


Candidate: TAK-981
Home » 2019 (Page 11)
Yearly Archives: 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cefamandole, セファマンドール ,цефамандол , سيفاماندول , 头孢孟多 ,
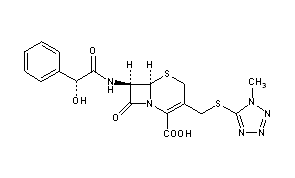
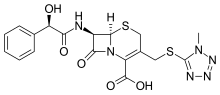

CAS Registry Number: 34444-01-4
CAS Name: (6R,7R)-7-[[(2R)-Hydroxyphenylacetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
Additional Names: 7-mandelamido-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid; 7-D-mandelamido-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic acid; 7-D-mandelamido-3-(1-methyl-1,2,3,4-tetrazole-5-thiomethyl)-D3-cephem-4-carboxylic acid; CMT
Manufacturers’ Codes: compd 83405
Molecular Formula: C18H18N6O5S2
Molecular Weight: 462.50
Percent Composition: C 46.74%, H 3.92%, N 18.17%, O 17.30%, S 13.87%
Literature References: Broad-spectrum semi-synthetic cephalosporin antibiotic. Prepn: C. W. Ryan, DE 2018600; idem, US3641021 (1970, 1972 to Lilly); J. M. Greene, DE 2312997; idem, US 3840531 (1973, 1974 to Lilly). Biological properties: W. E. Wick, D. A. Preston, Antimicrob. Agents Chemother. 1, 221 (1972). Antibacterial activity: S. Eykyn et al., ibid. 3, 657 (1973); H. C. Neu, ibid. 6, 177 (1974); A. D. Russell, J. Antimicrob. Chemother. 1, 97 (1975). Pharmacologic studies: B. R. Meyers et al.,Antimicrob. Agents Chemother. 9, 140 (1976); R. S. Griffith et al., ibid. 10, 814 (1976). Comprehensive description: R. H. Bishara, E. C. Rickard, Anal. Profiles Drug Subs. 9, 125-154 (1980).
Derivative Type: Nafate
CAS Registry Number: 42540-40-9
Trademarks: Bergacef (Bergamon); Cedol (Tiber); Cefam (Magis); Cefiran (Poli); Cemado (Francia); Cemandil (SIT); Fado (Errekappa); Kefadol (Lilly); Kefandol (Lilly); Lampomandol (AGIPS); Mandokef (Lilly); Mandol (Lilly); Mandolsan (San Carlo); Neocefal (Metapharma); Pavecef (IBP)
Molecular Formula: C19H17N6NaO6S2
Molecular Weight: 512.49
Percent Composition: C 44.53%, H 3.34%, N 16.40%, Na 4.49%, O 18.73%, S 12.51%
Properties: White, odorless needles, mp 190° (dec). uv max (H2O): 269 nm (e 10800). pKa 2.6-3.0. Sol in water, methanol. Practically insol in ether, chloroform, benzene, cyclohexane.
Melting point: mp 190° (dec)
pKa: pKa 2.6-3.0
Absorption maximum: uv max (H2O): 269 nm (e 10800)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Cephalosporins.
- Use:antibiotic
- Chemical name:[6R-[6α,7β(R*)]]-7-[(hydroxyphenylacetyl)amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
- Formula:C18H18N6O5S2
- MW:462.51 g/mol
- CAS-RN:34444-01-4
- InChI Key:OLVCFLKTBJRLHI-AXAPSJFSSA-N
- InChI:InChI=1S/C18H18N6O5S2/c1-23-18(20-21-22-23)31-8-10-7-30-16-11(15(27)24(16)12(10)17(28)29)19-14(26)13(25)9-5-3-2-4-6-9/h2-6,11,13,16,25H,7-8H2,1H3,(H,19,26)(H,28,29)/t11-,13-,16-/m1/s1
- EINECS:252-030-0
Derivatives
Formate monosodium salt (nafate)
- Formula:C19H17N6NaO6S2
- MW:512.50 g/mol
- CAS-RN:42540-40-9
- EINECS:255-877-4
- LD50:3915 mg/kg (M, i.v.);
2562 mg/kg (R, i.v.)
Cefamandole (INN, also known as cephamandole) is a second-generation broad-spectrumcephalosporinantibiotic. The clinically used form of cefamandole is the formateestercefamandole nafate, a prodrug which is administered parenterally. Cefamandole is no longer available in the United States.
The chemical structure of cefamandole, like that of several other cephalosporins, contains an N-methylthiotetrazole (NMTT or 1-MTT) side chain. As the antibiotic is broken down in the body, it releases free NMTT, which can cause hypoprothrombinemia (likely due to inhibition of the enzymevitamin K epoxide reductase)(vitamin K supplement is recommended during therapy) and a reaction with ethanol similar to that produced by disulfiram (Antabuse), due to inhibition of aldehyde dehydrogenase.
Cefamandole has a broad spectrum of activity and can be used to treat bacterial infections of the skin, bones and joints, urinary tract, and lower respiratory tract. The following represents cefamandole MIC susceptibility data for a few medically significant microorganisms.
- Escherichia coli: 0.12 – 400 μg/ml
- Haemophilus influenzae: 0.06 – >16 μg/ml
- Staphylococcus aureus: 0.1 – 12.5 μg/ml
CO2 is generated during the normal constitution of cefamandole and ceftazidime, potentially resulting in an explosive-like reaction in syringes.[2]
SYNTHESIS
US 3641021
US 3840531 US 3974153 US 3903278 US 2018600 US 2065621 DE 2018600 DE 2065621 DE 2730579
DE 2312997

SYN
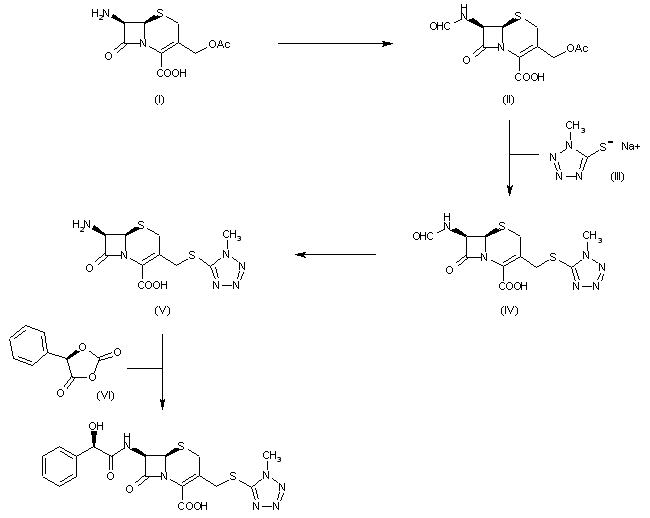
The formylation of 7-aminocephalosporanic acid (I) by the usual techniques produces 7-formamidocephalosporanic acid (II), which is then treated with the sodium salt of 1-methyl-1H-tetrazole-5-thiol (III) to yield 7-formamido-3-(1-methyl-1H-tetrazol-5-ylthio)methyl-3-cephem-4-carboxylic acid (IV). The resulting product (IV) is deformylated affording 7-amino-3-(1-methyl-1H-tetrazol-5-ylthio)methyl-3-cephem-4-carboxylic acid (V), which is finally acylated with anhydro-O-carboxymandelic acid (VI) using the usual techniques.
References
- ^ http://www.toku-e.com/Assets/MIC/Cefamandole%20sodium%20salt.pdf
- ^ Stork CM (2006). “Antibiotics, antifungals, and antivirals”. In Nelson LH, Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA. Goldfrank’s toxicologic emergencies. New York: McGraw-Hill. p. 847. ISBN 0-07-143763-0. Retrieved 2009-07-03.
-
- US 3 641 021 (Lilly; 8.2.1972; appl. 18.4.1969).
- DE 2 018 600 (Lilly; appl. 17.4.1970; USA-prior. 18.4.1969).
- DAS 2 065 621 (Lilly; appl. 17.4.1970; USA-prior. 18.4.1969).
- US 3 840 531 (Lilly; 8.10.1974; appl. 21.3.1972).
- US 3 903 278 (Smith Kline Corp.; 2.9.1975; prior. 4.11.1971).
- DOS 2 730 579 (Pierrel S.p.A.; appl. 6.7.1977; GB-prior. 10.7.1976).
-
preparation and/or purification via the trimethylsilyl-derivatives:
- DOS 2 711 095 (Lilly; appl. 14.3.1977; USA-prior. 17.3.1976).
-
purification:
- US 4 115 644 (Lilly; 19.9.1978; appl. 19.9.1978).
- DOS 2 839 670 (Lilly; appl. 12.9.1978; USA-prior. 19.9.1977).
-
crystalline sodium salt:
- US 4 054 738 (Lilly; 18.10.1977; appl. 22.12.1975).
- US 4 168 376 (Lilly; 18.9.1979; appl. 5.6.1978).
-
lithium salt:
- GB 1 546 757 (Lilly; appl. 10.4.1975; valid from 7.4.1976).
-
O-formyl-derivative:
- US 3 928 592 (Lilly; 23.12.1975; appl. 21.2.1974).
- GB 1 493 676 (Lilly; appl. 20.2.1975; USA-prior. 22.2.1974).
- GB 1 546 898 (Lilly; appl. 7.4.1976; USA-prior. 11.4.1975).
- DOS 2 506 622 (Lilly; appl. 17.2.1975; USA-prior. 22.2.1974).
-
crystalline sodium salt of O-formylcefamandole:
- US 4 006 138 (Lilly; 1.2.1977; appl. 11.4.1975).
-
complex of cefamandole sodium with 1,4-dioxane and water:
- US 3 947 414 (Lilly; 30.3.1976; appl. 23.12.1974).
-
complex of cefamandole sodium with ethyl l-(–)-lactate:
- US 3 947 415 (Lilly; 30.3.1976; appl. 23.12.1974).
 |
|
| Clinical data | |
|---|---|
| Trade names | former Mandol |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601206 |
| Pregnancy category |
|
| Routes of administration |
Intramuscular, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 75% |
| Elimination half-life | 48 minutes |
| Excretion | Mostly renal, as unchanged drug |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.047.285 |
| Chemical and physical data | |
| Formula | C18H18N6O5S2 |
| Molar mass | 462.505 g/mol g·mol−1 |
| 3D model (JSmol) | |
Tegaserod, テガセロド
Tegaserod
- Molecular FormulaC16H23N5O
- Average mass301.387 Da
- テガセロド
145158-71-0 cas
HTF 919 / HTF-919 / SDZ HTF 919 / SDZ-HTF-919
N’-[(E)-[(5-methoxy-1H-indol-3-yl)methylidene]amino]-N-pentylguanidine
(2E)-2-[(5-Methoxy-1H-indol-3-yl)methylene]-N-pentylhydrazinecarboximidamide [ACD/IUPAC Name]
(2E)-2-[(5-methoxy-1H-indol-3-yl)methylidene]-N’-pentylhydrazinecarboximidamide
(2E)-2-[(5-methoxy-1H-indol-3-yl)methylidene]-N-pentylhydrazinecarboximidamide
145158-71-0 [RN]
7606
Hydrazinecarboximidamide, 2-[(5-methoxy-1H-indol-3-yl)methylene]-N-pentyl-, (2E)
Sundaram Venkataraman, Srinivasulu Gudipati, Brahmeshwararao Mandava Venkata Naga, Goverdhan Banda, Radhakrishna Singamsetty, “Process for preparing form I of tegaserod maleate.” U.S. Patent US20050272802, issued December 08, 2005.US20050272802
Tegaserod maleate [USAN]
189188-57-6
Tegaserod
CAS Registry Number: 145158-71-0
CAS Name: 2-[(5-Methoxy-1H-indol-3-yl)methylene]-N-pentylhydrazinecarboximidamide
Molecular Formula: C16H23N5O
Molecular Weight: 301.39
Percent Composition: C 63.76%, H 7.69%, N 23.24%, O 5.31%
Literature References: Selective serotonin 5HT4-receptor partial agonist. Prepn: R. K. A. Giger, H. Mattes, EP 505322; eidem, US5510353 (1992, 1996 both to Sandoz); K.-H. Buchheit et al., J. Med. Chem. 38, 2331 (1995). Clinical pharmacology: S. Appel et al., Clin. Pharmacol. Ther. 62, 546 (1997); and pharmacokinetics: idem et al., J. Clin. Pharmacol. 37, 229 (1997). Clinical trial in irritable bowel syndrome: S. A. Müller-Lissner et al., Aliment. Pharmacol. Ther. 15, 1655 (2001); in female patients: J. Novick et al.,ibid. 16, 1877 (2002). Review of clinical efficacy: B. W. Jones et al., J. Clin. Pharm. Ther. 27, 343-352 (2002); of mechanism of action, efficacy and safety: M. Corsetti, J. Tack, Expert Opin. Pharmacother. 3, 1211-1218 (2002).
Properties: mp 155°.
Melting point: mp 155°
Derivative Type: Maleate
CAS Registry Number: 189188-57-6
Manufacturers’ Codes: SDZ-HTF-919
Trademarks: Zelmac (Novartis); Zelnorm (Novartis)
Molecular Formula: C16H23N5O.C4H4O4
Molecular Weight: 417.46
Percent Composition: C 57.54%, H 6.52%, N 16.78%, O 19.16%
Therap-Cat: Gastroprokinetic; in treatment of irritable bowel syndrome.
Keywords: Gastroprokinetic; Serotonin Receptor Agonist.
Tegaserod is a 5-HT4 agonist manufactured by Novartis and sold under the names Zelnorm and Zelmac for the management of irritable bowel syndrome and constipation.[1] Approved by the FDA in 2002, it was subsequently removed from the market in 2007 due to FDA concerns about possible adverse cardiovascular effects. Before then, it was the only drug approved by the United States Food and Drug Administration to help relieve the abdominal discomfort, bloating, and constipation associated with irritable bowel syndrome. Its use was also approved to treat chronic idiopathic constipation.[2]
In 2000, originator Novartis established an alliance with Bristol-Myers Squibb for the codevelopment and copromotion of tegaserod maleate, which is now available in more than 55 countries worldwide for the treatment of IBS with constipation. In 2015, Zelnorm was acquired by Sloan Pharma from Novartis.
Novartis’ brand name Zelnorm (tegaserod) had originally received approval from the US FDA in 2002 for the treatment of irritable bowel syndrome with constipation (IBS-C) [5, 8]. It was, however, voluntarily withdrawn from widespread use in the US market in 2007 after concerns arose over the possibility that tegaserod could potentially cause dangerous cardiovascular events in patients [5,8]. Since then, closer evaluations of the original data suggesting such cardiovascular risk have resulted in the limited reintroduction or ‘re-approval’ of tegaserod for treatment of IBS-C specifically in female patients less than 65 years of age and whom are considered to be at a lower risk of a cardiovascular event than the broader population . Zelnorm (tegaserod) by Sloan Pharma subsequently gained re-approval in April of 2019 [5]. Nevertheless, tegaserod remains un-approved in certain regions [7].
Despite the relative complications involved in its history of regulatory approval, ever since its first introduction in 2002 tegaserod remains the only therapy for IBS-C that possesses the unique mechanism of action of acting on serotonin-4 (5-HT(4)) receptors in smooth muscle cells and in the gastrointestinal wall to facilitate actions like esophageal relaxation, peristaltic gut movement, and natural secretions in the gut, among others
Mechanism of action
The drug functions as a motility stimulant, achieving its desired therapeutic effects through activation of the 5-HT4 receptors of the enteric nervous system in the gastrointestinal tract. It also stimulates gastrointestinal motility and the peristaltic reflex, and allegedly reduces abdominal pain.[3] Additionally, tegaserod is a 5-HT2B receptor antagonist.[4]
Withdrawal from market
On 30 March 2007, the United States Food and Drug Administration requested that Novartis withdraw Zelnorm from shelves.[5] The FDA alleges a relationship between prescriptions of the drug and increased risks of heart attack or stroke. An analysis of data collected on over 18,000 patients demonstrated adverse cardiovascular events in 13 of 11,614 patients treated with Zelnorm (a rate of 0.11%) as compared with 1 of 7,031 patients treated with placebo (a rate of 0.01%). Novartis alleges all of the affected patients had preexisting cardiovascular disease or risk factors for such, and further alleges that no causal relationship between tegaserod use and cardiovascular events has been demonstrated.[6] On the same day as the FDA announcement, Novartis Pharmaceuticals Canada announced that it was suspending marketing and sales of the drug in Canada in response to a request from Health Canada.[7] In a large cohort study based on a US health insurance database, no increase in the risk of cardiovascular events were found under tegaserod treatment.[8] Currently, tegaserod may only be used in emergency situations only with prior authorization from the FDA.[9]
Paper
The serotonin 5-HT4 receptor. 2. Structure-activity studies of the indole carbazimidamide class of agonists
J Med Chem 1995, 38(13): 2331
https://pubs.acs.org/doi/abs/10.1021/jm00013a010
PATENT
US 5510353
WO 2005105740
WO 2007119109
WO 2007126889
CN 103467358
WO 2006116953
Syn
PATENT
https://patents.google.com/patent/US20090306170A1/en

-
In a preferred embodiment of the first aspect of the present invention, the process of preparing tegaserod or a salt thereof comprises the steps of:
- (a) coupling S-methyl-isothiosemicarbazide or a salt thereof and 5-methoxy-indole-3-carboxaldehyde to form 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide:
-

-
and
- (b) reacting the 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide with n-pentyl amine to form tegaserod:
-

- [0013]
The skilled person will appreciate that:
-
- S-methyl-isothiosemicarbazide and salts thereof exist in two tautomeric forms:
-
-

-
- 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide exists in four tautomeric forms:
-
-

