
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TROXACITABINE троксацитабин , تروكساسيتابين , 曲沙他滨 ,
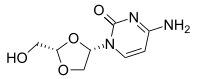
Troxacitabine
CAS 145918-75-8
- Molecular FormulaC8H11N3O4
- Average mass213.191 Da
Hmd-cytosine; NCGC00183848-01; Beta-L-Dioxolane-cytidine; 4-amino-1-[(2S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]pyrimidin-2-one; 2R(-)-cis-Hmd-cytosine, (-)-ODDC
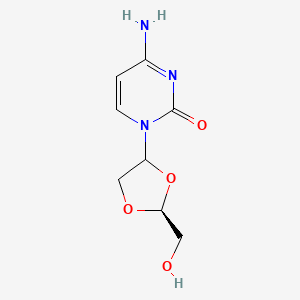
4-amino-1-[(2S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]pyrimidin-2-one
Troxacitabine (brand name Troxatyl) is a nucleoside analogue with anticancer activity. Its use is being studied in patients with refractory lymphoproliferative diseases.[1]
Troxacitabine (brand name Troxatyl) is a nucleoside analogue with anticancer activity. Its use is being studied in patients with refractory lymphoproliferative diseases.
| Investigated for use/treatment in leukemia (myeloid). |
PATENT
https://www.google.com/patents/WO1992018517A1?cl=en
WO 9218517
| Inventors | Yung-Chi Cheng, Chung K. Chu, Hea O. Kim, Kirupathevy Shanmuganathan |
| Applicant | Yale University, The University Of Georgia Research Foundation, Inc. |
SYNTHESIS
WO 2016030335

PATENT
WO 2016030335
PATENT
MACHINE TRANSLATED FROM CHINESE, BEAWARE OF FUNNY NAMES
PATENT
PATENT
CN 105503838
PAPER
In vitro optimization of non-small cell lung cancer activity with troxacitabine, L-1,3-dioxolane-cytidine, prodrugs
Journal of medicinal chemistry (2007), 50, (9), 2249-53.

l-1,3-Dioxolane-cytidine, a potent anticancer agent against leukemia, has limited efficacy against solid tumors, perhaps due to its hydrophilicity. Herein, a library of prodrugs were synthesized to optimize in vitro antitumor activity against non-small cell lung cancer. N4-Substituted fatty acid amide prodrugs of 10−16 carbon chain length demonstrated significantly improved antitumor activity over l-1,3-dioxolane-cytidine. These in vitro results suggest that the in vivo therapeutic efficacy of l-1,3-dioxolane-cytidine against solid tumors may be improved with prodrug strategies.
PAPER
- Kim, Hea O.; Schinazi, Raymond F.; Shanmuganathan, Kirupathevy; Jeong, Lak S.; Beach, J. Warren; Nampalli, Satyanarayana; Cannon, Deborah L.; Chu, Chung K.
- From Journal of Medicinal Chemistry (1993), 36(5), 519-28.
PAPER
- Jin, Haolun; Tse, Allan Tse; Evans, Colleen A.; Mansour, Tarek S.; Beels, Christopher M.; Ravenscroft, Paul; Humber, David C.; Jones, Martin F.; Payne, Jeremy J.; Ramsay, Michael V. J.
- From Tetrahedron: Asymmetry (1993), 4(2), 211-14
PAPER
- Belleau, Bernard R.; Evans, Colleen A.; Tse, H. L. Allan; Jin, Haolun; Dixit, Dilip M.; Mansour, Tarek S.
- From Tetrahedron Letters (1992), 33(46), 6949-52.
PAPER
http://pubs.acs.org/doi/pdf/10.1021/jm00089a007
J. Med. Chem. 1992,35,1987-1995 Asymmetric Synthesis of 1,3-Dioxolane-Pyrimidine Nucleosides and Their Anti-HIV Activity
 References
References
- Jump up^ Vose, Julie M.; Panwalkar, Amit; Belanger, Robert; Coiffier, Bertrand; Baccarani, Michele; Gregory, Stephanie A.; Facon, Thierry; Fanin, Renato; Caballero, Dolores; Ben-Yehuda, Dina; Giles, Francis (2007). “A phase II multicenter study of troxacitabine in relapsed or refractory lymphoproliferative neoplasms or multiple myeloma”. Leukemia & Lymphoma. 48 (1): 39–45. doi:10.1080/10428190600909578.
- Lee CK, Rowinsky EK, Li J, Giles F, Moore MJ, Hidalgo M, Capparelli E, Jolivet J, Baker SD: Population pharmacokinetics of troxacitabine, a novel dioxolane nucleoside analogue. Clin Cancer Res. 2006 Apr 1;12(7 Pt 1):2158-65. [PubMed:16609029 ]
- Quintas-Cardama A, Cortes J: Evaluation of the L-stereoisomeric nucleoside analog troxacitabine for the treatment of acute myeloid leukemia. Expert Opin Investig Drugs. 2007 Apr;16(4):547-57. [PubMed:17371201 ]
- Swords R, Giles F: Troxacitabine in acute leukemia. Hematology. 2007 Jun;12(3):219-27. [PubMed:17558697 ]
- Orsolic N, Giles FJ, Gourdeau H, Golemovic M, Beran M, Cortes J, Freireich EJ, Kantarjian H, Verstovsek S: Troxacitabine and imatinib mesylate combination therapy of chronic myeloid leukaemia: preclinical evaluation. Br J Haematol. 2004 Mar;124(6):727-38. [PubMed:15009060 ]
- Boivin AJ, Gourdeau H, Momparler RL: Action of troxacitabine on cells transduced with human cytidine deaminase cDNA. Cancer Invest. 2004;22(1):25-9. [PubMed:15069761 ]
- Kim TE, Park SY, Hsu CH, Dutschman GE, Cheng YC: Synergistic antitumor activity of troxacitabine and camptothecin in selected human cancer cell lines. Mol Pharmacol. 2004 Aug;66(2):285-92. [PubMed:15266019 ]
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2013011392 | METHOD FOR ASSESSING THE ABILITY OF A PATIENT TO RESPOND TO OR BE SAFELY TREATED BY A NUCLEOSIDE ANALOG BASED-CHEMOTHERAPY | 2010-11-19 | 2013-01-10 |
| US7927613 | Pharmaceutical co-crystal compositions | 2003-09-11 | 2011-04-19 |
| US7790905 | Pharmaceutical propylene glycol solvate compositions | 2003-12-29 | 2010-09-07 |
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C8H11N3O4 |
| Molar mass | 213.19 g/mol |
| 3D model (Jmol) | |
//////////////TROXACITABINE, троксацитабин , تروكساسيتابين , 曲沙他滨 , Hmd-cytosineM, NCGC00183848-01, Beta-L-Dioxolane-cytidine, 2R(-)-cis-Hmd-cytosine, (-)-ODDC
Astellas Pharma Inc. new Glucokinase Activator, ASP ? for Type 2 Diabetes
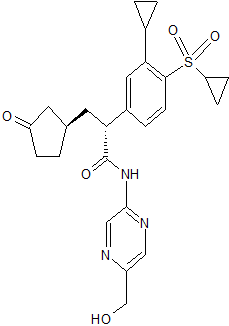
ASP ?
(2R)-2-(4-cyclopropanesulfonyl-3-cyclopropylphenyl)-N-[5-(hydroxymethyl)pyrazin-2-yl]-3-[(R)-3-oxocyclopentyl]propanamide
- Molecular Weight, 483.58
- [α]D20 −128.7 (c 1.00, MeOH);
- 1H NMR (DMSO-d6, 400 MHz) δ 11.07 (s, 1H), 9.20 (d, J = 1.4 Hz, 1H), 8.41 (d, J = 1.4 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.41 (dd, J = 8.2, 1.8 Hz, 1H), 7.15 (d, J = 1.8 Hz, 1H), 5.52 (t, J = 5.7 Hz, 1H), 4.56 (d, J = 6.0 Hz, 2H), 4.04 (t, J = 7.6 Hz, 1H), 3.03–2.97 (m, 1H), 2.79 (tt, J = 8.4, 5.1 Hz, 1H), 2.25–1.81 (m, 8H), 1.53–1.47 (m, 1H), 1.17–1.12 (m, 2H), 1.08–1.02 (m, 4H), 0.89–0.84 (m, 2H);
- 13C NMR (DMSO-d6, 101 MHz) δ 218.5, 171.8, 152.1, 147.3, 145.7, 143.2, 140.3, 138.2, 134.8, 129.0, 125.3, 125.1, 62.5, 49.9, 44.4, 38.4, 38.2, 34.8, 32.1, 29.1, 12.4, 10.8, 10.7, 5.8;
- FTIR (ATR, cm–1) 3544, 3257, 1727, 1692, 1546, 1507, 1363, 1285, 1149, 719;
- HRMS (ESI) m/z [M + Na]+ calcd for C25H29N3O5S 506.1726, found 506.1747.
- Anal. Calcd for C25H29N3O5S: C, 62.09; H, 6.04; N, 8.69. Found: C, 61.79; H, 6.19; N, 8.62.
![]()
| Inventors | Masahiko Hayakawa, Yoshiyuki Kido, Takahiro Nigawara, Mitsuaki Okumura, Akira Kanai, Keisuke Maki, Nobuaki Amino |
| Applicant | Astellas Pharma Inc. |
Synthesis

