
Home » 2021 (Page 22)
Yearly Archives: 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ROLUPERIDONE
- Molecular FormulaC22H23FN2O2
- Average mass366.429 Da
Roluperidone
CAS 359625-79-9
1937215-88-7 hcl
ролуперидон [Russian] [INN]
رولوبيريدون [Arabic] [INN]
罗鲁哌酮 [Chinese] [INN]
1H-Isoindol-1-one, 2-[[1-[2-(4-fluorophenyl)-2-oxoethyl]-4-piperidinyl]methyl]-2,3-dihydro-2-({1-[2-(4-Fluorophenyl)-2-oxoethyl]-4-piperidinyl}methyl)-1-isoindolinone
2-[[1- [2–fluorophenyl) -2-oxotyl] piperidine –4-yl] methyl] isoindrin-hydrochloride
CYR-101
UNII-4P31I0M3BF
MIN-101
SYN

Roluperidone (former developmental code names MIN-101, CYR-101, MT-210) is a 5-HT2A and σ2 receptor antagonist that is under development by Minerva Neurosciences for the treatment of schizophrenia.[1][2][3][4] One of its metabolites also has some affinity for the H1 receptor.[2] As of May 2018, the drug is in phase III clinical trials.[5]
Minerva Neurosciences (following the merger of Cyrenaic and Sonkei Pharmaceuticals ), under license from Mitsubishi Tanabe Pharma , is developing roluperidone (MIN-101, CYR-101, MT-210), a dual 5-HT2A /sigma 2 antagonist, as a modified-release formulation, for the potential oral treatment of schizophrenia. In December 2017, a phase III trial was initiated in patients with negative symptoms of schizophrenia. By March 2020, Minerva had filed an IND for apathy in dementia.
Schizophrenia is a complex, challenging, and heterogeneous psychiatric condition, affecting up to 0.7% of the world population according to the World Health Organization (WHO, 2006). Patients suffering with schizophrenia present with a range of symptoms, including: positive symptoms, such as delusions, hallucinations, thought disorders, and agitation; negative symptoms, such as mood flatness and lack of pleasure in daily life; cognitive symptoms, such as the decreased ability to understand information and make decisions, difficulty focusing, and decreased working memory function; and sleep disorders.
The etiology of schizophrenia is not fully understood. A major explanatory hypothesis for the pathophysiology of schizophrenia is the Dopamine (DA) hypothesis, which proposes that hyperactivity of DA transmission is responsible for expressed symptoms of the disorder. This hypothesis is based on the observation that drugs effective in treating schizophrenia share the common feature of blocking DA D2 receptors. However, these so-called typical antipsychotics are associated with a very high incidence of extrapyramidal symptoms (EPS). Furthermore, negative symptoms and cognitive impairment are considered relatively unresponsive to typical antipsychotics.
Most currently approved therapies for schizophrenia show efficacy primarily in the management of positive symptoms. An estimated 4.2 million people suffered from schizophrenia in 2012 in the United States and the five major European Union markets. Of those, an estimated 48% experienced predominantly negative symptoms and 80% suffered from cognitive impairment. In addition, about 50% of patients with schizophrenia experience sleep disorders, which can further exacerbate both positive and negative symptoms.
The introduction of the so-called atypical antipsychotics in the last decade represented a significant advance in the treatment of schizophrenia. Although these atypical antipsychotics differ widely in chemical structure and receptor-binding profiles, they share a characteristic of potent antagonism of the Serotonin (5-hydroxytryptamine) type 2 receptor (5-HT2A). A high 5-HT2A:D2 affinity ratio is thought to substantially reduce the liability for inducing EPS, compared with typical antipsychotics.
However, many patients are still treatment-noncompliant despite the advantage of atypical antipsychotics of tolerability. Although the risk of EPS is clearly lower with the atypical antipsychotics, the high doses required with some atypical antipsychotics are likely to result in an increased incidence of EPS and require concomitant medications such as antiparkinson drugs.
In addition to EPS, antipsychotic medications cause a broad spectrum of side effects including sedation, anticholinergic effects, prolactin elevation, orthostatic hypotension, weight gain, altered glucose metabolism, and QTc prolongation. These side effects can affect patients’ compliance with their treatment regimen. It should be noted that noncompliance with treatment regimen is a primary reason for relapse of the disease.
Although atypical antipsychotics offer advantages over typical antipsychotics in terms of symptom alleviation and side effect profile, these differences are generally modest. A certain population of patients still remains refractory to all currently available antipsychotics. Newer agents to address these issues continue to be sought.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Roluperidone hydrochloride | WFL7TF8DTP | 1937215-88-7 | NZKANSJXJCILHS-UHFFFAOYSA-N |
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001064670
Example 1: 2-[[1- [2–fluorophenyl) -2-oxotyl] piperidine –4-yl] methyl] isoindrin-hydrochloride (Compound 1 in Table 1)
a) tert-Butyl 4-aminomethylpiperidine-carpoxylate hydrochloride’salt
4-Aminomethylpiperidin 5. 71g as a starting material
Tert-Butyl 4-aminomethylbiperidine-power reportage was synthesized according to the method described in Synthetic Commun., 22 (16), 2357-2360 (1992). This compound was dissolved in 80 ml of ethyl acetate, 4N ethyl monoacetate hydrochloride was added, and the mixture was stirred. Precipitated solid
Was collected to obtain 10.27 g (yield 82%) of the indicated compound. At melting point 236-240.
Ή-NMR (DMS0-d 6 ): 8.00 (3H, s), 3. 92 (2H, br d, J = 12.6), 2.68 H, m), 1.77- 1. 65 (3H, m), 1.39 (9H, s), 1.02 (2H, m) b) 2-Bromomethylbenzoic acid etyl ester
2-Methylbenzoic acid etyl ester (2.00 g, 11.9 mmol) is dissolved in carbon tetrachloride (60 ml), and N-promosucciimide (2.56 g, 14.4 mmo 1) and a catalytic amount of benzoyl peroxide are added to the solution. In addition, heat reflux. After 1 hour, the reaction mixture was cooled to room temperature, hexan (40 m was added, the insoluble material was filtered off, and the filtrate was distilled off under reduced pressure to obtain 3.16 g of the indicated compound as a yellow oil. It was used for the next reaction without purification as it was.
c) tert-Butyl 4- (1-oxoisoindrin-2 -ylmethyl) piperidine-1 -carpoxylate
Add 3.15 g of the compound obtained in Example lb and the compound (3.00 g, 12. Ommol) obtained in Example la to dimethylformamide (30πΠ), and stir at room temperature with trietylamine (3.5 ml, 25 mmol). ) Is added and stirred at the same temperature for 17 hours. Water is added to the reaction mixture, and the mixture is extracted with a mixed solvent of etyl hexane vinegar. The organic layer is washed with 10% aqueous quenic acid solution, water, sodium bicarbonate solution, and saturated brine, and dried with magnesium sulfate. The insoluble material was filtered, the filtrate was distilled off under reduced pressure, and the obtained oil was purified by silicon gel column chromatography (etyl-hexan acetate). I got it as a thing.
Ή-NMR (CDC1 3 ): 7.85 (1H, d, J = 7.5), 7.4-7.6 (3Η, m),
4.41 (2H, s), 4.0-4.2 (2H, m), 3.4-3.6 (2H, m), 2.6-2.8 (2H, m), 1.8-2.0 (1H, m), 1.5 -1.7 (4H, m), to 45 (9H, s)
d) 2- (Piperidine -4 -Ilmethyl) Isondrin -1 -On Hydrochloride
The compound (1.6 lg, 4.87 mmol) obtained in Example 1c is dissolved in methylene chloride (5 ml) and ethanol (lm mixed solvent, and at room temperature, 4 standard ethyl acetate solvent (5 ml, 20 mmol) is added. Stir at warm temperature for 1 hour and filter the precipitated solid. The obtained solid was washed with ethanol acetate and then dried under reduced pressure to give the indicated compound 7260 ^ (yield 56%) as a colorless solid. ..
Ή-NMR (DMS0-d 6 ): 8. 83 (1H, brs), 8. 53 (1H, brs), 7. 4-7. 7 (4 Η, m), 4. 50 (2H, s), 3. 44 (2H, d, J = 7.2), 3. 2-3. 3 (2H, i), 2. 7-2.9 (2H, m), 1. 9-2.1 (1H) , m), 1. 6-1. 8 (2H, m), 1. 3-1. 5 (2H, m)
e) 2- [Π_ [2- (4-Fluo-mouth phenyl) -2-oxotil] Piperidin –4-yl] Methyl] Isoindrin-卜 on
Add the compounds obtained in Example Id (518 mg, 1. 94 mmo and 2-cloucet -4, -fluoroacetophenone (358 mg, 2.07 mmol) to dimethylform amamide (12 ml) with stirring at room temperature. Add trietylamine (575 1, 4. 13 mmol). After stirring at the same temperature for 4 hours, add water to the reaction solution and extract with ethyl acetate. The organic layer is washed with water and saturated saline and sodium sulfate. Dry with thorium. Filter the insoluble material and concentrate the filtrate under reduced pressure to obtain 0.70 g of orange oil. Add hexane to the obtained oil to solidify. Filter this. By drying under reduced pressure, 551 mg (yield 77%) of the notation compound was obtained as a pale yellow solid.
! H-NMR (CDC1 3 ): 8.0-8 . 1 (2H, m), 7. 85 (1H, d = 7.2), 7.4-7. 55 (3 Η, m), 7.1 2 ( 2H, t), 4. 41 (2H, s), 3. 73 (2H, s), 3.51 (2H, d, J = 7.5), 2. 9-3. 0 (2H, m) , 2. 1-2. 2 (2H, m), 1. 4-19.9 (5H, m)
f) 2- [Π- [2- (4 -Fluolophenyl) -2 -Oxoetyl] Piperidin –4-yl] Methyl] Isoindoline-Piol hydrochloride
The compound (550 mg, 1.5 Ommo 1) obtained in Example le was used as an etano.
Dissolve in (2 ml) and add 4 specified ethyl hydrochloride solvent (2 ml, 8 imol) at room temperature and stir at the same temperature for 15 minutes. Ethyl acetate (10 ml) is added to the reaction solution, and the precipitated solid is filtered. The obtained solid is washed with ethyl acetate and then dried under reduced pressure to obtain 364 mg of white powder. This was recrystallized from ethanol monoacetate to give 246 mg (yield 41%) of the notation compound as a colorless solid. At melting point 182-188.
Ή-NMR (DMS0-d 6 ): 9.93 (1H, brs), 8.0-8. 2 (2H, m), 7.4-7.7 (6 Η, m), 4. 9-5.1 (2H, m), 4.53 (2H, s), 2.9-3.6 (6H, m), 1.6-2.2 (5H, m)
PATENT
https://patents.google.com/patent/US7166617B2/en
Example 12-[[1-[2-(4-Fluorophenyl)-2-oxoethyl]piperidin-4-yl]methyl]isoindolin-1-one hydrochloride (Compound 1 in Table 1)a) tert-Butyl 4-aminomethylpiperidine-1-carboxylate hydrochloride
By using 4-aminomethylpiperidine 5.71 g as a starting material, tert-butyl 4-aminomethylpiperidine-1-carboxylate was prepared according to the method described in Synthetic Commun., 22(16), 2357–2360 (1992). The resulting compound was dissolved in 80 ml of ethyl acetate, and the solution was added with 4N hydrogen chloride-ethyl acetate and stirred. The precipitated solids were collected by filtration to obtain the title compound (10.27 g, yield: 82%).
Melting point: 236–240° C. 1H-NMR(DMSO-d6): 8.00(3H,s), 3.92(2H, br d, J=12.6), 2.68(4H, m), 1.77–1.65(3H, m), 1.39(9H, s), 1.02(2H, m)
b) 2-Bromomethylbenzoic acid ethyl ester
2-Methylbenzoic acid ethyl ester (2.00 g, 11.9 mmol) was dissolved in carbon tetrachloride (60 ml), and the solution was added with N-bromosuccinimide (2.56 g, 14.4 mmol) and a catalytic amount of benzoylperoxide and then heated under reflux. After one hour, the reaction mixture was cooled to room temperature and added with hexane (40 ml) to remove insoluble solids by filtration. The filtrate was evaporated under reduced pressure to obtain the title compound 3.16 g as yellow oil. the product was used in the next reaction without purification.
c) tert-Butyl 4-(1-oxoisoindolin-2-yl-methyl)piperidine-1-carboxylate
The compound obtained in Example 1b (3.15 g), and the compound obtained in Example 1a (3.00 g, 12.0 mmol) were added in dimethylformamide (30 ml). The mixture was added with triethylamine (3.5 ml, 25 mmol) with stirring at room temperature, and then stirring was continued for 17 hours at the same temperature. Water was added to the reaction mixture and extracted with a mixed solvent of ethyl acetate-hexane. The organic layer was washed with 10% aqueous citric acid solution, water, aqueous sodium bicarbonate solution, and then with saturated brine and the dried over magnesium sulfate. Insoluble solids were removed by filtration, and the filtrate was evaporated under reduced pressure. The resulting oil was purified by silica gel column chromatography (ethyl acetate-hexane) to obtain the title compound as yellow oil (yield: 41%)
1H-NMR(CDCl3): 7.85(1H,d,J=7.5), 7.4–7.6(3H,m), 4.41(2H,s), 4.0–4.2(2H,m), 3.4–3.6(2H,m), 2.6–2.8(2H,m), 1.8–2.0(1H,m), 1.5–1.7(4H,m), 1.45(9H,s)
d) 2-(Piperidin-4-yl-methyl)isoindolin-1-one hydrochloride
The compound obtained in Example 1c (1.61 g, 4.87 mmol) was dissolved in a mixed solvent of methylene chloride (5 ml) and ethanol (1 ml) and the solution was added with 4N hydrochloric acid in ethyl acetate (5 ml, 20 mmol) at room temperature. The mixture was stirred at the same temperature for 1 hour, and the precipitated solids were collected by filtration. The resulting solids were washed with ethyl acetate and then dried under reduced pressure to obtain the title compound as colorless solid (726 mg, yield: 56%).