-
- tegaserod exists in four tautomeric forms:
-
-

- [0017]
It is to be understood that where tautomeric forms occur, the present invention embraces all tautomeric forms and their mixtures, i.e. although S-methyl-isothio-semicarbazide and 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide are mostly defined for convenience by reference to one isothiosemicarbazide form only, and although tegaserod is mostly defined for convenience by reference to one guanidino form only, the invention is not to be understood as being in any way limited by the particular nomenclature or graphical representation employed.
- [0018]
When an S-methyl-isothiosemicarbazide salt is used in the process of the present invention, this may be an acid addition salt with acids, including but not limited to inorganic acids such as hydrohalogenic acids (for example, hydrofluoric, hydrochloric, hydrobromic or hydroiodic acid) or other inorganic acids (for example, nitric, perchloric, sulfuric or phosphoric acid), or organic acids such as organic carboxylic acids (for example, propionic, butyric, glycolic, lactic, mandelic, citric, acetic, benzoic, salicylic, succinic, malic or hydroxysuccinic, tartaric, fumaric, maleic, hydroxymaleic, mucic or galactaric, gluconic, pantothenic or pamoic acid), organic sulfonic acids (for example, methanesulfonic, trifluoromethanesulfonic, ethanesulfonic, 2-hydroxyethanesulfonic, benzenesulfonic, p-toluenesulfonic, naphthalene-2-sulfonic or camphorsulfonic acid) or amino acids (for example, ornithinic, glutamic or aspartic acid). Preferably the S-methyl-isothiosemicarbazide salt is a hydrohalide (such as the hydrofluoride, hydrochloride, hydrobromide, or hydroiodide) or a sulfonate (such as the methanesulfonate, benzenesulfonate, or p-toluenesulfonate). Preferably the S-methyl-isothiosemicarbazide salt is S-methyl-isothiosemicarbazide hydroiodide.
-
-
The following synthetic scheme demonstrates a preferred process of the present invention.
-
- [0032]
The invention is now demonstrated by the following non-limiting illustrative example.
-
EXAMPLE Step 1: Schiff’s Base Formation of 5-methoxy-indole-3-carboxaldehyde and S-methyl-isothiosemi-carbazide hydroiodide
-
- [0033]
5-Methoxy-indole-3-carboxaldehyde (1.5 g, 1 eq) and S-methyl-isothiosemicarbazide hydroiodide (3.99 g, 2 eq) in methanol (15 ml, 10 vol) were stirred in the presence of triethylamine (3 ml, 2 vol) at 25-30° C. for 2 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. and ethyl acetate (10.5 ml, 7 vol) was added to the residue to precipitate out the product. The product, 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemi-carbazide, was separated by filtration, washed with ethyl acetate (3 ml, 2 vol) and dried under vacuum at 45-50° C. The yield was almost quantitative (˜100%).
- [0033]
Step 2: Conversion of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide to 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod)
-
- [0034]
A solution of 1-((5-methoxy-1H-indol-3-yl)methylene)-S-methyl-isothiosemicarbazide (8.0 g, 1 eq) and n-pentyl amine (2.65 g, 1 eq) was refluxed in methanol (8 ml, 1 vol) at 66° C. for 4 hours. After completion of the reaction, the methanol was removed by distillation under reduced pressure at 45-50° C. to obtain tegaserod free base as a yellowish brown solid. Yield=97%. HPLC purity=95%.
- [0034]
Step 3: Conversion of 1-((5-methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (Tegaserod) to Tegaserod Maleate
- [0035]
1-((5-Methoxy-1H-indol-3-yl)methyleneamino)-3-pentyl-guanidine (55 g, 1 eq) was taken in methanol (357.5 ml, 6.5 vol) and stirred. To this reaction mixture was added at room temperature a solution of maleic acid (74.15 g, 3.5 eq) in water (137.5 ml, 2.5 vol) and the reaction mixture stirred for one hour at room temperature. The solid obtained was then filtered through a Buchner funnel and dried at 700 mmHg and 500° C. Yield=36.8 g, 48.42%. HPLC purity=99.45%.
Polymorphs
WO 2007084697
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007084697
EXAMPLES
PXRD:
EV 320 251 655 US Powder X-ray diffraction (“PXRD”) analysis using a SCINTAG powder X-ray diffϊactometer model X’TRA equipped with a solid-state detector. Copper radiation of λ=1.5418 A was used. The sample was introduced using a round standard aluminum sample holder with round zero background quartz plate in the bottom.
Thermal Gravimetric Analysis TTGA):
TGA/SDTA 85 r, Mettler Toledo , Sample weight 7-15 mg.
Heating rate: 100C/ min., in N2 stream: flow rate: 50 ml/min
Example 1 : Preparation of Tegaserod maleate Form B
To a mixture of 90 g MICHO and 63 g NaOH [47 %] was added a solution of 212 g AGPΗI dissolved in 566 mL of water at room temperature. The resultant reaction mixture was heated to 400C. After 3 hours, 522 mL of ethyl acetate was added and the reaction mixture was stirred for an additional hour. The organic phase was washed with water (3 x 450 mL), and vacuum filtered. After addition of 211 mL ethyl acetate and 870 mL of n-propanol, the mixture was heated to 600C and a solution of maleic acid (86.5 g in 180 mL water), at the same temperature, was added to the reaction mixture and stirred at the same temperature. After 2 hours the reaction mixture was cooled to about 100C and stirred for an additional hour. The resulting solid was filtered off, washed with n-propanol, and dried in a vacuum oven over night to give 195.8 g of tegaserod maleate Form B.
6
EV 320251 655 US
PATENT
Tegaserod maleate is an aminoguanidine indole 5HT4 agonist for the treatment of irritable bowel syndrome (IBS). Tegaserod maleate has the following structure:
According to the prescribing information (Physician’s Desk Reference, 57th Ed., at Page 2339), tegaserod as the maleate salt is a white to off-white crystalline powder and is slightly soluble in ethanol and very slightly soluble in water. Tegaserod maleate is available commercially as ZELNORM®, in which it is present as crystalline form.
Tegaserod maleate is disclosed in US patent No. 5,510,353 and in its equivalent EP 0 505 322 (example 13), and is reported to have a melting point of 1900C (table 1 example 13).
The literature (Buchheit K.H, et al., J.Med.Chem., 1995, 38, 2331) describes a general method for the condensation of amino guanidines with indole-3-carbadehydes in methanol in the presence of HCl (pH 3-4). The product obtained after solvent evaporation maybe converted to its hydrochloride salt by treatment of the methanolic solution with diethylether/HCl followed by recrystallization from
methanol/diethylether. Tegaserod base prepared according to this general method is characterized solely by a melting point of 155 0C (table 3 compound 5b). Additional Tegaserod maleate characterization was done by 1H and 13C-NMR according to the literature (Jing J. et. al., Guangdong Weiliang Yuansu Kexue, 2002, 9/2, 51).
WO 04/085393 discloses four crystalline forms of tegaserod maleate. The search report for WO 04/085393 further identifies WO 00/10526, and Drugs Fut. 1999, 24(1) which provides an overview for tegaserod maleate. Additional crystalline forms of tegaserod maleate are provided in WO 2005/058819, one of which is characterized by an X-ray Diffraction pattern having peaks at 15.7, 16.9, 17.2, 24.1, 24.6 and 25.2±0.2 two theta (designated as Form B in that PCT publication).
The solid state physical properties of tegaserod salt may be influenced by controlling the conditions under which tegaserod salt is obtained in solid Form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid may have therapeutic consequences since it imposes an upper limit on the rate at which an orally- administered active ingredient may reach the patient’s bloodstream. The rate of dissolution is also a consideration in
formulating syrups, elixirs and other liquid medicaments. The solid state Form of a compound may also affect its behavior on compaction and its storage stability.
These practical physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic Form of a substance. The polymorphic form may give rise to thermal behavior different from that of the amorphous material or another polymorphic Form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) and may be used to distinguish some polymorphic forms from others. A particular polymorphic Form may also give rise to distinct spectroscopic properties that may be detectable by powder X-ray crystallography, solid state C NMR spectrometry and infrared spectrometry.
The discovery of new polymorphic forms of a pharmaceutically useful compound provides a new opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic.
The polymorphic forms may further help in purification of tegaserod, particularly if they possess high crystallinity. In the event of metastability, a metastable polymorphic form may be used to prepare a more stable polymorph.
Hence, discovery of new polymorphic forms and new processes help in advancing a formulation scientist in preparation of tegaserod as an active pharmaceutical ingredient in a formulation.
The present invention provides an additional polymorphic form of a maleate salt of tegaserod.
Example 1 : Preparation of sesqui-tefiaserod maleate Foπn H2 through tegaserod base
To a mixture of AGPΗI (112.7 g) in 283 mL of water was added 5-MICHO (45 g) followed by NaOH (52.8 g, 47%) and stirred at room temperature. After three hours, 522 mL of ethyl acetate were added and the mixture stirred for an additional four hours. After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum. The resulting solution was heated to 60 0C and a solution of maleic acid (14.4 g) in 45 mL water was dropped during half hour, and the reaction mixture stirred at the same temperature for an additional two hours. The mixture was cooled to 100C during one hour, kept under stirring at the same temperature for 12 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of ethyl acetate and dried in a vacuum oven at 45°C for 16 hours to give 85% of the product.
Example 2: Preparation of sesqui-tegaserod maleate Form H2
45 gr MICHO were added to a 1 L reactor at RT. A solution of 112.7 gr of AGP HI and 283 ml water was added to the reactor. 52.8 gr of NaOH 47% were added to the mixture while stirring. The mixture was heated to 400C and stirred for 12 hrs. 522 ml of Ethyl Acetate were added and the mixture was stirred for 4 hrs.
After phase separation at 400C the organic phase was washed with water (3 x 218 ml), and filtrated under vacuum.
The mixture was heated to 600C and a mixture o 14.4 gr of Maleic Acid in 45 ml water was dropped during 5 min.
The mixture was stirred at 600C for 2 hrs.
The mixture was cooled to 100C during 1 hour, stirred at 100C for 13 hrs and then filtered under vacuum. The wet product was washed twice with 65 ml of n-Propanol. The wet product was dried in a vacuum oven at 45°C.
Yield: 71.2%
Example 3: Preparation of Tegaserod maleate Form B from Sesqui-tegaserod maleate Form H2
6.9 g of maleic acid were added to a slurry of Sesqui-Tegaserod maleate Form H2 (41.5 g) in 208 ml n-propanol at room temperature. The mixture was stirred for 5 hours at the same temperature, filtered and washed with n-propanol. After drying on vacuum oven at 450C for 15 hours the product was analyzed by XRD and found to be Form B (89% yield).
PATENT
https://patents.google.com/patent/WO2005058819A2/en


PATENT
The formation of hydrazones is catalyzed by both general acids and general bases. General base catalysis of dehydration of the tetrahedral intermediate involves nitrogen deprotonation concerted with elimination of hydroxide ion as shown in the Scheme (Sayer J.M., et al. J. Am. Chem. Soc. 1973, 95, 4277). R fast O I H h° NH2R’ R- -NHR’ R R

In many cases, the equilibrium constant for their formation in aqueous solution is high. The additional stability may be attributed to the participation of the atom adjacent to the nitrogen in delocalized bonding. – + RRC = N – NH2 ~*→- RRC – N = NH2
In order to obtain only the maleic salt, the product when using an acid halide (HA) or other acids has to first be converted into the free base, before the addition of maleic acid (Path a), which results in an additional step to the synthesis. On the other hand, the reaction of the present invention in the presence of organic or inorganic base results in the formation of tegaserod free base which gives only the maleate salt after the addition of maleic acid (Path b).


TGS

TGS-MA
EXAMPLES
HPLC method for detecting the level of the impurities:
Column: Atlantis dcl8(150*4.6),
Mobile phase: A.80% KH2PO4(0.02M) pH=5, 20% acetonitrile(ACN), B.100% ACN. Gradient: time 0= A: 100 B: 0, time 25 min= A:50%, B:50%, time 30 min= A:50%, B:50%, + 10 minutes of equilibration time. Wavelength= 225 nm
Sample concentration: 0.5 mg/mL
Temperature = 25°C
Example 1- Preparation of Tegaserod maleate in water with HCl.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a solution of NaHCO3 (10%) until pH 9, and heated to 65°C for 20 minutes. After cooling, 100 mL of EtOAc were added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.27 g of crude tegaserod maleate with a purity of 99.70% (by HPLC).
Example 2- Preparation of Tegaserod maleate in water with HCl in two steps. a. Preparation of Tegaserod free base.
To a mixture of AGP-HI (163.3 g, 0.6 mol) in 375 mL water was added 5-MICHO (52.5 g, 0.3 mol) followed by HCl (37%) until pH 4. The mixture was heated to reflux for 1 hour and then cooled to room temperature. To the resulting slurry was added a liter of a solution of NaHCO (10%) until pH 9, and heated to 65 °C for one hour. After cooling, 1500 mL of EtOAc were added, and the organic phase washed with water. The remaining organic phase was evaporated to dryness to give tegaserod free base with a purity of 87.42 % (by HPLC). b. Preparation of Tegaserod maleate. To a solution of 2 g of tegaserod free base in MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 10 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 1.09 g of crude tegaserod maleate with a purity of 96.81 % (by HPLC).
Example 3- Preparation of Tegaserod maleate in water with TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by TEA (11.0 mL, 0.08 mol) and stirred at room temperature. After one hour, 25 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 7.92 g of crude tegaserod maleate with a purity of 94 % (by HPLC).
Example 4- Preparation of Tegaserod maleate in water with NaHCO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.71 g of crude tegaserod maleate with a purity of 98 % (by HPLC) .
Example 5- Preparation of Tegaserod maleate in water with NaHCO3 in two steps. a. Preparation of Tegaserod free base. To a mixture of AGP-HI (32.66 g, 0.12 mol) in 300 mL water was added 5-MICHO (10.51 g, 0.06 mol) followed by NaHCO3(20.16 g, 0.24 mol) and heated to reflux for 1 hour. After cooling, 150 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 20.4 g of tegaserod free base (91.55%) purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 2g of the resulting tegaserod free base in 8 mL MeOH was added a solution of maleic acid (1.28 g, 0.011 mol) in 5 mL MeOH. The resulting solid was filtered off and washed with MeOH to give 2.1 g of crude tegaserod maleate with a purity of 99.63 % (by HPLC).
Example 6- Preparation of Tegaserod maleate in water with Na2CO3. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 100 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by Na2CO3 (4.24 g, 0.04 mol) and heated to reflux for 1 hour. After cooling, 50 mL of EtOAc was added, and the organic phase washed with water. A solution of maleic acid (3.48 g, 0.03 mol) in 100 mL EtOAc was added, and the resulting solid was filtered off and washed with EtOAc to give 6.48 g of crude tegaserod maleate with a purity of 98.2 % (by HPLC).
Example 7- Preparation of Tegaserod maleate in MeOH with TEA in two steps. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL MeOH was added 5-MICHO (3.50 g, 0.02 mol) followed by triethylamine (11.0 mL, 0.08 mol). After 1 h at room temperature the mixture was evaporated to dryness, and washed with water, giving 5.79 g of tegaserod free base (86.90 % purity by HPLC). b. Preparation of tegaserod maleate
To a solution of 2 g of the resulting tegaserod free base in 10 mL MeOH was added a solution of maleic acid (1.16 g, 0.01 mol) in water. The resulting solid was filtrated and washed with water to give 1.45 g of crude tegaserod maleate as a white solid (94.60 % purity by HPLC). Crystallization in MeOH improved the purity to 98.94% by HPLC.
Example 8- Preparation of Tegaserod maleate in IPA with K2CO3.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL IPA was added 5-MICHO (3.50 g, 0.02 mol) followed by K2CO3 (5.53g, 0.04 mol). After 22 h at room temperature the mixture was washed with brine. The organic phase was treated with a solution of maleic acid (3.48 g, 0.03 mol) in IPA. The resulting solid was filtrated and washed with IPA to give 3.26 g of a white solid (98.97% purity by HPLC).
Example 9- Preparation of Tegaserod maleate in TEA.
To a mixture of AGP-HI (10.88 g, 0.04 mol) and 5-MICHO (3.50 g, 0.02 mol) was added 11 mL of TEA (0.08 mol). After 2 h at room temperature 25 mL of EtOAc were added and the mixture was stirred for 1 h. The resulting solid was filtrated and washed with 25 mL EtOAc, to give 5.7 g of crude.
2 g of the residue was dissolved in 13 mL MeOH and treated with 7 mL of a solution of maleic acid (2.7 g, 0.023 mol) in water. The resulting solid was filtered and washed with water to give 1.5 g of tegaserod maleate (99.26 % purity by HPLC). Crystallization of the solid in MeOH improved the purity to 99.89%) by HPLC.
Example 10- Preparation of Tegaserod maleate in toluene/water with NaHCO3. a. Preparation of tegaserod free base To a mixture of AGP-HI (10.88 g, 0.04 mol) in 200 mL of water/toluene 1:1 was added 5-MICHO (3.50 g, 0.02 mol) followed by NaHCO3 (6.72 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.25 g of tegaserod free base was obtained (93.8 % purity by HPLC). b. Preparation of tegaserod maleate To a solution of 3 g of the product in 10 mL MeOH was added a solution of maleic acid (2.31 g, 0.02 mol) in 10 mL water. The resulting solid was filtered off and washed with a solution of MeOH / water to give 2.50 g of crude tegaserod maleate with a purity of 96.6 % (by HPLC).
Example 11- Preparation of Tegaserod maleate in water with NaOH. a. Preparation of tegaserod free base
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 3 hours 50 mL of EtOAc was added, and the organic phase washed with water and evaporated to dryness to give 5.6 g of tegaserod free base (98.80% purity by HPLC). b. Preparation of Tegaserod maleate.
To a solution of 1.6 g of tegaserod free base in 15 mL ethyl acetate was added a solution of maleic acid (0.7 g, 0.006 mol) in 5 mL ethyl acetate. The resulting solid was filtered off and washed with ethyl acetate to give 1.65 g of crude tegaserod maleate, with a purity of 99.87 % (by HPLC)
Example 12- Preparation of Tegaserod maleate in water with maleic acid. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.3 g, 0.08 mol) and heated to reflux for 1 hour. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.92 g of tegaserod maleate crude was obtained (92.4 % purity by HPLC).
Example 13- Preparation of Tegaserod maleate in methanol with maleic acid.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of methanol was added 5- MICHO (3.50 g, 0.02 mol) followed by maleic acid (9.29 g, 0.08 mol) and heated to reflux for 2 hours. After cooling, the solid was filtrated out of the mixture and washed with water. After drying 6.51 g of tegaserod maleate crude was obtained (97.4 % purity by HPLC).
Example 14- Preparation of Tegaserod maleate in water with NaOH in one pot. To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL of water was added 5-MICHO (3.50 g, 0.02 mol) followed by NaOH (2 g, 0.05 mol) and stirred at room temperature. After 4 hours a solution of maleic acid (4.35 g, 0.0375 mol) in 25 mL water was added, and the reaction mixture was stirred overnight. The resulting solid was filtered off and washed with water to give 7.87 g of crude tegaserod maleate (99.16% purity by HPLC).
Example 15- Preparation of Tegaserod maleate in water with NaOH in one pot.
To a mixture of AGP-HI (174.2 g, 0.64 mol) in 362 mL of water was added 5-MICHO (56.2 g, 0.32 mol) followed by NaOH (68.1 g, 47%) and stirred at room temperature. After 4.5 hours, 640 mL of EtOAc was added, and the organic phase washed with water, treated with active carbon and filtrated through hyper flow bed. A solution of maleic acid (44.57 g, 0.38 mol) in 415 mL ethyl acetate / water 97:3 was added, and the reaction mixture was heating to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate to give 121.4 g of crude tegaserod maleate (up to 99.88 % purity by HPLC).
Example 16- Preparation of Tegaserod maleate (from Tegaserod acetate).
To a solution of 8.2 g of tegaserod acetate in 15 mL ethyl acetate heated to 65 °C was added a solution of 3.3 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for an additional 2 hours, followed by cooling to room temperature and stirring overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45 °C for 15 hours, 9.18 g of tegaserod maleate were obtained. Tegaserod acetate is prepared according to Examples 19, 20 and 21 of U.S. Appl. No. 11/015,875 and PCT/US04/42822.
Example 19 of U.S. Appl. No. 11/015,875 reads as follows: A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at 20- 30 °C for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 20 of U.S. Appl. No. 11/015,875 reads as follows:
A slurry of tegaserod base amorphous (6 g) in 50 mL ethyl acetate was stirred at reflux for 24 hours. The solid was filtrated and washed with 15 mL of same solvent and dried in a vacuum oven at 40 °C for 16 hours.
Example 21 of U.S. Appl. No. 11/015,875 reads as follows:
To a slurry of tegaserod maleate Form A (15 g) in EtOAc (210 mL) and water (210 mL) was added 38.4 g of NaOH 47%. The mixture was stirred overnight and the resulting white solid was isolated by filtration and washed with 100 mL of water. Drying in vacuum oven at 40 °C for 16 hours gives 12.38 g (90% yield). Tegaserod acetate was characterized by H and C-NMR.
Example 17: General method for the preparation of Tegaserod maleate Form A from crystallization.
Tegaserod maleate (1 g) was combined with the appropriate solvent (5 mL), and heated to reflux. Then, additional solvent was added until complete dissolution. After the compound was dissolved, the oil bath was removed and the solution was cooled to room temperature. The solid was filtrated and washed with 5 mL of the same solvent and dried in a vacuum oven at 40 C for 16 hours.


Example 18: Preparation of Tegaserod maleate in water with p-TSOH.
To a mixture of AGP-HI (10.88 g, 0.04 mol) in 25 mL water was added 5-MICHO (3.50 g, 0.02 mol) followed by para-toluenesulfonic acid monohydrate (0.45 g, 0.0024 mol). The mixture was heated to reflux for 4 hour and then cooled to room temperature. The resulting solid was filtered off and washed with water to give 8.32 g of a white solid (84.74 % purity by HPLC).
Example 19: Preparation of Tegaserod maleate from Tegaserod Hemi-maleate hemihydrate
To a solution of 1.72 g of Tegaserod Hemi-maleate hemihydrate in 20 mL ethyl acetate at room temperature was added a solution of 0.134 g maleic acid in 5 ml ethyl acetate/water 95:5, and the mixture was stirred at the same temperature for overnight. The resulting solid was filtered off and washed with ethyl acetate/water 95:5. After drying on vacuum oven at 45°C for 15 hours, 1.68 g of tegaserod maleate were obtained. Tegaserod Hemi-maleate hemihydrate was prepared according to Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822. Example 23 of U.S. Appl. No. 11/015,875 and PCT/US04/42822 reads as follows: A solution of maleic acid (2.32 g in 22 mL ethyl acetate/water 97:3) was added to a mixture of tegaserod base in ethyl acetate, and the reaction mixture was heated to 65 °C and stirrer overnight. The resulting solid was filtered off and washed with water and ethyl acetate. Drying in vacuum oven at 40 °C for 16 hours gives 12.19 g of Tegaserod hemi-maleate hemihydrate. Depending on the base polymorph used a solution or slurry is obtained. When using amorphous tegaserod base, a solution is obtained, while when using any other base polymorph of tegaserod, a slurry is obtained.
PATENT
https://patents.google.com/patent/WO2009063247A1/en
Tegaserod, chemically named 2-[(5-methoxy-liϊ-indol-3-yl)methylene]-IV-pentylhydrazine- carboximidamide, is a selective serotonin 4 (5-HT4) receptor agonist, which can be used to treat gastrointestinal disorders such as heartburn, bloating, postoperative ileus, abdominal pain and discomfort, epigastric pain, nausea, vomiting, regurgitation, intestinal pseudoobstruction, irritable bowel syndrome and gastro-oesophageal reflux. Tegaserod as the maleate salt is marketed for the short-term treatment of irritable bowel syndrome in women whose primary bowel symptom is constipation.
Tegaserod, represented by the formula (I), was first described in US 5 510 353 as well as processes for its preparation. The maleate salt of tegaserod is also disclosed, but interestingly a method of manufacturing tegaserod maleate is not disclosed. The only characterizing data is the melting point which is disclosed as 1900C for the maleate salt and 124°C for the tegaserod base.

