
Home » 2024 (Page 2)
Yearly Archives: 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Deuruxolitinib


Deuruxolitinib
C17H18N6, 314.422
Fda approved Leqselvi, 7/25/2024, To treat severe alopecia areata
C-21543, CTP 543, CTP-543, CTP543
(3r)-3-(2,2,3,3,4,4,5,5-d8)cyclopentyl-3-(4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-1h-pyrazol-1-yl)propanenitrile
1h-pyrazole-1-propanenitrile, .beta.-(cyclopentyl-2,2,3,3,4,4,5,5-d8)-4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-, (.beta.r)-D8-ruxolitinib
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Deuruxolitinib phosphate | 8VJ43S4LCM | 2147706-60-1 | JFMWPOCYMYGEDM-NTVOUFPTSA-N |
unii
0CA0VSF91Y
Deuruxolitinib, sold under the brand name Leqselvi, is a medication used for the treatment of alopecia areata.[1] It is a Janus kinase inhibitor selective for JAK1 and JAK2.[2] Although the relative effectiveness of deuruxolitinib and another Janus kinase inhibitor—baricitinib—for alopecia areata may vary depending on the population studied, both drugs are more effective than alternative treatments.[3]
Deuruxolitinib was approved for medical use in the United States in July 2024.[1][4]
Medical uses
Deuruxolitinib is indicated for the treatment of adults with severe alopecia areata.[1]
Side effects
The FDA prescribing label for deuruxolitinib contains a boxed warning for serious infections; malignancies; cardiovascular death, myocardial infarction, and stroke; and thrombosis.[5]
Society and culture
Names
Deuruxolitinib is the international nonproprietary name[6] and the United States Adopted Name.[7]
SYN
20240108633METHOD FOR PREVENTING OR TREATING DISEASE OR CONDITION ASSOCIATED WITH ANTITUMOR AGENT
20240058345TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
2023553253重水素化JAK阻害剤による脱毛障害の治療のためのレジメン
20230390292REGIMENS FOR THE TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
20230322787PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS
1020230093504중수소화된 JAK 억제제를 이용한 탈모 장애의 치료를 위한 요법
WO/2023/018954TREATMENT OF JAK-INHIBITION-RESPONSIVE DISORDERS WITH PRODRUGS OF JAK INHIBITORS
2022171838TREATMENT OF ALOPECIA CAUSED BY DEUTERATED JAK INHIBITOR
2022171838TREATMENT OF ALOPECIA CAUSED BY DEUTERATED JAK INHIBITOR
20220226327Combination therapy comprising JAK pathway inhibitor and rock inhibitor
20220213105PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS
20220202834JAK inhibitor with a vitamin D analog for treatment of skin diseases
20210387991Deuterated JAK inhibitor and uses thereof SUN
WO/2020/163653PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS CONCERT
20200222408TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
2019516684Treatment of Hair Loss Disorders with Deuterated JAK Inhibitors
PATENT
US20210387991
USE OF COMPD NOT SYNTHESIS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US344953814&_cid=P12-M0XGHQ-19840-2
Example 1
Synthesis of Compound 10

| HRMS: Agilent 6530 Q-TOF LC/MS system with electrospray ionization in positive mode. The measured time-of-flight mass-to-charge ratio (m/z) is 333.22839 (theoretical value=333.22735). |
| Clinical data | |
|---|---|
| Trade names | Leqselvi |
| Other names | CTP-543 |
| License data | US DailyMed: Deuruxolitinib |
| Routes of administration | By mouth |
| Drug class | Janus kinase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1513883-39-0as phosphate: 2147706-60-1 |
| PubChem CID | 72704611as phosphate: 154572727 |
| DrugBank | DB18847 |
| ChemSpider | 115010950 |
| UNII | 0CA0VSF91Yas phosphate: 8VJ43S4LCM |
| KEGG | D11866as phosphate: D11867 |
| ChEMBL | ChEMBL4594381 |
| Chemical and physical data | |
| Formula | C17H18N6 |
| Molar mass | 306.373 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
References
King B, Mesinkovska N, Mirmirani P, Bruce S, Kempers S, Guttman-Yassky E, Roberts JL, McMichael A, Colavincenzo M, Hamilton C, Braman V, Cassella JV: Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata. J Am Acad Dermatol. 2022 Aug;87(2):306-313. doi: 10.1016/j.jaad.2022.03.045. Epub 2022 Mar 29. [Article]Yan T, Wang T, Tang M, Liu N: Comparative efficacy and safety of JAK inhibitors in the treatment of moderate-to-severe alopecia areata: a systematic review and network meta-analysis. Front Pharmacol. 2024 Apr 10;15:1372810. doi: 10.3389/fphar.2024.1372810. eCollection 2024. [Article]Barati Sedeh F, Michaelsdottir TE, Henning MAS, Jemec GBE, Ibler KS: Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis. Acta Derm Venereol. 2023 Jan 25;103:adv00855. doi: 10.2340/actadv.v103.4536. [Article]Sardana K, Bathula S, Khurana A: Which is the Ideal JAK Inhibitor for Alopecia Areata – Baricitinib, Tofacitinib, Ritlecitinib or Ifidancitinib – Revisiting the Immunomechanisms of the JAK Pathway. Indian Dermatol Online J. 2023 Jun 28;14(4):465-474. doi: 10.4103/idoj.idoj_452_22. eCollection 2023 Jul-Aug. [Article]FDA Approved Drug Products: LEQSELVI (deuruxolitinib) tablets, for oral use [Link]AJMC: FDA Approves Deuruxolitinib for Alopecia Areata [Link]
^ Jump up to:a b c d “Archived copy” (PDF). Archived (PDF) from the original on 29 July 2024. Retrieved 26 July 2024.
- ^ King, Brett; Mesinkovska, Natasha; Mirmirani, Paradi; Bruce, Suzanne; Kempers, Steve; Guttman-Yassky, Emma; et al. (August 2022). “Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata”. Journal of the American Academy of Dermatology. 87 (2): 306–313. doi:10.1016/j.jaad.2022.03.045. ISSN 1097-6787. PMID 35364216. S2CID 247866262.
- ^ SEDEH, Farnam Barati; MICHAELSDÓTTIR, Thorunn Elísabet; HENNING, Mattias Arvid Simon; JEMEC, Gregor Borut Ernst; IBLER, Kristina Sophie (25 January 2023). “Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis”. Acta Dermato-Venereologica. 103: 4536. doi:10.2340/actadv.v103.4536. ISSN 0001-5555. PMC 10391778. PMID 36695751.
- ^ “U.S. FDA Approves Leqselvi (deuruxolitinib), an Oral JAK Inhibitor for the Treatment of Severe Alopecia Areata” (Press release). Sun Pharmaceutical. 25 July 2024. Archived from the original on 26 July 2024. Retrieved 26 July 2024 – via PR Newswire.
- ^ http://www.leqselvi.com/&a=Prescribing Information
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
- ^ “Deuruxolitinib”. American Medical Association. Retrieved 27 July 2024.
Further reading
Passeron T, King B, Seneschal J, Steinhoff M, Jabbari A, Ohyama M, et al. (2023). “Inhibition of T-cell activity in alopecia areata: recent developments and new directions”. Frontiers in Immunology. 14: 1243556. doi:10.3389/fimmu.2023.1243556. PMC 10657858. PMID 38022501.
External links
- “Deuruxolitinib (Code C175770)”. NCI Thesaurus.
- “Deuruxolitinib Phosphate (Code C175771)”. NCI Thesaurus.
- Clinical trial number NCT04518995 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA1) (THRIVE-AA1)” at ClinicalTrials.gov
- Clinical trial number NCT04797650 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA2) (THRIVE-AA2)” at ClinicalTrials.gov
////Deuruxolitinib, alopecia areata, Leqselvi , approvals 2024, fda 2024, C-21543, CTP 543, CTP-543, CTP543, UNII-0CA0VSF91Y, WHO 11622
Zelatriazin


Zelatriazin,
C18H15F3N4O3, 392.3 g/mol
1929519-13-0
NBI-1065846 or TAK-041
Phase 2
(S)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)-N-(1-(4-(trifluoromethoxy)phenyl)ethyl)acetamide
Zelatriazin (NBI-1065846 or TAK-041) is a small-molecule agonist of GPR139. It was developed for schizophrenia and anhedonia in depression but trials were unsuccessful and its development was discontinued in 2023.[1][2][3][4][5][6][7]
SCHEME

