
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LEVALBUTEROL TARTRATE


LEVALBUTEROL TARTRATE
Levosalbutamol
cas 661464-94-4
4-[(1R)-2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl)phenol;(2R,3R)-2,3-dihydroxybutanedioic acid
MW 628.7, C30H48N2O12
- Xopenex HFA
- Levosalbutamol tartrate
- ADS4I3E22M
- UNII-ADS4I3E22M
- Levosalbutamol tartrate(levalbuterol) is the R-enantiomer of the short-acting β2-adrenergic receptor agonist salbutamol.
Levalbuterol Tartrate is the tartrate salt form of levalbuterol, the R-enantiomer of the short-acting beta-2 adrenergic receptor agonist albuterol, with bronchodilator activity. Levalbuterol selectively binds to beta-2 adrenergic receptors in bronchial smooth muscle, thereby activating intracellular adenyl cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cAMP). Increased cAMP levels cause relaxation of bronchial smooth muscle, relieve bronchospasms, improve mucociliary clearance and inhibit the release of mediators of immediate hypersensitivity from cells, especially from mast cells.
British patent document GB1298494A firstly discloses synthesis of levosalbutamol, which comprises the steps of carrying out crystallization resolution by using D- (+) -dibenzoyl tartaric acid, carrying out ester reduction reaction, and removing two benzyl protecting groups to obtain levosalbutamol, wherein the process route is as follows:
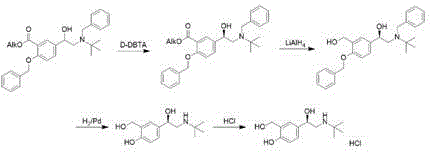
chinese patent CN1705634A, and using rhodium and chiral bidentate phosphine ligand combination, levosalbutamol can be obtained with good yield and good optical purity on a technical scale. The disadvantages are that the toxicity of the reagent is high, the hydrogenation risk is high, and the process route is as follows:
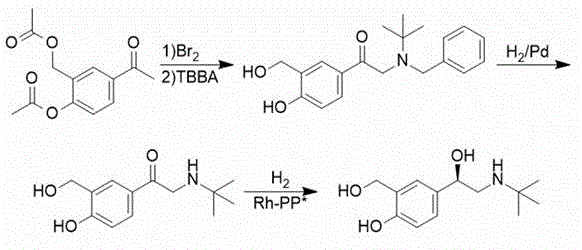
SCHEME

PATENTS
MX2012014342
IN2009MU01097
IN2007CH01847
US20040115136
CN1382685
PATENT
https://patents.google.com/patent/CN114539077A/en
The technical scheme adopted by the invention is as follows: 1) 1- (2, 2-dimethyl-4H-benzo [ d ] [1,3] dioxin-6-yl) ethanol and titanium dioxide are used as initial raw materials, a solvent-free system is adopted, and 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxin is synthesized through dehydration.
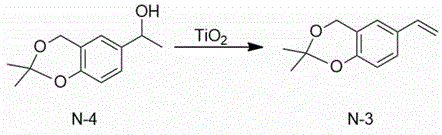
Then, the 2, 2-dimethyl-6-vinyl-4H-benzo [ D ] [1,3] dioxin is subjected to epoxidation under the combined action of 1,2:4, 5-di-O-isopropylidene-BETA-D-erythro-2, 3-dione-2, 6-pyranose (Shi’s Catalyst), Oxone and potassium hydroxide to obtain (R) -2, 2-dimethyl-6- (oxirane-2-yl) -4H-benzo [ D ] [1,3] dioxin.
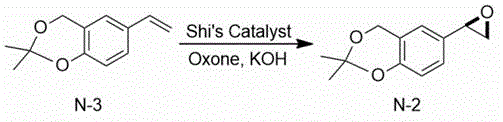
Reacting and condensing (R) -2, 2-dimethyl-6- (epoxy ethane-2-group) -4H-benzo [ D ] [1,3] dioxin and tert-butylamine in ethanol, and salifying with D- (+) -malic acid to obtain (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-group) ethanol D- (+) -malate.
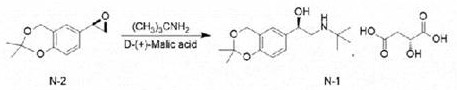
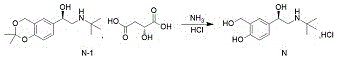
And (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate is subjected to hydroaminolysis and reacts with hydrogen chloride ethanol to prepare the levosalbutamol hydrochloride.
The invention discloses a novel method for synthesizing levalbuterol hydrochloride, wherein the synthesis of a key intermediate is novel without cyclization oxidation, the total yield is 85-90%, and the method is higher than that of the conventional method. The process is convenient to operate, the raw materials are economical, and the method is suitable for large-scale industrial production.
EXAMPLE 12 preparation of 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxine
A1000 mL flask was charged with 208g (1.0 mol) of 1- (2, 2-dimethyl-4H-benzo [ d ] [1,3] dioxin-6-yl) ethanol, which was accurately weighed, and stirring was started. Then slowly adding 16g (0.2 mol) of titanium dioxide, installing a water separator and a water flow pipe, starting heating until the internal temperature is kept at 120-130 ℃, and stirring for 12 hours. After the reaction is finished, the temperature is reduced to below 50 ℃, the water separator is removed, the reduced pressure distillation device is changed, and 120 ℃ (less than 100 Pa) fraction is collected to obtain 180.7g of 2, 2-dimethyl-6-vinyl-4H-benzo [ d ] [1,3] dioxin with the yield of 95%.
Mass spectrum: EI (m/z): 190; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,CDCl3)δ7.55(d,J=4Hz,1H),7.11(s,1H),6.87(d,J=4Hz,1H),6.65~6.60(m,1H),5.63~5.60(m,1H),5.19~5.5(m,1H),4.59(s,2H),1.49(s,6H)。
EXAMPLE 2 Synthesis of (R) -2, 2-dimethyl-6- (oxiran-2-yl) -4H-benzo [ d ] [1,3] dioxine
A clean 5000mL three-neck flask is taken, 180.5g (0.95 mol) of the compound
2, 2-dimethyl-6-vinyl-4H-benzo [ D ] [1,3] dioxin obtained in the example 1 is added, 2000mL of acetonitrile is added for dissolution, 1,2:4, 5-di-O-isopropylidene-BETA-D-erythro-2, 3-dione-2, 6-pyranose 49.1g (0.19 mol) is added, potassium monopersulfate (Oxone) 876g (1.43 mol) is added under stirring, a proper amount of potassium hydroxide is added after the addition is finished, the pH of the system is kept between 10 and 11, and the stirring reaction is continued at 25 ℃ for 8 to 12 hours. After the reaction, the mixture was slowly poured into 2000ml of purified water prepared in advance, stirred sufficiently for 30min, and then was allowed to stand for layering, and the organic layer was collected. 2000ml of dichloromethane is added for extraction, organic layers are combined and washed by saturated sodium chloride solution, the organic layer is dried by adding anhydrous sodium sulfate and concentrated to dryness to obtain 196g of crude colorless liquid with the yield of 100 percent.
Mass spectrum: EI (m/z): 207; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,CDCl3)δ7.25(s,1H),7.18(d,J=4Hz,1H),6.85(d,J=4Hz,1H),4.59(s,2H),3.85~3.81(m,1H),2.96~2.71(m,2H),1.49(s,6H)。
EXAMPLE 3 preparation of (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate salt
A clean 5000mL three-neck flask is taken, the compound (R) -2, 2-dimethyl-6- (oxiranyl-2-yl) -4H-benzo [ d ] [1,3] dioxin obtained in the example 2 is added, 196g (0.95 mol) of the clean 5000mL three-neck flask is taken, 1000mL of ethanol is added for dissolution, 80.4g (1.1 mol) of tert-butylamine is added, stirring is started, heating is carried out till reflux, reaction is carried out for 3H, and the progress of the reaction is detected by TLC. After the reaction is finished, 127g (0.95 mol) of D- (+) -malic acid is added in batches, and stirring and refluxing are continued for 2h after the addition is finished. And then cooling to 5-15 ℃, precipitating a large amount of solid, stirring for 3H, filtering, washing the filter cake with ethanol, collecting the filter cake, and drying to obtain (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate 372g of white solid with the yield of 94.7%.
Mass spectrum: ESI (m/z): 280.1, respectively; hydrogen nuclear magnetic resonance spectroscopy:1HNMR(400MHz,d-DMSO)δ7.25(s,1H),7.18(d,J=4Hz,1H),6.85(d,J=4Hz,1H),4.90~4.76(m,2H),4.59(s,2H),4.44~4.40(m,2H),3.65(br,2H),3.15~2.90(m,2H),2.77~2.52(m,2H),2.03(s,1H),1.50(s,6H),1.27(s,9H)。
EXAMPLE 4 preparation of L-salbutamol hydrochloride
A5000 mL beaker was charged with 372g of the compound (R) -2- (tert-butylamine) -1- (2, 2-dimethyl-4H-benzo [ D ] [1,3] dioxin-6-yl) ethanol D- (+) -malate salt obtained in example 3, 1500mL of purified water was added, and the mixture was stirred to dissolve it, followed by addition of 1500mL of dichloromethane and cooling in an ice bath. Slowly adding a proper amount of concentrated ammonia water under stirring to adjust the pH value of the water phase to 9-10, continuously stirring for 30min, and standing for layering. Separating and collecting organic layer, adding 1000ml of dichloromethane into water layer, stirring for 10min, standing and demixing. Separating and collecting organic layers, combining the organic layers, adding 2000ml of saturated sodium chloride solution into the organic layers, stirring for 30min, standing for layering, collecting the organic layers, adding a proper amount of anhydrous sodium sulfate, drying, filtering, washing with dichloromethane, and collecting filtrate.
And (3) carrying out rotary evaporation and concentration on the filtrate to about 1500mL, transferring the concentrated filtrate into a 5000mL three-neck bottle, and placing the three-neck bottle in an ice bath to cool the three-neck bottle to 5-15 ℃. About 110g of 30% hydrogen chloride ethanol solution is dropwise added under stirring, and after the dropwise addition is finished, 2000mL of methyl tertiary butyl ether is dropwise added under stirring, so that a large amount of white solid is precipitated. And after the addition is finished, continuously stirring for 3 hours at the temperature of 5-15 ℃, filtering, adding methyl tert-butyl ether into a filter cake for washing, collecting the filter cake, and drying to obtain 241.5g with the yield of 97.3%. Through HPLC analysis, the purity is 99.95%, and the isomer content is not detected, as shown in figures 1-4. The total yield of the four-step reaction is 87.5 percent.
