
Home » 2021 (Page 2)
Yearly Archives: 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CITICOLINE
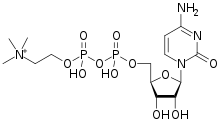
![[(2R,3S,4R,5R)-5-(4-amino-2-oxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[2-(trimethylazaniumyl)ethoxy]phosphoryl] phosphate.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=11583971&t=l)
CITICOLINE
- Molecular FormulaC14H26N4O11P2
- Average mass488.324 Da
5′-O-[Hydroxy({[2-(trimethylammonio)ethoxy]phosphinato}oxy)phosphoryl]cytidine
987-78-0[RN]
1-{5-O-[({Hydroxy[2-(trimethylammonio)ethoxy]phosphoryl}oxy)phosphinato]-β-D-ribofuranosyl}-4-imino-1,4-dihydro-2-pyrimidinol
213-580-7[EINECS], 2290
2-Pyrimidinol, 1,4-dihydro-1-[5-O-[hydroxy[[hydroxy[2-(trimethylammonio)ethoxy]phosphinyl]oxy]phosphinyl]-β-D-ribofuranosyl]-4-imino-, inner salt
CiticolineCAS Registry Number: 987-78-0
CAS Name: Cytidine 5¢-(trihydrogen diphosphate) P¢-[2-(trimethylammonio)ethyl] ester inner salt
Additional Names: choline cytidine 5¢-pyrophosphate (ester); cytidine diphosphate choline ester; CDP-choline
Trademarks: Difosfocin (Magis); Nicholin (Wyeth); Recognan (Asahi); Rexort (Hoechst); Somazina (Ferrer)
Molecular Formula: C14H26N4O11P2, Molecular Weight: 488.32
Percent Composition: C 34.43%, H 5.37%, N 11.47%, O 36.04%, P 12.69%
Literature References: Naturally occurring nucleotide; intermediate in the major pathway of lecithin biosynthesis. Identification: E. P. Kennedy, S. B. Weiss, J. Am. Chem. Soc.77, 250 (1955).Crystallization from yeast extract: I. Lieberman et al.,Science124, 81 (1956).Synthesis: E. P. Kennedy, J. Biol. Chem.222, 185 (1956); K. Kikugawa et al.,Chem. Pharm. Bull.19, 1011, 2466 (1971). Molecular structure: M. A. Viswamitra et al.,Nature258, 497 (1975). Series of articles on pharmacology and toxicology: Arzneim.-Forsch.33, 1009-1080 (1983). Acute toxicity: T. Grau et al.,ibid. 1033. Clinical trial in ischemic stroke: W. M. Clark et al.,Neurology49, 671 (1997).Review of biosynthesis, metabolism, pharmacology: G. B. Weiss, Life Sci.56, 637-660 (1995); and clinical experience: J. J. Secades, G. Frontera, Methods Find. Exp. Clin. Pharmacol.17, Suppl. B, 1-54 (1995).Properties: Amorphous, somewhat hygroscopic powder. [a]D25 +19.0° (c = 0.86 in H2O). uv max (pH 1): 280 nm (e 12800). Dissolves readily in water to form acidic soln. Practically insol in most organic solvents. pKa 4.4. LD50 in mice, rats (mg/kg): 4600 ±335, 4150 ±370 i.v.; both species 8 g/kg orally (Grau).
pKa: pKa 4.4Optical Rotation: [a]D25 +19.0° (c = 0.86 in H2O)
Absorption maximum: uv max (pH 1): 280 nm (e 12800)
Toxicity data: LD50 in mice, rats (mg/kg): 4600 ±335, 4150 ±370 i.v.; both species 8 g/kg orally (Grau)
Derivative Type: Sodium saltCAS Registry Number: 33818-15-4
Trademarks: Acticolin (Upsamedica); Brassel (Searle); Ceraxon (Ferrer); Neuroton (Berlin-Chemie); Sintoclar (Pulitzer)
Molecular Formula: C14H25N4NaO11P2, Molecular Weight: 510.31
Percent Composition: C 32.95%, H 4.94%, N 10.98%, Na 4.51%, O 34.49%, P 12.14%
Properties: White, crystalline, spongy, hygrosopic powder, dec 250°. [a]D20 +12.5° (c = 1.0 in H2O). Sol in water. Practically insol in alcohol.
Optical Rotation: [a]D20 +12.5° (c = 1.0 in H2O)
Therap-Cat: Neuroprotective. In treatment of ischemic stroke and head trauma.
Keywords: Neuroprotective.
Citicoline (INN), also known as cytidine diphosphate-choline (CDP-Choline) or cytidine 5′-diphosphocholine is an intermediate in the generation of phosphatidylcholine from choline, a common biochemical process in cell membranes. Citicoline is naturally occurring in the cells of human and animal tissue, in particular the organs.
Studies suggest that CDP-choline supplements increase dopamine receptor densities.[1] Intracerebroventricular administration of citicoline has also been shown to elevate ACTH independently from CRH levels and to amplify the release of other HPA axis hormones such as LH, FSH, GH and TSH in response to hypothalamic releasing factors.[2] These effects on HPA hormone levels may be beneficial for some individuals but may have undesirable effects in those with medical conditions featuring ACTH or cortisol hypersecretion including PCOS, type II diabetes and major depressive disorder.[3][4]
Citicoline was originally developed in Japan for stroke. Citicoline or its sodium salt was later introduced as a prescription drug in many European countries. In these countries it is now frequently prescribed for thinking problems related to circulation problems in the brain. In the US, citicoline is marketed as a dietary supplement. Citicoline or its sodium salt is used for Alzheimers disease and other types of dementia, head trauma, cerebrovascular disease such as stroke, age-related memory loss, Parkinsons disease, and glaucoma.
Citicoline sodium is chemically known as 5-0-[hydroxy({hydroxy[2-(trimethylammonio)ethoxy]phosphoryl}oxy)phosphoryl]cytidine sodium which is represented by formula I,
There are many process described in the art for the preparation of citicoline. Japanese patent 51028636 describes a process for the preparation of citicoline by neutralisation of Calcium salt of phosphorylcholine chloride with 98% H2SO4 to make phosphorylcholine chloride, which is further treated with cytidine-5-phosphate in presence of DCC and pyridine at 70 C to obtain citicoline hydrate. The drawback of this process is that citicoline is very unstable
in this harsh reaction condition such as formamide, 98% H2SO4 and high temperature of 70 C.
Chinese patent 1944661 describes an enzymatic process for the preparation of citicoline which involves twice pH adjustment to precipitatethe product,filtration of the solids, charcolisation, washing with pure water, eluting through chloride type ion exchange resin with water ethanol/alcohol reagents, desalting the eluate, decoloring and collecting the liquid, vacuum-concentration of the eluate by adding an alcohol solvent to get the solid to obtain the crude product and dissolving the crude product, microfiltering, ultrafiltering, adding an alcoholic solvent, to obtain the wet productand drying to obtain the final product. The primary disadvantage of this process is that the above reaction involves water and ethanol mixture for elution of ion exchange column and also vacuum concentration of water ethanol mixture which requires high energy, more time, leads to decomposition of product and also leads to the formation of more effluent hence it is not suitable for large scale production.
The primary disadvantage of this process is that the above reaction involves water and ethanol mixture for elution of ion exchange column and also vacuum concentration of water ethanol mixture which requires high energy, more time, leads to decomposition of product and also leads to the formation of more effluent hence it is not suitable for large scale production.
US20090286284 describes a microbial process for preparation of citicoline. This patent also discloses a process for purification of citicoline by passing through acidic cation exchange and anion exchange resin. The drawback of this process is that in this process citicoline is passed through cation /anion exchange resin in free form which is unstable and liable to formation of unwanted impurities. Therefore for the purification it needs very high volume of resin (200 times) and high volume (100 times) of solvent. This process further needs reconcentration of huge volume of solvents, which is time taking and energy consuming.
Chemical and Pharmaceutical Bulletin 1971, 19(5), 1011-16 describes a process for the preparation of citicoline by direct condensation of cytidine 5-
monophosphate and choline phosphate by using p-toluenesulfonyl chloride or methanesulfonyl chloride combined with DMF. After completion of reaction the mass was diluted with water, pH was adjusted with ammonia solution to 9.5 and product was purified by using Dowex-1 ion exchange resin by eluting with formic acid. Another Chemical and Pharmaceutical Bulletin 1971, 19(12), 2499-71 describes a process for the preparation of citicoline by direct condensation cytidine 5-monophosphate and choline phosphate in presence of thionyl chloride and DMF.The product obtained was further purified by using Dowex-1 ion exchange resin by eluting with formic acid.
Journal of Biological Chemistry, 1956, 185-191 describes a process for the preparation of citicoline by direct condensation5-cytidylic acid and phosphorylcholine in a mixture of water and pyridine in presence of DCC, stirred for few days by adding DCC in lots, after completion of reaction, reaction mass was diluted with water and filtered. The pH of the filtrate was adjusted 8-9 using 0.5N KOH, diluted further with water and passed through Dowex-1 formate resin by eluting with formic acid and water.
The drawbacks of these processes are that they use hazardous reagents such as p-toluenesulfonyl chloride, methanesulfonyl chloride, thionyl chloride etc. Hence they are not suitable for large scale production. Also, the prior art processes pass citicoline solution, without isolating it, to ion exchange resins for purification. During this process most of the inorganic impurities present along with citicoline or its salt pass through the column, thus making purification difficult.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
Journal of Chemical Research, 40(6), 358-360; 2016
An improved, three-step synthesis of cytidine diphosphate choline (CDP-choline) from cytidine was achieved in 68% overall yield. Selective phosphorylation of cytidine was accomplished by the use of morpholinophosphodichloridate to give cytidine-5′-phosphomorpholide, which was condensed with choline phosphate chloride in the presence of a catalytic amount of H2SO4 to give CDP-choline. The intermediates and products could be efficiently purified by recrystallisation, thus avoiding the use of chromatography at all stages. The reaction could be scaled up to 200 g in 64% overall yield, making this route attractive for industrial application.

Cytidine diphosphate choline (CDP-choline 1) is a nucleotide coenzyme and serves as a choline donor in the biosynthesis of lipids,1 lecithins,2 and sphingomyelin.3 It is a clinical drug for the treatment of several illnesses involving disturbance of the central nervous system, in particular, for regaining a patient’s consciousness and for treatment of neuropsychic symptoms occurring during skull traumas and brain surgery.4 Among various methods for the synthesis of CDP-choline in the literature, the current preferred method is via the condensation of cytidine-5’-phosphomorpholide (2) with choline phosphate chloride (3) under mild reaction conditions.5–7 Compound 2 was synthesised from 5’-cytidine monophosphate (4) and morpholine in the presence of DCC (N,N’-dicyclohexylcarbodiimide)8 or via the controlled hydrolysis of cytidine-5’-phosphodichloride (5) followed by P–N bond formation with morpholine (Scheme 1, route a).7 However, DCC is toxic and converted into urea which is difficult to separate from the mixture, thus leading to poor purity of product. Furthermore, phosphorylation with POCl3 always meets with side reactions from the 2’ or 3’ hydroxyls and detracts from the acceptance of this method in industry.9 In the context of ongoing projects on the synthesis of nucleoside drugs,10–14 herein we report the synthesis of CDP-choline via the selective phosphorylation of 6 wiResults and discussion Central to our approach for the synthesis of CDP-choline is the selective phosphorylation of 6 using sterically-hindered 7 as phosphorylation regent. 7 was synthesised by the direct phosphorylation of morpholine with POCl3 , a compound whose utility for the conversion of alcohols and amines into various phosphorylation derivatives.15 Due to the reactivity of three chloro atoms in POCl3 , gradually adding POCl3 to excess morpholine avoids the bifunctional reaction exclusively. After reaction, 7 could be purified by fractional distillation to yield as a colourless oil (b.p. 124–126 °C at 1.33 KPa). Due to the presence of the electron-donating morpholino group, 7 displays lower reactivity than POCl3 and could tolerate moisture and air better. Usefully, 7 could be synthesised on the 200 g scale and stored at 4 °C. The major concern of utilising 7 as phosphorylation reagent is its selectivity for the 5’ hydroxyl group. We therefore assessed the selectivity for 5’ hydroxylation using 6 and 7 in the presence of various organic bases. After phosphorylation, H2 O was added to destroy the excess of 7, and 2 was obtained in a single step. The solvent, the base, temperature and the ratio of substrates were evaluated and the results are summarised in Table 1
https://journals.sagepub.com/doi/pdf/10.3184/174751916X14628025243831
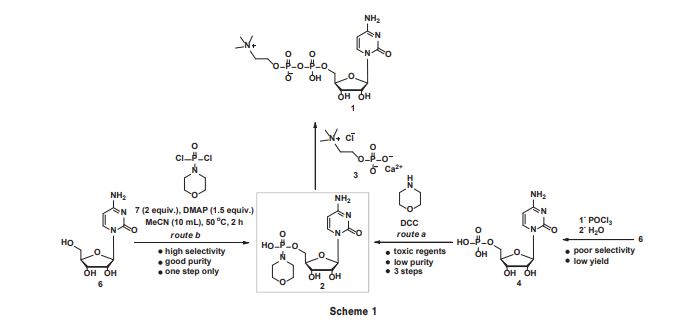

Cytidine-5’-phosphomorpholide (2): Cytidine (0.243 g, 1.0 mmol) and DMAP (0.183 g, 1.5 mmol) in MeCN (10 mL) were stirred slowly and cooled to 0 °C, and 7 (2.0 mmol) was added slowly. The mixture was heated to 50 °C and kept at this temperature for 2 h. The solvent was removed in vacuo and the residue was purified by recrystallisation from EtOH to give 2 as a white semi-solid (0.318 g); yield 81%; m.p. 62–64 °C; 1 H NMR (400 MHz, DMSO-d6 ) δ 8.43 (d, J = 7.6 Hz, 1H), 7.39 (s, 2H), 7.19 (d, J = 7.6 Hz, 1H), 5.77 (d, J = 2.8 Hz, 1H), 5.51 (d, J = 4.8 Hz, 1H), 5.18 (t, J = 5.2 Hz, 1H), 5.08 (d, J = 5.6 Hz, 1H), 3.76–3.71 (m, 1H), 3.61–3.56 (m, 1H), 3.45–3.42 (m, 4H), 3.03–2.99 (m, 4H); 13C NMR (100 MHz, DMSO-d6 ) δ 166.5, 157.8, 145.9, 141.6, 88.1, 86.2, 74.3, 70.7, 65.1, 65.0, 60.3, 60.2; HRMS calcd for C13H22N4 O8 P [M + H]+ 393.1170, found: 393.1172.
CDP-choline (1): 2 (0.392 g, 1.0 mmol) was added to MeOH (10 mL) followed by the addition of 3 (0.310 g, 1.2 mmol) and was stirred at room temperature for 10 min. Then 98% H2 SO4 (0.005 mL, 10 mol%) was added. The mixture was kept at 50 °C for 3 h. The solvent was removed in vacuo and the residue was purified by recrystallisation from EtOH to give 1 as a white solid (0.410 g); yield 85%. 1 H NMR (400 MHz, D2 O) δ 7.86 (s, 2H), 6.04 (d, J = 5.2 Hz, 1H), 5.91 (d, J = 5.2 Hz, 1H), 4.32 (brs, 2H), 4.26–4.22 (m, 2H), 4.18 (brs, 2H), 4.11 (t, J = 3.2 Hz, 1H), 3.60 (t, J = 2.4 Hz, 2H), 3.14 (s, 9H); 13C NMR (100 MHz, D2 O) δ 166.1, 157.7, 141.5, 96.6, 89.3, 82.6, 74.1, 69.3, 66.0, 65.9, 64.8, 59.9, 54.0; HRMS calcd for C14H27N4 O11P2 [M + H]+ 489.1146, found: 489.1140.
| 1H NMR | (400 MHz. D2O) δ 7.86 (s, 2H). 6.04 (d. J = 5.2 Hz, 111). 5.91 (d. J = 5.2 Hz. 1Hj, 4.32 (brs. 2H), 4.26-4.22 (m, 2H). 4.18 (brs, 2H), 4.11 (t. J = 3.2 Hz. 1H). 3.60 (t. J = 2.4 Hz. 2H), 3.14 (s, 9H) |
| 13C NMR | (100 MHz. D2O) δ 166.1. 157.7, 141.5,96.6.89.3,82.6,74.1.69.3,66.0, 65.9, 64.8. 59~9. 54.0 |
| HRMS | calcd for C14H27N4O11P2 (M + H]+ 489.1146. found: 489.1140 |
| State | white solid |
SYNKikugawa, Kiyomi; Ichino, MotonobuChemical & Pharmaceutical Bulletin (1971), 19, (5), 1011-16.https://www.jstage.jst.go.jp/article/cpb1958/19/5/19_5_1011/_pdf/-char/enCytidine diphosphate choline (CDP-choline), one of the nucleotide coenzymes, is known to be a precursor of phospholipid and play an important role in the living organisms. The coenzyme was synthesized in a fairly good yield by direct condensation of cytidine-5′ monophosphate (5′-CMP) and choline phosphate (P-choline) by the use of p-toluenesulfonyl chloride or methanesulfonyl chloride combined with dimethylformamide
Method B, with Methanesulfonyl Chloride and DMF: A mixture of 1.3g (11.5 mmole) of methanesulfonyl chloride and 3ml of DMF was added to the gummy mixture containing 10 mmole of P-choline (II). It was shaken at room temperature for 10 min, and 1.0g (3.1 mmole) of 5′-CMP (I) was added to the viscous solution. It was then stirred at room temperature for one hour. Paper chromatography and paper electrophoresis of the reaction mixture showed that CDP-choline (III) was a major reaction product. The separa tion, isolationand identification of the product (III) were same as in method A. Crystalline white powder of CDP-choline was obtained in a yield of 50.0%. Method C, with p-Toluenesulfonyl Chloride and HMPA: A mixture of 2.2g (11.5 mmole) of p-toluene sulfonyl chloride and 3ml of HMPA was added to the gummy mixture containing 10 mmole of P-choline (II). 5′-C1IP (I) (1.0g, 3.1 mmole) was reacted under the same condition as in method A, and isolation was performed similarly. Crystalline powder of CDP-choline (III) was obtained in a yield of about 10%. Method D according to the Morpholidate Method 6): 5′-CMP-Morpholidate (4-morpholine-N, N’-dicyclohexylcarboxamidinium salt) (1.28g, 2 mmole) was reacted with 8 mmole of P-choline (II) according to the method of Tanaka, et al. 6) Separation and isolation of the product were similarly performed as in method A. Crystalline powder of the authentic CDP-choline was obtained in a yield of 55%. CDP-Choline Monosodium Salt Monosodium salt of CDP-choline (III) was prepared from the product (III) obtained by method A. Thus, 200mg of CDP-choline (III) was dissolved in 1.0ml of water, and after the pH of the solution was adjusted to 6.0 with 2N NaOH, 3ml of ethanol was added. Crystallization occurred after standing at room temperature overnight to afford plates of 130 mg of CDP-choline monosodium salt. Determination of the Yield of CDP-Choline (III) in the Condensation with p-Toluenesulfonyl Chloride and DMF In the condensation reaction using p-toluenesulfonyl chloride and DMF, the effects of the amount of p-toluenesulfonyl chloride and the reaction temperature were examined. 5′-CMP (I) (1.0g, 3.1 mmole) was added to the mixture of 10 mmole of P-choline (II), 3ml of DMF and p-toluenesulfonyl chloride which were previously mixed and treated at room temperature for 10 min. The reaction mixtures were stirred at 25•‹for one hour with the varying amounts of p-toluenesulfonyl chloride of 1.5g (7.9 mmole), 1.9g (10 mmole), 2.2g (11.5 mmole), 3g (15.8 mmole), 4g (21.0 mmole) and 5g (26.3 mmole). The yields of the compound (III) estimated were 30, 50, 60, 49, 44 and 37% respectively.
SYN
Indian Pat. Appl., 2014MU00923

SYN
CN 111647636

Syn
Biotechnology and Bioengineering, 117(5), 1426-1435; 2020
https://onlinelibrary.wiley.com/doi/10.1002/bit.27291
Cytidine-5′-diphosphocholine (CDP-choline) is a widely used neuroprotective drug for multiple indications. In industry, CDP-choline is synthesized by a two-step cell culture/permeabilized cell biotransformation method because substrates often do not enter cells in an efficient manner. This study develops a novel one-step living cell fermentation method for CDP-choline production. For this purpose, the feasibility of Pichia pastoris as a chassis was demonstrated by substrate feeding and CDP-choline production. Overexpression of choline phosphate cytidylyltransferase and choline kinase enhanced the choline transformation pathway and improved the biosynthesis of CDP-choline. Furthermore, co-overexpression of ScHnm1, which is a heterologous choline transporter, highly improved the utilization of choline substrates, despite its easy degradation in cells. This strategy increased CDP-choline titer by 55-folds comparing with the wild-type (WT). Overexpression of cytidine-5′-monophosphate (CMP) kinase and CDP kinase in the CMP transformation pathway showed no positive effects. An increase in the ATP production by citrate stimulation or metabolic pathway modification further improved CDP-choline biosynthesis by 120%. Finally, the orthogonal optimization of key substrates and pH was carried out, and the resulting CDP-choline titer (6.0 g/L) at optimum conditions increased 88 times the original titer in the WT. This study provides a new paradigm for CDP-choline bioproduction by living cells.
SYN
Citicoline sodium is a chemically designate as Cytidine 5’-(trihydrogendiphosphate) P’-[2-(trimethylammonio) ethyl] ester monosodium salt, its molecular formula is C14H25N4NaO11P2 and molecular weight is 510.31(salt) and 488.32 (base- C14H26N4O11P2). It is a white crystalline, hygroscopic powder and readily soluble in water but practically insoluble in alcohol. Its melting point was 259 – 268°C and dissociation constant (Pka) was 4.4 [1]. Biopharmaceutical classification system (BCS) for Citicoline is Class – I (High solubility and High Permeability) [3]. Citicoline has a broad spectrum of therapeutic index, as a Neuroprotectant or Cerebroprotectant, in particular citicoline is useful the victims of ischemic stroke, head trauma and neurodegenerative disease. Citicoline is also used to treat unconsciousness resulting from cerebral thrombosis, hemorrhages, demyelinating diseases, cranial trauma and cerebropathies due to atherosclerosis [2]. Citicoline was originally developed in Japan for stroke. It was later introduced as a prescription drug in many European countries. In these countries it is now frequently prescribed for thinking problems related to circulation problems in the brain. In the US, citicoline is marketed as a dietary supplement [3]. Citicoline daily dosages may range from 250 mg to about 3000 mg and more preferably from 500 mg to about 2000 mg up to four or more times daily, duration of the treatment may vary from several weeks to several years, dosages may be varied over time depending on the severity of symptoms [4].

SYN
192/MUM/2012
The present invention discloses a novel, cost-effective process for preparing psychostimulant drug cytidinediphosphate-choline (CDP-Choline) commonly known as citicoline. The process comprises reacting cytidine 5-monophosphate with morpholine in presence of a coupling reagent and an organic solvent to form morpholidate compound; condensing morpholidate compound with calcium salt of phosphorylcholinehalide in presence of an acid to form citicoline calcium chloride; and purifying the citicoline calcium chlorideby passing through cationic and anionic resinsand eluting by water to form citicoline sodium of formula I.
Example:
(a) Preparation of citicoline calcium chloride:
S-Cytidine mono phosphate (1.25 kg)and morpholine (1.12 kg) were added into methanol {6.25 L) and DCC (1.50 kg) at 25 to 35 C.The reaction mixture was heated to 50 to 55 C and stirred for 7 hours. After completion of the reaction, the reaction mass was cooled to 25 to 35 C and the obtained reaction mass was added slowly to phosphoryl choline chloride calcium salt (1.9 kg) in methanol (8.75 L) solution. The pH was maintained to 3.8 to 4.2 using HC1 gas in IPA and stirred for 6 hours at 25 to 35 . The reaction mass was further heated to 45 to 5Q C. After completion of reaction the yeactkm mass was cooled and stirced for 1 hour. The product was filtered, washed with chilled methanol at 0 to 5 Cand suck dried to obtain citicoline calcium chloride.
Yield; 3.70-4.0 kg (b) Preparation of citicoline sodium:
The above obtained crude citicolinecalcium chloride was dissolved in water (6.25 L), filtered, washed with water and suck dried. Filtrate containing the product was re-filtered through Hyflo bed. The clear filtrate was eluted through column containing acidic cation exchange resins (12.5 L). The material was washed with water. The eluent was further passed through anion exchange resin (12.5 L). column and washed with water.
Complete aqueous solution after the passing through an-ion exchange resin was collected, pH of the solution was adjusted to 6.5 to 7.0 using 30 % sodium hydroxide solution (0.3Kg in 0.45L) and solution was concentrated using reverse osmosis. The solution was cooled to 25 to 35 C and charcoalated. The solution was filtered through hyflo bed at 25 to 35 C, washed with water. The solution was further filtered through ultra-filter at 25 to 35 C.
Clear filtrate and mixture of isopropanol and Methanol (1:1) (25 L) were stirred, the reaction mass was cooled to 0 to 5 C, and stirred for 2 hours. The product was filtered under nitrogen atmosphere, solid was washed with the mixture of IPA and methanol (1:1) (1.25 L) at 0 to 5 C and dried under vacuum below 95 C until moisture/LOD is less the 2.0%.
Yield: 1 to 1.2 kg

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
https://patents.google.com/patent/CN1944661A/enEmbodiment 1:With 30 kilograms of quick-frozen yeast, 3 kilograms of phosphorylcholines, 1 kilogram 5 ‘-cytidylic acid, 10 kilograms of glucose, 2 kilograms of potassium hydroxide, 800 kg of water are mixed back temperature adjustment to 25 ℃, PH=6 carries out 65 rev/mins of stirring reactions and it was fully reacted in 6 hours; Reaction solution is warming up to 50 ℃ of deactivations, carries out liquid-solid separation; Transfer PH=8.0, part basic protein and nucleic acid precipitation are carried out liquid-solid separation, and then are transferred PH=2.5, make the acidic protein precipitation, carry out liquid-solid separation, sediment separate out; Use Activated Carbon Adsorption Separation, PH=2.5 washs with pure water; Carry out wash-out with the molten reagent of ethanol alkali, elutriant carries out desalination, decolouring is handled, and collects liquid; The elutriant vacuum concentration; Concentrated solution adds 2 times of ethanol, crystallization, liquid-solid separate crude product; Dissolving crude product, ultrafiltration behind the micro-filtration adds 2 times of ethanol, crystallization, liquid-solid separate wet product, after the drying finished product.Embodiment 2:With 80 kilograms of quick-frozen yeast, 4 kilograms of phosphorylcholines, 4 kilogram 5 ‘-cytidylic acid, 16 kilograms of glucose, 4 kilograms of potassium hydroxide, 1100 kg of water are mixed back temperature adjustment to 30 ℃, and add 0.5 kilogram of MgSO 4Solution, PH=6 carries out 120 rev/mins of stirring reactions and it was fully reacted in 8 hours; Reaction solution is warming up to 70 ℃ of deactivations, carries out liquid-solid separation; Transfer PH=10, part basic protein and nucleic acid precipitation are carried out liquid-solid separation, and then are transferred PH=4, make the acidic protein precipitation, carry out liquid-solid separation, sediment separate out; Use Activated Carbon Adsorption Separation, PH=4 washs with pure water; Carry out wash-out with the molten reagent of ethanol alkali, elutriant carries out desalination, decolouring is handled, and collects liquid; The elutriant vacuum concentration; Concentrated solution adds 2 times of methyl alcohol, crystallization, liquid-solid separate crude product; Dissolving crude product, ultrafiltration behind the micro-filtration adds 2 times of methyl alcohol, crystallization, liquid-solid separate wet product, after the drying finished product.Embodiment 3:With 100 kilograms of quick-frozen yeast, 8 kilograms of phosphorylcholines, 5 kilogram 5 ‘-cytidylic acid, 20 kilograms of glucose, 5 kilograms of potassium hydroxide, 1500 kg of water are mixed back temperature adjustment to 40 ℃, and add 6 kilograms of MgSO 4Solution, PH=8 carries out 150 rev/mins of stirring reactions and it was fully reacted in 10 hours; Reaction solution is warming up to 90 ℃ of deactivations, carries out liquid-solid separation; Transfer PH=12.0, part basic protein and nucleic acid precipitation are carried out liquid-solid separation, and then are transferred PH=5.5, make the acidic protein precipitation, carry out liquid-solid separation, sediment separate out; Use Activated Carbon Adsorption Separation, PH=5.5 washs with pure water; Carry out wash-out with the molten reagent of ethanol alkali, elutriant carries out desalination, decolouring is handled, and collects liquid; The elutriant vacuum concentration; Concentrated solution adds 2 times of first and second alcoholic solution, crystallization, liquid-solid separate crude product; Dissolving crude product, ultrafiltration behind the micro-filtration adds 2 times of first and second alcoholic solution, crystallization, liquid-solid separate wet product, after the drying finished product.
PATENThttps://patents.google.com/patent/WO2013128393A1/enCiticoline (CDP-Choline), naturally occurring nucleotide, is a neuroprotective indicated for the treatment of ischemic stroke and head trauma in patients. Citicoline (CDP-Choline) is represented by formula (I).