WO 2006/116953 describes crystalline forms of the hydrobromide, dihydrogen phosphate and oxalate salts of tegaserod. Also claimed is a process for preparing the hydrochloride, hydrobromide, dihydrogen phosphate, tartrate, citrate, lactate, mesylate, oxalate, succinate, glutarate, adipate, salicylate, sulfate, mandelate, camphor sulfonate and hydrogen sulfate salts of tegaserod from a specific crystalline form of tegaserod base. Another process described is a method of preparing the dihydrogen phosphate, maleate, tartrate, citrate, mesylate, lactate, succinate, oxalate, hydrochloride, salicylate, glutarate, adipate, hydrobromide, sulfate and hydrogen sulfate from a hydrogen halide salt of tegaserod.
There are often major hurdles to overcome before an active pharmaceutical ingredient (API) can be formulated into a composition that can be marketed. For example, the rate of dissolution of an API that has poor aqueous solubility is often problematic. The aqueous solubility is a major influence on the bioavailability of the API such that a poorly soluble API can mean the API is not available to have a pharmaceutical effect on the body. The API can also cause problems during manufacture of a pharmaceutical composition. For example, flowability, compactability and stickiness are all factors affected by the solid state properties of an API.
It has thus always been an aim of the pharmaceutical industry to provide many forms of an API in order to mitigate the problems described above. Different salts, crystalline forms also known as polymorphs, solvates and amorphous forms are all forms of an API that can have different physiochemical and biological characteristics. Indeed, it has been discovered that the tegaserod maleate product on the market, Zelnorm , has been linked to an increase in heart problems in a proportion of individuals. One possible reason is that the maleate moiety reacts with the tegaserod, resulting over time in the production of a toxic impurity.
This impurity could be a contributor to the heart problems seen in some patients.
PATENT
https://patents.google.com/patent/WO2004085393A1/en
Figure 1 is a x-ray powder diffraction pattern of tegaserod maleate Form I. Figure 2 is a x-ray powder diffraction pattern of tegaserod maleate Form II. Figure 3 is a x-ray powder diffraction pattern of tegaserod maleate Form III. Figure 4 is a x-ray powder diffraction pattern of tegaserod maleate Form IV. x-Ray powder diffraction spectrum was measured on a Siemens D5000 x- ray powder diffractometer having a copper-Kα radiation.
The following examples further illustrate the invention.
Example 1 Tegaserod free base (10 gm) is dissolved in acetone (100 ml). Maleic acid (4 gm) is added to the solution and the contents are maintained for 1 hour at 25°C. The separated solid is filtered to give 12.5 gm of tegaserod maleate Form I.
Example 2 Tegaserod maleate Form II (5 gm) and acetone (70 ml) are mixed and refluxed for 1 hour and cooled to 25°C and filtered to give 4.8 gm of tegaserod maleate Form I.
Example 3 Tegaserod maleate Form I (10 gm) is dissolved in methanol (100 ml). Acetonitrile (150 ml) is added to the solution and the contents are heated to reflux. The contents are then cooled to 25°C and maintained for 30 minutes. The separated crystals are collected by filtration to give 9 gm of tegaserod maleate Form II.
Example 4 Tegaserod free base (10 gm) is dissolved in methanol (100 ml) and maleic acid (4 gm) is added to the solution. Then the contents are maintained for 30 minutes at 25°C. Then the separated solid is filtered to give 13 gm of tegaserod maleate Form III.
Example 5
Tegaserod maleate (5 gm) is dissolved in methanol (50 ml) and the solution is maintained at 25°C for 30 minutes. The separated crystals are collected by filtration to give 4.8 gm of tegaserod maleate Form III. Example 6 Tegaserod free base (10 gm) is dissolved in methanol (50 ml), maleic acid (4 gm) is added and the contents are refluxed for 30 minutes and then the resulting solution is cooled to 25°C. Methylene dichloride (200 ml) is added and the contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 13 gm of tegaserod maleate Form IV.
Example 7 Maleic acid (4 gm) is added to a solution of tegaserod free base (10 gm) in methanol (50 ml). The contents are maintained for 30 minutes at 25°C and isopropyl alcohol (150 ml) is mixed and contents are maintained for 30 minutes at 25°C. The separated solid is collected by filtration to give 12.5 gm of tegaserod maleate Form IV
CLIP
References
- ^ “New Data for Zelnorm”. Archived from the original on December 9, 2007. Retrieved March 30, 2007.
- ^ “FDA approves first treatment for women with irritable-bowel syndrome”. Archived from the original on February 5, 2007. Retrieved March 30, 2007.
- ^ Rossi, S. (2004). Australian Medicines Handbook. Adelaide: Health Communication Network. ISBN 0-9578521-4-2.
- ^ Beattie DT, Smith JA, Marquess D, et al. (November 2004). “The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo”. Br. J. Pharmacol. 143 (5): 549–60. doi:10.1038/sj.bjp.0705929. PMC 1575425. PMID 15466450.
- ^ “FDA Announces Discontinued Marketing of GI Drug, Zelnorm, for Safety Reasons”. FDA Press Release. 30 March 2007.
- ^ “Zelnorm” (PDF). Novartis. Archived from the original (PDF) on 2007-04-10. Retrieved 2007-03-30.
- ^ “Novartis suspends Canadian marketing and sales of Zelnorm in response to request from Health Canada”. Retrieved 2007-03-30.
- ^ Loughlin J, Quinn S, Rivero E, Wong J, Huang J, Kralstein J, Earnest DL, Seeger JD (2010). “Tegaserod and the Risk of Cardiovascular Ischemic Events: An Observational Cohort Study”. J Cardiovasc Pharmacol Ther. 15 (2): 151–7. doi:10.1177/1074248409360357. PMID 20200325.
- ^ http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm103223.htm
- Beattie DT, Smith JA, Marquess D, Vickery RG, Armstrong SR, Pulido-Rios T, McCullough JL, Sandlund C, Richardson C, Mai N, Humphrey PP: The 5-HT4 receptor agonist, tegaserod, is a potent 5-HT2B receptor antagonist in vitro and in vivo. Br J Pharmacol. 2004 Nov;143(5):549-60. Epub 2004 Oct 4. [PubMed:15466450]
- Talley NJ: Irritable bowel syndrome. Intern Med J. 2006 Nov;36(11):724-8. doi: 10.1111/j.1445-5994.2006.01217.x. [PubMed:17040359]
- Borman RA, Tilford NS, Harmer DW, Day N, Ellis ES, Sheldrick RL, Carey J, Coleman RA, Baxter GS: 5-HT(2B) receptors play a key role in mediating the excitatory effects of 5-HT in human colon in vitro. Br J Pharmacol. 2002 Mar;135(5):1144-51. doi: 10.1038/sj.bjp.0704571. [PubMed:11877320]
- Vickers AE, Zollinger M, Dannecker R, Tynes R, Heitz F, Fischer V: In vitro metabolism of tegaserod in human liver and intestine: assessment of drug interactions. Drug Metab Dispos. 2001 Oct;29(10):1269-76. [PubMed:11560869]
- FDA approves the reintroduction of Zelnorm™ (tegaserod) for Irritable Bowel Syndrome with Constipation (IBS-C) in women under 65 [Link]
- Tegaserod 2019 FDA Label [File]
- EMA Refusal Assessment Report for Zelnorm (Tegaserod) [File]
- FDA Joint Meeting of the Gastrointestinal Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee Briefing Document for Zelnorm (tegaserod maleate) [File]
Title
ANDRES: “Labelling of HTF919…” J.LABELLED COMP.RADIOPHARM., vol. 42, 1999, pages 1008-1009, XP002354977 *
BUCHHEIT K H ET AL: “THE SEROTONIN 5-HT4 RECEPTOR. 2. STRUCTURE-ACTIVITY STUDIES OF THE INDOLE CARBAZIMIDAMIDE CLASS OF AGONISTS” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 38, 1995, pages 2331-2338, XP000867864 ISSN: 0022-2623 cited in the application *
GRAUL A ET AL: “TEGASEROD MALEATE” DRUGS OF THE FUTURE, BARCELONA, ES, vol. 24, no. 1, 1999, pages 38-44, XP000874672 ISSN: 0377-8282 *
LALEZARI ET AL.: “Selective synthesis of …” J. HETEROCYCL. CHEM., vol. 8, 1971, pages 689-691, XP002354978 *
WAN ET AL.: “Improved synthesis of tegaserod maleate” CHINESE J. MED. CHEM., vol. 13, no. 1, 2003, pages 40-41, XP009057178 *
WO2004085393A1 *2003-03-252004-10-07Hetero Drugs LimitedNovel crystalline forms of tegaserod maleate
WO2006116953A1 *2005-05-022006-11-09Zentiva, A.S.A method for the preparation of tegaserod and slected salts thereof
WO2007084761A1 *2006-01-182007-07-26Teva Pharmaceutical Industries Ltd.Maleate salt of tegaserod and crystalline forms thereof
WO2007084761A1 *2006-01-182007-07-26Teva Pharmaceutical Industries Ltd.Maleate salt of tegaserod and crystalline forms thereof
WO2007120924A1 *2006-04-172007-10-25Teva Pharmaceutical Industries Ltd.Preparation of tegaserod maleate free of iodide
WO2007126889A1 *2006-03-272007-11-08Teva Pharmaceutical Industries Ltd.Preparation of tegaserod acetate
WO2007146717A3 *2006-06-122008-03-27Joginder S BajwaProcess for making salts of n-hydroxy-3-[4-[[[2-(2-methyl-1h-indol-3-yl)ethyl]amino]methyl]phenyl]-2e-2-propenamide
EP1955998A1 *2007-02-072008-08-13Chemo Ibérica, S.A.New addition salt of N-amino-N’-pentylguanidine, the process for its preparation and use thereof for obtaining tegaserod
CL2008000070A1 *2007-01-172008-07-25Lg Life Sciences LtdMaleic acid mono (3 – [({1 – [(2-amino-9 H -purin-9-yl) methyl] cyclopropyl} oxy) methyl] -8,8-dimethyl-3,7-dioxo-2,4 , 6-trioxa-3 lambda 5 -phosphanon-1-yl pivalate; pharmaceutical composition comprising said mono, and use to treat virus h
US5510353A *1991-03-221996-04-23Sandoz Ltd.Certain aminoguanidine compounds, pharmaceutical compositions containing them and their use in treating gastrointestinal motility disorders and disorders associated with cephalic pain
Family To Family Citations
WO2006116953A1 *2005-05-022006-11-09Zentiva, A.S.A method for the preparation of tegaserod and slected salts thereof
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Zelnorm, Zelmac |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 10% |
| Protein binding | 98% |
| Metabolism | Gastric and hepatic |
| Elimination half-life | 11 ± 5 hours |
| Excretion | Fecal and renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C16H23N5O |
| Molar mass | 301.39 g/mol g·mol−1 |
| 3D model (JSmol) | |
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5510353 | No | 1996-04-23 | 2013-04-26 |  |
References
-
- Buchheit, K.-H. et al.: J. Med. Chem. (JMCMAR) 38, 2331 (1995).
- US 5 510 353 (Novartis; 23.4.1996; GB-prior. 22.3.1991).
- EP 505 322 (Sandoz; GB-prior. 22.3.1991).
-
Preparation of 5-methoxyindole:
- Tsuji, Y. et al.: J. Org. Chem. (JOCEAH) 55 (2), 580 (1990).
- Jones, G.B. et al.: J. Org. Chem. (JOCEAH) 58 (20), 5558 (1993).
- Kondo, Y. et al.: J. Org. Chem. (JOCEAH) 62 (19), 6507 (1997).
- JP 3 024 055 (Kawaken Fine Chemicals; 1.2.1991; J-prior. 21.6.1989).
/////////Tegaserod, HTF 919, HTF-919, SDZ HTF 919, SDZ-HTF-919, テガセロド , Sloan Pharma, Novartis,
CCCCCNC(=N)N\N=C\C1=CNC2=C1C=C(OC)C=C2
BMS 986236
BMS-986236
CAS 2058035-15-5
MW C22 H25 N9 O
MF 431.49
1-(5-(4-(3-Hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
1H-Pyrazolo[3,4-b]pyridine-5-carbonitrile, 1-[5-[4-(3-hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl]-4-[(1-methylethyl)amino]-2-pyridinyl]-
1-[5-[4-(3-hydroxy-3-methylbutyl)triazol-1-yl]-4-(propan-2-ylamino)pyridin-2-yl]pyrazolo[3,4-b]pyridine-5-carbonitrile
|
The present invention generally relates to heteroaryl substituted aminopyridine compounds useful as kinase inhibitors, including the modulation of IRAK-4. Provided herein are heteroaryl substituted aminopyridine compounds, compositions comprising such compounds, and methods of their use. The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to kinase modulation and methods of inhibiting the activity of kinases, including IRAK-4 in a mammal.
|
PATENT
US2018186799
https://patentscope.wipo.int/search/en/detail.jsf?docId=US222843237&tab=PCTDESCRIPTION&maxRec=1000
PATENT
Gardner, D. S.; Santella, J. B.; Paidi, V. R.; Wu, H.; Duncia, J. V.; Nair, S. K.; Hynes, J. (BMS, USA). Heteroaryl Substituted Aminopyridine Compounds. PCT Int. Appl. WO/2016/210034 A1, 2016.
https://patents.google.com/patent/WO2016210034A1/en
Clip
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00023
Development of a Scalable Synthesis for the Potent Kinase Inhibitor BMS-986236; 1-(5-(4-(3-Hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
Pirama Nayagam Arunachalam†, Prakasam Kuppusamy†, Sivakumar Ganesan†, Suresh Krishnamoorthy†, Roshan Y. Nimje†, Lokesh Babu Jarugu†, Nanjundaswamy Kanikahalli Chikkananjaiah†, China Anki Reddy†, Prakash Anjanappa†, Murali Botlagunta†, Sridhar Vanteru†, Nageswararao Maddala†, Muniyappa Shankar†, Satheesh Nair†, John Hynes, Jr.‡, Joseph B. Santella, III‡, Percy H. Carter‡  , Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡
, Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡
, Richard Rampulla‡, Muthalagu Vetrichelvan*† , Anuradha Gupta† , Arun Kumar Gupta†, and Arvind Mathur‡† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Center, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bangalore-560 099, India
‡ Discovery Chemistry, Bristol-Myers Squibb, P.O. Box 5400, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.9b00023
*E-mail: muthalagu.vetrichelvan@syngeneintl.com.
A scalable route to 1-(5-(4-(3-hydroxy-3-methylbutyl)-1H-1,2,3-triazol-1-yl)-4-(isopropylamino)pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile (1, BMS-986236) was developed by incorporating an alternate azide intermediate following safety-driven processes. The newly developed process involved mitigating safety hazards and eliminating the column chromatography purification. The issue of trace metal contamination in the final API observed in the first-generation synthesis has been overcome.
1 (92.5 g, 73% yield, 99.5% purity by HPLC) as a cream-colored solid.
1H NMR (400 MHz, DMSO-d6) δ = 9.21–8.86 (m, 2H), 8.66 (s, 1H), 8.45–8.24 (m, 2H), 7.49 (s, 1H), 6.57 (d, J = 7.5 Hz, 1H), 4.33 (s, 1H), 3.83 (d, J = 7.0 Hz, 1H), 2.91–2.72 (m, 2H), 1.97–1.68 (m, 2H), 1.24 (d, J = 6.5 Hz, 12H).
13C NMR (100 MHz, DMSO) δ = 151.7, 150.8, 149.8, 147.9, 147.7, 143.7, 136.8, 136.3, 122.9, 118.9, 117.6, 116.0, 102.8, 99.4, 68.4, 43.6, 42.7, 29.2, 21.7, 20.2.
HRMS [M + H]+ calcd for C22H25N9O 432.2255, found 432.2259.



//////// BMS-986236, BMS 986236
CC(C)(O)CCc1cn(nn1)c2cnc(cc2NC(C)C)n4ncc3cc(cnc34)C#N
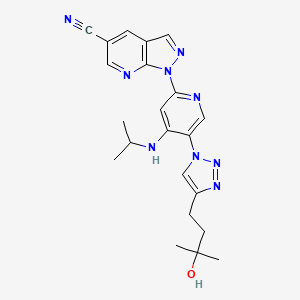
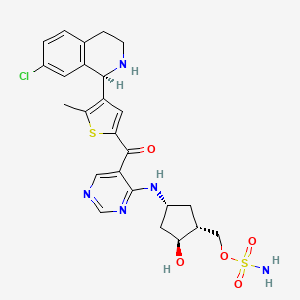
TAK-981

