
Home » 2017 (Page 14)
Yearly Archives: 2017
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves drug Xadago (Safinamide, сафинамид , سافيناميد , 沙非胺 , ) to treat Parkinson’s disease
Safinamide
- Molecular Formula C17H19FN2O2
- Average mass 302.343 Da
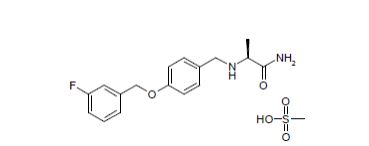
(+)-(S)-2-[[p-[(m-fluorobenzyl)oxy]benzyl]amino]propionamide monomethanesulfonate
Propanamide, 2-[[[4-[(3-fluorophenyl)methoxy]phenyl]methyl]amino]-, (2S)-, methanesulfonate
| Molecular Weight | 398.45 |
| Formula | C17H19FN2O2 ● CH4O3S |
CAS 202825-46-5 (Safinamide Mesylate)
Safinamide is a white to off-white, non-hygroscopic crystalline solid. It shows pH dependent solubility in aqueous buffers due to the secondary amine moiety, being soluble at acidic pH and practically insoluble at neutral pH.
It is freely soluble in de-ionized water, methanol and DMSO but practically insoluble in non-polar organic solvents.
Safinamide is chiral and possesses a single stereogenic centre.
Three crystalline forms are known. The anhydrous form selected for commercialisation is the most thermodynamically stable form, whilst the others are either not physiologically relevant or have very similar dissolution profiles. SOURCE EMA
Safinamide methanesulfonate was approved by European Medicine Agency (EMA) on Feb 22, 2015. It was developed by Newron and Zambon, then marketed as Xadago® by Zambon in EU.
FDA approved March 21, 2017,
- Chemistry Review(s) (PDF) for correct structure
- Chemistry Review(s) (PDF) for correct structure
Safinamide is a unique molecule with a novel dual mechanism of action based on the enhancement of the dopaminergic function (through potent reversible inhibition of MAO-B and of dopamine uptake) and inhibition of the excessive release of glutamate. It is indicated for the treatment of Parkinson’s disease (PD).
Xadago® is available as film-coated tablet for oral use, containing Eq. 50 mg/100 mg of free Safinamide. The recommended dose is 50 mg or 100 mg once daily.
March 21, 2017, Release
The U.S. Food and Drug Administration today approved Xadago (safinamide) tablets as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes. An “off” episode is a time when a patient’s medications are not working well, causing an increase in Parkinson’s symptoms, such as tremor and difficulty walking.
“Parkinson’s is a relentless disease without a cure,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to helping make additional treatments for Parkinson’s disease available to patients.”
An estimated 50,000 Americans are diagnosed with Parkinson’s disease each year, according to the National Institutes of Health, and about one million Americans have the condition. The neurological disorder typically occurs in people over age 60, though it can occur earlier, when cells in the brain that produce a chemical called dopamine become impaired or die. Dopamine helps transmit signals between the areas of the brain that produce smooth, purposeful movement – such as eating, writing, and shaving. Early symptoms of the disease are subtle and occur gradually. In some people, Parkinson’s disease progresses more quickly than in others.
The efficacy of Xadago in treating Parkinson’s disease was shown in a clinical trial of 645 participants who were also taking levodopa and were experiencing “off” time. Those receiving Xadago experienced more beneficial “on” time, a time when Parkinson’s symptoms are reduced, without troublesome uncontrolled involuntary movement (dyskinesia), compared to those receiving a placebo. The increase in “on” time was accompanied by a reduction in “off” time and better scores on a measure of motor function assessed during “on” time than before treatment.
In another clinical trial of 549 participants, the participants adding Xadago to their levodopa treatment had more “on” time without troublesome uncontrolled involuntary movement compared to those taking a placebo, and also had better scores on a measure of motor function assessed during “on” time than before treatment.
Certain patients should not take Xadago. These include patients who have severe liver problems, or who take a medicine used to treat a cough or cold called dextromethorphan. It also should not be taken by patients who take another medicine called a monoamine oxidase inhibitor (MAOI) because it may cause a sudden severe increase in blood pressure, or by those who take an opioid drug, St. John’s wort, certain antidepressants (such as serotonin-norepinephrine reuptake inhibitors, tricyclics, tetracyclics, and triazolopyridines), or cyclobenzaprine, because it may cause a life-threatening reaction called serotonin syndrome.
The most common adverse reactions observed in patients taking Xadago were uncontrolled involuntary movement, falls, nausea, and trouble sleeping or falling asleep (insomnia).
Serious, but less common, risks include the following: exacerbated high blood pressure (hypertension); serotonin syndrome when used with MAOIs, antidepressants, or opioid drugs; falling asleep during activities of daily living; hallucinations and psychotic behavior; problems with impulse control/compulsive behaviors; withdrawal-emergent hyperpyrexia (fever) and confusion; and retinal pathology.
The FDA granted approval of Xadago to Newron Pharmaceuticals.
Safinamide (INN; brand name Xadago) is a drug indicated for the treatment of Parkinson’s disease with monoamine oxidase B inhibiting and other methods of action.[2] It was approved in Europe in February 2015,[3] and in the United States on March 21, 2017[4]. It has also been tested for the use in patients with restless legs syndrome (RLS), but no study results have been published.

Medical uses
Safinamide has been approved by the European Medicines Agency for the treatment of adult patients with idiopathic Parkinson’s disease as add-on therapy to a stable dose of levodopa (L-dopa) alone or in combination with other Parkinson drugs in patients with mid-to-late-stage fluctuating disease.[5]
Contraindications
Safinamide is contraindicated in patients with severe liver impairment, with albinism, retinitis pigmentosa, severe diabetic neuropathy, uveitis and other disorders of the retina. Combination with other monoamine oxidase (MAO) inhibitors and pethidine is also contraindicated.[6]
Adverse effects
Common adverse events in clinical trials (in more than 1% of patients) included nausea, dizziness, tiredness, sleeplessness, orthostatic hypotension (low blood pressure), and headache. There was no significant difference in the occurrence of these effects between safinamide and placebo treated patients.[6][7]
In experiments with rats (but not in those with monkeys), retinopathies have been observed.[1][8]
Overdose
Expected overdose effects are hypertension (high blood pressure), orthostatic hypotension, hallucinations, psychomotor agitation, nausea, vomiting, and dyskinesia. In studies, a singe patient was suspected to have overdosed for a month; symptoms were confusion, drowsiness and mydriasis (dilation of the pupils) and subsided completely after the drug was discontinued. No specific antidote is available.[6]
Interactions
As a MAO inhibitor, safinamide can theoretically cause hypertensive crises, serotonin syndrome and other severe side effects when combined with other MAO inhibitors or with drugs that are known to interact with MAO inhibitors, such as pethidine, dextromethorphan, selective serotonin reuptake inhibitors (SSRIs), serotonin–noradrenaline reuptake inhibitors (SNRIs), tricyclic and tetracyclic antidepressants. An interaction with tyramine, a substance found in various foods, could be expected by the same reasoning but has been excluded in studies.[6]
Another theoretical interaction is with drugs with affinity to the transporter protein ABCG2 (also known as BCRP), such as pitavastatin, pravastatin, ciprofloxacin, methotrexat, and diclofenac; a study with the latter has shown no clinical relevance.[9] A study testing possible interactions with amidase inhibitors is part of the post-authorisation development plan.[1] There are no relevant interactions related to cytochrome P450 (CYP) liver enzymes, although one inactivation pathway of safinamide seems to be mediated by CYP3A4.[6]
Pharmacology
Mechanisms of action
Like the older antiparkinson drugs selegiline and rasagiline, safinamide is a selective monoamine oxidase B inhibitor, reducing degradation of dopamine; in contrast to the other two, its action is reversible. Safinamide also inhibits glutamate release[7][10] and dopamine reuptake.[11] Additionally, it blocks sodium and calcium channels,[10][12] the relevance of which for its antiparkinson action is however unknown.[6]
Pharmacokinetics
Safinamide is absorbed quickly and nearly completely from the gut and reaches highest blood plasma concentrations after 1.8 to 2.8 hours. There is no relevant first-pass metabolism; total bioavailability is 95%. The substance is bound to plasma proteins to 88–90%.[6]
The metabolism is not well understood. The principal step is mediated by amidases which have not been identified, and produces safinamide acid (NW-1153). Other relevant metabolites are O-debenzylated safinamide (NW-1199),[9] the N-dealkylated amine which is then oxidized to a carboxylic acid (NW-1689), and the glucuronide of the latter.[6][13] In tests with liver microsomes, dealkylation seemed to be mediated by CYP3A4, but other CYP enzymes appear to be involved as well. Safinamide acid binds to the organic anion transporter 3 (OAT3), but this has probably no clinical relevance. Safinamide itself transiently binds to ABCG2. No other transporter affinities have been found in preliminary studies.[6]
Safinamide is eliminated, mainly (>90%) in form of its metabolites, via the kidney, with an elimination half-life of 20 to 30 hours. Only 1.5% are found in the stool.[6]

Metabolism pathways of safinamide.[9][13] Enzymes: CYP = cytochrome P450, MAO-A = monoamine oxidase A, ALDH = aldehyde dehydrogenases, UGT = UDP-glucuronosyltransferases. Gluc = acyl glucuronide.
History
The compound was originally discovered at Farmitalia-Carlo Erba, which was acquired by Pharmacia in 1993. In 1995, Pharmacia merged with Upjohn. Safinamide was first disclosed in 1998.[14] In the course of a major restructuring in the same year, all rights for safinamide were transferred to the newly formed company Newron Pharmaceuticals, which developed the drug until it was sold to Merck KGaA in 2006.[15]
In 2007, a Phase III clinical trial was started, scheduled to run until 2011.[16] In October 2011 Merck, now Merck-Serono, announced that they would give all rights to develop the compound back to Newron because they wanted to prioritise other projects and had corrected their estimates for safinamide’s market potential downwards.[17]
The US Food and Drug Administration (FDA) refused to file Newron’s application in 2014 on formal grounds.[18] Newron re-applied in December 2014.[19] In spring 2015, the European Medicines Agency (EMA) approved the drug. Safinamide is the first antiparkinson medication to be approved for ten years.[8]
Research
Potential additional uses might be restless legs syndrome (RLS) and epilepsy.[20] They were being tested in Phase II trials in 2008, but no results are available.

(+)-(S)-2-[[p-[(m-fluorobenzyl)oxy]benzyl]amino]propionamide monomethanesulfonate
Propanamide, 2-[[[4-[(3-fluorophenyl)methoxy]phenyl]methyl]amino]-, (2S)-, methanesulfonate
| Molecular Weight | 398.45 |
| Formula | C17H19FN2O2 ● CH4O3S |
CAS 202825-46-5 (Safinamide Mesylate)
Safinamide is a white to off-white, non-hygroscopic crystalline solid. It shows pH dependent solubility in aqueous buffers due to the secondary amine moiety, being soluble at acidic pH and practically insoluble at neutral pH.
It is freely soluble in de-ionized water, methanol and DMSO but practically insoluble in non-polar organic solvents.
Safinamide is chiral and possesses a single stereogenic centre.
Three crystalline forms are known. The anhydrous form selected for commercialisation is the most thermodynamically stable form, whilst the others are either not physiologically relevant or have very similar dissolution profiles.SOURCE EMA
Safinamide methanesulfonate was approved by European Medicine Agency (EMA) on Feb 22, 2015. It was developed by Newron and Zambon, then marketed as Xadago® by Zambon in EU.
FDA approved March 21, 2017
Safinamide is a unique molecule with a novel dual mechanism of action based on the enhancement of the dopaminergic function (through potent reversible inhibition of MAO-B and of dopamine uptake) and inhibition of the excessive release of glutamate. It is indicated for the treatment of Parkinson’s disease (PD).
Xadago® is available as film-coated tablet for oral use, containing Eq. 50 mg/100 mg of free Safinamide. The recommended dose is 50 mg or 100 mg once daily.
SYNTHESIS
 |
|
| Safinamide has been obtained by reductocondensation of 4-(3-fluorobenzyloxy)benzaldehyde (I) with L-alaninamide (II) by means of sodium cyanoborohydride in methanol.EP 0400495; EP 0426816; JP 1992500215; US 5236957; US 5391577; US 5502079; WO 9014334 |
CLIP
http://pubs.rsc.org/en/content/articlehtml/2016/sc/c6sc00197a