Publication numberPriority datePublication dateAssigneeTitle
CN1413976A *2002-09-132003-04-30苏州君宁新药开发中心有限公司New process for preparing levo-albuterol
US20050261368A1 *2004-05-202005-11-24Valeriano MerliPreparation of levalbuterol hydrochloride
CN103951568A *2014-05-192014-07-30苏州弘森药业有限公司New process for synthesizing salbutamol and sulfate of salbutamol
CN104557572A *2014-12-302015-04-29上海默学医药科技有限公司Levalbuterol intermediate and levalbuterol hydrochloride synthesis method
CN110963929A *2019-11-262020-04-07安徽恒星制药有限公司Preparation method of salbutamol hydrochloride suitable for industrial production
CN113227113A *2018-12-202021-08-06帝斯曼知识产权资产管理有限公司Improved synthesis of epoxidation catalysts
CN113801029A *2020-06-162021-12-17盈科瑞(天津)创新医药研究有限公司Preparation method of levalbuterol hydrochloride
//////////Levosalbutamol, LEVALBUTEROL TARTRATE, Xopenex HFA, Levosalbutamol tartrate, ADS4I3E22M, UNII-ADS4I3E22M
Zilucoplan
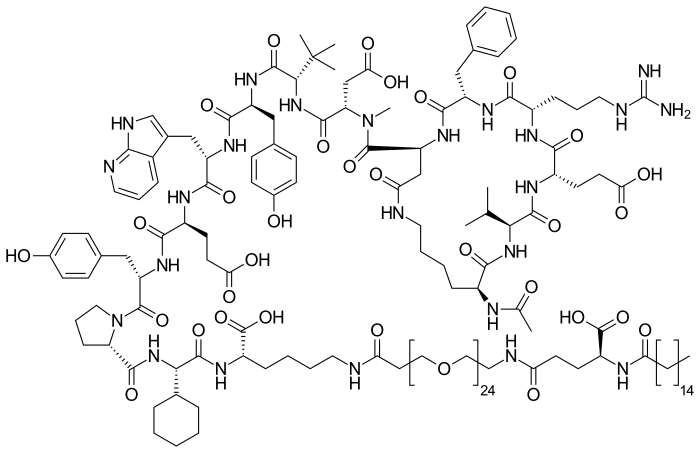
Zilucoplan
CAS 1841136-73-9
YG391PK0CC, RA101495, WHO 10602
3562 g/mol, C172H278N24O55
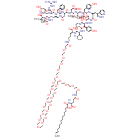
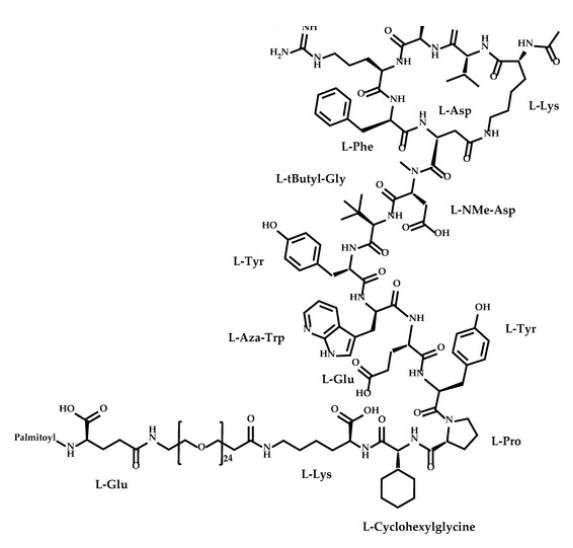
Zilucoplan lso designated as RA101495, is the active principle of Zilbrysq®, commercialized by UCB Pharma S.A. It is a 3.5 kDa synthetic macrocyclic peptide composed of 15 amino acid residues, including four unnatural amino acids [27]. The amino acid residues composition is: L-Lys, L-Val, L-Glu, L-Arg, L-Phe, L-Asp, L-L-NMe-Asp, L-tButyl-Gly, L-Tyr, L-7-aza-Trp, L-Glu, L-Tyr, L-Pro, L-Cyclohexyl-Gly, and L-Lys.
N2-ACETYL-L-LYSYL-L-VALYL-L-.ALPHA.-GLUTAMYL-L-ARGINYL-L-PHENYLALANYL-L-.ALPHA.-ASPARTYL-N-METHYL-L-.ALPHA.-ASPARTYL-3-METHYL-L-VALYL-L-TYROSYL-3-(1H-PYRROLO(2,3-B)PYRIDIN-3-YL)-L-ALANYL-L-.ALPHA.-GLUTAMYL-L-TYROSYL-L-PROLYL-(2S)-2-CYCLOHEXYLGLYCYL-N6-(3
POLY(OXY-1,2-ETHANEDIYL), ALPHA-(2-(((4S)-4-CARBOXY-1-OXO-4-(1-OXOHEXADECYL)BUTYL)AMINO)ETHYL)-OMEGA-HYDROXY-, 15-ETHER WITH N-ACETYL-L-LYSYL-L-VALYL-L-ALPHA-GLUTAMYL-L-ARGINYL-L-PHENYLALANYL-L-ALPHA-ASPARTYL-N-METHYL-L-ALPHA-ASPARTYL-3-METHYL-
(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,5S,8S,11S,14S,22S)-22-acetamido-11-benzyl-8-(3-carbamimidamidopropyl)-5-(2-carboxyethyl)-3,6,9,12,16,23-hexaoxo-2-propan-2-yl-1,4,7,10,13,17-hexazacyclotricosane-14-carbonyl]-methylamino]-3-carboxypropanoyl]amino]-3,3-dimethylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(1H-pyrrolo[2,3-b]pyridin-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]-2-cyclohexylacetyl]amino]-6-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[(4S)-4-carboxy-4-(hexadecanoylamino)butanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]hexanoic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Zilucoplan Sodium | Not Available | Not Available | FUSMWKLQHKXKHI-WHKBRXDJSA-J |
FDA 10/17/2023, Zilbrysq, To treat generalized myasthenia gravis in adults who are anti-acetylcholine receptor (AChR) antibody positive
Drug Trials Snapshot
Zilucoplan, sold under the brand name Zilbrysq, is a medication used for the treatment of generalized myasthenia gravis.[6][9][10] It is a complement inhibitor that is injected subcutaneously (under the skin).[6]
Zilucoplan is a cyclic peptide that binds to the protein complement component 5 (C5) and inhibits its cleavage into C5a and C5b.[11]
Zilucoplan was approved for medical use in the United States in October 2023,[6][12] in the European Union in December 2023,[7] and in Australia in July 2024.[1]
Zilucoplan is a 15 amino-acid, synthetic macrocyclic peptide with formula C172H278N24O55. Its sodium salt is used for the treatment of generalised myasthenia gravis (a disease that leads to muscle weakness and tiredness) in adults whose immune system produces antibodies against acetylcholine receptors. It has a role as a complement component 5 inhibitor and an immunosuppressive agent. It is a macrocycle, a homodetic cyclic peptide and a polyether. It is a conjugate acid of a zilucoplan(4-).
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11752190 | No | 2023-09-12 | 2035-06-12 |  |
| US11014965 | No | 2021-05-25 | 2035-06-12 | |
| US10435438 | No | 2019-10-08 | 2035-06-12 | |
| US10208089 | No | 2019-02-19 | 2035-06-12 | |
| US10106579 | No | 2018-10-23 | 2035-06-12 | |
| US10835574 | No | 2020-11-17 | 2035-06-12 | |
| US11535650 | No | 2022-12-27 | 2035-06-12 | |
| US10562934 | No | 2020-02-18 | 2035-06-12 | |
| US11965040 | No | 2024-04-23 | 2035-06-12 | |
PAPER
https://www.mdpi.com/2813-2998/3/2/18
References
- ^ Jump up to:a b c “Zilbrysq (zilucoplan)”. Therapeutic Goods Administration (TGA). 24 September 2024. Retrieved 12 October 2024.
- ^ “Therapeutic Goods (Poisons Standard—June 2024) Instrument 2024”. Federal Register of Legislation. 30 May 2024. Retrieved 10 June 2024.
- ^ “Zilbrysq (UCB Australia Pty Ltd T/A UCB Pharma Division of UCB Australia)”. Therapeutic Goods Administration (TGA). 13 September 2024. Retrieved 15 September 2024.
- ^ “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-08-13]”. Health Canada. 13 August 2024. Retrieved 15 August 2024.
- ^ “Regulatory Decision Summary for Zilbrysq”. Drug and Health Products Portal. 11 July 2024. Retrieved 27 December 2024.
- ^ Jump up to:a b c d e “Zilbrysq- zilucoplan injection, solution”. DailyMed. 19 July 2024. Retrieved 15 September 2024.
- ^ Jump up to:a b c d “Zilbrysq EPAR”. European Medicines Agency. 1 December 2023. Retrieved 11 December 2023.
- ^ “Zilbrysq Product information”. Union Register of medicinal products. 4 December 2023. Archived from the original on 11 December 2023. Retrieved 11 December 2023.
- ^ Howard JF, Kaminski HJ, Nowak RJ, Wolfe GI, Benatar MG, Ricardo A, et al. (April 2018). “RA101495, a subcutaneously administered peptide inhibitor of complement component 5 (C5) for the treatment of generalized myasthenia gravis (gMG): Phase 1 results and phase 2 design (S31. 006)”. Neurology. 90 (15 Supplement). doi:10.1212/WNL.90.15_supplement.S31.006. S2CID 56969245. Archived from the original on 22 February 2022. Retrieved 24 June 2021.
- ^ Howard JF, Vissing J, Gilhus NE, Leite MI, Utsugisawa K, Duda PW, et al. (May 2021). “Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody-Positive Generalized Myasthenia Gravis”. Expert Opinion on Investigational Drugs. 30 (5): 483–493. doi:10.1080/13543784.2021.1897567. hdl:11250/2770699. PMID 33792453. S2CID 232482753.
