
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Olverembatinib

Olverembatinib
1257628-77-5- 3-((1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)benzamide
- HQP1351
- 4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide
- HQP1351 is under investigation in clinical trial NCT03883100 (A Pivotal Study of HQP1351 in Patients of Chronic Myeloid Leukemia in Accelerated Phase With T315I Mutation).
- 4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide
- D-824
- GZD824
WeightAverage: 532.571
Monoisotopic: 532.219844002, Chemical FormulaC29H27F3N6O
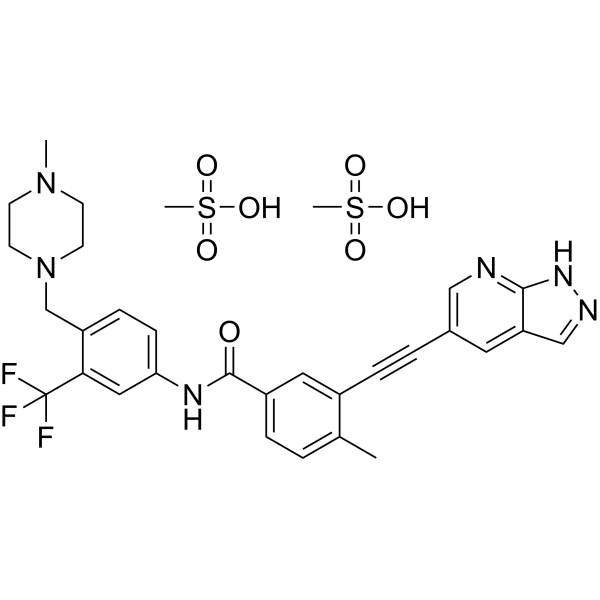
| Molecular Weight | 724.77 |
|---|---|
| Formula | C31H35F3N6O7S2 |
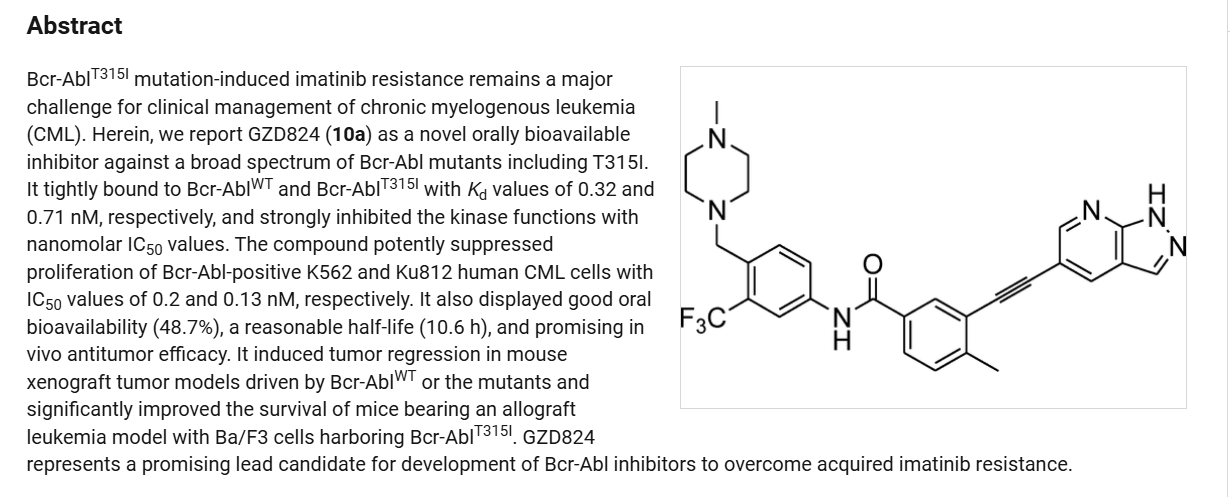
Olverembatinib (GZD824) dimesylate is a potent and orally active pan-Bcr-Abl inhibitor. Olverembatinib dimesylate potently inhibits a broad spectrum of Bcr-Abl mutants. Olverembatinib dimesylate strongly inhibits native Bcr-Abl and Bcr-AblT315I with IC50s of 0.34 nM and 0.68 nM, respectively. Olverembatinib dimesylate has antitumor activity. Olverembatinib (dimesylate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Olverembatinib is a BCR-ABL1 tyrosine kinase inhibitor developed by Ascentage Pharma. In 2021, it was approved in China “for the treatment of adult patients with TKI-resistant chronic-phase CML (CML-CP) or accelerated-phase CML (CML-AP) harbouring the T315I mutation”.[1][2][3]
SYN
Ren, Xiaomei;Pan, Xiaofen;Zhang, Zhang;Wang, Deping;Lu, Xiaoyun;Li, Yupeng;Wen, Donghai;Long, Huoyou;Luo, Jinfeng;Feng, Yubing;Zhuang, Xiaoxi;Zhang, Fengxiang;Liu, Jianqi;Leng, Fang;Lang, Xingfen;Bai, Yang;She, Miaoqin;Tu, Zhengchao;Pan, Jingxuan;Ding, Ke [Journal of Medicinal Chemistry,2013,vol. 56,# 3,p. 879 – 894]
https://pubs.acs.org/doi/10.1021/jm301581y
PATENT
CN 114163434
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN355399053&_cid=P10-MDPKRT-75688-1
| Example |
| The following examples further illustrate but do not limit the present invention. It should be noted that those skilled in the art can make various modifications and improvements without departing from the inventive concept of the present invention, all of which are included in the scope of protection of the present invention. |
| The specific conditions not disclosed in the experimental methods of the following examples can be selected according to conventional methods and conditions, or according to the product instructions. |
| Unless otherwise specified, “room temperature” in the following examples refers to 20°C to 25°C. The term “h” used herein refers to hours. |
| Example 1 |
| Step 1: |
| |
| Under nitrogen, N-methylpyrrolidone (137.6 g) was heated to 30-35°C to obtain the compound of Formula 1 (14.4 g, 1.3 eq) and the compound of Formula 2 (19.14 g, 1 eq). Bis(triphenylphosphate)palladium dichloride (0.46 g, 0.01 eq) and cuprous iodide (0.113 g, 0.01 eq) were added sequentially. Triethylamine (9.45 g, 1.5 eq) was then added under nitrogen. The reaction mixture was heated to 65-75°C and maintained at this temperature for 2 hours. The reaction process was monitored by liquid chromatography-mass spectrometry. The reaction was terminated when the content of the compound of Formula 2 was ≤0.1%. After completion of the reaction, the reaction solution was cooled to 35-45°C and N-acetyl-L-cysteine (1 g, 0.1 eq) was added directly. The reaction was stirred for 4-5 hours. The resulting product was cooled to room temperature, precipitated with water, centrifuged, and washed with pure water to obtain a crude filter cake. The crude filter cake was vacuum-dried and then slurried with a mixture of ethyl acetate and n-heptane (5 mL of the mixed solvent, wherein the volume ratio of ethyl acetate to n-heptane was 1:1) at a rate of 5 mL per gram of crude filter cake. The resulting slurry was vacuum-dried to yield the compound of Formula 3 with a yield of 85.97% and a purity of 98.2%. |
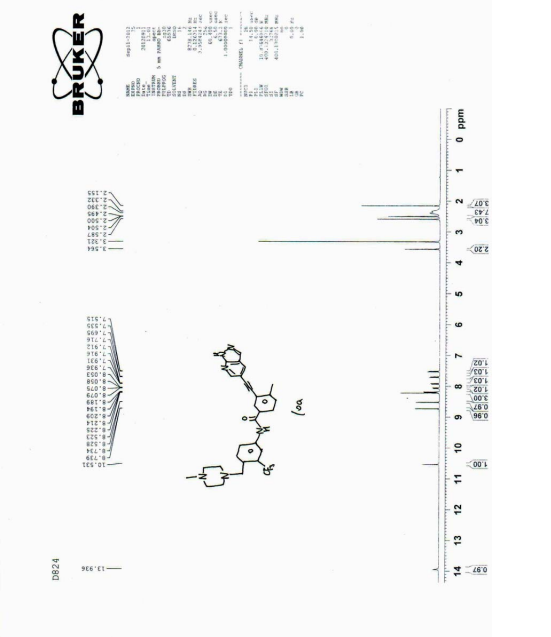
| The NMR data for the compound of Formula 3 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 8.93 (1H, d, J = 2.0 Hz); 8.63 (1H, d, J = 2.0 Hz); 8.49 (1H, s); 8.11 (1H, d, J = 2.0 Hz); 7.92 (1H, dd, J = 1.6 Hz; J = 8.0 Hz); 7.52 (1H, d, J = 8.0 Hz); 3.88 (3H, s); 2.59 (3H, s); 1.65 (9H, s). |
| Step 2: |
| |
| Under nitrogen, methanol (160 g) and water (50 g) were sequentially added to the compound of formula 3 (20 g, 1.0 eq). The reaction system was stirred at reflux for 18 hours with process control. The resulting product was cooled to room temperature and filtered to obtain a filter cake (no drying required). Recrystallization was performed by adding 10 times the mass of the filter cake in methanol. The resulting mixture was stirred at 60-70°C for 8-10 hours, then cooled to 40-50°C and subjected to a gradient cooling process at a cooling rate of 5°C per 1 to 1.5 hours to slowly form a solid precipitate. The resulting mixture was filtered, the filter cake was washed with methanol, and vacuum dried to obtain the compound of formula 4 in a 91% yield and 99.7% purity. |
| The NMR data for the compound of Formula 4 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 8.73 (1H, d, J = 2.0 Hz); 8.52 (1H, t, J = 2.0 Hz); 8.21 (1H, d, J = 2.0 Hz); 8.06 (1H, s); 7.86 (1H, dd, J1 = 2.0 Hz; J2 = 8.0 Hz); 7.49 (1H, dd, J1 = 1.6 Hz; J2 = 7.6 Hz); 3.86 (3H, s); 2.56 (3H, s). |
| Step 3: |
| |
| Under nitrogen, THF (448 mL), compound of formula 4 (29.1 g, 1 eq), and compound of formula 5 (24.6 g, 0.9 eq) were added, stirred, and cooled to -65°C to -60°C. At this temperature, potassium tert-butoxide (19 g x 3) was added in batches every 0.5 h. The reaction process was controlled by liquid phase detection. After 2 hours, the reaction temperature was raised to -5 to 0°C. The reaction solution was washed with purified water, stirred for 0.5-1 hour, washed with brine, and separated to obtain an organic phase. N-acetyl-L-cysteine (11.41 g, 0.7 eq) was added to the organic phase, stirred, washed with brine, neutralized, and concentrated under reduced pressure. The resulting filter cake was washed with purified water and made into a slurry. The resulting product was washed again with purified water and dried under vacuum to obtain compound of formula 6 with a yield of 88.2% and a purity of 98.6%. |
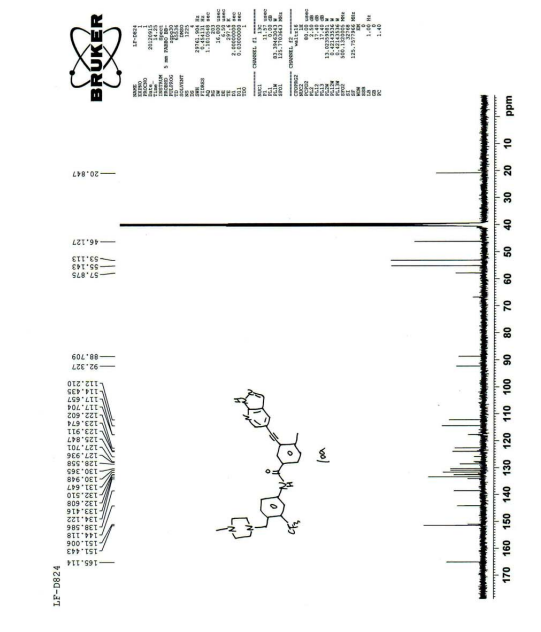
| The NMR data for the compound of formula 6 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 10.53 (1H, s); 8.75 (d, J = 2.0); 8.53 (d, J = 2.4); 8.24 (1H, s); 8.23 (d, J = 2.4); 8.21 (d, J = 1.6); 8.09 (dd, J1 = 1.6; J2 = 8.4); 7.94 (dd, J1 = 2.0; J2 = 8.0); 7.71 (d, J = 8.8); 7.53 (d, J = 8.0); 3.56 (2H, s); 2.59 (3H, s); 2.34-2.35 (8H, m), 2.16 (3H, s). |
| Its carbon spectrum data are 13 C NMR (100 MHz, d-DMSO): δ ppm: 20.38, 45.65, 52.64, 54.67, 57.41, 88.26, 91.86, 111.76, 113.98, 117.19, 122.14, 123.43, 127.35 (q), 124.30 (q), 128.10, 129.89, 130.49, 131.15, 132.02, 132.13, 132.93, 133.66, 138.15, 143.65, 150.55, 164.64. |
PATENT
CN 101885722
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84081329&_cid=P10-MDPKML-68458-1
| Example 23 |
| 3-((1H-pyrazolo[3,4-b]pyridine-5-substituted)ethynyl)-4-methyl-N-(4-((4-methylpiperazine-1-substituted)methyl)3-(trifluoromethyl)phenyl)benzamide (D824) |
| (3-((1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)m ethyl)-3-(trifluoromethyl)phenyl)benzamide) |

| The synthesis method is the same as in Example 1. |
| 1 HNMR (400MHz, d-DMSO), δ13.92 (s, 1H), 10.55 (s, 1H), 8.72 (d, J=2.0Hz, 1H), 8.52 (d, J=2.0Hz, 1H), 8.17 (m, 3H), 8.10 (d, J=8.0Hz, 1H), 7.92 (dd, J=8.0, 2.0Hz, 1H), 7.70 (d, J=8.8Hz, 1H), 7.53 (d, J=8.0Hz, 1H), 3.80 (s, 2H), 3.10 (brs, 8H), 2.71 (s, 3H), 2.57 (s, 3H). |
| MS(ESI), m/z: 533, (M + +H + ). |
SYN
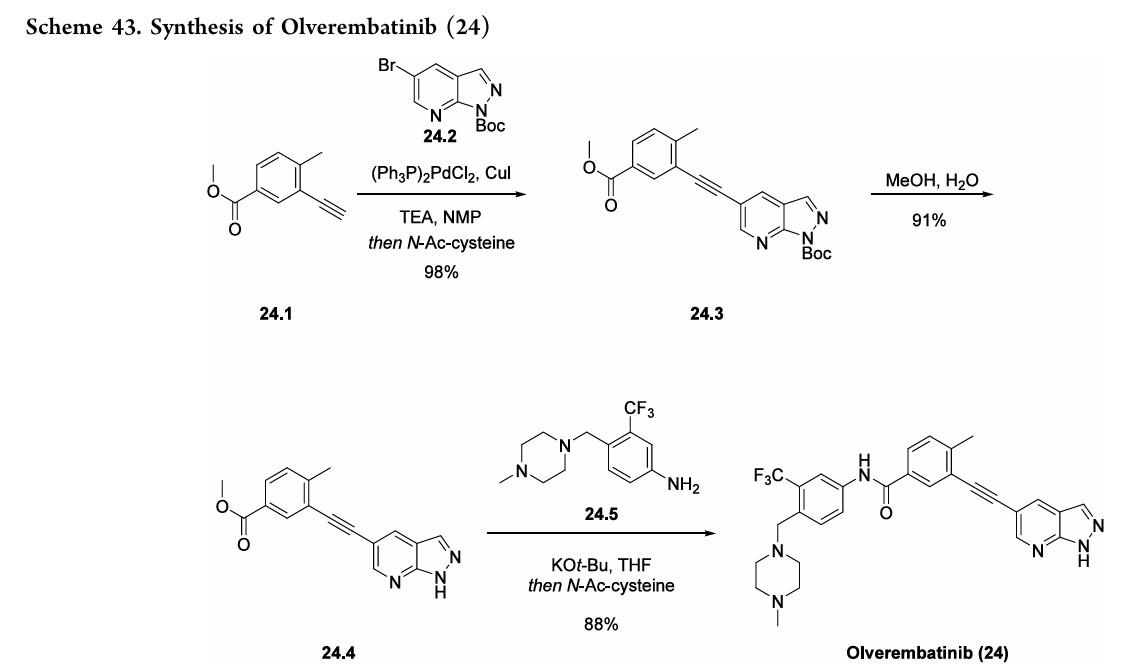
Olverembatinib(24) wasdeveloped by Ascentage Pharma as anorally available, third-generation
tyrosinekinase inhibitor (TKI) for the treatment of chronic myeloid leukemia (CML), acute myeloid leukemia, acute lymphoblastic leukemia (ALL), and solid tumors.167 It received its first approval inChina inNovember 2021 and was approved for use in adults with TKI-resistant CML chronicphaseandCML-acceleratephaseharboringtheT315I “gatekeeper” mutation.168 The current mainstay of CML
treatmentiscenteredaroundTKIs;however,resistancetoTKItherapy, often through BCR-ABL1 kinase domain point mutations, remains a challenge for early generation therapies.169Olverembatinibretainsitsefficacybyfunctioningasan ATP-bindingsiteinhibitorofwild-typeBCR-ABL1kinaseand broadly relatedmutants including T315I, which otherwise confers resistance against all first and second generation TKIs.168
Thesynthesisofolverembatinibhasbeenreportedinseveral patents,170−172 aswell as a journal article173 that details the divergentapproachtorelatedanalogues. Inarecentpatent,170 the synthesis of olverembatinib began with a Sonogashira coupling of commercially available alkyne 24.1 with
bromopyridine24.2toaffordester24.3in98%yield(Scheme43). Cleavage of the N-Boc group was accomplished by refluxingcarbamate24.3inaMeOHandwatermixturetogive pyrazole24.4 in91%yield. AfinalKOtBumediatedamide formation with aniline 24.5 resulted in the isolation of
olverembatinib(24) in88%yield.
(167) Dhillon, S. Olverembatinib: First approval. Drugs 2022, 82,
469−475.
(168) Braun, T. P.; Eide, C. A.; Druker, B. J. Response and resistance
to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530−542.
(169) Shoukier, M.; Kubiak, M.; Cortes, J. Review of new-generation
tyrosine kinase inhibitors for chronic myeloid leukemia. Curr. Oncol.
Rep. 2021, 23, 91.
(170) Wen, J.; Feng, J.; Wu, T.; Cai, M.; Teng, S. Preparation
method of alkynyl containing compound and its intermediate. China
Patent CN 114163434, 2022.
(171) Guo, M.; Wen, J.; Teng, S.; Wu, T.; Feng, J. Preparation of
(trifluoromethylphenyl)(pyrazolo[3,4-b]pyridinylethynyl)benzamide
derivative. China Patent CN 113292556, 2021.
(172) Ding, K.; Wang, D.; Pei, D.; Zhang, Z.; Shen, M.; Luo, K.;
Feng, Y. Heterocyclic alkynylbenzene derivatives as cancer cell line
inhibitors and their preparation, pharmaceutical compositions and use
in the treatment of cancer. China Patent CN 101885722, 2010.
(173) Ren, X.; Pan, X.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.;
Long, H.; Luo, J.; Feng, Y.; et al. Identification of GZD824 as an
orally bioavailable inhibitor that targets phosphorylated and non
phosphorylated breakpoint cluster region−abelson (Bcr-Abl) kinase
and overcomes clinically acquired mutation-induced resistance against
imatinib. J. Med. Chem. 2013, 56, 879−894.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Dhillon, Sohita (March 2022). “Olverembatinib: First Approval”. Drugs. 82 (4): 469–475. doi:10.1007/s40265-022-01680-9. PMID 35195876. S2CID 247027755.
- Jiang, Qian; Li, Zongru; Qin, Yazhen; Li, Weiming; Xu, Na; Liu, Bingcheng; Zhang, Yanli; Meng, Li; Zhu, Huanling; Du, Xin; Chen, Suning; Liang, Yang; Hu, Yu; Liu, Xiaoli; Song, Yongping; Men, Lichuang; Chen, Zi; Niu, Qian; Wang, Hengbang; Lu, Ming; Yang, Dajun; Zhai, Yifan; Huang, Xiaojun (18 August 2022). “Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial”. Journal of Hematology & Oncology. 15 (1): 113. doi:10.1186/s13045-022-01334-z. PMC 9389804. PMID 35982483.
- Jiang, Qian; Huang, Xiaojun; Chen, Zi; Niu, Qian; Shi, Dayu; Li, Zongru; Hou, Yue; Hu, Yu; Li, Weiming; Liu, Xiaoli; Xu, Na; Song, Yongping; Zhang, Yanli; Meng, Li; Hong, Zhenya; Liu, Bingcheng; Zeng, Shan; Men, Lichuang; Li, Yan; Chen, Suning; Xue, Mengxing; Zhu, Huanling; Li, He; Du, Xin; Lou, Jin; Zhang, Xiaohan; Liang, Yang; Dai, Yujun; Lu, Ming; Wang, Hengbang; Ji, Jiao; Yue, Changai; Yang, Dajun; Zhai, Yifan (5 November 2020). “Novel BCR-ABL1 Tyrosine Kinase Inhibitor (TKI) HQP1351 (Olverembatinib) Is Efficacious and Well Tolerated in Patients with T315I-Mutated Chronic Myeloid Leukemia (CML): Results of Pivotal (Phase II) Trials”. Blood. 136 (Supplement 1): 50–51. doi:10.1182/blood-2020-142142. S2CID 228875477.
| Clinical data | |
|---|---|
| Other names | GZD-824; GZD824 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1257628-77-5 |
| PubChem CID | 51038269 |
| IUPHAR/BPS | 10630 |
| DrugBank | DB16185 |
| ChemSpider | 29395146 |
| UNII | KV1M7Q3CBP |
| ChEMBL | ChEMBL2316582 |
| CompTox Dashboard (EPA) | DTXSID301352011 |
| Chemical and physical data | |
| Formula | C29H27F3N6O |
| Molar mass | 532.571 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
[1]. Ren X, Pan X, Zhang Z, Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-Abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem. 2013 Feb 14;56(3):879-94. [Content Brief]
//////////Olverembatinib, approvals 2021, china 2021, Ascentage Pharma, cancer, HQP1351, HQP 1351, D-824, D 824, KV1M7Q3CBP, GZD824
Dorzagliatin
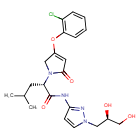
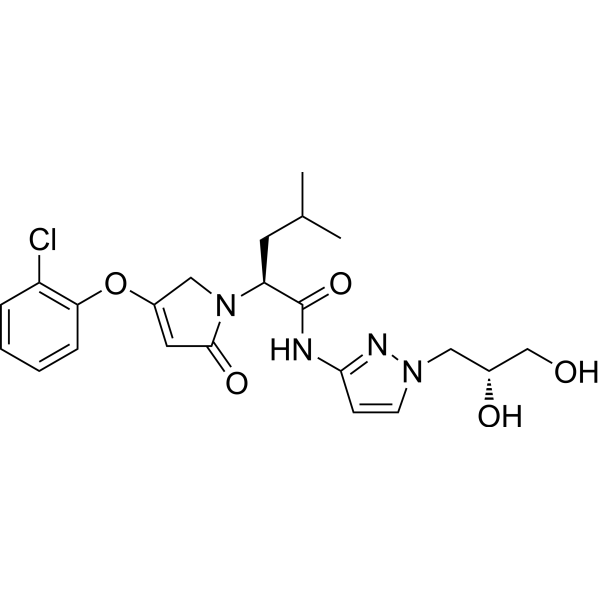
Dorzagliatin
- CAS 1191995-00-2
- HMS5552
- Sinogliatin
- HMS-5552
- MW 462.9 g/mol MF C22H27ClN4O5
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methylpentanamide
- RO5305552
- RO-5305552
- X59W6980E8
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methyl-pentanamide
- 1H-PYRROLE-1-ACETAMIDE, 4-(2-CHLOROPHENOXY)-N-(1-((2R)-2,3-DIHYDROXYPROPYL)-1H-PYRAZOL-3-YL)-2,5-DIHYDRO-.ALPHA.-(2-METHYLPROPYL)-2-OXO-, (.ALPHA.S)-
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM). CHINA 2022
Dorzagliatin is a glucokinase activator that is being developed to treat diabetes.[1] Unlike other diabetes drugs, it is intended to increase insulin sensitivity.[2]
Dorzagliatin is under investigation in clinical trial NCT03173391 (Long-term Efficacy and Safety of HMS5552 in T2DM).