1H-NMR(DMSO-d6): 8.83(1H,brs), 8.53(1H,brs), 7.4–7.7(4H,m), 4.50(2H,s), 3.44(2H,d,J=7.2), 3.2–3.3(2H,m), 2.7–2.9(2H,m), 1.9–2.1(1H,m), 1.6–1.8(2H,m), 1.3–1.5(2H,m)
e) 2-[[1-[2-(4-Fluorophenyl)-2-oxoethyl]piperidin-4-yl]methyl]isoindolin-1-one
The compound obtained in Example 1d (518 mg, 1.94 mmol) and 2-chloro-4′-fluoroacetophenone (358 mg, 2.07 mmol) was added to dimethylformamide (12 ml), and the solution was added with triethylamine (575 μl, 4.13 mmol) with stirring at room temperature. Stirring was continued at the same temperature for 4 hours, and then the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with water and then with saturated brine, and then dried over sodium sulfate. Insoluble solids were removed by filtration and the filtrate was evaporated under reduced pressure to obtain orange oil (0.70 g). The resulting oil was solidified by adding hexane, and the solids were collected by filtration and dried under reduced pressure to obtain the title compound as pale yellow solid (551 mg, yield: 77%).
1H-NMR(CDCl3): 8.0–8.1(2H,m), 7.85(1H,d=7.2), 7.4–7.55(3H,m), 7.12(2H,t), 4.41(2H,s), 3.73(2H,s), 3.51(2H,d,J=7.5), 2.9–3.0(2H,m), 2.1–2.2(2H,m), 1.4–1.9(5H,m)
f) 2-[[1-[2-(4-Fluorophenyl)-2-oxoethyl]piperidin-4-yl]methyl]isoindolin-1-one hydrochloride
The compound obtained in Example 1e (550 mg, 1.50 mmol) was dissolved in ethanol (2 ml), and the solution was added with 4N hydrochloric acid in ethyl acetate (2 ml, 8 mmol) at room temperature, and stirring was continued at the same temperature for 15 minutes. The reaction mixture was added with ethyl acetate (10 ml) and the precipitated solids were collected by filtration. The resulting solids were washed with ethyl acetate and then dried under reduced pressure to obtain white powder (364 mg). The product was recrystallized from ethanol-ethyl acetate to obtain the title compound as colorless solid (246 mg, yield: 41%)
Melting point: 182–188° C. 1H-NMR(DMSO-d6): 9.93(1H,brs), 8.0–8.2(2H,m), 7.4–7.7(6H,m), 4.9–5.1(2H,m), 4.53(2H,s), 2.9–3.6(6H,m), 1.6–2.2(5H, m)
PATENT
https://patents.google.com/patent/US9458130B2/en?oq=9%2c458%2c130+US
PATENT
WO-2020264486
Novel crystalline form of roluperidone HCL (designated as form 4) as 5-HT 2a receptor antagonist useful for treating schizophrenia.
Roluperidone has the chemical name 2-({ l-[2-(4-Fluorophenyl)-2-oxoethyl]-4-piperidinyl}methyl)-l-isoindolinone. Roluperidone has the following chemical structure:
[0003] Roluperidone is reported to be a drug candidate with equipotent affinities for 5-hydroxytryptamine-2A (5-HT2A) and sigma2 and, at lower affinity levels, al -adrenergic receptors. A pivotal Phase 3 clinical trial is ongoing with roluperidone as a monotherapy for negative symptoms in patients diagnosed with schizophrenia.
[0004] Roluperidone is known from U.S. Patent No. 7,166,617.
[0005] Solid state form of 2-((l-(2-(4-Fluorophenyl)-2-oxoethyl)piperidin-4-yl)methyl)isoindolin-l-o-ne monohydrochloride dihydrate is known from U.S. Patent No.9,458,130.
Examples
[00113] Roluperidone can be prepared according to the procedure described in U.S. Patent No. 7,166,617.
Example 1: Preparation of Roluperidone HC1
[00114] 2.02 grams of Roluperidone was dissolved in acetone (80 mL). 2.76 mL of HC1 (2M) was added to the solution. The obtained suspension was stirred for 21 hours at 10°C and then filtered over black ribbon filter paper under vacuum. Obtained solid was analyzed by PXRD.
References
- ^ Mestre TA, Zurowski M, Fox SH (April 2013). “5-Hydroxytryptamine 2A receptor antagonists as potential treatment for psychiatric disorders”. Expert Opinion on Investigational Drugs. 22 (4): 411–21. doi:10.1517/13543784.2013.769957. PMID 23409724.
- ^ Jump up to:a b Ebdrup BH, Rasmussen H, Arnt J, Glenthøj B (September 2011). “Serotonin 2A receptor antagonists for treatment of schizophrenia”. Expert Opinion on Investigational Drugs. 20 (9): 1211–23. doi:10.1517/13543784.2011.601738. PMID 21740279.
- ^ Köster LS, Carbon M, Correll CU (December 2014). “Emerging drugs for schizophrenia: an update”. Expert Opinion on Emerging Drugs. 19 (4): 511–31. doi:10.1517/14728214.2014.958148. PMID 25234340.
- ^ “Drug Development in Schizophrenia: Summary and Table”. Pharmaceutical Medicine. 28 (5): 265–271. 2014. doi:10.1007/s40290-014-0070-6. ISSN 1178-2595.
- ^ “Roluperidone – Minerva Neurosciences”. Adis Insight. Springer Nature Switzerland AG.
| Clinical data | |
|---|---|
| Other names | MIN-101; CYR-101; MT-210 |
| Routes of administration | By mouth |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 359625-79-9 |
| PubChemCID | 9799284 |
| DrugBank | DB13080 |
| ChemSpider | 7975049 |
| UNII | 4P31I0M3BF |
| KEGG | D11258 |
| CompTox Dashboard (EPA) | DTXSID10189512 |
| Chemical and physical data | |
| Formula | C22H23F2N2O2 |
| Molar mass | 385.435 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[show] | |
| InChI[show] |
/////////////////Roluperidone, PHASE 3, ролуперидон , رولوبيريدون , 罗鲁哌酮 , CYR 101, UNII-4P31I0M3BF , MIN 101,
C1CN(CCC1CN2CC3=CC=CC=C3C2=O)CC(=O)C4=CC=C(C=C4)F
GLUCAGON
glucagon
EMA……Ogluo (glucagon), a hybrid medicine for the treatment of severe hypoglycaemia in diabetes mellitus. Hybrid applications rely in part on the results of pre-clinical tests and clinical trials of an already authorised reference product and in part on new data.
On 10 December 2020, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product Ogluo, intended for the treatment of severe hypoglycaemia in diabetes mellitus. The applicant for this medicinal product is Xeris Pharmaceuticals Ireland Limited.
Ogluo will be available as 0.5 and 1 mg solution for injection. The active substance of Ogluo is glucagon, a pancreatic hormone (ATC code: H04AA01); glucagon increases blood glucose concentration by stimulating glycogen breakdown and release of glucose from the liver.
The benefits with Ogluo are its ability to restore blood glucose levels in hypoglycaemic subjects. The most common side effects are nausea and vomiting.
Ogluo is a hybrid medicine1 of GlucaGen/GlucaGen Hypokit; GlucaGen has been authorised in the EU since October 1962. Ogluo contains the same active substance as GlucaGen but is available as a ready-to-use formulation intended for subcutaneous injection.
The full indication is:
Ogluo is indicated for the treatment of severe hypoglycaemia in adults, adolescents, and children aged 2 years and over with diabetes mellitus.
Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
1 Hybrid applications rely in part on the results of pre-clinical tests and clinical trials for a reference product and in part on new data.
Glucagon is a peptide hormone, produced by alpha cells of the pancreas. It works to raise the concentration of glucose and fatty acids in the bloodstream, and is considered to be the main catabolic hormone of the body.[3] It is also used as a medication to treat a number of health conditions. Its effect is opposite to that of insulin, which lowers extracellular glucose.[4] It is produced from proglucagon, encoded by the GCG gene.
The pancreas releases glucagon when the amount of glucose in the bloodstream is too low. Glucagon causes the liver to engage in glycogenolysis: converting stored glycogen into glucose, which is released into the bloodstream.[5] High blood-glucose levels, on the other hand, stimulate the release of insulin. Insulin allows glucose to be taken up and used by insulin-dependent tissues. Thus, glucagon and insulin are part of a feedback system that keeps blood glucose levels stable. Glucagon increases energy expenditure and is elevated under conditions of stress.[6] Glucagon belongs to the secretin family of hormones.
Function
Glucagon generally elevates the concentration of glucose in the blood by promoting gluconeogenesis and glycogenolysis.[7] Glucagon also decreases fatty acid synthesis in adipose tissue and the liver, as well as promoting lipolysis in these tissues, which causes them to release fatty acids into circulation where they can be catabolised to generate energy in tissues such as skeletal muscle when required.[8]
Glucose is stored in the liver in the form of the polysaccharide glycogen, which is a glucan (a polymer made up of glucose molecules). Liver cells (hepatocytes) have glucagon receptors. When glucagon binds to the glucagon receptors, the liver cells convert the glycogen into individual glucose molecules and release them into the bloodstream, in a process known as glycogenolysis. As these stores become depleted, glucagon then encourages the liver and kidney to synthesize additional glucose by gluconeogenesis. Glucagon turns off glycolysis in the liver, causing glycolytic intermediates to be shuttled to gluconeogenesis.
Glucagon also regulates the rate of glucose production through lipolysis. Glucagon induces lipolysis in humans under conditions of insulin suppression (such as diabetes mellitus type 1).[9]
Glucagon production appears to be dependent on the central nervous system through pathways yet to be defined. In invertebrate animals, eyestalk removal has been reported to affect glucagon production. Excising the eyestalk in young crayfish produces glucagon-induced hyperglycemia.[10]
Mechanism of action
Metabolic regulation of glycogen by glucagon.
Glucagon binds to the glucagon receptor, a G protein-coupled receptor, located in the plasma membrane of the cell. The conformation change in the receptor activates G proteins, a heterotrimeric protein with α, β, and γ subunits. When the G protein interacts with the receptor, it undergoes a conformational change that results in the replacement of the GDP molecule that was bound to the α subunit with a GTP molecule. This substitution results in the releasing of the α subunit from the β and γ subunits. The alpha subunit specifically activates the next enzyme in the cascade, adenylate cyclase.
Adenylate cyclase manufactures cyclic adenosine monophosphate (cyclic AMP or cAMP), which activates protein kinase A (cAMP-dependent protein kinase). This enzyme, in turn, activates phosphorylase kinase, which then phosphorylates glycogen phosphorylase b (PYG b), converting it into the active form called phosphorylase a (PYG a). Phosphorylase a is the enzyme responsible for the release of glucose 1-phosphate from glycogen polymers. An example of the pathway would be when glucagon binds to a transmembrane protein. The transmembrane proteins interacts with Gɑβ𝛾. Gɑ separates from Gβ𝛾 and interacts with the transmembrane protein adenylyl cyclase. Adenylyl cyclase catalyzes the conversion of ATP to cAMP. cAMP binds to protein kinase A, and the complex phosphorylates phosphorylase kinase.[11] Phosphorylated phosphorylase kinase phosphorylates phosphorylase. Phosphorylated phosphorylase clips glucose units from glycogen as glucose 1-phosphate. Additionally, the coordinated control of glycolysis and gluconeogenesis in the liver is adjusted by the phosphorylation state of the enzymes that catalyze the formation of a potent activator of glycolysis called fructose 2,6-bisphosphate.[12] The enzyme protein kinase A (PKA) that was stimulated by the cascade initiated by glucagon will also phosphorylate a single serine residue of the bifunctional polypeptide chain containing both the enzymes fructose 2,6-bisphosphatase and phosphofructokinase-2. This covalent phosphorylation initiated by glucagon activates the former and inhibits the latter. This regulates the reaction catalyzing fructose 2,6-bisphosphate (a potent activator of phosphofructokinase-1, the enzyme that is the primary regulatory step of glycolysis)[13] by slowing the rate of its formation, thereby inhibiting the flux of the glycolysis pathway and allowing gluconeogenesis to predominate. This process is reversible in the absence of glucagon (and thus, the presence of insulin).
Glucagon stimulation of PKA also inactivates the glycolytic enzyme pyruvate kinase in hepatocytes.[14]
Physiology
Production
A microscopic image stained for glucagon
The hormone is synthesized and secreted from alpha cells (α-cells) of the islets of Langerhans, which are located in the endocrine portion of the pancreas. Production, which is otherwise freerunning, is suppressed/regulated by amylin, a peptide hormone co-secreted with insulin from the pancreatic β cells.[15] As plasma glucose levels recede, the subsequent reduction in amylin secretion alleviates its suppression of the α cells, allowing for glucagon secretion.
In rodents, the alpha cells are located in the outer rim of the islet. Human islet structure is much less segregated, and alpha cells are distributed throughout the islet in close proximity to beta cells. Glucagon is also produced by alpha cells in the stomach.[16]
Recent research has demonstrated that glucagon production may also take place outside the pancreas, with the gut being the most likely site of extrapancreatic glucagon synthesis.[17]
Regulation
Secretion of glucagon is stimulated by:
- Hypoglycemia
- Epinephrine (via β2, α2,[18] and α1[19] adrenergic receptors)
- Arginine
- Alanine (often from muscle-derived pyruvate/glutamate transamination (see alanine transaminase reaction).
- Acetylcholine[20]
- Cholecystokinin
- Gastric inhibitory polypeptide
Secretion of glucagon is inhibited by:
- Somatostatin
- Amylin [21]
- Insulin (via GABA)[22]
- PPARγ/retinoid X receptor heterodimer.[23]
- Increased free fatty acids and keto acids into the blood.[24]
- Increased urea production
- Glucagon-like peptide-1
Structure
Glucagon is a 29-amino acid polypeptide. Its primary structure in humans is: NH2–His–Ser–Gln–Gly–Thr–Phe–Thr–Ser–Asp–Tyr–Ser–Lys–Tyr–Leu–Asp–Ser–Arg–Arg–Ala–Gln–Asp–Phe–Val–Gln–Trp–Leu–Met–Asn–Thr–COOH.