US patent no. 3,666,748 discloses a process for preparing Citicoline sodium by reaction of 4- morpholino-N,N’-dicyclohexylcarboxamidine chloride salt of choline phosphormorpholidate (I) with cytidine-5-monophosphate in free form or its salts with base in a solvent such as o- chlorophenol, m-cresol, acetonitrile, pyridine and the like. The Citicoline thus obtained is purified through a column chromatograph packed with activated carbon followed by elution to get ammonium salt of citicoline, which is further converted to citicoline followed by citicoline sodium.US patent no. 3,787,392 discloses a process for preparing Citicoline by adding the acidic calcium phosporyl choline chloride tetra hydrate to the solution of morpholidiate cytidine 5- monophosphate and DCC in methanol followed by isolation and purification by means of chromatography column containing anion exchanger (Dowex 1×2 type formate form; 50-100 mesh) which is further converted to its sodium salt by neutralizing with sodium hydroxide. Further, US patent no. 3,803,125 discloses a process for preparing citicoline by reacting morpholidiate cytidine 5 ‘-monophosphate with calcium phosporyl choline chloride tetra hydrate in solvent system of an aliphatic alcohol or dialkyl ketone or dimethyl formamide at pH from 1 to 6.5. The product thus obtained is further isolated; purified by means of chromatography column containing anion exchanger; concentrated; and neutralized with aqueous solution of sodium hydroxide to get citicoline sodium.Example 1To a solution of calcium phosphoryl choline chloride tetra hydrate (50.0 gm) in water, a solution of oxalic acid in RO water (19.5 gm oxalic acid in 90 ml RO water) was added at 45- 50°C. The reaction mass was filtered and distilled out to get residue followed by addition of methanol. To the above solution, solution of morpholine and DCC in methanol was added. The temperature of the reaction was raised to 50-55°C and to this, solution of cytidine 5′- monophospahte in methanol (12.2 gm in 40 ml methanolic HCl and 20 ml methanol) was added and reaction was maintained. The pH 3.5 of reaction mixture was maintained by methanolic HCl. Reaction mass was cooled and IPA was added after completion of the reaction. The precipitated product, citicoline, was filtered and dried. The crude Citicoline (16.0 gm) was dissolved in water and treated with charcoal to get the purified Citicoline acid which on reaction with aqueous sodium hydroxide gave Citicoline Sodium with purity > 99%.Example 2To the solution of cytidine 5′-monophospahate (5′-CMP) (100 gm) in methanol (750 ml), solution of morpholine (75 gm) and DCC (100 gm) in methanol was added at room temperature. The temperature of the reaction was raised to 50-55°C for a time period of 3-7 hrs followed by cooling the reaction mass and filtered to get morpholidiate cytidine 5’- monophospahate in mother liquor. To this, solution of calcium phosphoryl choline chloride (200 gm) in methanol was added and the temperature of reaction mass was raised to 50-55°C and maintained at pH of 3.5 by methanolic HCl. The reaction mass was cooled and filtered to get crude Citicoline by adding IPA. Further, morpholidiate salt of oxalic acid (138.3 gm) was added to the solution of crude citicoline in methanol at 30-35°C followed by the addition of IPA to get the precipitated Citicoline, which is further treated with activated charcoal in water followed by filtration. To filtrate containing purified Citicoline, aqueous solution of sodium hydroxide was added at room temperature followed by addition of ethanol and the temperature of reaction mass was raised to 50-55°C. The precipitated product was filtered and dried where the purity of citicoline sodium is > 99% measured by HPLC. (265 gm).
ClaimsHide Dependent
We Claim:1. A process for preparing pure Citicoline (CDP-Choline), the process comprising:reacting a cytidine 5′-monophospahte or its amide salts with calcium phosphoryl choline chloride tetra hydrate or its amide salts in presence of dicyclohexyl carbodiimide (DCC) and a solvent,wherein a dicarboxylic acid or its salt is employed in the process to obtain citicoline with a purity of more than 99% measured by HPLC.2. The process as claimed in claim 1, further comprising preparing highly pure sodium salt of citicoline by reacting the pure citicoline with sodium hydroxide.3. The process as claimed in claim 1, wherein the dicarboxylic acid is used either in the form of free acid or its base salts.4. The process as claimed in any one of the preceding claims, wherein dicarboxylic acid is selected from the group consisting of oxalic acid, malonic acid, succininc acid and glutaric acid.5. The process as claimed in any one of the preceding claims, wherein the base of dicarboxylic acid is selected from the group consisting of organic bases such as amidates, amines or inorganic base such as alkali or alkaline earth metal.6. The process as claimed in claim 1, wherein the solvent is selected from the group consisting of aliphatic alcohols from C atoms, ketones such as acetone, methyl isobutyl ketone and the like or mixture thereof.7. The process as claimed in claim 1, wherein the solvent is methanol.8. The process as claimed in any of the preceding claims, wherein the dicarboxylic acid or its salts lessen the solubility of inorganic impurities such as calcium chloride, calcium hydroxide, unreacted choline phosphate, 5-CMP.
Patent
US3666748A *1967-12-181972-05-30Takeda Chemical Industries LtdMethod for production of cytidine (or deoxycytidine)-5{40 -diphosphate choline and intermediates thereforUS3787392A *1970-12-021974-01-22Boehringer Mannheim GmbhProcess for the preparation of nucleoside diphosphate estersFamily To Family CitationsCN102010454B *2010-12-022012-03-07胡建荣Citicoline sodium compound and new method thereofPublication numberPriority datePublication dateAssigneeTitleCN104031105A *2014-06-062014-09-10浙江天冉药物研究有限公司Method for preparing citicoline sodiumCN105732752A *2016-03-182016-07-06新乡学院Citicoline and synthetic method thereofCN106146590A *2016-06-292016-11-23陈建峰A kind of preparation method of C14H25N4NaO11P2CN110684066A *2019-05-222020-01-14广东金城金素制药有限公司Cytophosphocholine medicinal preparation and new application thereof in cerebral infarction acute-stage disturbance of consciousness
| Clinical data | |
|---|---|
| Trade names | Neurocoline |
| Other names | Cytidine diphosphate choline |
| AHFS/Drugs.com | International Drug Names |
| ATC code | N06BX06 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 987-78-0 |
| PubChem CID | 11583971 |
| ChemSpider | 13207 |
| UNII | 536BQ2JVC7 |
| KEGG | D00057 |
| ChEBI | CHEBI:16436 |
| ChEMBL | ChEMBL1618340 |
| CompTox Dashboard (EPA) | DTXSID9048431 |
| ECHA InfoCard | 100.012.346 |
| Chemical and physical data | |
| Formula | C14H27N4O11P2+ |
| Molar mass | 489.335 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
Use as a dietary supplement
Citicoline is available as a supplement in over 70 countries under a variety of brand names: Cebroton, Ceraxon, Cidilin, Citifar, Cognizin, Difosfocin, Hipercol, NeurAxon, Nicholin, Sinkron, Somazina, Synapsine, Startonyl, Trausan, Xerenoos, etc.[5] When taken as a supplement, citicoline is hydrolyzed into choline and cytidine in the intestine.[6] Once these cross the blood–brain barrier it is reformed into citicoline by the rate-limiting enzyme in phosphatidylcholine synthesis, CTP-phosphocholine cytidylyltransferase.[7][8]
Research
Memory and cognition
Studies have failed to confirm any potential benefits of citicoline for cognitive impairment.[9]
Ischemic stroke
Some preliminary research suggested that citicoline may reduce the rates of death and disability following an ischemic stroke.[10][11] However, the largest citicoline clinical trial to date (a randomised, placebo-controlled, sequential trial of 2298 patients with moderate-to-severe acute ischaemic stroke in Europe), found no benefit of administering citicoline on survival or recovery from stroke.[12] A meta-analysis of seven trials reported no statistically significant benefit for long-term survival or recovery.[13]
Vision
The effect of citicoline on visual function has been studied in patients with glaucoma, with possible positive effect for protecting vision.[14]
Mechanism of action

Enzymes involved in reactions are identified by numbers. See file description.
Neuroprotective effects
Citicoline may have neuroprotective effects due to its preservation of cardiolipin and sphingomyelin, preservation of arachidonic acid content of phosphatidylcholine and phosphatidylethanolamine, partial restoration of phosphatidylcholine levels, and stimulation of glutathione synthesis and glutathione reductase activity. Citicoline’s effects may also be explained by the reduction of phospholipase A2 activity.[15] Citicoline increases phosphatidylcholine synthesis.[16][17][18] The mechanism for this may be:
- By converting 1, 2-diacylglycerol into phosphatidylcholine
- Stimulating the synthesis of SAMe, which aids in membrane stabilization and reduces levels of arachidonic acid. This is especially important after an ischemia, when arachidonic acid levels are elevated.[19]
Neuronal membrane
The brain preferentially uses choline to synthesize acetylcholine. This limits the amount of choline available to synthesize phosphatidylcholine. When the availability of choline is low or the need for acetylcholine increases, phospholipids containing choline can be catabolized from neuronal membranes. These phospholipids include sphingomyelin and phosphatidylcholine.[15] Supplementation with citicoline can increase the amount of choline available for acetylcholine synthesis and aid in rebuilding membrane phospholipid stores after depletion.[20] Citicoline decreases phospholipase stimulation. This can lower levels of hydroxyl radicals produced after an ischemia and prevent cardiolipin from being catabolized by phospholipase A2.[21][22] It can also work to restore cardiolipin levels in the inner mitochondrial membrane.[21]
Cell signalling
Citicoline enhances cellular communication by increasing the availability of neurotransmitters, including acetylcholine, norepinephrine, and dopamine.[23] In simple terms, the choline component of citicoline is used to create acetylcholine, which is a primary executive neurotransmitter in the human brain. Clinical trials have found that citicoline supplementation improves attention, focus and learning in large part due to the increase in acetylcholine that results.[24]
Glutamate transport
Citicoline lowers increased glutamate concentrations and raises decreased ATP concentrations induced by ischemia. Citicoline also increases glutamate uptake by increasing expression of EAAT2, a glutamate transporter, in vitro in rat astrocytes. It is suggested that the neuroprotective effects of citicoline after a stroke are due in part to citicoline’s ability to decrease levels of glutamate in the brain.[25]
Pharmacokinetics
Citicoline is water-soluble, with more than 90% oral bioavailability.[20] Plasma levels peak one hour after oral ingestion, and a majority of the citicoline is excreted as CO2 in respiration, and again 24 hours after ingestion, where the remaining citicoline is excreted through urine.[26]
Side effects
Citicoline has a very low toxicity profile in animals and humans. Clinically, doses of 2000 mg per day have been observed and approved. Minor transient adverse effects are rare and most commonly include stomach pain and diarrhea.[17][unreliable medical source?] There have been suggestions that chronic citicoline use may have adverse psychiatric effects. However, a meta-analysis of the relevant literature does not support this hypothesis.[27][28] At most, citicoline may exacerbate psychotic episodes or interact with anti-psychotic medication.
Synthesis
In vivo
Phosphatidylcholine is a major phospholipid in eukaryotic cell membranes. Close regulation of its biosynthesis, degradation, and distribution is essential to proper cell function. Phosphatidylcholine is synthesized in vivo by two pathways
- The Kennedy pathway, which includes the transformation of choline to citicoline, by way of phosphorylcholine, to produce phosphatidylcholine when condensed with diacylglycerol.
- Phosphatidylcholine can also be produced by the methylation pathway, where phosphatidylethanolamine is sequentially methylated.[29]
References
- ^ Giménez R, Raïch J, Aguilar J (Nov 1991). “Changes in brain striatum dopamine and acetylcholine receptors induced by chronic CDP-choline treatment of aging mice”. British Journal of Pharmacology. 104 (3): 575–8. doi:10.1111/j.1476-5381.1991.tb12471.x. PMC 1908237. PMID 1839138.
- ^ Cavun S, Savci V (Oct 2004). “CDP-choline increases plasma ACTH and potentiates the stimulated release of GH, TSH and LH: the cholinergic involvement”. Fundamental & Clinical Pharmacology. 18 (5): 513–23. doi:10.1111/j.1472-8206.2004.00272.x. PMID 15482372. S2CID 33866107.
- ^ Benson S, Arck PC, Tan S, Hahn S, Mann K, Rifaie N, Janssen OE, Schedlowski M, Elsenbruch S (Jun 2009). “Disturbed stress responses in women with polycystic ovary syndrome”. Psychoneuroendocrinology. 34 (5): 727–35. doi:10.1016/j.psyneuen.2008.12.001. PMID 19150179. S2CID 13202703.
- ^ Florio P, Zatelli MC, Reis FM, degli Uberti EC, Petraglia F (2007). “Corticotropin releasing hormone: a diagnostic marker for behavioral and reproductive disorders?”. Frontiers in Bioscience. 12: 551–60. doi:10.2741/2081. PMID 17127316.
- ^ Single-ingredient Preparations (: Citicoline). In: Martindale: The Complete Drug Reference [ed.by Sweetman S], 35th Ed. 2007, The Pharmaceutical Press: London (UK); e-version. .
- ^ Wurtman RJ, Regan M, Ulus I, Yu L (Oct 2000). “Effect of oral CDP-choline on plasma choline and uridine levels in humans”. Biochemical Pharmacology. 60 (7): 989–92. doi:10.1016/S0006-2952(00)00436-6. PMID 10974208.
- ^ Alvarez XA, Sampedro C, Lozano R, Cacabelos R (Oct 1999). “Citicoline protects hippocampal neurons against apoptosis induced by brain beta-amyloid deposits plus cerebral hypoperfusion in rats”. Methods and Findings in Experimental and Clinical Pharmacology. 21 (8): 535–40. doi:10.1358/mf.1999.21.8.794835. PMID 10599052.
- ^ Carlezon WA, Pliakas AM, Parow AM, Detke MJ, Cohen BM, Renshaw PF (Jun 2002). “Antidepressant-like effects of cytidine in the forced swim test in rats”. Biological Psychiatry. 51 (11): 882–9. doi:10.1016/s0006-3223(01)01344-0. PMID 12022961. S2CID 21170398.
- ^ Gareri P, Castagna A, Cotroneo AM, Putignano S, De Sarro G, Bruni AC (2015). “The role of citicoline in cognitive impairment: pharmacological characteristics, possible advantages, and doubts for an old drug with new perspectives”. Clin Interv Aging. 10: 1421–9. doi:10.2147/CIA.S87886. PMC 4562749. PMID 26366063.
- ^ Warach S, Pettigrew LC, Dashe JF, Pullicino P, Lefkowitz DM, Sabounjian L, Harnett K, Schwiderski U, Gammans R (Nov 2000). “Effect of citicoline on ischemic lesions as measured by diffusion-weighted magnetic resonance imaging. Citicoline 010 Investigators”. Annals of Neurology. 48 (5): 713–22. doi:10.1002/1531-8249(200011)48:5<713::aid-ana4>3.0.co;2-#. PMID 11079534.
- ^ Saver JL (Fall 2008). “Citicoline: update on a promising and widely available agent for neuroprotection and neurorepair”. Reviews in Neurological Diseases. 5 (4): 167–77. PMID 19122569.
- ^ Dávalos A, Alvarez-Sabín J, Castillo J, Díez-Tejedor E, Ferro J, Martínez-Vila E, Serena J, Segura T, Cruz VT, Masjuan J, Cobo E, Secades JJ (Jul 2012). “Citicoline in the treatment of acute ischaemic stroke: an international, randomised, multicentre, placebo-controlled study (ICTUS trial)”. Lancet. 380 (9839): 349–57. doi:10.1016/S0140-6736(12)60813-7. hdl:10400.10/663. PMID 22691567. S2CID 134947.
- ^ Shi PY, Zhou XC, Yin XX, Xu LL, Zhang XM, Bai HY (2016). “Early application of citicoline in the treatment of acute stroke: A meta-analysis of randomized controlled trials”. J. Huazhong Univ. Sci. Technol. Med. Sci. 36 (2): 270–7. doi:10.1007/s11596-016-1579-6. PMID 27072975. S2CID 25352343.
- ^ Roberti G, Tanga L, Michelessi M, Quaranta L, Parisi V, Manni G, Oddone F (2015). “Cytidine 5′-Diphosphocholine (Citicoline) in Glaucoma: Rationale of Its Use, Current Evidence and Future Perspectives”. Int J Mol Sci. 16 (12): 28401–17. doi:10.3390/ijms161226099. PMC 4691046. PMID 26633368.
- ^ Jump up to:a b Adibhatla RM, Hatcher JF, Dempsey RJ (Jan 2002). “Citicoline: neuroprotective mechanisms in cerebral ischemia”. Journal of Neurochemistry. 80 (1): 12–23. doi:10.1046/j.0022-3042.2001.00697.x. PMID 11796739.
- ^ López-Coviella I, Agut J, Savci V, Ortiz JA, Wurtman RJ (Aug 1995). “Evidence that 5′-cytidinediphosphocholine can affect brain phospholipid composition by increasing choline and cytidine plasma levels”. Journal of Neurochemistry. 65 (2): 889–94. doi:10.1046/j.1471-4159.1995.65020889.x. PMID 7616250. S2CID 10184322.
- ^ Jump up to:a b Conant R, Schauss AG (Mar 2004). “Therapeutic applications of citicoline for stroke and cognitive dysfunction in the elderly: a review of the literature”. Alternative Medicine Review. 9 (1): 17–31. PMID 15005642.
- ^ Babb SM, Wald LL, Cohen BM, Villafuerte RA, Gruber SA, Yurgelun-Todd DA, Renshaw PF (May 2002). “Chronic citicoline increases phosphodiesters in the brains of healthy older subjects: an in vivo phosphorus magnetic resonance spectroscopy study”. Psychopharmacology. 161 (3): 248–54. doi:10.1007/s00213-002-1045-y. PMID 12021827. S2CID 28454793.
- ^ Rao AM, Hatcher JF, Dempsey RJ (Dec 1999). “CDP-choline: neuroprotection in transient forebrain ischemia of gerbils”. Journal of Neuroscience Research. 58 (5): 697–705. doi:10.1002/(sici)1097-4547(19991201)58:5<697::aid-jnr11>3.0.co;2-b. PMID 10561698.
- ^ Jump up to:a b D’Orlando KJ, Sandage BW (Aug 1995). “Citicoline (CDP-choline): mechanisms of action and effects in ischemic brain injury”. Neurological Research. 17 (4): 281–4. doi:10.1080/01616412.1995.11740327. PMID 7477743.
- ^ Jump up to:a b Rao AM, Hatcher JF, Dempsey RJ (Mar 2001). “Does CDP-choline modulate phospholipase activities after transient forebrain ischemia?”. Brain Research. 893 (1–2): 268–72. doi:10.1016/S0006-8993(00)03280-7. PMID 11223016. S2CID 37271883.
- ^ Adibhatla RM, Hatcher JF (Aug 2003). “Citicoline decreases phospholipase A2 stimulation and hydroxyl radical generation in transient cerebral ischemia”. Journal of Neuroscience Research. 73 (3): 308–15. doi:10.1002/jnr.10672. PMID 12868064. S2CID 17806057.
- ^ Secades JJ, Lorenzo JL (Sep 2006). “Citicoline: pharmacological and clinical review, 2006 update”. Methods and Findings in Experimental and Clinical Pharmacology. 28 Suppl B: 1–56. PMID 17171187.
- ^ Tardner, P. (2020-08-30). “The use of citicoline for the treatment of cognitive decline and cognitive impairment: A meta-analysis of pharmacological literature • International Journal of Environmental Science & Technology”. International Journal of Environmental Science & Technology. Retrieved 2020-08-31.
- ^ Hurtado O, Moro MA, Cárdenas A, Sánchez V, Fernández-Tomé P, Leza JC, Lorenzo P, Secades JJ, Lozano R, Dávalos A, Castillo J, Lizasoain I (Mar 2005). “Neuroprotection afforded by prior citicoline administration in experimental brain ischemia: effects on glutamate transport”. Neurobiology of Disease. 18 (2): 336–345. doi:10.1016/j.nbd.2004.10.006. PMID 15686962. S2CID 2818533.
- ^ Dinsdale JR, Griffiths GK, Rowlands C, Castelló J, Ortiz JA, Maddock J, Aylward M (1983). “Pharmacokinetics of 14C CDP-choline”. Arzneimittel-Forschung. 33 (7A): 1066–70. PMID 6412727.
- ^ Tardner, P. (2020-08-28). “Can Citicoline Cause Depression?: A review of the clinical literature • International Journal of Environmental Science & Technology”. International Journal of Environmental Science & Technology. Retrieved 2020-08-31.
- ^ Talih, Farid; Ajaltouni, Jean (2015). “Probable Nootropicinduced Psychiatric Adverse Effects: A Series of Four Cases”. Innovations in Clinical Neuroscience. 12 (11–12): 21–25. ISSN 2158-8333. PMC 4756795. PMID 27222762.
- ^ Fernández-Murray JP, McMaster CR (Nov 2005). “Glycerophosphocholine catabolism as a new route for choline formation for phosphatidylcholine synthesis by the Kennedy pathway”. The Journal of Biological Chemistry. 280 (46): 38290–6. doi:10.1074/jbc.M507700200. PMID 16172116.
//////////CITOCOLINE, CDP-choline, Neuroprotective, ischemic stroke, head trauma,
C[N+](C)(C)CCOP(=O)(O)OP(=O)([O-])OCC1C(C(C(O1)N2C=CC(=NC2=O)N)O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Povidone-iodine