Scheme 2 Synthesis and isolation of [18F]safinamide, [18F]FMT, and [18F]mFBG.
PATENT
Safinamide (NW- 1015, FCE-26743A, PNU- 151774E) is a sodium channel blocker, a calcium channel modulator, a monoamino oxidase B (MAO-B) inhibitor, a glutamate release inhibitor and a dopamine metabolism modulator. Safinamide is useful in the treatment of CNS disorders, in particular of epilepsy, Parkinson’s disease, Alzheimer’s disease, depression, restless legs syndrome and migraine (WO 90/ 14334, WO 2004/089353, WO 2005/ 102300 and WO 2004/062655). Ralfinamide (NW- 1029, FCE-26742A, PNU-0154339E) is a sodium channel blocker useful in the treatment of pain conditions, including chronic pain and neuropathic pain, migraine, bipolar disorders, depressions, cardiovascular, inflammatory, urogenital, metabolic and gastrointestinal disorders (WO 99/35125, WO 03/020273, WO 2004/062655, WO 2005/018627, WO 2005/070405, WO 2005/ 102300).
In particular, safinamide is specifically described in WO 90/ 14334. Safinamide, its R-enantiomer, their racemic mixture and their salts with pharmaceutically acceptable acids and the use thereof for the preparation of pharmaceutical compositions active as anti-epileptic, anti-Parkinson, neuroprotective, antidepressant, antispastic and/or hypnotic agents are specifically claimed in WO 90/ 14334. Ralfinamide is specifically described in WO 90/ 14334. Ralfinamide, its R- enantiomer, their racemic mixture and their salts with pharmaceutically acceptable acids and their use thereof for the preparation of pharmaceutical compositions active as anti-epileptic, anti-Parkinson, neuroprotective, antidepressant, antispastic and/or hypnotic agent are comprised by the claims of WO 90/ 14334.
Moreover, the use as analgesics of safinamide, ralfinamide, the respective R-enantiomers, the respective racemic mixtures and their salts with pharmaceutically acceptable acids is claimed in WO 99/035125. WO 2006/027052 A2 specifically discloses and claims the use of the single R-enantiomer of ralfinamide i.e., (R)-2-[4-(2- fluorobenzyloxy)benzylamino]propanamide (I’b), and its salts with pharmaceutically acceptable acids as a selective sodium and calcium channel modulator for the selective treatment of pathological affections wherein sodium or calcium channel mechanism(s) play(s) a pathological role, including pain, migraine, inflammatory processes affecting all body systems, disorders affecting skin and related tissue, disorders of the respiratory system, disorders of the immune and endocrinological systems, gastrointestinal, and urogenital disorders, wherein the therapeutical activity of said compound is substantially free from any MAO inhibitory side effect or exhibits significantly reduced MAO inhibitory side effect.
It has now been discovered that the large scale preparations of safinamide and ralfinamide according to the methods described in the prior art, contain two undesired impurities, i.e., respectively, (S)-2-[3-(3- fluorobenzyl)-4-(3-fluorobenzyloxy)-benzylamino]propanamide (Ha) and (S)- 2-[3-(2-fluorobenzyl)-4-(2-fluorobenzyloxy)-benzylamino]propanamide (lib), and their salt, in particular the respective methanesulfonates (lie) and (Hd)
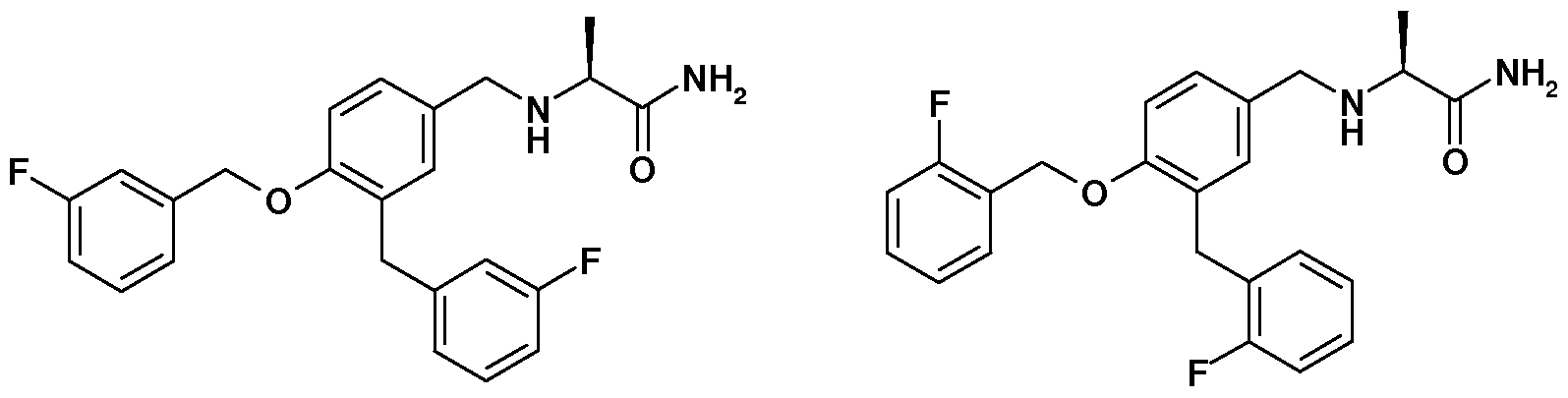
(Ha) (lib)
The same situation occurs with the preparation according the prior art methods for the R-enantiomers (I’a) and (I’b) of, respectively, safinamide and ralfinamide, the respective racemic mixtures (Ia, I’a) and (Ib, I’b), and the salts thereof with pharmaceutically acceptable acids, (I’c), (I’d) and the respective racemic mixtures (Ic, I’c) and (Id, I’d) in particular the methanesulfonates, which result to be contaminated by the respective R isomers (Il’a), (Il’b), (II’c), and (Il’d) of the above identified impurities (Ha), (lib), (lie) and (Hd) or the respective racemic mixtures (Ha, Il’a), (lib, Il’b), (Hc, II’c) and (Hd, Il’d).

