
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Taletrectinib
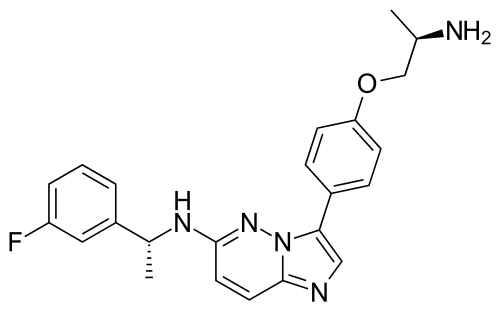
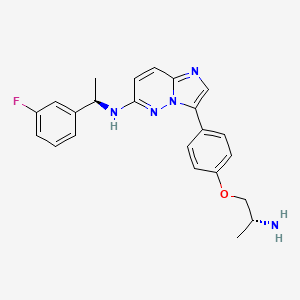
Taletrectinib
CAS 1505514-27-1
as salt: 1505515-69-4, Taletrectinib adipate
FDA 6/11/2025, Ibtrozi, To treat locally advanced or metastatic ROS1-positive non-small cell lung cancer ALSO CHINA 2024 APPROVED |
405.5 g/mol, C23H24FN5O, UNII-W4141180YD
3-[4-[(2R)-2-aminopropoxy]phenyl]-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine
Taletrectinib adipate
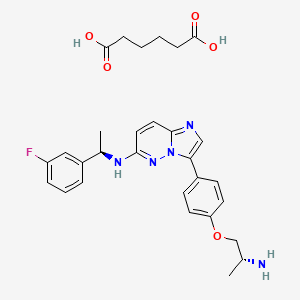
WeightAverage: 551.619
Monoisotopic: 551.254397378
Chemical FormulaC29H34FN5O5
DS-6051B, CAS 1505515-69-4,
6KLL51GNBG, 3-{4-[(2R)-2-aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine; hexanedioic acid
Taletrectinib, sold under the brand name Ibtrozi, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2] It is used as the salt, taletrectinib adipate.[1] Taletrectinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Taletrectinib was approved for medical use in the United States in June 2025.[3]
SYN
US20200062765
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289038418&_cid=P12-MCIHV1-02369-1
Example 1
tert-Butyl [(2R)-1-(4-bromophenoxy)propan-2-yl]carbamate (1)
Example 2
6-Fluoroimidazo[1,2-b]pyridazine methanesulfonate (2)
Example 3
tert-Butyl {(2R)-1-[4-(6-fluoroimidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate (3)
Example 4
tert-Butyl {(2R)-1-[4-(6-{[(1R)-1-(3-fluorophenyl)ethyl]amino}imidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate hydrochloride (4)
Example 5
3-{4-[(2R)-2-Aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethylimidazo[1,2-b]pyridazin-6-amine dihydrochloride (5)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023272701&_cid=P12-MCIHPU-95869-1
The NMR data for the crystalline form A of Compound 1 adipate are as follows: 1H NMR (500 MHz, DMSO) δ 1.13-1.14 (d, J=5.0 Hz, 3H) , 1.47-1.48 (d, J=5.0 Hz, 7H) , 2.15-2.18 (t, J=5.0 Hz, J=10.0 Hz, 4H) , 3.25-3.29 (m, 1H) , 3.79-3.83 (m, 2H) , 4.80-4.85 (m, 1H) , 6.76-6.77 (d, J=5.0 Hz, 1H) , 6.92-6.94 (d, J=10.0 Hz, 2H) , 7.01-7.05 (t, J=10.0 Hz, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.64-7.65 (d, J=5.0 Hz, 1H) , 7.72-7.76 (t, J=10.0 Hz, 4H) .
[0148]
The IR data for the crystalline form A of Compound 1 adipate are as follows: IR (cm -1) : 1701, 1628, 1612, 1586, 1463, 1333, 1246, 1110, 829, 821.
Example 5: Preparation and Characterization of Crystalline Form A of Compound 1 Free Base
[0212]
Compound 1 HCl (75.5 g) (e.g., obtained by using the method described in Example 5 of U.S. Application Publication No. 2020/0062765) was dissolved in ethanol (604 mL) at 50℃. Sodium hydroxide (68.1 g) was added to the above solution. The mixture was cooled to 1℃ in 1.5 hours and stirred for 18.5 hours. The mixture was then filtered, and the solid thus obtained was washed with a cooled mixture of ethanol (151 mL) and water (151 mL) and dried. The solid thus obtained was confirmed to be the crystalline form A of Compound 1 free base.
[0213]
The NMR data for the crystalline form A of Compound 1 free base are as follows: 1H NMR (500 MHz, DMSO) δ 1.09-1.10 (d, J=5.0 Hz, 3H) , 1.48-1.49 (d, J=5.0 Hz, 3H) , 3.16-3.20 (m, 1H) , 3.75-3.79 (m, 2H) , 4.82-4.86 (m, 1H) , 6.76-6.78 (d, J=10.0 Hz, 1H) , 6.92-6.94 (m, 2H) , 7.01-7.05 (m, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.62-7.63 (d, J=5.0 Hz, 1H) , 7.72-7.75 (m, 4H) .
[0214]
The IR data for the crystalline form A of Compound 1 free base are as follows: IR (cm -1) : 3350, 3247, 3055, 2961, 2923, 2864, 1611, 1586, 1349, 829, 819.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Taletrectinib is an oral, next-generation ROS1 TKI developed by Nuvation Bio Inc. for the treatment of ROS1-positive NSCLC. In 2024, the NMPA approved taletrectinib for adult patients with locally advanced or metastatic ROS1-positive NSCLC, regardless of prior ROS1TKI treatment [47]. Under an exclusive license agreement, Innovent Biologics will commercialize taletrectinib in China under the brand
name DOVBLERON®. Taletrectinib exerts its pharmacological action through the mechanism of selectively impeding the ROS1 receptor tyrosine kinase, which effectively disrupts the signaling cascades which are responsible for facilitating the growth and survival of cancer cells in ROS1-positive NSCLC. This inhibition of the ROS1 receptor tyrosine kinase is a key event in the drug’s mode of action, as it specifically targets the molecular processes that drive the progression of the disease in ROS1-positive NSCLC cases [48]. The NMPA granted approval founded on the data sourced from the crucial Phase 2 TRUST – I study. This study substantiated that patients administered with taletrectinib achieved sustained responses and extended PFS. Regarding safety, taletrectinib boasted a generally good tolerability. It presented an advantageous safety profile and favorable tolerability characteristics, as evidenced by the low incidences of dose reduction and treatment discontinuation triggered by adverse effects. [49]. Overall, taletrectinib represents a promising therapeutic option for patients with advanced ROS1-positive NSCLC, offering efficacy in both TKI-naïve and TKI-pretreated populations, including those with CNS metastases [50–52].
The synthesis of Taletrectinib, illustrated in Scheme 12, commences with Mitsunobu coupling of Tale-001 and Tale-002 to afford Tale-003, which then undergoes Suzuki coupling with Tale-004 constructing
Tale-005 [53]. Sequential acidolysis/deprotection of Tale-005 ultimately delivers Taletrectinib
[47] M. P´ erol, N. Yang, C.M. Choi, Y. Ohe, S. Sugawara, N. Yanagitani, G. Liu, F.G.M.
D. Braud, J. Nieva, M. Nagasaka, 1373P efficacy and safety of taletrectinib in
patients (pts) with ROS1+ non-small cell lung cancer (NSCLC): interim analysis of
global TRUST-II study, Ann. Oncol. 34 (2023) S788–S789.
[48] G. Harada, F.C. Santini, C. Wilhelm, A. Drilon, NTRK fusions in lung cancer: from
biology to therapy, Lung Cancer 161 (2021) 108–113.
[49] W. Li, A. Xiong, N. Yang, H. Fan, Q. Yu, Y. Zhao, Y. Wang, X. Meng, J. Wu, Z. Wang,
Y. Liu, X. Wang, X. Qin, K. Lu, W. Zhuang, Y. Ren, X. Zhang, B. Yan, C.M. Lovly,
C. Zhou, Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non-
small cell lung cancer: the phase II TRUST-I study, J. Clin. Oncol. 42 (2024)
2660–2670.
[50] M. Nagasaka, D. Brazel, S.I. Ou, Taletrectinib for the treatment of ROS-1 positive
non-small cell lung cancer: a drug evaluation of phase I and II data, Expert Opin
Investig Drugs 33 (2024) 79–84.
[51] S. Waliany, J.J. Lin, Taletrectinib: TRUST in the continued evolution of treatments
for ROS1 fusion-positive lung cancer, J. Clin. Oncol. 42 (2024) 2622–2627.
[52] M. Nagasaka, Y. Ohe, C. Zhou, C.M. Choi, N. Yang, G. Liu, E. Felip, M. P´ erol,
B. Besse, J. Nieva, L. Raez, N.A. Pennell, A. Dimou, F. Marinis, F. Ciardiello,
T. Seto, Z. Hu, M. Pan, W. Wang, S. Li, S.I. Ou, TRUST-II: a global phase II study of
taletrectinib in ROS1-positive non-small-cell lung cancer and other solid tumors,
Future Oncol. 19 (2023) 123–135.
[53] Y. Takeda, K. Yoshikawa, Y. Kagoshima, Y. Yamamoto, R. Tanaka, Y. Tominaga,
M. Kiga, Y. Hamada, Preparation of imidazo[1,2-b]pyridazine Derivatives as
Potent Inhibitors of ROS1 Kinase and NTRK Kinase, 2013. WO2013183578A1.