Povidone-iodine
PVP 1
UNII85H0HZU99M, BETADINE
CAS number 25655-41-8, Molecular Formula, (C6-H9-N-O)x-.x-I2, Molecular Weight, 364.9431
1-ethenylpyrrolidin-2-one;molecular iodine Povidone-Iodine
CAS Registry Number: 25655-41-8
CAS Name: 1-Ethenyl-2-pyrrolidinone homopolymer compd with iodine
Additional Names: 1-vinyl-2-pyrrolidinone polymers, iodine complex; iodine-polyvinylpyrrolidone complex; polyvinylpyrrolidone-iodine complex; PVP-I
Trademarks: Betadine (Purdue Frederick); Betaisodona (Mundipharma); Braunol (Braun Melsungen); Braunosan H (Braun Melsungen); Disadine D.P. (Stuart); Efodine (Fougera); Inadine (J & J); Isodine (Blair); Proviodine (Rougier); Traumasept (Wolff)
Literature References: An iodophor, q.v., prepd by Beller, Hosmer, US2706701; Hosmer, US2826532; Siggia, US2900305 (1955, 1958, and 1959, all to GAF). Prepn, history and use: Shelanski, Shelanski, J. Int. Coll. Surg.25, 727 (1956).
Properties: Yellowish-brown, amorphous powder with slight characteristic odor. Aq solns have a pH near 2 and may be made more neutral (but less stable) by the addition of sodium bicarbonate. Sol in alc, water. Practically insol in chloroform, carbon tetrachloride, ether, solvent hexane, acetone. Solns do not give the familiar starch test when freshly prepared.
Therap-Cat: Anti-infective (topical).
Therap-Cat-Vet: Anti-infective (topical).
Keywords: Antiseptic/Disinfectant; Halogens/Halogen Containing Compounds.
- An iodinated polyvinyl polymer used as topical antiseptic in surgery and for skin and mucous membrane infections, also as aerosol. The iodine may be radiolabeled for research purposes.
Povidone-iodine is a stable chemical complex of polyvinylpyrrolidone (povidone, PVP) and elemental iodine. It contains from 9.0% to 12.0% available iodine, calculated on a dry basis. This unique complex was discovered in 1955 at the Industrial Toxicology Laboratories in Philadelphia by H. A. Shelanski and M. V. Shelanski. During in vitro testing to demonstrate anti-bacterial activity it was found that the complex was less toxic in mice than tincture of iodine. Human clinical trials showed the product to be superior to other iodine formulations. Povidone-iodine was immediately marketed, and has since become the universally preferred iodine antiseptic.
Povidone-iodine (PVP-I), also known as iodopovidone, is an antiseptic used for skin disinfection before and after surgery.[1][2] It may be used both to disinfect the hands of healthcare providers and the skin of the person they are caring for.[2] It may also be used for minor wounds.[2] It may be applied to the skin as a liquid or a powder.[2]
Side effects include skin irritation and sometimes swelling.[1] If used on large wounds, kidney problems, high blood sodium, and metabolic acidosis may occur.[1] It is not recommended in women who are less than 32 weeks pregnant or are taking lithium.[2] Frequent use is not recommended in people with thyroid problems.[2] Povidone-iodine is a chemical complex of povidone, hydrogen iodide, and elemental iodine.[3] It contains 10% Povidone, with total iodine species equaling 10,000 ppm or 1% total titratable iodine.[3] It works by releasing iodine which results in the death of a range of microorganisms.[1]
Povidone-iodine came into commercial use in 1955.[4] It is on the World Health Organization’s List of Essential Medicines.[5] Povidone-iodine is available over the counter.[6] It is sold under a number of brand names including Betadine.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Wound area covered in povidone-iodine. Gauze has also been applied.
Povidone-iodine is a broad spectrum antiseptic for topical application in the treatment and prevention of wound infection. It may be used in first aid for minor cuts, burns, abrasions and blisters. Povidone-iodine exhibits longer lasting antiseptic effects than tincture of iodine, due to its slow absorption via soft tissue, making it the choice for longer surgeries. Chlorhexidine provides superior results with equivalent adverse events.[7]
Consequently, PVP-I has found broad application in medicine as a surgical scrub; for pre- and post-operative skin cleansing; for the treatment and prevention of infections in wounds, ulcers, cuts and burns; for the treatment of infections in decubitus ulcers and stasis ulcers; in gynecology for vaginitis associated with candidal, trichomonal or mixed infections. For these purposes PVP-I has been formulated at concentrations of 7.5–10.0% in solution, spray, surgical scrub, ointment, and swab dosage forms; however, use of 10% povidone-iodine though recommended, is infrequently used, as it is poorly accepted by health care workers and is excessively slow to dry.[8][9]
Because of these critical indications, only sterile povidone-iodine should be used in most cases. Non-sterile product can be appropriate in limited circumstances in which people have intact, healthy skin that will not be compromised or cut. The non-sterile form of Povidone iodine has a long history of intrinsic contamination with Burkholderia cepacia (aka Pseudomonas cepacia), and other opportunistic pathogens. Its ability to harbor such microbes further underscores the importance of using sterile products in any clinical setting. Since these bacteria are resistant to povidone iodine, statements that bacteria do not develop resistance to PVP-I,[10] should be regarded with great caution: some bacteria are intrinsically resistant to a range of biocides including povidone-iodine.[11]
Antiseptic activity of PVP-I is because of free iodine (I2) and PVP-I only acts as carrier of I2 to the target cells. Most commonly used 10% PVP-I delivers about 1-3 ppm of I2 in a compound of more than 31,600 ppm of total iodine atoms. All the toxic and staining effects of PVP-I is due to the inactive iodine only.
Eyes
A buffered PVP-I solution of 2.5% concentration can be used for prevention of neonatal conjunctivitis, especially if it is caused by Neisseria gonorrhoeae, or Chlamydia trachomatis. It is currently unclear whether PVP-I is more effective in reducing the number of cases of conjunctivitis in neonates over other methods.[12] PVP-I appears to be very suitable for this purpose because, unlike other substances, it is also efficient against fungi and viruses (including HIV and Herpes simplex).[13]
Pleurodesis
It is used in pleurodesis (fusion of the pleura because of incessant pleural effusions). For this purpose, povidone-iodine is equally effective and safe as talc, and may be preferred because of easy availability and low cost.[14]
Alternatives
There is strong evidence that chlorhexidine and denatured alcohol used to clean skin prior to surgery is better than any formulation of povidone-iodine[7]
Contraindications
PVP-I is contraindicated in people with hyperthyroidism (overactive thyroid gland) and other diseases of the thyroid, after treatment with radioiodine, and in people with dermatitis herpetiformis[why?] (Duhring’s disease).[15]
Side effects
The sensitization rate to the product is 0.7%.[16]
Interactions
The iodine in PVP-I reacts with hydrogen peroxide, silver, taurolidine and proteins such as enzymes, rendering them (and itself) ineffective. It also reacts with many mercury compounds, giving the corrosive compound mercury iodide, as well as with many metals, making it unsuitable for disinfecting metal piercings.[15]
Iodine is absorbed into the body to various degrees, depending on application area and condition of the skin. As such, it interacts with diagnostic tests of the thyroid gland such as radioiodine diagnostics, as well as with various diagnostic agents used on the urine and stool, for example Guaiacum resin.[15]
Structure
Structure of povidone-iodine complex.
Povidone-iodine is a chemical complex of the polymer povidone (polyvinylpyrrolidone) and triiodide (I3−).[17]
It is soluble in cold and mild-warm water, ethyl alcohol, isopropyl alcohol, polyethylene glycol, and glycerol. Its stability in solution is much greater than that of tincture of iodine or Lugol’s solution.
Free iodine, slowly liberated from the povidone-iodine (PVP-I) complex in solution, kills cells through iodination of lipids and oxidation of cytoplasmic and membrane compounds. This agent exhibits a broad range of microbiocidal activity against bacteria, fungi, protozoa, and viruses. Slow release of iodine from the PVP-I complex in solution minimizes iodine toxicity towards mammalian cells.
PVP-I can be loaded into hydrogels, which can be based on carboxymethyl cellulose (CMC), poly(vinyl alcohol) (PVA), and gelatin, or on crosslinked polyacrylamide. These hydrogels can be used for wound dressing. The rate of release of the iodine in the PVP-I is heavily dependent on the hydrogel composition: it increases with more CMC/PVA and decreases with more gelatin.
History
PVP-I was discovered in 1955, at the Industrial Toxicology Laboratories in Philadelphia by H. A. Shelanski and M. V. Shelanski.[18] They carried out tests in vitro to demonstrate anti-bacterial activity, and found that the complex was less toxic in mice than tincture of iodine. Human clinical trials showed the product to be superior to other iodine formulations.[19]
Following the discovery of iodine by Bernard Courtois in 1811, it has been broadly used for the prevention and treatment of skin infections, as well as the treatment of wounds. Iodine has been recognized as an effective broad-spectrum bactericide, and is also effective against yeasts, molds, fungi, viruses, and protozoans. Drawbacks to its use in the form of aqueous solutions include irritation at the site of application, toxicity, and the staining of surrounding tissues. These deficiencies were overcome by the discovery and use of PVP-I, in which the iodine is carried in a complexed form and the concentration of free iodine is very low. The product thus serves as an iodophor.
Research
Schematic of povidone-iodine complex wrapping a single wall carbon nanotube (black).[20]
Povidone-iodine has found application in the field of nanomaterials. A wound-healing application has been developed which employs a mat of single wall carbon nanotubes (SWNTs) coated in a monolayer of povidone-iodine.[20]
Research has previously found that the polymer polyvinylpyrrolidone (PVP, povidone) can coil around individual carbon nanotubes to make them water-soluble.[21]
References
- ^ Jump up to:a b c d World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. pp. 321–323. hdl:10665/44053. ISBN 9789241547659.
- ^ Jump up to:a b c d e f g British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 840. ISBN 9780857111562.
- ^ Jump up to:a b Encyclopedia of polymer science and technology (3 ed.). John Wiley & Sons. 2013. p. 728. ISBN 9780470073698. Archived from the original on 2017-01-13.
- ^ Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 68. ISBN 9780470015520. Archived from the original on 2017-01-13.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ “Povidone/iodine solution: Indications, Side Effects, Warnings – Drugs.com”. http://www.drugs.com. Archived from the original on 13 January 2017. Retrieved 11 January 2017.
- ^ Jump up to:a b Wade RG, Burr NE, McCauley G, Bourke G, Efthimiou O (September 2020). “The Comparative Efficacy of Chlorhexidine Gluconate and Povidone-iodine Antiseptics for the Prevention of Infection in Clean Surgery: A Systematic Review and Network Meta-analysis”. Annals of Surgery. Publish Ahead of Print. doi:10.1097/SLA.0000000000004076. PMID 32773627.
- ^ Slater K, Cooke M, Fullerton F, Whitby M, Hay J, Lingard S, et al. (September 2020). “Peripheral intravenous catheter needleless connector decontamination study-Randomized controlled trial”. American Journal of Infection Control. 48 (9): 1013–1018. doi:10.1016/j.ajic.2019.11.030. PMID 31928890.
- ^ Slater K, Fullerton F, Cooke M, Snell S, Rickard CM (September 2018). “Needleless connector drying time-how long does it take?”. American Journal of Infection Control. 46 (9): 1080–1081. doi:10.1016/j.ajic.2018.05.007. PMID 29880433. S2CID 46968733.
- ^ Fleischer W, Reimer K (1997). “Povidone-iodine in antisepsis–state of the art”. Dermatology. 195 Suppl 2 (Suppl 2): 3–9. doi:10.1159/000246022. PMID 9403248.
- ^ Rose H, Baldwin A, Dowson CG, Mahenthiralingam E (March 2009). “Biocide susceptibility of the Burkholderia cepacia complex”. The Journal of Antimicrobial Chemotherapy. 63 (3): 502–10. doi:10.1093/jac/dkn540. PMC 2640157. PMID 19153076.
- ^ Martin I, Sawatzky P, Liu G, Mulvey MR (February 2015). “Neisseria gonorrhoeae in Canada: 2009-2013”. Canada Communicable Disease Report. 41 (2): 35–41. doi:10.1002/14651858.CD001862.pub3. PMC 6457593.
- ^ Najafi Bi R, Samani SM, Pishva N, Moheimani F (2003). “Formulation and Clinical Evaluation of Povidone-Iodine Ophthalmic Drop”. Iranian Journal of Pharmaceuticical Research. 2 (3): 157–160.
- ^ Agarwal R, Khan A, Aggarwal AN, Gupta D (March 2012). “Efficacy & safety of iodopovidone pleurodesis: a systematic review & meta-analysis”. The Indian Journal of Medical Research. 135: 297–304. PMC 3361864. PMID 22561614.
- ^ Jump up to:a b c Jasek W, ed. (2007). Austria-Codex (in German) (62nd ed.). Vienna: Österreichischer Apothekerverlag. pp. 983–5. ISBN 978-3-85200-181-4.
- ^ Niedner R (1997). “Cytotoxicity and sensitization of povidone-iodine and other frequently used anti-infective agents”. Dermatology. 195 Suppl 2 (Suppl 2): 89–92. doi:10.1159/000246038. PMID 9403263.
- ^ Kutscher, Bernhard (2020). “Dermatologicals (D), 4. Antiseptics and Disinfectants (D08), Anti‐Acne Preparations (D10), and Other Dermatological Preparations (D11)”. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. pp. 1–22. doi:10.1002/14356007.w08_w03.
- ^ U.S. Patent 2,739,922
- ^ Sneader W (2005). Drug Discovery: A History. New York: John Wiley & Sons. p. 68. ISBN 978-0-471-89979-2.
- ^ Jump up to:a b Simmons TJ, Lee SH, Park TJ, Hashim DP, Ajayan PM, Linhardt RJ (2009). “Antiseptic Single Wall Carbon Nanotube Bandages” (PDF). Carbon. 47 (6): 1561–1564. doi:10.1016/j.carbon.2009.02.005. Archived from the original (PDF) on 2010-06-21.
- ^ Simmons TJ, Hashim D, Vajtai R, Ajayan PM (August 2007). “Large area-aligned arrays from direct deposition of single-wall carbon nanotube inks”. Journal of the American Chemical Society. 129 (33): 10088–9. doi:10.1021/ja073745e. PMID 17663555.
Further reading
- Wong RH, Hung EC, Wong VW, Wan IY, Ng CS, Wan S, Underwood MJ (2009). “Povidone-iodine wound irrigation: A word of caution”. Surgical Practice. 13 (4): 123–4. doi:10.1111/j.1744-1633.2009.00461.x. S2CID 71797553.
- Wong RH, Wong VW, Hung EC, Lee PY, Ng CS, Wan IY, Underwood MJ (2011). “Topical application of povidone-iodine before wound closure is associated with significant increase in serum iodine level”. Surgical Practice. 19 (3): 79–82. doi:10.1111/j.1744-1633.2011.00547.x. S2CID 70528331.
- Wong RH, Ng CS, Underwood MJ (May 2012). “Iodine pleurodesis–a word of caution”. European Journal of Cardio-Thoracic Surgery. 41 (5): 1209. doi:10.1093/ejcts/ezr137. PMID 22219431.
External links
“Povidone-iodine”. Drug Information Portal. U.S. National Library of Medicine.
| Povidone-iodine applied to an abrasion using a cotton swab. | |
| Clinical data | |
|---|---|
| Trade names | Betadine, Wokadine, Pyodine, others |
| Other names | polyvidone iodine, iodopovidone |
| AHFS/Drugs.com | Consumer Drug Information |
| License data | US DailyMed: Povidone-iodine |
| Routes of administration | Topical |
| ATC code | D08AG02 (WHO)D09AA09 (WHO) (dressing)D11AC06 (WHO)G01AX11 (WHO)R02AA15 (WHO)S01AX18 (WHO)QG51AD01 (WHO) |
| Legal status | |
| Legal status | US: OTC / Rx-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 25655-41-8 |
| PubChem CID | 410087 |
| DrugBank | DB06812 |
| ChemSpider | none |
| UNII | 85H0HZU99M |
| KEGG | D00863C08043 |
| ChEBI | CHEBI:8347 |
| ChEMBL | ChEMBL1201724 |
| CompTox Dashboard (EPA) | DTXSID8035712 |
| ECHA InfoCard | 100.110.412 |
| Chemical and physical data | |
| Formula | (C6H9NO)n·xI |
| Molar mass | variable |
| (what is this?) (verify) |
///////////Povidone-iodine, PVP 1, BETADINE
C=CN1CCCC1=O.II

NEW DRUG APPROVALS
ONE TIME
$10.00
4-Hydroxy-TEMPO, TEMPOL, MBM-02, MTS 01
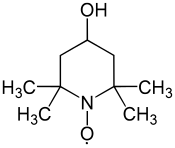
4-Hydroxy-TEMPO, TEMPOL, MBM-02, MTS 01
- Molecular FormulaC9H18NO2
- Average mass172.245 Da
2,2,6,6-Tetramethyl-4-hydroxypiperidinooxy
2,2,6,6-Tetramethyl-4-hydroxypiperidinooxy radical
2,2,6,6-Tetramethyl-4-piperidinol 1-oxyl
CAS 2226-96-2[RN]
4-hydroxy-1-oxyl-2,2,6,6-tetramethylpiperidine
4-Hydroxy-2,2,6,6-tetramethyl-1-piperidin-1-yloxy, free radical
4-Hydroxy-2,2,6,6-tetramethylpiperidine N-oxide
4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl
TEMPOLCAS Registry Number: 2226-96-2
CAS Name: 4-Hydroxy-2,2,6,6-tetramethyl-1-piperidinyloxy
Additional Names: 4-hydroxy-TEMPO; 4-hydroxy-2,2,6,6-tetramethyl piperidine N-oxide; 4-hydroxy-2,2,6,6-tetramethylpiperidinooxy
Molecular Formula: C9H18NO2, Molecular Weight: 172.24
Percent Composition: C 62.76%, H 10.53%, N 8.13%, O 18.58%
Literature References: Stable nitroxyl radical; water-soluble analogue of TEMPO, q.v. Functions as a membrane-permeable radical scavenger. Prepn: E. G. Rozantsev, Bull. Acad. Sci. USSR Div. Chem. Sci.12, 2085 (1964). Energy transfer studies: N. N. Quan, A. V. Guzzo, J. Phys. Chem.85, 140 (1981). IR conformation study: W. A. Bueno, L. Degrève, J. Mol. Struct.74, 291 (1981).Solid state NMR spectra: C. J. Groombridge, M. J. Perkins, J. Chem. Soc. Chem. Commun.1991, 1164. LC/MS/MS determn: I. D. Podmore, J. Chem. Res. Synop.2002, 574. Use as a phase transfer catalyst: X.-Y. Wang et al.,Synth. Commun.29, 157 (1999). Review of effects in animal models for shock, ischemia-reperfusion injury, and inflammation: C. Thiemermann, Crit. Care Med.31, S76-S84 (2003).
Properties: Crystals from ether + hexane, mp 71.5°. uv max (hexane): 240, 450-500 (e ~1800, ~5). uv max (ethanol): 242, 435-455 (e ~3800, ~10). Sol in water.
Melting point: mp 71.5°
Absorption maximum: uv max (hexane): 240, 450-500 (e ~1800, ~5); uv max (ethanol): 242, 435-455 (e ~3800, ~10)
Use: Spin label for EPR studies; phase transfer dehydration catalyst; antioxidant; inhibitor of olefin free radical polymerization.Topical PiperidineNitroxide MTS-01 is a topical gel containing a cell permeable hydrophilic piperidinenitroxide with potential radioprotective and antioxidant activity. As a stable, free radical compound, MTS-01 may be able to protect cells against the damaging effects of reactive oxygen species (ROS), upon exposure to ionizing radiation and oxidative stress. The topically applied MTS-01 may protect normal tissue from radiation-induced toxicity, such as radiation dermatitis, during radiation therapy.
4-Hydroxy-TEMPO is a member of aminoxyls and a member of piperidines. It has a role as a radical scavenger and a catalyst. It derives from a TEMPO.
4-Hydroxy-TEMPO or TEMPOL, formally 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, is a heterocyclic compound. Like the related TEMPO, it is used as a catalyst and chemical oxidant by virtue of being a stable aminoxyl radical. Its major appeal over TEMPO is that is less expensive, being produced from triacetone amine, which is itself made via the condensation of acetone and ammonia. This makes it economically viable on an industrial scale.[3]

Example synthesis of 4-Hydroxy-TEMPO from phorone, which is itself made from acetone and ammonia
In biochemical research, 4-hydroxy-TEMPO has been investigated as an agent for limiting reactive oxygen species. It catalyzes the disproportionation of superoxide, facilitates hydrogen peroxide metabolism, and inhibits Fenton chemistry.[4] 4-Hydroxy-TEMPO, along with related nitroxides, are being studied for their potential antioxidant properties.[5]
On an industrial-scale 4-hydroxy-TEMPO is often present as a structural element in hindered amine light stabilizers, which are commonly used stabilizers in plastics, it is also used as a polymerisation inhibitor, particularly during the purification of styrene.
It is a promising model substance to inhibit SARS-CoV-2 RNA-dependent RNA polymerase.[6]
SYN

SYN
Inorganica Chimica Acta, 370(1), 469-473; 2011
| IR | (KBr)vmax/cm-1: 3413 (m(O-H)) |
| Crystal Structure Data | Empirical formula C25H26NO8F6Cu; Formula weigh 646.02; T (K) 293(2); λ/Å 0.71073; Crystal system monoclinic; Space group P21/c; a (Å) 10.132(2); b (Å) 25.103(5); c (Å) 13.578(5); α (°) 90; β (°) 121.67(2); γ (°) 90; V (Å3) 2939.2(14); Z = 4 |
SYN
Bioorganic & Medicinal Chemistry Letters, 22(2), 920-923; 2012
SYN
https://pubs.acs.org/doi/10.1021/ol0712024
SYN
https://pubs.acs.org/doi/10.1021/es302157j
PAT
CN 113429392
SYN
Journal of the American Chemical Society, 138(29), 9069-9072; 2016
https://pubs.acs.org/doi/10.1021/jacs.6b05421
file:///C:/Users/Inspiron/Downloads/ja6b05421_si_001.pdf
| 1H NMR | (400 MHz, CDCl3) δH 3.89 (1H, tt, J = 11.4, 4.3 Hz, H4), 1.82.-.1.77 (2H, m, H3, H5), 1.43 (2H, t, J = 11.9 Hz, H3, H5), 1.14 (6H, s, 2 ×CH3), 1.07 (6H, s, 2 ×CH3); |
| 13C NMR | (100 MHz, CDCl3) δH 63.1, 47.5, 31.6, 20.6. |
| IR | (thin film, νmax / cm-1) 3407, 1472, 1376, 1174, 1161, 1066; |
| Rf | 0.22 (ethyl acetate / petroleum ether (1:1)); |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
Thursday, June 3, 2021
NIH researchers identify potential new antiviral drug for COVID-19
Compound targets essential viral enzyme and prevents replication in cells.https://www.nih.gov/news-events/news-releases/nih-researchers-identify-potential-new-antiviral-drug-covid-19 small spherical structures in the center of the image are SARS-CoV-2 virus particles. The string-like protrusions extending from the cells are cell projections or pseudopodium. NIAID
small spherical structures in the center of the image are SARS-CoV-2 virus particles. The string-like protrusions extending from the cells are cell projections or pseudopodium. NIAID
The experimental drug TEMPOL may be a promising oral antiviral treatment for COVID-19, suggests a study of cell cultures by researchers at the National Institutes of Health. TEMPOL can limit SARS-CoV-2 infection by impairing the activity of a viral enzyme called RNA replicase. The work was led by researchers at NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The study appears in Science.
“We urgently need additional effective, accessible treatments for COVID-19,” said Diana W. Bianchi, M.D., NICHD Director. “An oral drug that prevents SARS-CoV-2 from replicating would be an important tool for reducing the severity of the disease.”
The study team was led by Tracey A. Rouault, M.D., head of the NICHD Section on Human Iron Metabolism. It discovered TEMPOL’s effectiveness by evaluating a more basic question on how the virus uses its RNA replicase, an enzyme that allows SARS-CoV-2 to replicate its genome and make copies of itself once inside a cell.
Researchers tested whether the RNA replicase (specifically the enzyme’s nsp12 subunit) requires iron-sulfur clusters for structural support. Their findings indicate that the SARS-CoV-2 RNA replicase requires two iron-sulfur clusters to function optimally. Earlier studies had mistakenly identified these iron-sulfur cluster binding sites for zinc-binding sites, likely because iron-sulfur clusters degrade easily under standard experimental conditions.
Identifying this characteristic of the RNA replicase also enables researchers to exploit a weakness in the virus. TEMPOL can degrade iron-sulfur clusters, and previous research from the Rouault Lab has shown the drug may be effective in other diseases that involve iron-sulfur clusters. In cell culture experiments with live SARS-CoV-2 virus, the study team found that the drug can inhibit viral replication.
Based on previous animal studies of TEMPOL in other diseases, the study authors noted that the TEMPOL doses used in their antiviral experiments could likely be achieved in tissues that are primary targets for the virus, such as the salivary glands and the lungs.
“Given TEMPOL’s safety profile and the dosage considered therapeutic in our study, we are hopeful,” said Dr. Rouault. “However, clinical studies are needed to determine if the drug is effective in patients, particularly early in the disease course when the virus begins to replicate.”
The study team plans on conducting additional animal studies and will seek opportunities to evaluate TEMPOL in a clinical study of COVID-19.
NIH authors on the study include researchers from the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and the National Institute of Neurological Disorders and Stroke. Authors from the Pennsylvania State University are funded by NIH’s National Institute of General Medical Sciences.
About the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD): NICHD leads research and training to understand human development, improve reproductive health, enhance the lives of children and adolescents, and optimize abilities for all. For more information, visit https://www.nichd.nih.gov.
About the National Institutes of Health (NIH): NIH, the nation’s medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit www.nih.gov.
NIH…Turning Discovery Into Health®
Article
Maio N, et al. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science DOI: 10.1126/science.abi5224(link is external) (2021)
References
- ^ Zakrzewski, Jerzy; Krawczyk, Maria (1 January 2011). “Reactions of Nitroxides. Part XII [1]. – 2,2,6,6-Tetramethyl-1-oxyl- 4-piperidyl Chloroformate – A New Reactive Nitroxyl Radical. A One-pot Synthesis of 2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl N,N-Dialkyl-carbamates”. Zeitschrift für Naturforschung B. 66 (5). doi:10.1515/znb-2011-0509.
- ^ Jump up to:a b c d Sigma-Aldrich Co., 4-Hydroxy-TEMPO. Retrieved on 2015-08-24.
- ^ Ciriminna, Rosaria; Pagliaro, Mario (15 January 2010). “Industrial Oxidations with Organocatalyst TEMPO and Its Derivatives”. Organic Process Research & Development. 14 (1): 245–251. doi:10.1021/op900059x.
- ^ Wilcox, C. S.; Pearlman, A. (2008). “Chemistry and Antihypertensive Effects of Tempol and Other Nitroxides”. Pharmacological Reviews. 60 (4): 418–69. doi:10.1124/pr.108.000240. PMC 2739999. PMID 19112152.
- ^ Lewandowski, M; Gwozdzinski, K. (2017). “Nitroxides as Antioxidants and Anticancer Drugs”. International Journal of Molecular Sciences. 18 (11): 2490. doi:10.3390/ijms18112490. PMC 5713456.
- ^ Maio, N.; Lafont, B.A.P.; Sil, D.; Li, Y.; Bollinger, M.; Krebs, C. (2021). “Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets”. Science. 373 (6551): 236–241. doi:10.1126/science.abi5224.
| Names | |
|---|---|
| Preferred IUPAC name(4-Hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)oxyl | |
| Other namestempol; tanol; TMPN; 4-Oxypiperidol; nitroxyl 2; HyTEMPO | |
| Identifiers | |
| CAS Number | 2226-96-2 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:180664 |
| ChEMBL | ChEMBL607023 |
| ChemSpider | 121639 |
| ECHA InfoCard | 100.017.056 |
| PubChem CID | 137994 |
| UNII | U78ZX2F65X |
| CompTox Dashboard (EPA) | DTXSID4041280 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C9H18NO2 |
| Molar mass | 172.248 g·mol−1 |
| Appearance | Orange crystals |
| Melting point | 71–73 °C (160–163 °F; 344–346 K)[1] |
| Solubility in water | 629.3 g/l (20 °C) |
| Hazards | |
| GHS labelling: | |
| Pictograms | [2] |
| Signal word | Warning[2] |
| Hazard statements | H302, H315, H319, H335[2] |
| Precautionary statements | P261, P305+P351+P338[2] |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////////////4-Hydroxy-TEMPO, TEMPOL, MBM-02, MTS 01, ZJ 701, CORONA VIRUS, COVID 19
https://www.clinicaltrials.gov/ct2/show/NCT04729595
CC1(CC(CC(N1[O])(C)C)O)C