SYN
WO2016081736
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016081736&_cid=P21-M0X9BK-38013-1
Example 2: (S)-2-(4-oxobenzo[d][l,2,3]triazin-3(4H)-yl)-N-(l-(4-(trifluoromethoxy)phenyl)ethyl)acetamide
[0166] To a vial containing 2-(4-oxobenzo[d][l,2,3]triazin-3(4H)-yl)acetic acid (15 mg, 0.073 mmol), HOBT (15 mg, 0.095 mmol) and EDC (21 mg, 0.110 mmol) was added DMF (244 μΕ). After stirring at RT for 5 min, (S)- 1 -(4-(trifluoromethoxy)phenyl)ethanamine (18 mg, 0.088 mmol) and DIPEA (64, 0.366 mmol) were added. The reaction mixture was
allowed to stir at RT for 1 h then water was added (5 mL). The solid was filtered off and washed with water to yield the title compound as a white solid (20 mg, 71 % yield). XH NMR
(500 MHz, DMSO-i¾) δ ppm 1.40 (d, J=6.8 Hz, 3 H), 4.98 (quin, J=7.1 Hz, 1 H), 5.09 (s, 2
H), 7.33 (d, J=7.8 Hz, 2 H), 7.44 – 7.49 (m, 2 H), 7.93 – 7.98 (m, 1 H), 8.09 – 8.15 (m, 1 H),
8.21 – 8.29 (m, 2 H), 8.85 (d, J=7.8 Hz, 1 H); ESI-MS m/z [M+H]+ 393.9.
REF
Takeda Pharmaceutical Company Limited, WO2016081736
WO2022058791
Journal of Medicinal Chemistry (2021), 64(15), 11527-11542
Publication Name: Journal of Medicinal Chemistry, Publication Date: 2023-10-13, PMID: 37830160
DOI: 10.1021/acs.jmedchem.3c01034
PATENT
https://patents.google.com/patent/US9556130B2/en
Example 2(S)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)-N-(1-(4-(trifluoromethoxy)phenyl)ethyl)acetamide

To a vial containing 2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetic acid (15 mg, 0.073 mmol), HOBT (15 mg, 0.095 mmol) and EDC (21 mg, 0.110 mmol) was added DMF (244 μL). After stirring at RT for 5 min, (S)-1-(4-(trifluoromethoxy)phenyl)ethanamine (18 mg, 0.088 mmol) and DIPEA (64, 0.366 mmol) were added. The reaction mixture was allowed to stir at RT for 1 h then water was added (5 mL). The solid was filtered off and washed with water to yield the title compound as a white solid (20 mg, 71% yield). 1H NMR (500 MHz, DMSO-d6) δ ppm 1.40 (d, J=6.8 Hz, 3H), 4.98 (quin, J=7.1 Hz, 1H), 5.09 (s, 2H), 7.33 (d, J=7.8 Hz, 2H), 7.44-7.49 (m, 2H), 7.93-7.98 (m, 1H), 8.09-8.15 (m, 1H), 8.21-8.29 (m, 2H), 8.85 (d, J=7.8 Hz, 1H); ESI-MS m/z [M+H]+ 393.9.
PATENT
| Clinical data | |
|---|---|
| Other names | NBI-1065846; TAK-041 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1929519-13-0 |
| PubChem CID | 121349608 |
| Chemical and physical data | |
| Formula | C18H15F3N4O3 |
| Molar mass | 392.338 g·mol−1 |
References
- ^ Kamel, Amin; Bowlin, Steve; Hosea, Natalie; Arkilo, Dimitrios; Laurenza, Antonio (February 2021). “In Vitro Metabolism of Slowly Cleared G Protein–Coupled Receptor 139 Agonist TAK-041 Using Rat, Dog, Monkey, and Human Hepatocyte Models (HepatoPac): Correlation with In Vivo Metabolism”. Drug Metabolism and Disposition. 49 (2): 121–132. doi:10.1124/dmd.120.000246. PMID 33273044. S2CID 227282766.
- ^ Schiffer, Hans; Atienza, Josephine; Reichard, Holly; Mulligan, Victoria; Cilia, Jackie; Monenschein, Holger; Collia, Deanna; Ray, Jim; Kilpatrick, Gavin; Brice, Nicola; Carlton, Mark; Hitchcock, Steve; Corbett, Ged; Hodgson, Robert (18 May 2020). “S180. The Selective Gpr139 Agonist Tak-041 Reverses Anhedonia and Social Interaction Deficits in Rodent Models Related to Negative Symptoms in Schizophrenia”. Schizophrenia Bulletin. 46 (Supplement_1): S106–S107. doi:10.1093/schbul/sbaa031.246. PMC 7234360.
- ^ Yin, Wei; Han, David; Khudyakov, Polyna; Behrje, Rhett; Posener, Joel; Laurenza, Antonio; Arkilo, Dimitrios (August 2022). “A phase 1 study to evaluate the safety, tolerability and pharmacokinetics of TAK-041 in healthy participants and patients with stable schizophrenia”. British Journal of Clinical Pharmacology. 88 (8): 3872–3882. doi:10.1111/bcp.15305. PMC 9544063. PMID 35277995. S2CID 247407736.
- ^ Rabiner, Eugenii A.; Uz, Tolga; Mansur, Ayla; Brown, Terry; Chen, Grace; Wu, Jingtao; Atienza, Joy; Schwarz, Adam J.; Yin, Wei; Lewis, Yvonne; Searle, Graham E.; Dennison, Jeremy M. T. J.; Passchier, Jan; Gunn, Roger N.; Tauscher, Johannes (June 2022). “Endogenous dopamine release in the human brain as a pharmacodynamic biomarker: evaluation of the new GPR139 agonist TAK-041 with [11C]PHNO PET”. Neuropsychopharmacology. 47 (7): 1405–1412. doi:10.1038/s41386-021-01204-1. PMC 9117280. PMID 34675381.
- ^ Reichard, Holly A.; Schiffer, Hans H.; Monenschein, Holger; Atienza, Josephine M.; Corbett, Gerard; Skaggs, Alton W.; Collia, Deanna R.; Ray, William J.; Serrats, Jordi; Bliesath, Joshua; Kaushal, Nidhi; Lam, Betty P.; Amador-Arjona, Alejandro; Rahbaek, Lisa; McConn, Donavon J.; Mulligan, Victoria J.; Brice, Nicola; Gaskin, Philip L. R.; Cilia, Jackie; Hitchcock, Stephen (12 August 2021). “Discovery of TAK-041: a Potent and Selective GPR139 Agonist Explored for the Treatment of Negative Symptoms Associated with Schizophrenia”. Journal of Medicinal Chemistry. 64 (15): 11527–11542. doi:10.1021/acs.jmedchem.1c00820. PMID 34260228. S2CID 235908256.
- ^ Münster, Alexandra; Sommer, Susanne; Kúkeľová, Diana; Sigrist, Hannes; Koros, Eliza; Deiana, Serena; Klinder, Klaus; Baader-Pagler, Tamara; Mayer-Wrangowski, Svenja; Ferger, Boris; Bretschneider, Tom; Pryce, Christopher R.; Hauber, Wolfgang; von Heimendahl, Moritz (August 2022). “Effects of GPR139 agonism on effort expenditure for food reward in rodent models: Evidence for pro-motivational actions”. Neuropharmacology. 213: 109078. doi:10.1016/j.neuropharm.2022.109078. PMID 35561791. S2CID 248574904.
- ^ Taylor, Nick Paul (10 November 2023). “Neurocrine hit with one-two punch as Takeda and Xenon pacts deliver midphase flops”. Fierce Biotech. Retrieved 4 December 2023.
//////Zelatriazin, 1929519-13-0, NBI-1065846, TAK-041, Phase 2
Vorasidenib