PATENT
https://patents.google.com/patent/CN112062754A/en
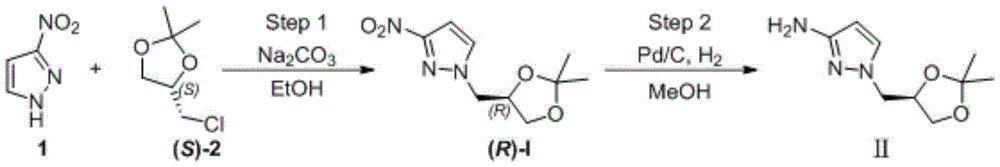
(R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is a very important medical intermediate for synthesizing Dorzagliatin. Dorzagliatin is a novel medicine for treating type 2 diabetes mellitus, and (R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is an essential intermediate in the synthetic process of the medicine, and along with the steady promotion of new Dorzagliatin medicines to the market, the demand of the chiral intermediate in the market is required to be rapidly increased.
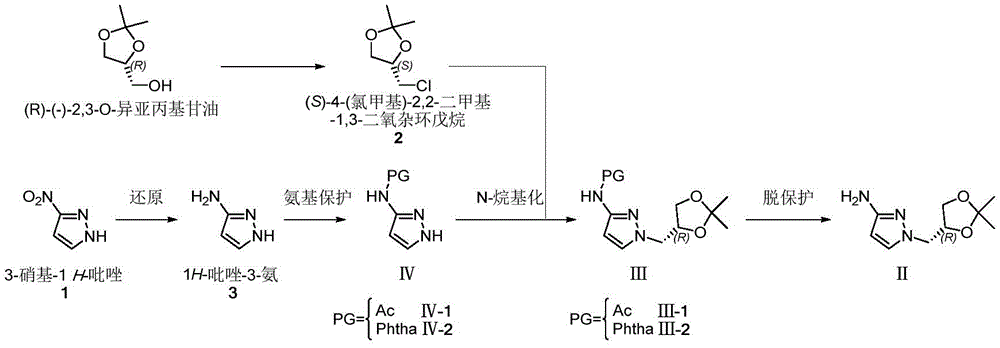
The main production method of the key chiral intermediate is shown as follows: reducing nitro in 3-nitro-1H-pyrazole substrate into amino, protecting free amino, carrying out N-alkylation reaction with (R) – (-) -2, 3-O-isopropylidene glycerol-OH derivative active intermediate, and deprotecting to obtain the final product. The synthetic route needs to be subjected to an N-protection process, so that route steps are added, and the cost is increased. The synthesis of N-protected substrate iv is reported: in the patent US2013203802, 1H-pyrazole-3-ammonia is protected by acetic anhydride, and in WO2017040757, N-acetyl-1H-pyrazole-3-ammonia is obtained by an N- (1-benzyl-1H-pyrazole-3-yl) acetamide debenzylation method; the protection of the N-benzoyl group of 1H-pyrazol-3-amine is reported in the patent US 6118008; in addition, WO2009106209, US2012095064, mention the phthalimide protection strategy of 1H-pyrazole-3-ammonia with phthalic anhydride.
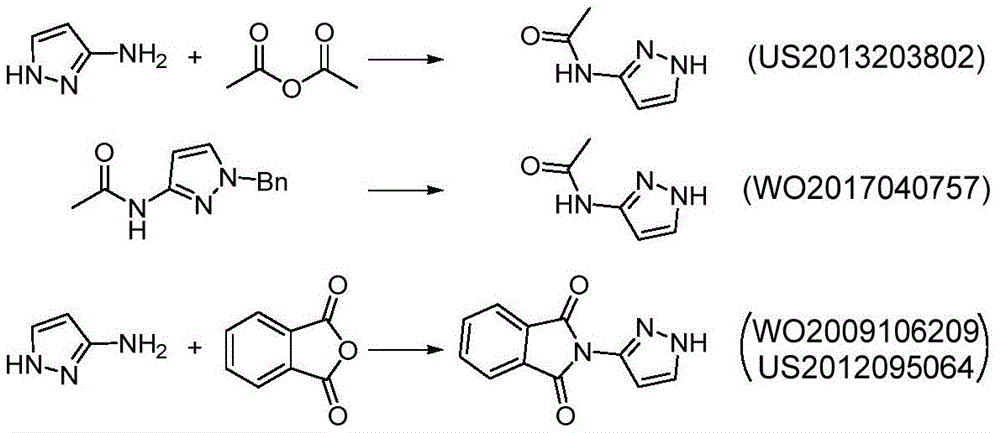
Example 1
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine
The first step is as follows: intermediate (R) -I preparation:
under the protection of nitrogen, 3-nitro-1H-pyrazole (1) (100.00g,0.884mol), ethanol (1.0L) and sodium carbonate (133.90g, 1.26mol) are sequentially added into a 3L reaction bottle, and the system is stirred for 0.5H at room temperature; (S) – (-) -4-chloromethyl-2, 2-dimethyl-1, 3-dioxolane ((S) -2) (126.84g, 0.842mol) was dissolved and diluted with 634ml of ethanol and then added dropwise to the reaction flask. After the dropwise addition, the temperature is raised to 50 ℃ and the reaction is stirred for 5 hours. Ethanol was distilled off under reduced pressure, and the residue was diluted with (1.0L) of water and then extracted twice with dichloromethane (500ml × 2); the organic phase was washed with water and then with saturated sodium chloride brine. Concentrating under reduced pressure to remove dichloromethane to obtain crude oily substance; the crude product was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate mixed system) to give 166.5g of a pale yellow oily product, with a yield of 87% and an ee value of 98% or more.
The second step is that: reducing nitro to obtain target product
A2L autoclave was charged with (R) -I substrate (150g, 0.66mol), methanol (750mL), Pd/C (0.75g, 0.5% W/W), and the mixture was subjected to nitrogen substitution three times, then hydrogen substitution three times, under a hydrogen-charging pressure of 2.0MPa, at a temperature of 50 ℃ for reaction for 8 hours. Filtering, filtering to remove Pd/C catalyst, concentrating the filtrate to remove methanol to obtain 123.70g of light yellow oily matter, wherein the yield is 95%, and the ee value is more than or equal to 98%.
Example 2
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by Raney-Ni reduction system
The first step is the same as in example 1.
The second step is that: reduction of nitro groups by Rany-Ni
The intermediate (R) -I (150g, 0.66mol) obtained in the first step was charged into a 2L reactor, and ethanol (1.2L) was added thereto and stirred, followed by adding Rany-Ni (75g) and stirring at room temperature for reaction for 15 hours. Filtering, filtering to remove the solid catalyst, and concentrating the filtrate to dryness to obtain 106.77g of light yellow oily substance with yield of 82% and ee value of more than or equal to 97%.
Example 3
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by hydrazine hydrate system
The first step is the same as in example 1.
The second step is that: A2L reaction flask was charged with intermediate (R) -I (150g, 0.66mol), ferric trichloride (528mg, 3.3mmol), and ethanol (1.2L), stirred, charged with hydrazine hydrate (39.5g, 0.79mol), and heated to reflux for 6 h. Ethanol was removed by concentration under reduced pressure, the residue was diluted with 750ml of water and extracted twice with ethyl acetate (250 ml. times.2). The organic phase was washed with water and then with saturated brine. The ethyl acetate is removed by concentration to obtain 110.7g of crude light yellow oily substance, the yield is 85 percent, and the ee value is more than or equal to 97 percent.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374J.Med.Chem.2024,67,4376−4418
Dorzagliatin(HuaTangNing).
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM).133 This first-in-class, small
molecule,oral,glucokinaseactivator(GKA)wasfirst approved in ChinainSeptember2022foradultpatientswithT2DMasa monotherapy and in combination with metformin (an antidiabetic medication).134 Expression of glucokinase is reduced for individuals with T2DM, thus GKAs such as dorzagliatin serve as a novel class of antidiabetic treatment options.135,136 Theinitialpatent thatdisclosesthesynthesisofdorzagliatin (18)began fromreadily availablematerials 3-aminopyrazole
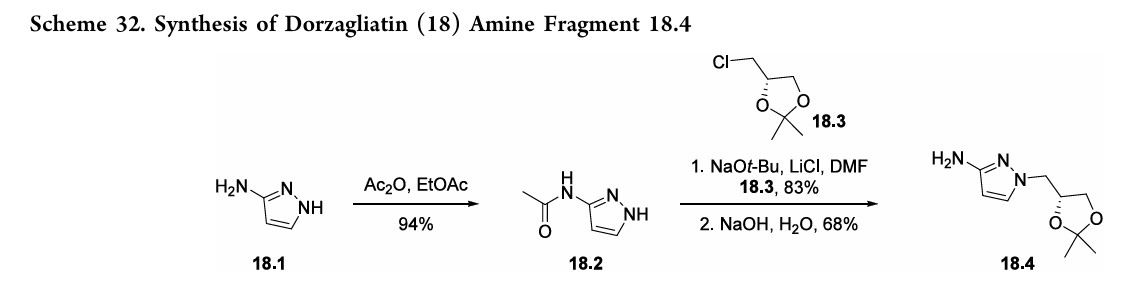
(18.1) and 2-chlorophenol (18.5). The synthetic strategy reliedonaconvergentamidecouplingofamine18.4(Scheme32) and carboxylic acid 18.9 (Scheme 33).137 A later disclosure provided an updated route toward amine 18.4 (Scheme 32), detailing the synthetic improvements with respect to yield and purity.138 This later disclosure also detailed the synthesis of dorzagliatinonmultikilogramscale fromtheamidationofacid18.9withamine18.4,yieldingover
10kgoftheactivepharmaceutical ingredient.Acetylationof3 aminopyrazole (18.1) with acetic anhydride provided the protectedpyrazole18.2(Scheme32). Subsequent alkylation with alkyl chloride 18.3 followed by base-mediated deprotectionyieldedamine18.4. The synthesis of acid 18.9 began with base-mediated
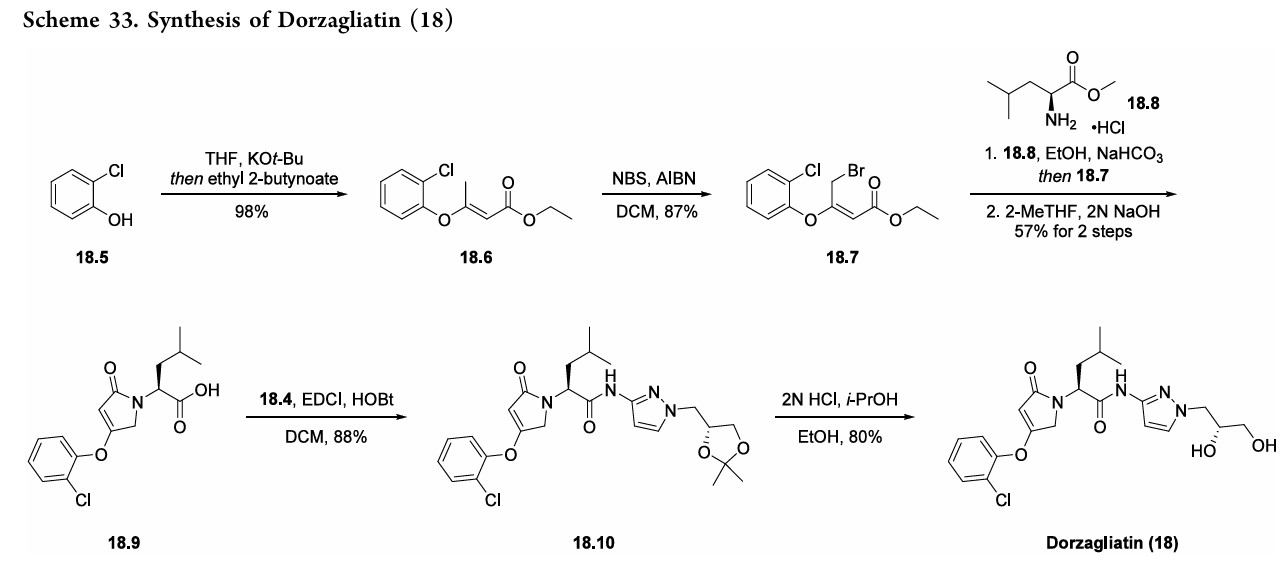
alkenylationof2-chlorophenol (18.5)withethyl 2-butynoate toprovideester18.6(Scheme33). Subsequentbromination withNBSandAIBNyieldsallylbromide18.7.Next,subjection
ofL-leucinemethylesterhydrochloride(18.8)tobaseresulted ina freeamine thatunderwent allylationwithbromide18.7. Acid 18.9was subsequently generated froma cyclization
condensation sequence and saponification reaction with NaOH. Final amidebondformationwas facilitatedbyEDCI andHOBt toprovideamide18.10, anddorzagliatin(18)was generatedonthemultikilogramscale followingacid-mediated acetonidedeprotectiontoreveal the1,2-diol.
(133) Syed, Y. Y. Dorzagliatin: First approval. Drugs 2022, 82,
1745−1750.
(134) Xu, H.; Sheng, L.; Chen, W.; Yuan, F.; Yang, M.; Li, H.; Li, X.;
Choi, J.; Zhao, G.; Hu, T.; et al. Safety, tolerability, pharmacokinetics,
and pharmacodynamics of novel glucokinase activator HMS5552:
results from a first-in-human single ascending dose study. Drug Des.
Devel. Ther. 2016, 10, 1619−26.
(135) Ren, Y.; Li, L.; Wan, L.; Huang, Y.; Cao, S. Glucokinase as an
emerging anti-diabetes target and recent progress in the development
of its agonists. J. Enzyme Inhib. Med. Chem. 2022, 37, 606−615.
(136) Toulis, K. A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.
H.; Tahrani, A. A. Glucokinase activators for type 2 diabetes:
Challenges and future developments. Drugs 2020, 80, 467−475.
(137) Berthel, S. J.; Brinkman, J. A.; Hayden, S.; Haynes, N.-E.;
Kester, R. F.; McDermott, L. A.; Qian, Y.; Sarabu, R.; Scott, N. R.;
Tilley, J. W. Pyrrolidinone as glucokinase activators and their
preparation, pharmaceutical compositions and use in the treatment
of metabolic disorders. WO 2009127546, 2009.
(138) Chen, J.; Ren, Y.; She, J.; Wang, L. Process for the preparation
of 1-([1,3]dioxolan-4-ylmethyl)-1h-pyrazol-3-ylamine. U.S. Patent US
20150315176, 2015.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Chow, Elaine; Wang, Ke; Lim, Cadmon K.P.; Tsoi, Sandra T.F.; Fan, Baoqi; Poon, Emily; Luk, Andrea O.Y.; Ma, Ronald C.W.; Ferrannini, Ele; Mari, Andrea; Chen, Li; Chan, Juliana C.N. (1 February 2023). “Dorzagliatin, a Dual-Acting Glucokinase Activator, Increases Insulin Secretion and Glucose Sensitivity in Glucokinase Maturity-Onset Diabetes of the Young and Recent-Onset Type 2 Diabetes”. Diabetes. 72 (2): 299–308. doi:10.2337/db22-0708. PMC 9871194.
- Zhu, Dalong; Li, Xiaoying; Ma, Jianhua; Zeng, Jiao’e; Gan, Shenglian; Dong, Xiaolin; Yang, Jing; Lin, Xiaohong; Cai, Hanqing; Song, Weihong; Li, Xuefeng; Zhang, Keqin; Zhang, Qiu; Lu, Yibing; Bu, Ruifang; Shao, Huige; Wang, Guixia; Yuan, Guoyue; Ran, Xingwu; Liao, Lin; Zhao, Wenjuan; Li, Ping; Sun, Li; Shi, Lixin; Jiang, Zhaoshun; Xue, Yaoming; Jiang, Hongwei; Li, Quanmin; Li, Zongbao; Fu, Maoxiong; Liang, Zerong; Guo, Lian; Liu, Ming; Xu, Chun; Li, Wenhui; Yu, Xuefeng; Qin, Guijun; Yang, Zhou; Su, Benli; Zeng, Longyi; Geng, Houfa; Shi, Yongquan; Zhao, Yu; Zhang, Yi; Yang, Wenying; Chen, Li (May 2022). “Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial”. Nature Medicine. 28 (5): 965–973.
- [1]. Zhu XX, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic β-cell function in patients with type 2 diabetes: A 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018 Sep;20(9):2113-2120. [Content Brief][2]. Wang P, et al. Effects of a Novel Glucokinase Activator, HMS5552, on Glucose Metabolism in a Rat Model of Type 2 Diabetes Mellitus. J Diabetes Res. 2017;2017:5812607. [Content Brief]
//////////Dorzagliatin, APPROVALS 22, CHINA 22, DIABETES, Hua Medicine, 1191995-00-2, HMS 5552, Sinogliatin, HMS-5552, RO 5305552, RO-5305552, X59W6980E8
Chiglitazar
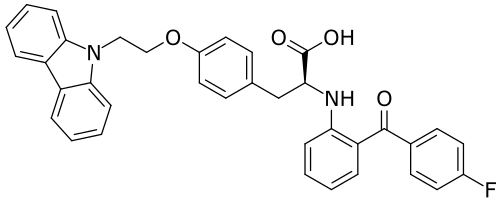
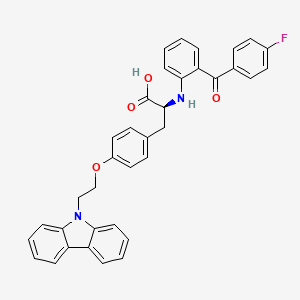
Chiglitazar
CAS 743438-45-1
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Chiglitazar sodium, (S)- | YN12H6OCV6 | 2390374-10-2 | RMVIEXHXRDCWBT-UCRKPPETSA-M |
- CS 038
- Carfloglitazar, (s)-
- E6EJV1J6Y0
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- C36H29FN2O4
- 572.6 g/mol
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- (2S)-3-(4-(2-CARBAZOL-9-YLETHOXY)PHENYL)-2-(2-(4-FLUOROBENZOYL)ANILINO)PROPANOIC ACID
- (2s)-3-(4-(2-carbazol-9-ylethoxy)phenyl)-2-(2-(4-fluorobenzoyl)anilino)propanoic acid
- Carfloglitazar, (s)-
- L-tyrosine, o-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-
- O-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-l-tyrosine
Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.
Chiglitazar (trade name Bilessglu) is a drug for the treatment of type 2 diabetes.[1] It is a peroxisome proliferator-activated receptor (PPAR) agonist.
In China, chiglitazar is approved for glycemic control in adult patients with type 2 diabetes when used in combination with diet and exercise.[2]
Chiglitazar is under investigation in clinical trial NCT06125587 (Chiglitazar/metformin in Non-obese Women With PCOS).
SYN
WO 2004048333
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004048333&_cid=P12-MDMUOB-48741-1
Example 15
Preparation of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-carbazolylethoxy)-phenyl]
-propionic acid (compound CS038)
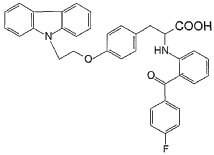
To a solution of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-bromoethoxy)-phenyl] -propionic acid methyl ester (0.25 g, 0.49 mmol) and carbazole (0.082 g, 0.49 mmol) in benzene (10 ml) is added tetrabutyl ammonium bromide (0.08 g) and 50% NaOH aqueous solution (0.084 g, 1.08 mmol), then the mixture is heated to reflux for 10 h. After cooled, benzene (30ml) is added, and the mixture is washed with water (3×30 ml). Then the solvent is evaporated under a vacuum. The crude product is purified by silica gel chromatography using CHCl3/MeOH (4:1) as eluent to give the title compound (0.10 g, 36%). HRMS calcd for C36H29FN204: 572.6357. Found: 572.6354. MA calcd for C36H29FN204: C, 75.51%; H, 5.11%; N, 4.89%. Found: C, 75.83%; H, 5.10%; N, 4.90%.
PATENT
US 10640465
https://patentscope.wipo.int/search/en/detail.jsf?docId=US249083802&_cid=P12-MDMUQY-52500-1
The pharmacological activity of the compound is described in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157. 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is able to selectively activate PPAR-α, PPAR-γ and PPAR-6, and can be used to treat the diseases associated with metabolic syndrome such as diabetes, hypertension, obesity, insulin resistance, hypertriglyceridemia, hyperglycemia, high cholesterol, arteries atherosclerosis, coronary heart disease, etc. A preparation method of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is disclosed in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157, and the synthetic route thereof is as follows:
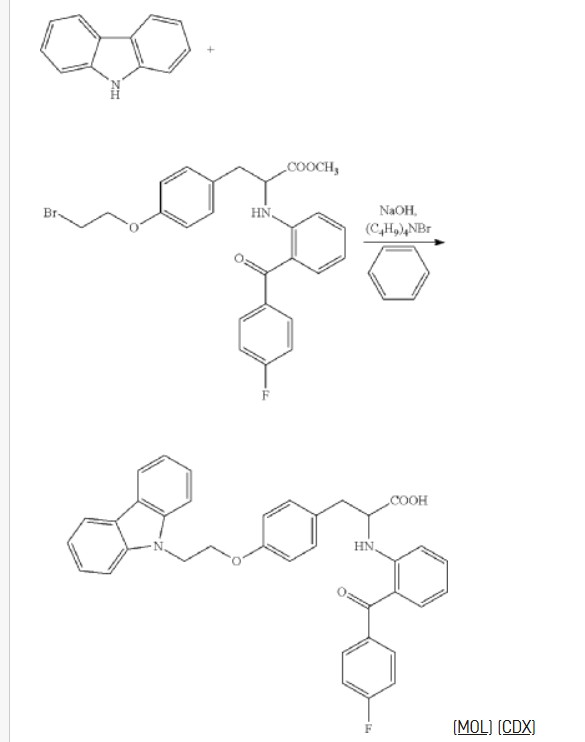
EXAMPLES
Example 1: Preparation of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic Acid
SYN
J. Med. Chem. 2024, 67, 4376−4418
Chiglitazar (Bilessglu). Chiglitazar (17), a novel nonthiazolidinedione pan-agonist of α, δ, and γ peroxisome proliferator-activated receptors (PPARs), has shown promise for the treatment of type 2 diabetes. 126 Type 2 diabetes impacts over 374 million patients worldwide and continues to
rise in incidence and prevalence globally. 127 Chiglitazar preferentially regulates expression of ANGPTL4 and PDK4 genes, which are involved in glucose and lipid metabolism. 128 Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
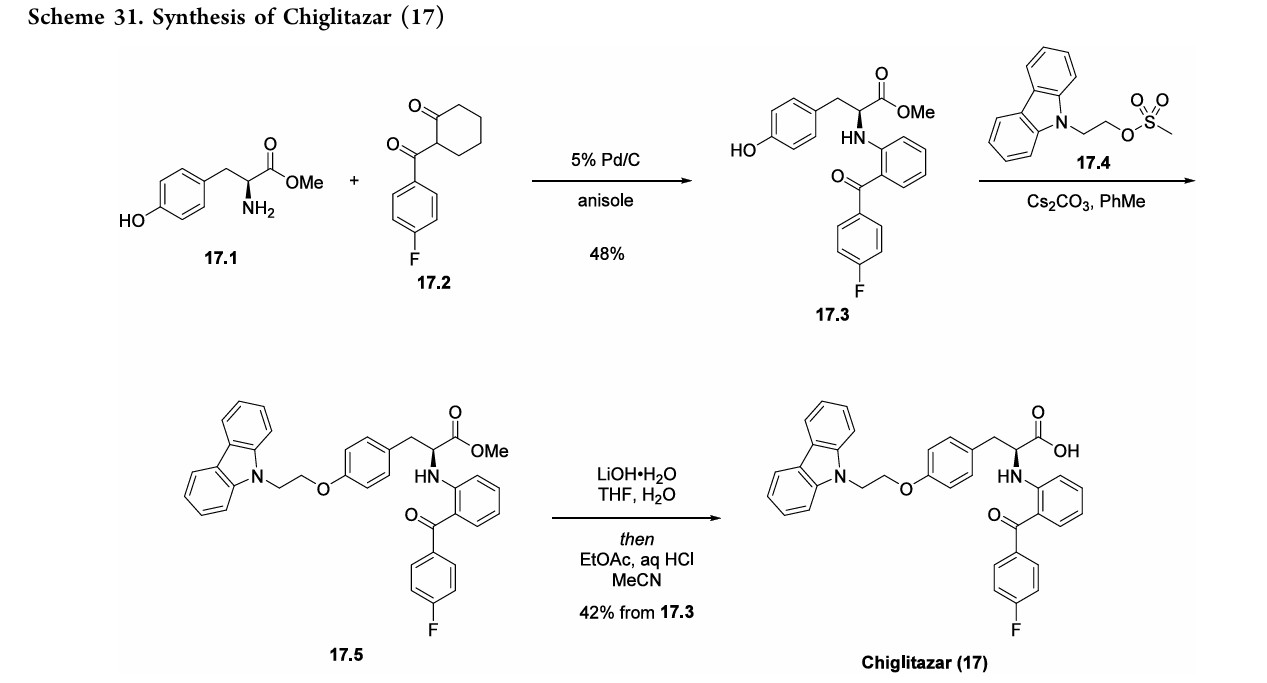
patients with type2 diabetes in October2021.129 Thesynthesisof17beganwithimineformationbetweenL
tyrosine methyl ester (17.1) and 2-(4-fluorobenzoyl) cyclohexanone(17.2)with tandemaromatizationunderPd/C catalysis to generate aniline derivative 17.3 (Scheme31).130,131 Alkylation of the phenol moiety of 17.3 with mesylate17.4furnishedphenyl alkyl etherderivative17.5.132
Hydrolysisof themethylester in17.5withlithiumhydroxide followedbyacidificationwithhydrochloricacidandrecrystal lization fromacetonitrile afforded chiglitazar (17) in 42% overall yield from17.3.Thisprocessdeliveredchiglitazar in 99.4%purityat24gscale.