PATENT
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by bradykinesia, rigidity, resting tremor, and ataxia. These symptoms are caused by decreased dopamine release in the striatum. Clinically, PD is defined by presence of Lewy bodies, intracellular neuronal inclusions in the substantia nigra and at other sites in the brain. Estimated prevalence of this disease is 100 to 200 per 100,000 population including males and females across the entire age group. Current treatment for PD comprises dopaminergic medications that include levodopa, dopamine agonists (DAs), monoamine oxidase-B (MAO-B) inhibitors. Figure 1 provides few examples of pharmaceutically important benzyloxy-benzylamine derivatives. Many of these benzyl oxy-benzylamines with various amine functions were studied and has been patented as sodium channel blockers. Among them, safinamide ((5)-N2– {4-[3- fluorobenzyl)oxy] benzyl}- alaninamide methanesulfonate) is a noted example which is under phase III clinical trials for treatment of Parkinson’s disease. Its mechanism of action is manifold which comprise MAO-B and dopamine uptake inhibition. Further, safinamide is believed to block voltage-dependent sodium channels, modulates calcium channels and reduction of glutamate release in the central nervous system. WOl 998003472 discloses serinamide, glycinamide, alaninamide and phenylalaninamide derivatives of a compound (I). These compounds (I) are useful for the treatment of neurological diseases.
EP2474521 discloses high purity degree (S)-2-[4-(3-fluorobenzyloxy)- benzylamino]propanamide (safinamide) or (S)-2-[4-(2-fluorobenzyloxy)- benzylamino]propanamide (ralfinamide) or a salt thereof with a pharmaceutically acceptable acid with a content of the respective impurity (S)-2-[3-(3-fluorobenzyl)-4-(3- fluorobenzyloxy)-benzylamino]propanamide or (S)-2-[3-(2-fluorobenzyl)-4-(2- fluorobenzyloxy)-benzylamino]propanamide.
US2009149544 relates to novel alpha- aminoamide derivatives, their pharmaceutically acceptable salts, solvates, and hydrates thereof. The application also provides compositions comprising a compound and the use of such compositions in methods of treating diseases and conditions that are beneficially treated by administering an inhibitor of monoamine oxidase type B (MAO-B) and/or a sodium (Na.sup.+) channel blocker, and/or a calcium (Ca.sup.2+) channel modulator.
The strategy employed in the art to prepare benzyloxy-benzylamine derivatives including safinamide or its analogue ralfinamide is chiral pool approach starting from L-alaniriamide and reductively aminating with 4-(3-fluorobenzyloxy) benzaldehyde. Although this method is very simple and straightforward, it suffers from several serious drawbacks, such as need to use toxic reagents such as sodium cyanoborohydride and further formation of toxic by-products such as hydrogen cyanide and sodium cyanide and other toxic impurities in large-scale production Importantly, the possibility of generating a range of safinamide analogues by means of the chiral-pool approach is limited in terms of the structure and stereochemistry of the products because of inadequacies in the availability of D-alaninamide and its analogues
Hence, the developments of newer methods for the preparation of compounds of formula (I) comprising safinamide and related analogues are highly desirable
Example 2: Synthesis of (R)-l-(benzyIoxy)propan-2-ol [(R)-compound 3]
To a solution of (7? benzyl glycidyl ether [fR)-compound 2] (4 g, 24.4 mmol) in dry THF (10 mL) at 0 °C, a pre-cooled solution of lithium aluminium hydride (1.4 g, 36.6 mmol) in anhydrous THF (10 mL) was added slowly with stirring under nitrogen. After 60 min, the reaction mixture was quenched with 1 ml of water and 1 ml of 15 % NaOH solution and the content was stirred for 15 min. The inorganic precipitate was filtered, washed with ethyl acetate and the solvent evaporated under reduced pressure. The residue was purified by a short filtration column to afford (-fl)-compound 3 as a colorless oil (3.8 g, 95%); [a]22D = -14.5 (c 2, CHC13); IR (CHC13): vmax3418, 3087, 3063, 3030, 2963, 2924, 1952, 1873, 1600, 1495, 1454, 1363, 1244, 1099, 1028, 918, 808, 698 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.13 (d, J = 6.3 Hz, 3H), 2.5 (bs, 1H), 3.23-3.32 (dd, J = 9.8, 1.3 Hz, 1H), 3.43-3.49 (dd, J = 9.45, 3.2 Hz, 1H), 3.91-4.03 (m, 1H), 4.55 (s, 2H), 7.25-7.37 (m, 5H); I3C NMR (50 MHz, CDC13): 5C 137.8 (C), 128.3 (CH, 2 carbons), 127.7 (CH, 3 carbons), 75.7 (CH2), 73.2 (CH2), 66.4 (CH), 18.6 (CH3); MS: m/z 189 [M+Na]+.
Example 3: Synthesis of (S)-((2-azidopropoxy)methyl)benzene [(S)- compound 4]
To a stirred solution of secondary alcohol ( )-compound 3 (3 g, 18.1 mmol) in dry dichloromethane (25 mL), Et3N (3.1 mL, 21.7 mmol) at 0 °C was added, followed by drop wise addition of mesyl chloride (1.8 mL, 21.7 mmol). The reaction mixture was stirred at 0°C for 2 hours, subsequently at room temperature for 3 hours under a nitrogen atmosphere. After completion of the reaction (indicated by TLC), the reaction mixture was diluted with dichloromethane and washed with a saturated solution of sodium bicarbonate (30 mL) and water (2 x 10 mL). The organic layer was separated, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give the O-mesyl compound (4.3 g; crude).
To a solution of the crude 0-mesyl compound (4 g, 16.37 mmol) in dry DMF (10 mL), sodium azide (1.6 g, 24.55 mmol) was added and the reaction mixture was heated at 60°C for 6 hours under nitrogen atmosphere. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with ethyl acetate (2 x 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield (¾)-compound 4 as a colorless oil. (2.8 g; 89%); [a]22D = +6.1 (c 1.3, CHC13); IR (CHC13): vmax 3394, 3032, 2977, 2864, 2500, 2104, 1724, 1641 , 1496, 1454, 1363, 1269, 1 101 , 913, 698 αη ‘,Ή NMR (200 MHz, CDC13): δΗ 1.20 (d, J = 6.7 Hz, 3H), 3.39-3.54 (m, 2H), 3.61-3.77 (m, 1H), 4.57 (s, 2H), 7.25-7.39 (m, 5H); 13C NMR (50 MHz, CDC13): 5C 137.8 (C), 128.4 (CH, 2 carbons), 127.7 (CH), 127.5 (CH, 2 carbons), 73.7 (CH2), 73.2 (CH2), 56.9 (CH), 16.1 (CH3);MS: m/z 214 [M+Na]+.
Example 4: Synthesis of (S)-N-(l-hydroxypropan-2-yl)-2-nitrobenzenesulfonamide [(S)- compound 5]
To a solution of ^-compound 4 (2.5 g, 13.1 mmol) in methanol (15 mL), trifluoroacetic acid (2 mL) and palladium hydroxide on activated carbon (0.05 g, 10-20 wt %) were added and the reaction mixture was stirred under hydrogen (60 psi) for 8 hours. After completion of the reaction (indicated by TLC), the catalyst was filtered over a plug of celite and the solvent was evaporated under reduced pressure to half of its volume which was basified with 2.5 M methanolic NaOH. Evaporation of the remaining solvent under reduced pressure was done followed by filtration of the residue through a short bed of basic alumina (eluent; MeOH) to obtain the amino alcohol as a pale brown oil (0.94 g, crude) which was subjected to the next reaction without further purification.
To a solution of amino alcohol (0.9 g, 1 1.98 mmol) in dry dichloromethane (5 mL), 2-nitrobenzenesulfonylchloride (3.2 g, 14.37 mmol) in dichloromethane (8 mL) and triethylamine (2.6 mL, 17.97 mmol) at 0 °C were slowly added under nitrogen atmosphere. The solution was stirred for 2 hours. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with dichloromethane (2 x 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 60:40) to yield (S)- compound 5 as a pale yellow oil (2.33 g, 75% ); [a]22D = +80.2 (c 2.1, CHClj); IR (CHC13): vmax 3546, 3367, 3022, 2883, 2401, 1594, 1542, 1412, 1362, 1216, 1170, 1 125, 1059, 971, 854, 668 cm“1; ]H NMR (200 MHz, CDC13): δΗ 1.13 (d, J = 6.5 Hz, 3H), 2.16 (bs, 1H), 3.45-3.70 (m, 3H), 5.61 (d, J = 6.6 Hz, 1H), 7.73-7.80 (m, 2H), 7.86-7.91 (m, 1H), 8.13-8.22 (m, 1H); 13C NMR (50 MHz, CDC13): 5C 147.8 (C), 134.4 (C), 133.7 (CH), 133.0 (CH), 130.9 (CH), 125.5 (CH), 66.2 (CH2), 52.5 (CH), 17.8 (CH3); MS: m/z 283 [M+Na]+.
Example 5: Synthesis of l-fluoro-3-(iodomethyl)benzene ( compound 7)
To a stirred solution of triphenyl phosphine (4.15 g, 15.85 mmol), imidazole (1.1 g, 15.85 mmol) in dry dichloromethane (20 mL), iodine (4.8 g, 19.02 mmol) at 0°C was added and the solution was stirred for 5 min. To this, 3-fluoro benzyl alcohol (compound 6) (2 g, 15.85 mmol) dissolved in dichloromethane (5 mL) was added drop wise over 10 min and the stirring was continued for 1 hour with exclusion of light. After completion of the reaction (indicated by TLC), the reaction mixture was quenched by addition of an aqueous Na2S203 solution (15 mL), then extracted with dichloromethane (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield compound 7 as a colorless oil (3.5 g, 95% ); (IR (CHC13): vmax 3460, 3060, 2965, 1695, 1613, 1593, 1482, 1446, 1259, 1 156, 1068, 944, 871, 782, 736, 686 cm“1 ; Ή NMR (200 MHz, CDC13): δΗ 4.42 (s, 2H), 6.89-6.99 (m, 1H), 7.05-7.17 (m, 2H), 7.21-7,29 (m, 1H); 13C NMR (50 MHz, CDC13): 6C 165.0 (C), 141.6 (C), 130.2 (CH), 124.4 (CH), 1 15.9 (CH), 1 14.7 (CH), 3.9 (C¾).
Example 6: Synthesis of (4-((3-flurobenzyl)oxy)phenyl)methanol (compound 8)
To a stirred solution of 4-(hydroxymethyl)phenol (1.57 g, 12.7 mmol) and K2C03 (8.8 g, 63.55 mmol) in dry acetonitrile (25 mL), compound 7 (3 g, 12.7 mmol) in acetonitrile was slowly added and the reaction mixture was heated at 70°C for 6 hours. After completion of the reaction (indicated by TLC), water (20 mL) was added to the reaction mixture, then extracted with ethylacetate (3 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 70:30) to yield compound 8 as a colorless solid (2.7 g, 91% ); mp 63-65 °C; IR (CHC13): vmax 3422, 3017, 1612, 1512, 1489, 1381, 1216, 1 174, 1020, 829, 668 cm“1; Ή NMR (200 MHz, CDC13): δΗ 4.61 (s, 2H), 5.06 (s, 2H), 6.91-6.98 (m, 2H), 7.00-7.06 (m, 1H), 7.12-7.20 (m, 2H), 7.25-7.37 (m, 3H); 13C NMR (50 MHz, CDC13): 5C 165.4 (C), 160.5 (C), 158.0 (C), 139.6 (C), 133.5 (CH), 130.2 (CH), 128.7 (CH, 2 carbons), 122.7 (CH), 1 14.8 (CH, 2 carbons), 1 13.9 (CH), 69.1 (CH2), 64.9 (CH2); MS: m/z 255 [M+Na]+.
Example 7: Synthesis of l-fluoro-3-((4-(iodomethyl)phenoxy)methyI)benzene (compound 9)
To a stirred solution of triphenyl phosphine (2.82 g, 10.8 mmol), imidazole (0.73 g, 10.76 mmol) in dry dichloromethane (20 mL), iodine (3.27 g, 12.9 mmol) at 0 °C was added and the solution was stirred for 5 min. To this, compound 8 (2.5 g, 10.8 mmol) dissolved in dichloromethane (5 mL) was added drop wise over 10 min and the stirring was continued for 1 hour with exclusion of light. After completion of the reaction (indicated by TLC), the reaction mixture was quenched by addition of an aqueous Na2S203 solution (15 mL), then extracted with dichloromethane (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield compound 9 as a colorless oil (3.4 g, 93%); IR (CHC13): vmax 3503, 3033, 2925, 2089, 1607, 1509, 1488, 1381, 1301, 1250, 1 155, 1079, 944, 869, 776, 684 cm“1; 1H NMR (200 MHz, CDC13): δΗ 4.47 (s, 2H), 5.04 (s, 2H), 6.85-6.91 (m, 2H), 6.96-7.02 (m, 1H), 7.05-7.12 (m, 1H), 7.16-7.20 (m, 1H), 7.29-7.40 (m, 3H).
,3C NMR (50 MHz, CDC13): 6C 165.4 (C), 160.5 (C), 158.1 (C), 131.9 (C), 130.2 (CH), 130.1 (CH, 2 carbons), 122.7 (CH), 1 15.1 (CH, 2 carbons), 1 14.7 (CH), 1 13.9 (CH), 69.2 (CH2), 6.33 (CH2).
Example 8: Synthesis of (S)-N-(4-((3-flurobenzyl)oxy)benzyl)-N-(l-hydroxypropan-2-yl)-2-nitrobenzenesulfonamide [(S)-compound 10]
To a stirred solution of (^-compound 5 (1 g, 3.8 mmol) and K2C03 (2.65 g, 19.2 mmol) in dry acetonitrile (25 mL), compound 9 (1.84 g, 5.4 mmol) in acetonitrile was slowly added and the reaction mixture was heated at 70°C for 72 hours. After completion of the reaction (indicated by TLC), water (20 mL) was added to the reaction mixture, then extracted with ethylacetate (3 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 80:20) to yield (¾)-compound 10 as a colorless oil (1.46 g, 80% ); [a]22D = +5.4 (c 1.5, CHC13); IR (CHC13): vmax 3445, 3020, 2928, 2400, 1613, 1544, 1512, 1453, 1371, 1216, 1 162, 1029, 852, 668 cm“1; 1H NMR (200 MHz, CDC13): δΗ 1.07 (d, J = 6.9 Hz, 3H), 1.91 (t, J = 5.2 Hz, 1H), 3.41-3.53 (m, 2H), 4.05-4.22 (m, 1H), 4.37-4.57 (m, 2H), 5.02 (m, 2H), 6.87 (d, J = 8.53 Hz, 2H), 6.97-7.12 (m, 2H), 7.20 (d, J = 7.2 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.47-7.67 (m, 3H), 7.89 (d, J = 8.09 Hz, 1H); 13C NMR (50 MHz, CDC13): 6C 165.5 (C), 160.6 (C), 158.4 (C), 147.7 (C), 139.6 (C), 134.1 (C), 133.4 (CH), 131.6 (CH), 131.4 (CH), 130.3 (CH), 129.7 (CH, 2 carbons), 124.1 (CH), 122.8 (CH), 115.1 (CH), 114. 9 (CH, 2 carbons), 114.0 (CH), 69.2 (CH2), 64.3 (CH2), 56.2 (CH), 46.9 (CH2), 15.4 (CH3); MS: m/z 497 [M+Na]+.
Example 9: Synthesis of (S)-2-(N-(4-((3-fluorobenzyl)oxy)benzyl)-2-nitrophenylsulfonamido) propanoic acid [(S)-compound 11]
A mixture of (S compound 10 (1.25 g, 2.6 mmol), TEMPO (0.028 g, 0.18 mmol), acetonitrile (20 mL), and sodium phosphate buffer (16 mL, 0.67 M, pH 6.7) was heated to 35°C. Next, sodium chlorite (0.47 g dissolved in 2 mL water, 7.9 mmol) and diluted bleach (4-6%, 0.09 mL diluted in 1 mL water) were added simultaneously over 1 hour. The reaction mixture was stirred at 35°C until the reaction was complete (3 hours, TLC), then cooled to room temperature. Water (30 mL) was added and the pH adjusted to 8 with 2 M NaOH. The reaction was quenched by pouring it into ice cold Na2S03 solution maintained at <20°C. After stirring for 30 min at room temperature, ethyl acetate (20 mL) was added and the stirring was continued for an additional 15 min. The organic layer was separated and discarded. More ethyl acetate (20 mL) was added, and the aqueous layer was acidified with 1 M HC1 to pH 3-4. The organic layer was separated, washed with water (2 x 15 mL), brine and concentrated under reduced pressure to afford the carboxylic acid (S -compound 1 1 (1.1 g, 85%); [ ]22ο = -20.4 (c 1.1, CHC13); IR (CHC13): vmax 3398, 3095, 1718, 1612, 1591, 1543, 1512, 1489, 1457, 1371, 1303, 1251, 1163, 1059, 900, 852, 831 , 778, 684 cm“1; 1H NMR (200 MHz, CDC13): 8H 1.44 (d, J = 7.3 Hz, 3H), 4.23 (d, J = 15.6 Hz, 1H), 4.64 (d, J = 15.6 Hz, 1H), 4.82-4.90 (q, J = 7.4 Hz, 1H), 4.92 (s, 2H), 6.68 (d, J = 8.6 Hz, 2H), 6.89-7.01 (m, 2H), 7.07-7.13 (m, 3H), 7.18-7.33 (m, 2H), 7.43-7.55 (m, 3H), 8.81 (bs, 1H); 13C NMR (50 MHz, CDC13): 5C 176.5 (CO), 165. 0 (C), 158.0 (C), 147.4 (C), 139.4 (C), 134.1 (C), 133.2 (CH), 131.4 (CH), 130.3 (CH), 129.9 (CH, 2 carbons), 128.4 (C), 124.1
(CH), 122.6 (CH), 1 15.0 (CH), 114.6 (CH, 2 carbons), 1 14.3 (CH), 1 13.8 (CH) 69.1 (CH2), 56.1 (CH), 49.0 (CH2), 16.8 (CH3); MS: m/z 51 1 [M+Na .
Example 10: Synthesis of (S)-2-(N-(4-((3-fluorobenzyI)oxy)benzyl)-2-nitrophenylsulfonamido) propanamide [(S)- compound 12]
To a solution of carboxylic acid (¾)-compound 1 1 (1 g, 2.04 mmol) and triethyl amine (0.34 mL, 2.4 mmol) in dry THF (20 mL), ethyl chloroformate (0.21 mL, 2.2 mmol) at 0 °C was added under nitrogen atmosphere. After 1 hour, ammonium hydroxide (25% w/v aqueous solution, 1.4 mL, 10.2 mmol) was added and the resulting reaction mixture was stirred at room temperature for 16 hours. After completion of the reaction, potassium carbonate (0.29 g, 2.1 mmol) was added and the reaction mixture was filtered, and washed with ethylacetate. The solvent was removed under reduced pressure and the crude product was subjected to column chromatography (silica gel, petroleum ether/EtOAc, 50:50) to obtain sulfonamide (Sj-compound 12 as a colorless oil (0.9 g, 91%); [a]22D = -32.1 (c 1.2, CHC13); IR (CHC13): vmax 3472, 1961 , 161 1, 1592, 1542, 1511, 1449, 1371, 1304, 1243, 1 163, 1060, 1029, 895, 852, 684 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.43 (d, J = 7.1 Hz, 3H), 4.44 (d, J = 15.4 Hz, 1H), 4.59 (d, J = 15.5 Hz, 1H), 4.60-4.71 (q, J= 7.0 Hz, 1 H), 5.01 (s, 2H), 5.50 (bs, 1H), 6.31 (bs, 1H), 6.78 (d, J = 8.71 Hz, 2H), 6.98-7.1 1 (m, 2H), 7.15-7.22 (m, 3H), 7.31-7.45 (m, 2H), 7.59-7.64 (m, 3H);13C NMR (50 MHz, CDC13): 5C 172.3 (CO), 165.5 (C), 158.2 (C), 147.5 (C), 139.6 (C), 139.4 (C), 133.6 (CH), 131.7 (CH), 130.5 (CH, 2 carbons),130.3 (CH), 128.1 (C), 124.2 (CH), 122.7 (CH), 1 15.1 (CH), 1 14.7 (CH, 2 carbons),1 14.4 (CH), 1 13.9 (CH), 69.0 (CH2), 55.7 (CH), 48.3 (CH2), 14.9 (CH3); MS: m/z 510 [M+Na]+.
Example 11: Synthesis of (S)-2-((4-((3-fluorobenzyl)oxy) benzyl) amino) propanamide [(S)-compound of formula I]
To a solution of sulfonamide (S)- compound 12 (0.8 g, 1.64 mmol), potassium carbonate (0.56 g, 4.9 mmol) in dry DMF (10 mL), thiophenol (0.2 mL, 1.9 mmol) was added. The reaction mixture was vigorously stirred for 6 hours. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with ethylacetate (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 60:40) to yield (S) -compound of formula I as a colorless solid (0.43 g, 86% ); mp 207-09 °C; [a]22D = +3.89 (c 1.55, CHC13); IR (CHC13): vmax 3341, 2970, 2927, 2853, 1648, 1592, 1512, 1489, 1445, 1406, 1384, 1254, 1176, 1 137, 1030, 953, 928, 829, 680 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.34 (d, J = 6.9 Hz, 3H), 2.49 (bs, 2H), 3.19-3.30 (q, J = 6.8 Hz, 1H), 3.63-3.78 (dd, J = 19.4, 3.9 Hz, 2H), 5.05 (s, 2H), 5.85 (bs, 1H), 6.95 (d, J = 8.7 Hz, 2H), 7.00-7.06 (m, 1H), 7.13-7.24 (m, 4H), 7.29-7.40 (m, 1H). 13C NMR (50 MHz, CDC13): 8C 178.3 (CO), 165.4 (C), 157.7 (C), 139.6 (C), 132.1 (C), 130.2 (CH), 129.3 (CH, 2 carbons), 122.7 (CH), 1 14.9 (CH, 2 carbons), 1 14.6 (CH), 1 13.9 (CH), 69.2 (CH2), 57.5 (CH), 51.9 (CH2), 19.6 (CH3); MS: m/z 302 [M]+, 325 [M+Na]+.
Example 12: Synthesis of (S)-Safinamide mesylate
To a stirred solution of (^-compound of formula I (0.1 g, 0.33 mmol) in ethylacetate (3 mL) at 70°C, methanesulfonic acid (0.02 mL, 0.33 mmol) was added and the reaction mixture was stirred for 2 hours. Subsequently, the temperature was lowered to 35°C and the stirring was continued for additional 1 hour. The solvent was evaporated under reduced pressure and the residue was filtered through a short bed of basic alumina [eluent: EtOAc/MeOH; (95:5)] to obtain safinamide mesylate as a white solid (0.11 g, 90%); mp 209-10 °C [lit.7mp 210]; [a]22D = +9.6 (c 1.1, AcOH); {lit.7 [a] D = +12.9 (c 1.1, AcOH)} ee >98% [The ee of safinamide mesylate was determined by chiral HPLC analysis; Chiralcel OD-RH (150 x 4.6 mm) column; eluent:
Methanol/ Acetonitrile/Buffer-TEAP, pH 3 (20: 10:70); flow rate 0.5 mL/min (780 psi); detector: 224 nm] [f¾)-isomer tR = 1 1.55 min, (SJ-isomer tR = 12.94 min].