- ^ Ricardo A, Arata M, DeMarco S, Dhamnaskar K, Hammer R, Fridkis-Hareli M, et al. (2015). “Preclinical Evaluation of RA101495, a Potent Cyclic Peptide Inhibitor of C5 for the Treatment of Paroxysmal Nocturnal Hemoglobinuria”. Blood. 126 (23): 939. doi:10.1182/blood.V126.23.939.939.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 22 December 2023. Archived from the original on 8 January 2023. Retrieved 27 December 2023.
- ^ Jump up to:a b “Zilbrysq: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 26 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Zilucoplan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Archived from the original on 17 October 2023. Retrieved 19 October 2023.
- ^ “EU/3/22/2650: Orphan designation for the treatment of myasthenia gravis”. European Medicines Agency. 15 September 2023. Archived from the original on 29 January 2023. Retrieved 24 September 2023.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
- Clinical trial number NCT04115293 for “Safety, Tolerability, and Efficacy of Zilucoplan in Subjects With Generalized Myasthenia Gravis (RAISE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zilbrysq |
| Other names | RA101495 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624002 |
| License data | US DailyMed: Zilucoplan |
| Pregnancy category | AU: D[1] |
| Routes of administration | Subcutaneous |
| Drug class | Complement inhibitor |
| ATC code | L04AJ06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[2][3][1]CA: ℞-only[4][5]US: ℞-only[6]EU: Rx-only[7][8] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1841136-73-9 |
| PubChem CID | 133083018 |
| DrugBank | DB15636 |
| ChemSpider | 71115966 |
| UNII | YG391PK0CC |
| KEGG | D12357 |
| ChEBI | CHEBI:229659 |
| Chemical and physical data | |
| Formula | C172H278N24O55 |
| Molar mass | 3562.229 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////zilucoplan, Zilbrysq, FDA 2023, APPROVALS 2023, EU 2023, EMA 2023, RA101495, RA 101495, WHO 10602
Labuxtinib
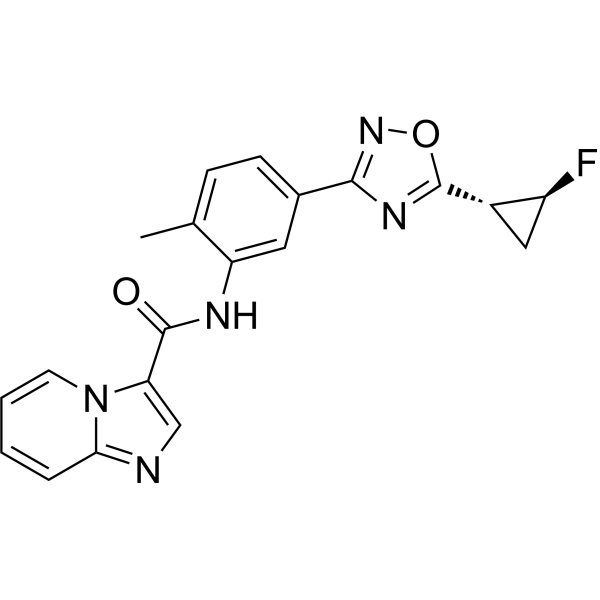
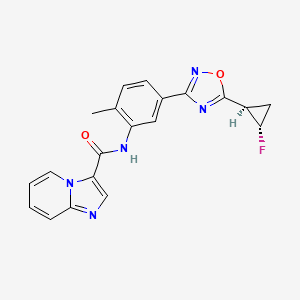
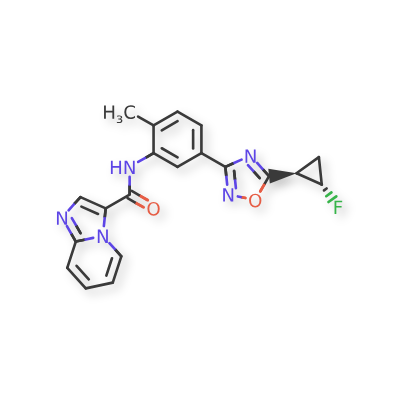
Labuxtinib
CAS 1426449-01-5
Labuxtinib, also known as EVT-8565072,
| Molecular Weight | 377.37 |
|---|---|
| Formula | C20H16FN5O2 |
- N-[5-[5-[(1R,2S)-2-fluorocyclopropyl]-1,2,4-oxadiazol-3-yl]-2-methylphenyl]imidazo[1,2-a]pyridine-3-carboxamide
- N-(5-(5-((1R,2S)-2-fluorocyclopropyl)-1,2,4-oxadiazol-3-yl)-2-methylphenyl)imidazo[1,2-a]pyridine-3-carboxamide
- QNX4G754W6
Labuxtinib is c-kit tyrosine kinase inhibitor.
Labuxtinib, also known as EVT-8565072, is a synthetic organic compound that acts as a tyrosine kinase inhibitor, specifically targeting c-KIT. It is a potential anti-cancer agent and is likely the INN (Proposed International Nonproprietary Name) for Third Harmonic Bio‘s candidate KIT inhibitor, THB335. The initial clinical lead, THB001, was discontinued due to hepatotoxicity, and THB335 is a follow-up molecule with structural modifications to address this issue.
SCHEME
SIDECHAIN
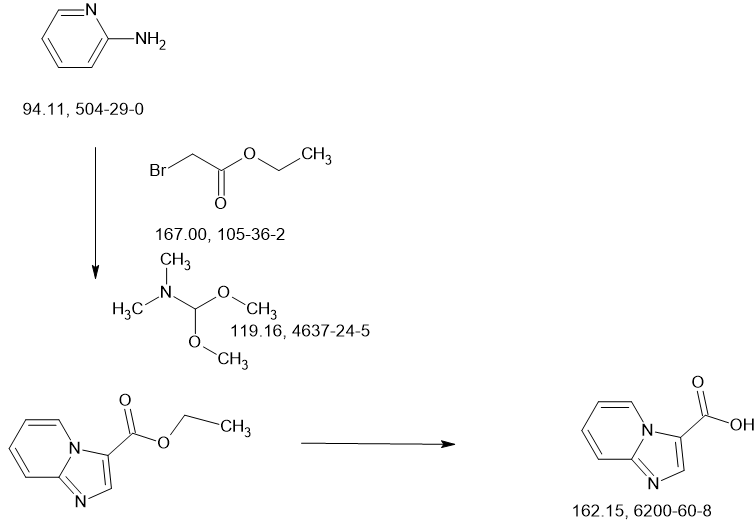
MAIN
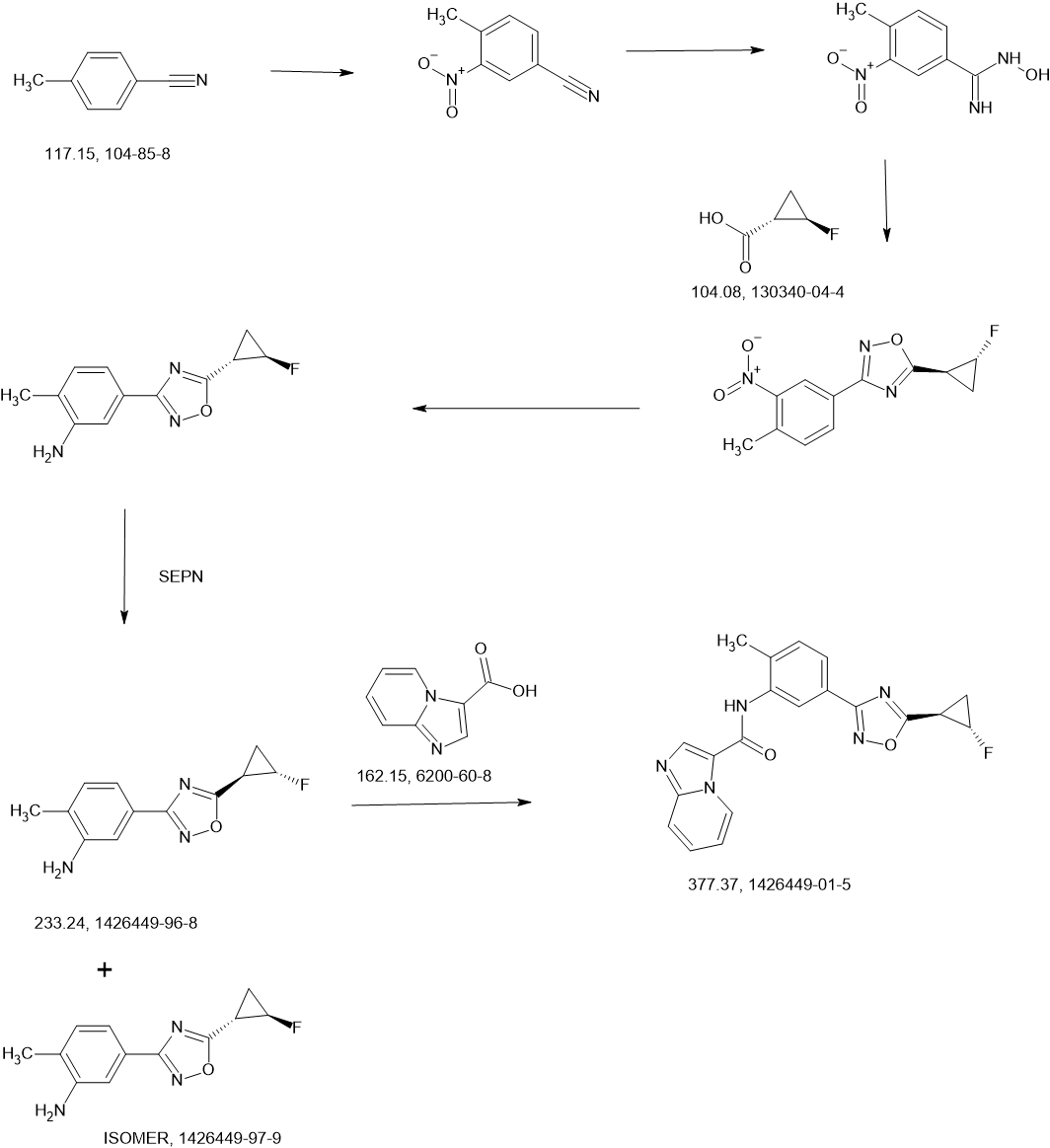
WO2013033070 F91
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013033070&_cid=P11-MBX2OL-77562-1
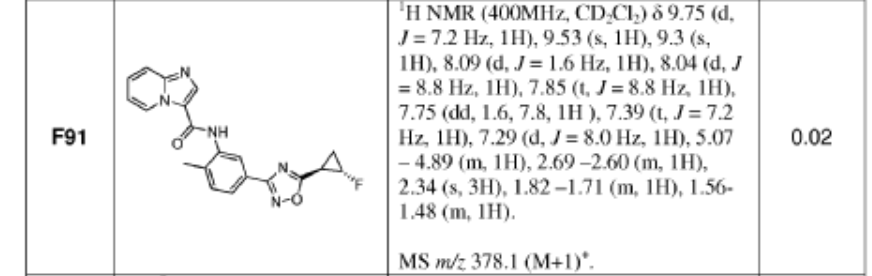
PATENT
WO2022109595 COMPD 1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022109595&_cid=P11-MBX2U2-81593-1
A. Compound 1
[0022] As defined above, a pharmaceutical composition of the present invention is a micronized powder comprising Compound 1. Compound 1 can be prepared according to example Fl 10 of WO 2013/033070 Al, which is incorporated by reference herein, as summarized in the
Scheme 1 provided below:
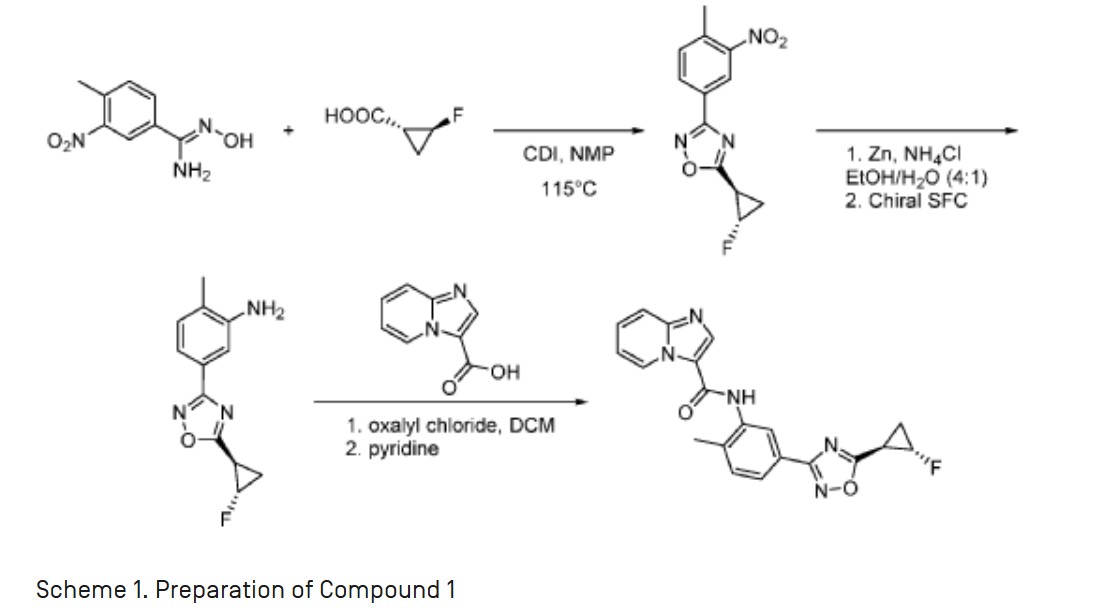
Scheme 1. Preparation of Compound 1
[0023] In some embodiments, the pharmaceutical composition is a micronized powder comprising dry microparticles of Compound 1. In some embodiments the microparticles of Compound 1 comprise amorphous Compound 1. In some embodiments, the microparticles of Compound 1 comprise a crystalline solid form of Compound 1. In some embodiments, the microparticles of Compound 1 comprise a crystalline free base solid form of Compound 1. In some embodiments, the microparticles of Compound 1 comprise a crystalline salt solid form of Compound 1.
[0024] In some embodiments, the crystalline solid form of Compound 1 is an anhydrate form. In some embodiments, the crystalline solid form of Compound 1 is a hydrate form. In some embodiments, the crystalline solid form of Compound l is a monohydrate. In some embodiments, the crystalline solid form of Compound l is a hemihydrate. In some embodiments, the crystalline solid form of Compound 1 is a dihydrate.