NEW DRUG APPROVALS
ONE TIME
$10.00
Rabeximod, ROB 803,

Rabeximod, ROB 803
C22H24ClN5O, 409.92
2-(9-chloro-2,3-dimethylindolo[3,2-b]quinoxalin-6-yl)-N-[2-(dimethylamino)ethyl]acetamide
CAS 872178-65-9UNII-J4D3K58W3Z, рабексимод , رابيكسيمود 雷贝莫德
6H-Indolo[2,3-b]quinoxaline-6-acetamide, 9-chloro-N-[2-(dimethylamino)ethyl]-2,3-dimethyl-
872178-65-9[RN]8866, J4D3K58W3Z
- OriginatorOxyPharma
- DeveloperCyxone; University of California
- ClassAcetamides; Anti-inflammatories; Disease-modifying antirheumatics; Heterocyclic compounds with 4 or more rings; Small molecules
- Mechanism of ActionCell differentiation modulators; Macrophage inhibitors
- Phase IICOVID 2019 infections; Rheumatoid arthritis
- 12 Oct 2021Cyxone terminates a phase-II trial in COVID-2019 infections in Slovakia (PO) (EudraCT2020-004571-41)
- 10 Aug 2021Cyxone completes a phase-II trial in COVID-2019 infections in Slovakia (PO) (EudraCT2020-004571-41)
- 23 Feb 2021Phase-II clinical trials in COVID-2019 infections in Slovakia (PO) (EudraCT2020-004571-41)
SYN
US 20050288296
https://patents.google.com/patent/US20050288296
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
WO 2014140321
https://patents.google.com/patent/WO2014140321A1/en Example 29-chloro-7V- [2-(dimethylamino)ethyl] -2,3-dimethyl-6H-Indolo [2,3-6] quinoxaline-6- acetamide

This compound was prepared as described in PCT/SE2005/000718 (WO 2005/123741), cf. “Compound E” at page 12 of said WO pamphlet.SYNWO 2005/123741https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005123741
Compound E
9-Chloro-2,3-dimerthyl-6-(N,N-dimethylaminoethylamino-2-oxoethyl)-6H-indolo- [2,3-b]quinoxaline (R1=Cl, R2=CH3, X=CO, Y=NH-CH2-CH2-R3; R3=NR5R6;
R5=R6=CH3)
Yield: 58%; 1H-NMR δ: 8.29 (d, 1H), 8.23 (t, 1H), 7.98 (s, 1H), 7.82 (s, 1H), 7.71
(dd, 1H), 7.61 (d, 1H), 5.09 (s, 2H), 3,16 (q, 2H), 2.47 (s, 6H), 2.28 (t, 2H), 2,12
(s, 6H);
SYN

Rabeximod is an orally administered compound for treatment of moderate or severe active rheumatoid arthritis that is currently undergoing phase II clinical testing in eight European countries.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
PATENT
The compound rabeximod has been described in European patent application publication EP1756111A1 later granted as EP1756111 B1. The preparation of rabex imod, as compound E, is specifically described in EP1756111A1 as a small-scale process without any description on how to develop a process that can be used for GMP and upscaled. Rabeximod was made in a 58% yield in a small-scale lab pro cess, but no parameters for scaling up have been disclosed.
The objective of the present invention is to provide a process that is suitable for large scale synthesis in good yield, with stable process parameters, and suitable for GMP production.
Experimental
The current process to manufacture Rabeximod involves several process steps as illustrated in below reaction scheme and as described in detail hereunder.
Manufacturing Process OXY001-01 Intermediate
Starting materials: 5-Chloroisatin (CIDO) and 4,5-Dimethyl-1 ,2-phenylenediamine (DAX)
Table 1: Overview Required Raw Materials and Quantities Step 1
Table 2: Raw Materials Specifications Step 1
Resulting Product (Intermediate): OXY001-01
Batch size: 13.03 kg of OXY001-01
Process description: 4,5-Dimethyl-1 ,2-phenylenediamine (1.1 equivalent) was added to acetic acid (4.7 volumes) in reactor (reactor was running under nitrogen at atmospheric pressure) and stirred up to 3 hours at moderate rate at +20 to +25 °C until clear dark brown solution was formed. 4, 5-Dimethyl-1 ,2-phenylenediamine so lution in acetic acid solution was transferred to intermediate feeding vessel. 5-chloro-isatin (1 .0 equivalent) was added to acetic acid (14.3 volumes) in reactor and stirred while jacket temperature of reactor was adjusted to approximately +150 °C to achieve a reflux temperature for active reflux of solvents. When reflux temperature was reached the 4, 5-Dimethyl-1 ,2-phenylenediamine solution in acetic acid was slowly added over 2-3 hours while distilling acetic acid (4.7 volumes) from the reaction mix ture. A fresh portion of acetic acid (4.7 volumes) was added to the reactor at about the same rate as distillation (4.7 volumes) occurred. After distillation the reaction mixture was stirred at reflux temperature for at least another 2 hours. The expected appearance of content in the reactor was a dark yellow to orange slurry. The reaction mixture was cooled to +65 to +70 °C and filtered using a Nutsche filter using Polyes ter filter cloth (27 pm) or similar as filter media. The filter cake was washed 3 times with fresh ethanol (3 x 4.2 volumes) and 1 time with water (1 x 4.2 volume). After washing the filter cake was dried at +40 to +45 °C for 12 hours and additionally in a vacuum tray dryer for 12 hours at +40 °C resulting in a yellow to orange/brown solid. An in-process control sample was taken and analysed for loss on drying (LOD). LOD should be < 2% (w/w). If the LOD is > 2%, the vacuum tray dryer step was repeated.
Theoretical yield: 18.62 kg
Yield: 70±5% (13.03±0.96kg)
Maximum volume: 216 L
Manufacturing Process OXY001-03 HCI Intermediate
CAC DMEN OXY001-03 HCI
Starting materials: Chloroacetyl chloride (CAC) and N,N-Dimethylethylene diamine (DMEN)
Table 3: Overview Required Raw Materials and Quantities Step 2
a) mol//mol of DMEN; b) kg/kg of DMEN; c) L/kg of DMEN
Table 4: Raw Materials Specifications Step 2
Resulting Product (Intermediate): OXY001-03 HCI
Batch size: 22.6 kg of OXY001 -03 HCI
Process description: Chloroacetyl chloride (1.03 equivalents) was dissolved in ethyl acetate (15 volumes) in reactor (reactor was running under nitrogen at atmos pheric pressure) at +20 °C. The solution was stirred and cooled down to +10 °C.
N,N-dimethylethylene diamine (1.00 equivalent) solution in ethyl acetate (1.0 volume) was slowly charged to the reactor when the temperature reached a range from +10 to +25 °C and at such a rate over 1-2 hours that the internal temperature did not exceed +25 °C. The slurry was stirred for 5 to 30 minutes at +20 to +25 °C and filtered using a Nutch filter using Polyamide filter cloth (25 pm) or similar as filter media. The product was washed 3 times on the filter with ethyl acetate (3 x 5 volumes) and dried on the filter for at least 16 hours and additionally in a vacuum tray dryer for 12 hours at +40 °C resulting in an off-white to beige solid.
Theoretical yield: 25.09 kg
Yield: 90±5% (22.6±1 .25 kg)
Maximum volume: 202 L
Manufacturing Process OXY001 Crude
– OXY001 Crude Starting materials: OXY001-01 and OXY001-03 HCI
Table 5: Overview Required Raw Materials and Quantities Step 3
Table 6: Raw/Intermediate Materials Specifications Step 3
Resulting Product: OXY001 Crude (crude rabeximod) Batch size: 11.38 kg of OXY001 Crude
Process description: OXY001-01 (1.0 equivalent) was dissolved in tetrahydrofuran (15.4 volumes) and 50% NaOH aqueous solution (8.0 equivalents in relation to OXY001 -01 ) in reactor (reactor was running under nitrogen at atmospheric pressure) and mixed at +55 to +60 °C up to approximately 1 hour until clear dark red solution was formed. Potassium iodide (0.81 equivalents) was added under vigorous stirring and mixed for 10 to 30 minutes at +55 to +60 °C. OXY001-03 HCI (2.0 equivalents) was added to the solution and mixed for at least 2 hours at +55 to +60 °C. Following completion of the reaction, the mixture was quenched with water (15.4 volumes) and tetrahydrofuran removed (15.4 volumes) by evaporation under reduced pressure. The slurry was cooled to +20 to +25 °C and stirred for 1 hour and filtered with a Nutch filter using Polyamide filter cloth (25 pm) or similar as filter media. Resulting cake was washed 3 times with water (3 x 5 volumes) until the pH of the filtrate was between 8-7 and dried on the filter at +40 to +45 °C for at least 12 hours by air suction and additionally in a vacuum tray dryer for 12 hours at +40 °C. Afterwards resulting ma terial was suspended in in tetrahydrofuran (25 volumes) at +45 to +50 °C for at least 1 hour. OXY001 Crude was isolated by filtration with a Nutch filter using Polyamide filter cloth (25 pm) or similar as filter media and washed 2 times on the filter with tetrahydrofuran (2 x 7 volumes). Resulting cake was dried on the filter at +40 to +45 °C for at least 12 hours and additionally in a vacuum tray dryer for 12 hours at +40 °C.
Theoretical yield: 18.96 kg
Yield: 60±5% (11 38±0.95 kg)
Maximum volume: 500 L
Purification of crude Rabeximod:
OXY001 crude (1 .0 equivalent) was dissolved in tetrahydrofuran (10 volumes), water (3 volume), and 2M HCI (1.4 volumes) mixture. The solution was clear filtered and heated to +50 °C. pH of mixture was adjusted to 10-12 by addition of 2M NaOH (1.3 volume). The formed slurry was cooled to +20 to +25 °C and diluted with water (12 volumes).
After stirring for at least 12 hours the slurry was filtered at +20 to +25 °C and washed on the filter with tetrahydrofuran:water (5:2) mixture (2×3 volumes). Rabeximod has a molecular weight of 409.92 g/mol and is isolated as a crystalline free base having a melting point of 259-261 °C.
Batch release results of batches used in Phase 2 and Phase 1 clinical studies are provided in Table 7.
Purity is equal to or above 98% as measured by HPLC.
Table 7: Batch release results of Rabeximod drug substance batches used in Phase 1 and phase 2 clinical studies
/////////////////Rabeximod, ROB 803, UNII-J4D3K58W3Z, рабексимод , رابيكسيمود 雷贝莫德 ,OXYPHARMA, PHASE 2, CYXONE
CC1=CC2=C(C=C1C)N=C3C(=N2)C4=C(N3CC(=O)NCCN(C)C)C=CC(=C4)Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
ZY 19489, MMV 253

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine

SYN
IN 201721031453
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
SYN
WO 2019049021
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019049021
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.

WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
2 4
Example-14: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1, 5-dimethyl-lH-pyrazol-3-yl)-5-(3-methylpiperazin-l-yl)pyrimidine-2,4-diamine
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
2 4
Example-15: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
2 4
Example-16: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
SYN
WO 2015165660
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015165660
Example 13
Synthetic scheme 1
Synthetic scheme 2
(R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
MHz, DMSO-d6) δ ppm 0.67 – 0.78 (m, 2 H) 1.00 (d, J=6.22 Hz, 3 H) 1.02 – 1.08 (m, 2 H) 1.96 – 2.10 (m, 1 H) 2.23 (s, 7 H) 2.30 – 2.38 (m, 4 H) 2.73 – 2.96 (m, 4 H) 3.33 (s, 3 H) 6.83 (s, 1 H) 7.67 (d, J=5.09 Hz, 1 H) 8.00 (s, 1 H) 8.03 (s, 1 H) 9.26 (s,1 H) MS (ES+), (M+H)+ = 466.45 for C21H32FN9.
SYN
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.

(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.


| Product vision |
|
| MoA |
|
| Key features |
|
| Challenges |
|
| Status |
|
| Next milestone |
|
| Previously |
|
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
////////////ZY 19489, MMV 253, Orphan Drug Designation, PHASE 1, ZYDUS CADILA, ANTIMALARIAL
Cn1nc(Nc2nc(Nc3cc(C4CC4)c(F)c(C)n3)ncc2N2C[C@@H](C)N(C)CC2)cc1C
CC1CN(CCN1C)C2=CN=C(N=C2NC3=NN(C(=C3)C)C)NC4=NC(=C(C(=C4)C5CC5)F)C
PILOCARPINE
PILOCARPINE
- Molecular FormulaC11H16N2O2
- Average mass208.257 Da
2(3H)-Furanone, 3-ethyldihydro-4-[(1-methyl-1H-imidazol-5-yl)methyl]-, (3S-cis)-
202-128-4[EINECS]92-13-7 CAS
54-71-7[RN]
(+)-pilocarpine
(3S,4R)-3-Ethyl-4-[(1-methyl-1H-imidazol-5-yl)methyl]dihydro-2(3H)-furanone
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Pilocarpine hydrochloride | 0WW6D218XJ | 54-71-7 | RNAICSBVACLLGM-GNAZCLTHSA-N |
| Pilocarpine nitrate | M20T465H6J | 148-72-1 | PRZXEPJJHQYOGF-GNAZCLTHSA-N |
PilocarpineCAS Registry Number: 92-13-7
CAS Name: (3S-cis)-3-Ethyldihydro-4-[(1-methyl-1H-imidazol-5-yl)methyl]-2(3H)-furanone
Trademarks: Ocusert Pilo (Cusi)
Molecular Formula: C11H16N2O2, Molecular Weight: 208.26
Percent Composition: C 63.44%, H 7.74%, N 13.45%, O 15.36%
Literature References: Cholinergic principle from Pilocarpus jaborandi Holmes, Rutaceae. Isoln: Petit, Polanovski, Bull. Soc. Chim. [3] 17, 557, 702 (1897). Structure: Jowett, J. Chem. Soc.77, 473, 851 (1900); 83, 438 (1903). Stereoisomeric with isopilocarpine: Polonovski, Polonovski, Bull. Soc. Chim. [4] 31, 1314 (1922). Has the cis configuration; isopilocarpine is trans: Zav’yalov, Dokl. Akad. Nauk SSSR82, 257 (1952). Absolute configuration: Hill, Barcza, Tetrahedron22, 2889 (1966). Synthesis: Preobrashenski et al.,Ber.66, 1187 (1933); Samokhvalov, Med. Prom. SSSR11, no. 2, 10 (1957); DeGraw, Tetrahedron28, 967 (1972); Link, Bernauer, Helv. Chim. Acta55, 1053 (1972). Stereoselective synthesis: A. Noordam et al.,Rec. Trav. Chim.98, 467 (1979). Review: Langenbeck, Angew. Chem.60, 297 (1948); van Rossum et al.,Experientia16, 373 (1960). Toxicity studies: Beccari, Boll. Chim. Farm.106, 8 (1967). Comprehensive description: A. A. Al-Badr, H. Y. Aboul-Enein, Anal. Profiles Drug Subs.12, 385-432 (1983). Clinical trial in Sjögren’s syndrome: F. B. Vivino et al., Arch. Intern. Med.159, 174 (1999); in radiation-induced xerostomia: J.-C. Horiot et al.,Radiother. Oncol.55, 233 (2000).
Properties: Oil or crystals, mp 34°. bp5 260° (partial conversion to isopilocarpine). [a]D18 +106° (c = 2). pK1 (20°) 7.15; pK2 (20°) 12.57. Sol in water, alcohol, chloroform; sparingly sol in ether, benzene. Almost insol in petr ether.
Melting point: mp 34°
Boiling point: bp5 260° (partial conversion to isopilocarpine)
pKa: pK1 (20°) 7.15; pK2 (20°) 12.57
Optical Rotation: [a]D18 +106° (c = 2)
Derivative Type: Hydrochloride
CAS Registry Number: 54-71-7
Trademarks: Akarpine (Akorn); Almocarpine (Ayerst); Isopto Carpine (Alcon); Pilogel (Alcon); Pilopine HS (Alcon); Pilostat (Bausch & Lomb); Salagen (MGI)
Molecular Formula: C11H16N2O2.HCl, Molecular Weight: 244.72
Percent Composition: C 53.99%, H 7.00%, N 11.45%, O 13.08%, Cl 14.49%
Properties: Hygroscopic crystals from alcohol, mp 204-205°. [a]D18 +91° (c = 2). Freely sol in water, alcohol. Practically insol in ether, chloroform. Keep well closed and protected from light.
Melting point: mp 204-205°
Optical Rotation: [a]D18 +91° (c = 2)
Derivative Type: Nitrate
CAS Registry Number: 148-72-1
Trademarks: Chibro Pilocarpine (Chibret); Licarpin (Allergan); Pilo (Novopharma); Pilofrin (Allergan); Pilagan (Allergan)
Molecular Formula: C11H16N2O2.HNO3, Molecular Weight: 271.27
Percent Composition: C 48.70%, H 6.32%, N 15.49%, O 29.49%
Properties: mp 173.5-174.0° (dec). Poisonous! [a]D +77 to +83° (c = 10). One gram dissolves in 4 ml water, 75 ml alcohol. Insol in chloroform, ether. Incompat. Silver nitrate, mercury bichloride, iodides, gold salts, tannin, calomel, KMnO4, alkalies.
Melting point: mp 173.5-174.0° (dec)
Optical Rotation: [a]D +77 to +83° (c = 10)
Derivative Type: Isopilocarpine
Additional Names: b-Pilocarpine
Properties: Hygroscopic oily liquid or prisms. bp10 261°. [a]D18 +50° (c = 2). pK1 (18°) 7.17. Miscible with water and alcohol; very sol in chloroform; less sol in benzene, ether. Almost insol in petr ether.
Boiling point: bp10 261°
pKa: pK1 (18°) 7.17
Optical Rotation: [a]D18 +50° (c = 2)
Derivative Type: Isopilocarpine hydrochloride hemihydrate
Molecular Formula: C11H16N2O2.HCl.½H2O, Molecular Weight: 253.73
Percent Composition: C 52.07%, H 7.15%, N 11.04%, O 15.76%, Cl 13.97%
Properties: Scales from alcohol + ether, mp 127°; when anhydr, mp 161°. [a]D18 +39° (c = 5). Sol in 0.27 part water; 2.1 parts alcohol.
Melting point: mp 127°; mp 161°
Optical Rotation: [a]D18 +39° (c = 5)
Derivative Type: Isopilocarpine nitrate
Molecular Formula: C11H16N2O2.HNO3, Molecular Weight: 271.27Percent Composition: C 48.70%, H 6.32%, N 15.49%, O 29.49%
Properties: Prisms from water, scales from alcohol, mp 159°. [a]D18 +39° (c = 2). Sol in 8.4 parts water, in 350 parts abs alcohol.
Melting point: mp 159°
Optical Rotation: [a]D18 +39° (c = 2)
Therap-Cat: Antiglaucoma agent; miotic; sialogogue.
Therap-Cat-Vet: Parasympathomimetic; miotic; gastric secretory stimulant.
Keywords: Antiglaucoma; Miotic; Sialagogue.
Pilocarpine is a muscarinic cholinergic agonist used on the eye to treat elevated intraocular pressure, various types of glaucoma, and to induce miosis. Also available orally to treat symptoms of dry mouth associated with Sjogren’s syndrome and radiotherapy.
Pilocarpine is a medication used to reduce pressure inside the eye and treat dry mouth.[1][3] As eye drops it is used to manage angle closure glaucoma until surgery can be performed, ocular hypertension, primary open angle glaucoma, and to bring about constriction of the pupil following its dilation.[1][4][5] However, due to its side effects it is no longer typically used in the long term management.[6] Onset of effects with the drops is typically within an hour and lasts for up to a day.[1] By mouth it is used for dry mouth as a result of Sjögren syndrome or radiation therapy.[7]
Common side effects of the eye drops include irritation of the eye, increased tearing, headache, and blurry vision.[1] Other side effects include allergic reactions and retinal detachment.[1] Use is generally not recommended during pregnancy.[8] Pilocarpine is in the miotics family of medication.[9] It works by activating cholinergic receptors of the muscarinic type which cause the trabecular meshwork to open and the aqueous humor to drain from the eye.[1]
Pilocarpine was isolated in 1874 by Hardy and Gerrard and has been used to treat glaucoma for more than 100 years.[10][11][12] It is on the World Health Organization’s List of Essential Medicines.[13] It was originally made from the South American plant Pilocarpus.[10]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
Pilocarpine hydrochloride, KSS-694, MGI-647, Pilobuc, Pilocar, Isopto carpine, Spersacarpin, Pilo, Isopto-pilocarpine, Pilocarpina lux, Pilogel, PilaSite(sustained release), Salagen, Pilopine HS
SYN
The alkylation of pilosine (I) with ethyl chloride (II) by means of LDA in THF gives trans-pilocarpine (III), which is isomerized with LDA in THF, yielding a mixture of cis- and trans-pilocarpine (IV). Finally, this mixture is resolved by crystallization with di-p-toluoyl tartaric acid.
SYN
Journal of Organic Chemistry, 58(1), 62-4; 1993
https://pubs.acs.org/doi/abs/10.1021/jo00053a016
SYN
Tetrahedron, 65(39), 8283-8296; 2009
SYN
Science of Synthesis, 20b, 987-1046; 2006
SYN
https://linkinghub.elsevier.com/retrieve/pii/S0040402008014002