(126) Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.;
Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a
novel peroxisome proliferator-activated receptor pan-agonist, in
patients with type 2 diabetes: a randomized, double-blind, placebo
controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571−1580.
(127) Chatterjee, S.; Khunti, K.; Davies, M. J. Type 2 diabetes.
Lancet 2017, 389, 2239−2251.
(128) Pan, D.-S.; Wang, W.; Liu, N.-S.; Yang, Q.-J.; Zhang, K.; Zhu,
J.-Z.; Shan, S.; Li, Z.-B.; Ning, Z.-Q.; Huang, L.; Lu, X.-P. Chiglitazar
preferentially regulates gene expression via configuration-restricted
binding and phosphorylation inhibition of PPARγ. PPAR Research
2017 2017, 2017, 1−16.
(129) Deeks, E. D. Chiglitazar: First approval. Drugs 2022, 82, 87−
92.
(130) Li, Z.; Lu, X.-P.; Liao, C.; Shi, L.; Liu, Z.; Ma, B. Substituted
arylalcanoic acid derivatives as PPAR pan agonists with potent
antihyperglycemic and antihyperlipidemic activity. WO 2004048333
A1, 2004.
(131) Sutter, M.; Sotto, N.; Raoul, Y.; Métay, E.; Lemaire, M.
Straightforward heterogeneous palladium catalyzed synthesis of aryl
ethers and aryl amines via a solvent free aerobic and non-aerobic
dehydrogenative arylation. Green Chem. 2013, 15, 347−352.
(132) Lu, X.; Li, Z.; Wang, X. Method for preparing phenylalanine
compound. U.S. Patent US 10640465 B2, 2020.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji L, Song W, Fang H, Li W, Geng J, Wang Y, et al. (August 2021). “Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial (CMAP)”. Science Bulletin. 66 (15): 1571–1580. Bibcode:2021SciBu..66.1571J. doi:10.1016/j.scib.2021.03.019. PMID 36654286. S2CID 233650336.
- Deeks ED (January 2022). “Chiglitazar: First Approval”. Drugs. 82 (1): 87–92. doi:10.1007/s40265-021-01648-1. PMID 34846697. S2CID 244716275.
| Clinical data | |
|---|---|
| Trade names | Bilessglu |
| Other names | Carfloglitazar |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 743438-45-1 |
| PubChem CID | 71402018 |
| ChemSpider | 57523239 |
| UNII | E6EJV1J6Y0 |
| ChEMBL | ChEMBL4650349 |
| CompTox Dashboard (EPA) | DTXSID00225352 |
| Chemical and physical data | |
| Formula | C36H29FN2O4 |
| Molar mass | 572.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Chiglitazar, Chipscreen Biosciences, CHINA 2021, DIABETES, CS 038, Carfloglitazar, (s)-, E6EJV1J6Y0,
Vamorolone

Vamorolone
CAS 13209-41-1
| Molecular Weight | 356.46 |
|---|---|
| Formula | C22H28O4 |
- Agamree
- 17,21-Dihydroxy-16alpha-methylpregna-1,4,9(11)-triene-3,20-dione
- VBP-15 free alcohol
- (8S,10S,13S,14S,16R,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-7,8,12,14,15,16-hexahydro-6H-cyclopenta[a]phenanthren-3-one
- (16alpha)-17,21-dihydroxy-16-methylpregna-1,4,9(11)-triene-3,20-dione
- (8S,10S,13S,14S,16R,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-7,8,12,14,15,16-hexahydro-6H-cyclopenta(a)phenanthren-3-one
- DTXCID601356317
- (1R,2R,3aS,3bS,9aS,11aS)-1-hydroxy-1-(2-hydroxyacetyl)-2,9a,11a-trimethyl-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,11H,11aH-cyclopenta(a)phenanthren-7-one
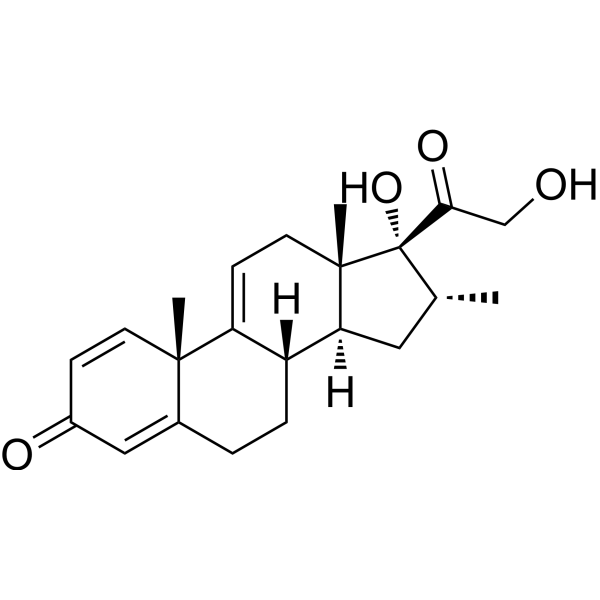
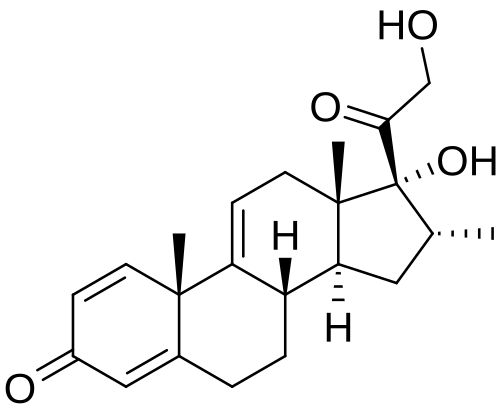
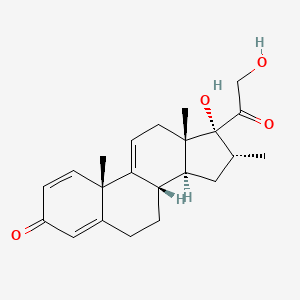
Vamorolone (VBP15) is a first-in-class, orally active dissociative steroidal anti-inflammatory agent and membrane-stabilizer. Vamorolone improves muscular dystrophy without side effects. Vamorolone shows potent NF-κB inhibition and substantially reduces hormonal effects.
Vamorolone, sold under the brand name Agamree, is a synthetic corticosteroid, which is used for the treatment of Duchenne muscular dystrophy.[4][5][6][7][8] It is taken by mouth.[1] It is a dual atypical glucocorticoid and antimineralocorticoid.[9]
The most common adverse reactions include cushingoid features, psychiatric disorders, vomiting, increased weight, and vitamin D deficiency.[10]
Vamorolone was approved for medical use in the United States in October 2023,[11][10] and in the European Union in December 2023.[2][3]
Vamorolone is a novel and fully synthetic glucocorticoid developed by Santhera Pharmaceuticals. It is used to manage inflammation and immune dysregulation in patients with Duchenne muscular dystrophy (DMD), a neuromuscular disorder characterized by the insidious regression of neuromuscular function and the most common form of muscular dystrophy in the United States. Corticosteroid therapy is the current standard of care for DMD despite relatively high rates of adverse effects. Vamorolone is positioned as having a more tolerable adverse effect profile than other corticosteroids owing to its unique receptor binding profile, thus providing an additional treatment option in patients for whom corticosteroid adverse effects are intolerable or otherwise unacceptable. Vamorolone was approved by the FDA in October 2023 for the management of DMD in patients ≥2 years of age. In December 2023, it was approved in the EU for the treatment of patients ≥4 years of age.
PATENT
https://patents.google.com/patent/WO2023016817A1/en
Vamorolone is currently produced from the commercially available 3TR (Tetraene acetate) – see Scheme 2. In step a, TMS imidazole, MeMgCI and THF are added to 3TR, with subsequent addition of CuAc2, H2O, DMPU, MeMgCI and THF in step b. Under treatment with peracetic acid in Toluene from compound 2 the intermediate 3 is formed in step c. After treatment with NaHSCO3 and TFA (step d), EtOAc and heptane (step e) and acetonitrile trituration (step f) HBr in CH2CI2 is added (step g) and MeOH (step h) is used for crystallization to form Acetyl- Vamorolone 4. Acetyl-Vamorolone is deacetylated with K2CO3 in MeOH, followed by HCI to obtain Vamorolone (step i). The synthesis is disclosed in Bioorganic & Medicinal Chemistry, Volume 21 , Issue 8, 15 April 2013, Pages 2241-2249.
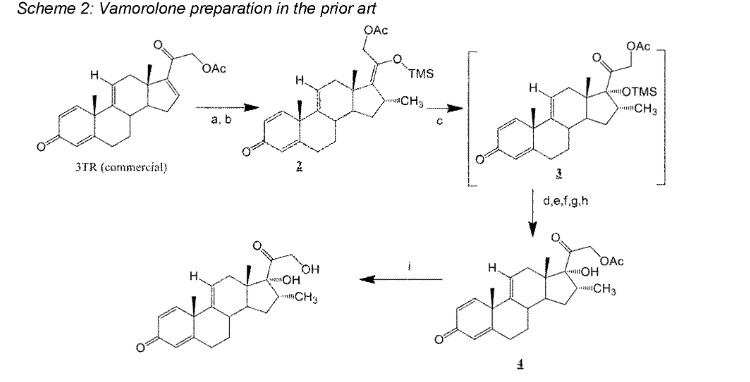
Example 2: Synthesis of the present invention
Scheme C: Route of Synthesis of Vamorolone from 8-DM

Vamorolone was synthesized in three synthetic steps from commercially available 8-DM.
The synthetic route started with the acetylation of 8-DM using acetic anhydride and catalytic DMAP in THF, followed by crystallization of 8-DM Acetate after aqueous quench. Then, a deoxygenation reaction converted 8-DM Acetate directly into Vamorolone Acetate. This deoxygenation proceeded via initial formation of an iodohydrin with excess aq. HI, followed by simultaneous I2 and H2O-elimination to give Vamorolone Acetate. During the reaction, partial de-acetylation occurred (20-25%) and therefore re-acetylation with acetic anhydride was necessary. After completed re-acetylation, Vamorolone Acetate was directly crystallized by addition of H2O. Finally, the acetate group is cleaved under basic conditions to give crude Vamorolone, which was recrystallized from iPrOH to obtain the pure product.
2.1 Acetylation

A 10 L glass dj ( double jacketed reactor )-reactor was charged with 8-DM (490 g, 1.32 mol, 1.0 eq.) and DMAP (16.1 g, 0.132 mmol, 0.10 eq.). THF (1.25 L, 2.5 vol.) was added at IT = 20-25 °C. Then, AC2O (201 g, 187 mL, 1.97 mol, 1.5 eq.) was added dropwise over 20-40 min, keeping IT below 30 °C during the addition. After complete addition, the reaction mixture was stirred at IT = 20-25 °C for 30 min. IPC control by LC/MS indicated >99% conversion of 8-DM to 8-DM Acetate.
The reaction mixture was quenched by dropwise addition of H2O (4.9 L, 10 vol.) over 30-45 min, keeping IT below 25 °C. The resulting aqueous suspension was aged at IT = 20-25 °C for 1 h. The product was filtered off, washed with H2O (3 x 0.5 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide 8-DM Acetate (539 g, 1.30 mol, 99% yield, >99% a/a, 98% w/w) as a white solid (cryst 1#1).
Analytical Data:
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of 8-DM relative to formation of 8- DM Acetate.
Detected mass: [M+1]= 373.19 for 8-DM and [M+1] = 415,19 8-DM Acetate. 2.2 Deoxygenation with HI

A 10 L glass dj-reactor was charged with 8-DM Acetate (500 g, 1.21 mol, 1.0 eq.). Toluene (2.5 L, 5 vol.) was added. The suspension was cooled to IT = 0-5 °C and then a solution of 57% aqueous HI (1 .08 kg, 637 mL, 4.83 mol, 4.0 eq.) in AcOH (1.25 L, 2.5 vol.) was added via peristaltic pump over 45-60 min, keeping IT below 5 °C during the addition. The resulting dark purple to brown solution was stirred at IT = 3-5 °C for 24 h. IPC control by LC/MS indicated >98% conversion of 8-DM Acetate/intermediate iodohydrin to Vamorolone Acetate/Vamorolone.
The reaction mixture was quenched by dropwise addition of 25% aq. Na2SO3 solution (2.0 L, 4 vol.) over 10-20 min, keeping IT below 15 °C. After complete addition, EtOAc (1.0 L, 2 vol.) was added and the biphasic mixture was warmed to IT = 15-20 °C. Stirring was stopped and the phases were separated (Organic Phase 1 and aqueous Phase 1 ; goal pH of the aqueous Phase 1 : 2; aqueous Phase 1 disposed). 25% aq. Na2SO3 solution (1.25 L, 2.5 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 2; goal pH aqueous Phase 2: 4-5; aqueous Phase 2 disposed). 25% aq. Na2SO3 solution (1.25 L, 2.5 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 3; goal pH aqueous Phase 3: 5-6; aqueous Phase 3 disposed). H2O (0.5 L, 1.0 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 4; goal pH aqueous Phase 4: 5-6; aqueous Phase 4 disposed).
A slight vacuum was applied to the double-jacketed reactor (100-150 mbar), containing Organic Phase 1 , and toluene was distilled off at 70 °C jacket temperature (ET) from the reaction mixture with continuous addition of MeCN, and the distillation continued until target residual toluene value has been reached (goal: less than 5% toluene according to 1 H-NMR of reaction mixture. Final volume in reactor after distillation: ca. 3.5 L (7.5 vol.).
Once toluene was removed, the vacuum was broken with N2 and resulting fine suspension cooled to IT = 20-25 °C. At this point, the amount of Vamorolone was assessed by IPC (typical ratio: Vamorolone Acetate to Vamorolone: 75:25; x = 25% a/a). DMAP (3.7 g, 0.0302 mol, 0.025 eq.) was added, followed by slow addition of AC2O (61.6 g, 57 mL, 0.603 mol, 0.5 eq.) over 5-10 min at IT = 20-25 °C. After complete addition of AC2O, the reaction mixture was stirred for 30 min at IT = 20-25 °C. IPC control by LC/MS indicated ≤ 2% a/a Vamorolone (ratio: Vamorolone Acetate to Vamorolone: 98.5:1.5).
The reaction mixture was quenched by slow addition of H2O (4.9 L, 10 vol.) over 15-30 min, keeping IT below 25 °C. The resulting aqueous suspension was cooled to IT = 0-5 °C and aged at this temperature for 2 h. The product was filtered off, washed with H2O/MeCN 4:1 (2 x 0.5 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone Acetate (301 g, 0.76 mol, 63% yield, 98% a/a, 98% w/w) as an off-white solid (cryst 1#1).
Over the course of the reaction, partial de-acetylation of Vamorolone Acetate to Vamorolone was observed (between 20-25% a/a). Therefore, after aq. workup and solvent switch to MeCN, the ratio of Vamorolone Acetate to Vamorolone was assessed by LC/MS (in % a/a), and the following amounts of DMAP and Ac2O were added: x = amount of Vamorolone in % a/a (e.g. x = 20% a/a)
DMAP eq. = (0.1 -x)/100 (e.g. 0.02 eq.)
Ac2O eq. = (2.0-x)/100 (e.g. 0.40 eq.)
Analytical Data
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of the sum of (8-DM Acetate + intermediate iodohydrin) relative to the sum of (Vamorolone Acetate + Vamorolone). Detected mass: [M+1] = 415,19 for 8-DM Acetate, [M+1]= 357,28 for Vamorolone, 399,20 for
Vamorolone Acetate and 543,12 for intermediate lodohydrin
2.3 De-Acetylation

A 10 L glass dj-reactor was charged with Vamorolone Acetate (280 g, 0.703 mol, 1.0 eq.). MeOH (1.54 L, 5.5 vol.) was added. The suspension was cooled to IT = 0-5 °C and then a solution of K2CO3 (107 g, 0.773 mol, 1.1 eq.) in H2O (0.7 L, 2.5 vol.) was added dropwise via peristaltic pump over 20-40 min, keeping IT below 10 °C during the addition. After complete addition, the reaction mixture was warmed IT = 20-25 °C and stirred for 5 h. IPC control by LC/MS indicated 99.3% conversion of Vamorolone Acetate to Vamorolone.
The reaction mixture was cooled to IT = 15-17 °C and quenched by dropwise addition of 1 M aq. HCI (950 mL, 0.95 mol, 1.35 eq.) over 20-40 min, keeping IT below 20 °C during the addition (goal pH: 5-6). The resulting aqueous suspension was aged at IT = 15-20 °C for 12 h. The product was filtered off, washed with H2O/MeOH 2:1 (3 x 0.3 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone (241.5 g, 0.68 mol, 96% yield, >99% a/a, 98% w/w) as a slightly yellow solid (crude 1#1).
Analytical Data
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of Vamorolone Acetate relative to formation of Vamorolone. 2.4 Recrystallization

A 10 L glass dj-reactor was charged with Vamorolone (230 g, 0.645 mol, 1.0 eq.). iPrOH (5 L, 22 vol.) was added. The suspension was heated to reflux (jacket temperature ET = 97 °C) and stirred until complete dissolution of Vamorolone occurred (10-15 min on this scale).
After complete dissolution, the clear yellow solution was slowly cooled to IT = 0-5 °C over the course of 12 h and then aged at IT = 0-5 °C for 1 h. The recrystallized product was filtered off, washed with cold iPrOH (2 x 250 mL), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone (201 g, 87% recovery, >99% a/a, 99% w/w) as an off white glimmery solid (cryst 1#1).
Iso-propanol (iPrOH) was found to the best solvent for recrystallization with excellent purity upgrading properties (by rejection of impurities), although a high dilution is necessary to completely dissolve the crude Vamorolone at reflux temperature. Higher concentrations for the recrystallization satisfactory results are obtainable using mixtures of isopropanol and water. Maximum solubility of Vamorolone was determined to be at reflux of a 80:20 (isopropanol : water) mixture.
PATENT
https://patents.google.com/patent/US20200281942A1/en
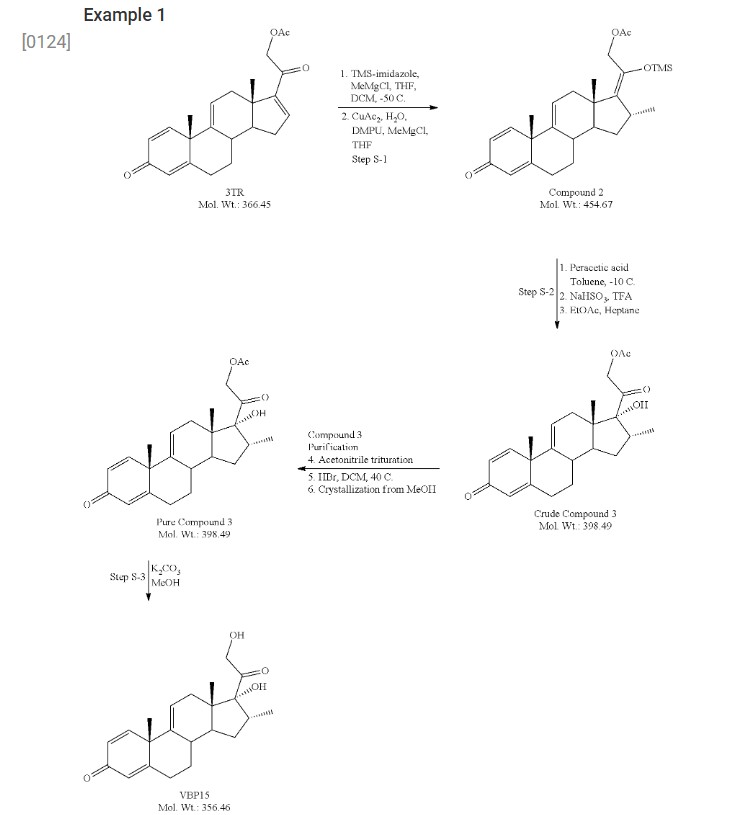
- [0124]
- [0125]3-TR (100 g, 273 mmol), dichloromethane (DCM, 500 mL) and tetrahydrofuran (THF, 400 mL) were charged to a reaction flask under nitrogen. To this was charged trimethylsilyl imidazole (TMS-imidazole, 65.3 g, 466 mmol, 1.7 eq). The resulting mixture was stirred at room temperature for 3 hours.
- [0126]In a separate flask, copper acetate monohydrate (5.4 g, 27 mmol), tetrahydrofuran (400 ml) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU, 53.3 g, 416 mmol) were combined and stirred at room temperature for approximately 3 hours. The blue mixture was subsequently cooled to −50° C., and to this was added methyl magnesium chloride solution (27 ml, 3.0 M in THF, 82 mmol) dropwise. After 30 minutes, the mixture had formed a deep blue, sticky “ball.”
- [0127]The 3-TR/TMS-imidazole mixture was cooled to −50° C. and to this was charged the copper acetate/DMPU solution above via canula. The residual sticky mass from the copper acetate/DMPU mixture was dissolved using DCM (50 mL) and also transferred.
- [0128]Methyl magnesium chloride (123.2 mL, 3.0 M solution in THF, 368 mmol) was added dropwise over 45 minutes to the combined reaction mixtures, which were then allowed to stir for 2 hours at −50° C. Subsequent HPLC analysis showed complete consumption of starting material. The mixture was allowed to warm to room temperature overnight, with stirring.
- [0129]Toluene (800 mL) was added to the mixture, followed by 5% acetic acid solution (600 mL). The aqueous layer was removed and discarded. The acetic acid wash was repeated. The organic layer was washed with brine (400 mL), 5% sodium bicarbonate solution (400 mL×2), followed by a brine wash (400 mL). The organic solution was dried over sodium sulfate, then concentrated to dryness under reduced pressure. The product was recovered as a viscous, light golden oil. Mass recovery was 146 grams (119% of theoretical).