PAPERS
Synthesis2014, 46, 1751-1756.

N2-{4-[(3-Fluorobenzyl)oxy]benzyl}-L-alaninamide [(S)-14] BASE FORM
PhSH (0.2 mL, 1.9 mmol) was added to a solution of sulfonamide (S)-13 (0.8 g, 1.64 mmol) and K2CO3 (0.56 g, 4.9 mmol) in anhyd DMF (10 mL), and the mixture was vigorously stirred for 6 h. When the reaction was complete (TLC), H2O (10 mL) was added and the mixture was extracted with EtOAc (2 × 20 mL). The organic layers were combined, washed with brine (2 × 10), dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by column chromatography [silica gel, PE–EtOAc(60:40)] to give a colorless solid; yield: 0.43 g (86%); mp 207–09 °C;
[α]D22 +3.89 (c 1.55, CHCl3).
IR (CHCl3): 3341, 2970, 2927, 2853, 1648, 1592, 1512, 1489, 1445,1406, 1384, 1254, 1176, 1137, 1030, 953, 928, 829, 680 cm–1.
1H NMR (200 MHz, CDCl3): δH = 1.34 (d, J = 6.9 Hz, 3 H), 2.49 (brs, 2 H), 3.19–3.30 (q, J = 6.8 Hz, 1 H), 3.71 (dd, J = 19.4, 3.9 Hz, 2H), 5.05 (s, 2 H), 5.85 (br s, 1 H), 6.95 (d, J = 8.7 Hz, 2 H), 7.00–7.06 (m, 1 H), 7.13–7.24 (m, 4 H), 7.29–7.40 (m, 1 H).
13C NMR (50 MHz, CDCl3): δC = 178.3 (CO), 165.4 (C), 157.7 (C),139.6 (C), 132.1 (C), 130.2 (CH), 129.3 (CH, 2 C), 122.7 (CH), 114.9 (CH, 2 C), 114.6 (CH), 113.9 (CH), 69.2 (CH2), 57.5 (CH),51.9 (CH2), 19.6 (CH3).
MS: m/z = 302 [M]+, 325 [M + Na]+.
(S)-Safinamide Mesylate (1)
MsOH (0.02 mL, 0.33 mmol) was added to a stirred solution of sulfonamide (S)-14 (0.1 g, 0.33 mmol) in EtOAc (3 mL) at 70 °C, and the mixture was stirred for 2 h. The temperature was then lowered to 35 °C, and the mixture was stirred for an additional 1 h. The solvent was evaporated under reduced pressure and the residue was filtered
through a short bed of basic alumina with elution by EtOAc–MeOH; (95:5) to give a white solid; yield: 0.11 g (90%);
mp 209–210 °C [Lit.7a 210 °C];
[α]D22 +9.6 (c 1.1, AcOH); {Lit.7 [α]D22+12.9 (c 1.1, AcOH)}.
Chiral HPLC: column: Chiralcel OD-RH (150 × 4.6 mm); eluent:MeOH–MeCN–buffer-TEAP (pH 3) (20:10:70); flow rate: 0.5mL/min (780 psi); detector: 224 nm [(R)-isomer: tR = 11.55 min;
(S)-isomer: tR = 12.94 min]; ee >98%.
7a) Pevarello, P.; Bonsignori, A.; Dostert, P.;
Heidempergher, F.; Pinciroli, V.; Colombo, M.; McArthur,
R. A.; Salvati, P.; Post, C.; Fariello, R. G.; Varasi, M. J. Med.
Chem. 1998, 41, 579.
PAPER

Chin. J. Pharmas.2012, 43, 161-163.

 …………….BASE
…………….BASE
 …………MESYLATE
…………MESYLATE
PAPER
J. Med. Chem. 2007, 50, 4909-4916.

(S)-2-[6-(3-Fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]-propionamide (21). The title compound was obtained using the same procedure described for the synthesis of (R)-2-[6-(3-fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]propionamide, starting from 6-(3-fluorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline (0.24 g, 0.95 mmol) and (R)-2-amino-1-methyl-2-oxoethyl-2-nitrobenzenesulfonate (0.52 g, 1.9 mmol). After column chromatography
purification using 99:1 DCM/MeOH as eluent, 0.075 g (24% yield) of the title compound was obtained as a pure white solid. Mp 153- 154 °C. 1H NMR (CDCl3) ä 1.35 (d, 3H, J ) 7.0), 2.67-2.97 (m, 4H), 3.28 (q, 1H, J ) 7.0), 3.64 (d, 1H, J ) 14.2), 3.77 (d, 1H, J ) 14.2), 5.05 (s, 2H), 5.36 (br, 1H), 6.74 (d, 1H, J ) 2.5), 6.79 (dd, 1H, J ) 8.5, 2.5), 6.97 (d, 1H, J ) 8.5), 6.99-7.06 (m, 1H), 7.06-7.24 (m, 3H), 7.30-7.40 (m, 1H).
J. Med. Chem.1998, 41, 579-590.

References
- “Summary of the risk management plan (RMP) for Xadago (safinamide)” (PDF). European Medicines Agency. January 2015.
- Fariello, RG (2007). “Safinamide”. Neurotherapeutics. 4 (1): 110–116. doi:10.1016/j.nurt.2006.11.011. PMID 17199024.
- “EPAR Summary for the Public for Xadago” (PDF). European Medicines Agency. February 2015.
- “After an odyssey of setbacks, FDA finally green-lights Newron’s Parkinson’s drug Xadago”. endpts.com. Retrieved 2017-03-21.
- Lawrence, Janna (2015-01-19). “Safinamide recommended for approval as Parkinson’s disease therapy”. The Pharmaceutical Journal. Royal Pharmaceutical Society. Retrieved 2015-01-19.
- Haberfeld, H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- H. Spreitzer (14 April 2014). “Neue Wirkstoffe – Safinamid”. Österreichische Apothekerzeitung (in German) (8/2014): 30.
- Klement, A (18 July 2016). “Xadago”. Österreichische Apothekerzeitung (in German) (15/2016): 10.
- “Summary of Product Characteristics for Xadago” (PDF). European Medicines Agency. 24 February 2015.
- ^ Jump up to:a b Caccia, C; Maj, R; Calabresi, M; Maestroni, S; Faravelli, L; Curatolo, L; Salvati, P; Fariello, RG (2006). “Safinamide: From molecular targets to a new anti-Parkinson drug”. Neurology. 67 (7 Suppl 2): S18–23. doi:10.1212/wnl.67.7_suppl_2.s18. PMID 17030736.
- Merck Serono: Vielversprechende Daten zur kognitiven Wirkung von Safinamid bei Parkinson im Frühstadium. (German) 8 June 2007.
- Pevarello, P; Bonsignori, A; Caccia, C; Amici, R; Salvati, P; Fariello, RG; McArthur, RA; Varasi, M (1999). “Sodium channel activity and sigma binding of 2-aminopropanamide anticonvulsants”. Bioorganic & Medicinal Chemistry Letters. 9 (17): 2521–2524. doi:10.1016/s0960-894x(99)00415-1.
- ^ Jump up to:a b Krösser, Sonja; Marquet, Anne; Gallemann, Dieter; Wolna, Peter; Fauchoux, Nicolas; Hermann, Robert; Johne, Andreas (2012). “Effects of ketoconazole treatment on the pharmacokinetics of safinamide and its plasma metabolites in healthy adult subjects”. Biopharmaceutics & Drug Disposition. 33 (9): 550. doi:10.1002/bdd.1822. PMID 23097240.
- Jump up^ Pevarello, P; Bonsignori, A; Dostert, P; Heidempergher, F; Pinciroli, V; Colombo, M; McArthur, RA; Varasi, M (1998). “Synthesis and Anticonvulsant Activity of a New Class of 2-[(Arylalkyl)amino]alkanamide Derivatives”. Journal of Medicinal Chemistry. 41 (4): 579–590. doi:10.1021/jm970599m. PMID 9484507.
- Jump up^ “Wichtigste Ergebnisse der Langzeitstudie mit Safinamid als Begleittherapie zu Levodopa bei Parkinson im fortgeschrittenen Stadium” [Major results from the long-term study of safinamide as add-on to levodopa for late-stage Parkinson] (in German). Merck KGaA. 4 November 2010.
- Jump up^ Study of Safinamide in Early Parkinson’s Disease as Add-on to Dopamine Agonist (MOTION)
- Jump up^ Merck Returns Rights for Safinamide to Newron, 21 October 2011.
- Jump up^ “Information about FDA Refusal to File” (PDF). Newron. 29 July 2014.
- “Information about FDA re-application” (PDF). Newron. 29 December 2014.
- Chazot, PL (2007). “Drug evaluation: Safinamide for the treatment of Parkinson’s disease, epilepsy and restless legs syndrome”. Current Opinion in Investigational Drugs. 8 (7): 570–579. PMID 17659477.
 |
|
| Clinical data | |
|---|---|
| Trade names | Xadago |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Protein binding | 88–90% |
| Metabolism | Amidases, glucuronidation |
| Biological half-life | 20–30 hrs |
| Excretion | 76% renal, 1.5% faeces |
| Identifiers | |
| Synonyms | EMD-1195686, PNU-15774E; (2S)-2-[[4-[(3-fluorophenyl)methoxy]phenyl] methylamino]propanamide |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.120.167 |
| Chemical and physical data | |
| Formula | C17H19FN2O2 |
| Molar mass | 302.34 g/mol |
| 3D model (Jmol) | |
//////////Xadago, safinamide, Newron Pharmaceuticals, FDA 2017, Parkinson’s disease, 133865-89-1 , сафинамид , سافيناميد, 沙非胺, EMD-1195686, ZP-034, FCE-28073(R-isomer), PNU-151774E, NW-1015, FCE-26743
C[C@H](NCC1=CC=C(OCC2=CC=CC(F)=C2)C=C1)C(N)=O
New paper on Trelagliptin succinate


Trelagliptin succinate, a novel once-weekly oral dipeptidyl peptidase-4 (DPP-4) inhibitor, was approved for the Japanese market on March 26, 2015
Trelagliptin exhibited a better potency against human DPP-4 than alogliptin and sitagliptin, along with its excellent selectivity and slow-binding properties that may partially contribute to its sustained efficacy. In phase III clinical studies, once-weekly oral trelagliptin provided long-term safety and efficacy in both monotherapy and combination with other antidiabetic medicines and was proved to be noninferior to its analogue alogliptin used once daily.
2-({6-[(3R)-3-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)-4-fluorobenzonitrile Monosuccinate (1)
Synthesis of Trelagliptin Succinate

An improved process for the synthesis of antidiabetic drug trelagliptin succinate through unprotected (R)-3-aminopiperidine was described. The impurity profile with different conditions of the key substitution was illustrated, and then the best reaction condition was identified. The optimizations also included the bromination of 4-fluoro-2-methylbenzonitrile so that the process became efficient and concise.
- 1.
Zhang, Z.; Wallace, M. B.; Feng, J.; Stafford, J. A.; Skene, R. J.; Shi, L.; Lee, B.; Aertgeerts, K.; Jennings, A.; Xu, R.; Kassel, D. B.; Kaldor, S. W.; Navre, M.; Webb, D. R.; Gwaltney, S. L.J. Med. Chem. 2011, 54, 510– 524, DOI: 10.1021/jm101016w
- 2.
Feng, J.; Gwaltney, S. L.; Dipeptidyl Peptidase Inhibitors. PCT Int. Appl. WO 2005095381, October 13, 2005.
- 3.
Grimshaw, C. E.; Jennings, A.; Kamran, R.; Ueno, H.; Nishigaki, N.; Kosaka, T.; Tani, A.; Sano, H.; Kinugawa, Y.; Koumura, E.; Shi, L.; Takeuchi, K. PLoS One 2016, 11, e0157509, DOI: 10.1371/journal.pone.0157509
4.
- 4.
Inagaki, N.; Onouchi, H.; Maezawa, H.; Kuroda, S.; Kaku, K. Lancet Diabetes Endocrinol.2015, 3, 191– 197, DOI: 10.1016/S2213-8587(14)70251-7
- 5.
Inagaki, N.; Sano, H.; Seki, Y.; Kuroda, S.; Kaku, K. J.Diabetes Investig. 2016, 7, 718– 726, DOI: 10.1111/jdi.12499
6.
Feng, J.; Gwaltney, S. L.; Dipeptidyl Peptidase Inhibitors. PCT Int. Appl. WO 2007035629, March 29, 2007.
//////////
NEW DRUG APPROVALS BLOG HITS 16 LAKH VIEWS IN 213 COUNTRIES

NEW DRUG APPROVALS BLOG HITS 16 LAKH VIEWS IN 213 COUNTRIES
Novartis Kisqali® (ribociclib, LEE011) receives FDA approval as first-line treatment for HR+/HER2- metastatic breast cancer in combination with any aromatase inhibitor