[0025] In some embodiments, the microparticles of Compound 1 comprise a crystalline solid form of Compound 1 disclosed in PCT/CN2020/090060, which is incorporated by reference herein.
PATENT
PATENT
WO2020228746 NOVARTIS
WO2013033070 IRM LLC
As of January 2025, Labuxtinib remains in preclinical/early clinical development, with no publicly disclosed Phase 1 data . THB’s strategy focuses on:
- Targeting KIT-driven malignancies: GISTs, mastocytosis, melanoma
- Addressing resistance: Overcoming mutations conferring resistance to imatinib/nilotinib .
///////////Labuxtinib, QNX4G754W6, EVT-8565072, EVT 8565072
Gepirone
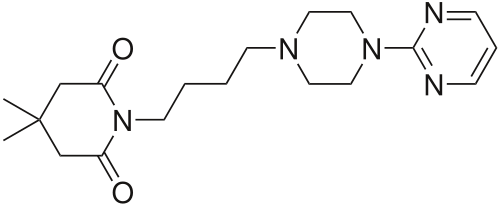
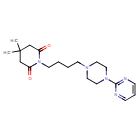
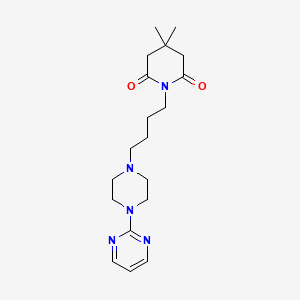
Gepirone
CAS 83928-76-1
BMY 13805, MJ 13805, ORG 13011, Gepirona,
JW5Y7B8Z18
FDA 9/22/2023, Gepirone is indicated for the treatment of major depressive disorder (MDD) in adults
Exxua |
Average: 359.474
Monoisotopic: 359.232125194
Chemical Formula
C19H29N5O2
4,4-dimethyl-1-{4-[4-(pyrimidin-2-yl)piperazin-1-yl]butyl}piperidine-2,6-dione
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Gepirone Hydrochloride | 80C9L8EP6V | 83928-66-9 | DGOCVISYYYQFEP-UHFFFAOYSA-N |
Gepirone, sold under the brand name Exxua, is a medication used for the treatment of major depressive disorder.[1] It is taken orally.[1]
Side effects of gepirone include dizziness, nausea, insomnia, abdominal pain, and dyspepsia (indigestion).[1] Gepirone acts as a partial agonist of the serotonin 5-HT1A receptor.[1][2] An active metabolite of gepirone, 1-(2-pyrimidinyl)piperazine, is an α2-adrenergic receptor antagonist.[1][3] Gepirone is a member of the azapirone group of compounds.[2]
Gepirone was synthesized by Bristol-Myers Squibb in 1986 and was developed and marketed by Fabre-Kramer Pharmaceuticals.[4] It was approved for the treatment of major depressive disorder in the United States in September 2023.[4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness.[5]
History
Gepirone was developed by Bristol-Myers Squibb in 1986,[5] but was out-licensed to Fabre-Kramer in 1993. The FDA rejected approval for gepirone in 2002 and 2004.[5] It was submitted for the preregistration (NDA) phase again in May 2007 after adding additional information from clinical trials as the FDA required in 2009. However, in 2012 it once again failed to convince the FDA of its qualities for treating anxiety and depression.[5] In December 2015, the FDA once again gave gepirone a negative review for depression due to concerns of efficacy.[12] However, in March 2016, the FDA reversed its decision and gave gepirone ER a positive review.[13] Gepirone ER was finally approved for the treatment of major depressive disorder in the United States in September 2023.[5]
SYN
Synthesis

Ormaza, V. A.; 1986, ES 8606333.
SYN
https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/gepirone
Gepirone (Exxua)
Gepirone, a selective and affinitive 5-hydroxytryptamine 1A (5-HT1A) agonist, received FDA approval on September 22, 2023, to treat major depressive disorder in adults [37]. Gepirone, an azapirone compound, is a pharmacological derivative of buspirone that exhibits specific activity on both pre- and post-synaptic 5-HT1A receptors. Despite the promising results observed in previous clinical trials for Gepirone, the need for frequent administration is a requirement due to its formulation as an immediate-release tablet and its short half-lives. Gepirone did not become a potential candidate for a new antidepressant until an extended-release formulation of it was developed [38–40].
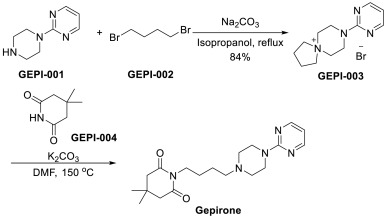
An efficient approach of Gepirone has been disclosed in Scheme 11 [41]. Substitution of 2-(piperazin-1-yl)pyrimidine (GEPI-001) with 1,4-dibromobutane (GEPI-002), followed by ring opening and addition, generated the final product Gepirone.
PATENT
https://patents.google.com/patent/WO2020148621A1/en
Gepirone (4,4-dimethyl-l-[4-[4-(2-pyrimidinyl)-l-piperazinyl]butyl]-2,6- piperidindione) is an antidepressant and anxiolytic medicament belonging to the azapirone group, currently at the pre-registration stage in the USA. Like other azapirones, gepirone is a selective partial agonist of the 5-HT1A receptor.
The prior art includes some synthesis strategies for the preparation of gepirone (I); they are mainly multi-step reactions which present various drawbacks such as economic inefficiency, low yield and low industrial applicability.
The synthesis of gepirone (I) is described in J. Med. Chem. 1988, 31, 1967-1971; WO 2012/016569; EP 0680961; Dier Junyi Daxue Xuebao, 26(2), 223-224; 2005; Patentschrift (CH), 682564, 15 October 1993; Heterocycles, 36(7), 1463-9, 1993; and Bioorganic & Medicinal Chemistry Letters, 14(7), 1709-1712, 2004.