- [0130]Compound 2 (92 g, 202 mmol) and toluene (1000 mL, 10.9 vol) were charged to a reaction flask under nitrogen and the solution was cooled to −10° C. A 32 wt % solution of peracetic acid in acetic acid (60 mL, 283 mmol, 1.4 eq) was added dropwise over about 30 min maintaining the temperature at −10° C. The reaction was held for approximately 20 h (HPLC showed 75% Cmpd 3, Cmpd 2 1.5%, 6% diastereomer; 5% epoxide). Starting at −10° C., a 20% aqueous solution of sodium bisulfite (920 mL, 10 vol) was added carefully via addition funnel, keeping the temperature below 10° C. Trifluoroacetic acid (16 mL, 202 mmol, 1 eq) was added and the mixture was held for 3 h at 0-5° C. to complete desilylation (endpoint by HPLC). The lower aqueous layer was drained, and the organic layer was washed with a saturated solution of sodium bicarbonate (3×250 mL), followed by water (1×250 mL), and brine (1×150 mL). The organic layer was then dried over Na2SO4, filtered and concentrated to a pasty solid (89 g). The residue was taken up in 1.5 vol of EtOAc and transferred to neat heptane (19 vol) to precipitate crude Cmpd 3 as an off-white solid (50 g, 62.5% yield; HPLC 79% Cmpd 3, 5.6% epoxide, 1.7% diastereomer). The crude Cmpd 3 (48.5 g) was triturated in hot acetonitrile (2 vol) at 60° C. for 4 h, and then gradually cooled to ambient temperature overnight. The mixture was filtered using the recycled filtrate to rinse and wash the wet cake. After drying, the recovery was 64.3% (31.2 g; HPLC 93.5% Cmpd 3, 3.3% epoxide). To remove the epoxide impurity, the 31 Cmpd 3 was dissolved in DCM (250 mL, 8 vol) and a solution of 48% HBr in water was added (7.5 mL). The mixture was heated at 40° C. for 1 h (HPLC<0.3% epoxide). The mixture was cooled and transferred to a separatory funnel. The lower aqueous layer (brown) was removed and the upper organic layer was washed with water (200 mL), saturated NaHCO3 (150 mL), and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated to a tan foam (32 g, ˜100% recovery). Methanol (64 mL, 2 vol) was added to the 32 g foam forming a slurry. To this was added a 1:1 solution of MeOH:water (60 mL, 2 vol) dropwise. The slurry cooled to slightly below ambient temperature and filtered using recycled filtrate to rinse and wash the wet cake. The solids were dried to constant weight, affording 26.1 g Cmpd 3 (81% recovery; HPLC 97.8%). The overall yield for Step 2 was 32.5%.
- [0131]Compound 3 (26 g, 65 mmol) and MeOH (156 mL, 6 vol) were mixed in a reaction flask and cooled to 0-5° C. A solution of K2CO3 (9.9 g, 72 mmol, 1.1 eq) in water (65 mL) was added dropwise, and the mixture was allowed to gradually warm to ambient temperature overnight. Analysis by HPLC showed 2.5% SM and another 5 mol % K2CO3 was added and the mixture stirred for another day (HPLC endpoint 1.1% Cmpd 3). The mixture was neutralized to pH 7 with 1.5 M HCl (53 mL) and ˜25% of the MeOH (30 g) was removed under vacuum to maximize recovery. After stirring for 2 days, the product was isolated by filtration using the recycled filtrate to aid transferring the wet cake to the funnel. The wet cake was dried under vacuum, affording 19.3 g VBP15 (83% yield) as an off-white powder. Analysis of the solids by HPLC showed 98.8% purity with 0.6% Cmpd 3 as the only major impurity.
- [0132]Power X-Ray Diffraction (pXRD)
- [0133]The solid samples were examined using X-ray diffractometer (Bruker D8 advance). The system is equipped with highly-parallel x-ray beams (Gobel Mirror) and LynxEye detector. The samples were scanned from 3 to 40°2θ, at a step size 0.02°2θ and a time per step of 19.70 seconds. The tube voltage and current were 45 kV and 40 mA, respectively. The sample was transferred from sample container onto zero background XRD-holder and gently ground.
Syn
EuropeanJournalofMedicinalChemistry265(2024)116124
Vamorolone (Agamree)
On October 26, 2023, Vamorolone, developed jointly by Santhera Pharmaceuticals and ReveraGen BioPharma, has received FDA approval to treat DMD in patients aged 2 years and older [1]. DMD is a prevalent neuromuscular disorder in childhood, ranking among the most common.
This condition is caused by mutations in the gene responsible for producing the dystrophin protein, which plays a crucial role in maintaining muscle integrity. Moreover, DMD is an X-linked genetic disorder [69]. Vamorolone is a novel steroidal anti-inflammatory and membrane-stabilizing agent that can be taken orally. The distinction between it and traditional corticosteroid drugs lies in its capacity to
specifically activate particular signaling pathways of corticosteroids. In individuals diagnosed with DMD, the primary mechanism through which corticosteroid drugs exhibit their effectiveness is by exerting
anti-inflammatory effects. However, the secondary activities of corticosteroids can lead to adverse effects that impact the overall well-being of patients. Vamorolone has the ability to decrease the occurrence of
adverse effects while still preserving the therapeutic effectiveness of corticosteroids in individuals with DMD [70].
Preparation of Vamorolone is depicted in Scheme 19, which began with commercially available steroid 3 TR VAMO-001 [71]. Copper catalyzed addition of VAMO-001 with trimethylsilyl chloride (TMSCl)
gave silyl enol ether VAMO-002. VAMO-002 was oxidized by peracetic acid in acetic acid to yield intermediate VAMO-003, which was deprotected and hydrolyzed to obtain Vamorolone.
[69] D. Duan, N. Goemans, S. Takeda, E. Mercuri, A. Aartsma-Rus, Duchenne muscular
dystrophy, Nat. Rev. Dis. Prim. 7 (2021) 13.
[70] M. Guglieri, P.R. Clemens, S.J. Perlman, E.C. Smith, I. Horrocks, R.S. Finkel, J.
K. Mah, N. Deconinck, N. Goemans, J. Haberlova, V. Straub, L.J. Mengle-Gaw, B.
D. Schwartz, A.D. Harper, P.B. Shieh, L. De Waele, D. Castro, M.L. Yang, M.
M. Ryan, C.M. McDonald, M. Tulinius, R. Webster, H.J. McMillan, N.L. Kuntz, V.
K. Rao, G. Baranello, S. Spinty, A.M. Childs, A.M. Sbrocchi, K.A. Selby,
M. Monduy, Y. Nevo, J.J. Vilchez-Padilla, A. Nascimento-Osorio, E.H. Niks, I.J.
M. de Groot, M. Katsalouli, M.K. James, J. van den Anker, J.M. Damsker,
A. Ahmet, L.M. Ward, M. Jaros, P. Shale, U.J. Dang, E.P. Hoffman, Efficacy and
safety of vamorolone vs placebo and prednisone among boys with duchenne
muscular dystrophy: a randomized clinical trial, JAMA Neurol. 79 (2022)
1005–1014.
[71] E.K.M. Reeves, E.P. Hoffman, K. Nagaraju, J.M. Damsker, J.M. McCall, VBP15:
preclinical characterization of a novel anti-inflammatory delta 9,11 steroid,
Bioorg. Med. Chem. 21 (2013) 2241–2249.

Syn
J. Med. Chem. 2025, 68, 2147−2182
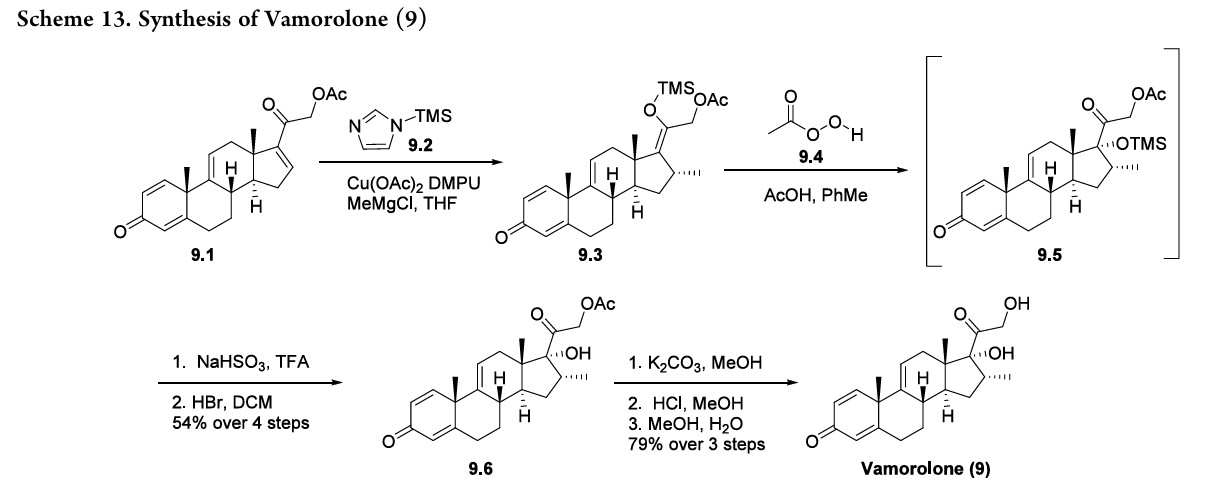
Vamorolone (Agamree). Developed by Santhera and ReveraGen BioPharma, the corticosteroid vamorolone (9) was approved for the treatment of Duchenne muscular dystrophy in October 2023.
70 Traditional corticosteroid treatment has been hampered by safety concerns including decreased bone mineral density and increased muscle atrophy. 71−73 Vamorolone is structurally distinct from other corticosteroids such as prednisone (Figure 3). 74 Removalofthe11βcarbonylmaintains binding to the glucocorticoid receptor but results in mineralocorticoid receptor antagonism; prednisone is a
mineralocorticoid receptor agonist. 75,76 This also results in decreased glucocorticoid receptor-drive transactivation, ultimately improving the safety profile of vamorolone as compared to other corticosteroid therapies. 74 The synthesis of vamorolone (9) as disclosed by ReveraGen BioPharma is summarized in Scheme 13. 77 Readily available steroid 9.1 was subjected to copper-catalyzed Michael addition.
Thein situ generated enolate was trapped using TMS-imidazole 9.2, providing the silyl enol ether 9.3. Treatment of crude 9.3 with peracetic acid 9.4 resulted in oxidized intermediate 9.5. Quenching of the peracetic acid and silyl deprotection afforded the protected steroid 9.6 in 54% yield from 9.1. Finally, K2CO3 mediated acetate deprotection of 9.6, neutralization and methanol/water crystallization provided vamorolone (9) in 79% yield over three steps.
(70) Keam, S. J. Vamorolone: first approval. Drugs 2024, 84, 111−
117.
(71) Hoffman, E. P.; Nader, G. A. Balancing muscle hypertrophy and
atrophy. Nat. Med. 2004, 10, 584−585.
(72) Hoffman, E. P.; Reeves, E.; Damsker, J.; Nagaraju, K.; McCall, J.
M.; Connor, E. M.; Bushby, K. Novel approaches to corticosteroid
treatment in Duchennemusculardystrophy.Phys.Med.Rehabil. Clin. N.
Am. 2012, 23, 821−828.
(73) Singh, A.; Schaeffer, E. K.; Reilly, C. W. Vertebral fractures in
Duchenne muscular dystrophy patients managed with Deflazacort. J.
Pediatr. Orthop. 2018, 38, 320−324.
(74) Liu, X.; Wang, Y.; Gutierrez, J. S.; Damsker, J. M.; Nagaraju, K.;
Hoffman, E. P.; Ortlund, E. A. Disruption of a key ligand-H-bond
network drives dissociative properties in vamorolone for Duchenne
muscular dystrophy treatment. Proc. Natl. Acad. Sci. U. S. A. 2020, 117,
24285−24293.
(75) Heier, C. R.; Yu, Q.; Fiorillo, A. A.; Tully, C. B.; Tucker, A.;
Mazala, D. A.; Uaesoontrachoon, K.; Srinivassane, S.; Damsker, J. M.;
Hoffman, E.P.; et al. Vamorolone targets dual nuclear receptors to treat
inflammation and dystrophic cardiomyopathy. Life Sci. Alliance 2019, 2,
No. e201800186.
(76)Boger, D.L.Thedifferenceasingleatomcanmake:synthesisand
design at the chemistry−biology interface. J. Org. Chem. 2017, 82,
11961−11980.
(77) Reeves, E. K. M.; Hoffman, E. P.; Nagaraju, K.; Damsker, J. M.;
McCall, J. M. VBP15: Preclinical characterization of a novel anti
inflammatory delta 9,11 steroid. Bioorg. Med. Chem. 2013, 21, 2241−
2249.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
PATENTS
Drugs 2024, 84, 111−117.
Nat. Med. 2004, 10, 584−585.
Clin. N.Am. 2012, 23, 821−828.
Pediatr. Orthop. 2018, 38, 320−324.
Proc. Natl. Acad. Sci. U. S. A. 2020, 117,24285−24293.
Life Sci. Alliance 2019, 2,No. e201800186.
J. Org. Chem. 2017, 82,11961−11980.
Bioorg. Med. Chem. 2013, 21, 2241−2249.
Neurol. 79 (2022)1005–1014
Bioorg. Med. Chem. 21 (2013) 2241–2249
Nat. Rev. Dis. Prim. 7 (2021) 13
Chemistry
Vamorolone is a synthetic corticosteroid and is also known by the chemical name 17α,21-dihydroxy-16α-methylpregna-1,4,9(11)-triene-3,20-dione or as 16α-methyl-9,11-dehydroprednisolone. It is a derivative of cortisol (hydrocortisone) and prednisolone (1,2-dehydrocortisol).
Anti-inflammatory drugs of the corticosteroid class show a carbonyl (=O) or hydroxyl (-OH) group on the C11 carbon of the steroid backbone. In contrast, vamorolone contains a Δ9,11 double bond between the C9 and C11 carbons. This change in structure has been shown to remove a molecular contact site with the glucocorticoid receptor, and leads to dissociative properties.[12]
History
In phase I clinical trials of adult volunteers, vamorolone was shown to be safe and well tolerated, with blood biomarker data suggesting possible loss of safety concerns of the corticosteroid class.[13]
In phase IIa dose-ranging clinical trial of 48 children with Duchenne muscular dystrophy (2 weeks on drug, 2 weeks off drug), vamorolone was shown to be safe and well tolerated, and showed blood biomarker data consistent with a myofiber membrane stabilization and anti-inflammatory effects, and possible loss of safety concerns.[14] These children continued on to a 24-week open-label extension study at the same doses, and this showed dose-dependent improvement of motor outcomes, with 2.0 and 6.0 mg/kg/day suggesting benefit.[15] These same children continued on a long-term extension study with dose escalations, and this suggested continued clinical improvement through 18-months treatment.[16]
Population pharmacokinetics (PK) of vamorolone was shown to fit to a 1-compartment model with zero-order absorption, with both adult men and young boys showing dose-linearity of PK parameters for the doses examined, and no accumulation of the drug during daily dosing. Apparent clearance averaged 2.0 L/h/kg in men and 1.7 L/h/kg in boys. Overall, vamorolone exhibited well-behaved linear PK, with similar profiles in healthy men and boys with DMD, moderate variability in PK parameters, and absorption and disposition profiles similar to those of classical glucocorticoids.[17] Exposure/response analyses have suggested that the motor outcome of time to stand from supine velocity showed the highest sensitivity to vamorolone, with the lowest AUC value providing 50% of maximum effect (E50 = 186 ng·h/mL), followed by time to climb 4 stairs (E50 = 478 ng·h/mL), time to run/walk 10 m (E50 = 1220 ng·h/mL), and 6-minute walk test (E50 = 1770 ng·h/mL). Week 2 changes of proinflammatory PD biomarkers showed exposure-dependent decreases. The E50 was 260 ng·h/mL for insulin-like growth factor-binding protein 2, 1200 ng·h/mL for matrix metalloproteinase 12, 1260 ng·h/mL for lymphotoxin α1/β2, 1340 ng·h/mL for CD23, 1420 ng·h/mL for interleukin-22-binding protein, and 1600 ng·h/mL for macrophage-derived chemokine/C-C motif chemokine 22.[18]
A trial titled “Efficacy and Safety of Vamorolone Over 48 Weeks in Boys With Duchenne Muscular Dystrophy” published in March 2024 found vamorolone (Agamree) at a dose of 6 mg/kg/d showed maintenance of improvement for all motor outcomes to week 48. There was also significant improvement in linear growth after crossover in the prednisone to vamorolone 6 mg/kg/d group, and quick reversal of prednisone-induced decline in bone turnover biomarkers in each crossover group.[19]
The US Food and Drug Administration (FDA) approved vamorolone based on evidence from a single clinical trial of 121 boys with DMD who were 4 to <7 years of age. The trial (Study 1) was conducted at 33 sites in 11 countries in Australia, Belgium, Canada, the Czech Republic, Spain, the United Kingdom, Greece, Israel, Netherlands, Sweden, and the United States.[10] In addition to Study 1, safety was also evaluated in a separate, open-label study of children with DMD aged 2 to <4 years (N=16) and children with DMD aged 7 to <18 years (N=16).[10]
Society and culture
Legal status
Santhera Pharmaceuticals signed an agreement with Catalyst Pharmaceuticals for the North American commercialization of vamorolone in July 2023.[20]
In October 2023, the FDA approved vamorolone (Agamree; Catalyst Pharmaceuticals) for the treatment of Duchenne muscular dystrophy.[11][21][22]
In October 2023, the Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Agamree, intended for the treatment of Duchenne muscular dystrophy.[2] The applicant for this medicinal product is Santhera Pharmaceuticals (Deutschland) GmbH.[2] Vamorolone was approved for medical use in the European Union in December 2023.[2][3]
Brand names
Vamorolone is the international nonproprietary name.[23]
Vamorolone is sold under the brand name Agamree.[1][2][3] Agamree (vamorolone) is a dissociative steroid that selectively binds to the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Vamorolone also inhibits mineralocorticoid receptor activation by aldosterone.[24]
References
- “Agamree- vamorolone kit”. DailyMed. 26 October 2023. Retrieved 20 November 2023.
- “Agamree EPAR”. European Medicines Agency. 12 October 2023. Retrieved 27 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Agamree Product information”. Union Register of medicinal products. 15 December 2023. Retrieved 26 December 2023.
- “Vamorolone – ReveraGen Biopharma”. AdisInsight. Springer Nature Switzerland AG. Archived from the original on 7 October 2017. Retrieved 2 July 2017.
- Reeves EK, Hoffman EP, Nagaraju K, Damsker JM, McCall JM (April 2013). “VBP15: preclinical characterization of a novel anti-inflammatory delta 9,11 steroid”. Bioorganic & Medicinal Chemistry. 21 (8): 2241–2249. doi:10.1016/j.bmc.2013.02.009. PMC 4088988. PMID 23498916.
- Heier CR, Damsker JM, Yu Q, Dillingham BC, Huynh T, Van der Meulen JH, et al. (October 2013). “VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects”. EMBO Molecular Medicine. 5 (10): 1569–1585. doi:10.1002/emmm.201302621. PMC 3799580. PMID 24014378.
- Dadgar S, Wang Z, Johnston H, Kesari A, Nagaraju K, Chen YW, et al. (October 2014). “Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy”. The Journal of Cell Biology. 207 (1): 139–158. doi:10.1083/jcb.201402079. PMC 4195829. PMID 25313409.
- Damsker JM, Conklin LS, Sadri S, Dillingham BC, Panchapakesan K, Heier CR, et al. (September 2016). “VBP15, a novel dissociative steroid compound, reduces NFκB-induced expression of inflammatory cytokines in vitro and symptoms of murine trinitrobenzene sulfonic acid-induced colitis”. Inflammation Research. 65 (9): 737–743. doi:10.1007/s00011-016-0956-8. PMID 27261270. S2CID 18698831.
- Heier CR, Yu Q, Fiorillo AA, Tully CB, Tucker A, Mazala DA, et al. (February 2019). “Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy”. Life Sci Alliance. 2 (1): e201800186. doi:10.26508/lsa.201800186. PMC 6371196. PMID 30745312.
- “Drug Trials Snapshots: Agamree”. U.S. Food and Drug Administration (FDA). 16 February 2024. Archived from the original on 18 February 2024. Retrieved 14 March 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “Drug Approval Package: Agamree”. U.S. Food and Drug Administration (FDA). 7 November 2023. Archived from the original on 13 November 2023. Retrieved 13 November 2023.
- Liu X, Wang Y, Gutierrez JS, Damsker JM, Nagaraju K, Hoffman EP, et al. (September 2020). “Disruption of a key ligand-H-bond network drives dissociative properties in vamorolone for Duchenne muscular dystrophy treatment”. Proceedings of the National Academy of Sciences of the United States of America. 117 (39): 24285–24293. Bibcode:2020PNAS..11724285L. doi:10.1073/pnas.2006890117. PMC 7533876. PMID 32917814.
- Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, et al. (June 2018). “Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes”. Steroids. 134: 43–52. doi:10.1016/j.steroids.2018.02.010. PMC 6136660. PMID 29524454.
- Conklin LS, Damsker JM, Hoffman EP, Jusko WJ, Mavroudis PD, Schwartz BD, et al. (October 2018). “Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug”. Pharmacological Research. 136: 140–150. doi:10.1016/j.phrs.2018.09.007. PMC 6218284. PMID 30219580.
- Hoffman EP, Schwartz BD, Mengle-Gaw LJ, Smith EC, Castro D, Mah JK, et al. (September 2019). “Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function”. Neurology. 93 (13): e1312 – e1323. doi:10.1212/WNL.0000000000008168. PMC 7011869. PMID 31451516.
- Smith EC, Conklin LS, Hoffman EP, Clemens PR, Mah JK, Finkel RS, et al. (September 2020). “Efficacy and safety of vamorolone in Duchenne muscular dystrophy: An 18-month interim analysis of a non-randomized open-label extension study”. PLOS Medicine. 17 (9): e1003222. doi:10.1371/journal.pmed.1003222. PMC 7505441. PMID 32956407.
- Mavroudis PD, van den Anker J, Conklin LS, Damsker JM, Hoffman EP, Nagaraju K, et al. (July 2019). “Population Pharmacokinetics of Vamorolone (VBP15) in Healthy Men and Boys With Duchenne Muscular Dystrophy”. Journal of Clinical Pharmacology. 59 (7): 979–988. doi:10.1002/jcph.1388. PMC 6548694. PMID 30742306.
- Li X, Conklin LS, van den Anker J, Hoffman EP, Clemens PR, Jusko WJ (October 2020). “Exposure-Response Analysis of Vamorolone (VBP15) in Boys With Duchenne Muscular Dystrophy”. Journal of Clinical Pharmacology. 60 (10): 1385–1396. doi:10.1002/jcph.1632. PMC 7494537. PMID 32434278.
- “Efficacy and Safety of Vamorolone Over 48 Weeks in Boys With Duchenne Muscular Dystrophy: A Randomized Controlled Trial”. PMID 38335499.
{{cite web}}: Missing or empty|url=(help) - Deswal P. “Santhera and Catalyst to market DMD drug vamorolone in North America”. Pharmaceutical Technology.
- “FDA Approves Vamorolone for Treatment of Duchenne Muscular Dystrophy in Patients Aged 2 Years and Older”. Pharmacy Times. 26 October 2023. Archived from the original on 27 October 2023. Retrieved 27 October 2023.
- “Santhera Receives U.S. FDA Approval of Agamree (vamorolone) for the Treatment of Duchenne Muscular Dystrophy” (Press release). Santhera Pharmaceuticals Holding AG. 27 October 2023. Archived from the original on 31 October 2023. Retrieved 13 November 2023 – via GlobeNewswire.