SYN
https://www.mdpi.com/1420-3049/26/12/3676/htm
Schmidt, Theresa et alFrom Molecules, 26(12), 3676; 2021
Figure 1. Structure of natural occurring pilocarpine (+)-1 and its enantiomer (–)-1.
Scheme 1. Reactions and conditions: (a) hν, Bengal rosa, 8 h, 20 °C, 76% (of 3) and 5% (of 4); (b) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 98%; (c) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 99%; (d) THF, Na, 25 °C, 15 h, 72%; (e) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 77% (of 6) and 19% (of 7); (f) HBr, reflux, 2 d, 83%; (g) HBr, reflux, 4 d, 4%.
Scheme 2. Reactions and conditions: (a) SOCl2, reflux, 3 h, quant.; (b) Hex-OH, reflux, 16 h, 98%; (c) Rh/Al2O3, H2 (1 at), THF, 5 d, quant.; (d) Lipase PS, pH = 7.0, 2 d, 22 °C, 48% (of (±)-16) and 42% (of (–)-17); (e) PLE, pH = 7.0, 22 °C, 2 d, 96%; (f) N-methylmorpholine, iBu-chloroformate, N,O-dimethylhydroxylamine hydrochloride, 23 °C, 1 d, 84% (of (+)-18) and 85% of (–)-18); (g) LiAlH4, Et2O, 23 °C, 30 min, 95% (of (+)-19) and 95% of (–)-19; (h) CH3NH2, TosMic, DCM, benzene, NEt3, 7 d, 23 °C, 59% (of (+1)-1 and 60% of (–)-1; Hex stands for n-hexyl.
(+)-Pilocarpine [(+)-1]
Following the procedure given for the synthesis of its enantiomer, (+)-1 (1.92 g, 59%) was obtained as a colorless oil; Rf = 0.60 (SiO2, DCM/MeOH/aq NH4OH (25%), 95:4:1); [α]D = +115.7° (c 0.6, CHCl3), ee > 99% (by HPLC, Chiralcel OC, n-hexane/ethanol, 3:7, 0.3 mL/min, UV-detection λ = 215 nm; tR = (+)-1 47.1 min, tR = (–)-1 = 52.32 min); IR (film), 1H-NMR, 13C-NMR and MS (ESI, MeOH) were identical to the enantiomer (vide supra); analysis calcd. for C11H16N2O2 (208.26): C 63.44, H 7.74, N 13.45; found: C 63.31, H 7.98, N 13.32
PAPERBy Fuerstner, AloisFrom e-EROS Encyclopedia of Reagents for Organic Synthesis, 1-7; 2001
| Clinical data | |
|---|---|
| Trade names | Isopto Carpine, Salagen, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608039 |
| Pregnancy category | AU: B3 |
| Routes of administration | Topical eye drops, by mouth |
| Drug class | Miotic (cholinergic)[1] |
| ATC code | N07AX01 (WHO) S01EB01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Elimination half-life | 0.76 hours (5 mg), 1.35 hours (10 mg)[2] |
| Excretion | urine |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 92-13-7 54-71-7 (hydrochloride) |
| PubChem CID | 5910 |
| IUPHAR/BPS | 305 |
| DrugBank | DB01085 |
| ChemSpider | 5699 |
| UNII | 01MI4Q9DI3 |
| KEGG | D00525 |
| ChEBI | CHEBI:8207 |
| ChEMBL | ChEMBL550 |
| CompTox Dashboard (EPA) | DTXSID1021162 |
| ECHA InfoCard | 100.001.936 |
| Chemical and physical data | |
| Formula | C11H16N2O2 |
| Molar mass | 208.261 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
Medical uses
Pilocarpine stimulates the secretion of large amounts of saliva and sweat.[14] It is used to prevent or treat dry mouth, particularly in Sjögren syndrome, but also as a side effect of radiation therapy for head and neck cancer.[15]
It may be used to help differentiate Adie syndrome from other causes of unequal pupil size.[16][17][clarification needed]
It may be used to treat a form of dry eye called aqueous deficient dry eye (ADDE)[18]
Surgery
Pilocarpine is sometimes used immediately before certain types of corneal grafts and cataract surgery.[19][20] In ophthalmology, pilocarpine is also used to reduce symptomatic glare at night from lights when the patient has undergone implantation of phakic intraocular lenses; the use of pilocarpine would reduce the size of the pupils, partially relieving these symptoms.[dubious – discuss] The most common concentration for this use is pilocarpine 1%.[citation needed] Pilocarpine is shown to be just as effective as apraclonidine in preventing intraocular pressure spikes after laser trabeculoplasty.[21]
Presbyopia
In 2021, the US Food and Drug Administration approved pilocarpine hydrochloride as an eyedrop treatment for presbyopia, age-related difficulty with near-in vision. Marketed as vuity, the effect lasts for 7 to 10 hours.[22]
Other
Pilocarpine is used to stimulate sweat glands in a sweat test to measure the concentration of chloride and sodium that is excreted in sweat. It is used to diagnose cystic fibrosis.[23]
Adverse effects
Use of pilocarpine may result in a range of adverse effects, most of them related to its non-selective action as a muscarinic receptor agonist. Pilocarpine has been known to cause excessive salivation, sweating, bronchial mucus secretion, bronchospasm, bradycardia, vasodilation, and diarrhea. Eye drops can result in brow ache and chronic use in miosis.
Pharmacology
Pilocarpine is a drug that acts as a muscarinic receptor agonist. It acts on a subtype of muscarinic receptor (M3) found on the iris sphincter muscle, causing the muscle to contract – resulting in pupil constriction (miosis). Pilocarpine also acts on the ciliary muscle and causes it to contract. When the ciliary muscle contracts, it opens the trabecular meshwork through increased tension on the scleral spur. This action facilitates the rate that aqueous humor leaves the eye to decrease intraocular pressure. Paradoxically, when pilocarpine induces this ciliary muscle contraction (known as an accommodative spasm) it causes the eye’s lens to thicken and move forward within the eye. This movement causes the iris (which is located immediately in front of the lens) to also move forward, narrowing the Anterior chamber angle. Narrowing of the anterior chamber angle increases the risk of increased intraocular pressure.[24]
Society and culture
Preparation
Plants in the genus Pilocarpus are the only known sources of pilocarpine, and commercial production is derived entirely from the leaves of Pilocarpus microphyllus (Maranham Jaborandi). This genus grows only in South America, and Pilocarpus microphyllus is native to several states in northern Brazil.[25]
Pilocarpine is extracted from the powdered leaf material in a multi-step process. First the material is treated with ethanol acidified with hydrochloric acid, and the solvents removed under reduced pressure. The resultant aqueous residue is neutralized with ammonia and put aside until the resin has completely settled. It is then filtered and concentrated by sugar solution to a small volume, made alkaline with ammonia, and finally extracted with chloroform. The solvent is removed under reduced pressure.[verification needed]
Cost
Pilocarpine is one of the lowest cost medications for glaucoma.[26]
Trade names
Pilocarpine is available under several trade names such as: Diocarpine (Dioptic), Isopto Carpine (Alcon), Miocarpine (CIBA Vision), Ocusert Pilo-20 and -40 (Alza), Pilopine HS (Alcon), Salagen (MGI Pharma), Scheinpharm Pilocarpine (Schein Pharmaceutical), Timpilo (Merck Frosst) and Vuity (Abbvie).
Research
Pilocarpine is used to induce chronic epilepsy in rodents, commonly rats, as a means to study the disorder’s physiology and to examine different treatments.[27][28] Smaller doses may be used to induce salivation in order to collect samples of saliva, for instance, to obtain information about IgA antibodies.
Veterinary
Pilocarpine is given in moderate doses (about 2 mg) to induce emesis in cats that have ingested foreign plants, foods, or drugs. One feline trial determined it was effective, even though the usual choice of emetic is xylazine.
References
- ^ Jump up to:a b c d e f g “Pilocarpine”. The American Society of Health-System Pharmacists. Archived from the original on 28 December 2016. Retrieved 8 December 2016.
- ^ Gornitsky M, Shenouda G, Sultanem K, Katz H, Hier M, Black M, Velly AM (July 2004). “Double-blind randomized, placebo-controlled study of pilocarpine to salvage salivary gland function during radiotherapy of patients with head and neck cancer”. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 98 (1): 45–52. doi:10.1016/j.tripleo.2004.04.009. PMID 15243470.
- ^ Tarascon Pocket Pharmacopoeia 2019 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. 2018. p. 224. ISBN 9781284167542.
- ^ World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 439. hdl:10665/44053. ISBN 9789241547659.
- ^ “Glaucoma and ocular hypertension. NICE guideline 81”. National Institute for Health and Care Excellence. November 2017. Retrieved 19 September 2019.
Ocular hypertension… alternative options include carbonic anhydrase inhibitors such as brinzolamide or dorzolamide, a topical sympathomimetic such as apraclonidine or brimonidine tartrate, or a topical miotic such as pilocarpine, given either as monotherapy or as combination therapy.
- ^ Lusthaus J, Goldberg I (March 2019). “Current management of glaucoma” (PDF). The Medical Journal of Australia. 210 (4): 180–187. doi:10.5694/mja2.50020. PMID 30767238. S2CID 73438590.
Pilocarpine is no longer routinely used for long term IOP control due to a poor side effect profile
- ^ Hamilton R (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 415. ISBN 9781284057560.
- ^ “Pilocarpine ophthalmic Use During Pregnancy | Drugs.com”. http://www.drugs.com. Archived from the original on 28 December 2016. Retrieved 28 December 2016.
- ^ British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 769. ISBN 9780857111562.
- ^ Jump up to:a b Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 98. ISBN 978-0-471-89979-2. Archived from the original on 2016-12-29.
- ^ Rosin A (1991). “[Pilocarpine. A miotic of choice in the treatment of glaucoma has passed 110 years of use]”. Oftalmologia (in Romanian). 35 (1): 53–5. PMID 1811739.
- ^ Holmstedt, B; Wassén, SH; Schultes, RE (January 1979). “Jaborandi: an interdisciplinary appraisal”. Journal of Ethnopharmacology. 1 (1): 3–21. doi:10.1016/0378-8741(79)90014-x. PMID 397371.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “Pilocarpine”. MedLinePlus. U.S. National Library of Medicine. Archived from the original on 2010-03-06.
- ^ Yang, WF; Liao, GQ; Hakim, SG; Ouyang, DQ; Ringash, J; Su, YX (1 March 2016). “Is Pilocarpine Effective in Preventing Radiation-Induced Xerostomia? A Systematic Review and Meta-analysis”. International Journal of Radiation Oncology, Biology, Physics. 94 (3): 503–11. doi:10.1016/j.ijrobp.2015.11.012. hdl:10722/229069. PMID 26867879.
- ^ Kanski JJ, Bowling B (2015-03-24). Kanski’s Clinical Ophthalmology E-Book: A Systematic Approach. Elsevier Health Sciences. p. 812. ISBN 9780702055744.
- ^ Bartlett JD, James SD (October 2013). “Drug Affect the Autonomous Nervous System”. Clinical Ocular Pharmacology. Elsevier. p. 118. ISBN 9781483193915.
- ^ Mannis, Mark J; Holland, Edward J (September 2016). “Chapter 33: Dry Eye”. Cornea E-Book. Elsevier Health Sciences. p. 388. ISBN 978-0-323-35758-6. OCLC 960165358.
- ^ Parker, Jack (2017). Descemet Membrane Endothelial Keratoplasty (DMEK): A Review (PDF) (Thesis). Leiden University.
- ^ Ahmed E, E A (2010). Comprehensive Manual of Ophthalmology. JP Medical Ltd. p. 345. ISBN 9789350251751.
- ^ Zhang L, Weizer JS, Musch DC (February 2017). “Perioperative medications for preventing temporarily increased intraocular pressure after laser trabeculoplasty”. The Cochrane Database of Systematic Reviews. 2 (2): CD010746. doi:10.1002/14651858.CD010746.pub2. PMC 5477062. PMID 28231380.
- ^ Bankhead, Charles (2021-11-01). “First Eye Drop Treatment for Presbyopia Wins FDA Approval”. http://www.medpagetoday.com. Retrieved 2021-11-02.
- ^ Prasad RK (2017-07-11). Chemistry and Synthesis of Medicinal Agents: (Expanding Knowledge of Drug Chemistry). BookRix. ISBN 9783743821415.
- ^ Shaarawy TM, Sherwood MB, Hitchings RA, Crowston JG (September 2014). “Lsser Peripheral Iridoplasty”. Glaucoma E-Book. Elsevier Health Sciences. p. 718. ISBN 9780702055416.
- ^ De Abreu IN, Sawaya AC, Eberlin MN, Mazzafera P (November–December 2005). “Production of Pilocarpine in Callus of Jaborandi (Pilocarpus microphyllus Stapf)”. In Vitro Cellular & Developmental Biology – Plant. Society for In Vitro Biology. 41 (6): 806–811. doi:10.1079/IVP2005711. JSTOR 4293939. S2CID 26058596.
- ^ Schwab, Larry (2007). Eye Care in Developing Nations. CRC Press. p. 110. ISBN 9781840765229.
- ^ Károly N (2018). Immunohistochemical investigations of the neuronal changes induced by chronic recurrent seizures in a pilocarpine rodent model of temporal lobe epilepsy (Thesis). University of Szeged. doi:10.14232/phd.9734.
- ^ Morimoto K, Fahnestock M, Racine RJ (May 2004). “Kindling and status epilepticus models of epilepsy: rewiring the brain”. Progress in Neurobiology. 73 (1): 1–60. doi:10.1016/j.pneurobio.2004.03.009. PMID 15193778. S2CID 36849482.
External links
- “Pilocarpine”. Drug Information Portal. U.S. National Library of Medicine.
CLIP
Firms Team Up To Sustain Natural Pilocarpine
Sustainable harvest is key to a new pharmaceutical chemicals venture
https://cen.acs.org/articles/93/i11/Firms-Team-Sustain-Natural-Pilocarpine.html
Last summer, Andrew Badrot bought a portfolio of plant-sourced pharmaceutical chemicals from Boehringer Ingelheim and acquired BI’s distribution rights for pilocarpine, a plant-derived glaucoma treatment.
For BI, the transactions were small ones. The German drugmaker had been exiting its private-label active pharmaceutical ingredients (API) business, scaling back to produce only the chemicals it uses to manufacture its own drugs.
But for Badrot the deals were potentially big. He leads the company that bought the businesses—Centroflora CMS, a joint venture between the Brazilian botanicals firm Centroflora and CMS Pharma, Badrot’s custom chemicals consultancy. Together, Centroflora and Centroflora CMS are committed to nurturing the natural source of pilocarpine, an alkaloid used medicinally for more than 100 years, and to expanding into other APIs neglected by larger firms.
Pilocarpine’s source, Pilocarpus microphyllus, better known as jaborandi, had been harvested vigorously in the wild by Merck KGaA, which in 1975 built a factory in Parnaíba in northern Brazil to extract pilocarpine. By the mid-1980s, however, jaborandi had been overharvested, and the government declared it a protected species. Merck began obtaining the leaves from a plantation in the northern Brazilian state of Maranhão.
Demand for the drug as a glaucoma treatment began to decline, and Merck eventually closed the plant. When the market for the drug revived with new indications as a dry-mouth remedy, the company saw an opportunity to sell the site and did so in 2002.
The buyer was Centroflora, which was founded in 1957 in São Paulo. The firm was interested in adding pilocarpine to its botanical extracts business, according to its chief executive, Peter Andersen, a native of Brazil whose coffee-trader father bought into Centroflora in 1983. Along with the purchase, Centroflora signed a deal for BI to distribute the drug.
The company wanted to revitalize natural harvesting of jaborandi and began working with the Brazilian government to promulgate sustainable practices in the field. Centroflora also worked closely with a German government agency, Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ), which promotes sustainable harvesting internationally and had been working in the north of Brazil for decades.
Centroflora’s distribution agreement with BI arose through connections at GIZ, according to Andersen. BI also had been Merck’s biggest customer for pilocarpine.
But ecological sustainability was only half of the problem, Andersen says. Centroflora also found itself dealing with middlemen who would collect the jaborandi from poor family farms in remote areas and pay them next to nothing. Establishing a direct supply channel was not easy.

“I can spend a few days telling you about that process,” he says. “Stories of difficult relationships and difficult moments. But in some cases we managed to hire some of the middlemen to work for us on a salary basis. They made less money, but they had a job.”
Today, farmers in Brazil are paid at least twice what they were paid by intermediaries, Andersen says.
Key to the process was a program Centroflora launched in 2004 called Partnerships for a Better World to train and certify growers, establish community associations to support growers, and maintain sustainable harvesting practices.
Centroflora is the leading supplier of pilocarpine. Its only competitor, Sourcetech, with a plant near São Paulo, accesses jaborandi from the plantation that supplied Merck, now owned by U.S.-based Quercegen.
Pilocarpine accounts for only about 5% of Centroflora’s $95 million in annual sales. The company produces a long list of botanical extracts, including nutritional supplements and herbal medicines such as acai, acerola, coffee powder, and powdered fruit.The company manufactures at four facilities in Brazil, including the former Merck plant, which is dedicated to pilocarpine. But Andersen sees the partnership with CMS as a route to increase phytochemical API manufacturing at that site.
“The facility has the capacity to produce 12 metric tons per year of alkaloids,” Andersen says. It currently makes less than three metric tons. “So there is a lot of space to produce more, and the idea is that we can do some of the APIs we got from Boehringer Ingelheim.”
Those include atropine, digoxin, homatropine, and dihydroergotamine mesylate. Centroflora CMS also obtained distribution rights to BI’s scopolamine N-butyl bromide. All are derived from botanicals harvested on farms around the world.
Badrot was vice president of strategy for Lonza’s exclusive synthesis division before starting CMS in 2010 to consult on manufacturing and mergers and acquisitions in the custom chemicals business. “But for me, the dream was to return to manufacturing APIs,” he says.
The phytochemicals portfolio, including some of the oldest APIs made by BI, for which CMS has done consulting work, seemed like an ideal reentry to manufacturing, according to Badrot. “They are niche products that maybe fly a bit under the radar,” he says. “They seemed to fit us well because we can give them some attention.”
Centroflora CMS’s first order of business, he says, is to establish manufacturing for the BI products, which BI will continue to make until then. Badrot says Centroflora is well suited to manufacture at least the digoxin and atropine, but decisions have not been finalized. The partners will likely use contract manufacturers for some of the products. And Badrot says Centroflora CMS seeks to replicate the kind of deal it has with BI.
“We are looking for other companies with APIs that represent 0–1% of sales, products that lack focus,” he says. “We would take them over.”
Badrot and Andersen say they are also interested in sharing the Partnerships for a Better World program with other companies involved in harvesting natural products. And Centroflora looks for other ways to support its supply chain. Last month, it was approved as a trading member of the Union for Ethical BioTrade, a nonprofit that promotes sustainable development and biodiversity. As a member, Centroflora commits to sustainable sourcing practices and will be required to undergo periodic audits.
Last year, Centroflora received government recognition for its efforts on both the environmental and social fronts. The National Confederation of Industry in Brazil named Centroflora’s jaborandi harvesting program one of the country’s 10 most sustainable business practices. And Banco do Brasil, the national bank, recognized the firm for its work to improve conditions for farmers in the northern forest region of the country.
As the joint venture starts to work with its new portfolio of phytochemicals, both Andersen and Badrot look back at the jaborandi success as the road forward, a template for fostering a plant-based API business that may inspire other companies.
For Andersen, Partnerships for a Better World is an essential foundation of trust for the ecological and socially responsible harvesting of botanicals in Brazil. “There were a lot of problems along the way,” he says. “But we are at peace with it today.”
////////////////PILOCARPINE, Pilocarpine hydrochloride, KSS-694, MGI-647, Pilobuc, Pilocar, Isopto carpine, Spersacarpin, Pilo, Isopto-pilocarpine, Pilocarpina lux, Pilogel, PilaSite(sustained release), Salagen, Pilopine HS
CC[C@H]1[C@@H](CC2=CN=CN2C)COC1=O

NEW DRUG APPROVALS
ONE TIME
$10.00
loteprednol etabonate
loteprednol etabonate
- Molecular FormulaC24H31ClO7
- Average mass466.952 Da
cas 82034-46-6
chloromethyl (8S,9S,10R,11S,13S,14S,17R)-17-ethoxycarbonyloxy-11-hydroxy-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthrene-17-carboxylate(11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxo-androsta-1,4-diene-17-carboxylic acid chloromethyl ester
(11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxoandrosta-1,4-diene-17-carboxylic Acid Chloromethyl Ester
(8S,9S,10R,11S,13S,14S,17R)-17-[(éthoxycarbonyl)oxy]-11-hydroxy-10,13-diméthyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodécahydro-3H-cyclopenta[a]phénanthrène-17-carboxylate de chlorométhyle
129260-79-3[RN]
17a-Ethoxycarbonyloxy-D’-cortienic Acid Chloromethyl Ester
82034-46-6[RN]
Androsta-1,4-diene-17-carboxylic acid, 17-((ethoxycarbonyl)oxy)-11-hydroxy-3-oxo-, chloromethyl ester, (11β,17α)-
Androsta-1,4-diene-17-carboxylic acid, 17-[(ethoxycarbonyl)oxy]-11-hydroxy-3-oxo-, chloromethyl ester, (11β,17α)-
Loteprednol Etabonate
CAS Registry Number: 82034-46-6
CAS Name: (11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxoandrosta-1,4-diene-17-carboxylic acid chloromethyl ester
Additional Names: chloromethyl 17a-ethoxycarbonyloxy-11b-hydroxyandrosta-1,4-diene-3-one-17b-carboxylate; 17a-ethoxycarbonyloxy-D¢-cortienic acid chloromethyl ester
Manufacturers’ Codes: CDDD-5604; HGP-1; P-5604
Trademarks: Alrex (Bausch & Lomb); Lotemax (Bausch & Lomb)
Molecular Formula: C24H31ClO7, Molecular Weight: 466.95
Percent Composition: C 61.73%, H 6.69%, Cl 7.59%, O 23.98%
Literature References: Ophthalmic corticosteroid. Prepn: N. S. Bodor, BE889563 (1981 to Otsuka); idem,US4996335 (1991). Physicochemical properties: M. Alberth et al.,J. Biopharm. Sci.2, 115 (1991). HPLC determn in plasma and urine: G. Hochhaus et al.,J. Pharm. Sci.81, 1210 (1992). NMR structural studies: S. Rachwal et al.,Steroids61, 524 (1996); idem et al., ibid. 63, 193 (1998). Metabolism and transdermal permeability: N. Bodor et al.,Pharm. Res.9, 1275 (1992). Evaluation of effect on intraocular pressure: J. D. Bartlett et al.,J. Ocul. Pharmacol.9, 157 (1993). Clinical trial in keratoconjunctivitis sicca: S. C. Pflugfelder et al.,Am. J. Ophthalmol.138, 444 (2004). Review of ophthalmic clinical studies: J. F. Howes, Pharmazie55, 178-183 (2000).
Properties: Crystals from THF + hexane, mp 220.5-223.5°. Soly at 25° (mg/ml): 0.0005 in water; 0.037 in 50% propylene glycol + water. Lipophilicity (log K): 3.04.
Melting point: mp 220.5-223.5°
Therap-Cat: Anti-inflammatory (topical).
Keywords: Glucocorticoid.
Research Code:HGP-1; CDDD-5604; P-5604Trade Name:Lotemax® / Alrex®MOA:CorticosteroidIndication:Acne rosacea; Superficial punctate keratitis; Postoperative inflammation and pain following ocular surgery; Iritis; Herpes zoster keratitis; Allergic conjunctivitis; CyclitisCompany:Bausch & Lomb (Originator)Sales:ATC Code:S01BA14
Loteprednol etabonate was approved by the U.S. Food and Drug Administration (FDA) on Mar 9, 1998. It was developed and marketed as Lotemax® by Bausch & Lomb.
Loteprednol etabonate is a corticosteroid used in ophthalmology. It is indicated for the treatment of steroid responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea and anterior segment of the globe such as allergic conjunctivitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis, cyclitis, selected infective conjunctivitides.
Lotemax® is available as drops for ophthalmic use, containing 0.5% of Loteprednol etabonate. The recommended dose is one to two drops into the conjunctival sac of the affected eyes four times daily.
Loteprednol (as the ester loteprednol etabonate) is a corticosteroid used to treat inflammations of the eye. It is marketed by Bausch and Lomb as Lotemax[1] and Loterex.
It was patented in 1980 and approved for medical use in 1998.[2]
Loteprednol Etabonate is the etabonate salt form of loteprednol, an ophthalmic analog of the corticosteroid prednisolone with anti-inflammatory activity. Loteprednol etabonate exerts its effect by interacting with specific intracellular receptors and subsequently binds to DNA to modify gene expression. This results in an induction of the synthesis of certain anti-inflammatory proteins while inhibiting the synthesis of certain inflammatory mediators. Loteprednol etabonate specifically induces phospholipase A2 inhibitory proteins (collectively called lipocortins), which inhibit the release of arachidonic acid, thereby inhibiting the biosynthesis of potent mediators of inflammation, such as prostaglandins and leukotrienes.
Loteprednol etabonate is an etabonate ester, an 11beta-hydroxy steroid, a steroid ester, an organochlorine compound, a steroid acid ester and a 3-oxo-Delta(1),Delta(4)-steroid. It has a role as an anti-inflammatory drug. It derives from a loteprednol.
Loteprednol Etabonate (LE) is a topical corticoid anti-inflammatory. It is used in ophthalmic solution for the treatment of steroid responsive inflammatory conditions of the eye such as allergic conjunctivitis, uveitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis, cyclitis, and selected infective conjunctivitides. As a nasal spray, it can be used for the treatment and management of seasonal allergic rhinitis. Most prescription LE products, however, tend to be indicated for the treatment of post-operative inflammation and pain following ocular surgery. A number of such new formulations that have been approved include Kala Pharmaceutical’s Inveltys – the first twice-daily (BID) ocular corticosteroid approved for this indication, designed specifically to enhance patient compliance and simplified dosing compared to all other similar ocular steroids that are dosed four times daily. Moreover, LE was purposefully engineered to be a ‘soft drug’, one that is designed to be active locally at the site of administration and then rapidly metabolized to inactive components after eliciting its actions at the desired location, thereby subsequently minimizing the chance for adverse effects.
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-09-28 | New dosage form | Lotemax | Postoperative inflammation and pain following ocular surgery | Gel | 0.5% | Bausch & Lomb | |
| 2011-04-15 | New dosage form | Lotemax | Postoperative inflammation and pain following ocular surgery | Ointment | 0.5% | Bausch & Lomb | |
| 1998-03-09 | First approval | Lotemax | Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis | Suspension/ Drops | 0.5% | Bausch & Lomb |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-11-26 | Marketing approval | 露达舒/Lotemax | Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis,Postoperative inflammation and pain following ocular surgery | Suspension | 滴眼剂,0.5%(2.5ml:12.5mg,5ml:25mg) | Bausch & Lomb | |
| 2011-11-05 | Marketing approval | 露达舒/Lotemax | Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis,Postoperative inflammation and pain following ocular surgery | Suspension | 滴眼剂,0.5%(2.5ml:12.5mg,5ml:25mg); 滴眼剂,0.5%(10ml:50mg,15ml:75mg) | Bausch & Lomb |
Reference:1. US4710495A / US4996335A.Route 2
Reference:1. CN103183714A.
SYN
doi:10.1016/0960-0760(91)90120-T doi: 10.1016/j.steroids.2011.01.006

| Clinical data | |
|---|---|
| Trade names | Lotemax |
| Other names | 11β,17α,Dihydroxy-21-oxa-21-chloromethylpregna-1,4-diene-3,20-dione 17α-ethylcarbonate |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Routes of administration | Eye drops |
| Drug class | Corticosteroid; glucocorticoid |
| ATC code | S01BA14 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | None |
| Protein binding | 95% |
| Metabolism | Ester hydrolysis |
| Metabolites | Δ1-cortienic acid and its etabonate |
| Onset of action | ≤2 hrs (allergic conjunctivitis) |
| Elimination half-life | 2.8 hrs |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 82034-46-6 |
| PubChem CID | 444025 |
| IUPHAR/BPS | 7085 |
| DrugBank | DB14596 |
| ChemSpider | 392049 |
| UNII | YEH1EZ96K6 |
| KEGG | D01689 |
| ChEBI | CHEBI:31784 |
| ChEMBL | ChEMBL1200865 |
| CompTox Dashboard (EPA) | DTXSID2046468 |
| ECHA InfoCard | 100.167.120 |
| Chemical and physical data | |
| Formula | C24H31ClO7 |
| Molar mass | 466.96 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 220.5 to 223.5 °C (428.9 to 434.3 °F) |
| Solubility in water | 0.0005 mg/mL (20 °C) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
Medical uses
Applications for this drug include the reduction of inflammation after eye surgery,[1] seasonal allergic conjunctivitis, uveitis,[3] as well as chronic forms of keratitis (e.g. adenoviral and Thygeson’s keratitis), vernal keratoconjunctivitis, pingueculitis, and episcleritis.[citation needed]
Contraindications
As corticosteroids are immunosuppressive, loteprednol is contraindicated in patients with viral, fungal or mycobacterial infections of the eye.[1][3][4]
Adverse effects
The most common adverse effects in patients being treated with the gel formulation are anterior chamber inflammation (in 5% of people), eye pain (2%), and foreign body sensation (2%).[5]
Interactions
Because long term use (more than 10 days) can cause increased intraocular pressure, loteprednol may interfere with the treatment of glaucoma. Following ocular administration, the drug is very slowly absorbed into the blood, therefore the blood level is limited to an extremely small concentration, and interactions with drugs taken by mouth or through any route other than topical ophthalmic are very unlikely.[1]
Pharmacology
Mechanism of action
Main article: Glucocorticoid § Mechanism of action
Pharmacokinetics
Neither loteprednol etabonate nor its inactive metabolites Δ1–cortienic acid and Δ1-cortienic acid etabonate are detectable in the bloodstream, even after oral administration. A study with patients receiving loteprednol eye drops over 42 days showed no adrenal suppression, which would be a sign of the drug reaching the bloodstream to a clinically relevant extent.[1]
Steroid receptor affinity was 4.3 times that of dexamethasone in animal studies.[1]
Retrometabolic drug design
Loteprednol etabonate was developed using retrometabolic drug design. It is a so-called soft drug, meaning its structure was designed so that it is predictably metabolised to inactive substances. These metabolites, Δ1-cortienic acid and its etabonate, are derivatives of cortienic acid, itself an inactive metabolite of hydrocortisone.[1][4][6]
- Cortisol, a naturally occurring corticosteroid, known as hydrocortisone when used as a drug
- Δ1-Cortienic acid, inactive metabolite of loteprednol
- Cortienic acid, inactive metabolite of hydrocortisone
Chemistry
Loteprednol etabonate is an ester of loteprednol with etabonate (ethyl carbonate). The pure chemical compound has a melting point between 220.5 °C (428.9 °F) and 223.5 °C (434.3 °F). Its solubility in water is 1:2,000,000,[4] therefore it is formulated for ophthalmic use as either an ointment, a gel, or a suspension.[7]
Loteprednol is a corticosteroid. The ketone side chain of classical corticosteroids such as hydrocortisone is replaced by a cleavable ester, which accounts for the rapid inactivation.[8] (This is not the same as the etabonate ester.)