contd…………………………..

PATENT
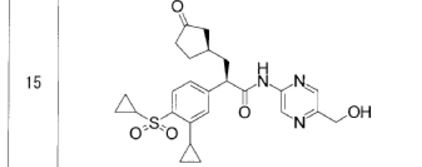
- PAPER
A Practical and Scalable Synthesis of a Glucokinase Activator via Diastereomeric Resolution and Palladium-Catalyzed C–N Coupling Reaction
 , Yasuhiro Morinaga†, Makoto Kasai†, Takao Hashimoto†, Yuji Takahama†, Atsushi Ohigashi†, Satoshi Yonishi‡, and Motohiro Akazome§
, Yasuhiro Morinaga†, Makoto Kasai†, Takao Hashimoto†, Yuji Takahama†, Atsushi Ohigashi†, Satoshi Yonishi‡, and Motohiro Akazome§
Here we describe the research and development of a process for the practical synthesis of glucokinase activator (R)-1 as a potential drug for treating type-2 diabetes. The key intermediate, chiral α-arylpropionic acid (R)-2, was synthesized in high diastereomeric excess through the diasteromeric resolution of 7 without the need for a chiral resolving agent. The counterpart 2-aminopyrazine derivative 3 was synthesized using a palladium-catalyzed C–N coupling reaction. This efficient process was demonstrated at the pilot scale and yielded 19.0 kg of (R)-1. Moreover, an epimerization process to obtain (R)-7 from the undesired (S)-7 was developed.
Hayakawa, M.; Kido, Y.; Nigawara, T.; Okumura, M.; Kanai, A.; Maki, K.; Amino, N. PCT Int. Appl. WO/2009/091014 A1 20090723,2009.
https://www.astellas.com/en/ir/library/pdf/3q2017_rd_en.pdf
///////////1174229-89-0, ASTELLAS, Glucokinase Activator, TYPE 2 DIABETES, PRECLINICAL, ASP ?, WO 2009091014, Masahiko Hayakawa, Yoshiyuki Kido, Takahiro Nigawara, Mitsuaki Okumura, Akira Kanai, Keisuke Maki, Nobuaki Amino, WO2009091014,
O=C(Nc1cnc(cn1)CO)[C@H](C[C@@H]2CC(=O)CC2)c3ccc(c(c3)C4CC4)S(=O)(=O)C5CC5
FDA approves first treatment Noctiva (Desmopressin acetate) nasal spray for frequent urination at night due to overproduction of urine