- World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
- “Agamree for the Treatment of Duchenne Muscular Dystrophy, US”. Clinicaltrials Arena. Retrieved 11 February 2025.
External links
Clinical trial number NCT03439670 for “A Study to Assess the Efficacy and Safety of Vamorolone in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
- [1]. Heier CR, et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. 2013 Oct;5(10):1569-85. [Content Brief][2]. Dillingham BC, et al. VBP15, a novel anti-inflammatory, is effective at reducing the severity of murine experimental autoimmune encephalomyelitis. Cell Mol Neurobiol. 2015 Apr;35(3):377-387. [Content Brief][3]. Heier CR, et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance. 2019 Feb 11;2(1). pii: e201800186. [Content Brief]
| Clinical data | |
|---|---|
| Trade names | Agamree |
| Other names | VBP; VBP-15; 17α,21-Dihydroxy-16α-methylpregna-1,4,9(11)-triene-3,20-dione |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624005 |
| License data | US DailyMed: Vamorolone |
| Routes of administration | By mouth |
| ATC code | H02AB18 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 13209-41-1 |
| PubChem CID | 3035000 |
| DrugBank | DB15114 |
| ChemSpider | 2299335 |
| UNII | 8XP29XMB43 |
| KEGG | D11000 |
| ChEBI | CHEBI:228304 |
| ChEMBL | ChEMBL2348780 |
| CompTox Dashboard (EPA) | DTXSID60927527 |
| ECHA InfoCard | 100.032.874 |
| Chemical and physical data | |
| Formula | C22H28O4 |
| Molar mass | 356.462 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////////Vamorolone, VBP 15, APPROVALS 2023, EMA 2023, FDA 2023, 8XP29XMB43, AGAMREE, EU 2023
Tebapivat
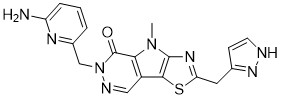
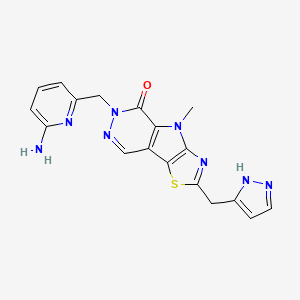
Tebapivat
CAS 2283422-04-6
WeightAverage: 392.44
Monoisotopic: 392.116778341
Chemical FormulaC18H16N8OS
10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
- AG946
- CS-0115951
- HY-135884
- ORG4KGP5ZS
- OriginatorAgios Pharmaceuticals
- ClassAntianaemics; Small molecules
- Mechanism of ActionPyruvate kinase stimulants
- Orphan Drug StatusYes – Myelodysplastic syndromes
- Phase IIAnaemia; Sickle cell anaemia
- 01 May 2025Phase-II clinical trials in Sickle cell anaemia in USA (PO) (NCT06924970)
- 01 May 2025Agios plans to initiate a phase II clinical trial for Sickle cell disease(PO) in mid-2025.
- 21 Feb 2025Agios Pharmaceuticals completes a phase I bioavailability trial (In volunteers) in USA (PO, capsule) (NCT06745271)
Tebapivat is under investigation in clinical trial NCT05490446 (A Study of Tebapivat (AG-946) in Participants With Anemia Due to Lower-risk Myelodysplastic Syndromes (LR-MDS)).
Tebapivat is an orally available activator of the red cell isoform of pyruvate kinase (PK-R; PKR), with potential to improve hemolytic anemia and related-symptoms in patients with pyruvate kinase deficiency (PKD). Upon oral administration, tebapivat binds to and activates PKR, thereby enhancing glycolytic pathway activity in red blood cells (RBCs), improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels. This may result in increased oxygen affinity, improved RBC deformability, decreased sickle RBC hemolysis, increased hemoglobin (Hb) levels and improved RBC membrane function. Mutations in PKR cause deficiency in PKR which prevents adequate RBC glycolysis, leading to a build-up of the upstream glycolytic intermediate 2,3-DPG and deficiency in the PKR product ATP.
SCHEME
COUPLER
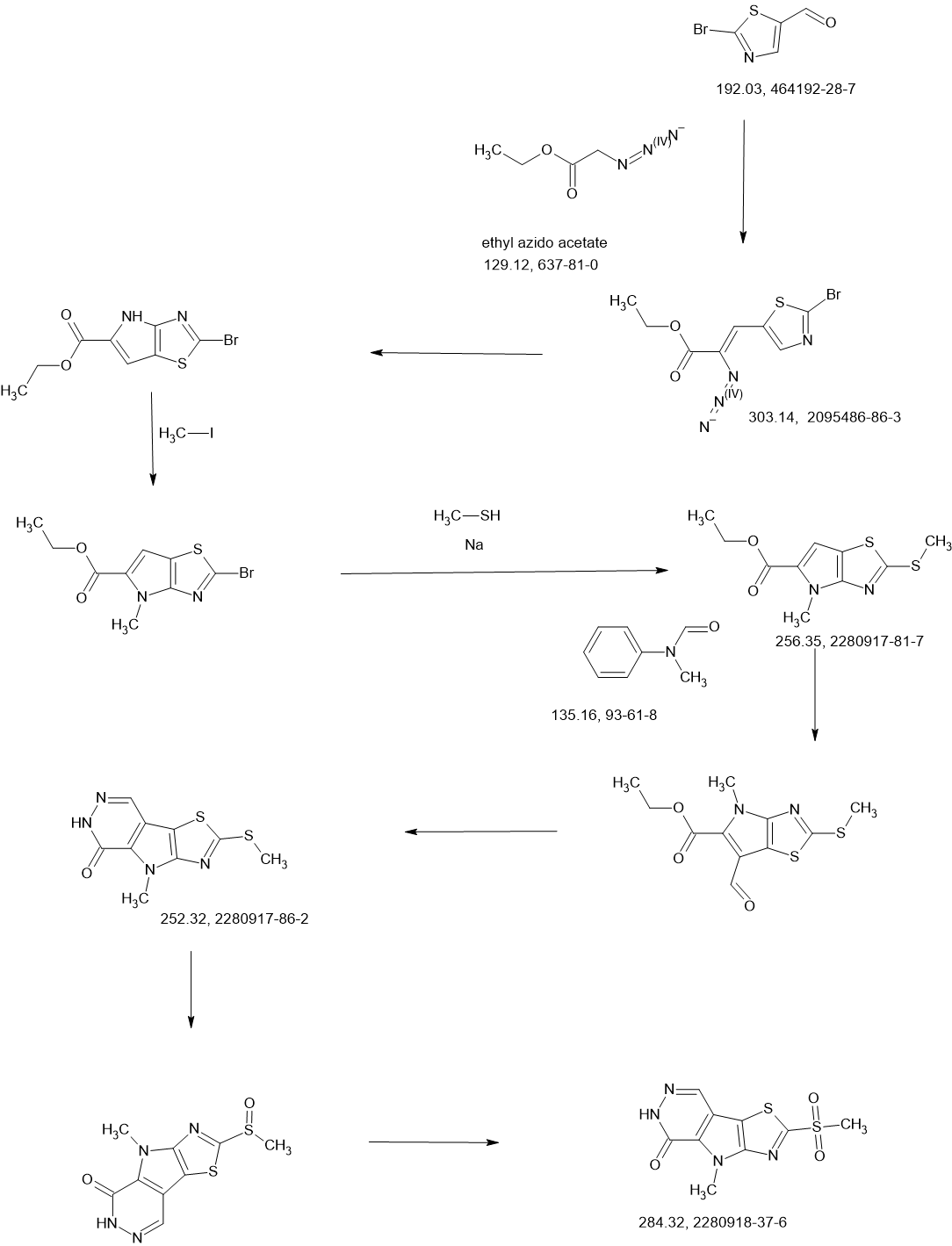
COUPLER
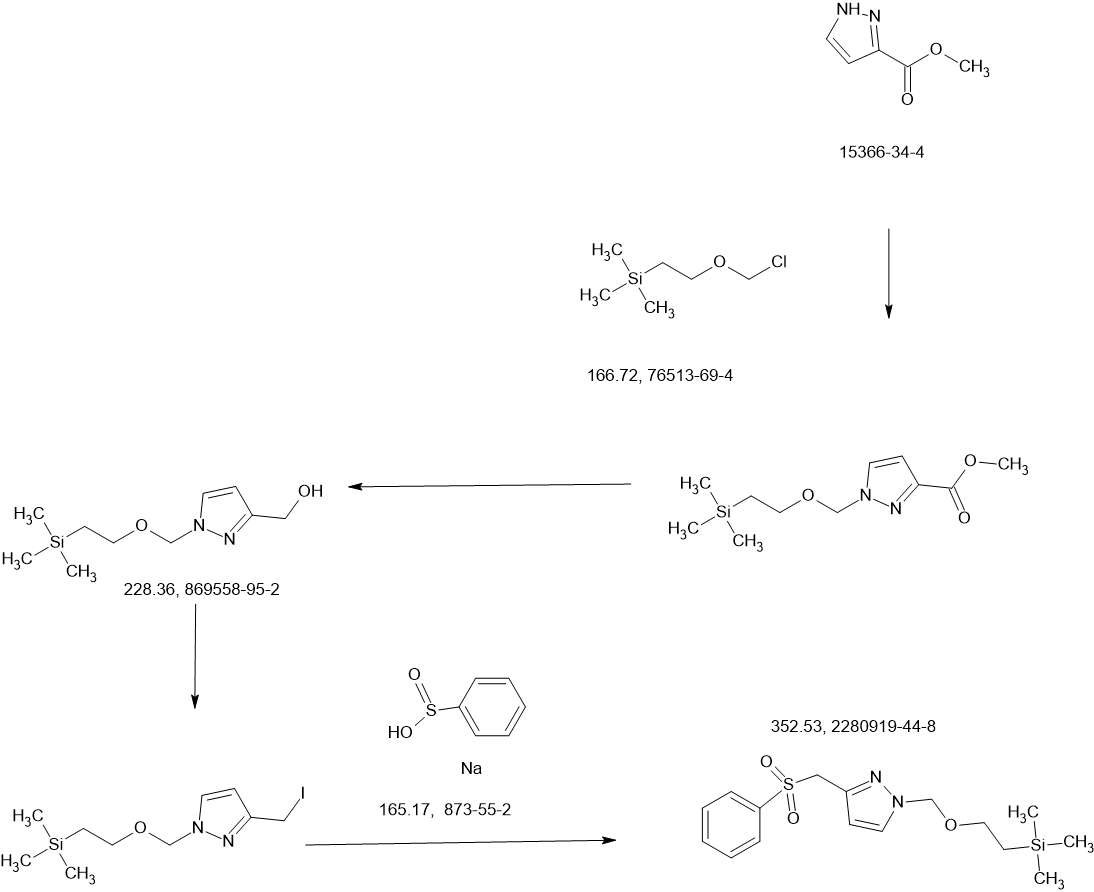
MAIN
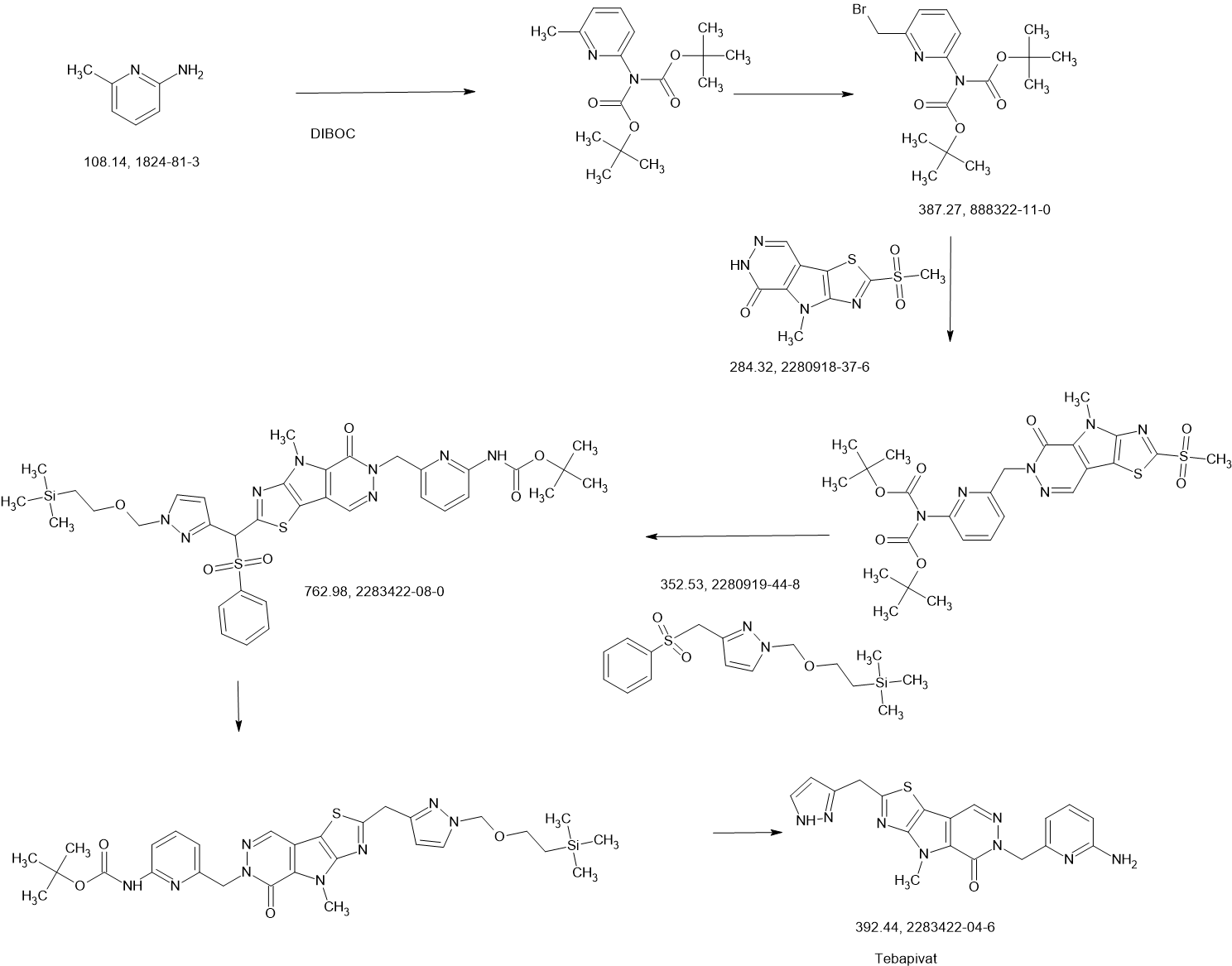
PATENT
Agios Pharmaceuticals, Inc.
WO2019035864
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019035864&_cid=P22-MDGSEF-03229-1
Example 8A. Synthesis of 2-((1H-pyrazol-3-yl)methyl)-6-((6-aminopyridin-2-yl)methyl)- 4-methyl-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one and 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrroIo[2,3-d]pyridazin-5(6H)-one


Step F. Synthesis of 6-((6-aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3- carbonyl)-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one To a solution of tert- butyl (6-((4-methyl-5-oxo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carbonyl)- 4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-6(5H)-yl)methyl)pyridin-2-yl)carbamate (20 mg, 0.03 mmol) in EtOH (1 mL) was added HCl (1 mL, 4 mol/L in dioxane). The mixture was stirred at 80 °C for lhr and cooled down. The precipitate was collected by filtration and neutralized with sat. NaHCO3, washed with water and dried to afford 5 mg of 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one. LC-MS (ESI): m/z 407 (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (s, 1H), 7.96 (s, 1H), 7.50 (s, 1H), 7.31-7.22 (m, 1H), 6.31 (d, 1H), 6.14 (d, 1H), 5.91 (s, 2H), 5.23 (s, 2H), 4.38 (s, 3H).
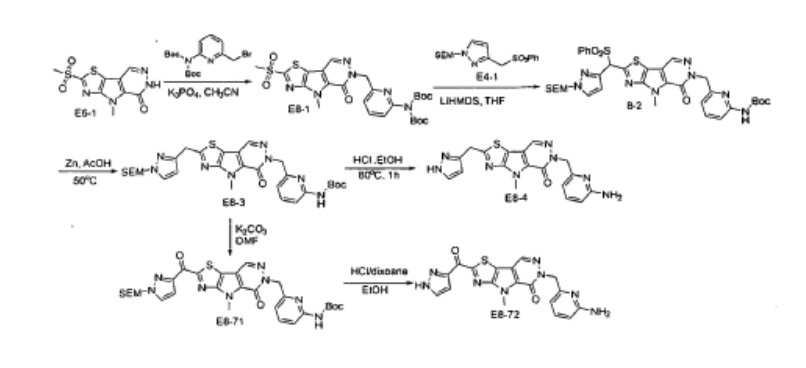
PATENT
WO2023091414
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023091414&_cid=P22-MDGSRV-15431-1
PATENT
WO2019035863
WO2019035865
WO2019035864
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Tebapivat, 2283422-04-6, AG946, CS-0115951, HY-135884, AG 946, CS 0115951, HY 135884, ORG4KGP5ZS, AGIOS, Orphan Drug, PHASE 2,
Tagtociclib
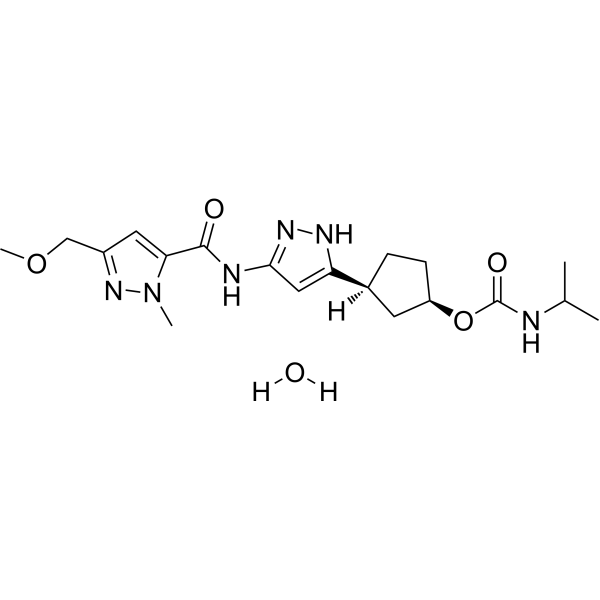
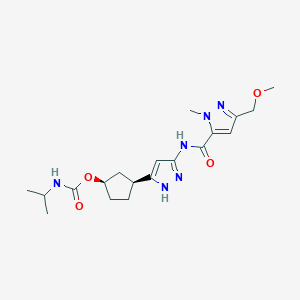
Tagtociclib (PF-07104091), 2460249-19-6, MW 404.5, C19H28N6O4
CAS 2733575-91-0 HYDRATE
| Molecular Weight HYDRATE | 422.48 |
|---|---|
| Formula | C19H30N6O5 |
[(1R,3S)-3-[3-[[5-(methoxymethyl)-2-methylpyrazole-3-carbonyl]amino]-1H-pyrazol-5-yl]cyclopentyl] N-propan-2-ylcarbamate
- (1R,3S)-3-[5-[[[3-(Methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl N-(1-methylethyl)carbamate
- (1R,3S)-3-{5-[3-(methoxymethyl)-1-methyl-1H-pyrazole-5carboxamido]-1H-pyrazol-3-yl}cyclopentyl (propan-2yl)carbamate
- (1R,3S)-3-(3-(3-(Methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamido)-1H-pyrazol-5-yl)cyclopentylisopropylcarbamate
- Carbamic acid, N-(1-methylethyl)-, (1R,3S)-3-[5-[[[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl ester
PF-07104091 hydrate is a potent and selective CDK2/cyclin E1 and GSK3β inhibitor, with Kis of 1.16 and 537.81 nM, respectively. PF-07104091 hydrate has anti-tumor activity for cyclin E1-amplified cancers. (patent WO2020157652A2).
- OriginatorPfizer
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 2 inhibitors
Phase IIBreast cancer; Solid tumours
Phase I/IINon-small cell lung cancer; Ovarian cancer; Small cell lung cancer
13 Sep 2024Efficacy, adverse events, pkarmacokinetics and pharmacodynamics data from a phase I/II trial in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
13 Sep 2024Pharmacodynamics data from a preclinical trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
05 Apr 2024Pharmacodynamics data form preclinical trial in Breast cancer and Ovarian cancer presented at the 115th Annual Meeting of the American Association for Cancer Research (AACR-2024)
Tegtociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 2 (CDK2), with potential antineoplastic activity. Upon administration, tegtociclib selectively targets, binds to and inhibits the activity of CDK2. This may lead to cell cycle arrest, the induction of apoptosis, and the inhibition of tumor cell proliferation. CDKs are serine/threonine kinases that are important regulators of cell cycle progression and cellular proliferation and are frequently overexpressed in tumor cells. CDK2/cyclin E complex plays an important role in retinoblastoma (Rb) protein phosphorylation and the G1-S phase cell cycle transition. CDK2/cyclin A complex plays an important role in DNA synthesis in S phase and the activation of CDK1/cyclin B for the G2-M phase cell cycle transition.
