
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
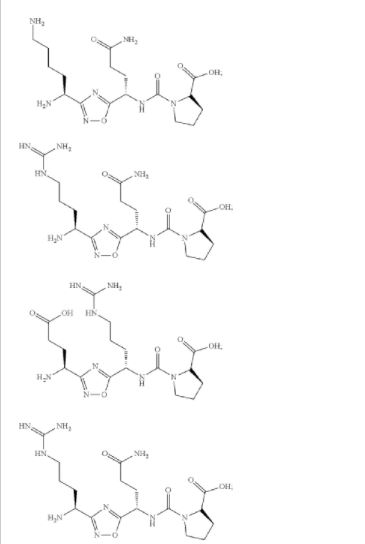
AUPM 170, CA 170, PD-1-IN-1
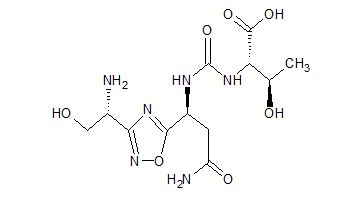



(2S,3R)-2-(3-((S)-3-amino-1-(3-((R)-1-amino-2-hydroxyethyl)-1,2,4-oxadiazol-5-yl)-3-oxopropyl)ureido)-3-hydroxybutanoic acid

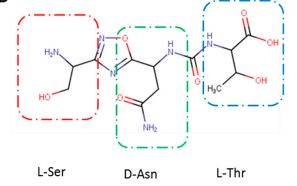
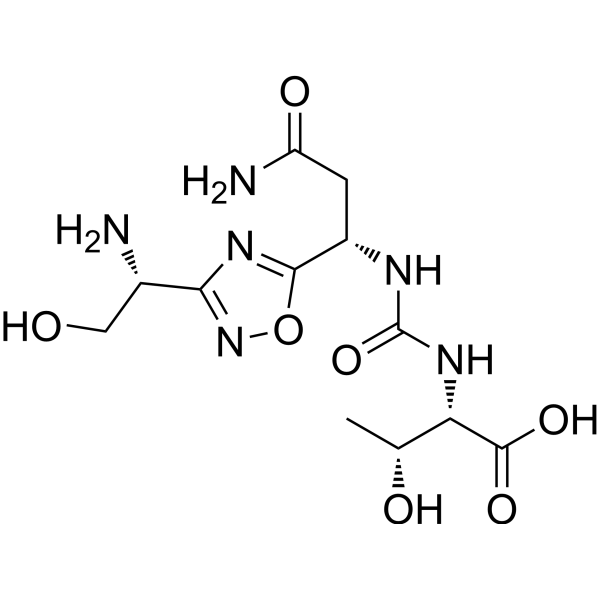
| Molecular Weight (MW) | 360.33 |
|---|---|
| Formula | C12H20N6O7 |
| CAS No. | 1673534-76-3 |
N-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-
AUPM 170, CA 170, AUPM-170, CA-170, PD-1-IN-1
Novel inhibitor of programmed cell dealth-1 (PD-1)
CA-170 (also known as AUPM170 or PD-1-IN-1) is a first-in-class, potent and orally available small molecule inhibitor of the immune checkpoint regulatory proteins PD-L1 (programmed cell death ligand-1), PD-L2 and VISTA (V-domain immunoglobulin (Ig) suppressor of T-cell activation (programmed death 1 homolog; PD-1H). CA-170 was discovered by Curis Inc. and has potential antineoplastic activities. CA-170 selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. Curis is currently investigating CA-170 for the treatment of advanced solid tumours and lymphomas in patients in a Phase 1 trial (ClinicalTrials.gov Identifier: NCT02812875).
References: www.clinicaltrials.gov (NCT02812875); WO 2015033299 A1 20150312.
Aurigene Discovery Technologies Limited INNOVATOR

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CURIS AND AURIGENE ANNOUNCE AMENDMENT OF COLLABORATION FOR THE DEVELOPMENT AND COMMERCIALIZATION OF CA-170
PRESS RELEASE
Curis and Aurigene Announce Amendment of Collaboration for the Development and Commercialization of CA-170
– Aurigene to fund and conduct a Phase 2b/3 randomized study of CA-170 in patients with non-squamous non-small cell lung cancer (nsNSCLC) –
– Aurigene to receive Asia rights for CA-170; Curis entitled to royalty payments in Asia –
LEXINGTON, Mass., February 5, 2020 /PRNewswire/ — Curis, Inc. (NASDAQ: CRIS), a biotechnology company focused on the development of innovative therapeutics for the treatment of cancer, today announced that it has entered into an amendment of its collaboration, license and option agreement with Aurigene Discovery Technologies, Ltd. (Aurigene). Under the terms of the amended agreement, Aurigene will fund and conduct a Phase 2b/3 randomized study evaluating CA-170, an orally available, dual
inhibitor of VISTA and PDL1, in combination with chemoradiation, in approximately 240 patients with nonsquamous
non-small cell lung cancer (nsNSCLC). In turn, Aurigene receives rights to develop and commercialize CA-170 in Asia, in addition to its existing rights in India and Russia, based on the terms of the original agreement. Curis retains U.S., E.U., and rest of world rights to CA-170, and is entitled to receive royalty payments on potential future sales of CA-170 in Asia.
In 2019, Aurigene presented clinical data from a Phase 2a basket study of CA-170 in patients with multiple tumor types, including those with nsNSCLC. In the study, CA-170 demonstrated promising signs of safety and efficacy in nsNSCLC patients compared to various anti-PD-1/PD-L1 antibodies.
“We are pleased to announce this amendment which leverages our partner Aurigene’s expertise and resources to support the clinical advancement of CA-170, as well as maintain our rights to CA-170 outside of Asia,” said James Dentzer, President and Chief Executive Officer of Curis. “Phase 2a data presented at the European Society for Medical Oncology (ESMO) conference last fall supported the potential for CA-170 to serve as a therapeutic option for patients with nsNSCLC. We look forward to working with our partner Aurigene to further explore this opportunity.”
“Despite recent advancements, patients with localized unresectable NSCLC struggle with high rates of recurrence and need for expensive intravenous biologics. The CA-170 data presented at ESMO 2019 from Aurigene’s Phase 2 ASIAD trial showed encouraging results in Clinical Benefit Rate and Prolonged PFS and support its potential to provide clinically meaningful benefit to Stage III and IVa nsNSCLC patients, in combination with chemoradiation and as oral maintenance” said Kumar Prabhash, MD, Professor of Medical Oncology at Tata Memorial Hospital, Mumbai, India.
Murali Ramachandra, PhD, Chief Executive Officer of Aurigene, commented, “Development of CA-170, with its unique dual inhibition of PD-L1 and VISTA, is the result of years of hard-work and commitment by many people, including the patients who participated in the trials, caregivers and physicians, along with the talented teams at Aurigene and Curis. We look forward to further developing CA-170 in nsNSCLC.”
About Curis, Inc.
Curis is a biotechnology company focused on the development of innovative therapeutics for the treatment of cancer, including fimepinostat, which is being investigated in combination with venetoclax in a Phase 1 clinical study in patients with DLBCL. In 2015, Curis entered into a collaboration with Aurigene in the areas of immuno-oncology and precision oncology. As part of this collaboration, Curis has exclusive licenses to oral small molecule antagonists of immune checkpoints including, the VISTA/PDL1 antagonist CA-170, and the TIM3/PDL1 antagonist CA-327, as well as the IRAK4 kinase inhibitor, CA- 4948. CA-4948 is currently undergoing testing in a Phase 1 trial in patients with non-Hodgkin lymphoma.
In addition, Curis is engaged in a collaboration with ImmuNext for development of CI-8993, a monoclonal anti-VISTA antibody. Curis is also party to a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are commercializing Erivedge® for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at http://www.curis.com.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene’s ROR-gamma inverse agonist AUR-101 is currently in phase 2 clinical development under a US FDA IND. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/
Curis with the option to exclusively license Aurigene’s orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field
Addressing immune checkpoint pathways is a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients.
Through its collaboration with Aurigene, Curis is now engaged in the discovery and development of the first ever orally bioavailable, small molecule antagonists that target immune checkpoint receptor-ligand interactions, including PD-1/PD-L1 interactions. In the first half of 2016, Curis expects to file an IND application with the U.S. FDA to initiate clinical testing of CA-170, the first small molecule immune checkpoint antagonist targeting PD-L1 and VISTA. The multi-year collaboration with Aurigene is focused on generation of small molecule antagonists targeting additional checkpoint receptor-ligand interactions and Curis expects to advance additional drug candidates for clinical testing in the coming years. The next immuno-oncology program in the collaboration is currently targeting the immune checkpoints PD-L1 and TIM3.
In November 2015, preclinical data were reported. Data demonstrated tha the drug rescued and sustained activation of T cells functions in culture. CA-170 resulted in anti-tumor activity in multiple syngeneic tumor models including melanoma and colon cancer. Similar data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
By August 2015, preclinical data had been reported. Preliminary data demonstrated that in in vitro studies, small molecule PD-L1 antagonists induced effective T cell proliferation and IFN-gamma production by T cells that were specifically suppressed by PD-L1 in culture. The compounds were found to have effects similar to anti-PD1 antibodies in in vivo tumor models
(Oral Small Molecule PD-L1/VISTAAntagonist)
Certain human cancers express a ligand on their cell surface referred to as Programmed-death Ligand 1, or PD-L1, which binds to its cognate receptor, Programmed-death 1, or PD-1, present on the surface of the immune system’s T cells. Cell surface interactions between tumor cells and T cells through PD-L1/PD-1 molecules result in T cell inactivation and hence the inability of the body to mount an effective immune response against the tumor. It has been previously shown that modulation of the PD-1 mediated inhibition of T cells by either anti-PD1 antibodies or anti-PD-L1 antibodies can lead to activation of T cells that result in the observed anti-tumor effects in the tumor tissues. Therapeutic monoclonal antibodies targeting the PD-1/PD-L1 interactions have now been approved by the U.S. FDA for the treatment of certain cancers, and multiple therapeutic monoclonal antibodies targeting PD-1 or PD-L1 are currently in development.
In addition to PD-1/PD-L1 immune regulators, there are several other checkpoint molecules that are involved in the modulation of immune responses to tumor cells1. One such regulator is V-domain Ig suppressor of T-cell activation or VISTA that shares structural homology with PD-L1 and is also a potent suppressor of T cell functions. However, the expression of VISTA is different from that of PD-L1, and appears to be limited to the hematopoietic compartment in tissues such as spleen, lymph nodes and blood as well as in myeloid hematopoietic cells within the tumor microenvironment. Recent animal studies have demonstrated that combined targeting/ blockade of PD-1/PD-L1 interactions and VISTA result in improved anti-tumor responses in certain tumor models, highlighting their distinct and non-redundant functions in regulating the immune response to tumors2.
As part of the collaboration with Aurigene, in October 2015 Curis licensed a first-in-class oral, small molecule antagonist designated as CA-170 that selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. CA-170 was selected from the broad PD-1 pathway antagonist program that the companies have been engaged in since the collaboration was established in January 2015. Preclinical data demonstrate that CA-170 can induce effective proliferation and IFN-γ (Interferon-gamma) production (a cytokine that is produced by activated T cells and is a marker of T cell activation) by T cells that are specifically suppressed by PD-L1 or VISTA in culture. In addition, CA-170 also appears to have anti-tumor effects similar to anti-PD-1 or anti-VISTA antibodies in multiple in vivo tumor models and appears to have a good in vivo safety profile. Curis expects to file an IND and initiate clinical testing of CA-170 in patients with advanced tumors during the first half of 2016.
 Jan 21, 2015
Jan 21, 2015
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About Immune Checkpoint Modulation and Programmed Death 1 Pathway
Modulation of immune checkpoint pathways has emerged as a highly promising therapeutic approach in a wide range of human cancers. Immune checkpoints are critical for the maintenance of self-tolerance as well as for the protection of tissues from excessive immune response generated during infections. However, cancer cells have the ability to modulate certain immune checkpoint pathways as a mechanism to evade the immune system. Certain immune checkpoint receptors or ligands are expressed by various cancer cells, targeting of which may be an effective strategy for generating anti-tumor activity. Some immune-checkpoint modulators, such as programmed death 1 (PD-1) protein, specifically regulate immune cell effector functions within tissues. One of the mechanisms by which tumor cells block anti-tumor immune responses in the tumor microenvironment is by upregulating ligands for PD-1, such as PD-L1. Hence, targeting of PD-1 and/or PD-L1 has been shown to lead to the generation of effective anti-tumor responses.
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
POSTER


WO2011161699, WO2012/168944, WO2013144704 and WO2013132317 report peptides or peptidomimetic compounds which are capable of suppressing and/or inhibiting the programmed cell death 1 (PD1) signaling pathway.
PATENT

Inventors
- SASIKUMAR, Pottayil Govindan Nair
- RAMACHANDRA, Muralidhara
- NAREMADDEPALLI, Seetharamaiah Setty Sudarshan
Priority Data
| 4011/CHE/2013 | 06.09.2013 | IN |
Example 4: Synthesis of Co
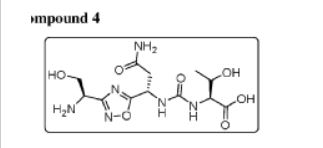
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using
instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.


REFERENCES
US20150073024
| WO2011161699A2 | 27 Jun 2011 | 29 Dec 2011 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| WO2012168944A1 | 21 Dec 2011 | 13 Dec 2012 | Aurigene Discovery Technologies Limited | Therapeutic compounds for immunomodulation |
| WO2013132317A1 | 4 Mar 2013 | 12 Sep 2013 | Aurigene Discovery Technologies Limited | Peptidomimetic compounds as immunomodulators |
| WO2013144704A1 | 28 Mar 2013 | 3 Oct 2013 | Aurigene Discovery Technologies Limited | Immunomodulating cyclic compounds from the bc loop of human pd1 |
http://www.curis.com/pipeline/immuno-oncology/pd-l1-antagonist
http://www.curis.com/images/stories/pdfs/posters/Aurigene_PD-L1_VISTA_AACR-NCI-EORTC_2015.pdf
References:
1) https://bmcimmunol.biomedcentral.com/articles/10.1186/s12865-021-00446-4
2) https://www.nature.com/articles/s42003-021-02191-1
3) https://www.esmoopen.com/article/S2059-7029(20)30108-3/fulltext
4) https://www.mdpi.com/1420-3049/24/15/2804
////////Curis, Aurigene, AUPM 170, CA 170, AUPM-170, CA-170, PD-L1, VISTA antagonist, PD-1-IN-1, phase 2, CANCER
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O

NEW DRUG APPROVALS
ONE TIME
$9.00
AUR 101
AUR 101
AUR101-201
ANTIINNFLAMATORY
AUR-101, a ROR gamma inverse agonist for autoimmune disorders like psoriasis
AUR-101 is an ROR-gammaT inverse agonist in phase II clinical development at Aurigene for the treatment of patients with moderate-to-severe chronic plaque-type psoriasis.
- DrugsAUR 101 (Primary)
- IndicationsPlaque psoriasis
- FocusAdverse reactions; First in man
- AcronymsINDUS
- SponsorsAurigene Discovery Technologies
- OriginatorAurigene Discovery Technologies
- ClassAntipsoriatics; Small molecules
- Mechanism of ActionNuclear receptor subfamily 1 group F member 3 inverse agonists
- Phase IIPsoriasis
- 28 Aug 2021No recent reports of development identified for phase-I development in Psoriasis(In volunteers) in Australia (PO, Tablet)
- 23 Apr 2021Aurigene Discovery Technologies plans a phase II INDUS-3 trial for Psoriasis in USA (PO) in May 2021 (NCT04855721)
- 15 Apr 2021Aurigene Discovery Technologies completes a phase II trial in Psoriasis in India (PO) (NCT04207801)
- CDSCO
- https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/Aurigene20.pdf
- NCT04207801
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
AURIGENE ANNOUNCES FIRST PATIENT DOSED WITH AUR101 IN PHASE II STUDY IN PATIENTS WITH MODERATE TO SEVERE PSORIASIS
PRESS RELEASE
Aurigene Announces First Patient Dosed with AUR101 in Phase II Study in Patients with Moderate to Severe Psoriasis
Bangalore, February 17, 2020 — Aurigene, a development stage biotechnology company, today announced dose administration for the first patient in INDUS-2, a Phase II double blind placebo-controlled three-arm study of AUR101 in patients with moderate to severe psoriasis. AUR101 is an oral small molecule inverse agonist of RORγ and has shown desirable pharmacodynamic modulation of IL-17 and acceptable safety in a completed Phase I human study conducted in Australia.
“The initiation of this Phase II study under a US FDA IND represents a significant milestone for Aurigene, as it marks the first program which Aurigene has led from the bench side to the clinic all by itself,” said Murali Ramachandra, PhD, Chief Executive Officer of Aurigene. “We look forward to producing important clinical data by the end of 2020 to guide our future development plans and demonstrating Aurigene’s unique expertise in conducting Proof-of-Concept studies in a quality and fast-paced manner.”
About AUR101-201 and the Phase II Study of AUR101 in Patients with Moderate to Severe Psoriasis
The purpose of the Phase II multi-center, blinded, placebo-controlled, three-arm study is to evaluate the clinical activity of AUR101 in patients with moderate to severe psoriasis. In two of the arms, AUR101 will be administered twice daily, at 400 mg PO BID and 600 mg PO BID, for 12 weeks. Patients in the third arm will receive matched blinded placebo in a double dummy fashion. The trial is listed at clinicaltrials.gov with identifier NCT04207801.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY,NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene has also submitted an IND to DCGI, India for a Phase IIb/III trial of CA-170, a dual inhibitor of PD-L1 and VISTA, in non-squamous NSCLC. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/.
CLIP
Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways
https://onlinelibrary.wiley.com/doi/10.1111/bjd.20090
M.A.X. Tollenaere,J. Hebsgaard,D.A. Ewald,P. Lovato,S. Garcet,X. Li,S.D. Pilger,M.L. Tiirikainen,M. Bertelsen,J.G. Krueger,H. Norsgaard,First published: 01 April 2021 https://doi.org/10.1111/bjd.20090Citations: 2Funding sources LEO Pharma A/S funded this study.Conflicts of interest M.A.X.T., J.H., D.A.E., P.L., S.D.P., M.L.T., M.B. and H.N. are employees of LEO Pharma. J.G.K. received grants paid to his institution from Novartis, Pfizer, Amgen, Lilly, Boehringer, Innovaderm, BMS, Janssen, AbbVie, Paraxel, LEO Pharma, Vitae, Akros, Regeneron, Allergan, Novan, Biogen MA, Sienna, UCB, Celgene, Botanix, Incyte, Avillion and Exicure; and personal fees from Novartis, Pfizer, Amgen, Lilly, Boehringer, Biogen Idec, AbbVie, LEO Pharma, Escalier, Valeant, Aurigene, Allergan, Asana, UCB, Sienna, Celgene, Nimbus, Menlo, Aristea, Sanofi, Sun Pharma, Almirall, Arena and BMS.Data Availability Statement The gene array dataset described in this publication has been deposited in NCBI’s Gene Expression Omnibus and is accessible through GEO Series accession number GSE158448 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158448).
CLOP
https://www.drugdiscoverychemistry.com/Anti-Inflammatories/16
10:35 Small Molecule Inhibitors of RORgamma and IRAK4 for the Treatment of Autoimmune Disorders
 Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Although biologics such as anti-TNFα antibody are fairly successful in the treatment of autoimmune disorders, there is significant unmet need due to heterogeneity in diseases and lack of response to established therapies in some patients. While biologics typically target one cytokine signaling pathway, small molecule therapeutics directed towards intracellular target(s) can interfere in the signaling from multiple cytokines potentially leading to improved response. Development of small molecule oral inhibitors of IRAK4 and RORgamma to target TLR/IL-R and Th17 pathway respectively will be discussed.
PATENT
2448/CHE/2015 15.05.2015 IN
PATENT
PATENT
This application claims the benefit of Indian provisional application number 5641/CHE/2013 filed on 06th December 2013 which hereby incorporated by reference.
PATENT
- KOTRABASAIAH UJJINAMATADA, Ravi
- PANDIT, Chetan
2049005-13-0
2-Quinolinecarboxamide, 6-(2,6-dimethyl-4-pyrimidinyl)-N-[[4-(ethylsulfonyl)phenyl]methyl]-5,6,7,8-tetrahydro-6-methyl-5-oxo-, (6S)-
Molecular Weight492.59, C26 H28 N4 O4 S

EXAMPLE
PATENT
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0223523419301011
2013239366 CA 170

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////////////////AUR 101, AURIGENE, ROR, IL-17, PHASE 2, CDSCO, Ravi Ujjinamatada, KOTRABASAIAH UJJINAMATADA Ravi, PANDIT Chetan, AUR101-201, plaque-type psoriasis


Ravi Ujjinamatada
JNJ-A07

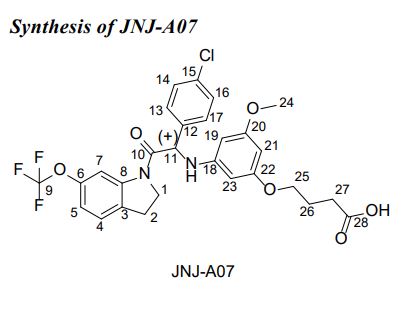

JNJ-A07
S + FORM
CAS 2135640-93-4 ROT (+)S
Butanoic acid, 4-[3-[[1-(4-chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]-, (+)-
(+)-4-[3-[[1-(4-Chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]butanoic acid
(+)-4-[3-([(1S)-1-(4-Chlorophenyl)-2-oxo-2-[6-(trifluoromethoxy)-2,3-dihydro-1H-indol-1-yl]ethyl]amino)-5-methoxyphenoxy]butanoic acidMolecular FormulaC28 H26 Cl F3 N2 O6Molecular Weight578.964
REF
Kaptein, S.J.F., Goethals, O., Kiemel, D. et al. A pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction. Nature (2021). https://doi.org/10.1038/s41586-021-03990-6

JNJ-018
CAS 2135640-91-2 +/-, R,S
CAS 2135640-92-3 ROT (-)R
Butanoic acid, 4-[3-[[1-(4-chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]-, (-)-
(-)-4-[3-[[1-(4-Chlorophenyl)-2-[2,3-dihydro-6-(trifluoromethoxy)-1H-indol-1-yl]-2-oxoethyl]amino]-5-methoxyphenoxy]butanoic acid
- Janssen (Originator)
- Katholieke Universiteit Leuven (Originator)
- NS4B Protease (Dengue Virus) Inhibitors
- Serine Protease NS3/Non-Structural Protein NS4B Protease (Dengue Virus) Interaction Inhibitors
A pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction
https://www.nature.com/articles/s41586-021-03990-6
https://www.nature.com/articles/s41586-021-03990-6#citeas
Abstract
Dengue virus causes approximately 96 million symptomatic infections annually, manifesting as dengue fever or occasionally as severe dengue1,2. There are no antiviral agents available to prevent or treat dengue. Here, we describe a highly potent dengue virus inhibitor (JNJ-A07) that exerts nanomolar to picomolar activity against a panel of 21 clinical isolates that represent the natural genetic diversity of known genotypes and serotypes. The molecule has a high barrier to resistance and prevents the formation of the viral replication complex by blocking the interaction between two viral proteins (NS3 and NS4B), thus revealing a previously undescribed mechanism of antiviral action. JNJ-A07 has a favourable pharmacokinetic profile that results in outstanding efficacy against dengue virus infection in mouse infection models. Delaying start of treatment until peak viraemia results in a rapid and significant reduction in viral load. An analogue is currently in further development.


