
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AZVUDINE
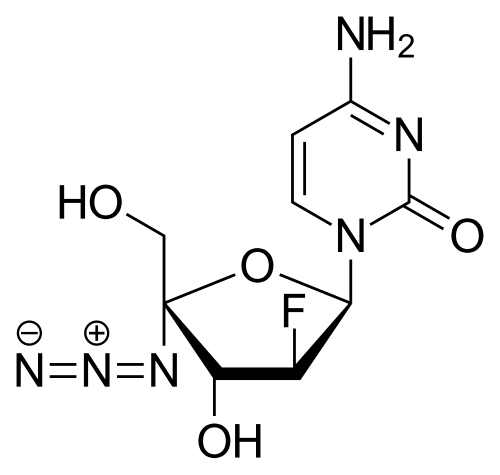
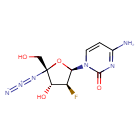
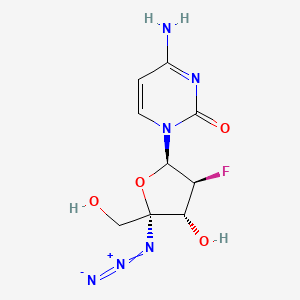
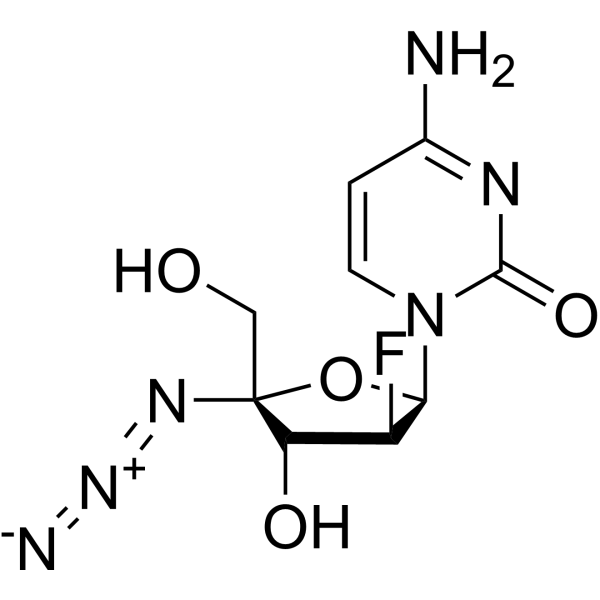
AZVUDINE
CAS
WeightAverage: 286.223
Monoisotopic: 286.082581021
Chemical FormulaC9H11FN6O4
- FNC
- HY-19314
- RO 0622
- RO-0622
- SB17040
IJ2XP0ID0K- DTXSID901027757
4-amino-1-[(2R,3S,4R,5R)-5-azido-3-fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one
- 2′-Deoxy-2′-beta-fluoro-4′-azidocytidine
- 2(1H)-Pyrimidinone, 4-amino-1-(4-c-azido-2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-
- 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-5-((imino-l,5-azanylidene)amino)tetrahydrofuran-2-yl)pyrimidin-2-one
- 4-amino-1-((2R,3S,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-5-((imino-l,5-azanylidene)amino)tetrahydrofuran-2-yl)pyrimidin-2-one
- 4-Amino-1-(4-c-azido-2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-2(1H)-pyrimidinone
- 4′-C-azido-2′-deoxy-2′-fluoro-beta-D-arabinocytidine
- 4-amino-1-[(2R,3S,4R,5R)-5-azido-3-fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one
- AZVUDINE [WHO-DD]
- 4-amino-1-((2R,3S,4R,5R)-5-azido-3-fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one
- 4-AMINO-1-(4-C-AZIDO-2-DEOXY-2-FLUORO-.BETA.-D-ARABINOFURANOSYL)-2(1H)-PYRIMIDINONE
- 2(1H)-PYRIMIDINONE, 4-AMINO-1-(4-C-AZIDO-2-DEOXY-2-FLUORO-.BETA.-D-ARABINOFURANOSYL)-
- 4′-C-Azido-2′-deoxy-2′-fluoro-b-D-arabinocytidine
Azvudine is an antiviral drug which acts as a reverse transcriptase inhibitor.[3] It was discovered for the treatment of hepatitis C[4] and has since been investigated for use against other viral diseases such as AIDS and COVID-19,[2][5] for which it was granted conditional approval in China.[6][7]
Azvudine was first discovered in 2007.[8] It costs 350 Chinese yuan per 7 days for COVID, as of November 2022.[9]
Azvudine is under investigation in clinical trial NCT04668235 (Study on Safety and Clinical Efficacy of AZVUDINE in COVID-19 Patients (Sars-cov-2 Infected)).
Azvudine (RO-0622) is a potent nucleoside reverse transcriptase inhibitor (NRTI), with antiviral activity on HIV, HBV and HCV. Azvudine exerts highly potent inhibition on HIV-1 (EC50s ranging from 0.03 to 6.92 nM) and HIV-2 (EC50s ranging from 0.018 to 0.025 nM). Azvudine inhibits NRTI-resistant viral strains. Azvudine is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
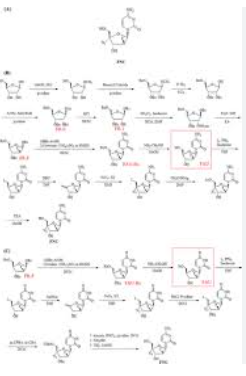
SYN
https://patents.google.com/patent/CN111892636A/en
According to the inventor’s research and understanding, at present, the synthesis of azvudine mainly includes the following methods according to different raw materials:
1. It is prepared by using a ribonucleotide as a raw material. The method requires a total of 15 steps of reaction to obtain the target product. The inventor’s research found that DAST is used as a fluorination reagent in the fluorination process of this method, which has large steric hindrance and is difficult to fluoride, and the route process is complicated and the route is long. Low cost, high cost, not suitable for industrial production.
2. It is prepared by using 1,3,5-O-tribenzoyl-2-deoxy-2-fluoro-D-arabinofuranoside as raw material. The inventors have found that the preparation raw materials are not easy to obtain, and the route involves uridine to In the conversion reaction of cytidine, the reaction route is further extended, thus limiting the further application of this method.
3. Using ribonucleotides as raw materials to synthesize, the inventors found that this method requires 12 steps to synthesize the target product, and DAST is also used as a fluorination reagent in the fluorination process, which has large steric hindrance and low fluorination efficiency.
4. It is prepared by using uracil nucleotide as raw material. The inventors have found that the raw material cost of this method is relatively high, it involves the conversion process of uridine to cytidine, and the reaction yield is not high.
To sum up, the inventors have found that the currently known processes for preparing azvudine have the following disadvantages: synthesizing uracil nucleotides with ribonucleotides as raw materials, and then converting uracil nucleotides into target products, synthesizing uracil nucleotides. The process routes all exceed 12 steps, and the fluorination reaction uses DAST (diethylaminosulfur trifluoride) reagent, which increases the difficulty of the reaction due to steric hindrance, reduces the yield, and brings difficulties to industrial production.
Example 1

Accurately weigh 4.86 g of cytidine (compound 1), dissolve it in 20 mL of ethanol, then add 4.52 g of benzoic anhydride, raise the temperature to 60° C., and stir overnight. After the reaction was completed, the solvent was removed under reduced pressure, and 100 mL of deionized water was added to wash and filter to obtain compound 2 (96.5% yield). The structural characterization is shown in Figure 1 .
Example 2

Weigh 3.5 g of compound 2, dissolve in 25 mL of pyridine, ice-water bath at 0°C, add 3.0 mL of TIPDS under nitrogen protection, react for 1 h, decompose the reaction mixture with water, remove pyridine under reduced pressure, extract with chloroform, and wash with saturated aqueous sodium bicarbonate solution , and dried over anhydrous sodium sulfate to obtain compound 3 (83.6% yield). The structural characterization is shown in Figure 2.
Example 3

Weigh 5.9 g of compound 3, dissolve it in 100 mL of tetrahydrofuran, add 2.82 g of trifluoromethanesulfonic anhydride (Tf 2 O), and react at room temperature for 2 h under nitrogen protection. Then, the reaction temperature was lowered to -20°C, 2.46 g of tetrabutylammonium fluoride (Bu 4 NF) was added, and the reaction was continued for 10 h. After the reaction was completed, tetrahydrofuran was removed under reduced pressure, extracted with chloroform, washed with saturated aqueous sodium bicarbonate solution, and dried over anhydrous sodium sulfate to obtain compound 4 (86.8% yield). The structural characterization is shown in Figure 3.
Example 4

5.9 g of compound 3 was weighed, dissolved in 100 mL of dichloromethane and 10 mL of anhydrous pyridine, cooled to -50°C under nitrogen protection, added with 1.61 g of DAST, and reacted for 12 h. After the reaction was completed, the solvent was removed under reduced pressure, extracted with chloroform, washed with saturated aqueous sodium bicarbonate solution, and dried over anhydrous sodium sulfate to obtain compound 4 (76.0% yield).
Example 5

Weigh 3.5g of compound 4, add 100mL of tetrahydrofuran, add 0.5g of imidazole, 0.5g of triphenylphosphine, slowly add 3.75g of tetrahydrofuran solution containing 10wt% iodine, stir at room temperature for 5h, the reaction is complete, remove the solvent under reduced pressure, Compound 5 was prepared (84% yield) and the structural characterization is shown in Figure 4 .
Example 6

Weigh 4.59g of compound 5, dissolve it in 100mL of methanol, add 0.5g of DBU, control the temperature to 60°C, react for 12h, cool to room temperature, add saturated aqueous sodium chloride solution, adjust the acidic pH=3 with 1M hydrochloric acid, extract with ethyl acetate, It was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain compound 6 (77.4% yield). The structural characterization is shown in Figure 5 .
Example 7

Weigh 3.3 g of compound 6, add 50 mL of DMF solution dissolved with 0.6 g of sodium azide, add 50 mL of DMF solution dissolved with 0.6 g of ICl, control the temperature to 0 ° C, and react for 12 h. After the reaction is completed, add sodium bisulfite until The color of iodine disappears completely. The solvent was removed under reduced pressure to obtain compound 7 (77% yield). The structural characterization is shown in FIG. 6 .
Example 8

Weigh 2.5 g of compound 7, dissolve it in 50 mL of DMF, add 0.65 g of benzoic acid, add 0.5 g of silver acetate, and stir at room temperature for 12 h. After the reaction was completed, the mixture was filtered, and the solvent was removed under reduced pressure to obtain compound 8 (71.2% yield). The structural characterization is shown in FIG. 7 .
Example 9

Weigh 4.82 g of compound 8, add 100 mL of methanol, 10 mL of deionized water, 3 mL of triethylamine, stir at room temperature for 5 h, and remove the solvent under reduced pressure to obtain compound 9 (88.7% yield). The structural characterization is shown in Figure 8.
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00166
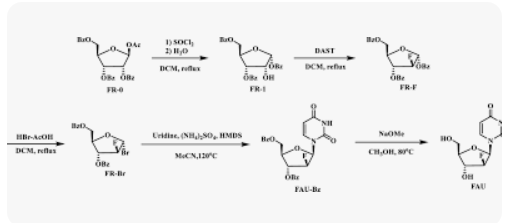
Azvudine was approved for the treatment of adult HIV-1 infection in China in 2021, and it was approved for conditional marketing for the treatment of SARS-CoV-2 in China in 2022. In this work, we describe a fully continuous flow synthesis of 2′-deoxy-2′-fluoroarabinoside, a key intermediate for azvudine. The process was accomplished via six chemical transformations, including chlorination, hydrolysis, fluorination, bromination, condensation, and deprotection in six sequential continuous flow devices. Under the optimized process conditions, the total yield was 32.3% with a total residence time of 156 min. Compared with batch conditions, the total yield was doubled, the total reaction time was shortened 16 times, and the E factor was reduced 1.63 times.
1,3,5-Tri-O-benzoyl-D-ribofuranose (FR-1)
To a stirred solution of O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (FR-0,
1.008 g, 2.0 mmol) in anhydrous dichloromethane (DCM, 10 mL) was added
dropwise thionyl chloride (0.713 g,6 mmol) at 0 oC, and the resulting mixture
allowed to stir at room temperature for 14 h. The solution was evaporated,
dissolved with toluene (5 mL× 3), and then evaporated. To the residue was
added DCM (10 mL) and water (5 mL), and and continued stirring at room
temperature for 2 h. The solution was then washed with saturated aqueous
NaHCO3 (15 mL) and dried over Na2SO4, filtered and evaporated. DCM (2 mL)
and n-Hexane (20 mL) was added to the residue and the mixture was stirred at
room. A white solid was collected by filtration to give FR-1 (yield: 47.8%).
2-Deoxy-2-fluoro-1,3,5-tri-O-benzoyl-D-ribofuranose (FR-F)
To a stirred solution of FR-1 (0.924 g, 2.0 mmol) in anhydrous DCM (10 mL) was
added dropwise DAST (0.976 g, 6.0 mmol) for 30 min, and the resulting mixture
allowed to stir at 40 °C (16 h). The reaction was cooled and quenched with
saturated aqueous NaHCO3 (15 mL). The solution was extracted with DCM and
water, and then washed with saturated aqueous NaHCO3 (20 mL). This was then
dried over Na2SO4, filtered and evaporated. The crude product was purified by
silica gel column chromatography (ethyl acetate: petroleum ether =1:10) to
obtain a white solid (yield: 41.2%).
α-D-Arabinofuranosyl bromide, 2-deoxy-2-fluoro-, 3,5-dibenzoate (FR-Br)
To a stirred solution of FR-F (0.928 g, 2.0 mmol) in anhydrous DCM (10 mL) was
added dropwise 33.3% HBr-HOAc (1.47 g, 6.0 mmol) at 0 oC, and the resulting
mixture were allowed to stir at room temperature for 7 h. The reaction was
quenched with saturated aqueous NaHCO3 (10 mL), dried over Na2SO4, filtered
and evaporated to yield the product (FR-Br) as a brown oil (yield: 99.8%).
1-(2-deoxy-2-fluoro-3,5-di-O-benzoyl-β-D-arabino-furanosyl)uracil (FAU-Bz)
A mixture of uracil (0.336 g, 3 mmol) and (NH4)2SO4 (10 mg, 0.075 mmol) in
hexamethyldisilazane (HMDS) (6 mL) was refluxed under nitrogen for 5 h. To
the silylated uracil solution was added a solution of FR-Br in dry acetonitrile (10
mL) and the mixture was refluxed under nitrogen for 5 h. The solution was
evaporated, extracted with DCM (20 mL) and washed with saturated aqueous
NaHCO3 (15 mL). This was then dried over Na2SO4, filtered and evaporated.
Ethyl acetate (10 mL) and petroleum ether (50 mL) was added to the residue
and the mixture was stirred at room temperature. A light yellow solid was
collected by filtration to give FAU-Bz (yield: 83.2%).
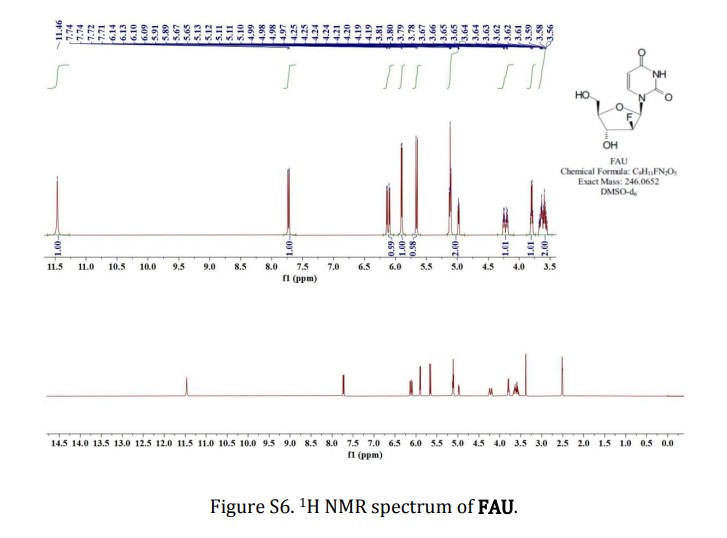
2′-deoxy-2′-fluoro- arabinoside (FAU)
To a solution of FUA-Bz (0.908 g, 2.0 mmol) in anhydrous methanol (MeOH, 10
mL) was added NH3-MeOH (5 mL), stirred at room temperature for 15 h and
evaporated to dryness under reduced pressure. White solid
SYN
: J. Med. Chem. 2024, 67, 4376−4418
Azvudine (1). Azvudine (1) is an antiviral manufactured by China-based Genuine Biotech. It was
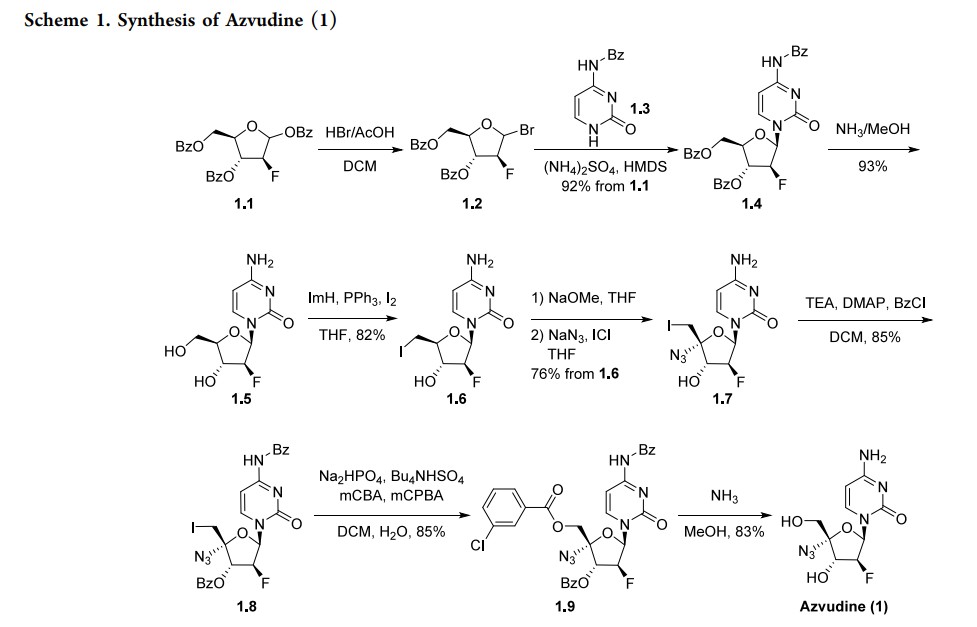
approved in China in 2021 as a first-in-class treatment for human immunodeficiency virus (HIV). It has a dual function, acting as a reverse transcriptase inhibitor and targeting the viral infectivity factor/apolipoprotein B mRNA-editing enzyme, catalytic subunit 3G (Vif/A3G) protein−protein interaction.6 Azvudine has activity against both wild-type and drug-resistant strains of HIV due to the presence of a 3′-hydroxy group and substitution in the 4′-position of the ribose core.7 Due to its known antiviral activity, azvudine was repurposed as a treatment for COVID-19 and approved for this indication inChina in 2022. It acts as an RNA-dependent RNA polymerase (RdRp) inhibitor, the same mechanism as the previously approved molnupiravir and remdesivir. In addition to its antiviral activity, concentration of the drug in the thymus has suggested immune-targeting activity; this dual function is unique among RdRp inhibitors.8 Several syntheses of azvudine have been reported in the scientific and patent literature. Scheme 1 highlights a 100 g scale synthesis from a patent filed by Shandong University.9 Other syntheses are similar, containing the furanose functional group manipulations in 1.5−1.8, though these routes differ in choice of nucleobase and protecting group strategy, and were reported on a smaller scale.10−12 The synthesis began from benzoyl-protected fluoro-furanose 1.1. Bromination with HBr in acetic acid followed by displacement of the bromide 1.2 with protected cytosine 1.3 yielded intermediate 1.4. Deprotection of the benzoyl groups with ammonia in MeOH formed diol 1.5, and a Mitsunobu reaction converted the
primary alcohol to alkyl iodide 1.6. Elimination of the iodide with sodium methoxide followed by addition of sodium azide and iodine monochloride across the resulting alkene produced substitution in the 4′-position of the ribose core.7 Due to its known antiviral activity, azvudine was repurposed as a treatment for COVID-19 and approved for this indication in China in 2022. It acts as an RNA-dependent RNA polymerase (RdRp) inhibitor, the same mechanism as the previously approved molnupiravir and remdesivir. In addition to its antiviral activity, concentration of the drug in the thymus has suggested immune-targeting activity; this dual function is unique among RdRp inhibitors.8 Several syntheses of azvudine have been reported in the scientific and patent literature. Scheme 1 highlights a 100 g scale synthesis from a patent filed by Shandong University.9 Other syntheses are similar, containing the furanose functional group manipulations in 1.5−1.8, though these routes differ in choice of nucleobase and protecting group strategy, and were reported on a smaller scale.10−12 The synthesis began from
benzoyl-protected fluoro-furanose 1.1. Bromination with HBr in acetic acid followed by displacement of the bromide 1.2 with protected cytosine 1.3 yielded intermediate 1.4. Deprotection of the benzoyl groups with ammonia in MeOH formed diol 1.5, and a Mitsunobu reaction converted the primary alcohol to alkyl iodide 1.6. Elimination of the iodide with sodium methoxide followed by addition of sodium azide
and iodine monochloride across the resulting alkene producedazide 1.7. Both the alcohol and amine were reprotected with benzoyl chloride, and the iodide was displaced with metachlorobenzoic acid in an oxidative nucleophilic substitution reaction to yield penultimate intermediate 1.9. All protecting
groups were then removed with ammonia in MeOH to yield
azvudine (1).
(6) Sun, L.; Peng, Y.; Yu, W.; Zhang, Y.; Liang, L.; Song, C.; Hou, J.;
Qiao, Y.; Wang, Q.; Chen, J.; et al. Mechanistic insight into
antiretroviral potency of 2’-deoxy-2’-beta-fluoro-4’-azidocytidine
(FNC) with a long-lasting effect on HIV-1 prevention. J. Med.
Chem. 2020, 63, 8554−8566.
(7) Chang, J. 4’-Modified nucleosides for antiviral drug discovery:
achievements and perspectives. Acc. Chem. Res. 2022, 55, 565−578.
(8) Zhang, J.-L.; Li, Y.-H.; Wang, L.-L.; Liu, H.-Q.; Lu, S.-Y.; Liu, Y.;
Li, K.; Liu, B.; Li, S.-Y.; Shao, F.-M.; Wang, K.; Sheng, N.; Li, R.; Cui,
J.-J.; Sun, P.-C.; Ma, C.-X.; Zhu, B.; Wang, Z.; Wan, Y.-H.; Yu, S.-S.;
Che, Y.; Wang, C.-Y.; Wang, C.; Zhang, Q.; Zhao, L.-M.; Peng, X.-Z.;
Cheng, Z.; Chang, J.-B.; Jiang, J.-D. Azvudine is a thymus-homing
anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Signal
Transduct. Target Ther. 2021, 6, 414.
(9) Chen, X.; Wang, Z.; Yu, H.; Liu, X. Preparation method of
azvudine and its intermediates. China Patent CN 115960147, 2023.
(10) Smith, D. B.; Kalayanov, G.; Sund, C.; Winqvist, A.; Maltseva,
T.; Leveque, V. J.-P.; Rajyaguru, S.; Pogam, S. L.; Najera, I.;
Benkestock, K.; Zhou, X.-X.; Kaiser, A. C.; Maag, H.; Cammack, N.;
Martin, J. A.; Swallow, S.; Johansson, N. G.; Klumpp, K.; Smith, M.
The design, synthesis, and antiviral activity of monofluoro and
difluoro analogues of 4’-azidocytidine against hepatitis C virus
replication: The discovery of 4’-azido-2’-deoxy-2’-fluorocytidine and4’-azido-2’-dideoxy-2’,2’-difluorocytidine. J. Med. Chem. 2009, 52,
2971−2978.
(11) Wang, Q.; Hu, W.; Wang, S.; Pan, Z.; Tao, L.; Guo, X.; Qian,
K.; Chen, C. H.; Lee, K. H.; Chang, J. Synthesis of new 2’-deoxy-2’-
fluoro-4’-azido nucleoside analogues as potent anti-HIV agents. Eur. J.
Med. Chem. 2011, 46, 4178−83.
(12) Deng, W.; Jiang, S.; Yu, T.; Zhai, D. Synthesis method of
azvudine. China Patent CN 111892636, 2020.
Medical uses
In July 2021, azvudine became conditionally approved in China for the following indication: “to treat high-viral-load cases of HIV-1, in combination with a nucleoside reverse-transcriptase inhibitor and a non-nucleoside reverse-transcriptase inhibitor”. The approval text describes it as a dual reverse transcriptase and Vif inhibitor.[10]
In July 2022, azvudine received emergency conditional approval for COVID-19 in adults.[11] It is believed to work by inhibiting the RNA-dependent RNA polymerase (RdRp) enzyme in the SARS-CoV-2 virus.[12][13]
Adverse effects
According to the manufacturer, phase II trials of azvudine in combination with doravirine and tenofovir disoproxil fumarate in HIV patients found an adverse effect profile similar to, but milder, than lamivudine combined with the two drugs. Very common (> 10%) side effects include dizziness, elevated liver enzymes, vomiting, and elevated alkaline phosphatase. Common (> 1%) side effects include nausea, elevated blood lipids, fever, insomnia, tiredness, and diarrhea. Detailed numbers are provided by Genuine in the slides and the medication package insert.[14][15] A boxed warning is present at the beginning of the Chinese package insert, describing a risk of “decrease in absolute neutrophil count, increase in total bilirubin, increase in glutathione aminotransferase, and increase in blood glucose”.[15]
The small (n=10) open-label pilot study for azvudine used alone in COVID reported no adverse events.[16]
Non-human models
Azvudine is found to be mutagenic in in vitro in the Ames test, CHL test, and in vitro in the mice micronucleus test.[17]
Azvudine is toxic to the reproductive system of rats and rabbit. The minimum reproductive NOAEL found for males is 5.0 mg/kg/d and for females 0.5 mg/kg/d. It is excreted in rat breast milk; the NOAEL for rat pups is 1.5 mg/kg/d.[17]
Azvudine is mainly toxic to the immune system, bone marrow, and digestive system of model animals. The chronic NOAELs are 0.5 mg/kg/d (rat, 3 months), 0.3 mg/kg/d (rat, 26 weeks), and 0.1 mg/kg/d (beagle dog, 1 month and 39 weeks).[17] For comparison, the chronic human dose for HIV treatment is 0.05 mg/kg/d, using the reference 3 mg dose and an average Chinese body mass of 59.5 kg (2014).
History
Azvudine was first found in literature in a patent filed by Chang Jun-biao of Zhengzhou University.[8] It received its current name in 2009, when researchers at Roche independently discovered it as a Hep C RNA polymerase inhibitor in vitro.[4] In the following years, Chinese scientists tested it in vitro for a number of targets, most importantly HBV (human and duck) and HTLV-1, two viruses with a reverse transcriptase.[18][19][20]
It was first proposed as an HIV treatment in 2011, when in vitro tests by the Chang group provided positive results.[21] In 2014, its oral pharmokinetics in rats was elucidated.[1] A phase II study (NCT04109183) was finished in March 2019 by Genuine Biotech. In August 2020, the Chang group found that the substance inhibits vif in vitro.[22] In the same month, China’s drug regulator (NMPA) decided to fast-track the approval process, labelling it a first-in-class medication.[14] In July 2021, NMPA granted conditional approval for HIV-1.[7] It was included in the 2021 HIV treatment recommendnation by the Chinese Medical Association and Chinese CDC, published October that year.[14] Curiously, no full results of the trial have been made available for this study in any journal detailing the experiment design as of December 2022.[23] Parts of the results are shown on the drug monograph as well as a 2022 slides deck produced by Genuine for the NHSA available on the latter’s website.[14]
Azvudine was found to inhibit some coronaviruses in vitro around 2020, leading to an interest in its use in COVID. An open-label pilot study on mild and moderate cases was performed in 2020, with mildly positive results.[16] A phase III trial was performed in 2022 in China. In July 2022, China’s drug regulator granted conditional approval for it to be used to treat COVID-19, following a local phase III trial.[6] Initially, no detailed description of the said trial was published in any journals, but state media quoted some numbers from the developer: “40% clinical improvement in 7 days by FNC group, compared to 11% in control”.[7] It is unclear how such “improvement” is defined.