SCHEME
COUPLER
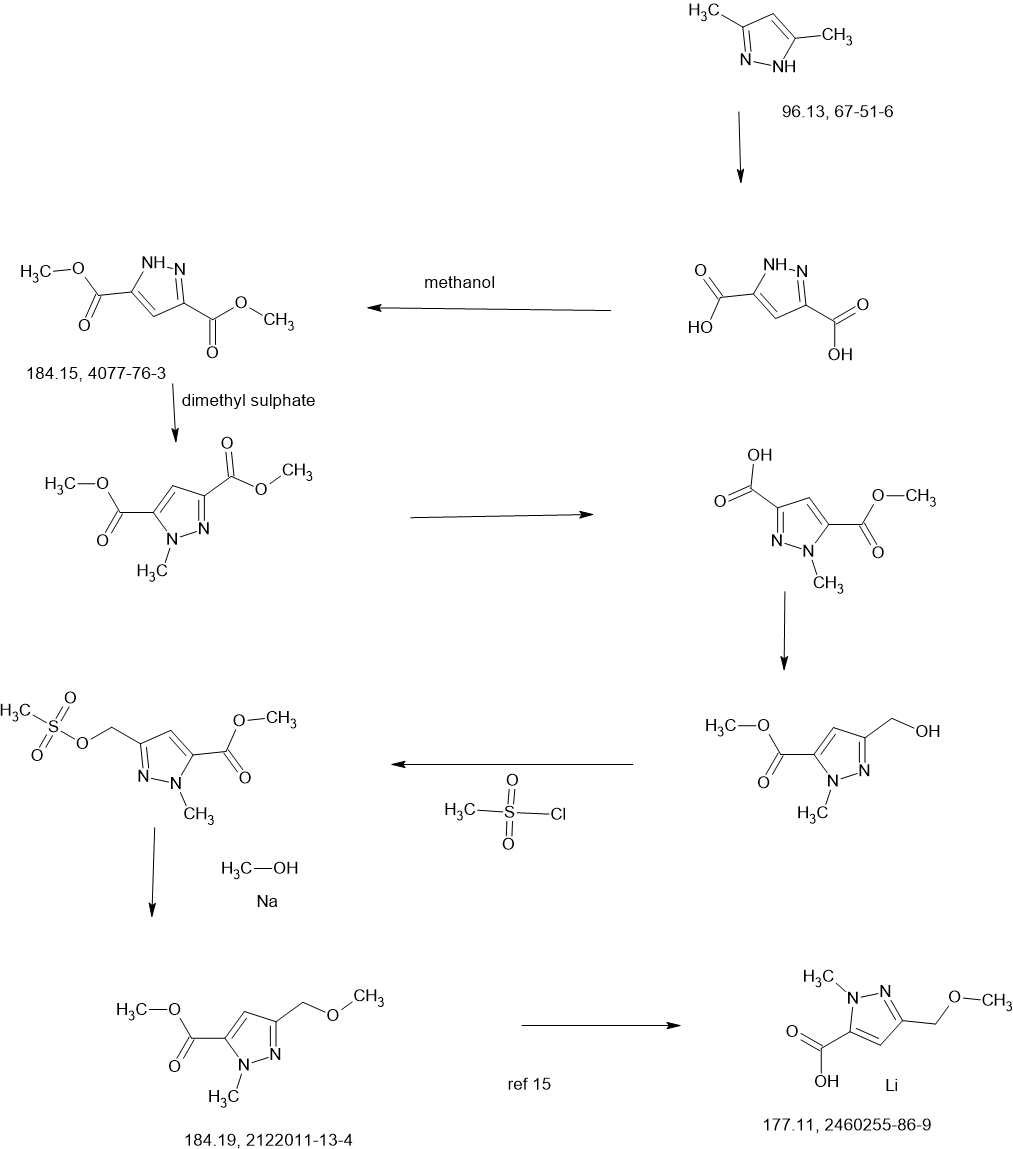
MAIN
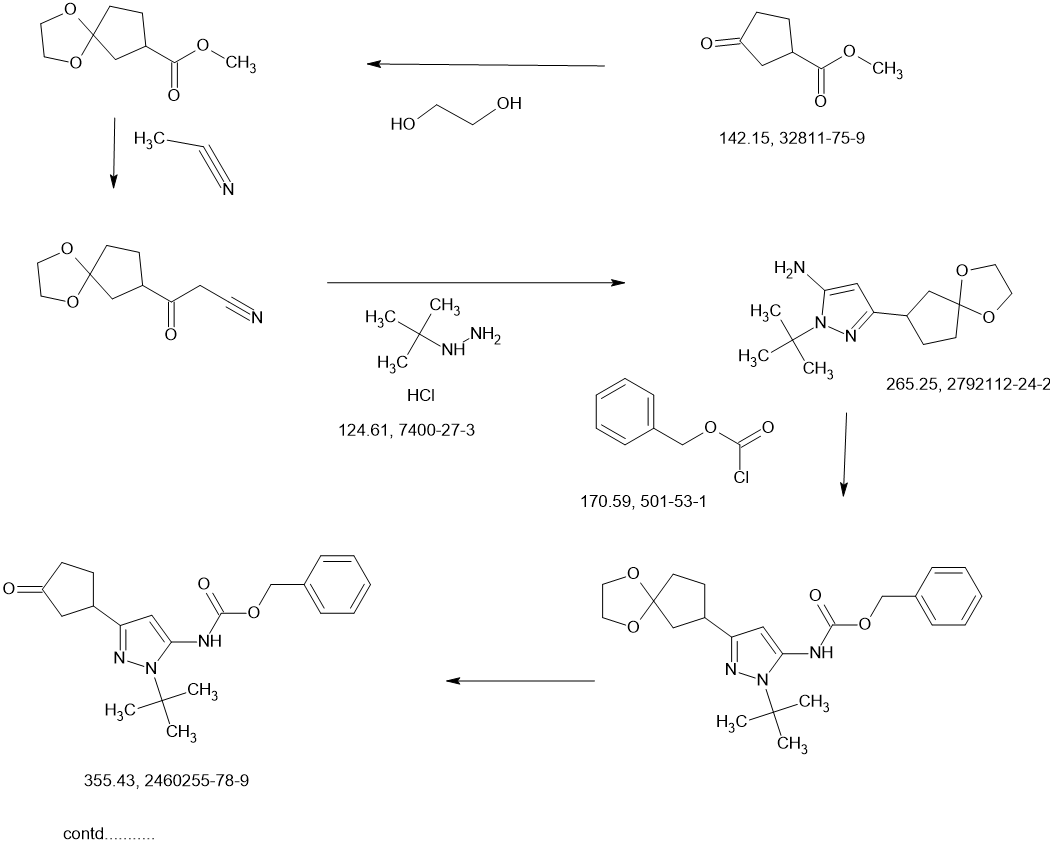
CONTD………….
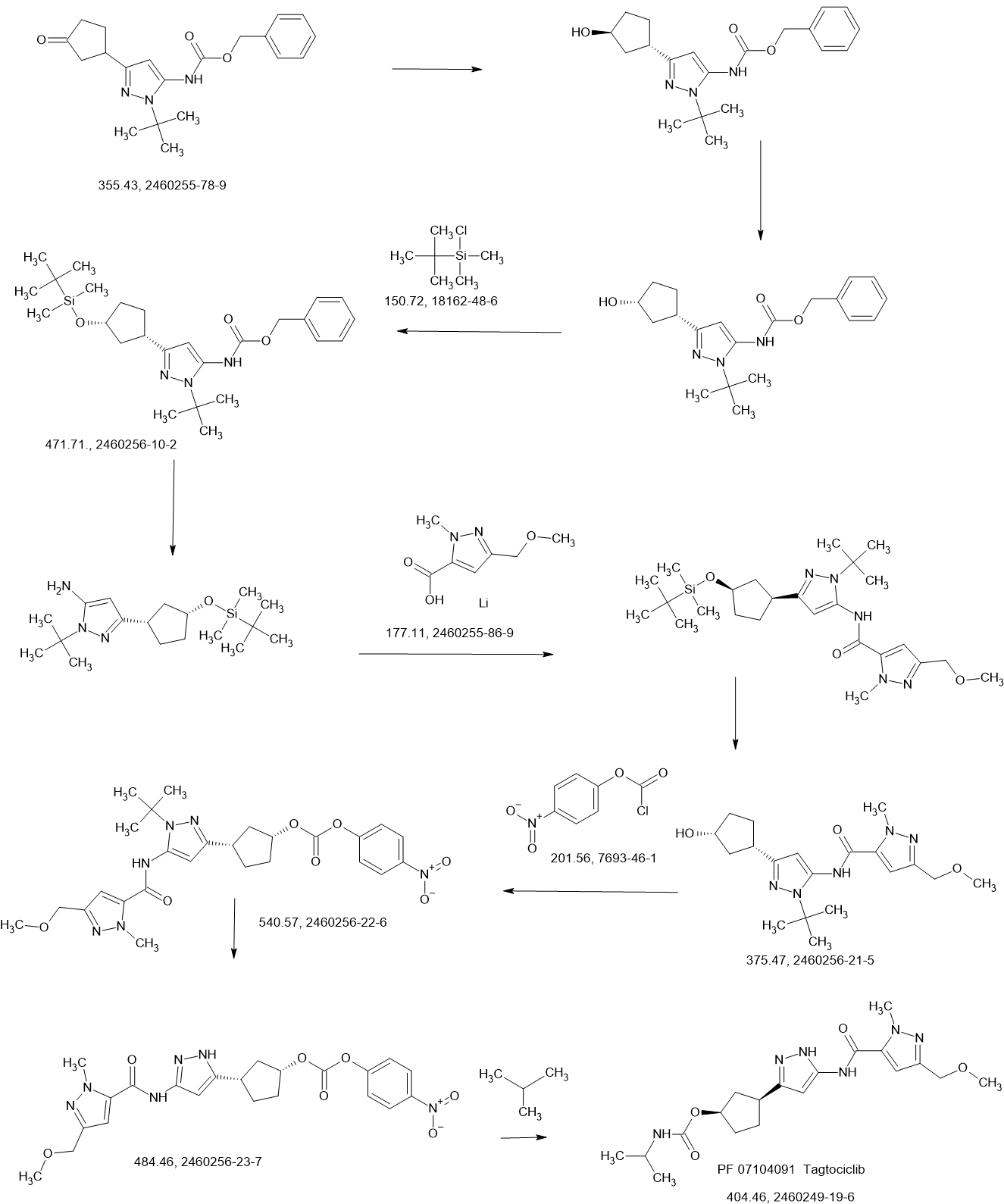
PATENTS
WO2022018596 78%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022018596&_cid=P22-MDFCVG-44044-1
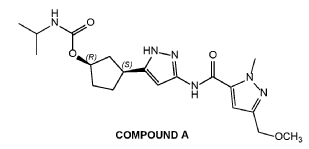
COMPOUND A was prepared as described in Example 13 of U.S. Patent No.
11,014,911.
Preparation of Intermediate 1: benzyl {1-tert-butyl-3-[(1S,3R)-3-hvdroxycvclopentyl]1H-pyrazol-5-yl)carbamate; and Intermediate 2: benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycvclopentyl1-1H-pyrazol-5-yl)carbamate.
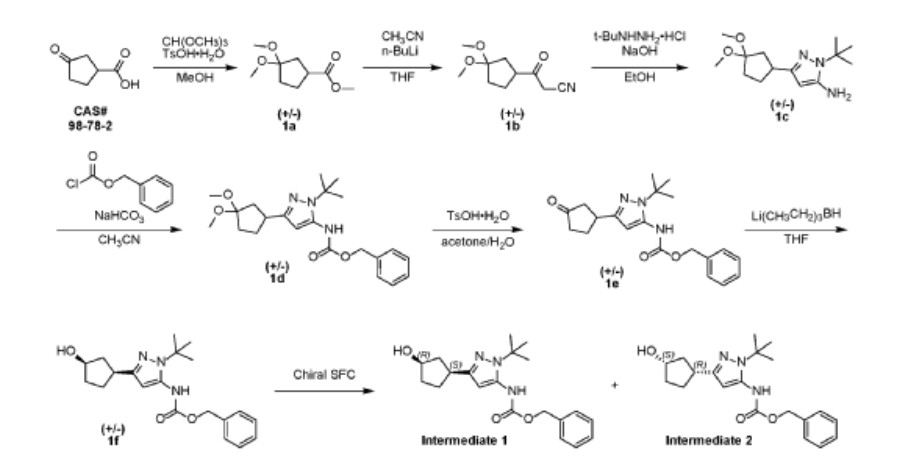
Two parallel reactions, each containing a solution of (±)-3- oxocyclopentanecarboxylic acid (CAS#98-78-2, 900 g, 7.02 mol) in methanol (5 L) at 13 °C were each treated with trimethyl orthoformate (4.47 kg, 42.15 mol, 4.62 L) and 4- toluenesulfonic acid monohydrate (26.72 g, 140.5 mmol). The mixtures were stirred at 13 °C for 25 hours. Each batch was quenched separately with sat. aq NaHCO3 (1 L), then the two batches were combined and concentrated under vacuum to remove most of the methanol. The residue was diluted with ethyl acetate (4 L), and the layers separated. The aqueous layer was further extracted with ethyl acetate (2 x 1 L). The combined organic layers were washed with sat. aq NaCI (3 x 1 L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give (±)-methyl 3,3- dimethoxycyclopentanecarboxylate (1a, 2.5 kg, 13.28 mol, 94%) as a light yellow oil. 1H NMR (400MHz, CHLOROFORM -d) δ = 3.67 (s, 3H), 3.20 (s, 3H), 3.19 (s, 3H), 2.94- 2.82 (m, 1 H), 2.16-2.00 (m, 2H), 1.99-1.76 (m, 4H).
A solution of n-butyllithium (3.44 L of a 2.5 M solution in hexanes, 8.6 mol) was added to a reactor containing THF (3 L) at -65 °C. Anhydrous acetonitrile (453 mL, 353 g, 8.61 mol) was added dropwise, slowly enough to maintain the internal temperature below -55 °C. The mixture was stirred for an additional 1 hour at -65 °C. A solution of (±)-methyl 3,3-dimethoxycyclopentanecarboxylate (1a, 810 g, 4.30 mol) in THF (1 L) was then added dropwise, slowly enough to maintain the internal temperature below -50 °C. After stirring for an additional hour at -65 °C, the reaction was quenched with water (4 L), neutralized with aq HCI (1 M) to pH 7, and extracted with ethyl acetate (3 x 3L). The combined organic layers were washed with sat. aq NaCI (2 x 3L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 722 g, 3.66 mol, 85%) as a red oil, which was used without further purification.
Solid sodium hydroxide (131.4 g, 3.29 mol total) was added in portions to a suspension of tert-butylhydrazine hydrochloride (409.4 g, 3.29 mol) in ethanol (3 L) at 16-25 °C. Stirring was continued at 25 °C for 1 hour. A solution of crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 540 g, 2.74 mol) in ethanol was added at 25 °C, then the mixture was heated to 75 °C internal and stirred for 30 hours. The reaction was filtered, and the filtrate concentrated under vacuum to give crude product as a red oil. This product was combined with crude from three more identically-prepared batches (each starting with 540 g 1b; 2.16 kg, 10.96 mol total for the 4 batches), and purified by silica gel chromatography (eluting with 0-35% ethyl acetate in petroleum ether), affording (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 1.60 kg, 5.98 mol, 54% yield) as a red oil. 1H NMR (CHLOROFORM -d) δ = 5.41 (s, 1 H), 3.50 (br. s., 2H), 3.22 (s, 3H), 3.20 (s, 3H), 3.13 (tt, J=7.9, 9.6 Hz, 1H), 2.25 (dd, J=8.0, 13.3 Hz, 1H), 2.09-2.00 (m, 1H), 1.99-1.91 (m, 1H), 1.83 (dd, J=10.8, 12.8 Hz, 2H), 1.78-1.68 (m, 1H), 1.60 (s, 9H).
Benzyl chloroformate (563.6 mL, 676.3 g, 3.96 mol) was added to a chilled (0-5 °C) solution of (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 530 g, 1.98 mol) in acetonitrile (3.5 L). The mixture was stirred at 23 °C for 2 hours, and then solid sodium hydrogen carbonate (532.9 g, 6.34 mol) was added in portions. Stirring was continued at 23 °C for 26 hours. The resulting suspension was filtered and the filtrate concentrated under vacuum to give crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) as a red oil, which was used in the next step without further purification.
A solution of the crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) in acetone (2 L) and water (2 L) at 18 °C was treated with 4-toluenesulfonic acid monohydrate (48.75 g, 256.3 mmol). The mixture was heated to 60 °C internal for 20 hours. After concentration under vacuum to remove most of the acetone, the aqueous residue was extracted with dichloromethane (3 x 3 L). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated under vacuum to a crude red oil. This crude product was combined with crude from two other identically-prepared batches (each derived from 1.98 mol 1c, 5.94 mol total for the 3 batches), and purified by silica gel chromatography (eluting with 0- 50% ethyl acetate in petroleum ether) to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.6 kg) as a yellow solid. This solid was stirred in 10:1 petroleum ether/ethyl acetate (1.5 L) at 20 °C for 18 hours. The resulting suspension was filtered, the filter cake washed with petroleum ether ( 2 x 500 mL), and the solids dried under vacuum to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.4 kg, 3.9 mol, 66% combined for the three batches). 1H NMR (DMSO–d6) δ = 9.12 (br. s., 1H), 7.56-7.13 (m, 5H), 6.03 (s, 1 H), 5.12 (s, 2H), 3.41-3.27 (m, 1H), 2.48-2.39 (m, 1H), 2.34-2.10 (m, 4H), 1.98-1.81 (m, 1 H), 1.48 (s, 9H).
A solution of (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 320 g, 0.900 mol) in THF (1.5 L) was degassed under vacuum and purged with dry nitrogen (3 cycles), then cooled to -65 °C internal. A solution of lithium triethylborohydride (1.0 M in THF, 1.80 L, 1.80 mol) was added dropwise at a rate which maintained the internal temperature below -55 °C, then stirring was continued at -65 °C for 1.5 hours. The reaction mixture was quenched with sat. aq NaHCO3 (1.5 L) at -40 to -30 °C. Hydrogen peroxide (30% aqueous, 700 g) was added to the mixture dropwise, while the internal temperature was maintained at -10 to 0 °C. The mixture was stirred at 10 °C for 1 hour, then extracted with ethyl acetate (3 x 2 L). The combined organic layers were washed with sat. aq Na2SO3 (2 x 1 L) and sat. aq NaCI (2 x 1 L). The organics were dried over magnesium sulfate, filtered, and concentrated under vacuum to a crude yellow oil. The crude product from this batch was combined with crude from three other, identically-prepared batches (each starting from 0.900 mol 1 e, for a total of 3.60 mol) for purification. Before chromatography, the combined mixture showed ~3.3:1 cis/trans ratio by NMR. The combined crude product was purified twice by silica gel chromatography, eluting with 0-50% ethyl acetate in dichloromethane), affording (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 960 g) as a light yellow solid, which was further purified by trituration, as described below.
A previous batch of 1f had been obtained from smaller-scale reactions, starting from a total of 120 g 1e (0.34 mol). The columned product from this batch was combined with the columned product from the batch above (which had been derived from 3.60 mol 1 e, for a total of 3.94 mol 1e used for all the combined batches), suspended in 10:1 dichloromethane/methanol (1.5 L), and stirred at 20 °C for 16 hours. The suspension was filtered, and the filter cake washed with petroleum ether (2 x 500 mL). The solids were dried under vacuum to give clean (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 840 g, 2.35 mol, 60% total yield for all the combined batches) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.07 (br. s., 1 H), 7.45-7.27 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.5 Hz, 1 H), 4.21-4.07 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.92-1.78 (m, 1 H), 1.78-1.62 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.43 (m, 1 H). MS: 358 [M+H]+.
The enantiomers of (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 700 g, 1.96 mol) were separated by chiral SFC.
The product from the first-eluting enantiomer peak (310 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 1 , 255 g, 713 mmol, 36%, >99% ee) as a white solid. 1H NMR (400MHz, DMSO -d6) δ = 9.08 (br. s., 1 H), 7.58-7.20 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.19-4.09 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.91-1.79 (m, 1 H), 1.79-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D +3.76 (c 1.0, MeOH). Chiral purity: >99% ee, retention time 3.371 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
The product from the second-eluting enantiomer peak (300 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 2, 255 g, 713 mmol, 36%, 94% ee) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.08 (br. s., 1 H), 7.55-7.19 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.23-4.07 (m, 1 H), 2.88 (quin, J=8.7 Hz, 1 H), 2.23-2.14 (m, 1 H), 1.90-1.79 (m, 1 H), 1.77-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1 .47 (s, 9H), 1.52-1 .44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D -2.43 (c 1 .0, MeOH). Chiral purity: 94% ee, retention time 3.608 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
A sample of the second-eluting enantiomer from a previous batch with [α]D -3.1 (c 1.1, MeOH) and 96% ee was crystalized from dichloroethane/pentane. A crystal structure was obtained by small-molecule X-ray crystallography, which showed (1R,3S) geometry. The absolute stereochemistry of Intermediate 2 was thus assigned (1R,3S) based on its comparable optical rotation and order of elution in the analytical method. Intermediate 1, the enantiomer of Intermediate 2, was thus assigned (1S,3R) stereochemistry.
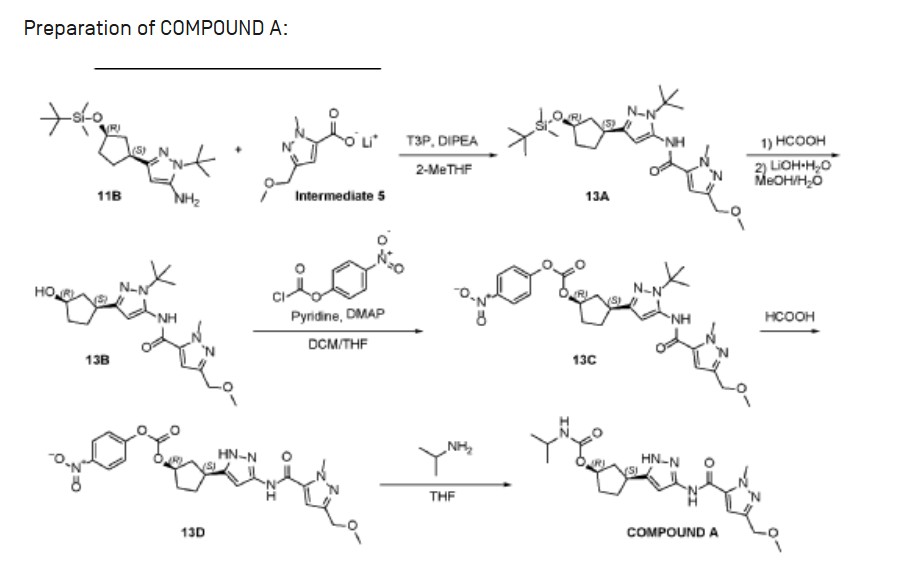
Propylphosphonic anhydride (T3P®, 50 wt% solution in EtOAc, 50.3 g, 79.1 mmol) was added to a room temperature (26 °C) solution of 1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-amine (11 B, 8.90g, 26.4 mmol), lithium 3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxylate (Intermediate 5, 5.83 g,
34.3 mmol), and diisopropylethyl amine (10.2 g, 79.1 mmol) in 2-methyltetrahydrofuran (100.0 mL). The resulting mixture was stirred at this temperature for 18 hours. After concentrating the mixture to dryness, the residue was dissolved in dichloromethane (150 mL), and the solution washed sequentially with water (2 x 30 mL), sat. aq NaHCO3 (2 x 30 mL) and sat. aq NaCI (30 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated to give crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert- butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H- pyrazole-5-carboxamide (13A, 12.9 g, 100%) as an oil. MS: 490 [M+H]+.
The crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]- 1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13A, 12.9 g,
26.3 mmol) was dissolved in formic acid (80 mL) and stirred at room temperature (27 °C) for 30 minutes. The mixture was concentrated to dryness, and the residue
dissolved in methanol (80 mL). A solution of lithium hydroxide monohydrate (3.43 g, 81.8 mmol) in water (15 mL) was added, and the mixture stirred at room temperature (27 °C) for 1 hour. The mixture was concentrated to dryness, and the residue was purified by silica gel chromatography (eluting with 0-80% ethyl acetate in petroleum ether) to give N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 78%) as a yellow gum. MS: 376 [M+H]+.
A solution of N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 21 mmol) in dichloromethane (80 mL) and THF (80 mL) was treated with DMAP (260 mg, 2.13 mmol), pyridine (5.06 g, 63.9 mmol), and 4-nitrophenyl chloroformate (8.59 g, 42.6 mmol). The resulting yellow suspension was stirred at room temperature for 18 hours. The reaction mixture was concentrated to dryness and purified by silica gel chromatography (eluting with 0-45% ethyl acetate in petroleum ether) to give (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 92%) as a light brown gum. MS: 541 [M+H]+.
A solution of (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 19.6 mmol) in formic acid (80 mL) was stirred at 70 °C for 18 hours. The solution was concentrated to dryness. The residue was dissolved in dichloromethane (150 mL) and the solution neutralized with sat. aq NaHCO3. The organic layer was washed with water (30 mL) and sat. aq NaCI (30 mL), dried over sodium carbonate, filtered, and concentrated to give crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 8.5 g, 90%, 86% pure by LCMS) as a light yellow glass. MS: 485 [M+H]+.
