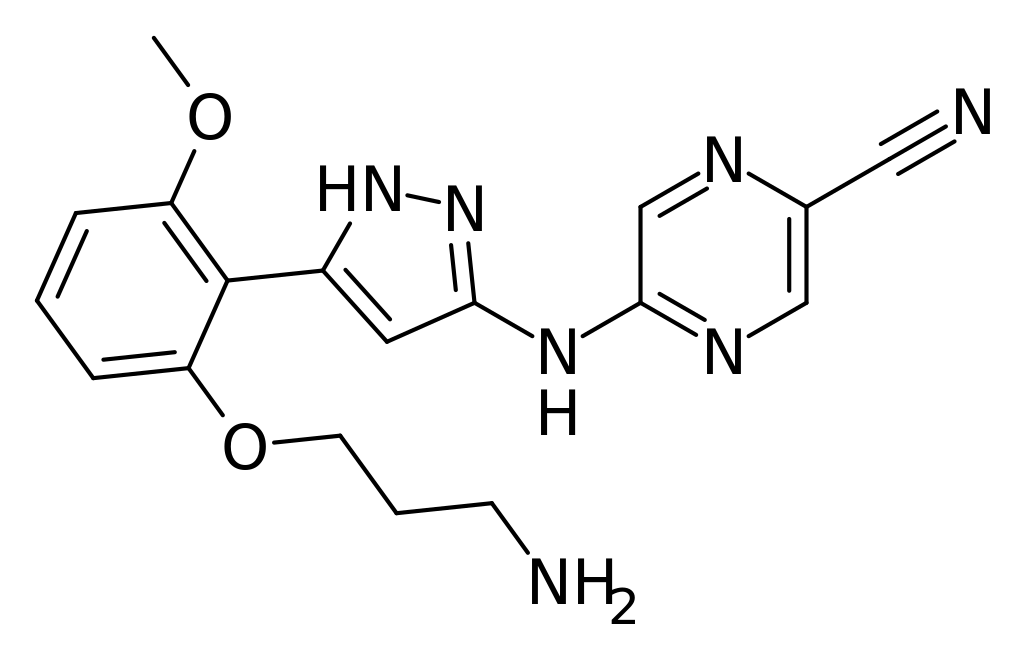
Prexasertib
Captisol® enabled prexasertib; CHK1 Inhibitor II; LY 2606368; LY2606368 MsOH H2O
5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-1H-pyrazol-3-ylamino)pyrazine-2-carbonitrile
2-Pyrazinecarbonitrile, 5-[[5-[2-(3-aminopropoxy)-6-methoxyphenyl]-1H-pyrazol-3-yl]amino]-
| Name |
Prexasertib |
| Lab Codes |
LY-2606368 |
| Chemical Name |
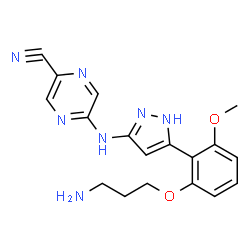
5-({5-[2-(3-aminopropoxy)-6-methoxyphenyl]-1H-pyrazol-3-yl}amino)pyrazine-2-carbonitrile |
| Chemical Structure |
 |
| Molecular Formula |
C18H19N7O2 |
| UNII |
UNII:820NH671E6 |
| Cas Registry Number |
1234015-52-1 |
| OTHER NAMES |
прексасертиб [Russian] [INN]
بريكساسيرتيب [Arabic] [INN]
|
| Originator |
Array BioPharma |
| Developer |
Eli Lilly, National Cancer Institute |
| Mechanism Of Action |
Checkpoint kinase inhibitors, Chk-1 inhibitors |
| Who Atc Codes |
L01X-E (Protein kinase inhibitors) |
| Ephmra Codes |
L1H (Protein Kinase Inhibitor Antineoplastics) |
| Indication |
Breast cancer, Ovarian cancer, Solid tumor, Head and neck cancer, Leukemia, Neoplasm Metastasis, Colorectal Neoplasms, Squamous Cell Carcinoma |


 2100300-72-7 CAS
2100300-72-7 CAS

Prexasertib mesylate hydrate
CAS#: 1234015-57-6 (mesylate hydrate)
Chemical Formula: C19H25N7O6S
Molecular Weight: 479.512, CODE LY-2940930
LY-2606368 (free base)

Prexasertib mesylate ANHYDROUS
CAS#: 1234015-55-4 (mesylate)
Chemical Formula: C19H23N7O5S
Molecular Weight: 461.497