The scientific article published in J. Med. Chem. 1988, 31, 1967-1971 describes the synthesis of gepirone (I) from N-bromobutyl phthalimide (2) and l-(pyrimidin-2- yl)piperazine (3) in the presence of potassium carbonate to give the intermediate 2-(4-(4- (pyrimidin-2-yl)piperazin-l-yl)butyl)isoindoline-l,3-dione (4), from which the phthalimide protecting group is removed with hydrazine hydrate. The compound 4-(4-(pyrimidin-2-yl)piperazin- 1 -yl)butan- 1 -amine (5) thus synthesised is used in the reaction with 4,4-dimethyldihydro-2H-pyran-2,6(3H)-dione (6) to obtain gepirone (I) (Scheme 1).
SCHEME 2
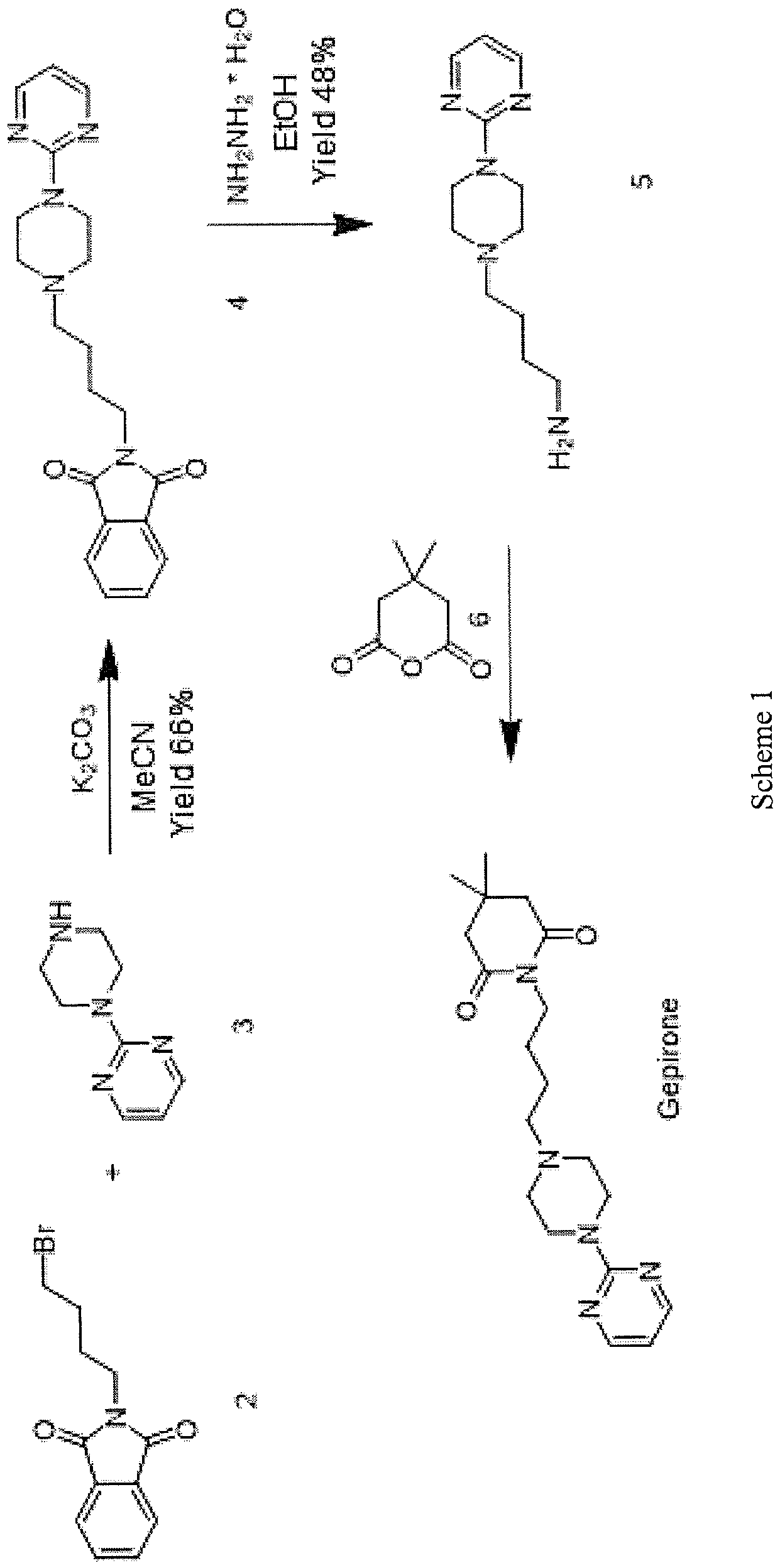
In WO 2012/016569, gepirone (I) is synthesised in four synthesis steps from 1- (pyrimidin-2-yl)pipetazine (3) and 4-((tert-butoxycarbonyl)amino)butanoic acid (7) with the use of condensing agents, such as HATU, and strong reducing agents such as lithium aluminium hydride. The use of condensing agents makes the process practically unusable on an industrial scale because of their high economic impact and the formation of countless by-products which are difficult to remove during the work-up step (Scheme 2).
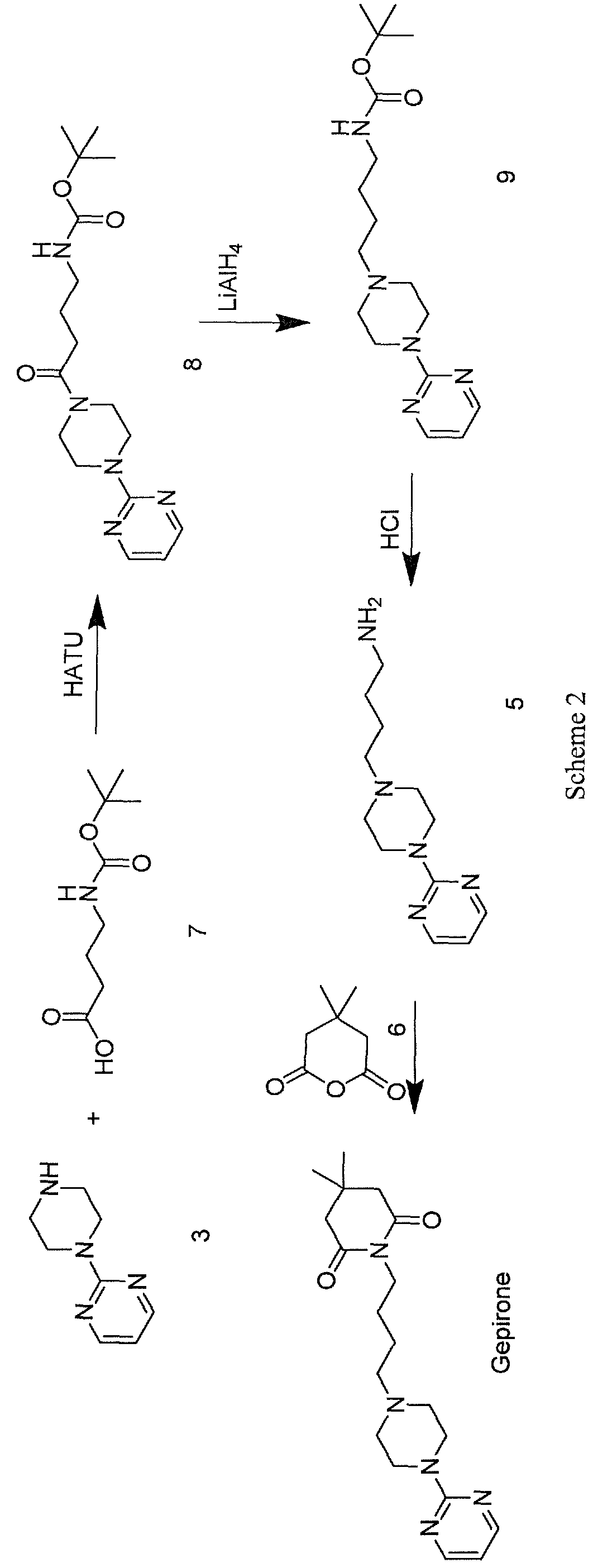
A further approach for the synthesis of gepirone is described in the literature (Ί). This strategy, disclosed in EP 0680961, initially involves synthesising (i) a spiranic intermediate (8-(pyrimidin-2-yl)- 5 ,8-diazaspiro [4,5] decan- 5 -ium bromide) (11) from 1- (pyrimidin-2-yl)piperazine (3) and (ii) 1,4-dibromobutane (10), then opening the spiranic compound (11) with the use of potassium 4,4-dimethyl-2,6-dioxopiperidin-l-ide (13), a secondary amine characterised by a high level of nucleophilicity (Scheme 3)
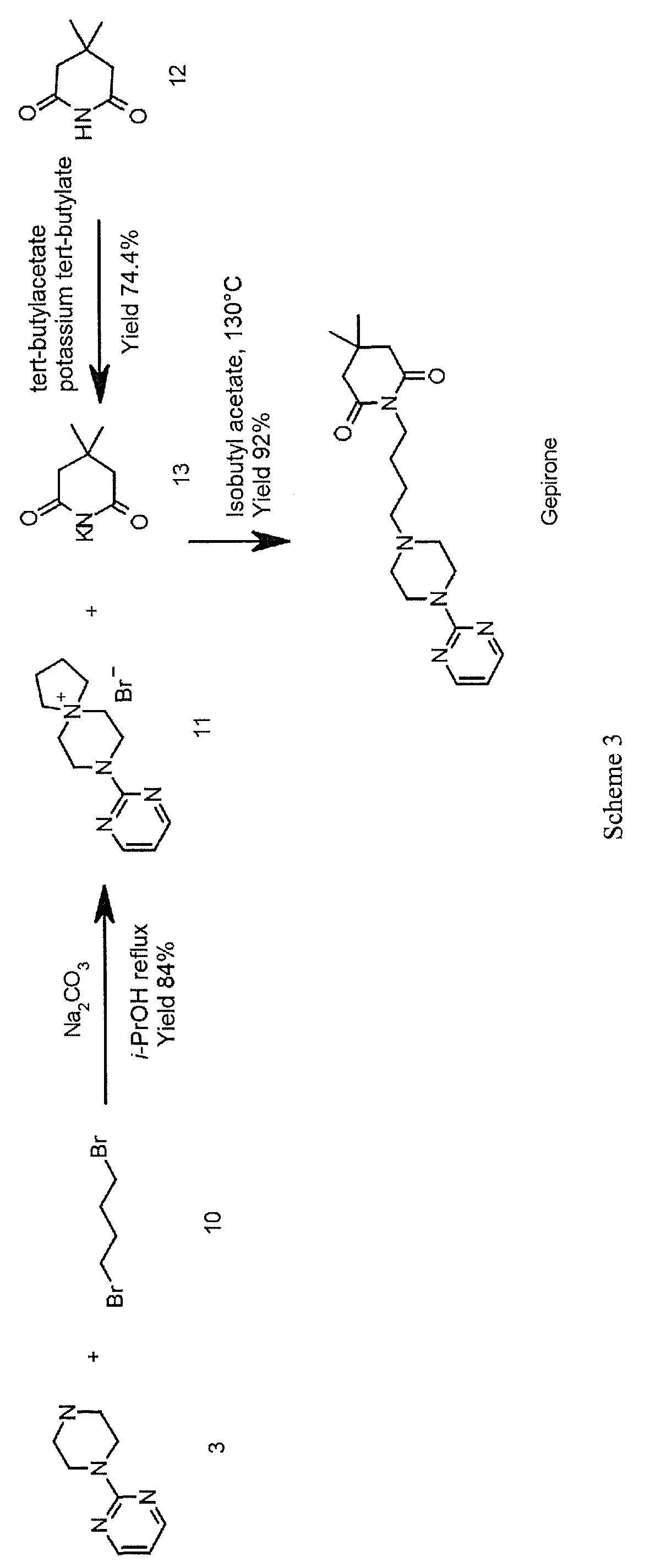
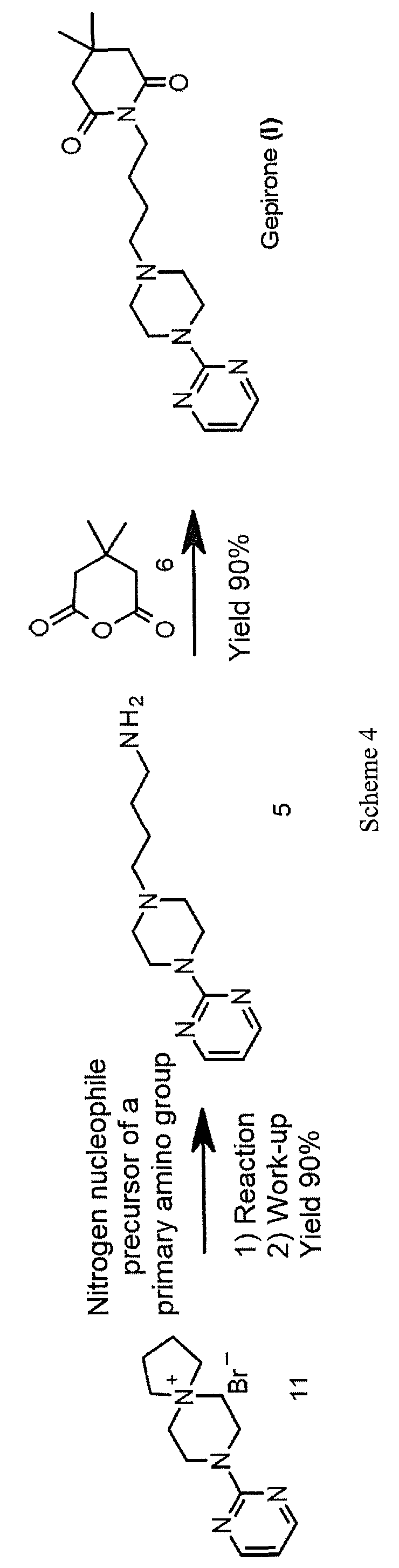
A novel approach to the synthesis of gepirone (I) has now been found, which involves opening spiranic derivative (11) to give 4-(4-(pyrimidin-2-yl)piperazin-l- yl)butan- 1 -amine (5), using suitable nitrogen nucleophile precursors of a primary amino group having the following characteristics: moderate nucleophilicity, so as to prevent reaction by-products, and easy generation of a primary amino group by means of a mild work-up (Scheme 4).
synthesis of gepirone (I) from
8-(pyrimidin-2-yl)-5,8-diazaspiro[4,5]decan-5-ium bromide (11), which is commercially available or easily obtainable by well-known procedures, such as those described in US 4351939.
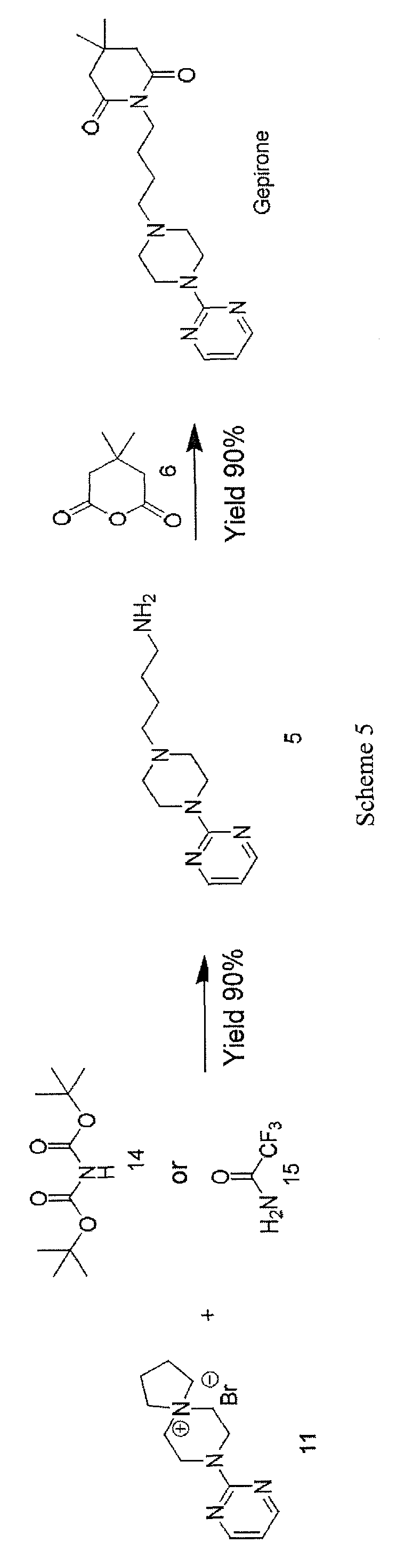