Four phase III clinical trials investigated azvudine’s efficacy and safety in adults with mild-to-moderate COVID-19. The findings indicate that azvudine may reduce the time to eliminate detectable levels of virus (viral load) and improve symptoms faster than standard treatment. In trials, it was reported to be safe with few side effects. However, some studies produced inconsistent results in terms of symptom improvement and severe illness prevention. Additionally, the studies tended to use a smaller number of participants than other major COVID-19 drug trials.[12][13]
Society and culture
Genuine owns two different tradenames for this medication: 双新艾克 (literally “dual new AIDS inhibitor”) for HIV use[14] and 捷倍安 (literally “fast extra safe”) for COVID use.[7] No generics are available.
Geniune holds one patent related to the drug: the original 2007 patent on the entire class of 2′-fluorine-4′-substituted nucleotides, purchased from Zhengzhou University.[8] Two other Chinese patents on synthesizing the drug are found on Google Patents, but the owners do not appear to be connected to Geniune.[24] Roche held one 2002 patent, CNA028118480A (CN1516590A), over the broader class of 4′-substituted nucleotides. The patent was voided in 2019 after Riboscience, its new holder, stopped paying fees.[25]
References
- Peng Y, Cheng T, Dong L, Zhang Y, Chen X, Jiang J, et al. (September 2014). “Quantification of 2′-deoxy-2′-β-fluoro-4′-azidocytidine in rat and dog plasma using liquid chromatography-quadrupole time-of-flight and liquid chromatography-triple quadrupole mass spectrometry: Application to bioavailability and pharmacokinetic studies”. Journal of Pharmaceutical and Biomedical Analysis. 98: 379–386. doi:10.1016/j.jpba.2014.06.019. PMID 24999865.
- Liu Y, Liu B, Zhang Y, Peng Y, Huang C, Wang N, et al. (July 2017). “Intestinal absorption mechanisms of 2′-deoxy-2′-β-fluoro-4′-azidocytidine, a cytidine analog for AIDS treatment, and its interaction with P-glycoprotein, multidrug resistance-associated protein 2 and breast cancer resistance protein”. European Journal of Pharmaceutical Sciences. 105: 150–158. doi:10.1016/j.ejps.2017.05.009. PMID 28487144. S2CID 4252337.
- Wang RR, Yang QH, Luo RH, Peng YM, Dai SX, Zhang XJ, et al. (2014). “Azvudine, a novel nucleoside reverse transcriptase inhibitor showed good drug combination features and better inhibition on drug-resistant strains than lamivudine in vitro”. PLOS ONE. 9 (8): e105617. Bibcode:2014PLoSO…9j5617W. doi:10.1371/journal.pone.0105617. PMC 4140803. PMID 25144636.
- Smith DB, Kalayanov G, Sund C, Winqvist A, Maltseva T, Leveque VJ, et al. (May 2009). “The design, synthesis, and antiviral activity of monofluoro and difluoro analogues of 4′-azidocytidine against hepatitis C virus replication: the discovery of 4′-azido-2′-deoxy-2′-fluorocytidine and 4′-azido-2′-dideoxy-2′,2′-difluorocytidine”. Journal of Medicinal Chemistry. 52 (9): 2971–2978. doi:10.1021/jm801595c. PMID 19341305.
- Harrison C (April 2020). “Coronavirus puts drug repurposing on the fast track”. Nature Biotechnology. 38 (4): 379–381. doi:10.1038/d41587-020-00003-1. PMID 32205870.
- Ye Y (July 2022). “China approves first homegrown COVID antiviral”. Nature. doi:10.1038/d41586-022-02050-x. PMID 35883009. S2CID 251104078.
- “首个国产抗新冠口服药附条件获批上市” [First domestic oral anti-Covid drug conditionally approved]. 新华网. 证券日报. 2022-07-26. Archived from the original on 2022-08-09. Retrieved 2022-07-26.
- Chang J, Bao X, Wang Q, Guo X, Wang W, Qi X. Preparation of 2′-fluoro-4′-substituted nucleoside analogs as antiviral agents. 20070807. Chinese Patent Application No: CN 2007-10137548. Chinese Patent No: CN 101177442A, 20080514.
- “新冠口服药阿兹夫定片线上开售, 每瓶售价350元” [Oral COVID drug Azvudine tablet available online at 350 yuan/bottle]. Xinmin Evening News. 19 November 2022. Retrieved 28 December 2022 – via Beijing Daily (repost).
- “国家药监局附条件批准阿兹夫定片上市” [NMPA conditionally approvals azvudine tablets]. http://www.nmpa.gov.cn (in Chinese). 2021-07-21.
- “国家药监局应急附条件批准河南真实生物科技有限公司阿兹夫定片增加新冠肺炎治疗适应症注册申请” [NMPA grants emergency conditional approval for additional indication registration of azvudine tablets (Hebei Genuine Biotech Co., Ltd.)]. http://www.nmpa.gov.cn. 2022-07-25.
- Zhu KW (2023). “Efficacy and safety evaluation of Azvudine in the prospective treatment of COVID-19 based on four phase III clinical trials”. Frontiers in Pharmacology. 14: 1228548. doi:10.3389/fphar.2023.1228548. PMC 10484631. PMID 37693894.
- Wang Y, Xie H, Wang L, Fan J, Zhang Y, Pan S, Zhou W, Chen Q, Liu X, Wu A, Zhang H, Wang J, Tian X (February 2024). “Effectiveness of azvudine in reducing mortality of COVID-19 patients: a systematic review and meta-analysis”. Virology Journal. 21 (1): 46. doi:10.1186/s12985-024-02316-y. PMC 10893615. PMID 38395970.
- Genuine Biotech (July 11, 2022). “阿兹夫定片(双新艾克)” [Azvudine Tablets (Shuāngxīnàikè)]. NHSA.gov.cn. Archived from the original on 2022-09-06.
- “阿兹夫定片说明书” [Azvudine Tablets, Monograph] (PDF). WUXU DATA. Retrieved 2023-01-03.
- Ren Z, Luo H, Yu Z, Song J, Liang L, Wang L, et al. (October 2020). “A Randomized, Open-Label, Controlled Clinical Trial of Azvudine Tablets in the Treatment of Mild and Common COVID-19, a Pilot Study”. Advanced Science. 7 (19): e2001435. doi:10.1002/advs.202001435. PMC 7404576. PMID 35403380.
- Drug Review Center (China) (June 30, 2022). “阿兹夫定片 (CXHS2000016-17) 申请上市技术审评报告” [Azvudine tabs (CHXS2000016-17) request for marketing technical review report] (PDF). WUXU DATA. Retrieved 2023-01-03.
- Wang Q, Liu X, Wang Q, Zhang Y, Jiang J, Guo X, et al. (April 2011). “FNC, a novel nucleoside analogue inhibits cell proliferation and tumor growth in a variety of human cancer cells”. Biochemical Pharmacology. 81 (7): 848–855. doi:10.1016/j.bcp.2011.01.001. PMID 21219886.
- Zheng L, Wang Q, Yang X, Guo X, Chen L, Tao L, et al. (2012). “Antiviral activity of FNC, 2′-deoxy-2′-β-fluoro-4′-azidocytidine, against human and duck HBV replication”. Antiviral Therapy. 17 (4): 679–687. doi:10.3851/IMP2094. PMID 22452880. S2CID 25576607.
- Zhou Y, Zhang Y, Yang X, Zhao J, Zheng L, Sun C, et al. (2012). “Novel nucleoside analogue FNC is effective against both wild-type and lamivudine-resistant HBV clinical isolates”. Antiviral Therapy. 17 (8): 1593–1599. doi:10.3851/IMP2292. PMID 22910281. S2CID 29382902.
- Wang Q, Hu W, Wang S, Pan Z, Tao L, Guo X, et al. (September 2011). “Synthesis of new 2′-deoxy-2′-fluoro-4′-azido nucleoside analogues as potent anti-HIV agents”. European Journal of Medicinal Chemistry. 46 (9): 4178–4183. doi:10.1016/j.ejmech.2011.06.020. PMC 3164908. PMID 21745701.
- Sun L, Peng Y, Yu W, Zhang Y, Liang L, Song C, et al. (August 2020). “Mechanistic Insight into Antiretroviral Potency of 2′-Deoxy-2′-β-fluoro-4′-azidocytidine (FNC) with a Long-Lasting Effect on HIV-1 Prevention”. Journal of Medicinal Chemistry. 63 (15): 8554–8566. doi:10.1021/acs.jmedchem.0c00940. PMID 32678592. S2CID 220631451.
- Li G, Wang Y, De Clercq E (April 2022). “Approved HIV reverse transcriptase inhibitors in the past decade”. Acta Pharmaceutica Sinica. B. 12 (4): 1567–1590. doi:10.1016/j.apsb.2021.11.009. PMC 9279714. PMID 35847492.
- Google Patents Search, “阿兹夫定” (with quotes), CN114149475A, CN111892636A.
- Guokr.com (10 August 2022). “真实生物的真实面目”. Huxiu.com. Retrieved 30 December 2022.
Further reading
- Zhu KW (2023). “Efficacy and safety evaluation of Azvudine in the prospective treatment of COVID-19 based on four phase III clinical trials”. Frontiers in Pharmacology. 14: 1228548. doi:10.3389/fphar.2023.1228548. PMC 10484631. PMID 37693894.
| Clinical data | |
|---|---|
| Trade names | 捷倍安, 双新艾克 |
| Other names | 2′-Deoxy-2′-β-fluoro-4′-azidocytidine (FNC), RO-0622 |
| Legal status | |
| Legal status | US: Investigational drugCN: Conditional use Rx |
| Pharmacokinetic data | |
| Bioavailability | 83% (rat, dog)[1] |
| Metabolism | liver (CYP3A)[2] |
| Elimination half-life | 4 hours (dog)[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1011529-10-4 |
| PubChem CID | 24769759 |
| DrugBank | DB16407 |
| ChemSpider | 24717759 |
| UNII | IJ2XP0ID0K |
| ChEMBL | ChEMBL519846 |
| Chemical and physical data | |
| Formula | C9H11FN6O4 |
| Molar mass | 286.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////AZVUDINE, Genuine Biotech, APPROVALS 2022, CHINA 2022, FNC, HY 19314, RO 0622, RO-0622, SB17040, IJ2XP0ID0K, DTXSID901027757



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Sontigidomide
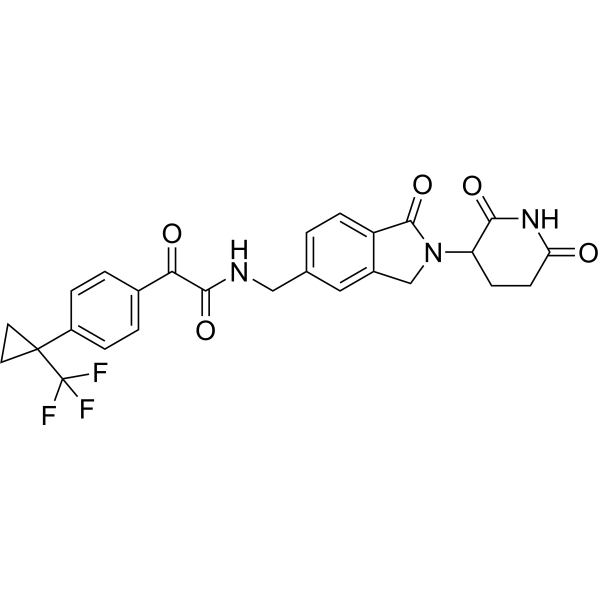
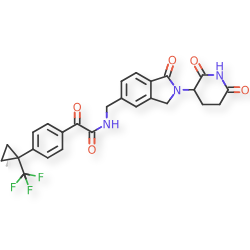
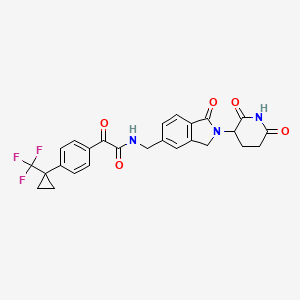
Sontigidomide
CAS 2560577-69-5
| Molecular Weight | 513.47 |
|---|---|
| Formula | C26H22F3N3O5 |
N-[[2-(2,6-Dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α-oxo-4-[1-(trifluoromethyl)cyclopropyl]benzeneacetamide
enzeneacetamide, N-[[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α-oxo-4-[1-(trifluoromethyl)cyclopropyl]-
FDD2NVW84X, Sontigidomida
Sontigidomide (Compound 5) is an antineoplastic compound. Sontigidomide inhibits MOLM-13 cell proliferation more than 80% at 1 μM (3 days).
SCHEME
COUPLER………….
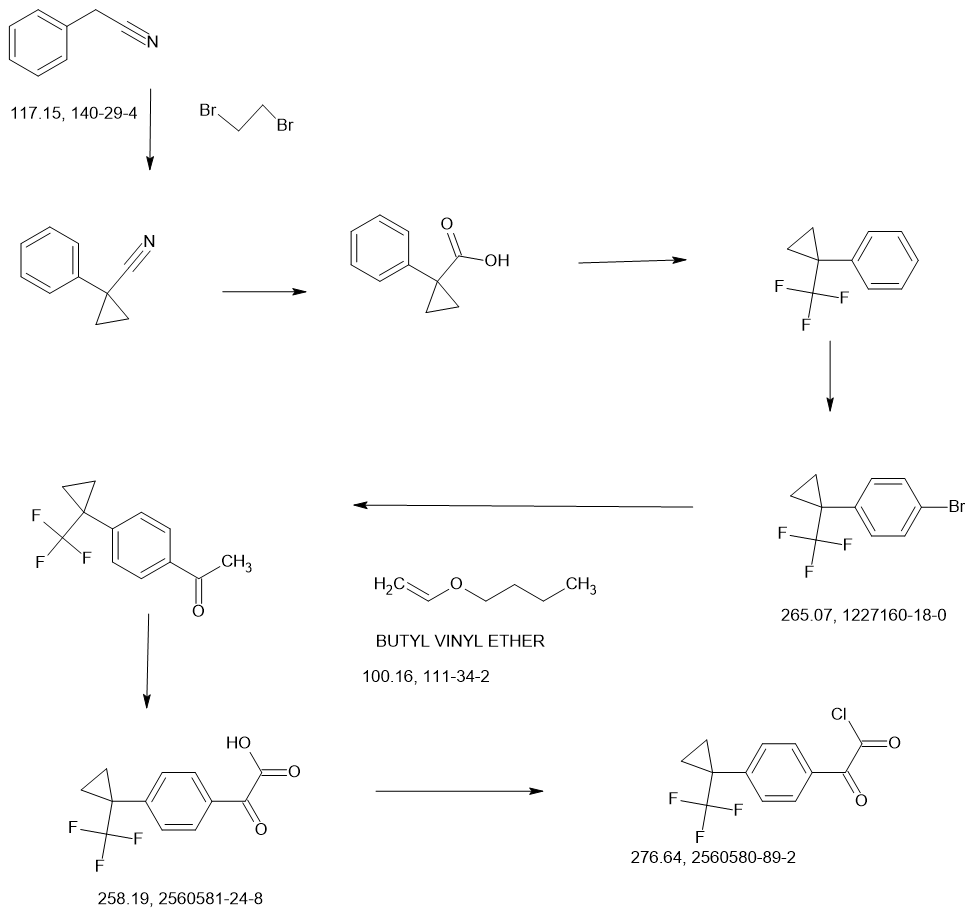
MAIN……….
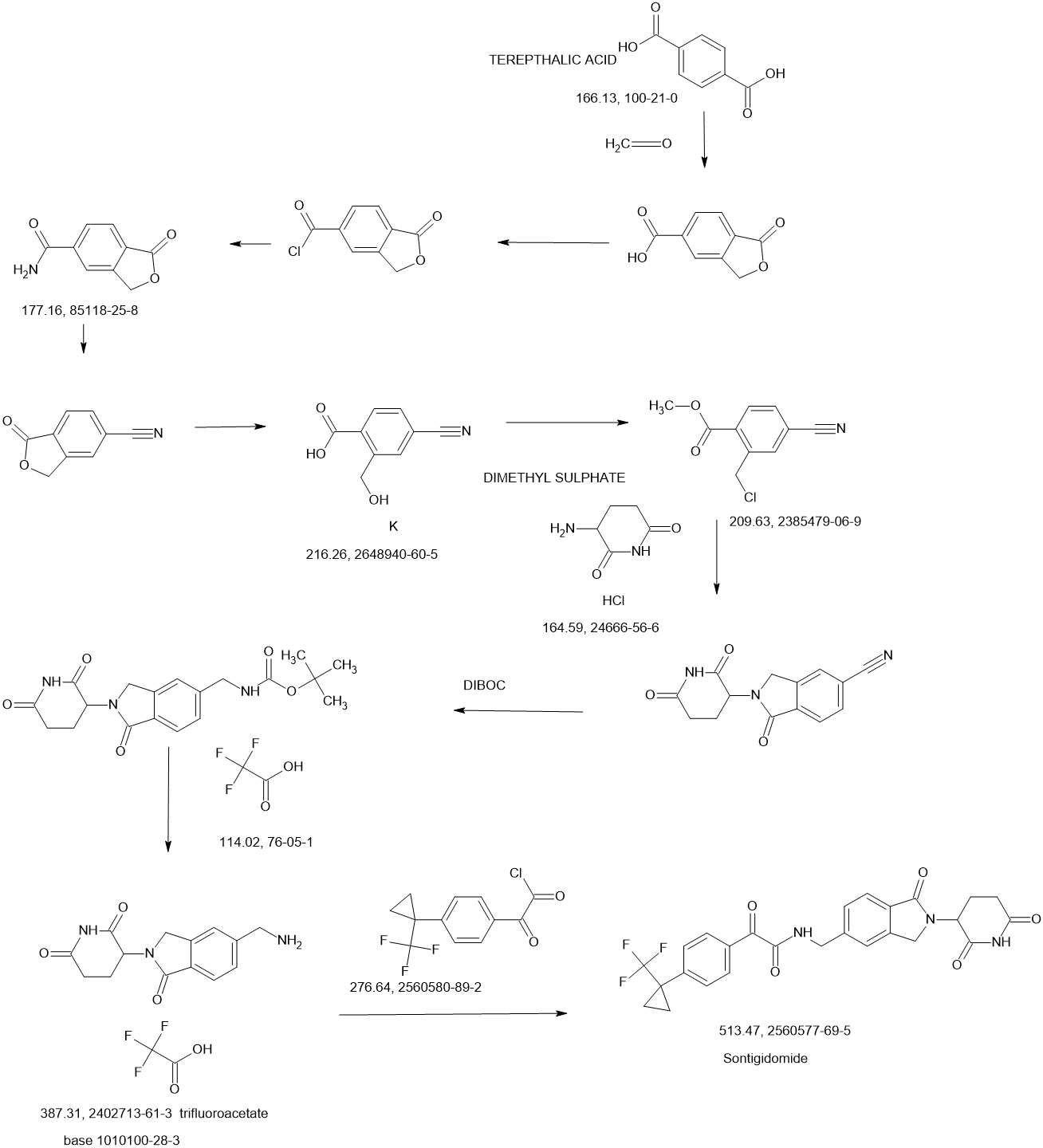
PATENTS
WO2023070120 BioTheryX, Inc.
PATENT
US20200369679
https://patentscope.wipo.int/search/en/detail.jsf?docId=US311579044&_cid=P20-MD87Y5-18242-1
Example 5
Compound I-5: N-((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)methyl)-2-oxo-2-(4-(1-(trifluoromethyl)cyclopropyl)phenyl)acetamide
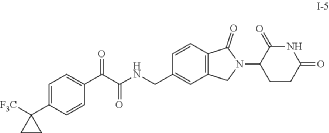
Compound I-5 was synthesized as shown in Scheme 5.
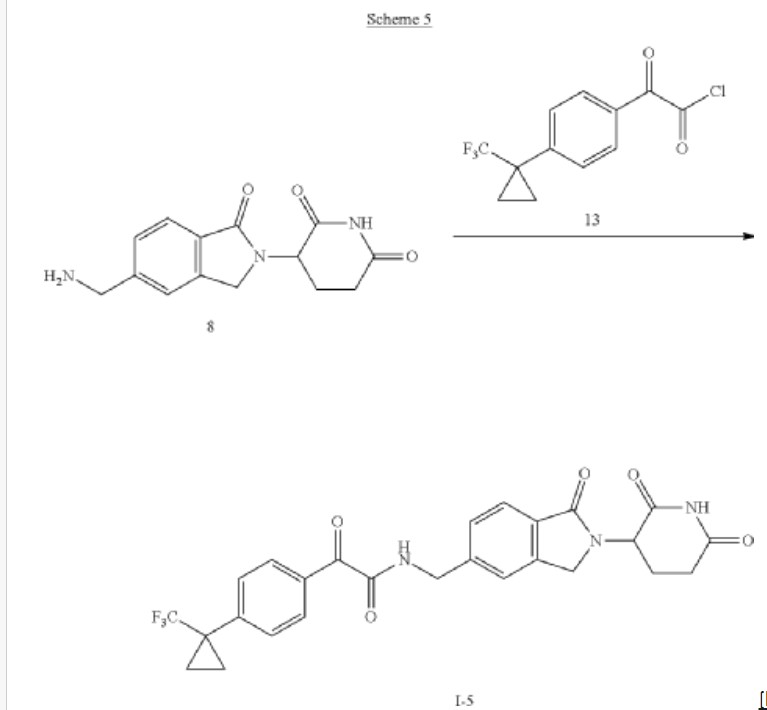
To a solution of 3-(5-(aminomethyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 8 (80 mg, 0.258 mmol) in DCM (4 mL) at 0° C. was added TEA (52.2 mg, 0.516 mmol). After stirring for 2 min, 2-oxo-2-(4-(1-(trifluoromethyl)cyclopropyl)phenyl)acetyl chloride 13 (71.3 mg, 0.258 mmol) was added and the mixture was stirred at RT for 2 h. After concentration, the residue was purified using prep-HPLC eluting with ACN/H 2O (0.1% TFA) from 10% to 95% to afford compound I-5 (16.1 mg) in 12% yield. MS (ESI) m/z: 514.0 [M+H] +; 1H NMR (400 MHz, DMSO-d 6) δ 10.98 (s, 1H), 9.57 (t, J=6.0 Hz, 1H), 8.03-8.01 (m, 2H), 7.74-7.48 (m, 7H), 5.13-5.09 (m, 1H), 4.59-4.57 (m, 2H), 4.49-4.31 (m, 2H), 2.95-2.87 (m, 1H), 2.63-2.58 (m, 1H), 2.45-2.38 (m, 1H), 2.03-1.99 (m, 1H), 1.43-1.40 (m, 2H), 1.24-1.21 (m, 2H).
- Ketoamides for treating malignancyPublication Number: WO-2023070120-A1Priority Date: 2021-10-22
- Protein-targeting compounds and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: US-2020369679-A1Priority Date: 2019-05-24
- Compounds targeting proteins and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: WO-2020242960-A1Priority Date: 2019-05-24
- Compounds targeting proteins and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: AU-2020283744-A1Priority Date: 2019-05-24
- Targeted protein compound, its pharmaceutical composition and therapeutic applicationPublication Number: CN-114502543-APriority Date: 2019-05-24
- Compounds targeting proteins and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: EP-3976623-A1Priority Date: 2019-05-24
- Protein targeting compounds, pharmaceutical compositions thereof and therapeutic applications thereofPublication Number: KR-20220023343-APriority Date: 2019-05-24
- Protein-targeting compounds and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: US-11345712-B2Priority Date: 2019-05-24Grant Date: 2022-05-31
- Protein-targeting compounds and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: US-2022298172-A1Priority Date: 2019-05-24
////////Sontigidomide, FDD2NVW84X, CANCER, Sontigidomida
..
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Keverprazan
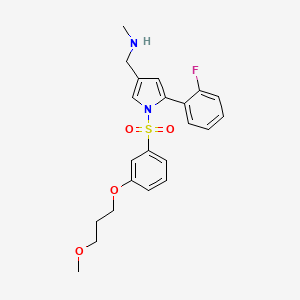
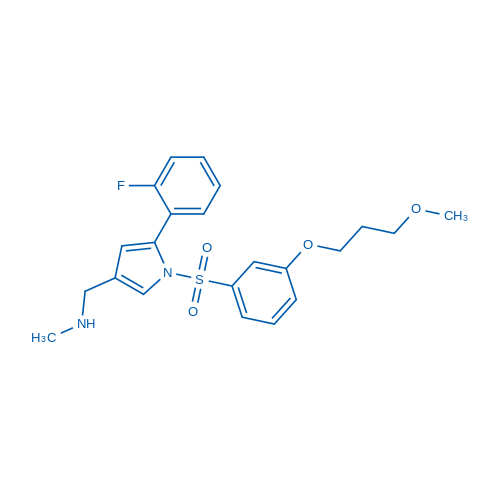
- P-CAB agent 2
- keverprazan
- 1978371-23-1
- Keprason
- SOC12UY3ZP
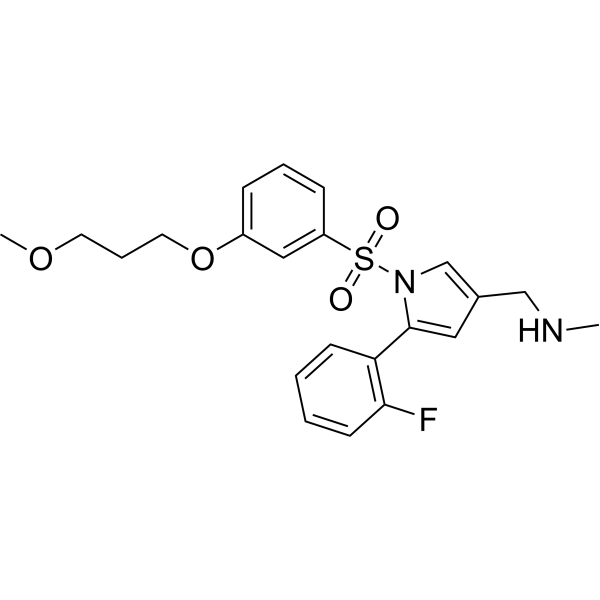
Keverprazan
432.5 g/mol
1-[5-(2-fluorophenyl)-1-[3-(3-methoxypropoxy)phenyl]sulfonylpyrrol-3-yl]-N-methylmethanamine
C22H25FN2O4S
- 1H-Pyrrole-3-methanamine, 5-(2-fluorophenyl)-1-[[3-(3-methoxypropoxy)phenyl]sulfonyl]-N-methyl-
- 5-(2-Fluorophenyl)-1-[[3-(3-methoxypropoxy)phenyl]sulfonyl]-N-methyl-1H-pyrrole-3-methanamine
- Keverprazan Hydrochloride: First ApprovalPublication Name: DrugsPublication Date: 2023-04-19PMID: 37074491DOI: 10.1007/s40265-023-01865-w
- Efficacy of keverprazan for duodenal ulcer: A phase II randomized, double‐blind, parallel‐controlled trialPublication Name: Journal of Gastroenterology and HepatologyPublication Date: 2022-09-16PMID: 36068945DOI: 10.1111/jgh.16000
- The efficacy and safety of keverprazan, a novel potassium‐competitive acid blocker, in treating erosive oesophagitis: a phase <scp>III</scp>, randomised, double‐blind multicentre studyPublication Name: Alimentary Pharmacology & TherapeuticsPublication Date: 2022-05-03PMID: 35505467DOI: 10.1111/apt.16959
- Potassium-competitive acid blockers – are they the next generation of proton pump inhibitors?Publication Name: World Journal of Gastrointestinal Pharmacology and TherapeuticsPublication Date: 2018-12-13PMCID: PMC6305499PMID: 30595950DOI: 10.4292/wjgpt.v9.i7.63
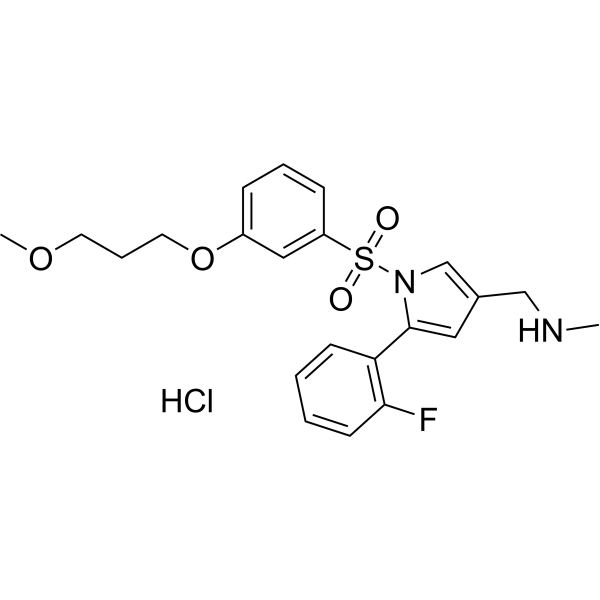
CAS 2209911-80-6
| Molecular Weight | 468.97 |
|---|---|
| Formula | C22H26ClFN2O4S |
Jiangsu Carephar Pharmaceuticals, is
a potassium ion competitive acidblocker (P-CAB) that was approved inFebruary2023inChina for thetreatment of refluxesophagitis or duodenal ulcer inadults
P-CAB agent 2 hydrochloride is a potent and orally active potassium-competitive acid blocker and a gastric acid secretion inhibitor. P-CAB agent 2 hydrochloride inhibits H+/K+-ATPase activity with an IC50 value of <100 nM. P-CAB agent 2 hydrochloride inhibits the hERG potassium channel with an IC50 value of 18.69 M. P-CAB agent 2 hydrochloride shows no acute toxicity and inhibits histamine (HY-B1204)-induced gastric acid secretion.