Vorasidenib
6-(6-chloropyridin-2-yl)-N2,N4-bis[(2R)-1,1,1-trifluoropropan-2-yl]-1,3,5-triazine-2,4-diamine
CAS 1644545-52-7, C14H13ClF6N6, 414.74
FDA APPROVED, 8/6/2024, Voranigo, To treat Grade 2 astrocytoma or oligodendroglioma
UNII 789Q85GA8P
- AG 881
- AG-881
- AG881
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Vorasidenib citrate | X478M962XG | 2316810-02-1 | YOUTVRFNJAAFNS-DLVAHKFUSA-N |
| Vorasidenib citrate anhydrous | W4XG3EQK7B | 2316810-00-9 | OCEHQNOYRLHJCI-WPRTUUMNSA-N |
Vorasidenib, sold under the brand name Voranigo, is an anti-cancer medication used for the treatment of certain forms of glioma.[1][2] Vorasidenib acts to inhibit the enzymes isocitrate dehydrogenase-1 (IDH1) and isocitrate dehydrogenase-2 (IDH2).[1][2]
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2]
Vorasidenib was approved for medical use in the United States in August 2024.[2][3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation.[2]
Medical uses
Vorasidenib is indicated for the treatment of people aged twelve years of age and older with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation, following surgery including biopsy, sub-total resection, or gross total resection.[2]
Side effects
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2] The most common grade 3 or 4 laboratory abnormalities include increased alanine aminotransferase, increased aspartate aminotransferase, GGT increased, and decreased neutrophils.[2]
History
Efficacy was evaluated in 331 participants with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation following surgery enrolled in INDIGO (NCT04164901), a randomized, multicenter, double-blind, placebo-controlled trial.[2] Participants were randomized 1:1 to receive vorasidenib 40 mg orally once daily or placebo orally once daily until disease progression or unacceptable toxicity.[2] Isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation status was prospectively determined by the Life Technologies Corporation Oncomine Dx Target Test.[2] Participants randomized to placebo were allowed to cross over to vorasidenib after documented radiographic disease progression.[2] Participants who received prior anti-cancer treatment, including chemotherapy or radiation therapy, were excluded.[2]
Society and culture
Legal status
Vorasidenib was approved for medical use in the United States in August 2024.[2]
The FDA granted the application for vorasidenib priority review, fast track, breakthrough therapy, and orphan drug designations.[2]
SYN
WO/2024/161041NOVEL COMPOUNDS THAT CAN BE USED AS THERAPEUTIC AGENTS
20240254118PRMT5 INHIBITORS AND USES THEREOF
118359585共晶体、其药物组合物以及涉及其的治疗方法
WO/2024/148437USE OF PCLX-001 OR PCLX-002 AS A RADIOSENSITIZER
20240238424HETEROBIFUNCTIONAL COMPOUNDS AND METHODS OF TREATING DISEASE
1020240097895CD73 화합물
WO/2024/137852PRMT5 INHIBITORS AND USES THEREOF
2024057088THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE
20240116928CD73 COMPOUNDS
117586228Preparation method of triazine medicine
20240041892THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE
117529323Therapeutically active compounds and methods of use thereof
WO/2024/006929CD73 COMPOUNDS
PATENT
https://patents.google.com/patent/US10028961B2/en
Step 3: Preparation of 6-(6-chloropyridin-2-yl)-N2,N4-bis((R)-1,1,1-trifluoro propan-2-yl)-1,3,5-triazine-2,4-diamine
A mixture of 2,4-dichloro-6-(6-chloro-pyridin-2-yl)-1,3,5-triazine (0.27 g, 1.04 mol), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (0.39 g, 2.6 mol), and potassium carbonate (0.43 g, 3.1 mol) in dry 1,4-dioxane (2.5 mL) was stirred under the atmosphere of N2 at 50° C. for 36 hr then at 100° C. for another 36 hr until the reaction was complete. The resulting mixture was filtered through Celite and the cake was washed with EtOAc. The filtrate was concentrated and the residue was purified by standard methods to give the desired product.

1H NMR (400 MHz, CDCl3) δ 8.32 (m, 1H), 7.80 (m, 1H), 7.48 (d, J=7.9 Hz, 1H), 5.61 (m, 1.5H), 5.25 (m, 0.5H), 5.09 (m, 0.5H), 4.88 (m, 1.5H), 1.54-1.26 (m, 6H). LC-MS: m/z 415 (M+H)+.
The procedure set forth in Example 10 was used to produce the following compounds using the appropriate starting materials.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.29-8.16 (m, 1H), 7.72 (d, J=7.6 Hz, 1H), 7.41 (d, J=7.9 Hz, 1H), 5.70-5.13 (m, 2H), 5.09-4.71 (m, 2H), 1.34 (m, 6H). LC-MS: m/z 415 (M+H)+.Compound 6-(6-Chloropyridin-2-yl)-N2—((R)-1,1,1-trifluoropropan-2-yl)-N4—((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.41-8.23 (m, 1H), 7.83 (s, 1H), 7.51 (d, J=6.2 Hz, 1H), 5.68-5.20 (m, 2H), 5.18-4.81 (m, 2H), 1.48-1.39 (m, 6H). LC-MS: m/z 415 (M+H)+.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis(1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.29-8.16 (m, 1H), 7.72 (d, J=7.6 Hz, 1H), 7.41 (d, J=7.9 Hz, 1H), 5.70-5.13 (m, 2H), 5.09-4.71 (m, 2H), 1.34 (m, 6H). LC-MS: m/z 415 (M+H)+.

| Clinical data | |
|---|---|
| Trade names | Voranigo |
| License data | US DailyMed: Vorasidenib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1644545-52-7 |
| PubChem CID | 117817422 |
| IUPHAR/BPS | 10663 |
| DrugBank | DB17097 |
| ChemSpider | 64835242 |
| UNII | 789Q85GA8P |
| KEGG | D11834 |
| ChEMBL | ChEMBL4279047 |
| Chemical and physical data | |
| Formula | C14H13ClF6N6 |
| Molar mass | 414.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
^ Jump up to:a b c “Voranigo- vorasidenib citrate tablet, film coated”. DailyMed. 9 August 2024. Retrieved 15 August 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves vorasidenib for Grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation”. U.S. Food and Drug Administration (FDA). 6 August 2024. Archived from the original on 7 August 2024. Retrieved 7 August 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Servier’s Voranigo (vorasidenib) Tablets Receives FDA Approval as First Targeted Therapy for Grade 2 IDH-mutant Glioma” (Press release). Servier Pharmaceuticals. 6 August 2024. Archived from the original on 7 August 2024. Retrieved 7 August 2024 – via PR Newswire.
Further reading
- Mellinghoff IK, Lu M, Wen PY, Taylor JW, Maher EA, Arrillaga-Romany I, et al. (March 2023). “Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial”. Nature Medicine. 29 (3): 615–622. doi:10.1038/s41591-022-02141-2. PMC 10313524. PMID 36823302.
- Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA, Maher EA, Janku F, et al. (August 2021). “Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-in-Human Phase I Trial”. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research. 27 (16): 4491–4499. doi:10.1158/1078-0432.CCR-21-0611. PMC 8364866. PMID 34078652.
- Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, et al. (August 2023). “Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma”. The New England Journal of Medicine. 389 (7): 589–601. doi:10.1056/NEJMoa2304194. PMID 37272516.
- Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. (April 2018). “Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers”. ACS Medicinal Chemistry Letters. 9 (4): 300–305. doi:10.1021/acsmedchemlett.7b00421. PMC 5900343. PMID 29670690.
External links
Clinical trial number NCT04164901 for “Study of Vorasidenib (AG-881) in Participants With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation (INDIGO)” at ClinicalTrials.gov
- Clinical trial number NCT02481154 for “Study of Orally Administered AG-881 in Patients With Advanced Solid Tumors, Including Gliomas, With an IDH1 and/or IDH2 Mutation” at ClinicalTrials.gov
- Clinical trial number NCT03343197 for “Study of AG-120 and AG-881 in Subjects With Low Grade Glioma” at ClinicalTrials.gov
////////Vorasidenib, Voranigo, FDA 2024, APPROVALS 2024, AG 881, AG-881, AG881
Arbemnifosbuvir, AT-752, PD160572


Arbemnifosbuvir, AT-752, 1998705-63-7, PD160572
E9V7VHK36U INN 12706
C24H33FN7O7P 581.5 g/mol

SYN
propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-[2-amino-6-(methylamino)purin-9-yl]-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate
L-ALANINE, N-((P(R),2’R)-2-AMINO-2′-DEOXY-2′-FLUORO-N,2′-DIMETHYL-P-PHENYL-5′ -ADENYLYL)-, 1-METHYLETHYL ESTER
N-((P(R),2’R)-2-AMINO-2′-DEOXY-2′-FLUORO-N,2′-DIMETHYL-P-PHENYL-5′ -ADENYLYL)-L-ALANINE 1-METHYLETHYL ESTER
WO2022040473 Atea Pharmaceuticals, Inc.
CN113784721
US20160257706
WO2022076903 US10874687
PATENT
US20160257706
https://patentscope.wipo.int/search/en/detail.jsf?docId=US177601863&_cid=P11-M0VTE4-38538-1