A room temperature (27 °C) solution of crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 1.7 g, 3.5 mmol) and 2-propylamine (1.04 g, 17.5 mmol) in THF (30 mL) was stirred for 6 hours. The solution was concentrated to dryness, and the residue was combined with the residue from a second batch which had been derived from 1.7 g, 3.5 mmol 13D (total 6.27 mmol 13D consumed for the combined two batches) to give 3.2 g crude product. This product was purified by preparative HPLC on a Phenomenex Gemini C18 250*50mm*10 pm column, eluting with 15-45% water (0.05% ammonium
hydroxide v/v) in acetonitrile. After lyophilization, (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2 -ylcarbamate (COMPOUND A, 2.06 g, 78%) was obtained as a white crystalline solid monohydrate. MS: 405 [M+H]+. 1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1 H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [α]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
PATENT
WO2020157652 EX 13
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020157652&_cid=P22-MDFD2U-50269-1
Example 13: (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}-amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate
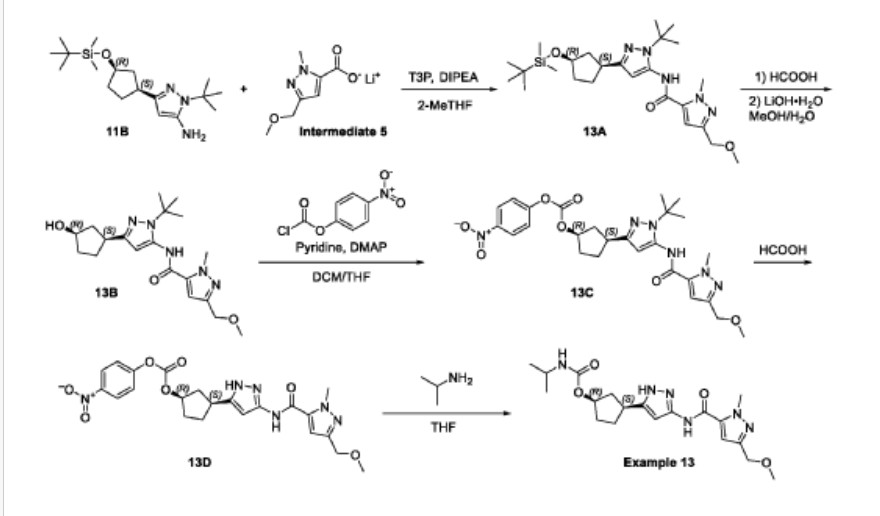
(1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate (Example 13, 2.06 g, 78%) was obtained as a white crystalline solid found to be a monohydrate (Form 1) based on elemental analysis. MS: 405 [M+H]+.1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [a]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
The white crystalline solid from above (500 mg) was recrystallized from 9: 1 H2O/CH3CN (2 mL) by heating until dissolved and then allowing the resulting solution to stand at room temperature for 18 h. During the 18 h time period, larger crystals of monohydrate (Form 1) formed. Single crystal X-ray diffraction of a selected crystal from this material provided the structure in FIG.1.
PATENTS
WO2022018667
WO2022174031
WO2022137106
[1]. Douglas Carl BEHENNA, et al. Cdk2 inhibitors. WO2020157652A2.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Tagtociclib, PF-07104091, 2460249-19-6, Tegtociclib, XBD0JF5EHJ, PF 07104091
SPIROBUDIFEN
SPIROBUDIFEN
cas 1305319-70-3
413.3 g/mol, C20H22Cl2O5
Butyl 3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl carbonate Butyl 3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl carbonate
- Butyl (3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro(4.5)dec-3-en-4-yl) carbonate
- butyl [3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl] carbonate
Spirobudifen is an oxaspiro compound that is 1-oxaspiro[4.5]dec-3-en-2-one substituted by 2,4-dichlorophenyl and (butoxycarbonyl)oxy groups at positions 3 and 4, respectively. It is an acaricide from Zhejiang Udragon Bioscience. It is a dichlorobenzene, an oxaspiro compound, an organochlorine acaricide and a carbonate ester.
SCHEME
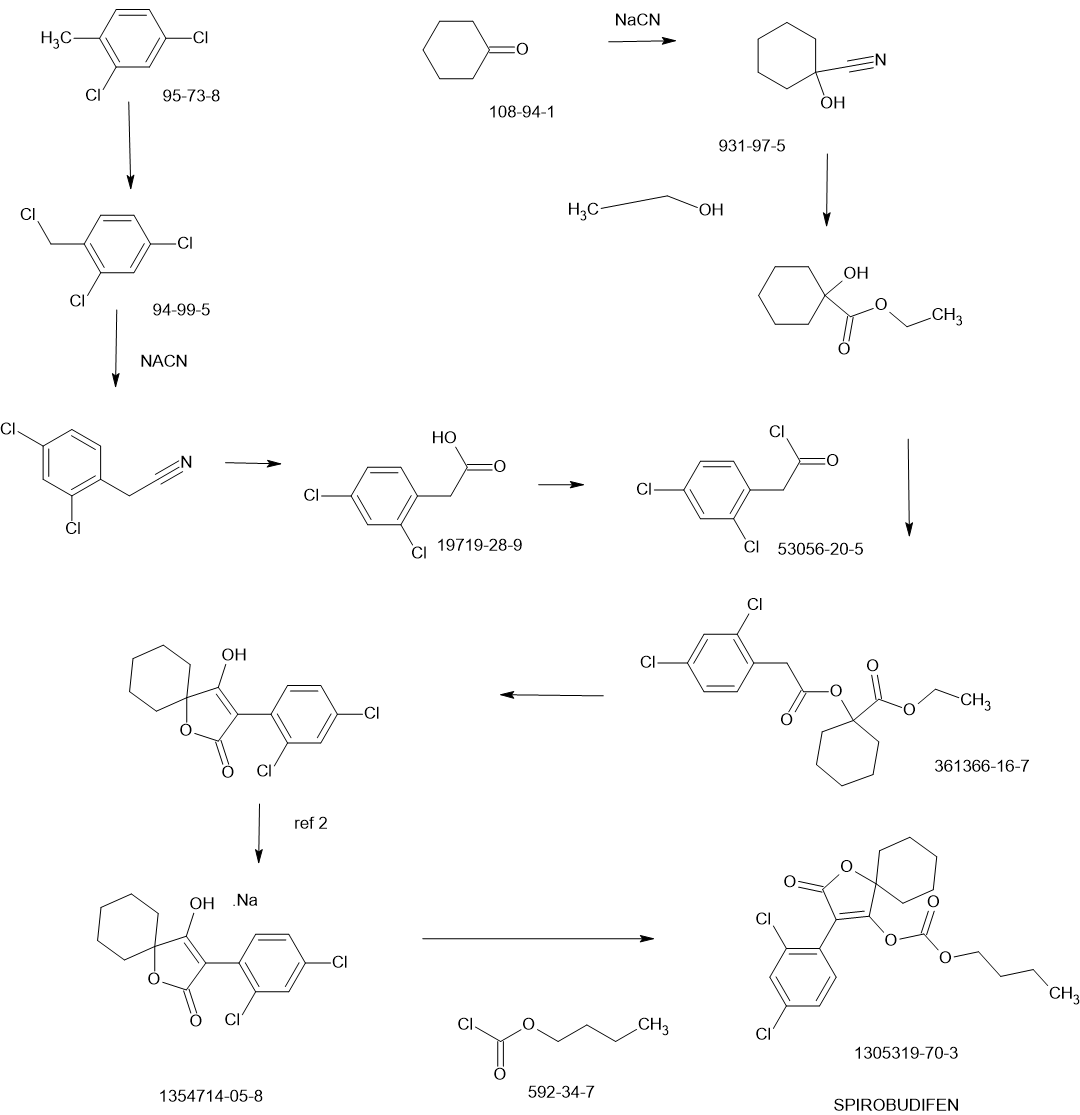
PATENTS
CN112745286
CN102060818
Xiandai Nongyao (2012), 11(1), 15-21
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////SPIROBUDIFEN, 1305319-70-3
Hetrombopag Olamine
Hetrombopag Olamine, RAFUTROMBOPAG OLAMINE
- Hetrombopag diolamine
- SHR8735 olamine
- Hetrombopag ethanolamine
- SHR-8735 olamine
580.6 g/mol, C29H36N6O7, V45T2I862X
2-aminoethanol;5-[2-hydroxy-3-[[5-methyl-3-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4-yl]diazenyl]phenyl]furan-2-carboxylic acid
CAS 1257792-42-9
1257792-41-8 (free acid) 1257792-41-8 (ethanolamine) 1257792-42-9 (olamine)
Jiangsu Hengrui Pharmaceutical, was approved in China in June 2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemia who have not responded well to other treatments
Hetrombopag Olamine is the orally active ethanolamine salt of hetrombopag, a small-molecule, nonpeptide thrombopoietin receptor (TPO-R or CD110) agonist, with megakaryopoiesis-stimulating activity. Upon oral administration, hetrombopag targets, binds to and stimulates the transmembrane domain of the platelet TPO-R, a member of the hematopoietin receptor superfamily. Activation of TPO-R leads to the proliferation and differentiation of cells in the megakaryocytic lineage and an increase in platelet production. This may prevent or treat chemotherapy-induced thrombocytopenia.
- OriginatorJiangsu Hengrui Medicine Co.
- DeveloperAtridia; Jiangsu Hengrui Medicine Co.
- ClassAntianaemics; Antihaemorrhagics; Aza compounds; Carboxylic acids; Furans; Pyrazolones; Small molecules; Tetrahydronaphthalenes
- Mechanism of ActionThrombopoietin receptor agonists
- Orphan Drug StatusYes – Thrombocytopenia
- MarketedAplastic anaemia; Idiopathic thrombocytopenic purpura
- Phase IIIThrombocytopenia
- No development reportedUnspecified
- 07 Dec 2024Efficacy and adverse events data from a phase-III trial in Aplastic anaemia presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
- 31 Jul 2024Phase-III clinical trials in Thrombocytopenia in China (PO) (NCT06507436)
- 25 Jul 2024Jiangsu Hengrui Medicine plans a phase III trial in Thrombocytopenia (PO) in July 2024 (NCT06507436)
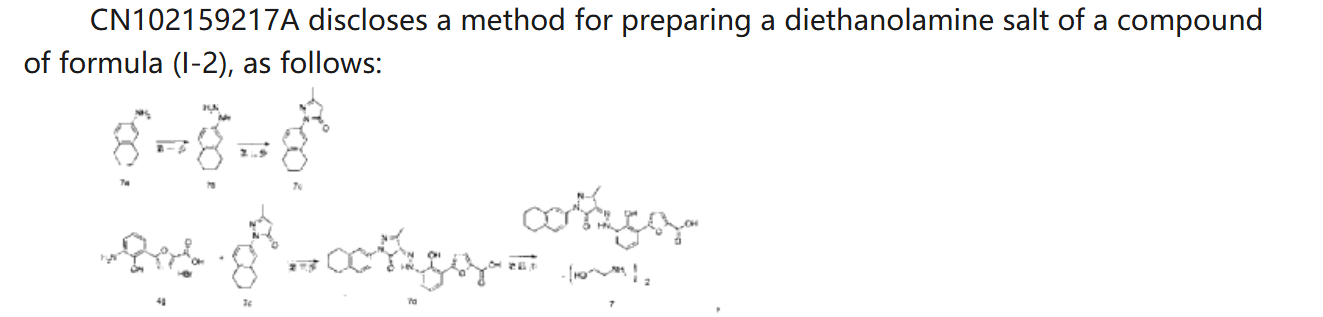
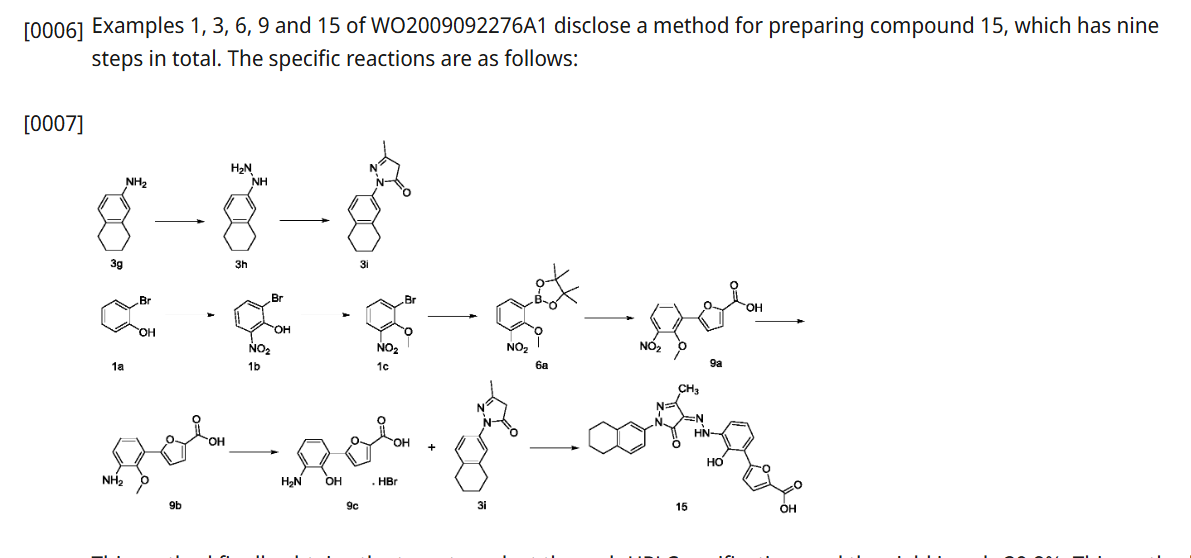
SYN
CN 113929668
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN349207982&_cid=P21-MDCUSL-44897-1
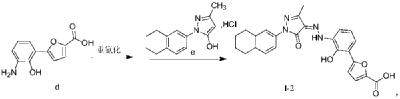
| Example 1. Synthesis of 5-(2-carbonyl-2,3-dihydrobenzoxazol-7-yl)furan-2-carboxylic acid |
| |
| Add purified water to the batching barrel, add 4.0kg of compound a under stirring, then add 10L of hydrochloric acid, stir, pump the material into a 50L reactor, add 10L of purified water to the batching barrel and pump it into the reactor. Turn on stirring, start cooling, the temperature drops to -5~2°C, start adding sodium nitrite aqueous solution (6.4L purified water, 1840g sodium nitrite), keep the temperature in the reactor no higher than 5°C during the process; after adding, continue stirring for 10~20min; add 800g of urea, continue stirring for 10~20min, the obtained diazonium salt solution is ready for use, and the temperature in the whole process is kept no higher than 5°C. |
| 44kg of acetone was pumped into a 200L reactor, and 15.0kg of compound b and 463.5g of copper chloride dihydrate were added in sequence under stirring. The temperature was raised to 30-35°C, and the obtained diazonium salt solution was added. The temperature was maintained at 30-40°C during the period. After the addition was completed, the temperature was maintained at 30-40°C and the reaction was continued with stirring for 1-1.5h. 120.0L of purified water was added, the temperature was raised to 40-45°C, and stirring was continued for a period of time. Filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. 80L of purified water was added to the reactor, stirring was started, and the filter cake was added. Sodium hydroxide aqueous solution was added to the reactor to adjust the pH, the pH value was maintained at 8-10 for a period of time, and the filtrate was pumped into the reactor, and the filter was pressed into the material barrel through the filter press. Then 10L of purified water was pumped into the reactor and filtered into the material barrel. The material in the material barrel was pumped into the reactor, and then ethyl acetate was pumped in, stirred, and allowed to stand for 30-40 minutes. The aqueous phase was separated and collected, and the aqueous phase was pumped into the reactor, and the pH was adjusted to 3-4 with hydrochloric acid solution, and the filter cake was washed with purified water until the filtrate was neutral, and then the filter cake was collected. The filter cake was dried to obtain compound c. The yield of this step was 3.59 kg, and the yield was 55%. |
| Example 2: Synthesis of 5-(3-amino-2-hydroxyphenyl)furan-2-carboxylic acid |
| |
| Purified water was pumped into the 50L reactor, stirring was started, 3.53kg of sodium hydroxide was added, and compound c obtained in the previous step was added. Under nitrogen protection, the reaction mixture was heated to reflux in the reactor for reaction. After the reaction, the reaction solution was cooled, the temperature was lowered to 0-10°C, and hydrochloric acid solution was added to adjust the pH value to 5-6. The filter cake was filtered, and the filtrate was washed with purified water until neutral, and then filtered again to collect the filter cake. The filter cake was dried to obtain compound d. The yield in this step was 2.78kg, with a yield of 90%. |
| Example 3. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| Purified water was added to the batching barrel, and compound d was added in sequence under stirring, and then 6.3L hydrochloric acid was added, and the materials were pumped into a 200L reactor. Purified water was added to the batching barrel again, and then pumped into the reactor. Stirring was started, and the temperature was lowered to -5 to 2°C. Sodium nitrite aqueous solution (sodium nitrite to compound d molar ratio is 1:1) was added, and the internal temperature was kept at no more than 5°C during the process. After the addition was completed, stirring was continued; urea was added, and stirring was continued to obtain a diazonium salt solution for use, and the internal temperature was kept at no more than 5°C during the whole process. |
| Add 36L purified water and 4000g sodium hydroxide to the batching barrel, stir to dissolve, and set aside. Take 26kg of the above sodium hydroxide aqueous solution, add compound e (the molar ratio of compound e to compound d is 0.9:1), stir, and add the resulting solution to the diazonium salt solution, keeping the temperature not exceeding 8°C. Add the above-prepared sodium hydroxide aqueous solution dropwise, adjust the pH to 8-10, and keep the temperature at 5-10°C for 3-4h. Add hydrochloric acid solution dropwise to adjust the pH to 2-3, keep the temperature not exceeding 25°C, filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. Pump 48.0kg of tetrahydrofuran aqueous solution (22.5kg tetrahydrofuran, 25.5L purified water) into the reactor, add the above-obtained filter cake, beat, filter, wash the filter cake with tetrahydrofuran aqueous solution, wash the filter cake with purified water, filter again, and collect the filter cake. Dry the filter cake. |
| Ethyl acetate was pumped into the reactor, and the above-obtained materials were added to the reactor for slurrying, and the filter cake was washed with ethyl acetate, and the filter cake was washed until no obvious droplets flowed out of the mirror, and the filter cake was collected and dried to obtain the compound of formula (I-2). The yield in this step was 5.34 kg, and the yield was 97.5%. |
| Example 4. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the equivalent of compound e was adjusted from 0.9 in Example 3 to the current 0.95, other conditions remained unchanged). |
| Comparative Example 1: Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazole-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the step of adding urea was changed to starch potassium iodide test paper to indicate the reaction endpoint, and other conditions remained unchanged). |
| Test Example 1: Effect of urea on the preparation process of the compound of formula (I-2) |
| HPLC conditions: |
| Chromatographic column: Welch Ultimate |
| Flow rate: 1.0ml/min |
| Injection volume: 10 μl |
| Detector: UV detector |
| Detection wavelength: 251nm |
| Mobile phase: 0.1% trifluoroacetic acid aqueous solution was used as mobile phase A, acetonitrile was used as mobile phase B, and elution was performed at a ratio of 50%/50% of mobile phase A/mobile phase B. |
PATENT
EP 2441457
PATENT
WO 2010142137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010142137&_cid=P21-MDCUXF-51461-1
PATENT
WO 2018133818
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018133818&_cid=P21-MDCUYN-53075-1
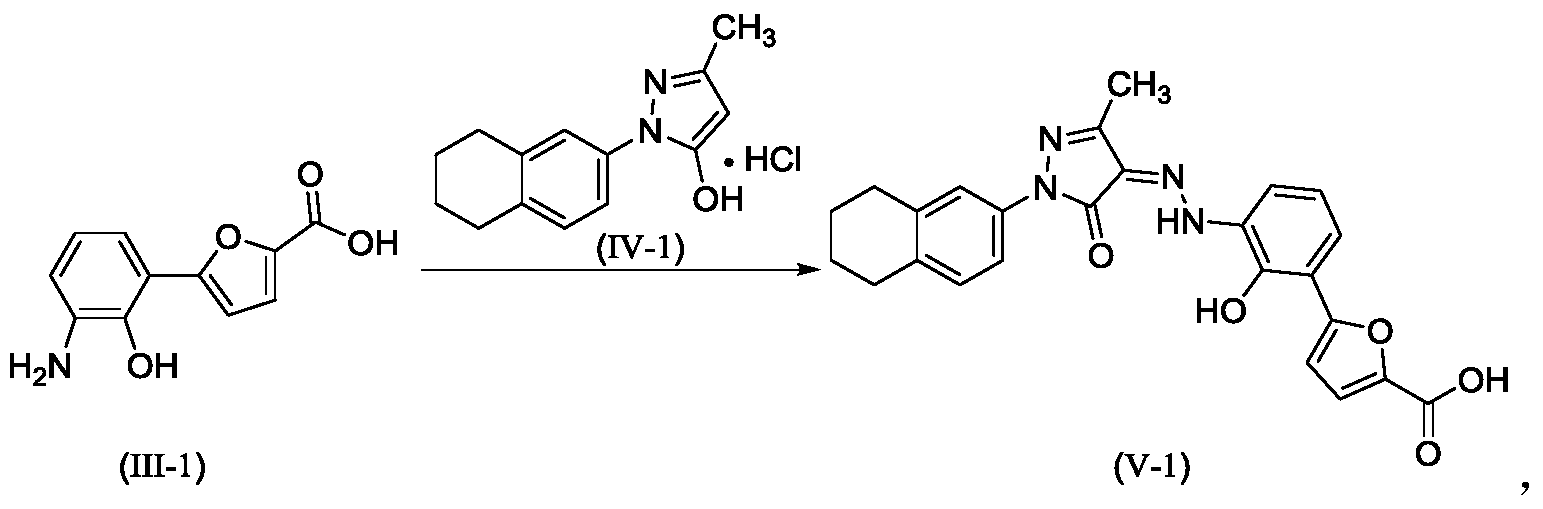
Example 1. Preparation of 3-methyl-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-5-ol hydrochloride
[0107]
[0108](5,6,7,8-tetrahydronaphthalene-2-yl)hydrazine hydrochloride (1.3 kg, prepared according to the method in patent application WO2009092276A1) and ethyl acetoacetate (1.17 L) were added to ethyl acetate (5.2 L). The mixture was heated under reflux for 2 hours. The reaction solution was cooled to room temperature, then cooled to 0-5°C, stirred for 1 hour, filtered, and the solid was washed with a small amount of ethyl acetate to obtain a white solid product (1.4 kg, yield 81%).
[0109]
[0110]Example 2. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid (V-1)
[0111]
[0112]Step 1: Synthesis of intermediate (II-1)
[0113]Purified water (14.80 kg), 7-aminobenzo[d]oxazol-2(3H)-one (2.00 kg, prepared according to the method in patent application WO2005016898A2), and hydrochloric acid (5.33 kg) were added to the reaction kettle, the temperature was raised to 40-45°C, stirred for 10 min, cooled to -3-5°C, and sodium nitrite aqueous solution (sodium nitrite 940 g, water 3.20 kg) was added dropwise, the internal temperature was kept at no more than 5°C, the end point was controlled by starch potassium iodide test paper, and stirring was continued for 15 min;
[0114]Add acetone (28L) to the reactor, then add furoic acid (4.57kg) and cupric chloride dihydrate (232g), stir at 35-40℃ until dissolved, add diazonium salt solution dropwise, keep the internal temperature at 35-40℃, and continue stirring for 1.5h. Add purified water (60L), heat to 35-40℃ and stir for 30min. Filter, wash the filter cake with 45-50℃ purified water. Add the filter cake to purified water (40kg), adjust the pH to 8-9 with 15% sodium hydroxide aqueous solution, filter, adjust the pH of the filtrate to 3-4 with 6mol/L hydrochloric acid, filter, wash the filter cake with purified water, and dry to obtain a solid (1.63kg, yield 50%).
[0115]Step 2: Synthesis of intermediate (III-1)
[0116]The product from the previous step (1.4 kg) and 15% aqueous sodium hydroxide solution (9.7 kg) were heated to reflux under argon protection and reacted for 28 hours. The reaction solution was poured into ice water (5-6 kg), and hydrochloric acid (6N, 3 L) was slowly added to adjust the pH value to 5-6. The temperature was maintained below 20°C. During this period, ethyl acetate was added to eliminate bubbles. The mixture was filtered, washed with purified water, and dried to obtain a solid (1.18 kg, yield 94%).
[0117]Step 3: Synthesis of intermediate (V-1)
[0118]Add the product of the previous step (1.10kg), purified water (27.5kg), and hydrochloric acid (2.92kg) to the reactor in sequence, stir and dissolve, cool to -4 to -1°C, add sodium nitrite aqueous solution (346g sodium nitrite, 5.5kg water), and continue to react for 15min after the addition is completed. Cool to -8 to -5°C. Dissolve sodium hydroxide (1.48kg) in purified water (13.2kg) to obtain a 10% sodium hydroxide aqueous solution. Add 5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-2H-pyrazole-3-ol hydrochloride (1.26kg) to the above sodium hydroxide aqueous solution (10kg) to dissolve, and add the resulting solution to the diazonium salt solution at once, keeping the temperature not higher than 10°C. Add the remaining 10% sodium hydroxide aqueous solution, adjust the pH to 8 to 9, naturally heat to 8 to 12°C for reaction, and react for 4h. Add 6N hydrochloric acid, adjust pH=2-3, keep the temperature not more than 20°C, filter, and wash the filter cake with water until pH=6-7. Add the filter cake to 50% tetrahydrofuran aqueous solution (19kg), slurry at room temperature for 2h, filter, wash with 50% tetrahydrofuran aqueous solution, wash with water, and dry. Add ethyl acetate (20kg) to the solid, slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, add the solid to ethyl acetate (20kg), slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, and dry to obtain a solid (2.18kg, yield 95%, purity 99.5%).