Prexasertib dihydrochloride
1234015-54-3. MW: 438.3169
LY2606368 is a small-molecule Chk-1 inhibitors invented by Array and being developed by Eli Lilly and Company. Lilly is responsible for all clinical development and commercialization activities. Chk-1 is a protein kinase that regulates the tumor cell’s response to DNA damage often caused by treatment with chemotherapy. In response to DNA damage, Chk-1 blocks cell cycle progression in order to allow for repair of damaged DNA, thereby limiting the efficacy of chemotherapeutic agents. Inhibiting Chk-1 in combination with chemotherapy can enhance tumor cell death by preventing these cells from recovering from DNA damage.
Originator Array BioPharma; Eli Lilly
Developer Eli Lilly; National Cancer Institute (USA)
Class Antineoplastics; Nitriles; Pyrazines; Pyrazoles; Small molecules
Mechanism of Action Checkpoint kinase 1 inhibitors; Checkpoint kinase 2 inhibitors
Highest Development Phases
- Phase II Breast cancer; Ovarian cancer; Small cell lung cancer; Solid tumours
- Phase I Acute myeloid leukaemia; Colorectal cancer; Head and neck cancer; Myelodysplastic syndromes; Non-small cell lung cancer
Most Recent Events
- 10 Apr 2017 Eli Lilly completes a phase I trial for Solid tumours (Late-stage disease, Second-line therapy or greater) in Japan (NCT02514603)
- 10 Mar 2017 Phase-I clinical trials in Solid tumours (Combination therapy, Metastatic disease, Inoperable/Unresectable) in USA (IV) (NCT03057145)
- 22 Feb 2017 Khanh Do and AstraZeneca plan a phase H trial for Solid tumour (Combination therapy, Metastatic disease, Inoperable/Unresectable) in USA (NCT03057145)
Prexasertib (LY2606368) is a small molecule checkpoint kinase inhibitor, mainly active against CHEK1, with minor activity against CHEK2. This causes induction of DNA double-strand breaks resulting in apoptosis. It is in development by Eli Lilly. [1]
A phase II clinical trial for the treatment of small cell lung cancer is expected to be complete in December 2017.[2]
an aminopyrazole compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, that inhibits Chkl and is useful for treating cancers characterized by defects in deoxyribonucleic acid (DNA) replication, chromosome segregation, or cell division.
Chkl is a protein kinase that lies downstream from Atm and/or Atr in the DNA damage checkpoint signal transduction pathway. In mammalian cells, Chkl is phosphorylated in response to agents that cause DNA damage including ionizing radiation (IR), ultraviolet (UV) light, and hydroxyurea. This phosphorylation which activates Chkl in mammalian cells is dependent on Atr. Chkl plays a role in the Atr dependent DNA damage checkpoint leading to arrest in S phase and at G2M. Chkl phosphorylates and inactivates Cdc25A, the dual-specificity phosphatase that normally dephosphorylates cyclin E/Cdk2, halting progression through S-phase. Chkl also phosphorylates and inactivates Cdc25C, the dual specificity phosphatase that dephosphorylates cyclin B/Cdc2 (also known as Cdkl) arresting cell cycle progression at the boundary of G2 and mitosis (Fernery et al, Science, 277: 1495-1, 1997). In both cases, regulation of Cdk activity induces a cell cycle arrest to prevent cells from entering mitosis in the presence of DNA damage or unreplicated DNA. Various inhibitors of Chkl have been reported. See for example, WO 05/066163,
WO 04/063198, WO 03/093297 and WO 02/070494. In addition, a series of aminopyrazole Chkl inhibitors is disclosed in WO 05/009435.
However, there is still a need for Chkl inhibitors that are potent inhibitors of the cell cycle checkpoints that can act effectively as potentiators of DNA damaging agents. The present invention provides a novel aminopyrazole compound, or a pharmaceutically acceptable salt thereof or solvate of the salt, that is a potent inhibitor of Chkl . The compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, potently abrogates a Chkl mediated cell cycle arrest induced by treatment with DNA damaging agents in tissue culture and in vivo. Furthermore, the compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, of the present invention also provides inhibition of Chk2, which may be beneficial for the treatment of cancer. Additionally, the lack of inhibition of certain other protein kinases, such as CDKl, may provide a -2- therapeutic benefit by minimizing undesired effects. Furthermore, the compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, of the present invention inhibits cell proliferation of cancer cells by a mechanism dependent on Chkl inhibition.
WO 2010077758
Preparation 8
tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate
A solution of tert-butyl 3-(2-(3-(5-bromopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate (0.378 g, 0.730 mmol) and zinc cyanide (0.10 g, 0.870 mmol) in DMF (10 mL) is degassed with a stream of nitrogen for one hour and then -25- heated to 80 0C. To the reaction is added Pd(Ph3P)4 (0.080 g, 0.070 mmol), and the mixture is heated overnight. The reaction is cooled to room temperature and concentrated under reduced pressure. The residue is purified by silica gel chromatography (CH2Cl2/Me0H) to give 0.251 g (73%) of the title compound.
Preparation 12 tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate

A 5 L flange-neck round-bottom flask equipped with an air stirrer rod and paddle, thermometer, pressure-equalizing dropping funnel, and nitrogen bubbler is charged with 5-(5-(2-hydroxy-6-methoxy-phenyl)-lH-pyrazol-3-ylamino)-pyrazine-2-carbonitrile (47.0 g, 152 mmol) and anhydrous THF (1.2 L). The stirred suspension, under nitrogen, is cooled to 0 0C. A separate 2 L 3 -necked round-bottom flask equipped with a large -28- magnetic stirring bar, thermometer, and nitrogen bubbler is charged with triphenylphosphine (44.0 g; 168 mmol) and anhydrous THF (600 mL). The stirred solution, under nitrogen, is cooled to 0 0C and diisopropylazodicarboxylate (34.2 g; 169 mmol) is added and a milky solution is formed. After 3-4 min, a solution of7-butyl-N-(3- hydroxypropyl)-carbamate (30.3 g, 173 mmol) in anhydrous THF (100 mL) is added and the mixture is stirred for 3-4 min. This mixture is then added over 5 min to the stirred suspension of starting material at 0 0C. The reaction mixture quickly becomes a dark solution and is allowed to slowly warm up to room temperature. After 6.5 h, more reagents are prepared as above using PPh3 (8 g), DIAD (6.2 g) and carbamate (5.4 g) in anhydrous THF (150 mL). The mixture is added to the reaction mixture, cooled to -5 0C and left to warm up to room temperature overnight. The solvent is removed in vacuo. The resulting viscous solution is loaded onto a pad of silica and product is eluted with ethyl acetate. The concentrated fractions are separately triturated with methanol and resulting solids are collected by filtration. The combined solids are triturated again with methanol (400 mL) and then isolated by filtration and dried in vacuo at 50 0C overnight to give 31.3 g of desired product. LC-ES/MS m/z 466.2 [M+ 1]+.
Example 2
5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile dihydrogen chloride salt
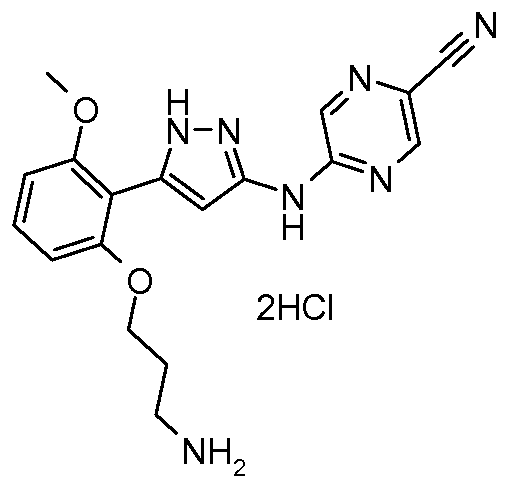
A 5 L flange-neck, round-bottom flask equipped with an air stirrer rod and paddle, thermometer, and air condenser with bubbler attached, is charged with tert-bvXyl 3-(2-(3- (5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3-methoxyphenoxy)propylcarbamate (30.9 g, 66.3 mmol) and ethyl acetate (3 L). The mechanically stirred yellow suspension is cooled to just below 10 0C. Then hydrogen chloride from a lecture bottle is bubbled in -29- vigorously through a gas inlet tube for 15 min with the ice-bath still in place. After 5 h the mixture is noticeably thickened in appearance. The solid is collected by filtration, washed with ethyl acetate, and then dried in vacuo at 60 0C overnight to give 30.0 g. 1H NMR (400 MHz, DMSO-d6) δ 2.05 (m, 2H), 2.96 (m, 2H), 3.81 (s, 3H), 4.12 (t, J = 5.8 Hz, 2H), 6.08 (br s, 3H), 6.777 (d, J = 8.2 Hz, IH), 6.782 (d, J = 8.2 Hz, IH), 6.88 (br s, IH), 7.34 (t, J = 8.2 Hz, IH), 8.09 (br s, IH), 8.55 (br s, IH), 8.71 (s, IH), 10.83 (s, IH), 12.43 (br s, IH). LC-ES/MS m/z 366.2 [M+lf.
Example 3 5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile
5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile dihydrogen chloride salt (3.0 g, 6.84 mmol) is suspended in 200 mL of CH2Cl2. 1 N NaOH is added (200 mL, 200 mmol). The mixture is magnetically stirred under nitrogen at room temperature for 5 h. The solid is collected by filtration and washed thoroughly with water. The filter cake is dried in vacuo at 50 0C overnight to give 2.26 g (90%) of the free base as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.81 (m, 2H), 2.73 (t, J = 6.2 Hz, 2H), 3.82 (s, 3H), 4.09 (t, J = 6.2 Hz, 2H), 6.76 (t, J = 8.2 Hz, 2H), 6.93 (br s, IH), 7.31 (t, J = 8.2 Hz, IH), 8.52 (br s, IH), 8.67 (s, IH). LC- MS /ES m/z 366.2 [M+ 1]+.
Example 4
5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile methanesulfonic acid salt -30-
5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (1.0 g, 2.74 mmol) is suspended in MeOH (100 mL). A I M solution of methanesulfonic acid in MeOH (2.74 mL, 2.74 mmol) is added to the mixture dropwise with stirring. The solid nearly completely dissolves and is sonicated and stirred for 15 min, filtered, and concentrated to 50 mL. The solution is cooled overnight at -15 0C and the solid that forms is collected by filtration. The solid is dried in a vacuum oven overnight to give 0.938 g (74%) of a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.97 (m, 2H), 2.28 (s, 3H), 2.95 (m, 2H), 3.79 (s, 3H), 4.09 (t, J = 5.9 Hz, 2H), 6.753 (d, J = 8.4 Hz, IH), 6.766 (d, J = 8.4 Hz, IH), 6.85 (br s, IH), 7.33 (t, J = 8.4 Hz, IH), 7.67 (br s, 3H), 8.49 (br s, IH), 8.64 (s, IH), 10.70 (s, IH), 12.31 (s, IH). LC-ES/MS m/z 366.2 [M+l]+.
Preparation 18 tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate
5-(5-(2-Hydroxy-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (618 g, 1.62 mol) is slurried in tetrahydrofuran (6.18 L, 10 volumes) and chilled to -5 to 0 0C with an acetone/ice bath. Triethylamine (330 g, 3.25 mol) is added through an addition funnel over 30 – 40 min at -5 to 5 0C. The resulting slurry is stirred at -5 to 5 0C for 60 – 90 min. The insoluble triethylamine hydrochloride is filtered and the solution of the phenol ((5-(2-hydroxy-6-methoxyphenyl)-lH-pyrazol-3- ylamino)pyrazine-2-carbonitrile) collected in an appropriate reaction vessel. The cake is rinsed with THF (1.24 L). The THF solution of the phenol is held at 15 to 20 0C until needed.
Triphenylphosphine (1074 g, 4.05 mol) is dissolved at room temperature in THF (4.33 L). The clear colorless solution is cooled with an acetone/ice bath to -5 to 5 0C. Diisopropylazodicarboxylate (795 g, 3.89 mol) is added dropwise through an addition funnel over 40 – 60 min, keeping the temperature below 10 0C. The resulting thick white slurry is cooled back to -5 to 0 0C. tert-Butyl 3-hydroxypropylcarbamate (717g, 4.05 moles) is dissolved in a minimum of THF (800 mL). The tert-butyl 3- hydroxypropylcarbamate/THF solution is added, through an addition funnel, over 20 – 30 -35- min at -5 to 5 0C to the reagent slurry. The prepared reagent is stirred in the ice bath at -5 to 0 0C until ready for use.
The prepared reagent slurry (20%) is added to the substrate solution at 15 to 20 0C. The remaining reagent is returned to the ice bath. The substrate solution is stirred at ambient for 30 min, then sampled for HPLC. A second approximately 20% portion of the reagent is added to the substrate, stirred at ambient and sampled as before. Addition of the reagent is continued with monitoring for reaction completion by HPLC. The completed reaction is concentrated and triturated with warm methanol (4.33 L, 50 – 60 0C) followed by cooling in an ice bath. The resulting yellow precipitate is filtered, rinsed with cold MeOH (2 L), and dried to constant weight to provide 544 g (72%) of the title compound, mp 214 – 216 0C; ES/MS m/z 466.2 [M+l]+.
Example 5
2-Pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3- yl]amino] monomesylate monohydrate (Chemical Abstracts nomenclature)

tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate (1430 g, 3.07 mol) is slurried with acetone (21.5 L) in a 30 L reactor. Methanesulfonic acid (1484 g, 15.36 mol) is added through an addition funnel in a moderate stream. The slurry is warmed to reflux at about 52 0C for 1 to 3 h and monitored for reaction completion by HPLC analysis. The completed reaction is cooled from reflux to 15 to 20 0C over 4.5 h. The yellow slurry of 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3-yl]amino] dimesylate salt is filtered, rinsed with acetone (7 L) and dried in a vacuum oven. The dimesylate salt, (1608 g, 2.88 mol) is slurried in water (16 L). Sodium hydroxide (aqueous 50%, 228 g, 2.85 mol) is slowly poured into the slurry. The slurry is -36- heated to 60 0C and stirred for one hour. It is then cooled to 16 0C over 4 h and filtered. The wet filter cake is rinsed with acetone (4 L) and dried to constant weight in a vacuum oven at 40 0C to provide 833 g (94%) of 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3- aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3-yl]amino] monomesylate monohydrate. mp 222.6 0C; ES/MS m/z 366.2 [M+l]+.
Example 5a
2-Pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3- yl] amino] monomesylate monohydrate (Chemical Abstracts nomenclature)
Crude 2-pyrazinecarbonitrile, 5 -[ [5 – [- [2-(3 -aminopropyl)-6-methoxyphenyl]- IH- pyrazol-3-yl] amino] monomesylate monohydrate is purified using the following procedure. The technical grade 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6- methoxyphenyl]-lH-pyrazol-3-yl] amino] mono mesylate mono hydrate (1221 g, 2.55 mol) is slurried in a solvent mixture of 1: 1 acetone/water (14.7 L). The solid is dissolved by warming the mixture to 50 – 55 0C. The solution is polish filtrated while at 50 – 55 0C through a 0.22μ cartridge filter. The solution is slowly cooled to the seeding temperature of about 42 – 45 0C and seeded. Slow cooling is continued over the next 30 – 60 min to confirm nucleation. The thin slurry is cooled from 38 to 15 0C over 3 h. A vacuum distillation is set up and the acetone removed at 110 – 90 mm and 20 – 30 0C. The mixture is cooled from 30 to 15 0C over 14 h, held at 15 0C for 2 h, and then filtered. The recrystallized material is rinsed with 19: 1 water/acetone (2 L) and then water (6 L) and dried to constant weight in a vacuum oven at 40 0C to provide 1024 g (83.9%) of the title compound, mp 222.6 0C; ES/MS m/z 366.2 [M+l]+. X-ray powder diffraction (XRPD) patterns may be obtained on a Bruker D8
Advance powder diffractometer, equipped with a CuKa source (λ=l.54056 angstrom) operating at 40 kV and 40 mA with a position-sensitive detector. Each sample is scanned between 4° and 35° in °2Θ ± 0.02 using a step size of 0.026° in 2Θ ± 0.02 and a step time of 0.3 seconds, with a 0.6 mm divergence slit and a 10.39 mm detector slit. Primary and secondary Soller slits are each at 2°; antiscattering slit is 6.17 mm; the air scatter sink is in place. -37-
Characteristic peak positions and relative intensities:
Differential scanning calorimetry (DSC) analyses may be carried out on a Mettler- Toledo DSC unit (Model DSC822e). Samples are heated in closed aluminum pans with pinhole from 25 to 350 0C at 10 °C/min with a nitrogen purge of 50 mL/min. Thermogravimetric analysis (TGA) may be carried out on a Mettler Toledo TGA unit (Model TGA/SDTA 85Ie). Samples are heated in sealed aluminum pans with a pinhole from 25 to 350 0C at 10 0C /min with a nitrogen purge of 50 mL/min.
The thermal profile from DSC shows a weak, broad endotherm form 80 – 1400C followed by a sharp melting endotherm at 222 0C, onset (225 0C, peak). A mass loss of 4% is seen by the TGA from 25 – 140 0C.
PATENT
US 20110144126
WO 2017015124
WO 2017100071
WO 2017105982
WO 2016051409
PATENT
WO 2017100071
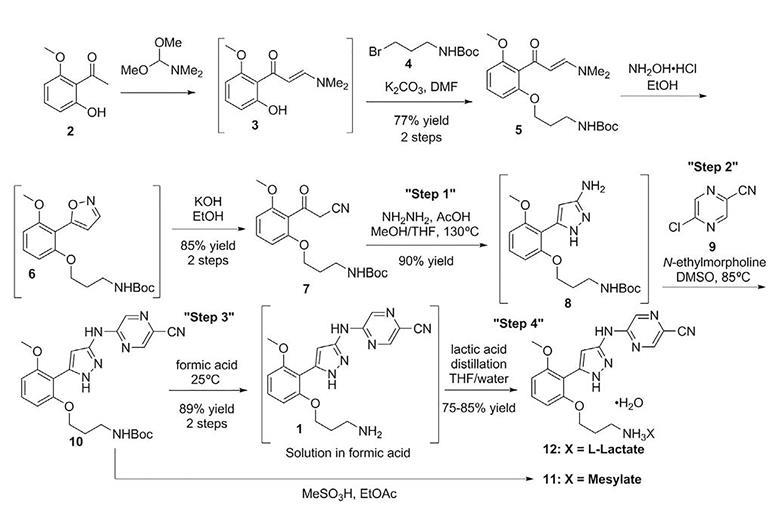
Preparation 1
tert-Butyl (E)-(3-(2-(3-(dimethylamino)ac^’loyl)-3-me1hoxyphenox50propyl)carbamate

L _l H
Combine l-(2-hydroxy-6-methox>’phenyl)e1han-l-one (79.6 kg, 479 mol) and 1,1-<iimethoxy-N,N-dimemylmethanamino (71.7 kg, 603.54 mol) with DMF (126 kg). Heat to 85-90 °C for 12 hours. Cool the reaction mixture containing intermediate (E)-3-(dimethylamino)-l-(2-hydroxy-6-methoxyphenyl)prop-2-en-l-one (mp 84.74 °C) to ambient temperature and add anhydrous potassium phosphate (136 kg, 637.07 mol) and tert-butyl (3-bromopropyl)carbamate (145 kg, 608.33 mol). Stir the reaction for 15 hours at ambient temperature. Filter the mixture and wash the filter cake with ΜΓΒΕ (3 χ , 433 kg, 300 kg, and 350 kg). Add water (136 kg) and aqueous sodium chloride (25% solution, 552 kg) to the combined MTBE organic solutions. Separate the organic and aqueous phases. Back-extract the resulting aqueous phase with MTBE (309 kg) and add the MTBE layer to the organic solution. Add an aqueous sodium chloride solution (25% solution, 660 kg) to the combined organic extracts and separate the layers. Concentrate the organic extracts to 1,040 kg – 1,200 kg and add water (400 kg) at 30-35 °C to the residue. Cool to ambient temperature and collect material by filtration as a wet cake to give the title product (228.35 kg, 90%). ES/MS (m/z): 379.22275 (M+l).
Preparation 2
tert-Butyl (3-(2-(2-cyanoacetyl)-3-methoxyphenoxy)propyl)carbamate
“9 o