Medical uses
Taletrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[1][2]
Adverse effects
The FDA prescribing information for taletrectinib includes warnings and precautions for hepatotoxicity, interstitial lung disease/pneumonitis, QTc interval prolongation, hyperuricemia, myalgia with creatine phosphokinase elevation, skeletal fractures, and embryo-fetal toxicity.[1][3]
History
The efficacy of taletrectinib to treat ROS1-positive non-small cell lung cancer was evaluated in participants with locally advanced or metastatic, ROS1-positive non-small cell lung cancer enrolled in two multi-center, single-arm, open-label clinical trials, TRUST-I (NCT04395677) and TRUST-II (NCT04919811).[3] The efficacy population included 157 participants (103 in TRUST-I; 54 in TRUST-II) who were naïve to treatment with a ROS1 tyrosine kinase inhibitor (TKI) and 113 participants (66 in TRUST-I; 47 in TRUST-II) who had received one prior ROS1 tyrosine kinase inhibitor.[3] Participants may have received prior chemotherapy for advanced disease.[3] The US Food and Drug Administration (FDA) granted the application for taletrectinib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Taletrectinib was approved for medical use in the United States in June 2025.[3][4]
Names
Taletrectinib is the international nonproprietary name.[5]
Taletrectinib is sold under the brand name Ibtrozi.[3][4]
References
- ^ Jump up to:a b c d e f g “Prescribing Information for NDA 219713, Supplement 000” (PDF). Drugs@FDA. U.S. Food and Drug Administration. April 2025. Retrieved 14 June 2025.
- ^ Jump up to:a b Khan I, Sahar A, Numra S, Saha N, Nidhi, Parveen R (April 2025). “Efficacy and safety of taletrectinib for treatment of ROS1 positive non-small cell lung cancer: A systematic review”. Expert Opinion on Pharmacotherapy. 26 (6): 765–772. doi:10.1080/14656566.2025.2487150. PMID 40170301.
- ^ Jump up to:a b c d e f g h “FDA approves taletrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 11 June 2025. Retrieved 13 June 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “U.S. Food and Drug Administration Approves Nuvation Bio’s Ibtrozi (taletrectinib), a Next-Generation Oral Treatment for Advanced ROS1-Positive Non-Small Cell Lung Cancer”. Nuvation Bio (Press release). 12 June 2025. Retrieved 13 June 2025.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT04395677 for “A Study of AB-106 in Subjects With Advanced NSCLC Harboring ROS1 Fusion Gene” at ClinicalTrials.gov
- Clinical trial number NCT04919811 for “Taletrectinib Phase 2 Global Study in ROS1 Positive NSCLC (TRUST-II)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ibtrozi |
| License data | US DailyMed: Taletrectinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1505514-27-1as salt: 1505515-69-4 |
| PubChem CID | 72202474as salt: 72694302 |
| DrugBank | DB18711 |
| ChemSpider | 114934673as salt: 88297530 |
| UNII | W4141180YDas salt: 6KLL51GNBG |
| KEGG | D12363as salt: D12364 |
| ChEMBL | ChEMBL4650989as salt: ChEMBL4650361 |
| Chemical and physical data | |
| Formula | C23H24FN5O |
| Molar mass | 405.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Taletrectinib, FDA 2025, APPROVALS 2025, Ibtrozi, CANCER, AB-106, DS-6051a, UNII-W4141180YD, DS 6051B, APPROVALS 2024, CHINA 2024, Nuvation Bio Inc
Olgotrelvir
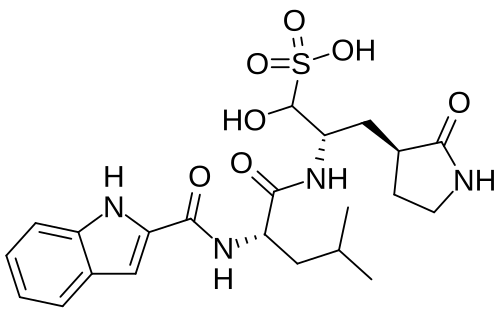
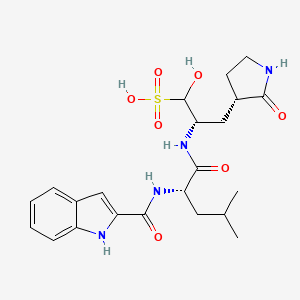
Olgotrelvir
STI-1558, HY-156655, CS-0887294, STI 1558, HY 156655, CS 0887294
Cas 2763596-71-8
494.6 g/mol, C22H30N4O7S, ZP3BDH359D
C22H30N4O7S 3-Pyrrolidinepropaney, α-hydroxy-β-[[(2S)-2-[(1H-indol-2-ylcarbonyl)amino]-4-methyl-1-oxopentyl]amino]-2-oxo-, (βS,3S)-
- (2S)-2-[(S)-2-(1H-Indole-2-carboxamido)-4-methylpentanamido]-1-hydroxy-3-[(S)-2-oxopyrrolidin-3-yl]propane-1-sulfonic acid
- 3-Pyrrolidinepropanesulfonic acid, alpha-hydroxy-beta-[[(2S)-2-[(1H-indol-2-ylcarbonyl)amino]-4-methyl-1-oxopentyl]amino]-2-oxo-, (betaS,3S)-
Olgotrelvir sodium, C22H30N4O7S.Na, CAS 2763596-71-8
3-Pyrrolidinepropanesulfonic acid, α-hydroxy-β-[[(2S)-2-[(1H-indol-2-ylcarbonyl)amino]-4-methyl-1-oxopentyl]amino]-2-oxo-, sodium salt (1:1), (βS,3S)-
Olgotrelvir (STI-1558) is an experimental antiviral medication being studied as a potential treatment for COVID-19. It is believed to work by inhibiting the SARS-CoV-2 main protease (Mpro), a key enzyme that SARS-CoV-2 needs to replicate,[1][2][3][4] and by blocking viral entry.[2][5]
SCHEME
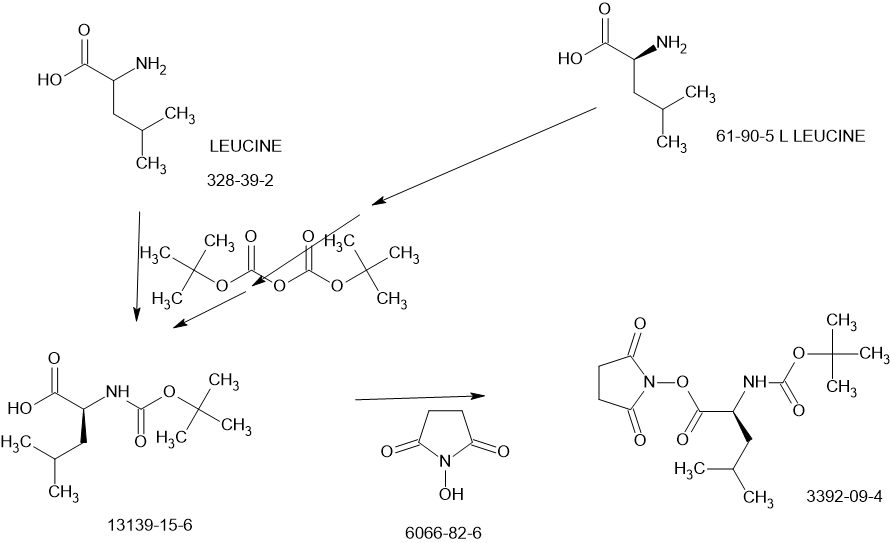
Main
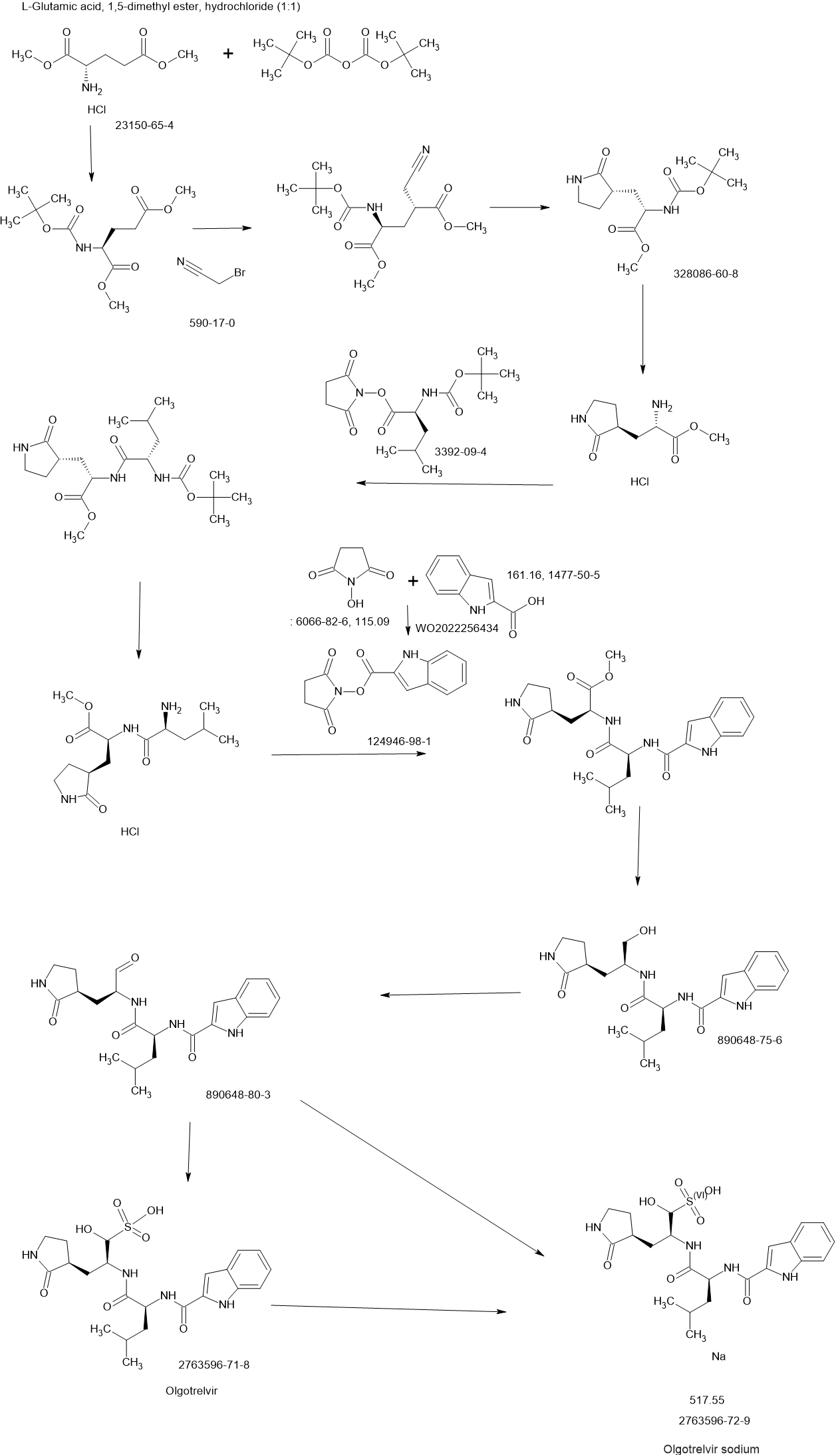
PATENT
US20230322668 – PROTEASE INHIBITORS AS ANTIVIRALS
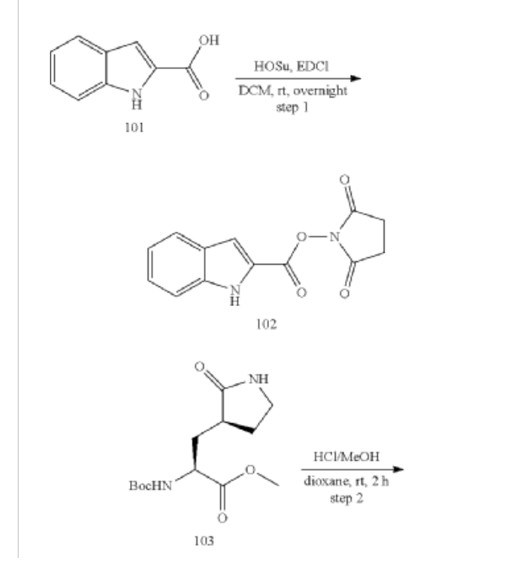
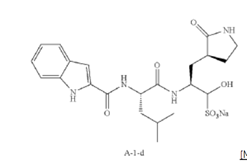
Example S1: Synthesis of Compounds A-1-a, A-1-b, A-1-c and A-1-d