2-(4-Chlorophenyl)-1-(6-(trifluoromethoxy)indolin-1-yl)-ethanone (1)
127 A mixture of 6-(trifluoromethoxy)indoline ([CAS 959235-95-1], 2 g, 9.84 mmol), 2-(4-chlorophenyl)acetic acid 128 ([CAS 1878-66-6], 1.85 g, 10.8 mmol), HATU (5.6 g, 14.8 mmol) and diisopropylethylamine (4.9 mL, 29.5
129 mmol) in DMF (40 mL) was stirred at room temperature for 12 h. Water was added and the precipitate was
130 filtered off. The residue was taken up with EtOAc. The organic solution was washed with a 10 % aqueous
131 solution of K2CO3, brine, dried over MgSO4, filtered, and the solvent was evaporated under reduced pressure. 132 The residue was purified by chromatography on silica gel (15-40 pm, 80 g, heptane/EtOAc gradient 90/10 to 133 60/40). The pure fractions were combined and the solvent was concentrated under reduced pressure to give 2-(4-
134 chlorophenyl)-1-(6-(trifluoromethoxy)indolin-1-yl)-ethanone 1 (3 g, yield: 86 %).
135 1 H NMR (400 MHz, DMSO-d6) d ppm 7.99 (s, 1 H), 7.37 – 7.41 (m, 2 H), 7.29 – 7.34 (m, 3 H), 6.97 (dd, J = 8.1, 1.3 Hz, 1 H), 4.25 (t, J = 8.6 Hz, 2 H), 3.88 (s, 2 H), 3.18 (t, J = 8.5 Hz, 2 H); 13
136 C NMR (101 MHz, 137 CHLOROFORM-d) δ ppm 168.91, 148.65, 148.63, 144.05, 133.16, 132.26, 130.63, 129.54, 128.93, 124.87, 120.50 (q, J=257.2 Hz), 116.38, 110.83, 77.26, 48.86, 42.52, 27.59; LC-MS: [M+H]+
138 728; purity 99 % (method LCMS2); Melting Point: 116-131 °C (DSC peak: 120.2 °C); HRMS (ESI+) m/z: [M]+ 139 calcd for C17H13ClF3NO2,
140 356.0660; found, 356.0657
141 2-Bromo-2-(4-chlorophenyl)-1- (6-(trifluoromethoxy)indolin-1-yl)ethanone (2)
142 At -78 °C, under nitrogen flow, LiHMDS (1.5 M in THF, 11.2 mL, 16.9 mmol) was added dropwise to a mixture 143 of 1 (3 g, 8.43 mmol) in THF (50 mL). The mixture was stirred for 15 min at -78 °C and a solution of N
144 bromosuccinimide (1.65 g, 9.3 mmol) in THF (30 mL) was added dropwise. After stirring for 2 h at -78 °C, the 145 reaction was quenched with a saturated aqueous solution of NH4Cl. The mixture was extracted with EtOAc. The 146 organic layer was separated, dried over MgSO4, filtered, and the solvent was evaporated under reduced pressure
147 to give 2-bromo-2-(4-chlorophenyl)-1- (6-(trifluoromethoxy)indolin-1-yl)ethanone 2 (3.6 g, yield: 98 %) as an 148 oil. The compound was used without further purification in the next step.
149 1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 8.19 (s), 7.52 – 7.57 (m), 7.34 – 7.39 (m), 7.17 (d, J=8.2 Hz), 6.92 (dd, J=8.2, 1.1 Hz), 5.56 (s), 4.37 (td, J=10.1, 6.5 Hz), 4.09 (td, J=10.1, 6.7 Hz), 3.12 – 3.31 (m); 13
150 C NMR
151 (101 MHz, CHLOROFORM-d) δ ppm 164.90 (s), 148.68 (d, J=2.2 Hz), 143.75 (s), 135.46 (s), 133.99 (s), 152 130.52 (s), 129.79 (s), 129.10 (s), 125.01 (s), 117.20 (s), 120.47 (q, J=257.2 Hz), 111.36 (s), 48.88 (s), 46.61 (s), 27.65 (s); LC-MS: [M+H]+ 436; purity 100 % (method LCMS2); HRMS (ESI+) m/z: [M]+ 153 calcd for
154 C17H13O2NBrClF3, 433.9765; found, 433.9764
155 tert-Butyl 4-(3-amino-5-methoxyphenoxy)butanoate (3) 156 To a mechanically stirred solution of tert-butyl 4-bromobutanoate ([CAS 110661- 5 91-1], 42.3 g, 0.19 mol) in
157 DMF (600 mL) was added in portions a solid mixture of 3-amino-5-methoxyphenol ([CAS 162155-27-3], 26.4 158 g, 0.19 mol) and Cs2CO3 (123.6 g, 0.379 mol). The reaction mixture was stirred at 60 °C for 65 h, and allowed to
159 reach room temperature. The mixture was poured out into water (2.5 L). The product was extracted with Et2O (2 160 x). The combined organic layers were washed with brine, dried over MgSO4, and filtered. The solvent was
161 evaporated under reduced pressure, and then co-evaporated with toluene. The residue was purified by normal 162 phase HPLC (Stationary phase: silica gel 60A 25-40 pm (Merck), Mobile phase: gradient EtOAc/heptane 20/80 163 to 60/40), yielding tert-butyl 4-(3-amino-5-methoxyphenoxy)butanoate 3 as an oil (27 g, yield: 50 %).
164 1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 5.89 – 5.92 (m), 5.86 (d, J=2.2 Hz), 3.92 (t, J=6.2 Hz), 3.73 (s), 3.66 (br s), 2.40 (t, J=7.4 Hz), 1.98 – 2.08 (m), 1.45 (s); 13
165 C NMR (101 MHz, CHLOROFORM-d) δ ppm 172.61 166 (s), 161.69 (s), 161.02 (s), 148.35 (s), 94.33 (s), 93.89 (s), 91.52 (s), 80.35 (s), 66.74 (s), 55.17 (s), 32.07 (s), 28.13 (s), 24.78 (s); LC-MS: [M+H]+ 282; purity 94 % (method LCMS2); HRMS (ESI+) m/z: [M]+
167 calcd for 168 C15H24O4N, 282.1700; found, 282.1695
169 tert-Butyl 4-(3-((1-(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-
170 methoxyphenoxy)butanoate (4)
171 A mixture of 2 (3.6 g, 8.3 mmol), 3 (2.3 g, 8.3 mmol) and diisopropylethylamine (1.7 ml, 9.94 mmol) in CH3CN 172 (80 mL) was stirred at 70 °C for 4 h. The mixture was concentrated under reduced pressure, diluted with EtOAc,
173 and washed with 1 N aqueous HCl and water. The organic phase was separated, dried over MgSO4, filtered, and 174 the solvent was evaporated under reduced pressure. The compound was purified by flash chromatography on 175 silica gel (15-40 pm, 120 g, heptane/EtOAc 80/20). The pure fractions were combined and evaporated to dryness
176 to give, after crystallization from diisopropyl ether, tert-butyl 4-(3-((1-(4-chlorophenyl)-2-oxo-2-(6-
177 (trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-methoxyphenoxy)butanoate 4 (2.6 g, yield: 49 %).
178 1 H NMR (400 MHz, DMSO-d6) d ppm 8.03 (s, 1 H), 7.55 (d, J = 8.6 Hz, 2 H), 7.43 (d, J = 8.6 Hz, 2 H), 7.33 (d, 179 J = 8.1 Hz, 1 H), 7.01 (dd, J = 8.1, 1.5 Hz, 1 H), 6.44 (d, J = 8.8 Hz, 1 H), 5.94 (d, J = 2.0 Hz, 2 H), 5.75 (t, J = 180 2.0 Hz, 1 H), 5.55 (d, J = 8.8 Hz, 1 H), 4.51 (td, J = 10.3, 6.5 Hz, 1 H), 4.04 (td, J = 10.3, 7.3 Hz, 1 H), 3.84 (t, J 6 181 = 6.3 Hz, 2 H), 3.62 (s, 3 H), 3.09 – 3.23 (m, 2 H), 2.31 (t, J = 7.3 Hz, 2 H), 1.86 (quin, J = 6.8 Hz, 2 H), 1.39 (s, 9 H); 13
182 C NMR (101 MHz, CHLOROFORM-d) δ ppm 172.57, 168.84, 161.66, 161.02, 148.65, 148.63, 147.68,
183 143.79, 135.66, 134.48, 129.58, 129.42, 129.38, 124.99, 116.92, 120.50 (q, J=257.2 Hz), 111.13, 93.02, 92.72, 91.06, 80.38, 77.25, 66.79, 59.74, 55.17, 48.31, 32.09, 28.15, 27.64, 24.77; LC-MS: [M+H]+
184 635; purity: 98 % (method LCMS3); Melting Point: 109-125 °C (DSC peak: 116.1 °C); HRMS (ESI+) m/z: [M]+ 185 calcd for 186 C32H34ClF3N2O6, 635.2130; found, 635.2127 187 (+)-4-(3-((1-(4-Chlorophenyl)-2-oxo2-(6-(trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-
188 methoxyphenoxy)butanoic acid (JNJ-A07) 189 A solution of 4 (2.4 g, 3.8 mmol) in 4 M HCl in dioxane (24 mL) was stirred at 5 °C for 3 h and at room 190 temperature for 3 h. The precipitate was filtered off and dried to afford 4-(3-((1-(4-chlorophenyl)-2-oxo2-(6- 191 (trifluoromethoxy)indolin-1-yl)ethyl)amino)-5-methoxyphenoxy)butanoic acid as an HCl salt (racemic JNJ192 A07, 2 g, 0.8 eq. HCl, 0.07 eq. H2O). This salt was neutralized prior to chiral separation by dissolving it in 193 EtOAc and treating this solution with 1 N aqueous NaOH and evaporation of the organic layer under reduced 194 pressure.
The enantiomers were separated via preparative chiral SFC (Stationary phase: Chiralcel® OD-H 5 pm 195 250 x 30 mm, Mobile phase: 50 % CO2, 50 % iPrOH (+ 0.3 % iPrNH2)) and further purified via preparative 196 achiral SFC (Stationary phase: Cyano® 6 pm 150 x 21.2 mm, Mobile phase: 80 % CO2, 20 % MeOH (+ 0.3 % 197 iPrNH2)). The product fractions were combined and evaporated under reduced pressure. Each enantiomer was 198 taken up with EtOAc and washed with 1 N aqueous HCl. The organic layers were separated, dried over MgSO4, 199 filtered, and the solvent was evaporated under reduced pressure. The first eluted enantiomer was solidified from 200 diethyl ether/diisopropyl ether to give the epimer of JNJ-A07 (616 mg, yield: 28 %).
The second eluted
201 enantiomer was solidified from diethyl ether/diisopropyl ether to give JNJ-A07 (715 mg, yield: 32 %).
202 203 Epimer of JNJ-A07:
204 1 H NMR (500 MHz, DMSO-d6) δ ppm 12.12 (br s, 1 H), 8.04 (br s, 1 H), 7.55 (br d, J = 8.2 Hz, 2 H), 7.44 (br d, 205 J = 8.5 Hz, 2 H), 7.34 (br d, J = 7.9 Hz, 1 H), 7.01 (br d, J = 7.6 Hz, 1 H), 6.45 (br s, 1 H), 5.95 (br d, J = 10.1 206 Hz, 2 H), 5.76 (s, 1 H), 5.57 (br s, 1 H), 4.47 – 4.57 (m, 1 H), 3.99 – 4.11 (m, 1 H), 3.85 (br t, J = 6.3 Hz, 2 H), 3.62 (s, 3 H), 3.08 – 3.27 (m, 2 H), 2.34 (br t, J = 7.3 Hz, 2 H), 1.87 (quin, J = 6.7 Hz, 2 H); 13
207 C NMR (101 MHz, 208 DMSO-d6) δ ppm 174.56, 169.79, 161.46, 160.71, 149.08, 147.64, 144.48, 137.28, 132.91, 131.95, 130.56, 209 128.89, 126.34, 120.58 (d, J=256.0 Hz), 116.69, 109.52, 93.08, 92.80, 90.23, 66.65, 58.69, 55.20, 48.65, 30.57, 27.48, 24.72; LC-MS: [M+H]+ 579; purity: 100 % (method LCMS1);
Chiral SFC: [M+H]+ 210 579; chiral purity 100 % (method SFC1); [a]D20 211 : -48.5° (589 nm, c 0.27 w/v %, DMF, 20 °C); Melting Point: 62-80 °C (DSC peak: 70.6 °C); HRMS (ESI+) m/z: [M]+ 212 calcd for C28H27O6N2ClF3, 579.1504; found, 579.1501 213 214
JNJ-A07: 215 1 H NMR (500 MHz, DMSO-d6) δ ppm 12.12 (brs, 1 H), 8.04 (br s, 1 H), 7.55 (br d, J = 8.2 Hz, 2 H), 7.44 (br d, 216 J = 8.2 Hz, 2 H), 7.34 (br d, J = 7.9 Hz, 1 H), 7.01 (br d, J = 7.9 Hz, 1 H), 6.45 (br s, 1 H), 5.95 (br d, J = 10.1 217 Hz, 2 H), 5.76 (br s, 1 H), 5.57 (s, 1 H), 4.46 – 4.59 (m, 1 H), 3.99 – 4.10 (m, 1 H), 3.85 (br t, J = 6.1 Hz, 2 H), 3.62 (s, 3 H), 3.09 – 3.27 (m, 2 H), 2.34 (br t, J = 7.3 Hz, 2 H), 1.87 (br t, J = 6.8 Hz, 2 H); 13
218 C NMR (101 MHz,
219 DMSO-d6) δ ppm 174.53 (C28), 169.79 (C10), 161.47 (C20), 160.72 (C22), 149.08 (C18), 147.65 (C6), 144.48 220 (C8), 137.29 (C12), 132.92 (C15), 131.95 (C3), 130.56 (C13, C17), 128.89 (C14, C16), 126.34 (C4), 120.58 (q, 221 J = 255.1 Hz, C9), 116.67 (C5), 109.51 (C7), 93.11 (C23), 92.81 (C21), 90.26 (C19), 66.66 (C25), 58.70 (C11), 55.21 (C24), 48.67 (C1), 30.57 (C27), 27.49 (C2), 24.72 (C26); LC/MS: [M+H]+
222 579; purity 100 % (method LCMS1); Chiral SFC: [M+H]+ 579; chiral purity 100 % (method SFC1); [a]D20
223 : +42.9° (589 nm, c 0.28 w/v %, 224 DMF, 20 °C); Melting point: 62-78 °C (DSC peak: 71.3 °C) ; HRMS (ESI+) m/z calcd for C28H27O6N2ClF3 [M]+ 225 , 579.1504, found 579.1500; Elemental analysis requires C, 58.09 %; H, 4.53 %; N, 4.84 % found C, 226 58.60 %; H, 4.59 %; N, 4.80 %
CLIP
https://www.bioworld.com/articles/512333-potent-selective-pan-serotype-dengue-inhibitor-developed
Blocking the interaction between two dengue virus (DENV) nonstructural proteins, NS3 and NS4B, with a newly developed small-molecule inhibitor resulted in potent antiviral activity in mouse models, according to an international collaborative study led by scientists at the University of Leuven (KU Leuven), CD3 the Centre for Drug Design and Discovery in Leuven, and Janssen Pharmaceutica in Beerse, Belgium.
This protein interaction represents a promising new target for the development of pan-serotype DENV inhibitors with a high barrier to resistance, with the potency of the inhibition warranting further development of these compounds, the authors reported in the October 6, 2021, edition of Nature.
“This is the first study to show that blocking the NS3/NS4B interaction has potent antiviral activity in mice warranting the further development of such inhibitors,” said study co-leader Johan Neyts, professor of virology at KU Leuven.
Dengue is currently among the leading threats to global public health, with an estimated 96 million individuals developing dengue disease, which is probably an underestimation.
In addition, the incidence of dengue has increased approximately 30-fold over the past 50 years. DENV is now endemic in the subtropical regions of 128 countries, with an estimated 4 billion people at risk of infection, predicted to increase to 6 billion by 2080.
This dengue upsurge is driven by various factors, most notably rapid urbanization and the spread of the Aedes mosquito vectors due to climate change.
The DENV has four serotypes that are further classified into genotypes, which are increasingly co-circulating in endemic regions. Antibodies to infection with one serotype can lead to a more severe second infection with a different serotype increases the risk of potentially life-threatening severe dengue.
The DENV vaccine Dengvaxia (Sanofi-Pasteur), which has been approved in several countries for individuals aged at least 9 years, is only recommended for those with previous DENV exposure.
Moreover, there are currently no available antiviral agents for dengue prevention or treatment, while development of pan-serotype DENV inhibitors has proven challenging.
“The major developmental challenge has been to obtain ultrapotent antivirals that also have equipotent activity against the four DENV serotypes,” Neyts told BioWorld Science.
Such drugs should lower viral loads during an ongoing infection, thereby reducing dengue-associated morbidity and mortality, as well as transmission.
In their new Nature study, researchers co-led by Neyts, Patrick Chaltin, managing director of CD3 the Centre for Drug Design and Discovery, and Marnix Van Loock, R&D Lead Emerging Pathogens, Janssen Global Public Health at Janssen Pharmaceutica, identified potential new DENV inhibitors using large-scale cell-based anti-DENV-2 screening.
“We screened tens of thousands of molecules and interesting hits were further optimized to eventually obtain JNJ-A07 and other ultrapotent and selective analogues, with roughly 2,000 analogues being synthesized and tested,” said Neyts.
Notably, the promising small molecule JNJ-A07 was demonstrated to have nanomolar to picomolar activity against a panel of 21 clinical isolates representing the natural genetic diversity of known DENV genotypes and serotypes.
The molecule was then shown to have a high barrier to resistance “by months of culturing the dengue virus in suboptimal concentrations of the inhibitor,” Neyts said.
JNJ-A07 was then shown to prevent formation of the viral replication complex by blocking the interaction between the nonstructural proteins NS3 and NS4B, thereby revealing a previously undescribed mechanism of antiviral action.
JNJ-A07 was further demonstrated to have a favorable pharmacokinetic (PK) profile resulting in outstanding efficacy against DENV infection in mouse models.
“JNJ-A07’s favorable PK profile resulted from optimization of the ADME [absorption, distribution metabolism and excretion] properties of the analogues within this chemical series,” Janssen’s Van Loock told BioWorld Science.
“This enabled us to administer the compound [twice daily] in mice and assess its efficacy, which resulted in a significantly reduced viral load and protected against mortality in a mouse lethal challenge model.”
However, “additional research will be required in preclinical models, to understand how these findings reflect those in humans, as currently no translational models are available to assess the potential effect in humans,” noted Van Loock.
Delaying treatment commencement until peak viremia had developed was shown to result in a rapid and significant reduction in viral load in the mouse models of infection.
This is an important finding, as “one wants an antiviral effect that is independent of how much [viral] replication is ongoing,” Van Loock said.
“In these mice, the reduction in viral load was also very pronounced if the treatment was initiated on the day of peak viral load, when the effect was quantified 24 hours later.”
On safety, said Neyts, as JNJ-A07 and its analogues “target specific viral proteins that have no homologues in eukaryotic cells, we expect a considerable safety window, with these agents being very well tolerated.” The safety and potency of DENV inhibition established in this study justifies the further development of these novel antivirals, with an analogue being currently in further development.
Further development will include “using our know-how to also develop drugs against the other member of the flavivirus family to which DENV belongs, including Japanese encephalitis, Zika, yellow fever, West Nile virus, et cetera,” said Neyts.
Meanwhile, “Janssen has moved the compound into clinical development and continues to work closely in this regard with teams at KU Leuven and elsewhere,” said Van Loock.
“We will be sharing information about progress of the compound’s clinical development during the American Society of Tropical Medicine and Hygiene meeting this November.”
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////PatentWO2017167951https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017167951
- IN201827040889
- US2020299235
- US2019112266
- US10689340
Due to the presence of said chiral carbon atom, a “compound of formula (I)” can be the (R)-enantiomer, the (S)-enantiomer, the racemic form, or any possible combination of the two individual enantiomers in any ratio. When the absolute (R)-or (S)-configuration of an enantiomer is not known, this enantiomer can also be identified by indicating whether the enantiomer is dextrorotatory (+)- or levorotatory (-)- after measuring the specific optical rotation of said particular enantiomer.
In an aspect the present invention relates to a first group of compound of formula (I) wherein the compounds of formula (I) have the (+) specific rotation.
In a further aspect the present invention relates to a second ground of compounds of formula (I) wherein the compounds of formula (I) have the (-) specific rotation.
Example 4: synthesis of 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)-indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoic acid (Compound 4) and chiral separation into Enantiomers 4A and 4B.
Synthesis of intermediate 4a:
A mixture of 6-(trifluoromethoxy)indoline [CAS 959235-95-1] (2 g, 9.84 mmol), 2-(4-chlorophenyl)acetic acid [CAS 1878-66-6] (1 .85 g, 10.8 mmol), HATU (5.6 g, 14.8 mmol) and diisopropylethylamine (4.9 ml_, 29.5 mmol) in DMF (40 ml_) was stirred at room temperature for 12 h. Water was added and the precipitate was filtered off. The residue was taken up with EtOAc. The organic solution was washed with a 10% aqueous solution of K2CO3, brine, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on silica gel (15-40 μιτι, 80 g, heptane/EtOAc gradient 90/10 to 60/40). The pure fractions were combined and the solvent was concentrated under reduced pressure to give 2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)-ethanone 4a (3 g).
Synthesis of intermediate 4b:
At -78°C, under N2 flow, LiHMDS 1 .5 M in THF (1 1 .2 ml_, 16.9 mmol) was added dropwise to a mixture of 2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)-ethanone 4a (3 g, 8.43 mmol) in THF (50 ml_). The mixture was stirred for 15 min at -78°C and a solution of /V-bromosuccinimide (1 .65 g, 9.3 mmol) in THF (30 ml_) was added dropwise. After stirring for 2 h at -78°C, the reaction was quenched with a saturated solution of NH CI. The mixture was extracted with EtOAc. The organic layer was separated, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure to give 2-bromo-2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)ethanone 4b (3.6 g). The compound was used as such in the next step.
Synthesis of intermediate 4c:
A mixture of 2-bromo-2-(4-chlorophenyl)-1 -(6-(trifluoromethoxy)indolin-1 -yl)-ethanone 4b (3.6 g, 8.3 mmol), terf-butyl 4-(3-amino-5-methoxyphenoxy)-butanoate 1a (2.3 g, 8.3 mmol) and diisopropylethylamine (1 .7 mL, 9.94 mmol) in CH3CN (80 mL) was stirred at 70°C for 4 h. The mixture was concentrated under reduced pressure, diluted with EtOAc, and washed with 1 N HCI and water. The organic phase was separated, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The compound was purified by flash chromatography on silica gel (15-40 μιτι, 120 g, heptane/EtOAc 80/20). The pure fractions were combined and evaporated to dryness to give, after crystallization from diisopropyl ether, te/t-butyl 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoro-methoxy)indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoate 4c (2.6 g).
Synthesis of Compound 4 and chiral separation into Enantiomers 4A and 4B: A solution of terf-butyl 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)-indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoate 4c (2.4 g, 3.8 mmol) in 4M HCI in dioxane (24 mL) was stirred at 5°C for 3 h and at room temperature for 3h. The precipitate was filtered off and dried to afford 4-(3-((1 -(4-chlorophenyl)-2-oxo-2-(6-(trifluoromethoxy)indolin-1 -yl)ethyl)amino)-5-methoxyphenoxy)butanoic acid as an HCI salt (Compound 4, 2 g, 0.8 equiv. HCI, 0.07 equiv. H2O). Compound 4 (2 g, HCI salt) was neutralized prior to chiral separation by treatment of a solution of Compound 4 (HCI salt) in ethylacetate with 1 N NaOH and evaporation of the organic layer under reduced pressure. The enantiomers were separated via Preparative Chiral SFC (Stationary phase: Chiralcel® OD-H 5 μηη 250 x 30 mm, Mobile phase: 50% CO2, 50% iPrOH (+ 0.3% iPrNH2)) and further purified via Preparative achiral SFC (Stationary phase: Cyano® 6 μιτι 150×21 .2mm, Mobile phase: 80% CO2, 20% MeOH (+ 0.3% iPrNH2)). The product fractions were combined and evaporated under reduced pressure. The two enantiomers were taken up with EtOAc and washed with 1 N HCI. The organic layers were separated, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The first eluted enantiomer was solidified from ether/diisopropyl ether to give Enantiomer 4A (616 mg). The second eluted enantiomer was solidified from ether/diisopropyl ether to give Enantiomer 4B (715 mg).
It is also possible to separate the enantiomers starting from the HCI salt of the racemate using the same conditions for chiral separation.
Compound 4:
1H NMR (500 MHz, DMSO-c/6) δ ppm 1 .87 (quin, J=6.9 Hz, 2 H) 2.34 (t, J=7.3 Hz, 2 H) 3.07 – 3.28 (m, 2 H) 3.62 (s, 3 H) 3.85 (t, J=6.5 Hz, 2 H) 4.04 (td, J=10.5, 7.1 Hz, 1 H) 4.52 (td, J=10.3, 6.5 Hz, 1 H) 5.57 (s, 1 H) 5.76 (t, J=2.2 Hz, 1 H) 5.90 – 6.00 (m, 2 H) 7.01 (dd, J=8.2, 1 .6 Hz, 1 H) 7.33 (d, J=8.2 Hz, 1 H) 7.41 – 7.48 (m, 2 H) 7.55 (d, J=8.5 Hz, 2 H) 8.03 (s, 1 H)
LC/MS (method LC-B): Rt 2.70 min, MH+ 579
Melting point: 150°C
Enantiomer 4A:
1H NMR (500 MHz, DMSO-c/6) δ ppm 1 .87 (quin, J=6.7 Hz, 2 H) 2.34 (br t, J=7.3 Hz, 2 H) 3.08 – 3.27 (m, 2 H) 3.62 (s, 3 H) 3.85 (br t, J=6.3 Hz, 2 H) 3.99 -4.1 1 (m, 1 H) 4.47 – 4.57 (m, 1 H) 5.57 (br s, 1 H) 5.76 (s, 1 H) 5.95 (br d, J=10.1 Hz, 2 H) 6.45 (br s, 1 H) 7.01 (br d, J=7.6 Hz, 1 H) 7.34 (br d, J=7.9 Hz, 1 H) 7.44 (br d, J=8.5 Hz, 2 H) 7.55 (br d, J=8.2 Hz, 2 H) 8.04 (br s, 1 H) 12.12 (br s, 1 H) LC/MS (method LC-A): Rt 2.95 min, MH+ 579
[a]D20: -48.5° (c 0.27, DMF)
Chiral SFC (method SFC-A): Rt 1 .13 min, MH+ 579, chiral purity 100%.
Enantiomer 4B:
1H NMR (500 MHz, DMSO-c/6) δ ppm 1 .87 (br t, J=6.8 Hz, 2 H) 2.34 (br t, J=7.3 Hz, 2 H) 3.09 – 3.27 (m, 2 H) 3.62 (s, 3 H) 3.85 (br t, J=6.1 Hz, 2 H) 3.99 -4.10 (m, 1 H) 4.46 – 4.59 (m, 1 H) 5.57 (s, 1 H) 5.76 (br s, 1 H) 5.95 (br d, J=10.1 Hz, 2 H) 6.45 (br s, 1 H) 7.01 (br d, J=7.9 Hz, 1 H) 7.34 (br d, J=7.9 Hz, 1 H) 7.44 (br d, J=8.2 Hz, 2 H) 7.55 (br d, J=8.2 Hz, 2 H) 8.04 (br s, 1 H) 12.12 (br s, 1 H) LC/MS (method LC-A): Rt 2.94 min, MH+ 579
[a]D20: +42.9° (c 0.28, DMF)
Chiral SFC (method SFC-A): Rt 2.13 min, MH+ 579, chiral purity 100%.
Patent
WO2021094563
The compounds of formula I according to the present invention may be synthesized according to methods described in the art, as disclosed in WO 2016/180696. The compounds of formula II according to the present invention may be prepared according to methods described in the art, as disclosed in WO2017/167951.
Compound (b) of the present invention was tested in AG129 mouse viremia model. The synthesis of compound (b) is described in WO 2017/167951, under Example 4.
compound (b)
PATENT
WO 2018215316
The compounds of formula (I) of the present invention all have at least one asymmetric carbon atom as indicated in the figure below by the carbon atom labelled with * :
Ref
https://doi.org/10.1038/s41586-021-03990-6
////////////////////JNJ-A07, DENGUE, VIRUS, PRECLINICAL

NEW DRUG APPROVALS
ONE TIME
$10.00
XL 114, AUR 104 and XL 102, AUR 102 (NO CONCLUSIONS, ONLY PREDICTIONS)

XL 114
FOR BOTH, JUST PREDICTION
PREDICTIONS
or


N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
![(2S)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117943691&t=l)

(2S)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
CAS 2305027-62-5
C12 H20 N6 O7, 360.32Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-, (2S,3ξ)-N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
ALSO SEE


![(2S,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://drugapprovalsint.com/wp-content/uploads/2021/10/str1-10.jpg)
1673534-76-3C12 H20 N6 O7, 360.32
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]
(2S,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acidN-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
CAS 1673534-76-3
PD-1-IN-1 free base, EX-A1918, CS-6240, NSC-799645, CA-170 (AUPM-170)|PDL1 inhibitor, HY-101093, PD-1-IN-1
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O
XL 114, AUR 104
A novel covalent inhibitor of FABP5 for cancer therapy
XL 102, AUR 102
A potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7)
NO CONCLUSIONS, ONLY PREDICTIONS
PREDICTIONS MORE
![(2R,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155529077&t=l)

(2R,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
C12H20N6O7, 360.32
![(2S,3S)-2-[[(1S)-3-Amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155339943&t=l)

(2S,3S)-2-[[(1S)-3-amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
XL102, AUR 102
XL102 is a potent, selective and orally bioavailable covalent inhibitor of CDK7, which is an important regulator of the cellular transcriptional and cell cycle machinery. CDK7 helps regulate cell cycle progression, with overexpression observed in multiple cancers, such as breast, prostate and ovarian cancers. In preclinical studies, XL102 revealed potent anti-proliferative activity, induced cell death in a large panel of cancer cell lines and caused tumor growth inhibition and regression in xenograft models, demonstrating its potential as a targeted antitumor agent.
In late 2020, Exelixis exercised its option to in-license XL102 (formerly AUR102) from Aurigene per the companies’ July 2019 collaboration, option and license agreement. Exelixis has assumed responsibility for the future clinical development, manufacturing and commercialization of XL102. Aurigene retains limited development and commercial rights for India and Russia.
SYN