Desmopressin acetate
March 3, 2017
The U.S. Food and Drug Administration today approved Noctiva (desmopressin acetate) nasal spray for adults who awaken at least two times per night to urinate due to a condition known as nocturnal polyuria (overproduction of urine during the night). Noctiva is the first FDA-approved treatment for this condition.
“Today’s approval provides adults who overproduce urine at night with the first FDA-approved therapeutic option to help reduce the number of times a night they wake up to urinate,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Division of Bone, Reproductive, and Urologic Products in the FDA’s Center for Drug Evaluation and Research. “It is important to know that Noctiva is not approved for all causes of night-time urination, so patients should discuss their symptoms with their health care provider who can determine the underlying cause of the night-time urination and whether Noctiva is right for them.”
Nocturia (wakening at night to urinate) is a symptom that can be caused by a wide variety of conditions, such as congestive heart failure, poorly controlled diabetes mellitus, medications, or diseases of the bladder or prostate. Before considering Noctiva, health care providers should evaluate each patient for possible causes for the nocturia, and optimize the treatment of underlying conditions that may be contributing to the night-time urination. Because Noctiva is approved only for adults with nocturia caused by nocturnal polyuria, health care providers should confirm overproduction of urine at night with a 24-hour urine collection, if one has not been obtained previously. Health care providers should also be mindful of underlying conditions that can cause nocturia, but that make treatment with Noctiva unsafe, such as excessive drinking of fluids or symptomatic congestive heart failure.
Noctiva is taken daily, approximately 30 minutes before going to bed. It works by increasing the absorption of water through the kidneys, which leads to less urine production.
Noctiva’s efficacy was established in two 12-week, randomized, placebo-controlled trials in 1,045 patients 50 years of age and older with nocturia due to nocturnal polyuria. Although these trials showed a small reduction in the average number of night-time urinations with Noctiva compared to placebo, more patients treated with Noctiva were able to at least halve their number of night-time urinations, and patients treated with Noctiva had more nights with one or fewer night-time urinations.
Noctiva is being approved with a boxed warning and a Medication Guide because it can cause low sodium levels in the blood (hyponatremia). Severe hyponatremia can be life-threatening if it is not promptly diagnosed and treated, leading to seizures, coma, respiratory arrest or death. Health care providers should make sure the patient’s sodium level is normal before starting Noctiva, and should check sodium levels within one week and approximately one month after starting treatment and periodically thereafter. The lower Noctiva dose is recommended as the starting dose for those who may be at risk for hyponatremia, such as the elderly. Noctiva should not be used in patients at increased risk of severe hyponatremia, such as those with excessive fluid intake, those who have illnesses that can cause fluid or electrolyte imbalances, certain patients with kidney damage, and in those using certain medicines, known as loop diuretics or glucocorticoids.
Noctiva should also not be used in patients with symptomatic congestive heart failure or uncontrolled hypertension because fluid retention can worsen these underlying conditions. Use of Noctiva should be discontinued temporarily in patients with certain nasal conditions such as colds or allergies until those conditions have resolved.
Noctiva is also not recommended for the treatment of nocturia in pregnant women. Nocturia is usually related to normal changes in pregnancy that do not require treatment with Noctiva. Noctiva should not be used in children.
The most common side effects of Noctiva in clinical trials included nasal discomfort, cold symptoms (nasopharyngitis), nasal congestion, sneezing, high or increased blood pressure, back pain, nose bleeds, bronchitis and dizziness.
Although there are other FDA-approved medications that also contain desmopressin, none of those medications are approved to treat nocturia.
Noctiva is marketed by Milford, Pennsylvania-based Renaissance Lakewood, LLC for Serenity Pharmaceuticals, LLC.
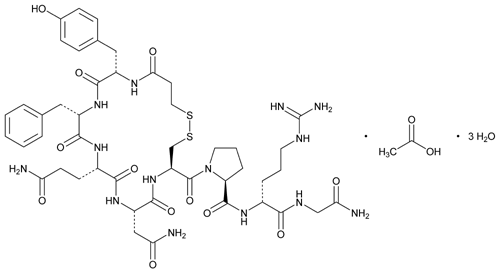
C48H68N14O14S2 C48H68N14O14S2·xH2O
(anhydrous) ![]()
![]()
![]()
![]()
![]() 1129.27
1129.27![]()
![]()
![]() [62288-83-9].
[62288-83-9].
1-(3-Mercaptopropionic acid)-8-D-arginine-vasopressin monoacetate (salt).

oxopentan-2-yl]-1-[4-(2-amino-2-oxoethyl)-7-(3-amino-3-oxopropyl)-10-benzyl-13-[(4-hydroxyphenyl)methyl]-3,6,9,12,15-pentaoxo-18,19-dithia-2,5,8,11,14-pentazacycloicosane-1-carbonyl]pyrrolidine-2-carboxamide;
Synonyms: 3-MERCAPTOPROPIONYL-TYR-PHE-GLN-ASN-CYS-PRO-D-ARG-GLY-NH2 ACETATE SALT;DDAVP ACETATE;[DEAMINO-CYS1,D-ARG8]-VASOPRESSIN ACETATE SALT;DESMOPRESSIN MONOACETATE;DESMORESSIN ACETATE;Mpr-Tyr-Phe-Gln-Asn-Cys-Pro-D-Arg-Gly-NH2(S-S:1-5);DESMOPRESSIN ACETATE;DESMOPRESSIN ACETATE SALT;
The Molecular Weight of Desmopressin Acetate(62288-83-9): 1129.27




Analytica Chimica Acta (2006), 572, (2), 197-204
Abstract
A monolithic column was prepared using l-phenylalanine as template and a covalent approach through the formation of Schiff base with o-phthalaldehyde (OPA). OPA, allylmercaptan, l-phenylalanine, and triethylamine were stirred at first, then methacrylic acid, 2-vinylpyridine, ethyleneglycol dimethacrylate, α,α-azobisisobutyronitrile, and 1-propanol were added to the reaction mixture. The resulting material was introduced into a capillary column. Following thermal polymerization, the template was then extracted with a mixture of HCl and methanol. The column was employed for the capillary electrochromatographic separation of oligopeptides. A capillary column of 75 (50) cm × 75 μm ID with a mobile phase of phosphate buffer (pH 7.0, 40 mM)/methanol (5%, v/v), an applied voltage of +15 kV, and detection at 214 nm, could baseline separate angiotensin I, angiotensin II, [Sar1, Thr8] angiotensin, oxytocin, vasopressin, tocinoic acid, β-casomorphin bovine, β-casomorphin human, and FMRF amide within 20 min. The separation behavior of the templated polymer was also compared with that of the non-templated polymer. As a result, it can be concluded that the electrochromatographic separation of this set of peptides was mediated by a combination of electrophoretic migration and chromatographic retention involving hydrophobic, hydrogen bonding, electrostatic as well as the Schiff base formation with OPA in the cavity of the templated polymer.
PATENT
CN 101372504
WO 2010119450
IN 2009CH00794
CN 103102395
CN 103467574
CN 105131079
CN 104761619
Desmopressin acetate is a structural analogue of natural arginine vasopressin, which is the result of two changes in the chemical structure of natural hormones. The structure is as follows:
M $ a-Tyr-Phe-Gln-Asn-C such as -Pro-D-Arg-GIy-N
Desmopressin acetate has a good hemostatic effect and does not produce side effects of pressurization. Mainly used to treat central diabetes insipidus, hemophilia and therapeutic control of bleeding and preoperative bleeding prevention. Good results and small side effects.
In the existing synthetic method of desmopressin acetate, liquid phase synthesis to produce more waste, the reaction time is long, each coupling an amino acid need to be purified, post-processing cumbersome, low yield, is not conducive to Industrial production.
Solid phase synthesis method, Chinese Patent CN 101372505, CN103992389 using Sieber Amide Resin or Rink Amide AM Resin one by one coupling to obtain linear peptide resin, and then solid-phase oxidation resin, cleavage and purification of desmopressin acetate. Chinese Patent CN103102395, CN102863513 Using Sieber Amide Resin or Rink AM Resin, linear peptide resin was obtained by coupling one by one, and liquid desulfurization was obtained after lysis to obtain desmopressin.
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8765152 | Pharmaceutical or neutraceutical formulation | 2010-02-25 | 2014-07-01 |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| US005726287 | Title not available | ||||
| US005990273 | Title not available | ||||
| US20060276626 | May 2, 2006 | Dec 7, 2006 | Avi Tovi | Methods for the production of peptide derivatives | |
| WO2004092202A1 | Apr 5, 2004 | Oct 28, 2004 | Novetide, Ltd. | Process for production of cyclic peptides |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102863513A * | Sep 12, 2012 | Jan 9, 2013 | 无锡市凯利药业有限公司 | Preparation method of desmopressin acetate |
Ramizol