US20230026438 – PROTEASE INHIBITORS AS ANTIVIRALS
WO2022256434 – PROTEASE INHIBITORS AS ANTIVIRALS
Example S1: Synthesis of Compounds A-1-a, A-1-b, A-1-c and A-1-d
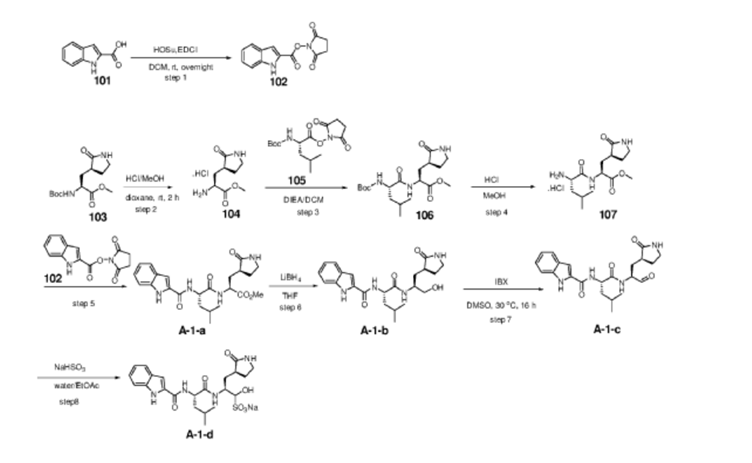
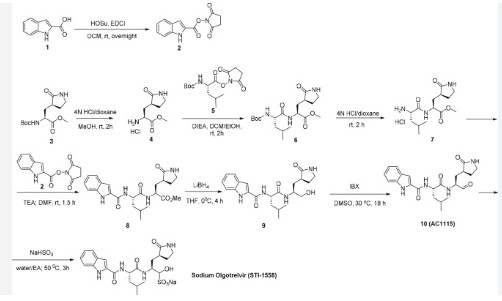
[00224] To a dichloromethane (2.5 L) solution of 1H-indole-2-carboxylic acid (compound 101) (200 g, 1.24 mol) and N-hydroxy succinimide (157.1 g, 1.37 mol) was added EDCI (286 g, 1.49 mmol) at 0℃. After stirring at room temperature overnight, the solvent was removed under reduced pressure. The resulting solid was triturated with deionized water, and the solid was collected and dried under reduced pressure to give the compound 102 as a light-brown solid (310 g, 96%).
1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.49 – 7.35 (m, 3H), 7.19 (t, J = 7.4 Hz, 1H), 2.92 (s, 4H).
[00225] To a stirred mixture of methyl (2S)-2-{[(tert-butoxy)carbonyl]amino}-3-[(3S)-2- oxopyrrolidin-3-yl]-propanoate (compound 103) (500 g, 1748.24 mmol) in MeOH (200 mL) was
added 4M HCl in 1,4-dioxane (2000 mL) at room temperature. The mixture was stirred at rt for 2 h. LCMS indicated completion of the reaction. The reaction mixture was concentrated under reduced pressure to afford methyl (2S)-2-amino-3-[(3S)-2-oxopyrrolidin-3-yl]propanoate hydrochloride salt (compound 104) (389 g, 1721 mmol, 98%) as a light-yellow solid, which was used for next step without further purification. LCMS= [M+H]+: 187.1.
[00226] To a stirred mixture of methyl (2S)-2-amino-3-[(3S)-2-oxopyrrolidin-3-yl]propanoate hydrochloride (389 g, 1721 mmol) (compound 104) and DIEA (866.162 mL, 5240.94 mmol) in DCM (1800 mL) and EtOH (500 mL) was added 2,5-dioxopyrrolidin-1-yl (2R)-2-{[(tert-butoxy)carbonyl]amino}-4-methyl-pentanoate (compound 105) (573.66 g, 1746.98 mmol) at room temperature. The reaction mixture was stirred at room temperature for 2 h. LCMS indicated completion of the reaction. The reaction mixture was successively washed with water (1.0 L x 2), 0.5 M HCl (1.1 L), sat. NaHCO3 (1 L) and water (1 L). The organic layer was separated, dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford the compound 106 (700 g, 1752.23 mmol, >99%) as a light-yellow solid, which was used for next step without further purification. LCMS = [M+H]+: 400.3.
(400 MHz, DMSO-d6) δ 8.32 (d, J = 8.0 Hz, 1H), 7.62 (s, 1H), 6.88 (d, J = 8.0 Hz, 1H), 4.40 – 4.28 (m, 1H), 3.94 (dd, J = 15.1, 8.1 Hz, 1H), 3.74 – 3.52 (m, 3H), 3.15 (t, J = 8.8 Hz, 1H), 3.06 (dd, J = 16.4, 9.2 Hz, 1H), 2.33 (t, J = 9.2 Hz, 1H), 2.14 – 2.00 (m, 2H), 1.68 – 1.51 (m, 3H), 1.42 – 1.34 (m, 11H), 0.87 (dd, J = 11.4, 6.6 Hz, 6H).
[00227] A mixture of methyl (2S)-2-[(2S)-2-{[(tert-butoxy)carbonyl]amino}-4-methylpentanamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanoate (compound 106) (590 g, 1476.88 mmol) in HCl/dioxane (3 L) was stirred at room temperature for 2 h. LC-MS indicated completion of the reaction. The reaction mixture was concentrated under reduced pressure to give compound 107 as a yellow solid (490 g, 99%), which was used for next step without further purification.
LCMS = [M+H]+: 300.2.
[00228] To a stirred mixture of methyl (S)-2-((S)-2-amino-4-methylpentanamido)-3-((S)-2-oxopyrrolidin-3-yl)propanoate hydrochloride (compound 107) (418 g, 1235 mmol) and TEA (519.020 mL, 3734.03 mmol) in DMF (2500 mL) at room temperature was added 2,5-dioxopyrrolidin-1-yl 1H-indole-2-carboxylate (compound 102) (353 g, 1369.15 mmol) . The reaction mixture was stirred for 1.5 h. LCMS indicated that the reaction was complete. EtOAc (6 L) was added into the reaction mixture, which was then washed with brine (6 L x 6). The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated down under reduced
pressure. Compound A-1-a was obtained as an off-white solid (414 g. Y: 76%), which was used for next step without further purification. LCMS = [M+H]+: 443.3. 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 8.54 (t, J = 12.2 Hz, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.24 (t, J = 10.3 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 4.65 – 4.50 (m, 1H), 4.44 – 4.28 (m, 1H), 3.72 – 3.55 (s, 3H), 3.19 – 3.06 (m, 2H), 2.36 (ddd, J = 13.8, 10.3, 4.0 Hz, 1H), 2.16 – 2.03 (m, 2H), 1.79 – 1.49 (m, 5H), 0.92 (dt, J = 14.4, 7.2 Hz, 6H).
[00229] To a stirred solution of methyl (S)-2-((S)-2-(1H-indole-2-carboxamido)-4-methylpentanamido)-3-((S)-2-oxopyrrolidin-3-yl)propanoate (compound A-1-a) (500 g, 1131 mmol) in THF (20 L) LiBH4 (74 g, 3393 mmol) was added portionwise at 0 ℃. The reaction mixture was stirred at 0 ℃ for 4 h. After reaction was completed (monitored by LCMS), the reaction mixture was quenched with sat. aqueous NH4Cl until no more gas formed. The mixture was washed with brine (5 L x 4), organic layer was collected, dried over anhydrous sodium sulfate, filtered, and concentrated down in vacuum. The resulting residue was purified by silica column chromatography (DCM : MeOH = 15 : 1) to give the desired product compound A-1-b (310 g, 66%) as a white solid. LCMS = [M+H]+: 415.2.
NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.52 (s, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.26 (d, J = 1.4 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H), 4.67 (t, J = 5.6 Hz, 1H), 4.50 (td, J = 9.7, 5.0 Hz, 1H), 3.80 (s, 1H), 3.40 – 3.28 (m, 1H), 3.28 – 3.20 (m, 1H), 3.15 – 2.99 (m, 2H), 2.33 – 2.20 (m, 1H), 2.12 (dt, J = 17.8, 9.4 Hz, 1H), 1.86 – 1.75 (m, 1H), 1.75 – 1.64 (m, 2H), 1.56 (ddd, J = 19.3, 9.6, 6.9 Hz, 2H), 1.45 – 1.35 (m, 1H), 0.91 (dd, J = 15.6, 6.3 Hz, 6H).