TAK-981
C25 H28 Cl N5 O5 S2, 578.103
[(1R,2S,4R)-4-[(5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl)amino]-2-hydroxycyclopentyl]methyl sulfamate
[(1R,2S,4R)-4-[[5-[4-[(1R)-7-Chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methyl-thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate
Sulfamic acid, [(1R,2S,4R)-4-[[5-[[4-[(1R)-7-chloro-1,2,3,4-tetrahydro-1-isoquinolinyl]-5-methyl-2-thienyl]carbonyl]-4-pyrimidinyl]amino]-2-hydroxycyclopentyl]methyl ester
| CAS 1858276-04-6 FREE
CAS 1858279-63-6 HYDRATE |
|
| MW | 578.103 |
- Originator Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Small ubiquitin-related modifier protein inhibitors
- Phase I Lymphoma; Solid tumours
- 01 Oct 2018 Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) and and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in USA (IV) (NCT03648372)
- 03 Sep 2018 Takeda Oncology plans a phase I trial for Solid tumours (Late-stage disease, Metastatic disease) and Lymphoma (Refractory metastatic disease, Second-line therapy or greater) in September 2018 (IV) (NCT03648372)
- 03 Sep 2018 Preclinical trials in Lymphoma in USA (IV) prior to September 2018 (NCT03648372)
Takeda is evaluating TAK-981, a SUMO-Activating Enzyme (SAE) inhibitor, in early clinical trials for the treatment of adult patients with advanced or metastatic solid tumors or with relapsed or refractory lymphomas.
Small ubiquitin-like modifier (SUMO) is a member of the ubiquitin-like protein (Ubl) family that is covalently conjugated to cellular proteins in a manner similar to Ub-conjugation (Kerscher, O., Felberbaum, R., and Hochstrasser, M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 22: 159-80). Mammalian cells express three major isoforms: SUMO l , SUM02 and SUM03. SUM02 and SUM03 share -95% amino acid sequence homology but have -45% sequence homology with SUMO l (Kamitani, T., Kito, K., Nguyen, H. P., Fukuda-Kamitani, T., and Yeh, E. T. 1998. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 273( 18): 1 1349-53). SUMO proteins can be conjugated to a single lysine residue of a protein (monosumoylation) or to a second SUMO protein that is already conjugated to a protein forming a SUMO chain (polysumoylation). Only SUM02/3 can form such chains because they possess internal consensus SUMO modification sites (Tatham, M. H., Jaffray, E., Vaughan, O. A., Desterro, J. M., Botting, C. H., Naismith, J. H., Hay, R. T. 2001. Polymeric chains of SUMO-2 and SUM 0-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 276(38):35368-74). An additional isoform, SUM04, is found in kidney, lymph node and spleen cells, but it is not known whether SUM04 can be conjugated to cellular proteins.
[0003] SUMO l , SUM02 and SUM03 are activated in an ATP-dependent manner by the SUMO-activating enzyme (SAE). SAE is a heterodimer that consists of SAE 1 (SUMO-activating enzyme subunit 1) and SAE2 (UBA2). SAE, like other El activating enzymes, uses ATP to adenylate the C-terminal glycine residue of SUMO. In a second step, a thioester intermediate is then formed between the C-terminal glycine of SUMO and a cysteine residue in SAE2. Next, SUMO is transferred from the El to the cysteine residue of the SUMO conjugating enzyme (E2), UBC9. Unlike the Ub pathway that contains many E2 enzymes, Ubc9 is currently the only known conjugating enzyme for SUMO and functions with SUMOl , SUM02 and SUM03 proteins. SUMO proteins are then conjugated to the target protein, either directly or in conjunction with an E3 ligase, through isopeptide bond formation with the epsilon amino group of a lysine side chain on a target protein. Several SUMO E3 ligases, including PIAS (protein inhibitor of activated signal transducer and activator of transcription protein) proteins and Ran-binding protein 2 (RanBP2), and polycomb 2 (Pc2), have been identified (Johnson, E. S., and Gupta, A. A. 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 106(6):735-44; Pichler, A., Gast, A., Seeler, J. S., Dejean, A.; Melchior, F. 2002. The nucleoporin RanBP2 has SUMOl E3 ligase activity. Cell. 108(1): 109-20; Kagey, M. H., Melhuish, T. A., and Wotton, D. 2003. The polycomb protein Pc2 is a SUMO E3. Cell. 1 13(1): 127- 37). Once attached to cellular targets, SUMO modulates the function, subcellular localization, complex formation and/or stability of substrate proteins (Miiller, S., Hoege, C, Pyrowolakis, G., and Jentsch, S. 2001. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2(3):202-10). SUMO- conjugation is reversible through the action of de-sumoylating enzymes called SENPs (Hay, R. T. 2007. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17(8):370-6) and the SUMO proteins can then participate in additional conjugation cycles.
[0004] SAE-initiated SUMO-conjugation plays a major role in regulating diverse cellular processes, including cell cycle regulation, transcriptional regulation, cellular protein targeting, maintenance of genome integrity, chromosome segregation, and protein stability (Hay, R. T. 2005. SUMO: a history of modification. Mol Cell. 18( 1): 1 -12; Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18(17):2046-59). For example, SUMO- conjugation causes changes in the subcellular localization of RanGAPl by targeting it to the nuclear pore complex (Mahajan, R., Delphin, C., Guan, T., Gerace, L., and Melchior, F. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAPl to nuclear pore complex protein RanBP2. Cell. 88(1):97- 1070). Sumoylation counteracts ubiquitination and subsequently blocks the degradation of Ι Β, thereby negatively regulating NF-κΒ activation (Desterro, J. M., Rodriguez, M. S., Hay, R. T. 1998. SUMO- 1 modification of IkappaB alpha inhibits NF-kappaB activation. Mol Cell. 2(2):233-9). Sumoylation has been reported to play an important role in transcription exhibiting both repressive and stimulatory effects. Many of the transcriptional nodes that are modulated play important roles in cancer. For example, sumoylation stimulates the transcriptional activities of transcription factors such as p53 and HSF2 (Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., and Hay, R. T. 1999. SUMO- 1 modification activates the transcriptional response of p53. EMBO J. 18(22):6455-61 ; Goodson, M. L., Hong, Y., Rogers, R., Matunis, M. J., Park-Sarge, O. K., Sarge, K. D. 2001. Sumo- 1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J Biol Chem. 276(21 ): 18513-8). In contrast, SUMO-conjugation represses the transcriptional activities of transcription factors such as LEF (Sachdev, S., Bruhn, L., Sieber, H., Pichler, A., Melchior, F., Grosschedl, R. 2001. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15(23):3088- 103) and c-Myb (Bies, J., Markus, J., and Wolff, L. 2002. Covalent attachment of the SUMO- 1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. / Biol Chem. 277( 1 1):8999-9009). Thus, SUMO-conjugation controls gene expression and growth control pathways that are important for cancer cell survival.
[0005] Altered expression of SAE pathway components have been noted in a variety of cancer types: (Moschos, S. J., Jukic, D. M., Athanassiou, C., Bhargava, R., Dacic, S., Wang, X., Kuan, S. F., Fayewicz, S. L., Galambos, C., Acquafondata, M., Dhir, R., and Becker, D. 2010. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum Pathol. 41(9): 1286-980); including multiple myeloma (Driscoll, J. J., Pelluru, D., Lefkimmiatis, K., Fulciniti, M., Prabhala, R. H., Greipp, P. R., Barlogie, B., Tai, Y. T., Anderson, K. C, Shaughnessy, J. D. Jr., Annunziata, C. M., and Munshi, N. C. 2010. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 1 15(14):2827-34); and breast cancer (Chen, S. F., Gong, C, Luo, M., Yao, H. R., Zeng, Y. J., and Su, F. X. 201 1. Ubc9 expression predicts chemoresistance in breast cancer. Chin J Cancer. 30(9):638-44), In addition, preclinical studies indicate that Myc-driven cancers may be especially sensitive to SAE inhibition (Kessler, J. D., Kahle, K. T., Sun, T., Meerbrey, K. L., Schlabach, M. R., Schmitt, E. M., Skinner, S. O., Xu, Q., Li, M. Z., Hartman, Z. C, Rao, M., Yu, P., Dominguez-Vidana, R., Liang, A. C, Solimini, N. L., Bernardi, R. J., Yu, B., Hsu, T., Golding, I., Luo, J., Osborne, C. K., Creighton, C. J., Hilsenbeck, S. G., Schiff, R., Shaw, C. A., Elledge, S. J., and Westbrook, T. F. 2012. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 335(6066):348-53; Hoellein, A., Fallahi, M., Schoeffmann, S., Steidle, S., Schaub, F. X., Rudelius, M., Laitinen, I., Nilsson, L., Goga, A., Peschel, C, Nilsson, J. A., Cleveland, J. L., and Keller, U. 2014. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood. 124( 13):2081 -90). Since SUMO-conjugation regulates essential cellular functions that contribute to the growth and survival of tumor cells, targeting SAE could represent an approach to treat proliferative disorders such as cancer.
[0006] SAE inhibitors may also be applicable for the treatment of other diseases and conditions outside of oncology. For example, SUMO modifies proteins that play important roles in neurodegenerative diseases (Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C, Slepko, N., Hies, K., Lukacsovich, T., Zhu, Y. Z., Cattaneo, E., Pandolfi, P. P., Thompson, L. M., Marsh, J. L. 2004. SUMO modification of Huntington and Huntington’s disease pathology. Science. 304(5667): 100-4); Dorval, V., and Fraser, P. E. 2006. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 281 ( 15):9919-24; Ballatore, C, Lee, V. M., and Trojanowski, J. Q. 2007. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 8(9):663-72). Sumoylation also has been reported to play important role in pathogenic viral infection, inflammation and cardiac function (Lee, H. R., Kim, D. J., Lee, J. M., Choi, C. Y., Ahn, B. Y., Hayward, G. S., and Ahn, J. H. 2004. Ability of the human cytomegalovirus ΓΕ1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. / Virol. 78(12):6527-42; Liu, B., and Shuai, K. 2009. Summon SUMO to wrestle with inflammation. Mol Cell. 35(6):731-2; Wang, J., and Schwartz, R. J. 2010. Sumoylation and regulation of cardiac gene expression. Circ Rei. l07( l): 19-29). [0007] It would be beneficial therefore to provide new SAE inhibitors that possess good therapeutic properties, especially for the treatment of proliferative, inflammatory, cardiovascular and neurodegenerative disorders.
PATENT
WO 2016004136

https://patents.google.com/patent/WO2016004136A1/en
Example 133: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxy-cyclopentyl]methyl sulfamate I-263a

Step 1: 7-Chloro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-l,2,3,4-tetrahydroisoquinoline
[00714] An oven-dried 2-neck 250 mL round bottom flask under nitrogen was charged with THF (40 mL) and cooled to -74 °C . Added 2.50 M ra-BuLi in hexane (6.92 mL, 17.3 mmol). Added a solution of Int-1 (4.00 g, 16.0 mmol) in THF (60 mL) slowly keeping the internal temperature less than -70 °C . Stirred with cooling 5 min. A second oven-dried 250 mL round bottom flask under nitrogen was charged with THF (60 mL) and Int-50 (2.04 g, 12.4 mmol) and the resulting solution was cooled to 0 °C . Added boron trifluoride diethyl ether complex ( 1.71 mL, 13.6 mmol) slowly and cooled to -30 °C . The contents of the first flask were transferred via cannula to the second flask. Reaction was quenched with saturated aqueous NaHC03 and warmed to rt. Water was added, and the mixture was extracted three times with EtOAc. Combined organic portions were washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. Residue was purified via flash column chromatography eluting with a hexane / EtOAc gradient (0 to 100% EtOAc) to afford the title compound as a white solid ( 1.88g, 45%). Ή NMR (400 MHz, Chloroform-d) δ 7.17 – 7.01 (m, 2H), 6.83 – 6.61 (m, 2H), 5.92 (s, 1H), 5.09 (s, 1H), 4.17 – 4.04 (m, 2H), 4.03 – 3.92 (m, 2H), 3.37 – 3.25 (m, 1H), 3.13 – 2.91 (m, 2H), 2.82 – 2.69 (m, 1H), 2.46 (s, 3H). LCMS: (AA) M+l 336.1
Step 2: ieri-Butyl 7-chIoro-l-[5-(l,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3,4-dihydroisoquinoIine -2(lH)-carboxyIate [00715] A 50 mL round bottom flask under nitrogen was charged with 7-chloro-l -[5-(l ,3-dioxolan-2- yl)-2-methyl-3-thienyl]- l ,2,3,4-tetrahydroisoquinoline (5.67 g, 16.9 mmol) and DCM ( 100 mL), to which was added triethylamine (4.71 mL, 33.8 mmol), di-ieri-butyldicarbonate (4.61 g, 21.1 mmol), and N,N-dimethylaminopyridine (23 mg, 0.18 mmol). Reaction was stirred for 1 h at rt and then poured into saturated NaHC03 solution. Mixture was extracted three times with DCM, and the combined organic portions were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford 6.96g (95%) of the title compound. LCMS: (AA) M+ l 436.1
Step 3: tert-Butyl 7-chloro-l-(5-formyl-2-methyl-3-thienyl)-3,4-dihydroisoquinoline -2(1H)- carboxylate
[00716] A 1 L round bottom flask was charged with ferf-butyl 7-chloro-
1 -[5-( 1 ,3-dioxolan-2-yl)-2-methyl-3-thienyl]-3 ,4-dihydroisoquinoline-2( 1 H)-carboxylate (7.30 g, 16.7 mmol), methanol (200 mL), and water (20 mL), to which was added a solution of 12M HC1 (4.00 mL, 130 mmol) in methanol (200 mL), and the reaction was stirred at rt for 1 h. Reaction was quenched via addition of 50mL of saturated NaHC03 and stirred for 5 min. Methanol was removed in vacuo, and the resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.55g, 70%). Ή NMR (400 MHz, Chloroform-d) δ 9.67 (s, 1 H), 7.27 – 7.15 (m, 2H), 7.12 (s, 1 H), 6.98 – 6.94 (m, 1 H), 6.34 (m, l H), 4.15 (s, 1 H), 3.18 – 3.06 (m, 1 H), 3.05 – 2.93 (m, 1H), 2.82 – 2.73 (m, 1 H), 2.69 (s, 3H), 1.50 (s, 9H). LCMS: (AA) M+Na 414.2
Step 4: tert-Butyl 7-chIoro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyI]-2-methyl-3-thienyl}- 3,4-dihydroisoquinoline-2(lH)-carboxylate
[00717] An oven-dried 500 mL 3-neck round bottom flask under nitrogen was charged with 4-chloro- 5-iodopyrimidine (4.08 g, 17.0 mmol) and 2-methyltetrahydrofuran ( 150 mL). An addition funnel containing a solution of rert-butyl 7-chloro- l -(5-formyl-2-methyl-3-thienyl)-3,4- dihydroisoquinoline-2(l H)-carboxylate (4.75 g, 12.1 mmol) in 2-methyltetrahydrofuran (50 mL) was attached, and the contents of the reaction flask were cooled to -75 °C . 2.50 M n-BuLi in hexane ( 14.1 mL, 35.2 mmol) was added in small portions keeping the internal temperature less than -70 °C , at which point the contents of addtion funnel were added in a single portion. Upon completion of addition, the reaction was quenched by adding 20 mL of saturated NaHC03 in small portions and warmed to rt. The aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (4.85g, 79%). LCMS: (AA) M+Na 528.1
Step 5: tert-Butyl 7-chloro-l-{5-[(4-chloropyrimidin-5-yl)(hydroxy)methyl]-2-methyl-3-thienyl}- 3,4- dihydroisoquinoline-2(lH)-carboxylate
[00718] A 1 L round bottom flask was charged with fe/Y-butyl 7-chloro- l – { 5-[(4-chloropyrimidin-5- yl)(hydroxy)methyl]-2-methyl-3-thienyl}-3,4-dihydroisoquinoline-2(l H)-carboxylate (4.85 g, 9.58 mmol) and DCM (300 mL). Manganese (IV) oxide (14.2 g, 163 mmol) was added and the reaction was stirred at rt for 18 h. Mixture was filtered through Celite, and the filter cake was rinsed with hot EtOAc. Filtrate was concentrated in vacuo to afford the title compound (4.47g , 93%). Ή NMR (400 MHz, Chloroform-d) δ 9.09 (s, 1 H), 8.70 (s, 1 H), 7.24 – 7.16 (m, 1 H), 7.16
– 7.07 (m, 1 H), 7.00 – 6.90 (m, 2H), 6.32 (s, 1 H), 4.28 – 3.97 (m, 1H), 3.14 – 2.89 (m, 2H), 2.78
– 2.65 (m, 4H), 1 .53 – 1.43 (m, 9H).
Step 6: tert-Butyl (lR)-7-chloro-l-[5-[4-[[(lR,3R,4S)-3-(hydroxymethyl)-4-triisopropylsiIyloxy- cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dihydro-lH-isoquinoline-2- carboxylate
[00719] A 1 L round bottom flask under nitrogen was charged with iert-butyl 7-chloro- l – { 5-[(4- chloropyrimidin-5-yl)carbonyI]-2-methyl-3-thienyl }-3,4-dihydroisoquinoline-2( l H)-carboxylate (4.47 g, 8.86 mmol), DMF (20.0 mL, 258 mmol), Int-259 (3.06 g, 10.6 mmol), and triethylamine (3.09 mL, 22.2 mmol) and the mixture was stirred at rt for 18 h. Reaction mixture was poured into water and saturated NaHC03, and then extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a 70/30 to 60/40 hexane/EtOAc gradient to afford 0.56g of first-eluting diastereomer 1 (not pictured), 4.3 l g of a mixture of diastereomers, and 1.1 lg ( 17%) of second-eluting diastereomer 2 (the title compound). The mixture of diastereomers thus obtained was resubjected to the described chromatography conditions two additional times to afford a total of 2.62 g of the desired diastereomer. Ή NMR (400 MHz, Methanol-d4) δ 8.54 – 8.46 (m, 2H), 7.27 – 7.19 (m, 2H), 7.09 – 6.99 (m, 2H), 6.37 (s, 1H), 4.87 – 4.75 (m, 1H), 4.38 – 4.29 (m, 1H), 4.20 – 4.09 (m, 1H), 3.66 – 3.52 (m, 2H), 3.28- 3.14 (m, 2H), 3.02 – 2.89 (m, 1 H), 2.89 – 2.78 (m, 1 H), 2.68 (s, 3H), 2.54 – 2.41 (m, 1 H), 2.22 – 2.09 (m, 2H), 1.86 – 1.73 (m, 1H), 1.50 (s, 8H), 1.39 – 1.23 (m, 2H), 1.15 – 1.04 (m, 20H).
LCMS: (AA) M+ 1 755.3
Step 7: tert-Butyl (lR)-7-chloro-l-[2-methyl-5-[4-[[(lR,3R,4S)-3-(sulfamoyloxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3-thienyl]-3,4-dihydro-lH- isoquinoline-2-carboxylate [00720] A solution of ie/t-butyl (lR)-7-chloro-l-[5-[4-[[( lR,3R,4S)-3-(hydroxymethyl)-4- triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-2-methyl-3-thienyl]-3,4-dih lH-isoquinoline-2-carboxylate (2.46 g, 3.26 mmol) in 2-methyltetrahydrofuran (25 mL), and DMF (25 mL) was cooled to 0 °C. Triethylamine ( 1.82 mL, 13.0 mmol) and chlorosulfonamide (1.50 g, 13.0 mmol) were added and the reaction was stirred for 10 min. Added methanol (0.53 mL, 13.0 mmol) and stirred for 15 min. Reaction mixture was poured into saturated NaHC03, extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a hexane / EtOAc gradient to afford the title compound (2.41g, 89%). Ή NMR (400 MHz, Methanol-d4) δ 8.58 – 8.45 (m, 2H), 7.29 – 7.17 (m, 2H), 7.1 1 – 6.98 (m, 2H), 6.36 (s, 1 H), 4.84 – 4.73 (m, 1H), 4.44 – 4.33 (m, 1H), 4.21 – 4.08 (m, 4H), 3.27- 3.17 (m, 1 H),3.02 – 2.89 (m, 1 H), 2.88 – 2.78 (m, 1 H), 2.67 (s, 3H), 2.57 – 2.47 (m, 1 H), 2.41 – 2.30 (m, 1 H), 2.23 – 2.13 (m, 1 H), 1.87- 1.78 (m, 1 H), 1.50 (s, 9H), 1.43 – 1 .33 (m, 1 H), 1 .17 – 1.04 (m, 20H). LCMS: (AA) M+l 834.3
Step 8: [(lR,2S,4R)-4-[[5-[4-[(lR)-7-Chloro-l,2,3,4-tetrahydroisoquinolin-l-yl]-5-methyl- thiophene-2-carbonyl]pyrimidin-4-yI]aniino]-2-hydroxy-cyclopentyl]methyl sulfamate
[00721] A solution of f«?r/-butyl ( l R)-7-chloro- l -[2-methyl-5-[4-[[( l R,3R,4S)-3-
(sulfamoyloxymethyl)-4-triisopropylsilyloxy-cyclopentyl]amino]pyrimidine-5-carbonyl]-3- thienyl]-3,4-dihydro- l H-isoquinoline-2-carboxylate (2.41 g, 2.89 mmol) in CH3CN ( 10 mL) was cooled in an ice bath to + 1 °C . Phosphoric acid ( 10 mL, 200 mmol) was added dropwise and the reaction was stirred with ice bath cooling for 60 min. The mixture was warmed to rt and stirred for an additional 3 h. Reaction was poured into a stirring mixture of 50 mL water and 50 mL EtOAc, and the the pH was adjusted to ~9 by slowly adding 200 mL of saturated NaHC03 with stirring. Resulting aqueous mixture was extracted three times with EtOAc, and then the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated in vacuo. The residue was subjected to flash column chromatography eluting with a gradient that began with 100% DCM and increased in polarity to 80% DCM / 20% methanol / 2% ammonium hydroxide gradient to afford the title compound (1.50 g, 90%). Ή NMR (400 MHz, Methanol-d4) δ 8.61 (s, 1H), 8.52 (s, 1 H), 7.27 (s, 1 H), 7.18 – 7.13 (m, 2H), 6.73 – 6.68 (m, 1 H), 5.23 (s, 1H), 4.81 – 4.70 (m, 1 H), 4.26 – 4.10 (m, 3H), 3.29 – 3.23 (m, 2H), 3.1 1 – 2.96 (m, 2H), 2.87 – 2.76 (m, 1H), 2.60 (s, 3H), 2.55 – 2.42 (m, 1 H), 2.33 – 2.19 (m, 1H), 2.18 – 2.07 (m, 1H), 1.95 – 1.81 (m, 1H), 1.47 – 1.35 (m, 1 H). LCMS: (AA) M+l 580.0
CLIP