Example 1. Preparation of isopropyl ((((R,S)-(2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-phenoxy-phosphoryl)-L-alaninate
Step 1. Preparation of ((2R,3R,4R,5R)-3-(benzoyloxy)-5-bromo-4-fluoro-4-methyltetrahydrofuran-2-yl)methyl benzoate (2)
Step 2. Preparation of (2R,3R,4R,5R)-5-(2-amino-6-chloro-9H-purin-9-yl)-2-(benzoyloxymethyl)-4-fluoro-4-methyltetrahydrofuran-3-yl benzoate (3)
Step 3. Preparation of (2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)-4-methyltetrahydrofuran-3-ol (4)
Step 4. Preparation of isopropyl ((((R,S)-(2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-phenoxy-phosphoryl)-L-alaninate (5)
PATENT
/////Arbemnifosbuvir, AT-752, 1998705-63-7, PD160572, E9V7VHK36U INN 12706
Zelicapavir


Zelicapavir, Enanta Pharmaceuticals
Alternative Names: EDP-938; EP 023938
cas 2070852-76-3
RSV-IN-7
549.5 g/mol, C27H22F3N7O3
UNII U4OI721DMD
(3S)-3-[[5-[3-morpholin-4-yl-5-(trifluoromethyl)pyridin-2-yl]-1,3,4-oxadiazol-2-yl]amino]-5-phenyl-1,3-dihydro-1,4-benzodiazepin-2-one
SYN
New England Journal of Medicine (2022), 386(7), 655-666
WO2022157327
WO2018152413
WO2019067864
WO2017015449
PATENT
WO2018152413
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018152413&_cid=P20-M0V26A-30323-1
Step 5 : (<Sf)-3-((5-(3-morpholino-5-(trifluoromethyl)pyridin-2-yl)-1.3.4-oxadiazol-2-

To a mixture of (5)-2-(3-morpholino-5-(trifluoromethyl)picolinoyl)-N-2-oxo-5-phenyl-2,3-dihydro-lH-benzo[e] [l,4]diazepin-3-yl)hydrazine-l-carboxamide (1.4 kg, 1 eq.) in DCM (11.2 L) in a flask was charged with 4A-MS (1.4 kg) and stirred at 20±5 °C for 2hrs. Then, it was cooled to 0°C, charged with triethylamine (0.62 Kg, 2.5 eq.) and stirred for 10 min. /^-Toluenesulfonyl chloride (0.7 kg, 1.5 eq.) in DCM (1.4 L) solution was dropwise added to the reaction mixture with maintaining below 5°C and stirred at at 0±5 °C for 5 hrs. The reaction mixture was filtered and washed with DCM (2 X 4.2 L). The filtrate was treated with water (4.2 L) at 0°C and stirred between 0 and 10°C for 5 min. After separation, the organic phase was washed with 5% aqueous NaHCC solution (7 L), water (7 L) and brine (7 L) successively and separated. The DCM layer was concentrated in vacuo at below 30°C to leave ~7L of organic layer. MTBE (7 L) was added to organic layer and concentrated in vacuo to leave ~ 7 L of organic layer (This step was repeated once). The organic layer was charged with water (7 L) and stirred at 20±5 °C for 4 hrs. The solid was filtered and washed with MTBE (3 X 2.1 L) and purified water (2.8 L). The wet cake was stirred with ethyl acetate (7 L) for 12 hrs, charged with n-heptane (14 L) and stirred at 20±5 °C for 5 hrs. The solid was filtered, washed with n-heptane (2 X 2.8 L) and dried under vacuum at ambient temperature to provide the title compound (0.776 kg, 99.6% purity by HPLC, 97.8%
chiral purity by chiral HPLC) as a pale yellowish solid. LC-MS(ESI, m/z): 550.17 [M+H]+;
¾ NMR: ( DMSO-c 6400 MHz): δ 10.98 (br-s, 1H), 9.40 (d, J=8.0 Hz, 1H), 8.69 (br-d, J=4.0 Hz, 1H), 7.89 (d, J=4.0 Hz, 1H), 7.68 (dt, J=8.0 and 4.0 Hz, 1H), 7.56-7.51 (m, 3H), 7.49-7.45 (m, 2H), 7.38-7.35 (m, 2H), 7.29 (br-t, J=8.0 Hz, 1H)
5.22 (d, J=8.0 Hz, 1H), 3.75-3.72 (m, 4H), 3.09-3.07 (m, 4H); 13C (DMSO-c¾, 100 MHz): δ 167.3, 167.0, 162.8, 156.4, 147.2, 139.2, 138.7, 138.4, 138.3, 138.0, 132.30, 130.7, 130.5, 129.5, 128.4, 126.2, 124.5, 123.4, 121.5, 71.8, 65.9, 51.0.
SCHEME

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US368999603&_cid=P20-M0V2BF-36596-1
Example 253
Example 160 was prepared using a procedure similar to that used to prepare Example 152 where methyl 5-cyano-3-morpholinopicolinate was used in place of ethyl 2-morpholino-4-(trifluoromethyl)benzoate. ESI-MS m/z: 507.2 [M+H] +. 1H NMR (400 MHz, DMSO-d 6) δ 3.02-3.04 (m, 4H), 3.71-3.73 (m, 4H), 5.19-5.21 (d, J=8.0 Hz, 1H), 7.26-7.30 (m, 1H), 7.34-7.36 (m, 2H), 7.44-7.55 (m, 5H), 7.65-7.70 (m, 1H), 8.13 (s, 1H), 8.72 (s, 1H), 9.42-9.45 (m, 1H), 10.98 (s, 1H).
//////////////Zelicapavir, EDP-938, EP 023938, EDP 938, RSV-IN-7, ENANTA
Palopegteriparatide

Palopegteriparatide
Yorvipath , FDA 2024, 8/9/2024, To treat hypoparathyroidism
- G2N64C3385
- 2222514-07-8
- Palopegteriparatide
- UNII-G2N64C3385
- ACP-014
- Mpeg 40000-teriparatide
- Palopegteriparatide [INN]
- Transcon parathyroid hormone (1-34)
- Transcon pth (1-34)
- Palopegteriparatide [USAN]
- TransCon PTH
- WHO 11060


Palopegteriparatide, sold under the brand name Yorvipath, is a hormone replacement therapy used for the treatment of hypoparathyroidism.[1][2] It is a transiently pegylated parathyroid hormone.[4] It is a parathyroid hormone analog.[1]
Palopegteriparatide was approved for medical use in the European Union in November 2023,[2] and in the United States in August 2024.[1][5]
Medical uses
Palopegteriparatide is indicated for the treatment of adults with hypoparathyroidism.[1][2]
Adverse effects
The US Food and Drug Administration (FDA) prescription label for palopegteriparatide includes warnings for a potential risk of risk of unintended changes in serum calcium levels related to number of daily injections and total delivered dose, serious hypocalcemia and hypercalcemia (blood calcium levels that are too high), osteosarcoma (a rare bone cancer) based on findings in rats, orthostatic hypotension (dizziness when standing), and a risk of a drug interaction with digoxin (a medicine for certain heart conditions).[5]
History
The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism.[5] Prior to randomization, all participants underwent an approximate four-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-corrected serum calcium concentration between 7.8 and 10.6 mg/dL, a magnesium concentration ≥1.3 mg/dL and below the upper limit of the reference range, and a 25(OH) vitamin D concentration between 20 to 80 ng/mL.[5] During the double-blind period, participants were randomized to either palopegteriparatide (N = 61) or placebo (N= 21), at a starting dose of 18 mcg/day, co-administered with conventional therapy (calcium and active vitamin D).[5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels.[5] At the end of the trial, 69% of the participants in the palopegteriparatide group compared to 5% of the participants in the placebo group were able to maintain their calcium level in the normal range, without needing active vitamin D and high doses of calcium (calcium dose ≤ 600 mg/day).[5]
The FDA granted the application for palopegteriparatide orphan drug and priority review designations.[5]
Society and culture
Legal status
In September 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yorvipath, intended for the treatment of chronic hypoparathyroidism in adults.[4][6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.[4] Palopegteriparatide was approved for medical use in the European Union in November 2023.[2]
Palopegteriparatide was granted an orphan drug designation by the US Food and Drug Administration (FDA) in 2018,[7] and by the EMA in 2020.[8]
Brand names
Palopegteriparatide is the international nonproprietary name.[9][10]
Palopegteriparatide is sold under the brand name Yorvipath.[2]
References
- ^ Jump up to:a b c d e “Yorvipath injection, solution”. DailyMed. 14 August 2024. Retrieved 5 September 2024.
- ^ Jump up to:a b c d e f “Yorvipath EPAR”. European Medicines Agency. 19 October 2020. Archived from the original on 10 December 2023. Retrieved 11 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Yorvipath Product information”. Union Register of medicinal products. 20 November 2023. Archived from the original on 26 November 2023. Retrieved 11 December 2023.
- ^ Jump up to:a b c “Yorvipath: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h “FDA approves new drug for hypoparathyroidism, a rare disorder”. U.S. Food and Drug Administration (FDA) (Press release). 9 August 2024. Archived from the original on 13 August 2024. Retrieved 13 August 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Ascendis Pharma Receives Positive CHMP Opinion for TransCon PTH (palopegteriparatide) for Adults with Chronic Hypoparathyroidism”. Ascendis Pharma (Press release). 14 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ “TransCon Parathyroid Hormone (mPEG conjugated parathyroid hormone 1-34) Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ “EU/3/20/2350”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
External links
- Palopegteriparatide Global Substance Registration System
- Palopegteriparatide NCI Thesaurus
- Clinical trial number NCT04701203 for “A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH Administered Daily in Adults With Hypoparathyroidism (PaTHway)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Yorvipath |
| Other names | ACP-014, TransCon PTH |
| License data | US DailyMed: Palopegteriparatide |
| Routes of administration | Subcutaneous |
| Drug class | Hormonal agent |
| ATC code | H05AA05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| CAS Number | 2222514-07-8 |
| UNII | G2N64C3385 |
| KEGG | D12395 |
//////Palopegteriparatide, APPRoVALS 2024, FDA 2024, Yorvipath, hypoparathyroidism, UNII-G2N64C3385, ACP-014, TransCon PTH, WHO 11060
Aneratrigine