[00230] To a stirred solution of N-((S)-1-(((S)-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)-1H-indole-2-carboxamide (compound A-1-b) (8.3 g, 20 mmol) in DMSO (60 mL) was added 2-iodoxybenzoic acid (IBX) (11.2 g, 40 mmol) at room temperature. The reaction mixture was stirred at 30 ℃ for 18 h, and LCMS indicated completion of the reaction. The reaction mixture was diluted with EtOAc (300 mL) and filtered. The filtrate was washed with mixture of brine and sat. aqueous NaHCO3 (1:1 to 5:1, 200 mL x 5). The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and concentrated down at rt to afford crude product. THF (40 mL) was added, and the mixture was stirred overnight at room temperature. The resulting solid was collected and dried under vacuum to yield the desired product N-((S)-4-methyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)-propan-2-yl)amino)pentan-2-yl)-1H-indole-2-carboxamide (compound A-1-c) as a white solid (2.5 g, 31%). LCMS = [M+H]+:
413.2. 1H NMR (400 MHz, CDCl3) δ 9.75 (s, 1H), 9.49 (s, 1H), 8.64 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.14-7.05 (m, 2H), 7.01 (s, 1H), 6.34 (s, 1H), 4.90 (s, 1H), 4.34 (s, 1H), 3.27–3.22 (m, 2H), 2.43 (s, 1H), 2.30 (s, 1H), 2.01-1.96 (m, 1H), 1.94-1.91 (m, 1H) 1.88 – 1.65 (m, 4H), 1.00-0.98 (m, 6H).
[00231] To a stirred solution of N-((S)-4-methyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)pentan-2-yl)-1H-indole-2-carboxamide (compound A-1-c) (31 g, 75.25 mmol) in EtOAc (300 mL) at room temperature was added a solution of NaHSO3 (27.56 mg, 72.73 mmol) in water (100 mL). The reaction mixture was heated at 50 ℃ for 3 h. After completion of reaction (monitored by LCMS), the organic layer was separated and removed. The aqueous layer was washed with EtOAc (100 mL x 5), concentrated down to remove remaining EtOAc, and then lyophilized to provide the desired product sodium (2S)-2-((S)-2-(1H-indole-2-carboxamido)-4-methylpentanamido)-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propane-1-sulfonate (compound A-1-d) as off-white solid (32 g, 85%). LCMS = [M-Na+2H]+: 495.2. 1H NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 8.45 (dd, J = 20.7, 8.2 Hz, 1H), 7.72 (dd, J = 48.9, 9.2 Hz, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.50 – 7.38 (m, 2H), 7.25 (dd, J = 5.1, 1.4 Hz, 1H), 7.18 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 5.43 (dd, J = 50.7, 5.9 Hz, 1H), 4.57 – 4.41 (m, 1H), 4.33 – 4.03 (m, 1H), 4.01 – 3.82 (m, 1H), 3.19 – 2.92 (m, 2H), 2.29 – 2.08 (m, 2H), 2.06 – 1.90 (m, 1H), 1.83 – 1.51 (m, 5H), 1.00 – 0.83 (m, 6H).
PAPER
https://www.sciencedirect.com/science/article/pii/S2666634023004026
Mechanism of action
Olgotrelvir is a prodrug that first converts to its active form, AC1115.[2][5] AC1115 is believed to work by inhibiting the SARS-CoV-2 main protease (also known as 3C-like protease). This protein is a crucial enzyme responsible for cleaving viral polyproteins into functional subunits essential for viral replication. By binding to the active site of the protease, the drug prevents this cleavage process, effectively halting viral assembly and impeding the virus’s ability to produce future virions.[1][2][3][5]
Olgotrelvir also appears to inhibit cathepsin L (CTSL),[2][5] a protein implicated in facilitating viral entry of SARS-CoV-2 into the host cell.[2][5][6]
Clinical trials
In September 2023, the drug’s developer, Sorrento Therapeutics, announced top-line data that olgotrelvir had met its primary endpoints in a phase III clinical trial that enrolled 1,212 patients with mild or moderate COVID-19. The drug appeared to shorten the recovery time of 11 COVID-19 symptoms in olgotrelvir-treated patients by 2.4 days on average compared to patients in the placebo group. The drug was also shown to reduce the viral load at day 4 in treated patients compared to the placebo group. Side effects were mostly mild and infrequent, with the most common being nausea (1.5% vs. 0.2%) and skin rash (3.3% vs. 0.3%), which occurred more often in the olgotrelvir group.[7][8][9]
References
- ^ Jump up to:a b Tong X, Keung W, Arnold LD, Stevens LJ, Pruijssers AJ, Kook S, et al. (November 2023). “Evaluation of in vitro antiviral activity of SARS-CoV-2 Mpro inhibitor pomotrelvir and cross-resistance to nirmatrelvir resistance substitutions”. Antimicrobial Agents and Chemotherapy. 67 (11): e0084023. doi:10.1128/aac.00840-23. PMC 10649086. PMID 37800975.
Other examples of Mpro inhibitors in late-stage development include STI-1558, currently in the phase 3 clinical trial in adult subjects with mild or moderate COVID-19 (NCT05716425).
- ^ Jump up to:a b c d e f Hackett DW (26 June 2023). “Second Generation Oral Mpro Inhibitor for COVID-19 Treatment Proceeds in Phase 3 Study”. Precision Vaccinations. Retrieved 27 December 2023.
- ^ Jump up to:a b “Coronavirus disease 2019 (COVID-19) emerging treatments”. BMJ Best Practice US. Archived from the original on 27 December 2023. Retrieved 27 December 2023.
- ^ Janin YL (September 2023). “On the origins of SARS-CoV-2 main protease inhibitors”. RSC Medicinal Chemistry. 15 (1): 81–118. doi:10.1039/D3MD00493G. ISSN 2632-8682. PMC 10809347. PMID 38283212. S2CID 264103864.
- ^ Jump up to:a b c d e Mao L, Shaabani N, Zhang X, Jin C, Xu W, Argent C, et al. (January 2024). “Olgotrelvir, a dual inhibitor of SARS-CoV-2 Mpro and cathepsin L, as a standalone antiviral oral intervention candidate for COVID-19”. Med (New York, N.Y.). 5 (1): 42–61.e23. doi:10.1016/j.medj.2023.12.004. PMID 38181791.