Credit: Tien Nguyen/C&EN
Presenter: Steven Paul Langston, associate director at Takeda Pharmaceuticals International
Target: Sumo activating enzyme
Disease: Solid tumors
Reporter’s notes: Langston gave the last talk of the morning session, placing him in the “precarious position of being between you and lunch,” he said. Takeda acquired this drug development program, falling under the umbrella of immuno-oncology, along with Millenium Pharmaceuticals in 2008. The team targeted a pathway known as SUMOylation, a protein post translation modification that is implicated in a number of cellular processes including immune response. In SUMOylation, enzymes attach a small protein to another protein. They found that inhibiting this pathway activates a type I interferon response in immune cells. How the molecule, TAK-981, inhibits this pathway is quite complicated, Langston said. TAK-981 forms an adduct with a small ubiquitin like modifier (SUMO) to inhibit a SUMO activating enzyme that catalyzes SUMOylation. While the synthesis of TAK-981 is fairly short, it requires a nonideal chiral chromatography separation after the first step. TAK-981 is in Phase I clinical trials as an intravenous infusion for patients with metastatic solid tumors or lymphomas.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2018311239 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2018-03-16 | |
| US9962386 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2017-04-17 | |
| US9683003 | HETEROARYL COMPOUNDS USEFUL AS INHIBITORS OF SUMO ACTIVATING ENZYME | 2015-06-30 | 2016-01-14 |
//////////TAK-981, TAK 981, Phase I, Lymphoma, Solid tumours, TAKEDA,
Cc3sc(cc3[C@@H]1NCCc2ccc(Cl)cc12)C(=O)c5cncnc5N[C@@H]4C[C@H](COS(N)(=O)=O)[C@@H](O)C4
BIIB-095

BIIB-095
ROTATION (+)
1493790-64-9 CAS free form,
1493772-48-7 cas Hcl salt
cas 1493790-65-0, 1496563-32-6 ,SULPHATE ???
cas 1496563-31-5 SULFATE 1;1
cas 1496563-32-6 SULFATE HYDRATE 1;1;1
(2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one
1,7-Diazaspiro[4.4]nonan-6-one, 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)phenyl]-2-pyrimidinyl]-, (2R,5S)-
C20 H21 F3 N4 O, 390.40
- Originator Biogen
- Class Analgesics
- Mechanism of Action Nav1.7 voltage-gated sodium channel inhibitors
- Phase I Neuropathic pain
- 29 Mar 2018 Phase-I clinical trials in Neuropathic pain (In volunteers) in United Kingdom (PO) (NCT03454126)
- 05 Mar 2018 Biogen plans a phase I trial for Pain, including Neuropathic pain (In volunteers) in USA (PO) (NCT03454126)
- 05 Mar 2018 Preclinical trials in Neuropathic Pain in USA (PO), before March 2018
In March 2018, a randomized, double blind, placebo controlled, single and multiple-ascending dose, dose-escalation phase I study ( NCT03454126; 255HV101; 2017-003982-90) was initiated in the UK in healthy subjects (expected n = 80) to evaluate the safety, tolerability and pharmacokinetics of BIIB-095. At that time, the trial was expected to complete in December 2018
Biogen is developing BIIB-095, a voltage-gated sodium channel 1.7 inhibitor, for the potential oral treatment of neuropathic pain [2027279], [2027426]. In March 2018, a phase I trial was initiated in healthy subjects
Biogen is developing oral agent BIIB-095 for the treatment of chronic pain, including neuropathic pain. A phase I clinical trial is under way in healthy volunteers.
The compound was first claimed in WO2013175205 , for treating schizophrenia, assigned to subsidiary Convergence Pharmaceuticals Limited , naming some of the inventors. This might present the structure of BIIB-095 , a voltage-gated sodium channel 1.7 inhibitor, being developed by Biogen for the oral treatment of neuropathic pain; in March 2018, a phase I trial was initiated in healthy subjects.
PATENT
WO2013175205

CONTD………………

INTERMEDIATE

WO 2013175206
US 20150119404
https://patents.google.com/patent/US20150119404
Patent
WO-2019067961
Novel salts (citrate, mesylate, hydrosulfate, saccharinate and oxalate) forms of 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one, processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating diseases and conditions mediated by modulation of voltage-gated sodium channels.
Voltage-gated sodium channels are responsible for the initial phase of the action potential, which is a wave of electrical depolarisation usually initiated at the soma of the neuron and propagated along the axon to the terminals. At the terminals, the action potential triggers the influx of calcium and the release of neurotransmitter. Drugs, such as lidocaine, that block voltage-gated sodium channels are used as local anaesthetics. Other sodium channel blockers, such as lamotrigine and carbamazepine are used to treat epilepsy. In the latter case, partial inhibition of voltage-gated sodium channels reduces neuronal excitability and reduces seizure propagation. In the case of local anaesthetics, regional block of sodium channels on sensory neurons prevents the conduction of painful stimuli. A key feature of these drugs is their state-dependent mechanism of action. The drugs are thought to stabilise an inactivated conformation of the channel that is adopted rapidly after the channel opens. This inactivated state provides a refractory period before the channel returns to its resting (closed) state ready to be reactivated. As a result, state-dependent sodium channel blockers inhibit the firing of neurons at high frequency, for example in response to painful stimuli, and will help to prevent repetitive firing during periods of prolonged neuronal depolarisation that might occur, for example, during a seizure. Action potentials triggered at lower frequencies, for example in the heart, will not be significantly affected by these drugs, although the safety margin differs in each case, since at high enough concentrations each of these drugs is capable of blocking the resting or open states of the channels.
The voltage-gated sodium channel family is made up of 9 subtypes, four of which are found in the brain, NaV1.1 , 1.2, 1.3 and 1.6. Of the other subtypes, NaV1.4 is found only in skeletal muscle, NaV1.5 is specific to cardiac muscle, and NaV1.7, 1.8, and 1.9 are found
predominantly in sensory neurons. The hypothesised binding site for state-dependent sodium channel blockers is the local anaesthetic (LA) binding site in the inner vestibule of the pore on transmembrane S6 of domain IV. Critical residues are located in a highly conserved region among the different subtypes, thus presenting a challenge for the design of new subtype selective drugs. Drugs such as lidocaine, lamotrigine and carbamazepine do not distinguish between the subtypes. However, selectivity can be achieved, and can be further enhanced functionally, as a result of the different frequencies at which the channels operate.
Drugs that block voltage-gated sodium channels in a state-dependent manner are also used in the treatment of bipolar disorder, either to reduce symptoms of mania or depression, or as mood stabilisers to prevent the emergence of mood episodes. Clinical and preclinical evidence also suggests that state-dependent sodium channel blockers may help to reduce the symptoms of schizophrenia. For example, lamotrigine has been shown to reduce symptoms of psychosis induced by ketamine in healthy human volunteers, and furthermore, studies in patients suggest that the drug can augment the antipsychotic efficacy of some atypical antipsychotic drugs, such as clozapine or olanzapine. It is hypothesised that efficacy in these psychiatric disorders may result in part from a reduction of excessive glutamate release. The reduction in glutamate release is thought to be a consequence of sodium channel inhibition in key brain areas, such as the frontal cortex. However, interaction with voltage-gated calcium channels may also contribute to the efficacy of these drugs.
WO 2013/175205 (Convergence Pharmaceuticals Limited) describes (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one hydrochloride, sulfuric acid salt and sulfuric acid salt hydrate which are claimed to be modulators of voltage-gated sodium channels. The object of the invention is to identify alternative salts of said compound which have advantageous properties.
Example 1
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of citric acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 30 minutes a seed of citrate salt (0.1 wt%) was added and the mixture allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a very thick suspension (white) that required mobilisation with 20 ml additional ethanol and a further maturation period of 2 hours at ambient temperature. Filtration was carried out under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 6.0 g of crystalline white solid (91 % yield).
H NMR (400MHz, DMSO-D6): δΗ 1.90-2.05 (2H, m), 2.10-2.20 (2H, m,), 2.20-2.30 (1 H, m), -2.50 (1 H, m, partially masked by solvent)), 2.55-2.68 (4H, m), 2.56 (3H, s), 2.79 (3H, s),
3.28-3.40 (2H, m), 4.79 (1 H, t, J= 8.0 Hz), 7.92 (2H, d, J = 8.4 Hz), 8.03 (1 H, s), 8.45 (2H, d, J= 8.8Hz) ppm, (exchangeables not reported)
Characterisation of Example 1
The XRPD of Example 1 is presented in FIG. 1 and the DSC/TGA of Example 1 is presented in FIG. 2. The citrate salt of Example 1 displayed the following characteristics:
1 endotherm onset: 171.82°C
peak maximum: 174.55°C
There was an endotherm post the main endotherm.
There was no weight reduction until ca 168°C had been reached. The weight reduction commenced with the start of the main endotherm and coincided with the endotherm post the main endotherm which indicated that this thermal event was the onset of compound decomposition and loss of citric acid. Thermal events >220°C were due to compound decomposition.
The XPRD data in FIG. 1 demonstrated that under different extremes of humidity indicate a stable crystalline form of the citrate salt of Example 1 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 2 which show clear transitions and no evidence of solvates.
Aqueous solubility of the citrate salt (Example 1) = 22mg/ml (25°C).
Example 2
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one ) salt (E2)
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of methanesulfonic acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 10 minutes nucleation and gradual crystallisation was noted to afford a thick mixture. Additional ethanol was added (10 ml) to mobilise the suspension that was then allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a thin, mobile suspension (white) that was filtered under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 4.0 g of crystalline white solid (72% yield).
H NMR (400MHz, DMSO-D6): δΗ 2.1-2.45 (4H, m), 2.27 (3H, s), 2.50-2.75 (2H, m), 2.61 (3H, s), 2.86 (3H, s), 3.35-3.50 (2H, m), 5.20 (1 H, t, J = 8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.17 (1 H, s), 8.51 (2H, d, J = 8.4Hz), 9.45 (1 H, br), 10.16 (1 H, br) ppm.
Characterisation of Example 2
The XRPD of Example 2 is presented in FIG. 3 and the DSC/TGA of Example 2 is presented in FIG. 4. The DSC thermograph of the methanesulfonate (mesylate) (Example 2) displayed the following characteristics:
One distinct endotherm onset: 247.34°C
peak maximum: 250.34°C
The TGA thermograph showed no weight reduction until ca 250°C had been reached. The weight reduction commenced with the start of the main endotherm and indicated that this thermal event was the onset of compound decomposition. There is no evidence of entrapped solvents or water.
The XPRD data in FIG. 3 demonstrated that under different extremes of humidity indicate a stable crystalline form of the mesylate salt of Example 2 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 4 which show clear transitions and no evidence of solvates.
Aqueous solubility of the mesylate salt (Example 2) = 65mg/ml (25°C).
Example 3
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one hydrosulfate single crystals: 25.0 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluorome
one hydrosulfate was added to 4 mL vial. 1.000 mL of anhydrous EtOH was added, and the sample was filtered. Anhydrous hexanes were added dropwise until the solution neared the precipitation point. The vial was sealed and left undisturbed for 24 hr, after which time a crop of single crystals was evident. The sample was sent for single crystal analysis and confirmed as the anhydrous (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate form (FIGs. 5A-5B).
Example 4
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one freebase: 8.00 g of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate (JM Lot R-2017-4323 D 301) was added to a 1 L Erlenmeyer flask and suspended and stirred vigorously in 400 mL of THF. 20% K2C03 (250 mL) was added and dissolved. The mixture was transferred to 1 L sep. funnel. 100 mL EtOAc was added and the aqueous and organic layers were separated. The aqueous layer was re-extracted with 50 mL of EtOAc and the combined organics were back-extracted with brine (100 mL) and water (100 mL). Due to fairly poor separation, a significant quantity of MgSCU was required to dry the solution. The solution was reduced via Rotavap (45 °C) to -50 mL, transferred to a 100 mL RB flask, reduced down to -10 mL, transferred to 20 mL scintillation vial and continued to be reduced to a thick oil. The oil was left on the Rotavap for another hour and a “wet” solid was obtained. Loosened solids on the bottom of the vial were left on the Rotavap for 1 hr with no heat applied to obtain a chunky solid. The contents was transferred to a mortar and pestle, ground to powder and fine granules, placed back in a 20 mL scintillation vial and left on a Rotavap overnight to obtain a dry solid (5.1 g). The XRPD pattern of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase is shown in FIG. 6.
Example 5
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one saccharinate: 199.7 mg of (2R,5S)-7-Methyl-2-(4-
methyl-6-(4-(trifluoromethyl)phenyl)pyrirnidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one free base (0.5115 mmol) was dissolved in 4.2 mL of 2-Me-THF. 98.1 mg of saccharin (0.5106 mmol) was dissolved in 4.2 mL of 2-Me-THF. Saccharin was added to the freebase, and after 15 seconds the mixture began to precipitate and solidify. 10 mL of 2-Me-THF was added and stirred at max rpm as to provide a thick white suspension in 10 min. The suspension was filtered, air dried under vacuum for 10 min on frit, then dried under a stream of nitrogen for 30 min resulting in 215 mg of white solid product. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one saccharinate is shown in FIG. 7.
Example 6
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one oxalate: 403 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase was dissolved in 4.03 mL EtOH. 1.000 mL of this solution was added to a 4 mL vial. 23.8 mg of oxalic acid was dissolved in 1.000 mL of EtOH and added dropwise to the stirring (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase solution. After 5 min, a white precipitate was evident and 2.000 mL of EtOH was added to the slurry to aid stirring. The resulting suspension was stirred overnight. The following day the suspension was filtered and dried on a frit under vacuum for 10 min yielding 106 mg of white solid. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one oxalate is shown in FIG. 8.
Example 7
The single crystal structural information and refinement parameters for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate are shown in Table 1.
Table 1.
Largest peak, hole / e A-3 0.363, -0.264
The most prominent XRPD diffraction peaks were (2Θ): 7.8±0.2°, 8.1±0.2°, 12.6±0.2°, 14.3±0.2°, 16.5±0.2°, 18.5±0.2°, 19.6±0.2°, 24.8±0.2° and 25.3±0.2°.
PATENTS
US2018360833NOVEL PYRIMIDINYL-DIAZOSPIRO COMPOUNDS2018-06-27
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017304303 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2017-07-11 | |
| US9737536 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-05-25 | 2016-09-15 |
| US2016184306 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-02-15 | 2016-06-30 |
| US9309254 | NOVEL COMPOUNDS | 2013-05-22 | 2015-04-30 |
| US9376445 | NOVEL COMPOUNDS | 2013-05-22 | 2015-06-18 |
////////////////BIIB-095, BIIB095, BIIB 095, PHASE 1
CC1=NC(=NC(=C1)C2=CC=C(C=C2)C(F)(F)F)C3CCC4(N3)CCN(C4=O)C
VNRX-7145
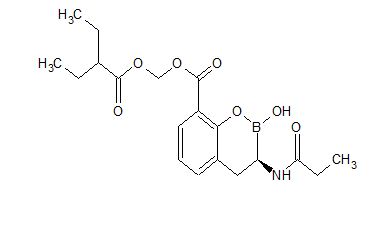

CAS 1842399-68-1
MF C19 H26 B N O7
MW 391.22
2H-1,2-Benzoxaborin-8-carboxylic acid, 3,4-dihydro-2-hydroxy-3-[(1-oxopropyl)amino]-, (2-ethyl-1-oxobutoxy)methyl ester, (3R)-
The VNRX-7145 combination is now in Phase I studies to treat resistant urinary tract infections.
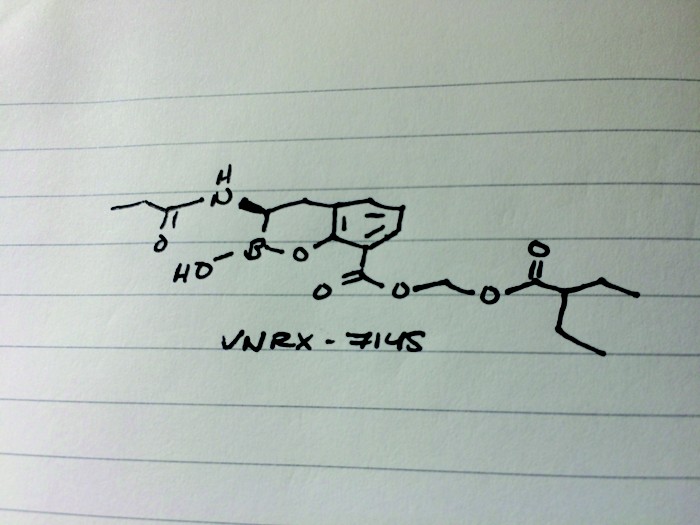
VNRX-7145
PATENT
WO 2015191907
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015191907
ntibiotics are the most effective drugs for curing bacteria-infectious diseases clinically. They have a wide market due to their advantages of good antibacterial effect with limited side effects. Among them, the beta-lactam class of antibiotics (for example, penicillins, cephalosporins, and carbapenems) is widely used because they have a strong bactericidal effect and low toxicity.
[0005] To counter the efficacy of the various beta-lactams, bacteria have evolved to produce variants of beta-lactam deactivating enzymes called beta-lactamases, and in the ability to share this tool inter- and intra-species. These beta-lactamases are categorized as“serine” or“metallo” based, respectively, on presence of a key serine or zinc in the enzyme active site. The rapid spread of this mechanism of bacterial resistance can severely limit beta-lactam treatment options in the hospital and in the community.
SCHEME 1
SCHEME 2
SCHEME 3
[00390] Alternatively, (II) can be obtained by treatment of (I) with hydrochloric acid (around 3-5 Molar in dioxane) in an alcohol solvent such as methanol, ethanol, or n-butanol at a temperature between room temperature and 120 ºC (SCHEME 4).
SCHEME 4
SCHEME 5
EXAMPLE 62: (R)-2-Hydroxy-3-propionylamino-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid
Step 1. Synthesis of 2-Methoxy-3-[2-propionylamino-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-benzoic acid tert-butyl ester.
[00540] Prepared from [(1S)-2-(3-tert-butoxycarbonyl-2-methoxy-phenyl)-1-chloro-ethyl]boronic acid (+) pinanediol ester and propionic acid following the procedure in Step 2 of Example 1. The crude product was purified by flash chromatography on silica gel (25-100% EtOAc/Hexane). ESI-MS m/z 486 (MH)+.
Step 2. Synthesis of (R)-2-Hydroxy-3-propionylamino-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid.
[00541] Prepared from 2-Methoxy-3-[2-propionylamino-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-benzoic acid tert-butyl ester following the procedure described in Step 3 of Example 1. The crude product was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 264 (MH)+
CLIP
Candidate: VNRX-7145