P-CAB agent 2 is a potent and orally active potassium-competitive acid blocker and a gastric acid secretion inhibitor. P-CAB agent 2 inhibits H+/K+-ATPase activity with an IC50 value of <100 nM. P-CAB agent 2 inhibits the hERG potassium channel with an IC50 value of 18.69 M. P-CAB agent 2 shows no acute toxicity and inhibits histamine (HY-B1204)-induced gastric acid secretion[1].
REF
https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c02079?ref=PDF
KeverprazanHydrochloride. Keverprazan hydrochloride(4),developedby Jiangsu CarepharPharmaceuticals,is
apotassiumioncompetitiveacidblocker (P-CAB) thatwas approvedinFebruary2023inChina forthetreatmentofrefluxesophagitisorduodenal ulcer inadults.36Those illnesses arecaused by gastric acid entering the esophagus, leading toregurgitation and heartburn.37 Current treatments mainly
employ acid-suppressing therapies such as proton pumpinhibitors (PPIs, e.g., lansoprazole); however, PPIs do notacidicenvironments.38Toaddress those issues,P-CABshavebeen developed as a new class of acid suppressants.39
Keverprazan, a novel P-CAB, reversibly binds to gastricH+,K+-ATPase in competition against K+ ions, therebyinhibitingtheenzyme.40Itprovidesstableandsustainedgastricacidinhibitioneffectat20mgdose,whilebeingsafeandwelltoleratedupto60mginasingle-ascendingdosestudy.41Theoriginal synthesisof keverprazanhydrochloridedevelopedbyQinetal. isillustratedinScheme7.42TheroutebeganwithSN2alkylationof3-nitrophenol4.1withalkylbromide4.2
toformphenylether4.3.Thenitrogroupwasreducedtoaniline 4.4underRaney-Ni-mediatedhydrogenation.Aniline4.4wasconverted to sulfonyl chloride 4.5 via copper-mediated
sulfonylationin55%yield.Thesulfonyl chloridewascoupledwithpyrrolederivative4.6toformsulfonamide4.7in60%yield.
Finally, reductiveaminationofthealdehydewithmethylamine and sodium cyanoborohydride, followed by HCl salt formation,43furnishedkeverprazanhydrochloride(4)in18%yield.
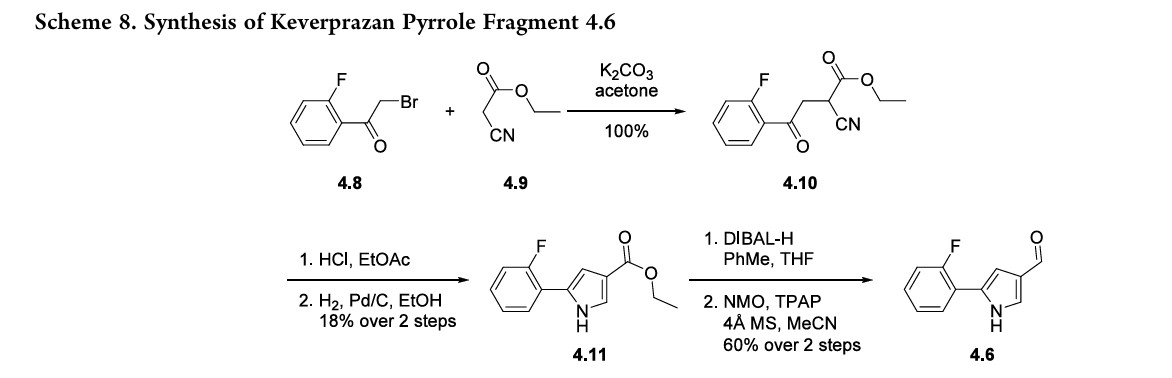
The synthesis of pyrrole fragment 4.6was reported byArikawaetal.andillustratedinScheme8.39SN2displacementofα-bromoketone 4.8 with ethyl cyanoacetate 4.9 affordedintermediate 4.10. Treatment of 4.10 with anhydrous HCl in EtOAc followed by hydrogenation effected cyclization to form pyrazole 4.11. The ester was reduced to the corresponding
alcohol with DIBAL-H,andthealcoholoxidized to aldehyde4.6 with NMO and TPAP.
(36) Kang, C.Keverprazan Hydrochloride: first approval. Drugs 2023,
83, 639−643.
(37) Chen, S.; Liu, D.; Chen, H.; Liao, A.; Li, F.; Liu, C.; Li, X.; Li, S.;
Zhang, Y.; Wang, Y.; et al. The efficacy and safety of keverprazan, a
novel potassium-competitive acid blocker, in treating erosive
oesophagitis: a phase III, randomised, double-blind multicentre
study. Aliment. Pharmacol. Ther. 2022, 55, 1524−1533.
(38)Tan,N.-d.; Liu, X.-w.; Liu, C.-x.; Li, S.-b.; Chen, H.-h.; Li, X.; Wu,
H.; Liao, A.-J.; Zhen, Y.-b.; Shen, P.-z.; et al. Efficacy of keverprazan for
duodenal ulcer: a phase II randomized, double-blind, parallel
controlled trial. J. Gastroenterol. Hepatol. 2022, 37, 2060−2066.
(39) Arikawa, Y.; Nishida, H.; Kurasawa, O.; Hasuoka, A.; Hirase, K.;
Inatomi, N.; Hori, Y.; Matsukawa, J.; Imanishi, A.; Kondo, M.; et al.
Discovery of a novel pyrrole derivative 1-[5-(2-fluorophenyl)-1
(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine fuma
rate (TAK-438) as a potassium-competitive acid blocker (P-CAB). J.
Med. Chem. 2012, 55, 4446−4456.
(40) Parsons, M. E.; Keeling, D. J. Novel approaches to the
pharmacological blockade of gastric acid secretion. Expert Opin. Invest.
Drugs 2005, 14, 411−421.
(41) Zhou,S.;Xie, L.; Zhou, C.; Zhao, Y.; Wang,L.;Ding,S.;Chen, J.;
Zhu, B.; Su, M.; Shao, F. Keverprazan, a novel potassium-competitive
acid blocker: single ascending dose safety, tolerability, pharmacoki
netics, pharmacodynamics and food effect in healthy subjects. Eur. J.
Pharm. Sci. 2023, 190, No. 106578.
(42) Qin, Y.; Su, M.; Jin, Q.; Chen, T.; Jiang, J. Pyrrole sulfonyl
derivative, and preparation method and medical use thereof. EP
3248963 B1, 2019.
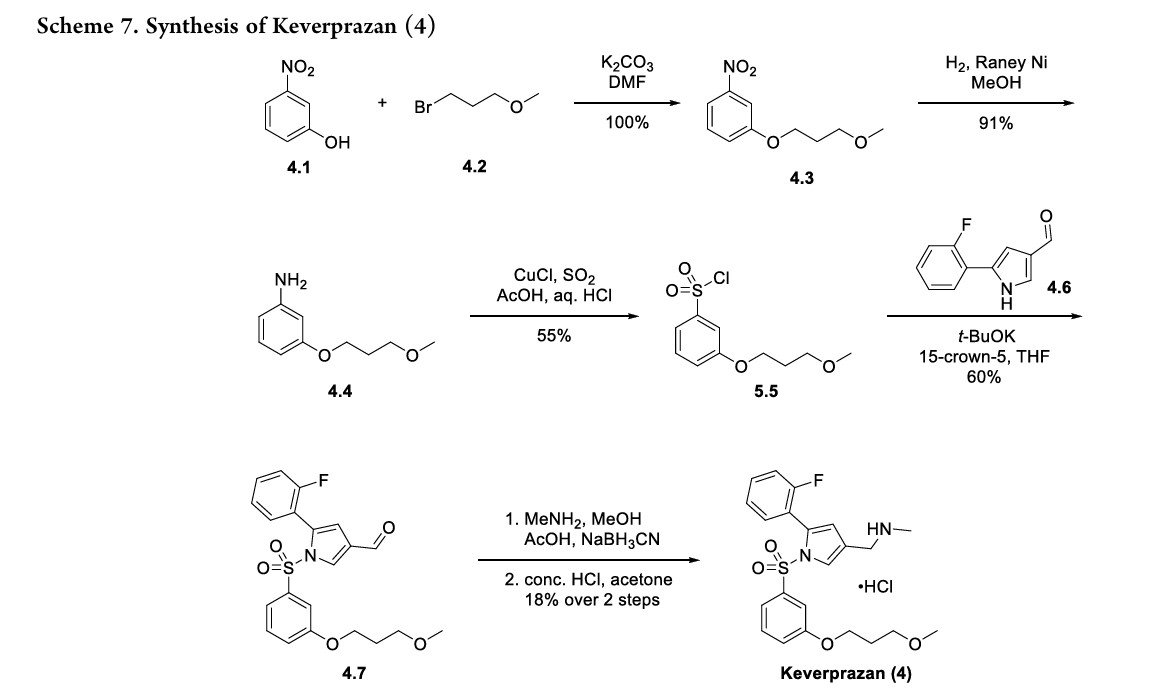
SYN
WO2016119505
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016119505&_cid=P12-MD5D4X-02931-1
[0042]Preparation of 1-(5-(2-fluorophenyl)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine (EXP 1)
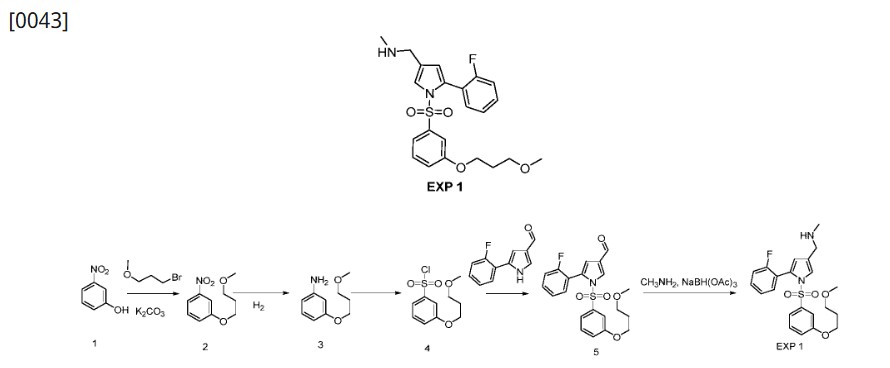
1) Preparation of 1-(3-methoxypropoxy)-nitrobenzene (Compound 2)
[0045]3-Nitrophenol (Compound 1, 1.0 g, 7.19 mmol), potassium carbonate (2.9 g, 21.6 mmol) and 1-bromo-3-methoxypropane (1.65 g, 10.79) were dissolved in anhydrous DMF (20 mL), stirred at 90 °C overnight, added with water (50 mL), and extracted with ethyl acetate (50 mL x 3). The organic phases were combined, dried, and concentrated to give a yellow solid 1-(3-methoxypropoxy)-nitrobenzene (Compound 2, 1.5 g, yield 100%).
[0046]2) Preparation of 1-(3-methoxypropoxy)-aniline (Compound 3)
[0047]1-(3-methoxypropoxy)-nitrobenzene (Compound 2, 1.0 g, 4.74 mmol) and Raney Ni (100 mg) were dissolved in anhydrous methanol (20 mL) and stirred overnight at room temperature under a hydrogen atmosphere. The mixture was filtered and the filtrate was dried to obtain solid 1-(3-methoxypropoxy)-aniline (Compound 3, 0.80 g, 91% yield).
[0048]3) Preparation of 1-(3-methoxypropoxy)-benzenesulfonyl chloride (Compound 4)
[0049]At 0°C, sodium nitrite (571 mg, 8.29 mmol) was added to acetic acid (10 mL) and aqueous hydrochloric acid solution (2N, 10 mL) of 1-(3-methoxypropoxy)-aniline (compound 3, 1.0 g, 5.52 mmol) in batches. After the addition was completed, the mixture was stirred at 0°C for 25 min to obtain solution I. At 0°C, cuprous chloride (190 mg, 1.1 mmol) was added to an acetic acid solution of sulfur dioxide (10 mL, 2N) to obtain solution II. At 0°C, solution I was added dropwise to solution II, and after the addition was completed, the mixture was naturally warmed to room temperature and stirred for 3 hours. The mixture was extracted with ethyl acetate (150 mL x 3), the organic phases were combined, dried, concentrated, and column chromatography (petroleum ether: ethyl acetate = 20:1) was performed to obtain 1-(3-methoxypropoxy)-benzenesulfonyl chloride (compound 4, 800 mg, yield 55%) as a yellow oil.
[0050]4) Preparation of 5-(2-fluorophenyl)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-carbaldehyde (Compound 5)
[0051]At 0°C, t-BuOK (233 mg, 2.08 mmol) was added to a solution of 5-(2-fluorophenyl)-1H-pyrrole-3-carboxaldehyde (200 mg, 1.04 mmol) in anhydrous THF (5 mL), and the mixture was stirred at 0°C for 30 min. After the reaction was completed, 15-crown-5 (542 mg, 2.08 mmol) and 1-(3-methoxypropoxy)-benzenesulfonyl chloride (compound 4, 412 mg, 2.08 mmol) were added respectively. After the addition was completed, the mixture was naturally heated to room temperature and stirred for 90 min. After the reaction was completed, the mixture was quenched with ice water (50 g) and extracted with ethyl acetate (50 mL x 3). The organic phases were combined, dried, concentrated, and purified by column chromatography (petroleum ether:ethyl acetate=8:1) to give a yellow solid 5-(2-fluorobenzene)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-carbaldehyde (260 mg, yield 60%).
[0052]5) Preparation of 1-(5-(2-fluorophenyl)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrol-3-yl)-N-methylmethanamine (EXP 1)
[0053]5-(2-Fluorobenzene)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-carbaldehyde (Compound 5, 500 mg, 1.19 mmol), acetic acid (144 mg, 2.39 mmol), and methylamine alcohol solution (1 mL) were dissolved in 3 mL of anhydrous methanol and stirred at room temperature for 4 h. NaBH
3 CN (212 mg, 3.59 mmol) was added. The mixture was stirred for 60 min. Ice water (30 g) was added to quench the reaction and the mixture was extracted with ethyl acetate (50 mL x 3). The organic phases were combined, dried, concentrated, and subjected to column chromatography to obtain 1-(5-(2-fluorobenzene)-1-((3-(3-methoxypropoxy)phenyl)sulfonyl)-1H-pyrrole-3-yl)-N-methylmethanamine (EXP 1, 100 mg, 20%) as a white solid.
[0054]
HPLC:99.4%;MS(ESI)m/z:[M+H] +=433.0; 1H-NMR(400MHz,DMSO-d6)δ:8.72(s,1H),7.78(d,1H),7.46-7.55(m,2H),7.21-7.32(m,3H),6.85-7.11(m,2H),6.83-6.85(m,1H),6.44(d,1H),3.95-4.02(m,4H),3.47(t,2H),3.32(s,3H),2.52(m,3H),1.94(t,2H)ppm。
[References]
[1] YinLin Qin, et al. Pyrrole sulfonyl derivative, and preparation method and medical use thereof. WO2016119505A1.
///////////KEVERPRAZAN, Keverprazan, CHINA 2023, APPROVAL 2023, Jiangsu Carephar Pharmaceuticals, ULCER
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Risevistinel
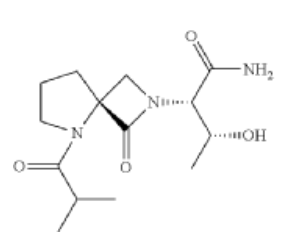
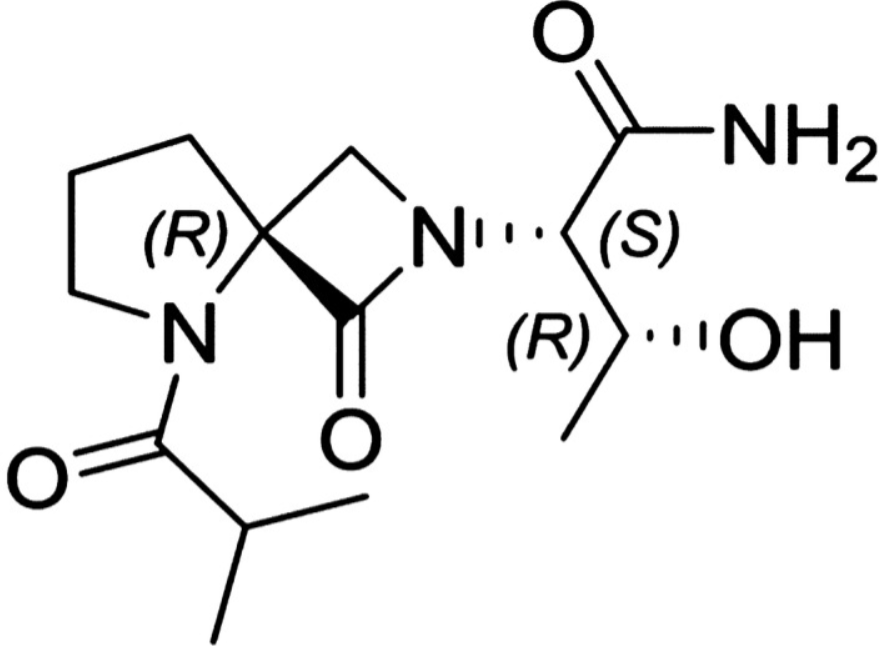
Risevistinel
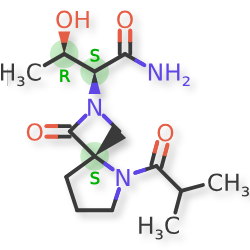
NYX-783 CAS 2591344-26-0, UNII-52TU5MZG22
NYX 2925, 2012536-16-0, X062KF5ZV3
C14H23N3O4, 297.35
UNII-52TU5MZG22,
X062KF5ZV3
(αS,4R)-α-[(1R)-1-Hydroxyethyl]-5-(2-methyl-1-oxopropyl)-1-oxo-2,5-diazaspiro[3.4]octane-2-acetamide
2,5-Diazaspiro[3.4]octane-2-acetamide, α-[(1R)-1-hydroxyethyl]-5-(2-methyl-1-oxopropyl)-1-oxo-, (αS,4S)-
(2S,3R)-3-hydroxy-2-[(4S)-5-(2-methylpropanoyl)-3-oxo-2,5-diazaspiro[3.4]octan-2-yl]butanamide
- NYX-783, NYX 2925
- CS-0113907
- HY-135741
- NYX-2925
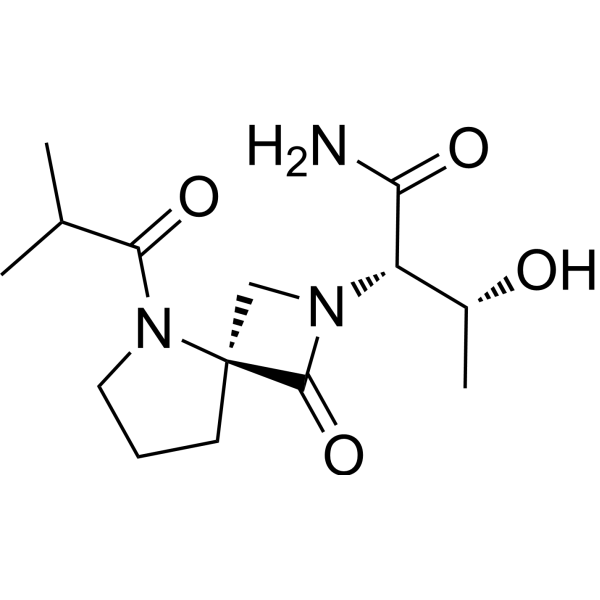
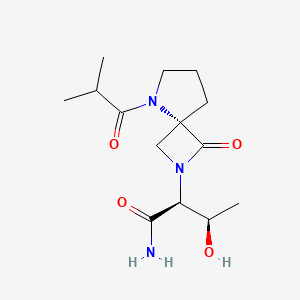
Risevistinel (NYX-783) is a positive allosteric modulator of N-methyl-D-aspartate (NMDA) receptor. Nevadistinel can be used to inhibit cognitive impairment associated with neurodegenerative diseases, such as mild cognitive impairment, mild Alzheimer’s disease, Parkinson’s disease, Lewy body disease.
NYX-2925 is an N-methyl-D-aspartate receptor (NMDAR) modulator, and at low concentrations of endogenous agonist (glycine or D-serine) and in the presence of glutamate, NYX-2925 partially activates the NMDAR. NYX-2925 appears to act at a binding site that is distinct from NMDAR agonists or antagonists studied to date, such as D-cycloserine, ketamine, MK-801, or kynurenic acid. The mode of action of NYX-2925 seems to be distinct from that of all existing and emerging drugs that are indicated for the treatment of neuropathic pain. While current medications target individual elements of pain signal transmission or modulation, NYX-2925 can modulate multiple synaptic relays within pain circuits.
NYX-2925 is under investigation in clinical trial NCT04146896 (Efficacy and Safety of NYX-2925 in Subjects With Neuropathic Pain Associated With Diabetic Peripheral Neuropathy (DPN)).
SCHEME
COUPLER
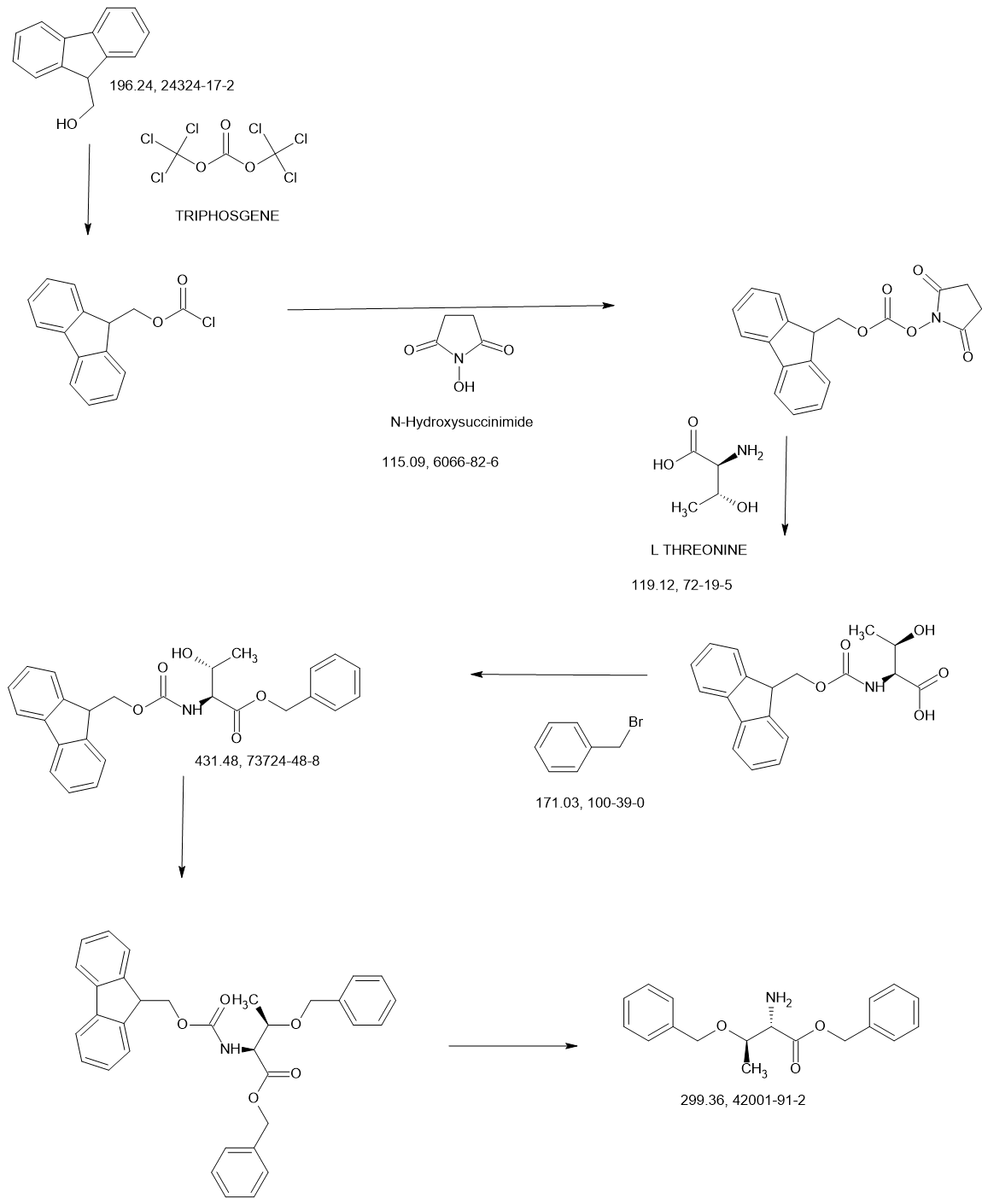
MAIN
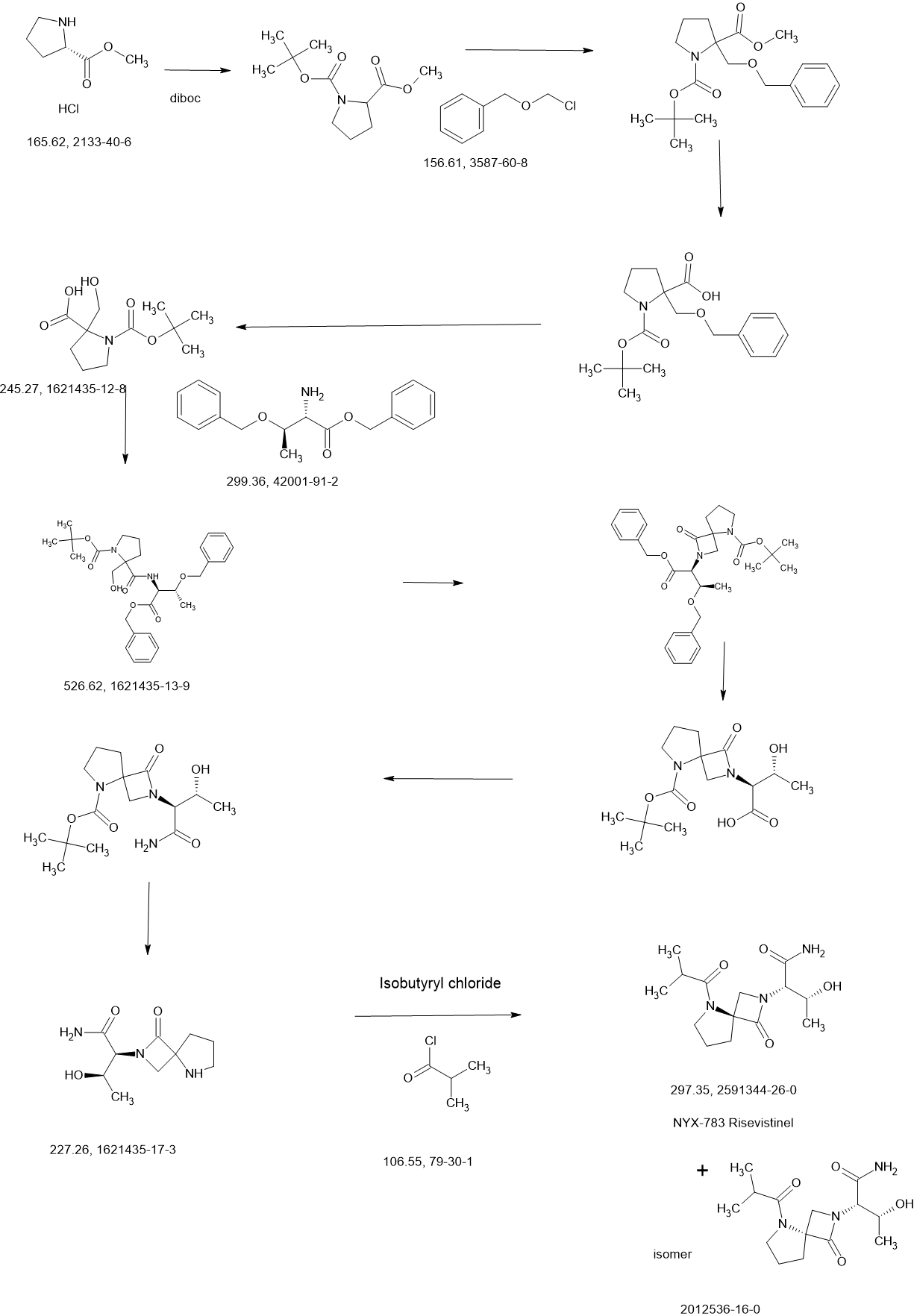
REF
US20210139489 Aptinyx Inc.