- ^ Berdowska I, Matusiewicz M (October 2021). “Cathepsin L, transmembrane peptidase/serine subfamily member 2/4, and other host proteases in COVID-19 pathogenesis – with impact on gastrointestinal tract”. World Journal of Gastroenterology. 27 (39): 6590–6600. doi:10.3748/wjg.v27.i39.6590. PMC 8554394. PMID 34754154.
- ^ Jiang R, Han B, Xu W, Zhang X, Peng C, Dang Q, et al. (June 2024). “Olgotrelvir as a Single-Agent Treatment of Nonhospitalized Patients with Covid-19”. NEJM Evidence. 3 (6): EVIDoa2400026. doi:10.1056/EVIDoa2400026. PMID 38804790.
- ^ Sherman AC, Baden LR (June 2024). “How To Measure Benefit in a Changing Pandemic – Olgotrelvir for SARS-CoV-2”. NEJM Evidence. 3 (6): EVIDe2400144. doi:10.1056/EVIDe2400144. PMID 38804789.
- ^ “Sorrento Announces Phase 3 Trial Met Primary Endpoint and Key Secondary Endpoint in Mild or Moderate COVID-19 Adult Patients Treated with Ovydso (Olgotrelvir), an Oral Mpro Inhibitor as a Standalone Treatment for COVID-19” (Press release). BioSpace. 12 September 2023. Retrieved 27 December 2023.
| Clinical data | |
|---|---|
| Trade names | Ovydso |
| Other names | STI-1558, HY-156655, CS-0887294 |
| Routes of administration | By mouth |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2763596-71-8 |
| PubChem CID | 166157331 |
| UNII | ZP3BDH359D |
| KEGG | D12777 |
| Chemical and physical data | |
| Formula | C22H30N4O7S |
| Molar mass | 494.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////Olgotrelvir, STI-1558, HY-156655, CS-0887294, STI 1558, HY 156655, CS 0887294, ZP3BDH359D
Acoltremon
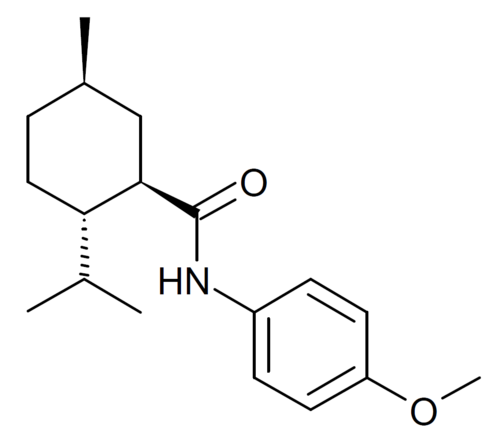
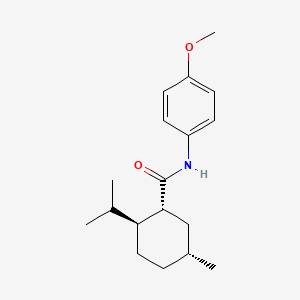
Acoltremon
CAS 68489-09-8
WeightAverage: 289.419
Monoisotopic: 289.204179113
Chemical FormulaC18H27NO2
FDA 2025, 5/28/2025, To treat the signs and symptoms of dry eye disease
Tryptyr |
WS 12
(1R,2S,5R)-N-(4-methoxyphenyl)-5-methyl-2-(propan-2-yl)cyclohexane-1-carboxamide
Fema No. 4681
N-(4-methoxyphenyl)-p-menthanecarboxamide
- OriginatorInstituto de Neurociencias de Alicante
- DeveloperAlcon; AVX Pharma
- ClassCyclohexanes; Ethers; Eye disorder therapies; Small molecules
- Mechanism of ActionTRPM8 protein stimulants
- RegisteredDry eyes
- 30 May 2025Alcon plans to launch Acoltremon for Dry eyes in USA in the third quarter of 2025
- 28 May 2025Registered for Dry eyes in USA (Ophthalmic) – First global approval
- 05 May 2025FDA assigns PDUFA action date of 30/05/2025 for Acoltremon for Dry eyes
Acoltremon sold under the brand name Tryptyr, is a medication used for the treatment of dry eye syndrome.[1]
PATENT
US 217370
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023114986&_fid=RU437402572
https://patentscope.wipo.int/search/en/detail.jsf?docId=US193167995&_cid=P11-MCE7BB-27500-1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012032209&_fid=US193167995
Medical uses
Acoltremon was approved for medical use in the United States in May 2025, for the treatment of signs and symptoms associated with dry eye disease.[2]
Pharmacology
Acoltremon acts as a potent and selective activator (opener) of the TRPM8 calcium channel, which is responsible for the sensation of coldness produced by menthol.[3] It is slightly less potent as a TRPM8 activator compared to icilin, but is a much more selective TRPM8 ligand when compared to menthol.[4]
Society and culture
Legal status
Acoltremon was approved for medical use in the United States in May 2025.[5]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/217370s000lbl.pdf
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 29 May 2025. Archived from the original on 3 March 2025. Retrieved 29 May 2025.
- ^ Ma S, Gisselmann G, Vogt-Eisele AK, Doerner JF, Hatt H (October 2008). “Menthol derivative WS-12 selectively activates transient receptor potential melastatin-8 (TRPM8) ion channels”. Pakistan Journal of Pharmaceutical Sciences. 21 (4): 370–378. PMID 18930858.
- ^ Kühn FJ, Kühn C, Lückhoff A (February 2009). “Inhibition of TRPM8 by icilin distinct from desensitization induced by menthol and menthol derivatives”. The Journal of Biological Chemistry. 284 (7): 4102–4111. doi:10.1074/jbc.M806651200. PMID 19095656.
- ^ “Alcon Announces FDA Approval of Tryptyr (acoltremon ophthalmic solution) 0.003% for the Treatment of the Signs and Symptoms of Dry Eye Disease” (Press release). Alcon. 28 May 2025. Archived from the original on 29 May 2025. Retrieved 29 May 2025 – via Business Wire.
External links
- Clinical trial number NCT05285644 for “Study Evaluating the Safety and Efficacy of AR-15512 (COMET-2)” at ClinicalTrials.gov
- Clinical trial number NCT05360966 for “Study Evaluating the Safety and Efficacy of AR-15512 (COMET-3)” at ClinicalTrials.gov
| molecular structure | |
| 3D representation | |
| Clinical data | |
|---|---|
| Trade names | Tryptyr |
| Other names | AVX-012, WS-12 |
| License data | US DailyMed: Acoltremon |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68489-09-8 |
| PubChem CID | 11266244 |
| DrugBank | DB19202 |
| ChemSpider | 9441255 |
| UNII | 1L7BVT4Z4Z |
| KEGG | D13125 |
| ChEMBL | ChEMBL2441929 |
| CompTox Dashboard (EPA) | DTXSID10460636 |
| Chemical and physical data | |
| Formula | C18H27NO2 |
| Molar mass | 289.419 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Beck B, et al. Prospects for prostate cancer imaging and therapy using high-affinity TRPM8 activators. Cell Calcium. 2007 Mar;41(3):285-94. [Content Brief][2]. Ma S, et al. Menthol derivative WS-12 selectively activates transient receptor potential melastatin-8 (TRPM8) ion channels. Pak J Pharm Sci. 2008 Oct;21(4):370-8. [Content Brief]
///////Acoltremon, FDA 2025, APPROVALS 2025, WS-12, WS 12, Fema No. 4681, Tryptyr, 1L7BVT4Z4Z, AR-15512
NERIGLIATIN