Aneratrigine
2097163-74-9
5-chloro-2-fluoro-4-[4-fluoro-2-[methyl-[2-(methylamino)ethyl]amino]anilino]-N-(1,3-thiazol-4-yl)benzenesulfonamide
5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
| Benzenesulfonamide, 5-chloro-2-fluoro-4-[[4-fluoro-2-[methyl[2-(methylamino)ethyl]amino]phenyl]amino]-N-4-thiazolyl- |
C19H20ClF2N5O2S2 488.0 g/mol
UNII 6A5ZY5LT78
WHO
SYN

Assignee: Daewoong Pharmaceutical Co., Ltd.
World Intellectual Property Organization, WO2017082688
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017082688&_cid=P11-M0UEPF-95506-1
Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 1) Preparation of tert-butyl (1-(2-amino-5-fluorophenyl)pyridin-3-yl)(methyl)carbamate
2,4-Difluoro-1-nitrobenzene (2.0 g, 12.6 ng/mol) and tert-butyl methyl (pyridin-3-yl)carbamate (2.5 g, 1.0 eq.) were dissolved in DMF (20 mL), and K2C03 ( 2.6 g , 1.5 eq .) was added. The internal temperature was maintained at 60–70 ° C and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by TLC when the reaction solution turned deep yellow. After cooling to room temperature, ethyl acetate (EA)/H20 was added, stirred, and the layers were separated. MgS04 was added to the separated organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was dissolved in EtOH (10 mL) and distilled water (10 mL), and then Na 2 S 2 0 4 (13.0 g, 6 eq.) was added. After stirring for 2 hours while maintaining the internal temperature at 60 to 70 ° C, the completion of the reaction was confirmed by TLC when the yellow color of the reaction solution lightened and became almost colorless. After cooling to room temperature, distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was separated by column chromatography (n-Hexane/EA = 3/1) to obtain the title compound (2.0 g, 51. ).
1H NMR (MeOD): 6.73(m, 1H), 6.57(t, 1H), 3.23(m, 1H), 3.10(m, 2H), 2.94(m, 1H), 2.91(s, 3H), 2.25( m, 1H), 1.99(m, 1H)
Step 2) Preparation of tert-butyl thiazol-4-ylcarbamate
Thiazole-4-carboxylic acid (5.0 g, 38.8 vol) was dissolved in t-Bu0H (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added. The internal temperature was maintained at 90–100 ° C, and the mixture was stirred for 3 days. The completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and the solution was washed with EA (100 mL).
It was extracted twice. MgSQ 4 was added to the organic layer, stirred, dried, and filtered.
After concentrating the filtrate under reduced pressure, the residue was added to a small amount of EA, slurried, and the resulting solid was filtered to obtain the white title compound (4.0 g, 51.5%).
1H NMR (MeOD): 8.73(s, 1H), 7.24(s, 1H), 1.52(s, 9H)
Step 3) Preparation of tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate
Step 2) The tert-Butyl thiazol-4-ylcarbamate (4.0 g, 20.0 ng ol) prepared in the reaction vessel was placed in a reaction vessel and the interior was replaced with nitrogen gas. After dissolving in THF (32 mL), it was cooled to _78 ° C using dry ice— acetone. After cooling, LiHMDS (22.4 mL, 1.5 eq.) was slowly added and the reaction mass was stirred for 30 minutes. 4-Bromo-5-chloro-2-fluorobenzenesulfonyl chloride (6.0 g, 1.0 eq.) was dissolved in THF (10 mL) and slowly added to the reaction mixture. The reaction mass was stirred overnight and the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was crystallized from THF/n-hexane to obtain the title compound (4.4 g, 59.0%).
1H NMR (MeOD): 9.00(s, 1H), 8.22(d, 1H), 7.90(d, 1H), 7.78(s, 1H), 1.35(s, 9H)
Step 4) Preparation of tert-butyl (l-(2-((4-(N-(tert-butyloxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)-2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate
Tert-butyl (1-(2-amino-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate (0.5 g, 1.1 ng ol) prepared in Step 1) and tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate (0.9 g, 1.2 eq.) prepared in Step 3) were dissolved in 1,4-dioxane (10 mL). Pd(OAc) 2 (0.03 g, 0.1 eq), rac-BINAP (0.19 g, 0.2 eq.), Cs 2 C0 3 (1.5 g, 3.0 eq.) were added to the reaction solution. After reacting at 120 ° C for 30 minutes using a microwave initiator, the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL).
MgS0 4 was added to the organic layer, stirred, filtered and dried. The filtrate was concentrated under reduced pressure, and the residue was separated by column chromatography (EA/n-Hexane = 1/1). This was repeated twice to obtain the title compound (2.0 g, 88.2%).
1H NMR (MeOD): 8.95(s, 1H), 7.94(d, 1H), 7.65(s, 1H), 7.14(t, 1H), 6.70(d, 1H), 6.64(t, 1H), 6.07( d, 1H)ᅳ 3.40(m, 1H), 3.28(m, 2H), 3.16(m, 1H), 2.64(s, 3H), 2.06(m, 1H), 1.89(m, 1H), 1.41(s , 9H), 1.36(s, 9H)
Step 5) Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 4) was prepared by adding 1.25 M HCl in MeOH (15 mL) to tert-butyl (1-(2-((4-(Ν-(tert-butoxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)—2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl) (methyl)carbamate (2.0 g, 2.9 µl). The mixture was heated to 40–50 ° C and stirred overnight, and the completion of the reaction was confirmed by TLC. The product was concentrated, and methylene chloride (15 mL) was added to the residue, which was stirred for 1 hour, and the resulting solid was filtered to obtain the title compound (0.9 g, 58.8%).
1H 證 (MeOD): 8.73(s, 1H), 7.75(d, 1H), 7.12(t, 1H), 7.00(s, 1H), 6.69(d, 1H), 6.67(t, 1H), 6.05( d, 1H), 3.73(m, 1H) , 3.54(m, 1H), 3.45(m, 1H), 3.38(m, 1H), 3.26(m, 1H), 2.63(s, 3H) , 2.31(m , 1H), 1.96(m, 1H)
PATENTS
0002705578SODIUM CHANNEL BLOCKER
20180346459Substituted benzenesulfonamides as sodium channel blockers
2018533606ナトリウムチャネル遮断剤
3375782SODIUM CHANNEL BLOCKER
108349963SODIUM CHANNEL BLOCKER
1020170056461SODIUM CHANNEL BLOCKER