Credit: Tien Nguyen/C&EN
Presenter: Christopher John Burns, president and chief executive officer of VenatoRx Pharmaceuticals
Target: β-lactamases
Disease: Resistant urinary tract infections
Reporter’s notes: Having unveiled an antibacterial candidate at last spring’s first time disclosures session, Burns was back with another, this time the molecule can be taken orally. Both VenatoRx (pronounced Ven-a-tor-ix) compounds resuscitate the activity of β-lactam drugs, which make up more than 60% of all antibiotics prescribed. Unfortunately, many bacteria have grown resistant to these antibiotics. The new compounds rescue the old antibacterials by inhibiting β-lactamases, enzymes that chew up the antibiotics. To test the activity of new β-lactamase-targeting compounds, the researchers settled on several “sentinel” bacteria strains. Then to find a candidate with oral bioavailability, the team focused on molecules with low polarity and low molecular weight. They found VNRX-7145, developed as a prodrug in which esterases in the liver clip off the tips of the molecule to reveal the active drug. VNRX-5133, disclosed at last year’s meeting, had to be delivered intravenously along with another IV-antibiotic Cefepime, and targeted serine and metallo β-lactamases. The new oral candidate VNRX-7145 inhibits serine β-lactamases with Ceftibuten as its partner. The VNRX-7145 combination is now in Phase I studies to treat resistant urinary tract infections.
////////////VNRX-7145, VNRX7145, VNRX 7145, Phase I, VenatoRx
CCC(CC)C(=O)OCOC(=O)c1cccc2C[C@H](NC(=O)CC)B(O)Oc12
CCC(CC)C(=O)OCOC(=O)c1cccc2C[C@H](NC(=O)CC)B(O)Oc12
Acefylline
Acefylline
- Molecular FormulaC9H10N4O4
- Average mass238.200 Da
(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetic acid
1,2,3,6-Tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic Acid
1,3-Dimethylxanthine-7-acetic acid
211-490-2 [EINECS]
652-37-9 [RN]
7-(Carboxymethyl)theophylline
7H-Purine-7-acetic acid, 1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-
CAS Registry Number: 652-37-9
CAS Name: 1,2,3,6-Tetrahydro-1,3-dimethyl-2,6-dioxopurine-7-acetic acid
Additional Names: carboxymethyltheophylline; 7-theophyllineacetic acid
Molecular Formula: C9H10N4O4
Molecular Weight: 238.20
Percent Composition: C 45.38%, H 4.23%, N 23.52%, O 26.87%
Literature References: Prepn: DE 352980 (1922 to E. Merck); Frdl. 14, 1320; S. M. Ride et al., Pharmazie 32, 672 (1977). Prepn of salts: J. Baisse, Bull. Soc. Chim. Fr. 1949, 769; M. Milletti, F. Virgili, Chimica 6, 394 (1951), C.A. 46, 8615h (1952). GC determn in urine: J. Zuidema, H. Hilbers, J. Chromatogr. 182, 445 (1980). HPLC determn in serum and pharmacokinetics: S. Sved et al.,Biopharm. Drug Dispos. 2, 177 (1981).
Properties: Crystals from water, mp 271°.
Melting point: mp 271°
Derivative Type: Sodium salt
CAS Registry Number: 837-27-4
Molecular Formula: C9H9N4NaO4
Molecular Weight: 260.18
Percent Composition: C 41.55%, H 3.49%, N 21.53%, Na 8.84%, O 24.60%
Properties: Silky needles, mp >300°.
Melting point: mp >300°
Derivative Type: Compd with piperazine
Additional Names: Acefylline piperazine; acepifylline
Trademarks: Dynaphylline (Welcker-Lyster); Etaphylline (Delalande); Etafillina (Delalande)
Properties: Undefined mixture of the 1:1 and 2:1 salts; contains 75-78% theophylline acetic acid and 22-25% anhydrous piperazine.
Therap-Cat: Bronchodilator.
Keywords: Bronchodilator; Xanthine Derivatives.
Acefylline (INN),[1] also known as acetyloxytheophylline, is a stimulant drug of the xanthine chemical class. It acts as an adenosine receptor antagonist. It is combined with diphenhydramine in the pharmaceutical preparation etanautine to help offset diphenhydramine induced drowsiness.[2]
Synthesis
DE 352980 (1922 to E. Merck); Frdl. 14, 1320; S. M. Ride et al., Pharmazie 32, 672 (1977).
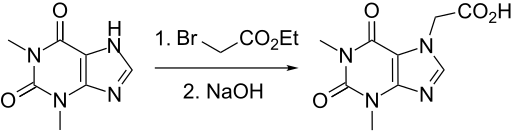
Acefylline
- Use:cardiotonic, diuretic, antispasmodic, bronchodilator
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid
- Formula:C9H10N4O4
- MW:238.20 g/mol
- CAS-RN:652-37-9
- EINECS:211-490-2
- LD50:1180 mg/kg (M, i.p.); 2733 mg/kg (M, p.o.)
Acepifylline
- Use:
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid compd. with piperazine
- Formula:C9H10N4O4 • xC4H10N2
- MW:unspecified
- CAS-RN:18833-13-1
- EINECS:242-614-3
Acefylline heptaminol
- Use:
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid compd. with 6-amino-2-methyl-2-heptaminol (1:1)
- Formula:C9H10N4O3 • C8H19NO
- MW:367.45 g/mol
- CAS-RN:59989-20-7
- EINECS:262-012-4
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names (Rec. INN): List 21” (PDF). World Health Organization. Retrieved 29 December 2016.
- ^ Zuidema, Jan. (1978). “Biofarmaceutische en farmacokinetische aspecten van theofylline en acefylline”. Thesis (doctoral)–Universiteit van Amsterdam. References
Baisse, J.: Bull. Soc. Chim. Fr. (BSCFAS) 1949, 769.
DE 352 980 (E. Merck; 1922).
 |
|
 |
|
| Clinical data | |
|---|---|
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| ECHA InfoCard | 100.010.447 |
| Chemical and physical data | |
| Formula | C9H10N4O4 |
| Molar mass | 238.20 g/mol g·mol−1 |
| 3D model (JSmol) | |
////////Acefylline
LHC 165

LHC165
3-[5-amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]benzo[f][1,7]naphthyridin-8-yl]propanoic acid
C29H32F2N3O7P, 603.56 g/mol
CAS 1258595-14-0
5-Amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]benzo[f][1,7]naphthyridine-8-propanoic acid
Benzo[f][1,7]naphthyridine-8-propanoic acid, 5-amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]-
- Originator Novartis
- Class Antineoplastics
- Mechanism of Action
- Undefined mechanism
- Phase I Solid tumours
- 31 Jan 2018 Phase-I clinical trials in Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Italy, Japan (Intratumoural) (NCT03301896)
- 31 Jan 2018 Phase-I clinical trials in Solid tumours (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Monotherapy, Second-line therapy or greater) in USA, Japan, Italy, Belgium (Intratumoural) (NCT03301896)
- 10 Oct 2017 Novartis plans a phase I trial for Solid tumours (Monotherapy, Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Canada, France, Germany, Italy, South Korea and Spain in November 2017 (Intratumoural) (NCT03301896)
PATENT
PATENT
US 20110053893
PATENT
WO 2011130379
PATENT
Scheme (III)
Scheme (IV)
Scheme (V)
Example 19 (Table 1: Compound 19): Synthesis of ‘3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo[f][ 1, 7]naphthyridin-8-yl)propanoic acid (19)
Scheme 6
Step 1: (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3)
[517] To a solution of tert-butyl 5-bromo-2-chlorophenylcarbamate (6-1) (1.0 equiv.) in acetonitrile (0.3 M) and EtOH (0.5 M) was added K2C03 (2.0 equiv.). The reaction was degassed and flushed with N , then added (E)-ethyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)acrylate (6-2) (1.2 equiv.) and Pd(PPh3)4 (0.1 equiv.). The reaction was flushed again with N2 and stirred at 100 °C overnight. After cooling to room temperature, hexane was added, and the mixture was filtered through a pad of silica, eluting with EA/Hex (1 : 1) until the product was completely eluted. The filtrate was concentrated and purified on Combiflash, eluting with 0-15% EA in Hex to give (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) as a white solid.
Step 2: ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4)
[518] To a solution of (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) (1.0 equiv.) in ethyl acetate/ethanol (1 : 1 , 0.3 M) was added Wilkinson’s catalyst (0.10 equiv.).
Hydrogen gas was introduced via a ballon, and the reaction was stirred at room temperature for 24 hours. The mixture was filtered through a pad of celite, washing with dichloromethane. The filtrate was concentrated in vacuo and purified by Combiflash using 0-10% ethyl acetate in hexane to give ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) as a solid.
Step 3: ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5)
[519] A solution of ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) (1 .0 equiv.), 4,4,4,,4′,5,5,5′,5′-octamethyl-2,2′-bi(l ,3,2-dioxaborolane) (2.0 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.05 equiv.), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (0.20 equiv.), and potassium acetate (2.0 equiv.) in 1 ,4-dioxane (0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was concentrated in vacuo. The crude material was purified by Combiflash using 0-50% ethyl acetate in hexane to afford ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) as a brown oil. The product was stored at -20°C and used within a month of synthesis.
Step 4: l-bromo-4-(methoxymethoxy)-2-methylbenzene (6-7)
[520] To a solution of 4-bromo-3-methylphenol (6-6) (1.0 equiv.) in DMF (0.5 M) at 0 °C was added portionwise 60% wt NaH (1.5 equiv.). The addition was controlled such that internal reaction temperature never went above 10 °C. The reaction was stirred at room temperature for 45 minutes, then a solution of chloro(methoxy)methane (1.2 equiv.) in DMF (3 M) was added dropwise via additional funnel. The reaction was stirred at room temperature for 3.5 hours, and then quenched by pouring into ice. The resulting mixture was stirred at room temperature for 1 hour. Ether was added, and the two layers were separated. The aqueous layer was extracted (lx) with ether. The combined organic layers were washed with water (2x), brine, dried over MgS04, and concentrated to give 1 -bromo-4-(methoxymethoxy)-2-methylbenzene (6-7) as a colorless oil. The crude material was used in the next step without further purification.
Step 5: triethylf (4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane
[521] A solution of l -bromo-4-(methoxymethoxy)-2-methylbenzene (1.0 equiv.), triethylamine (5.0 equiv.) in DMF (0.5 M) was degassed and flushed with nitrogen. To the reaction was added TES-acetylene (1.05 equiv.), Cul (0.098 equiv.), and Pd(PPh3)2Cl2 (0.098 equiv.). The reaction was heated to 60 °C and stirred overnight. After cooling to room temperature, water and ether were added. The layers were separated, and the organic layer was washed with water (2x). The organic layer was separated and passed through a pad of silica (packed with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated to give triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane as a black oil. The crude material was used in the next step without further purification.
Step 6: l-ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8)
[522] To a solution of triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane (1.0 equiv.) at
0 °C was slowly added tetrabutylammonium fluoride (1M solution in THF, 0.20 equiv.). At this
point, the ice-bath was removed and the reaction mixture was allowed to stir at room temperature for 45 minutes. The reaction mixture was then passed through a pad of silica (packed with hexane) and eluted with 20% EtOAc in Hexanes to remove insoluble salts. The crude product was then purified by Combiflash using 0-10% EtOAc in Hexanes to give 1 -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) as a slightly brown liquid.
Step 7: 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10)
[523] A solution of l -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) (1 .0 equiv.), 3,5-dichloropicolinonitrile (6-9) (0.90 equiv.), Cul (0.10 equiv.), and Pd(PPh3)2CI2 (0.10 equiv.), and triethylamine (5.0 equiv.) in DMF (0.25 M) was degassed and flushed with nitrogen. The reaction mixture was then heated to 60 °C and stirred overnight. After cooling to room temperature, water was added. The mixture was extracted with EA (2x). The combined organic layers were washed with 10% aq NH4OH (2x), brine, and concentrated. The crude material was filtered through a pad of silica (wetted with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated. The resulting solids were washed in hot ether and filtered to give a yellow solid, which was used in the next step without further purification. The filtrate was concentrated and purified by Combiflash using 0- 10% EtOAc in Hexanes to give 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) as a yellow solid.
Step 8: ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-ben∑o fJfl, 7J
naphthyridin-8-yl)propanoate (6-11)
[524] A solution of 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) (1 .0 equiv.), ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) (1.25 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.10 equiv.), dicyclohexyl(2′,6′-dimethoxybiphenyl-2-yl)phosphine (0.20 equiv.), and sodium bicarbonate (3.0 equiv.) in «-butanol /H20 (5: 1 , 0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was diluted with ethyl acetate and water. The two phases were separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgS04, and concentrated in vacuo. The crude material was purified by flash chromatography on a COMBIFLASH® system (1SCO) using 0-40% ethyl acetate in DCM first to remove the impurity, then 0-4% MeOH in DCM to give ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl) propanoate (6-11). Further purification was accomplished by precipitating and washing in hot ether.
Step 9: ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fl[l ]naphthyridin-8-yl)propanoate (6-12)
[525] A solution of ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-11) (1.0 equiv.) in EtOH/THF (3: 1 , 0.16 M) was flushed with nitrogen. Then, 10% wt Pd/C (0.20 equiv. by weight) was added. The reaction was flushed with hydrogen (2x) and stirred under a hydrogen balloon. After 24 hours, the reaction was filtered through a pad of celite, washing with 5%MeOH in DCM. The filtrate was checked for the presence of starting material using LCMS. The hydrogenation reaction was repeated until no more
of the alkyne starting material or alkene intermediate was detected. The crude product was purified by Combiflash using 0-4% eOH in DCM to give ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-12) as a white solid.
Step 10: ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fl[l ]naphthyridin-8-yl)propanoate (6-13)
[526] Ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-12) (1 .0 equiv.) was dissolved in EtOH (0.2 M), then added a solution of 4M HC1 in dioxane (0.2 M). The product precipitated out as a yellow salt. After stirring for 3 hours, the reaction was poured into a stirring solution of ether. The mixture was stirred for 10 minutes, then filtered and washed with ether. Ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-13) was obtained as a yellow solid which was dried on vacuum overnight (bis-HCl salt). Alternatively, the crude product was purified by Combiflash using 0-5% MeOH in DCM to give the free base.
Step 11: ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f] [1 , 7]naphthyridin-8-yl)propanoate ( 6-15)
[527] To a solution of ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ [ l ,7]naphthyridin-8-yl)propanoate (6-13) (1.0 equiv.) dissolved in DMF (0.14 M) was added a solution of diethyl 3-(2-bromoethoxy)-l ,l -difluoropropylphosphonate (6-14: described in Example 7 – Step 1) (1 .3 equiv.) in DMF (0.7 M) and cesium carbonate (4 equiv.). The reaction was stirred at 60 °C. After 1.5 hours (or until reaction is complete by LCMS), DCM (2 volume equivalent) was added to the reaction. The solids (inorganic) were filtered, and the filtrate was concentration. The crude product was purified by Combiflash using 0-5%MeOH in DCM to give ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[fJ
[1 ,7]naphthyridin-8-yl)propanoate (6-15) as an oil which upon standing became a white solid.
Step 12: 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphompropoxy)ethoxy)-2-methylphenethyl)be o[f]
[1, 7]naphthyridin-8-yl)propanoic acid (19)
[528] To a solution of ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-15) (1.0 equiv.) in DCM (0.16 M) at 0 °C was added slowly TMSBr (10 equiv.). The reaction was stirred at room temperature overnight. Additional TMSBr (5.0 equiv.) was added at 0 °C, and the reaction was again stirred at room temperature overnight. The solvent was removed by evaporation and the crude orange solids dried on hi-vac briefly. The solids were suspended in EtOH (0.5 M) and added 2.5 N
NaOH (10.0 equiv.). The reaction was stirred at 80 °C for 3 hours. After cooling to room temperature, the mixture was adjusted to pH 9 to 10 and directly purified on RP-HPLC using a CI 8 column, eluting with 10-40% 95:5 (MeCN/5mM NH4OAc) in l OmM NH4OAc (pH 9) gradient. The fractions containing the product were combined and concentrated in vacuo. The resulting white gel was dissolved in refluxing 1 :1 EtOH/water (0.04 M) with the addition of a few drops of ammonium hydroxide. While hot, the mixture was slowly poured into a stirring hot solution of acetone (0.009
M) preheated at 50 °C. The acetone suspension was slowly cooled to room temperature for 15 minutes with continued stirring, and then sat in an ice bath for 10 minutes. The solids were filtered and washed successively with acetone (2x) and ether (2x). The solids were dried on hi-vac overnight to give the 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo [fj[l ,7]naphthyridin-8-yl)propanoic acid (19) as a solid. Ή NMR (Dimethylsulfoxide-d6): δ 9.02 (s, 1 H), 8.82 (s, 1H), 8.55 (d, 1H, J = 8.4 Hz), 7.58 (s, 1H), 7.48 (d, 1 H, J = 8.4 Hz), 7.07 (d, 1H, J = 8.4 Hz), 6.75 (s, 1 H), 6.68 (d, 1H, J = 8.4 Hz), 4.03-4.00 (m, 2H), 3.72-3.68 (m, 4H), 3.16-3.12 (m, 2H), 3.03-2.96 (m, 4H), 2.67-2.64 (m, 2H), 2.33-2.32 (m, 2H), 2.26 (s, 3H). LRMS [M+H] = 604.2
PATENT
US 20120237546
PATENT
WO 2012031140
PATENT
Toll-like receptors (TLRs) are pattern recognition receptors which play an essential role in the innate immunity, by recognizing invasion of microbial pathogens and initiating intracellular signal transduction pathways to trigger expression of genes, the products of which can control innate immune responses. Specifically, Toll like receptor (TLR) agonists activate innate immune cells through the TLR-MyD88-NFk and IRF3/7 pathways. TLR7, TLR8, and TLR9 belong to a subfamily of TLRs based on their genomic structure, sequence similarities, and homology. TLR7, TLR8, and TLR9 are located in intracellular endolysosomal compartments and show a unique pattern of cell type-specific expression that is thought to be responsible for different pathogen response profiles.
Small molecule agonists of TLR7 and/or TLR8 have been reported and shown to activate innate immune responses by inducing selected cytokine biosynthesis, the induction of co-stimulatory molecules, and by increased antigen-presenting capacity. Such compounds include imidazoquinoline amine derivatives (U.S. Patent No. 4689338), imidazopyridine amine derivative (U.S. Patent No. 5446153), imidazonaphthyridine derivative (U.S. Patent No.
6194425), oxazoloquinoline amine derivatives (U.S. Patent No. 61 10929); thiazoloquinoline amine derivatives (U.S. Patent No. 61 10929), selenazoloquinoline amine derivatives (U.S. Patent No. 61 10929), pyrazolopyridine derivatives (U.S. Patent No. 9145410), and
benzonaphthyridine amine derivatives (U.S. Patent Nos. 8466167 and 9045470).
The synthetic TLR7 agonist, Imiquimod (1 -(2-methylpropyl)-1 H-imidazo[ 4,5-c]quinolin-4-amine) is FDA-approved in a cream formulation for the topical treatment of cutaneous basal cell carcinoma, actinic keratosis and genital warts, and has limited activity against cutaneous melanoma and breast tumors (J. Immunol. 2014, 193(9) : 4722^1-731 ). Systemic administration of Imiquimod, and structurally similar Resiquimod, is limited by cytokine- mediated adverse effects including severe flu-like symptoms (Expert Opin. Emerging Drugs (2010), 15:544-555). Consequently, Imiquimod is used exclusively in topical applications and is not used to treat deep, non-cutaneous tumors such as melanoma or solid tumors.
An injectable lipid modified imidazoquinoline (TLR7/8 dual agonist) that forms a tissue depot with gradual, sustained release which allows for local TLR triggering activity without systemic cytokine release has been reported (J. Immunol. 2014, 193(9): 4722^731 ). However, this compound was shown to be ineffective for large tumors and in addition the serum concentration of this compound 24 hours post subcutaneous administration decreased by approximately 50% (Journal for ImmunoTherapy of Cancer, 2014, 2:12). Therefore, there remains a need for intratumor administration of a TLR7 agonist with prolonged sustained release, which may benefit the treatment of large tumors.
clip
Candidate: LHC165

Credit: Tien Nguyen/C&EN
Presenter: Alex Cortez, senior Investigator I at the Genomics Institute of the Novartis Research Foundation
Target: Toll-like receptor 7 (TLR7)
Disease: Solid tumors
Reporter’s notes: Cortez shared another story in the realm of immuno-oncology, although the program that yielded this compound actually started in the world of vaccines. Cortez’s team had been focusing on vaccine adjuvants, small molecules that turn on the immune system to enhance a vaccine’s effect. They developed one such class of compound that activates toll-like receptor 7 (TLR7), a protein in the immune system that recognizes dangerous-looking molecules and can trigger the release of infection-clearing proteins. After observing TLR7 agonists’ ability to induce an immune response with vaccines, the researchers wondered whether the molecules could also be effective in immuno-oncology.
They found that LHC165 adsorbed to aluminum hydroxide reduced tumor growth in mice and, intriguingly, showed signs of an abscopal effect, in which untreated tumors shrink concurrently with treated tumors. The implication is that if the immune system recognizes one tumor site, it can recognize others. As with several of the candidates presented throughout the day, LHC165 bears a phosphate group and is injected into the tumor. It’s currently in Phase I trials in patients with advanced malignancies, which means they’ve already tried second and third line therapies, as a single agent and in combination with the checkpoint inhibitor PDR001.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9597326 | BENZONAPTHYRIDINE COMPOSITIONS AND USES THEREOF | 2011-04-13 | 2013-05-16 |
| US9950062 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2010-09-01 | 2012-09-20 |
| US9517263 | BENZONAPHTHYRIDINE-CONTAINING VACCINES | 2010-06-10 | 2012-10-18 |
| US2015225432 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2015-04-24 | 2015-08-13 |
| US9315530 | ADSORPTION OF IMMUNOPOTENTIATORS TO INSOLUBLE METAL SALTS | 2011-09-01 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016213776 | ADSORPTION OF IMMUNOPOTENTIATORS TO INSOLUBLE METAL SALTS | 2016-04-07 | 2016-07-28 |
| US2012177681 | Formulation of immunopotentiators | 2011-09-01 | 2012-07-12 |
| US9045470 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2011-03-03 | |
| US2018169204 | COMBINATION VACCINES WITH LOWER DOSES OF ANTIGEN AND/OR ADJUVANT | 2018-02-02 | |
| US9375471 | ADJUVANTED FORMULATIONS OF BOOSTER VACCINES | 2013-03-08 | 2013-09-12 |
//////LHC165, LHC 165, LHC -165, Phase I, Solid tumours, novartis
O=P(O)(O)C(F)(F)CCOCCOc4ccc(CCc1cc2c3ccc(CCC(=O)O)cc3nc(N)c2nc1)c(C)c4
CC1=C(C=CC(=C1)OCCOCCC(F)(F)P(=O)(O)O)CCC2=CN=C3C(=C2)C4=C(C=C(C=C4)CCC(=O)O)N=C3N
AB 680
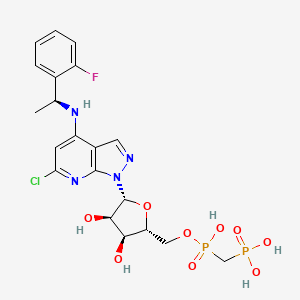
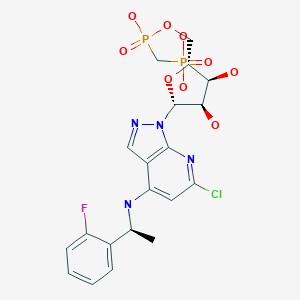