The polypeptide has a molecular mass of 3485 daltons.[25] Glucagon is a peptide (nonsteroid) hormone.
Glucagon is generated from the cleavage of proglucagon by proprotein convertase 2 in pancreatic islet α cells. In intestinal L cells, proglucagon is cleaved to the alternate products glicentin, GLP-1 (an incretin), IP-2, and GLP-2 (promotes intestinal growth).[26]
Pathology
Abnormally elevated levels of glucagon may be caused by pancreatic tumors, such as glucagonoma, symptoms of which include necrolytic migratory erythema,[27] reduced amino acids, and hyperglycemia. It may occur alone or in the context of multiple endocrine neoplasia type 1[28]
Elevated glucagon is the main contributor to hyperglycemic ketoacidosis in undiagnosed or poorly treated type 1 diabetes. As the beta cells cease to function, insulin and pancreatic GABA are no longer present to suppress the freerunning output of glucagon. As a result, glucagon is released from the alpha cells at a maximum, causing rapid breakdown of glycogen to glucose and fast ketogenesis.[29] It was found that a subset of adults with type 1 diabetes took 4 times longer on average to approach ketoacidosis when given somatostatin (inhibits glucagon production) with no insulin. Inhibiting glucagon has been a popular idea of diabetes treatment, however some have warned that doing so will give rise to brittle diabetes in patients with adequately stable blood glucose.[citation needed]
The absence of alpha cells (and hence glucagon) is thought to be one of the main influences in the extreme volatility of blood glucose in the setting of a total pancreatectomy.
History
In the 1920s, Kimball and Murlin studied pancreatic extracts, and found an additional substance with hyperglycemic properties. They described glucagon in 1923.[30] The amino acid sequence of glucagon was described in the late 1950s.[31] A more complete understanding of its role in physiology and disease was not established until the 1970s, when a specific radioimmunoassay was developed.[citation needed]
Etymology
Kimball and Murlin coined the term glucagon in 1923 when they initially named the substance the glucose agonist.[32]
References
- ^ Jump up to:a b c GRCh38: Ensembl release 89: ENSG00000115263 – Ensembl, May 2017
- ^ “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Voet D, Voet JG (2011). Biochemistry (4th ed.). New York: Wiley.
- ^ Reece J, Campbell N (2002). Biology. San Francisco: Benjamin Cummings. ISBN 978-0-8053-6624-2.
- ^ Orsay J (2014). Biology 1: Molecules. Examkrackers Inc. p. 77. ISBN 978-1-893858-70-1.
- ^ Jones BJ, Tan T, Bloom SR (March 2012). “Minireview: Glucagon in stress and energy homeostasis”. Endocrinology. 153 (3): 1049–54. doi:10.1210/en.2011-1979. PMC 3281544. PMID 22294753.
- ^ Voet D, Voet JG (2011). Biochemistry (4th ed.). New York: Wiley.
- ^ HABEGGER, K. M., HEPPNER, K. M., GEARY, N., BARTNESS, T. J., DIMARCHI, R. & TSCHÖP, M. H. (2010). “The metabolic actions of glucagon revisited”. Nature Reviews. Endocrinology. 6 (12): 689–697. doi:10.1038/nrendo.2010.187. PMC 3563428. PMID 20957001.
- ^ Liljenquist JE, Bomboy JD, Lewis SB, Sinclair-Smith BC, Felts PW, Lacy WW, Crofford OB, Liddle GW (January 1974). “Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men”. The Journal of Clinical Investigation. 53 (1): 190–7. doi:10.1172/JCI107537. PMC 301453. PMID 4808635.
- ^ Leinen RL, Giannini AJ (1983). “Effect of eyestalk removal on glucagon induced hyperglycemia in crayfish”. Society for Neuroscience Abstracts. 9: 604.
- ^ Yu Q, Shuai H, Ahooghalandari P, Gylfe E, Tengholm A (July 2019). “Glucose controls glucagon secretion by directly modulating cAMP in alpha cells”. Diabetologia. 62 (7): 1212–1224. doi:10.1007/s00125-019-4857-6. PMC 6560012. PMID 30953108.
- ^ Hue L, Rider MH (July 1987). “Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues”. The Biochemical Journal. 245 (2): 313–24. doi:10.1042/bj2450313. PMC 1148124. PMID 2822019.
- ^ Claus TH, El-Maghrabi MR, Regen DM, Stewart HB, McGrane M, Kountz PD, Nyfeler F, Pilkis J, Pilkis SJ (1984). “The role of fructose 2,6-bisphosphate in the regulation of carbohydrate metabolism”. Current Topics in Cellular Regulation. 23: 57–86. doi:10.1016/b978-0-12-152823-2.50006-4. ISBN 9780121528232. PMID 6327193.
- ^ Feliú JE, Hue L, Hers HG (August 1976). “Hormonal control of pyruvate kinase activity and of gluconeogenesis in isolated hepatocytes”. Proceedings of the National Academy of Sciences of the United States of America. 73 (8): 2762–6. Bibcode:1976PNAS…73.2762F. doi:10.1073/pnas.73.8.2762. PMC 430732. PMID 183209.
- ^ Zhang, Xiao-Xi (2016). “Neuroendocrine Hormone Amylin in Diabetes”. World J Diabetes. 7 (9): 189–197. doi:10.4239/wjd.v7.i9.189. PMC 4856891. PMID 27162583.
- ^ Unger RH, Cherrington AD (January 2012). “Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover”. The Journal of Clinical Investigation. 122(1): 4–12. doi:10.1172/JCI60016. PMC 3248306. PMID 22214853.
- ^ Holst JJ, Holland W, Gromada J, Lee Y, Unger RH, Yan H, Sloop KW, Kieffer TJ, Damond N, Herrera PL (April 2017). “Insulin and Glucagon: Partners for Life”. Endocrinology. 158(4): 696–701. doi:10.1210/en.2016-1748. PMC 6061217. PMID 28323959.
- ^ Layden BT, Durai V, Lowe WL (2010). “G-Protein-Coupled Receptors, Pancreatic Islets, and Diabetes”. Nature Education. 3 (9): 13.
- ^ Skoglund G, Lundquist I, Ahrén B (November 1987). “Alpha 1- and alpha 2-adrenoceptor activation increases plasma glucagon levels in the mouse”. European Journal of Pharmacology. 143 (1): 83–8. doi:10.1016/0014-2999(87)90737-0. PMID 2891547.
- ^ Honey RN, Weir GC (October 1980). “Acetylcholine stimulates insulin, glucagon, and somatostatin release in the perfused chicken pancreas”. Endocrinology. 107 (4): 1065–8. doi:10.1210/endo-107-4-1065. PMID 6105951.
- ^ Zhang, Xiao-Xi (2016). “Neuroendocrine Hormone Amylin in Diabetes”. World J Diabetes. 7 (9): 189–197. doi:10.4239/wjd.v7.i9.189. PMC 4856891. PMID 27162583.
- ^ Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, Liu S, Wendt A, Deng S, Ebina Y, Wheeler MB, Braun M, Wang Q (January 2006). “Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system”. Cell Metabolism. 3 (1): 47–58. doi:10.1016/j.cmet.2005.11.015. PMID 16399504.
- ^ Krätzner R, Fröhlich F, Lepler K, Schröder M, Röher K, Dickel C, Tzvetkov MV, Quentin T, Oetjen E, Knepel W (February 2008). “A peroxisome proliferator-activated receptor gamma-retinoid X receptor heterodimer physically interacts with the transcriptional activator PAX6 to inhibit glucagon gene transcription”. Molecular Pharmacology. 73 (2): 509–17. doi:10.1124/mol.107.035568. PMID 17962386. S2CID 10108970.
- ^ Johnson LR (2003). Essential Medical Physiology. Academic Press. pp. 643–. ISBN 978-0-12-387584-6.
- ^ Unger RH, Orci L (June 1981). “Glucagon and the A cell: physiology and pathophysiology (first two parts)”. The New England Journal of Medicine. 304 (25): 1518–24. doi:10.1056/NEJM198106183042504. PMID 7015132.
- ^ Orskov C, Holst JJ, Poulsen SS, Kirkegaard P (November 1987). “Pancreatic and intestinal processing of proglucagon in man”. Diabetologia. 30 (11): 874–81. doi:10.1007/BF00274797 (inactive 2020-10-11). PMID 3446554.
- ^ John AM, Schwartz RA (December 2016). “Glucagonoma syndrome: a review and update on treatment”. Journal of the European Academy of Dermatology and Venereology. 30 (12): 2016–2022. doi:10.1111/jdv.13752. PMID 27422767. S2CID 1228654.
- ^ Oberg K (December 2010). “Pancreatic endocrine tumors”. Seminars in Oncology. 37 (6): 594–618. doi:10.1053/j.seminoncol.2010.10.014. PMID 21167379.
- ^ Fasanmade OA, Odeniyi IA, Ogbera AO (June 2008). “Diabetic ketoacidosis: diagnosis and management”. African Journal of Medicine and Medical Sciences. 37 (2): 99–105. PMID 18939392.
- ^ Kimball C, Murlin J (1923). “Aqueous extracts of pancreas III. Some precipitation reactions of insulin”. J. Biol. Chem. 58 (1): 337–348.
- ^ Bromer W, Winn L, Behrens O (1957). “The amino acid sequence of glucagon V. Location of amide groups, acid degradation studies and summary of sequential evidence”. J. Am. Chem. Soc. 79 (11): 2807–2810. doi:10.1021/ja01568a038.
- ^ “History of glucagon – Metabolism, insulin and other hormones – Diapedia, The Living Textbook of Diabetes”. http://www.diapedia.org. Archived from the original on 2017-03-27. Retrieved 2017-03-26.