1,3,5-Tris[(1E)-2′-(4′′-benzoic acid)vinyl]benzene] (Ramizol™)
TSB-007
| Allan James Mckinley, Thomas V Riley, Nigel Lengkeek, Scott Stewart, Ramiz Boulos | |
| Applicant | The University Of Western Australia |

1,3,5-Tris[(1E)-2′-(4′′-benzoic acid)vinyl]benzene] (Ramizol™) is a potent and non-toxic synthetic antimicrobial agent, and we now establish that it is also a potent inhibitor of reactive oxygen species (ROS) generation, with similar antioxidant activity to α-tocopherol (Vitamin E), which is a standard antioxidantdrug.
Ramizol, useful for treating bacterial infections such as Gram positive bacterial infection. Boulos & Cooper Pharmaceuticals could be seen to have ramizol in preclinical development for treating Clostridium difficile associated diseases. preparation of ramizol that was first described by the inventor Dr Ramiz Boulos, one of the company’s founding directors and CEO, in WO2011075766 as TSB-007 (claim 3, page 71) – said family of patenting having been originally assigned to the University of Western Australia and from whom Dr Boulos is reported to have acquired the rights to said intellectual property in late 2012 (ramizol having seemingly been previously being developed by the University with the name NAL-135B for treating Gram positive bacterial infections).

Professor Ramiz Boulos with a vial of Ramizol
A scientific paper released today in the Journal of Antibiotics presents the pre-clinical development of Ramizol®, a first generation drug belonging to a new class of styrylbenzene antibiotics with a novel mechanism of action.
The research was undertaken by Australian company Boulos & Cooper Pharmaceuticals in partnership with the University of South Australia, Flinders University, Eurofins Panlabs and Micromyx LLC. The study found that over 99.9% of the drug, administered orally, stays in the gastrointestinal tract where it can reach the bacteria in the colon at high enough concentrations to yield a therapeutic effect.
Chief Executive Officer of Boulos & Cooper Pharmaceuticals, Dr Ramiz Boulos, said “this new class of antibiotics has antioxidant properties and can be manufactured for a low cost; benefits that will be felt by the end-user”.
The new antibiotic has low frequency of resistance and shows promise as a monotherapy for the treatment of Clostridium difficile associated disease. Dr Boulos stated “we are very excited about these results given the unforgiving nature of Clostridium difficile infections”. He added “In a world where there are few treatment options, we are desperate for new antibiotics to fight intractable infections”.
The company expects to start Phase I clinical trials in 2017.
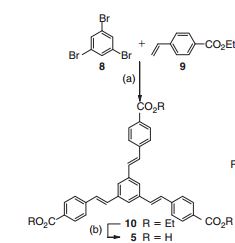
1,3,5-Tris[(1E)-20 -(400-benzoic acid)vinyl]benzene……………….recrystallised from THF/H2O and dried to give the triacid as a pale brown powder.
1 H NMR (500.1 MHz, d6-DMSO): d 7.49 (m, 6H, vinyl CH), 7.76 (d, J 8.5, 6H, ArH), 7.88 (s, 3H, core ArH), 7.98 (d, J 8.5, 6H, ArH);
13C NMR (125.8 MHz, d6-DMSO): d [ppm] 125.0, 126.5, 128.4, 129.7, 129.9, 130.50, 137.6, 141.3, 167.1;
IR (KBr): n [cm1 ] 3067, 3026, 1684 (nC¼O), 1604, 1566, 1420, 1384, 1312, 1286, 1179;
HR-EIþ-MS: C33H24O6 requires 516.1573 amu, found 516.1564;
EIþ-MS: MI ¼ C33H24O6; m/z: 516.1 (100%) ¼ MIþ, 472.1 (11.3%) ¼ [MI CO2] þ.
The Synthesis of Fluorescent DNA Intercalator Precursors through Efficient Multiple Heck Reactions
Nigel A. Lengkeek A , Ramiz A. Boulos A , Allan J. McKinley A , Thomas V. Riley C , Boris Martinac B and Scott G. Stewart A D
A M313, Chemistry, School of Biomedical, Biomolecular and Chemical Science, University of Western Australia, 35 Stirling Highway, Crawley, WA, 6009, Australia.
B Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool Street, Darlinghurst, Sydney, NSW 2010, Australia.
C M502, Microbiology and Immunology, School of Biomedical, Biomolecular and Chemical Sciences, The University of Western Australia, 35 Stirling Hwy, Nedlands, WA 6009, Australia.
D Corresponding author. Email: sgs@cyllene.uwa.edu.au
Australian Journal of Chemistry 64(3) 316-323 http://dx.doi.org/10.1071/CH10374
PATENT
PATENT
WO-2017027933
Compounds with antimicrobial properties have attracted great interest in recent times as a result of an increase in the prevalence of infections caused by Gram-positive bacteria, resulting in serious or fatal diseases. Furthermore, the regular use of broad spectrum antibiotic formulas has led to the increased occurrence of bacterial strains resistant to some antimicrobial formulations.
Novel antimicrobial compounds have the potential to be highly effective against these types of treatment-resistant bacteria. The pathogens, having not previously been exposed to the antimicrobial formulation, may have little to no resistance to the treatment.
International patent application WO 2012/075766 describes a series of novel aryl compounds and their use as antimicrobials to treat bacterial infections or diseases. The chemical synthesis of a therapeutic drug has a direct effect on its cost, dosing regimens and popularity. Drugs with complicated or expensive chemical synthesis will find it challenging to reach the market, notwithstanding their efficacy. Further, syntheses amenable to application at commercial scales are highly advantageous. The development of an efficient and large-scale synthesis of a therapeutic drug is critical for its drug developmental pathway, and highly commercially advantageous.
1H NMR PREDICT