////////////Aneratrigine, DAEWOONG
Seladelpar


Seladelpar
cas 851528-79-5
C21H23F3O5S, 444.47
fda approved 8/14/2024, To treat primary biliary cholangitis (PBC), Livdelzi
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Seladelpar lysine | N1429130KR | 928821-40-3 | WTKSWPYGZDCUNQ-JZXFCXSPSA-N |
- (+)-MBX-8025
- MBX 8025
- MBX-8025
- MBX8025
- RWJ-800025
- ((4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENYL)OXY)ACETIC ACID
- (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)SULFANYL)-2-METHYLPHENOXY)ACETIC ACID PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR (PPAR) AGONIST,ANTIHYPERLIPIDAEMIC
- (R)-2-(4-((2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)-THIO)-2-METHYLPHENOXY)ACETIC ACID
- ACETIC ACID, (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)-
- ACETIC ACID, (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)- ((4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENYL)OXY)ACETIC ACID
- ACETIC ACID, 2-(4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)-
- Seladelpar
Seladelpar, sold under the brand name Livdelzi, is a medication used for the treatment of primary biliary cholangitis.[1] It is used as the lysine dihydrate salt.[1] It is a PPARδ receptor agonist.[1][2][3] The compound was licensed from Janssen Pharmaceutica NV.[4]
Seladelpar was approved for medical use in the United States in August 2024.[1][5]
Seladelpar is a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. Seladelpar is a single enantiomer of the R-configuration.5 On August 14, 2024, seladelpar was granted accelerated approval by the FDA for the treatment of primary biliary cholangitis,6 which is a condition associated with aberrant bile acid metabolism. Seladelpar works to block bile acid synthesis.1
Medical uses
Seladelpar is indicated for the treatment of primary biliary cholangitis in combination with ursodeoxycholic acid in adults who have an inadequate response to ursodeoxycholic acid, or as monotherapy in people unable to tolerate ursodeoxycholic acid.[1]
Clinically, Seladelpar reduces pruritus and IL-31 in patients with primary biliary cholangitis.[6]
- compound 3r [PMID: 17524639]
- Bioorg Med Chem Lett. 2007 Jul 15;17(14):3855-9. doi: 10.1016/j.bmcl.2007.05.007. Epub 2007 May 10.
- 10.1016/j.bmcl.2007.05.007
Drug Discovery, Johnson and Johnson Pharmaceutical Research and Development, LLC, 8 Clarke Drive, Cranbury, NJ 08512, USA
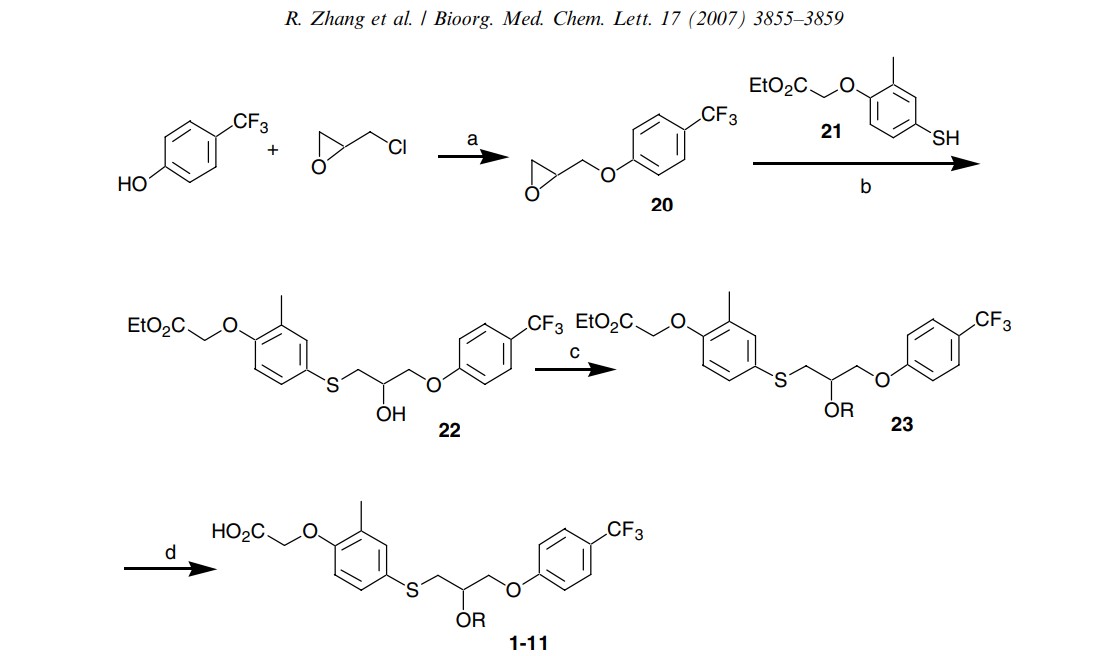
Scheme 1. Reagents and condition: (a) Cs2CO3, dioxane, 100 C 80%; (b) TBAF (cat), THF, 85%; (c) NaH, RI, THF or DMF for esters of 2–5, 8–9, 10–80%; iPr2NEt, RBr or MOMCl, THF for esters of 6–7, 58–79%; ADDP, Ph3P, phenol, CH2Cl2 for esters of 10–11, 68–73%; (d) LiOH, H2O, THF, 90–95%.
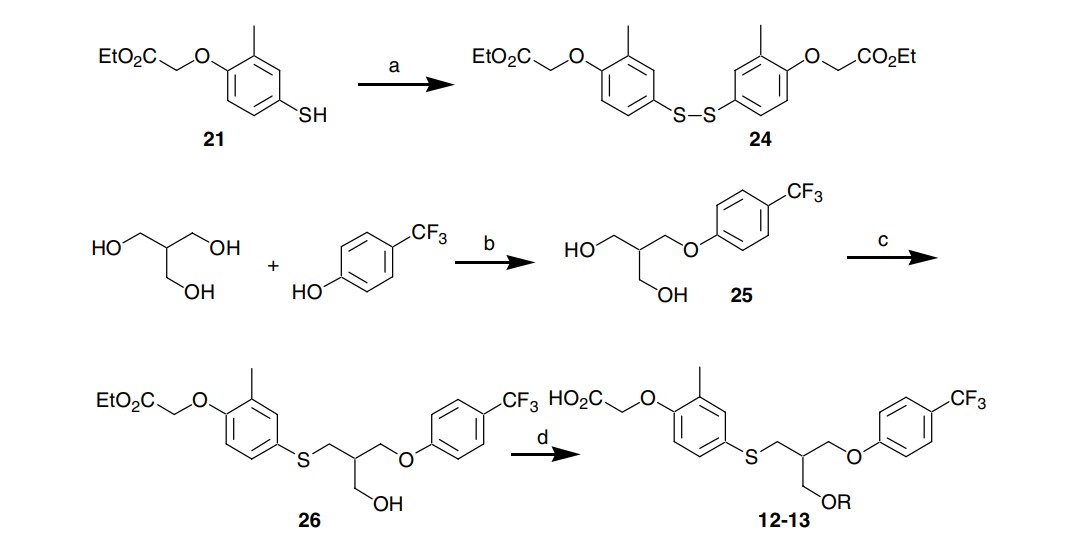
Scheme 2. Reagents: (a) Ba(MnO4)2, CH2Cl2, 89%; (b) DIAD, Ph3P, DMF, THF, 17%; (c) n-Bu3P, 24, Py, 55%; (d) i—NaHMDS, EtOTf, THF for the ethyl ester of 12, 47%; DIAD, Ph3P, para-trifluoromethylphenol for the ethyl ester of 13, 79%; ii—LiOH, H2O, THF, 84–88%.
References
- ^ Jump up to:a b c d e f “Livdelzi- seladelpar lysine capsule”. DailyMed. 14 August 2024. Retrieved 5 September 2024.
- ^ Billin AN (October 2008). “PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home”. Expert Opinion on Investigational Drugs. 17 (10): 1465–1471. doi:10.1517/13543784.17.10.1465. PMID 18808307. S2CID 86564263.
- ^ Bays HE, Schwartz S, Littlejohn T, Kerzner B, Krauss RM, Karpf DB, et al. (September 2011). “MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin”. The Journal of Clinical Endocrinology and Metabolism. 96 (9): 2889–2897. doi:10.1210/jc.2011-1061. PMID 21752880.
- ^ “Targeting Mixed Dyslipidemia and Metabolic Syndrome”. Metabolex, Inc. 2005. Archived from the original on 17 October 2006.
- ^ “Gilead’s Livdelzi (Seladelpar) Granted Accelerated Approval for Primary Biliary Cholangitis by U.S. FDA” (Press release). Gilead. 14 August 2024. Retrieved 15 August 2024 – via Business Wire.
- ^ Kremer AE, Mayo MJ, Hirschfield GM, Levy C, Bowlus CL, Jones DE, et al. (July 2024). “Seladelpar treatment reduces IL-31 and pruritus in patients with primary biliary cholangitis”. Hepatology. 80 (1): 27–37. doi:10.1097/HEP.0000000000000728. PMC 11191048.
| Clinical data | |
|---|---|
| Trade names | Livdelzi |
| Other names | MBX-8025; RWJ-800025 |
| License data | US DailyMed: Seladelpar |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 851528-79-5 |
| PubChem CID | 11236126 |
| DrugBank | DB12390 |
| ChemSpider | 9411171 |
| UNII | 7C00L34NB9 |
| KEGG | D11256 |
| ChEMBL | ChEMBL230158 |
| CompTox Dashboard (EPA) | DTXSID001045332 |
| Chemical and physical data | |
| Formula | C21H23F3O5S |
| Molar mass | 444.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////////////Livdelzi, Seladelpar, (+)-MBX-8025, MBX 8025, MBX-8025, MBX8025, RWJ-800025, FDA 2024, APPROVALS 2024
Zevotrelvir, EDP 235
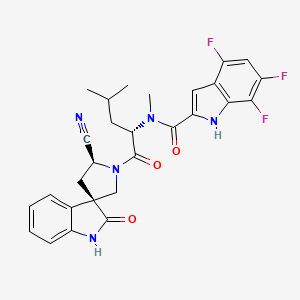
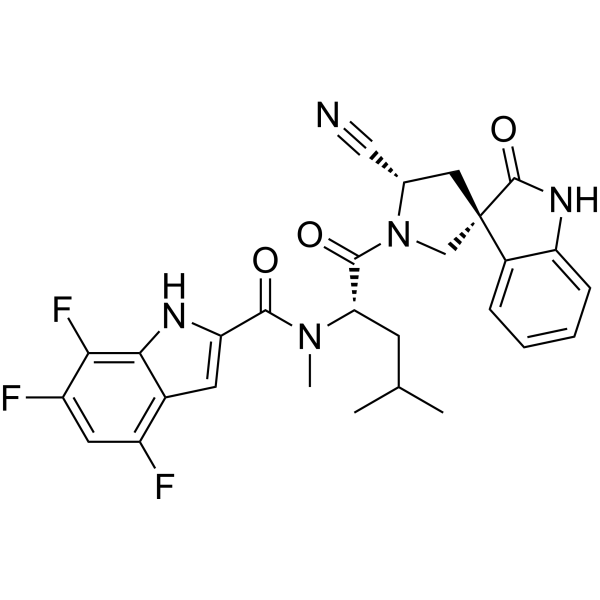
Zevotrelvir, EDP 235
cas 2773516-53-1
N-[(2S)-1-[(2′S,3R)-2′-cyano-2-oxospiro[1H-indole-3,4′-pyrrolidine]-1′-yl]-4-methyl-1-oxopentan-2-yl]-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide
C28H26F3N5O3, 537.5
Zevotrelvir (Compound 52) is a coronavirus inhibitor with IC50 ranges of <0.1 μM and <0.1mM for 229E hCoV and SARS-CoV-23C-like (3CL) proteases, respectively. Zevotrelvir has the potential to study viral infections.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses. The genomic RNA of CoVs has a 5′-cap structure and 3′-poly-A tail and contains at least 6 open reading frames (ORFs). The first ORF (ORF 1a/b) directly translates two polyproteins: pp1a and pp1ab. These polyproteins are processed by a 3C-Like protease (3CLpro), also known as the main protease (Mpro), into 16 non-structural proteins. These non-structural proteins engage in the production of subgenomic RNAs that encode four structural proteins, namely envelope, membrane, spike, and nucleocapsid proteins, among other accessory proteins. As a result, it is understood that 3C-Like protease has a critical role in the coronavirus life cycle.
3CLpro is a cysteine protease involved in most cleavage events within the precursor polyprotein. Active 3CLpro is a homodimer containing two protomers and features a Cys-His dyad located in between domains I and II.3CLpro is conserved among coronaviruses and several common features are shared among the substrates of 3CLpro in different coronaviruses. As there is no human homolog of 3CLpro, it is an ideal antiviral target. Although compounds have been reported to inhibit 3CLpro activity, they have not been approved as coronavirus therapies. (Refer to
WO 2004101742 A2, US 2005/0143320 Al, US 2006/0014821 Al, US 2009/0137818
Al, WO 2013049382 A2, WO 2013166319 A1, WO2018042343, WO2018023054, WO 2022013684, WO 2021252644, WO2022020711, WO 2022020242, US 11,174,231 B1, US 11,124,497 B1, WO 2005113580, and WO2006061714).
There is a need in the art for novel therapeutic agents that treat, ameliorate or prevent SARS-CoV-2 infection. The present invention provides the process of novel compounds which act in inhibiting or preventing SARS-CoV-2 viral replication and thus are used in the treatment of COVID-19 (see PCT/US21/60247).
Synthesis of substituted spirooxindole and its intermediate has been previously published (Refer to PCT/US21/60247, WO2019086142, WO 2020221811, WO2020221826, J. Med. Chem.2012, 55, 9069). However, the scale-up using previous process is very challenging due to the safety concern associated with certain intermediates, instability of certain intermediates as well as lack of purification process other than column chromatograph. Thus, there is a strong need for developing a safe and efficient processes for the large-scale preparation of these novel substituted spirooxindole derivatives.
SYNTHESIS