ABOUT Fatty acid-binding proteins (FABPs)
Fatty acid-binding proteins (FABPs) are involved in binding and storing hydrophobic ligands such as long-chain fatty acids, as well as transporting them to the appropriate compartments in the cell. Epidermal fatty acid-binding protein (FABP5) is an intracellular lipid-binding protein that is abundantly expressed in adipocytes and macrophages. Previous studies have revealed that the FABP5 expression level is closely related to malignancy in various types of cancer. However, its precise functions in the metabolisms of cancer cells remain unclear. Here, we revealed that FABP5 knockdown significantly induced downregulation of the genes expression, such as hormone-sensitive lipase (HSL), monoacylglycerol lipase (MAGL), elongation of long-chain fatty acid member 6 (Elovl6), and acyl-CoA synthetase long-chain family member 1 (ACSL1), which are involved in altered lipid metabolism, lipolysis, and de novo FA synthesis in highly aggressive prostate and breast cancer cells. Moreover, we demonstrated that FABP5 induced inflammation and cytokine production through the nuclear factor-kappa B signaling pathway activated by reactive oxygen species and protein kinase C in PC-3 and MDA-MB-231 cells. Thus, FABP5 might regulate lipid quality and/or quantity to promote aggressiveness such as cell growth, invasiveness, survival, and inflammation in prostate and breast cancer cells. In the present study, we have revealed for the first time that high expression of FABP5 plays a critical role in alterations of lipid metabolism, leading to cancer development and metastasis in highly aggressive prostate and breast cancer cells.
Fatty acid-binding protein, epidermal is a protein that in humans is encoded by the FABP5 gene
Function
This gene encodes the fatty acid binding protein found in epidermal cells, and was first identified as being upregulated in psoriasis tissue. Fatty acid binding proteins are a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. It is thought that FABPs roles include fatty acid uptake, transport, and metabolism.[6]
The phytocannabinoids (THC and CBD) inhibit endocannabinoid anandamide (AEA) uptake by targeting FABP5, and competition for FABPs may in part or wholly explain the increased circulating levels of endocannabinoids reported after consumption of cannabinoids.[7] Results show that cannabinoids inhibit keratinocyte proliferation, and therefore support a potential role for cannabinoids in the treatment of psoriasis.[8]
Interactions
FABP5 has been shown to interact with S100A7.[
ABOUT CD47/SIRPa axis
CD47/SIRPa axis is established as a critical regulator of myeloid cell activation and serves as an immune checkpoint for macrophage mediated phagocytosis. Because of its frequent upregulation in several cancers, CD47 contributes to immune evasion and cancer progression. CD47 regulates phagocytosis primarily through interactions with SIRPla expressed on macrophages. Blockade of SIRPla/CD47 has been shown to dramatically enhance tumor cell phagocytosis and dendritic cells maturation for better antigen presentation leading to substantially improved antitumor responses in preclinical models of cancer (M. P. Chao et al. Curr Opin Immunol. 2012 (2): 225-232). Disruption of CD47-SIRPa interaction is now being evaluated as a therapeutic strategy for cancer with the use of monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys.
CD47 is expressed on virtually all non-malignant cells, and blocking the CD47 or the loss of CD47 expression or changes in membrane distribution can serve as markers of aged or damaged cells, particularly on red blood cells (RBC). Alternatively, blocking SIRPa also allows engulfment of targets that are not normally phagocytosed, for those cells where pre-phagocytic signals are also present. CD47 is a broadly expressed transmembrane glycoprotein with a single Ig-like domain and five membrane- spanning regions, which functions as a cellular ligand for SIRPa with binding mediated through the NH2-terminal V-like domain of SIRPa. SIRPa is expressed primarily on myeloid cells, including macrophages, granulocytes, myeloid dendritic cells (DCs), mast cells, and their precursors, including hematopoietic stem cells.
CD47 is also constitutively upregulated on a number of cancers such as Non-Hodgkin Lymphoma (NHL), Acute myeloid leukemia (AML), breast, colon, glioblastoma, glioma, ovarian, bladder and prostate cancers, etc. Overexpression of CD47 by tumor cells, which efficiently helps them to escape immune surveillance and killing by innate immune cells. However, in most of the tumor types, blockade of the CD47-SIRPa interaction as a single agent may not be capable of inducing significant phagocytosis and antitumor immunity, necessitating the need to combine with other therapeutic agents. The concomitant engagement of activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors (collectively known as “eat-me” signals) may be necessary for exploiting the maximum potential of the CD-47-SIPRa pathway blockade.
The role of engagement of prophagocytic receptors is proved by inefficiency to trigger phagocytosis either by anti-CD47 F(ab) fragments, single chain variable fragments of CD-47 or non-Fc portion- containing SIRPa proteins in blocking of the CD47-SIRPa interaction. When activating prophagocytic receptors are engaged, as evident in the case of using Fc portion-containing blocking anti-CD47 antibodies, CD47- SIRPa blockade is able to trigger more efficient phagocytosis. Combining CD47-SIRPa blocking agents with therapeutic antibodies (Fc-containing) targeting tumor antigens stimulate activating Fc receptors (FcRs) leading to efficient phagocytosis. The Fc portion of therapeutic antibody targeting tumor antigen also induces antibody-dependent cellular cytotoxicity (ADCC), which also adds to the therapeutic efficacy. Hence antibodies selected from the group consisting of rituximab, herceptin, trastuzumab, alemtuzumab, bevacizumab, cetuximab and panitumumab, daratumumab due to its tumor targeting nature and ADCC, can trigger more efficient phagocytosis.
Earlier approaches to disrupt CD47- SIRPa interaction utilized monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys fused to Fc fragment. However, a concern with this approach is that CD47 is highly expressed on both hematopoietic and non-hematopoietic normal cells. Hence along with tumor cells CD47-SIRPa blocking agents containing Fc-portion may also target many normal cells potentially leading to their elimination by macrophages. The interaction of blocking antibodies with normal cells is considered as a major safety issue resulting in anemia, thrombocytopenia, and leukopenia. These agents may also affect solid tissues rich in macrophages such as liver, lung, and brain. Hence it may be ideal to block the CD47- SIRPa interaction by agents devoid of Fc portion, such as small
molecules, peptides, Fab fragments etc. while activating prophagocytic receptors in tumor cells by appropriate combinations to induce efficient phagocytosis of tumor cells.
Apart from Fc Receptors, a number of other prophagocytic receptors are also reported to promote engulfment of tumor cells in response to CD47-SIRPa blockade by triggering the phagocytosis. These include receptors for SLAMF7, Mac-l, calreticulin and possibly yet to identified receptors. B cell tumor lines such as Raji and other diffuse large B cell lymphoma express SLAMF7 and are implicated in triggering prophagocytic signals during CD47-SIRPa blockade.
Therapeutic agents known to activate prophagocytic receptors are also therefore ideal partners for use in combination with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These agents include proteasome inhibitors (bortezomib, ixazomib and carfilzomib), Anthracyclines (Doxorubicin, Epirubicin, Daunorubicin, Idarubicin, Mitoxantrone) Oxaliplatin, Cyclophosphamide, Bleomycin, Vorinostat, Paclitaxel, 5-Fluorouracil, Cytarabine, BRAF inhibitory drugs (Dabrafenib, Vemurafenib), PI3K inhibitor, Docetaxel, Mitomycin C, Sorafenib, Tamoxifen and oncolytic viruses.
Apart from the specific agents known to have effect on‘eat me’ signals other agents including Abiraterone acetate, Afatinib, Aldesleukin, Aldesleukin, Alemtuzumab, Anastrozole, Axitinib, Belinostat, Bendamustine, Bicalutamide, Blinatumomab, Bosutinib, Brentuximab, Busulfan, Cabazitaxel, Capecitabine, Carboplatin, Carfilzomib, Carmustine, Ceritinib, Clofarabine, Crizotinib, Dacarbazine, Dactinomycin, Dasatinib, Degarelix, Denileukin, Denosumab, Enzalutamide, Eribulin, Erlotinib, Everolimus, Exemestane, Exemestane, Fludarabine, Fulvestrant, Gefitinib, Goserelin, Ibritumomab, Imatinib, Ipilimumab, Irinotecan, Ixabepilone, Lapatinib, Lenalidomide, Letrozole, Leucovorin, Leuprolide, Lomustine, Mechlorethamine, Megestrol, Nelarabine, Nilotinib, Nivolumab, Olaparib, Omacetaxine, Palbociclib, Pamidronate, Panitumumab, Panobinostat, Pazopanib, Pegaspargase, Pembrolizumab, Pemetrexed Disodium, Pertuzumab, Plerixafor, Pomalidomide, Ponatinib, Pralatrexate, Procarbazine, Radium 223, Ramucirumab, Regorafenib, rIFNa-2b, Romidepsin, Sunitinib, Temozolomide, Temsirolimus, Thiotepa, Tositumomab, Trametinib, Vinorelbine, Methotrexate, Ibrutinib, Aflibercept, Toremifene, Vinblastine, Vincristine, Idelalisib, Mercaptopurine and Thalidomide could potentially have effect on‘eat me’ signal pathway on combining with CD-47-SIRPa blocking agents.
In addition to the therapeutic agents mentioned above, other treatment modalities that are in use in cancer therapy also activate prophagocytic receptors, and thus can be combined with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These include Hypericin-based photodynamic therapy (Hyp-PDT), radiotherapy, High-hydrostatic pressure, Photofrin-based PDT and Rose Bengal acetate -based PDT.
However, there is an unmet need for combining small molecule CD-47-SIRPa pathway inhibitors with agents capable of stimulating activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors, or combining with other treatment modalities that are in use in cancer therapy to activate prophagocytic receptors for exploiting the maximum potential of the CD-47- SIRPa pathway blockade.
CLIP
Exelixis In-Licenses Second Anti-Cancer Compound from Aurigene Following FDA Acceptance of Investigational New Drug Application for Phase 1 Clinical Trial in Non-Hodgkin’s Lymphoma
– Robust preclinical data support Exelixis’ clinical development of XL114, with phase 1 trial in Non-Hodgkin’s lymphoma expected to begin in the coming months –
– Exelixis will make an option exercise payment of $10 million to Aurigene –
https://www.businesswire.com/news/home/20211014005549/en/Exelixis-In-Licenses-Second-Anti-Cancer-Compound-from-Aurigene-Following-FDA-Acceptance-of-Investigational-New-Drug-Application-for-Phase-1-Clinical-Trial-in-Non-Hodgkin%E2%80%99s-LymphomaOctober 14, 2021 08:00 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today announced that Exelixis has exercised its exclusive option under the companies’ July 2019 agreement to in-license XL114 (formerly AUR104), a novel anti-cancer compound that inhibits the CARD11-BCL10-MALT1 (CBM) signaling pathway, which promotes lymphocyte survival and proliferation. Exelixis has now assumed responsibility for the future clinical development, commercialization and global manufacturing of XL114. Following the U.S. Food and Drug Administration’s (FDA) recent acceptance of its Investigational New Drug (IND) application, Exelixis will soon initiate a phase 1 clinical trial evaluating XL114 monotherapy in patients with Non-Hodgkin’s lymphoma (NHL). At the American Association of Cancer Research Annual Meeting in April of this year, Aurigene presented preclinical data (Abstract 1266) demonstrating that XL114 exhibited potent anti-proliferative activity in a large panel of cancer cell lines ranging from hematological cancers to solid tumors with excellent selectivity over normal cells. In addition, oral dosing of XL114 resulted in significant dose-dependent tumor growth inhibition in diffuse large B-cell lymphoma (DLBCL) and colon carcinoma models.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline”
XL114 is the second molecule that Exelixis in-licensed from Aurigene under the companies’ July 2019 collaboration, option and license agreement. Exelixis previously exercised its option to in-license XL102, a potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7), from Aurigene in December 2020 and initiated a phase 1 trial of XL102 as a single agent and in combination with other anti-cancer agents in patients with advanced or metastatic solid tumors in January 2021.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline,” said Peter Lamb, Ph.D., Executive Vice President, Scientific Strategy and Chief Scientific Officer, Exelixis. “XL114 has shown potent anti-proliferative activity in lymphoma cell lines that have aberrant activation of the CBM signaling pathway and may have a differentiated profile and potential as a best-in-class molecule that could improve outcomes for patients with Non-Hodgkin’s lymphoma and other hematologic cancers.”
XL114 was identified to have anti-proliferative activity in cell lines with constitutive activation of CBM signaling, including activated B-cell-like DLBCL (ABC-DLBCL), mantle cell lymphoma and follicular lymphoma cell lines. Further characterization of XL114 in cell-based assays demonstrated a functional role in B-cell (BCR) signaling pathways. Additionally, XL114 showed dose-dependent tumor growth inhibition in an ABC-DLBCL mouse xenograft tumor model. In preclinical development, XL114 also demonstrated a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. While the precise molecular mechanism underlying XL114’s function in repressing BCR signaling and MALT1 activation has yet to be characterized, the fatty acid-binding protein 5 (FABP5) has been identified as a prominent XL114-binding target.
“XL114 is the second molecule that Exelixis has opted to in-license under our July 2019 agreement, underscoring the significant potential of our approach to the discovery and preclinical development of innovative cancer therapies that target novel mechanisms of action,” said Murali Ramachandra, Ph.D., Chief Executive Officer, Aurigene. “Exelixis has a track record of success in the clinical development and commercialization of anti-cancer therapies that provide patients with important new treatment options, and we are pleased that the continued advancement of XL114 will be supported by the company’s extensive clinical, regulatory and commercialization infrastructure.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to obtain an exclusive license from Aurigene to three preexisting programs, including the compounds now known as XL102 and XL114. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for an additional upfront payment of $2.5 million per program. The collaboration was expanded in 2021 to include three additional early discovery programs. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all nine programs. Exelixis may exercise its option for a program at any time up until the first IND for the program becomes effective. Having exercised options on two programs thus far (XL102 and XL114), if and when Exelixis exercises a future option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. To exercise its option for XL114, Exelixis will make an option exercise payment to Aurigene of $10 million. Once Exelixis exercises its option for a program, Aurigene will be eligible for clinical development, regulatory and sales milestones, as well as royalties on future potential sales of the compound. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene Discovery Technologies Limited is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY, NSEIFSC: DRREDDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the U.S. and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of the Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. In November 2020, the company was named to Fortune’s 100 Fastest-Growing Companies list for the first time, ranking 17th overall and the third-highest biopharmaceutical company. For more information about Exelixis, please visit www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.

Dinesh Chikkanna
Director, Medicinal Chemistry Aurigene Discovery Technologies

Murali Ramachandra
CEO at Aurigene Discovery Technologies

CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1266
Abstract 1266: Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapyDinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar and Murali Ramachandra
DOI: 10.1158/1538-7445.AM2021-1266 Published July 2021
Proceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Dysregulated fatty acid metabolism is thought to be a hallmark of cancer, wherein fatty acids function both as an energy source and as signals for enzymatic and transcriptional networks contributing to malignancy. Fatty acid-binding protein 5 (FABP5) is an intracellular protein that facilitates transport of fatty acids and plays a role in regulating the expression of genes associated with cancer progression such as cell growth, survival, and metastasis. Overexpression of FABP5 has been reported to contribute to an aggressive phenotype and a poor survival correlation in several cancers. Therefore, inhibition of FABP5 is considered as a therapeutic approach for cancers. Phenotypic screening of a library of covalent compounds for selective sensitivity of cancer cells followed by medicinal chemistry optimization resulted in the identification of AUR104 with desirable properties. Chemoproteomic-based target deconvolution revealed FABP5 as the cellular target of AUR104. Covalent adduct formation with Cys43 of FABP5 by AUR104 was confirmed by mass spectrometry. Target occupancy studies using a biotin-tagged AUR104 demonstrated potent covalent binding to FABP5 in both cell-free and cellular conditions. Ligand displacement assay with a fluorescent fatty acid probe confirmed the competitive binding mode of AUR104 with fatty acids. Binding at the fatty acid site and covalent bond formation with Cys43 were also demonstrated by crystallography. Furthermore, AUR104 showed a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. AUR104 exhibited potent anti-proliferative activity in a large panel of cell lines derived from both hematological and solid cancers with a high degree of selectivity over normal cells. Anti-proliferative activity in lymphoma cell lines correlated with inhibition of MALT1 pathway activity, cleavage of RelB/Bcl10 and secretion of cytokines, IL-10 and IL-6. AUR104 displayed desirable drug-like properties and dose-dependent oral exposure in pharmacokinetic studies. Oral dosing with AUR104 resulted in dose-dependent anti-tumor activity in DLBCL (OCI-LY10) and NSCLC (NCI-H1975) xenograft models. In a repeated dose MTD studies in rodents and non-rodents, AUR104 showed good tolerability with an exposure multiple of >500 over cellular EC50 for up to 8 hours. In summary, we have identified a novel covalent FABP5 inhibitor with optimized properties that showed anti-tumor activity in in vitro and in vivo models with acceptable safety profile. The data presented here strongly support clinical development of AUR104.
Citation Format: Dinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar, Murali Ramachandra. Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapy [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1266.
Patent
US20200147054 – COMBINATION OF SMALL MOLECULE CD-47 INHIBITORS WITH OTHER ANTI-CANCER AGENTS
Muralidhara Ramachandra
Pottayil Govindan Nair Sasikumar
Girish Chandrappa Daginakatte
Kiran Aithal Balkudru
PATENT
WO 2020095256
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020095256
Example- 1: The synthetic procedures for the preparation of compounds described in the present invention were described in co-pending Indian provisional patent application 201841001438 dated 12* Jan 2018, which is converted as PCT application
PCT/IB2019/050219, the contents of which are hereby incorporated by reference in their entirety.
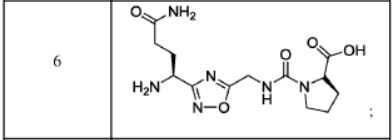
PATENT
WO 2018178947https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018178947&tab=PCTDESCRIPTION
PATENT
WO 2019138367
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019138367
PATENT
WO 2019073399
https://patents.google.com/patent/WO2019073399A1/en
Priority to IN201741036169
Example 4 of WO 2015/033299
PATENT
https://patents.google.com/patent/BR112020014202A2/en
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
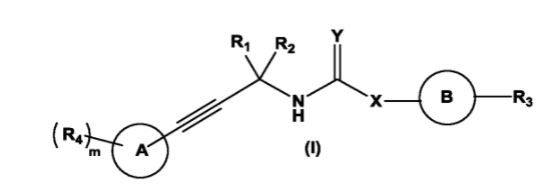
Patent
Example 1
(((S)-4-amino-1-(3-((S)-1,5-diaminopentyl)-1,2,4-oxadiazol-5-yl)-4-oxobutyl)carbamoyl)-L-proline (Compound 1)
Synthesis of Compound 1 b
Synthesis of Compound 1C
Synthesis of Compound 1d
Synthesis of Compound 1f
Synthesis of Compound 1g
Synthesis of Compound 1h
Synthesis of Compound 1i
Synthesis of compound 1j
Synthesis of Compound 1
Example 2
(S)-4-(3-((S)-1-amino-4-guanidinobutyl)-1,2,4-oxadiazol-5-yl)-4-(3-((S)-1-carboxy-2-phenylethyl) ureido)butanoic acid (Compound 7)
Synthesis of Compound 2b
Synthesis of Compound 2c
Synthesis of Compound 2d
Synthesis of Compound 2f
Synthesis of Compound 2g
Synthesis of Compound 2h
Synthesis of Compound 2i
Synthesis of Compound 2j
Synthesis of Compound 7
PATENT
WO 2015/033299
https://patents.google.com/patent/WO2015033299A1/en?oq=WO+2015%2f033299
Pottayil Govindan Nair SasikumarMuralidhara RamachandraSeetharamaiah Setty Sudarshan Naremaddepalli
Example 1: Synthesis of Compound 1

Step la:

Ethylchloroformate (1.5 g, 13.78 mniol) and N-Methylmorpholine ( 1.4 g, 13.78 mmol) were added to a solution of compound la (3 g, 11.48 mmol) in THE (30 mL) arid stirred at -20 °C. After 20 min. Liquid ammonia (0.77 g, 45.92 mmol) was added to the active mixed anhydride formed in- situ and stirred at 0-5 °C for 20 min. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOs, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to get 2.9 g of compound lb (Yield: 96.3%). LCMS: 261.0 ( Vi+H ; .
Step lb:

1 b 1cTrifluroacetic anhydride (9.7 g, 46.0 mmol) was added to a solution of compound lb (8 g, 30.7 mmol) in pyridine (24.3 g, 307.0 mmol) and stirred at room temperature for 3 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCO?,, citric acid, brine solution, dried over Na2-S04 and evaporated under reduced pressure to afford 7 g of compound lc (Yield: 94.0%). LCMS: 187.2 (M-¾u )+.
Step lc:

1 c 1dHydroxylamine hydrochloride (3 g, 43.37 mmol) and potassium carbonate (6 g, 43.37 mmol) were added to a solution of compound lc (7 g, 28.91 mmol) in EtOH (70 m L) and stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with brine solution, dried over Na2S04 and evaporated under reduced pressure. The crude compound was purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to get 4.2 g of compound Id (Yield: 52.8%). LCMS: 276.4 (M+H)+.Step Id:

Deoxo-Fluor® (1.83 g, 8.3 mmol) was added to a solution of Fmoc-Asn(Trt)-OH (4.5 g, 7.5 mmol) in CH2Q2 (50 mL) and stirred at 0 °C for 3 h. Then CH2CI2 was evaporated and triturated with hexane, decanted and evaporated under vacuum to get the corresponding acid fluoride. NMM (1.17 g, 1 1.6 mmol) and compound Id (1.6 g, 5.8 mmol) in THF were added to the acid fluoride and stirred at room temperature for 12 h. Then THF was evaporated and sodium acetate (0.72 g, 8.7 mmol) was added followed by EtOH (50 mL). The reaction mixture was stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOa, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure, which was further purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to afford 2.8 g of compound le (Yield: 44.4%). LCMS: 836.4 (M+Hf .Step le:
Ph3

To compound le (2.3 g, 2.7 mmol) in CH2CI2 (10 mL) diethyiarnine (10 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get gummy residue. The crude compound was purified by neutral alumina column chromatography (Eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to get 1.4 g of If (Yield: 90 %). LCMS: 636.5 (M+Na)+.

1f 1To a solution of compound If (0.45 g) in CH2CI2 (5 mL), trifluoroacetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred for 3 h at room temperature to remove the acid sensitive protecting groups. The resulting solution was concentrated in vacuum to afford 0.29 g of crude compound 1 which was purified using prep-HPLC method described under experimental conditions. \H NMR (DMSQ-d6, 400 MHz): δ 2.58 (m, 2H), 3.53 (m, 3H), 3.91 (t, 1H), 4.36 (t, 1H), 6.91 (s, 1H), 7.45 (s, 1H); 1 C NMR (DMSO-de, 400 MHz): δ 20.85, 45.71 , 50.23, 65.55, 171.03, 171 .41, 181.66. LCMS: 216.2 (Μ+ΗΓ; HPLC: tR = 13.1 min.Example 2: Synthesis of Co

Step 2a:

1f2a
The urea linkage was carried out by the coupling compound If (2.7 g, 4.39 mmoi) in THF (30 mL) at room temperature with compound 2b (1.67 g, 4.39 mmoi). The coupling was initiated by the addition of TEA (0.9 g, 8.78 mmoi) in THF (10 m L) and the resultant mixture was stirred at room temperature. After completion of 20 h, THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get compound 2a, which was further purified by silica gel column chromatography (Fluent: 0-50% ethyl acetate in hexane) to afford 3.46 g of compound 2a (Yield: 92.10%). LCMS 857.4 (M+H)+.

2aTo a solution of compound 2a (0.22 g, 0.25 mmol) in 0¾ί¾ (5 m L), trifluoroaeetic acid (5 mL) and catalytic amount of triisopropyisilane were added and stirred for 3h at room, temperature. The resulting solution was concentrated under reduced pressure to obtain 0.35 g of crude compound. The crude solid material was purified using preparative- HPLC method described under experimental conditions. LCMS: 347.1 (M+H)+; HPLC: tR = 12.9 min.
Synthesis of

2bTo the compound H-Ser(tBu)-OiBu (2 g, 9.2 mmol) in C I I■■(.‘{■ (20 mL), triethylamine (1.39 g, 13.8 mmol) was added and the solution was stirred at room temperature for 5-10 min. To this mixture, solution of 4-Nitrophenyl chioro formate (2.22 g, 11.04 mmol) in CH2CI2 was added and the resultant mixture was stirred at room temperature for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction, reaction mixture was diluted with CH2CI2 and washed with water and 5.0 M citric acid solution, dried over Na2SC>4 and evaporated under reduced pressure to get crude compound 2b, which was further purified by silica gel column chromatography (Eiuent: 0-20% ethyl acetate in hexane) to yield 2.1 g (58.9%) of 2b.Example 3: Synthesis of Compound 3

The compound was synthesised using similar procedure as depicted in Example 1 (compound 1) and D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH (compound la, Example 1) and Fmoc-D- Asn(trt)-OH in place of Fmoc-Asn(trt)-OH to yield 0.15 g crude material of the title compound 3. LCMS: 230.1 (M+H)+.Example 4: Synthesis of Co
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using

instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.Example 5: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)- OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.Example 6: Synthesis of Compound 6

The compound was synthesised using similar procedure as depicted in Example 2 by using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.2 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 375.1 (M+H)+, HPLC: tR = 1.84 min.Example 7: Synthesis of Compound 7

Step 7a:

1f7aThe compound 7a was synthesised using similar procedure as for compound 2a (Example 2, step 2a) using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-OtBu to get crude material which was further purified by silica gel column chromatography (Eluent: 0-50% ethyl acetate in he ane) to get 2.0 g of compound 7a (Yield: 74 %). LCMS: 829.2 (M+H)+.Step 7b:

7a 7bTo a solution of compound 7a (0.35 g, 4.0 mmol) in THF (5 mL) was added lithium hydroxide (0.026 g, 0.63 mmol) at 0 °C and the mixture was stirred for 2 h at room temperature. The completion of the reaction was confirmed by TLC analysis. THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to afford 7b, which was further purified by silica gel column chromatography (Eluent: 0-5% methanol in DCM) to get 0.3 g of product 7b (Yield: 86.7%). LCMS 815.2 (M+H)+.
Step 7c:

7b 7Compound 7b (0.295 g, 0.39 mmol) was anchored to Rink amide resin (0.7 g, 0.55 mmol/g) using HOBT (0.072 g, 0.54 mmol) and DIC (0.068 g, 0.54 mmol) method in DMF (10 mL). The resin was stirred for 12 h at room temperature. The resin was washed with DCM, DMF and DCM and dried. The target compound was cleaved from the rink amide resin using TFA (5 mL) and catalytic amount of TIPS. The resin was allowed to remain at room temperature for 2 h with occasional stirring. After 2 h, TFA and TIPS were evaporated under nitrogen atmosphere and the resulting residue was washed with diethyl ether to yield 0.1 g crude material of the title compound 7. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 360.0 (M+H)+, HPLC: tR = 13.88 min.Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324&tab=FULLTEXT
Patenthttps://patents.google.com/patent/WO2019067678A1/enPATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324
PATENThttps://patents.google.com/patent/WO2018073754A1/en
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
PAPERSScientific Reports (2019), 9(1), 1-19. https://www.nature.com/articles/s41598-019-48826-6
Chemical structures of PD-L1 inhibitors developed by Aurigene (Aurigene-1) and Bristol-Meyers Squibb (BMSpep-57, BMS-103, and BMS-142). Chemical structures were generated using ChemDraw Professional 15. PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
L-threonine’ mentioned in compound of formula (I) thereof can be represented by any one of the following formulae:
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020289477-A1 | Conjoint therapies for immunomodulation | 2017-11-06 | |
| WO-2019073399-A1 | CRYSTALLINE FORMS OF 1,2,4-OXADIAZOLE SUBSTITUTED IN POSITION 3 | 2017-10-11 | |
| AU-2018341583-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| WO-2019061324-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 | |
| WO-2019067678-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020247766-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| US-2020061030-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| WO-2018073754-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| US-2020361880-A1 | 1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators | 2015-03-10 | |
| EP-3041827-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2018-04-18 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3363790-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-02-19 |
| US-10173989-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2019-01-08 |
| US-10590093-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-03-17 |
| US-2015073024-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2017101386-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018072689-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2019144402-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2020199086-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-9771338-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2017-09-26 |
| WO-2015033299-A1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 |
////////////Investigational New Drug Application, Phase 1, Clinical Trial, Non-Hodgkin’s Lymphoma, XL 114, AUR 104, aurigene, Exelixis
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
https://patentscope.wipo.int/search/en/result.jsf?inchikey=HFOBENSCBRZVSP-WHFCDURNSA-N

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT


The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
XL 102
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
Exelixis Forward-Looking Statements
This press release contains forward-looking statements, including, without limitation, statements related to: Exelixis’ and Aurigene’s plans to present preclinical data in support of the continued development of AUR102 in a poster as part of the 32nd ENA Symposium; the potential for AUR102 to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma; the potential for AUR102 to be the subject of an Investigational New Drug filing later in 2020; Exelixis’ potential future financial and other obligations under the exclusive collaboration, option and license agreement with Aurigene; and Exelixis’ plans to reinvest in its business to maximize the potential of the company’s pipeline, including through targeted business development activities and internal drug discovery. Any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements and are based upon Exelixis’ current plans, assumptions, beliefs, expectations, estimates and projections. Forward-looking statements involve risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in the forward-looking statements as a result of these risks and uncertainties, which include, without limitation: the availability of data at the referenced times; the level of costs associated with Exelixis’ commercialization, research and development, in-licensing or acquisition of product candidates, and other activities; uncertainties inherent in the drug discovery and product development process; Exelixis’ dependence on its relationship with Aurigene, including Aurigene’s adherence to its obligations under the exclusive collaboration, option and license agreement and the level of Aurigene’s assistance to Exelixis in completing clinical trials, pursuing regulatory approvals or successfully commercializing partnered compounds in the territories where they may be approved; the continuing COVID-19 pandemic and its impact on Exelixis’ research and development operations; complexities and the unpredictability of the regulatory review and approval processes in the U.S. and elsewhere; Exelixis’ and Aurigene’s continuing compliance with applicable legal and regulatory requirements; Exelixis’ and Aurigene’s ability to protect their respective intellectual property rights; market competition; changes in economic and business conditions; and other factors affecting Exelixis and its product pipeline discussed under the caption “Risk Factors” in Exelixis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission (SEC) on August 6, 2020, and in Exelixis’ future filings with the SEC. All forward-looking statements in this press release are based on information available to Exelixis as of the date of this press release, and Exelixis undertakes no obligation to update or revise any forward-looking statements contained herein, except as required by law.
Exelixis, the Exelixis logo, CABOMETYX, COMETRIQ and COTELLIC are registered U.S. trademarks. MINNEBRO is a registered Japanese trademark.
PIROXICAM

PIROXICAM
- Molecular FormulaC15H13N3O4S
- Average mass331.346 Da
1,1-Dioxyde de 4-hydroxy-2-méthyl-N-(2-pyridinyl)-2H-1,2-benzothiazine-3-carboxamide
13T4O6VMAM
252-974-3[EINECS]
2H-1,2-Benzothiazine-3-carboxamide, 4-hydroxy-2-methyl-N-2-pyridinyl-, 1,1-dioxide
36322-90-4[RN]37134
-Hydroxy-2-methyl-3-(pyrid-2-yl-carbamoyl)-2H-1,2-benzothiazine 1,1-dioxide
Piroxicam
CAS Registry Number: 36322-90-4
CAS Name: 4-Hydroxy-2-methyl-N-2-pyridinyl-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide
Additional Names: 3,4-dihydro-2-methyl-4-oxo-N-2-pyridyl-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide
Manufacturers’ Codes: CP-16171
Trademarks: Artroxicam (Coli); Baxo (Toyama); Bruxicam (Bruschettini); Caliment (Apotex); Erazon (Krka); Feldene (Pfizer); Flogobene (Farge); Geldene (Pfizer); Improntal (Kabi); Larapam (Lagap); Pirkam (DAK); Piroflex (Lagap); Reudene (ABC); Riacen (Chiesi); Roxicam (Gramon); Roxiden (Pulitzer); Sasulen (Andreu); Solocalm (Microsules); Zunden (Luitpold)Molecular Formula: C15H13N3O4S
Molecular Weight: 331.35
Percent Composition: C 54.37%, H 3.95%, N 12.68%, O 19.31%, S 9.68%
Literature References: Non-steroidal anti-inflammatory with long half-life. Prepn (keto form): J. Lombardino, DE1943265; idem,US3591584 (1970, 1971 to Pfizer).Synthesis and biological properties: J. Lombardino, E. Wiseman, J. Med. Chem.15, 848 (1972); J. Lombardino et al.,ibid.16, 493 (1973). Pharmacology: E. Wiseman et al.,Arzneim.-Forsch.26, 1300 (1976). Evaluation of ulcerogenic effects: G. Palacios et al.,Methods Find. Exp. Clin. Pharmacol.9, 353 (1987). Clinical pharmacology: L. Martinez et al.,ibid.10, 729 (1988). Review:eidem, in Pharmacological and Biochemical Properties of Drug Substancesvol. 3, M. E. Goldberg, Ed. (Am. Pharm. Assoc., Washington, DC, 1981) pp 324-346. Review of pharmacology and therapeutic efficacy: R. N. Brogden et al.,Drugs22, 165-187 (1981); eidem,ibid.28, 292-323 (1984). Symposium on clinical efficacy and safety: Am. J. Med.81, Suppl. 5B, 1-55 (1986). Comprehensive description: M. Mihalic et al.,Anal. Profiles Drug Subs.15, 509-531 (1986).
Properties: Crystals from methanol, mp 198-200°. pKa 6.3 (2:1 dioxane-water). LD50 orally in mice: 360 mg/kg (Wiseman).
Melting point: mp 198-200°
pKa: pKa 6.3 (2:1 dioxane-water)
Toxicity data: LD50 orally in mice: 360 mg/kg (Wiseman)
Derivative Type: Cinnamic acid ester
CAS Registry Number: 87234-24-0
Additional Names: Piroxicam cinnamate; cinnoxicam
Manufacturers’ Codes: SPA-S-510
Trademarks: Sinartrol (SPA); Zelis (Proter); Zen (Prophin)
Molecular Formula: C24H19N3O5S
Molecular Weight: 461.49
Percent Composition: C 62.46%, H 4.15%, N 9.11%, O 17.33%, S 6.95%
Derivative Type: Compd with b-cyclodextrinCAS Registry Number: 121696-62-6
Trademarks: Brexin (Chiesi); Cicladol (Master); Cycladol (Promedica)
Molecular Formula: C57H83N3O39S
Molecular Weight: 1466.33
Percent Composition: C 46.69%, H 5.71%, N 2.87%, O 42.55%, S 2.19%
Therap-Cat: Anti-inflammatory.
Keywords: Anti-inflammatory (Nonsteroidal); Thiazinecarboxamides.
- EINECS:252-974-3
- LD50:250 mg/kg (M, p.o.);
216 mg/kg (R, p.o.);
108 mg/kg (dog, p.o.)
Piroxicam is a nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class used to relieve the symptoms of painful inflammatory conditions like arthritis.[3][4] Piroxicam works by preventing the production of endogenous prostaglandins] which are involved in the mediation of pain, stiffness, tenderness and swelling.[3] The medicine is available as capsules, tablets and (not in all countries) as a prescription-free gel 0.5%.[5] It is also available in a betadex formulation, which allows a more rapid absorption of piroxicam from the digestive tract.[3] Piroxicam is one of the few NSAIDs that can be given parenteral routes.
It was patented in 1968 by Pfizer and approved for medical use in 1979.[6] It became generic in 1992,[7] and is marketed worldwide under many brandnames.[1]
Medical uses
It is used in the treatment of certain inflammatory conditions like rheumatoid and osteoarthritis, primary dysmenorrhoea, postoperative pain; and act as an analgesic, especially where there is an inflammatory component.[3] The European Medicines Agency issued a review of its use in 2007 and recommended that its use be limited to the treatment of chronic inflammatory conditions, as it is only in these circumstances that its risk-benefit ratio proves to be favourable.[5][8]
Adverse effects
See also: Nonsteroidal anti-inflammatory drug
As with other NSAIDs the principal side effects include: digestive complaints like nausea, discomfort, diarrhoea and bleeds or ulceration of the stomach, as well as headache, dizziness, nervousness, depression, drowsiness, insomnia, vertigo, hearing disturbances (such as tinnitus), high blood pressure, oedema, light sensitivity, skin reactions (including, albeit rarely, Stevens–Johnson syndrome and toxic epidermal necrolysis) and rarely, kidney failure, pancreatitis, liver damage, visual disturbances, pulmonary eosinophilia and alveolitis.[5] Compared to other NSAIDs it is more prone to causing gastrointestinal disturbances and serious skin reactions.[5]
In October 2020, the U.S. Food and Drug Administration (FDA) required the drug label to be updated for all nonsteroidal anti-inflammatory medications to describe the risk of kidney problems in unborn babies that result in low amniotic fluid.[9][10] They recommend avoiding NSAIDs in pregnant women at 20 weeks or later in pregnancy.[9][10]
Mechanism of action
See also: Nonsteroidal anti-inflammatory drug
Piroxicam is an NSAID and, as such, is a non-selective COX inhibitor possessing both analgesic and antipyretic properties.[5]
Chemical properties
Piroxicam exists as alkenol tautomer in organic solvents and as zwitterionic form in water.[11]
History
The project that produced piroxicam began in 1962 at Pfizer; the first clinical trial results were reported in 1977, and the product launched in 1980 under the brand name “Feldene”.[7][12] Major patents expired in 1992[7] and the drug is marketed worldwide under many brandnames.[1]
NMR
SYN
https://pubs.acs.org/doi/10.1021/jp1084444
Influence of Structure on the Spectroscopic Properties of the Polymorphs of Piroxicam
SYN
https://www.sciencedirect.com/science/article/abs/pii/S092420310400058X?via%3
PATENT