13C NMR PREDICT


REFERENCES
N. A. Lengkeek, R. A. Boulos, A. J. McKinley, T. V. Riley, B. Martinac and S. G. Stewart, Aust. J. Chem., 2011, 64, 316–323
http://pubs.rsc.org/en/content/articlehtml/2013/ra/c3ra40658j#cit11
/////////////Ramizol, PHASE 1, TSB-007
OC(=O)c4ccc(/C=C/c3cc(/C=C/c1ccc(cc1)C(=O)O)cc(/C=C/c2ccc(cc2)C(=O)O)c3)cc4
Oxymetazoline, оксиметазолин , أوكسيميتازولين , 羟甲唑啉
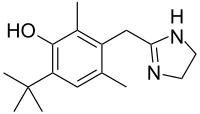
Oxymetazoline
1491-59-4 CAS NUMBER
- Molecular FormulaC16H24N2O
- Average mass260.375 Da
Oxymetazoline is a selective α1 adrenergic receptor agonist and α2 adrenergic receptorpartial agonist. It is a topical decongestant, used in the form of oxymetazoline hydrochloride. It was developed from xylometazoline at E. Merck Darmstadt by Fruhstorfer in 1961.[1]Oxymetazoline is generally available as a nasal spray.

Oxymetazoline HCl; CAS 2315-02-8
Oxymetazoline hydrochloride is a vasoconstrictor. Chemically it is 3-[(4,5-dihydro-1H-imidazol-2-yl)methyl]6-(1,1,-dimethylethyl)-2,4-dimethylphenolmono-hydrochloride. Its molecular weight is 296.8 for the hydrochloride salt and 260.4 for the free base. It is freely soluble in water and ethanol and has a partition coefficient of 0.1 in octanol/water. Its structural formula is:
 |
Medical uses
Oxymetazoline is available over-the-counter as a topical decongestant in the form of oxymetazoline hydrochloride in nasal sprays such as Afrin, Operil, Dristan, Dimetapp, oxyspray, Facimin, Nasivin, Nostrilla, Sudafed OM, Vicks Sinex, Zicam, SinuFrin, and Mucinex Full Force.[2]
Due to its vasoconstricting properties, oxymetazoline is also used to treat nose bleeds[3][4]and eye redness due to minor irritation (marketed as Visine L.R. in the form of eye drops).[5]
Company:
Allergan
Approval Status:
Approved January 2017
Specific Treatments:
facial erythema associated with rosacea
Therapeutic Areas
Dermatology
Find Related Trials for The Following Conditions
Rosacea
General Information
Rhofade (oxymetazoline hydrochloride) is an alpha1A adrenoceptor agonist. Oxymetazoline acts as a vasoconstrictor.
Rhofade is spccifically indicated for the topical treatment of persistent facial erythema associated with rosacea in adults.
Rhofade is supplied as a cream for topical administration. Apply a pea-sized amount of Rhofade cream, once daily in a thin layer to cover the entire face (forehead, nose, each cheek, and chin) avoiding the eyes and lips. Wash hands immediately after applying Rhofade cream.

Side effects and special considerations
Rebound congestion
It is recommended that oxymetazoline not be used for more than three days, as rebound congestion, or rhinitis medicamentosa, may occur.[6] Patients who continue to use oxymetazoline beyond this point may become dependent on the medication to relieve their chronic congestion.
Effects of benzalkonium chloride
Some studies have found that benzalkonium chloride, a common additive to oxymetazoline nasal sprays, may damage nasal epithelia and exacerbate rhinitis medicamentosa. However, the majority of studies find benzalkonium chloride to be a safe preservative.[7]
Use in pregnancy
The Food and Drug Administration places oxymetazoline in category C, indicating risk to the fetus cannot be ruled out. While it has been shown that a single dose does not significantly alter either maternal or fetal circulation,[8] this subject has not been studied extensively enough to draw reliable conclusions.
Overdose
If accidentally ingested, standard methods to remove unabsorbed drugs should be considered.[clarification needed] There is no specific antidote for oxymetazoline, although its pharmacological effects may be reversed by α adrenergic antagonists such as phentolamine. In the event of a possibly life-threatening overdose (such as a hypertensive crisis), benzodiazepines should be considered to decrease the likelihood of seizures and convulsions, as well as reduce anxiety and to lower blood pressure. In children, oxymetazoline may produce profound central nervous system depression due to stimulation of central α2receptors and imidazoline receptors, much like clonidine.
Pharmacology
Mechanism of action
Oxymetazoline is a sympathomimetic that selectively agonizes α1 and, partially, α2 adrenergic receptors.[9] Since vascular beds widely express α1 receptors, the action of oxymetazoline results in vasoconstriction. In addition, the local application of the drug also results in vasoconstriction due to its action on endothelial postsynaptic α2 receptors; systemic application of α2 agonists, in contrast, causes vasodilation because of centrally-mediated inhibition of sympathetic tone via presynaptic α2 receptors.[10] Vasoconstriction of vessels results in relief of nasal congestion in two ways: first, it increases the diameter of the airway lumen; second, it reduces fluid exudation from postcapillary venules.[11] It can reduce nasal airway resistance (NAR) up to 35.7% and nasal mucosal blood flow up to 50%.[12]
Pharmacokinetics
Imidazolines are sympathomimetic agents, with primary effects on α adrenergic receptors and little if any effect on β adrenergic receptors. Oxymetazoline is readily absorbed orally. Effects on α receptors from systemically absorbed oxymetazoline hydrochloride may persist for up to 7 hours after a single dose. The elimination half-life in humans is 5–8 hours. It is excreted unchanged both by the kidneys (30%) and in feces (10%).
History
The oxymetazoline brand Afrin was first sold as a prescription medication in 1966. After finding substantial early success as a prescription medication, it became available as an over-the-counter drug in 1975. Schering-Plough did not engage in heavy advertising until 1986.[13]From the late 1980s to mid 1990s, Afrin featured in many notable television advertisements. Some of these commercials showed men, women, and children using other brands of nasal sprays, and then standing upside down or hanging upside down from playground equipment to prevent their nasal spray from dripping out. This was juxtaposed with Afrin users having no problems.
Society and culture
Brand names
Brand names include Afrin, Dristan, Nasivin, Nezeril, Nostrilla, Logicin, Vicks Sinex, Visine L.R., Sudafed OM, Zicam, SinuFrin and Mucinex Sinus-Max.