AB 680
C20H24ClFN4O9P2, 580.827 g/mol
Cas 2105904-82-1
1H-Pyrazolo[3,4-b]pyridin-4-amine, 6-chloro-N-[(1S)-1-(2-fluorophenyl)ethyl]-1-[5-O-[hydroxy(phosphonomethyl)phosphinyl]-β-D-ribofuranosyl]-
[[(2R,3S,4R,5R)-5-[6-chloro-4-[[(1S)-1-(2-fluorophenyl)ethyl]amino]pyrazolo[3,4-b]pyridin-1-yl]-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]methylphosphonic acid
[({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
- Originator C
- Class Antineoplastics; Small molecules
- Mechanism of Action 5-nucleotidase inhibitors; Adenosine A2 receptor antagonists
- Phase I Cancer
- 19 Nov 2018 Arcus Biosciences plans to initiate a clinical trial in Cancer in first half of 2019
- 16 Oct 2018 Phase-I clinical trials in Cancer (In volunteers) in Australia (IV) (NCT03677973)
- 30 Sep 2018 Preclinical pharmacodynamics data in Cancer presented at 4th CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference (CRI-CIMT-EATI-AACR – 2018)
Clip
Credit: Tien Nguyen/C&EN
Presenter: Kenneth V. Lawson, senior scientist at Arcus Biosciences
Target: Ecto-5’-nucleotidase (CD73)
Disease: Cancer
Reporter’s notes: In the first talk of the day, Lawson introduced the idea of cancer drugs that target the host’s immune system. “Checkpoint inhibitors changed the way we think of treating cancer,” he said. These drugs successfully disrupt the binding interaction between a protein and a checkpoint protein that stops immune T cells from killing cancer cells. As a result, these drugs turn immune cells loose to attack tumor cells. But the drugs work only in about 30-40% of patients—an issue pharmaceutical companies like Arcus hope to address with new immunotherapies that can be taken in combination with checkpoint inhibitors.
Lawson’s team set out to inhibit an enzyme commonly found in tumors called CD73, the second of two enzymes which break down extracellular adenosine trisphosphate (ATP) to adenosine. Adenosine then binds to immunosuppressive receptors on immune cells and shuts them down. Yet developing a small molecule inhibitor of CD73 proved challenging, Lawson said. After striking out with high-throughput screening, the team turned to CD73’s natural substrate for inspiration. However, the molecule possessed more than one phosphate group, which is notoriously a liability for drug molecules because small molecules with such negative changes struggle to cross cell membranes. The team’s goal was to remove the phosphate groups, Lawson says, but things didn’t exactly go according to plan. After showing the audience a series of compounds from structure-activity relationship (SAR) studies—slides no medicinal chemistry talk would be complete without—Lawson revealed the structure of their final clinical compound AB680 as the sound of people flipping notebook sheets rippled across the room. Synthesized in 34% overall yield, the candidate ultimately included two phosphate groups—a feature that surprised audience members.
Tests revealed that AB680 can be given intravenously but the compound also showed moderate oral bioavailability. Lawson suggested a possible route for how the molecule might pass from the digestive tract to the bloodstream, a paracellular mechanism by which molecules cross the epithelium by passing through the space between cells. AB680 showed “extraordinary potency,” inhibiting CD73 in human T-cells at a concentration of 0.008 nM. The compound has a 4 day half-life, which means it could be dosed every two weeks, coinciding with the dosing schedule for patients who receive a checkpoint inhibitor. AB680 is currently in Phase 1 clinical trials with healthy patients.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US204141996&tab=PCTDESCRIPTION&maxRec=1000
|
Purinergic signaling, a type of extracellular signaling mediated by purine nucleotides and nucleosides such as ATP and adenosine, involves the activation of purinergic receptors in the cell and/or in nearby cells, resulting in the regulation of cellular functions. Most cells have the ability to release nucleotides, which generally occurs via regulated exocytosis (see Praetorius, H. A.; Leipziger, J. (1 Mar. 2010) Ann Rev Physiology 72(1): 377-393). The released nucleotides can then be hydrolyzed extracellularly by a variety of cellular membrane-bound enzymes referred to as ectonucleotidases.
|
Example 92
Synthesis of [({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
PATENT

////////////////ARCUS, AB 680, AB680, AB-680, PHASE 1
https://www.arcusbio.com/wp-content/uploads/2018/04/AACR_AB680_1756_final_90x42-abstract-4886.pdf
Fc1ccccc1[C@H](C)Nc4cc(Cl)nc3c4cnn3[C@@H]2O[C@H](COP(=O)(O)CP(=O)(O)O)[C@@H](O)[C@H]2O
CC(C1=CC=CC=C1F)NC2=CC(=NC3=C2C=NN3C4C(C(C(O4)COP(=O)(CP(=O)(O)O)O)O)O)Cl
Solriamfetol hydrochloride, ソルリアムフェトル塩酸塩 , солриамфетол , سولريامفيتول , 索安非托 ,
Solriamfetol hydrochloride
FDA APPROVED 2019/3/20, Sunosi
ソルリアムフェトル塩酸塩; R228060, R 228060
| Formula |
C10H14N2O2. HCl
|
|---|---|
| CAS |
178429-65-7 HCL
|
| Mol weight |
230.6913
|
(2R)-2-Amino-3-phenylpropyl carbamate
(2R)-2-Amino-3-phenylpropylcarbamat
10117
178429-62-4 [RN] FREE FORM
Benzenepropanol, β-amino-, carbamate (ester), (βR)- [
солриамфетол [Russian] [INN]
سولريامفيتول [Arabic] [INN]
索安非托 [Chinese] [INN]
JZP-110
Originator SK Holdings
- Developer Jazz Pharmaceuticals plc; SK biopharmaceuticals
- Class Carbamates; Sleep disorder therapies; Small molecules
- Mechanism of Action Adrenergic uptake inhibitors; Dopamine uptake inhibitors
- Orphan Drug Status Yes – Narcolepsy
- Registered Hypersomnia
- Discontinued Depressive disorders
- 26 Mar 2019 Discontinued – Phase-I for Depressive disorders (Adjunctive treatment) in USA (PO) (Jazz Pharmaceuticals pipeline, March 2019)
- 20 Mar 2019 Registered for Hypersomnia (excessive daytime sleepiness) in patients with obstructive sleep apnoea and narcolepsy in USA (PO) – First global approval
- 20 Mar 2019 US FDA approves solriamfetol to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea(OSA)
- New Drug Application (NDA): 211230
Company: JAZZ PHARMA IRELAND LTD
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Solriamfetol hydrochloride | K7RO88SP7A | 178429-65-7 | KAOVAAHCFNYXNJ-SBSPUUFOSA-N |
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1]
Common side effects include headache, nausea, anxiety, and trouble sleeping.[1] It is a norepinephrine–dopamine reuptake inhibitor(NDRI). It is derived from phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[2]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]Aerial ran two Phase II trials of the drug in narcolepsy[4] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticalspaid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double digit royalties to SK.[3][5]
In March 2019 the FDA accepted SK’s and Jazz’ NDA for use of solriamfetol to treat excessive sleepiness in people with narcolepsy or obstructuve sleep apnea; the drug has an orphan designation for narcolepsy.[3][6]
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[6]
Research
Solriamfetol had also been tested in animal models of depression, but as of 2017 that work had not been advanced to clinical trials.[7]
PATENT
WO 9607637
https://patents.google.com/patent/WO1996007637A1/e
Organic alkyl carbamates have been effectively used for controlling various central nervous system (CNS) disorders. For example, U.S. Pat. Nos . 2,884,444, 2,937,119 and 3,313,697 disclose function of carbamate in CNS disorders, especially as antiepileptic and centrally acting muscle relaxant.
Phenylethylamine derivatives, one important class of therapeutical medicines useful for managing CNS diseases, have been used mainly to treat obesity, narcolepsy, minimal brain dysfunction and mild depression.
Recent design of pharmacologically useful compounds has been based on amino acids or the derivatives thereof, which is mainly attributable to the fact that many of the compounds found in biological systems come from amino acids or the derivatives thereof. In addition, in most cases, the function of a pharmaceutically useful compound is effected after it binds to an enzyme or receptor, which may trigger the regulatory mechanisms of the enzyme or receptor.
REACTION SCHEME I
REACTION SCHEME II
REACTION SCHEME III
EXAMPLE I
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
In a 500 mL RB flask equipped with a mechanical stirrer and a dropping funnel, D-phenylalaninol (45.4 g, 300 mmol) was dissolved in 220 mL of distilled water, and cooled in an ice-bath. The pH of the solution was adjusted with 50 % sodium hydroxide to 14. Benzyl chloroformate (49.3 mL, 345 mmol) was charged into the dropping funnel and added slowly to the well stirred solution over 0.5 hr. After the completion of the addition, the reaction mixture was stirred for 1 hr. at 0 *C. The product precipitated from the reaction mixture as a white solid. It was collected by filtration and washed completely with distilled water. After being dried in vacuo, the solid thus obtained weighed 104 grams without any further purification: 99.8% Yield.
Melting point = 90 – 92 *C
[α]D20 = + 43.4 (c = 1.0, EtOH)
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.35; H, 6.71; N,4.91
EXAMPLE II
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
carbamate
In a 500 mL RB flask, N-benzyloxycarbonyl-D- phenylalaninol (13.56 g, 50 mmol) was charged with antipyrine (11.29 g, 60 mmol) in 250 mL of dry THF under a nitrogen atmosphere. The reaction mixture was cooled in an ice-bath and phosgene (30.3 mL of 1.93 M solution in toluene, 58.5 mmol) was added quickly while vigorously stirring. After stirring for 1 hr. , the formation of a corresponding chloroformate from the starting material was monitored by TLC. The chloroformate solution thus prepared, was slowly added to a well stirred and ice-chilled aqueous ammonium hydroxide solution (75 mL, 28-30 %, 1,190 mmol) via cannula over 0.5 hr. The resulting reaction mixture was stirred for an extra 0.5 hr. The organic phase separated was collected. The aqueous phase was extracted twice with methylene chloride (100 mL). The combined organic phase was washed with brine (50 mL), dried over sodium sulfate, and concentrated to yield 17.8 g (113%) of foamy solid. It was purified a flash column chromatography to give 14.8 g of the title compound, white solid: 94% Yield.
Melting point = 121 – 125 *C
[α]D20 = + 28.6 (c = 2.0, EtOH)
Analysis calc. : C, 65.84; H, 6.14; N, 8.53
Found: C, 66.68; H, 6.21; N, 7.80
EXAMPLE III
Preparation of D-Phenylalaninol carbamate hydrochloric
acid salt In a 160 mL Parr reactor, N-benzyloxycarbonyl-D-phenylalaninol carbamate (9.43 g) was added with 75 mL of anhydrous methanol and 10 % palladium on charcoal (0.32 g). Then, the reactor was closed and purged with hydrogen for 1 in. The reaction was completed in 2 hrs . under 40 psi pressure of hydrogen at 45 #C. The catalyst was filtered off. Thereafter, the organic layer was concentrated into 5.97 g (102 %) of pale yellow thick liquid. The liquid was poured in 50 mL of anhydrous THF and cooled to 0 “C. Anhydrous hydrogen chloride gas was then purged through the solution with slowly stirring for
0.5 hr. 50 mL of anhydrous ether was added, to give a precipitate. Filtration with THF-ether (1:1) mixture provided 6.1 g of the title compound as a white solid: 88 % Yield.
Melting point = 172 – 174 “C
[α]D20 = – 12.9 (c = 2.0, H20)
Analysis calc. : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.90; H, 6.60; N, 12.15; Cl ,
15.52
EXAMPLE IV
Preparation of N-benzyloxγcarbonyl-L-Phenγlalaninol
The title compound was prepared in the same manner as that of Example I, except that (L)-phenylalaninol was used as the starting material.
Melting point = 90 – 92 *C
[α]D20 = – 42.0 (c = 1.0, EtOH)
Analysis calc. : C, 71.56; H, 6.71; N,4.91
Found: C, 70.98; H, 6.67; N,4.95
EXAMPLE V
Preparation of -N-benzyloxycarbonyl-L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-L-phenylalaninol was used as the starting material.
Melting point = 121 – 128 ‘C
[α]D20 = – 28.9 (c = 2.0, EtOH)
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.45; H, 6.15; N, 8.32
EXAMPLE VI
Preparation of L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-L-phenylalaninol carbamate was used as the starting material.
Melting point = 175 – 177 *C [α]D20 = + 13.1 (c = 1.0, H20)
Analysis calc : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.95; H, 6.58; N, 12.09; Cl , 15.37
EXAMPLE VII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
The title compound was prepared in the same manner as that of Example I, except that (D,L)-phenylalaninol was used as the starting material.
Melting point = 72 – 75 #C
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.37; H, 6.74; N,4.84
EXAMPLE VIII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-D,L-phenylalaninol was used as the starting material.
Melting point = 130 – 133 *C
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.85; H, 6.14; N, 8.49 EXAMPLE IX
Preparation of D,L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-D,L-phenylalaninol carbamate was used as the starting material.
Melting point = 163 – 165 *C
Analysis calc: C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.92; H, 6.56; N, 11.95; Cl , 15.82
PATENT
US 20050080268
PATENT
WO 2018133703
https://patents.google.com/patent/WO2018133703A1/en
Excessive daytime sleepiness (Excessive Daytime Sleepiness, EDS) or pathological somnolence refers to excessive daytime sleep and wakefulness associated with various sleep disorders. These disorders can be the basis for a sleep disorder or sleep have side effects caused by some other medical conditions. Excessive daytime sleep, also known as narcolepsy, sleep clinics is seen mainly in patients with disease that affects 12% of the general population. EDS patients may be manifested as mental distress, poor work or school performance, increasing the risk of accidents, the impact of EDS can debilitating, even life-threatening.
R228060, also known JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor, originally developed by R & D, SK biopharmaceutical, 2014 Sir ownership of the pharmaceutical compound. R228060 has the potential to treat narcolepsy and sleep apnea syndrome, in three multi-center study in two global reached the primary endpoint, and achieved positive results, significantly improved adult obstructive sleep apnea patients excessive sleepiness in patients with narcolepsy and excessive sleep problems.
R228060 chemical name is O- carbamoyl – (D) – phenylalaninol, as shown in the structural formula of formula (I):