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750708&_cid=P11-MD3X0E-31059-1
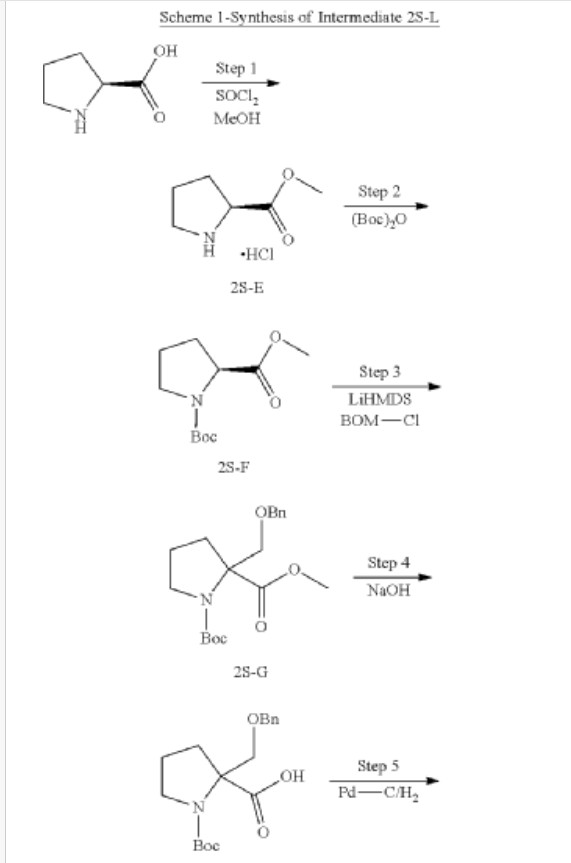
Synthesis of methyl pyrrolidine-2-carboxylate (2S-E)
| 1H-NMR: (500 MHz, DMSO-d 6): δ 4.15-4.13 (m, 1H), 3.65 (s, 3H), 3.35-3.30 (m, 2H), 2.23-2.15 (m, 1H), 1.86-1.78 (m, 3H), 1.41 (s, 9H); |
Synthesis of 1-tert-butyl 2-methyl pyrrolidine-1,2-dicarboxylate (2S-F)
| 1H-NMR: (400 MHz, DMSO-d 6): δ 4.15-4.13 (m, 1H), 3.65 (s, 3H), 3.35-3.30 (m, 2H), 2.23-2.15 (m, 1H), 1.86-1.78 (m, 3H), 1.41 (s, 9H); |
Synthesis of 1-tert-butyl 2-methyl 2-((benzyloxy) methyl) pyrrolidine-1,2-dicarboxylate (2S-G)
| 1H-NMR: (500 MHz, DMSO-d 6): δ 7.36-7.22 (m, 5H), 4.59-4.48 (m, 2H), 4.02-3.88 (m, 1H), 3.63 (s, 3H), 3.49-3.35 (m, 2H), 3.34-3.30 (m, 1H), 2.31-2.23 (m, 1H), 2.04-1.89 (m, 2H), 1.82-1.78 (m, 1H); |
Synthesis of 2-((benzyloxy) methyl)-1-(tert-butoxycarbonyl) pyrrolidine-2-carboxylic acid (2S-H)
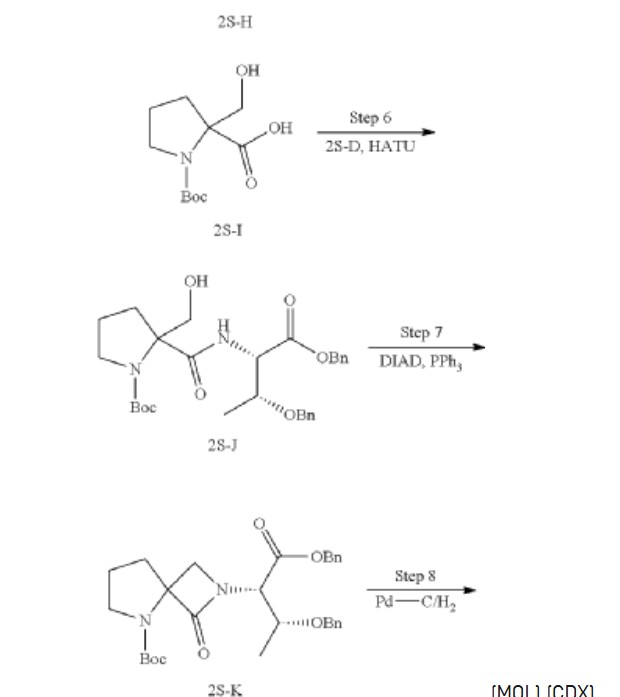
Synthesis of 1-(tert-butoxycarbonyl)-2-(hydroxymethyl) pyrrolidine-2-carboxylic acid (2S-I)
| 1H-NMR: (400 MHz, DMSO-d 6): δ 4.66 (br s, 1H), 3.96-3.83 (m, 1H), 3.63-3.59 (m, 1H), 3.49-3.41 (m, 1H), 3.34-3.25 (m, 1H), 2.30-2.17 (m, 1H), 1.95-1.72 (m, 3H), 1.38 (s, 9H). |
Synthesis of tert-butyl 2-(((2S,3R)-1,3-bis (benzyloxy)-1-oxobutan-2-yl) carbamoyl)-2-(hydroxymethyl) pyrrolidine-1-carboxylate (2S-J)
Synthesis of tert-butyl 2-((2S,3R)-1,3-bis (benzyloxy)-1-oxobutan-2-yl)-1-oxo-2,5-diazaspiro [3.4] octane-5-carboxylate (2S-K)
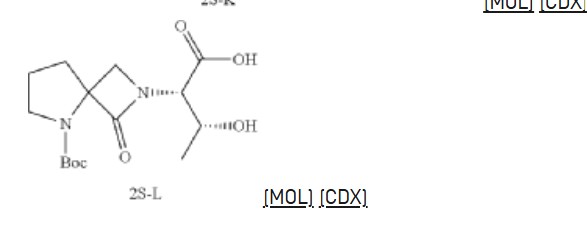
Synthesis of (2S,3R)-2-(5-(tert-butoxycarbonyl)-1-oxo-2,5-diazaspiro [3.4] octan-2-yl)-3-hydroxybutanoic acid (2S-L)
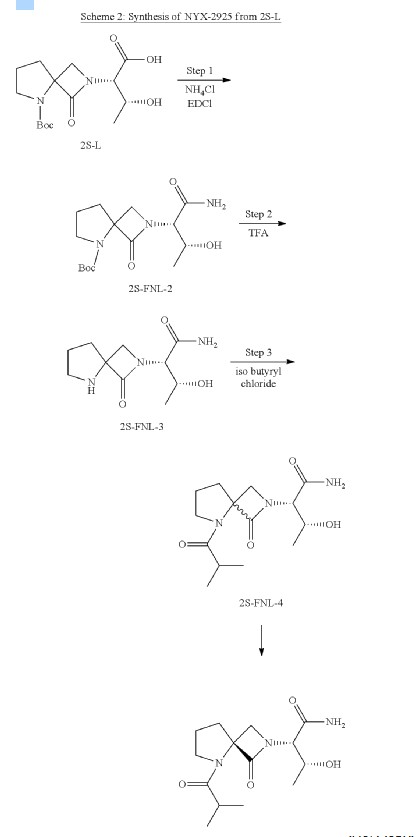
Synthesis of tert-butyl 2-((2S,3R)-1-amino-3-hydroxy-1-oxobutan-2-yl)-1-oxo-2,5-diazaspiro [3.4] octane-5-carboxylate (2S-FNL-2)
Synthesis of (2S,3R)-3-hydroxy-2-(1-oxo-2,5-diazaspiro [3.4] octan-2-yl) butanamide (2S-FNL-3)
| 1H-NMR: (400 MHz, D 2O): δ 4.33-4.29 (m, 2H), 4.09 (d, 1H), 3.95 (d, 1H), 3.57-3.48 (m, 2H), 2.51-2.46 (m, 2H), 2.25-2.19 (m, 2H), 1.31 (d, 3H); |
Synthesis of (2S,3R)-3-hydroxy-2-(5-isobutyryl-1-oxo-2,5-diazaspiro [3.4] octan-2-yl) butanamide (NYX-2925)
PATENT
WO2022086858
WO2021021996
//////Risevistinel, NYX 783, UNII-52TU5MZG22, Aptinyx Inc, NYX 2925, CS 0113907, HY 135741, NYX-2925
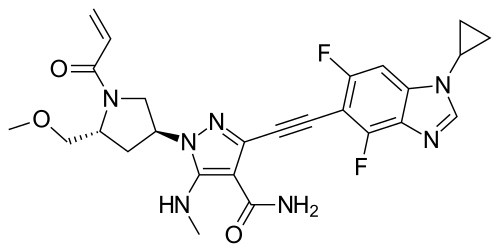
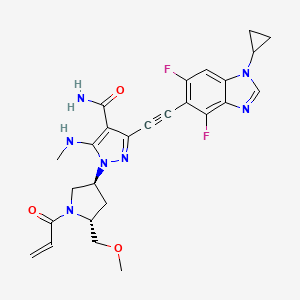
Rezatapopt
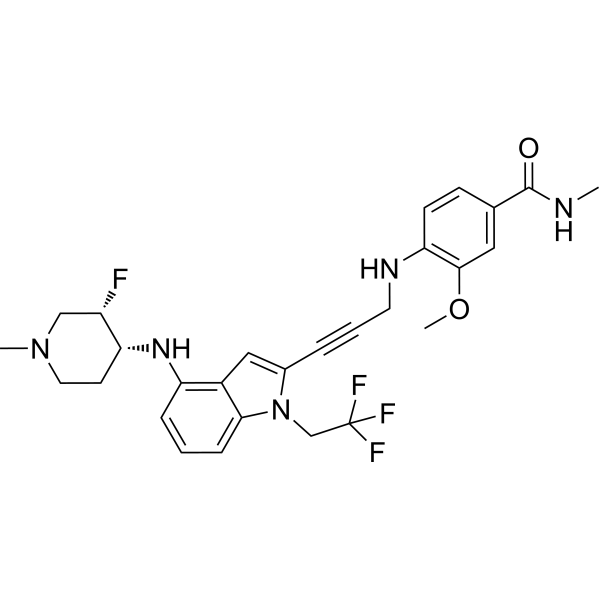
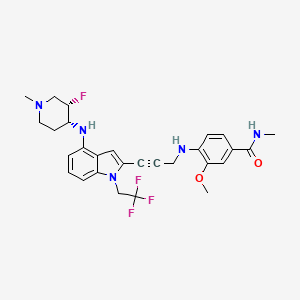
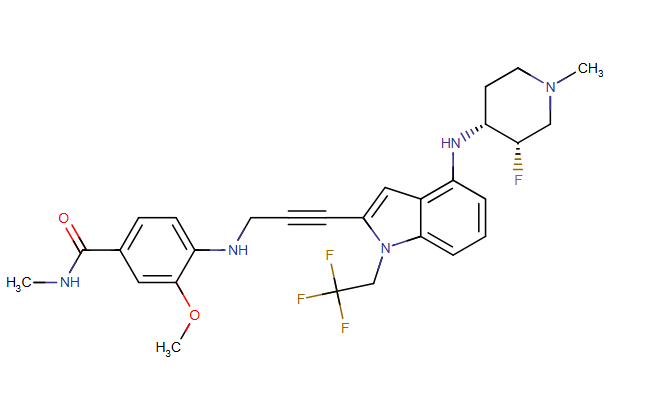
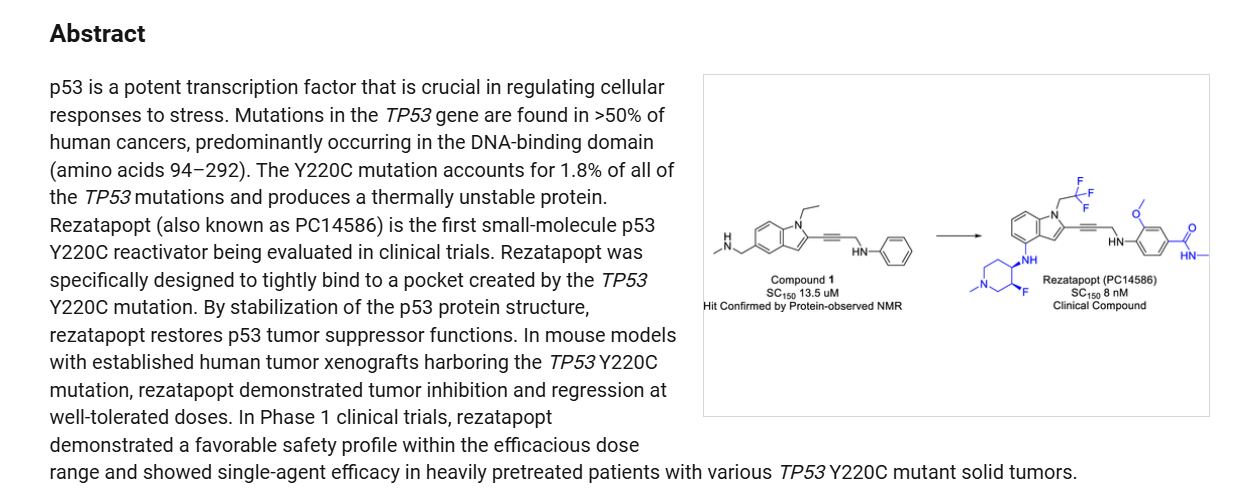
Rezatapopt, PC 14586
CAS 2636846-41-6
| Molecular Weight | 545.57 |
|---|---|
| Synonyms | PC14586 |
| Formula | C28H31F4N5O2 |
| CAS No. | 2636846-41-6 |
4-[3-[4-[[(3S,4R)-3-fluoro-1-methylpiperidin-4-yl]amino]-1-(2,2,2-trifluoroethyl)indol-2-yl]prop-2-ynylamino]-3-methoxy-N-methylbenzamide
- 4-[3-[4-[[(3S,4R)-3-fluoro-1-methylpiperidin-4-yl]amino]-1-(2,2,2-trifluoroethyl)indol-2-yl]prop-2-ynylamino]-3-methoxy-N-methylbenzamide
- Benzamide, 4-[[3-[4-[[(3S,4R)-3-fluoro-1-methyl-4-piperidinyl]amino]-1-(2,2,2-trifluoroethyl)-1H-indol-2-yl]-2-propyn-1-yl]amino]-3-methoxy-N-methyl-
Rezatapopt (PC14586) is an orally active antineoplastic agent. Rezatapopt binds to a pocket created by the TP53 Y220C mutation. Rezatapopt restores p53 tumor suppressor functions by stabilization of the p53 protein structure. Rezatapopt demonstrates tumor inhibition and regression in mouse models with established human tumor xenografts harboring the TP53 Y220C mutation.
SCHEME
COUPLER
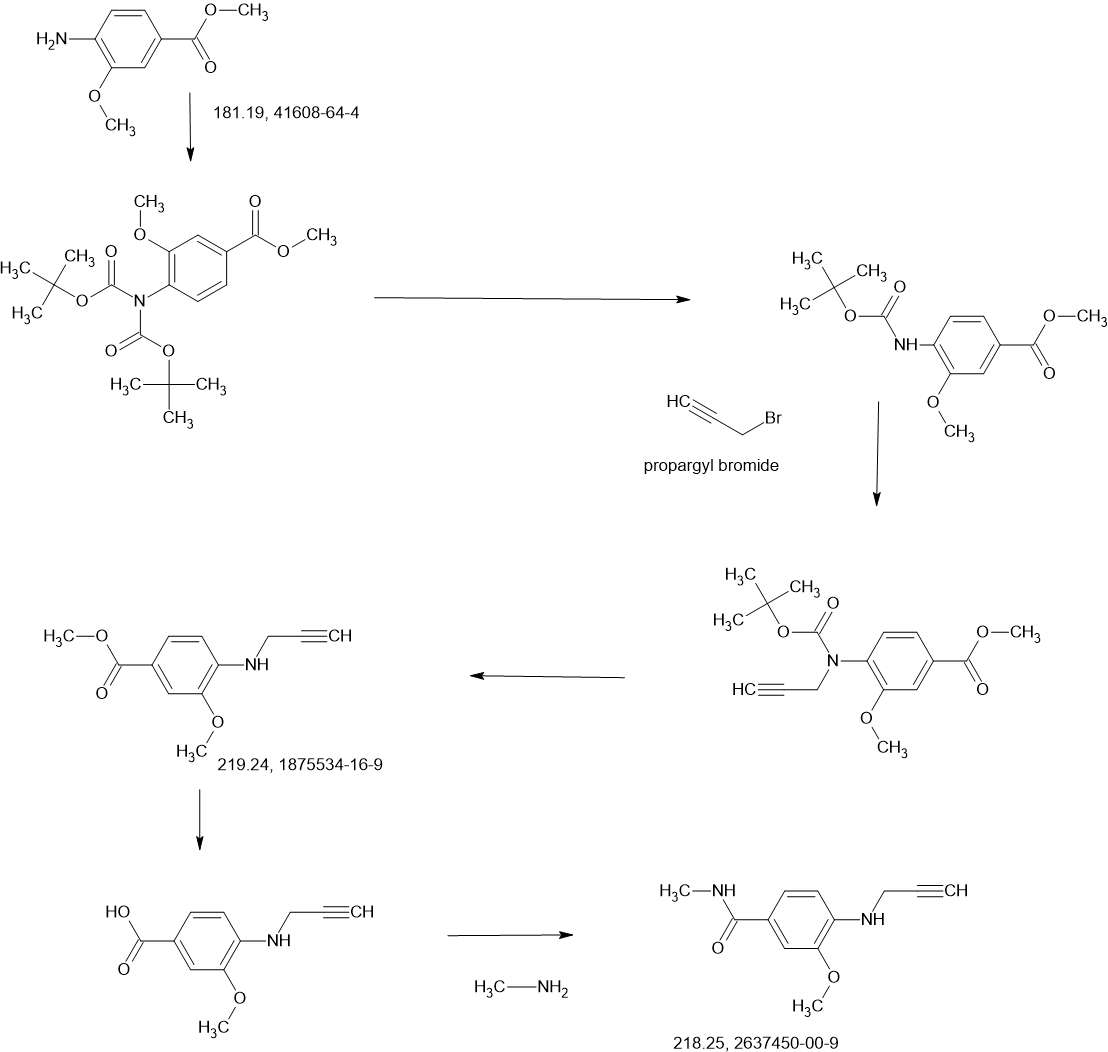
COUPLER
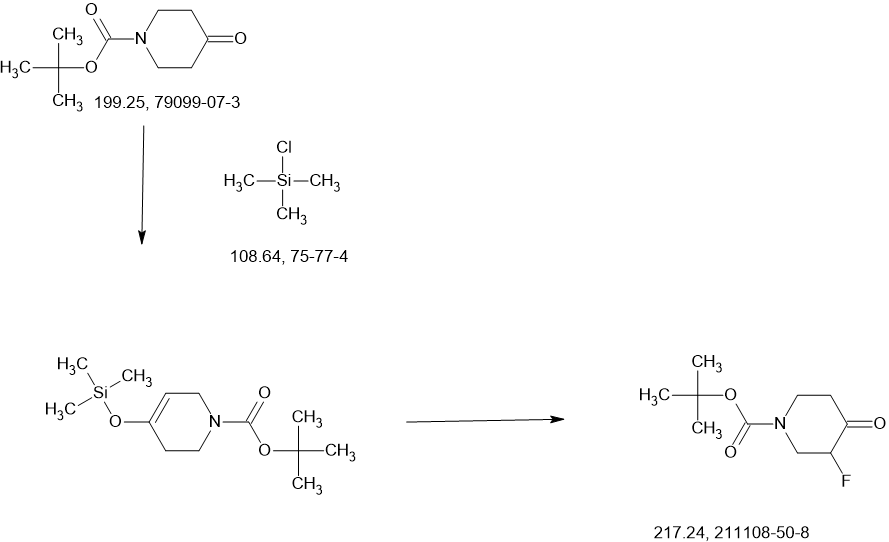
MAIN
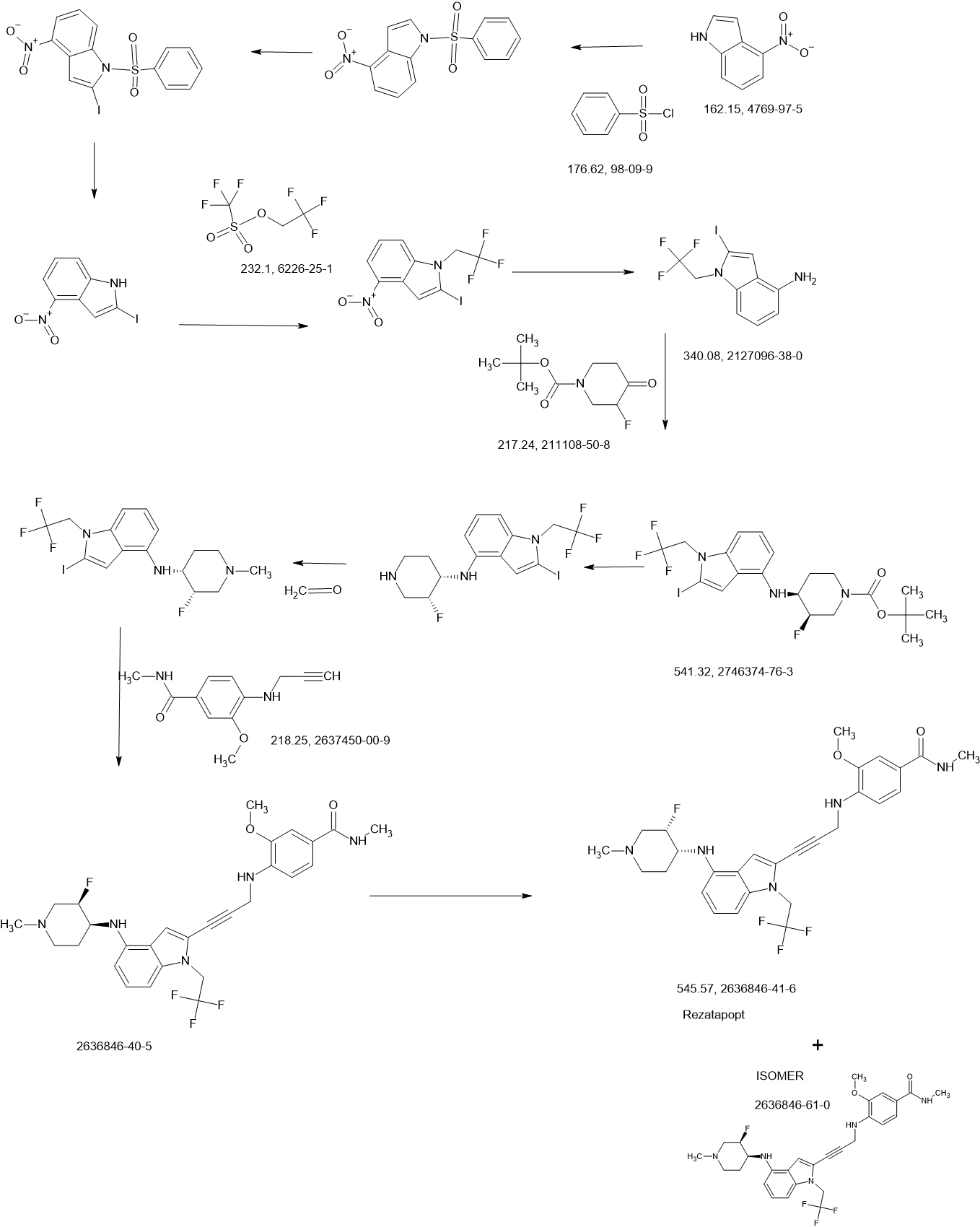
REF
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00379
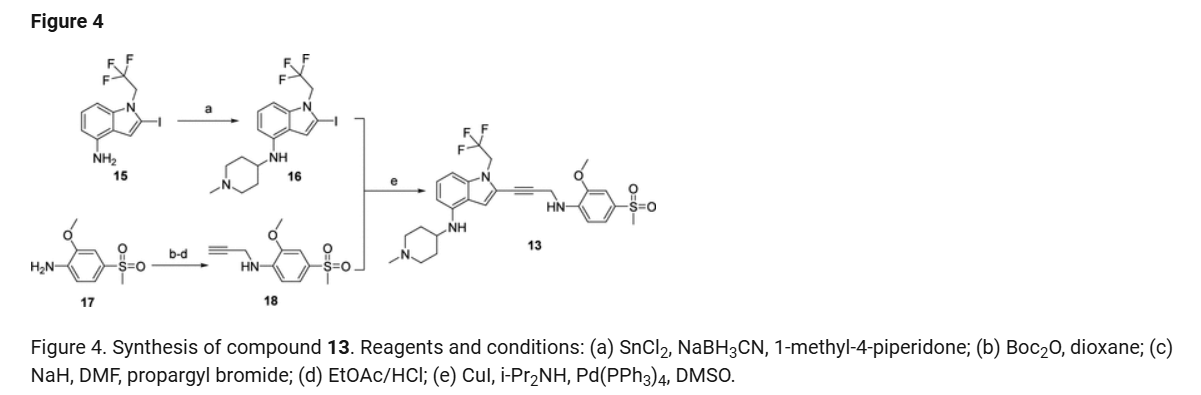
2-Iodo-1-(2,2,2-trifluoroethyl)-1H-indol-4-amine 15 was prepared from 4-nitroindole as described in
WO2017143291. 1
H NMR (400 MHz, dimethylsulfoxide [DMSO]-d6) δ ppm 9.19–10.88 (m, 2 H), 7.63
(d, J=8.34 Hz, 1 H), 7.16–7.25 (m, 1 H), 7.04–7.14 (m, 2 H), 5.14–5.33 (m, 2 H). LCMS (ES+
, m/z):
340.9 [(M+H)+
].