[0120]Example 3. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid ethanolamine salt (1:2)
[0121]
[0122]Preparation of crude product
[0123]The compound of formula (V-1) (1.8 kg) was suspended in a tetrahydrofuran/ethanol (14.5 kg, V/V = 2:1) mixed solvent at room temperature, stirred for 0.5 h, cooled to 10-15 ° C, and a tetrahydrofuran ethanol solution of ethanolamine (479.6 g) (tetrahydrofuran 91 g and ethanol 41 g) was added dropwise. The mixture was naturally heated to room temperature and reacted for 20 h. Filtered, washed with a tetrahydrofuran/ethanol (V/V = 2:1) mixed solvent, washed with ethyl acetate, filtered, and dried to obtain a dark red solid (1.73 kg, yield 76%, purity 99.7%).
[0124]
1H-NMR(500MHz,D 2O+NaOH)δ7.725-7.741(d,1H),7.298-7.316(d,3H),7.183-7.198(d,1H),7.131-7.149(m,2H),6.612-6.643(t,1H),3.574-3.596(t,4H),2.759-2.778(br,4H),2.698-2.721(t,4H),2.428(s,3H),1.772(br,4H).
SYN
J.Med.Chem.2024,67,4376−4418
HetrombopagOlamine (Hengqu).
Hetrombopag olamine (6), an oral nonpeptide thrombopoietin receptor
(TpoR)agonistdevelopedby JiangsuHengruiPharmaceutical, was approved in China in June2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemiawhohave not responded well to other treatments.46Hetrombopag, like other TpoR agonists, increases platelet production by binding to the transmembranedomainofTpoRinprogenitorcells, inducing
megakaryocytes.Theeffectisadditivewiththeactionofnative thrombopoietin, whichbinds to the extracellular domainof TpoR.Hetrombopag is structurallyrelatedtoeltrombopag, a previously approvedTpoR, withmodifications to enhance potencyandminimizetoxicity.46−48InaPhaseIIIclinicaltrial, ITPpatients demonstratedadurableplatelet response, with reducedbleedingriskanduseof rescuetherapycomparedto
placebo.49 Akilo-scale, chromatography-freesynthesisofhetrombopag has been reported by researchers at Jiangsu Hengrui Pharmaceutical in the Chinese-language patent literature (Scheme 12).50,51 Commercially available aniline 6.1 was coupledwith furoic acid (6.2) using aMeerwein arylation reaction togive intermediate6.3.This process first involves diazotizationof the anilineusing sodiumnitrite andhydrochloricacid.Ureawasusedtoquenchtheresidualnitrousacid, animprovement thatultimatelygavetheproductwithhigher purity and lower levels of specific impurities; the crude
diazoniumsalt solutionwas carried forwarddirectlywithout furthermanipulation.Furoicacid(6.2)inacetonewastreated withcopper(II)chloridedihydratefollowedbyadditionofthe
diazonium salt solution to affect the arylation. The crude productwaspurifiedbyacid−baseextractionandisolatedby filtrationtoprovide6.3 in55%yield.Basichydrolysisof the
cycliccarbamateunveiledthefreeanilineandphenolmoieties in arene 6.4. Nucleophilic attack of the enolate anion of pyrazolone 6.5 (see Scheme 13) on the diazoniumsalt of aniline6.4 formed the central hydrazonemoiety ina JappKlingemann-like reaction. The crude product was triturated withethylacetatetorapidlyprovidehetrombopagfreebasein
97.5%yield.TreatmentwithethanolamineinTHFandEtOH thengeneratedhetrombopagolamine (6) in76%yieldand 99.7%purity.51 Pyrazolone intermediate6.5was synthesized in two steps
(Scheme 13).52,53 5,6,7,8-Tetrahydronaphthalen-2-yl amine (6.6)was converted to the diazoniumion and reduced in situ to the corresponding hydrazine 6.7 using stannous chloridedihydrate.Condensationof thehydrazinewithethyl acetoacetate in ethyl acetate and in situ cyclization gave pyrazolone6.5.While the synthesis fromaniline6.1 to the activepharmaceutical ingredient(API)6wasreportedonthe
kilo-scale, thesynthesisofpyrazolone6.5wasreportedonlyon gram-scale
(46) Syed, Y. Y. Hetrombopag: First approval. Drugs 2021, 81, 1581−1585.
(47) Xie, C.; Zhao, H.; Bao, X.; Fu, H.; Lou, L. Pharmacological characterization of hetrombopag, a novel orally active human thrombopoietin receptor agonist. J. Cell. Mol. Med. 2018, 22, 5367−5377.
(48) Zheng, L.; Liang, M.-z.; Zeng, X.-l.; Li, C.-z.; Zhang, Y.-f.; Chen, X.-y.; Zhu, X.; Xiang, A.-b. Safety, pharmacokinetics and pharmacodynamics of hetrombopag olamine, a novel TPO-R agonist, in healthy individuals. Basic Clin. Pharmacol. Toxicol. 2017, 121, 414−422.
(49) Mei, H.; Liu, X.; Li, Y.; Zhou, H.; Feng, Y.; Gao, G.; Cheng, P.; Huang, R.; Yang, L.; Hu, J.; Hou, M.; Yao, Y.; Liu, L.; Wang, Y.; Wu, D.; Zhang, L.; Zheng, C.; Shen, X.; Hu, Q.; Liu, J.; Jin, J.; Luo, J.; Zeng, Y.; Gao, S.; Zhang, X.; Zhou, X.; Shi, Q.; Xia, R.; Xie, X.; Jiang, Z.; Gao, L.; Bai, Y.; Li, Y.; Xiong, J.; Li, R.; Zou, J.; Niu, T.; Yang, R.;
Hu, Y. A multicenter, randomized phase III trial of hetrombopag: a novel thrombopoietin receptor agonist for the treatment of immune thrombocytopenia. J. Hematol. Oncol. 2021, 14, 37.
(50) Shi, A.; Diao, A.; Du, Y. Preparation of bicyclic substituted pyrazolone azo derivatives. China Patent CN 113929668, 2022.
(51) Diao, A.; Gao, X.; Bian, L. Method for preparing bicyclo substituted pyrazolone azo derivatives and intermediates. WO 2018133818, 2018.
(52) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Preparation of pyrazole derivatives as thrombopoietin receptor agonists. WO 2010142137, 2010.
(53) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Salts of bicyclo substituted pyrazolon azo derivatives, preparation method and use
thereof. European Patent EP 2441457, 2014.

.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Hetrombopag Olamine, CHINA 2021, APPROVALS 2021, Hetrombopag diolamine, SHR 8735 olamine, Hetrombopag ethanolamine, SHR-8735 olamine, V45T2I862X, RAFUTROMBOPAG OLAMINE
SOVESUDIL
SOVESUDIL
PHP-201; AMA 0076, C23O3R93BM
CAS 1333400-14-8
| Molecular Weight | 407.44 |
|---|---|
| Formula | C23H22FN3O3 |
Sovesudil (PHP-201) is a potent, ATP-competitive, locally acting Rho kinase (ROCK) inhibitor with IC50s of 3.7 and 2.3 nM for ROCK-I and ROCK-II, respectively. Sovesudil lowers intraocular pressure (IOP) without inducing hyperemia.
SCHEME
PATENTS
Bioorganic & Medicinal Chemistry Letters (2013), 23(23), 6442-6446
https://www.sciencedirect.com/science/article/abs/pii/S0960894X13011141
Figure 2. Synthetic scheme for synthesis of compounds 10–35. (a) H2SO4, MeOH, 60 C, 16 h; (b) NBS, AIBN, CCl4, reflux, 16 h; (c) Boc2NH, t-BuOK, DMF, rt, 16 h; (d) DCM/TFA
(50:1), 0 C ? rt, 4 h; (e) NaOH, MeOH, 50 C, 2 h; (f) HATU, DMAP, NEt3, DMA, 30 C, 16 h; (g) Pd(dppf)Cl2, Na2CO3, H2O, DMF, 100 C; 16 h; (h) ROH, TBTU, HOBT, DIEA, DMF,
rt, 16 h or ROH, DCC, DMAP, DCM, rt, 16 h or Me2C@C(Cl)NMe2, THF or DCM, rt, followed by ROH, 16 h; (i) DCM/TFA (7:1), 30 C, 16 h or HCl(g) in DCM, 30 C, 16 h.
PATENT
WO2011107608
- [1]. Van de Velde S, et al. AMA0076, a novel, locally acting Rho kinase inhibitor, potently lowers intraocular pressure in New Zealand white rabbits with minimal hyperemia. Invest Ophthalmol Vis Sci. 2014;55(2):1006-1016. Published 2014 Feb 18. [Content Brief][2]. Ha A, et al. Sovesudil (locally acting rho kinase inhibitor) for the treatment of normal-tension glaucoma: the randomized phase II study [published online ahead of print, 2021 Jul 28]. Acta Ophthalmol. 2021;10.1111/aos.14949. [Content Brief]
////////SOVESUDIL, 1333400-14-8, PHP 201, AMA 0076, C23O3R93BM
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Contezolid
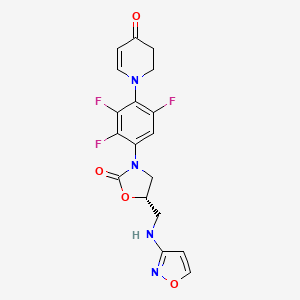

Contezolid
- MRX-I
- 1112968-42-9
- MRX-1
- B669M62ELP
- (5S)-5-[(1,2-oxazol-3-ylamino)methyl]-3-[2,3,5-trifluoro-4-(4-oxo-2,3-dihydropyridin-1-yl)phenyl]-1,3-oxazolidin-2-one
- 4(1H)-Pyridinone, 2,3-dihydro-1-(2,3,6-trifluoro-4-((5S)-5-((3-isoxazolylamino)methyl)-2-oxo-3-oxazolidinyl)phenyl)-
- (5S)-5-[(1,2-oxazol-3-ylamino)methyl]-3-[2,3,5-trifluoro-4-(4-oxo-2,3-dihydropyridin-1-yl)phenyl]-1,3-oxazolidin-2-one
- 4(1H)-Pyridinone, 2,3-dihydro-1-[2,3,6-trifluoro-4-[(5S)-5-[(3-isoxazolylamino)methyl]-2-oxo-3-oxazolidinyl]phenyl]-
- 1-{2,3,6-trifluoro-4-[(5S)-5-{[(1,2-oxazol-3-yl)amino]methyl}-2-oxo-1,3-oxazolidin-3-yl]phenyl}-1,2,3,4-tetrahydropyridin-4-one
- 5-((isoxazol-3-ylamino)methyl)-3-(2,3,5-trifluoro-4-(4-oxo-3,4-dihydropyridin-1(2H)-yl)phenyl)oxazolidin-2-one
- コンテゾリド;
WeightAverage: 408.337
Monoisotopic: 408.104539468
Chemical FormulaC18H15F3N4O4
Shanghai MicuRx Pharmaceutical Co. Ltd
Contezolid was approved for use by the National Medical Products Administration (NMPA) of China in 2021
- OriginatorMicuRx Pharmaceuticals
- ClassAntibacterials; Oxazolidinones; Skin disorder therapies
- Mechanism of ActionProtein synthesis inhibitors
- Phase IIIDiabetic foot; Skin and soft tissue infections
- No development reportedGram-positive infections
- 28 Jan 2025No recent reports of development identified for phase-I development in Gram-positive-infections(In volunteers) in China (IV)
- 28 Jan 2025No recent reports of development identified for phase-I development in Gram-positive-infections(In volunteers) in China (PO)
- 29 Nov 2024Phase-III clinical trials in Skin and soft tissue infections in China (IV), prior to November 2024
Contezolid (trade name Youxitai) is an antibiotic of the oxazolidinone class.[1][2] It is effective against Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pyogenes, Streptococcus agalactiae, and other bacteria.[3]
In 2021, it was approved by the National Medical Products Administration of China for the treatment of complicated skin and soft tissue infections (cSSTI).[3][4]
A prodrug of contezolid, contezolid acefosamil, which is formulated for IV administration[5] is in Phase III clinical trials for diabetic foot infection.[6]

Chemical structure of contezolid acefosamil
SYN
https://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.2c00191
Abstract

New oral antibiotic contezolid (CZD) is effective against Gram-positive infections but unsuitable for intravenous (IV) administration due to its modest solubility. To address the medical need for an IV form of CZD, its isoxazol-3-yl phosphoramidate derivatives have been explored, and contezolid acefosamil (CZA, 8), the first representative of a novel O-acyl phosphoramidate prodrug class, has been identified. CZA exhibits high aqueous solubility (>200 mg/mL) and good hydrolytic stability at media pH suitable for IV administration. CZA rapidly converts into the active drug CZD in vivo. In a pharmacokinetic (PK) rat model, the exposure of active drug CZD after IV administration of the prodrug CZA was similar to or higher than that from the IV administration of CZD. The prodrug CZA is bioequivalent to or better than CZD in several preclinical infection models. CZA is likewise active upon its oral administration. To date, CZA has been evaluated in Phase 1 and Phase 2 clinical trials in the USA. It is advancing into further clinical studies including step-down therapy with in-hospital intravenous CZA administration followed by outpatient oral CZD treatment.
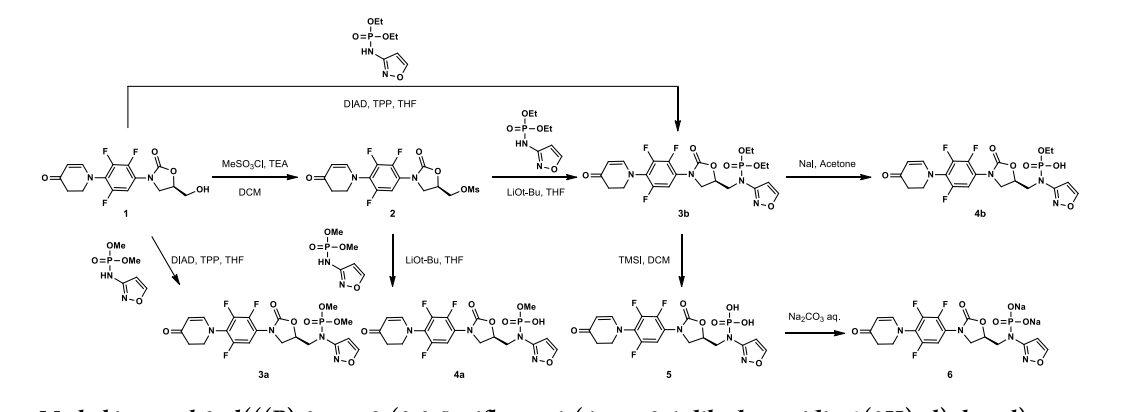
SYN
Contezolid (Youxitai). Contezolid (4), also referred to as MRX-I, is an orally administered oxazolidinone
antibacterial agent developed by Shanghai MicuRx Pharmaceutical Co. Ltd. Contezolid was developed to overcome the myelosuppression and monoamine oxidase (MAO) inhibition limitations of the structurally similar linezolid. 32 Contezolid is used to treat complicated skin and soft tissue infections arising
from multidrug-resistant Gram-positive bacterial infections including methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pyogenes, Streptococcus agalactiae, and vancomycin-resistant enterococci.3334 Contezolid was approved for use by the National Medical Products Administration (NMPA) of China in 2021.
As with most antibacterial oral therapies, high 35 dosage is required; the drug is given twice daily for 7−14 days.36,37
The synthesis of contezolid builds on prior research from other groups.
A sequence developed by Pharmaciawith a facile SN38began Ar reaction between polyfluorinated nitro
benzene 4.1 and piperidine-4-one 4.2 to furnish 4.3 in good yield (Scheme 9). Silyl enol ether formation afforded 4.4, which was subjected to Tsuji’s 39 method to give the α,βunsaturated ketone in excellent yield. Subsequent reduction of the nitro group gave aryl amine 4.5. Treatment of 4.5 with isobutyl chloroformate gave carbamate 4.6, which was treated with optically pure epoxide 4.7 to give xazolidinone 4.8. 38Mesylation of the free alcohol and displacement with N-Bocaminoisoxazole 4.9 afforded the Boc-protected contezolid 4.10. Simple acidic removal of the Boc group provided contezolid 4.
(32) Wang, W.; Voss, K. M.; Liu, J.; Gordeev, M. F. Nonclinical
evaluation of antibacterial oxazolidinones contezolid and contezolid
acefosamil with low serotonergic neurotoxicity. Chem. Res. Toxicol.
2021, 34, 1348−1354.
(33) Hoy, S. M. Contezolid: First approval. Drugs 2021, 81, 1587−
1591.
(34) MicuRx Pharmaceuticals. China NMPA approves MicuRx’s
contezolid for treatment of drug-resistant bacterial infection. http://www.
micurx.com/703.html (accessed 2023-06).
(35) MSD Pharmaceuticals. Usual dosages of commonly prescribed
antibiotics. https://www.msdmanuals.com/en-jp/professional/
multimedia/table/usual-dosages-of-commonly-prescribed-antibioticsa
(accessed 2023-06).
(36) Barbachyn, M. R.; Hutchinson, D. K.; Brickner, S. J.; Cynamon,
M. H.; Kilburn, J. O.; Klemens, S. P.; Glickman, S. E.; Grega, K. C.;
Hendges, S. K.; Toops, D. S.; et al. Identification of a novel
oxazolidinone (U-100480) with potent antimycobacterial activity. J.
Med. Chem. 1996, 39, 680−685.
(37) Im, W. B.; Choi, S. H.; Park, J. Y.; Choi, S. H.; Finn, J.; Yoon, S.
H. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone
antibacterial agent. Eur. J. Med. Chem. 2011, 46, 1027−1039.
(38) Manninen, P. R.; Brickner, S. J. Preparation of N-aryl-5R
hydroxymethyl-2-oxazolidinones from N-aryl carbamates: N-phenyl
(5R)-hydroxymethyl-2-oxazolidinone. Organic Synth 2005, 81, 112.
(39) Tsuji, J.; Minami, I.; Shimizu, I. A novel palladium-catalyzed
preparative method of α,β-unsaturated ketones and aldehydes from
saturated ketones and aldehydes via their silyl enol ethers. Tetrahedron
Lett. 1983, 24, 5635−5638.
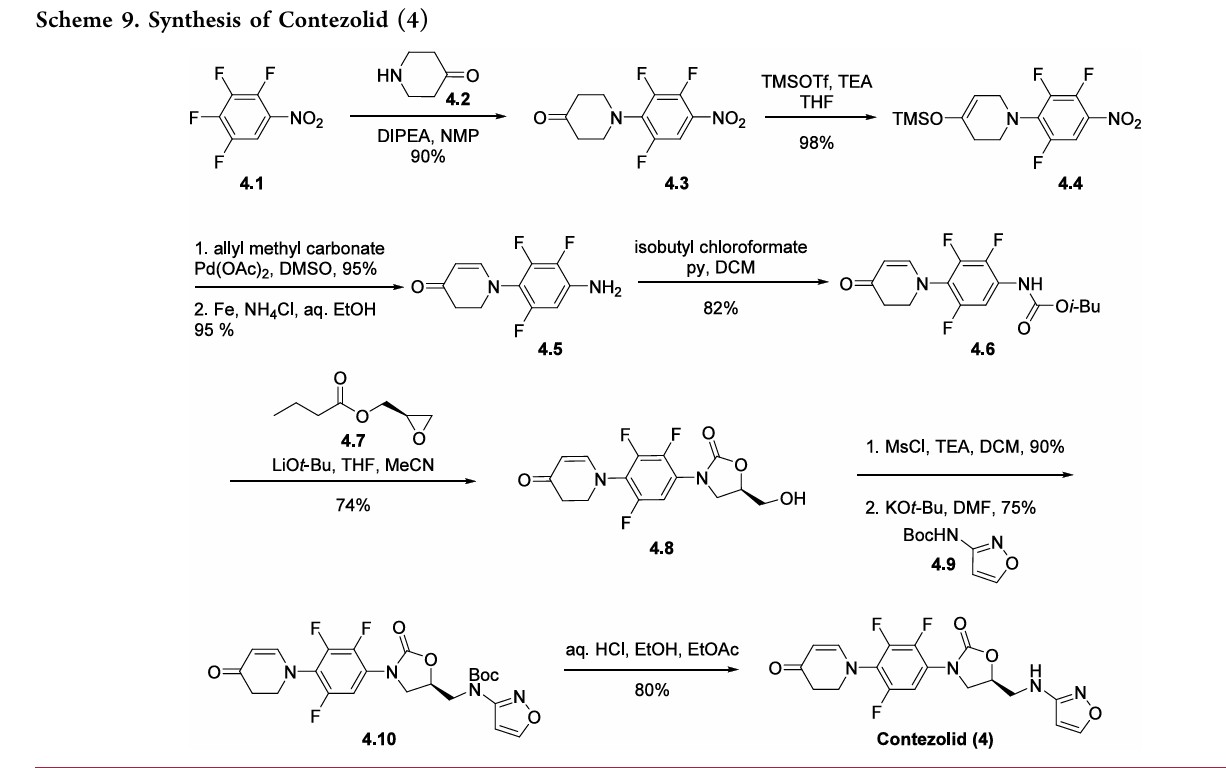
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Trade names | Youxitai |
| Other names | MRX-I |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1112968-42-9 |
| PubChem CID | 25184541 |
| IUPHAR/BPS | 10795 |
| DrugBank | DB12796 |
| ChemSpider | 34217570 |
| UNII | B669M62ELP |
| KEGG | D11297 |
| ChEMBL | ChEMBL3287379 |
| CompTox Dashboard (EPA) | DTXSID901353186 |
| Chemical and physical data | |
| Formula | C18H15F3N4O4 |
| Molar mass | 408.337 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Gordeev MF, Yuan ZY (June 2014). “New Potent Antibacterial Oxazolidinone (MRX-I) with an Improved Class Safety Profile”. Journal of Medicinal Chemistry. 57 (11): 4487–4497. doi:10.1021/jm401931e. PMID 24694071.
- Zhao X, Huang H, Yuan H, Yuan Z, Zhang Y (May 2022). “A Phase III multicentre, randomized, double-blind trial to evaluate the efficacy and safety of oral contezolid versus linezolid in adults with complicated skin and soft tissue infections”. The Journal of Antimicrobial Chemotherapy. 77 (6): 1762–1769. doi:10.1093/jac/dkac073. PMID 35265985.
- Hoy SM (September 2021). “Contezolid: First Approval”. Drugs. 81 (13): 1587–1591. doi:10.1007/s40265-021-01576-0. PMC 8536612. PMID 34365606.
- Mak E (3 June 2021). “Micurx wins China approval for antibacterial contezolid”. BioWorld.
- Liu J, Wang W, Wang C, Zhang L, Zhang X, Liu S, et al. (July 2022). “Discovery of Antibacterial Contezolid Acefosamil: Innovative O-Acyl Phosphoramidate Prodrug for IV and Oral Therapies”. ACS Medicinal Chemistry Letters. 13 (7): 1030–1035. doi:10.1021/acsmedchemlett.2c00191. PMC 9290071. PMID 35859881.
- “Contezolid acefosamil by MicuRx Pharmaceuticals for Diabetic Foot Infection (DFI): Likelihood of Approval”. GlobalData. 31 May 2023 – via Pharmaceutical Technology.
/////////Contezolid, CHINA 2021, APPROVALS 2021, MRX-I, 1112968-42-9, MRX 1, B669M62ELP, コンテゾリド ,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}