External links
- PDBe-KB provides an overview of all the structure information available in the PDB for Human Glucagon
| GCG | ||
|---|---|---|
| Available structuresPDBHuman UniProt search: PDBe RCSBshowList of PDB id codes | ||
| Identifiers | ||
| Aliases | GCG, GLP1, glucagon, GRPP, GLP-1, GLP2 | |
| External IDs | OMIM: 138030 HomoloGene: 136497 GeneCards: GCG | |
| hideGene location (Human)Chr.Chromosome 2 (human)[1]Band2q24.2Start162,142,882 bp[1]End162,152,404 bp[1] | ||
| hideRNA expression patternMore reference expression data | ||
| showGene ontology | ||
| Orthologs | ||
| Species | Human | Mouse |
| Entrez | 2641 | n/a |
| Ensembl | ENSG00000115263 | n/a |
| UniProt | P01275 | n/a |
| RefSeq (mRNA) | NM_002054 | n/a |
| RefSeq (protein) | NP_002045 | n/a |
| Location (UCSC) | Chr 2: 162.14 – 162.15 Mb | n/a |
| PubMed search | [2] | n/a |
| Wikidata | ||
| View/Edit Human |
///////////GLUCAGON, DIABETES, PEPTIDE, HORMONE
Vonicog alfa
>>von Willebrand factor<<<
MIPARFAGVLLALALILPGTLCAEGTRGRSSTARCSLFGSDFVNTFDGSMYSFAGYCSYL
AGGCQKRSFSIIGDFQNGKRVSLSVYLGEFFDIHLFVNGTVTQGDQRVSMPYASKGLYLE
TEAGYYKLSGEAYGFVARIDGSGNFQVLLSDRYFNKTCGLCGNFNIFAEDDFMTQEGTLT
SDPYDFANSWALSSGEQWCERASPPSSSCNISSGEMQKGLWEQCQLLKSTSVFARCHPLV
DPEPFVALCEKTLCECAGGLECACPALLEYARTCAQEGMVLYGWTDHSACSPVCPAGMEY
RQCVSPCARTCQSLHINEMCQERCVDGCSCPEGQLLDEGLCVESTECPCVHSGKRYPPGT
SLSRDCNTCICRNSQWICSNEECPGECLVTGQSHFKSFDNRYFTFSGICQYLLARDCQDH
SFSIVIETVQCADDRDAVCTRSVTVRLPGLHNSLVKLKHGAGVAMDGQDIQLPLLKGDLR
IQHTVTASVRLSYGEDLQMDWDGRGRLLVKLSPVYAGKTCGLCGNYNGNQGDDFLTPSGL
AEPRVEDFGNAWKLHGDCQDLQKQHSDPCALNPRMTRFSEEACAVLTSPTFEACHRAVSP
LPYLRNCRYDVCSCSDGRECLCGALASYAAACAGRGVRVAWREPGRCELNCPKGQVYLQC
GTPCNLTCRSLSYPDEECNEACLEGCFCPPGLYMDERGDCVPKAQCPCYYDGEIFQPEDI
FSDHHTMCYCEDGFMHCTMSGVPGSLLPDAVLSSPLSHRSKRSLSCRPPMVKLVCPADNL
RAEGLECTKTCQNYDLECMSMGCVSGCLCPPGMVRHENRCVALERCPCFHQGKEYAPGET
VKIGCNTCVCRDRKWNCTDHVCDATCSTIGMAHYLTFDGLKYLFPGECQYVLVQDYCGSN
PGTFRILVGNKGCSHPSVKCKKRVTILVEGGEIELFDGEVNVKRPMKDETHFEVVESGRY
IILLLGKALSVVWDRHLSISVVLKQTYQEKVCGLCGNFDGIQNNDLTSSNLQVEEDPVDF
GNSWKVSSQCADTRKVPLDSSPATCHNNIMKQTMVDSSCRILTSDVFQDCNKLVDPEPYL
DVCIYDTCSCESIGDCACFCDTIAAYAHVCAQHGKVVTWRTATLCPQSCEERNLRENGYE
CEWRYNSCAPACQVTCQHPEPLACPVQCVEGCHAHCPPGKILDELLQTCVDPEDCPVCEV
AGRRFASGKKVTLNPSDPEHCQICHCDVVNLTCEACQEPGGLVVPPTDAPVSPTTLYVED
ISEPPLHDFYCSRLLDLVFLLDGSSRLSEAEFEVLKAFVVDMMERLRISQKWVRVAVVEY
HDGSHAYIGLKDRKRPSELRRIASQVKYAGSQVASTSEVLKYTLFQIFSKIDRPEASRIA
LLLMASQEPQRMSRNFVRYVQGLKKKKVIVIPVGIGPHANLKQIRLIEKQAPENKAFVLS
SVDELEQQRDEIVSYLCDLAPEAPPPTLPPHMAQVTVGPGLLGVSTLGPKRNSMVLDVAF
VLEGSDKIGEADFNRSKEFMEEVIQRMDVGQDSIHVTVLQYSYMVTVEYPFSEAQSKGDI
LQRVREIRYQGGNRTNTGLALRYLSDHSFLVSQGDREQAPNLVYMVTGNPASDEIKRLPG
DIQVVPIGVGPNANVQELERIGWPNAPILIQDFETLPREAPDLVLQRCCSGEGLQIPTLS
PAPDCSQPLDVILLLDGSSSFPASYFDEMKSFAKAFISKANIGPRLTQVSVLQYGSITTI
DVPWNVVPEKAHLLSLVDVMQREGGPSQIGDALGFAVRYLTSEMHGARPGASKAVVILVT
DVSVDSVDAAADAARSNRVTVFPIGIGDRYDAAQLRILAGPAGDSNVVKLQRIEDLPTMV
TLGNSFLHKLCSGFVRICMDEDGNEKRPGDVWTLPDQCHTVTCQPDGQTLLKSHRVNCDR
GLRPSCPNSQSPVKVEETCGCRWTCPCVCTGSSTRHIVTFDGQNFKLTGSCSYVLFQNKE
QDLEVILHNGACSPGARQGCMKSIEVKHSALSVELHSDMEVTVNGRLVSVPYVGGNMEVN
VYGAIMHEVRFNHLGHIFTFTPQNNEFQLQLSPKTFASKTYGLCGICDENGANDFMLRDG
TVTTDWKTLVQEWTVQRPGQTCQPILEEQCLVPDSSHCQVLLLPLFAECHKVLAPATFYA
ICQQDSCHQEQVCEVIASYAHLCRTNGVCVDWRTPDFCAMSCPPSLVYNHCEHGCPRHCD
GNVSSCGDHPSEGCFCPPDKVMLEGSCVPEEACTQCIGEDGVQHQFLEAWVPDHQPCQIC
TCLSGRKVNCTTQPCPTAKAPTCGLCEVARLRQNADQCCPEYECVCDPVSCDLPPVPHCE
RGLQPTLTNPGECRPNFTCACRKEECKRVSPPSCPPHRLPTLRKTQCCDEYECACNCVNS
TVSCPLGYLASTATNDCGCTTTTCLPDKVCVHRSTIYPVGQFWEEGCDVCTCTDMEDAVM
GLRVAQCSQKPCEDSCRSGFTYVLHEGECCGRCLPSACEVVTGSPRGDSQSSWKSVGSQW
ASPENPCLINECVRVKEEVFIQQRNVSCPQLEVPVCPSGFQLSCKTSACCPSCRCERMEA
CMLNGTVIGPGKTVMIDVCTTCRCMVQVGVISGFKLECRKTTCNPCPLGYKEENNTGECC
GRCLPTACTIQLRGGQIMTLKRDETLQDGCDTHFCKVNERGEYFWEKRVTGCPPFDEHKC
LAEGGKIMKIPGTCCDTCEEPECNDITARLQYVKVGSCKSEVEVDIHYCQGKCASKAMYS
IDINDVQDQCSCCSPTRTEPMQVALHCTNGSVVYHEVLNAMECKCSPRKCSK
Vonicog alfa
ボニコグアルファ (遺伝子組換え) ;
フォン・ヴィレブランド因子;
| Formula | C9712H15373N2737O3032S210 |
|---|---|
| CAS | 109319-16-6 |
| Mol weight | 225723.1487 |
JAPAN 2020, APPROVED 2020/3/25, VONVENDI
Anticoagulant, Hemostatic, Replenisher (von Willebrand factor)
Active Substance
General information Recombinant von Willebrand Factor (rVWF) is co-expressed with recombinant Factor VIII (rFVIII) in Chinese hamster ovary (CHO) cells as part of the ADVATE (Centrally authorised product) manufacturing process. The rVWF protein is separated from the FVIII and further purified.
Structural formula
Vonicog alfa is expressed as a 2813 amino acid pro-VWF molecule. The pro-VWF is composed of A, B, C and D repeats, which contain various functional domains that have been identified. The mature VWF monomer is a 2050 amino acid protein. Every monomer contains a number of specific domains with a specific function. Elements of note are: • The D’/D3 domain, which binds to Factor VIII • The A1 domain, which binds to: Platelet gp1b-receptor, Heparin, Collagen • The A3 domain, which binds to collagen • The C1 domain, in which the RGD domain binds to platelet integrin αIIbβ3 when this is activated • The “cysteine knot” domain Monomers of pro-VWF are subsequently N-glycosylated, arranged into dimers via a C-terminal disulfide bond in the endoplasmic reticulum and into multimers by crosslinking of N-terminal cysteine residues via disulfide bonds.
Figure 1. Structure of Von Willebrand Factor Monomer/Dimer

After reduction of disulfide bonds in electrophoretic analysis, rVWF appears as a single predominant band having an apparent molecular weight of approximately 260 kDa. In low resolution agarose gel electrophoresis, rVWF shows a characteristic ladder of bands also known as multimers. In this analysis, rVWF contains as many distinct bands as VWF detectable in normal human plasma or VWF isolated from human plasma but in addition, has a zone with unresolved bands in the ultra-high molecular weight range. Highresolution electrophoresis shows a single band for all multimer levels without any satellite bands, as rVWF has never been exposed to ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) cleavage.

Vonicog alfa, sold under the brand names Vonvendi and Veyvondi, is a medicine used to control bleeding in adults with von Willebrand disease (an inherited bleeding disorder).[5][4][6] It is a recombinant von Willebrand factor.[5][4]
The most common adverse reactions are generalized itching, vomiting, nausea, dizziness, and vertigo.[5]
Vonicog alfa should not be used in the treatment of Hemophilia A.[4]
In the UK it is available only via a named patient access program.[7]
Vonicog alfa was approved for medical use in the United States in December 2015, in the European Union in August 2018, and in Australia in April 2020.[3][5][4][8] It was granted orphan drug designations in both the United States and the European Union.[4][1]
Medical uses
Vonicog alfa is indicated in adults with von Willebrand Disease (VWD), when desmopressin (DDAVP) treatment alone is ineffective or not indicated for the
Adverse effects
The following side effects may occur during treatment with vonicog alfa: hypersensitivity (allergic) reactions, thromboembolic events (problems due to the formation of blood clots in the blood vessels), development of inhibitors (antibodies) against von Willebrand factor, causing the medicine to stop working and resulting in a loss of bleeding control.[4] The most common side effects with vonicog alfa (which may affect up to 1 in 10 patients) are dizziness, vertigo (a spinning sensation), dysgeusia (taste disturbances), tremor, rapid heartbeat, deep venous thrombosis (blood clot in a deep vein, usually in the leg), hypertension (high blood pressure), hot flush, vomiting, nausea (feeling sick), pruritus (itching), chest discomfort, sensations like numbness, tingling, pins and needles at the site of infusion, and an abnormal reading on the electrocardiogram (ECG).[4]
References
- ^ Jump up to:a b c “Veyvondi Australian prescription medicine decision summary”. Therapeutic Goods Administration (TGA). 29 April 2020. Retrieved 16 August 2020.
- ^ “Vonvendi 650 IU powder and solvent for solution for injection – Summary of Product Characteristics (SmPC)”. (emc). 7 May 2020. Retrieved 16 August 2020.
- ^ Jump up to:a b “Vonvendi”. U.S. Food and Drug Administration (FDA). 9 May 2018. Archived from the original on 23 April 2019. Retrieved 15 April 2020.
- ^ Jump up to:a b c d e f g h i j “Veyvondi EPAR”. European Medicines Agency (EMA). 20 September 2018. Retrieved 27 March 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d “Vonvendi (von willebrand factor- recombinant kit”. DailyMed. 13 February 2019. Retrieved 27 March 2020.
- ^ “Veyvondi-epar product information” (PDF). European Medicines Agency.
- ^ “Vonicog alfa”. Specialist Pharmacy Service. 15 January 2020. Retrieved 27 March 2020.
- ^ “Vonvendi”. U.S. Food and Drug Administration (FDA). 13 April 2018. STN: 125577. Retrieved 27 March 2020.
Further reading
- AusPAR: Vonicog alfa. Therapeutic Goods Administration (TGA) (Report). October 2020.
External links
- “Vonicog alfa”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Vonvendi, Veyvondi |
| Other names | BAX-111 |
| AHFS/Drugs.com | Monograph |
| License data | EU EMA: by INNUS DailyMed: Vonvendi |
| Pregnancy category | AU: B2[1] |
| Routes of administration | Intravenous |
| Drug class | Hemostatic |
| ATC code | B02BD10 (WHO) B02BD06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]UK: POM (Prescription only) [2]US: ℞-only [3]EU: Rx-only [4]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 109319-16-6 |
| DrugBank | DB12872 |
| UNII | 5PKM8P0G5I |
| KEGG | D08681 |
| Chemical and physical data | |
| Formula | C9712H15373N2737O3032S210 |
| Molar mass | 225725.54 g·mol−1 |
General References
- Singal M, Kouides PA: Recombinant von Willebrand factor: a first-of-its-kind product for von Willebrand disease. Drugs Today (Barc). 2016 Dec;52(12):653-664. doi: 10.1358/dot.2016.52.12.2570978. [PubMed:28276537]
- Brown R: Recombinant von Willebrand factor for severe gastrointestinal bleeding unresponsive to other treatments in a patient with type 2A von Willebrand disease: a case report. Blood Coagul Fibrinolysis. 2017 Oct;28(7):570-575. doi: 10.1097/MBC.0000000000000632. [PubMed:28379876]
- Gill JC, Castaman G, Windyga J, Kouides P, Ragni M, Leebeek FW, Obermann-Slupetzky O, Chapman M, Fritsch S, Pavlova BG, Presch I, Ewenstein B: Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015 Oct 22;126(17):2038-46. doi: 10.1182/blood-2015-02-629873. Epub 2015 Aug 3. [PubMed:26239086]
- Lenting PJ, Christophe OD, Denis CV: von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015 Mar 26;125(13):2019-28. doi: 10.1182/blood-2014-06-528406. Epub 2015 Feb 23. [PubMed:25712991]
- Chung MC, Popova TG, Jorgensen SC, Dong L, Chandhoke V, Bailey CL, Popov SG: Degradation of circulating von Willebrand factor and its regulator ADAMTS13 implicates secreted Bacillus anthracis metalloproteases in anthrax consumptive coagulopathy. J Biol Chem. 2008 Apr 11;283(15):9531-42. doi: 10.1074/jbc.M705871200. Epub 2008 Feb 8. [PubMed:18263586]
- Boston Children’s Hospital [Link]
- EMA [Link]
- FDA application [Link]
- National Institute for Health Research [Link]
- Hemophilia [Link]
////////Vonicog alfa, JAPAN 2020, APPROVALS 2020,, VONVENDI, BAX 111,
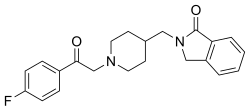
DOFETILIDE

Dofetilide
115256-11-6[RN]
6756
b-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidide
Methanesulfonamide, N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-
MFCD00869707 [MDL number]
- Molecular FormulaC19H27N3O5S2
- Average mass441.565 Da
- UK68798UNII:R4Z9X1N2NDUNII-R4Z9X1N2NDXelideβ-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidideдофетилидدوفيتيليد多非利特
N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-methanesulfonamideNCGC00164549-01PB0478000SMR000466333Tikosyn (TN)Research Code:UK-68798Trade Name:Tikosyn®MOA:Atrial potassium channel blockerIndication:Atrial flutter; Atrial fibrillationStatus:ApprovedCompany:Pfizer (Originator)Sales:ATC Code:C01BD04
INDIA 31/7/2020 APPROVED CDSCO
Dofetilide was first approved by the U.S. Food and Drug Administration (FDA) on Oct 1, 1999, then approved by European Medicine Agency (EMA) on Nov 29, 1999. It was developed and marketed as Tikosyn® by Pfizer.
Dofetilide is a selective blocker of delayed rectifier outward potassium current (IKr). It is indicated for the maintenance of normal sinus rhythm (delay in time to recurrence of atrial fibrillation/atrial flutter [AF/AFl]) in patients with atrial fibrillation/atrial flutter of greater than one week duration who have been converted to normal sinus rhythm.
Tikosyn® is available capsule for oral use, containing 0.125, 0.25 or 0.5 mg of free Dofetilide. The recommended dose is 500 µg orally twice daily.
Dofetilide is a class III antiarrhythmic agent.[1] It is marketed under the trade name Tikosyn by Pfizer, and is available in the United States in capsules containing 125, 250, and 500 µg of dofetilide. It is not available in Europe[2] or Australia.[3] In the United States it is only available by mail order or through specially trained local pharmacies.[4]
Medical uses
Dofetilide is used for the maintenance of sinus rhythm in individuals prone to the occurrence of atrial fibrillation and flutter arrhythmias, and for chemical cardioversion to sinus rhythm from atrial fibrillation and flutter.[5][6]
Based on the results of the Danish Investigations of Arrhythmias and Mortality on Dofetilide (“DIAMOND”) study,[7] dofetilide does not affect mortality in the treatment of patients post-myocardial infarction with left ventricular dysfunction, however it was shown to decrease all-cause readmissions as well as CHF-related readmissions.[8] Because of the results of the DIAMOND study, some physicians use dofetilide in the suppression of atrial fibrillation in individuals with LV dysfunction, however use appears limited: After initially receiving marketing approval in Europe in 1999, Pfizer voluntarily withdrew this approval in 2004 for commercial reasons[2] and it is not registered in other first world countries.