Combine ethanol (1044 kg), hydroxyl amino hydrochloride (30 kg, 431.7 mol), and terr-butyl (E)-(3-(2-(3-(^me%lamino)acryloyl)-3-
methoxyphenoxy)propyl)carbamate (228.35 kg, 72% as a wet water solid, 434.9 mol) to form a solution. Heat the solution to 35 – 40 °C for 4-6 hours. Cool the reaction to ambient temperature and concentrate to a residue. Add MTBE (300 kg) to the residue and concentrate the solution to 160 kg – 240 kg. Add MTBE (270 kg) and concentrate the solution. Add MTBE (630 kg), water (358 kg), and sodium chloride solution (80 kg, 25% aqueous) and stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes. Separate the aqueous layer. Add water (360 kg) and sodium chloride solution (82 kg, 25% sodium chloride) to the organic phase. Stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes. Separate the aqueous portion. Add sodium chloride solution (400 kg, 25 % aqueous) to the organic portion. Stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes at ambient temperature. Separate the aqueous portion. Concentrate the organic portion to 160 kg – 240 kg at 40 °C. Add ethanol (296 kg) to the organic portion. Concentrate the solution to 160 kg to 240 kg at 40 °C to provide an intermediate of tert-butyl (3-(2-(isoxazol-5-yl)-3-methox>’phenoxy)propyl)carbamate. Add ethanol (143 kg) and water (160 kg) to the concentrated solution. Add potassium hydroxide (31.8 kg) at 40 °C. Add ethanol (80 kg) and adjust the temperature to 45-50 °C. Stir for 4-6 hours at 45-50 °C and concentrate to 160 kg – 240 kg at 40 °C. Add water to the concentrate (160 kg) and acetic acid (9.0 kg) drop-wise to adjust the pH to 10-12 while mamtaining the temperature of the solution at 25 to 35 °C. Add ethyl acetate (771 kg) and acetic acid drop-wise to adjust the pH to 5-7 while maintaining the temperature of the solution at 25-35 °C. Add sodium chloride solution (118 kg, 25% aqueous solution). Stir the mixture for 20 minutes at ambient temperature. Let the solution stand for 30 minutes at ambient temperature. Separate Ihe aqueous portion. Heat the organic portion to 30-35 °C. Add water (358 kg) drop-wise. Stir the solution for 20 minutes while maintaining the temperature at 30 to 35 °C. Let the mixture stand for 30 minutes and separate the aqueous portion. Wash the organic portion with sodium chloride solution (588 kg, 25% aqueous) and concentrate the organic portion to 400 kg – 480 kg at 40-50 °C. Heat the concentrated solution to 50 °C to form a solution. Maintain the solution at 50 °C and add M-heptane (469 kg) drop-wise. Stir the solution for 3 hours at 50 °C before slowly cooling to ambient temperature to crystallize the product. Stir at ambient temperature for 15 hours and filter the crystals. Wash the crystals with ethanol/«-heptane (1 :2, 250 kg) and dry at 45 °C for 24 hours to provide the title compound (133.4 kg, 79.9%), rap. 104.22 °C,
Example 1
5-(5-(2-(3-Ammopropoxy)-6-memoxyphenyl)-lH-pyrazol-3-ylammo)pyrazine-2- carbonitrile (S)-lactate monohydrate