References
- Jump up^ German Patent 1,117,588
- Jump up^ “Oxymetazoline: Drug Information Provided by Lexi-Comp: Merck Manual Professional”. Merck.com. Retrieved 2013-04-15.
- Jump up^ Katz, Robert I.; Hovagim, Alec R.; Finkelstein, Harvey S.; Grinberg, Yair; Boccio, Remigio V.; Poppers, Paul J. (1990). “A comparison of cocaine, lidocaine with epinephrine, and oxymetazoline for prevention of epistaxis on nasotracheal intubation”. Journal of Clinical Anesthesia. 2 (1): 16–20. doi:10.1016/0952-8180(90)90043-3. PMID 2310576.
- Jump up^ Krempl, G. A.; Noorily, A. D. (1995). “Use of oxymetazoline in the management of epistaxis”. The Annals of otology, rhinology, and laryngology. 104 (9 Pt 1): 704–6.PMID 7661519.
- Jump up^ “VISINE® Original Red Eye Drops | VISINE® products”. Visine.com. Retrieved2013-04-15.
- Jump up^ Ramey, J. T.; Bailen, E; Lockey, R. F. (2006). “Rhinitis medicamentosa”. Journal of investigational allergology & clinical immunology. 16 (3): 148–55. PMID 16784007.
- Jump up^ Marple, B; Roland, P; Benninger, M (2004). “Safety review of benzalkonium chloride used as a preservative in intranasal solutions: An overview of conflicting data and opinions”. Otolaryngology – Head and Neck Surgery. 130 (1): 131–41.doi:10.1016/j.otohns.2003.07.005. PMID 14726922.
- Jump up^ Rayburn, W. F.; Anderson, J. C.; Smith, C. V.; Appel, L. L.; Davis, S. A. (1990).“Uterine and fetal Doppler flow changes from a single dose of a long-acting intranasal decongestant”. Obstetrics and gynecology. 76 (2): 180–2. PMID 2196495.
- Jump up^ Westfall Thomas C, Westfall David P, “Chapter 6. Neurotransmission: The Autonomic and Somatic Motor Nervous Systems” (Chapter). Brunton LL, Lazo JS, Parker KL: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11e: http://www.accessmedicine.com/content.aspx?aID=954433.
- Jump up^ Biaggioni Italo, Robertson David, “Chapter 9. Adrenoceptor Agonists & Sympathomimetic Drugs” (Chapter). Katzung BG: Basic & Clinical Pharmacology, 11e: http://www.accessmedicine.com/content.aspx?aID=4520412.
- Jump up^ Widdicombe, John (1997). “Microvascular anatomy of the nose”. Allergy. 52 (40 Suppl): 7–11. doi:10.1111/j.1398-9995.1997.tb04877.x. PMID 9353554.
- Jump up^ Bende, M.; Löth, S. (2007). “Vascular effects of topical oxymetazoline on human nasal mucosa”. The Journal of Laryngology & Otology. 100 (3): 285–8.doi:10.1017/S0022215100099151. PMID 3950497.
- Jump up^ Dougherty, Phillip H. (20 October 1986). “Advertising; Afrin Goes After Users Of Nasal Decongestants”. The New York Times. The New York Times Company. Retrieved 2015-03-30.
 |
|
| Clinical data | |
|---|---|
| Trade names | Afrin, Ocuclear, Drixine |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Dependence liability |
Moderate |
| Routes of administration |
Intranasal |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Kidney (30%), fecal (10%) |
| Biological half-life | 5–6 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.014.618 |
| Chemical and physical data | |
| Formula | C16H24N2O |
| Molar mass | 260.375 g·mol−1 |
| 3D model (Jmol) | |
| Melting point | 301.5 °C (574.7 °F) |
/////////Oxymetazoline, оксиметазолин , أوكسيميتازولين , 羟甲唑啉 , Rhofade, oxymetazoline hydrochloride, alpha1A adrenoceptor agonist, vasoconstrictor
CC1=CC(=C(C(=C1CC2=NCCN2)C)O)C(C)(C)C.Cl
FDA approves Odactra for house dust mite allergies

March 1, 2017
Release
The U.S. Food and Drug Administration today approved Odactra, the first allergen extract to be administered under the tongue (sublingually) to treat house dust mite (HDM)-induced nasal inflammation (allergic rhinitis), with or without eye inflammation (conjunctivitis), in people 18 through 65 years of age.
“House dust mite allergic disease can negatively impact a person’s quality of life,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research. “The approval of Odactra provides patients an alternative treatment to allergy shots to help address their symptoms.”
House dust mite allergies are a reaction to tiny bugs that are commonly found in house dust. Dust mites, close relatives of ticks and spiders, are too small to be seen without a microscope. They are found in bedding, upholstered furniture and carpeting. Individuals with house dust mite allergies may experience a cough, runny nose, nasal itching, nasal congestion, sneezing, and itchy and watery eyes.
Odactra exposes patients to house dust mite allergens, gradually training the immune system in order to reduce the frequency and severity of nasal and eye allergy symptoms. It is a once-daily tablet, taken year round, that rapidly dissolves after it is placed under the tongue. The first dose is taken under the supervision of a health care professional with experience in the diagnosis and treatment of allergic diseases. The patient is to be observed for at least 30 minutes for potential adverse reactions. Provided the first dose is well tolerated, patients can then take Odactra at home. It can take about eight to 14 weeks of daily dosing after initiation of Odactra for the patient to begin to experience a noticeable benefit.
The safety and efficacy of Odactra was evaluated in studies conducted in the United States, Canada and Europe, involving approximately 2,500 people. Some participants received Odactra, while others received a placebo pill. Participants reported their symptoms and the need to use symptom-relieving allergy medications. During treatment, participants taking Odactra experienced a 16 to 18 percent reduction in symptoms and the need for additional medications compared to those who received a placebo.
The most commonly reported adverse reactions were nausea, itching in the ears and mouth, and swelling of the lips and tongue. The prescribing information includes a boxed warning that severe allergic reactions, some of which can be life-threatening, can occur. As with other FDA-approved allergen extracts administered sublingually, patients receiving Odactra should be prescribed auto-injectable epinephrine. Odactra also has a Medication Guide for distribution to the patient.
Odactra is manufactured for Merck, Sharp & Dohme Corp., (a subsidiary of Merck and Co., Inc., Whitehouse Station, N.J.) by Catalent Pharma Solutions Limited, United Kingdom.
(sublingually) to treat house dust mite (HDM)-induced nasal inflammation (allergic rhinitis), with or without eye inflammation (conjunctivitis), in people 18 through 65 years of age
/////////////Odactra, Merck, Sharp & Dohme Corp, Catalent Pharma Solutions Limited, United Kingdom, FDA 2017, approves, house dust mite allergies
Award for me, 100 Most Impactful Health care Leaders, Global listing