It has clinical advantages over other class III antiarrhythmics in chemical cardioversion of atrial fibrillation, and maintenance of sinus rhythm, and does not have the pulmonary or hepatotoxicity of amiodarone, however atrial fibrillation is not generally considered life-threatening, and dofetilide causes an increased rate of potentially life-threatening arrhythmias in comparison to other therapies.[9]
Contraindications
Prior to administration of the first dose, the corrected QT (QTc) must be determined. If the QTc is greater than 440 msec (or 500 msec in patients with ventricular conduction abnormalities), dofetilide is contraindicated. If heart rate is less than 60 bpm, the uncorrected QT interval should be used. After each subsequent dose of dofetilide, QTc should be determined and dosing should be adjusted. If at any time after the second dose of dofetilide the QTc is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), dofetilide should be discontinued. [4]
Adverse effects
Torsades de pointes is the most serious side effect of dofetilide therapy. The incidence of torsades de pointes is 0.3-10.5% and is dose-related, with increased incidence associated with higher doses. The majority of episodes of torsades de pointes have occurred within the first three days of initial dosing. Patients should be hospitalized and monitored for the first three days after starting dofetilide.[10]
The risk of inducing torsades de pointes can be decreased by taking precautions when initiating therapy, such as hospitalizing individuals for a minimum of three days for serial creatinine measurement, continuous telemetry monitoring and availability of cardiac resuscitation.
Pharmacology
Mechanism of action
Dofetilide works by selectively blocking the rapid component of the delayed rectifier outward potassium current (IKr).[11]
This causes the refractory period of atrial tissue to increase, hence its effectiveness in the treatment of atrial fibrillation and atrial flutter.
Dofetilide does not affect dV/dTmax (the slope of the upstroke of phase 0 depolarization), conduction velocity, or the resting membrane potential.

Dofetilide synthesis
There is a dose-dependent increase in the QT interval and the corrected QT interval (QTc). Because of this, many practitioners will initiate dofetilide therapy only on individuals under telemetry monitoring or if serial EKG measurements of QT and QTc can be performed.
Pharmacokinetics
Peak plasma concentrations are seen two to three hours after oral dosing when fasting. Dofetilide is well absorbed in its oral form, with a bioavailability of >90%. Intravenous administration of dofetilide is not available in the United States. [12]
The elimination half-life of dofetilide is roughly 10 hours; however, this varies based on many physiologic factors (most significantly creatinine clearance), and ranges from 4.8 to 13.5 hours. Due to the significant level of renal elimination (80% unchanged, 20% metabolites), the dose of dofetilide must be adjusted to prevent toxicity due to impaired renal function.[13]
Dofetilide is metabolized predominantly by CYP3A4 enzymes predominantly in the liver and GI tract. This means that it is likely to interact with drugs that inhibit CYP3A4, such as erythromycin, clarithromycin, or ketoconazole, resulting in higher and potentially toxic levels of dofetilide. [14]
Metabolism
A steady-state plasma level of dofetilide is achieved in 2–3 days.
80% of dofetilide is excreted by the kidneys, so the dose of dofetilide should be adjusted in individuals with chronic kidney disease, based on creatinine clearance.
In the kidneys, dofetilide is eliminated via cation exchange (secretion). Agents that interfere with the renal cation exchange system, such as verapamil, cimetidine, hydrochlorothiazide, itraconazole, ketoconazole, prochlorperazine, and trimethoprim should not be administered to individuals taking dofetilide.
About 20 percent of dofetilide is metabolized in the liver via the CYP3A4 isoenzyme of the cytochrome P450 enzyme system. Drugs that interfere with the activity of the CYP3A4 isoenzyme can increase serum dofetilide levels. If the renal cation exchange system is interfered with (as with the medications listed above), a larger percentage of dofetilide is cleared via the CYP3A4 isoenzyme system.
History
After its initial US FDA approval, due to the pro-arrhythmic potential it was only made available to hospitals and prescribers that had received education and undergone specific training in the risks of treatment with dofetilide; however, this restriction was subsequently removed in 2016. [15
SYN


REF
Reference:1. US5079248A / US4959366A.
2. J. Med. Chem. 1990, 33, 1151-1155.
SYN
SYN
SYN
SYN
EP 0245997; JP 1987267250; US 4959366; US 5079248
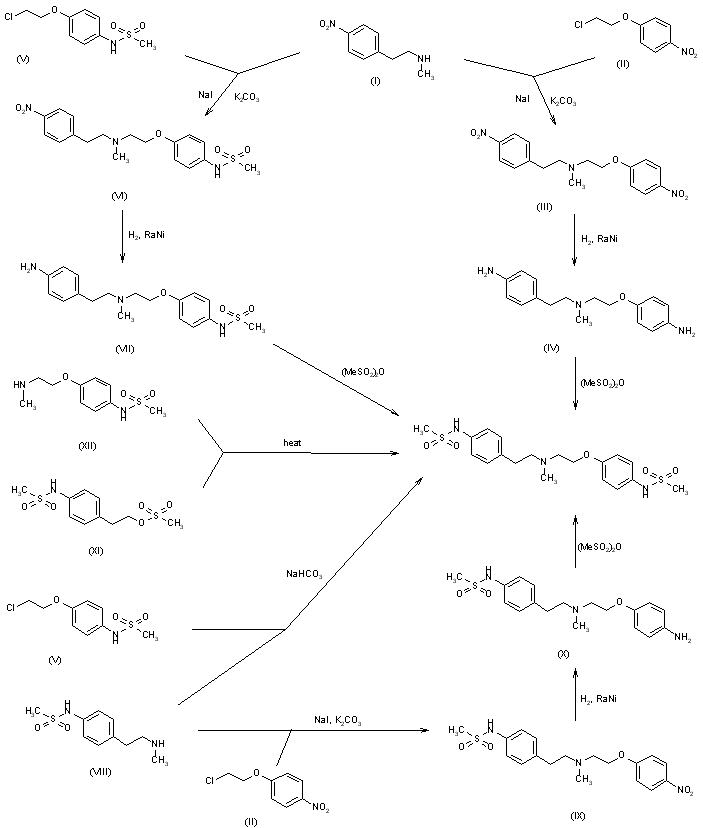
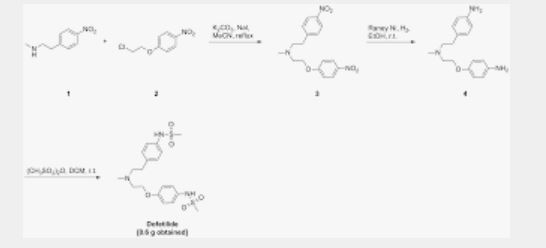
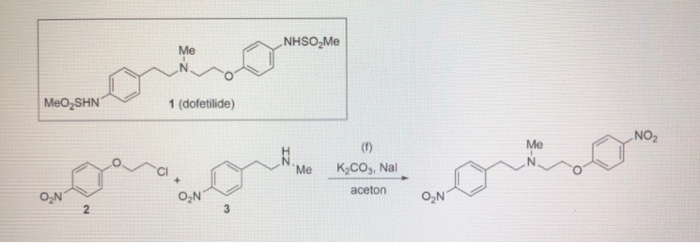
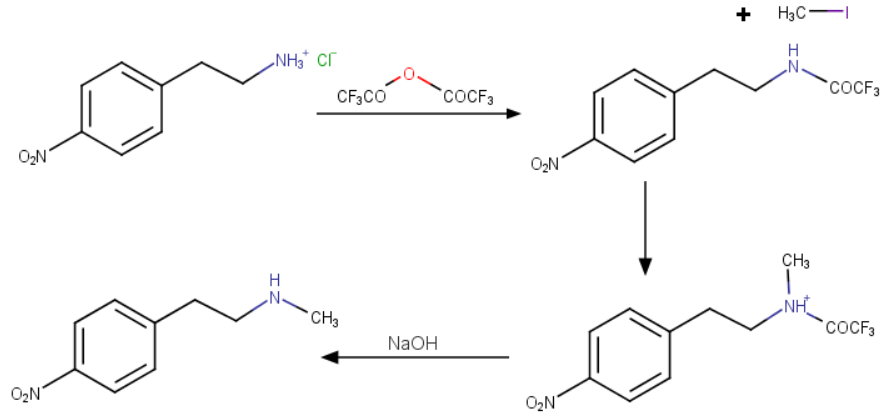
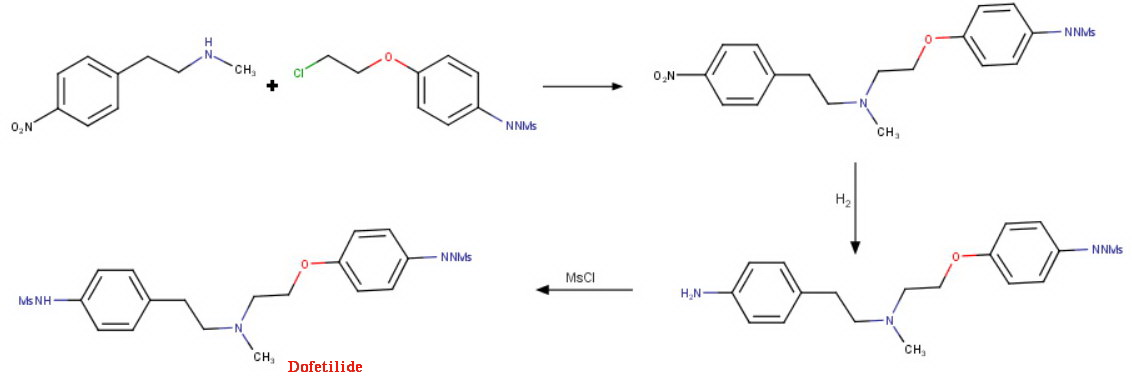
This compound can be prepared by several related ways: 1) The condensation of N-methyl-2-(4-nitrophenyl)ethylamine (I) with 4-(2-chloroethoxy)nitrobenzene (II) by means of NaI and K2CO3 in refluxing acetonitrile gives 1-(4-nitrophenoxy)-5-(4-nitrophenyl)-3-methyl-3-azapentane (III), which is reduced with H2 over Pd/C in ethanol, yielding the corresponding diamino derivative (IV). Finally, this compound is acylated with methanesulfonyl anhydride in dichloromethane. 2) The condensation of (I) with N-[4-(2-chloroethoxy)phenyl]methanesulfonamide (V) with NaI and K2CO3 as before gives 1-[4-(methanesulfonamide)phenoxy]-3-methyl-5-(4-nitrophenyl)-3-azapentane (VI), which is reduced with H2 over Pd/C as before, yielding the corresponding amino derivative (VII). Finally, this compound is acylated with methanesulfonyl anhydride as usual. 3) The condensation of (II) with N-[4-[2-(methylamino)ethyl]phenyl]methanesulfonamide (VIII) with NaI and K2CO3 as usual gives 1-[4-(methanesulfonamido)phenyl]-3-methyl-5-(4-nitrophenoxy)-3-azapentane (IX), which is reduced with H2 and RaNi to the corresponding amino derivative (X). Finally, this compound is acylated with methanesulfonyl chloride and pyridine. 4) By condensation of N-[4-[2-(methanesulfonyloxy)ethyl]phenyl]methanesulfonamide (XI) with N-[4-[2-(methylamino)ethoxy]phenyl]methanesulfonamide (XII) in refluxing ethanol. 5) By condensation of (V) with (VIII) by means of NaHCO3.
References
- ^ Lenz TL; Hilleman DE (July 2000). “Dofetilide, a new class III antiarrhythmic agent”. Pharmacotherapy. 20 (7): 776–86. doi:10.1592/phco.20.9.776.35208. PMID 10907968.
- ^ Jump up to:a b Wathion, Noel (2004-04-13). “Public Statement on Tikosyn (dofetilide): Voluntary Withdrawal of the Marketing Authorisation in the European Union” (PDF). European Agency for the Evaluation of Medicinal Products.
- ^ Australian Medicines Handbook 2014
- ^ Jump up to:a b TIKOSYN® (dofetilide). Pfizer. <http://www.tikosyn.com/>.
- ^ Banchs JE; Wolbrette DL; Samii SM; et al. (November 2008). “Efficacy and safety of dofetilide in patients with atrial fibrillation and atrial flutter”. J Interv Card Electrophysiol. 23(2): 111–5. doi:10.1007/s10840-008-9290-6. PMID 18688699. S2CID 25162347.
- ^ Lenz TL; Hilleman DE (November 2000). “Dofetilide: A new antiarrhythmic agent approved for conversion and/or maintenance of atrial fibrillation/atrial flutter”. Drugs Today. 36 (11): 759–71. doi:10.1358/dot.2000.36.11.601530. PMID 12845335.
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. The New England Journal of Medicine. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Torp-Pedersen C; ller M; Mø Bloch-Thomsen PE; et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. N. Engl. J. Med. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Micromedex Drugdex drug evaluations micromedex.com
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med 1999; 341:857.
- ^ Roukoz H; Saliba W (January 2007). “Dofetilide: a new class III antiarrhythmic agent”. Expert Rev Cardiovasc Ther. 5 (1): 9–19. doi:10.1586/14779072.5.1.9. PMID 17187453. S2CID 11255636.
- ^ 1Rasmussen HS, Allen MJ, Blackburn KJ, et al. Dofetilide, a novel class III antiarrhythmic agent. J Cardiovasc Pharmacol 1992; 20 Suppl 2:S96.
- ^ “Dofetilide.” Lexicomp. Wulters Kluwer Health, n.d. Web. <online.lexi.com>.
- ^ Walker DK, Alabaster CT, Congrave GS, et al. Significance of metabolism in the disposition and action of the antidysrhythmic drug, dofetilide. In vitro studies and correlation with in vivo data. Drug Metab Dispos 1996; 24:447.