Combine a THJF solution (22%) of fcrt-butyl (3-(2-(2-cyanoacetyl)-3-memoxyphenoxy)propyl)carbamate (1.0 eqv, this is define as one volume) with hydrazine (35%, 1.5 eqv), acetic acid (glacial, 1.0 eqv), water (1 volume based on the THF solution) and methanol (2 volumes based on the THF solution). This is a continuous operation. Heat the resulting mixture to 130 °C and 1379 kPa with a rate of V/Q = 70 minutes, tau = 60. Extract the solution with toluene (4 volumes), water (1 volume), and sodium carbonate (10% aqueous, 1 eqv). Isolate Ihe toluene layer and add to DMSO (0.5 volumes). Collect a solution of the intermediate compound tert-butyl (3-(2-(3-amino-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate (26.59 kg, 91%) in 10 days, mp = 247.17 °C as a DMSO solution (3 volumes of product). N-Eftylmorpholine (1.2 eqv) and 5-chloropyrazine-2-carbonitrile (1.15 eqv) in 2 volumes of DMSO is combined in a tube reactor at 80 °C, V/Q = 3 and tau = 170 minutes at ambient pressure. Add the product stream to methanol (20 vol). As a continuous process, filter the mixture and wash with methanol followed by MTBE. Air dry the material on the filter to give tert-butyl (3-(2-(3-((5-cyanopyrazm-2-yl)arnino)-lH-pyrazol-5-yl)-3-methox>’phenoxy) propyl)carbamate in a continuous fashion (22.2 kg, 88.7%, 8 days). Dissolve a solution of fcrt-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in formic acid (99%, 142 kg) at ambient temperature and agitate for 4 hours to provide an intermediate of 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile formate. Dilute the solution with water (55 kg), (S)-lactic acid (30%, 176 kg) and distill the resulting mixture until < 22 kg formic acid remains. Crystallize the resulting residue from THF and wash with a THF -water (0.5% in THF) solution. Dry the wet cake at 30 °C at >10% relative humidity to give the title product as a white to yellow solid (24.04 kg, 85-90%), mp. 157 °C.
Alternate Preparation Example 1
5-(5-(2-(3-Ammopropoxy)-6-memoxyphenyl)-lH-pyrazol-3-ylammo)pyrazine-2- carbonitrile (S)-lactate monohydrate
Add 5-({3-[2-(3-aminopropoxy)-6-methoxyphenyl]-lH-pyrazol-5-yl}ammo)pyrazine-2-carbonitrile (4.984 g, 13.33 mmol, 97.7 wt%) to n-PrOH (15.41 g, 19.21 mL) to form a slurry. Heat the slurry to 60 °C. Add (S)-lactic acid (1.329 g, 14.75 mmol) to water (19.744 mL) and add this solution to the slurry at 58 °C. Heat the solution to 60 °C and add n-PrOH (21.07 g, 26.27 mL). Seed the solution with 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)ammo)pyrazme-2-carbom^ (S)-lactate monohydrate (48.8 mg, 0.1 mmol) and cool the solution to 40 °C over 35 minutes. Add H-PrOH (60.5 mL) to the slurry at 40 °C via a syringe pump over 2 hours and maintain the temperature at 40 °C. Once complete, air cool the slurry to ambient temperature for 2 hours, the cool the mixture in ice-water for 2 hours. Filter the product, wash the wet cake with 6:1 (v/v) rc-PrOH : H20 (15 mL), followed by n-PrOH (15 mL) and dry the wet cake for 20 minutes. Dry the solid overnight at 40 °C in vacuo to give the title compound as a white to yellow solid (5.621 g, 89.1%), m.p. 157 °C.
Crystalline Example 1
Crystalline 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3- ylamino)pyrazine-2-carbonitrile (S)-lactate monohydrate Prepare a slurry having 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3 -y lamino)py razine-2-carbonitrile (368 mg, 1.0 mmol) in a 10:1 THF-water (5 mL) solution and stir at 55 °C. Add (S)-lactic acid (110 mg, 1.22 mmol) dissolved in THF (1 mL). A clear solution forms. Stir for one hour. Reduce Ihe temperature to 44 °C and stir until an off-white precipitate forms. Filter the material under vacuum, rinse with THF, and air dry to give the title compound (296 mg, 80%).
X-Ray Powder Diffraction, Crystalline Example 1 Obtain the XRPD patterns of the crystalline solids on a Bruker D4 Endeavor X-ray powder diffractometer, equipped with a CuKa source (λ = 1.54060 A) and a Vantec detector, operating at 35 kV and 50 mA. Scan the sample between 4 and 40° in 2Θ, with a step size of 0.0087° in 2Θ and a scan rate of 0.5 seconds/step, and with 0.6 mm divergence, 5.28mm fixed anti-scatter, and 9.5 mm detector slits. Pack the dry powder on a quartz sample holder and obtain a smooth surface using a glass slide. It is well known in the crystallography art that, for any given crystal form, the relative intensities of the diffraction peaks may vary due to preferred orientation resulting from factors such as crystal morphology and habit. Where the effects of preferred orientation are present, peak intensities are altered, but the characteristic peak positions of the polymorph are unchanged. See, e.g. The U. S. Pharmacopeia 35 – National Formulary 30 Chapter <941> Characterization of crystalline and partially crystalline solids by XRPD Official December 1, 2012-May 1, 2013. Furthermore, it is also well known in the
crystallography art that for any given crystal form the angular peak positions may vary slightly. For example, peak positions can shift due to a variation in the temperature or humidity at which a sample is analyzed, sample displacement, or the presence or absence of an internal standard. In the present case, a peak position variability of ± 0.2 in 2Θ will take into account these potential variations without hindering the unequivocal identification of the indicated crystal form Confirmation of a crystal form may be made based on any unique combination of distinguishing peaks (in units of ° 2Θ), typically the more prominent peaks. The crystal form diffraction patterns, collected at ambient temperature and relative humidity, were adjusted based on NIST 675 standard peaks at 8.85 and 26.77 degrees 2-theta,
Characterize a prepared sample of crystalline 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)- lH-pyrazol-3-ylamino)pyrazine-2-carbonitrile (S)-lactate monohydrate by an XPRD pattern using CuKa radiation as having diffraction peaks (2-theta values) as described in Table 1 below. Specifically the pattern contains a peak at 12.6 in
combination with one or more of the peaks selected from the group consisting of 24.8, 25.5, 8.1, 6.6, 12.3, and 16.3 with a tolerance for the diffraction angles of 0.2 degrees.
PATENT
WO 2017105982
Example 1
5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile S)-lactate monohydrate