Spiranic derivative (11) initially undergoes selective opening by suitable nitrogen nucleophile precursors of a primary amino group, such as di-tert-butyl iminodicarboxylate (14) and 2,2,2-trifluoroacetamide (15), in the presence of an organic and/or inorganic base. The opening of spiranic ring (11), followed by a simple, mild acid- base work-up, produces, in a single high-yield synthesis step, the intermediate 4-(4-(pyrimidin-2-yl)piperazin- 1 -yl)butan- 1 -amine (5), which is converted to gepirone (I) by reaction with 4,4-dimethyldihydro-2H-pyran-2,6(3H)-dione (6) (Scheme 5).
Example 1 – 4-(4-(pyrimidin-2-yl)piperazin- 1 -yl)butan- 1 -amine (5)
10.0 g of 8-(pyrimidin-2-yl)-5,8-diazaspiro[4,5]decan-5-ium bromide (11) (0.0334 moles), obtained according to US 4423049, is suspended in xylene (150 mL). 21.78 g of caesium carbonate (0.0668 moles) is then added. The resulting mixture is heated to 130°C and left under stirring for 60 minutes. 12.7 g of di-tert-butyl iminodicarboxylate (0.0584 moles) is then added and left under stirring until the reaction is complete. The mixture is cooled to about 80°C and filtered under vacuum, and the solid filtrate is washed with xylene (100 mL). 50 mL of 37% HC1 is added to the organic phase, and the resulting mixture is left under stirring for 10 min. The phases are then separated, and the organic phase is washed with a mixture of 50 mL of water and 5 mL of 37% HC1. 130 mL of dichloromethane is added to the aqueous acid phase and basified with 30% NaOH until pH = 13 is reached. The resulting mixture is left under stirring for 10 min., and the phases are separated. The aqueous phase is re-extracted with 200 mL of dichloromethane, and the combined organic phases are washed with 300 mL of water and 50 mL of brine, dried on sodium sulphate, filtered, and finally concentrated under vacuum to give 7.8 g of 4-(4-(pyrimidin-2-yl)piperazin-l-yl)butan-l-amine (5) (orange oil; yield 99%).
1H NMR (400 MHz, chloroform-d) d 8,17 (d, J = 4,7 Hz, 2H), 6,34 (t, J = 4,7 Hz, 1H), 3,76 – 3,63 (m, 4H), 2,60 (t, J = 6,9 Hz, 2H), 2,46 – 2,32 (m, 4H), 2,32 – 2,21 (m, 2H), 1,53 – 1,24 (m, 6H).
13C NMR (101 MHz, chloroform-d) d 161,55, 157,55, 109,65, 58,48, 53,02, 43,57, 42,01, 31,63, 24,18.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am “EXXUA (gepirone) extended-release tablets, for oral use” (PDF). Mission Pharmacal Company. U.S. Food and Drug Administration. 2023. Archived from the original (PDF) on 28 September 2023. Retrieved 28 September 2023.
- ^ Jump up to:a b c Kishi T, Meltzer HY, Matsuda Y, Iwata N (August 2014). “Azapirone 5-HT1A receptor partial agonist treatment for major depressive disorder: systematic review and meta-analysis” (PDF). Psychological Medicine. 44 (11): 2255–2269. doi:10.1017/S0033291713002857. PMID 24262766. S2CID 20830020. Archived from the original (PDF) on 18 February 2019.
- ^ Jump up to:a b Halbreich U, Montgomery SA (1 November 2008). Pharmacotherapy for Mood, Anxiety, and Cognitive Disorders. American Psychiatric Pub. pp. 375–. ISBN 978-1-58562-821-6.