CN 101210013
https://patents.google.com/patent/CN101210013A/enIn the glassed steel reaction vessels of 2000L, add first ethyl ester thing 140Kg, dimethylbenzene 1500L, silica gel 10Kg.Be warming up to 100 ℃ of amino pyrrole 52Kg of adding 2-, continue to be warming up to the solvent refluxing temperature, keep refluxing slowly, steam the ethanol of reaction generation and the mixture of dimethylbenzene simultaneously, TLC follows the tracks of reaction, and reaction in 4.5-5 hour finishes.Underpressure distillation, the control temperature in the kettle is no more than 70 ℃, when the system volume be about cumulative volume 1/3 the time stop distillation, be cooled to normal temperature, stir 6-8h and filter, be i.e. crude product.Crude product adds methyl alcohol 1500L and adds the 15Kg gac, refluxes 30 minutes, filters, and is cooled to normal temperature, stirs 6-8h, methyl alcohol drip washing, 60-70 ℃ is dried by the fire 3-5h, measure product 140.5Kg, yield 85%.Press Cp2005 version standard detection, outward appearance; Off-white color, content 〉=99%.Methanol mother liquor reclaims methyl alcohol to overall 1/3 o’clock, and cooling stirring at normal temperature 6-8h filters and collects product, oven dry measure product 10Kg, yield 5.7%, this product meet the Cp2005 version and require to add up to yield.Add up to yield 90.7%.PAPER Bulletin of the Korean Chemical Society, 26(11), 1771-1775; 2005

SYN



| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 504-29-0 | C5H6N2 | 2-aminopyridine | 2-Pyridinamine |
| 79-04-9 | C2H2Cl2O | chloroacetyl chloride | Acetyl chloride, chloro- |
| 29209-30-1 | C11H11NO5S | 3,4-dihydro-2-methyl-4-oxo-2H-1,2-benzothiazine-3-carboxylic acid methyl ester 1,1-dioxide | 2H-1,2-Benzothiazine-3-carboxylic acid, 3,4-dihydro-2-methyl-4-oxo-, methyl ester, 1,1-dioxide |
| 29209-29-8 | C10H9NO5S | 3-methoxycarbonyl-4-oxo-3,4-dihydro-2H-1,2-benzothiazine 1,1-dioxide | 2H-1,2-Benzothiazine-3-carboxylic acid, 3,4-dihydro-4-oxo-, methyl ester, 1,1-dioxide |
- Drebushchak, V. A.; Journal of Thermal Analysis and Calorimetry 2006, V84(3), P643-649
- Gehad, G. Mohamed; Vibrational Spectroscopy 2004, V36(1), P97-104
- Pajula, Katja; Molecular Pharmaceutics 2010, V7(3), P795-804
- Wassvik, Carola M.; European Journal of Pharmaceutical Sciences 2006, V29(3-4), P294-305
- Wassvik, Carola M.; Journal of Medicinal Chemistry 2008, V51(10), P3035-3039
- Zayed, M. A.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2004, V60A(12), P2843-2852
- Zia-ur-Rehman, Muhammad; Bulletin of the Korean Chemical Society 2005, V26(11), P1771-1775
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Stulzer, H. K.; Pharmaceutical Chemistry Journal 2008, V42(4), P215-219 CAPLUS
- Drebushchak, V. A.; Journal of Thermal Analysis and Calorimetry 2006, V86(2), P303-309
- Hughes, Laura D.; Journal of Chemical Information and Modeling 2008, V48(1), P220-232
- Laban, Gunter; DD 260398 A3 1988
- Svoboda, Jiri; Collection of Czechoslovak Chemical Communications 1986, V51(5), P1133-9
- (26) Perillo, Isabel A.; Journal of Heterocyclic Chemistry 1983, V20(1), P155-60
- Zak, Bohumil; CS 276217 B6 1992 CAPLUS
- Dalla Croce, Piero; Journal of Chemical Research, Synopses 1986, (4), P150-1
- Vemavarapu, Chandra; Powder Technology 2009, V189(3), P444-453
- Sanghavi, N. M.; Indian Journal of Technology 1989, V27(2), P93-5
- “PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
- Mohamed, Gehad G.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2004, V60A(13), P3141-3154
- Zayed, M. A.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2006, V64A(1), P216-232
- Habibi-Yangjeh, Aziz; Bulletin of the Korean Chemical Society 2008, V29(4), P833-841
- Mahlin, Denny; Molecular Pharmaceutics 2011, V8(2), P498-506
- Kozjek, Franc; Acta Pharmaceutica Jugoslavica 1985, V35(4), P275-81
- Laban, Gunter; DD 258532 A3 1988
- Caira, Mino R.; Journal of Pharmaceutical Sciences 1998, V87(12), P1608-1614
- Mohamed, Gehad G.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2005, V62A(4-5), P1165-1171
- Lin, Yannan; Journal of Pharmaceutical and Biomedical Analysis 2010, V51(4), P979-984
References
- ^ Jump up to:a b c Drugs.com Drugs.com international listings for piroxicamPage accessed July 3, 2015
- ^ https://www.ema.europa.eu/documents/psusa/piroxicam-list-nationally-authorised-medicinal-products-psusa/00002438/202004_en.pdf
- ^ Jump up to:a b c d e f g Brayfield, A, ed. (14 January 2014). “Piroxicam”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 June 2014.
- ^ “TGA Approved Terminology for Medicines, Section 1 – Chemical Substances” (PDF). Therapeutic Goods Administration, Department of Health and Ageing, Australian Government. July 1999: 97.
- ^ Jump up to:a b c d e Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. pp. 665, 673–674. ISBN 978-0-85711-084-8.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 519. ISBN 9783527607495.
- ^ Jump up to:a b c Lombardino, JG; Lowe, JA 3rd (2004). “The role of the medicinal chemist in drug discovery–then and now”. Nat Rev Drug Discov. 3 (10): 853–62. doi:10.1038/nrd1523. PMID 15459676. S2CID 11225541.. See: [1] Box 1: Discovery of piroxicam (1962–1980)
- ^ “COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) OPINION FOLLOWING AN ARTICLE 31(2) REFERRAL PIROXICAM CONTAINING MEDICINAL PRODUCTS” (PDF). European Medicines Agency. London, UK: European Medicines Agency. 20 September 2007. Retrieved 24 June 2014.
- ^ Jump up to:a b “FDA Warns that Using a Type of Pain and Fever Medication in Second Half of Pregnancy Could Lead to Complications”. U.S. Food and Drug Administration (FDA) (Press release). 15 October 2020. Retrieved 15 October 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “NSAIDs may cause rare kidney problems in unborn babies”. U.S. Food and Drug Administration. 21 July 2017. Retrieved 15 October 2020. This article incorporates text from this source, which is in the public domain.
- ^ Ivanova D, Deneva V, Nedeltcheva D, Kamounah FS, Gergov G, Hansen PE, Kawauchi S, Antonov L (2015). “Tautomeric transformations of piroxicam in solution: a combined experimental and theoretical study”. RSC Advances. 5 (40): 31852–31860. doi:10.1039/c5ra03653d.
- ^ Weintraub M, Jacox RF, Angevine CD, Atwater EC (1977). “Piroxicam (CP 16171) in rheumatoid arthritis: a controlled clinical trial with novel assessment techniques”. Journal of Rheumatology. 4 (4): 393–404. PMID 342691.
Further reading
- Dean L (2019). “Piroxicam Therapy and CYP2C9 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 30742401. Bookshelf ID: NBK537367.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Pronunciation | /paɪˈrɒksɪˌkæm/ |
| Trade names | Feldene, others[1] |
| Other names | Piroksikam, piroxikam |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a684045 |
| Pregnancy category | AU: C |
| Routes of administration | By mouth |
| ATC code | M01AC01 (WHO) M02AA07 (WHO), S01BC06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only)US: ℞-onlyEU: Rx-only [2] |
| Pharmacokinetic data | |
| Protein binding | 99%[3] |
| Metabolism | Liver-mediated hydroxylation and glucuronidation[3] |
| Elimination half-life | 50 hours[3] |
| Excretion | Urine, faeces |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 36322-90-4 |
| PubChem CID | 54676228 |
| IUPHAR/BPS | 7273 |
| DrugBank | DB00554 |
| ChemSpider | 10442653 |
| UNII | 13T4O6VMAM |
| KEGG | D00127 |
| ChEBI | CHEBI:8249 |
| ChEMBL | ChEMBL527 |
| CompTox Dashboard (EPA) | DTXSID5021170 |
| ECHA InfoCard | 100.048.144 |
| Chemical and physical data | |
| Formula | C15H13N3O4S |
| Molar mass | 331.35 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
///////////PIROXICAM

NEW DRUG APPROVALS
ONE TIME
$10.00
PIRACETAM

Piracetam
- ATC:N06BX03
- MW:142.16 g/mol
- CAS-RN:7491-74-9
- InChI Key:GMZVRMREEHBGGF-UHFFFAOYSA-N
- InChI:InChI=1S/C6H10N2O2/c7-5(9)4-8-3-1-2-6(8)10/h1-4H2,(H2,7,9)
- EINECS:231-312-7
- LD50:9200 mg/kg (M, i.v.); 2 g/kg (M, p.o.)
CAS Registry Number: 7491-74-9
CAS Name: 2-Oxo-1-pyrrolidineacetamide
Additional Names: 2-pyrrolidoneacetamide; 2-pyrrolidinoneacetamide; 2-ketopyrrolidine-1-ylacetamide; 1-acetamido-2-pyrrolidinone
Manufacturers’ Codes: UCB-6215
Trademarks: Avigilen (Riemser); Axonyl (Pfizer); Cerebroforte (Azupharma); Encetrop (Alpharma); Gabacet (Sanofi-Synthelabo); Geram (UCB); Nootrop (UCB); Nootropil (UCB); Nootropyl (UCB); Norzetam (UCB); Normabraïn (UCB); Piracebral (Hexal); Piracetrop (Holsten); Sinapsan (Rodleben)Molecular Formula: C6H10N2O2
Molecular Weight: 142.16
Percent Composition: C 50.69%, H 7.09%, N 19.71%, O 22.51%
Literature References: Prepn: H. Morren, NL6509994; eidem,US3459738 (1966, 1969 both to U.C.B.). Pharmacology: Giurgea et al.,Arch. Int. Pharmacodyn. Ther.166, 238 (1967); Giurgea, Moyersoons, ibid.188, 401 (1970); Giurgea et al.,Psychopharmacologia20, 160 (1971). Metabolism and biochemical studies: Gobert, J. Pharm. Belg.27, 281 (1972). Clinical studies: W. J. Oosterveld, Arzneim.-Forsch.30, 1947 (1980); G. Chouinard et al.,Psychopharmacol. Bull.17, 129 (1981); in dyslexia: M. Di Ianni et al.,J. Clin. Psychopharmacol.5, 272 (1985).Properties: Crystals from isopropanol, mp 151.5-152.5°.
Melting point: mp 151.5-152.5°
Therap-Cat: Nootropic.
Keywords: Nootropic.
Piracetam is in the racetams group, with chemical name 2-oxo-1-pyrrolidine acetamide. It is a derivative of the neurotransmitter GABA[5] and shares the same 2-oxo-pyrrolidone base structure with pyroglutamic acid. Piracetam is a cyclic derivative of GABA (gamma-aminobutyric acid). Related drugs include the anticonvulsants levetiracetam and brivaracetam, and the putative nootropics aniracetam and phenylpiracetam.Piracetam is a drug marketed as a treatment for myoclonus[3] and a cognitive enhancer.[4] Evidence to support its use is unclear, with some studies showing modest benefits in specific populations and others showing minimal or no benefit.[5][6] Piracetam is sold as a medication in many European countries. Sale of piracetam is not illegal in the United States, although it is not regulated nor approved by the FDA so it must be marketed as a dietary supplement.[4]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Efficacy
Dementia
A 2001 Cochrane review concluded that there was not enough evidence to support piracetam for dementia or cognitive problems.[6] A 2005 review found some evidence of benefit in older subjects with cognitive impairment.[5] In 2008, a working group of the British Academy of Medical Sciences noted that many of the trials of piracetam for dementia were flawed.[7]
There is no good evidence that piracetam is of benefit in treating vascular dementia.[8]
Depression and anxiety
Some sources suggest that piracetam’s overall effect on lowering depression and anxiety is higher than on improving memory.[9] However, depression is reported to be an occasional adverse effect of piracetam.[10]
Other
Piracetam may facilitate the deformability of erythrocytes in capillary which is useful for cardiovascular disease.[5][3]
Peripheral vascular effects of piracetam have suggested its use potential for vertigo, dyslexia, Raynaud’s phenomenon and sickle cell anemia.[5][3] There is no evidence to support piracetam’s use in sickle cell crisis prevention[11] or for fetal distress during childbirth.[12] There is no evidence for benefit of piracetam with acute ischemic stroke,[13] though there is debate as to its utility during stroke rehabilitation.[14][15]
Anti-vasospasm
Piracetam has been found to diminish erythrocyte adhesion to vascular wall endothelium, making any vasospasm in the capillary less severe. This contributes to its efficacy in promoting microcirculation, including to the brain and kidneys.[5][3]
Side effects
Symptoms of general excitability, including anxiety, insomnia, irritability, headache, agitation, nervousness, tremor, and hyperkinesia, are occasionally reported.[10][16][17] Other reported side effects include somnolence, weight gain, clinical depression, weakness, increased libido, and hypersexuality.[10]
According to a 2005 review, piracetam has been observed to have the following side effects: hyperkinesia, weight gain, nervousness, somnolence, depression and asthenia.[5]
Piracetam reduces platelet aggregation as well as fibrinogen concentration, and thus is contraindicated to patients suffering from cerebral hemorrhage.[5][3]
Toxicity
Piracetam does not appear to be acutely toxic at the doses used in human studies.[6][18][19]
The LD50 for oral consumption in humans has not been determined.[20] The LD50 is 5.6 g/kg for rats and 20 g/kg for mice, indicating extremely low acute toxicity.[21] For comparison, in rats the LD50 of vitamin C is 12 g/kg and the LD50 of table salt is 3 g/kg.
Mechanisms of action
Piracetam’s mechanism of action, as with racetams in general, is not fully understood. The drug influences neuronal and vascular functions and influences cognitive function without acting as a sedative or stimulant.[5] Piracetam is a positive allosteric modulator of the AMPA receptor, although this action is very weak and its clinical effects may not necessarily be mediated by this action.[22] It is hypothesized to act on ion channels or ion carriers, thus leading to increased neuron excitability.[20] GABA brain metabolism and GABA receptors are not affected by piracetam[23]
Piracetam improves the function of the neurotransmitter acetylcholine via muscarinic cholinergic (ACh) receptors[citation needed], which are implicated in memory processes.[24] Furthermore, piracetam may have an effect on NMDA glutamate receptors, which are involved with learning and memory processes. Piracetam is thought to increase cell membrane permeability.[24][25] Piracetam may exert its global effect on brain neurotransmission via modulation of ion channels (i.e., Na+, K+).[20] It has been found to increase oxygen consumption in the brain, apparently in connection to ATP metabolism, and increases the activity of adenylate kinase in rat brains.[26][27] Piracetam, while in the brain, appears to increase the synthesis of cytochrome b5,[28] which is a part of the electron transport mechanism in mitochondria. But in the brain, it also increases the permeability of some intermediates of the Krebs cycle through the mitochondrial outer membrane.[26]
Piracetam inhibits N-type calcium channels. The concentration of piracetam achieved in central nervous system after a typical dose of 1200 mg (about 100 μM)[29] is much higher than the concentration necessary to inhibit N-type calcium channels (IC50 of piracetam in rat neurons was 3 μM).[30]
History
Piracetam was first made some time between the 1950s and 1964 by Corneliu E. Giurgea.[31] There are reports of it being used for epilepsy in the 1950s.[32]
Society and culture
In 2009 piracetam was reportedly popular as a cognitive enhancement drug among students.[33]
Legal status
Piracetam is an uncontrolled substance in the United States meaning it is legal to possess without a license or prescription.[34]
Regulatory status
In the United States, piracetam is not approved by the Food and Drug Administration.[1] Piracetam is not permitted in compounded drugs or dietary supplements in the United States.[35] Nevertheless, it is available in a number of dietary supplements.[4]
In the United Kingdom, piracetam is approved as a prescription drug Prescription Only Medicine (POM) number is PL 20636/2524[36] for adult with myoclonus of cortical origin, irrespective of cause, and should be used in combination with other anti-myoclonic therapies.[37]
In Japan piracetam is approved as a prescription drug.[38]
Piracetam has no DIN in Canada, and thus cannot be sold but can be imported for personal use in Canada.[39]
In Hungary, piracetam was a prescription-only medication, but as of 2020, no prescription is required and piracetam is available as an over-the-counter drug under the name Memoril Mite, and is available in 600 mg pills.

According to the literature reports, the synthetic route of piracetam can be divided into four synthetic methods: α-pyrrolidone method, glycine method, succinic anhydride method and one-step synthesis method:[0009] I. α-pyrrolidone method, 2-pyrrolidone is a lactam, which can react with a strong base (sodium hydride or potassium hydride, sodium methoxide) to generate pyrrolidone metal salt, which can be further combined with halogenated ester or halogen Substitute amide reaction to generate N-alkylated product.[0010] In 1966, a method for preparing piracetam by reacting pyrrolidone and chloroacetamide in 1,4-dioxane with sodium hydrogen as a strong base was reported. The specific synthetic route is shown in Scheme 1:[0011]

[0012] In this process, due to the high price of dioxane, industrial production is still difficult. On the basis of the above process, Xu Yungen used dimethyl sulfoxide as the solvent and sodium methoxide as the acid binding agent to synthesize piracetam in the presence of the phase transfer catalyst benzyltriethylammonium chloride. Due to the difficulty of solvent recovery, the cost of this route is relatively high.[0013] In 1981, Zhou Renxing et al. used sodium methoxide as a strong base to extract methanol in toluene by fractional distillation to convert pyrrolidone into the corresponding sodium salt, and then react with ethyl chloroacetate. The resulting ethyl pyrrolidone ethyl acetate was subjected to ammonolysis. Piracetam can be produced. The specific synthetic route is shown in Scheme 2.[00141

[0015] Because the ammonolysis is carried out in a methanol solution of ammonia, the calculated amount of ethanol generated during the ammonolysis contaminates the methanol solution of ammonia used, which affects the recycling of the methanol solution of ammonia, and is therefore not conducive to process production.[0016] 2. Glycine method, glycine and its derivatives can be used as starting materials for the synthesis of pyroacetamide. Glycine can be prepared by γ-chlorination butylation, amination and cyclization.[0017] According to a British patent report in 1979, glycine trimethylsilyl ester was first condensed with γ-chlorobutyryl chloride, and the corresponding acid chloride was subjected to ammonolysis, and finally cyclized to produce piracetam. The specific synthesis method is as Scheme 3 Shown[0018]

[0019] In this type of synthesis route, some raw materials are not easily available, which restricts industrial production.[0020] 3. Succinic acid method, succinic acid is heated and dehydrated to generate succinic anhydride, succinic anhydride then reacts with glycine to generate an aminolysis product, and the aminolysis product is reduced by sodium tetrafluoroborate, and piracetam can be synthesized by aminolysis , The specific synthetic route is shown in SCheme4. [0021]

[0022] Because sodium tetrafluoroborate is used as a reducing agent, it is expensive, and it is difficult to expand the scale of industrial production. Succinimide generates sodium salt under the action of metal sodium, and its sodium salt reacts with chloroacetamide to generate N-alkylated product. The alkylated product can be electrolytically reduced to obtain piracetam. Since electrolytic reduction is still in the research stage in our country, the production cost of this method is relatively high.[0023] 4. One-step synthesis method, using ethyl 4-chloro-n-butyrate in the presence of sodium bicarbonate, using anhydrous ethanol as a solvent, and glycinamide hydrochloride under heating and refluxing to obtain piracetam in one step, The specific synthetic route is shown in S Cheme5.[0024]

[0025] In this route, glycinamide hydrochloride is very easy to absorb moisture and agglomerate to affect the reaction rate, and the reaction is not easy to control, so it is difficult to achieve industrial production.
SYN

SYN
http://www.cjph.com.cn/EN/abstract/abstract373.shtml
With absolute ethanol as the solvent, ethyl 4-chloro-n-butanoate and glycinamide hydrochloride were refluxed for 20 h in the presence of sodium bicarbonate to obtain central stimulant piracetam. After recrystallization from isopropanol, the yield was about 58% with a purity of 99.6%.
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 79-07-2 | C2H4ClNO | 2-chloroacetamide | Acetamide, 2-chloro- |
| 105-39-5 | C4H7ClO2 | ethyl chloroacetate | Acetic acid, chloro-, ethyl ester |
| 61516-73-2 | C8H13NO3 | ethyl 2-oxo-1-pyrrolidineacetate | 1-Pyrrolidineacetic acid, 2-oxo-, ethyl ester |
| 616-45-5 | C4H7NO | 2-pyrrolidone | 2-Pyrrolidinone |
PATENT
https://patents.google.com/patent/CN104478779A/zh