SnCl2.2H2O (398.11 mg, 1.76 mmol, 0.20 eq.) was added to a solution of 2-iodo-1-(2,2,2-trifluoroethyl)-
1H-indol-4-amine 15 (3.00 g, 8.82 mmol, 1.00 eq.) and 1-methylpiperidin-4-one (1.20 g, 10.61 mmol,
1.20 eq.) in MeOH (10.00 mL). The mixture was stirred at 25 °C for 3 hours (h), and then NaBH3CN
(2.77 g, 44.1 mmol, 5.00 eq.) was added, stirring at 25 °C for 69 h. Thin-layer chromatography (TLC)
indicated that the starting material was consumed, and the reaction mixture was filtered. The filtrate was
poured into H2O (200 mL) and extracted with ethyl acetate ([EtOAc] 200 mL2). The combined organic layers were washed with H2O (200 mL), dried over Na2SO4, and concentrated under reduced pressure to give a residue. The crude material was purified by flash column chromatography (Silica gel, petroleum ether (PE) : EtOAc = 0:1) and then by preparative high performance chromatography ([prep-HPLC] column: Phenomenex Luna C18 10040mm5 um; mobile phase: [H2O (0.2% Formic acid-acetonitrile [ACN])]; gradient: 10%–50% acetonitrile over 8.0 minutes) to yield 2-iodo-N-(1-methylpiperidin-4-yl)-1- (2,2,2-trifluoroethyl)-1H-indol-4-amine 16 (2.50 g, 5.72 mmol, 64.93% yield) as a light-brown solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 8.24 (br s, 1 H, formic acid salt), 7.17 (s, 1 H), 6.85–6.95 (m, 1 H), 6.78 (br d, J = 7.99 Hz, 1 H), 6.16 (br d, J = 7.63 Hz, 1 H), 5.44 (br d, J = 2.62 Hz, 1 H), 4.99 (q, J = 8.54 Hz, 2 H), 3.33 (br s, 1 H), 2.85 (br d, J = 9.66 Hz, 2 H), 2.25 (br s, 3 H), 2.07–2.18 (m, 2 H), 1.94 (br d, J = 12.04 Hz, 2 H), 1.46–1.58 (m, 2 H). LCMS (ES+, m/z): 438.0 [(M+H)+]. Boc2O (26.03 g, 119.26 mmol, 6.00 eq.) was added to a solution of 2-methoxy-4-(methylsulfonyl)aniline 17 (4.00 g, 19.88 mmol, 1.00 eq.) in dioxane (40.00 mL) at 25 o C (room temperature). The reaction mixture was stirred at 110 °C for 16 h. TLC and LCMS indicated that the reaction was completed, and it was concentrated in vacuo. The residue was purified by column chromatography (SiO2, PE/EtOAc = 10/1 to 1:1) to yield tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)carbamate (6.00 g, 19.92 mmol, 72.6% purity, 72% yield) as a yellow gum. 1 H NMR (400 MHz, DMSO-d6) δ ppm 8.33 (s, 1 H), 8.03 (d, J = 8.38 Hz, 1 H), 7.47 (dd, J = 8.38, 2.00 Hz, 1 H), 7.44 (d, J = 2.00 Hz, 1 H), 3.91 (s, 3 H), 3.18 (s, 3 H), 1.47 (s, 9 H). LCMS (ES+, m/z): 324.1 [(M+Na)+ ]. NaH (867.27 mg, 60% purity, 21.69 mmol, 3.00 eq.) was added in portions at 0 °C to a mixture of tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)carbamate (3.00 g, 7.23 mmol, 1.00 eq.) in dimethylformamide ([DMF] 30.00 mL) and stirred at 0 °C for 0.5 h. 3-Bromoprop-1-yne (3.23 g, 21.69 mmol, 3.00 eq.) was added to the reaction mixture, stirring at 0 °C for 2.5 h. TLC (Plate 1: PE : EtOAc = 1:1) and LCMS indicated that the starting material was consumed, and the product was detected. The reaction mixture was poured into a saturated solution of NH4Cl (200 mL) at 0 o C and was extracted with EtOAc (200 mL3). The combined organic phase was dried over Na2SO4, filtered, and concentrated
in vacuo. The residue was purified by column chromatography (SiO2, PE : EtOAc = 5:1 to 1:2) to give
tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)(prop-2-yn-1-yl)carbamate (3.00 g, 8.85 mmol,
74% purity, 90% yield) as a light-yellow gum.
1
H NMR (400 MHz, DMSO-d6) δ ppm 7.53–7.56 (m, 1 H), 7.46–7.53 (m, 2 H), 4.10–4.51 (m, 2 H), 3.90
(s, 3 H), 3.27 (s, 3 H), 3.17 (t, J = 2.32 Hz, 1 H), 1.27–1.39 (m, 9 H). LCMS (ES+
, m/z): 283.9 [(M+H-tBu)+].
A solution of 4M HCl/EtOAc (20.00 mL) was added to the solution of tert-butyl (2-methoxy-4-
(methylsulfonyl)phenyl)(prop-2-yn-1-yl)carbamate (3.00 g, 6.54 mmol, 1.00 eq.) in EtOAc (1.00 mL).
The reaction mixture was stirred at 25 °C for 2 h. TLC indicated that the starting material was consumed
completely. The reaction mixture was concentrated in vacuo to yield 2-methoxy-4-(methylsulfonyl)-N-
(prop-2-yn-1-yl)aniline 18 (1.80 g, 7.53 mmol, 85.3% yield, HCl salt) as a yellow solid.
1
H NMR (400 MHz, DMSO-d6) δ ppm 7.38 (dd, J = 8.40, 1.60 Hz, 1 H), 7.22 (d, J = 1.60 Hz, 1 H), 6.75
(d, J = 8.80 Hz, 1 H), 3.99 (d, J = 2.4 Hz, 2 H), 3.87 (s, 3 H) 3.10 (s, 3 H), 3.08 (t, J = 2.31 Hz, 1 H).
LCMS (ES+
, m/z): 240.1 [(M+H)+
].
i-Pr2NH (2.08 g, 20.58 mmol, 2.91 mL, 10 eq.), CuI (392.02 mg, 2.06 mmol, 1 eq), 2-iodo-N-(1-
methylpiperidin-4-yl)-1-(2,2,2-trifluoroethyl)-1H-indol-4-amine 16 (0.9 g, 2.06 mmol, 1 eq.) and
Pd(PPh3)4 (475.71 mg, 411.67 μmol, 0.2 eq.) was added to a solution of 2-methoxy-4-(methylsulfonyl)-N-
(prop-2-yn-1-yl)aniline 18 (622.16 mg, 2.47 mmol, 1.2 eq.) in DMSO (10 mL) at 45 °C under N2. The
reaction mixture was stirred at 45 °C for 1 h. TLC (DCM/MeOH=10:1, Rf = 0.3) indicated that the
starting material was consumed completely. It was poured into ethylenediaminetetraacetic acid ([EDTA]
20 mL) and stirred for 1 h, then extracted with EtOAc (40 mL3). The combined organic phase was washed with brine (40 mL), dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, PE : EtOAc = 1:1 to dichloromethane (DCM) / MeOH = 10:1, Rf = 0.3), then by prep-HPLC (column: Phenomenex Luna(2) C18 25050 10u; mobile
phase: [water (0.1% trifluoroacetic acid)-ACN]; B%: 30%–50%, 20 min) to yield compound 13 (0.6 g,
1.09 mmol, 53.08% yield, 99.9% purity) as a light-yellow solid.
1 H NMR (400 MHz, DMSO-d6) δ ppm 1.41–1.54 (m, 2 H), 1.91 (br d, J = 11.00 Hz, 2 H), 1.95–2.08 (m,
2 H) 2.17 (s, 3 H), 2.68–2.80 (m, 2 H), 3.10 (s, 3 H), 3.20–3.29 (m, 1 H), 3.89 (s, 3 H), 4.36 (d,
J = 6.24 Hz, 2 H), 4.92 (q, J = 9.09 Hz, 2 H), 5.49 (d, J = 7.95 Hz, 1 H), 6.15 (d, J = 7.83 Hz, 1 H),
6.50 (t, J = 6.24 Hz, 1 H), 6.68 (d, J = 8.19 Hz, 1 H), 6.89 (d, J = 8.44 Hz, 1 H), 6.99 (t, J = 8.01 Hz,
1 H), 7.09 (s, 1 H), 7.25 (d, J = 1.83 Hz, 1 H), 7.39 (dd, J = 8.31, 1.83 Hz, 1 H). LCMS (ES+, m/z):
549.3 [(M+H)+
]
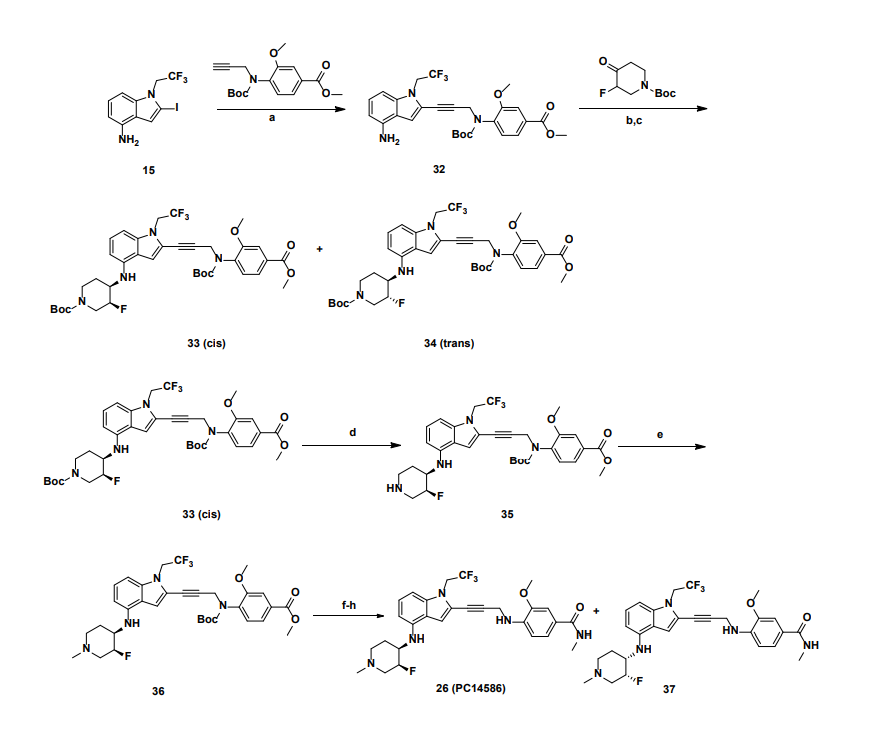
a
Reagents and conditions: (a) Pd(PPh3)4, CuI, diisopropylamine, DMSO, 20 °C, 1 h; (b) TMSCl, DMF, 0 °C, 0.5 h;
(c) BH3.THF, 0 °C, 0.5 h; (d) EtOAc/HCl, 20 °C, 1 h; (e) 10 eq. (CH2O)n, NaBH3CN, MeOH, 20 °C, 16 h; f)
LiOH.H2O, MeOH, 40 °C, 12 h; g) MeNH3Cl, HOBT, EDCI, TEA, DCM, RT, 16 h; h) Chiral SFC separation
PATENTS
WO2023016434 36%
WO2021061643
US20230024905
WO2023016434 Jacobio Pharmaceuticals Co., Ltd.
WO2023225477 PMV Pharmaceuticals, Inc.
US20230024905 PMV Pharmaceuticals, Inc.
WO2021061643 PMV Pharmaceuticals, Inc.
WO2021262483, PMV Pharmaceuticals, Inc.
WO2023196993 PMV Pharmaceuticals, Inc.
WO2021262484 WO2021262541
- [1]. Li Sujing, et al. Heteroarylalkyne compounds for targeting mutant of p53 and their preparation. World Intellectual Property Organization, WO2023016434 A1. 2023-02-16.[2]. Vu BT, et al. Discovery of Rezatapopt (PC14586), a First-in-Class, Small-Molecule Reactivator of p53 Y220C Mutant in Development. ACS Med Chem Lett. 2024 Nov 4;16(1):34-39. [Content Brief][3]. Spiegelberg D, et al. Targeting mutant p53: Evaluation of novel anti-p53R175H monoclonal antibodies as diagnostic tools. Sci Rep. 2025 Jan 6;15(1):1000. [Content Brief][4]. Schram A M, et al. 691TiP PYNNACLE phase II trial of rezatapopt (PC14586) in solid tumors with a TP53 Y220C mutation[J]. Annals of oncology, 2024, 35: S535-S536.
//////////Rezatapopt, PC 14586, 5W59S33KC9
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Resigratinib
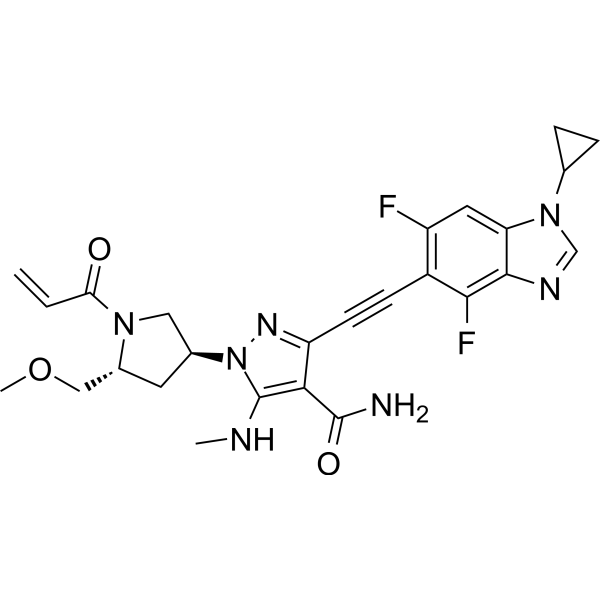
Resigratinib, KIN 3248
CAS 2750709-91-0
C26H27F2N7O3
523.5 g/mol
3-[2-(1-cyclopropyl-4,6-difluorobenzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-prop-2-enoylpyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
- 3-[2-(1-Cyclopropyl-4,6-difluoro-1H-benzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(1-oxo-2-propen-1-yl)-3-pyrrolidinyl]-5-(methylamino)-1H-pyrazole-4-carboxamide
- 3-[2-(1-cyclopropyl-4,6-difluorobenzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-prop-2-enoylpyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
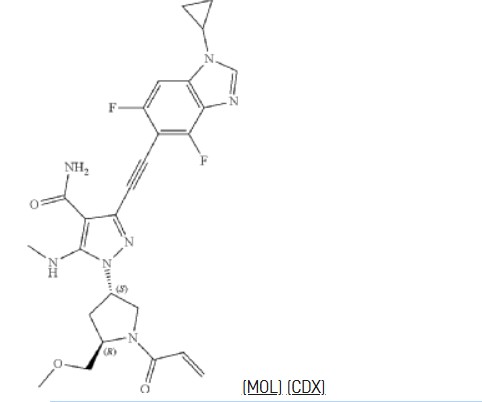
Resigratinib (KIN-3248) is an experimental anticancer medication which acts as a fibroblast growth factor receptor inhibitor (FGFRi) and is in early stage human clinical trials.[1][2][3]
KIN-3248 is a small molecule that targets and inhibits oncogenic fibroblast growth factor receptors (FGFRs). It was designed to mainly target FGFR2 and FGFR3 alterations, which act as oncogenic drivers in 10-20% of cholangiocarcinoma and 20-35% of urothelial cancers, respectively. While effective, disease progression may occur 6 to 8 months after treatment with currently approved FGFR inhibitors is started, and this effect is usually associated with on-target resistance mutations in the kinase domain of FGFR. Therefore, the broad inhibition of FGFR isoforms may be effective against different types of tumors. The safety, tolerability, pharmacokinetics, and preliminary efficacy of KIN-3248 are currently being evaluated in adults with advanced tumors harboring FGFR2 and/or FGFR3 gene alterations. In February 2023, Kinnate Biopharma received Fast Track designation from the FDA for KIN-3248 to treat unresectable, locally advanced or metastatic cholangiocarcinoma (CCA).
SCHEME
COUPLER
COUPLER
MAIN
CONTINUED………….
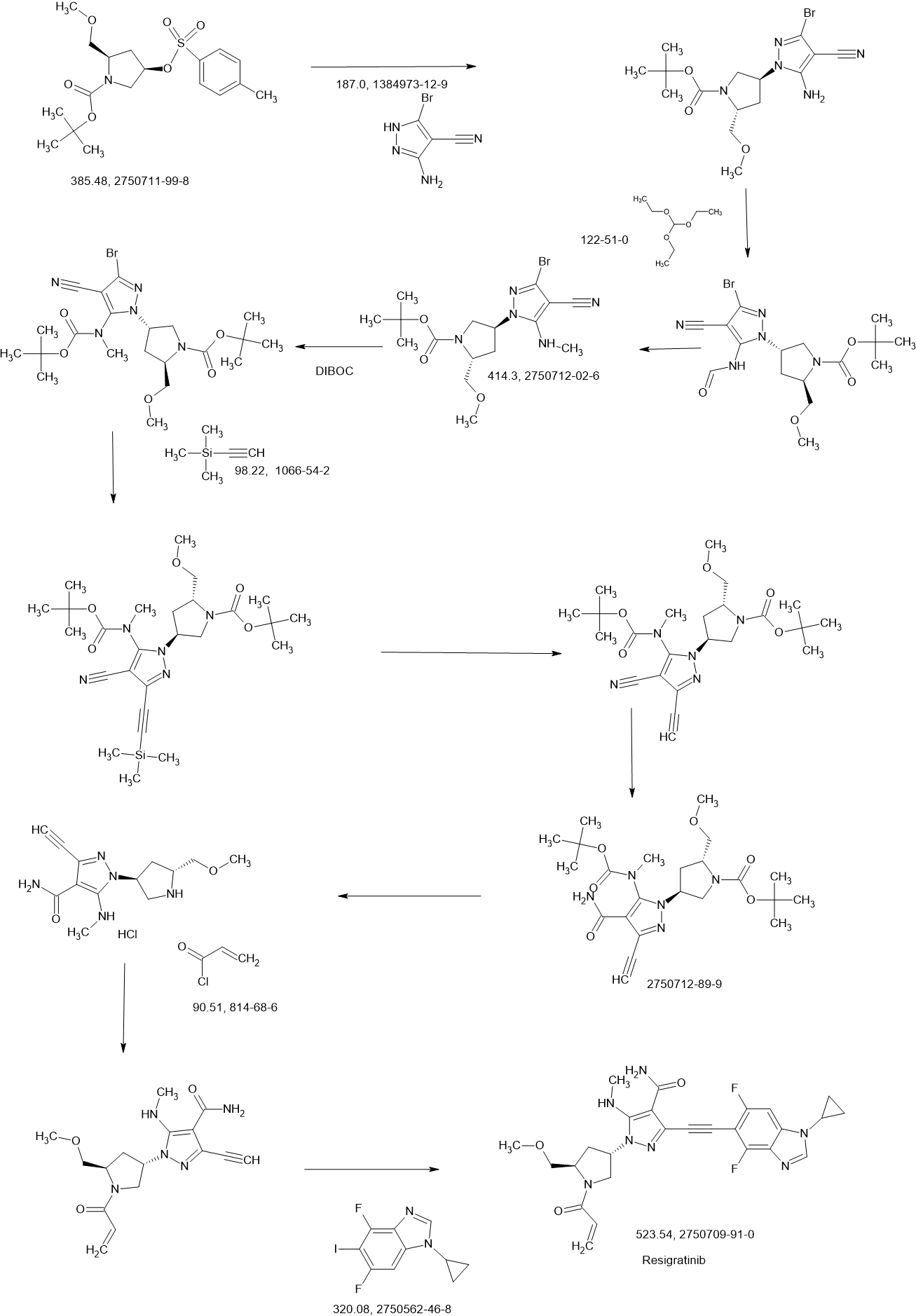
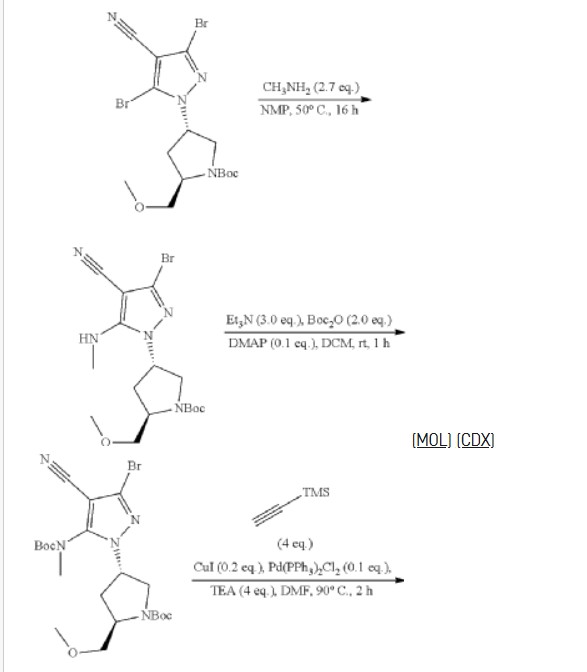
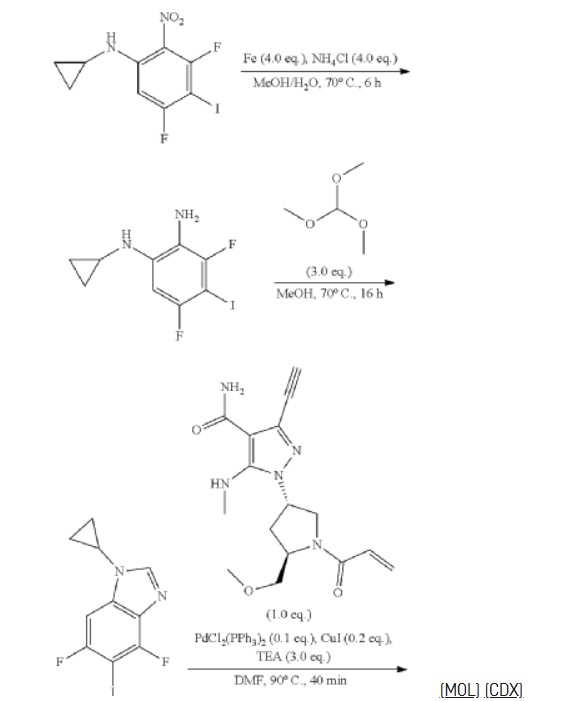
REF
https://patents.google.com/patent/US11345681B1/en
Example 78
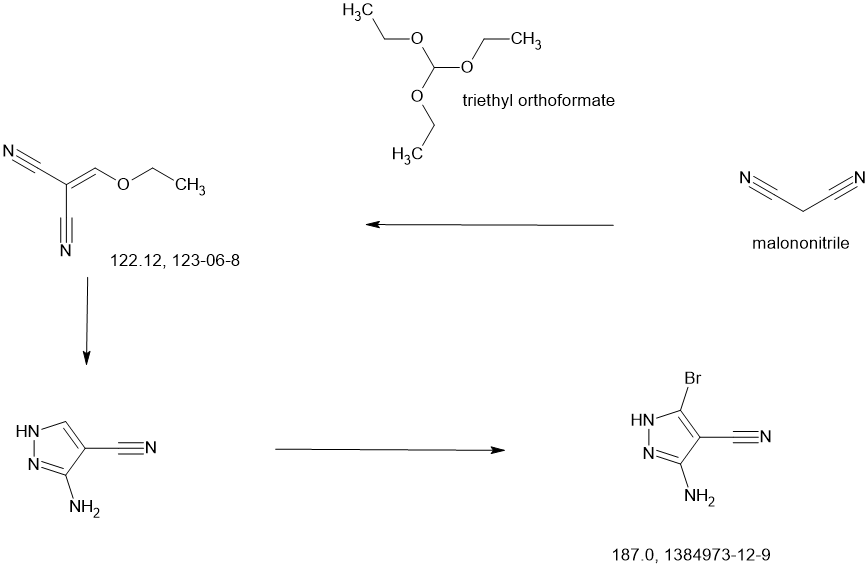
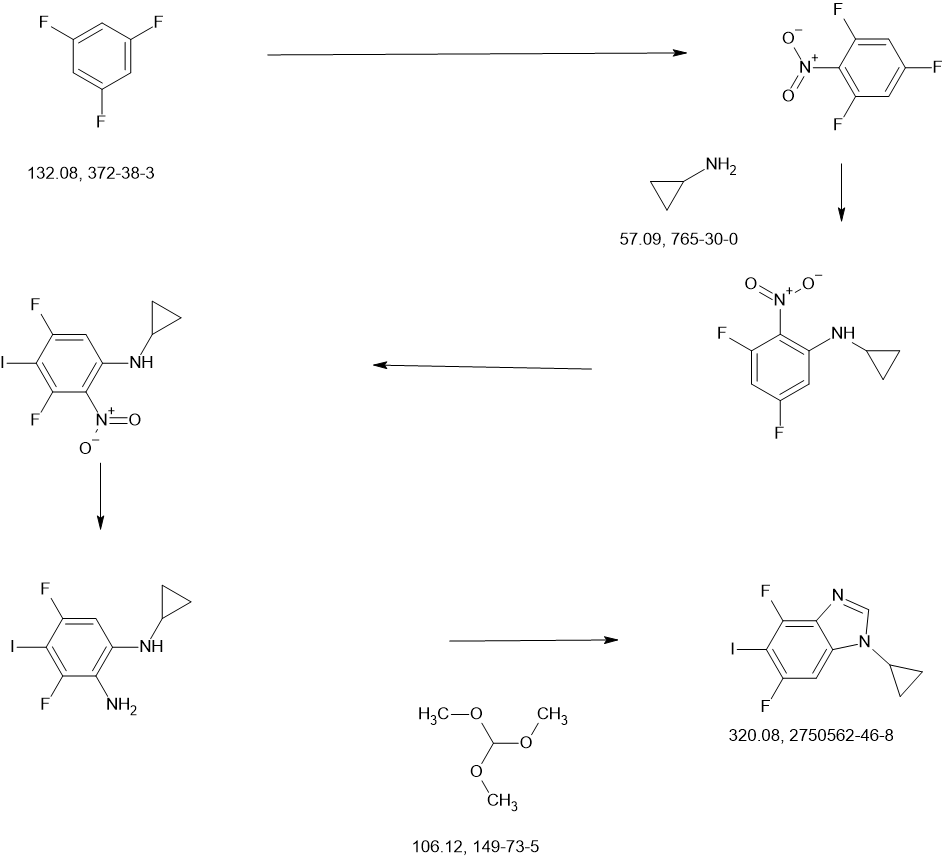
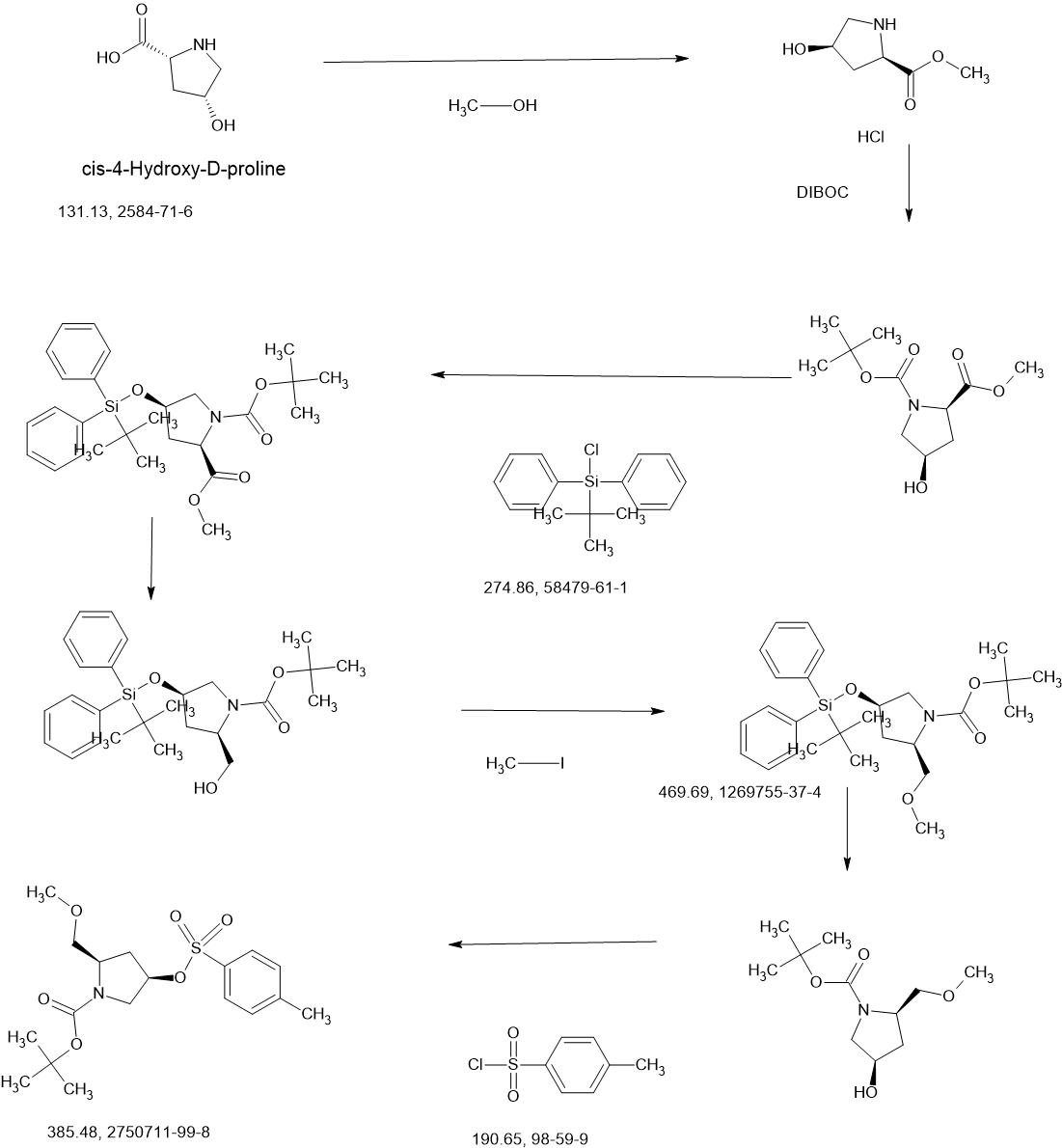
3-[2-(1-Cyclopropyl-4,6-difluoro-1,3-benzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
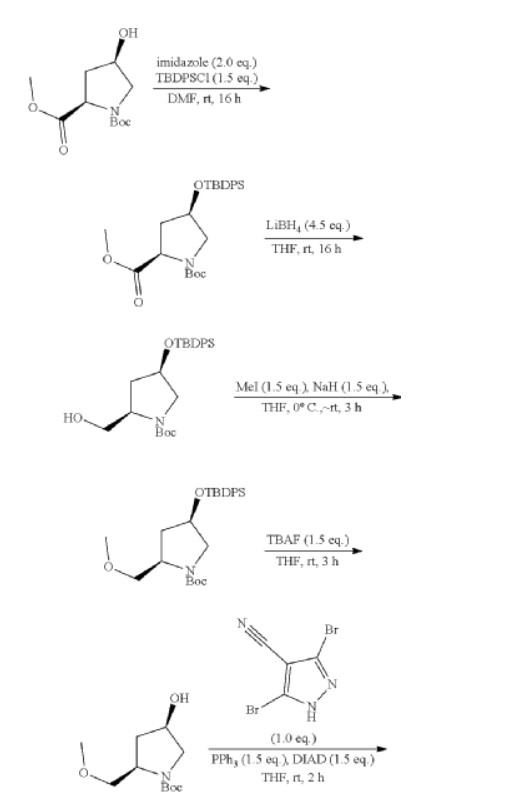


| Step 1: 1-(Tert-butyl) 2-methyl (2R,4R)-4-((tert-butyldiphenylsilyl)oxy)pyrrolidine-1,2-dicarboxylate |
| Step 2: Tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(hydroxymethyl)pyrrolidine-1-carboxylate |
| Step 3: Tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 5: Tert-butyl (2R)-4-(3,5-dibromo-4-cyanopyrazol-1-yl)-2-methoxymethyl)pyrrolidine-1-carboxylate |
| Step 6: Tert-butyl (2S,4R)-4-[3-bromo-4-cyano-5-(methylamino)pyrazol-1-yl]-2-(methoxymethyl)pyrrollidine-1-carboxylate |
| Step 7: (2R,4S)-4-[3-bromo-5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyanopyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 8: Tert-butyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-[2-(trimethylsilyl)ethynyl]pyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 9: Tert-butyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 10: Tert-butyl (2R,4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-carbamoyl-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 11: 3-Ethynyl-1-[(3S,5R)-5-(methoxymethyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide dihydrochloride |
| Step 12: 3-Ethynyl-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide |
| Step 17: 3-[2-(1-Cyclopropyl-4,6-difluoro-1,3-benzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide |
PATENT
WO2021247969 Kinnate Biopharma Inc EG78
WO2023107980 solid state forms, Kinnate Biopharma Inc
WO2023107979 FGFR kinase inhibitor, Kinnate Biopharma Inc
References
- Franovic A, Mohan A, Uryu S, Wu Q, Jiang P, Miller N, et al. (February 2022). “Activity of KIN-3248, a next-generation pan-FGFR inhibitor, against acquired FGFR-gatekeeper and molecular-brake drug resistance mutations”. Journal of Clinical Oncology. 40 (4_suppl): 461. doi:10.1200/JCO.2022.40.4_suppl.461.