- ^ Jump up to:a b c d e “Gepirone – Fabre-Kramer Pharmaceuticals”. AdisInsight. Springer Nature Switzerland AG. Archived from the original on 11 April 2023. Retrieved 28 September 2023.
- ^ Jump up to:a b c d e Becker Z (28 September 2023). “Decades long regulatory odyssey ends with FDA nod for Fabre-Kramer’s depression med Exxua”. Fierce Pharma.
- ^ Firth S (30 November 2015). “Controversial Antidepressant Comes Up for FDA OK — Again”. MedPage Today.
- ^ Kirsch I (2014). “Antidepressants and the Placebo Effect”. Zeitschrift für Psychologie. 222 (3): 128–134. doi:10.1027/2151-2604/a000176. PMC 4172306. PMID 25279271.
- ^ “FDA Rules Favorably On Efficacy Of Travivo (Gepirone ER) For Treatment Of Major Depressive Disorder”. Fabre-Kramer Pharmaceuticals, Inc. Cision PR Newswire. 17 March 2016.
- ^ “Gepirone”. Drugs and Lactation Database. National Institute of Child Health and Human Development. 2006. PMID 37856644. Retrieved 11 December 2023.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 494–. ISBN 978-1-58562-309-9.
- ^ Kaur Gill A, Bansal Y, Bhandari R, Kaur S, Kaur J, Singh R, et al. (July 2019). “Gepirone hydrochloride: a novel antidepressant with 5-HT1A agonistic properties”. Drugs of Today. 55 (7): 423–437. doi:10.1358/dot.2019.55.7.2958474. PMID 31347611. S2CID 198911377.
- ^ “Gepirone ER”. Adis Insight. Archived from the original on 6 August 2016. Retrieved 13 January 2016.
- ^ “FDA Rules Favorably On Efficacy Of Travivo (Gepirone ER) For Treatment Of Major Depressive Disorder” (Press release). 17 March 2016. Archived from the original on 24 September 2017. Retrieved 23 January 2018.
- ^ Jump up to:a b Fabre LF, Brown CS, Smith LC, Derogatis LR (May 2011). “Gepirone-ER treatment of hypoactive sexual desire disorder (HSDD) associated with depression in women”. The Journal of Sexual Medicine. 8 (5): 1411–1419. doi:10.1111/j.1743-6109.2011.02216.x. PMID 21324094.
- ^ Jump up to:a b Fabre LF, Clayton AH, Smith LC, Goldstein I, Derogatis LR (March 2012). “The effect of gepirone-ER in the treatment of sexual dysfunction in depressed men”. The Journal of Sexual Medicine. 9 (3): 821–829. doi:10.1111/j.1743-6109.2011.02624.x. PMID 22240272.
- Robinson DS, Sitsen JM, Gibertini M: A review of the efficacy and tolerability of immediate-release and extended-release formulations of gepirone. Clin Ther. 2003 Jun;25(6):1618-33. doi: 10.1016/s0149-2918(03)80159-5. [Article]
- Jenkins SW, Robinson DS, Fabre LF Jr, Andary JJ, Messina ME, Reich LA: Gepirone in the treatment of major depression. J Clin Psychopharmacol. 1990 Jun;10(3 Suppl):77S-85S. doi: 10.1097/00004714-199006001-00014. [Article]
- Yocca FD: Neurochemistry and neurophysiology of buspirone and gepirone: interactions at presynaptic and postsynaptic 5-HT1A receptors. J Clin Psychopharmacol. 1990 Jun;10(3 Suppl):6S-12S. [Article]
- FDA Approved Drug Products: EXXUA (gepirone) extended-release tablets, for oral use [Link]
- Gepirone US Patent Application Publication [Link]
- Fabre-Kramer Pharmaceuticals Announces FDA Approval of EXXUA™, the First and Only Oral Selective 5HT1a Receptor Agonist for the Treatment of Major Depressive Disorder in Adults [Link]
| Clinical data | |
|---|---|
| Trade names | Exxua |
| Other names | BMY-13805; MJ-13805; ORG-13011 |
| Routes of administration | By mouth[1] |
| ATC code | N06AX19 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Pharmacokinetic data | |
| Bioavailability | 14–17%[1] |
| Protein binding | 72%[1] |
| Metabolism | CYP3A4[1] |
| Metabolites | 3′-OH-gepirone; 1-(2-Pyrimidinyl)piperazine[1] |
| Elimination half-life | IRTooltip Instant release: 2–3 hours ERTooltip Modified-release dosage: 5 hours[1] |
| Excretion | Urine: 81%[1] Feces: 13%[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 83928-76-1 83928-66-9 |
| PubChem CID | 55191 |
| DrugBank | DB12184DBSALT002148 |
| ChemSpider | 49836 49835 |
| UNII | JW5Y7B8Z1880C9L8EP6V |
| KEGG | D04314 |
| ChEBI | CHEBI:135990 |
| ChEMBL | ChEMBL284092 ChEMBL1204187 |
| CompTox Dashboard (EPA) | DTXSID90232813 |
| Chemical and physical data | |
| Formula | C19H29N5O2 |
| Molar mass | 359.474 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////Gepirone, FDA 2023, APPROVALS 2023, Exxua, BMY 13805, MJ 13805, ORG 13011, BMY-13805, MJ-13805, ORG-13011, Gepirona, JW5Y7B8Z18
HEXASODIUM PHYTATE





HEXASODIUM PHYTATE
cas 34367-89-0
myo-Inositol, 1,2,3,4,5,6-hexakis(dihydrogen phosphate) sodium salt (1:6)
hexasodium;[2,3,4,5,6-pentakis[[hydroxy(oxido)phosphoryl]oxy]cyclohexyl] hydrogen phosphate
free form RN: 83-86-3
C6H12Na6O24P6, 791.93
- Inositol, hexakis(dihydrogen phosphate) hexasodium salt, myo– (8CI)
- myo-Inositol, hexakis(dihydrogen phosphate), hexasodium salt (9CI)
- Hexasodium fytate
- Hexasodium phytate
- SNF 472
- UNII-ZBX50UG81V
- CSL-525; Hexasodium phytate; Myo-inositol hexaphosphate; SNF-472
x Na salt
14306-25-3
C6H18O24P6.xNa
free form : 83-86-3
myo-Inositol, 1,2,3,4,5,6-hexakis(dihydrogen phosphate), sodium salt
| (1R,2R,3S,4S,5R,6S)-CYCLOHEXANE-1,2,3,4,5,6-HEXAYL-HEXAKIS(DIHYDROGEN PHOSPHATE) |
- Inositol, hexakis(dihydrogen phosphate) sodium salt, myo– (8CI)
- myo-Inositol, hexakis(dihydrogen phosphate), sodium salt (9CI)
- Inositol hexaphosphate sodium salt
- Phytic acid sodium salt
- Sodium inositol hexaphosphate
- Sodium phytate
- OriginatorLaboratoris Sanifit
- DeveloperCSL Vifor; Laboratoris Sanifit
- ClassAntineoplastics; Calcium regulators; Cardiovascular therapies; Phosphates; Small molecules; Sodium compounds; Sugar alcohols
- Mechanism of ActionUndefined mechanism
- Orphan Drug StatusYes – Peripheral arterial disorders; Calciphylaxis
- Phase IIICalciphylaxis; Peripheral arterial disorders
- 09 Nov 2022Phase-III clinical trials in Peripheral arterial disorders in USA (IV) (CSL Behring pipeline, November 2022)
- 24 Oct 2022Sanifit Therapeutics completes a phase III trial in Calciphylaxis in Belgium, Poland, United Kingdom, Germany, Spain, USA (IV) (NCT04195906)
- 28 Sep 2022Hexasodium fytate is still in phase III trials for Calciphylaxis in USA (IV) (NCT04195906)
- You need to be a logged in or subscribed to view this
Hexasodium phytate (also known as SNF472) is a compound being developed as a potential treatment for calciphylaxis, a condition causing skin damage and tissue death in patients with end-stage renal disease. It works by inhibiting the formation and growth of hydroxyapatite crystals, which are implicated in calciphylaxis.
What it is:Hexasodium phytate is the hexasodium salt of myo-inositol hexaphosphate (IP6), a naturally occurring substance found in foods like beans and grains.
- How it works:It binds to hydroxyapatite crystals, the main component of vascular calcification, and prevents their growth, potentially disrupting the calciphylaxis process.
- Why it’s used:Hexasodium phytate is being investigated as a treatment for calciphylaxis, a serious complication of end-stage renal disease characterized by skin and tissue damage due to calcification of small blood vessels.
- Mechanism:It is believed to work by inhibiting the formation and growth of calcium-phosphate crystals (hydroxyapatite) in the blood vessels, thus preventing the calcification that leads to calciphylaxis.
- Clinical trials:Clinical trials have demonstrated the safety and potential efficacy of hexasodium phytate in reducing hydroxyapatite crystallization in patients undergoing hemodialysis, providing a basis for its use in treating calciphylaxis.
- Intravenous administration:It is administered intravenously during dialysis sessions to achieve supra-physiological plasma concentrations, which are thought to be necessary for its therapeutic effect.
- Benefits:It has shown promise in preclinical studies and clinical trials, potentially improving wound healing, pain, and health-related quality of life in patients with calciphylaxis.
- Active development:It is currently in active development as a novel experimental drug for the treatment of calciphylaxis and other related conditions.
Phytic acid is a major phosphorus storage compound of most seeds and cereal grains. It has the strong ability to chelate multivalent metal ions, especially zinc, calcium, and iron. Phytic acid is also considered to be a natural antioxidant and is suggested to have potential functions of reducing lipid peroxidation and as a preservative in foods. Clathrin-associated adaprot complex AP-2 has it been suggested may act as one of the receptor sites for Phytic acid. Both in vivo and in vitro experiments have demonstrated striking anticancer (preventive as well as therapeutic) effects of Phytic acid.