![]()
- Approved based on a first-line Phase III trial that met its primary endpoint of progression-free survival (PFS) at interim analysis due to superior efficacy compared to letrozole alone[1]
- At this interim analysis, Kisqali plus letrozole reduced risk of disease progression or death by 44% over letrozole alone, and demonstrated tumor burden reduction with a 53% overall response rate[1]
- Kisqali plus letrozole showed treatment benefit across all patient subgroups regardless of disease burden or tumor location[1]
- At a subsequent analysis with additional follow-up and progression events, a median PFS of 25.3 months for Kisqali plus letrozole and 16.0 months for letrozole alone was observed[2]
Basel, March 13, 2017 – The US Food and Drug Administration (FDA) has approved Kisqali®(ribociclib, formerly known as LEE011) in combination with an aromatase inhibitor as initial endocrine-based therapy for treatment of postmenopausal women with hormone receptor positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) advanced or metastatic breast cancer.
Kisqali is a CDK4/6 inhibitor approved based on a first-line Phase III trial that met its primary endpoint early, demonstrating statistically significant improvement in progression-free survival (PFS) compared to letrozole alone at the first pre-planned interim analysis[1]. Kisqali was reviewed and approved under the FDA Breakthrough Therapy designation and Priority Review programs.
“Kisqali is emblematic of the innovation that Novartis continues to bring forward for people with HR+/HER2- metastatic breast cancer,” said Bruno Strigini, CEO, Novartis Oncology. “We at Novartis are proud of the comprehensive clinical program for Kisqali that has led to today’s approval and the new hope this medicine represents for patients and their families.”
The FDA approval is based on the superior efficacy and demonstrated safety of Kisqali plus letrozole versus letrozole alone in the pivotal Phase III MONALEESA-2 trial. The trial, which enrolled 668 postmenopausal women with HR+/HER2- advanced or metastatic breast cancer who received no prior systemic therapy for their advanced breast cancer, showed that Kisqali plus an aromatase inhibitor, letrozole, reduced the risk of progression or death by 44 percent over letrozole alone (median PFS not reached (95% CI: 19.3 months-not reached) vs. 14.7 months (95% CI: 13.0-16.5 months); HR=0.556 (95% CI: 0.429-0.720); p<0.0001)[1].
More than half of patients taking Kisqali plus letrozole remained alive and progression free at the time of interim analysis, therefore median PFS could not be determined[1]. At a subsequent analysis with additional 11-month follow-up and progression events, a median PFS of 25.3 months for Kisqali plus letrozole and 16.0 months for letrozole alone was observed[2]. Overall survival data is not yet mature and will be available at a later date.
“In the MONALEESA-2 trial, ribociclib plus letrozole reduced the risk of disease progression or death by 44 percent over letrozole alone, and more than half of patients (53%) with measurable disease taking ribociclib plus letrozole experienced a tumor burden reduction of at least 30 percent. This is a significant result for women with this serious form of breast cancer,” said Gabriel N. Hortobagyi, MD, Professor of Medicine, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center and MONALEESA-2 Principal Investigator. “These results affirm that combination therapy with a CDK4/6 inhibitor like ribociclib and an aromatase inhibitor should be a new standard of care for initial treatment of HR+ advanced breast cancer.”
Kisqali is taken with or without food as a once-daily oral dose of 600 mg (three 200 mg tablets) for three weeks, followed by one week off treatment. Kisqali is taken in combination with four weeks of any aromatase inhibitor[1].
Breast cancer is the second most common cancer in American women[3]. The American Cancer Society estimates more than 250,000 women will be diagnosed with invasive breast cancer in 2017[3]. Up to one-third of patients with early-stage breast cancer will subsequently develop metastatic disease[4].
Novartis is committed to providing patients with access to medicines, as well as resources and support to address a range of needs. The Kisqali patient support program is available to help guide eligible patients through the various aspects of getting started on treatment, from providing educational information to helping them understand their insurance coverage and identify potential financial assistance options. For more information, patients and healthcare professionals can call 1-800-282-7630.
The full prescribing information for Kisqali can be found at https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf(link is external).
About Kisqali® (ribociclib)
Kisqali (ribociclib) is a selective cyclin-dependent kinase inhibitor, a class of drugs that help slow the progression of cancer by inhibiting two proteins called cyclin-dependent kinase 4 and 6 (CDK4/6). These proteins, when over-activated, can enable cancer cells to grow and divide too quickly. Targeting CDK4/6 with enhanced precision may play a role in ensuring that cancer cells do not continue to replicate uncontrollably.
Kisqali was developed by the Novartis Institutes for BioMedical Research (NIBR) under a research collaboration with Astex Pharmaceuticals.
About the MONALEESA Clinical Trial Program
Novartis is continuing to assess Kisqali through the robust MONALEESA clinical trial program, which includes two additional Phase III trials, MONALEESA-3 and MONALEESA-7, that are evaluating Kisqali in multiple endocrine therapy combinations across a broad range of patients, including premenopausal women. MONALEESA-3 is evaluating Kisqali in combination with fulvestrant compared to fulvestrant alone in postmenopausal women with HR+/HER2- advanced breast cancer who have received no or a maximum of one prior endocrine therapy. MONALEESA-7 is investigating Kisqali in combination with endocrine therapy and goserelin compared to endocrine therapy and goserelin alone in premenopausal women with HR+/HER2- advanced breast cancer who have not previously received endocrine therapy.
About Novartis in Advanced Breast Cancer
For more than 25 years, Novartis has been at the forefront of driving scientific advancements for breast cancer patients and improving clinical practice in collaboration with the global community. With one of the most diverse breast cancer pipelines and the largest number of breast cancer compounds in development, Novartis leads the industry in discovery of new therapies and combinations, especially in HR+ advanced breast cancer, the most common form of the disease.
Kisqali® (ribociclib) Important Safety Information
Kisqali® (ribociclib) can cause a heart problem known as QT prolongation. This condition can cause an abnormal heartbeat and may lead to death. Patients should tell their healthcare provider right away if they have a change in their heartbeat (a fast or irregular heartbeat), or if they feel dizzy or faint. Kisqali can cause serious liver problems. Patients should tell their healthcare provider right away if they get any of the following signs and symptoms of liver problems: yellowing of the skin or the whites of the eyes (jaundice), dark or brown (tea-colored) urine, feeling very tired, loss of appetite, pain on the upper right side of the stomach area (abdomen), and bleeding or bruising more easily than normal. Low white blood cell counts are very common when taking Kisqali and may result in infections that may be severe. Patients should tell their healthcare provider right away if they have signs and symptoms of low white blood cell counts or infections such as fever and chills. Before taking Kisqali, patients should tell their healthcare provider if they are pregnant, or plan to become pregnant as Kisqali can harm an unborn baby. Females who are able to become pregnant and who take Kisqali should use effective birth control during treatment and for at least 3 weeks after the last dose of Kisqali. Do not breastfeed during treatment with Kisqali and for at least 3 weeks after the last dose of Kisqali. Patients should tell their healthcare provider about all of the medicines they take, including prescription and over-the-counter medicines, vitamins, and herbal supplements since they may interact with Kisqali. Patients should avoid pomegranate or pomegranate juice, and grapefruit or grapefruit juice while taking Kisqali. The most common side effects (incidence >=20%) of Kisqali when used with letrozole include white blood cell count decreases, nausea, tiredness, diarrhea, hair thinning or hair loss, vomiting, constipation, headache, and back pain. The most common grade 3/4 side effects in the Kisqali + letrozole arm (incidence >2%) were low neutrophils, low leukocytes, abnormal liver function tests, low lymphocytes, and vomiting. Abnormalities were observed in hematology and clinical chemistry laboratory tests.
Please see the Full Prescribing Information for Kisqali, available at https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf(link is external).
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, cost-saving generic and biosimilar pharmaceuticals and eye care. Novartis has leading positions globally in each of these areas. In 2016, the Group achieved net sales of USD 48.5 billion, while R&D throughout the Group amounted to approximately USD 9.0 billion. Novartis Group companies employ approximately 118,000 full-time-equivalent associates. Novartis products are sold in approximately 155 countries around the world. For more information, please visit http://www.novartis.com.
Novartis is on Twitter. Sign up to follow @Novartis and @NovartisCancer at http://twitter.com/novartis(link is external) and http://twitter.com/novartiscancer (link is external)
For Novartis multimedia content, please visit www.novartis.com/news/media-library
For questions about the site or required registration, please contact media.relations@novartis.com
References
[1] Kisqali (ribociclib) Prescribing information. East Hanover, New Jersey, USA: Novartis Pharmaceuticals Corporation; March 2016.
[2] Novartis Data on File
[3] American Cancer Society. How Common Is Breast Cancer? Available at https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html(link is external). Accessed January 23, 2017.
[4] O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. The Oncologist. 2005;10(Suppl 3):20-29.
рибоциклиб , ريبوسيكليب , 瑞波西利
Ribociclib « New Drug Approvals
////////////////Novartis, Kisqali®, ribociclib, LEE011, FDA 2017, HR+/HER2- metastatic breast cancer, рибоциклиб , ريبوسيكليب , 瑞波西利
GDC 0994, Ravoxertinib
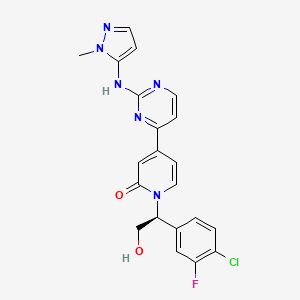
GDC 0994
GDC-0994; Ravoxertinib; 1453848-26-4; GDC0994; UNII-R6AXV96CRH; R6AXV96CRH, RG7842; RG-7842; RG 7842
CAS 1453848-26-4
1-[(1S)-1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl]-4-[2-[(2-methylpyrazol-3-yl)amino]pyrimidin-4-yl]pyridin-2-one
| Molecular Formula: | C21H18ClFN6O2 |
|---|---|
| Molecular Weight: | 440.863 g/mol |
PHASE 1
Ravoxertinib also known as GDC-0994 and RG7842, is an orally available inhibitor of extracellular signal-regulated kinase (ERK), with potential antineoplastic activity. Upon oral administration, GDC-0994 inhibits both ERK phosphorylation and activation of ERK-mediated signal transduction pathways. This prevents ERK-dependent tumor cell proliferation and survival. The mitogen-activated protein kinase (MAPK)/ERK pathway is upregulated in a variety of tumor cell types and plays a key role in tumor cell proliferation, differentiation and survival.
GDC-0994 is an ERK inhibitor invented by Array under a collaboration agreement with Genentech. Array has received certain clinical milestones and is entitled to additional potential clinical and commercial milestones and royalties on product sales under the agreement. ERK is a key protein kinase in the RAS/RAF/MEK/ERK pathway, which regulates several key cellular activities including proliferation, differentiation, migration, survival and angiogenesis. Inappropriate activation of this pathway has been shown to occur in many cancers. GDC-0994 is currently advancing in a Phase 1 trial in patients with solid tumors.


| Applicants: | ARRAY BIOPHARMA INC. [US/US]; 3200 Walnut Street Boulder, Colorado 80301 (US). GENENTECH, INC. [US/US]; 1 DNA Way South San Francisco, California 94080-4990 (US) |
| Inventors: | BLAKE, James F.; (US). CHICARELLI, Mark Joseph; (US). GARREY, Rustam Ferdinand; (US). GAUDINO, John; (US). GRINA, Jonas; (US). MORENO, David A.; (US). MOHR, Peter J.; (US). REN, Li; (US). SCHWARZ, Jacob; (US). CHEN, Huifen; (US). ROBARGE, Kirk; (US). ZHOU, Aihe; (US) |
- OriginatorArray BioPharma
- DeveloperGenentech
- ClassAntineoplastics; Small molecules
- Mechanism of ActionExtracellular signal-regulated MAP kinase inhibitors; Mitogen activated protein kinase 3 inhibitors; Mitogen-activated protein kinase 1 inhibitors
- Phase ISolid tumours
Most Recent Events
- 29 Nov 2016Pharmacodynamics data from a preclinical trial in Solid tumours presented at the 28th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics (EORTC-NCI-AACR-2016)
- 29 Nov 2016Adverse events, efficacy, pharmacokinetics and pharmacodynamics data from a phase I trial in Solid tumours presented at the 28th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics
- 16 Jul 2016No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy, Second-line therapy or greater) in USA
FREE FORM

(S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one,
GDC-0994 benzenesulfonate salt

CAS 1817728-45-2, C21 H18 Cl F N6 O2 . C6 H6 O3 S
GDC-0994 as a light yellow solid,
mp 197.7 °C;

1H NMR (600 MHz, DMSO-d6): 9.93, (s, 1H), 8.65 (d, J = 5.2 Hz, 1H), 7.95 (d, J = 7.27 Hz, 1H), 7.63 (m, 2H), 7.62 (d, J = 1.5 Hz, 1H), 7.58 (t, J = 8.2 Hz, 1H), 7.55 (d, J = 5.2 Hz, 1H), 7.44 (dd, J = 10.6, 1.9 Hz, 1H), 7.33 (m, 3H), 7.18 (d, J = 2.0 Hz, 1H), 7.17 (d, J = 2.1 Hz, 1H), 6.90 (dd, J = 7.3, 2.1 Hz, 1H), 6.48 (d, J = 2.2 Hz, 1H), 5.99 (dd, J = 8.1, 5.5 Hz, 1H), 4.17 (dd, J = 11.9, 8.2 Hz, 1H), 4.05 (dd, J = 11.9, 5.5 Hz, 1H), 3.78 (s, 3H).

13C NMR (150 MHz, DMSO-d6): 161.60, 161.14, 160.02, 159.79, 157.02 (d, J = 245 Hz), 148.0, 146.49, 139.53 (d, J = 6.0 Hz), 139.04, 136.96, 136.39, 130.66, 128.42, 127.59, 125.38, 124.99 (d, J = 3.0 Hz), 118.72 (d, J = 18.0 Hz), 117.29, 116.05 (d, J = 22.5 Hz), 109.75, 102.79, 98.77, 60.64, 58.68, 35.29.