Loteprednol etabonate
Chemical synthesis
References
- ^ Jump up to:a b c d e f g Haberfeld H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 488. ISBN 9783527607495.
- ^ Jump up to:a b Loteprednol Professional Drug Facts.
- ^ Jump up to:a b c Dinnendahl V, Fricke U (2008). Arzneistoff-Profile (in German). 6 (22 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ^ “Highlights of Prescribing Information: Lotemax” (PDF). 2012.
- ^ Bodor N, Buchwald P (2002). “Design and development of a soft corticosteroid, loteprednol etabonate”. In Schleimer RP, O’Byrne PM, Szefler SJ, Brattsand R (eds.). Inhaled Steroids in Asthma. Optimizing Effects in the Airways. Lung Biology in Health and Disease. 163. Marcel Dekker, New York. pp. 541–564.
- ^ “Loteprednol (Professional Patient Advice)”. Retrieved October 4, 2018.
- ^ Pavesio CE, Decory HH (April 2008). “Treatment of ocular inflammatory conditions with loteprednol etabonate”. The British Journal of Ophthalmology. 92 (4): 455–9. doi:10.1136/bjo.2007.132621. PMID 18245274. S2CID 25873047.
- ^ Druzgala P, Hochhaus G, Bodor N (February 1991). “Soft drugs–10. Blanching activity and receptor binding affinity of a new type of glucocorticoid: loteprednol etabonate”. The Journal of Steroid Biochemistry and Molecular Biology. 38 (2): 149–54. doi:10.1016/0960-0760(91)90120-T. PMID 2004037. S2CID 27107845.
Further reading
- Stewart R, Horwitz B, Howes J, Novack GD, Hart K (November 1998). “Double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammation. Loteprednol Etabonate Post-operative Inflammation Study Group 1”. Journal of Cataract and Refractive Surgery. 24 (11): 1480–9. doi:10.1016/s0886-3350(98)80170-3. PMID 9818338. S2CID 24423725.
////////////loteprednol etabonate
CCOC(=O)OC1(CCC2C1(CC(C3C2CCC4=CC(=O)C=CC34C)O)C)C(=O)OCCl

NEW DRUG APPROVALS
ONE TIME
$10.00
Regdanvimab

| (Heavy chain) QITLKESGPT LVKPTQTLTL TCSFSGFSLS TSGVGVGWIR QPPGKALEWL ALIDWDDNKY HTTSLKTRLT ISKDTSKNQV VLTMTNMDPV DTATYYCARI PGFLRYRNRY YYYGMDVWGQ GTTVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKRVEPKS CDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH EALHNHYTQK SLSLSPGK (Light chain) ELVLTQPPSV SAAPGQKVTI SCSGSSSNIG NNYVSWYQQL PGTAPKLLIY DNNKRPSGIP DRFSGSKSGT SATLGITGLQ TGDEADYYCG TWDSSLSAGV FGGGTELTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADGSPVK AGVETTKPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H97, H155-H211, H231-L215, H237-H’237, H240-H’240, H272-H332, H378-H436, H’22-H’97, H’155-H’211, H’231-L’215, H’272-H’332, H’378-H’436, L22-L89, L138-L197, L’22-L’89, L’138-L’197) |
>Regdanvimab light chain: ELVLTQPPSVSAAPGQKVTISCSGSSSNIGNNYVSWYQQLPGTAPKLLIYDNNKRPSGIP DRFSGSKSGTSATLGITGLQTGDEADYYCGTWDSSLSAGVFGGGTELTVLGQPKAAPSVT LFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASS YLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
>Regdanvimab heavy chain: QITLKESGPTLVKPTQTLTLTCSFSGFSLSTSGVGVGWIRQPPGKALEWLALIDWDDNKY HTTSLKTRLTISKDTSKNQVVLTMTNMDPVDTATYYCARIPGFLRYRNRYYYYGMDVWGQ GTTVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHT FPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPC PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVY TLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSK LTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Regdanvimab
レグダンビマブ;
EMA APPROVED, 2021/11/12, Regkirona
Treatment of adults with coronavirus disease 2019 (COVID-19)
MONOCLONAL ANTIBODY, ANTI VIRAL, PEPTIDE
CAS: 2444308-95-4, CT-P59
Regdanvimab, sold under the brand name Regkirona, is a human monoclonal antibody used for the treatment of COVID-19.[1] The antibody is directed against the spike protein of SARS-CoV-2. It is developed by Celltrion.[2][3] The medicine is given by infusion (drip) into a vein.[1][4]
The most common side effects include infusion-related reactions, including allergic reactions and anaphylaxis.[1]
Regdanvimab was approved for medical use in the European Union in November 2021.[1]
Regdanvimab is a monoclonal antibody targeted against the SARS-CoV-2 spike protein used to treat patients with COVID-19 who are at risk of progressing to severe COVID-19.
Regdanvimab (CT-P59) is a recombinant human IgG1 monoclonal antibody directed at the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.4 It blocks the interaction between viral spike proteins and angiotensin-converting enzyme 2 (ACE2) that allows for viral entry into the cell, thereby inhibiting the virus’ ability to replicate. Trials investigating the use of regdanvimab as a therapeutic candidate for the treatment of COVID-19 began in mid-2020.1,3 It received its first full approval in South Korea in September 2021,3 followed by the EU in November 2021.5
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5.
Celltrion’s Monoclonal Antibody Treatment regdanvimab, Approved by the European Commission for the Treatment of COVID-19
- The European Commission (EC) granted marketing authorisation for Celltrion’s regdanvimab following positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) last week (11/11/2021)
- Celltrion continues to discuss supply agreements with regulatory agencies and contractors in more than 30 countries in Europe, Asia and LATAM to accelerate global access to regdanvimab
- The use of regdanvimab across the Republic of Korea is rapidly increasing to address the ongoing outbreaks
November 14, 2021 08:04 PM Eastern Standard Time
INCHEON, South Korea–(BUSINESS WIRE)–Celltrion Group announced today that the European Commission (EC) has approved Regkirona (regdanvimab, CT-P59), one of the first monoclonal antibody treatments granted marketing authorisation from the European Medicines Agency (EMA). The EC granted marketing authorisation for adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The decision from the EC follows a positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on November 11th, 2021.1
“Today’s achievement, coupled with CHMP positive opinion for regdanvimab, underscores our ongoing commitment to addressing the world’s greatest health challenges,” said Dr. HoUng Kim, Ph.D., Head of Medical and Marketing Division at Celltrion Healthcare. “Typically, the recommendations from the CHMP are passed on to the EC for rapid legally binding decisions within a month or two, however, given the unprecedented times, we have received the EC approval within a day. As part of our global efforts to accelerate access, we have been communicating with the governments and contractors in 30 countries in Europe, Asia and LATAM. We will continue working with all key stakeholders to ensure COVID-19 patients around the world have access to safe and effective treatments.”
Monoclonal antibodies are proteins designed to attach to a specific target, in this case the spike protein of SARS-CoV-2, which works to block the path the virus uses to enter human cells. The EC approval is based on the global Phase III clinical trial involving more than 1,315 people to evaluate the efficacy and safety of regdanvimab in 13 countries including the U.S., Spain, and Romania. Data showed regdanvimab significantly reduced the risk of COVID-19 related hospitalisation or death by 72% for patients at high-risk of progressing to severe COVID-19.
Emergency use authorisations are currently in place in Indonesia and Brazil, and the monoclonal antibody treatment is fully approved in the Republic of Korea. In the U.S., regdanvimab has not yet been approved by the Food and Drug Administration (FDA), but the company is in discussion with the FDA to submit applications for an Emergency Use Authorisation (EUA).
As of November 12th, 2021, more than 22,587 people have been treated with regdanvimab in 129 hospitals in the Republic of Korea.
Notes to Editors:
About Celltrion Healthcare
Celltrion Healthcare is committed to delivering innovative and affordable medications to promote patients’ access to advanced therapies. Its products are manufactured at state-of-the-art mammalian cell culture facilities, designed and built to comply with the US FDA cGMP and the EU GMP guidelines. Celltrion Healthcare endeavours to offer high-quality cost-effective solutions through an extensive global network that spans more than 110 different countries. For more information please visit: https://www.celltrionhealthcare.com/en-us.
About regdanvimab (CT-P59)
CT-P59 was identified as a potential treatment for COVID-19 through screening of antibody candidates and selecting those that showed the highest potency in neutralising the SARS-CoV-2 virus. In vitro and in vivo pre- clinical studies showed that CT-P59 strongly binds to SARS-CoV-2 RBD and significantly neutralise the wild type and mutant variants of concern. In in vivo models, CT-P59 effectively reduced the viral load of SARS-CoV-2 and inflammation in lung. Results from the global Phase I and Phase II/III clinical trials of CT-P59 demonstrated a promising safety, tolerability, antiviral effect and efficacy profile in patients with mild-to-moderate symptoms of COVID-19.2 Celltrion also has recently commenced the development of a neutralising antibody cocktail with CT-P59 against new emerging variants of SARS-CoV-2.
Medical uses
In the European Union, regdanvimab is indicated for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.[1]
Society and culture
Names
Regdanvimab is the proposed international nonproprietary name (pINN).[5]
Legal status
In March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on regdanvimab.[6][7] In October 2021, the EMA started evaluating an application for marketing authorization for the monoclonal antibody regdanvimab (Regkirona) to treat adults with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID 19.[8] The applicant is Celltrion Healthcare Hungary Kft.[8] The European Medicines Agency (EMA) concluded that regdanvimab can be used for the treatment of confirmed COVID-19 in adults who do not require supplemental oxygen therapy and who are at high risk of progressing to severe COVID-19.[4]
In November 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for regdanvimab (Regkirona) for the treatment of COVID-19.[9][10] The company that applied for authorization of Regkirona is Celltrion Healthcare Hungary Kft.[10] Regdanvimab was approved for medical use in the European Union in November 2021.[1]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | Regkirona |
| Other names | CT-P59 |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous infusion |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 2444308-95-4 |
| DrugBank | DB16405 |
| UNII | I0BGE6P6I6 |
| KEGG | D12241 |
- Tuccori M, Ferraro S, Convertino I, Cappello E, Valdiserra G, Blandizzi C, Maggi F, Focosi D: Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020 Jan-Dec;12(1):1854149. doi: 10.1080/19420862.2020.1854149. [Article]
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5. [Article]
- Syed YY: Regdanvimab: First Approval. Drugs. 2021 Nov 1. pii: 10.1007/s40265-021-01626-7. doi: 10.1007/s40265-021-01626-7. [Article]
- EMA Summary of Product Characteristics: Regkirona (regdanvimab) concentrate for solution for intravenous infusion [Link]
- EMA COVID-19 News: EMA recommends authorisation of two monoclonal antibody medicines [Link]
- EMA CHMP Assessment Report: Celltrion use of regdanvimab for the treatment of COVID-19 [Link]
- Protein Data Bank: Crystal Structure of COVID-19 virus spike receptor-binding domain complexed with a neutralizing antibody CT-P59 [Link]
References
- ^ Jump up to:a b c d e f g “Regkirona EPAR”. European Medicines Agency. Retrieved 12 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Celltrion Develops Tailored Neutralising Antibody Cocktail Treatment with CT-P59 to Tackle COVID-19 Variant Spread Using Its Antibody Development Platform” (Press release). Celltrion. 11 February 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ “Celltrion Group announces positive top-line efficacy and safety data from global Phase II/III clinical trial of COVID-19 treatment candidate CT-P59” (Press release). Celltrion. 13 January 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ Jump up to:a b “EMA issues advice on use of regdanvimab for treating COVID-19”. European Medicines Agency. 26 March 2021. Retrieved 15 October 2021.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 660–1.
- ^ “EMA starts rolling review of Celltrion antibody regdanvimab for COVID-19” (Press release). European Medicines Agency (EMA). 24 February 2021. Retrieved 4 March 2021.
- ^ “EMA review of regdanvimab for COVID-19 to support national decisions on early use” (Press release). European Medicines Agency (EMA). 2 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b “EMA receives application for marketing authorisation Regkirona (regdanvimab) treating patients with COVID-19”. European Medicines Agency. 4 October 2021. Retrieved 15 October 2021.
- ^ “Regkirona: Pending EC decision”. European Medicines Agency. 11 November 2021. Retrieved 11 November 2021.
- ^ Jump up to:a b “COVID-19: EMA recommends authorisation of two monoclonal antibody medicines”. European Medicines Agency (EMA) (Press release). 11 November 2021. Retrieved 11 November 2021.
Further reading
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. (January 2021). “A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein”. Nature Communications. 12 (1): 288. doi:10.1038/s41467-020-20602-5. PMC 7803729. PMID 33436577.
External links
- “Regdanvimab”. Drug Information Portal. U.S. National Library of Medicine.
///////////Regdanvimab, Regkirona, MONOCLONAL ANTIBODY, ANTI VIRAL, EU 2021, APPROVALS 2021, EMA 2021, COVID 19, CORONAVIRUS, PEPTIDE, レグダンビマブ , CT-P59, CT P59