Example 1[0036] A method for synthesizing piracetam, which includes the following steps:[0037] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 70°C, a methanol solution of sodium methoxide (28.4% (w/w); 114.0 g; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0038] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. When the temperature of the reaction system drops to 60°C, a toluene solution of 58 mL (0.66 mol) of methyl chloroacetate is slowly added dropwise, and the reaction temperature is controlled to 80-100°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain α-pyrrolidone methyl acetate, and measure its content by HPLC (area normalization method). [C18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1 . OmL/min; detection wavelength is 205nm; injection volume is 20yL][0039] Preparation of Piracetam: Put about 130 mL of methanol in a 500 mL three-necked flask, and vent ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 10 h, allowed to cool, filtered with suction, and the filter cake was dried.[0040] The purification of piracetam: 25.50g crude piracetam and 100mL isopropanol were sequentially added in a 500mL three-necked flask, heated to reflux for 40min, activated carbon was added, reflux stirring, hot filtration, and the resulting properties were all white As a powdery solid, the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.85 g of a white solid with a yield of 81.76% (calculated as α-pyrrolidone, the same below).Example 2[0042] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 100°C, a methanol solution of sodium methoxide (28.4% (w/w)); 114. Og; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the addition is complete, add toluene, increase the temperature, and distill at normal pressure until the distillate is completely distilled out, and the reaction is complete.[0043] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. When the temperature of the reaction system drops to 60°C, a mixed solution of 63 mL (0.72 mol) of methyl chloroacetate and 30 mL of toluene is slowly added dropwise, and the reaction temperature is controlled to 80-100°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1 .OmL/ min; detection wavelength is 205nm; injection volume is 20 μL][0044] Preparation of Piracetam: Put about 130 mL of methanol in a 250 mL three-necked flask, and ventilate ammonia to saturation. The obtained ammonia/methanol solution was mixed with 50.0 g of α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 12 hours, allowed to cool, filtered with suction, and the filter cake was dried.[0045] Purification of piracetam: 25.50g crude piracetam and 75mL methanol were sequentially added to a 500mL three-necked flask, heated to reflux for 40min, added activated carbon 0.5g, refluxed for 1h, hot filtered, magnetically stirred Under the conditions, the activated carbon was filtered out, and the properties were all white powdery solids, and the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 21.02g of white solids with a yield of 82.42%.Embodiment 3[0047] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionating column is connected with a thermometer, a condenser and a 1000 mL receiving bottle. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 70°C, a methanol solution of sodium methoxide (28.4% (w/w)); 114. Og; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0048] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. A mixed solution of 79 mL (0.90 mol) of methyl chloroacetate and 50 mL of toluene was slowly added dropwise, and the reaction temperature was controlled to 70-90°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C 18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1.0mL/min; The detection wavelength is 205nm; The injection volume is 20 μL)[0049] Preparation of Piracetam: Put about 130 mL of methanol in a 250 mL three-necked flask, and vent ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 14h, allowed to cool, filtered with suction, and the filter cake was dried.[0050] Purification of piracetam: 25.50g crude piracetam and 125mL ethanol were sequentially added in a 500mL three-necked flask, heated to reflux for 40min, added activated carbon 0.5g, refluxed for 1h, hot filtered, magnetically stirred Activated carbon was filtered off under conditions to obtain white powdery solids in all properties, and the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.24 g of white solids with a yield of 79.37%.Example 4[0052] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 60°C, a methanol solution of sodium methoxide (28.4% (w/w); 114.0 g; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0053] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. A mixed solution of 105 mL (1.20 mol) of methyl chloroacetate and 70 mL of toluene was slowly added dropwise, and the reaction temperature was controlled to be 60~70°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C 18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1.0mL/min; The detection wavelength is 205nm; The injection volume is 20 μL)[0054] Preparation of Piracetam: Put about 130 mL of methanol in a 500 mL three-necked flask, and ventilate ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 16h, allowed to cool, filtered with suction, and the filter cake was dried.[0055] The purification of piracetam: 25.50g crude piracetam and 100mL methanol were sequentially added into a 500mL three-necked flask, heated to reflux for 40min, added activated carbon, refluxed for dissolution, hot filtered, and the properties were all white powders The solid, the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.69 g of a white solid, with a yield of 81. 13%.[0056] Chemical analysis of the white crystals synthesized in each of the foregoing examples, and the obtained physical property values are as follows, thereby confirming that the synthesized product is piracetam.[0057] Melting point: 151.6-152. (TC[0058] ESI-MS m / z: 165. 06 [M + Na] +[0059] 1H-NMR (400MHz, DMS〇-d6, ppm) δ : 7. 38 (s, 1H), 7. 09 (s, 1H), 3. 74 (s, 2H), 3. 36 (t, J =7. 08Hz, 2H), 2. 23 (t, J = 7. 84Hz, 2H), I. 93 (m, 2H).[0060] 13C-NMR(100MHz, DMS0-d6, ppm) δ : 17. 80, 30. 42, 45. 28, 47. 74, 170. 21,174. 90.
PATENTCN110903230A *2019-12-042020-03-24Beijing Yuekang Kechuang Pharmaceutical Technology Co., Ltd.An industrialized preparation method of Pramiracetam sulfate
PATENTCN104478779A2015-04-01New synthetic method of nootropic drug Piracetam
References
- ^ Jump up to:a b “Piracetam”. DrugBank database.
- ^ Leaflet of Piracetam.
- ^ Jump up to:a b c d e “Nootropil Tablets 1200 mg”. (emc). 15 February 2017. Retrieved 14 April 2019.
- ^ Jump up to:a b c Cohen, Pieter A.; Zakharevich, Igor; Gerona, Roy (25 November 2019). “Presence of Piracetam in Cognitive Enhancement Dietary Supplements”. JAMA Internal Medicine. 180 (3): 458–459. doi:10.1001/jamainternmed.2019.5507. PMC 6902196. PMID 31764936.
- ^ Jump up to:a b c d e f g h i Winblad B (2005). “Piracetam: a review of pharmacological properties and clinical uses”. CNS Drug Reviews. 11 (2): 169–82. doi:10.1111/j.1527-3458.2005.tb00268.x. PMC 6741724. PMID 16007238.
- ^ Jump up to:a b c Flicker, L; Grimley Evans, G (2001). “Piracetam for dementia or cognitive impairment”. The Cochrane Database of Systematic Reviews (2): CD001011. doi:10.1002/14651858.CD001011. PMID 11405971.
- ^ Horne G, et al. (May 2008). Brain science, addiction and drugs(PDF) (Report). Academy of Medical Sciences. p. 145. ISBN 978-1-903401-18-7.
- ^ Farooq MU, Min J, Goshgarian C, Gorelick PB (September 2017). “Pharmacotherapy for Vascular Cognitive Impairment”. CNS Drugs(Review). 31 (9): 759–776. doi:10.1007/s40263-017-0459-3. PMID 28786085. S2CID 23271739.
Other medications have been considered or tried for the treatment of VCI or VaD. These include […] piracetam. There is no convincing evidence about the efficacy of these medications in the treatment of VCI.
- ^ Malykh AG, Sadaie MR (February 2010). “Piracetam and piracetam-like drugs: from basic science to novel clinical applications to CNS disorders”. Drugs. 70 (3): 287–312. doi:10.2165/11319230-000000000-00000. PMID 20166767. S2CID 12176745.
- ^ Jump up to:a b c Nootropil®. Arzneimittel-Kompendium der Schweiz. 2013-09-12. Retrieved 2013-10-27.
- ^ Al Hajeri A, Fedorowicz Z (February 2016). “Piracetam for reducing the incidence of painful sickle cell disease crises”. The Cochrane Database of Systematic Reviews. 2: CD006111. doi:10.1002/14651858.CD006111.pub3. PMC 7390168. PMID 26869149.
- ^ Hofmeyr, GJ; Kulier, R (13 June 2012). “Piracetam for fetal distress in labour”. The Cochrane Database of Systematic Reviews (6): CD001064. doi:10.1002/14651858.CD001064.pub2. PMC 7048034. PMID 22696322.
- ^ Ricci S, Celani MG, Cantisani TA, Righetti E (September 2012). “Piracetam for acute ischaemic stroke”. The Cochrane Database of Systematic Reviews (9): CD000419. doi:10.1002/14651858.CD000419.pub3. PMC 7034527. PMID 22972044.
- ^ Zhang J, Wei R, Chen Z, Luo B (July 2016). “Piracetam for Aphasia in Post-stroke Patients: A Systematic Review and Meta-analysis of Randomized Controlled Trials”. CNS Drugs. 30 (7): 575–87. doi:10.1007/s40263-016-0348-1. PMID 27236454. S2CID 22955205.
- ^ Yeo SH, Lim ZI, Mao J, Yau WP (October 2017). “Effects of Central Nervous System Drugs on Recovery After Stroke: A Systematic Review and Meta-Analysis of Randomized Controlled Trials”. Clinical Drug Investigation. 37 (10): 901–928. doi:10.1007/s40261-017-0558-4. PMID 28756557. S2CID 6520934.
- ^ Chouinard G, Annable L, Ross-Chouinard A, Olivier M, Fontaine F (1983). “Piracetam in elderly psychiatric patients with mild diffuse cerebral impairment”. Psychopharmacology. 81 (2): 100–106. doi:10.1007/BF00429000. PMID 6415738. S2CID 32702769.
- ^ Hakkarainen H, Hakamies L (1978). “Piracetam in the treatment of post-concussional syndrome. A double-blind study”. European Neurology. 17 (1): 50–55. doi:10.1159/000114922. PMID 342247.
- ^ Koskiniemi M, Van Vleymen B, Hakamies L, Lamusuo S, Taalas J (March 1998). “Piracetam relieves symptoms in progressive myoclonus epilepsy: a multicentre, randomised, double blind, crossover study comparing the efficacy and safety of three dosages of oral piracetam with placebo”. Journal of Neurology, Neurosurgery, and Psychiatry. 64 (3): 344–348. doi:10.1136/jnnp.64.3.344. PMC 2169975. PMID 9527146.
- ^ Fedi M, Reutens D, Dubeau F, Andermann E, D’Agostino D, Andermann F (May 2001). “Long-term efficacy and safety of piracetam in the treatment of progressive myoclonus epilepsy”. Archives of Neurology. 58 (5): 781–786. doi:10.1001/archneur.58.5.781. PMID 11346373.
- ^ Jump up to:a b c Gouliaev AH, Senning A (May 1994). “Piracetam and other structurally related nootropics”. Brain Research. Brain Research Reviews. 19 (2): 180–222. doi:10.1016/0165-0173(94)90011-6. PMID 8061686. S2CID 18122566.
- ^ “Piracetam Material Safety Sheet” (PDF). Spectrum.
- ^ Ahmed AH, Oswald RE (March 2010). “Piracetam defines a new binding site for allosteric modulators of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors”. Journal of Medicinal Chemistry. 53 (5): 2197–203. doi:10.1021/jm901905j. PMC 2872987. PMID 20163115.
- ^ Giurgea CE (January 1982). “The nootropic concept and its prospective implications”. Drug Development Research. 2 (5): 441–446. doi:10.1002/ddr.430020505. ISSN 1098-2299. S2CID 145059666.
- ^ Jump up to:a b Winnicka K, Tomasiak M, Bielawska A (2005). “Piracetam–an old drug with novel properties?”. Acta Poloniae Pharmaceutica. 62(5): 405–9. PMID 16459490.
- ^ Müller WE, Eckert GP, Eckert A (March 1999). “Piracetam: novelty in a unique mode of action”. Pharmacopsychiatry. 32 (Suppl 1): 2–9. doi:10.1055/s-2007-979230. PMID 10338102.
- ^ Jump up to:a b Grau M, Montero JL, Balasch J (1987). “Effect of Piracetam on electrocorticogram and local cerebral glucose utilization in the rat”. General Pharmacology. 18 (2): 205–11. doi:10.1016/0306-3623(87)90252-7. PMID 3569848.
- ^ Nickolson VJ, Wolthuis OL (October 1976). “Effect of the acquisition-enhancing drug piracetam on rat cerebral energy metabolism. Comparison with naftidrofuryl and methamphetamine”. Biochemical Pharmacology. 25 (20): 2241–4. doi:10.1016/0006-2952(76)90004-6. PMID 985556.
- ^ Tacconi MT, Wurtman RJ (1986). “Piracetam: physiological disposition and mechanism of action”. Advances in Neurology. 43: 675–85. PMID 3946121.
- ^ Yeh HH, Yang YH, Ko JY, Chen SH (July 2006). “Rapid determination of piracetam in human plasma and cerebrospinal fluid by micellar electrokinetic chromatography with sample direct injection”. J Chromatogr A. 1120 (1–2): 27–34. doi:10.1016/j.chroma.2005.11.071. PMID 16343512.
- ^ Bravo-Martínez J, Arenas I, Vivas O, Rebolledo-Antúnez S, Vázquez-García M, Larrazolo A, García DE (October 2012). “A novel CaV2.2 channel inhibition by piracetam in peripheral and central neurons”. Exp Biol Med (Maywood). 237 (10): 1209–18. doi:10.1258/ebm.2012.012128. PMID 23045722.
- ^ Li JJ, Corey EJ (2013). Drug Discovery: Practices, Processes, and Perspectives. John Wiley & Sons. p. 276. ISBN 9781118354469.
- ^ Schmidt D, Shorvon S (2016). The End of Epilepsy?: A History of the Modern Era of Epilepsy Research 1860-2010. Oxford University Press. p. 69. ISBN 9780198725909.
- ^ Medew J (1 October 2009). “Call for testing on ‘smart drugs'”. Fairfax Media. Retrieved 29 May 2014.
- ^ “Erowid Piracetam Vault: Legal Status”.
- ^ Jann Bellamy (26 September 2019). “FDA proposes ban on curcumin and other naturopathic favorites in compounded drugs”. Science-Based Medicine.
- ^http://www.mhra.gov.uk/home/groups/spcpil/documents/spcpil/con1547788739542.pdf
- ^ “Nootropil Tablets 800 mg”. (emc).
- ^ “UCB’s piracetam approved in Japan”. The Pharma Letter. 25 November 1999.
- ^ “Guidance Document on the Import Requirements for Health Products under the Food and Drugs Act and its Regulations (GUI-0084)”. Health Canada / Health Products and Food Branch Inspectorate. 1 June 2010. Retrieved 15 December 2019.
- UCB Pharma Limited (2005). “Nootropil 800 mg & 1200 mg Tablets and Solution”. electronic Medicines Compendium. Datapharm Communications. Archived from the original on 7 December 2006. Retrieved 8 December 2005.
External links
Gouliaev AH, Senning A (May 1994). “Piracetam and other structurally related nootropics”. Brain Research. Brain Research Reviews. 19 (2): 180–222. doi:10.1016/0165-0173(94)90011-6. PMID 8061686. S2CID 18122566.
| Clinical data | |
|---|---|
| Trade names | Breinox, Dinagen, Lucetam, Nootropil, Nootropyl, Oikamid, Piracetam and many others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth, parenteral, or vaporized |
| ATC code | N06BX03 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: UnscheduledUK: POM (Prescription only)US: Unscheduled (Not permitted as drug or supplement[1]) |
| Pharmacokinetic data | |
| Bioavailability | ~100% |
| Onset of action | Swiftly following administration. Food delays time to peak concentration by 1.5 h approximately to 2–3 h since dosing.[2] |
| Elimination half-life | 4–5 h |
| Excretion | Urinary |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 7491-74-9 |
| PubChem CID | 4843 |
| IUPHAR/BPS | 4288 |
| DrugBank | DB09210 |
| ChemSpider | 4677 |
| UNII | ZH516LNZ10 |
| KEGG | D01914 |
| ChEMBL | ChEMBL36715 |
| CompTox Dashboard (EPA) | DTXSID5044491 |
| ECHA InfoCard | 100.028.466 |
| Chemical and physical data | |
| Formula | C6H10N2O2 |
| Molar mass | 142.158 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 152 °C (306 °F) |
| showSMILES | |
| showInChI | |
| (verify) |
///////////UCB 6215, Nootropic, PIRACETAM

NEW DRUG APPROVALS
ONE TIME
$10.00
MEROPENEM

Meropenem
CAS number96036-03-2
IUPAC Name(4R,5S,6S)-3-{[(3S,5S)-5-(dimethylcarbamoyl)pyrrolidin-3-yl]sulfanyl}-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
WeightAverage: 383.463
Monoisotopic: 383.151491615
Chemical FormulaC17H25N3O5S
- Antibiotic SM 7338
- ICI 194660
- SM 7338
CAS Registry Number: 96036-03-2
CAS Name: (4R,5S,6S)-3-[[(3S,5S)-5-[(Dimethylamino)carbonyl]-3-pyrrolidinyl]thio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
Additional Names: (1R,5S,6S)-2-[(3S,5S)-5-(dimethylaminocarbonyl)pyrrolidin-3-ylthio]-6-[(R)-1-hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid
Molecular Formula: C17H25N3O5S
Molecular Weight: 383.46
Percent Composition: C 53.25%, H 6.57%, N 10.96%, O 20.86%, S 8.36%
Literature References: Carbapenem antibiotic. Prepn: M. Sunagawa et al.,EP126587; M. Sunagawa, US4943569 (1984, 1990 both to Sumitomo).
Structure-activity study: M. Sunagawa et al.,J. Antibiot.43, 519 (1990).Crystal structure: K. Yanagi et al.,Acta Crystallogr.C48, 1737 (1992).HPLC determn in serum and bronchial secretions: M. Ehrlich et al., J. Chromatogr. B751, 357 (2001). Pharmacokinetics: R. Wise et al.,Antimicrob. Agents Chemother.34, 1515 (1990).Series of articles on antimicrobial activity, metabolism: J. Antimicrob. Chemother.24, Suppl. A, 1-320 (1989); and clinical performance: ibid.36, Suppl. A, 1-223 (1995).Review of clinical experience in intensive care: M. Hurst, H. M. Lamb, Drugs59, 653-680 (2000).
Derivative Type: Trihydrate
CAS Registry Number: 119478-56-7
Manufacturers’ Codes: ICI-194660; SM-7338
Trademarks: Meronem (AstraZeneca); Meropen (Sumitomo); Merrem (AstraZeneca)
Properties: White to pale yellow crystalline powder. Sparingly sol in water; very slightly sol in hydrated ethanol. Practically insol in acetone, ether.
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Carbapenems.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Meropenem sodium | Not Available | 211238-34-5 | UBQRNADYCUXRBD-NACOAMSHSA-N |
| Meropenem trihydrate | FV9J3JU8B1 | 119478-56-7 | CTUAQTBUVLKNDJ-OBZXMJSBSA-N |
International/Other BrandsAronem (ACI) / Aropen (Aristopharma) / Carbanem (Sanofi-Aventis) / Erope (Lincoln) / Fulspec (Acme) / I-penam (Incepta) / Merenz (Admac) / Merofit (FHC) / Meronem (AstraZeneca) / Meronis (Neiss) / Meropen (Swiss Parenterals) / Merotec (Zuventus) / Merrem I.V. (AstraZeneca) / Monan (AstraZeneca) / Ropenem (Drug International) / Zeropenem (Sanofi-Aventis)
Synthesis Reference
Yoon Seok Song, Sung Woo Park, Yeon Jung Yoon, Hee Kyoon Yoon, Seong Cheol Moon, Byung Goo Lee, Soo Jin Choi, Sun Ah Jun, “METHOD FOR PREPARING MEROPENEM USING ZINC POWDER.” U.S. Patent US20120065392, issued March 15, 2012.
SYN
Carbapenem antibiotic. Prepn: M. Sunagawa et al., EP 126587; M. Sunagawa, US 4943569 (1984, 1990 both to Sumitomo). Structure-activity study: M. Sunagawa et al., J. Antibiot. 43, 519 (1990).

SYN
https://patents.google.com/patent/WO2012062035A1/enCarbapenem, a type of β-lactam antibiotic, is known for its broad spectrum of antibacterial activity and strong antibacterial activity, such as meropenem (Me r0 p e nem), imine South (Imipenem) and Biabenem, etc., play an important role in the cure of severe infections.

Meropenem Imipenem For the synthetic methods of the Peinan type, the previous studies have mainly synthesized the corresponding Peinan side chain compound and the parent nucleus MAP, respectively, and then condensed and removed the protecting group to obtain the Peinan product. Such as US patentsUSP4933333, starting from 4-acetoxyazetidinone (4AA), obtained a matrix MAP after several steps of reaction. The mother nucleus is then condensed and deprotected from the side chain to obtain meropenem. However, this method is cumbersome, the synthesis step is long, and the total yield is low, and the noble metal catalyst is inevitably used in the synthesis of the compound (9).

MAP (10) Meropenem The Chinese invention patent document CN200810142137.5 has introduced a method for synthesizing meropenem.

(XII) (I)(TBD S = Si (CH 3 ) 2 C (CH 3) 3; PNB = p-N0 2 -C 6 H 4 CH 2; PNZ = 2 -C 6 H 4 CH 2 OCO N0 p-) This method of Scheme Short, easy to operate, easy to get raw materials, but there are some areas for improvement.

Example 11) (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy)ethyl]-4-[(2,S, 4’R)- 1- (allyl Synthesis of oxycarbonylxiaodimethylaminocarbonylpyrrolidinothio]-2-azetidinone (II) In a 500 ml reaction flask, add 22.6 g (0.075 mol) of (3S,4S)-3-[( R) l-(tert-Butyldimethylsilyloxy)ethyl]-4-[(R)-1-carbonylethyl]-2-azetidinone (IV), 17.1 g (0.083 mol) Dicyclohexylcarbodiimide (DCC) in 100 ml of acetone and 0.76 g of 4-dimethylaminopyridine (DMAP), 20.3 g (0.078 mol) of (2S, 4R)-2-dimethylamine was added dropwise with stirring. A solution of carbonyl-4-mercapto (i-propoxycarbonyl)pyrrolidine (V) in 125 ml of acetone was reacted at room temperature for 14 hours. Filtration, collecting the filtrate, concentrating, adding 200 ml of toluene thereto, using 200 ml of a 5 % acetic acid solution, 200 ml of a saturated sodium hydrogencarbonate solution and 150 ml of saturation Washed with brine, dried over anhydrous magnesium sulfate and evaporated to dryness <mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj 4-[(2,8, 4, ) small (propoxycarbonyl dimethyl dimethylaminocarbonyl)pyrrolidinyl]-2-azetidinone (II), directly without further treatment Invest in the next step.1H-NMR (400 MHz, CDC 13): </ RTI> <RTIgt; m), 2.816-2.849 (lH, s), 2.935-2.953 (3H, m), 3.027-079 (3H, d), 3.378-3.401 (lH, m), 3.792-3.796 (1H, d), 3.807- 3.953 (lH, m), 4.042-4.160 (3H, m), 4.492-4.570 (2H, m), 4.670-4.739 (lH, m), 5.164-5.295 (1H, m), 5.807-5.921 (lH, m ), 6.214(1H, s). Example 22) (31,48)-3-[(1 )-1-(tert-butyldimethylsilyloxy)ethyl]-4-[(2,8,4,1 )- 1- (allyl Synthesis of oxycarbonyl-1-dimethylaminocarbonylpyrrolidinothio]-1-(zincpropoxyl)-2-azetidinone (III) In a 1000 ml reaction flask, add 34.8 g (0.064) Mol) (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy)ethyl]-4-[(2,S, 4,R)-1-(allyl Oxycarbonyl-1-pyrimidinylcarbonyl)pyrrolidinylthio]-2-azetidinone (11), 15.0 ml of triethylamine and 350 ml of toluene, control temperature below -10 °C, add 18.9 g (0.128 mol) p-nitrobenzyl chloroacetate (VI), heated to 0 ° C (-20 ° 5 ° C can be) reaction l ~ 3h. Then slowly add 250 ml of ice water and stir for 10 min. The layers were static and the organic phase was washed three times with saturated sodium bicarbonate solution, 200 ml each time. Dry over anhydrous magnesium sulfate, filtered, and evaporated to dryness to give white crystals, 4,7g (0.0622mol, yield 97.3%) (3R, 4S)-3-[(R) small (tert-butyldimethylsilyloxy)ethyl ]-4-[(2,S, 4,R)-1-(allyloxycarbonyldimethyldimethylaminocarbonyl)pyrrolidinylsulfur]sodium (sweetoxypropanoyl)-2-azetidinone (III), the product was directly put into the next step without further purification.Mp: 33-34 °C1H-NMR (300 MHz, CDC 13):0.819(9H, s), 1.167(3H, d), 1.188(4H, d), 1.693(5H, s), 1.850-1.926(1H, m), 2.631-2.700(1H, m), 2.941-2.960( 3H,d), 3.029-3.080(3H,d), 3.357-3.433(lH, m), 3.506-3.545(2H, m), 3.918-3.968(1H, m), 4.054-4.123 (2H, m), 4.270-4.291(lH, m), 4.391(lH,s), 4.518-4.568(2H, m), 4.588-4.779(3H, m), 5.178-5.416(3H, m), 5.861-5.982(2H,m ). Example 33) (5R,6S,8R,2’S, 4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl) -1- dimethylaminocarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one- Synthesis of 2-carboxylate ![]() In a 500 ml reaction flask, 40; 7 g (0.0622 mol) of (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy) was added. Ethyl]-4-[(2,S,4,R)-1-(indolyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinylsulfate]small (sweetoxypropanoyl)-2-nitrogen Heterocyclic butanone (III) and 150 ml of toluene, 22 ml of trimethyl phosphite (furrowing lg of hydroquinone) were added under nitrogen. After reacting at 60 ° C for 16 hours, the solvent was evaporated under reduced pressure. It was recrystallized by adding 300 ml of ethyl acetate, and the solid was collected, and vacuum-dried at 40 ° C to obtain 32.8 g (0.0528 mol, yield: 85.0%) (5R, 6S, 8R, 2’S, 4,S)-[(R)- 1-(tert-Butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-ene Propoxycarbonyl ethoxy) small azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (oxime).1H-NMR (300 MHz, CDC 13):0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1 H, m), 2.97-3.11(6H, m ), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m). Example 44) (5R, 6S, 8R, 2, S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyrrolidinyl Synthesis of thio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (Vffl) at room temperature , in a 2000ml reaction flask, add 32.8g (0.0528mol) (5R,6S,8R,2’S,4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl] 3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-indolyloxycarbonylethoxy)-1-azabicyclo[3.2.0 -Hept-2-ene-7-one-2-carboxylate (W), 27.4 ml of acetic acid, 41.3 g of fluorohydrogenamine and 1000 ml of dichloromethane, stirred at room temperature for 48 h. After completion of the reaction, 500 ml of a saturated aqueous solution of sodium hydrogencarbonate was added to the reaction mixture, and the mixture was stirred for 10 minutes, and the methylene chloride layer was separated and dried over anhydrous magnesium sulfate to give a white solid (26.2 g (0.0517 mol, yield 98.0). %) (5R, 6S, 8R, 2’S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyr Rhodium thio] -6-(l-allyloxycarbonylethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (ring The product was directly charged to the next step without further purification.1H-NMR (300 MHz, CDC 13):1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m) ; 4.25(2H, m), 4.47-4.87 (5H, m), 5.15-5.50 (4H, m), 5.94 (2H, m). Example 55) (5R,6S,8R,2,S,4,S)-3-[4-dimethylaminocarbonyl)pyrrolidinyl]-6-(l-hydroxyethyl)-1-aza Synthesis of bicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) (5R, 6S, 8R, 2’S, 4’S) was added. – [(R)-l-(hydroxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-allyloxy Carbonyl ethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (VDI), 21.3 g (0.152 mol) dimethylcyclohexane The ketone and 550 ml of ethyl acetate were heated to 30 ° C, and a solution of 1.0 g (0.865 mmol) of tetratriphenylphosphine palladium in 150 ml of dichloromethane was added dropwise thereto, and the mixture was reacted at room temperature for 3 h under nitrogen atmosphere. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, the aqueous layer was washed with ethyl acetate, and then, 500 ml of tetrahydrofuran was added dropwise with stirring in an ice bath, and the crystals were stirred, and the crystals were collected and dried in vacuo to give pale yellow crystals of 13.4 g (0.0352 md, Yield 68.1%) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl) 1-Azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (I)-Meropectin.IR max KBr cm- 1 : 1755, 1627, 1393, 1252, 1130NMR (D20, 300Hz): 1.25 (3H, d), 1.81-1.96 (1H, m), 2.96 (3H, s), 3.03 (3H, s), 3.14-3.20 (3H, m), 3.31-3.41 (2H, m), 3.62- 3.72 (1H, m), 3.90-4.00 (1H, m), 4.14-4.26 (2H, m), 4.63 (1H, t). Example 6 6) (5R,6S,8R,2’S,4’S)-3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(l-hydroxyethyl)-1-azabicyclo[ Synthesis of 3.2.0]-hept-2-en-7-one-2-carboxylate (I)21.3 g (0.152 mol) of dimethylcyclohexanedione in Example 5 was replaced with 45.1 g (0.155 mol) of tributyltin hydride, and 0.125 g (0.108 mmol) of tetrakistriphenylphosphine palladium was added dropwise, and the other amount was added. And the same method, the obtained 16.2g (0.0426mol, 82.5%) (5R,6S,8R,2’S,4’S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl Sulfur]-6-(l-hydroxyethyl)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (1) ~ meropenem. Example 7 7) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-hydroxyethyl)-1- Synthesis of azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) of (5R, 6S, 8R, 2, S, 4’S)-[(R)-l-(hydroxy)ethyl]-3-[4-(1-allyl was added) Oxycarbonyl-1-ylaminocarbonylcarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)azaabicyclo[3. 2.]-hept-2-ene-7- Ketone-2-carboxylate 01), 6.0 g (0.0387 mol) of N, N-dimethylbarbituric acid and 500 ml of dichloromethane, and 6.0 g (5.2 mmol) of tetratriphenylphosphine was added dropwise thereto. A solution of palladium in 100 ml of dichloromethane was reacted at room temperature for 5 h under nitrogen. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, and the aqueous layer was washed with ethyl acetate. THF was evaporated and evaporated, and the crystals were evaporated, and crystals were collected, and the crystals were dried in vacuo to give 15.7 g (0.0413 mol, yield: 80.1%). 5R, 6S, 8R, 2,S,4,S) – 3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl)-1-azabicyclo [3. 2. 0] -Hept-2-ene-7-keto-2-carboxylic acid trihydrate (I)-Meropectin.
In a 500 ml reaction flask, 40; 7 g (0.0622 mol) of (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy) was added. Ethyl]-4-[(2,S,4,R)-1-(indolyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinylsulfate]small (sweetoxypropanoyl)-2-nitrogen Heterocyclic butanone (III) and 150 ml of toluene, 22 ml of trimethyl phosphite (furrowing lg of hydroquinone) were added under nitrogen. After reacting at 60 ° C for 16 hours, the solvent was evaporated under reduced pressure. It was recrystallized by adding 300 ml of ethyl acetate, and the solid was collected, and vacuum-dried at 40 ° C to obtain 32.8 g (0.0528 mol, yield: 85.0%) (5R, 6S, 8R, 2’S, 4,S)-[(R)- 1-(tert-Butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-ene Propoxycarbonyl ethoxy) small azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (oxime).1H-NMR (300 MHz, CDC 13):0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1 H, m), 2.97-3.11(6H, m ), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m). Example 44) (5R, 6S, 8R, 2, S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyrrolidinyl Synthesis of thio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (Vffl) at room temperature , in a 2000ml reaction flask, add 32.8g (0.0528mol) (5R,6S,8R,2’S,4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl] 3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-indolyloxycarbonylethoxy)-1-azabicyclo[3.2.0 -Hept-2-ene-7-one-2-carboxylate (W), 27.4 ml of acetic acid, 41.3 g of fluorohydrogenamine and 1000 ml of dichloromethane, stirred at room temperature for 48 h. After completion of the reaction, 500 ml of a saturated aqueous solution of sodium hydrogencarbonate was added to the reaction mixture, and the mixture was stirred for 10 minutes, and the methylene chloride layer was separated and dried over anhydrous magnesium sulfate to give a white solid (26.2 g (0.0517 mol, yield 98.0). %) (5R, 6S, 8R, 2’S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyr Rhodium thio] -6-(l-allyloxycarbonylethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (ring The product was directly charged to the next step without further purification.1H-NMR (300 MHz, CDC 13):1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m) ; 4.25(2H, m), 4.47-4.87 (5H, m), 5.15-5.50 (4H, m), 5.94 (2H, m). Example 55) (5R,6S,8R,2,S,4,S)-3-[4-dimethylaminocarbonyl)pyrrolidinyl]-6-(l-hydroxyethyl)-1-aza Synthesis of bicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) (5R, 6S, 8R, 2’S, 4’S) was added. – [(R)-l-(hydroxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-allyloxy Carbonyl ethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (VDI), 21.3 g (0.152 mol) dimethylcyclohexane The ketone and 550 ml of ethyl acetate were heated to 30 ° C, and a solution of 1.0 g (0.865 mmol) of tetratriphenylphosphine palladium in 150 ml of dichloromethane was added dropwise thereto, and the mixture was reacted at room temperature for 3 h under nitrogen atmosphere. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, the aqueous layer was washed with ethyl acetate, and then, 500 ml of tetrahydrofuran was added dropwise with stirring in an ice bath, and the crystals were stirred, and the crystals were collected and dried in vacuo to give pale yellow crystals of 13.4 g (0.0352 md, Yield 68.1%) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl) 1-Azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (I)-Meropectin.IR max KBr cm- 1 : 1755, 1627, 1393, 1252, 1130NMR (D20, 300Hz): 1.25 (3H, d), 1.81-1.96 (1H, m), 2.96 (3H, s), 3.03 (3H, s), 3.14-3.20 (3H, m), 3.31-3.41 (2H, m), 3.62- 3.72 (1H, m), 3.90-4.00 (1H, m), 4.14-4.26 (2H, m), 4.63 (1H, t). Example 6 6) (5R,6S,8R,2’S,4’S)-3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(l-hydroxyethyl)-1-azabicyclo[ Synthesis of 3.2.0]-hept-2-en-7-one-2-carboxylate (I)21.3 g (0.152 mol) of dimethylcyclohexanedione in Example 5 was replaced with 45.1 g (0.155 mol) of tributyltin hydride, and 0.125 g (0.108 mmol) of tetrakistriphenylphosphine palladium was added dropwise, and the other amount was added. And the same method, the obtained 16.2g (0.0426mol, 82.5%) (5R,6S,8R,2’S,4’S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl Sulfur]-6-(l-hydroxyethyl)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (1) ~ meropenem. Example 7 7) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-hydroxyethyl)-1- Synthesis of azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) of (5R, 6S, 8R, 2, S, 4’S)-[(R)-l-(hydroxy)ethyl]-3-[4-(1-allyl was added) Oxycarbonyl-1-ylaminocarbonylcarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)azaabicyclo[3. 2.]-hept-2-ene-7- Ketone-2-carboxylate 01), 6.0 g (0.0387 mol) of N, N-dimethylbarbituric acid and 500 ml of dichloromethane, and 6.0 g (5.2 mmol) of tetratriphenylphosphine was added dropwise thereto. A solution of palladium in 100 ml of dichloromethane was reacted at room temperature for 5 h under nitrogen. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, and the aqueous layer was washed with ethyl acetate. THF was evaporated and evaporated, and the crystals were evaporated, and crystals were collected, and the crystals were dried in vacuo to give 15.7 g (0.0413 mol, yield: 80.1%). 5R, 6S, 8R, 2,S,4,S) – 3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl)-1-azabicyclo [3. 2. 0] -Hept-2-ene-7-keto-2-carboxylic acid trihydrate (I)-Meropectin.
ClaimsHide Dependent
Rights requesta synthetic method of meropenem, characterized in that the specific reaction route of the synthetic method

