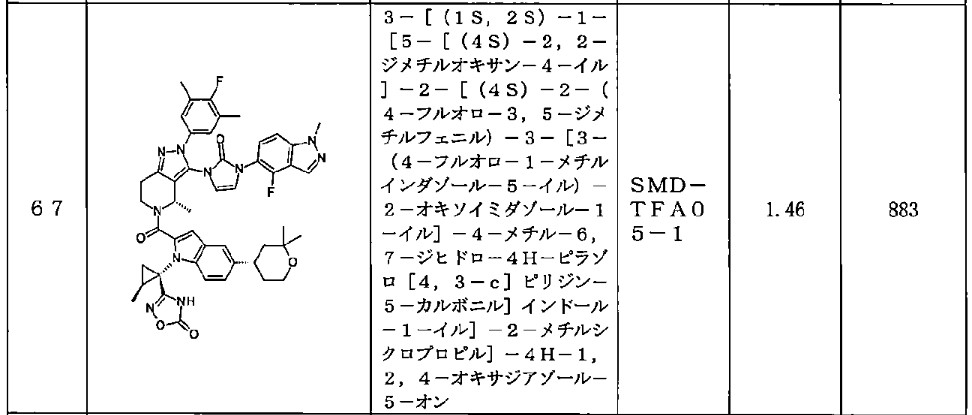
<Example Compound 67>
Main cycle isomer
1 H-NMR (600 MHz, CDCl
3 ) δ: 11.32 (1H, s), 8.13 (1H, d, J
HF=0.7 Hz), 7.59 (1H, d, J =8.6 Hz), 7.52 (1H, s), 7.48 (1H, dd, J =8.9 Hz, J
HF =6.9 Hz ), 7.28 (1H, d, J =8.9 Hz), 7.26 (1H, dd, J =8.6, 1.7 Hz), 7.16 (2H, d, J
HF =6.1Hz), 6.70 (1H, s), 6.61 (1H, dd, J = 3.0Hz,
JHF =1.1Hz), 6.31 (1H, d, J = 3.0Hz), 5.79 (1H, q, J = 6.7Hz), 4.4 7 (1H, dd, J=13.5, 5.2Hz), 4.12 (3H, s), 3.88 (1H, m), 3.83 (1 H, m), 3.60 (1H, ddd, J = 13.5, 12.9, 3.6Hz), 3.15 (1H, ddd, J = 15.8, 12.9, 5.2Hz), 3.04 (1H, m), 3.00 (1H, m), 2.29 (6H, d, J
HF =1.1Hz), 1.91 (1H, dd, J = 6.1, 5.8Hz), 1.79-1.76 ( 2H, m), 1.74 (1H, m), 1.65 (1H, m), 1.57 (3H, d, J=6.7 Hz), 1.60-1.55 (1H, m), 1.52 (1H, dd, J=9.5, 5.8Hz ), 1.34 (3H, s), 1.28 (3H, s), 1.20 (3H, d, J=6.0Hz).
[0437] Parainversion isomer
1 H-NMR (600 MHz, CDCl
3 ) δ: 11.27 (1H, s), 8.04 (1H, s), 7.55 (1H, d, J = 8.7 Hz), 7.52 (1H, s), 7.25-7.22 (2H, m), 7.12 (1H, d, J = 8.8 Hz), 7.06 (2H, d, J
HF =6.0Hz), 6.71 (1H, s), 6.47 (1H, m), 6.08 ( 1H, d, J=3.0Hz), 5.26 (1H, q, J=6.6Hz), 4. 87 (1H, dd, J = 13.1, 4.8Hz), 4.07 (3H, s), 3 .90-3.80 (2H, m), 3.39 (1H, ddd, J = 13.1, 1 2.2, 4.6Hz), 3.08-2.97 (3H, m), 2.25 (6H, s), 1.79-1.73 (3H, m), 1.67 (3H, d, J=6.6H z), 1.64 (1H, m), 1.45-1.37 (2H, m), 1.34 ( 3H, s), 1.28 (3H, s), 1.06 (3H, d, J=6.0Hz).