NEW DRUG APPROVALS
ONE TIME
$10.00
Diroximel fumarate

Diroximel fumarate
ジロキシメルフマル酸エステル;
| Formula |
C11H13NO6
|
|---|---|
| CAS | 1577222-14-0 |
| Mol weight |
255.224
|
2021/11/15 EMA APPROVED, VUMERITY
Treatment of multiple sclerosis
Diroximel fumarate, sold under the brand name Vumerity, is a medication used for the treatment of relapsing forms of multiple sclerosis (MS).[1][3][4]
Diroximel fumarate was approved for medical use in the United States in October 2019,[5] and in the European Union in November 2021.[2]
History
This drug was formulated by Alkermes in collaboration with Biogen.[6]
Society and culture
Legal status
Diroximel fumarate was approved for medical use in the United States in October 2019.[5]
On 16 September 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vumerity, intended for the treatment of adults with relapsing remitting multiple sclerosis.[7] The applicant for this medicinal product is Biogen Netherlands B.V.[7] It was approved for medical use in the European Union in November 2021.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
US 8669281
https://patents.google.com/patent/US8669281B1/en
PATENT
WO 2014152494
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152494
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (14)
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate 14 was synthesized following general procedure 1 (1.03 g, 35 %).
1H NMR (400 MHz, DMSO): δ 6.81 (2H, dd, J = 15.8 Hz); 4.36 (2H, t, J = 5.3 Hz); 3.84 (2H, t, J = 5.1 Hz); 3.80 (3H, s); 2.73 (4H, s). [M+H]+ = 256.07.
General Procedure 1
To a mixture of monomethyl fumarate (MMF) (1.0 equivalent) and HBTU (1.5 equivalents) in DMF (25 ml per g of MMF) was added Hünigs base (2.0 equivalents). The dark brown solution was stirred for 10 minutes, where turned into a brown suspension, before addition of the alcohol (1.0 – 1.5 equivalents). The reaction was stirred for 18 hours at room temperature. Water was added and the product extracted into ethyl acetate three times. The combined organic layers were washed with water three times, dried with magnesium sulphate, filtered and concentrated in vacuo at 45 ºC to give the crude product. The crude product was purified by silica chromatography and in some cases further purified by trituration with diethyl ether to give the clean desired ester product. All alcohols were either commercially available or made following known literature procedures.
As an alternative to HBTU (N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1 -yl)uronium hexafluorophosphate), any one of the following coupling reagents can be used: EDCI/HOBt (N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride/hydroxybenzotriazole hydrate); COMU ((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate); TBTU (O-(benzotriazol-1 -yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TATU (O-(7-azabenzotriazole-1-yl)-1,1 ,3,3-tetramethyluronium tetrafluoroborate); Oxyma (ethyl (hydroxyimino)cyanoacetate); PyBOP ((benzotriazol-1 -yloxy)tripyrrolidinophosphonium hexafluorophosphate); HOTT (5-(1-oxido-2-pyridyl)-N,N,N’,N’-tetramethylthiuronium hexafluorophosphate); FDPP (pentafluorophenyl diphenylphosphinate); T3P (propylphosphonic anhydride); DMTMM (4-(4,6-dimethoxy-1,3,5-triazin-2-y1)-4-methylmorpholinium tetrafluoroborate); PyOxim ([ethyl
cyano(hydroxyimino)acetato-O2]tri-1-pyrrolidinylphosphonium hexafluorophosphate); TSTU (N,N,N’,N’-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate); TDBTU (O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TPTU (O-(2-oxo-1(2H)pyridyl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TOTU (O-[(ethoxycarbonyl)cyanomethylenamino]-N,N,N’,N’-tetramethyluronium tetrafluoroborate); IIDQ (isobutyl 1,2-dihydro-2-isobutoxy- 1-quinolinecarboxylate); or PyCIU
(chlorodipyrrolidinocarbenium hexafluorophosphate),
As an alternative to Hünig’s base (diisopropylethylamine), any one of the following amine bases can be used: triethylamine; tributylamine; triphenylamine; pyridine; lutidine (2,6-dimethylpyridine); collidine (2,4,6-trimethylpyridine); imidazole; DMAP (4-(dimethylamino)pyridine); DABCO (1 ,4-diazabicyclo[2.2.2]octane); DBU (1 ,8-
diazabicyclo[5.4.0]undec-7-ene); DBN (1,5-diazabicyclo[4.3.0]non-5-ene); or proton sponge® (N,N,N’,N’-tetramethyl-1 ,8-naphthalenediamine).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
PATENT
WO 2016124960
PATENT
WO 2017108960
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017108960
Example 3b: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester
Procedure A:
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (3 g; 20.96 mmol) and maleic acid anhydride (2.26 g; 23.1 mmol) in toluene (10 mL) were heated to 60°C under stirring for 29 hours. The temperature was raised to 80°C and heated for another 19 hours. Acetyl chloride (0.3 mL; 4.2 mmol) was added and heating (80°C) was continued for 24 hours. The reaction mixture was cooled to RT. The biphasic system was separated, the upper layer was discarded. The lower layer (viscous oil) crystallized. The crystallized compound was suspended in acetone (50 mL) and stirred for 15 minutes before being filtrated off. The product was dried at 50°C for 5 hours and 8 mbar to yield the 1st crop (1.65 g). The mother liquor was evaporated and the obtained oil/solid was suspended in acetone (5 mL) and stirred overnight at RT. The product was filtrated off and dried at 50°C for 5 hours and 8 mbar to yield the 2nd crop (1.41 g). The mother liquor was evaporated and the obtained oil/solid was suspended in a mixture of diethylether/acetone (5 mL/1 mL) and stirred overnight at RT. The product was filtrated off and dried at 8mbar/50°C for 3 hours (3rd crop, 0.37 g).<a name=”
Yield: 3.43 g (68% of theory)
Purity: 1st crop 96.8 area%; 2nd crop 96.0 area-%; 3rd crop 85.4 area-% (HPLC/UV, method A, λ=200nm; tr: 3.8 min.)
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.61 (s, 4 H) 3.66 (t, J=5.47 Hz, 2 H) 4.23 (t, J=5.47 Hz, 2 H) 6.51 – 6.72 (m, 2 H) 6.60 (s, 1 H) 6.63 (s, 1 H) 13.21 (br s, 1 H)
Procedure B:
Reaction performed in a reactor (Mettler Toledo, Optimax):
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (20 g; 0.14 mol) and maleic acid anhydride (15 g; 0.15 mol) in toluene (70 mL) were heated to 80°C under stirring (150 rpm) for 29 hours. Acetyl chloride (2 mL; 0.03 mol) was added and heating (80°C) was continued overnight. Stirring speed was raised to 200 rpm) after 15.5 hours (at 80°C) (product precipitated upon raising stirring speed. The reaction mixture was cooled to 20°C within 1 hour, directly after highering stirring speed. The reaction mixture was stirred for 4 hours, before being filtrated off. The filtrated precipitate was washed with toluene (30 mL) and then with heptane (70 mL), the product was dried at 60°C and 18 mbar. The crude product (26.26 g) with -90% purity was suspended in a mixture of acetone (30 mL)/heptane (30 mL) and stirred at RT for 2 days. The product was filtrated off, washed with heptane (30 mL) and dried at 50°C and 7 mbar.
Yield: 24.12 g (72% of theory)
Purity: 97.4 area-% at 200 nm
Procedure C:
a) Ethylene carbonate (8.89 g; 0.1 mol), succinimide (10 g; 0.1 mol) and sodium carbonate (0.53 g, 5 mmol) were heated to 100°C, the temperature was hold overnight. The product was cooled down yielding a brownish solid (13.73 g) which was grinded in a mortar.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g, 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (33 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (0.5 mL; 7 mmol) was added and heating<a name=”
(80°C) was continued overnight. Heating was stopped and after stirring for another 2 hours the product was filtered off. The product was dried for 2 hours at 60°C and 8 mbar, yielding 15.82 g of crude product.
purity: 63 area-% at 200nm; 80 area-% at 220 nm
Procedure D
a) Ethylene carbonate (44.43 g; 0.5 mol), succinimide (50 g; 0.5 mol) and sodium carbonate (2.67 g; 25 mmol) were heated to 100°C. The reaction mixture was stirred at 100°C for overnight. The mixture was cooled to RT, yielding 72.4 g of the raw product.
40 g of the raw product were suspended in ethylacetate (40 mL) and heated to reflux for 30 minutes. The turbid mixture was cooled to RT and left stirring O/N. The product was filtrated off and dried under vacuum at RT to yield 29.19 g.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g; 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (30 mL) were heated to 80°C under stirring. Acetyl chloride (0.5 mL; 7 mmol) was added after 19 hours and heating (80°C) was continued overnight. Heating was stopped and stirring was continued for 2 days. The product was filtrated off and dried at 23 mbar and 60°C.
purity: 82 area% at 200 nm; 91 area-% at 220 nm
Procedure E:
a) Succinimide (500 g; 5.0 mol), ethylene carbonate (444.34 g; 5.0 mol) and sodium carbonate (26.74 g; 0.25 mol) were mixed and slowly heated to 130°C under stirring for 7 hours. The product was distilled via vacuum distillation to yield the product as colourless substance (628.14 g; 87% of theory)
b) The distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (150 g; 1.05 mol) from sequence a) and maleic acid anhydride (102.76 g; 1.05 mol) in toluene (350 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (7 mL; 0.01 mol) was added and heating (80°C) was continued. After 6 hours, the reaction mixture was cooled to 20°C within 30 minutes. The product was filtrated off and washed with toluene (200 mL), yielding 221.8 g of a white crystalline product (crude product).
purity: 91 area% at 200 nm; 92 area-% at 220 nm<a name=”
Procedure F:
a) Ethylene carbonate (9.78 g; 0.11 mol), succinimide (10 g; 0.10 mol) and triethylamine (0.7 mL; 5mmol) were heated to 98°C. The reaction mixture was stirred at this temperature overnight. The mixture was cooled to RT, yielding a colourless liquid, which crystallizes upon standing at RT to a colorless solid (14.89 g).
b) The crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione from sequence a) (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (25 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene and dried at 50°C and 8 mbar for 3 hours. Yield: 6.52 g (77%)purity: 93 area% at 200 nm; 94 area-% at 220 nm
Procedure F’
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in a reactor, succinimide (173.07 g, 1.747 mol) and Et3N (12.2 mL, 87.350 mmol) were added and the reaction mixture was warmed up to 90°C and stirred for 24h. Reaction mixture was cooled to 50°C, 500 mL of acetone was added, followed by addition of maleic anhydride (164.19 g, 1.674 mol) and Et3N (10.15 mL, 72.772 mmol). Reaction mixture was stirred at 50-55°C for 4h, cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid.
Yield: 274 g (65%)
Purity: 97.23 area % at 200 nm
Procedure F”
(Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (250 g, 1.036 mol) was suspended in acetone (500 mL) in 1-L reactor, acetyl chloride (5.53 mL, 77.736 mmol) was added drop wise at 20-25°C and reaction mixture was warmed up to 50-55°C and stirred for 20h. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone <a name=”(2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II).
Yield: 231.3 g (92.5%)
Purity: 99.47 area % at 200 nm
Summary:
Procedure B and E, using distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, showed purities of -90-91 area-% of the crude product, ongoing crystallization of the target compound could improve the purity to -97% also shown in procedure A. Distillation of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione needs harsh conditions (Ex. 3a; procedure A). Using the crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, produced with Na2CO3 lead to low product purities of 63 area-% (procedure C).
Crystallization of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (procedure D) lead to product purities comparable to procedure A, B and E with distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, but crystallization is compounded by a significant product loss of – 25%.
The raw 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione could be used without any disadvantageous impact on product quality by substituting Na2CO3 with triethylamine as shown in procedure F with a purity of 93 area-%.
Procedure G
Two experiments were performed in parallel:
Each with 1 g (7 mmol) 1-(2-hydroxy-ethyl)-pyrrolidine-2,5-dione and 0.75 g (7.7 mmol) maleic acid anhydride in 6 mL acetonitrile in screw capped vials. To one of the reaction mixtures was given 0.1 mL triethylamine. Both mixtures were stirred at RT. Samples were taken and investigated by NMR (in DMSO).
product formation after 1 hour (quantified by NMR):
mixture without triethylamine: 0%
mixture with triethylamine: 55%<a name=”
product formation after 2 hours:
mixture without triethylamine: 0%
mixture with triethylamine: 71 %
Procedure H (isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for -24 hours. The reaction was cooled to RT, first a biphasic layer was observed, then the product solidified (sticking to glass wall and stirrer). The product was filtrated off after 2.5 hours of stirring, washed with toluene (50 mL) and dried under vacuum. The dried product was milled and suspended again in toluene (60 mL) at RT, after 30 minutes the product was filtrated off and dried under atmospheric conditions to yield 7.24 g of the cis intermediate (86% of theory). The intermediate product was suspended in toluene (30 mL) and heated to 80°C, acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for 5 hours. The reaction mixture was cooled to RT and stirred for 2 hours. The product was filtrated off, washed with toluene (30 mL) and dried at 50°C and 8 mbar O/N.
purity: 95.6 area-% at 200nm; (0.2% of Impurity I)
Procedure H (without isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene (30 mL) and dried at 50°C and 8 mbar for 3 hours.
purity: 93.2 area-% at 200nm; (1.3% of Impurity I)
Procedure I (scale-up without cis isolation)
Maleic acid (959.09 g; 9.8 mol) was added to a reactor under stirring, which was already loaded with toluene (7 L), then 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione<a name=”
(1400 g; 9.8 mol) was added. Then the mixture was heated to 76°C within ~1 h (up to ~50°C the mixture is a suspension with the tendency of conglomeration of solids, very difficult consistency) at 50°C a turbid solution resulted. Stirring was continued at 80°C for 2 days. Acetyl chloride (138 mL; 1.96 mol) was added under enhanced stirring at 80°C. After -5-10 minutes a crystalline precipitate was formed, which transformed into a pasty/syrupy solid, sticking to reactor walls (difficult handling). Heating was continued overnight (reaction completed after 5 hours as IPC showed). Mixture is still an emulsion, seeding was added and the product precipitated. Stirring at 80°C was continued for ~2 hours then the mixture was cooled to RT. The solid was filtrated off and dried at 50°C and 12 mbar overnight to yield 1818.74 g of the product.
purity: 96.34 area-% at 218 nm; (1.5% of Impurity I)
Procedure J:
2L flask (reaction volume ~1 L): Succinimide (460 g; 4.6 mol), ethylene carbonate (450 g; 5.1 mol) and triethylamine (32 mL; 0.23 mol) were heated to 85°C under stirring overnight. Temperature was raised to 95°-97C and heating was continued O/N. The mixture was cooled to 50°C. Acetonitrile (1600 mL) was charged into a 10 L reactor. To the reaction mixture was added acetonitrile (1000 mL) at 50°C and the solution was transferred to the reactor (reactor T ~22°C), triethylamine (35 mL) was added, then maleic acid anhydride (500.81 g; 5.1 mol). The mixture was heated to 55°C for 5.5 hours. A part of the solvent was distilled off (~1200 mL). Then toluene (1200 mL) was added. The mixture was heated to 90°C. The mixture was cooled to 50°C. At 60°C (clear solution), seeding was added ~300 mg, after -3 minutes a suspension resulted. The mixture was further cooled down to 20°C within 10 hours and kept on stirring O/N. The white crystalline product was filtrated off, washed with toluene (1000 mL) and dried at 55°C and 9 mbar for 2 h to yield 908.99 g (81% yield).
905 g of the isolated, crystallized product was suspended in acetonitrile (2.9 L). Acetyl chloride (23 mL) was added and the mixture was heated to 80°C (clear, colorless solution) for 4 hours. Toluene (1000 mL) was added and the mixture was cooled to RT within 2 hours (linear). The mixture was further cooled to 0°C within 60 minutes. The <a name=”product was filtrated off and washed with toluene (1000 mL). The product was dried overnight at 9 mbar and 50°C.
Yield: 742.06 g (66%)
purity: 99.9 area% at 200 nm
Summary:
Isolation of the cis intermediate leads to a significantly lower content of impurities, in particular of Impurity I. Toluene as solvent leads to disadvantageous conditions regarding consistency of the reaction mixture (procedure H). The use of acetonitrile or acetone (procedure I/F) leads to improved reaction conditions and product quality.
Example 3c
Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) from ethylene carbonate and succinimide (without isolation of
intermediates)
Procedure
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in an 1-L reactor, succinimide (173.07 g, 1.747 mol) and Et3N (24.4 mL, 0.175 mol) were added and the reaction mixture was warmed up to 90-92°C and stirred for 24h. Distillation column<a name=”
was set up on the reactor and the remaining Et3N was distilled off. Reaction mixture was cooled to 40-45°C, 500 mL of acetone was added, followed by addition of maleic anhydride (184 g, 1.878 mol) and Et3N (10.96 mL, 78.615 mmol). Reaction was stirred at 40°C for 6h (precipitation occurred after 3h), cooled to 20-25°C and acetyl chloride (20.86 mL, 0.293 mol) was added drop wise. Reaction mixture was then warmed up to 50-55°C and stirred for 20h. Orange solution crystallized upon seeding. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid. Yield: 352.8 g (83.7%)
Purity: 99.69 area % at 200 nm
Example 4: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester
Procedure A:
The starting material (E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (5 g; 20 mmol) was suspended in dichloromethane (60 mL) and cooled to 0°C, triethylamine (3.16 mL; 22.8 mmol) was added, resulting a clear solution. To this solution methylchloroformate (3.3 mL; 20.7 mmol) was carefully added within 30 minutes via syringe (reaction very exothermic). After 15 min of stirring at 0°C, DMAP (0.25 g; 2.1 mmol) was added into the reaction mixture at 0°C, stirring was continued for 3 hours at 0°C. The reaction mixture was poured into water (200 mL) and additional dichloromethane (100 mL) was added. The organic layer was separated and the aqueous layer was extracted once again with dichloromethane (50 mL). The combined organic layers were washed with brine (50 mL). The solvent was evaporated at 52°C. To the brown oil, which solidified, was added acetone (20 mL) and the mixture was stirred overnight. The product was filtrated off (white solid, part I) (2.73 g) and to the mother <a name=”liquor silica was added, the mixture was evaporated. Acetone (50 mL) was added and silica was filtrated off. The solvent was evaporated and diethylether (30 mL) was added to the solid, the mixture was stirred for ~1 hour. The product was filtrated off (part II) (1.6 g).
Overall yield: 4.33 g (82%)
Purity: part I 100 area-% at 200 nm; part II 97.96 area-% at 200 nm
Procedure A’
(E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) (200 g, 0.829 mol) was suspended in acetone (2000 mL) in 3-L reactor at 20-25°C and cooled to 0°C. Et3N (150.31 mL, 1.078 mol) was added drop wise at 0-5°C. Into resulting solution, methyl chloroformate (83.27 mL, 1.072 mol) was added drop wise at 0-5°C. Reaction mixture was warmed up to 45°C and stirred for 2h. Upon completion, reaction mixture was cooled to 20-25°C and water (600 mL) was added drop wise with maintaining the temperature at 20-25°C resulting with off white to yellowish solution. pH was adjusted to 7 with 1M HCl. One more volume of water was added and pH corrected if needed. Part of acetone from the reaction mixture (5 volumes or 1000 mL) was distilled off under diminished pressure and reactor walls were washed with 1 more volume of water (200 mL), thus resulting in a solution of acetone/water mixture 1:1 (total 10 volumes). Reaction mixture was gradually cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold water (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (Formula I).
Yield: 183.7 g (86.8%)
Purity: 100.00 area % at 200 nm
Crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (170 g) was suspended in acetone (850 mL) at 20-25°C and warmed up to 50°C resulting with colorless solution. Water (850 mL) was added in portions at 50°C and solution was cooled gradually. Crystallization started at 32°C. Reaction mixture was stirred at crystallization temperature for 30 minutes and cooled further to 0°C, stirred at 0°C for 2h and resulting <a name=”white suspension was filtered off and solid was washed with cold water (2×170 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate.
Yield: 152.5 g (89.7%)
Purity: 100.00 area % at 200 nm
Procedure B:
The starting material (5 g, 20 mmol) was suspended in toluene (25 mL). Acetyl chloride (0.29 mL) and methanol (2.5 mL) were added, the reaction mixture was heated to 55°C and stirred for 3 hours. The reaction mixture was poured into water (100 mL) and extracted with ethylacetate (100 mL). The organic layer was separated and dried over sodium sulfate. The solvent was evaporated (crude product 4.7 g, main impurities dimetylfumarate (13%) and fumaric acid (1%) (HPLC at 200 nm)).
Yield: 4.7 g (88%)
Purity: 82.1 area-% at 200 nm
Procedure C:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form A; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (without isolation of (Z)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (30 g; 0.12 mol) was suspended in dichloromethane (DCM, 160 mL) and cooled to 0°C, triethylamine (TEA, 19 mL; 0.14 mol) was added, resulting a clear solution. To this solution methyl chloroformate (19.74 mL; 0.12 mol) was added carefully within 30 minutes via syringe. Stirring was continued for ~2 hours. Water (200 mL) was added to the reaction mixture and stirring was continued for 5-10 minutes. The organic layer was separated and the aqueous layer was washed with another portion of DCM (100 mL). The combined organic layers were dried over sodium sulfate, before being evaporated. To the crude product was added acetone (50 mL) and the mixture was stirred for 3 hours before being filtered off. The product was washed with heptane (50 mL) and dried at 50°C and 21 mbar for 1 h.<a name=”
Yield: 20.52 g (65%)
Purity: 98.7 area-% at 220 nm; (0.3% of Impurity I)
XRPD diffraction peaks: 7.1, 11.6, 13.5, 13.7, 16.3, 16.7, 18.0, 18.4, 21.1, 22.1, 23.1, 23.9, 24.4, 25.5, 27.0, 27.5, 28.0, 28.6, 30.8, 31.2, 31.9, 32.3, 33.7, 34.2, 34.4, 34.9, 35.1, 35.7, 36.0, 36.8, 38.3, 40.1, 40.5, 41.7, 42.4, 43.0, 43.4, 45.0, 45.3, 46.2, 46.4, 47.0, 48.6, 49.4, 49.9, 52.0 + 0.2 degrees two theta.
The Form A according to Procedure C showed a habitus as depicted in Figure 7a
Procedure D:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A The starting material (10 g; 41.5 mmol) was suspended in toluene (70 mL) at 23°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added. Methyl chloroformate (6.58 mL; 41.5 mmol) was slowly added within -30 minutes. After stirring for 2 hours water (40 mL) was added and shortly after acetone (110 mL), stirring was continued for ~2 minutes. The organic layer was separated and washed with brine (15 mL). After drying over sodium sulfate, the solvent was evaporated, yielding a slightly grey solid as crude product (9.42 g). The raw product was suspended in acetone (20 mL) and heptane (20 mL). The mixture was heated to reflux for 15 minutes resulting in a clear solution with just a small amount of solid. The mixture was cooled to RT and stirred overnight (precipitation started at 45°C, cooling: flask left in cooling oil bath ~lh to RT). The resulting product showed polymorphic form A.
Yield: 7.83 g (74%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure D showed a habitus as depicted in Figure 7b<a name=”
Procedure E:
The starting material (1 g; 4.15 mmol) was suspended in dichloromethane (50 mL) at RT. Methyl chloroformate (0.64 mL; 8.3 mmol) was added and stirring was continued overnight, in process control by HPLC showed no conversion.
Procedure F:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (7 g; 0.03 mol) and Na2CO3 were suspended in ethylacetate (50 mL). To the suspension was added methyl chloroformate (3.37 mL; 0.04 mol) in one portion. The reaction mixture was heated to 70°C. The temperature was kept for 15.5 h. The reaction mixture was cooled to 20°C and ethyl acetate (70 mL) was added to the white suspension. The solids were filtrated off and the ethyl acetate layer was washed with water (40 mL), dried over Na2S04 and evaporated to yield 6.4 g of the white crystalline crude product.
The crude product was suspended in a mixture of ethylacetate (10 mL) and heptane (10 mL). The suspension was heated to reflux for 30 minutes, then cooled to 23°C and stirred overnight. The product was filtrated off and dried at 8 mbar and 50°C overnight.
Yield: 5.62 g (75%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure E showed a habitus as depicted in Figure 7c
Procedure G:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form B; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B
(A) 9 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methylester was heated to 115°C. The melted compound was stirred for -20 minutes and then dropped into a precooled mortar (0°C).<a name=”
Purity of form B: 98.8 area-% at 200nm
XRPD-pattern: (Figure 4)
(B) 3.00 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester (form A) was suspended in 150 mL of dibutyl ether. Suspension was heated to 120° C while mixing. Solution was left at 25°C for 2 days. Crystallized material was filtered and dried at 23 °C at 12 mbar.
XRPD-pattern: (Figure 4′)
A measure of the relative volume change of a solid as a response to pressure change is called compressibility. An API should exhibit good compressibility which is dependent on the polymorphic state.
Experimental data:
The compressibility of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester form B and form A was assessed using a die and a flat-faced punch fitted on a TA-XT2 Texture analyser (Stable Micro Systems Ltd., Godalming, UK). 200 mg of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester sample is compressed in a steel mould (with the rate of displacement 0.03 mm/s). Cyclic procedure (similar to tapping) was performed: compressing, then retracting, relaxation for 15 s and then repeated compressive steps (altogether 10 steps). Each step exerts 0.2 MPa pressure on to the sample. Sample density is calculated by dividing the weight by the sample volume for each cycle. Maximum density is reached within 10 steps. Measurements were performed in duplicates for each sample, results are expressed as an average of duplicate measurements.
Results:
<a name=”
Form B of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester exhibits a higher density at compression, indicating superior compressibility compared to form A.
Procedure H:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form C; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form C
(E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (10 g; 41.5 mmol) was suspended in dichloromethane (DCM; 100 mL) and cooled to 0°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added, resulting a clear solution. To the reaction mixture was added methyl chloroformate (6.58 mL; 41.5 mmol) within 30 minutes via a syringe pump. After 15 min of stirring at 0°C, DMAP (0.51 g; 4 mmol) was added into the reaction mixture at 0°C. The resulting solution was stirred at 0°C for 2.5 hours, then the cold suspension was poured into water (70 mL), the reactor was washed with further DCM (20 mL), which was added also to the DCM/water mixture. The organic layer was separated and washed with HCl (32% aq) (5 mL) in water (60 mL), then with water (50 mL) and finally with brine (50 mL). To the obtained deep red to brown solution was added silica (40-63 um) and the mixture was stirred for 5 minutes, before being filtered off to yield a colorless solution, which was evaporated to yield a colorless oil (crude product). The obtained oil was dissolved in a mixture of ethyl acetate/heptane (1/4) (20 mL). The mixture was stirred for 2 days before being filtered off. The product was dried under vacuum.
Yield: 2.87 g (26%)
Purity: 90.9 area-% at 200 nm
XRPD diffraction peaks: 11.2, 11.8, 13.0, 13.6, 13.6, 16.8, 18.1, 19.6, 20.6, 21.2, 21.5, 22.3, 23.2, 23.7, 24.3, 24.4, 25.2, 25.6, 26.5, 27.6, 28.4, 29.1, 30.3, 31.1, 32.0, 33.1, 33.8, 36.1, 36.7, 37.5, 38.4, 38.9, 41.6, 42.5, 43.2, 44.8, 46.5, 48.7, 49.6, 49.9 + 0.2 degrees two theta.<a name=”
Procedure I:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form D; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form D
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B (1 g) was suspended in acetonitrile (3 mL). The suspension was stirred for 7 days in a closed screw cap vials followed by slow evaporation of the solvent under ambient conditions within 3 days.
Purity of form D: 96.3 area-% at 200 nm
XRPD diffraction peaks: 6.9, 11.7, 13.6, 13.9, 16.4, 16.9, 18.2, 20.9, 21.3, 22.3, 23.3, 24.0, 24.6, 25.7, 27.5, 27.7, 31.0, 31.3, 32.1, 32.4, 33.9, 35.3, 35.7, 38.4, 41.9, 42.7, 43.1, 43.6, 44.4, 46.5, 48.9 + 0.2 degrees two theta.
Procedure J:
The starting material (obtained via isolation of (Z)-But-2-enedioic acid mono-[2-(2, 5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (400 g; 1.7 mol) and Na2CO3 (264 g; 2.5 mol) were suspended in ethylacetate (2.7 L). To the suspension was added methyl chloroformate (193 mL; 2.5 mol) at 20°C. The reaction mixture was heated to 45°C within 90 minutes (linear heated). The mixture was kept on stirring for 5.5 hours. Ethylacetate (4 L) was added to the white suspension (at 45 °C). The suspension was stirred for 15 minutes before being filtrated off (45 °C suspension). The reactor was rinsed with another portion of ethylacetate (1 L). The filtrated solids were discarded. To the ethylacetate solution was added a mixture of HClaq (32%) (50 mL) and water (1 L) and the mixture was vigorously stirred for 10 minutes (at 35°C). Then the ethylacetate layer was separated (at ~35°C). The ethylacetate layer was transferred back to the reactor and stirred over sodium sulfate for 30 minutes, sodium sulfate was filtrated off and the ethylacetate layer was reduced to 900 mL. The suspension was transferred into a 3 L flask, equipped with a KPG stirrer and reflux condenser. The mixture was heated to reflux (stirring speed 160 rpm), the suspension was stirred until a clear solution was obtained (-30 minutes). Then heptane (550 mL) was added dropwise within 30 minutes<a name=”
under reflux conditions. Then the mixture (still solution) was slowly cooled to RT. The mixture was stirred O/N. The product was filtrated off and the filter cake was rinsed with heptane (500 mL) to yield the crystalline product (362.56 g; 86%).
purity: 99.8 area% at 218 nm (no Impurity I).
Alternative Procedure: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo- pyrrolidin-1-yl) -ethyl ester methyl ester
Procedure A
Monomethylfumarate (20 g, 0.15 mmol) was suspended in dry dichloromethane (400 mL) at RT, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (32.42 g, 0.17 mol), N-(2-hydroxyethyl)succinimide (21.57 g, 0.15 mol) and dimethylaminopyridine (0.94 g, 7.7 mmol) were added. The solution was stirred O/N at RT. The formed yellow solution was diluted with dichloromethane (300 mL) and washed twice with water (2×500 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure. To the crude product was added methyl tert. butyl ether (850 mL) and the reaction mixture was refluxed for 2.5 hours, cooled to RT, then filtrated and heated to reflux again for ~2 hours. After cooling to RT, the mixture was stored at ~5°C for 4 days. The white precipitate was filtrated off and washed with isopropylacetate (25 mL). The crystalline product was dried at 50°C and 7 mbar.
Yield: 10.8 g (28%)
Procedure B
Monomethylfumarate (1.5 g; 11.5 mmol) was suspended in dry DCM (30 mL) at 0°C. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (2.47 g; 12.8 mmol), N- (2-hydroxyethyl)succinimide (1.62 g; 11.3 mmol) and DMAP (0.07 g; 0.6 mmol) were added. The solution was stirred overnight at RT. The formed yellow solution was diluted with DCM (50 mL) and washed with water twice (2×35 mL). The organic layer<a name=”
was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-heptane:ethyl acetate 1:1->1:2). The final product showed polymorphic form A. The form A according to alternative Procedure B showed a prismatic habitus as depicted in Figure 7d
Yield: 2.3 g (78%)
Purity: 99.5 area-% at 200 nm
Example 5: Kinetic investigations
Monomethyl maleate was prepared in analogy to WO 2014/197860. Samples of 13.2 grams of monomethyl maleate in 50 mL of toluene and 0.1 equivalents of the isomerization catalyst were reacted at 80°C. Samples were taken after the given times and analyzed by HPLC at 200 nm. The absorbance ratio of monomethyl fumarate (3.8 min.) to monomethyl maleate (2.8 min.) was taken as conversion parameter. The results are shown in Figure 1. As it can be seen from Figure 1 the conversion of monomethyl maleate to monomethyl fumarate in the presence of is TMS (trimethylsilylchloride) is advantageously enhanced compared to the one in the presence of AcCl (acetyl chloride).
Example 6: Yield determination
Six samples of 13.2 g (0.1 mol) monomethyl maleate were diluted with toluene (50 mL) and 0.1 eq of the isomerization catalyst (trimethylsilylchloride or acetyl chloride) were added, three samples with trimethylsilylchloride and three samples with acetyl chloride. The resulting reaction mixtures were heated to the temperatures of 45 °C, 51°C and 80°C. After 22 hours the reaction mixtures were cooled to room temperature, the product was filtrated off and dried at 50°C/8-16 mbar overnight. The results are shown in Figure 2 As it can be seen from Figure 2 the isolated yields of the conversion of monomethyl maleate to monomethyl fumarate in the presence of TMS (trimethylsilylchloride) is at any
PATENT
CN 110698442
PATENT
WO 2021053476
PATENT
IN 201921037120
PATENT
WO 2021074842
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021074842
The drug compound having the adopted name “Diroximel fumarate” has chemical name: 2-(2,5-Dioxopyrrolidin-l-yl)ethyl methyl fumarate as below.
Diroximel fumarate is an investigational, novel oral fumarate with a distinct chemical structure and developed by Alkermes pic, for the treatment of relapsing-remitting multiple sclerosis (RRMS) and is currently under review by U.S. Food and Drug Administration. Biogen, under an exclusive license from Alkermes, intends to market Diroximel fumarate under the brand name VUMERITY™.
US 8669281 B1 first disclosed Diroximel fumarate, its preparation, composition and use thereof for treating multiple sclerosis. US 10080733 B2 further discloses the crystalline solid form of Diroximel fumarate having an X-ray powder diffraction pattern comprising 2Q peaks at 11.6, 21.0, 24.3, 27.4, and 27.9 ±0.2 2Q.
WO 2017/108960 A1 also discloses various alternative synthetic approaches to make Diroximel fumarate and crystalline solid forms thereof, designated as Polymorphic forms A to D.
Hence, there remains a need for alternate solid forms of Diroximel fumarate and preparative processes thereof, exhibiting desired bioavailability and stability. Hence, it is desirable to provide a viable solid form of Diroximel fumarate. The known processes for the preparation of Diroximel fumarate are not viable at industrial scale due to the use of expensive reagents and catalyst such as coupling agents disclosed in US 8669281 Bl, with very low yields. Hence, there remains a need for the improved process to make Diroximel fumarate.
In another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of monomethyl fumarate with (2,5-dioxopyrrolidin-l-yl)ethanol in the presence of an acid halide.
n another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid with methylation agent selected from the group consisting of 2,2-dimethoxypropane, trimethyl orthoformate and dimethyl carbonate.
Diroximel Fumarate
Example-7: Preparation of l-(2-hydroxyethyl)pyrrolidine-2,5-dione
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 0 °C; methyl /ert-butyl ether (300 mL) was added and the resulting mixture was stirred for 30 minutes at the same temperature. The solid was filtered and dried under vacuum for 5 minutes. The solid was combined with ethyl acetate (100 mL) at 0 °C and stirred at the same temperature for 30 minutes. The solid was filtered and dried in rotatory vacuum dryer at 40 °C for 30 minutes to obtain 142.5 g of the title compound as off-white solid with HPLC purity of 99.6%.
Example-8: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (4.0 g) and dichloromethane (40 mL) at 5 °C, Oxalyl chloride (5.85 g) was added slowly in 10 minutes, then a drop of DMF was added at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (5.06 g) and dichloromethane (35 mL), diisopropylethylamine (DIPEA) (9.93 g) was added and cooled the reaction mixture to 5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane was slowly added to this later mixture at 5 °C for 20 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with saturated ammonium chloride solution and the organic layer was separated. Organic layer was washed with 10% citric acid solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (15 mL) at 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with chilled acetone (3 mL) and then with cyclohexane (4 mL). The wet solid was dried at 40 °C under vacuum to obtain 3.3 g of the title compound with HPLC purity of 99.95 %
Example-9: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (100.0 g) and dichloromethane (1000 mL) at 5 °C, Oxalyl chloride (117 g) was added slowly in 15 minutes, then catalytic DMF (1 mL) was added slowly at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (110 g) and dichloromethane (900 mL), diisopropylethylamine (DIPEA) (139 g) was added and cooled the reaction mixture to -5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane (100 mL) was slowly added to this later mixture at – 5 °C for 60 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with water and the organic layer was separated. Organic layer was washed with 10% citric acid solution, 10% NaHC03 solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (400 mL) at 27 °C. The reaction mixture was heated to 45 °C and stirred at the same temperature for 1 hour. The mixture was cooled to 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with methanol (200 mL). The wet solid was dried at 45 °C under vacuum to obtain 120 g of the title compound with HPLC purity of 99.97 %
Example-10: Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 50 °C and triethylamine was removed by evaporation under vacuum. The reaction mixture was heated to 90 °C to distill out the traces of triethylamine under vacuum. The reaction mixture was cooled to 40 °C. Acetone (300 mL), maleic anhydride (106.2 g) and triethylamine (6.31 mL) were added. The resulting mixture was stirred at 40 °C for 6 hours. The mixture was cooled to 20 °C and acetyl chloride (12 mL) was added slowly over a period of 30 minutes. The mixture was slowly heated to 50 °C and stirred for 20 hours at the same temperature followed 2 hours at 0 °C. The solid was filtered and washed with cold acetone (2 x 120 mL).The wet solid was dried at 40 °C for 2 hours to obtain 194.2 g of the title compound as white solid with HPLC purity of 99.55%
Example-11: Preparation of Diroximel fumarate
Diroximel Fumarate
To a mixture of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid (5 g) and 2,2-dimethoxypropane (50 mL) at 29 °C, concentrated hydrochloric acid (1 mL) and water (5 mL) were added and stirred at the same temperature for 17 hours at the same temperature. The pH of the reaction mixture was adjusted to 7 with a saturated aqueous solution of NaHCCh and the solvent was evaporated completely at 40 °C. To the resultant solid, water (50 mL) was added at 29 °C and stirred for 15 minutes. The solid was filtered and dried under vacuum at 29 °C for 5 hours. The resultant solid was combined with methyl /er/-butyl ether (50 mL) at 29 °C and stirred for 20 hours at the same temperature. The solid obtained was filtered and washed with diethyl ether (20 mL). The wet solid was dried under vacuum for 3 hours at 29 °C to obtain 3.3 g of the title compound as white solid with HPLC purity of 98.11%
Example-12: Preparation of Amorphous solid dispersion of Diroximel fumarate with Copovidone
Diroximel fumarate (100 mg) and Copovidone (500 mg) were dissolved in acetone (30 mL) at 30 °C. The clear solution was filtered to make it particle free and the solvent was evaporated in a rotavapor at 45 °C under reduced pressure to obtain the title amorphous solid dispersion. The solid dispersion (100 mg) obtained was combined with Syloid (500 mg) and ground for 20 minutes to obtain the admixture of title compound. XRPD: Amorphous.
Example-13: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and methyl tert. butyl ether (30 mL) was added to the clear solution. A suspension of crystalline Diroximel fumarate seed (0.25 g) in methyl tert. butyl ether (10 m) was added at 40 °C and stirred the mixture at the same temperature for 30 minutes. Methyl tert. butyl ether (280 mL) was added slowly for 2 hours at 41 °C. The mixture was cooled to 25 °C in 3
hours and then to 0 °C in 1 hour. The mixture was stirred at 0 °C for 1 hour and the solid was filtered. The wet solid was washed with methyl tert. butyl ether (40 mL) and dried at 42 °C for 6 hours to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.776 pm, Dv (50) 31.292 pm & Dv (90) 133.437 pm
Example-14: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and DM water (100 mL) was added to the clear solution. A crystalline Diroximel fumarate seed (0.20 g) was added at 42 °C and stirred the mixture at the same temperature for 10 minutes. DM water (100 mL) was added slowly at 41 °C. The mixture was cooled to 28 °C in 1 hour. The mixture was stirred at 28 °C for 2 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 8.59 pm, Dv (50) 61.08 pm & Dv (90) 187.07 pm
Example-15: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 45 °C and DM water (400 mL) was added to the clear solution. The mixture was cooled to 30 °C in 1 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.22 pm, Dv (50) 45.5 pm &
Dv (90) 136.7 pm
Example-16: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in Isopropyl acetate (360 mL) at 55 °C and cooled to 28 °C. A crystalline Diroximel fumarate seed (0.25 g) was added at 28 °C and cool to 5 °C. The mixture was stirred for 1 hour at the same temperature and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.3 pm, Dv (50) 43.18 pm & Dv (90) 133.56 pm
PATENT
IN 201941042131
References
- ^ Jump up to:a b “Vumerity- diroximel fumarate capsule”. DailyMed. Retrieved 1 February 2021.
- ^ Jump up to:a b c “Vumerity EPAR”. European Medicines Agency. 14 September 2021. Retrieved 24 November 2021.
- ^ Wang Y, Bhargava P (July 2020). “Diroximel fumarate to treat multiple sclerosis”. Drugs of Today. 56 (7): 431–437. doi:10.1358/dot.2020.56.7.3151521. PMID 32648853. S2CID 220471534.
- ^ Kourakis S, Timpani CA, de Haan JB, Gueven N, Fischer D, Rybalka E (October 2020). “Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility?”. Pharmaceuticals (Basel, Switzerland). 13 (10): 306. doi:10.3390/ph13100306. PMC 7602023. PMID 33066228.
- ^ Jump up to:a b “Drug Approval Package: Vumerity”. U.S. Food and Drug Administration (FDA). 21 April 2020. Retrieved 1 February 2021.
- ^ “Diroximel fumarate”.
- ^ Jump up to:a b “Vumerity: Pending EC decision”. European Medicines Agency. 15 September 2021. Retrieved 17 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Diroximel fumarate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Vumerity |
| Other names | ALKS-8700 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620002 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H13NO6 |
| Molar mass | 255.226 g·mol−1 |
| 3D model (JSmol) | |
/////////Diroximel fumarate, EU 2021, EMA 2021, APPROVALS 2021, VUMERITY, ジロキシメルフマル酸エステル , K0N0Z40J3W, RDC-5108, дироксимела фумарат , ديروكسيميل فومارات , 富马地罗昔美 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
AVIPTADIL