Solid Form different chemicals, can cause varying their solubility and stability, and thus affects the absorption and bioavailability of the drug, and can lead to differences in clinical efficacy. Improve the candidate compound has a solubility by salt way become an important means of drug development. Compared to the free form of the drug, suitable pharmaceutically acceptable salts can improve the solubility of the drug type, increased physical and chemical stability, and also to improve the drug-salt having a melting point, hygroscopicity, crystal type and other physical properties, further development of the pharmaceutical dosage form It plays an important role. Patent Document WO1996007637A1 discloses R228060 hydrochloride and its preparation method, and other characteristics of the obtained having a melting point of 172-174 deg.] C as a white solid, the solid was not given in the text data. Further, the present inventors found no other relevant R228060 hydrochloride polymorph or patent literature. Accordingly, the present need in the art to develop a comprehensive system R228060 hydrochloride polymorph, found to be suitable to the development of crystalline form. The present inventors after many experiments, found that polymorph CS1 R228060 hydrochloride CS2 and a melting point polymorph, Form CS1 and CS2 is Form 183 ℃, much higher than the melting point disclosed in prior art solid. It provides a better alternative preparation of pharmaceutical preparations containing R228060 is, has very important implications for drug development.
PATENT
WO 2019027941
(i?)-2-amino-3-phenylpropyl carbamate (APC) is a phenylalanine analog that has been demonstrated to be useful in the treatment of a variety of disorders, including excessive daytime sleepiness, cataplexy, narcolepsy, fatigue, depression, bipolar disorder, fibromyalgia, and others. See, for example, US Patent Nos. 8,232,315; 8,440,715; 8,552,060; 8,623,913; 8,729,120; 8,741,950; 8,895,609; 8,927,602; 9,226,910; and 9,359,290; and U.S. Publication Nos. 2012/0004300 and 2015/0018414. Methods for producing APC (which also has other names) and related compounds can be found in US Patent Nos. 5,955,499; 5,705,640; 6,140,532 and 5,756,817. All of the above patents and applications are hereby incorporated by reference in their entireties for all purposes.
EXAMPLE 1
Synthesis of Compounds
Compound 8 (110CR002)
1 B 110CR002
[0083] tert- utyl (if)-(l-(Carbamothioyloxy)-3-phenylpropan-2-yl)carbamate (IB): A
60% dispersion of sodium hydride (0.36 g, 4.78 mmol, 1.2 equiv) in mineral oil was added in portions to compound 1A (1.0 g. 3.98 mmol, 1 equiv) in THF (20 mL) at 0 °C. After stirring for 1 hour, carbon disulfide (0.191 g, 4.78 mmol, 1.2 equiv) was added at 0 °C. After an additional hour of stirring, methyl iodide (0.3 mL, 4.78 mmol, 1.2 equiv) was added and the reaction was warmed to room temperature. After stirring two additional hours, concentrated ammonium hydroxide (1.6 mL, 7.98 mmol, 2 equiv) was added and the reaction was stirred overnight at room temperature. The reaction was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give crude compound IB. The solid was triturated in diethyl ether (20 mL) to give compound IB (0.17 g, 14% yield) as a light yellow solid.
[0084] (R)-0-(2-Amino-3-phenylpropyl) carbamothioate dihydrochloride (110CR002):
4M HCI in dioxane (0.68 mL, 2.74 mmol, 5 equiv) was added to neat compound IB (0.17 g, 0.548 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR003 (140 mg, 93% yield, 96.9% purity) as a white solid.
Compound 9 (110CR003)
Scheme 2
2A 2B 110CR003
[0085] (R)-2-((ter^Butoxycarbonyl)amino)-3-phenylpropyl sulfamate (2B): A solution of sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of compound 2 A (1.0 g, 3.98 mmol, 1 equiv) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for 4 hours, additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C. The reaction was stirred at room temperature overnight, at which point LCMS indicated a 3 :2 mixture of product to starting material. Additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C and the reaction was stirred at room temperature for an additional 6 hours. LCMS indicated a 4: 1 mixture of product to starting material. The reaction was quenched with saturated sodium bicarbonate (5 mL) and stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The product still contained unreacted starting material which could not be easily separated. Sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of crude compound 2B (0.9 g) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for two hours, the reaction was quenched with saturated sodium bicarbonate (5 mL) and the reaction was stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (Redisep 24 g silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 2B (0.37 g, 28% yield) as a white solid.
[0086] (R)-2-Amino-3-phenylpropyl sulfamate hydrochloride (110CR003): 4M HC1 in dioxane (1.4 mL, 5.6 mmol, 5 equiv) was added to neat compound 2B (0.37 g, 1.12 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under a vacuum at room temperature for two hours to give compound 110CR003 (250 mg, 84% yield, 97.8% purity) as a white solid.
Com ound 3 (110CR007)
[0087] (Benzyl (R)-(l-phenyl-3-ureidopropan-2-yl)carbamate) (3B): Concentrated hydrochloric acid (0.06 mL, 0.68 mmol, 0.12 equiv) was added to a solution of benzyl (ft)-(l -amino-3-phenylpropan-2-yl)carbamate ( 1.5 g, 5.28 mmol, 1 equiv) and urea (1.26 g, 21.21 mmol, 4 equiv) in toluene (150 mL) under nitrogen. After refluxing overnight, LCMS indicated the reaction was complete. The reaction was concentrated under reduced pressure, diluted with water (150 mL) and stirred for 30 minutes. The resulting solid was filtered and washed with water (25 mL) to give crude compound 3B (1.4 g, 4.27 mmol, 80% yield) as a white solid, which was used sequentially.
[0088] ((R)-l-(2-mino-3-phenylpropyl)urea) (3C): Compound 3B (0.5 g, 1.5 mmol, 1 equiv) and 10% palladium on carbon (0.09 g) in methanol (60 mL) was hydrogenated at 30 psi for 1 hour at which time LC-MS determined that the reaction was incomplete. The solution was filtered and fresh catalyst (0.09 g) was added. The solution was hydrogenated at 30 psi for an additional 45 minutes resulting in complete conversion. Two identical scale reactions were run for 105 minutes each, both resulting in complete conversion. The three runs were combined and filtered through celite, which was washed with methanol (50 mL). The filtrate was concentrated under reduced pressure to give crude compound 3C (0.9 g), which was used sequentially.
[0089] (R)-l-(2-Amino-3-phenylpropyI)urea hydrochloride (110CR007): Compound 3C (0.88 g, 4.58 mmol, 1 equiv) was dissolved diethyl ether (10 mL) and 4 N HCl in dioxane (2.31 mL, 9.27 mmol, 2 equiv) was added. The reaction was stirred overnight and then concentrated under reduced pressure to give crude 110CR007 as a white solid. The material was twice recrystallized from 10% methanol in ethanol (30 mL) to give 110CR007 (0.163 g, 16 % yield, 93.7 % purity) as a white solid.
Compound 4 (110CR009)
Scheme 4
[0090] Ethyl (R^)-4-((tert-butoxycarbonyI)amino)-5-phenylpent-2-enoate (4B): A solution of compound 4A (4.0 g, 16.1 mmol, 1 equiv) and ethyl (triphenylphos-phoranylidene)acetate (5.6 g, 16.1 mmol, 1 equiv) in dichloromethane (40 mL) was stirred at room temperature overnight. The reaction was concentrated under reduce pressure to remove the organic solvent and the resulting residue was purified on an AnaLogix automated system (40 g Sorbtech silica gel column), eluting with gradient of 50 to 100% ethyl acetate in heptanes, to give compound 4B (4.8 g, 94% yield) as a white solid.
[0091] (R^E)-4-((te *i-ButoxycarbonyI)amino)-5-phenylpent-2-enoic acid (4C): Lithium hydroxide (1.4 g, 60 mmol, 4 equiv) in water (15 mL) was added to compound 4B (4.8 g, 15 mmol, 1 equiv) in THF (60 mL) at room temperature and the reaction was stirred overnight. After 16 hours, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers was washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4C (4.2 g, 97% yield) as a light cream solid, which was used subsequently.
[0092] Methyl (R E)-4-((½ -i-butoxycarbonyl)amino)-5-phenylpent-2-enoate (4D1):
Isobutyl chloro formate (1.3 mL, 10 mmol, 1 equiv) in THF (4 mL) was added dropwise to a solution of compound 4C (3.0 g, 10 mmol, 1 equiv) and N-methyl-morpholine (1.1 mL, 10 mmol, 1 equiv) in THF (12 mL) at -15 °C. After 30 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 2M Ammonia in methanol (5 mL, 10 mmol, 1 equiv) was added dropwise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring, the reaction was warmed to room
temperature and stirred overnight. The reaction mixture was concentrated at reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (50 mL) and washed with water (100 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (80 g Sorbtech silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 4D1 (1.1 g, 35 % yield) as a white solid.
[0093] Methyl (S)-4-((te^-butoxycarbonyl)amino)-5-phenylpentanoate (4D2): A mixture of compound 4D1 (1.1 g, 3.6 mmol, 1 equiv) and 10% palladium on carbon (0.33 g, 50% wet) in methanol (40 mL) was hydrogenated at 40 psi at room temperature for 4 hours. The mixture was filtered through celite, which was washed with methanol (100 mL). The filtrate was concentrated under reduced pressure to give compound 4D2 (1.1 g, 99% yield) as a white solid.
[0094] (S)-4-((ii? i-Butoxycarbonyl)amino)-5-phenylpentanoic acid (4D3): Lithium hydroxide (73 mg, 3 mmol, 1.5 equiv) in water (1 mL) was added to compound 4B (0.6 g, 2 mmol, 1 equiv) in THF (9 mL) at room temperature. After stirring overnight, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (3 x 25 mL). The combined organic layers was washed with saturated brine (25 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4D3 (0.56 g, 98% yield) as a white solid, which was used subsequently.
[0095] tert-Butyl (S)-(5-amino-5-oxo-l-phenylpentan-2-yl)carbamate (4E): Isobutyl chloroformate (0.23 mL, 1.8 mmol, 1 equiv) in THF (0.5 mL) was added drop-wise to a solution of compound 4C (0.54 g, 1.8 mmol, 1 equiv) and N-methylmorpholine (0.2 mL, 1.8 mmol, 1 equiv) in THF (1 mL) at -15 °C. After 20 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 0.4M Ammonia in THF (9 mL, 3.6 mmol, 2 equiv) was added drop-wise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring the reaction was warmed to room temperature and stirred overnight. The reaction mixture was concentrated under reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (25 mL) and washed with water (25 mL). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 x 25 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under
reduced pressure to give compound 4E (0.5 g, 93% yield) as a white solid, which was used subsequently.
[0096] (S)-4-Amino-5-phenylpentanamide hydrochloride (110CR009): 4M HC1 in dioxane (6 mL, 25 mmol, 10 equiv) was added to compound 4E (0.73 g, 1.12 mmol, 1 equiv) After stirring overnight at room temperature, the reaction was diluted with diethyl ether (20 mL) and stirred for 6 hours. The resulting suspension was filtered and the solid was washed with diethyl ether (20 mL). The filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR009 (340 mg, 60% yield, 97.9 % purity) as a white solid.
Compound 10 (110CR012)
[0097] tert-Butyl (R)-(l-(carbamoylthio)-3-phenyIpropan-2-yI)carbamate (5B):
Compound 5 A (0.15 g, 0.56 mmol, 1 equiv) was dissolved in THF (8 mL) and sparged with nitrogen for 15 minutes. Trichloroacetyl isocyanate (0.1 mL, 0.84 mmol, 1.5 equiv) was added and the solution stirred for 3 hours, at which point TLC (30% ethyl acetate in heptane) indicated absence of starting material. The reaction was cooled to 0°C and concentrated ammonium hydroxide (0.15 mL) was added. After stirring overnight at room temperature, TLC indicated that the reaction was complete. The reaction was washed with a 10% ammonium hydroxide (10 mL). The organic layer was concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (12 g silica gel column), eluting with a gradient of 0 to 30% ethyl acetate in heptane, to give compound 5B. This reaction was repeated an additional two times 0.15 g and 0.18 g). The products were to give compound 5B (0.35 g, 1.12 mmol, 62.2% yield) as a white solid.
[0098] (R)-S-(2-Amino-3-phenylpropyl) carbamothioate hydrochloride (110CR012):
Compound 5B (0.35 g, 1.12 mmol, 1 equiv) was dissolved in 4N HCI in dioxane (2 mL). The reaction was stirred for two hours and then concentrated under reduced pressure to give crude 110CR012 as a white solid. The material was triturated in diethyl ether (15 mL) to give 110CR012 (0.215 g, 78 % yield, 98.0 % purity) as a white solid.
References
- ^ Jump up to:a b “SUNOSI™ (solriamfetol) Tablets, for Oral Use. Full Prescribing Information” (PDF). Jazz Pharmaceuticals. 2019. Retrieved 21 March2019.
- ^ Abad, VC; Guilleminault, C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC 5344488. PMID 28424564.
- ^ Jump up to:a b c d Ji-young, Sohn (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan, SS; Guilleminault, C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID 26558298.
- ^ Garde, Damian (January 14, 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:a b “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ de Biase, S; Nilo, A; Gigli, GL; Valente, M (August 2017). “Investigational therapies for the treatment of narcolepsy”. Expert Opinion on Investigational Drugs. 26 (8): 953–963. doi:10.1080/13543784.2017.1356819. PMID 28726523.
 |
|
| Clinical data | |
|---|---|
| Trade names | Sunosi |
| Synonyms | SKL-N05, ADX-N05, ARL-N05, and JZP-110; (R)-2-amino-3-phenylpropylcarbamate hydrochloride |
| Routes of administration |
By mouth |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | ~95% |
| Protein binding | 13.3–19.4% |
| Metabolism | negligible |
| Elimination half-life | ~7.1 h |
| Excretion | urine (95% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C10H14N2O2 |
| Molar mass | 194.234 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////Solriamfetol hydrochloride, Solriamfetol, ソルリアムフェトル塩酸塩; солриамфетол , سولريامفيتول , 索安非托 , JZP-110, Orphan Drug, fda 2019, R228060, R 228060
UPDATE MAR 2022
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1] It is derived from d-phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[3] It is a norepinephrine–dopamine reuptake inhibitor (NDRI). Common side effects include headache, nausea, anxiety, and trouble sleeping.[1]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of eleven countries in Asia to Aerial Pharma in 2011.[4]
Synthetic Description
Reference: Choi, Yong-Moon; Kim, Min Woo. Process for preparing O-carbamoyl amino alcohols by treatment of amino alcohols with cyanates in the presence of acid. US 20050080268. (2005).
SYN
https://www.researchgate.net/figure/Synthesis-of-solriamfetol-173_fig37_344079894

SYN
Cite this article
Yin, Z., Hu, W., Zhang, W. et al. Tailor-made amino acid-derived pharmaceuticals approved by the FDA in 2019. Amino Acids 52, 1227–1261 (2020). https://doi.org/10.1007/s00726-020-02887-4
Solriamfetol (Sunosi™) Solriamfetol (Sunosi™) (173), formerly known as JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor (DNRI) (Fig. 23). It was discovered by SK Biopharmaceuticals and developed by Jazz Pharmaceuticals (Markham 2019c). The afnity of solriamfetol for these monoamine transporters dopamine transporter (DAT, Ki=14.2 μM), norepinephrine transporter (NET, Ki=3.7 μM), and serotonin transporter (SERT, Ki=81.5 μM) was lower than that of cocaine in transfected cells and inhibits dopamine and norepinephrine reuptake with low potency (IC50=2.9 and 4.4 μM, respectively) (Baladi et al. 2018). In 2019, US FDA approved solriamfetol for using as an oral drug to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea (OSA). It was granted as an orphan drug (Schweitzer et al. 2019). The systematic name of solriamfetol is (R)-2-amino3-phenylpropylcarbamate hydrochloride, which contains a phenylalanine (171)-derived (R)-2-amino-3-phenylpropan1-ol (172) moiety (Fig. 23). Some alkyl carbamates have been introduced for controlling various central nervous system (CNS) disorders. Phenylethylamine derivatives are one of the important class of therapeutical medicines, useful for managing CNS diseases. After an intensive research, these two skeletons were combined to produce solriamfetol (173) as a drug for the treatment of CNS disorder, especially for depression. The compound 174 with a (S) carbon center showed almost no activity at all, which the racemic compound 175 displayed a half potency of the activity (Fig. 24) (Yang and Gao 2019; Choi and Byun 1996). Solriamfetol (173) was discovered and patented by SK Biopharmaceuticals in 1996 (Choi and Byun 1996). The synthesis of solriamfetol using (D)-phenylalaninol (176) as a starting material is highlighted in Scheme 21. (D)-Phenylalaninol (176) was frst converted to Cbz-protected D-phenylalaninol 177 by reacting with benzyl chloroformate. Carbamoylation of 177 with phosgene followed by ammonolysis with excess of concentrated ammonium hydroxide aqueous solation aforded (D)-O-carbamoyl-N-benzyloxycarbonylphenylalaninol 178. Hydrogenolysis removal of the Cbz protection group gave solriamfetol 173 which was treated

with HCl (gas) to provide (D)-O-carbamoylphenylalaninol hydrochloride salt. In 2020, the Zhang lab reported a method of Ni-catalyzehd asymmetric hydrogenation of 2-amidoacrylates for making solriamfetol (173) (Hu et al. 2020). In this method, o-methoxybenzoyl chloride reacted with glycine methyl ester hydrochloride 179 under a base condition and then hydrolysised in the presence of NaOH to aford desired o-methoxyhippuric acid 180. The one-step construction of oxazolone 181 was accomplished by cyclization and condensation of 180 with benzaldehyde in acetic anhydride and PPh3. Oxazolone 181 was then treated with MeOH and NaOMe to aford 2-amidoacrylate 182. Hydrogenation of 182 using Ni salt and ligand (S)-DM-MeO-BIPHEP gave product 183 in 92% ee. The reduction of 183 with LiBH4 followed by hydrolysised in the presence of NaOH provided intermediate (D)-phenylalaninol 184. Then, (D)-phenylalaninol 184 was reacted with NaOCN yielded solriamfetol (173) in 91% ee (Scheme 22). As a general comment related to this and other chiral compounds discussed here, we would like to emphasize the growing awareness about the Self-Disproportionation of Enantiomers (SDE) phenomenon and the problems related to accurate determination of the stereochemical outcome of enantioselective catalytic reactions (Han et al. 2018, 2019b, 2011a; Soloshonok et al. 2017; Sorochinsky et al. 2013c,
2013d). It was demonstrated that the SDE phenomenon is ubiquitous, being manifested virtually by all types of chiral compounds subjected to physicochemical phase transfer under totally achiral conditions (Han et al. 2019b; Sorochinsky et al. 2013c, d). One of the most frequent cases is a separation of more and less enantiomerically enriched fractions as compared with the original enantiomeric purity of a chiral compound. Consequently, to ensure the accuracy in the %ee determination, it was suggested to perform SDE tests, in particular, under the conditions of achiral column chromatography (Soroshinsky et al. 2013c) and sublimation (Han et al. 2011a).
(Soroshinsky et al. 2013c) Sorochinsky AE, Katagiri T, Ono T, Wzorek A, Aceña JL, Soloshonok VA (2013c) Optical purifcations via self-disproportionation of enantiomers by achiral chromatography; case study of a series of α-CF3-containing secondary alcohols. Chirality 25:365–368
SYN
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 63-91-2 | C9H11NO2 | D-Phenylalanine | |
| 5267-64-1 | C9H13NO | D-Phenylalaninol | |
| 75-44-5 | CCl2O | phosgene |
SYN
European Journal of Medicinal Chemistry
Volume 205, 1 November 2020, 112667
Solriamfetol (Sunosi). Solriamfetol is a norepinephrine-dopamine reuptake inhibitor (NDRI) that was developed by SK Biopharmaceuticals [124]. This drug was approved by the FDA for the treatment of EDS associated with obstructive sleep apnea (OSA) and narcolepsy [125]. In 12-week Phase III trials in this patient population, solriamfetol (75 mg or 150 mg once daily) could provide significantly great improvement [126]. Furthermore, solriamfetol was evaluated in more than 900 adults with EDS, and was maintained its effect relative to placebo after six months of use. The specific mechanism is still unknown, possibly through inhibiting dopamine and norepinephrine reuptakes [127,128].
A single-step kilogram-scale synthetic approach to solriamfetol is depicted in Scheme 21 [129]. D-Phenylalaninol 136 reacted with sodium cyanate in the presence of acid to give solriamfetol (XVI) in 89% yield.
[124] P.J. Strollo, J. Hedner, N. Collop, D.G. Lorch, D. Chen, L.P. Carter, Y. Lu, L. Lee, J. Black, J.L. Pepin, S. Redline, Solriamfetol for the treatment of excessive sleepiness in OSA: a placebo-controlled randomized withdrawal study, Chest 155 (2019) 364-374.
[125] A. Markham, Solriamfetol: first global approval, Drugs 79 (2019) 785-790.
[126] A. Malhotra, C. Shapiro, J.L. Pepin, J. Hedner, M. Ahmed, N. Foldvary-Schaefer, P.J. Strollo, G. Mayer, K. Sarmiento, M. Baladi, P. Chandler, L. Lee, R. Schwab, Long-term study of the safety and maintenance of efficacy of solriamfetol (JZP-110) in the treatment of excessive sleepiness in participants with narcolepsy or obstructive sleep apnea, Sleep 43 (2020) 220-230.
[127] M.J. Thorpy, C. Shapiro, G. Mayer, B.C. Corser, H. Emsellem, G. Plazzi, D. Chen, L.P. Carter, H. Wang, Y. Lu, J. Black, Y. Dauvilliers, A randomized study of solriamfetol for excessive sleepiness in narcolepsy, Ann. Neurol. 85 (2019) 359-370.
[128] P.K. Schweitzer, R. Rosenberg, G.K. Zammit, M. Gotfried, D. Chen, L.P. Carter, H. Wang, Y. Lu, J. Black, A. Malhotra, K.P. Strohl, Solriamfetol for excessive sleepiness in obstructive sleep apnea (TONES 3). A randomized controlled trial, Am. J. Respir. Crit. Care Med. 199 (2019) 1421-1431.
[129] Y.M. Choi, M.W. Kim, Process of preparing o-carbamoyl compounds in the presence of active amine group, 2005. WO2005033064.

Pharmacology
Pharmacodynamics
Solriamfetol is a norepinephrine–dopamine reuptake inhibitor (NDRI).[1] It binds to the dopamine transporter and the norepinephrine transporter with affinities (Ki) of 14.2 μM and 3.7 μM, respectively).[1] It inhibits the reuptake of dopamine and norepinephrine with IC50 values of 2.9 μM and 4.4 μM, respectively.[1] It has weak affinity for the serotonin transporter (Ki = 81.5 μM) and does not appreciably inhibit serotonin reuptake (IC50 > 100 μM).[1] Solriamfetol has no appreciable affinity for a variety of other targets, including the dopamine, serotonin, adrenergic, GABA, adenosine, histamine, orexin, benzodiazepine, and acetylcholine receptors.[1]
Pharmacokinetics
The elimination half-life of solriamfetol is about 7.1 hours.[1]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of eleven countries in Asia to Aerial Pharma in 2011.[4] Aerial ran two Phase II trials of the drug in narcolepsy[5] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticals paid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double-digit royalties to SK.[4][6]
In 2019, solriamfetol was approved in the United States to improve wakefulness in adults with narcolepsy or obstructive sleep apnea (OSA).[7][8] It was granted orphan drug designation.[9]
The U.S. Food and Drug Administration (FDA) approved solriamfetol based primarily on evidence from five clinical trials (Trial 1/NCT02348593, Trial 2/NCT02348606, Trial 3/NCT02348619, Trial 4/NCT02348632, Trial 5 NCT01681121) of 622 patients with narcolepsy or obstructive sleep apnea (OSA).[7] The trials were conducted in Canada, Europe, and the United States.[7]
Solriamfetol was approved for medical use in the European Union in January 2020.[2]
Society and culture
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[10]
Legal status
In the United States, solriamfetol is a Schedule IV controlled substance,[1] meaning that it has an accepted medical use and a low potential for abuse, but that abuse may lead to physical or psychological dependence.[11] A prescription is required, and can only be refilled up to five times in a six-month period.[12] In countries of the European Union, a prescription is required.[2]
Research
A case report of solriamfetol for the treatment of attention deficit hyperactivity disorder (ADHD) exists.[13]
References
- ^ Jump up to:abcdefghijklmn “Sunosi – solriamfetol tablet, film coated”. DailyMed. 16 October 2019. Retrieved 24 November 2019.
- ^ Jump up to:abc “Sunosi EPAR”. European Medicines Agency (EMA). 12 November 2019. Retrieved 26 September 2020.
- ^ Abad VC, Guilleminault C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC5344488. PMID28424564.
- ^ Jump up to:abc Ji-young S (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan SS, Guilleminault C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID26558298. S2CID7951307.
- ^ Garde D (14 January 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:abc “Drug Trials Snapshots: Sunosi”. U.S. Food and Drug Administration (FDA). 16 April 2019. Archived from the original on 28 September 2019. Retrieved 24 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Sunosi”. U.S. Food and Drug Administration (FDA). 29 April 2019. Retrieved 24 November 2019.This article incorporates text from this source, which is in the public domain.
- ^ “Solriamfetol Orphan Drug Approval”. U.S. Food and Drug Administration (FDA). Retrieved 24 November 2019.This article incorporates text from this source, which is in the public domain.
- ^ “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ 21 U.S.C.§ 812 – Schedules of controlled substances
- ^ “Manuals – Practitioner’s Manual – Section V”. Retrieved 2014-01-07
- ^ Naguy A, El-Sheshaie A, Elsori DH, Alamiri B (April 2021). “Solriamfetol for attention deficit hyperactivity disorder”. CNS Spectr: 1–2. doi:10.1017/S1092852921000328. PMID33870884.
External links
- “Solriamfetol”. Drug Information Portal. U.S. National Library of Medicine.
- * “Solriamfetol hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
//////////////////
N[C@@H](COC(N)=O)CC1=CC=CC=C1