19F NMR (282 MHz, DMSO-d6) −115.86 (dd, J = 10.6, 7.8).
HRMS calcd for C21H18ClFN6O2 [M + H] 441.1242, found 441.1245.
PATENT
Example 39

(S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5- yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one
[00398] Step A: (S)-1-(2-(tert-Butyldimethylsiloxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-(methylsulfonyl)pyrimidin-4-yl)pyridine-2(1H)-one (47 mg, 0.087 mmol), 2-methyl pyrazole-3 -amine (0.175 mmol, 2.0 equivalents) and anhydrous DMF (3.0 mL) were added to a 25 mL round bottomed flask equipped with a stirring bar. The flask was capped with a rubber septum and flushed with nitrogen. Under a blanket of nitrogen, sodium hydride (8.5 mg, 60% dispersion in mineral oil) was added in one portion. The flask was flushed with
nitrogen, capped and stirred at room temperature. The reaction progress was monitored by LCMS, and after 30 minutes, the starting material was consumed. The reaction mixture was quenched by the addition of water (0.5 mL) and ethyl acetate (15 mL). The contents of the round bottomed flask were transferred to a 125 mL separatory funnel, and the reaction flask was rinsed several times with additional ethyl acetate. Crude (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one was partitioned between ethyl acetate and water (80 mL/30 mL). The ethyl acetate layer was washed once with brine, dried (MgSO4), filtered and concentrated to give crude (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one. The crude was taken directly into the deprotection step.
[00399] Step B: Crude (S)-1-(2-((tert-butyldimethylsilyl)oxy)-1-(4-chloro-3-fluorophenyl)ethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (48 mg) was dissolved in ethyl acetate (4 mL) and treated dropwise slowly (over 2 minutes) with an ethyl acetate solution (1.0 mL, which had been saturated with HCl gas). The reaction stirred at room temperature for 15 minutes, after which time LCMS indicated complete consumption of the starting material. The reaction mixture was concentrated to an oily residue and purified by prep RP HPLC to yield (S)-1-(1-(4-chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (20.8 mg, 54.6% yield) as a lyophilized powder. 1H NMR (400 MHz, (CD3)2SO) δ 9.58 (s, 1H), 8.60 (d, J = 5.1 Hz, 1H), 7.91 (t, J = 9.0 Hz, 1H),7.58 (t, J = 8.1 Hz, 1H), 7.52-7.41 (m, 2H), 7.37 (d, J = 1.8 Hz, 1H), 7.14 (dd, J = 10.7,5.1 Hz 2H), 6.86 (dd, J = 7.3, 1.8 Hz, 1H), 6.27(d, J = 1.7 Hz, 1H), 5.97 (dd, J = 7.7, 5.7 Hz, 1H), 5.31(t, J = 5.2 Hz, 1H), 4.15 (m, 1H), 4.10-3.95 (m,1H), 3.69 (s, 3H); LCMS m/z 441 (M+H)+.
PAPER
Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development
The extracellular signal-regulated kinases ERK1/2 represent an essential node within the RAS/RAF/MEK/ERK signaling cascade that is commonly activated by oncogenic mutations in BRAF or RAS or by upstream oncogenic signaling. While targeting upstream nodes with RAF and MEK inhibitors has proven effective clinically, resistance frequently develops through reactivation of the pathway. Simultaneous targeting of multiple nodes in the pathway, such as MEK and ERK, offers the prospect of enhanced efficacy as well as reduced potential for acquired resistance. Described herein is the discovery and characterization of GDC-0994 (22), an orally bioavailable small molecule inhibitor selective for ERK kinase activity.
PATENT
WO 2015154674
https://www.google.com/patents/WO2015154674A1?cl=pt
(a) contacting 4-bromo-1-chloro-2-fluorobenzene with a metallating agent in an aprotic organic solvent to afford an organomagnesium compound, which is reacted with 2-chloro-N-methoxy-N-methylacetamide to afford 2-chloro-1- (4-chloro-3-fluorophenyl) ethanone (II) ;
(b) contacting II with sodium formate and formic acid in aqueous ethanol to afford 1- (4-chloro-3-fluorophenyl) -2-hydroxyethanone (III)
(c) contacting III with a ketoreductase to afford (R) -1- (4-chloro-3-fluorophenyl) ethane-1, 2-diol (IV) ;
(d) contacting IV with a silyl chloride (Ra) 3SiCl and at least one base in a non-polar aprotic solvent to afford (V) , and subsequently adding sulfonylchloride RbS (O) 2Cl to afford VI, wherein Ra is independently in each occurrence C1-6 alkyl or phenyl and Rb is selected from C1-4 alkyl or phenyl, optionally substituted with 1 to 3 groups independently selected from C1-3 alkyl, halogen, nitro, cyano, or C1-3 alkoxy;
(e) contacting 4- (2- (methylsulfonyl) pyrimidin-4-yl) pyridin-2 (1H) -one (VII) with a strong base in an organic solvent and subsequently adding VI to afford XI;
(f) treating XI with an oxidizing agent to afford I;
(g) treating 1-methyl-1H-pyrazol-5-amine with a strong base in an aprotic solvent at reduced temperature and adding the compound of formula I to afford IX; and,
(h) contacting IX with a de-silylating agent to afford VIII.
PAPER
Development of a Practical Synthesis of ERK Inhibitor GDC-0994
 , Nicholas Wong†, Hans Iding‡, Vera Jost‡, Haiming Zhang†, Stefan G. Koenig†, C. Gregory Sowell†, and Francis Gosselin†
, Nicholas Wong†, Hans Iding‡, Vera Jost‡, Haiming Zhang†, Stefan G. Koenig†, C. Gregory Sowell†, and Francis Gosselin†
The process development of a synthetic route to manufacture ERK inhibitor GDC-0994 on multikilogram scale is reported herein. The API was prepared as the corresponding benzenesulfonate salt in 7 steps and 41% overall yield. The synthetic route features a biocatalytic asymmetric ketone reduction, a regioselective pyridone SN2 reaction, and a safe and scalable tungstate-catalyzed sulfide oxidation. The end-game process involves a telescoped SNAr/desilylation/benzenesulfonate salt formation sequence. Finally, the development of the API crystallization allowed purging of process-related impurities, obtaining >99.5A% HPLC and >99% ee of the target molecule.
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016136150 | COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK | 2015-11-13 | 2016-05-19 |
| US2016122316 | SERINE/THREONINE KINASE INHIBITORS | 2016-01-12 | 2016-05-05 |
| US2015111869 | USE OF A COMBINATION OF A MEK INHIBITOR AND AN ERK INHIBITOR FOR TREATMENT OF HYPERPROLIFERATIVE DISEASES | 2014-08-29 | 2015-04-23 |
| US2015051209 | COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF MEK | 2014-08-05 | 2015-02-19 |
| US2014249127 | SERINE/THREONINE KINASE INHIBITORS | 2014-02-14 | 2014-09-04 |
| US8697715 | Serine/threonine kinase inhibitors | 2013-03-01 | 2014-04-15 |
///////////GDC 0994, Ravoxertinib, 1453848-26-4, GDC0994, UNII-R6AXV96CRH, R6AXV96CRH, RG7842, RG-7842, RG 7842, PHASE 1
CN1C(=CC=N1)NC2=NC=CC(=N2)C3=CC(=O)N(C=C3)C(CO)C4=CC(=C(C=C4)Cl)F
TROXACITABINE троксацитабин , تروكساسيتابين , 曲沙他滨 ,
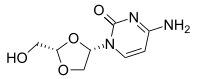
Troxacitabine
CAS 145918-75-8
- Molecular FormulaC8H11N3O4
- Average mass213.191 Da
Hmd-cytosine; NCGC00183848-01; Beta-L-Dioxolane-cytidine; 4-amino-1-[(2S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]pyrimidin-2-one; 2R(-)-cis-Hmd-cytosine, (-)-ODDC
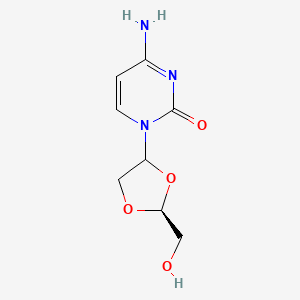
4-amino-1-[(2S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]pyrimidin-2-one
Troxacitabine (brand name Troxatyl) is a nucleoside analogue with anticancer activity. Its use is being studied in patients with refractory lymphoproliferative diseases.[1]
Troxacitabine (brand name Troxatyl) is a nucleoside analogue with anticancer activity. Its use is being studied in patients with refractory lymphoproliferative diseases.
| Investigated for use/treatment in leukemia (myeloid). |
PATENT
https://www.google.com/patents/WO1992018517A1?cl=en
WO 9218517
| Inventors | Yung-Chi Cheng, Chung K. Chu, Hea O. Kim, Kirupathevy Shanmuganathan |
| Applicant | Yale University, The University Of Georgia Research Foundation, Inc. |
SYNTHESIS
WO 2016030335

PATENT
WO 2016030335
PATENT
MACHINE TRANSLATED FROM CHINESE, BEAWARE OF FUNNY NAMES
PATENT
PATENT
CN 105503838
PAPER
In vitro optimization of non-small cell lung cancer activity with troxacitabine, L-1,3-dioxolane-cytidine, prodrugs
Journal of medicinal chemistry (2007), 50, (9), 2249-53.

l-1,3-Dioxolane-cytidine, a potent anticancer agent against leukemia, has limited efficacy against solid tumors, perhaps due to its hydrophilicity. Herein, a library of prodrugs were synthesized to optimize in vitro antitumor activity against non-small cell lung cancer. N4-Substituted fatty acid amide prodrugs of 10−16 carbon chain length demonstrated significantly improved antitumor activity over l-1,3-dioxolane-cytidine. These in vitro results suggest that the in vivo therapeutic efficacy of l-1,3-dioxolane-cytidine against solid tumors may be improved with prodrug strategies.
PAPER
- Kim, Hea O.; Schinazi, Raymond F.; Shanmuganathan, Kirupathevy; Jeong, Lak S.; Beach, J. Warren; Nampalli, Satyanarayana; Cannon, Deborah L.; Chu, Chung K.
- From Journal of Medicinal Chemistry (1993), 36(5), 519-28.
PAPER
- Jin, Haolun; Tse, Allan Tse; Evans, Colleen A.; Mansour, Tarek S.; Beels, Christopher M.; Ravenscroft, Paul; Humber, David C.; Jones, Martin F.; Payne, Jeremy J.; Ramsay, Michael V. J.
- From Tetrahedron: Asymmetry (1993), 4(2), 211-14
PAPER
- Belleau, Bernard R.; Evans, Colleen A.; Tse, H. L. Allan; Jin, Haolun; Dixit, Dilip M.; Mansour, Tarek S.
- From Tetrahedron Letters (1992), 33(46), 6949-52.
PAPER
http://pubs.acs.org/doi/pdf/10.1021/jm00089a007
J. Med. Chem. 1992,35,1987-1995 Asymmetric Synthesis of 1,3-Dioxolane-Pyrimidine Nucleosides and Their Anti-HIV Activity
 References
References
- Jump up^ Vose, Julie M.; Panwalkar, Amit; Belanger, Robert; Coiffier, Bertrand; Baccarani, Michele; Gregory, Stephanie A.; Facon, Thierry; Fanin, Renato; Caballero, Dolores; Ben-Yehuda, Dina; Giles, Francis (2007). “A phase II multicenter study of troxacitabine in relapsed or refractory lymphoproliferative neoplasms or multiple myeloma”. Leukemia & Lymphoma. 48 (1): 39–45. doi:10.1080/10428190600909578.
- Lee CK, Rowinsky EK, Li J, Giles F, Moore MJ, Hidalgo M, Capparelli E, Jolivet J, Baker SD: Population pharmacokinetics of troxacitabine, a novel dioxolane nucleoside analogue. Clin Cancer Res. 2006 Apr 1;12(7 Pt 1):2158-65. [PubMed:16609029 ]
- Quintas-Cardama A, Cortes J: Evaluation of the L-stereoisomeric nucleoside analog troxacitabine for the treatment of acute myeloid leukemia. Expert Opin Investig Drugs. 2007 Apr;16(4):547-57. [PubMed:17371201 ]
- Swords R, Giles F: Troxacitabine in acute leukemia. Hematology. 2007 Jun;12(3):219-27. [PubMed:17558697 ]
- Orsolic N, Giles FJ, Gourdeau H, Golemovic M, Beran M, Cortes J, Freireich EJ, Kantarjian H, Verstovsek S: Troxacitabine and imatinib mesylate combination therapy of chronic myeloid leukaemia: preclinical evaluation. Br J Haematol. 2004 Mar;124(6):727-38. [PubMed:15009060 ]
- Boivin AJ, Gourdeau H, Momparler RL: Action of troxacitabine on cells transduced with human cytidine deaminase cDNA. Cancer Invest. 2004;22(1):25-9. [PubMed:15069761 ]
- Kim TE, Park SY, Hsu CH, Dutschman GE, Cheng YC: Synergistic antitumor activity of troxacitabine and camptothecin in selected human cancer cell lines. Mol Pharmacol. 2004 Aug;66(2):285-92. [PubMed:15266019 ]
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2013011392 | METHOD FOR ASSESSING THE ABILITY OF A PATIENT TO RESPOND TO OR BE SAFELY TREATED BY A NUCLEOSIDE ANALOG BASED-CHEMOTHERAPY | 2010-11-19 | 2013-01-10 |
| US7927613 | Pharmaceutical co-crystal compositions | 2003-09-11 | 2011-04-19 |
| US7790905 | Pharmaceutical propylene glycol solvate compositions | 2003-12-29 | 2010-09-07 |
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C8H11N3O4 |
| Molar mass | 213.19 g/mol |
| 3D model (Jmol) | |
//////////////TROXACITABINE, троксацитабин , تروكساسيتابين , 曲沙他滨 , Hmd-cytosineM, NCGC00183848-01, Beta-L-Dioxolane-cytidine, 2R(-)-cis-Hmd-cytosine, (-)-ODDC
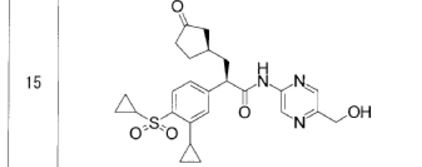
Astellas Pharma Inc. new Glucokinase Activator, ASP ? for Type 2 Diabetes
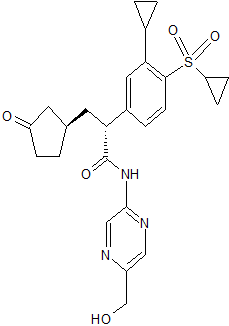
ASP ?
(2R)-2-(4-cyclopropanesulfonyl-3-cyclopropylphenyl)-N-[5-(hydroxymethyl)pyrazin-2-yl]-3-[(R)-3-oxocyclopentyl]propanamide
- Molecular Weight, 483.58
- [α]D20 −128.7 (c 1.00, MeOH);
- 1H NMR (DMSO-d6, 400 MHz) δ 11.07 (s, 1H), 9.20 (d, J = 1.4 Hz, 1H), 8.41 (d, J = 1.4 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.41 (dd, J = 8.2, 1.8 Hz, 1H), 7.15 (d, J = 1.8 Hz, 1H), 5.52 (t, J = 5.7 Hz, 1H), 4.56 (d, J = 6.0 Hz, 2H), 4.04 (t, J = 7.6 Hz, 1H), 3.03–2.97 (m, 1H), 2.79 (tt, J = 8.4, 5.1 Hz, 1H), 2.25–1.81 (m, 8H), 1.53–1.47 (m, 1H), 1.17–1.12 (m, 2H), 1.08–1.02 (m, 4H), 0.89–0.84 (m, 2H);
- 13C NMR (DMSO-d6, 101 MHz) δ 218.5, 171.8, 152.1, 147.3, 145.7, 143.2, 140.3, 138.2, 134.8, 129.0, 125.3, 125.1, 62.5, 49.9, 44.4, 38.4, 38.2, 34.8, 32.1, 29.1, 12.4, 10.8, 10.7, 5.8;
- FTIR (ATR, cm–1) 3544, 3257, 1727, 1692, 1546, 1507, 1363, 1285, 1149, 719;
- HRMS (ESI) m/z [M + Na]+ calcd for C25H29N3O5S 506.1726, found 506.1747.
- Anal. Calcd for C25H29N3O5S: C, 62.09; H, 6.04; N, 8.69. Found: C, 61.79; H, 6.19; N, 8.62.
![]()
| Inventors | Masahiko Hayakawa, Yoshiyuki Kido, Takahiro Nigawara, Mitsuaki Okumura, Akira Kanai, Keisuke Maki, Nobuaki Amino |
| Applicant | Astellas Pharma Inc. |
Synthesis