- Harding JJ, Perez CA, Kato S, Sharma M, Garmezy B, Quah CS, et al. (February 2023). “First in human (FIH) phase 1/1b study evaluating KIN-3248, a next-generation, irreversible pan-FGFR inhibitor (FGFRi), in patients (pts) with advanced cholangiocarcinoma (CCA) and other solid tumors harboring FGFR2 and/or FGFR3 gene alterations”. Journal of Clinical Oncology. 41 (4_suppl): TPS637-TPS637. doi:10.1200/JCO.2023.41.4_suppl.TPS637. S2CID 256257314.
- Wang Z, Anderson KS (2022). “Therapeutic Targeting of FGFR Signaling in Head and Neck Cancer”. Cancer Journal (Sudbury, Mass.). 28 (5): 354–362. doi:10.1097/PPO.0000000000000615. PMC 9523489. PMID 36165723.
| Identifiers | |
|---|---|
| CAS Number | 2750709-91-0 |
| PubChem CID | 162381323 |
| UNII | W728TB393W |
| Chemical and physical data | |
| Formula | C26H27F2N7O3 |
| Molar mass | 523.545 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- [1]. Tyhonas JS, et al. Discovery of KIN-3248, An Irreversible, Next Generation FGFR Inhibitor for the Treatment of Advanced Tumors Harboring FGFR2 and/or FGFR3 Gene Alterations. J Med Chem. 2024 Feb 8;67(3):1734-1746. [Content Brief][2]. Balasooriya ER, et al. The Irreversible FGFR Inhibitor KIN-3248 Overcomes FGFR2 Kinase Domain Mutations. Clin Cancer Res. 2024 May 15;30(10):2181-2192. [Content Brief]
/////////Resigratinib, Pan-FGFR Inhibitor KIN-3248, KIN 3248, Pan-fibroblast Growth Factor Receptor Inhibitor KIN-3248, W728TB393W
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Sebetralstat
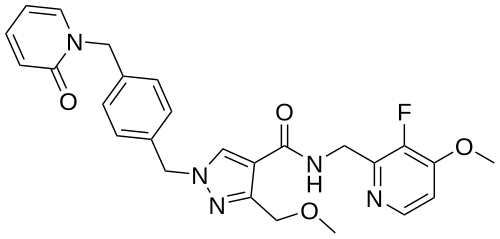
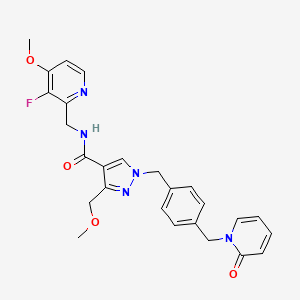
Sebetralstat, KVD 900
CAS 1933514-13-6
491.5 g/mol
FDA 7/3/2025, Ekterly, To treat acute attacks of hereditary angioedema
N-[(3-fluoro-4-methoxypyridin-2-yl)methyl]-3-(methoxymethyl)-1-[[4-[(2-oxopyridin-1-yl)methyl]phenyl]methyl]pyrazole-4-carboxamide
Sebetralstat, sold under the brand name Ekterly, is a medication used for the treatment of hereditary angioedema.[1] Sebetralstat is a plasma kallikrein inhibitor.[1]
Sebetralstat was approved for medical use in the United States in July 2025.[1][2]
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00921
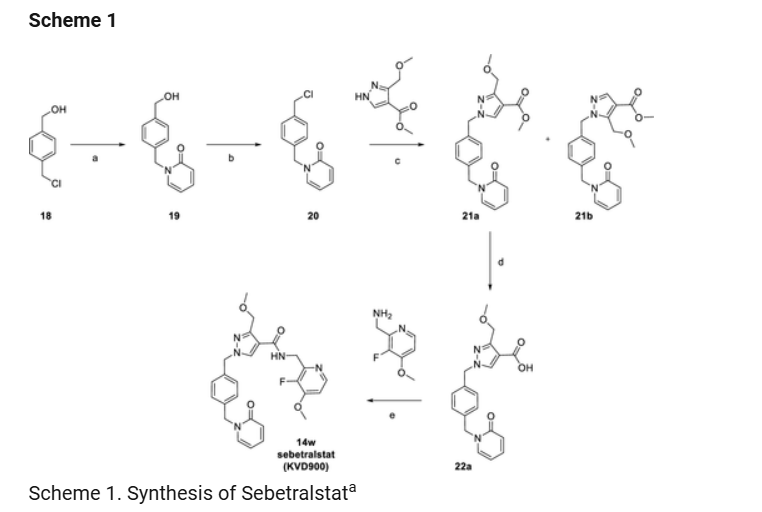
aReagents and conditions: (a) 2-Hydroxypyridine (1.2 equiv), K2CO3 (3.0 equiv), acetone, 50 °C, 18 h, 78%; (b) methanesulfonyl chloride (1.3 equiv), Et3N, (1.4 equiv), dichloromethane, rt, 18h, 93%; (c) methyl 3-(methoxymethyl)-1H-pyrazole-4-carboxylate (0.83 equiv), K2CO3 (2.0 equiv), DMF, 60 °C, 18 h, 54%; (d) NaOH (3.0 equiv), THF-MeOH-H2O, rt, 18 h, 34%; (e) 22a (1.0 equiv), C-(3-fluoro-4-methoxy-pyridin-2-yl)-methylamine (1.0 equiv), HATU (1.1 equiv), Et3N (6.0 equiv), dichloromethane, rt, 4 h, 64%.
Synthesis of Sebetralstat
1-(4-Hydroxymethyl-benzyl)-1H-pyridin-2-one (19)
4-(Chloromethyl)benzyl alcohol 18 (5.0 g, 31.9 mmol) was added to a solution of potassium carbonate (13.2 g, 96 mmol) and 2-hydroxypyridine (3.6 g, 38.3 mmol) in acetone (250 mL). The reaction mixture was heated at 50 °C for 18 h and then concentrated in vacuo. The residue was partitioned between dichloromethane (300 mL) and water (300 mL). The organic layer was separated, and the aqueous layer was extracted with dichloromethane (2 × 300 mL). The combined organic layers were washed with brine (300 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica (0–10% methanol in dichloromethane) to afford 19 (5.4 g, 25.1 mmol, 78% yield) as a white solid. MS (ESI) m/z 216.0 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 7.76 (dd, J = 6.8, 2.1 Hz, 1H), 7.41 (ddd, J = 9.0, 6.6, 2.1 Hz, 1H), 7.34–7.21 (m, 4H), 6.41 (dd, J = 9.1, 1.3 Hz, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.15 (t, J = 5.7 Hz, 1H), 5.07 (s, 2H), 4.46 (d, J = 5.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.4, 141.9, 140.0, 139.0, 135.7, 127.5, 126.6, 119.8, 105.4, 62.6, 50.8.
1-(4-Chloromethyl-benzyl)-1H-pyridin-2-one (20)
A reaction flask containing 1-(4-hydroxymethyl-benzyl)-1H-pyridin-2-one (19) (8.45 g, 39.3 mmol), dry dichloromethane (80 mL), and triethylamine (7.66 mL, 55.0 mmol) was cooled in an ice–water bath. Methanesulfonyl chloride (3.95 mL, 51.0 mmol) was added to the reaction at 0 °C, and ice–water bath cooling continued. After 15 min, the ice–water bath was removed and stirring continued at room temperature overnight. The reaction mixture was partitioned between dichloromethane (100 mL) and saturated aqueous ammonium chloride solution (100 mL). The aqueous layer was extracted with further dichloromethane (2 × 50 mL), and the combined organic layers were washed with brine (50 mL), dried over sodium sulfate, filtered, and concentrated to afford 20 (8.65 g, 36.6 mmol, 93% yield) as a pale yellow solid. MS (ESI) m/z 234.1 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 7.79 (ddd, J = 6.8, 2.1, 0.7 Hz, 1H), 7.49–7.39 (m, 1H), 7.40 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 6.42 (ddd, J = 9.2, 1.3, 0.7 Hz, 1H), 6.24 (td, J = 6.7, 1.4 Hz, 1H), 5.09 (s, 2H), 4.73 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.4, 140.1, 139.1, 137.6, 136.9, 129.0, 127.9, 119.9, 105.5, 50.8, 45.8.
Methyl 3-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylate (21a) and Methyl 5-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylate (21b)
Methyl 3-(methoxymethyl)-1H-pyrazole-4-carboxylate (2.11 g, 11.77 mmol; CAS No. 318496-66-1) was added to a solution of potassium carbonate (3.25 g, 23.54 mmol) and 1-(4-chloromethyl-benzyl)-1H-pyridin-2-one 20 (3.30 g, 14.12 mmol) in N,N-dimethylformamide (5 mL) and heated at 70 °C for 3 h. The reaction mixture was diluted with ethyl acetate (50 mL) and washed with brine (2 × 100 mL), and the organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography (120 g column, 0–100% (10% ethanol in ethyl acetate) in isohexanes to afford two regioisomers: 21a (2.03 g, 5.47 mmol, 47% yield) as an off-white solid and 21b (350 mg, 0.92 mmol, 8% yield). 21a MS (ESI) m/z 368.1 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.76 (dd, J = 6.8, 2.2 Hz, 1H), 7.41 (ddd, J = 8.9, 6.5, 2.1 Hz, 1H), 7.25 (d, J = 1.2 Hz, 4H), 6.40 (dt, J = 9.1, 1.0 Hz, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.30 (s, 2H), 5.07 (s, 2H), 4.49 (s, 2H), 3.72 (s, 3H), 3.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.2, 161.8, 150.5, 140.6, 139.6, 137.6, 136.3, 135.6, 128.5, 128.4, 120.3, 111.8, 106.0, 66.0, 58.0, 55.1, 51.5, 51.2. 21b MS (ESI) m/z 368.1 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (s, 1H), 7.76 (dd, J = 6.8, 2.1 Hz, 1H), 7.41 (ddd, J = 8.9, 6.6, 2.1 Hz, 1H), 7.28–7.21 (m, 2H), 7.17 (d, J = 8.2 Hz, 2H), 6.43–6.36 (m, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.35 (s, 2H), 5.06 (s, 2H), 4.78 (s, 2H), 3.75 (s, 3H), 3.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.4, 161.8, 142.4, 140.9, 140.5, 139.6, 137.4, 136.2, 128.3, 120.3, 112.8, 106.0, 61.7, 58.2, 53.0, 51.7, 51.2.
3-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylic acid (22a)
To methyl 3-(methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylate 21a (3.77 g, 10.26 mmol) in tetrahydrofuran (5 mL) and methanol (5 mL) was added 2 M aqueous sodium hydroxide solution (15.39 mL, 30.80 mmol), and the reaction mixture was stirred at room temperature overnight. The reaction was acidified with 1 M aqueous HCl solution (50 mL) and extracted with ethyl acetate (50 mL). The organic layer was washed with brine (50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo to afford 22a (1.22 g, 3.45 mmol, 34% yield) as a white solid. MS (ESI) m/z 354.2 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 1H), 8.32 (s, 1H), 7.76 (ddd, J = 6.8, 2.1, 0.7 Hz, 1H), 7.41 (ddd, J = 8.9, 6.6, 2.1 Hz, 1H), 7.30–7.20 (m, 4H), 6.40 (ddd, J = 9.1, 1.4, 0.7 Hz, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.29 (s, 2H), 5.07 (s, 2H), 4.50 (s, 2H), 3.22 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.3, 161.8, 150.5, 140.6, 139.6, 137.6, 136.4, 135.6, 128.5, 128.4, 120.3, 113.0, 106.0, 66.0, 58.0, 55.1, 51.2.
3-Methoxymethyl-1-[4-(2-oxo-2H-pyridin-1-ylmethyl)-benzyl]-1H-pyrazole-4-carboxylic Acid (3-Fluoro-4-methoxy-pyridin-2-ylmethyl)-amide (14w)
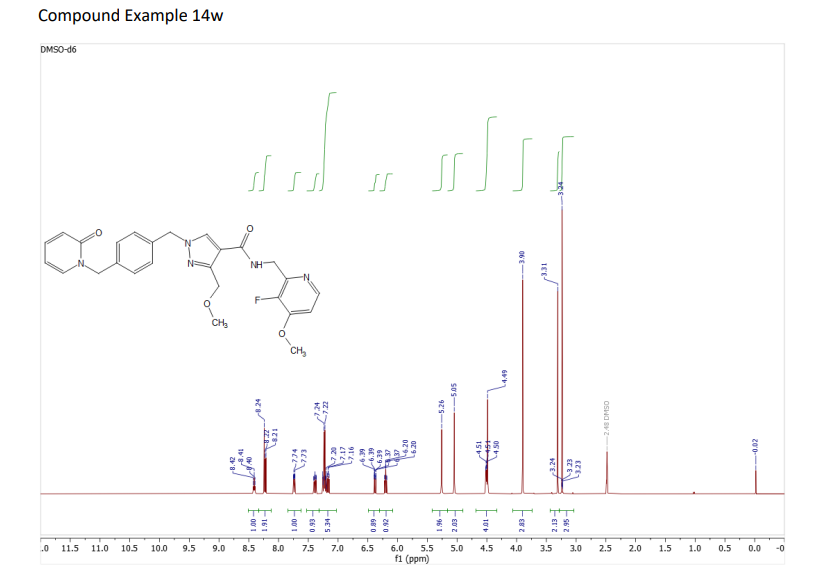
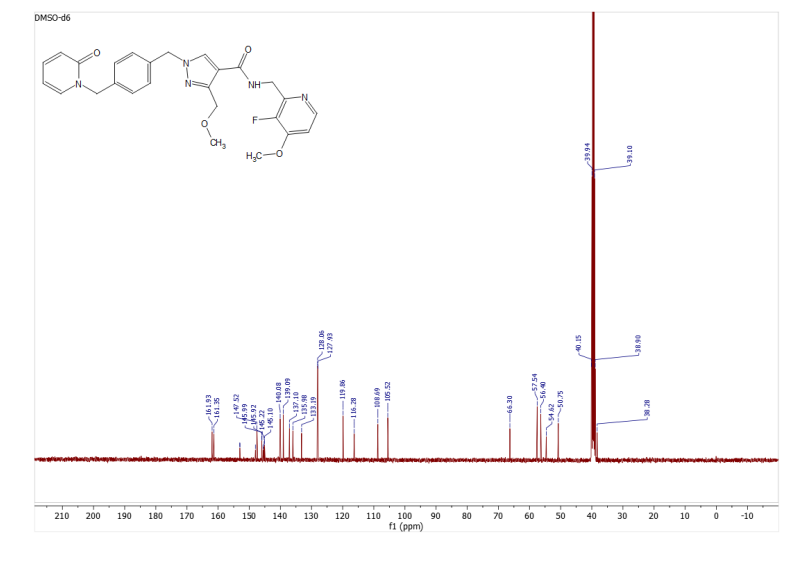
3-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylic acid 22a (75 mg, 0.212 mmol), C-(3-fluoro-4-methoxy-pyridin-2-yl)-methylamine (49 mg, 0.212 mmol; CAS No. 1256812-75-5), and HATU (89 mg, 0.233 mmol) were suspended in anhydrous dichloromethane (3 mL) to which triethylamine (177 μL, 1.270 mmol) was added, sonicated, and then left to stir at room temperature for 4 h. The solvent was removed under reduced pressure, and the resulting residue was quenched with saturated aqueous ammonium chloride solution (5 mL). An off-white solid resulted, which was sonicated, filtered under reduced pressure, washed with water, and dried in a vacuum oven at 40 °C overnight. The residue was purified by chromatography eluting with 1% NH3 in MeOH/dichloromethane to afford 14w as a white solid (67 mg, 64% yield). MS (ESI) m/z 492.0 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.42 (t, J = 5.4 Hz, 1H), 8.29–8.21 (m, 2H), 7.75 (ddd, J = 0.7, 2.1, 6.8 Hz, 1H), 7.41 (ddd, J = 2.1, 6.6, 8.9 Hz,1H), 7.28–7.17 (m, 5H), 6.39 (ddd, J = 0.7, 1.4, 9.2 Hz, 1H), 6.22 (td, J = 1.4, 6.7 Hz, 1H), 5.28 (s, 2H), 5.07 (s, 2H), 4.57–4.46 (m, 4H), 3.92 (s, 3H), 3.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.9, 161.3, 153.0 (JC–F = 8.7 Hz), 147.5, 146.8 (JC–F = 253.5 Hz), 146.0 (JC–F = 7.2 Hz), 145.2 (JC–F = 11.6 Hz), 140.1, 139.1, 137.1, 136.0, 133.2, 128.1, 127.9, 119.9, 116.3, 108.7, 105.5, 66.3, 57.5, 56.4, 54.6, 50.7, 38.3.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US232820883&_cid=P22-MCYEU3-92408-1
Example 41
3-Fluoro-4-methoxy-pyridine-2-carbonitrile
(3-Fluoro-4-methoxy-pyridin-2-ylmethyl)-carbamic acid tert-butyl ester
C-(3-Fluoro-4-methoxy-pyridin-2-yl)-methylamine hydrochloride salt
3-Methoxymethyl-1-[4-(2-oxo-2H-pyridin-1-ylmethyl)-benzyl]-1H-pyrazole-4-carboxylic acid (3-fluoro-4-methoxy-pyridin-2-ylmethyl)-amide
Medical uses
Sebetralstat is indicated for the treatment of acute attacks of hereditary angioedema.[1]
Pharmacology
Sebetralstat is a plasma kallikrein inhibitor that contains the unusual 2-pyridone heterocycle.[3]
Society and culture
Legal status
Sebetralstat was approved for medical use in the United States in July 2025.[1] The US Food and Drug Administration granted the application for sebetralstat orphan drug designation.[4]
Names
Sebetralstat is the international nonproprietary name.[5]
Sebetralstat is sold under the brand name Ekterly.[1]
References
- ^ Jump up to:a b c d e f g “Ekterly- sebetralstat tablet”. DailyMed. 7 July 2025. Retrieved 9 July 2025.
- ^ “KalVista Pharmaceuticals Announces FDA Approval of Ekterly (sebetralstat), First and Only Oral On-demand Treatment for Hereditary Angioedema” (Press release). Kalvista. 7 July 2025. Retrieved 9 July 2025 – via Business Wire.
- ^ Davie RL, Edwards HJ, Evans DM, Hodgson ST, Stocks MJ, Smith AJ, et al. (October 2022). “Sebetralstat (KVD900): A Potent and Selective Small Molecule Plasma Kallikrein Inhibitor Featuring a Novel P1 Group as a Potential Oral On-Demand Treatment for Hereditary Angioedema”. Journal of Medicinal Chemistry. 65 (20): 13629–13644. doi:10.1021/acs.jmedchem.2c00921. PMC 9620001. PMID 36251573.
- ^ “Sebetralstat Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Retrieved 9 July 2025.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
External links
- “Sebetralstat (Code – C184930)”. EVS Explore.