Orforglipron (LY-3502970) is an oral, non-peptide, small-molecule GLP-1 receptor agonist developed as a weight loss drug by Eli Lilly and Company.[1] It was discovered by Chugai Pharmaceutical Co., then was licensed to Lilly in 2018.[1]
Orforglipron is easier to produce than existing peptide GLP-1 agonists and is expected to be cheaper.[2]
Mechanism
Orforglipron is a small-molecule, partial GLP-1 receptor agonist affecting the activity of cyclic adenosine monophosphate (cAMP); its effects are similar to the actions of glucagon-like peptide-1 (GLP-1) for reducing food intake and lowering blood glucose levels.[1][3]
Clinical trials
The results of Phase I safety and Phase II ascending-dose clinical trials enrolling people with obesity or type 2 diabetes were published in 2023.[4][5]
Orforglipron has a half-life of 29 to 49 hours across the doses tested and is taken once per day by mouth without food or water restrictions.[3]
Safety and dosing trials showed that the incidence of adverse events in orforglipron-treated participants was 62–89%, mostly from gastrointestinal discomfort (44–70% with orforglipron, 18% with placebo) having mild to moderate severity.[6] The most common side effects of orforglipon are diarrhea, nausea, upset stomach, and constipation.[1][6]
The ability of orforglipron to reduce blood sugar levels and body weight was judged favorable compared to dulaglutide.[6]
Phase III ACHIEVE-1 trial
In April 2025, results from a Phase III clinical trial involving 559 people with type 2 diabetes who took an oral orforglipron pill, injectable dulaglutide or a placebo daily for 40 weeks showed that orforglipron produced a reduction in blood glucose levels by 1.3 to 1.6 percentage points from a starting level of 8%.[1][7]
More than 65% of participants taking the highest dose of orforglipron achieved a reduction of hemoglobin A1C level by more than or equal to 1.5 percentage points, bringing them into the non-diabetic range as defined by the American Diabetes Association.[1] People taking the highest dose of the pill lost 8% of their weight, or around 16 lb (7.3 kg), on average after 40 weeks.[1][8]
Side effects were similar to those seen with other GLP-1 agonists, and no significant liver problems were observed.[1]
References
- ^ Jump up to:a b c d e f g h “Lilly’s oral GLP-1, orforglipron, demonstrated statistically significant efficacy results and a safety profile consistent with injectable GLP-1 medicines in successful Phase 3 trial” (Press release). Eli Lilly. April 17, 2025. Retrieved April 18, 2025.
- ^ Sidik S (2023). “Beyond Ozempic: brand-new obesity drugs will be cheaper and more effective”. Nature. 619 (7968): 19. Bibcode:2023Natur.619…19S. doi:10.1038/d41586-023-02092-9. PMID 37369789.
- ^ Jump up to:a b Kokkorakis M, Chakhtoura M, Rhayem C, et al. (January 2025). “Emerging pharmacotherapies for obesity: A systematic review”. Pharmacological Reviews. 77 (1): 100002. doi:10.1124/pharmrev.123.001045. PMID 39952695.
- ^ Pratt E, Ma X, Liu R, et al. (June 2023). “Orforglipron (LY3502970), a novel, oral non-peptide glucagon-like peptide-1 receptor agonist: A Phase 1b, multicentre, blinded, placebo-controlled, randomized, multiple-ascending-dose study in people with type 2 diabetes”. Diabetes, Obesity & Metabolism. 25 (9): 2642–2649. doi:10.1111/dom.15150. PMID 37264711. S2CID 259022851.
- ^ Wharton S, Blevins T, Connery L, et al. (June 2023). “Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults with Obesity”. The New England Journal of Medicine. 389 (10): 877–888. doi:10.1056/NEJMoa2302392. PMID 37351564.
- ^ Jump up to:a b c Frias J, et al. (2023). “Efficacy and safety of oral orforglipron in patients with type 2 diabetes: a multicentre, randomised, dose-response, phase 2 study”. The Lancet. 402 (10400): 472–83.
- ^ Constantino AK (April 17, 2025). “Eli Lilly’s weight loss pill succeeds in first late-stage trial on diabetes patients”. CNBC. Retrieved April 17, 2025.
- ^ Kolata G (April 17, 2025). “Daily Pill May Work as Well as Ozempic for Weight Loss and Blood Sugar”. The New York Times. ISSN 0362-4331. Retrieved April 17, 2025.
External links
- What to Know About Eli Lilly’s Daily Pill for Weight Loss, The New York Times, April 17, 2025
| Above: molecular structure of orforglipron Below: 3D representation of an orforglipron molecule | |
| Clinical data | |
|---|---|
| Other names | LY-3502970 |
| Routes of administration | Oral |
| ATC code | None |
| Pharmacokinetic data | |
| Elimination half-life | 29–49 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2212020-52-3 |
| PubChem CID | 137319706 |
| ChemSpider | 71117507 |
| UNII | 7ZW40D021M |
| ChEMBL | ChEMBL4446782 |
| Chemical and physical data | |
| Formula | C48H48F2N10O5 |
| Molar mass | 882.974 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////////Orforglipron, LY-3502970, LY 3502970, OWL833, OWL 833

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}