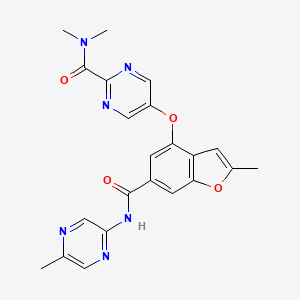
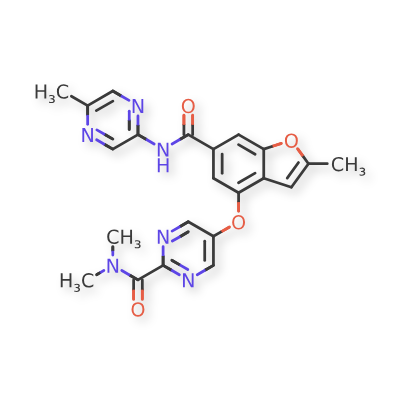
PF 04937319, NERIGLIATIN
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43, MF C22 H20 N6 O4
CAS 1245603-92-2
2-Pyrimidinecarboxamide, N,N-dimethyl-5-[[2-methyl-6-[[(5-methyl-2-pyrazinyl)amino]carbonyl]-4-benzofuranyl]oxy]-
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide
- N,N-Dimethyl-5-({2-Methyl-6-[(5-Methylpyrazin-2-Yl)carbamoyl]-1-Benzofuran-4-Yl}oxy)pyrimidine-2-Carboxamide
- 2-Pyrimidinecarboxamide, N,N-dimethyl-5-[[2-methyl-6-[[(5-methyl-2-pyrazinyl)amino]carbonyl]-4-benzofuranyl]oxy]-
- 7E99B9ZM19
Pfizer Inc. clinical candidate currently in Phase 2 development.
SCHEME
REF
MedChemComm (2011), 2(9), 828-839 81%
WO2010103437
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
Multiple dose study of PF-04937319 in patients with type 2 diabetes (NCT01272804)
Phase 2 study to evaluate safety and efficacy of investigational drug – PF04937319 in patients with type 2 diabetes (NCT01475461)
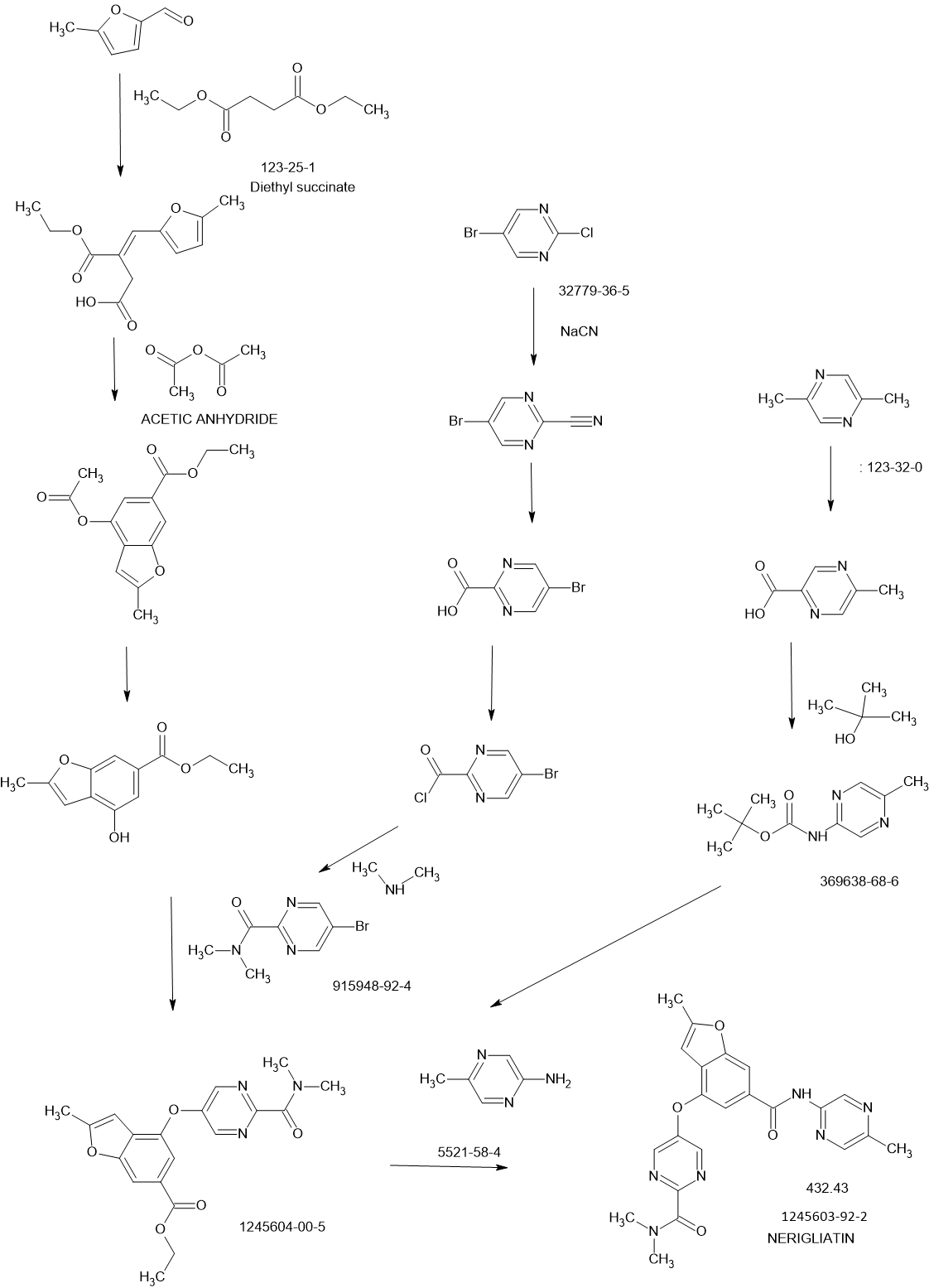
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
Jeffrey A. Pfefferkorn,*a et al
*Corresponding authors
aPfizer Worldwide Research & Development, Eastern Point Road, Groton
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
Med. Chem. Commun., 2011,2, 828-839
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes, NERIGLIATIN, 7E99B9ZM19


AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
//////////
Nerandomilast



Nerandomilast
CAS 1423719-30-5
C20H25ClN6O2S
| Molecular Weight | 448.97 |
|---|---|
| Formula | C20H25ClN6O2S |
fda 2025, approvals 2025, Jascayd,10/7/2025, To treat idiopathic pulmonary fibrosis
[1-[[(5R)-2-[4-(5-chloropyrimidin-2-yl)piperidin-1-yl]-5-oxo-6,7-dihydrothieno[3,2-d]pyrimidin-4-yl]amino]cyclobutyl]methanol
Cyclobutanemethanol, 1-[[(5R)-2-[4-(5-chloro-2-pyrimidinyl)-1-piperidinyl]-6,7-dihydro-5-oxidothieno[3,2-d]pyrimidin-4-yl]amino]-
1-[[(5R)-2-[4-(5-Chloro-2-pyrimidinyl)-1-piperidinyl]-6,7-dihydro-5-oxidothieno[3,2-d]pyrimidin-4-yl]amino]cyclobutanemethanol
Nerandomilast (BI 1015550) is an investigational oral medication being studied for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a preferential inhibitor of phosphodiesterase 4B (PDE4B) and has shown potential in slowing lung function decline in patients with IPF.
Key points about nerandomilast:
- Mechanism of Action:Nerandomilast inhibits PDE4B, an enzyme that plays a role in inflammation and fibrosis.
- Clinical Trials:Phase 3 clinical trials have shown that nerandomilast can slow lung function decline in patients with IPF and PPF.
- Efficacy:The trials demonstrated that nerandomilast led to a smaller decline in forced vital capacity (FVC), a measure of lung function, compared to placebo.
- Safety:Diarrhea was the most frequent adverse event, but serious adverse events were balanced across treatment groups.
- Progressive Fibrosing ILDs:Nerandomilast is also being investigated in other progressive fibrosing interstitial lung diseases (ILDs) beyond IPF.
- FDA Designation:Nerandomilast received Breakthrough Therapy Designation from the FDA for the treatment of IPF.
- Not a Cure:While nerandomilast can slow disease progression, it does not cure pulmonary fibrosis.
- Not Yet Approved:Nerandomilast is still an investigational drug and is not yet approved for use.
Nerandomilast (BI 1015550) is an orally active inhibitor of PDE4B with an IC50 value of 7.2 nM. Nerandomilast has good safety and potential applications in inflammation, allergic diseases, pulmonary fibrosis, and chronic obstructive pulmonary disease (COPD).
SCHEME


1H NMR (400 MHz, DMSO-D6) 1.57–1.84 (m, 2H), 1.96 (br d, J = 12.5 Hz, 2H), 2.10–2.21 (m, 2H), 2.24–
2.41 (m, 2H), 2.82–2.98 (m, 2H), 3.06 (br t, J = 11.7 Hz, 2H), 3.13–3.27 (m, 2H), 3.36–3.47 (m, 1H), 3.71 (d, J =
5.64 Hz, 2H), 4.70 (br d, J = 12.5 Hz, 2H), 4.84 (t, J = 5.7 Hz, 1H), 7.35 (s, 1H), 8.85 (s, 2H).


1H NMR (DMSO-d6, 400 MHz) 1.87–1.92 (m, 2H), 2.12–2.17 (m, 2H), 3.08 (ddd, J = 12.8, 12.8, 2.8 Hz,
2H), 3.21 (m, 1H), 3.34–3.42 (m, 2H), 8.47 (br, 2H), 8.19 (s, 2H).
PATENT
US20150045376
WO2013026797
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00309

A robust and scalable synthesis process for Nerandomilast (1, BI 1015550), a selective PDE4B inhibitor with potential therapeutic properties for the treatment of respiratory diseases, was developed and implemented at a pilot plant on a multikilogram scale. Key aspects of the process include the efficient synthesis of intermediate (1-((2-chloro-6,7-dihydrothieno[3,2-d]pyrimidin-4-yl)amino)cyclobutyl)methanol (4) by means of a regioselective SNAr reaction between (1-aminocyclobutyl)methanol (6) and 2,4-dichloro-6,7-dihydrothieno[3,2-d]pyrimidine (5), a new convergent synthesis of 5-chloro-2-(piperidin-4-yl)pyrimidine (3) by means of a Suzuki coupling, and a highly enantioselective sulfide oxidation to give chiral nonracemic (R)-2-chloro-4-((1-(hydroxymethyl)cyclobutyl)amino)-6,7-dihydrothieno[3,2-d]pyrimidine 5-oxide (2).