SCHEME

contd………

References
WO2022129148
EP4015494
CN114874473
iScience (2022), 25(3), 103950
CN111718463
CN110483240
Uzbekskii Khimicheskii Zhurnal (1995), (5-6), 72-75 JP61056142
////////HEXASODIUM PHYTATE, Hexasodium fytate, Hexasodium phytate, SNF 472, calciphylaxis, UNII-ZBX50UG81V, CSL-525, Hexasodium phytate, Myo-inositol hexaphosphate, SNF-472, ORPHAN DRUG
Ibuzatrelvir



Ibuzatrelvir
PF-07817883
CAS 2755812-39-4
| Molecular Weight | 489.49 |
|---|---|
| Formula | C21H30F3N5O5 |
- Ibuzatrelvir
- N-(Methoxycarbonyl)-3-methyl-L-valyl-(4R)-N-[(1S)-1-cyano-2-((3S)-2-oxopyrrolidin-3-yl)ethyl]-4-(trifluoromethyl)-L-prolinamide
- PF 07817883
- methyl N-[(2S)-1-[(2S,4R)-2-[[(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl]-4-(trifluoromethyl)pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]carbamate
- KZ2X7QH2VT
Ibuzatrelvir (development code PF-07817883) is an experimental antiviral drug being developed by Pfizer for the treatment of COVID-19.[1] It is a second-generation improvement over nirmatrelvir which has a similar chemical structure.[2] One of the disadvantages of nirmatrelvir is that it has low metabolic stability and must be given in combination with ritonavir (as Paxlovid) to limit its metabolic degradation in the body.[3] Ibuzatrelvir incorporates modifications to the chemical structure of nirmatrelvir that give it enhanced oral bioavailability, so it does not require coadministration with ritonavir.[3]
Ibuzatrelvir (PF-07817883), a second-generation, orally bioavailable, is SARS-CoV-2 main protease (Mpro and 3CLpro) inhibitor with improved metabolic stability. Ibuzatrelvir has demonstrated pan-human coronavirus antiviral activity and off-target selectivity profile in vitro and in preclinical animal studies. Ibuzatrelvir is well tolerated with a safety profile similar to placebo and prevents viral infection and transmission. Ibuzatrelvir can be used to inhibit COVID-19.
SCHEME
SIDECHAIN

MAIN

PATENT
WO2021250648 PFIZER
WO2023215910
PAPER
The Pfizer scientists described ibuzatrelvir’s medicinal chemistry campaign in a Journal of Medicinal Chemistry paper that was published in April 2024 (DOI: 10.1021/acs .jmedchem.3c02469).
https://pubs.acs.org/doi/10.1021/jacsau.4c00508
Ibuzatrelvir (1) was recently disclosed and patented by Pfizer for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It has received fast-track status from the USA Food and Drug Administration (FDA) and has entered phase III clinical trials as a possible replacement for Paxlovid. Like nirmatrelvir (2) in Paxlovid, this orally active drug candidate is designed to target viral main proteases (Mpro) through reversible covalent interaction of its nitrile warhead with the active site thiol of the chymotrypsin-like cysteine protease (3CL protease). Inhibition of Mpro hinders the processing of the proteins essential for viral replication in vivo. However, ibuzatrelvir apparently does not require ritonavir (3), which is coadministered in Paxlovid to block human oxidative metabolism of nirmatrelvir. Here, we report the crystal structure of the complex of ibuzatrelvir with the active site of SARS-CoV-2 Mpro at 2.0 Å resolution. In addition, we show that ibuzatrelvir also potently inhibits the Mpro of Middle East respiratory syndrome-related coronavirus (MERS-CoV), which is fortunately not widespread but can be dangerously lethal (∼36% mortality). Co-crystal structures show that the binding mode of the drug to both active sites is similar and that the trifluoromethyl group of the inhibitor fits precisely into a critical S2 substrate binding pocket of the main proteases. However, our results also provide a rationale for the differences in potency of ibuzatrelvir for these two proteases due to minor differences in the substrate preferences leading to a weaker H-bond network in MERS-CoV Mpro. In addition, we examined the reversibility of compound binding to both proteases, which is an important parameter in reducing off-target effects as well as the potential immunogenicity. The crystal structures of the ibuzatrelvir complexes with Mpro of SARS-CoV-2 and of MERS-CoV will further assist drug design for coronaviral infections in humans and animals.


General Boc-Deprotection and Coupling Procedure
This procedure was based on a literature procedure.1
The Boc-protected building block (1.0
equiv) was dissolved in 50/50 TFA/DCM and stirred for 1 h at room temperature. The reaction
mixture was then concentrated in vacuo and co-evaporated with DCM (5 × 5 mL). In a separate
RBF the carboxylic acid building block (1.0 equiv) and HATU (1.0 equiv) were dissolved in
DMF. HOAt (0.6 M in DMF) (0.1 equiv) and DIPEA (3.0 equiv) were added and the reaction
mixture was left to incubate at room temperature for 10 mins, as it turned yellow. The previously
concentrated Boc-deprotected building block was dissolved in DMF and added dropwise to the
incubating solution. The reaction mixture was capped under a blanket of argon and stirred at room
temperature for 2–3 h. The reaction mixture was diluted with 5 mL each of water and ethyl acetate
and the layers separated. The aqueous layer was extracted further with ethyl acetate (3 × 5 mL),
and all ethyl acetate layers combined and washed with sat. aq. NaHCO3 (10 mL), 1 M HCl (10
mL), water (2 × 10 mL) and brine (10 mL). It was then dried over Na2SO4, filtered, and
concentrated in vacuo to furnish the product.
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-
(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (1) Ibuzatrelvir
This known compound was synthesized according to the General Boc-Deprotection and
Coupling Procedure with building blocks 7 and 8. The characterization data matches the literature
report (IPN: WO2021250648A1). The crude material was obtained as a dark yellow sticky residue
that was then purified with flash column chromatography with an eluent of 92:8 EtOAc:MeOH.
The desired compound had an Rf
= 0.40 and was visible with KMnO4 stain. After concentration
of desired fractions, 1 was isolated as a clear, colorless oil that solidified to a white solid (0.051 g,
53%) This compound was isolated and used for all experiments as a mixture of diastereomers in a
ratio of about 2:1 and rotamers present, with only the major set of resonances reported, which are
for the desired isomer. It can be separated using high performance liquid chromatography (HPLC)
methods, as listed in the HPLC Separation of Ibuzatrelvir Diastereomers section.
IR (DCM cast film, vmax / cm–1) 3292, 3053, 2959, 2909, 2875, 1695, 1643, 1550, 1443, 1401,
1370, 1332, 1270, 1236, 1200, 1164, 1130
1H NMR (500 MHz, CDCl3) δH 8.32 (1H, d, J = 7.6 Hz), 6.22 (1H, br), 5.74 (1H, d, J = 9.3 Hz),
4.96 – 4.87 (1H, m), 4.54 (1H, dd, J = 8.6, 3.6 Hz), 4.30 (1H, d J = 9.9 Hz), 3.99 – 3.88 (2H, m),
3.65 (3H, s), 3.42 – 3.26 (2H, m), 2.66 – 2.57 (1H, m), 2.52 – 2.43 (1H, m), 2.40 – 2.28 (3H, m),
1.97 – 1.88 (1H, m), 1.84 – 1.75 (2H, m), 0.99 (9H, s)
13C {1H} NMR (125 MHz, CDCl3) δC 179.1, 171.4, 171.1, 156.9, 126.1 (q, J = 276.3 Hz), 118.3,
59.4, 58.9, 52.4, 47.3, 42.4 (q, J = 29.5 Hz), 40.4, 39.1 37.5, 35.6, 34.2, 28.2, 28.0, 26.3
SR: [α]D
26 = –35.71 (c = 0.21, DCM)
HRMS: (ESI) Calcd for C21H30F3N5NaO5 [M + Na]+
512.2091, found 512.2088



References
- ^ Allerton CM, Arcari JT, Aschenbrenner LM, Avery M, Bechle BM, Behzadi MA, et al. (August 2024). “A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19”. Journal of Medicinal Chemistry. 67 (16): 13550–13571. doi:10.1021/acs.jmedchem.3c02469. PMC 11345836. PMID 38687966.
- ^ Chen P, Van Oers TJ, Arutyunova E, Fischer C, Wang C, Lamer T, et al. (August 2024). “A Structural Comparison of Oral SARS-CoV-2 Drug Candidate Ibuzatrelvir Complexed with the Main Protease (Mpro) of SARS-CoV-2 and MERS-CoV”. JACS Au. 4 (8): 3217–3227. doi:10.1021/jacsau.4c00508. PMC 11350714. PMID 39211604.
- ^ Jump up to:a b Brewitz L, Schofield CJ (July 2024). “Fixing the Achilles Heel of Pfizer’s Paxlovid for COVID-19 Treatment”. Journal of Medicinal Chemistry. 67 (14): 11656–11661. doi:10.1021/acs.jmedchem.4c01342. PMC 11284777. PMID 38967233.
| Clinical data | |
|---|---|
| Other names | PF-07817883 |
| Routes of administration | Oral |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2755812-39-4 |
| PubChem CID | 163362000 |
| DrugBank | 111 |
| ChemSpider | 128942571 |
| UNII | KZ2X7QH2VT |
| Chemical and physical data | |
| Formula | C21H30F3N5O5 |
| Molar mass | 489.496 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Owen, et al. Preparation of peptidomimetic nitriles as SARS-CoV-2 3CL protease inhibitors and methods for the treatment of COVID-19. World Intellectual Property Organization, WO2021250648 A1. 2021-12-16.[2]. Mahta Mortezavi, et al. Virologic Response and Safety After Oral Administration of Ibuzatrelvir, a Novel SARS-CoV-2 Mpro Inhibitor, in Non-Hospitalized Adults With Symptomatic COVID-19. European Congress of Clinical Microbiology and Infectious Disease (ECCMID) 2024; 2024 April 27-30.[3]. Westberg M, et al. An orally bioavailable SARS-CoV-2 main protease inhibitor exhibits improved affinity and reduced sensitivity to mutations[J]. Sci Transl Med. 2024 Mar 13;16(738):eadi0979.[4]. Allerton CMN, et al. A Second-Generation Oral SARS-CoV-2 Main Protease Inhibitor Clinical Candidate for the Treatment of COVID-19[J]. J Med Chem. 2024 Apr 30. [Content Brief]
////Ibuzatrelvir, PF 07817883, PF-07817883, PF07817883, KZ2X7QH2VT

{kind=link}
{kind=link}
{kind=link}