The reaction steps are as follows:1) The compound of the formula (IV) and the compound of the formula (V) are dissolved in an organic solvent and then subjected to a condensation reaction to obtain a compound of the formula (Π), the reaction time is 2 to 24 hours, and the reaction temperature is 0 to 40 ° C. ;2) The compound of the formula (Π) and the compound of the formula (VI) are dissolved in toluene, ethyl acetate or tetrahydrofuran and reacted with a base to form a compound of the formula (III), and the reaction time is ! ~ 3 hours, the reaction temperature is -20~5 °C;3) The compound of the formula (III) is dissolved in cyclohexanyl, n-glyoxime, n-octyl, toluene or xylene, and a Wittig ring-closing reaction is carried out under the action of an organophosphorus reagent to obtain a compound of the formula (VD), the organophosphorus reagent Is triphenylphosphine, tri-n-butylphosphine, triethyl phosphite or trimethyl phosphite;4) The compound of the formula (VII) is dissolved in methanol, tetrahydrofuran, acetone, n-pentane, n-hexane, diethyl ether, acetonitrile, dichloromethane, chloroform or ethyl acetate to hydrolyze the silyl ether bond under the action of an acid to obtain a formula (W). a compound; the acid is dilute hydrochloric acid, hydrofluoric acid, tetrabutylammonium fluoride, benzyltributylammonium fluoride, hydrofluoric hinge or vinegar The acid, the molar ratio of the acid to the compound of the formula ![]() is 5 to 15: 1; the temperature of the hydrolysis reaction is 0 to 40 ° C, and the reaction time is 8 to 24 hours;5) a compound of the formula
is 5 to 15: 1; the temperature of the hydrolysis reaction is 0 to 40 ° C, and the reaction time is 8 to 24 hours;5) a compound of the formula ![]() dissolved in one or more of methanol, ethanol, tert-butanol, isobutanol, isopropanol, tetrahydrofuran, dioxanthene, acetone, dichloromethane, chloroform and water After the solvent is formed, the allylic group is hydrogenated by a palladium catalyst to obtain the target product (1). The molar ratio of the palladium catalyst to the compound of the formula 1) is 0.0001 to 0.5:1; the reaction temperature is 0 to 40 ° C. , the reaction time is 2~24h.2. A method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (IV) to the compound of the formula (V) is 1.05 to 1.0: 1, the condensing agent and The molar ratio of the compound of the formula (IV) is 1.50 to 1.05:1.The method for synthesizing meropenem according to claim 1 or 2, wherein the condensing agent is a carbodiimide reagent or hydrazine, Ν’-carbonyldiimidazole; and the organic solvent is acetone. , acetonitrile, toluene, tetrahydrofuran, chloroform or dimethylformamide.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (VI) to the compound of the formula (VI) is from 1.5 to 2.5:1, the base and the The molar ratio of the compound of the formula (VI) is from 1.2 to 2:1.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the organophosphorus reagent to the compound of formula (III) in step 3) is 2-8: 1; The reaction temperature is 25 to 100 £ ^, and the reaction time is 10 to 24 hours.The method for synthesizing meropenem according to claim 3, wherein the carbodiimide reagent is dicyclohexylcarbodiimide, diisopropylcarbodiimide or 1-( 3-dimethylaminopropyl)-3-ethylcarbodiimide.7. A method for synthesizing meropenem according to claim 1, wherein the base in step 2) is an inorganic base or an organic base; when it is an inorganic base, it is sodium hydroxide, sodium carbonate or Sodium bicarbonate; when it is an organic base, it is pyridine, triethylamine, diisopropylethylamine or 2,6-lutidine.The method for synthesizing meropenem according to claim 1, wherein the palladium catalyst is palladium acetate, palladium chloride, palladium nitrate, bistriphenylphosphine palladium chloride or tetrakistriphenylphosphine. palladium.9. A method for synthesizing meropenem according to claim 1, wherein the protecting group acceptor in step 5) is morpholine, dimethylcyclohexanedione, tributyltin hydride, N, N-dimethylbarbituric acid, -ethylhexanoic acid or hexanoic acid.
dissolved in one or more of methanol, ethanol, tert-butanol, isobutanol, isopropanol, tetrahydrofuran, dioxanthene, acetone, dichloromethane, chloroform and water After the solvent is formed, the allylic group is hydrogenated by a palladium catalyst to obtain the target product (1). The molar ratio of the palladium catalyst to the compound of the formula 1) is 0.0001 to 0.5:1; the reaction temperature is 0 to 40 ° C. , the reaction time is 2~24h.2. A method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (IV) to the compound of the formula (V) is 1.05 to 1.0: 1, the condensing agent and The molar ratio of the compound of the formula (IV) is 1.50 to 1.05:1.The method for synthesizing meropenem according to claim 1 or 2, wherein the condensing agent is a carbodiimide reagent or hydrazine, Ν’-carbonyldiimidazole; and the organic solvent is acetone. , acetonitrile, toluene, tetrahydrofuran, chloroform or dimethylformamide.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (VI) to the compound of the formula (VI) is from 1.5 to 2.5:1, the base and the The molar ratio of the compound of the formula (VI) is from 1.2 to 2:1.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the organophosphorus reagent to the compound of formula (III) in step 3) is 2-8: 1; The reaction temperature is 25 to 100 £ ^, and the reaction time is 10 to 24 hours.The method for synthesizing meropenem according to claim 3, wherein the carbodiimide reagent is dicyclohexylcarbodiimide, diisopropylcarbodiimide or 1-( 3-dimethylaminopropyl)-3-ethylcarbodiimide.7. A method for synthesizing meropenem according to claim 1, wherein the base in step 2) is an inorganic base or an organic base; when it is an inorganic base, it is sodium hydroxide, sodium carbonate or Sodium bicarbonate; when it is an organic base, it is pyridine, triethylamine, diisopropylethylamine or 2,6-lutidine.The method for synthesizing meropenem according to claim 1, wherein the palladium catalyst is palladium acetate, palladium chloride, palladium nitrate, bistriphenylphosphine palladium chloride or tetrakistriphenylphosphine. palladium.9. A method for synthesizing meropenem according to claim 1, wherein the protecting group acceptor in step 5) is morpholine, dimethylcyclohexanedione, tributyltin hydride, N, N-dimethylbarbituric acid, -ethylhexanoic acid or hexanoic acid.
SYN

Reference: Nadenik, Peter; Storm, Ole; Kremminger, Peter. Meropenem intermediate in crystalline form. WO 2005118586. (Assignee Sandoz AG, Switz)
SYN 2

Reference: Nishino, Keita; Koga, Teruyoshi. Improved process for producing carbapenem compound. WO 2007111328. (Assignee Kaneka Corporation, Japan)
SYN 3

Reference: Manca, Antonio; Monguzzi, Riccardo Ambrogio. Process for synthesizing carbapenem using Raney nickel. EP 2141167. (Assignee ACS Dobfar S.p.A., Italy)
SYN 4

Reference: Tseng, Wei-Hong; Chang, Wen-Hsin; Chang, Chia-Mao; Yeh, Chia-Wei; Kuo, Yuan-Liang. Improved process for the preparation of carbapenem using carbapenem intermediates and recovery of carbapenem. EP 2388261. (Assignee Savior Lifetec Corp., Taiwan)
STR5

Reference: Gnanaprakasam, Andrew; Ganapathy, Veeramani; Syed Ibrahim, Shahul Hameed; Karthikeyan, Murugesan; Sivasamy, Thangavel; Michael, Sekar Jeyaraj; Arulmoli, Thangavel; Das, Gautam Kumar. Preparation of meropenem trihydrate. WO 2012160576. (Assignee Sequent Anti Biotics Private Limited, India)
SYN 6
Reference: Gnanprakasam, Andrew; Ganapathy, Veeramani; Syed Ibrahim, Shahul Hameed; Karthikeyan, Murugesan; Sivasamy, Thangavel; Sekar, Jeyaraj; Arulmoli, Thangavel. Preparation of meropenem trihydrate. IN 2011CH01780. (Assignee Sequent Scientific Limited, India)
SYN7

Reference: Senthikumar, Udayampalayam Palanisamy; Sureshkumar, Kanagaraj; Babu, Kommoju Nagesh; Sudhan, Henry Syril; Kamaraj, Ponraj Pravin; Suresh, Thangaiyan. An improved process for the preparation of carbapenem antibiotic. WO 2013150550. (Assignee Orchid Chemicals & Pharmaceuticals Limited, India)
SYN 8

Reference: Ong, Winston Zapanta; Nowak, Pawel Wojciech; Kim, Jinsoo; Enlow, Elizabeth M.; Bourassa, James; Cu, Yen; Popov, Alexey; Chen, Hongming. Meropenem derivatives and uses thereof. WO 2014144285. (Assignee Kala Pharmaceuticals, Inc., USA)
SYN9

Reference: Cookson, James; McNair, Robert John; Satoskar, Deepak Vasant. Preparation of a carbapenem antibiotic by hydrogenation in the presence of a heterogeneous catalyst. WO 2015145161. (Assignee Johnson Matthey Public Limited Company, UK)
SYN 10

Reference: Gruenewald, Elena; Weidlich, Stephan; Jantke, Ralf. Process for the deprotection of a carbapenem by heterogeneous catalytic hydrogenation with hydrogen in the presence of an organic amine. WO 2018010974. (Assignee Evonik Degussa GmbH, Germany)
SYN 11

Some improvements in total synthesis of meropenem; Hu, Lai-Xing; Liu, Jun; Jin, Jie; Zhongguo Yiyao Gongye Zazhi; Volume 31; Issue 7; Pages 290-292; Journal; 2000
synhttps://www.researchgate.net/figure/Synthesis-of-MRPD-starting-from-meropenem_fig9_283306781

//////////////////////////
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Meropenem is an ultra-broad spectrum injectable antibiotic used to treat a wide variety of infections, including meningitis and pneumonia. It is a beta-lactam and belongs to the subgroup of carbapenem, similar to imipenem and ertapenem. Meropenem was originally developed by Sumitomo Pharmaceuticals. It is marketed outside Japan by AstraZeneca with the brand names Merrem and Meronem. Other brand names include Zwipen (India, Marketed by Nucleus) Mepem (Taiwan) Meropen (Japan, Korea) and Neopenem (NEOMED India) . It gained FDA approval in July 1996. It penetrates well into many tissues and body fluids including the cerebrospinal fluid, bile, heart valves, lung, and peritoneal fluid.
Meropenem, sold under the brandname Merrem among others, is an intravenous β-lactam antibiotic used to treat a variety of bacterial infections.[1] Some of these include meningitis, intra-abdominal infection, pneumonia, sepsis, and anthrax.[1]
Common side effects include nausea, diarrhea, constipation, headache, rash, and pain at the site of injection.[1] Serious side effects include Clostridium difficile infection, seizures, and allergic reactions including anaphylaxis.[1] Those who are allergic to other β-lactam antibiotics are more likely to be allergic to meropenem as well.[1] Use in pregnancy appears to be safe.[1] It is in the carbapenem family of medications.[1] Meropenem usually results in bacterial death through blocking their ability to make a cell wall.[1] It is more resistant to breakdown by β-lactamase producing bacteria.[1]
Meropenem was patented in 1983.[2] It was approved for medical use in the United States in 1996.[1] It is on the World Health Organization’s List of Essential Medicines.[3] The World Health Organization classifies meropenem as critically important for human medicine.[4]
Medical uses
The spectrum of action includes many Gram-positive and Gram-negative bacteria (including Pseudomonas) and anaerobic bacteria. The overall spectrum is similar to that of imipenem, although meropenem is more active against Enterobacteriaceae and less active against Gram-positive bacteria. It works against extended-spectrum β-lactamases, but may be more susceptible to metallo-β-lactamases.[5] Meropenem is frequently given in the treatment of febrile neutropenia. This condition frequently occurs in patients with hematological malignancies and cancer patients receiving anticancer drugs that suppress bone marrow formation. It is approved for complicated skin and skin structure infections, complicated intra-abdominal infections and bacterial meningitis.
In 2017 the FDA granted approval for the combination of meropenem and vaborbactam to treat adults with complicated urinary tract infections.[6]
Administration
Meropenem is administered intravenously as a white crystalline powder to be dissolved in 5% monobasic potassium phosphate solution. Dosing must be adjusted for altered kidney function and for haemofiltration.[7]
As with other ß-lactams antibiotics, the effectiveness of treatment depends on the amount of time during the dosing interval that the meropenem concentration is above the minimum inhibitory concentration for the bacteria causing the infection.[8] For ß-lactams, including meropenem, prolonged intravenous administration is associated with lower mortality than bolus intravenous infusion in persons with whose infections are severe, or caused by bacteria that are less sensitive to meropenem, such as Pseudomonas aeruginosa.[8][9]
Side effects
The most common adverse effects are diarrhea (4.8%), nausea and vomiting (3.6%), injection-site inflammation (2.4%), headache (2.3%), rash (1.9%) and thrombophlebitis (0.9%).[10] Many of these adverse effects were observed in severely ill individuals already taking many medications including vancomycin.[11][12] Meropenem has a reduced potential for seizures in comparison with imipenem. Several cases of severe hypokalemia have been reported.[13][14] Meropenem, like other carbapenems, is a potent inducer of multidrug resistance in bacteria.
Pharmacology
Mechanism of action
Meropenem is bactericidal except against Listeria monocytogenes, where it is bacteriostatic. It inhibits bacterial cell wall synthesis like other β-lactam antibiotics. In contrast to other beta-lactams, it is highly resistant to degradation by β-lactamases or cephalosporinases. In general, resistance arises due to mutations in penicillin-binding proteins, production of metallo-β-lactamases, or resistance to diffusion across the bacterial outer membrane.[10] Unlike imipenem, it is stable to dehydropeptidase-1, so can be given without cilastatin.
In 2016, a synthetic peptide-conjugated PMO (PPMO) was found to inhibit the expression of New Delhi metallo-beta-lactamase, an enzyme that many drug-resistant bacteria use to destroy carbapenems.[15][16]
Society and culture

Meropenem vial
Trade names
| Country | Name | Maker |
|---|---|---|
| India | Inzapenum | Dream India |
| Aurobindo Pharma | ||
| Penmer | Biocon | |
| Meronir | Nirlife | |
| Merowin | Strides Acrolab | |
| Aktimer | Aktimas Biopharmaceuticals | |
| Neopenem | Neomed | |
| Mexopen | Samarth life sciences | |
| Meropenia | SYZA Health Sciences LLP | |
| Ivpenem | Medicorp Pharmaceuticals | |
| Merofit | ||
| Lykapiper | Lyka Labs | |
| Winmero | Parabolic Drugs | |
| Bangladesh | ||
| Meroject | Eskayef Pharmaceuticals Ltd. | |
| Merocon | Beacon Pharmaceuticals | |
| Indonesia | Merofen | Kalbe |
| Brazil | Zylpen | Aspen Pharma |
| Japan, Korea | Meropen | |
| Australia | Merem | |
| Taiwan | Mepem | |
| Germany | Meronem | |
| Nigeria | Zironem | Lyn-Edge Pharmaceuticals |
| US | Meronem | AstraZeneca |
| … | Merosan | Sanbe Farma |
| Merobat | Interbat | |
| Zwipen | ||
| Carbonem | ||
| Ronem | Opsonin Pharma, BD | |
| Neopenem | ||
| Merocon | Continental | |
| Carnem | Laderly Biotech | |
| Penro | Bosch | |
| Meroza | German Remedies | |
| Merotrol | Lupin) | |
| Meromer | Orchid Chemicals | |
| Mepenox | BioChimico | |
| Meromax | Eurofarma | |
| Ropen | Macter | |
| mirage | adwic | |
| Meropex | Apex Pharma Ltd. | |
| Merostarkyl | Hefny Pharma Group[17] |
References
- ^ Jump up to:a b c d e f g h i j “Meropenem”. The American Society of Health-System Pharmacists. Retrieved 8 December 2017.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 497. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). Critically important antimicrobials for human medicine (6th revision ed.). Geneva: World Health Organization. hdl:10665/312266. ISBN 9789241515528.
- ^ AHFS Drug Information (2006 ed.). American Society of Health-System Pharmacists. 2006.
- ^ Commissioner, Office of the (24 March 2020). “Press Announcements – FDA approves new antibacterial drug”. http://www.fda.gov.
- ^ Bilgrami, I; Roberts, JA; Wallis, SC; Thomas, J; Davis, J; Fowler, S; Goldrick, PB; Lipman, J (July 2010). “Meropenem dosing in critically ill patients with sepsis receiving high-volume continuous venovenous hemofiltration” (PDF). Antimicrobial Agents and Chemotherapy. 54 (7): 2974–8. doi:10.1128/AAC.01582-09. PMC 2897321. PMID 20479205.
- ^ Jump up to:a b Yu Z, Pang X, Wu X, Shan C, Jiang S (2018). “Clinical outcomes of prolonged infusion (extended infusion or continuous infusion) versus intermittent bolus of meropenem in severe infection: A meta-analysis”. PLOS ONE. 13 (7): e0201667. Bibcode:2018PLoSO..1301667Y. doi:10.1371/journal.pone.0201667. PMC 6066326. PMID 30059536.
- ^ Vardakas KZ, Voulgaris GL, Maliaros A, Samonis G, Falagas ME (January 2018). “Prolonged versus short-term intravenous infusion of antipseudomonal β-lactams for patients with sepsis: a systematic review and meta-analysis of randomised trials”. Lancet Infect Dis. 18 (1): 108–120. doi:10.1016/S1473-3099(17)30615-1. PMID 29102324.
- ^ Jump up to:a b Mosby’s Drug Consult 2006 (16 ed.). Mosby, Inc. 2006.
- ^ Erden, M; Gulcan, E; Bilen, A; Bilen, Y; Uyanik, A; Keles, M (7 March 2013). “Pancytopenýa and Sepsýs due to Meropenem: A Case Report” (PDF). Tropical Journal of Pharmaceutical Research. 12 (1). doi:10.4314/tjpr.v12i1.21.
- ^ “Meropenem side effects – from FDA reports”. eHealthMe.
- ^ Margolin, L (2004). “Impaired rehabilitation secondary to muscle weakness induced by meropenem”. Clinical Drug Investigation. 24(1): 61–2. doi:10.2165/00044011-200424010-00008. PMID 17516692. S2CID 44484294.
- ^ Bharti, R; Gombar, S; Khanna, AK (2010). “Meropenem in critical care – uncovering the truths behind weaning failure”. Journal of Anaesthesiology Clinical Pharmacology. 26 (1): 99–101.
- ^ “New molecule knocks out superbugs’ immunity to antibiotics”. newatlas.com. 20 January 2017. Retrieved 2017-01-25.
- ^ K., Sully, Erin; L., Geller, Bruce; Lixin, Li; M., Moody, Christina; M., Bailey, Stacey; L., Moore, Amy; Michael, Wong; Patrice, Nordmann; M., Daly, Seth (2016). “Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) restores carbapenem susceptibility to NDM-1-positive pathogens in vitro and in vivo”. Journal of Antimicrobial Chemotherapy. 72 (3): 782–790. doi:10.1093/jac/dkw476. PMC 5890718. PMID 27999041.
- ^ “Hefny Pharma Group”. hefnypharmagroup.info. Retrieved 2018-05-22.
External links
- “Meropenem”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Merrem, others |
| AHFS/Drugs.com | Monograph |
| Pregnancy category | AU: B2 |
| Routes of administration | Intravenous |
| ATC code | J01DH02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | Approximately 2% |
| Elimination half-life | 1 hour |
| Excretion | Renal |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 119478-56-7 |
| PubChem CID | 441130 |
| DrugBank | DB00760 |
| ChemSpider | 389924 |
| UNII | FV9J3JU8B1 |
| KEGG | D02222 |
| ChEBI | CHEBI:43968 |
| ChEMBL | ChEMBL127 |
| PDB ligand | MEM (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID7045526 |
| ECHA InfoCard | 100.169.299 |
| Chemical and physical data | |
| Formula | C17H25N3O5S |
| Molar mass | 383.46 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4888344A *1986-07-301989-12-19Sumitomo Pharmaceuticals Company, LimitedCarbapenem compound in crystalline form, and its production and useCN101348486A *2008-08-292009-01-21深圳市海滨制药有限公司Preparation of meropenemCN101962383A *2010-11-122011-02-02上海巴迪生物医药科技有限公司Synthesis method of meropenemFamily To Family CitationsJPS6475488A *1987-09-171989-03-22Sumitomo PharmaProduction of beta-lactam compound* Cited by examiner, † Cited by third party
Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsCN101962383A *2010-11-122011-02-02上海巴迪生物医药科技有限公司Synthesis method of meropenemCN102250096B *2011-09-052016-04-06江西华邦药业有限公司A kind of preparation method of meropenemCN104072523B *2014-07-142017-10-24上海上药新亚药业有限公司The preparation method of BiapenemCN108191869A *2018-01-222018-06-22重庆天地药业有限责任公司The purification process of Meropenem
PublicationPublication DateTitleEP0007973B11984-02-01Process for the preparation of thienamycin and intermediatesUS4631150A1986-12-23Process for the preparation of penemsWO2012062035A12012-05-18Synthesis method for meropenemWO2010022590A12010-03-04Method for preparation of meropenemUS4443373A1984-04-17Process for the production of antibiotic penemsWO2008035153A22008-03-27Process for the preparation of beta-lactam antibioticEP0167154B11990-01-03Process for preparing 4-acetoxy-3-hydroxyethylazetizin-2-one derivativesKR101059339B12011-08-24Method for preparing carbapenem compound for oral administrationKR100886347B12009-03-03Process for stereoselective preparation of 4-BMA using a chiral auxiliaryUS4841043A1989-06-20Stereoselective synthesis of 1-β-alkyl carbapenem antibiotic intermediatesUS4772683A1988-09-20High percentage beta-yield synthesis of carbapenem intermediatesJP2000344774A2000-12-12Production of carbapenem compoundAU745980B22002-04-11Titanium catalyzed preparation of carbapenem intermediatesUS5700930A1997-12-234-substituted azetidinones as precursors to 2-substituted-3-carboxy carbapenem antibiotics and a method of producing themJP2002338572A2002-11-27Method for producing carbapenemsJP3684339B22005-08-17Method for producing carbapenem compoundsEP0066301B11986-01-22Intermediates for the preparation of thienamycin and process for preparing the sameWO2001053305A12001-07-26Processes for the preparation of carbapenem derivativesAU737502B22001-08-23Preparation of beta-methyl carbapenem intermediatesJP3213734B22001-10-02New β-lactam compoundsJP2004107289A2004-04-08Method for producing vinyl sulfide compoundJPH085853B21996-01-24Lactam compound and its manufacturing methodJPH0827168A1996-01-30Carbapenem intermediate fieldEP0204440A11986-12-10Azetidine derivatives productionWO1994021638A11994-09-29Process for the preparation of condensed carbapeneme derivatives
ApplicationPriority dateFiling dateTitleCN 2010105416652010-11-122010-11-12Synthesis method of meropenemCN201010541665.52010-11-12
Nmrhttps://www.researchgate.net/figure/1HNMR-spectra-of-meropenem-hydrolysis-catalyzed-by-NDM-1-Ecoli-cells-Only-1H-signals-of_fig3_272515470


NMRNMR spectra monitoring meropenem hydrolysis catalyzed by NDM-1. a¹H NMR spectrum of hydrolyzed meropenem recorded before and 6 or 20 min after NDM-1 addition to the reaction system. b Part of a ROESY spectrum of the hydrolysis product. Diagonal and cross peaks are shown in blue and red, respectively. Proton signal assignments are labeled beside the peaks. The chemical shifts of H2, H1, H5, and H10 are highlighted by dashed linesSEEhttps://www.mdpi.com/1420-3049/23/11/2738/htm
Figure 1. FT-IR spectra of unirradiated and irradiated (25 kGy) meropenem.
Figure 2. Raman spectra of unirradiated and irradiated (A-25 kGy) meropenem.
Figure 6. XRPD diffractograms of unirradiated and irradiated (25 kGy) meropenem.
Figure 7. Differential scanning calorimetry (DSC) curves of non-irradiated and irradiated (A-25 kGy, B-400 kGy) meropenem. The arrows indicate the changes in the DSC spectrum after irradiation.
Figure 9. FT-IR spectra of unirradiated and irradiated (400 kGy) meropenem. The arrows indicate the changes in the FT-IR spectrum after irradiation.
Figure 10. Raman spectra of unirradiated and irradiated (400 kGy) meropenem. The arrow indicates the change in the Raman spectrum after irradiation.
//////////////Meropenem, Merrem, intravenous β-lactam antibiotic, bacterial infections, meningitis, intra-abdominal infection, pneumonia, sepsis, anthrax, Antibiotic SM 7338, ICI 194660, SM 7338, ANTIBACTERIALS
[H][C@]1([C@@H](C)O)C(=O)N2C(C(O)=O)=C(S[C@@H]3CN[C@@H](C3)C(=O)N(C)C)[C@H](C)[C@]12[H]

NEW DRUG APPROVALS
ONE TIME
$10.00
RTS,S/AS01, RTS,S Mosquirix
Sequence:
1MMAPDPNANP NANPNANPNA NPNANPNANP NANPNANPNA NPNANPNANP51NANPNANPNA NPNANPNANP NANPNANPNA NPNKNNQGNG QGHNMPNDPN101RNVDENANAN NAVKNNNNEE PSDKHIEQYL KKIKNSISTE WSPCSVTCGN151GIQVRIKPGS ANKPKDELDY ENDIEKKICK MEKCSSVFNV VNSRPVTNME201NITSGFLGPL LVLQAGFFLL TRILTIPQSL DSWWTSLNFL GGSPVCLGQN251SQSPTSNHSP TSCPPICPGY RWMCLRRFII FLFILLLCLI FLLVLLDYQG301MLPVCPLIPG STTTNTGPCK TCTTPAQGNS MFPSCCCTKP TDGNCTCIPI351PSSWAFAKYL WEWASVRFSW LSLLVPFVQW FVGLSPTVWL SAIWMMWYWG401PSLYSIVSPF IPLLPIFFCL WVYI
RTS,S/AS01 (RTS,S)
RTS,S/AS01, Mosquirix
Cas 149121-47-1
203-400-Antigen CS (Plasmodium falciparum strain NF54 reduced), 203-L-methionine-204-L-methionine-205-L-alanine-206-L-proline-207-L-aspartic acid-210-L-alanine-211-L-asparagine-313-L-asparagine-329-L-glutamic acid-330-L-glutamine-333-L-lysine-336-L-lysine-339-L-isoleucine-373-L-glutamic acid-396-L-arginine-397-L-proline-398-L-valine-399-L-threonine-400-L-asparagine-, (400→1′)-protein with antigen (hepatitis B virus subtype adw small surface reduced) (9CI)
Other Names
- Malaria vaccine RTS,S
- Mosquirix
- RTS,S
Protein Sequence
Sequence Length: 424