- Clinical trial number NCT05259917 for “A Phase III, Crossover Trial Evaluating the Efficacy and Safety of KVD900 (Sebetralstat) for On-Demand Treatment of Angioedema Attacks in Adolescent and Adult Patients With Hereditary Angioedema (HAE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ekterly |
| Other names | KVD-900, KVD900 |
| License data | US DailyMed: Sebetralstat |
| Routes of administration | By mouth |
| ATC code | B06AC08 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1933514-13-6 |
| PubChem CID | 121365142 |
| IUPHAR/BPS | 11947 |
| DrugBank | DB18305 |
| ChemSpider | 115006749 |
| UNII | O5ZD2TU2B7 |
| KEGG | D12396 |
| ChEMBL | ChEMBL5095248 |
| Chemical and physical data | |
| Formula | C26H26FN5O4 |
| Molar mass | 491.523 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////Sebetralstat, FDA 2025, APPROVALS 2025, Ekterly, angioedema, KVD 900, O5ZD2TU2B7
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Redafamdastat
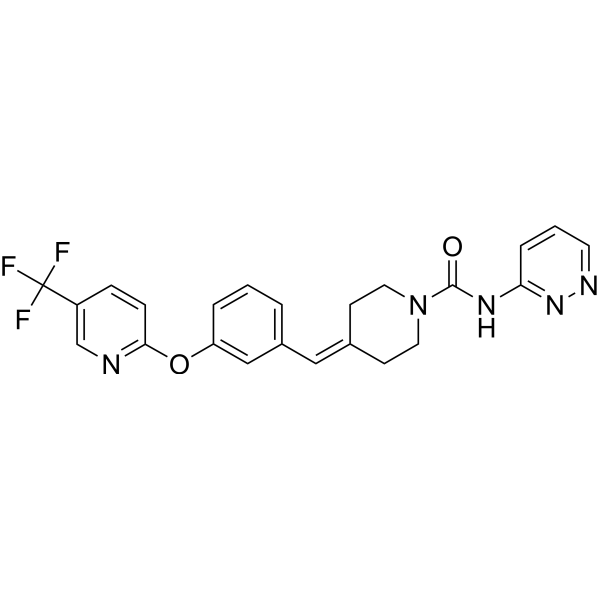
Redafamdastat
cas 1020315-31-4, PF 04457845
JZP-150; JZP150; PF-04457845; PF-4457845; PF04457845; PF4457845, Q7119045
WeightAverage: 455.441
Monoisotopic: 455.156909393
Chemical FormulaC23H20F3N5O2
- 1-Piperidinecarboxamide, N-3-pyridazinyl-4-[[3-[[5-(trifluoromethyl)-2-pyridinyl]oxy]phenyl]methylene]-
- N-pyridazin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methylidene]piperidine-1-carboxamide
Redafamdastat (INNTooltip International Nonproprietary Name; developmental code names JZP-150, PF-04457845) is an inhibitor of the enzyme fatty acid amide hydrolase (FAAH), with an IC50Tooltip half-maximal inhibitory concentration of 7.2 nM, and both analgesic and anti-inflammatory effects in animal studies comparable to those of the cyclooxygenase inhibitor naproxen.[1] It was being developed by Jazz Pharmaceuticals for the treatment of alcoholism, pain, and post-traumatic stress disorder (PTSD) and reached phase 2 clinical trials.[2][3] However, development of the drug was discontinued in December 2023.[2]
SCHEME
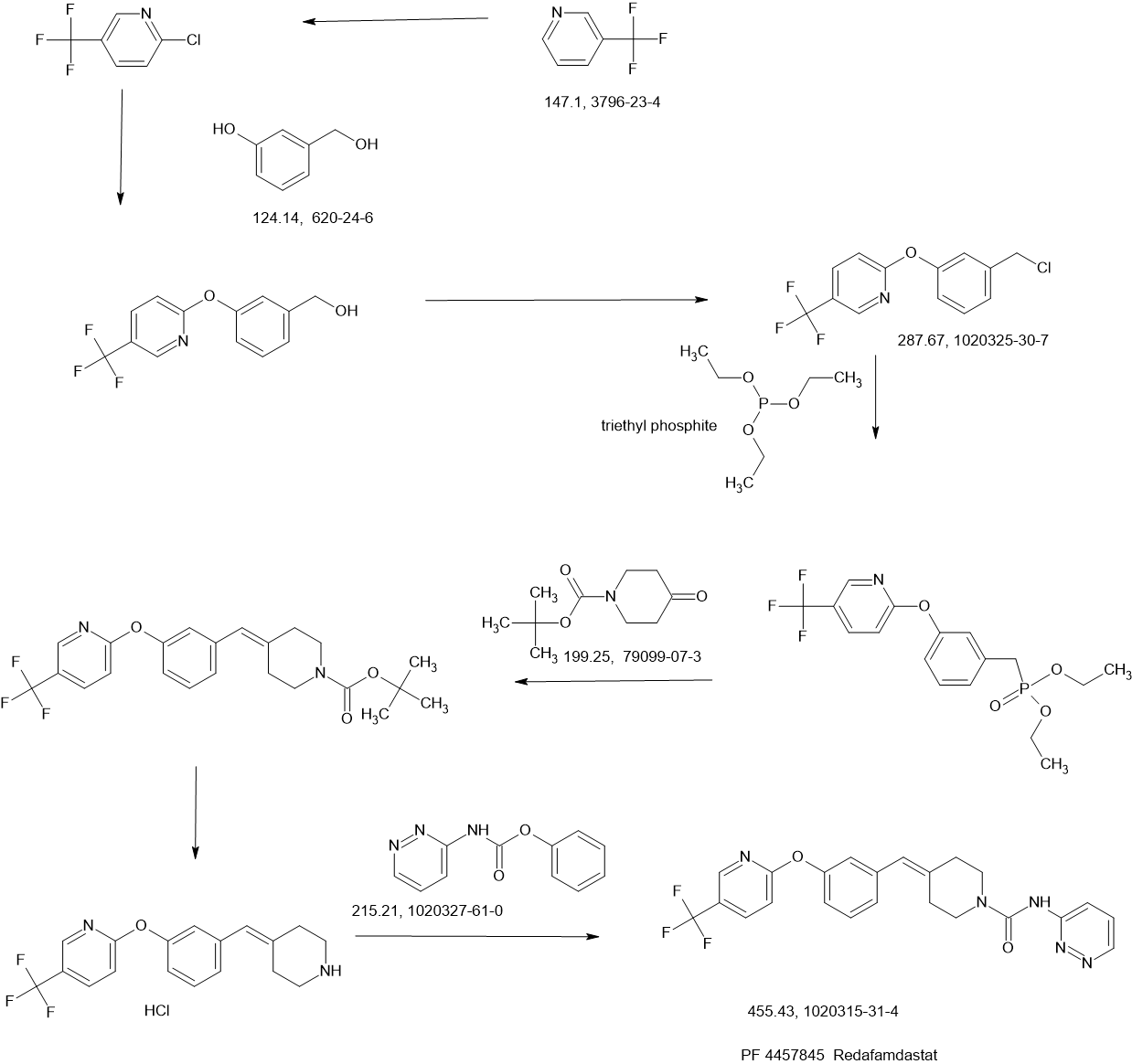
PAPER
ACS Medicinal Chemistry Letters (2011), 2(2), 91-96 86%
PATENT
WO2008047229 86%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008047229&_cid=P11-MCY7EC-33944-1
Example 1a
Synthesis of N-pyridin-3-yl-4-(3-(f5-(trifluoromethyl)pyridin-2-ylloxy)benzylidene)piperidine-1-carboxamide
Phenyl pyridin-3-ylcarbamate
To a stirred solution of 3-aminopyridine (51.7 g, 0.549 moles) in THF (900 mL) at -10 0C was added pyridine (52.1 g, 0.659 moles) in a stream over a 10 min period, followed by the dropwise addition of phenyl chloroformate (90 g, 0.575 moles) over a 20 min period. The reaction tempature increased to 5 0C. A precipitate formed during the addition. The resulting suspension was stirred at temperatures reaching ambient temperature over the next 3 h. The reaction mixture was partitioned between water (2 L) and EtOAc (1.5 L). The aqueous portion was extracted with EtOAc (1 L). The combined organic portions were dried (MgSO4) and concentrated in vacuo to a damp solid residue. This was suspended in
EtOAc:ether (1 :1 , 600 ml_). The resulting suspension was stirred at -10 0C for 2 h and filtered. The solid was rinsed with EtOAσether (1 :1 , 100 ml.) and pressed dry under suction. Further drying in vacuo at 35 0C for 7 h provided 104 g (88%) of product. Analysis, Calcd for Ci2H10N2O2: C, 67.28; H, 4.71 ; N, 13.08. Found: C, 67.15; H, 4.76; N, 12.87.
Step i
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol
3-Hydroxymethyl-phenol (5.00 g, 40.3 mmol, from Lancaster Synthesis), 2-chloro-5-trifluoromethyl-pyridine (7.31 g, 40.3 mmol, from TCI America) and potassium carbonate (6.96 g, 50.3 mmol) were suspended in dimethylformamide (80 mL) and heated to 95 0C. After stirring for 16 h, the solvent was distilled off in vacuo at 65 0C, and a residue was partitioned between water and heptane/ethyl acetate (1 :1 ). The organic layer was separated and the aqueous was extracted again with heptane/ethyl acetate (1 :1 ). The combined organic layer was dried over sodium sulfate, filtered and concentrated to give a residue. The residue was purified by silica gel chromatography (10-60%, EtOAc:heptane) to afford the desired product (5.70 g, 53% yield) as a light yellow oil. 1H NMR (400 MHz, CDCI3) δ ppm 4.73 (s, 2 H) 7.02 (dt, J=8.66, 0.57 Hz, 1 H) 7.04 – 7.11 (m, J=8.06, 2.40, 0.50, 0.50 Hz, 1 H) 7.15 – 7.19 (m, 1 H) 7.25 (ddd, J=8.39, 1.60, 0.80 Hz, 1 H) 7.42 (t, J=7.87 Hz, 1 H) 7.90 (ddd, J=8.67, 2.55, 0.50 Hz, 1 H) 8.43 (td, J=1.68, 0.84 Hz, 1 H).
Step 2
2-(3-Chloromethyl-phenoxy)-5-trifluoromethyl-pyridine
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol from Step 1 (4.68 g, 17.4 mmol), in
dichloromethane (46 mL), was cooled to 0 0C, and treated dropwise with thionyl chloride (1.40 mL, 19.1 mmol). The reaction mixture was allowed to warm to ambient temperature and was stirred for 30 rηjn. Toluene (10 mL) was added and the mixture was concentrated by evaporation to form a residue. The residue was evaporated again from toluene and dried under high vacuum to afford the desired product (4.88 g, 98% yield) as an oil. 1H NMR (400 MHz, CDCI3) δ ppm 4.60 (s, 2 H) 7.03 (d, J=8.70 Hz, 1 H) 7.11 (ddd, J=8.09, 2.35, 0.94 Hz, 1 H) 7.20 (t, J=2.03 Hz, 1 H) 7.26 – 7.31 (m, 1 H) 7.42 (t, J=7.88 Hz, 1 H) 7.91 (dd, J=8.67, 2.53 Hz, 1 H) 8.44 (dd, J=1.51 , 0.90 Hz, 1 H).
Step 3
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester
2-(3-Chloromethyl-phenoxy)-5-trifluoromethyl-pyridine (4.88 g, 17.0 mmol) from Step 2 was treated neat with triethylphosphite (4.36 mL, 25.4 mmol) and heated to 150 0C. After 6 h, the reaction mixture was cooled, treated with an additional 0.5 mL triethylphosphite (2.9 mmol) and reheated to 150 0C. After 6 h, the reaction mixture was removed from the heat and slowly treated with heptane (about 60 mL) while stirring to afford a white solid. The solid was collected by filtration, washed with heptane and dried in a vacuum oven for 16 h at 45 0C to afford a white powder (5.99 g, 91% yield). MS (APCI) M+1= 390.1 ; 1H NMR (400 MHz, CDCI3) δ ppm 1.26 (t, J=7.02 Hz, 6 H) 3.18 (d, J=21.83 Hz, 2 H) 3.99 – 4.10 (m, 4 H) 7.01 (d, J=8.58 Hz, 1 H) 7.03 – 7.08 (m, 1 H) 7.12 (q, J=2.21 Hz, 1 H) 7.19 – 7.24 (m, 1 H) 7.38 (t, J=7.90 Hz, 1 H) 7.90 (dd, J=8.58, 2.53 Hz, 1 H) 8.43 (dd, J=1.66, 0.88 Hz, 1 H).
Step 4
4-[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1 -carboxylic acid tert-butyl ester
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester (2.3 g, 6.0 mmol) from Step 3 and 1 ,4,7,10, IS-pentaoxacyclopentadecane (15-Crown-5, 0.03 ml_, 0.15 mmol) were combined in THF (10 ml_). The mixture was cooled to 0 °C and sodium hydride (240 mg, 60% dispersion in mineral oil, 6.0 mmol) was added. The reaction was warmed to room temperature, stirred for 30 minutes and then cooled back to 0 0C. A solution of 4-oxo-piperidine-1-carboxylic acid tert-butyl ester (1.2 g, 6.0 mmol) in THF (6 ml_) was added and the reaction was warmed to room temperature. After 16 hours, water was added and the layers were separated. The aqueous layer was extracted with EtOAc (2X200 mL) and the combined organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to a thick oil. Treatment of the oil with hot isopropyl ether (45 mL) provided the title compound as a white solid (1.88 g). 1H NMR (400 MHz, CD3OD) δ ppm 1.46 (s, 9 H) 2.34 (td, J=5.85, 1.18 Hz, 2 H) 2.46 (td, J=5.87, 1.07 Hz, 2 H) 3.37 – 3.44 (m, 2 H) 3.45 – 3.57 (m, 2 H) 6.41 (s, 1 H) 6.92 – 7.04 (m, 2 H) 7.06 – 7.17 (m, 2 H) 7.31 – 7.54 (m, 1 H) 8.08 (ddd, J=8.74, 2.59, 0.56 Hz, 1 H) 8.42 (td, J=1.73, 0.90 Hz, 1 H).
Step 5
2-(3-Piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride
4-[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1-carboxylic acid tert-butyl ester (1.35 g, 3.11 mmol) from Step 4 was dissolved in CH2CI2 (30 mL) and treated with HCI in diethyl ether (10 mL, 2.0 M, 20 mmol). After 16 hours the reaction was concentrated in vacuo to form a residue and the residue was suspended in acetonitrile (10 mL) to yield a solid. Filtration of the solid provided the title compound as a white solid (1.1 g). 1H NMR (400 MHz, CD3OD) δ ppm 2.62 (td, J=6.11 , 0.91 Hz, 2 H) 2.67 – 2.81 (m, 2 H) 3.14 – 3.21 (m, 2 H) 3.22 – 3.29 (m, 2 H) 6.56 (s, 1 H) 6.99 – 7.09 (m, 2 H) 7.10 – 7.18 (m, 2 H) 7.42 (t, J=7.91 Hz, 1 H) 8.09 (ddd, J=8.74, 2.60, 0.33 Hz, 1 H) 8.41 (td, J=1.63, 0.74 Hz, 1 H).
Step 6
2-(3-Piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride (800 mg, 2.16 mmol, from Step 5), phenyl pyridin-3-ylcarbamate (508 mg, 2.37 mmol) and diisopropylethylamine (0.75 mL, 4.52 mmol) were combined in acetonitrile (10 mL) and stirred at room temperature. After 16 hours, the reaction was concentrated forming a residue and the residue was partitioned between EtOAc and water. The organic layer was separated, washed with 5% NaOH (aq), dried over anhydrous sodium sulfate, filtered and concentrated. Treatment of the residue with hot isopropyl ether and purified from isopropyl ether/methanol provided the title compound as a white solid (574 mg). MS (APCI 10V) AP+ 455.3, 376.2, 335.2, AP- 453.2; 1H NMR (400 MHz, CD3OD) δ ppm 2.46 (td, J=5.86, 0.97 Hz, 2 H) 2.58 (td, J=5.82, 1.16 Hz, 2 H) 3.51 – 3.60 (m, 2 H) 3.61 – 3.70 (m, 2 H) 6.46 (s, 1 H) 6.98 – 7.07 (m, 2 H) 7.09 – 7.19 (m, 2 H) 7.34 (ddd, J=8.41 , 4.81 , 0.65 Hz, 1 H) 7.40 (td, J=7.69, 0.74 Hz, 1 H) 7.91 (ddd, J=8.38, 2.58, 1.44 Hz, 1 H) 8.08 (ddd, J=8.73, 2.61 , 0.55 Hz, 1 H) 8.16 (dd, J=4.84, 1.06 Hz, 1 H) 8.43 (td, J=1.74, 0.91 Hz, 1 H) 8.58 (d, J=1.88 Hz, 1 H).
Example 1b
Large scale synthesis of N-pyridin-3-yl-4-(3-{[5-(trifluoromethyl)pyridin-2-ylloxy)benzylidene)piperidine-1- carboxamide
Step 1 : Preparation of r3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyll-methanol
To a solution of S-trifluoromethyl^-chloro-pyridine (150.0 g, 0.826 mol) in DMF (1.9 L) was added 3-hydroxy-phenyl-methanol (112.5 g, 0.906 mol) and of potassium carbonate (171.0 g, 1.237 mol). The solids were washed into the flask with 100 mL of DMF. The stirred mixture was heated to 95-105 0C for 5 h. It was cooled to ambient temperature and then poured into 5 L of stirred ice-water. The mixture was extracted with etheπhexane (2:1 , 1.5 L, 1.0 L). The combined organic layers were dried over magnesium sulfate and concentrated in vacuo to dryness to give the product (222.5 g, 100%).
Step 2: Preparation of 2-(3-chloromethyl-phenoxy)-5-trifluoromethylpyridine
To a solution of [3-(5-trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol (281.0 g, 1.044 moles) in dichloromethane (2.0 L) at -5 0C was added dropwise over a 25 min period thionyl chloride (136.6 g, 1.148 mol). A few minutes into the addition, a white substance separated but this went into solution several minutes later. The reaction was stirred at ambient temperature for 1 h and then was concentrated in vacuo to near dryness (357 g). 200 mL of toluene was added to the residue and the solution was again concentrated in vacuo to near dryness. 200 mL of toluene was added and some solid (-8 g) was filtered off. The filtrate was concentrated in vacuo to -390 g of dark yellow liquid.
Step 3: Preparation of [3-(5-trifluoromethyl-pyridin-2-yloxy)-benzyll-phosphonic acid diethyl ester
A solution of 2-(3-chloromethyl-phenoxy)-5-trifluoromethylpyridine (-298 g, -1.036 mol) containing some toluene in triethyl phosphite (267.0 g, 1.551 mol) was heated to 135 °C-140 0C for 7 h. Boiling began at -110 0C and continued throughout the reaction. The solution was left standing at ambient temperature overnight and it solidified. The solid was suspended in etheπhexane (1 :2, 450 mL), and the suspension was stirred at ambient temperature for 3 h and filtered. The solid was rinsed with etherhexane (1 :2, 150 mL) and pressed dry under suction. Further drying in vacuo at 32 0C for 7 h provided 286.3 g (71 % – 2 steps from crude chloride) of product. The filtrate was concentrated in vacuo to remove the low boiling solvents. Triethyl phosphite (36.0 g, 0.217 mol) was added and the solution was heated to 130 0C for 2 h. The reaction was cooled to 100 0C and 300 mL of heptane was added slowly. A solid separated. As the temperature decreased to -30 0C, 150 mL of ether was added. The resulting suspension was left standing at ambient temperature overnight and was filtered. The solid was rinsed with etherheptane (1 :2, 75 mL) and pressed dry under suction. Further drying in vacuo at 32 0C for 7 h afforded and additional 35.7 g (9%) of product. Total yield = 322 g (80%). Anal. Calcd for C17H19 F3NO4P (389.31 ) : C, 52.45; H, 4.92; N, 3.60; F, 14.64; P, 7.96. Found : C, 52.73; H, 5.04; N, 3.58; F, 14.35; P, 7.74; chloride, <0.10%.
Step 4: Preparation of 4-f3-(5-trifluoromethyl-pyridin-2-yloxy)-benzylidenel-piperidine-1-carboxylic acid tert-butyl ester
To a stirred mixture of [3-(5-trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester (155.7 g, 0.40 mol) in tetrahydrofuran (800 mL) at -10 0C was added dropwise over a 5 min period 1.0 M tBuOK in tetrahydrofuran (420.0 mL, 0.42 mol). The temperature rose to -3 °C during the addition. The resulting red mixture was stirred between -6 0C and -10 0C for 2.5 h. A solution of tert-butyl 4-oxopiperidine-1-carboxylate (79.7 g, 0.40 mol) in tetrahydrofuran (300 mL) was added dropwise over a 5 min period. The temperature rose to 2 0C. The resulting red mixture was stirred at temperatures reaching 21 0C over the next 16 h. TLC showed product with no phosphonate present. The mixture was poured into 3.5 L of stirred ice-water. The resulting suspension was stirred at ambient temperature for 2.5 h and then was extracted with successive 1.0 L and 0.6 L portions of dichloromethane. The combined extracts were washed with 500 mL of brine, dried over magnesium sulfate and concentrated in vacuo to a thick semi solid residue. 250 mL of methyl t-butyl ether was added. The suspension was stirred at -10 0C for 2 h and filtered. Drying in vacuo at 25 CC for 66 h provided 85 g (49%) of product. The filtrate was concentrated in vacuo to a damp solid residue. This was taken up in 100 mL of methyl t-butyl ether. To the stirred suspension was added 300 mL of heptane and the resulting suspension was stirred at -10 0C for 2 h. The solid was filtered off, rinsed with 50 mL of methyl t-butyl etherheptane (1 :3) and pressed dry under suction. Further drying in vacuo at 34 °C for 6 h provided an additional 34.2 g (19.5%) of product. Total yield = 119.2 g (68.5%).
Step 5: Preparation of 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine, hydrochloride
To a mixture of 4-[3-(5-trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1-carboxylic acid fert-butyl ester (312 g, 0.718 mol) in ethyl acetate (2.8 L) at 0 0C to -5 0C was added streamwise over a 20 min period, 4.0 M hydrogen chloride in dioxane (800 mL, 3.2 mol). No significant temperature change was noted. The resulting suspension was stirred at temperatures reaching 22 0C over the next 17 h. The suspension was filtered. The solid was washed with EtOAc (500 mL) and pressed as dry as possible under suction. The damp solid was dried in vacuo at 33 0C for 7 h to afford 225 g (84%) of product.
Step 6: Preparation of N-pyridin-3-yl-4-(3-(f5-(trifluoromethyl)pyridin-2-ylloxy}benzylidene)piperidine-1-carboxamide
To a mixture of 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine (80.0 g, 0.216 mol) and phenyl pyridin-3-ylcarbamate (48.6 g, 0.227 mol) in acetonitrile (650 mL) was added dropwise diisopropylethyl amine (55.8 g, 0.432 mol). A solution formed after -45 min of stirring. The slightly turbid solution was stirred at ambient temperature for 18 h. TLC showed a prominent product spot with traces of both starting materials and two other fast moving spots. The solution was concentrated in vacuo to a viscous oil. This was partitioned between dichloromethane (600 mL) and water (500 mL). The aqueous layer was extracted with 200 mL of dichloromethane. The combined organic layers were washed with successive portions of 500 mL of 5% sodium hydroxide, and 200 mL of water, then dried over magnesium sulfate and concentrated in vacuo to 139.5 g of a viscous oil. This was dissolved in 350 ml_ of warm (50 0C) methyl t-butyl ether. Soon after a solution formed, solid began separating. The crystallizing mixture was kept at -10 0C for 4 h and filtered. The solid was rinsed with 60 ml_ of methyl t-butyl ether and pressed dry under suction. Further drying in vacuo at 28 0C for 16 h and then at 35 °C for 6 h provided 93.2 g (95%) of product.
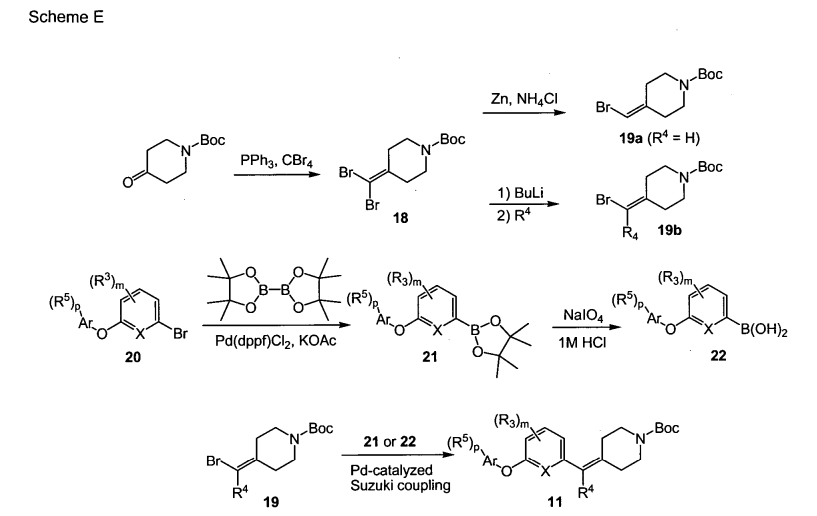
PAPER
ACS Medicinal Chemistry Letters (2011), 2(2), 91-96
https://pubs.acs.org/doi/10.1021/ml100190t
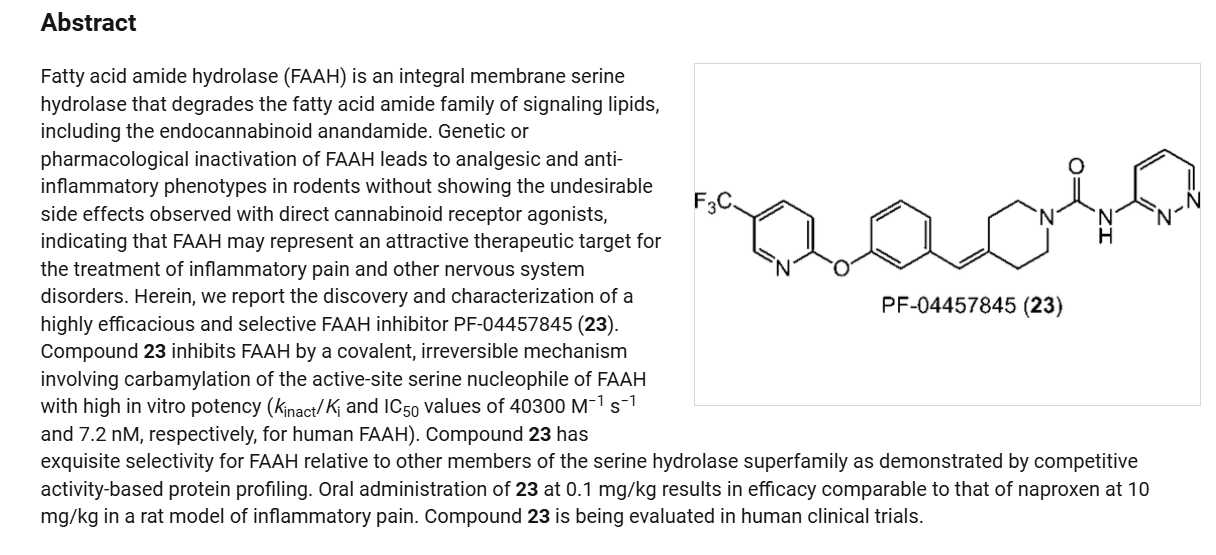
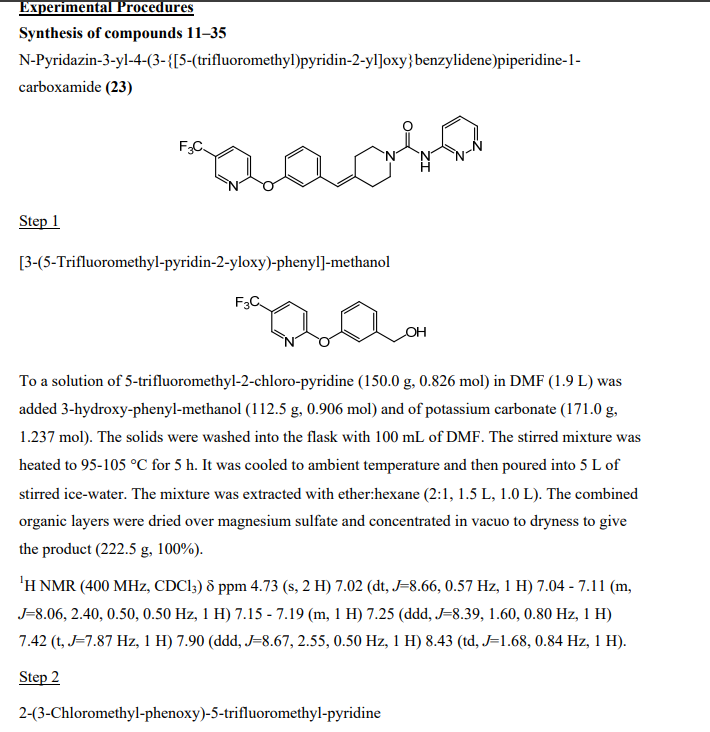




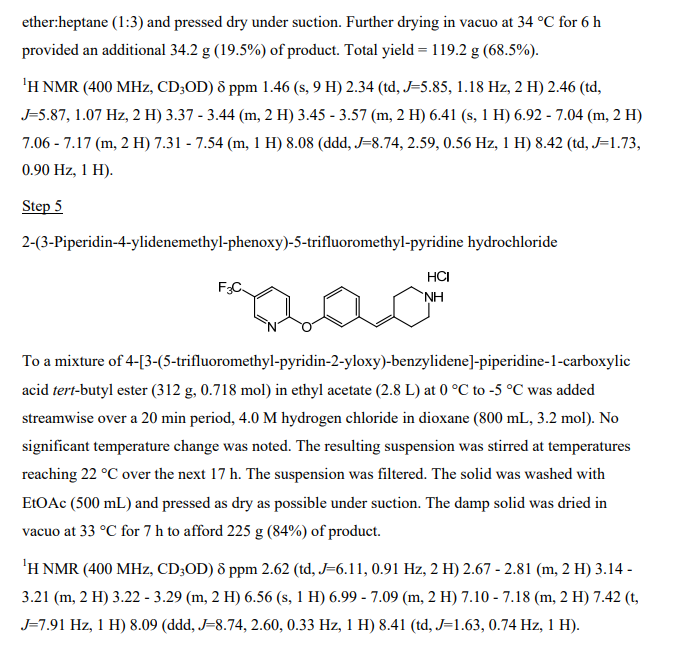

PAPER
Science (Washington, DC, United States) (2017), 356(6342), 1084-1087
Pfizer Products Inc.WO2008047229
References
- ^ Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, et al. (February 2011). “Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor”. ACS Medicinal Chemistry Letters. 2 (2): 91–96. doi:10.1021/ml100190t. PMC 3109749. PMID 21666860.