Combine a THF solution (22%) of i<?ri-butyl (3-(2-(2-cyanoacetyl)-3-methoxyphenoxy)propyl)carbamate (1.0 eqv, this is define as one volume) with hydrazine (35%, 1.5 eqv), acetic acid (glacial, 1.0 eqv), water (1 volume based on the THF solution) and methanol (2 volumes based on the THF solution). As this is a continuous operation, grams or kg is irrelevant in this processing methodology. Heat the resulting mixture to 130 °C and 1379 kPa with a rate of V/Q = 70 minutes (where V refers to the volume of the reactor and Q refers to flow rate), tau = 60. Extract the solution with toluene (4 volumes), water (1 volume), and sodium carbonate (10% aqueous, 1 eqv). Isolate the toluene layer and add to DMSO (0.5 volumes). Collect a solution of the intermediate compound i<?ri-butyl (3-(2-(3-amino- lH-pyrazol-5-yl)-3-methoxyphenoxy)
propyl)carbamate (26.59 kg, 91%) in 10 days, mp = 247.17 °C as a DMSO solution (3 volumes of product). N-ethylmorpholine (1.2 eqv) and 5-chloropyrazine-2-carbonitrile (1.15 eqv) in 2 volumes of DMSO is combined in a tube reactor at 80 °C, V/Q = 3 and tau = 170 minutes at ambient pressure. Add the product stream to methanol (20 vol). As a continuous process, filter the mixture and wash with methanol followed by MTBE. Air dry the material on the filter to give i<?ri-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in a continuous fashion (22.2 kg, 88.7%, 8 days). Dissolve a solution of i<?ri-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in formic acid (99%, 142 kg) at ambient temperature and agitate for 4 hours to provide an intermediate of 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile formate. Dilute the solution with water (55 kg), (S)-lactic acid (30%, 176 kg) and distill the resulting mixture until < 22 kg formic acid remains. Crystallize the resulting residue from THF and wash with a THF -water (0.5% in THF) solution. Dry the wet cake at 30 °C at >10% relative humidity to give the title product as a white to yellow solid (24.04 kg, 85-90%), m.p. 157 °C.
Alternate Preparation Example 1
5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (S)-lactate monohydrate
Add 5-({3-[2-(3-aminopropoxy)-6-methoxyphenyl]-lH-pyrazol-5-yl}amino)pyrazine-2-carbonitrile (4.984 g, 13.33 mmol, 97.7 wt%) to n-PrOH (15.41 g, 19.21 mL) to form a slurry. Heat the slurry to 60 °C. Add (S)-lactic acid (1.329 g, 14.75 mmol) to water (19.744 mL) and add this solution to the slurry at 58 °C. Heat the solution to 60 °C and add n-PrOH (21.07 g, 26.27 mL). Seed the solution with 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile (S)-lactate monohydrate (48.8 mg, 0.1 mmol) and cool the solution to 40 °C over 35 minutes. Add ft-PrOH (60.5 mL) to the slurry at 40 °C via a syringe pump over 2 hours and maintain the temperature at 40 °C. Once complete, air cool the slurry to ambient temperature for 2 hours, then cool the mixture in ice-water for 2 hours. Filter the product, wash the wet cake with 6:1 (v/v) n-PrOH : H20 (15 mL), followed by n-PrOH (15 mL)
and dry the wet cake for 20 minutes. Dry the solid overnight at 40 °C in vacuo to give the title compound as a white to yellow solid (5.621 g, 89.1%), m.p. 157 °C.
Clip
Kilogram-scale prexasertib monolactate monohydrate synthesis under continuous-flow CGMP conditions
- Kevin P. Cole1,*,
- Jennifer McClary Groh1,
- Martin D. Johnson1,
- Christopher L. Burcham1,
- Bradley M. Campbell1,
- William D. Diseroad2,
- Michael R. Heller1,
- John R. Howell1,
- Neil J. Kallman1,
- Thomas M. Koenig3,
- Scott A. May1,
- Richard D. Miller1,
- David Mitchell1,
- David P. Myers1,
- Steven S. Myers1,
- Joseph L. Phillips1,
- Christopher S. Polster1,
- Timothy D. White1,
- Jim Cashman4,
- Declan Hurley5,
- Robert Moylan5,
- Paul Sheehan5,
- Richard D. Spencer6,
- Kenneth Desmond7,
- Paul Desmond7,
- Olivia Gowran7
1Small Molecule Design and Development, Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN 46285, USA.
2Technical Services/Manufacturing Science, Eli Lilly and Company, Indianapolis, IN 46285, USA.
3Technical Services/Manufacturing Science, Eli Lilly and Company, Greenfield, IN 46140, USA.
4Quality Assurance, Eli Lilly and Company, Dunderrow, Kinsale, County Cork, Ireland.
5Technical Services/Manufacturing Science, Eli Lilly and Company, Dunderrow, Kinsale, County Cork, Ireland.
6Process Engineering, Eli Lilly and Company, Dunderrow, Kinsale, County Cork, Ireland.
7Quality Control, Eli Lilly and Company, Dunderrow, Kinsale, County Cork, Ireland.
- ↵*Corresponding author. Email: k_cole@lilly.com
Science 16 Jun 2017:
Vol. 356, Issue 6343, pp. 1144-1150
DOI: 10.1126/science.aan0745
science 2017, 356, 1144
Kilogram-Scale Prexasertib Monolactate Monohydrate Synthesis under Continuous-Flow CGMP Conditions