- ^ “Information for Tikosyn (dofetilide)”. US Food and Drug Administration. 2016-03-09.
DofetilideCAS Registry Number: 115256-11-6CAS Name:N-[4-[2-[Methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]methanesulfonamideAdditional Names: 1-(4-methanesulfonamidophenoxy)-2-[N-(4-methanesulfonamidophenethyl)-N-methylamino]ethaneManufacturers’ Codes: UK-68798Trademarks: Tikosyn (Pfizer)Molecular Formula: C19H27N3O5S2Molecular Weight: 441.56Percent Composition: C 51.68%, H 6.16%, N 9.52%, O 18.12%, S 14.52%Literature References: Potassium channel blocker. Prepn: J. E. Arrowsmith et al.,EP245997; P. E. Cross et al.,US4959366 (1987, 1990 both to Pfizer); idemet al.,J. Med. Chem.33, 1151 (1990). HPLC determn in urine: D. K. Walker et al.,J. Chromatogr.568, 475 (1991). Mechanism of action study: D. Carmeliet, J. Pharmacol. Exp. Ther.262, 809 (1992). Review of pharmacology and pharmacokinetics: H. S. Rasmussen et al.,ibid.20, Suppl. 2, S96-S105 (1992). Clinical trial in atrial fibrillation and flutter: B. L. Norgaard et al.,Am. Heart J.137, 1062 (1999); in congestive heart failure: C. Torp-Pedersen et al.,N. Engl. J. Med.341, 857 (1999).Properties: Crystals from ethyl acetate/methanol (10:1), mp 147-149° (Cross); from hexane/ethyl acetate, mp 151-152° (Arrowsmith). Also reported as white crystalline solid, mp 161° (Rasmussen). pKa 7.0, 9.0, 9.6. Distribution coefficient (pH 7.4): 0.96. Sol in 0.1M NaOH, acetone, 0.1M HCl; very slightly sol in water, propan-2-ol.Melting point: mp 147-149° (Cross); mp 151-152° (Arrowsmith); mp 161° (Rasmussen)pKa: pKa 7.0, 9.0, 9.6Therap-Cat: Antiarrhythmic (class III).Keywords: Antiarrhythmic; Potassium Channel Blocker.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601235 |
| ATC code | C01BD04 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 96% (oral) |
| Protein binding | 60% -70% |
| Elimination half-life | 10 hours |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 115256-11-6 |
| PubChem CID | 71329 |
| IUPHAR/BPS | 2604 |
| DrugBank | DB00204 |
| ChemSpider | 64435 |
| UNII | R4Z9X1N2ND |
| KEGG | D00647 |
| ChEBI | CHEBI:4681 |
| ChEMBL | ChEMBL473 |
| CompTox Dashboard (EPA) | DTXSID5046433 |
| ECHA InfoCard | 100.166.441 |
| Chemical and physical data | |
| Formula | C19H27N3O5S2 |
| Molar mass | 441.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc1ccc(cc1)CCN(CCOc2ccc(cc2)NS(=O)(=O)C)C)C | |
| InChI[hide]InChI=1S/C19H27N3O5S2/c1-22(13-12-16-4-6-17(7-5-16)20-28(2,23)24)14-15-27-19-10-8-18(9-11-19)21-29(3,25)26/h4-11,20-21H,12-15H2,1-3H3 Key:IXTMWRCNAAVVAI-UHFFFAOYSA-N |
////////////DOFETILIDE, 2020 APPROVALS, INDIA 2020, UK 68798, UNII:R4Z9X1N2ND, дофетилид , دوفيتيليد ,多非利特 , TIKOSYN
VILDAGLIPTIN

VILDAGLIPTIN
- Molecular FormulaC17H25N3O2
- Average mass303.399 Da
- (2S)-1-{2-[(3-hydroxyadamantan-1-yl)amino]acetyl}pyrrolidine-2-carbonitrile
(2S)-1-[N-(3-Hydroxyadamantan-1-yl)glycyl]pyrrolidine-2-carbonitrile(2S)-1-[N-(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)glycyl]pyrrolidine-2-carbonitrile274901-16-5[RN]2-Pyrrolidinecarbonitrile, 1-[2-[(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)amino]acetyl]-, (2S)-(-)-(2S)-1-[[(3-Hydroxytricyclo[3.3.1.1[3,7]]dec-1-yl)amino]acetyl]pyrrolidine-2-carbonitrile
(2S)-1-[N-(3-Hydroxyadamantan-1-yl)glycyl]-2-pyrrolidinecarbonitrile
Vildagliptin was approved by the European Medicines Agency (EMA) on Sep 26, 2007, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 20, 2010, following by China Food and Drug Administration (CFDA) on Aug 15, 2011. It was developed and marketed as Galvus® by Novartis in EU.
Vildagliptin is a potent selective inhibitor of dipeptidyl peptidase-4 (DPP-4) that improves glycaemic control by increasing islet α-cell and β-cell responsiveness to glucose. It is used to reduce hyperglycemia in type 2 diabete.
Galvus®is available as film-coated tablet for oral use, containing 50 mg free Vildagliptin. The recommended dose of vildagliptin is 100 mg, administered as one dose of 50 mg in the morning and one dose of 50 mg in the evening.Drug Name:VildagliptinResearch Code:LAF-237; DSP-7238; NVP-LAF-237Trade Name:Galvus® / Jalra® / Xiliarx® / Equa®MOA:Dipeptidyl peptidase-4 (DPP-4) inhibitorIndication:Type 2 diabetesStatus:ApprovedCompany:Novartis (Originator)Sales:$1,140 Million (Y2015); 
$1,224 Million (Y2014);
$1,200 Million (Y2013);
$910 Million (Y2012);
$677 Million (Y2011);ATC Code:A10BH02
Approved Countries or AreaUpdate Date:2015-07-29
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2008-11-19 | Marketing approval | Xiliarx | Type 2 diabetes | Tablet | 50 mg | Novartis | |
| 2008-11-19 | Marketing approval | Jalra | Type 2 diabetes | Tablet | 50 mg | Novartis | |
| 2007-09-26 | Marketing approval | Galvus | Type 2 diabetes | Tablet, Film coated | 50 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-01-20 | Marketing approval | Equa | Type 2 diabetes | Tablet | 50 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-08-15 | Marketing approval | 佳维乐/Galvus | Type 2 diabetes | Tablet | 50 mg | Novartis | |
| 2011-08-15 | Marketing approval | 佳维乐/Galvus | Type 2 diabetes | Tablet | 50 mg | Novartis |
Vildagliptin, previously identified as LAF237, is a new oral anti-hyperglycemic agent (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. Vidagliptin subsequently acts by inhibiting the inactivation of glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP) by DPP-4. This inhibitory activity ultimately results in a two-fold action where GLP-1 and GIP are present to potentiate the secretion of insulin by beta cells and suppress glucagon secretion by alpha cells in the islets of Langerhans in the pancreas. It is currently in clinical trials in the U.S. and has been shown to reduce hyperglycemia in type 2 diabetes mellitus. While the drug is still not approved for use in the US, it was approved in Feb 2008 by European Medicines Agency for use within the EU and is listed on the Australian PBS with certain restrictions.
Vildagliptin, sold under the brand name Galvus among others, is an oral anti-hyperglycemic agent (anti-diabetic drug) of the dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs. Vildagliptin inhibits the inactivation of GLP-1[2][3] and GIP[3] by DPP-4, allowing GLP-1 and GIP to potentiate the secretion of insulin in the beta cells and suppress glucagon release by the alpha cells of the islets of Langerhans in the pancreas.
Vildagliptin has been shown to reduce hyperglycemia in type 2 diabetes mellitus.[2]
Combination with metformin
The European Medicines Agency has also approved a combination of vildagliptin and metformin, vildagliptin/metformin (Eucreas by Novartis) as an oral treatment for type-2 diabetes.[4]
Adverse effects
Adverse effects observed in clinical trials include nausea, hypoglycemia, tremor, headache and dizziness. Rare cases of hepatoxicity have been reported.[5]
There have been case reports of pancreatitis associated with DPP-4 inhibitors. A group at UCLA reported increased pre-cancerous pancreatic changes in rats and in human organ donors who had been treated with DPP-4 inhibitors.[6][7] In response to these reports, the United States FDA and the European Medicines Agency each undertook independent reviews of all clinical and preclinical data related to the possible association of DPP-4 inhibitors with pancreatic cancer. In a joint letter to the New England Journal of Medicines, the agencies stated that “Both agencies agree that assertions concerning a causal association between incretin-based drugs and pancreatitis or pancreatic cancer, as expressed recently in the scientific literature and in the media, are inconsistent with the current data. The FDA and the EMA have not reached a final conclusion at this time regarding such a causal relationship. Although the totality of the data that have been reviewed provides reassurance, pancreatitis will continue to be considered a risk associated with these drugs until more data are available; both agencies continue to investigate this safety signal.”[8]
PATENT
https://patents.google.com/patent/US20080167479A1/en
- Vildagliptin is an active pharmaceutical substance with an empirical formula of C17H25N3O2 and a molecular weight of 303.40 g/mol. Vildagliptin is the international common accepted name for (2S)-1-[[(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)amino]acetyl]-2-pyrrolidine carbonitrile and has the structure of formula (I).
- [0003]Vildagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor and is disclosed in U.S. Pat. No. 6,166,063 (“the ‘063 patent”), the disclosure of which is incorporated herein by reference. The ‘063 patent discloses a synthesis of vildagliptin using the synthetic process represented in Scheme 1.
- [0004]Vildagliptin can exist as the (2S) and (2R) enantiomers. The stereoisomer with the desired biological activity is the (2S) enantiomer. Accordingly, it is desirable to synthesize (2S)-vildagliptin with high stereochemical purity. A process that yields vildagliptin with a high enantiomeric purity is disclosed in International Patent Publication WO 2004/092127, the disclosure of which is incorporated herein by reference. This reference discloses compositions containing from 95% to 99.99% of (2S)-vildagliptin.
- [0069]This example illustrates the synthesis of the compound of formula (I) in accordance with embodiments of the invention.
- [0070]Into a 100 mL rounded reaction vessel were charged 3 g (17.37 mmol) of 1-chloroacetyl-2-cyanopyrrolidine, 3.22 g (19.82 mmol) of 1-amino-3-adamantanol, 2.78 g (20.1 mmol) of potassium carbonate, and 30 mL isopropyl acetate. The mixture was refluxed for 4 h, cooled to room temperature, and the salts were filtered and washed with acetonitrile. The mother liquors were evaporated to dryness to obtain an oil which was aged in MEK from which a white solid crystallizes at 0-5° C. The solid was filtered washing the cake with MEK and dried at 40° C. in a vacuum oven until constant weight.
- [0071]Yield: 36%. Assay: 99.21%. HPLC purity: 97.55% of vildagliptin (measured according to Example 2). HPLC chiral purity: more than 99.99% of vildagliptin (measured according to Example 7).
- [0072]These results demonstrate that a compound of formula (I) comprising less than 0.01% of (2R)-1-[N-(3-hydroxytricyclo[3.3.1.13,7]dec-1-yl)glycyl]-2-pyrrolidinecarbonitrile (i.e., (2R)-vildagliptin).
Patent
https://patents.google.com/patent/WO2015145467A1/en
Vildagliptin is chemically known as (S)-l-[2-(3-Hydroxyadamantan-l-ylamino) acetyl] pyrrolidine-2-carbonitrile and exist as (2S) and (2R) enantiomers. The stereoisomer with the desired biological activity is the (2S) enantiomer, represented by the following structure:

U.S. Patent No. 6,166,063 (“the Ό63 patent”) discloses new class of Dipeptidyl peptidase 4 (DPP-4) inhibitors such as vildagliptin. The ‘063 patent further discloses a process for the preparation of vildagliptin by acylation of L-prolinamide with chloroacetyl chloride in the presence of a base in dichloromethane or tetrahydrofuran as solvent, filtration and subsequent dehydration with trifluoroacetic anhydride (TFAA) to provide (S) -1- (2- chloroacetyl) pyrrolidin-2-carbonitrile. The carbonitrile intermediate is isolated by distilling out the solvent, co-distillation with ethyl acetate, partitioning between water and ethyl acetate, extraction of the resulting aqueous layer with ethyl acetate followed by aqueous washings of the organic layer and concentrating to obtain carbonitrile intermediate as yellow solid. This is later reacted with about 2 moles of l-aminoadamantane-3-ol in the presence of about 4 moles of potassium carbonate in dichloromethane (DCM) or tetrahydrofuran (THF) for 6 days. Finally, the obtained crude vildagliptin is subjected to chromatography employing SIMS/Biotage Flash chromatography system providing vildagliptin with melting point of 138°C-140°C. The disclosed process is schematically represented as follows:

Amide Carbonitrile
A similar process is described in J. Med. Chem. 2003, 46, 2774-2789, where acylation of L-prolinamide with chloroacetyl chloride is carried out in the presence of potassium carbonate in tetrahydrofuran as solvent and subsequent dehydration with TFAA to provide (S) -1- (2-chloroacetyl) pyrrolidin-2 -carbonitrile. The carbonitrile intermediate was isolated by adding ethyl acetate, distillation of the solvent, partitioning between water and aqueous sodium bicarbonate, extraction of the resulting aqueous layer with ethyl acetate followed by aqueous washings of the organic layer and concentrating to obtain carbonitrile intermediate as yellow- white solid which was reacted with about 2-3 moles of 1- aminoadamantane-3-ol in the presence of about 3 moles of potassium carbonate in DCM or THF for 1-3 days followed by purification from a mixture of ethyl acetate and isopropanol provided Vildagliptin as a white solid.
U.S. Patent No. 6,011,155 discloses a process for the preparation of (S) -1- (2- bromooacetyl) pyrrolidin-2-carbonitrile by acylation of L-prolinamide with bromoacetyl bromide in the presence of triethyl amine and catalytic amount of DMAP in DCM as solvent wherein the resulting (S)-l -(2 -bromoacetyl) pyrrolidin-2-carboxamide is isolated and subsequently dehydrated with TFAA to obtain the carbonitrile intermediate as dark yellow solid.