At award function for my award “100 Most Impactful Health care Leaders Global listing”, conferred on me at Taj lands end, Mumbai, India on 14 Feb 2014 by World Health Wellness congress and awards
FDA approves Xermelo (telotristat ethyl) for carcinoid syndrome diarrhea

Telotristat ethyl
Molecular Formula, C27-H26-Cl-F3-N6-O3,
Molecular Weight, 574.9884,
RN: 1033805-22-9
UNII: 8G388563M
LX 1032
(2S)-2-Amino-3-[4-[2-amino-6-[[(1R)-1-[4-chloro-2-(3-methylpyrazol-1-yl)phenyl]-2,2,2-trifluoroethyl]oxy]pyrimidin-4-yl]phenyl]propionic acid ethyl ester
Ethyl-4-(2-amino-6-{(1R)-1-[4-chlor-2-(3-methyl-1H-pyrazol-1-yl)phenyl]-2,2,2-trifluorethoxy}-4-pyrimidinyl)-L-phenylalaninat

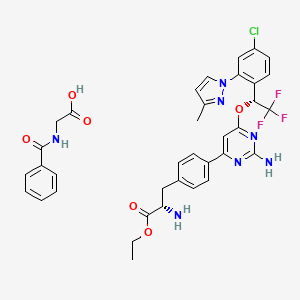
CAS: 1137608-69-5 (etiprate), LX 1606
Chemical Formula: C36H35ClF3N7O6
Molecular Weight: 754.16
- LX 1032 hippurate
- LX 1606



Carcinoid syndrome is a cluster of symptoms sometimes seen in people with carcinoid tumors. These tumors are rare, and often slow-growing. Most carcinoid tumors are found in the gastrointestinal tract. Carcinoid syndrome occurs in less than 10 percent of patients with carcinoid tumors, usually after the tumor has spread to the liver. The tumors in these patients release excess amounts of the hormone serotonin, resulting in diarrhea. Complications of uncontrolled diarrhea include weight loss, malnutrition, dehydration, and electrolyte imbalance.
“Today’s approval will provide patients whose carcinoid syndrome diarrhea is not adequately controlled with another treatment option,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research.
Xermelo, in a regimen with SSA therapy, is approved in tablet form to be taken orally three times daily with food. Xermelo inhibits the production of serotonin by carcinoid tumors and reduces the frequency of carcinoid syndrome diarrhea.
The safety and efficacy of Xermelo were established in a 12-week, double-blind, placebo-controlled trial in 90 adult participants with well-differentiated metastatic neuroendocrine tumors and carcinoid syndrome diarrhea. These patients were having between four to 12 daily bowel movements despite the use of SSA at a stable dose for at least three months. Participants remained on their SSA treatment, and were randomized to add placebo or treatment with Xermelo three times daily. Those receiving Xermelo added on to their SSA treatment experienced a greater reduction in average bowel movement frequency than those on SSA and placebo. Specifically, 33 percent of participants randomized to add Xermelo on to SSA experienced an average reduction of two bowel movements per day compared to 4 percent of patients randomized to add placebo on to SSA.
The most common side effects of Xermelo include nausea, headache, increased levels of the liver enzyme gamma-glutamyl transferase, depression, accumulation of fluid causing swelling (peripheral edema), flatulence, decreased appetite and fever. Xermelo may cause constipation, and the risk of developing constipation may be increased in patients whose bowel movement frequency is less than four bowel movements per day. Patients treated with a higher than recommended dosage of Xermelo developed severe constipation in clinical trials. One patient required hospitalization and two other patients developed complications of either intestinal perforation or intestinal obstruction. Patients should be monitored for severe constipation. If a patient experiences severe constipation or severe, persistent or worsening abdominal pain, they should discontinue Xermelo and contact their healthcare provider.
The FDA granted this application fast track designation and priority review. The drug also received orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
Xermelo is manufactured by Woodlands, Texas-based Lexicon Pharmaceuticals, Inc.
SYNTHESIS…….WO 2011100285

5.67. Synthesis of (S)-2-Amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yll- phenyll-2,2,2-trifluoro-ethoxy)-pyrimidin-4-yl)-phenyll-propionic acid ethyl ester

The title compound was prepared stepwise, as described below:
Step 1: Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-5°C (ice water bath) over 10 minutes. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHC03 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -70°C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added.
Tetrabutylamonium fluoride (1M, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 10 hours at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCI (100 ml) was added. The mixture was kept at 45-50°C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHC03 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). !H-NMR (300 MHz, CDC ): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (1M in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2-methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and 1-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2 hours. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 hours, and further stirred at -32 °C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4 hours. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol 28.2 g (70 %; 99-100 % e.e.). !H-NMR (400 MHz, CDCIs) δ (ppm) 5.48 (m, 1H), 7.40 (d, 1H), 7.61 (d, 2H).
Step 3: Synthesis of R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll-2.2.2-trifluoro-ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65 g, 54.06 mmol), 3-methylpyrazole (5.33 g, 65 mmol), Cul (2.06 g, 10.8 mmol), 2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 130°C (oil bath temperature) for 12 hours. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). i-H-NMR (400 MHz, CDC ): δ (ppm) 2.30(s, 3H), 4.90(m, 1H), 6.20(s, 1H), 6.84(d, 1H), 7.20(s, 1H), 7.30(d, 1H), 7.50(d, 1H).
Step 4: Synthesis of (S)-2-Amino-3- 4-(2-amino-6-fR-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll^^^-trifluoro-ethoxyl-pyrimidin^-yll-phenvD-propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert-butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and CS2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 100°C (oil bath temperature) for 12-24 hours. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 60°C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCI (200 ml). The mixture was heated at 35-40 °C for 12 hours. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a 1L beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58 °C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-60°C) (2 x 200 ml) and dried to give crude (S)-2-amino-3-[4-(2-amino-6-[R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}^ propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %.
To anhydrous ethanol (400 ml) was added SOC (22 ml, 306 mmol) dropwise at 0-5°C.
Crude acid (26.8 ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-45°C for 6-12 hours. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2C03 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}-propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. !H-NMR (400 MHz, CDsOD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
SYNTHESIS OF INTERMEDIATE
WO 2009048864

https://google.com/patents/WO2009048864A1?cl=en
6.15. Preparation of 6SV3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2- (fert-butoxycarbonylamino)propanoic Acid Using the Lithium Salt of (S)-2-(te^-butoxycarbonylamino)-3-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)propanoic Acid