Graphical depiction of circumsporozoite (CSP) and RTS,S structures. CSP comprises an N-terminal region containing a signal peptide sequence and Region I that binds heparin sulfate proteoglycans and has embedded within it a conserved five amino acid (KLKQP) proteolytic cleavage site sequence; a central region containing four-amino acid (NANP/NVDP) repeats; and a C-terminal region containing Region II [a thrombospondin (TSP)-like domain] and a canonical glycosylphosphatidylinositol (GPI) anchor addition sequence. The region of the CSP included in the RTS,S vaccine includes the last 18 NANP repeats and C-terminus exclusive of the GPI anchor addition sequence. Hepatitis B virus surface antigen (HBsAg) monomers self-assemble into virus-like particles and approximately 25% of the HBsAg monomers in RTS,S are genetically fused to the truncated CSP and serve as protein carriers. The CSP fragment in RTS,S contains three known T-cell epitopes: a highly variable CD4 + T-cell epitope before the TSP-like domain (TH2R), a highly variable CD8 + T-cell epitope within the TSP-like domain (TH3R), and a conserved “universal” CD4 + T cell epitope (CS.T3) at the C-terminus. (Figure courtesy of a recent publication16 and open access,
PATENTWO 2009080715
https://patents.google.com/patent/WO2009080715A2/tr
XAMPLES
Example 1Recipe for component for a single pediatric dose of RTS, S malaria vaccine (2 vial formulation)Component AmountRTS,S 25μgNaCl 2.25mgPhosphate buffer (NaZK2) 1OmMMonothioglycerol 125μgWater for Injection Make volume to 250 μLThe above is prepared by adding RTS, S antigen to a mix of Water for Injection, NaCl 150OmM, phosphate buffer (NaZK2) 50OmM (pH 6.8 when diluted x 50) and an aqueous solution of monothioglycerol at 10%. Finally pH is adjusted to 7.0 ± 0.1.This may be provided as a vial together with a separate vial of adjuvant, for example a liposomal formulation of MPL and QS21Component Amount l,2-di-oleoyl-5/?-glycero-3-phosphocholine (DOPC) 500 μgCholesterol 125 μgMPL 25 μgQS21 25 μgNaCl 2.25mg Phosphate buffer (NaZK2) 1 OmMWater for Injection Make volume to250 μLFor administration the adjuvant formulation is added to the component formulation, for example using a syringe, and then shaken. Then the dose is administered in the usual way. The pH of the final liquid formulation is about 6.6 +/- 0.1.Example IAA final pediatric liquid formulation (1 vial) according to the invention may be prepared according to the following recipe.Component AmountRTS,S 25μgNaCl 4.5mgPhosphate buffer (NaZK2) 1OmMMonothioglycerol 125μg1 ,2-di-oleoyl-5/?-glycero-3-phosphocholine (DOPC) 500 μgCholesterol 125 μgMPL 25 μgQS21 25 μgWater for Injection Make volume to500 μLThe pH of the above liquid formulation is either adjusted to 7.0 +/- 0.1 (which is favorable for antigen stability, but not favorable at all for the MPL stability), or to 6.1 +/- 0.1 (which is favorable for MPL stability, but not favorable at all for RT S, S stability). Therefore this formulation is intended for rapid use after preparation.The above is prepared by adding RTS, S antigen to a mix of Water for Injection, NaCl 150OmM, phosphate buffer (NaZK2) 50OmM (pH 6.8 when diluted x 50) and an aqueous solution of monothioglycerol at 10%. Then a premix of liposomes containing MPL with QS21 is added, and finally pH is adjusted. Example IBA final adult dose (1 vial formulation) for the RTS, S according to the invention may be prepared as follows:Component AmountRTS,S 50μgNaCl 4.5mgPhosphate buffer (NaZK2) 1OmMMonothioglycerol 250μg1 ,2-di-oleoyl-5/?-glycero-3-phosphocholine (DOPC) 1000 μgCholesterol 250 μgMPL 50 μgQS21 50 μgWater for Injection Make volume to500 μLExample 1CExample 1C may prepared by putting Example 1, IA or IB in an amber vial, for example flushed with nitrogen before filing.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
WHO recommends groundbreaking malaria vaccine for children at risk
Historic RTS,S/AS01 recommendation can reinvigorate the fight against malaria6 October 2021https://www.who.int/news/item/06-10-2021-who-recommends-groundbreaking-malaria-vaccine-for-children-at-risk
The World Health Organization (WHO) is recommending widespread use of the RTS,S/AS01 (RTS,S) malaria vaccine among children in sub-Saharan Africa and in other regions with moderate to high P. falciparum malaria transmission. The recommendation is based on results from an ongoing pilot programme in Ghana, Kenya and Malawi that has reached more than 800 000 children since 2019.
“This is a historic moment. The long-awaited malaria vaccine for children is a breakthrough for science, child health and malaria control,” said WHO Director-General Dr Tedros Adhanom Ghebreyesus. “Using this vaccine on top of existing tools to prevent malaria could save tens of thousands of young lives each year.”
Malaria remains a primary cause of childhood illness and death in sub-Saharan Africa. More than 260 000 African children under the age of five die from malaria annually.
In recent years, WHO and its partners have been reporting a stagnation in progress against the deadly disease.
“For centuries, malaria has stalked sub-Saharan Africa, causing immense personal suffering,” said Dr Matshidiso Moeti, WHO Regional Director for Africa. “We have long hoped for an effective malaria vaccine and now for the first time ever, we have such a vaccine recommended for widespread use. Today’s recommendation offers a glimmer of hope for the continent which shoulders the heaviest burden of the disease and we expect many more African children to be protected from malaria and grow into healthy adults.”
WHO recommendation for the RTS,S malaria vaccine
Based on the advice of two WHO global advisory bodies, one for immunization and the other for malaria, the Organization recommends that:
WHO recommends that in the context of comprehensive malaria control the RTS,S/AS01 malaria vaccine be used for the prevention of P. falciparum malaria in children living in regions with moderate to high transmission as defined by WHO. RTS,S/AS01 malaria vaccine should be provided in a schedule of 4 doses in children from 5 months of age for the reduction of malaria disease and burden.
Summary of key findings of the malaria vaccine pilots
Key findings of the pilots informed the recommendation based on data and insights generated from two years of vaccination in child health clinics in the three pilot countries, implemented under the leadership of the Ministries of Health of Ghana, Kenya and Malawi. Findings include:
- Feasible to deliver: Vaccine introduction is feasible, improves health and saves lives, with good and equitable coverage of RTS,S seen through routine immunization systems. This occurred even in the context of the COVID-19 pandemic.
- Reaching the unreached: RTS,S increases equity in access to malaria prevention.
- Data from the pilot programme showed that more than two-thirds of children in the 3 countries who are not sleeping under a bednet are benefitting from the RTS,S vaccine.
- Layering the tools results in over 90% of children benefitting from at least one preventive intervention (insecticide treated bednets or the malaria vaccine).
- Strong safety profile: To date, more than 2.3 million doses of the vaccine have been administered in 3 African countries – the vaccine has a favorable safety profile.
- No negative impact on uptake of bednets, other childhood vaccinations, or health seeking behavior for febrile illness. In areas where the vaccine has been introduced, there has been no decrease in the use of insecticide-treated nets, uptake of other childhood vaccinations or health seeking behavior for febrile illness.
- High impact in real-life childhood vaccination settings: Significant reduction (30%) in deadly severe malaria, even when introduced in areas where insecticide-treated nets are widely used and there is good access to diagnosis and treatment.
- Highly cost-effective: Modelling estimates that the vaccine is cost effective in areas of moderate to high malaria transmission.
Next steps for the WHO-recommended malaria vaccine will include funding decisions from the global health community for broader rollout, and country decision-making on whether to adopt the vaccine as part of national malaria control strategies.
Financial support
Financing for the pilot programme has been mobilized through an unprecedented collaboration among three key global health funding bodies: Gavi, the Vaccine Alliance; the Global Fund to Fight AIDS, Tuberculosis and Malaria; and Unitaid.
Note to editors:
- The malaria vaccine, RTS,S, acts against P. falciparum, the most deadly malaria parasite globally, and the most prevalent in Africa.
- The Malaria Vaccine Implementation Programme is generating evidence and experience on the feasibility, impact and safety of the RTS,S malaria vaccine in real-life, routine settings in selected areas of Ghana, Kenya and Malawi.
- Pilot malaria vaccine introductions are led by the Ministries of Health of Ghana, Kenya and Malawi.
- The pilot programme will continue in the 3 pilot countries to understand the added value of the 4th vaccine dose, and to measure longer-term impact on child deaths.
- The Malaria Vaccine Implementation Programme is coordinated by WHO and supported by in-country and international partners, including PATH, UNICEF and GSK, which is donating up to 10 million doses of the vaccine for the pilot.
- The RTS,S malaria vaccine is the result of 30 years of research and development by GSK and through a partnership with PATH, with support from a network of African research centres.
- The Bill & Melinda Gates Foundation provided catalytic funding for late-stage development of RTS,S between 2001 and 2015.
RTS,S/AS01 (trade name Mosquirix) is a recombinant protein-based malaria vaccine. In October 2021, the vaccine was endorsed by the World Health Organization (WHO) for “broad use” in children, making it the first malaria vaccine candidate, and first vaccine to address parasitic infection, to receive this recommendation.[3][4][5]
The RTS,S vaccine was conceived of and created in the late 1980s by scientists working at SmithKline Beecham Biologicals (now GlaxoSmithKline (GSK) Vaccines) laboratories in Belgium.[6] The vaccine was further developed through a collaboration between GSK and the Walter Reed Army Institute of Research in the U.S. state of Maryland[7] and has been funded in part by the PATH Malaria Vaccine Initiative and the Bill and Melinda Gates Foundation. Its efficacy ranges from 26 to 50% in infants and young children.
Approved for use by the European Medicines Agency (EMA) in July 2015,[1] it is the world’s first licensed malaria vaccine and also the first vaccine licensed for use against a human parasitic disease of any kind.[8] On 23 October 2015, WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) jointly recommended a pilot implementation of the vaccine in Africa.[9] This pilot project for vaccination was launched on 23 April 2019 in Malawi, on 30 April 2019 in Ghana, and on 13 September 2019 in Kenya.[10][11]
Background
Main article: Malaria vaccine
Potential malaria vaccines have been an intense area of research since the 1960s.[12] SPf66 was tested extensively in endemic areas in the 1990s, but clinical trials showed it to be insufficiently effective.[13] Other vaccine candidates, targeting the blood-stage of the malaria parasite’s life cycle, have also been insufficient on their own.[14] Among several potential vaccines under development that target the pre-erythrocytic stage of the disease, RTS,S has shown the most promising results so far.[15]
Approval history
The EMA approved the RTS,S vaccine in July 2015, with a recommendation that it be used in Africa for babies at risk of getting malaria. RTS,S was the world’s first malaria vaccine to get approval for this use.[16][8] Preliminary research suggests that delayed fractional dosing could increase the vaccine’s efficacy up to 86%.[17][18]
On 17 November 2016, WHO announced that the RTS,S vaccine would be rolled out in pilot projects in three countries in sub-Saharan Africa. The pilot program, coordinated by WHO, will assess the extent to which the vaccine’s protective effect shown in advanced clinical trials can be replicated in real-life settings. Specifically, the programme will evaluate the feasibility of delivering the required four doses of the vaccine; the impact of the vaccine on lives saved; and the safety of the vaccine in the context of routine use.[19]
Vaccinations by the ministries of health of Malawi, Ghana, and Kenya began in April and September 2019 and target 360,000 children per year in areas where vaccination would have the highest impact. The results are planned to be used by the World Health Organization to advise about a possible future deployment of the vaccine.[10][11][20] In 2021 it was reported that the vaccine together with other anti-malaria medication when given at the most vulnerable season could reduce deaths and illness from the disease by 70%.[21][22]
Funding
RTS,S has been funded, most recently, by the non-profit PATH Malaria Vaccine Initiative (MVI) and GlaxoSmithKline with funding from the Bill and Melinda Gates Foundation.[23] The RTS,S-based vaccine formulation had previously been demonstrated to be safe, well tolerated, immunogenic, and to potentially confer partial efficacy in both malaria-naive and malaria-experienced adults as well as children.[24]
Components and mechanism
The RTS,S vaccine is based on a protein construct first developed by GlaxoSmithKline in 1986. It was named RTS because it was engineered using genes from the repeat (‘R’) and T-cell epitope (‘T’) of the pre-erythrocytic circumsporozoite protein (CSP) of the Plasmodium falciparum malaria parasite together with a viral surface antigen (‘S’) of the hepatitis B virus (HBsAg).[7] This protein was then mixed with additional HBsAg to improve purification, hence the extra “S”.[7] Together, these two protein components assemble into soluble virus-like particles similar to the outer shell of a hepatitis B virus.[25]
A chemical adjuvant (AS01, specifically AS01E) was added to increase the immune system response.[26] Infection is prevented by inducing humoral and cellular immunity, with high antibody titers, that block the parasite from infecting the liver.[27]
The T-cell epitope of CSP is O-fucosylated in Plasmodium falciparum[28][29] and Plasmodium vivax,[30] while the RTS,S vaccine produced in yeast is not.
References
- ^ Jump up to:a b “Mosquirix H-W-2300”. European Medicines Agency (EMA). Retrieved 4 March 2021.
- ^ “RTS,S Malaria Vaccine: 2019 Partnership Award Honoree”. YouTube. Global Health Technologies Coalition. Retrieved 6 October 2021.
- ^ Davies L (6 October 2021). “WHO endorses use of world’s first malaria vaccine in Africa”. The Guardian. Retrieved 6 October2021.
- ^ Drysdale C, Kelleher K. “WHO recommends groundbreaking malaria vaccine for children at risk” (Press release). Geneva: World Health Organization. Retrieved 6 October 2021.
- ^ Mandavilli A (6 October 2021). “A ‘Historical Event’: First Malaria Vaccine Approved by W.H.O.” New York Times. Retrieved 6 October 2021.
- ^ “HYBRID PROTEIN BETWEEN CS FROM PLASMODIUM AND HBsAG”.
- ^ Jump up to:a b c Heppner DG, Kester KE, Ockenhouse CF, Tornieporth N, Ofori O, Lyon JA, et al. (March 2005). “Towards an RTS,S-based, multi-stage, multi-antigen vaccine against falciparum malaria: progress at the Walter Reed Army Institute of Research”. Vaccine. 23 (17–18): 2243–50. doi:10.1016/j.vaccine.2005.01.142. PMID 15755604. Archived from the original on 23 July 2018.
- ^ Jump up to:a b Walsh F (24 July 2015). “Malaria vaccine gets ‘green light'”. BBC News. Archived from the original on 21 July 2020. Retrieved 25 July 2015.
- ^ Stewart S (23 October 2015). “Pilot implementation of first malaria vaccine recommended by WHO advisory groups” (Press release). Geneva: World Health Organization. Archived from the original on 19 September 2021.
- ^ Jump up to:a b Alonso P (19 June 2019). “Letter to partners – June 2019”(Press release). Wuxi: World Health Organization. Retrieved 22 October 2019.
- ^ Jump up to:a b “Malaria vaccine launched in Kenya: Kenya joins Ghana and Malawi to roll out landmark vaccine in pilot introduction” (Press release). Homa Bay: World Health Organization. 13 September 2019. Retrieved 22 October 2019.
- ^ Hill AV (October 2011). “Vaccines against malaria”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 366 (1579): 2806–14. doi:10.1098/rstb.2011.0091. PMC 3146776. PMID 21893544.
- ^ Graves P, Gelband H (April 2006). Graves PM (ed.). “Vaccines for preventing malaria (SPf66)”. The Cochrane Database of Systematic Reviews (2): CD005966. doi:10.1002/14651858.CD005966. PMC 6532709. PMID 16625647.
- ^ Graves P, Gelband H (October 2006). Graves PM (ed.). “Vaccines for preventing malaria (blood-stage)”. The Cochrane Database of Systematic Reviews (4): CD006199. doi:10.1002/14651858.CD006199. PMC 6532641. PMID 17054281.
- ^ Graves P, Gelband H (October 2006). Graves PM (ed.). “Vaccines for preventing malaria (pre-erythrocytic)”. The Cochrane Database of Systematic Reviews (4): CD006198. doi:10.1002/14651858.CD006198. PMC 6532586. PMID 17054280.
- ^ “First malaria vaccine receives positive scientific opinion from EMA”. European Medicines Agency. 24 July 2015. Retrieved 24 July 2015.
- ^ Birkett A (16 September 2016). “A vaccine for malaria elimination?”. PATH.
- ^ Regules JA, Cicatelli SB, Bennett JW, Paolino KM, Twomey PS, Moon JE, et al. (September 2016). “Fractional Third and Fourth Dose of RTS,S/AS01 Malaria Candidate Vaccine: A Phase 2a Controlled Human Malaria Parasite Infection and Immunogenicity Study”. The Journal of Infectious Diseases. 214 (5): 762–71. doi:10.1093/infdis/jiw237. PMID 27296848.
- ^ “Malaria: The malaria vaccine implementation programme (MVIP)”.
- ^ “WHO | MVIP countries: Ghana, Kenya and Malawi”.
- ^ Chandramohan D, Zongo I, Sagara I, Cairns M, Yerbanga RS, Diarra M, et al. (September 2021). “Seasonal Malaria Vaccination with or without Seasonal Malaria Chemoprevention”. The New England Journal of Medicine. 385 (11): 1005–1017. doi:10.1056/NEJMoa2026330. PMID 34432975.
- ^ Roxby P (26 August 2021). “Trial suggests malaria sickness could be cut by 70%”. BBC News. Archived from the original on 3 October 2021. Retrieved 26 August 2021.
- ^ Stein R (18 October 2011). “Experimental malaria vaccine protects many children, study shows”. Washington Post.
- ^ Regules JA, Cummings JF, Ockenhouse CF (May 2011). “The RTS,S vaccine candidate for malaria”. Expert Review of Vaccines. 10 (5): 589–99. doi:10.1586/erv.11.57. PMID 21604980. S2CID 20443829.
- ^ Rutgers T, Gordon D, Gathoye AM, Hollingdale M, Hockmeyer W, Rosenberg M, De Wilde M (September 1988). “Hepatitis B Surface Antigen as Carrier Matrix for the Repetitive Epitope of the Circumsporozoite Protein of Plasmodium Falciparum”. Nature Biotechnology. 6 (9): 1065–1070. doi:10.1038/nbt0988-1065. S2CID 39880644.
- ^ RTS,S Clinical Trials Partnership (July 2015). “Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial”. Lancet. 386 (9988): 31–45. doi:10.1016/S0140-6736(15)60721-8. PMC 5626001. PMID 25913272.
- ^ Foquet L, Hermsen CC, van Gemert GJ, Van Braeckel E, Weening KE, Sauerwein R, et al. (January 2014). “Vaccine-induced monoclonal antibodies targeting circumsporozoite protein prevent Plasmodium falciparum infection”. The Journal of Clinical Investigation. 124 (1): 140–4. doi:10.1172/JCI70349. PMC 3871238. PMID 24292709.
- ^ Swearingen KE, Lindner SE, Shi L, Shears MJ, Harupa A, Hopp CS, et al. (April 2016). “Interrogating the Plasmodium Sporozoite Surface: Identification of Surface-Exposed Proteins and Demonstration of Glycosylation on CSP and TRAP by Mass Spectrometry-Based Proteomics”. PLOS Pathogens. 12 (4): e1005606. doi:10.1371/journal.ppat.1005606. PMC 4851412. PMID 27128092.
- ^ Lopaticki S, Yang AS, John A, Scott NE, Lingford JP, O’Neill MT, et al. (September 2017). “Protein O-fucosylation in Plasmodium falciparum ensures efficient infection of mosquito and vertebrate hosts”. Nature Communications. 8 (1): 561. Bibcode:2017NatCo…8..561L. doi:10.1038/s41467-017-00571-y. PMC 5601480. PMID 28916755.
- ^ Swearingen KE, Lindner SE, Flannery EL, Vaughan AM, Morrison RD, Patrapuvich R, et al. (July 2017). “Proteogenomic analysis of the total and surface-exposed proteomes of Plasmodium vivax salivary gland sporozoites”. PLOS Neglected Tropical Diseases. 11 (7): e0005791. doi:10.1371/journal.pntd.0005791. PMC 5552340. PMID 28759593.
Further reading
- Wilby KJ, Lau TT, Gilchrist SE, Ensom MH (March 2012). “Mosquirix (RTS,S): a novel vaccine for the prevention of Plasmodium falciparum malaria”. The Annals of Pharmacotherapy. 46 (3): 384–93. doi:10.1345/aph.1Q634. PMID 22408046.
- Asante KP, Abdulla S, Agnandji S, Lyimo J, Vekemans J, Soulanoudjingar S, et al. (October 2011). “Safety and efficacy of the RTS,S/AS01E candidate malaria vaccine given with expanded-programme-on-immunisation vaccines: 19 month follow-up of a randomised, open-label, phase 2 trial”. The Lancet. Infectious Diseases. 11 (10): 741–9. doi:10.1016/S1473-3099(11)70100-1. PMID 21782519.
External links
| Vaccine description | |
|---|---|
| Target | P. falciparum; to a lesser extent Hepatitis B |
| Vaccine type | Protein subunit |
| Clinical data | |
| Trade names | Mosquirix |
| Routes of administration | intramuscular injection (0.5 mL)[1] |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |

A poster advertising trials of the RTS,S vaccine[2]
A malaria vaccine is a vaccine that is used to prevent malaria. The only approved vaccine as of 2021, is RTS,S, known by the brand name Mosquirix.[1] It requires four injections.[1]
Research continues with other malaria vaccines. The most effective malaria vaccine is R21/Matrix-M, with a 77% efficacy rate shown in initial trials, and significantly higher antibody levels than with the RTS,S vaccine.[2] It is the first vaccine that meets the World Health Organization‘s (WHO) goal of a malaria vaccine with at least 75% efficacy.[3][2]
Approved vaccines
RTS,S
Main article: RTS,S
RTS,S (developed by PATH Malaria Vaccine Initiative (MVI) and GlaxoSmithKline (GSK) with support from the Bill and Melinda Gates Foundation) is the most recently developed recombinant vaccine. It consists of the P. falciparum circumsporozoite protein (CSP) from the pre-erythrocytic stage. The CSP antigen causes the production of antibodies capable of preventing the invasion of hepatocytes and additionally elicits a cellular response enabling the destruction of infected hepatocytes. The CSP vaccine presented problems in the trial stage, due to its poor immunogenicity. RTS,S attempted to avoid these by fusing the protein with a surface antigen from hepatitis B, hence creating a more potent and immunogenic vaccine. When tested in trials an emulsion of oil in water and the added adjuvants of monophosphoryl A and QS21 (SBAS2), the vaccine gave protective immunity to 7 out of 8 volunteers when challenged with P. falciparum.[4]
RTS,S/AS01 (commercial name Mosquirix),[5] was engineered using genes from the outer protein of P. falciparum malaria parasite and a portion of a hepatitis B virus plus a chemical adjuvant to boost the immune response. Infection is prevented by inducing high antibody titers that block the parasite from infecting the liver.[6] In November 2012, a Phase III trial of RTS,S found that it provided modest protection against both clinical and severe malaria in young infants.[7]
As of October 2013, preliminary results of a Phase III clinical trial indicated that RTS,S/AS01 reduced the number of cases among young children by almost 50 percent and among infants by around 25 percent. The study ended in 2014. The effects of a booster dose were positive, even though overall efficacy seems to wane with time. After four years reductions were 36 percent for children who received three shots and a booster dose. Missing the booster dose reduced the efficacy against severe malaria to a negligible effect. The vaccine was shown to be less effective for infants. Three doses of vaccine plus a booster reduced the risk of clinical episodes by 26 percent over three years, but offered no significant protection against severe malaria.[8]
In a bid to accommodate a larger group and guarantee a sustained availability for the general public, GSK applied for a marketing license with the European Medicines Agency (EMA) in July 2014.[9] GSK treated the project as a non-profit initiative, with most funding coming from the Gates Foundation, a major contributor to malaria eradication.[10]
On 24 July 2015, Mosquirix received a positive opinion from the European Medicines Agency (EMA) on the proposal for the vaccine to be used to vaccinate children aged 6 weeks to 17 months outside the European Union.[11][12][1] A pilot project for vaccination was launched on 23 April 2019, in Malawi, on 30 April 2019, in Ghana, and on 13 September 2019, in Kenya.[13][14]
In October 2021, the vaccine was endorsed by the World Health Organization for “broad use” in children, making it the first malaria vaccine to receive this recommendation.[15][16][17]
Agents under development
A completely effective vaccine is not available for malaria, although several vaccines are under development. Multiple vaccine candidates targeting the blood-stage of the parasite’s life cycle have been insufficient on their own.[18] Several potential vaccines targeting the pre-erythrocytic stage are being developed, with RTS,S the only approved option so far.[19][7]
R21/Matrix-M
The most effective malaria vaccine is R21/Matrix-M, with 77% efficacy shown in initial trials. It is the first vaccine that meets the World Health Organization’s goal of a malaria vaccine with at least 75% efficacy.[3] It was developed through a collaboration involving the University of Oxford, the Kenya Medical Research Institute, the London School of Hygiene & Tropical Medicine, Novavax, the Serum Institute of India, and the Institut de Recherche en Sciences de la Santé in Nanoro, Burkina Faso. The R21 vaccine uses a circumsporozoite protein (CSP) antigen, at a higher proportion than the RTS,S vaccine. It includes the Matrix-M adjuvant that is also utilized in the Novavax COVID-19 vaccine.[20]
A Phase II trial was reported in April 2021, with a vaccine efficacy of 77% and antibody levels significantly higher than with the RTS,S vaccine. A Phase III trial is planned with 4,800 children across four African countries. If the vaccine is approved, over 200 million doses can be manufactured annually by the Serum Institute of India.[2]
Nanoparticle enhancement of RTS,S
In 2015, researchers used a repetitive antigen display technology to engineer a nanoparticle that displayed malaria specific B cell and T cell epitopes. The particle exhibited icosahedral symmetry and carried on its surface up to 60 copies of the RTS,S protein. The researchers claimed that the density of the protein was much higher than the 14% of the GSK vaccine.[21][22]
PfSPZ vaccine
Main article: PfSPZ Vaccine
The PfSPZ vaccine is a candidate malaria vaccine developed by Sanaria using radiation-attenuated sporozoites to elicit an immune response. Clinical trials have been promising, with trials taking place in Africa, Europe, and the US protecting over 80% of volunteers.[23] It has been subject to some criticism regarding the ultimate feasibility of large-scale production and delivery in Africa, since it must be stored in liquid nitrogen.
The PfSPZ vaccine candidate was granted fast track designation by the U.S. Food and Drug Administration in September 2016.[24]
In April 2019, a phase 3 trial in Bioko was announced, scheduled to start in early 2020.[25]
saRNA vaccine against PMIF
A patent was published in February 2021 for a Self-amplifying RNA (saRNA) vaccine that targets the protein PMIF, which is produced by the plasmodium parasite to inhibit the body’s T-cell response. The vaccine has been tested in mice and is described as, “probably the highest level of protection that has been seen in a mouse model” according to Richard Bucala, co-inventor of the vaccine. There are plans for phase one tests in humans later in 2021.[26]
Other developments
- SPf66 is a synthetic peptide based vaccine developed by Manuel Elkin Patarroyo team in Colombia, and was tested extensively in endemic areas in the 1990s. Clinical trials showed it to be insufficiently effective, with 28% efficacy in South America and minimal or no efficacy in Africa.[27]
- The CSP (Circum-Sporozoite Protein) was a vaccine developed that initially appeared promising enough to undergo trials. It is also based on the circumsporozoite protein, but additionally has the recombinant (Asn-Ala-Pro15Asn-Val-Asp-Pro)2-Leu-Arg(R32LR) protein covalently bound to a purified Pseudomonas aeruginosa toxin (A9). However at an early stage a complete lack of protective immunity was demonstrated in those inoculated. The study group used in Kenya had an 82% incidence of parasitaemia whilst the control group only had an 89% incidence. The vaccine intended to cause an increased T-lymphocyte response in those exposed, this was also not observed.[citation needed]
- The NYVAC-Pf7 multi-stage vaccine attempted to use different technology, incorporating seven P.falciparum antigenic genes. These came from a variety of stages during the life cycle. CSP and sporozoite surface protein 2 (called PfSSP2) were derived from the sporozoite phase. The liver stage antigen 1 (LSA1), three from the erythrocytic stage (merozoite surface protein 1, serine repeat antigen and AMA-1) and one sexual stage antigen (the 25-kDa Pfs25) were included. This was first investigated using Rhesus monkeys and produced encouraging results: 4 out of the 7 antigens produced specific antibody responses (CSP, PfSSP2, MSP1 and PFs25). Later trials in humans, despite demonstrating cellular immune responses in over 90% of the subjects, had very poor antibody responses. Despite this following administration of the vaccine some candidates had complete protection when challenged with P.falciparum. This result has warranted ongoing trials.[citation needed]
- In 1995 a field trial involving [NANP]19-5.1 proved to be very successful. Out of 194 children vaccinated none developed symptomatic malaria in the 12-week follow up period and only 8 failed to have higher levels of antibody present. The vaccine consists of the schizont export protein (5.1) and 19 repeats of the sporozoite surface protein [NANP]. Limitations of the technology exist as it contains only 20% peptide and has low levels of immunogenicity. It also does not contain any immunodominant T-cell epitopes.[28]
- A chemical compound undergoing trials for treatment of tuberculosis and cancer—the JmJc inhibitor ML324 and the antitubercular clinical candidate SQ109—is potentially a new line of drugs to treat malaria and kill the parasite in its infectious stage. More tests still need to be carried out before the compounds would be approved as a viable treatment.[29]
Considerations
The task of developing a preventive vaccine for malaria is a complex process. There are a number of considerations to be made concerning what strategy a potential vaccine should adopt.
Parasite diversity
P. falciparum has demonstrated the capability, through the development of multiple drug-resistant parasites, for evolutionary change. The Plasmodium species has a very high rate of replication, much higher than that actually needed to ensure transmission in the parasite’s life cycle. This enables pharmaceutical treatments that are effective at reducing the reproduction rate, but not halting it, to exert a high selection pressure, thus favoring the development of resistance. The process of evolutionary change is one of the key considerations necessary when considering potential vaccine candidates. The development of resistance could cause a significant reduction in efficacy of any potential vaccine thus rendering useless a carefully developed and effective treatment.[30]
Choosing to address the symptom or the source
The parasite induces two main response types from the human immune system. These are anti-parasitic immunity and anti-toxic immunity.
- “Anti-parasitic immunity” addresses the source; it consists of an antibody response (humoral immunity) and a cell-mediated immune response. Ideally a vaccine would enable the development of anti-plasmodial antibodies in addition to generating an elevated cell-mediated response. Potential antigens against which a vaccine could be targeted will be discussed in greater depth later. Antibodies are part of the specific immune response. They exert their effect by activating the complement cascade, stimulating phagocytic cells into endocytosis through adhesion to an external surface of the antigenic substances, thus ‘marking’ it as offensive. Humoral or cell-mediated immunity consists of many interlinking mechanisms that essentially aim to prevent infection entering the body (through external barriers or hostile internal environments) and then kill any micro-organisms or foreign particles that succeed in penetration. The cell-mediated component consists of many white blood cells (such as monocytes, neutrophils, macrophages, lymphocytes, basophils, mast cells, natural killer cells, and eosinophils) that target foreign bodies by a variety of different mechanisms. In the case of malaria both systems would be targeted to attempt to increase the potential response generated, thus ensuring the maximum chance of preventing disease.[citation needed]
- “Anti-toxic immunity” addresses the symptoms; it refers to the suppression of the immune response associated with the production of factors that either induce symptoms or reduce the effect that any toxic by-products (of micro-organism presence) have on the development of disease. For example, it has been shown that Tumor necrosis factor-alpha has a central role in generating the symptoms experienced in severe P. falciparum malaria. Thus a therapeutic vaccine could target the production of TNF-a, preventing respiratory distress and cerebral symptoms. This approach has serious limitations as it would not reduce the parasitic load; rather it only reduces the associated pathology. As a result, there are substantial difficulties in evaluating efficacy in human trials.
Taking this information into consideration an ideal vaccine candidate would attempt to generate a more substantial cell-mediated and antibody response on parasite presentation. This would have the benefit of increasing the rate of parasite clearance, thus reducing the experienced symptoms and providing a level of consistent future immunity against the parasite.
Potential targets
See also: PfSPZ Vaccine
| Parasite stage | Target |
|---|---|
| Sporozoite | Hepatocyte invasion; direct anti-sporozite |
| Hepatozoite | Direct anti-hepatozoite. |
| Asexual erythrocytic | Anti-host erythrocyte, antibodies blocking invasion; anti receptor ligand, anti-soluble toxin |
| Gametocytes | Anti-gametocyte. Anti-host erythrocyte, antibodies blocking fertilisation, antibodies blocking egress from the mosquito midgut. |
By their very nature, protozoa are more complex organisms than bacteria and viruses, with more complicated structures and life cycles. This presents problems in vaccine development but also increases the number of potential targets for a vaccine. These have been summarised into the life cycle stage and the antibodies that could potentially elicit an immune response.
The epidemiology of malaria varies enormously across the globe, and has led to the belief that it may be necessary to adopt very different vaccine development strategies to target the different populations. A Type 1 vaccine is suggested for those exposed mostly to P. falciparum malaria in sub-Saharan Africa, with the primary objective to reduce the number of severe malaria cases and deaths in infants and children exposed to high transmission rates. The Type 2 vaccine could be thought of as a ‘travellers’ vaccine’, aiming to prevent all cases of clinical symptoms in individuals with no previous exposure. This is another major public health problem, with malaria presenting as one of the most substantial threats to travellers’ health. Problems with the available pharmaceutical therapies include costs, availability, adverse effects and contraindications, inconvenience and compliance, many of which would be reduced or eliminated entirely if an effective (greater than 85–90%) vaccine was developed.[citation needed]
The life cycle of the malaria parasite is particularly complex, presenting initial developmental problems. Despite the huge number of vaccines available, there are none that target parasitic infections. The distinct developmental stages involved in the life cycle present numerous opportunities for targeting antigens, thus potentially eliciting an immune response. Theoretically, each developmental stage could have a vaccine developed specifically to target the parasite. Moreover, any vaccine produced would ideally have the ability to be of therapeutic value as well as preventing further transmission and is likely to consist of a combination of antigens from different phases of the parasite’s development. More than 30 of these antigens are being researched[when?] by teams all over the world in the hope of identifying a combination that can elicit immunity in the inoculated individual. Some of the approaches involve surface expression of the antigen, inhibitory effects of specific antibodies on the life cycle and the protective effects through immunization or passive transfer of antibodies between an immune and a non-immune host. The majority of research into malarial vaccines has focused on the Plasmodium falciparum strain due to the high mortality caused by the parasite and the ease of a carrying out in vitro/in vivo studies. The earliest vaccines attempted to use the parasitic circumsporozoite protein (CSP). This is the most dominant surface antigen of the initial pre-erythrocytic phase. However, problems were encountered due to low efficacy, reactogenicity and low immunogenicity.[citation needed]
- The initial stage in the life cycle, following inoculation, is a relatively short “pre-erythrocytic” or “hepatic” phase. A vaccine at this stage must have the ability to protect against sporozoites invading and possibly inhibiting the development of parasites in the hepatocytes (through inducing cytotoxic T-lymphocytes that can destroy the infected liver cells). However, if any sporozoites evaded the immune system they would then have the potential to be symptomatic and cause the clinical disease.
- The second phase of the life cycle is the “erythrocytic” or blood phase. A vaccine here could prevent merozoite multiplication or the invasion of red blood cells. This approach is complicated by the lack of MHC molecule expression on the surface of erythrocytes. Instead, malarial antigens are expressed, and it is this towards which the antibodies could potentially be directed. Another approach would be to attempt to block the process of erythrocyte adherence to blood vessel walls. It is thought that this process is accountable for much of the clinical syndrome associated with malarial infection; therefore a vaccine given during this stage would be therapeutic and hence administered during clinical episodes to prevent further deterioration.
- The last phase of the life cycle that has the potential to be targeted by a vaccine is the “sexual stage”. This would not give any protective benefits to the individual inoculated but would prevent further transmission of the parasite by preventing the gametocytes from producing multiple sporozoites in the gut wall of the mosquito. It therefore would be used as part of a policy directed at eliminating the parasite from areas of low prevalence or to prevent the development and spread of vaccine-resistant parasites. This type of transmission-blocking vaccine is potentially very important. The evolution of resistance in the malaria parasite occurs very quickly, potentially making any vaccine redundant within a few generations. This approach to the prevention of spread is therefore essential.
- Another approach is to target the protein kinases, which are present during the entire lifecycle of the malaria parasite. Research is underway on this, yet production of an actual vaccine targeting these protein kinases may still take a long time.[31]
- Report of a vaccine candidate capable to neutralize all tested strains of Plasmodium falciparum, the most deadly form of the parasite causing malaria, was published in Nature Communications by a team of scientists from the University of Oxford in 2011.[32] The viral vector vaccine, targeting a full-length P. falciparum reticulocyte-binding protein homologue 5 (PfRH5) was found to induce an antibody response in an animal model. The results of this new vaccine confirmed the utility of a key discovery reported from scientists at the Wellcome Trust Sanger Institute, published in Nature.[33] The earlier publication reported P. falciparum relies on a red blood cell surface receptor, known as ‘basigin’, to invade the cells by binding a protein PfRH5 to the receptor.[33] Unlike other antigens of the malaria parasite which are often genetically diverse, the PfRH5 antigen appears to have little genetic diversity. It was found to induce very low antibody response in people naturally exposed to the parasite.[32] The high susceptibility of PfRH5 to the cross-strain neutralizing vaccine-induced antibody demonstrated a significant promise for preventing malaria in the long and often difficult road of vaccine development. According to Professor Adrian Hill, a Wellcome Trust Senior Investigator at the University of Oxford, the next step would be the safety tests of this vaccine. At the time (2011) it was projected that if these proved successful, the clinical trials in patients could begin within two to three years.[34]
- PfEMP1, one of the proteins known as variant surface antigens (VSAs) produced by Plasmodium falciparum, was found to be a key target of the immune system’s response against the parasite. Studies of blood samples from 296 mostly Kenyan children by researchers of Burnet Institute and their cooperators showed that antibodies against PfEMP1 provide protective immunity, while antibodies developed against other surface antigens do not. Their results demonstrated that PfEMP1 could be a target to develop an effective vaccine which will reduce risk of developing malaria.[35][36]
- Plasmodium vivax is the common malaria species found in India, Southeast Asia and South America. It is able to stay dormant in the liver and reemerge years later to elicit new infections. Two key proteins involved in the invasion of the red blood cells (RBC) by P. vivax are potential targets for drug or vaccine development. When the Duffy binding protein (DBP) of P. vivax binds the Duffy antigen (DARC) on the surface of RBC, process for the parasite to enter the RBC is initiated. Structures of the core region of DARC and the receptor binding pocket of DBP have been mapped by scientists at the Washington University in St. Louis. The researchers found that the binding is a two-step process which involves two copies of the parasite protein acting together like a pair of tongs which “clamp” two copies of DARC. Antibodies that interfere with the binding, by either targeting the key region of the DARC or the DBP will prevent the infection.[37][38]
- Antibodies against the Schizont Egress Antigen-1 (PfSEA-1) were found to disable the parasite ability to rupture from the infected red blood cells (RBCs) thus prevent it from continuing with its life cycle. Researchers from Rhode Island Hospital identified Plasmodium falciparum PfSEA-1, a 244 kd malaria antigen expressed in the schizont-infected RBCs. Mice vaccinated with the recombinant PfSEA-1 produced antibodies which interrupted the schizont rupture from the RBCs and decreased the parasite replication. The vaccine protected the mice from lethal challenge of the parasite. Tanzanian and Kenyan children who have antibodies to PfSEA-1 were found to have fewer parasites in their blood stream and milder case of malaria. By blocking the schizont outlet, the PfSEA-1 vaccine may work synergistically with vaccines targeting the other stages of the malaria life cycle such as hepatocyte and RBC invasion.[39][40]
Mix of antigenic components
Increasing the potential immunity generated against Plasmodia can be achieved by attempting to target multiple phases in the life cycle. This is additionally beneficial in reducing the possibility of resistant parasites developing. The use of multiple-parasite antigens can therefore have a synergistic or additive effect.
One of the most successful vaccine candidates in clinical trials[which?][when?] consists of recombinant antigenic proteins to the circumsporozoite protein.[41] (This is discussed in more detail below.)[where?]
Delivery system
The selection of an appropriate system is fundamental in all vaccine development, but especially so in the case of malaria. A vaccine targeting several antigens may require delivery to different areas and by different means in order to elicit an effective response. Some adjuvants can direct the vaccine to the specifically targeted cell type—e.g. the use of Hepatitis B virus in the RTS,S vaccine to target infected hepatocytes—but in other cases, particularly when using combined antigenic vaccines, this approach is very complex. Some methods that have been attempted include the use of two vaccines, one directed at generating a blood response and the other a liver-stage response. These two vaccines could then be injected into two different sites, thus enabling the use of a more specific and potentially efficacious delivery system.
To increase, accelerate or modify the development of an immune response to a vaccine candidate it is often necessary to combine the antigenic substance to be delivered with an adjuvant or specialised delivery system. These terms are often used interchangeably in relation to vaccine development; however in most cases a distinction can be made. An adjuvant is typically thought of as a substance used in combination with the antigen to produce a more substantial and robust immune response than that elicited by the antigen alone. This is achieved through three mechanisms: by affecting the antigen delivery and presentation, by inducing the production of immunomodulatory cytokines, and by affecting the antigen presenting cells (APC). Adjuvants can consist of many different materials, from cell microparticles to other particulated delivery systems (e.g. liposomes).
Adjuvants are crucial in affecting the specificity and isotype of the necessary antibodies. They are thought to be able to potentiate the link between the innate and adaptive immune responses. Due to the diverse nature of substances that can potentially have this effect on the immune system, it is difficult to classify adjuvants into specific groups. In most circumstances they consist of easily identifiable components of micro-organisms that are recognised by the innate immune system cells. The role of delivery systems is primarily to direct the chosen adjuvant and antigen into target cells to attempt to increase the efficacy of the vaccine further, therefore acting synergistically with the adjuvant.
There is increasing concern that the use of very potent adjuvants could precipitate autoimmune responses, making it imperative that the vaccine is focused on the target cells only. Specific delivery systems can reduce this risk by limiting the potential toxicity and systemic distribution of newly developed adjuvants.
Studies into the efficacy of malaria vaccines developed to date[when?] have illustrated that the presence of an adjuvant is key in determining any protection gained against malaria. A large number of natural and synthetic adjuvants have been identified throughout the history of vaccine development. Options identified thus far for use combined with a malaria vaccine include mycobacterial cell walls, liposomes, monophosphoryl lipid A and squalene.
History
Individuals who are exposed to the parasite in endemic countries develop acquired immunity against disease and death. Such immunity does not however prevent malarial infection; immune individuals often harbour asymptomatic parasites in their blood. This does, however, imply that it is possible to create an immune response that protects against the harmful effects of the parasite.
Research shows that if immunoglobulin is taken from immune adults, purified and then given to individuals who have no protective immunity, some protection can be gained.[42]
Irradiated mosquitoes
In 1967, it was reported that a level of immunity to the Plasmodium berghei parasite could be given to mice by exposing them to sporozoites that had been irradiated by x-rays.[43] Subsequent human studies in the 1970s showed that humans could be immunized against Plasmodium vivax and Plasmodium falciparum by exposing them to the bites of significant numbers of irradiated mosquitos.[44]
From 1989 to 1999, eleven volunteers recruited from the United States Public Health Service, United States Army, and United States Navy were immunized against Plasmodium falciparum by the bites of 1001–2927 mosquitoes that had been irradiated with 15,000 rads of gamma rays from a Co-60 or Cs-137 source.[45] This level of radiation is sufficient to attenuate the malaria parasites so that, while they can still enter hepatic cells, they cannot develop into schizonts nor infect red blood cells.[45] Over a span of 42 weeks, 24 of 26 tests on the volunteers showed that they were protected from malaria.[46]
References
- ^ Jump up to:a b c d e “Mosquirix: Opinion on medicine for use outside EU”. European Medicines Agency (EMA). Archived from the original on 23 November 2019. Retrieved 22 November 2019.
- ^ Jump up to:a b c “Malaria vaccine becomes first to achieve WHO-specified 75% efficacy goal”. EurekAlert!. 23 April 2021. Retrieved 24 April2021.
- ^ Jump up to:a b Roxby P (23 April 2021). “Malaria vaccine hailed as potential breakthrough”. BBC News. Retrieved 24 April 2021.
- ^ “RTS,S malaria candidate vaccine reduces malaria by approximately one-third in African infants”. malariavaccine.org. Malaria Vaccine Initiative Path. Archived from the original on 23 March 2013. Retrieved 19 March 2013.
- ^ “Commercial name of RTS,S”. Archived from the original on 5 April 2012. Retrieved 20 October 2011.
- ^ Foquet L, Hermsen CC, van Gemert GJ, Van Braeckel E, Weening KE, Sauerwein R, et al. (January 2014). “Vaccine-induced monoclonal antibodies targeting circumsporozoite protein prevent Plasmodium falciparum infection”. The Journal of Clinical Investigation. 124 (1): 140–4. doi:10.1172/JCI70349. PMC 3871238. PMID 24292709.
- ^ Jump up to:a b Agnandji ST, Lell B, Fernandes JF, Abossolo BP, Methogo BG, Kabwende AL, et al. (December 2012). “A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants”. The New England Journal of Medicine. 367 (24): 2284–95. doi:10.1056/NEJMoa1208394. PMID 23136909.
- ^ Borghino D (27 April 2015). “Malaria vaccine candidate shown to prevent thousands of cases”. http://www.gizmag.com. Retrieved 11 June 2016.
- ^ “GSK announces EU regulatory submission of malaria vaccine candidate RTS,S” (Press release). GSK. 24 July 2014. Archived from the original on 4 December 2016. Retrieved 30 July 2015.
- ^ Kelland K (7 October 2013). “GSK aims to market world’s first malaria vaccine”. Reuters. Retrieved 9 December 2013.
- ^ “First malaria vaccine receives positive scientific opinion from EMA” (Press release). European Medicines Agency (EMA). 24 July 2015. Retrieved 30 July 2015.
- ^ “GSK’s malaria candidate vaccine, Mosquirix (RTS,S), receives positive opinion from European regulators for the prevention of malaria in young children in sub-Saharan Africa” (Press release). GSK. 24 July 2015. Archived from the original on 28 July 2015. Retrieved 30 July 2015.
- ^ Alonso P (19 June 2019). “Letter to partners – June 2019”(Press release). Wuxi: World Health Organization. Retrieved 22 October 2019.
- ^ “Malaria vaccine launched in Kenya: Kenya joins Ghana and Malawi to roll out landmark vaccine in pilot introduction” (Press release). Homa Bay: World Health Organization. 13 September 2019. Retrieved 22 October 2019.
- ^ Davies L (6 October 2021). “WHO endorses use of world’s first malaria vaccine in Africa”. The Guardian. Retrieved 6 October2021.
- ^ “WHO recommends groundbreaking malaria vaccine for children at risk” (Press release). World Health Organization. Retrieved 6 October 2021.
- ^ Mandavilli A (6 October 2021). “A ‘Historical Event’: First Malaria Vaccine Approved by W.H.O.” The New York Times. Retrieved 6 October 2021.
- ^ Graves P, Gelband H (October 2006). “Vaccines for preventing malaria (blood-stage)”. The Cochrane Database of Systematic Reviews (4): CD006199. doi:10.1002/14651858.CD006199. PMC 6532641. PMID 17054281.
- ^ Graves P, Gelband H (October 2006). “Vaccines for preventing malaria (pre-erythrocytic)”. The Cochrane Database of Systematic Reviews (4): CD006198. doi:10.1002/14651858.CD006198. PMC 6532586. PMID 17054280.
- ^ Lowe D (23 April 2021). “Great Malaria Vaccine News”. Science Translational Medicine. Retrieved 24 April 2021.
- ^ “Researcher’s nanoparticle key to new malaria vaccine”. Research & Development. 4 September 2014. Retrieved 12 June2016.
- ^ Burkhard P, Lanar DE (2 December 2015). “Malaria vaccine based on self-assembling protein nanoparticles”. Expert Review of Vaccines. 14 (12): 1525–7. doi:10.1586/14760584.2015.1096781. PMC 5019124. PMID 26468608.
- ^ “Nature report describes complete protection after 10 weeks with three doses of PfSPZ- CVac” (Press release). 15 February 2017.
- ^ “SANARIA PfSPZ VACCINE AGAINST MALARIA RECEIVES FDA FAST TRACK DESIGNATION” (PDF). Sanaria Inc. 22 September 2016. Archived from the original (PDF) on 23 October 2016. Retrieved 23 January 2017.
- ^ Butler D (April 2019). “Promising malaria vaccine to be tested in first large field trial”. Nature. doi:10.1038/d41586-019-01232-4. PMID 32291409.
- ^ “First vaccine to fully immunize against malaria builds on pandemic-driven RNA tech”. academictimes.com. 25 February 2021. Retrieved 1 March 2021.
- ^ Graves P, Gelband H (April 2006). “Vaccines for preventing malaria (SPf66)”. The Cochrane Database of Systematic Reviews(2): CD005966. doi:10.1002/14651858.CD005966. PMC 6532709. PMID 16625647.
- ^ Ratanji KD, Derrick JP, Dearman RJ, Kimber I (April 2014). “Immunogenicity of therapeutic proteins: influence of aggregation”. Journal of Immunotoxicology. 11 (2): 99–109. doi:10.3109/1547691X.2013.821564. PMC 4002659. PMID 23919460.
- ^ Reuters Staff (15 January 2021). “South African scientists discover new chemicals that kill malaria parasite”. Reuters. Retrieved 2 February 2021.
- ^ Kennedy DA, Read AF (December 2018). “Why the evolution of vaccine resistance is less of a concern than the evolution of drug resistance”. Proceedings of the National Academy of Sciences of the United States of America. 115 (51): 12878–12886. doi:10.1073/pnas.1717159115. PMC 6304978. PMID 30559199.
- ^ Zhang VM, Chavchich M, Waters NC (March 2012). “Targeting protein kinases in the malaria parasite: update of an antimalarial drug target”. Current Topics in Medicinal Chemistry. 12 (5): 456–72. doi:10.2174/156802612799362922. PMID 22242850. Archived from the original on 30 May 2013. Retrieved 23 March2020.
- ^ Jump up to:a b Douglas AD, Williams AR, Illingworth JJ, Kamuyu G, Biswas S, Goodman AL, et al. (December 2011). “The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody”. Nature Communications. 2 (12): 601. Bibcode:2011NatCo…2..601D. doi:10.1038/ncomms1615. PMC 3504505. PMID 22186897.
- ^ Jump up to:a b Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, et al. (November 2011). “Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum”. Nature. 480 (7378): 534–7. Bibcode:2011Natur.480..534C. doi:10.1038/nature10606. PMC 3245779. PMID 22080952.
- ^ Martino M (21 December 2011). “New candidate vaccine neutralizes all tested strains of malaria parasite”. fiercebiotech.com. FierceBiotech. Retrieved 23 December 2011.
- ^ Parish T (2 August 2012). “Lifting malaria’s deadly veil: Mystery solved in quest for vaccine”. Burnet Institute. Retrieved 14 August2012.
- ^ Chan JA, Howell KB, Reiling L, Ataide R, Mackintosh CL, Fowkes FJ, et al. (September 2012). “Targets of antibodies against Plasmodium falciparum-infected erythrocytes in malaria immunity”. The Journal of Clinical Investigation. 122 (9): 3227–38. doi:10.1172/JCI62182. PMC 3428085. PMID 22850879.
- ^ Mullin E (13 January 2014). “Scientists capture key protein structures that could aid malaria vaccine design”. fiercebiotechresearch.com. Retrieved 16 January 2014.
- ^ Batchelor JD, Malpede BM, Omattage NS, DeKoster GT, Henzler-Wildman KA, Tolia NH (January 2014). “Red blood cell invasion by Plasmodium vivax: structural basis for DBP engagement of DARC”. PLOS Pathogens. 10 (1): e1003869. doi:10.1371/journal.ppat.1003869. PMC 3887093. PMID 24415938.
- ^ Mullin E (27 May 2014). “Antigen Discovery could advance malaria vaccine”. fiercebiotechresearch.com. Retrieved 22 June2014.
- ^ Raj DK, Nixon CP, Nixon CE, Dvorin JD, DiPetrillo CG, Pond-Tor S, et al. (May 2014). “Antibodies to PfSEA-1 block parasite egress from RBCs and protect against malaria infection”. Science. 344(6186): 871–7. Bibcode:2014Sci…344..871R. doi:10.1126/science.1254417. PMC 4184151. PMID 24855263.
- ^ Plassmeyer ML, Reiter K, Shimp RL, Kotova S, Smith PD, Hurt DE, et al. (September 2009). “Structure of the Plasmodium falciparum circumsporozoite protein, a leading malaria vaccine candidate”. The Journal of Biological Chemistry. 284 (39): 26951–63. doi:10.1074/jbc.M109.013706. PMC 2785382. PMID 19633296.
- ^ “Immunoglobulin Therapy & Other Medical Therapies for Antibody Deficiencies”. Immune Deficiency Foundation. Retrieved 30 September 2019.
- ^ Nussenzweig RS, Vanderberg J, Most H, Orton C (October 1967). “Protective immunity produced by the injection of x-irradiated sporozoites of plasmodium berghei”. Nature. 216 (5111): 160–2. Bibcode:1967Natur.216..160N. doi:10.1038/216160a0. PMID 6057225. S2CID 4283134.
- ^ Clyde DF (May 1975). “Immunization of man against falciparum and vivax malaria by use of attenuated sporozoites”. The American Journal of Tropical Medicine and Hygiene. 24 (3): 397–401. doi:10.4269/ajtmh.1975.24.397. PMID 808142.
- ^ Jump up to:a b Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, et al. (April 2002). “Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites”. The Journal of Infectious Diseases. 185 (8): 1155–64. doi:10.1086/339409. PMID 11930326.
- ^ Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, et al. (April 2002). “Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites”. The Journal of Infectious Diseases. 185 (8): 1155–64. doi:10.1086/339409. PMID 11930326.
Further reading
- Good MF, Levine MA, Kaper JB, Rappuoli R, Liu MA (2004). New Generation Vaccines. New York, N.Y: Marcel Dekker. ISBN 978-0-8247-4071-9.
- Hoffman SL, Doolan DL, Richie TL (January 2004). “Malaria: a complex disease that may require a complex vaccine.”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New Generation Vaccines (3rd ed.). CRC Press. pp. 1763–1790. ISBN 978-0-429-15186-6.
- Good M, Kemp D. “Overview of Vaccine Strategies for Malaria”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). ibid (3rd ed.). CRC Press. ISBN 978-0-429-15186-6.
- Saul A. “Malaria Transmission-Blocking Vaccines”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New Generation Vaccines (3rd ed.). CRC Press. ISBN 978-0-429-15186-6.
- Heppner DG, Cummings JF, Ockenhouse CF, Kester KE, Cohen J, Ballou WR (2004). “Adjuvanted RTS, S and other protein-based pre-erythrocytic stage malaria vaccines.”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New generation vaccines (3rd ed.). CRC Press. pp. 851–60. ISBN 978-0-429-15186-6.
- Stanisic DI, Martin LB, Good MF, Anders RF. “Plasmodium falciparum Asexual Blood Stage Vaccine Candidates: Current Status.”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New Generation Vaccines (3rd ed.). CRC Press. ISBN 978-0-429-15186-6.
- The Jordan Report
- “Case studies: Potential malaria vaccine” (Press release). GlaxoSmithKline. 21 August 2009. Archived from the original on 27 July 2009. Retrieved 27 November 2009.
- “World’s largest malaria vaccine trial now underway in seven African countries” (Press release). GlaxoSmithKline. 3 November 2009. Archived from the original on 10 November 2009. Retrieved 27 November 2009.
- Abdulla S, Oberholzer R, Juma O, Kubhoja S, Machera F, Membi C, et al. (December 2008). “Safety and immunogenicity of RTS,S/AS02D malaria vaccine in infants” (PDF). The New England Journal of Medicine. 359 (24): 2533–44. doi:10.1056/NEJMoa0807773. PMID 19064623.
- Aponte JJ, Aide P, Renom M, Mandomando I, Bassat Q, Sacarlal J, et al. (November 2007). “Safety of the RTS,S/AS02D candidate malaria vaccine in infants living in a highly endemic area of Mozambique: a double blind randomised controlled phase I/IIb trial”. Lancet. 370 (9598): 1543–51. doi:10.1016/S0140-6736(07)61542-6. PMID 17949807. S2CID 19372191.
- Bejon P, Lusingu J, Olotu A, Leach A, Lievens M, Vekemans J, et al. (December 2008). “Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age”. The New England Journal of Medicine. 359 (24): 2521–32. doi:10.1056/NEJMoa0807381. PMC 2655100. PMID 19064627.
- Delves PJ, Roitt IM (2001). Roitt’s essential immunology. Oxford: Blackwell Science. ISBN 978-0-632-05902-7.
- Gurunathan S, Klinman DM, Seder RA (2000). “DNA vaccines: immunology, application, and optimization*”. Annual Review of Immunology. 18: 927–74. doi:10.1146/annurev.immunol.18.1.927. PMID 10837079.
- Schwartz L, Brown GV, Genton B, Moorthy VS (January 2012). “A review of malaria vaccine clinical projects based on the WHO rainbow table”. Malaria Journal. 11: 11. doi:10.1186/1475-2875-11-11. PMC 3286401. PMID 22230255.
- Waters A (February 2006). “Malaria: new vaccines for old?”. Cell. 124 (4): 689–93. doi:10.1016/j.cell.2006.02.011. PMID 16497579.
External links
| Screened cup of malaria-infected mosquitoes which will infect a volunteer in a clinical trial | |
| Vaccine description | |
|---|---|
| Target | Malaria |
| Vaccine type | Protein subunit |
| Clinical data | |
| Trade names | Mosquirix |
| Routes of administration | Intramuscular[1] |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 149121-47-1 |
| ChemSpider | none |
//////////////RTS,S/AS01, Mosquirix, malaria vaccine, gsk, VACCINE, RTS,S, APPROVALS 2021

NEW DRUG APPROVALS
ONE TIME
$10.00
REACH, Registration, Evaluation, Authorisation and Restriction of Chemicals
DRUG REGULATORY AFFAIRS INTERNATIONAL

REACH
https://ec.europa.eu/environment/chemicals/reach/reach_en.htm#:~:text=REACH%20(EC%201907%2F2006),authorisation%20and%20restriction%20of%20chemicals.
REACH (EC 1907/2006)aims to improve the protection of human health and the environment through the better and earlier identification of the intrinsic properties of chemical substances. This is done by the four processes of REACH, namely the registration, evaluation, authorisation and restriction of chemicals. REACH also aims to enhance innovationand competitiveness of the EU chemicals industry.
“No data no market”: the REACH Regulation places responsibility on industry to manage the risks from chemicals and to provide safety information on the substances. Manufacturers and importers are required to gather information on the properties of their chemical substances, which will allow their safe handling, and to register the information in a central database in theEuropean Chemicals Agency (ECHA)in Helsinki. The Agency is the central point in the REACH system: it manages the databases necessary to operate the system, co-ordinates the in-depth evaluation of suspicious chemicals and is…
View original post 4,663 more words
Tisotumab vedotin

Tisotumab vedotin
チソツマブベドチン (遺伝子組換え)Immunoglobulin G1, anti-(human blood-coagulation factor III) (human monoclonal HuMax-TF heavy chain), disulfide with human monoclonal HuMax-TF κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
- HuMax-TF-ADC
- Immunoglobulin G1, anti-(human tissue factor) (human monoclonal HuMax-TF heavy chain), disulfide with human monoclonal HuMax-TF κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
Protein Sequence
Sequence Length: 1324, 448, 448, 214, 214multichain; modified (modifications unspecified)
| Formula | C6418H9906N1710O2022S44.(C68H106N11O15)n |
|---|---|
| Efficacy | Antineoplastic |
| Disease | Cervical cancer |
| Comment | Antibody-drug conjugateCAS:1418731-10-8 |
- HuMax-TF-ADC
- Tisotumab vedotin
- Tisotumab vedotin [WHO-DD]
- UNII-T41737F88A
- WHO 10148
US FDA APPROVED 2021/9/20 , TIVDAK

FDA grants accelerated approval to tisotumab vedotin-tftv for recurrent or metastatic cervical cancer……….. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-tisotumab-vedotin-tftv-recurrent-or-metastatic-cervical-cancer
On September 20, 2021, the Food and Drug Administration granted accelerated approval to tisotumab vedotin-tftv (Tivdak, Seagen Inc.), a tissue factor-directed antibody and microtubule inhibitor conjugate, for adult patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy.
Approval was based on innovaTV 204, an open-label, multicenter, single-arm clinical trial (NCT03438396). Efficacy was evaluated in 101 patients with recurrent or metastatic cervical cancer who had received no more than two prior systemic regimens in the recurrent or metastatic setting, including at least one prior platinum-based chemotherapy regimen. Sixty-nine percent of patients had received bevacizumab as part of prior systemic therapy. Patients received tisotumab vedotin-tftv 2 mg/kg every 3 weeks until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed objective response rate (ORR) as assessed by an independent review committee (IRC) using RECIST v1.1 and duration of response (DOR). The ORR was 24% (95% CI: 15.9%, 33.3%) with a median response duration of 8.3 months (95% CI: 4.2, not reached).
The most common adverse reactions (≥25%), including laboratory abnormalities, were hemoglobin decreased, fatigue, lymphocytes decreased, nausea, peripheral neuropathy, alopecia, epistaxis, conjunctival adverse reactions, hemorrhage, leukocytes decreased, creatinine increased, dry eye, prothrombin international normalized ratio increased, activated partial thromboplastin time prolonged, diarrhea, and rash. Product labeling includes a boxed warning for ocular toxicity.
The recommended dose is 2 mg/kg (up to a maximum of 200 mg for patients ≥100 kg) given as an intravenous infusion over 30 minutes every 3 weeks until disease progression or unacceptable toxicity.
View full prescribing information for Tivdak.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
A fully human monoclonal antibody specific for tissue factor conjugated to the microtubule-disrupting agent monomethyl auristatin E (MMAE) via a protease-cleavable valine-citrulline linker.
Tisotumab vedotin, sold under the brand name Tivdak is a human monoclonal antibody used to treat cervical cancer.[1]
Tisotumab vedotin was approved for medical use in the United States in September 2021.[1][2]
Tisotumab vedotin is the international nonproprietary name (INN).[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto

NEW DRUG APPROVALS
one time
$10.00
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208s000lbl.pdf
- ^ “Seagen and Genmab Announce FDA Accelerated Approval for Tivdak (tisotumab vedotin-tftv) in Previously Treated Recurrent or Metastatic Cervical Cancer”. Seagen. 20 September 2021. Retrieved 20 September 2021 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 159–60. hdl:10665/331046.
External links
- “Tisotumab vedotin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03438396 for “A Trial of Tisotumab Vedotin in Cervical Cancer” at ClinicalTrials.gov
- Clinical trial number NCT03245736 for “Tisotumab Vedotin Continued Treatment in Patients With Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02001623 for “Tisotumab Vedotin (HuMax-TF-ADC) Safety Study in Patients With Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02552121 for “Tisotumab Vedotin (HuMax-TF-ADC) Safety Study in Patients With Solid Tumors” at ClinicalTrials.gov
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Tissue factor (TF) |
| Clinical data | |
| Trade names | Tivdak |
| Other names | Tisotumab vedotin-tftv |
| License data | US DailyMed: Tisotumab_vedotin |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1418731-10-8 |
| UNII | T41737F88A |
| KEGG | D11814 |
//////////Tisotumab vedotin, チソツマブベドチン (遺伝子組換え) , FDA 2021, APPROVALS 2021, Antineoplastic, CERVICAL CANCER, CANCER, MONOCLONAL ANTIBODY, UNII-T41737F88A, WHO 10148

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}