U.S. Patent application No. 2008/0167479 discloses preparation of Vildagliptin with high chemical and enantiomeric purities wherein (S) -1- (2-chloroacetyl) pyrrolidin-2- carbonitrile is prepared in one step process by acylation of prolinamide with chloroacetyl chloride in a mixture of isopropyl acetate and DMF followed by dehydration with cyanuric chloride to obtain the carbonitrile intermediate as an oil which was crystallized from isopropanol. The resulting carbonitrile intermediate is reacted with l-aminoadamantane-3- ol in the presence of alkali metal carbonates such as potassium carbonate and an optional additive such as I in a solvent comprising at least an ester or ether or nitrile solvent and purification of vildagliptin from methyl ethyl ketone or from a mixture of isopropanol and methyl t-butyl ether.
PCT Publication No. 2010/022690 discloses a process for the preparation of vildagliptin wherein (S)-l -(2-chloroacetyl) pyrrolidin-2-carboxamide intermediate is isolated as a trialkylamine hydrohalide salt in two fractions and. dehydrated with TFAA to obtain (S)-l- (2-chloroacetyl) pyrrolidin-2-carbonitrile as light yellow powder after crystallization from heptane. The resulting carbonitrile intermediate is then reacted with 3-amino-l- adamantanol in the presence of alkali metal carbonate base and an alkali metal iodide as a catalyst in a mixture of organic ketones, ester and polar aprotic solvents. The crude product was subjected to multiple crystallizations in order to achieve high chemical purity of vildagliptin. This publication also disclosed final crystallization of vildagliptin from 2- butanone, toluene, 2-methyl tetrahydrofuran, isopropyl acetate, dimethyl carbonate, isopropanol. This process adds an extra step of isolation of the said carboxamide intermediate, uses mixture of solvents in the preparation of vildagliptin and to multiple crystallizations which makes the process uneconomical on large scale.
PCT Publication No. 2011/101861 discloses a process for the preparation of vildagliptin wherein (S)-l-(2-chloroacetyl) pyrrolidin-2-carboxamide and (S)-l-(2-chloroacetyl) pyrrolidin-2-carbonitrile intermediates are isolated as solids after purification and drying. Further, (S)-l-(2-chloroacetyl) pyrrolidin-2-carbonitrile is then converted to vildagliptin by reacting it with l-aminoadamantane-3-ol in the presence of potassium carbonate and KI in a suitable ether solvent like THF and purifying the obtained vildagliptin from a mixture of ethyl acetate and methanol. This publication also provided an alternate process for the preparation of vildagliptin by reacting 2-(3-hydroxyadamantan-l-yl amino) acid or derivative thereof with pyrrolidine-2-carbonitrile and various solvents from which vildagliptin may be crystallized such as ethyl acetate, 2-butanone, or mixture of ethyl acetate-methanol, ethyl acetate-isopropanol, methanol-DCM, ethyl acetate-cyclohexane and 2-butanone-methyl t-butyl ether.
U.S. Patent No. 7,375,238 discloses a one-pot process for the preparation of vildagliptin without isolation of the carboxamide and carbonitrile intermediates and further involves preparation of Vildagliptin by using potassium carbonate and potassium iodide (KI) as catalysts in 2-butanone solvent. Purification of the crude vildagliptin was carried out from a mixture of isopropanol and methyl t-butyl ether in the presence of 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU) base and final recrystallization from 2-butanone afforded pure vildagliptin. This process suffers from certain draw backs such as use of mixture of solvents for the acylation and condensation reactions; use of base and expensive additive such as KI in the condensation reaction.
PCT Publication No. 2011/012322 discloses a process wherein the (S) -1- (2-chloroacetyl) pyrrolidin-2-carbonitrile intermediate is isolated, purified and reacted with 1- aminoadamantane-3-ol in the presence of a phase transfer catalyst, optionally an inorganic base and a solvent selected from nitrile, ketone, ether, ester and mixtures thereof in a two phase reaction system wherein the first phase consist of a liquid phase and the second phase consists of an inorganic base. The final purification of vildagliptin was carried out in 2- butanone solvent.
PCT Publication No. 2013/179300 discloses preparation of vildagliptin from organic solvents such as aromatic hydrocarbons, aliphatic hydrocarbons, halogenated hydrocarbons, ethers, nitrile, dialkyl formamides, dialkylacetamides, dialkyl sulfoxides in the presence of organic or inorganic base. The resulting crude vildagliptin was purified by acid-base treatment and crystallization from a solvent selected from aliphatic hydrocarbons, aromatic hydrocarbons, ketones, esters, nitrile, ether, cyclic ether and alcohol or mixtures thereof.
PCT Publication No. 2012/022994 involves conversion of racemic vildagliptin to (S)- enantiomer via formation of vildagliptin adducts and final purification from ethyl acetate or mixture of ethyl acetate with 1% water.
U.S. Application No. 2006/0210627 discloses crystalline Form A of vildagliptin and its preparation from 2-butanone, isopropanol, acetone or a mixture of isopropanol-ethyl acetate in the presence of DBU base. This publication also discloses amorphous vildagliptin and its preparation by lyophilization from a water solution.
PCT Publication No. 2014/102815 disclosed a process for the preparation of vildagliptin by isolating the carboxamide and carbonitrile intermediates after crystallization and drying. The resulting carbonitrile intermediate is reacted with l-aminoadamantane-3-ol in the presence of organic base or inorganic base in nitrile, ester or alcohol solvent.
IN 3965 MUM/2013 publication discloses a process for the preparation of vildagliptin by preparing and crystallizing (S) -1- (2-chloroacetyl) pyrrolidin-2-carbonitrile intermediate and reacting it with l-aminoadamantane-3-ol in the presence of a potassium carbonate, optionally in presence of suitable catalyst such as KI in ketone solvent or in mixture of ketone with non polar solvents.
C.N. publication No. 102617434 discloses a one pot process for the preparation of Vildagliptin by reacting salt of pyrrolidine carbonitrile such as TFA salt with haloacetyl halide in the presence of a base followed by insiru reaction with l-aminoadamantane-3-ol in the presence of tertrabutyl ammonium iodide in halogenated hydrocarbon or ether as solvent to get vildagliptin which is further crystallized from ethyl acetate-petroleum ether.
C.N. publication No. 103804267 discloses a process for the preparation of vildagliptin by reacting (S)-l -(2 -haloacetyl) pyrrolidin-2-carbonitrile with l-aminoadamantane-3-ol in a mixed system of an organic solvent and water in the presence of a base and phase transfer catalyst followed by crystallization of the obtained crude vildagliptin.
C.N. publication No. 103787944 disclosed dehydration of-1- (2-chloroacetyl) -2- (S) – pyrrolidine carboxamide in the presence of a dehydrating agent and an acid-binding agent in an organic solvent followed by crystallization from mixture of isopropyl ether and ethyl acetate to provide l-(2-chloroacetyl)-2-(S)-pyrrolidine carbonitrile as white or pale yellow solid powder.
Furthermore, several techniques are known in the art for the purification of vildagliptin such as chromatography (US 6,166,063); or acid-base purification (IN 61 /MUM/2012 publication) or via formation of inorganic salt complexes (WO 2011/042765); or by solvent crystallizations such as mixture of ethyl acetate and isopropanol (J. Med. Chem. 2003, 46, 2774-2789); isopropanol and MTBE in the presence of DBU base and final recrystallization from 2-butanone (US 7,375,238); methyl ethyl ketone or from a mixture of isopropanol and MTBE (US 2008/0167479); acetone, 2-butanone, cyclohexanone, ethyl acetate, isopropyl acetate or dimethyl carbonate (IN 61 /MUM/2012 publication); 2- butanone (WO 2011/012322); aliphatic hydrocarbons, aromatic hydrocarbons, ketones, esters, nitrile, ether, cyclic ether and alcohol or mixtures thereof (WO 2013/179300); or from ethyl acetate or mixture of ethyl acetate with 1% water (WO 2012/022994).
Most of the processes known in the art for synthesizing vildagliptin are associated with one or more of the following disadvantages:
a) use of toxic TFAA for dehydration which is costly and environmentally harmful, b) lengthy and time consuming condensation process,
c) conventional solvents used in the condensation stage are costly, volatile, flammable, toxic, causing adverse health effects, in, addition to this potentially unsafe peroxide forming solvents such as THF were used, which process is more costlier than the process not having such elements,
d) purification of vildagliptin by chromatographic purification or by formation of inorganic salt complexes or by multiple crystallizations which are tedious, labor intensive, uses high amounts of solvents, require precise monitoring and time consuming and hence not viable for commercial scale operations.
Therefore, the present invention fulfills the need in the art and provides simple, industrially feasible and scalable processes for the preparation and purification of vildagliptin that circumvent disadvantages associated with the prior art process, proved to be advantageous from environmental and industrial point of view and also fulfill purity criteria. These processes allow the final product to be produced in a higher yield and purity by minimizing number of processing steps and reducing the number of solvent usage which is very practical for scale-up production, especially in terms of operating efficiency.
The new processes has a further advantage in recovering the expensive 1- aminoadamantane-3-ol from the reaction mixture and recycling in a simple manner that avoids use of inorganic salt complexes, which is economical and applicable on an industrial scale.
EXAMPLE 1: Preparation of (2S)- 1 -(Chloroacetyl)-2-pyrrolidinecarbonitrile.
To a solution of L-Prolinamide (100 gms) dissolved in DCM (1000 mL) was added triethyl amine (88.6 gms) and DMAP (1.07 gms) at 25-30°C under N2 atmosphere and stirred for 15 min at 25-30°C. This solution was added to a solution of chloroacetyl chloride (98.9 gms) in DCM (500 mL) under N2 atmosphere at -5 to 0°C over 2-3 hr. Raised the reaction mass temperature to 0-5°C and stirred for lhr. After reaction completion, charged phosphorus oxy chloride (201.5 gms) to the reaction mass at 0-5 °C, heated the reaction mass temperature to reflux and stirred for 6hr at same temperature. After reaction completion, allowed to cool to 10-20°C and added DM water (500 mL). Aqueous layer was separated and the organic layer was washed with DM water. To the organic layer DM water (300 mL) was added at 25-30°C and adjusted the reaction mass pH to 6.5-7.5 with -500 mL of sodium bicarbonate solution (-40 g of NaHC03 dissolved in 500 mL of DM Water). Separated the aqueous layer and concentrated the organic layer under vacuum at temperature of 30-40°C to get residual mass. Charged isopropanol (100 mL) and distilled out solvent completely under vacuum at <50°C. The resulting residue was allowed to cool to 30-40°C and charged isopropanol (500 mL). Heated the reaction mass temperature to 40- 45°C, stirred for 30 min at 40-45°C, allowed to cool to 0-5°C, stirred for 2 hr, filtered and washed wet cake with chilled isopropanol (100 mL), dried at 40-45°C for 6 hr to provide 115 gms of (2S)-l-(CMoroace1yl)-2-pyrrolidinecarbonitrile.
HPLC Purity: 99.86%.
Example 2: Preparation of Vildagliptin
To (2S)-l-(Chloroacetyl)-2 -Pyrrolidine carbonitrile (100 gms) dissolved in DM Water (500 mL), charged l-aminoadamantane-3-ol (242.2 g) at 25-35°C. Heated the reaction mass temperature to 40-45°C and stirred for 8-10 hr at 40-45°C. After reaction completion, allowed to cool to 25-30°C and charged DM water (700 mL) and DCM (600 mL). Separated the organic layer and extracted the aqueous layer with DCM. The total organic layer was concentrated under vacuum at temperature 30-40°C to get residual mass. Ethyl acetate (100 mL) was added to the residual mass and distilled completely under vacuum at <50°C. Charged ethyl acetate (500 mL) and refluxed for 1 hr. Allowed to cool to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (100 mL) then dried at 50-55°C for 6 hr to provide 130 gms of crude vildagliptin.
HPLC Purity: 99.56%.
Dimer impurity content: <0.32%;
R-isomer content (by chiral HPLC): <0.2%;
l-aminoadamantane-3-ol content (by GC): 0.56%.
EXAMPLE 3: Preparation of Vildagliptin (using K2C03 and KI)
To l-aminoadamantane-3-ol (19.4 g) taken in DM Water (50 mL), added potassium carbonate (8.0 gms), potassium iodide (0.1 gm) and stirred for 15 mins at 25-35°C. (2S)-1- (Chloroacetyl)-2-Pyrrolidine carbonitrile (10 gms) was added at 25-35°C and stirred for 15 mins at 25-35°C. Raised the reaction mass temperature to 40-45°C and stirred for 4 hr at 40-45°C. After reaction completion, cooled to 25-30°C and charged DCM (50 mL). Separated the organic layer and extracted the aqueous layer with DCM. The total organic layer was washed with DM water and the resulting organic layer was concentrated under vacuum at temperature <40°C to get residual mass. Charged ethyl acetate (70 mL) to above residual mass and refluxed for 1 hr. Cooled to 25-30°C and stirred for 2 hr. Filtered the reaction mass and wash wet cake with ethyl acetate (10 mL). Suck dried for 30 min, dried initially at 25-35°C for 1 hr and then at 50-55°C for 6 hr to provide 12 gms of crude vildagliptin.
HPLC Purity: 99.11%
Dimer impurity content: 0.50%; R-isomer content (by chiral HPLC): not detected
1- aminoadamantane-3-ol content (by GC): 2.09%.