contd…………………………..

PATENT
- PAPER
A Practical and Scalable Synthesis of a Glucokinase Activator via Diastereomeric Resolution and Palladium-Catalyzed C–N Coupling Reaction
, Yasuhiro Morinaga†, Makoto Kasai†, Takao Hashimoto†, Yuji Takahama†, Atsushi Ohigashi†, Satoshi Yonishi‡, and Motohiro Akazome§
Here we describe the research and development of a process for the practical synthesis of glucokinase activator (R)-1 as a potential drug for treating type-2 diabetes. The key intermediate, chiral α-arylpropionic acid (R)-2, was synthesized in high diastereomeric excess through the diasteromeric resolution of 7 without the need for a chiral resolving agent. The counterpart 2-aminopyrazine derivative 3 was synthesized using a palladium-catalyzed C–N coupling reaction. This efficient process was demonstrated at the pilot scale and yielded 19.0 kg of (R)-1. Moreover, an epimerization process to obtain (R)-7 from the undesired (S)-7 was developed.
Hayakawa, M.; Kido, Y.; Nigawara, T.; Okumura, M.; Kanai, A.; Maki, K.; Amino, N. PCT Int. Appl. WO/2009/091014 A1 20090723,2009.
https://www.astellas.com/en/ir/library/pdf/3q2017_rd_en.pdf
///////////1174229-89-0, ASTELLAS, Glucokinase Activator, TYPE 2 DIABETES, PRECLINICAL, ASP ?, WO 2009091014, Masahiko Hayakawa, Yoshiyuki Kido, Takahiro Nigawara, Mitsuaki Okumura, Akira Kanai, Keisuke Maki, Nobuaki Amino, WO2009091014,
O=C(Nc1cnc(cn1)CO)[C@H](C[C@@H]2CC(=O)CC2)c3ccc(c(c3)C4CC4)S(=O)(=O)C5CC5
FDA approves first treatment Noctiva (Desmopressin acetate) nasal spray for frequent urination at night due to overproduction of urine

Desmopressin acetate
March 3, 2017
The U.S. Food and Drug Administration today approved Noctiva (desmopressin acetate) nasal spray for adults who awaken at least two times per night to urinate due to a condition known as nocturnal polyuria (overproduction of urine during the night). Noctiva is the first FDA-approved treatment for this condition.
“Today’s approval provides adults who overproduce urine at night with the first FDA-approved therapeutic option to help reduce the number of times a night they wake up to urinate,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Division of Bone, Reproductive, and Urologic Products in the FDA’s Center for Drug Evaluation and Research. “It is important to know that Noctiva is not approved for all causes of night-time urination, so patients should discuss their symptoms with their health care provider who can determine the underlying cause of the night-time urination and whether Noctiva is right for them.”
Nocturia (wakening at night to urinate) is a symptom that can be caused by a wide variety of conditions, such as congestive heart failure, poorly controlled diabetes mellitus, medications, or diseases of the bladder or prostate. Before considering Noctiva, health care providers should evaluate each patient for possible causes for the nocturia, and optimize the treatment of underlying conditions that may be contributing to the night-time urination. Because Noctiva is approved only for adults with nocturia caused by nocturnal polyuria, health care providers should confirm overproduction of urine at night with a 24-hour urine collection, if one has not been obtained previously. Health care providers should also be mindful of underlying conditions that can cause nocturia, but that make treatment with Noctiva unsafe, such as excessive drinking of fluids or symptomatic congestive heart failure.
Noctiva is taken daily, approximately 30 minutes before going to bed. It works by increasing the absorption of water through the kidneys, which leads to less urine production.
Noctiva’s efficacy was established in two 12-week, randomized, placebo-controlled trials in 1,045 patients 50 years of age and older with nocturia due to nocturnal polyuria. Although these trials showed a small reduction in the average number of night-time urinations with Noctiva compared to placebo, more patients treated with Noctiva were able to at least halve their number of night-time urinations, and patients treated with Noctiva had more nights with one or fewer night-time urinations.
Noctiva is being approved with a boxed warning and a Medication Guide because it can cause low sodium levels in the blood (hyponatremia). Severe hyponatremia can be life-threatening if it is not promptly diagnosed and treated, leading to seizures, coma, respiratory arrest or death. Health care providers should make sure the patient’s sodium level is normal before starting Noctiva, and should check sodium levels within one week and approximately one month after starting treatment and periodically thereafter. The lower Noctiva dose is recommended as the starting dose for those who may be at risk for hyponatremia, such as the elderly. Noctiva should not be used in patients at increased risk of severe hyponatremia, such as those with excessive fluid intake, those who have illnesses that can cause fluid or electrolyte imbalances, certain patients with kidney damage, and in those using certain medicines, known as loop diuretics or glucocorticoids.
Noctiva should also not be used in patients with symptomatic congestive heart failure or uncontrolled hypertension because fluid retention can worsen these underlying conditions. Use of Noctiva should be discontinued temporarily in patients with certain nasal conditions such as colds or allergies until those conditions have resolved.
Noctiva is also not recommended for the treatment of nocturia in pregnant women. Nocturia is usually related to normal changes in pregnancy that do not require treatment with Noctiva. Noctiva should not be used in children.
The most common side effects of Noctiva in clinical trials included nasal discomfort, cold symptoms (nasopharyngitis), nasal congestion, sneezing, high or increased blood pressure, back pain, nose bleeds, bronchitis and dizziness.
Although there are other FDA-approved medications that also contain desmopressin, none of those medications are approved to treat nocturia.
Noctiva is marketed by Milford, Pennsylvania-based Renaissance Lakewood, LLC for Serenity Pharmaceuticals, LLC.
C48H68N14O14S2 C48H68N14O14S2·xH2O
(anhydrous) ![]()
![]()
![]()
![]()
![]() 1129.27
1129.27![]()
![]()
![]() [62288-83-9].
[62288-83-9].
1-(3-Mercaptopropionic acid)-8-D-arginine-vasopressin monoacetate (salt).

oxopentan-2-yl]-1-[4-(2-amino-2-oxoethyl)-7-(3-amino-3-oxopropyl)-10-benzyl-13-[(4-hydroxyphenyl)methyl]-3,6,9,12,15-pentaoxo-18,19-dithia-2,5,8,11,14-pentazacycloicosane-1-carbonyl]pyrrolidine-2-carboxamide;
Synonyms: 3-MERCAPTOPROPIONYL-TYR-PHE-GLN-ASN-CYS-PRO-D-ARG-GLY-NH2 ACETATE SALT;DDAVP ACETATE;[DEAMINO-CYS1,D-ARG8]-VASOPRESSIN ACETATE SALT;DESMOPRESSIN MONOACETATE;DESMORESSIN ACETATE;Mpr-Tyr-Phe-Gln-Asn-Cys-Pro-D-Arg-Gly-NH2(S-S:1-5);DESMOPRESSIN ACETATE;DESMOPRESSIN ACETATE SALT;
The Molecular Weight of Desmopressin Acetate(62288-83-9): 1129.27




Analytica Chimica Acta (2006), 572, (2), 197-204
Abstract
A monolithic column was prepared using l-phenylalanine as template and a covalent approach through the formation of Schiff base with o-phthalaldehyde (OPA). OPA, allylmercaptan, l-phenylalanine, and triethylamine were stirred at first, then methacrylic acid, 2-vinylpyridine, ethyleneglycol dimethacrylate, α,α-azobisisobutyronitrile, and 1-propanol were added to the reaction mixture. The resulting material was introduced into a capillary column. Following thermal polymerization, the template was then extracted with a mixture of HCl and methanol. The column was employed for the capillary electrochromatographic separation of oligopeptides. A capillary column of 75 (50) cm × 75 μm ID with a mobile phase of phosphate buffer (pH 7.0, 40 mM)/methanol (5%, v/v), an applied voltage of +15 kV, and detection at 214 nm, could baseline separate angiotensin I, angiotensin II, [Sar1, Thr8] angiotensin, oxytocin, vasopressin, tocinoic acid, β-casomorphin bovine, β-casomorphin human, and FMRF amide within 20 min. The separation behavior of the templated polymer was also compared with that of the non-templated polymer. As a result, it can be concluded that the electrochromatographic separation of this set of peptides was mediated by a combination of electrophoretic migration and chromatographic retention involving hydrophobic, hydrogen bonding, electrostatic as well as the Schiff base formation with OPA in the cavity of the templated polymer.
PATENT
CN 101372504
WO 2010119450
IN 2009CH00794
CN 103102395
CN 103467574
CN 105131079
CN 104761619
Desmopressin acetate is a structural analogue of natural arginine vasopressin, which is the result of two changes in the chemical structure of natural hormones. The structure is as follows:
M $ a-Tyr-Phe-Gln-Asn-C such as -Pro-D-Arg-GIy-N
Desmopressin acetate has a good hemostatic effect and does not produce side effects of pressurization. Mainly used to treat central diabetes insipidus, hemophilia and therapeutic control of bleeding and preoperative bleeding prevention. Good results and small side effects.
In the existing synthetic method of desmopressin acetate, liquid phase synthesis to produce more waste, the reaction time is long, each coupling an amino acid need to be purified, post-processing cumbersome, low yield, is not conducive to Industrial production.
Solid phase synthesis method, Chinese Patent CN 101372505, CN103992389 using Sieber Amide Resin or Rink Amide AM Resin one by one coupling to obtain linear peptide resin, and then solid-phase oxidation resin, cleavage and purification of desmopressin acetate. Chinese Patent CN103102395, CN102863513 Using Sieber Amide Resin or Rink AM Resin, linear peptide resin was obtained by coupling one by one, and liquid desulfurization was obtained after lysis to obtain desmopressin.
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8765152 | Pharmaceutical or neutraceutical formulation | 2010-02-25 | 2014-07-01 |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| US005726287 | Title not available | ||||
| US005990273 | Title not available | ||||
| US20060276626 | May 2, 2006 | Dec 7, 2006 | Avi Tovi | Methods for the production of peptide derivatives | |
| WO2004092202A1 | Apr 5, 2004 | Oct 28, 2004 | Novetide, Ltd. | Process for production of cyclic peptides |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102863513A * | Sep 12, 2012 | Jan 9, 2013 | 无锡市凯利药业有限公司 | Preparation method of desmopressin acetate |
Ramizol

1,3,5-Tris[(1E)-2′-(4′′-benzoic acid)vinyl]benzene] (Ramizol™)
TSB-007
| Allan James Mckinley, Thomas V Riley, Nigel Lengkeek, Scott Stewart, Ramiz Boulos | |
| Applicant | The University Of Western Australia |

1,3,5-Tris[(1E)-2′-(4′′-benzoic acid)vinyl]benzene] (Ramizol™) is a potent and non-toxic synthetic antimicrobial agent, and we now establish that it is also a potent inhibitor of reactive oxygen species (ROS) generation, with similar antioxidant activity to α-tocopherol (Vitamin E), which is a standard antioxidantdrug.
Ramizol, useful for treating bacterial infections such as Gram positive bacterial infection. Boulos & Cooper Pharmaceuticals could be seen to have ramizol in preclinical development for treating Clostridium difficile associated diseases. preparation of ramizol that was first described by the inventor Dr Ramiz Boulos, one of the company’s founding directors and CEO, in WO2011075766 as TSB-007 (claim 3, page 71) – said family of patenting having been originally assigned to the University of Western Australia and from whom Dr Boulos is reported to have acquired the rights to said intellectual property in late 2012 (ramizol having seemingly been previously being developed by the University with the name NAL-135B for treating Gram positive bacterial infections).