- ^ Jump up to:a b “JZP 150”. AdisInsight. 26 December 2023. Retrieved 16 August 2024.
- ^ “A Study of JZP150 in Adults With Posttraumatic Stress Disorder – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov.
- [1]. Johnson DS, et al. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011 Feb 10;2(2):91-96. [Content Brief][2]. Ahn K, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011 Jul;338(1):114-24. [Content Brief][3]. Buntyn RW, et al. Inhibition of Endocannabinoid-Metabolizing Enzymes in Peripheral Tissues Following Developmental Chlorpyrifos Exposure in Rats. Int J Toxicol. 2017 Jan 1:1091581817725272. [Content Brief]
////////Redafamdastat, PF 04457845, JZP-150, JZP150, PF-04457845, PF-4457845, PF04457845, PF4457845, Q7119045
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
ARAZASETRON BESYLATE
ARAZASETRON BESYLATE
R-Azasetron besylate, SENS-401
Cas 2025360-91-0
C17H20ClN3O3.C6H6O3S, 507.99, UXP39EQ477
2H-1,4-Benzoxazine-8-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-, benzenesulfonate (1:1)
N-[(3R)-1-azabicyclo[2.2.2]octan-3-yl]-6-chloro-4-methyl-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-8-carboxamide; benzenesulfonic acid
X HCL SALT, CAS , 2139305-21-6, Name2H-1,4-Benzoxazine-8-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-6-chloro-3,…
BASE: 2025360-90-9
.HCL SALT CAS, 2566443-39-6
Name 2H-1,4-Benzoxazine-8-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-6-chloro-3,…
BASE: 2025360-90-9
Base CHIRAL 2025360-90-9
- N-(3R)-1-Azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-2H-1,4-benzoxazine-8-carboxamide
(R)-Azasetron besylate (SENS-401) is an orally active calcineurin inhibitor. (R)-Azasetron besylate reduces Cisplatin (HY-17394)-induced hearing loss and cochlear damage.
Arazasetron Besylate is the besylate salt form of the R-enantiomer of azasetron, a benzamide derivative and selective serotonin (5-hydroxytryptamine; 5-HT) receptor and calcineurin antagonist, with potential antinauseant and antiemetic, and otoprotective activities. Upon administration, arazasetron selectively binds to and inhibits 5-HT subtype 3 receptors (5-HT3R) located peripherally on vagus nerve terminals and centrally in the chemoreceptor trigger zone (CTZ) of the area postrema, which may result in suppression of nausea and vomiting. R-azasetron also targets and inhibits the activation of calcineurin, thereby preventing inner ear lesions, nerve degeneration, induction of apoptosis and sensory hair loss. This may prevent hearing loss. Calcineurin activation plays a key role in structural degeneration, swelling, synaptic uncoupling and the induction of apoptosis in the inner ear leading to hair cell loss and hearing loss.
SCHEME
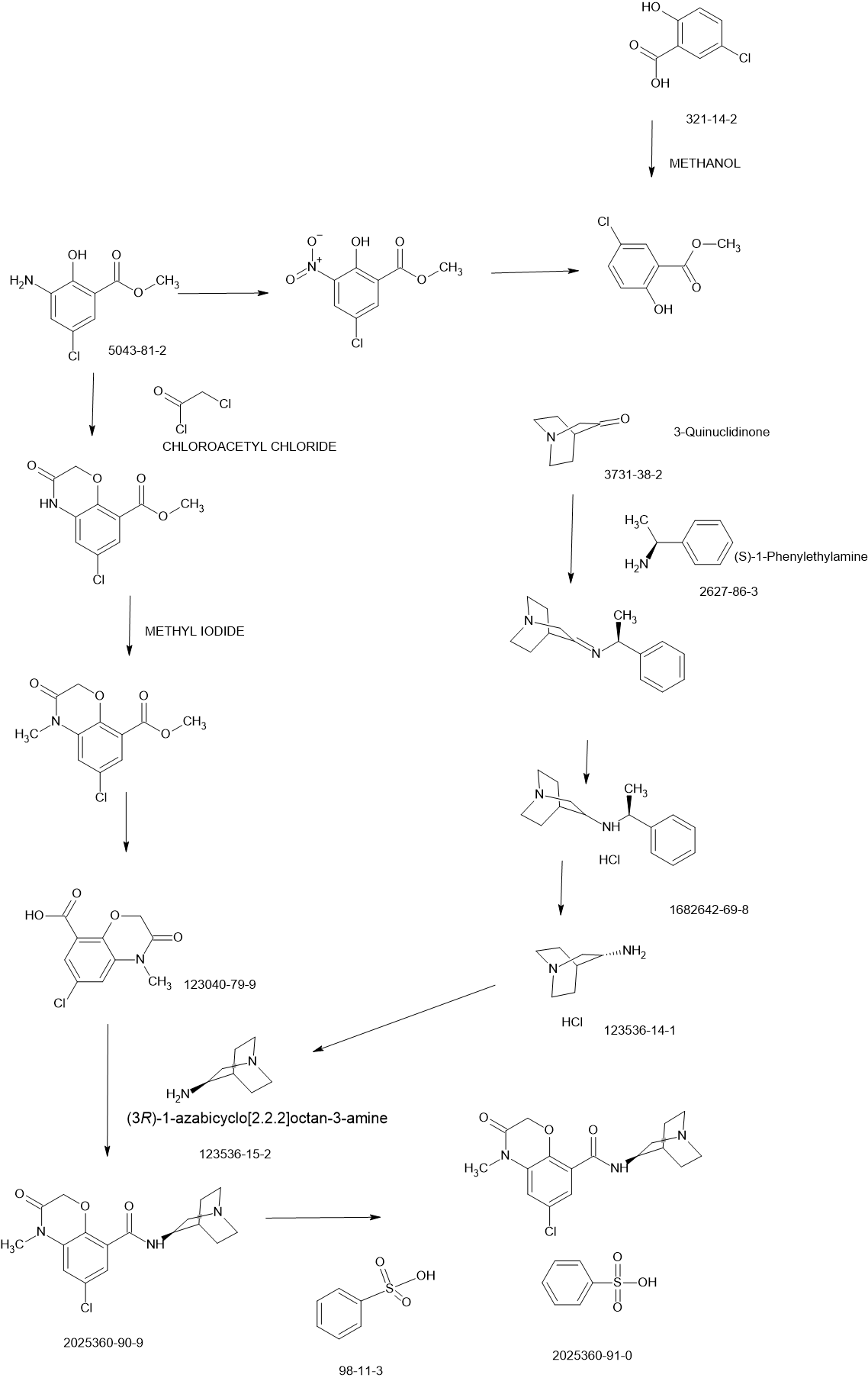
PATENTS
WO2017178645
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017178645&_cid=P22-MCX71S-95827-1
PATENTS
US20190083503
https://patentscope.wipo.int/search/en/detail.jsf?docId=US239434911&_cid=P22-MCX73M-97768-1
PATENTS
WO2010/133663
CN101786963
CN104557906
WO201717864
WO2021014014
WO2023175078
WO2023122719
Base WO2017178645
WO2021014014
WO2023175078
//////////ARAZASETRON BESYLATE, R-Azasetron besylate, UXP39EQ477, SENS-401, SENS 401
Sunvozertinib
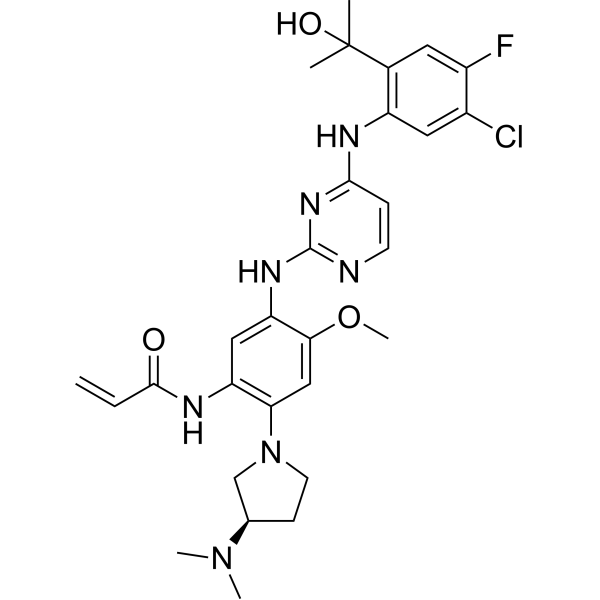
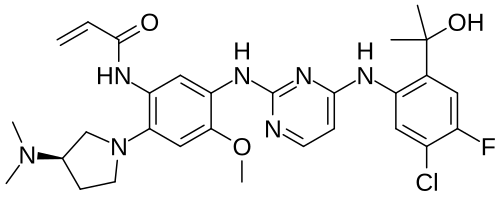
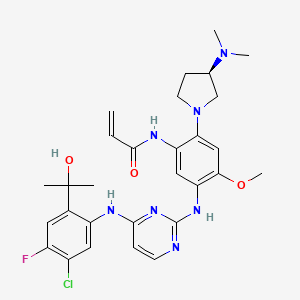
Sunvozertinib
CAS 2370013-12-8
DZD9008, 584.1 g/mol, C29H35ClFN7O3, A-1801, L1Q2K5JYO8
N-[5-[[4-[5-chloro-4-fluoro-2-(2-hydroxypropan-2-yl)anilino]pyrimidin-2-yl]amino]-2-[(3R)-3-(dimethylamino)pyrrolidin-1-yl]-4-methoxyphenyl]prop-2-enamide
FDA Zegfrovy, 7/2/2025
To treat locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations, as detected by an FDA-approved test, with disease progression on or after platinum-based chemotherapy |
Sunvozertinib (DZD9008) is a potent ErbBs (EGFR, Her2, especially mutant forms) and BTK inhibitor. Sunvozertinib shows IC50s of 20.4, 20.4, 1.1, 7.5, and 80.4 nM for EGFR exon 20 NPH insertion, EGFR exon 20 ASV insertion, EGFR L858R and T790M mutations, and Her2 Exon20 YVMA, and EGFR WT A431, respectively (patent WO2019149164A1, example 52).
Sunvozertinib, sold under the brand name 舒沃哲, among others is an anti-cancer medication used for the treatment of non-small-cell lung cancer.[2][3] It is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor.[2][4]
Sunvozertinib was approved for medical use in the United States in July 2025.[1]
Medical uses
In the US, sunvozertinib is indicated for the treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[1]
Side effects
The US FDA prescribing information for sunvozertinib includes warnings and precautions for interstitial lung disease/pneumonitis, gastrointestinal adverse reactions, dermatologic adverse reactions, ocular toxicity, and embryo-fetal toxicity.[1]
History
Sunvozertinib is being developed by Dizal Pharmaceutical.[5] In China, it was conditionally approved in 2023 for the treatment of NSCLC and full approval is contingent on results of phase 3 clinical trials.[6] In the United States, it has been designated by the Food and Drug Administration as a breakthrough therapy for patients with locally advanced or metastatic NSCLCs with an EGFR exon 20 insertion mutation.[7]
Efficacy was evaluated in WU-KONG1B (NCT03974022), a multinational, open-label, dose randomization trial.[1] Eligible participants had locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations with disease progression on or after platinum-based chemotherapy.[1] The primary efficacy population was in 85 participants who received sunvozertinib 200 mg orally once daily with food until disease progression or intolerable toxicity.[1]
The US Food and Drug Administration granted the application for sunvozertinib priority review and breakthrough therapy designations.[1]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019149164&_cid=P10-MCWSEU-52423-1
[1286]
(R) -N- (5- (4- (5-chloro-4-fluoro-2- (2-hydroxypropan-2-yl) phenylamino) pyrimidin-2-ylamino) -2- (3- (dimethylamino) pyrrolidin-1-yl) -4-methoxyphenyl) acrylamide
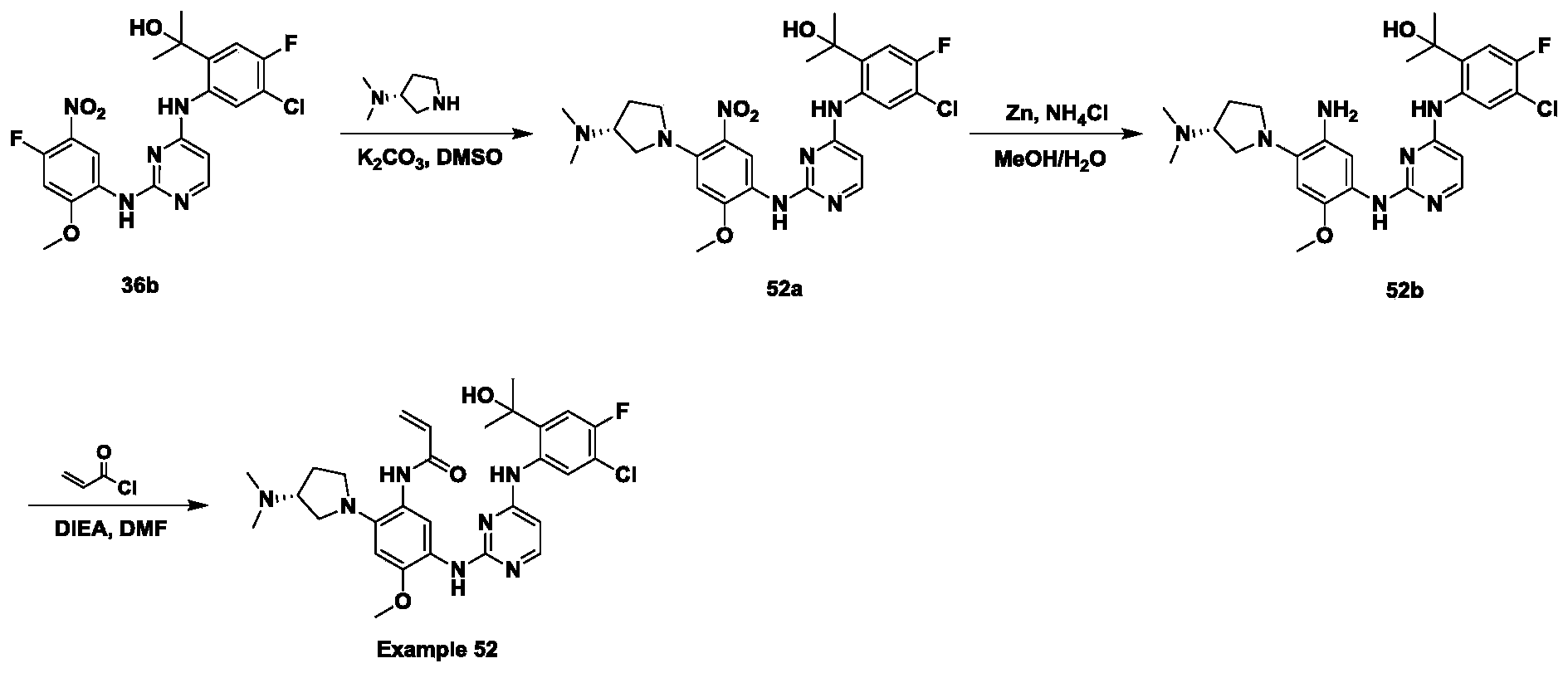
Procedure for the preparation of compound 52a:
[1289]
To a solution of compound 36b (200 mg, 0.429 mmol) and K 2CO 3(119 mg, 0.858 mmol) in DMSO (3 mL) was added (R) -N, N-dimethylpyrrolidin-3-amine (59 mg, 0.515 mmol) . The reaction mixture was stired at 22-30℃ for 4 h, then 50℃ for 1 h while color changed from brown to deep orange. The reaction mixture was added drop wise into H 2O (40 mL) under ice water bath. The precipitated solid was collected by filtration and washed with H 2O (15 mL × 3) , the filter cake was dissolved with CH 2Cl 2(20 mL) , dried over Na 2SO 4and concentrated in vacuum to give compound 52a (210 mg, 87.4%yield) as an orange solid.
[1290]
LCMS: R t=0.677 min in 5-95AB_220&254. lcm chromatography (MERCK RP18 2.5-2mm) , MS (ESI) m/z=559.9 [M+H] +.
[1291]
1H NMR (400MHz, Methanol-d 4) δ 8.42 (s, 1H) , 7.99 (d, J=7.6 Hz, 1H) , 7.96 (d, J=5.6 Hz, 1H) , 7.24 (d, J=10.8 Hz, 1H) , 6.50 (s, 1H) , 6.15 (d, J=5.6 Hz, 1H) , 3.96 (s, 3H) , 3.54 (dt, J=6.4, 10.4 Hz, 1H) , 3.36 (t, J=9.2 Hz, 1H) , 3.28 -3.24 (m, 1H) , 3.09 (dd, J=6.8, 10.0 Hz, 1H) , 2.93 -2.81 (m, 1H) , 2.33 (s, 6H) , 2.30 -2.25 (m, 1H) , 1.97 -1.82 (m, 1H) , 1.59 (d, J=3.6 Hz, 6H) .
[1292]
Procedure for the preparation of compound 52b:
[1293]
To a solution of compound 52a (210 mg, 0.375 mmol) in 5 mL MeOH/H 2O=5/1 (v/v) was added Zn (147 mg, 2.25 mmol) and NH 4Cl (120 mg, 2.25 mmol) . The resulting mixture was heated at 90℃ for 2 h while color changed from orange to brown. The reaction mixture was filtered, and the filtrate was concentrated in vacuum to give the crude residue, which was dissolved with CH 2Cl 2(20 mL) , washed with water (15 mL×3) , then dried over Na 2SO 4and concentrated in vacuum to give compound 52b (125 mg, 63%yield) as a brown solid.
[1294]
LCMS: R t=0.629 min in 5-95AB_220&254. lcm chromatography (MERCK RP18 2.5-2mm) , MS (ESI) m/z=530.1 [M+H] +.
[1295]
1H NMR (400MHz, CDCl 3) δ 8.85 (s, 1H) , 8.15 (d, J=7.2 Hz, 1H) , 8.03 (d, J=6.0 Hz, 1H) , 7.87 (s, 1H) , 7.43 (s, 1H) , 7.08 (d, J=10.4 Hz, 1H) , 6.66 (s, 1H) , 6.05 (d, J=5.6 Hz, 1H) , 3.81 (s, 3H) , 3.20 -3.11 (m, 2H) , 3.06 -2.97 (m, 2H) , 2.91 -2.83 (m, 1H) , 2.28 (s, 6H) , 2.17 -2.09 (m, 1H) , 1.92 -1.81 (m, 1H) , 1.65 (s, 6H) .
[1296]
Procedure for the preparation of Example 52:
[1297]
To a solution of compound 52b (125 mg, 0.236 mmol) and DIEA (46 mg, 0.354 mmol) in DMF (1.5 mL) was added acryloyl chloride (21 mg, 0.236 mmol) in ice water bath. The resulting mixture was stirred at 5-10℃ for 15 min. The reaction was quenched by H 2O (0.1 mL) and then filtered, the filtrate was purified by pre-HPLC directly (Column: Xtimate C18 150*25mm*5um; Condition: 35-65%B (A: 0.04%NH 3·H 2O+10mM NH 4HCO 3, B: CH 3CN) ; Flow Rate: 30 ml/min) and then lyophilized to give Example 52 (14.5 mg, 10.5%yield) as an off-white solid.
[1298]
LCMS: R t=2.028 min in 10-80CD_3min_220&254. lcm chromatography (XBrige Shield RP18 2.1*50mm, 5um) , MS (ESI) m/z=584.3 [M+H] +.
[1299]
1H NMR (400MHz, CDCl 3) δ 9.62 (s, 1H) , 9.43 (br s, 1H) , 8.59 (br s, 1H) , 8.08 (d, J=5.2 Hz, 1H) , 7.53 (d, J=6.8 Hz, 1H) , 7.47 (br s, 1H) , 7.14 (d, J=10.8 Hz, 1H) , 6.75 (s, 1H) , 6.41 -6.31 (m, 3H) , 5.77 (t, J=5.4 Hz, 1H) , 3.86 (s, 3H) , 3.15 -3.02 (m, 4H) , 2.97 -2.84 (m, 1H) , 2.30 (s, 6H) , 2.21 -2.14 (m, 1H) , 1.99 -1.94 (m, 1H) , 1.72 (s, 6H) .
PATENT
PAPER
https://www.mdpi.com/1420-3049/29/7/1448
Sunvozertinib, a novel TKI manufactured by Dizal Pharmaceuticals, represents an advancement arising from the need to overcome resistance mechanisms and thereby replaces previous generations of EGFR inhibitors. As marketed under the proprietary name DZD9008, this therapeutic entity embodies an innovative modality aimed at addressing NSCLC characterized by distinct mutations within the EGFR gene [24]. The pharmacological efficacy of sunvozertinib is predicated upon its specific capacity to inhibit EGFR with Exon 20 mutations, alongside targeting human epidermal growth factor receptor 2 (HER2) with Exon 20 insertions. This targeted inhibition is of significance due to the diminished responsiveness of cancer cells with these mutations to earlier generations of EGFR inhibitors. By competitively obstructing the ATP-binding site of these mutated tyrosine kinases, sunvozertinib effectively hinders the proliferative signaling pathways, exhibiting potent anti-tumor activity (ClinicalTrials.gov Identifier: NCT03974022). Preclinical models have demonstrated sunvozertinib’s capacity to inhibit tumor growth, especially in NSCLC models harboring Exon 20 insertions. Moreover, its substantiated ability to traverse the blood–brain barrier has provided favorable support for its prospective efficacy in managing central nervous system metastases. In clinical settings, sunvozertinib has shown promising efficacy, with ongoing trials further assessing its utility as a targeted intervention for individuals presenting specific EGFR mutations. The toxicity profile has been comparable to other EGFR inhibitors, with manageable side effects that do not significantly diminish its therapeutic value [25].
The preparation of sunvozertinib is shown in Scheme 1 [26]. SUNV-001 and SUNV-002 engage in nucleophilic substitution reactions under alkaline conditions, yielding the formation of SUNV-003 through a subsequent nucleophilic substitution process involving SUNV-004, ultimately leading to the generation of SUNV-005. SUNV-005 and SUNV-006 consecutively engage in nucleophilic substitution reactions, culminating in the formation of SUNV-007. The nitro moiety present in SUNV-007 undergoes a reduction process to yield an amino functional group, through the utilization of hydrogen gas as the reducing agent and platinum carbon as the catalytic mediator, ultimately affording the formation of SUNV-008. The amino moiety present in SUNV-008 and the acyl chloride functionality of SUNV-009 engage in a condensation reaction, leading to the formation of the amide compound SUNV-010. SUNV-010 is subjected to an elimination reaction in alkaline environments, leading to the formation of sunvozertinib.
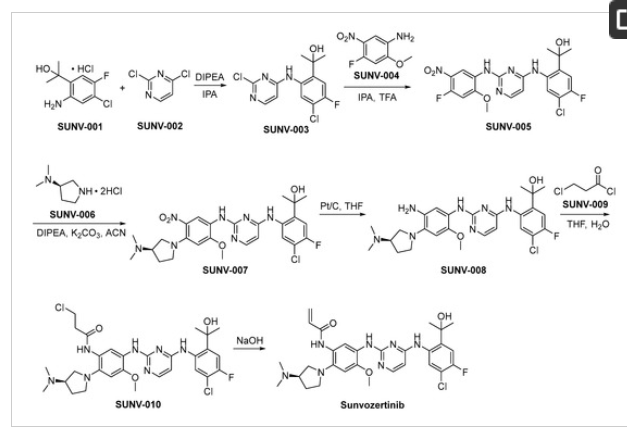
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to sunvozertinib for metastatic non-small cell lung cancer with EGFR exon 20 insertion mutations”. U.S. Food and Drug Administration (FDA). 2 July 2025. Retrieved 7 July 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b Wang M, Yang JC, Mitchell PL, Fang J, Camidge DR, Nian W, et al. (2022). “Sunvozertinib, a Selective EGFR Inhibitor for Previously Treated Non–Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations”. Cancer Discovery. 12 (7): 1676–1689. doi:10.1158/2159-8290.CD-21-1615. PMC 9262839. PMID 35404393.
- ^ Wang M, Fan Y, Sun M, Wang Y, Zhao Y, Jin B, et al. (2024). “Sunvozertinib for patients in China with platinum-pretreated locally advanced or metastatic non-small-cell lung cancer and EGFR exon 20 insertion mutation (WU-KONG6): Single-arm, open-label, multicentre, phase 2 trial”. The Lancet Respiratory Medicine. 12 (3): 217–224. doi:10.1016/S2213-2600(23)00379-X. PMID 38101437.
- ^ Hidetoshi Hayashi (2024). “Sunvozertinib: the next candidate of TKI for NSCLC”. The Lancet Respiratory Medicine. 12 (3): 185–186. doi:10.1016/S2213-2600(23)00419-8. PMID 38101435.
- ^ “ASH: With high tumor response, AstraZeneca spinout Dizal explores FDA path and US partner for PTCL drug”. Fierce Biotech. 11 December 2023.
- ^ Dhillon S (2023). “Sunvozertinib: First Approval”. Drugs. 83 (17): 1629–1634. doi:10.1007/s40265-023-01959-5. PMID 37962831.
- ^ “FDA Grants Breakthrough Therapy Designation to Sunvozertinib in EGFR Exon20+ NSCLC”. targetedonc.com. 9 April 2024.
External links
- Clinical trial number NCT03974022 for “Assessing an Oral EGFR Inhibitor, Sunvozertinib in Patients Who Have Advanced Non-small Cell Lung Cancer with EGFR or HER2 Mutation (WU-KONG1)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | 舒沃哲, Zegfrovy |
| Routes of administration | Oral |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1]Rx in China |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2370013-12-8 |
| PubChem CID | 139377809 |
| DrugBank | DB18925 |
| UNII | L1Q2K5JYO8 |
| Chemical and physical data | |
| Formula | C29H35ClFN7O3 |
| Molar mass | 584.09 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
//////////Sunvozertinib, DZD9008, DZD 9008, FDA 2025, APPROVALS 2025, A-1801, A 1801, L1Q2K5JYO8

{kind=link}
{kind=link}
{kind=link}
{kind=link}