https://patents.google.com/patent/US11352363B1/en

PATENT
SYN
[1]. Guoqiang Wang, et al. Novel spiropyrrolidine derived antiviral drugs. Patent CN114524821A.
1.20230295175PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
2.WO/2023/177854PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
3.WO/2022/109363NOVEL SPIROPYRROLIDINE DERIVED ANTIVIRAL AGENTS
Enanta Pharmaceuticals, Inc.
WO2023177854




Example 15. Preparation of Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))

DMF (760 kg, 8V) was added into the reaction at 0 °C (-5~5 °C) followed by compound (j) (63 kg, 1.05 eq) and N-Methylmorpholine (56 kg, 2 eq), HATU
(106 kg, 1.0 eq) and Compound (m-1) (100 kg, 1.0 eq). The reactor was rinsed with DMF (190 kg, 2V) under and warmed up to 25 °C (20~30 °C) and stirred for 5 h (3~6 h) at 25 °C (20~30 °C). After that, additional HATU (0.1 eq) was added and the reaction mixture was stirred for 16-24 h.25% Ammonium hydroxide (38 kg) was added to the reaction mixture at 25 °C (20~30 °C) and stirred for 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was then added to water (5000 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (500 kg, 5 V). The cake was dissolved in ethyl acetate (1350 kg, 15 V) and washed with 10% sodium chloride solution (500 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (660 kg, 5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (137 kg, 2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and the wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 16. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF solution of Compound (m-2) (1 kg, 1.0 eq.) was added to a reactor at around 0-10oC. Compound (l) (600 g, 1.0 eq.), NMM (3.00 eq., 850 g) and HATU (1.00 eq., 1.06 kg) was added to the reactor while maintaining the temperature at 0-10oC; The reaction was warmed to 20±5oC, and stirred for at least 6 hours at 20±5oC. HATU (0.20 eq., 210 g) was added to the reactor at 20±5oC and stirred for at least 6 hours at 20±5oC.25% Ammonium hydroxide (390 g, 1.0 eq) was added to the reaction mixture at 20 °C and stirred for 2 h (1~3 h) at 20 °C. EtOAc (14.0 V) and water (14 V) was added at around 25oC over 20 minutes, and the
solution was stirred for at least 30 min. Aqueous phase was extracted with EtOAc for three times and the organic phase was combined, and washed with 10% aq. NaCl for three times at 20±5oC. The organic phase was concentrated to 6 V then EtOH (7.0 V) was charged. The EtOAc-EtOH solvent swap was repeated for three times and concentrated to 5 V before water (7.0 v) was added at 20±5oC. The mixture was cooled to 0-10oC and stirred for 1 h before being filtered. The filter cake was dissolved in ethyl acetate (15 V) and washed with 10% sodium chloride solution for three times. The organic layer was concentrated to 2-3V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in about 70-75% yield over two steps.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 17. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (10.0 v) was added to a reactor at 25 °C followed by Compound (l) (4.4 kg, 1.0 eq.), NMM (3.0 eq.) Compound (m-3) (1.0 eq.) and HATU (1.0 eq) at 20-25oC. The reaction mixture was stirred for at least 12 hours at 20-25 °C. Once reaction was complete, aqueous ammonium hydroxide (1.0 eq.) was to the reaction system at 20-25 °C, then stirred for at least 2 hours at 20-25oC. The reaction mixture was then added to water (220 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (22 kg, 5 V). The cake was dissolved in ethyl acetate (135 g, 15 V) and washed with 10% sodium chloride solution (22 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45 ℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H). Example 18. Preparation of N-((S)-1-((3R,5’S)-5′-cyano-2-oxospiro[indoline-3,3′-pyrrolidin]-1′-yl)-4-methyl-1-oxopentan-2-yl)-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide toluene solvate (Compound (I))
(I))