Professor Ramiz Boulos with a vial of Ramizol
A scientific paper released today in the Journal of Antibiotics presents the pre-clinical development of Ramizol®, a first generation drug belonging to a new class of styrylbenzene antibiotics with a novel mechanism of action.
The research was undertaken by Australian company Boulos & Cooper Pharmaceuticals in partnership with the University of South Australia, Flinders University, Eurofins Panlabs and Micromyx LLC. The study found that over 99.9% of the drug, administered orally, stays in the gastrointestinal tract where it can reach the bacteria in the colon at high enough concentrations to yield a therapeutic effect.
Chief Executive Officer of Boulos & Cooper Pharmaceuticals, Dr Ramiz Boulos, said “this new class of antibiotics has antioxidant properties and can be manufactured for a low cost; benefits that will be felt by the end-user”.
The new antibiotic has low frequency of resistance and shows promise as a monotherapy for the treatment of Clostridium difficile associated disease. Dr Boulos stated “we are very excited about these results given the unforgiving nature of Clostridium difficile infections”. He added “In a world where there are few treatment options, we are desperate for new antibiotics to fight intractable infections”.
The company expects to start Phase I clinical trials in 2017.
1,3,5-Tris[(1E)-20 -(400-benzoic acid)vinyl]benzene……………….recrystallised from THF/H2O and dried to give the triacid as a pale brown powder.
1 H NMR (500.1 MHz, d6-DMSO): d 7.49 (m, 6H, vinyl CH), 7.76 (d, J 8.5, 6H, ArH), 7.88 (s, 3H, core ArH), 7.98 (d, J 8.5, 6H, ArH);
13C NMR (125.8 MHz, d6-DMSO): d [ppm] 125.0, 126.5, 128.4, 129.7, 129.9, 130.50, 137.6, 141.3, 167.1;
IR (KBr): n [cm1 ] 3067, 3026, 1684 (nC¼O), 1604, 1566, 1420, 1384, 1312, 1286, 1179;
HR-EIþ-MS: C33H24O6 requires 516.1573 amu, found 516.1564;
EIþ-MS: MI ¼ C33H24O6; m/z: 516.1 (100%) ¼ MIþ, 472.1 (11.3%) ¼ [MI CO2] þ.
The Synthesis of Fluorescent DNA Intercalator Precursors through Efficient Multiple Heck Reactions
Nigel A. Lengkeek A , Ramiz A. Boulos A , Allan J. McKinley A , Thomas V. Riley C , Boris Martinac B and Scott G. Stewart A D
A M313, Chemistry, School of Biomedical, Biomolecular and Chemical Science, University of Western Australia, 35 Stirling Highway, Crawley, WA, 6009, Australia.
B Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, Sydney, NSW 2010, Australia.
C M502, Microbiology and Immunology, School of Biomedical, Biomolecular and Chemical Sciences, The University of Western Australia, 35 Stirling Hwy, Nedlands, WA 6009, Australia.
D Corresponding author. Email: sgs@cyllene.uwa.edu.au
Australian Journal of Chemistry 64(3) 316-323 http://dx.doi.org/10.1071/CH10374
PATENT
PATENT
WO-2017027933
Compounds with antimicrobial properties have attracted great interest in recent times as a result of an increase in the prevalence of infections caused by Gram-positive bacteria, resulting in serious or fatal diseases. Furthermore, the regular use of broad spectrum antibiotic formulas has led to the increased occurrence of bacterial strains resistant to some antimicrobial formulations.
Novel antimicrobial compounds have the potential to be highly effective against these types of treatment-resistant bacteria. The pathogens, having not previously been exposed to the antimicrobial formulation, may have little to no resistance to the treatment.
International patent application WO 2012/075766 describes a series of novel aryl compounds and their use as antimicrobials to treat bacterial infections or diseases. The chemical synthesis of a therapeutic drug has a direct effect on its cost, dosing regimens and popularity. Drugs with complicated or expensive chemical synthesis will find it challenging to reach the market, notwithstanding their efficacy. Further, syntheses amenable to application at commercial scales are highly advantageous. The development of an efficient and large-scale synthesis of a therapeutic drug is critical for its drug developmental pathway, and highly commercially advantageous.
1H NMR PREDICT


13C NMR PREDICT


REFERENCES
N. A. Lengkeek, R. A. Boulos, A. J. McKinley, T. V. Riley, B. Martinac and S. G. Stewart, Aust. J. Chem., 2011, 64, 316–323
http://pubs.rsc.org/en/content/articlehtml/2013/ra/c3ra40658j#cit11
/////////////Ramizol, PHASE 1, TSB-007
OC(=O)c4ccc(/C=C/c3cc(/C=C/c1ccc(cc1)C(=O)O)cc(/C=C/c2ccc(cc2)C(=O)O)c3)cc4
Oxymetazoline, оксиметазолин , أوكسيميتازولين , 羟甲唑啉
Oxymetazoline
1491-59-4 CAS NUMBER
- Molecular FormulaC16H24N2O
- Average mass260.375 Da
Oxymetazoline is a selective α1 adrenergic receptor agonist and α2 adrenergic receptorpartial agonist. It is a topical decongestant, used in the form of oxymetazoline hydrochloride. It was developed from xylometazoline at E. Merck Darmstadt by Fruhstorfer in 1961.[1]Oxymetazoline is generally available as a nasal spray.

Oxymetazoline HCl; CAS 2315-02-8
Oxymetazoline hydrochloride is a vasoconstrictor. Chemically it is 3-[(4,5-dihydro-1H-imidazol-2-yl)methyl]6-(1,1,-dimethylethyl)-2,4-dimethylphenolmono-hydrochloride. Its molecular weight is 296.8 for the hydrochloride salt and 260.4 for the free base. It is freely soluble in water and ethanol and has a partition coefficient of 0.1 in octanol/water. Its structural formula is:
 |
Medical uses
Oxymetazoline is available over-the-counter as a topical decongestant in the form of oxymetazoline hydrochloride in nasal sprays such as Afrin, Operil, Dristan, Dimetapp, oxyspray, Facimin, Nasivin, Nostrilla, Sudafed OM, Vicks Sinex, Zicam, SinuFrin, and Mucinex Full Force.[2]
Due to its vasoconstricting properties, oxymetazoline is also used to treat nose bleeds[3][4]and eye redness due to minor irritation (marketed as Visine L.R. in the form of eye drops).[5]
Company:
Allergan
Approval Status:
Approved January 2017
Specific Treatments:
facial erythema associated with rosacea
Therapeutic Areas
Dermatology
Find Related Trials for The Following Conditions
Rosacea
General Information
Rhofade (oxymetazoline hydrochloride) is an alpha1A adrenoceptor agonist. Oxymetazoline acts as a vasoconstrictor.
Rhofade is spccifically indicated for the topical treatment of persistent facial erythema associated with rosacea in adults.
Rhofade is supplied as a cream for topical administration. Apply a pea-sized amount of Rhofade cream, once daily in a thin layer to cover the entire face (forehead, nose, each cheek, and chin) avoiding the eyes and lips. Wash hands immediately after applying Rhofade cream.

Side effects and special considerations
Rebound congestion
It is recommended that oxymetazoline not be used for more than three days, as rebound congestion, or rhinitis medicamentosa, may occur.[6] Patients who continue to use oxymetazoline beyond this point may become dependent on the medication to relieve their chronic congestion.
Effects of benzalkonium chloride
Some studies have found that benzalkonium chloride, a common additive to oxymetazoline nasal sprays, may damage nasal epithelia and exacerbate rhinitis medicamentosa. However, the majority of studies find benzalkonium chloride to be a safe preservative.[7]
Use in pregnancy
The Food and Drug Administration places oxymetazoline in category C, indicating risk to the fetus cannot be ruled out. While it has been shown that a single dose does not significantly alter either maternal or fetal circulation,[8] this subject has not been studied extensively enough to draw reliable conclusions.
Overdose
If accidentally ingested, standard methods to remove unabsorbed drugs should be considered.[clarification needed] There is no specific antidote for oxymetazoline, although its pharmacological effects may be reversed by α adrenergic antagonists such as phentolamine. In the event of a possibly life-threatening overdose (such as a hypertensive crisis), benzodiazepines should be considered to decrease the likelihood of seizures and convulsions, as well as reduce anxiety and to lower blood pressure. In children, oxymetazoline may produce profound central nervous system depression due to stimulation of central α2receptors and imidazoline receptors, much like clonidine.
Pharmacology
Mechanism of action
Oxymetazoline is a sympathomimetic that selectively agonizes α1 and, partially, α2 adrenergic receptors.[9] Since vascular beds widely express α1 receptors, the action of oxymetazoline results in vasoconstriction. In addition, the local application of the drug also results in vasoconstriction due to its action on endothelial postsynaptic α2 receptors; systemic application of α2 agonists, in contrast, causes vasodilation because of centrally-mediated inhibition of sympathetic tone via presynaptic α2 receptors.[10] Vasoconstriction of vessels results in relief of nasal congestion in two ways: first, it increases the diameter of the airway lumen; second, it reduces fluid exudation from postcapillary venules.[11] It can reduce nasal airway resistance (NAR) up to 35.7% and nasal mucosal blood flow up to 50%.[12]
Pharmacokinetics
Imidazolines are sympathomimetic agents, with primary effects on α adrenergic receptors and little if any effect on β adrenergic receptors. Oxymetazoline is readily absorbed orally. Effects on α receptors from systemically absorbed oxymetazoline hydrochloride may persist for up to 7 hours after a single dose. The elimination half-life in humans is 5–8 hours. It is excreted unchanged both by the kidneys (30%) and in feces (10%).
History
The oxymetazoline brand Afrin was first sold as a prescription medication in 1966. After finding substantial early success as a prescription medication, it became available as an over-the-counter drug in 1975. Schering-Plough did not engage in heavy advertising until 1986.[13]From the late 1980s to mid 1990s, Afrin featured in many notable television advertisements. Some of these commercials showed men, women, and children using other brands of nasal sprays, and then standing upside down or hanging upside down from playground equipment to prevent their nasal spray from dripping out. This was juxtaposed with Afrin users having no problems.
Society and culture
Brand names
Brand names include Afrin, Dristan, Nasivin, Nezeril, Nostrilla, Logicin, Vicks Sinex, Visine L.R., Sudafed OM, Zicam, SinuFrin and Mucinex Sinus-Max.


References
- Jump up^ German Patent 1,117,588
- Jump up^ “Oxymetazoline: Drug Information Provided by Lexi-Comp: Merck Manual Professional”. Merck.com. Retrieved 2013-04-15.
- Jump up^ Katz, Robert I.; Hovagim, Alec R.; Finkelstein, Harvey S.; Grinberg, Yair; Boccio, Remigio V.; Poppers, Paul J. (1990). “A comparison of cocaine, lidocaine with epinephrine, and oxymetazoline for prevention of epistaxis on nasotracheal intubation”. Journal of Clinical Anesthesia. 2 (1): 16–20. doi:10.1016/0952-8180(90)90043-3. PMID 2310576.
- Jump up^ Krempl, G. A.; Noorily, A. D. (1995). “Use of oxymetazoline in the management of epistaxis”. The Annals of otology, rhinology, and laryngology. 104 (9 Pt 1): 704–6.PMID 7661519.
- Jump up^ “VISINE® Original Red Eye Drops | VISINE® products”. Visine.com. Retrieved2013-04-15.
- Jump up^ Ramey, J. T.; Bailen, E; Lockey, R. F. (2006). “Rhinitis medicamentosa”. Journal of investigational allergology & clinical immunology. 16 (3): 148–55. PMID 16784007.
- Jump up^ Marple, B; Roland, P; Benninger, M (2004). “Safety review of benzalkonium chloride used as a preservative in intranasal solutions: An overview of conflicting data and opinions”. Otolaryngology – Head and Neck Surgery. 130 (1): 131–41.doi:10.1016/j.otohns.2003.07.005. PMID 14726922.
- Jump up^ Rayburn, W. F.; Anderson, J. C.; Smith, C. V.; Appel, L. L.; Davis, S. A. (1990).“Uterine and fetal Doppler flow changes from a single dose of a long-acting intranasal decongestant”. Obstetrics and gynecology. 76 (2): 180–2. PMID 2196495.
- Jump up^ Westfall Thomas C, Westfall David P, “Chapter 6. Neurotransmission: The Autonomic and Somatic Motor Nervous Systems” (Chapter). Brunton LL, Lazo JS, Parker KL: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11e: http://www.accessmedicine.com/content.aspx?aID=954433.
- Jump up^ Biaggioni Italo, Robertson David, “Chapter 9. Adrenoceptor Agonists & Sympathomimetic Drugs” (Chapter). Katzung BG: Basic & Clinical Pharmacology, 11e: http://www.accessmedicine.com/content.aspx?aID=4520412.
- Jump up^ Widdicombe, John (1997). “Microvascular anatomy of the nose”. Allergy. 52 (40 Suppl): 7–11. doi:10.1111/j.1398-9995.1997.tb04877.x. PMID 9353554.
- Jump up^ Bende, M.; Löth, S. (2007). “Vascular effects of topical oxymetazoline on human nasal mucosa”. The Journal of Laryngology & Otology. 100 (3): 285–8.doi:10.1017/S0022215100099151. PMID 3950497.
- Jump up^ Dougherty, Phillip H. (20 October 1986). “Advertising; Afrin Goes After Users Of Nasal Decongestants”. The New York Times. The New York Times Company. Retrieved 2015-03-30.
 |
|
| Clinical data | |
|---|---|
| Trade names | Afrin, Ocuclear, Drixine |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Dependence liability |
Moderate |
| Routes of administration |
Intranasal |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Kidney (30%), fecal (10%) |
| Biological half-life | 5–6 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.014.618 |
| Chemical and physical data | |
| Formula | C16H24N2O |
| Molar mass | 260.375 g·mol−1 |
| 3D model (Jmol) | |
| Melting point | 301.5 °C (574.7 °F) |
/////////Oxymetazoline, оксиметазолин , أوكسيميتازولين , 羟甲唑啉 , Rhofade, oxymetazoline hydrochloride, alpha1A adrenoceptor agonist, vasoconstrictor
CC1=CC(=C(C(=C1CC2=NCCN2)C)O)C(C)(C)C.Cl