AVIPTADIL
- Molecular FormulaC147H237N43O43S
37221-79-7[RN]
6J2WVD66KR
L-Asparagine, L-histidyl-L-seryl-L-α-aspartyl-L-alanyl-L-valyl-L-phenylalanyl-L-threonyl-L-α-aspartyl-L-asparaginyl-L-tyrosyl-L-threonyl-L-arginyl-L-leucyl-L-arginyl-L-lysyl-L-glutaminyl-L-met hionyl-L-alanyl-L-valyl-L-lysyl-L-lysyl-L-tyrosyl-L-leucyl-L-asparaginyl-L-seryl-L-isoleucyl-L-leucyl-
Vasoactive intestinal octacosapeptide
Invicorp (aviptadil + phentolamine)
(2S)-4-amino-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-5-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]propanoyl]amino]-3-methylbutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-carboxypropanoyl]amino]-4-oxobutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxybutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-4-methylpentanoyl]amino]-5-carbamimidamidopentanoyl]amino]hexanoyl]amino]-5-oxopentanoyl]amino]-4-methylsulfanylbutanoyl]amino]propanoyl]amino]-3-methylbutanoyl]amino]hexanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-3-methylpentanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoic acid

Aviptadil Acetate
CAS#: 40077-57-4 (free base)
Chemical Formula: C155H253N43O51S
Exact Mass:
Molecular Weight: 3567.039
H-His-Ser-Asp-Ala-Val-Phe-Thr-Asp-Asn-Tyr-Thr-Arg-Leu-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Asn-Ser-Ile-Leu-Asn-NH2 tetraacetic acid.
Aviptadil had been in phase II clinical trials for the treatment of pulmonary arterial hypertension and idiopathic pulmonary fibrosis. But these researches were discontinued in 2011.
In 2006, Orphan Drug Designations were granted in the E.U. for the treatment of pulmonary arterial hypertension, and sarcoidosis and acute lung injury in 2006, and 2008, respectively.
The compound was co-developed by Lung Rx (subsidiary of United Therapeutics) and Mondobiotech.
Aviptadil (INN) is an injectable synthetic formulation of human vasoactive intestinal peptide (VIP).[1] VIP was discovered in 1970, and has been used to treat various inflammatory conditions, such as acute respiratory distress syndrome (ARDS), asthma and chronic obstructive pulmonary disease (COPD).
| Clinical data | |
|---|---|
| Trade names | RLF-100 / Zyesamiô |
| AHFS/Drugs.com | International Drug Names |
| ATC code | none |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 40077-57-4 |
| PubChem CID | 16132300 |
| ChemSpider | 17288959 |
| UNII | A67JUW790C |
| KEGG | D12127 |
| ChEMBL | ChEMBL2106041 |
| CompTox Dashboard (EPA) | DTXSID7048584 |
| Chemical and physical data | |
| Formula | C147H237N43O43S |
| Molar mass | 3326.83 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
Regulatory history
ARDS in COVID-19
Studies have found that aviptadil may be beneficial for severely ill patients with COVID-19 related ARDS.[2] ACTIV-3, a trial examining aviptadil acetate (Zyesami), is recruiting patients as of 2 July 2021.[3] A separate trial is examining inhaled aviptadil for patients with high risk for ARDS, is ongoing as of 21 May 2021.[4] A trial for intravenous aviptadil for the same indication concluded in February 2021.[5]
U.S.-Israeli NeuroRx Inc partnered with Relief Therapeutics to develop aviptadil in the United States. In June 2020, the U.S. Food and Drug Administration granted fast-track designation to aviptadil for treatment of respiratory distress in COVID-19.[6] In September 2020, NeuroRX submitted a request for an Emergency Use Authorization to the US FDA for its use in patients in intensive care.[7] May 2021: NRx Pharmaceuticals Announces Positive Results for ZYESAMI™ (Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure.[8]
Jan, 2021: Zuventus healthcare Ltd seeks approval for Aviptadil from India’s drug controller for emergency use in COVID-19 treatment. Mumbai’s Zuventus healthcare Ltd. has got the nod to conduct Phase 3 clinical trials of Aviptadil injectable formulation. The SEC noted that Zuventus had presented revised Phase 3 clinical trial protocol before the committee, and after “detailed deliberation”, it recommended grant of permission of Phase 3 trials with the drug.[9] [10]
Aviptadil/phentolamine combination for Erectile Dysfunction (ED)
October 2000 UK (Invicorp): Aviptadil, an injectable formulation of vasoactive intestinal polypeptide (VIP) in combination with the adrenergic drug phentolamine is approved as an effective alternative therapy for erectile dysfunction (ED) patients. 1 dose intracavernosal injection contains 25 micrograms aviptadil and 2 mg of phentolamine mesilate for the treatment of erectile dysfunction. Aviptadil dose used for treatment of erectile dysfunction is far lesser as compared to dose used for the treatment of ARDS.[11][12]
Vasoactive intestinal peptide (VIP)
Vasoactive intestinal peptide (VIP) is a 28-residue amino acid peptide first characterized in 1970 that was initially isolated from porcine duodenum. A member of the secretin/glucagon hormone superfamily. VIP was initially discovered owing to its potent vasodilatory effects (as its name implies). VIP is widely distributed in the central and peripheral nervous system as well as in the digestive, respiratory, reproductive, and cardiovascular systems as a neurotransmitter and neuroendocrine releasing factor. These effects contribute to an extensive range of physiological and pathological processes related to development, growth, and the control of neuronal, epithelial, and endocrine cell function.[13]
VIP Receptors
VIP acts on two receptors – VPAC1 and VPAC2, which are class B of G-protein-coupled receptors (GPCRs).VPAC1 is mainly present in the lung and T-lymphocytes, whereas VPAC2 is mainly seen in the smooth muscle,mast cells and the basal parts of the lung mucosa.[14]
Expression of VIP
VIP is produced in the neurons in the central and peripheral nervous systems. VIP is mainly localized in the myenteric and submucosal neurons and nerve terminals in the GI tract. Endogenous VIP is released by numerous stimuli such as acetylcholine (ACh), ATP, serotonin (5-HT), substance P (SP), GLP-2 from at least two populations of VIP-positive nerves: cholinergic and non-cholinergic VIP-releasing nerves. In guinea pig small intestine, most VIP-positive nerves in the mucosa and submucosa are non-cholinergic secretomotor neurons and well colocalized with neuronal nitric oxide synthase (nNOS) in human colonic circular muscles. VIP is also expressed in immune cells, such as activated T cells and therefore present in lymphoid tissues including Peyer’s patches, the spleen, and lymph nodes, in addition to the VIP-ergic innervation in lymphoid tissues. Beside the neuronal source, VIP is also expressed and released from endocrine organs – Heart, Thyroid, Kidney and GI tracts.[15]
Localization of VIP
- VIP is highly localised in lungs (70%) and binds with alveolar type II (AT II) cells via VPAC1.[2] The biological (vasodilator) activity of vasoactive intestinal peptide (VIP) was discovered in the lungs before the peptide was isolated and chemical identity characterized from intestine. Although VIP levels are consideralbly high in the brain or gut:VIP is localized in key sites in the lung, has potent activities on its major functions, and appears to play an important role in pulmonary physiology and disease.[16]
- The principal localization of VIP-containing neurons in the tracheobronchial tree is in the smooth muscle layer, around submucosal mucous glands and in the walls of pulmonary and bronchial arteries. Immunoreactive VIP is also present in neuronal cell bodies forming microganglia that provide a source of intrinsic innervation of pulmonary structures.[16]
Vasoactive Intestinal Peptide (VIP) and SARS-CoV-2
VIP is highly localised in lungs and binds with alveolar type II (AT II) cells via VPAC1 receptor. AT II cells constitute only 5% of pulmonary epithelium. Angiotensin Converting Enzyme 2 (ACE 2) surface receptors arepresent in AT II cells. AT II cells produces surfactant and plays an important role in the maintenance of type 1epithelial cells. SARS-CoV-2 enters into AT II cells by binding to ACE 2 surface receptors with its spike protein. SARS CoV-2 attack mainly type II cells (not type I alveolar cells) and results in the death of alveolar type II (AT 11) cells which produces surfactant, resulting in[2]
- Profound defect in oxygenation
- Leading to hypoxia
Mechanism of action of Aviptadil
- Pulmonary alveolar type II Cells have a high concentration of ACE 2 receptors on their cell membrane
- Investigators have confirmed that the SARS-CoV family of viruses selectively attack pulmonary Alveolar Type II (ATII) cells because of their ACE2 receptors, in contrast to other pulmonary epithelial cells.
- SARS-CoV Viruses bind to ACE2 receptors in order to enter the cell. Viral replication and rupture liberates inflammatory cytokines and destroys surfactant production
- VIP binds uniquely to receptors on Alveolar Type II cells in the lung, the same cells that bind the SARS-CoV-2 virus via their ACE2 receptors
- VIP is heavily concentrated in the lung and binds specifically to VIP receptors on alveolar type II cells. VIP exerts a broad anti-cytokine effect on immune system cells
- VIP specifically upregulates surfactant production via upregulation of C-Fos protein and protects type II cells from cytokine
- Upregulating the production of surfactant, the loss of which is increasingly implicated in COVID-19 respiratory failure [17]
Aviptadil a synthetic form of VIP results in rapid clinical recovery in patients with SARS-CoV-2 infection.[2]
Effect of Aviptadil on Lungs in COVID-19
Preservation of Pulmonary Tissue
Preserving surfactant production in the lung and in protecting type 2 alveolar cells. Significantly delayed the onset of edematous lung injury, effective in preventing ischemia-reperfusion injury, Prevents NMDA-induced caspase-3 activation in the Lung.[18]
Inhibits alveolar epithelial cell Apoptosis
VIP is a proven inhibitor of activation-induced perforin, as well as of granzyme B and therefore actively contributes to the reduction of deleterious proinflammatory and cell death-inducing processes, particularly in the lungs. Aviptadil restores barrier function at the endothelial/alveolar interface and thereby protects the lung and other organs from failure.[18]
VIP Promotes synthesis of pulmonary surfactant
Studies have demonstrated that VIP binds on type II cells and increases the incorporation of methyl-choline into phosphatidylcholine – the major component of the pulmonary surfactants by enhancing the activity of the enzyme choline-phosphate cytidylyltransferase. VIP upregulates C-Fos protein expression in cultured type II alveolar cells, which is instrumental in promoting synthesis of pulmonary surfactant phospholipids (Li 2007) and induces surfactant protein A expression in ATII cells through activation of PKC/c-Fos pathway.[18]
VIP decreases Pulmonary Inflammation
Anti-cytokine effect- Inhibits IL-6,TNF-α production and inhibit NF-kB activation. Protects against HCl-induced pulmonary edema.[18]
Pharmacokinetic Properties
Half-life: Its plasma half-life of elimination is 1 to 2 minutes.[2] Metabolism/Distribution: After injection of 1 µg radioactively labelled Aviptadil as bolus to patients a very rapid tissue distribution was observed Within 30 min about 45% of the radioactivity was found in the lungs Over an observation period of 24 hrs only minimal activity was detected in the GI tract & almost no activity was found in the liver or spleen Radioactivity in the lungs decreased within four hours to 25% and within 24 hours to 10% Apparent volume of distribution: Aviptadil has a volume of distribution of 14 ml/kg.[2] Tissue Distribution:Aviptadil binds to its receptors in discrete locations within the gastrointestinal, respiratory, and genital tracts. Aviptadil is localized on respiratory epithelium, smooth muscles of the airways, blood vessels and alveolar walls. Elimination:After injection of radiolabelled Aviptadil radioactivity was almost completely eliminated by the kidneys, 35% within 4 hours, and 90% within 24 hours
Justification for Aviptadil use in the treatment of ARDS
COVID-19-related death is primarily caused by Acute Respiratory Distress Syndrome (ARDS). The trigger for ARDS is widely attributed to a cytokine storm in the lungs, in which the virus causes release of inflammatory cytokines. As a result, alveolae of the lungs fill with fluid and become impermeable to oxygen, even in the setting of mechanical ventilation. SARS-CoV-2 is known to cause respiratory failure, which is the hallmark of Acute COVID-19. Tragically, survival of patients with COVID-19 who progress to Acute Respiratory Distress is dismal. There is an urgent need for a treatment approach that goes right into the heart of the matter – the alveolar type 2 cells which are vulnerable entry points and hosts for the SARS-CoV-2 virus.[19]
Aviptadil-Evidence from Studies in ARDS
Phase III Study-Increased Recovery and Survival in Patients With COVID-19 Respiratory Failure Following Treatment with Aviptadil
A multicenter, randomized, placebo-controlled trial in 196 patients with PCR+ COVID-19 receiving intensive care at 10 U.S. hospitals – 6 tertiary care and 4 regional hospitals to determine whether intravenous aviptadil (synthetic VIP) is superior to placebo in achieving recovery from respiratory failure and survival at 60 days post treatment. Primary, prespecified endpoint was “alive and free from respiratory failure at day 60.” Across all patients and sites of care, patients treated with aviptadil were significantly more likely to be alive and free from respiratory failure at 60 days, compared to those treated with placebo (P=.02) and demonstrated improvement in survival alone (P<.001). Advantages in survival for aviptadil-treated patients were seen in both the subgroup classified as 2 on the National Institute of Allergy and Infectious Disease (NIAID) ordinal scale (58.6% vs. 0%; p=.001) and the NIAID=3 subgroup (83.1% vs. 62.8%; p=.03). Among patients who recovered successfully, those treated with Aviptadil had a median 10-day reduction in length of hospital stay compared to placebo patients (P=.025). Treatment with aviptadil demonstrates multi-dimensional efficacy in improving the likelihood of recovery from respiratory failure and survival to 60 days, and markedly reduced hospital stay in critically ill patients with respiratory failure caused by COVID-19.[20]
Case report: Rapid Clinical Recovery from Critical COVID-19 Pneumonia with Aviptadil
A 54 year old man with double lung transplant presented with headache, fever and productive cough. COVID-19 infection was confirmed by positive RT-PCR of nasopharyngeal swab. The patient required only supportive care for 3 days and was discharged home. Two weeks later he presented with worsening dyspnea, fever and severe hypoxemia requiring high flow O2 and ICU admission. Chest CT showed diffuse bilateral consolidations. He had markedly elevated inflammatory markers. He was treated with dexamethasone and tocilizumab without improvement. He was not a candidate for Remdesivir due to chronic kidney disease. Convalescent plasma was not available, Pro-BNP level was normal; echocardiogram showed preserved biventricular function. He received Aviptadil, a total of three doses, per an open label access under an emergency use approved by USFDA. Rapid improvement in oxygenation and radiologic findings were noticed. No adverse effects were recorded. Patient was transferred out of the ICU 24 hours following the third dose and discharged home on room air 15 days later. This case report of lung transplant recipient with critical COVID-19 pneumonia treated with Aviptadil demonstrates rapid clinical and radiologic improvement.This is consistent with that VIP protects ATII cells, ameliorating the inflammation and improving oxygenation in critical COVID-19 pneumonia.[21]
Posology and method of administration
Aviptadil intravenous infusion is administered by infusion pump in escalating doses for 3 successive days
- Day 1 : Aviptadil 0.166 mcg/kg/hr (equivalent to 1 vial of Aviptadil Injection)
- Day 2 : Aviptadil 0.332 mcg/kg/hr (equivalent to 2 vials of Aviptadil Injection)
- Day 3 : Aviptadil 0.498 mcg/kg/hr (equivalent to 3 vials of Aviptadil Injection)
Duration of infusion depends on the patient’s body weight
- Body weight < 60 kg – 14 hour infusions of Aviptadil at escalating doses on 3 successive days
- Body weight 60 – 90 kg – 12 hour infusions of Aviptadil at escalating doses on 3 successive days
- Body weight > 90 kg – 10 hour infusions of Aviptadil at escalating doses on 3 successive days
Undesirable Effects
Gastrointestinal Disorders – Diarrhea, Vascular disorders – Hypotension, cutaneous flushing, facial flushing & Infusion related reactions[20]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Keijzers GB (April 2001). “Aviptadil (Senatek)”. Current Opinion in Investigational Drugs. 2 (4): 545–9. PMID 11566015. Archived from the original on 2010-09-02. Retrieved 2020-04-01.
- ^ Jump up to:a b c d e f Raveendran, A; Al Dhuhli, K.; Harish Kumar, G. (2021). “Role of Aviptadil in COVID-19”. BMH Medical Journal. 8 (2): 77-83.
- ^ National Institute of Allergy and Infectious Diseases (NIAID) (2021-06-25). “A Multicenter, Adaptive, Randomized, Blinded Controlled Trial of the Safety and Efficacy of Investigational Therapeutics for Hospitalized Patients With COVID-19”. International Network for Strategic Initiatives in Global HIV Trials (INSIGHT), University of Copenhagen, Medical Research Council, Kirby Institute, Washington D.C. Veterans Affairs Medical Center, AIDS Clinical Trials Group.
- ^ Leuppi, Jörg (2021-05-20). “Inhaled Aviptadil for the Treatment of COVID-19 in Patients at High Risk for ARDS: A Randomized, Placebo Controlled, Multicenter Trial”. Clinicaltrials.gov.
- ^ NeuroRx, Inc. (2021-02-23). “ZYESAMI (Aviptadil) for the Treatment of Critical COVID-19 With Respiratory Failure”. Lavin Consulting, LLC.
- ^ “Critically ill COVID-19 patients make quick recovery with treatment RLF-100”. New York Post. 2 August 2020. Retrieved 3 August 2020.
- ^ NeuroRx. “NeuroRx submits request for Emergency Use Authorization for RLF-100™ (aviptadil) in the treatment of patients with Critical COVID-19 and Respiratory Failure who have exhausted approved therapy”. http://www.prnewswire.com. Retrieved 2020-09-24.
- ^ Pharmaceuticals, NRx. “NRx Pharmaceuticals Announces Positive Results for ZYESAMI™ (Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure”. http://www.prnewswire.com.
- ^ Das, Sohini (2021-01-25). “Dr Reddy’s, Zuventus get nod to conduct Covid-19 trials on repurposed drugs”. Business Standard India.
- ^ SECmeeting, e COVID-19. “Recommendations of the SECmeeting to examine COVID-19 related proposals under accelerated approval process made in its 140thmeeting held on 18.01.2021 & 19.01.2021 at CDSCO, HQ New Delhi” (PDF). CDSCO. Retrieved 1 July 2021.
- ^ Keijzers, GB (April 2001). “Aviptadil (Senatek)”. Current Opinion in Investigational Drugs. 2 (4): 545–9. PMID 11566015.
- ^ Procivni, Aviptadil/phentolamine mesilate. “Scientific discussion” (PDF).
- ^ Iwasaki, M; Akiba, Y; Kaunitz, JD (2019). “Recent advances in vasoactive intestinal peptide physiology and pathophysiology: focus on the gastrointestinal system”. F1000Research. 8: 1629. doi:10.12688/f1000research.18039.1. PMC 6743256. PMID 31559013.
- ^ Mathioudakis, A; Chatzimavridou-Grigoriadou, V; Evangelopoulou, E; Mathioudakis, G (January 2013). “Vasoactive intestinal Peptide inhaled agonists: potential role in respiratory therapeutics”. Hippokratia. 17 (1): 12–6. PMC 3738270. PMID 23935337.
- ^ Iwasaki, M; Akiba, Y; Kaunitz, JD (2019). “Recent advances in vasoactive intestinal peptide physiology and pathophysiology: focus on the gastrointestinal system”. F1000Research. 8: 1629. doi:10.12688/f1000research.18039.1. PMC 6743256. PMID 31559013.
- ^ Jump up to:a b Said, Sami I. (June 1988). “Vasoactive Intestinal Peptide in the Lung”. Annals of the New York Academy of Sciences. 527 (1 Vasoactive In): 450–464. Bibcode:1988NYASA.527..450S. doi:10.1111/j.1749-6632.1988.tb26999.x. PMID 2898912. S2CID 26804295.
- ^ Javitt, Jonathan C (2020-07-25). “Vasoactive Intestinal Peptide treats Respiratory Failure in COVID-19 by rescuing the Alveolar Type II cell”. doi:10.22541/au.159569209.99474501. S2CID 221509046.
- ^ Jump up to:a b c d Javitt, Jonathan C (2020-05-13). “Perspective: The Potential Role of Vasoactive Intestinal Peptide in treating COVID-19”. doi:10.22541/au.158940764.42332418. S2CID 219771946.
- ^ “Relief Therapeutics and NeuroRx Announce Final Manufacturing Validation of RLF-100 for Phase 2b/3 Clinical Trial in Patients with COVID-19 Associated Acute Respiratory Distress Syndrome”. GlobeNewswire News Room. 2020-05-14.
- ^ Jump up to:a b Youssef, Jihad G.; Lee, Richard; Javitt, Jonathan; Lavin, Philip; Lenhardt, Rainer; Park, David J; Perez Fernandez, Javier; Morganroth, Melvin; Jayaweera, Dushyantha (2021). “Increased Recovery and Survival in Patients With COVID-19 Respiratory Failure Following Treatment with Aviptadil: Report #1 of the ZYESAMI COVID-19 Research Group”. SSRN 3830051.
- ^ Beshay, S.; Youssef, J.G.; Zahiruddin, F.; Al-Saadi, M.; Yau, S.; Goodarzi, A.; Huang, H.; Javitt, J. (April 2021). “Rapid Clinical Recovery from Critical COVID-19 Pneumonia with Vasoactive Intestinal Peptide Treatment”. The Journal of Heart and Lung Transplantation. 40 (4): S501. doi:10.1016/j.healun.2021.01.2036. PMC 7979412. S2CID 232282732.
//////////AVIPTADIL, RLF 100, DK 1000
CCC(C)C(C(=O)NC(CC(C)C)C(=O)NC(CC(=O)N)C(=O)O)NC(=O)C(CO)NC(=O)C(CC(=O)N)NC(=O)C(CC(C)C)NC(=O)C(CC1=CC=C(C=C1)O)NC(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)C(C(C)C)NC(=O)C(C)NC(=O)C(CCSC)NC(=O)C(CCC(=O)N)NC(=O)C(CCCCN)NC(=O)C(CCCNC(=N)N)NC(=O)C(CC(C)C)NC(=O)C(CCCNC(=N)N)NC(=O)C(C(C)O)NC(=O)C(CC2=CC=C(C=C2)O)NC(=O)C(CC(=O)N)NC(=O)C(CC(=O)O)NC(=O)C(C(C)O)NC(=O)C(CC3=CC=CC=C3)NC(=O)C(C(C)C)NC(=O)C(C)NC(=O)C(CC(=O)O)NC(=O)C(CO)NC(=O)C(CC4=CN=CN4)N

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}