Ethyl acetate (630 kg, 10 V) was added into reactor (R1) followed by Compound (n) (70 kg). Make sure the water content was less than 0.20% (w/w). The reaction was cooled to 0 °C (-5 – 5°C) and then triethylamine (89.6 kg) was added followed by trifluoroacetic anhydride (92.4 kg) at 0 °C (-5 – 5°C). The reaction was stirred for 1 h (0.5~2 h) at 0 °C (-5 – 5°C). Once the reaction was complete, the reaction mixture was added slowly to 0.2 N aqueous HCl solution (700 kg) over 1 h at 0 °C (-5~5 °C). The resulting solution was stirred for 30 min at 0 °C (-5~5 °C) and the organic layer was separated.1% aqueous ammonium hydroxide (700 kg) was added to the organic layer and stirred at 20 °C for 30 min (15~25 °C). The organic layer was separated and washed with 10% brine for four times. Then the organic layer was separated and distilled to 2-3 V. Toluene-EtOAc swap was performed until precipitate was observed at 3-4 V. Then Toluene (5-6 V) was added and the slurry was stirred at 50 oC for 2 h. Then the solution was cooled down to 20 oC over 1-2 h and stirred for 10 hr (6~14 hr) at 20 °C (15~25 °C). The reaction mixture was filtered and the wet cake was rinsed with toluene (120 kg, 2V). The wet cake was then dried at 50˚C (45~55 °C) for 48 hr to provide desired compound (o) as a white solid in 80-85% yield.
1H NMR (400 MHz, Acetone-d6) δ 11.17 (s, 1H), 9.65 (s, 1H), 7.02 (dd, J = 13.7, 7.3 Hz, 2H), 6.94 (dd, J = 6.0, 3.5 Hz, 1H), 6.92 – 6.85 (m, 2H), 6.81 (t, J = 7.5 Hz, 1H), 5.56 (dd, J = 9.4, 5.6 Hz, 1H), 5.21 (t, J = 8.3 Hz, 1H), 4.25 (d, J = 10.7 Hz, 1H), 3.99 (d, J = 10.6 Hz, 1H), 3.43 (s, 3H), 2.79 – 2.61 (m, 2H), 1.93 (ddd, J = 14.4, 9.5, 5.1 Hz, 1H), 1.79 (ddd, J = 14.2, 8.7, 5.6 Hz, 1H), 1.64 (dpd, J = 8.7, 6.6, 5.1 Hz, 1H), 0.98 (dd, J = 18.5, 6.6 Hz, 6H).
US20230103494
CN114524821
SCHEME

MAIN

////////Zevotrelvir, EDP 235
O=C1[C@@]2(CN([C@@H](C2)C#N)C([C@H](CC(C)C)N(C)C(C3=CC4=C(F)C=C(F)C(F)=C4N3)=O)=O)C5=CC=CC=C5N1
Afimetoran
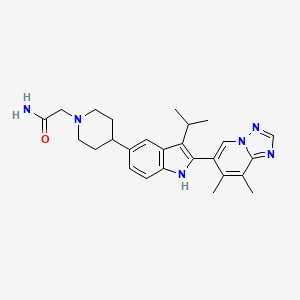

Afimetoran BMS-986256, WHO 11516
cas 2171019-55-7
2-[4-[2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-propan-2-yl-1H-indol-5-yl]piperidin-1-yl]acetamide
C26H32N6O,444.583, phase 1
Afimetoran is an immunomodulator and an antagonist of toll-like receptors 7 and 8.1,2 It is also is under investigation in clinical trial NCT04269356 (Study to Assess the Way the Body Absorbs, Distributes, Breaks Down and Eliminates Radioactive BMS-986256 in Healthy Male Participants).

Ref
WO2018005586
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018005586&_cid=P20-M0RQ0D-09010-1
The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to TLR modulation, such as inflammatory and autoimmune diseases, and methods of inhibiting the activity of TLRs in a mammal.
Toll/IL-1 receptor family members are important regulators of inflammation and host resistance. The Toll-like receptor family recognizes molecular patterns derived from infectious organisms including bacteria, fungi, parasites, and viruses (reviewed in Kawai, T. et al., Nature Immunol., 11:373-384 (2010)). Ligand binding to the receptor induces dimerization and recruitment of adaptor molecules to a conserved cytoplasmic motif in the receptor termed the Toll/IL-1 receptor (TIR) domain. With the exception of TLR3, all TLRs recruit the adaptor molecule MyD88. The IL-1 receptor family also contains a cytoplasmic TIR motif and recruits MyD88 upon ligand binding (reviewed in Sims, J.E. et al., Nature Rev. Immunol., 10:89-102 (2010)).
Toll-like receptors (TLRs) are a family of evolutionarily conserved, transmembrane innate immune receptors that participate in the first-line defense. As pattern recognition receptors, the TLRs protect against foreign molecules, activated by pathogen associated molecular patterns (PAMPs), or from damaged tissue, activated by danger associated molecular patterns (DAMPs). A total of 13 TLR family members have been identified, 10 in human, that span either the cell surface or the endosomal compartment. TLR7-9 are among the set that are endosomally located and respond to single-stranded RNA (TLR7and TLR8) or unmethylated single-stranded DNA containing cytosine-phosphate-guanine (CpG) motifs (TLR9).
Activation of TLR7/8/9 can initiate a variety of inflammatory responses (cytokine production, B cell activation and IgG production, Type I interferon response). In the case of autoimmune disorders, the aberrant sustained activation of TLR7/8/9 leads to worsening of disease states. Whereas overexpression of TLR7 in mice has been shown to exacerbate autoimmune disease, knockout of TLR7 in mice was found to be protective against disease in lupus-prone MRL/lpr mice. Dual knockout of TLR7 and 9 showed further enhanced protection.
As numerous conditions may benefit by treatment involving modulation of cytokines, IFN production and B cell activity, it is immediately apparent that new compounds capable of modulating TLR7 and/or TLR8 and/or TLR9 and methods of using these compounds could provide substantial therapeutic benefits to a wide variety of patients.
The present invention relates to a new class of [1,2,4]triazolo[1,5-a]pyridinyl substituted indole compounds found to be effective inhibitors of signaling through TLR7/8/9. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
EXAMPLE 15
2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-isopropyl-1H-indol-5-yl) piperidin-1-yl)acetamide
To a reaction flask were added
6-(3-isopropyl-5-(piperidin-4-yl)-1H-indol-2-yl)-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyrid ine, 2 HCl (47.66 g, 104 mmol), DCE (220 mL), DBU (62.4 mL, 414 mmol), and 2-bromoacetamide (17.14 g, 124 mmol). The reaction flask was capped. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, diluted with water, and stirred for 30 minutes then filtered. The solid was recrystallized using ethanol to afford 2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a] pyridin-6-yl)-3-isopropyl-1H-indol-5-yl)piperidin-1-yl)acetamide (42.3 g, 93 mmol,
90% yield) as a white solid. LCMS MH+: 445. HPLC Ret. Time 1.20 min. Method QC-ACN-TFA-XB. 1HNMR (400 MHz, DMSO-d6) δ 10.97-10.86 (m, 1H), 8.78-8.69 (m, 1H), 8.54-8.40 (m, 1H), 7.64-7.49 (m, 1H), 7.30-7.21 (m, 2H), 7.17-7.09 (m, 1H), 7.06-6.93 (m, 1H), 2.99-2.82 (m, 5H), 2.62-2.54 (m, 4H), 2.24-2.12 (m, 5H), 1.92-1.72 (m, 4H), 1.37-1.29 (m, 6H).
ACS Medicinal Chemistry Letters (2022), 13(5), 812-818 83%
References
- Bristol-Myers Squibb: Investor Series [Link]
- Bristol-Myers Squibb: Investor Series [Link]
- MedKoo Biosciences: Afimetoran [Link]
//////////////Afimetoran, BMS-986256, BMS 986256, WHO 11516, phase 1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}