EXAMPLE 4; Preparation of Vildagliptin (using K2HP04 buffer and KI)
·
To l-aminoadamantane-3-ol (19.4 g) taken in DM Water (100 mL), added K2HP04 (10.1 gms), potassium iodide (0.1 gm) and stirred for 15 rnins at 25-35°C. (2S)-l-(Chloroacetyl)-
2- Pyrrolidine carbonitrile (10 gms) was added at 25-35°C and stirred for 15 mins at 25- 35°C. Raised the reaction mass temperature to 40-45°C and stirred for 8-10 hr at 40-45°C. After reaction completion, cooled to 25-30°C and filtered the reaction mass to remove salts. The resulting filtrate was extracted with DCM, and the resulting organic layer was concentrated initially by atmospheric distillation and later under vacuum at temperature 30- 40°C to get residual mass. Charged ethyl acetate (50 mL) to above residual mass and refluxed for 1 hr. Cooled to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed the wet cake with ethyl acetate (10 mL). Suck dried for 30 min, dried initially at 25-35°C for 1 hr and then at 50-55°C for 6 hr to provide 12 gms of crude vildagliptin.
HPLC Purity: 96.54%
Dimer impurity content: 2.55%;
R-isomer content (by chiral HPLC): not detected
l-aminoadamantane-3-ol content (by GC): 0.86%.
Example 5: Purification of Vildagliptin.
Vildagliptin crude (100 gms) was dissolved in isopropanol (900 mL) by heating to 50-55°C and stirred for 30 min. Filtered the reaction mass over hyflo bed (10 gms) at 50-55°C and washed the hyflo bed with hot isopropanol (100 mL). Distilled out solvent under vacuum at
35-40°C up to 4 volumes remains and allowed to cool to 20-25°C and stirred for 1 hr at same temperature. Further, allowed to cool to 5-10°C, stirred for 2 hrs, filtered and washed with isopropanol (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to provide 80 gms of pure vildagliptin.
HPLC Purity: 99.89%;
Dimer impurity content: <0.1 %;
R-isomer content (by chiral HPLC): not detected
l-aminoadarnantane-3-ol content (by GC): 0.06%.
The purified vildagliptin (I) was analyzed by powder X-ray diffraction (PXRD) and is set forth in Figure. 01.
EXAMPLE 6: Preparation of Vildagliptin To a solution of L-Prolinamide (100 gms) dissolved in DCM (1000 mL) was added triethyl amine (88.6 gms) and DMAP (1.07 gms) at 25-30°C under N2 atmosphere and stirred for 15 min at 25-30°C. This solution was added to a solution of chloroacetyl chloride (118.7 gms) in DCM (500 mL) under N2 atmosphere at -5 to 0°C over 2-4 hr. Heated the reaction mass temperature to 10-15°C and stirred until reaction completion, charged phosphorus oxychloride (201.5 gms) to the reaction mass at 0-5°C, heated the reaction mass temperature to reflux and stirred for 6hr at same temperature. After reaction completion, allowed to cool to 5-15°C and slowly added DM water (500 mL). Aqueous layer was separated and the organic layer was washed with DM water. To the organic layer, DM water (300 mL) was added at 25-30°C and adjusted the reaction mass pH to 6.5-7.5 with -200 mL of sodium bicarbonate solution (-16 g of NaHC03 dissolved in 200 mL of DM Water). Separated the aqueous layer and concentrated the organic layer under vacuum at temperature of 30-40°C to get residual mass. The residual mass was dissolved in DM Water (640 mL), charged l-aminoadamantane-3-ol (310.6 g) at 25-35°C. Heated the reaction mass temperature to 40-45 °C and stirred for 9 hr at the same temperature. After reaction completion, allowed to cool to 25-30°C and charged DM water (900 mL) and DCM (1280 mL). Separated the organic layer and extracted the aqueous layer with DCM. The aqueous layer was separated and kept aside for l-aminoadamantane-3-ol recovery. The total organic layer was treated with P.S. 133 carbon, stirred for 30 rnins at 25-30°C and filtered over hyflo bed. The resulting filtrate was concentrated under, vacuum at temperature 30-40°C to get residual mass. To the residual mass, charged ethyl acetate (128 mL) and distilled completely under vacuum at 30-40°C to get semi solid mass. Charged ethyl acetate (640 mL) to the obtained semi solid and refluxed for 1 hr. The reaction mass was allowed to cool to 25-30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (128 mL) to obtain wet cake. Again charged ethyl acetate (512 mL) to the obtained wet cake and refluxed for 1 hr. The reaction mass was allowed to cool to 25- 30°C and stirred for 2 hr. Filtered the reaction mass and washed with ethyl acetate (128 mL) and then dried at 50-55°C for 6 hr to provide 175 gms of crude vildagliptin.
HPLC Purity: 99.66%.
Dimer impurity content: <0.2%;
R-isomer content (by chiral HPLC) : <0.1 %;
l-aminoadamantane-3-ol content (by GC): <0.7%.
DSC: 150.12°C.
EXAMPLE 7: Purification of Vildagliptin. Vildagliptin crude (100 gms) was dissolved in isopropanol (1100 mL) by heating to 50- 55°C and stirred for 30 min. Filtered the solution over hyflo bed at 50-55°C and wash with hot isopropanol (100 mL). Distilled out solvent under vacuum at <55°C up to 5 volumes remains and allowed to cool to 20-25 °C and stirred for 1 hr at same temperature. Further allowed to cool to 10-15 °C, stirred for 2 hrs, filtered and washed with chilled isopropanol (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to provide 80 gms of pure vildagliptin. HPLC Purity: >99.8%;
Dimer impurity content: <0.1%;
R-isomeri content (by chir‘al HPLC) : <0.1%;
l-aminoadamantane-3-ol content (by GC): <0.1%.
DSC: 151.92°C.
Example 8: Recovery of l-aminoadamantane-3-ol of formula (IV).
To aqueous layer (1700 mL) from example 1, 50% C.S.lye (435 mL) was added to adjust the pH to 13.0-14.0 at 25-35°C and stirred for 15 mins at 25-35°C. Raised the reaction mass temperature to 60-70°C and stirred for 3 hrs. Cooled to 25-35°C and added DCM (1700 mL), stirred for 15 min and separated the organic layer. The aqueous layer was extracted with DCM and the total organic layer was distilled out completely under vacuum at <40°C to get semisolid mass. Charged ethyl acetate (150 mL) and distilled out solvent completely under vacuum at <50°C to get semisolid material. Charged ethyl acetate (400 mL), stirred for 30 min at 40-45°C and cooled to 25-35°C. Further allowed to cool to 0- 5°C, stirred for 2hr, filtered the reaction mass at 5-10°C and washed with ethyl acetate (100 mL). The wet product was dried at 50-55°C under vacuum for 8 hr to obtain 140 gms of 1- aminoadamantane-3-ol.
Purity by GC: 99.8 %.PATENTS AND PAPERS
Reference:1. WO2004092127A1.
2. WO0034241A1.
3. J. Med. Chem. 2003, 46, 2774-2789.
4. WO2010022690A2.
5. WO2011012322A2.
6. WO2011101861A1.
Reference:1. Beilstein J. Org. Chem. 2008, 4, 20.
Reference:1. WO2011101861A1.
Reference:1. WO2011101861A1.
Reference:1. WO2011101861A1.
Reference:1. WO2012004210A1.
SYN

PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0040403917309176
An original synthesis of vildagliptin ((S)-1-[2-(3-hydroxyadamantan-1-ylamino)acetyl]pyrrolidine-2-carbonitrile), a powerful DPP-4 inhibitor, was developed. Vildagliptin was assembled from 3-amino-1-adamantanol, glyoxylic acid and l-prolinamide in a 4-step reaction sequence with the isolation of only two intermediates. The procedure is competitive with existing protocols, leading to vildagliptin in 63% overall yield.



PAPER
A Facile and Economical Method to Synthesize Vildagliptin
Author(s): Yu Deng, Anmin Wang, Zhu Tao, Yingjie Chen, Xinmei Pan, Xiangnan Hu
Journal Name: Letters in Organic Chemistry
Volume 11 , Issue 10 , 2014
DOI : 10.2174/1570178611666140922121805

A mild and economical method to prepare vildagliptin had been reported with a good yield. In this paper, vildagliptin was synthesized from L-proline and 3-amino-1-adamantanol through chloride acetylation, amination, dehydration and substitution. The total yield of the target compound was 59%.
References
- ^ WHO International Working Group for Drug Statistics Methodology (August 27, 2008). “ATC/DDD Classification (FINAL): New ATC 5th level codes”. WHO Collaborating Centre for Drug Statistics Methodology. Archived from the original on May 6, 2008. Retrieved September 5, 2008.
- ^ Jump up to:a b Ahrén, B; Landin-Olsson, M; Jansson, PA; Svensson, M; Holmes, D; Schweizer, A (2004). “Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes”. The Journal of Clinical Endocrinology and Metabolism. 89 (5): 2078–84. doi:10.1210/jc.2003-031907. PMID 15126524.
- ^ Jump up to:a b Mentlein, R; Gallwitz, B; Schmidt, WE (1993). “Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum”. European Journal of Biochemistry / FEBS. 214 (3): 829–35. doi:10.1111/j.1432-1033.1993.tb17986.x. PMID 8100523.
- ^ “EU approves Novartis’s Eucreas diabetes drug”. Reuters. February 25, 2008.
- ^ “Galvus” (PDF). http://www.ema.europa.eu. Retrieved July 29, 2018.
- ^ Matveyenko AV, Dry S, Cox HI, et al. (July 2009). “Beneficial endocrine but adverse exocrine effects of sitagliptin in the human islet amyloid polypeptide transgenic rat model of type 2 diabetes: interactions with metformin”. Diabetes. 58 (7): 1604–15. doi:10.2337/db09-0058. PMC 2699878. PMID 19403868.
- ^ Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC (July 2013). “Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors”. Diabetes. 62 (7): 2595–604. doi:10.2337/db12-1686. PMC 3712065. PMID 23524641.
- ^ Egan, Amy G.; Blind, Eberhard; Dunder, Kristina; De Graeff, Pieter A.; Hummer, B. Timothy; Bourcier, Todd; Rosebraugh, Curtis (2014). “Pancreatic Safety of Incretin-Based Drugs — FDA and EMA Assessment — NEJM” (PDF). New England Journal of Medicine. 370 (9): 794–7. doi:10.1056/NEJMp1314078. PMID 24571751.
External links
- “Vildagliptin”. Drug Information Portal. U.S. National Library of Medicine.
- Banting and Best Diabetes Centre at UT vildagliptin – Vildagliptin
- Banting and Best Diabetes Centre at UT dpp4 – About DPP-4
- The race to get DPP-4 inhibitors to market – Forbes.com
- Merck’s March Madness – Forbes.com
| Clinical data | |
|---|---|
| Trade names | Galvus, Xiliarx, Jalra, others |
| Other names | LAF237 |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by INN |
| Pregnancy category | Not recommended |
| Routes of administration | By mouth |
| ATC code | A10BH02 (WHO) A10BD08 (WHO) (with metformin)[1] |
| Legal status | |
| Legal status | UK: POM (Prescription only)EU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 85% |
| Protein binding | 9.3% |
| Metabolism | Mainly hydrolysis to inactive metabolite; CYP450 not appreciably involved |
| Elimination half-life | 2 to 3 hours |
| Excretion | Kidney |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 274901-16-5 |
| PubChem CID | 6918537 |
| IUPHAR/BPS | 6310 |
| DrugBank | DB04876 |
| ChemSpider | 5293734 |
| UNII | I6B4B2U96P |
| KEGG | D07080 |
| ChEMBL | ChEMBL142703 |
| CompTox Dashboard (EPA) | DTXSID80881091 |
| ECHA InfoCard | 100.158.712 |
| Chemical and physical data | |
| Formula | C17H25N3O2 |
| Molar mass | 303.406 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Solubility in water | Freely Soluble in water mg/mL (20 °C) |
| SMILES[hide]N#C[C@H]4N(C(=O)CNC13CC2CC(C1)CC(O)(C2)C3)CCC4 | |
| InChI[hide]InChI=1S/C17H25N3O2/c18-9-14-2-1-3-20(14)15(21)10-19-16-5-12-4-13(6-16)8-17(22,7-12)11-16/h12-14,19,22H,1-8,10-11H2/t12?,13?,14-,16?,17?/m0/s1 Key:SYOKIDBDQMKNDQ-XWTIBIIYSA-N |
////////VILDAGLIPTIN, LAF 237,NVP LAF 237, ビルダグリプチン , GALVUS, EQUA, NOVARTIS, DIABETES
OC12CC3CC(C1)CC(C3)(C2)NCC(=O)N1CCC[C@H]1C#N
Reference:
[1]. Japan, PMDA.
[2]. Drug@EMA, EMEA/H/C/000771 Galvus : EPAR – Scientific Discussion.
[3]. Postgrad. Med. J. 2008, 84, 524-531.
[4]. Diabetes Obes. Metab. 2011, 13, 7-18.
[5]. Diabetes Metab. 2012, 38, 89-101.
[6]. Formulary 2008, 43, 122-124, 131-134.
[7]. Br. J. Diabetes Vasc. Dis. 2008, 8, S10-S18.
[8]. Drugs 2011, 71, 1441-1467.
[9]. The relevance of off-target polypharmacology; John Wiley & Sons, Inc., 2012.
[10]. Int. J. Clin. Pract. Suppl. 2008, 62, 8-14.
[11]. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 479-486.
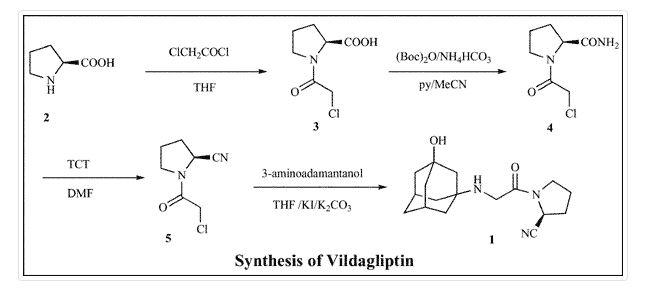
A Facile and Economical Method to Synthesize Vildagliptin
Author(s): Yu Deng, Anmin Wang, Zhu Tao, Yingjie Chen, Xinmei Pan and Xiangnan Hu
Volume 11 , Issue 10 , 2014
Page: [780 – 784]Pages: 5
DOI: 10.2174/1570178611666140922121805
Letters in Organic Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}