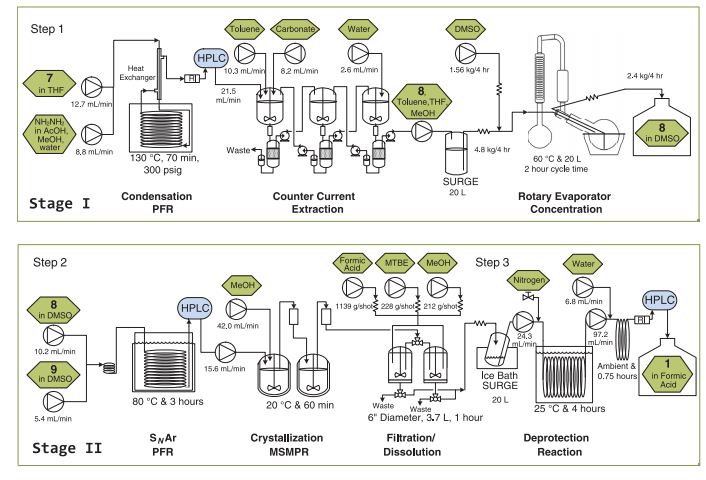
A multidisciplinary team from Eli Lilly reports the development and implementation of eight continuous unit operations for the synthesis of ca. 3 kg API per day under CGMP conditions (K. P. Cole et al., Science 2017, 356, 1144). The recent drive toward more potent APIs that have a low annual demand (<100 kg) has made continuous synthesis a viable alternative to traditional batch processes with advantages which include reducing equipment footprint and worker exposure. In this report the authors describe the enablement of three continuous synthetic steps followed by a salt formation, using surge tanks between steps to allow each step to be taken offline if online PAT detects a loss in reaction performance. A combination of MSMPRs (mixed-suspension, mixed-product removal) vessels, plug-flow reactors, and dissolve-off filters were used to perform the chemistry, with an automated 20 L rotary evaporator used to concentrate process streams and perform solvents swaps. This paper gives an excellent account of the potential solutions to continuous API synthesis and is well worth a read for anyone contemplating such methodology.
Integrated flow synthesis and purification process for prexasertib meets high industry standards
Eli Lilly has taken an important step away from traditional batch process drug manufacturing by using an industry-first continuous process to make a compound for phase I and II clinical trials. Workers at Lilly’s Kinsale site in Ireland, did three steps involved in producing cancer drug candidate prexasertib continuously, under current good manufacturing practice (CGMP) standards that ensure safety for human consumption.
Continuous processing relies on chemical and physical changes happening as substances flow through pipes. Isolated steps of this type are already well-established in the pharmaceutical industry. However, Lilly principal research scientist Kevin Cole stresses that a series including reaction and purification steps like this has not been demonstrated before. And the company wants to go much further.
‘We envision entire synthetic routes consisting of many reaction and separation unit operations being executed simultaneously in flow, with heavy reliance on design space understanding, process analytical technologies and process modelling to ensure quality,’ Cole says. ‘We think this will drastically change the environment for pharmaceutical manufacturing.’
In batch processes different chemical reaction and purification steps are typically done in large, costly vessels. However, this can be uneconomical when small amounts of drug molecules are needed for early stage clinical trials and, because drugs are getting more potent, increasingly in mainstream production.
By contrast, small volume continuous flow processing runs in more compact equipment in fume hoods. Flow systems can adapt to different processes, with cheap parts that can either be dedicated to specific drugs or readily replaced. The US Food and Drug Administration (FDA) has also been promoting continuous manufacturing because it integrates well with advanced process analytical technology. This helps pharmaceutical companies make high quality drugs with less FDA oversight.
Lilly chose prexasertib as its test case for such a process because it’s challenging to make. It is a chain of three aromatic rings, and one challenge comes because its central ring is formed using hydrazine. Hydrazine is used as a component in rocket fuel, and is also highly toxic. A second challenge comes from prexasertib itself, which, as a potent kinase inhibitor, is toxic to healthy cells, as well as cancerous ones, even at low doses. Lilly therefore wants to minimise its workers’ exposure.
Feeding the plant
Cole and his colleagues at Lilly’s labs in Indianapolis, US, have developed flow processes for three of the seven steps involved in prexasertib production. They start with the hydrazine step, which they could safely speed up by super-heating in the continuous process. After aqueous workup purification the solution of the two-ring intermediate solution runs into a ‘surge tank’. From there the solution flows intermittently into a rotary evaporator that removes solvents to concentrate it.
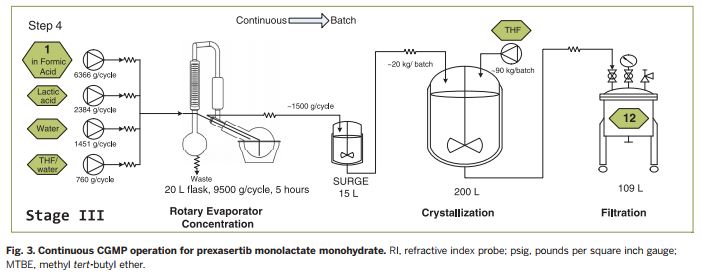
The second continuous flow step adds the third of prexasertib’s rings. In this case, the Lilly team purified the intermediate by crystallising it and filtering it out, washing away impurities. They could then redissolve the pure intermediate in formic acid, which also removes a protecting group, giving the desired prexasertib molecule. Automating this was probably the hardest part, Cole says. ‘Development of a predictive filtration model, equipment design and identification of formic acid as the solvent were keys to success,’ he explains. The final flow step then starts converting prexasertib to its final lactate salt form.

After developing the processes and systems in Indianapolis, Lilly shipped them to be equipped in an existing facility at its Kinsale manufacturing site at the cost of €1 million (£870,000). Once the prexasertib system was installed, the company was able to make 3kg of raw material per day for clinical trials. Cole describes the level of manual intervention needed as ‘moderate’.
Klavs Jensen from the Massachusetts Institute of Technology calls the paper describing the work ‘terrific’. ‘This work marks an important milestone in the continuous manufacturing of pharmaceuticals by demonstrating the feasibility of producing a modern kinase inhibitor under CGMP conditions,’ he says.
Likewise, Brahim Benyahia from Loughborough University, UK, calls this achievement ‘very interesting’. ‘The paper is another example that demonstrates the benefits and feasibility of the integrated continuous approach in pharma,’ he says.
Cole adds that Lilly has several other similar projects in advanced stages of development intended for the €35 million small-volume continuous plant it recently built in Kinsale. ‘We are committed to continuous manufacturing as well as full utilisation of our new facility,’ he says.
Correction: This article was updated on 16 June 2017 to clarify the chronology of the completion of the Kinsale, Ireland plant
References
REFERENCES
1: Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A, Stancato LF. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017 Mar 7. pii: clincanres.2876.2016. doi: 10.1158/1078-0432.CCR-16-2876. [Epub ahead of print] PubMed PMID: 28270495.
2: Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017 Jan 30. pii: molcanther.0352.2016. doi: 10.1158/1535-7163.MCT-16-0352. [Epub ahead of print] PubMed PMID: 28138028.
3: Ghelli Luserna Di Rorà A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C, Abbenante MC, Paolini S, Martinelli G. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget. 2016 Aug 16;7(33):53377-53391. doi: 10.18632/oncotarget.10535. PubMed PMID: 27438145; PubMed Central PMCID: PMC5288194.
REFERENCES
1: Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017 Jan 30. pii: molcanther.0352.2016. doi: 10.1158/1535-7163.MCT-16-0352. [Epub ahead of print] PubMed PMID: 28138028.
2: Ghelli Luserna Di Rorà A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C, Abbenante MC, Paolini S, Martinelli G. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget. 2016 Aug 16;7(33):53377-53391. doi: 10.18632/oncotarget.10535. PubMed PMID: 27438145; PubMed Central PMCID: PMC5288194.
3: King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther. 2015 Sep;14(9):2004-13. doi: 10.1158/1535-7163.MCT-14-1037. PubMed PMID: 26141948.
4: Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol. 2016 May 20;34(15):1764-71. doi: 10.1200/JCO.2015.64.5788. PubMed PMID: 27044938.
////////////Prexasertib, прексасертиб , بريكساسيرتيب , 普瑞色替 , PHASE 2, LY-2606368, LY 2606368
| N#CC1=NC=C(NC2=NNC(C3=C(OC)C=CC=C3OCCCN)=C2)N=C1 |

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














 Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.
Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.












 Moxalactam synthesis
Moxalactam synthesis



































































{kind=link}