- [1]. Pouzet P A, et al. Piperidino-dihydrothienopyrimidine sulfoxides and their use for treating COPD and asthma. United States. US9150586.[2]. Herrmann FE, et al. BI 1015550 is a PDE4B Inhibitor and a Clinical Drug Candidate for the Oral Treatment of Idiopathic Pulmonary Fibrosis. Front Pharmacol. 2022 Apr 20;13:838449. [Content Brief]
//////////Nerandomilast, BI 1015550, I5DGT51IB8, fda 2025, approvals 2025, Jascayd,
Nedometinib



Nedometinib
CAS 2252314-46-6
NFX-179, K5T4I78IYZ
| Molecular Weight | 470.24 |
|---|---|
| Formula | C17H16FIN4O3 |
2-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1-methylpyrrolo[2,3-b]pyridine-3-carboxamide
- EN300-27122249
- 1H-Pyrrolo[2,3-b]pyridine-3-carboxamide, 2-[(2-fluoro-4-iodophenyl)amino]-N-(2-hydroxyethoxy)-1-methyl-
- 2-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1-methylpyrrolo[2,3-b]pyridine-3-carboxamide
Nedometinib (NFX-179) is a specific MEK1 inhibitor with an IC50 of 135 nM. Nedometinib inhibits p-ERK, MAPK. Nedometinib exerts anticancer activity against squamous cell carcinoma. Nedometinib can be used for research in dermatosis, neurofibromatosis.
Nedometinib is a topical gel formulation composed of an inhibitor of mitogen-activated protein kinase kinase (MAP2K; MAPKK; MEK), with potential antineoplastic activity. Upon topical administration, nedometinib penetrates into the dermis of the skin where it specifically targets, binds to and inhibits the catalytic activity of MEK, thereby inhibiting the activation of MEK-dependent effector proteins including extracellular signal-regulated kinase (ERK) and inhibits the proliferation of tumor cells in which the RAS/RAF/MEK/ERK signaling pathway is overactivated. The threonine/tyrosine protein kinase MEK plays a key role in the RAS/RAF/MEK/ERK signaling pathway, which is frequently upregulated in a variety of tumor cell types and regulates key cellular activities including cell growth, proliferation, survival, differentiation and apoptosis. Rapid degradation of NFX-179 upon reaching the systemic circulation minimizes side effects caused by systemic exposure.
SCHEME

PATENTS
US11161845, https://patentscope.wipo.int/search/en/detail.jsf?docId=US295432044&_cid=P20-MC8HLL-16550-1
Example 2: 2-((2-Fluoro-4-iodophenyl)amino)—N-(2-hydroxyethoxy)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide
2-((2-Fluoro-4-iodophenyl)amino)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carbonyl chloride
Alternative 1 for the preparation of 2-((2-Fluoro-4-iodophenyl)amino)—N-(2-hydroxyethoxy)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide
Alternative 2 for the Preparation of 2-((2-Fluoro-4-iodophenyl)amino)—N-(2-hydroxyethoxy)-1-methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide
PATENTS
WO2018213810
Science Translational Medicine (2023), 15(717), eade1844
WO2018213810, Nflection Therapeutics, Inc.
WO2023096935
WO2022262797
WO2020106303
WO2020106304
WO2020106303
WO2020106307
WO2020106304
WO2018213810
WO2020106304
WO2020106303
REF
/////////Nedometinib, NFX-179, NFX 179, K5T4I78IYZ, EN300-27122249
Modoflaner



Modoflaner ‘
| Molecular Weight | 715.23 |
|---|---|
| Formula | C23H10F12IN3O2 |
| CAS No. | 1331922-53-2 |
6-fluoro-N-[2-fluoro-3-[[4-(1,1,1,2,3,3,3-heptafluoropropan-2-yl)-2-iodo-6-(trifluoromethyl)phenyl]carbamoyl]phenyl]pyridine-3-carboxamide
- 3-Pyridinecarboxamide, 6-fluoro-N-(2-fluoro-3-(((2-iodo-4-(1,2,2,2-tetrafluoro-1-(trifluoromethyl)ethyl)-6-(trifluoromethyl)phenyl)amino)carbonyl)phenyl)-
- 6-fluoro-N-[2-fluoro-3-[[4-(1,1,1,2,3,3,3-heptafluoropropan-2-yl)-2-iodo-6-(trifluoromethyl)phenyl]carbamoyl]phenyl]pyridine-3-carboxamide
- 6-Fluoro-N-(2-fluoro-3-((2-iodo-4-(perfluoropropan-2-yl)-6-(trifluoromethyl)phenyl)carbamoyl)phenyl)nicotinamide
E583FHZ8C9
Modoflaner is an isophenylamide insecticide. Modoflaner may act through allosteric regulation of gamma-aminobutyric acid-gated chloride channels.
SCHEME

PATENT
WO2019059412
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019059412&_cid=P20-MC71WG-08056-1
SYN

Modoflaner is another isophthalamide insecticide developed by Mitsui Chemicals Agriculture Co., Ltd. in Japan. Its structure is similar to broflanilide and cyproflanilide created in China, except that it introduces iodine and fluoropyridine structures. It is speculated that the mechanism of action of modoflaner is mainly through allosteric regulation of
γ -aminobutyric acid-gated chloride ion channels, which is similar to isoxazoline insecticides and acaricides such as mivolana and eumivolana. Indoor bioassay studies have shown that modoflaner has a killing rate of more than 70% (6 days) against Spodoptera litura, Plutella xylostella and Laodelphax
striatum at a concentration of 100 mg/L. It has a killing rate of 95% (48 hours) against adult
Ctenocephalides felis at a dose of 0.04 μg/
cm2 or 0.0064 mg/ L . It has a killing rate of 90% (48 hours)
against nymphs of American flower ticks, adults of
Ixodes ricinus and adults of
R. sanguineus at a dose of 0.2 μg/cm2. It can prevent female adults of R. sanguineus from laying eggs or hatching eggs after 7 days of in vitro injection at a dose of 0.032 μg/tick. The creation idea and synthetic route of Modoflaner are shown in Figure 2. The synthetic order of iodination and amidation deserves further study.
//////////Modoflaner, E583FHZ8C9
MIVORILANER



MIVORILANER
1414642-93-5
| Molecular Formula | C22H17Cl2F6N3O3S |
| Molecular Weight | 588.35 |
- 3-[(5S)-5-(3,5-Dichloro-4-fluorophenyl)-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-[(2,2-difluoroethyl)amino]-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxamide (ACI)
- 3-[(5S)-5-(3,5-dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4,5-dihydroisoxazol-3-yl]-N-[2-[(2,2-difluoroethyl)amino]-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxamide
- ITABH 19-01
- LY 3116151
- WHO 11674
- XN7QGY28HM
- HI-154
1-[(5S)-5-(3,5-dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-(2,2-difluoroethylamino)-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-3-carboxamide
- (S)-3-(5-(3,5-Dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4,5-dihydroisoxazol-3-yl)-N-(2-((2,2-difluoroethyl)amino)-2-oxoethyl)-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxamide
- 1-[(5S)-5-(3,5-dichloro-4-fluorophenyl)-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-(2,2-difluoroethylamino)-2-oxoethyl]-5,6-dihydro-4H-cyclopenta[c]thiophene-3-carboxamide
MIVORILANER is a small molecule drug with a maximum clinical trial phase of I and has 1 investigational indication.
Mivorilaner, an antineoplastic, can be used for the research of veterinary medicine
SCHEME

PATENT
WO2012155676
(S)-3-[5-(3,5-dichloro-4-fluoro-phenyl)-5-trifluoromethyl-4,5-dihydro-isoxazol-3-yl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxylic acid [(2,2-difluoro-ethylcarbamoyl)-methyl]-amide
3 g of 3-[5-(3,5-dichloro-4-fluoro-phenyl)-5-trifluoromethyl-4,5-dihydro-isoxazol-3-yl]-5,6-dihydro-4H-cyclopenta[c]thiophene-1-carboxylic acid [(2,2-difluoro-ethylcarbamoyl)-methyl]-amide is separated by SFC separation to give desired product (1.4 g, 93%). SFC conditions are as follows: Instrument: Thar 350 Column: AD 250 mm*50 mm, 10 um Mobile phase: A: Supercritical CO2, B: EtOH, A:B=60:40 at 240 ml/min Column Temp: 38° C. Nozzle Pressure: 100 Bar Nozzle Temp: 60° C. Evaporator Temp: 20° C. Trimmer Temp: 25° C. Wavelength: 220 nm. 1H NMR (CDCl3, 400 MHz): δ 7.56 (d, J=6.0, 2H), 6.64 (brs, 1H), 6.40 (brs, 1H), 6.03-5.73 (m, 1H), 4.15 (d, J=5.2, 2H), 4.01 (d, J=17.2, 1H), 3.74-3.65 (m, 1H), 3.62 (d, J=17.2, 1H), 2.97 (t, J=7.6, 2H), 2.89 (t, J=7.6, 2H), 2.56 (m, 2H).
WO2012158396
(WO2012155676, Example 245).
/////////MIVORILANER, ITABH 19-01, LY 3116151, XN7QGY28HM, WHO 11674, HI-154

{kind=link}
{kind=link}
{kind=link}
{kind=link}