During preparation of compound 7, the isolation of the free acid can be optionally omitted. Thus, an aqueous solution of the lithium salt of compound 7 in 100 ml water, prepared from 5.0 g of Boc-Tyr-OMe (4, 17 mmol), was mixed 2-amino-4,6- dichloropyrimidine (3.3 g, 1.2 eq), potassium bicarbonate (5.0 g, 3 eq), bis(triphenylphosphine)palladium(II) dichloride (60 mg, 0.5 mol%), and 100 ml ethanol. The resulting mixture was heated at 700C for 5 hours. Additional 2-amino-4,6- dichloropyrimidine (1.1 g, 0.4 eq) was added and heating was continued at 7O0C for an additional 2 hours. HPLC analysis showed about 94% conversion. Upon cooling and filtration, the filtrate was analyzed by HPLC against a standard solution of compound 8. The assay indicated 3.9 g compound 8 was contained in the solution (59% yield from compound 4).
6.16. Alternative Procedure for Preparation of (S)-3-(4-f2-Amino-6- chloropyrimidin-4-yl)phenyl)-2-(fe^-butoxycarbonylamino)propanoic Acid Using Potassium Carbonate as Base

The boronic acid compound 11 (Ryscor Science, Inc., North Carolina, 1.0 g, 4.8 mmol) and potassium carbonate (1.32 g, 2 eq) were mixed in aqueous ethanol (15 ml ethanol and 8 ml water). Di-ter£-butyldicarbonate (1.25 g, 1.2 eq) was added in one portion. After 30 minutes agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6- dichloropyrimidine (1.18 g, 1.5 eq) and the catalyst bis(triphenylphosphine)palladium(II) dichloride (34 mg, 1 mol%) were added and the resulting mixture was heated at 65-700C for 3 hours. HPLC analysis showed complete consumption of compound 12. After concentration and filtration, HPLC analysis of the resulting aqueous solution against a standard solution of compound 8 showed 1.26 g compound 8 (67% yield).
6.17. Alternative procedure for preparation of (5)-3-(4-(2-Amino-6-

The boronic acid compound 11 (10 g, 48 mmol) and potassium bicarbonate (14.4 g, 3 eq) were mixed in aqueous ethanol (250 ml ethanol and 50 ml water). Oi-tert- butyldicarbonate (12.5 g, 1.2 eq) was added in one portion. HPLC analysis indicated that the reaction was not complete after overnight stirring at room temperature. Potassium carbonate (6.6 g, 1.0 eq) and additional di-te/t-butyldicarbonate (3.1 g, 0.3 eq) were added. After 2.5 hours agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6-dichloropyrimidine (11.8 g, 1.5 eq) and the catalyst bis(triphenylphosphine)-palladium(II) dichloride (0.34 g, 1 mol%” were added and the resulting mixture was heated at 75-8O0C for 2 hours. HPLC analysis showed complete consumption of compound 12. The mixture was concentrated under reduced pressure and filtered. The filtrate was washed with ethyl acetate (200 ml) and diluted with 3 : 1 THF/MTBE (120 ml). This mixture was acidified to pH about 2.4 by 6 N hydrochloric acid. The organic layer was washed with brine and concentrated under reduced pressure. The residue was precipitated in isopropanol, filtered, and dried at 500C under vacuum to give compound 8 as an off-white solid (9.0 g, 48% yield). Purity: 92.9% by HPLC analysis. Concentration of the mother liquor yielded and additional 2.2 g off-white powder (12% yield). Purity: 93.6% by HPLC analysis
PATENT
https://www.google.com/patents/WO2013059146A1?cl=en
This invention is directed to solid pharmaceutical dosage forms in which an active pharmaceutical ingredient (API) is (S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-l-(4-chloro-2-(3- methyl-lH-pyrazol-l-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate
(telotristat):

or a pharmaceutically acceptable salt thereof. The compound, its salts and crystalline forms can be obtained by methods known in the art. See, e.g., U.S. patent no. 7,709,493.
PATENT
http://www.google.co.in/patents/WO2008073933A2?cl=en
6.19. Synthesis of (S)-2-Amino-3-r4-q-amino-6-{R-l-r4-chloro-2-(3-methyl- Pyrazol-l-yl)-phenyll-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyll- propionic acid ethyl ester

The title compound was prepared stepwise, as described below: Step 1 : Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-50C (ice water bath) over 10 min. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHCO3 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -700C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added. Tetrabutylamonium fluoride (IM, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 1Oh at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2- trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCl (100 ml) was added. The mixture was kept at 45-500C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHCO3 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford 1- (2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (IM in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2- methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2h. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 h, and further stirred at -32°C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4h. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro- phenyl)-2,2,2-trifiuoro-ethanol 28.2 g (70 %; 99-100 % e.e.). 1H-NMR (400 MHz, CDCl3) δ (ppm) 5.48 (m, IH), 7.40 (d, IH), 7.61 (d, 2H). Step 3: Synthesis of R-l-r4-chloro-2-(3-methyl-pyrazol-l-vπ-phenyl1-2.2.2-trifluoro- ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65g, 54.06 mmol), 3- methylpyrazole (5.33 g, 65 mmol), CuI (2.06 g, 10.8 mmol), K2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 1300C (oil bath temperature) for 12 h. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-I- [4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). 1H-NMR (400 MHz, CDCl3): δ (ppm) 2.30(s, 3H), 4.90(m, IH), 6.20(s, IH), 6.84(d, IH), 7.20(s, IH), 7.30(d, IH), 7.50(d, IH).
Step 4: Synthesis of (S)-2-Amino-3- r4-(2-amino-6- (R-I- r4-chloro-2-(3-methyl- pyrazol- 1 -ylVphenyl~|-2,2.,2-trifluoro-ethoxy| -pyrimidin-4-yl)-phenyU -propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert- butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and Cs2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 1000C (oil bath temperature) for 12-24 h. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 600C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCl (200 ml). The mixture was heated at 35-400C for 12h. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a IL beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58°C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-600C) (2 x 200 ml) and dried to give crude (S)-2- amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro- ethoxy}-pyrimidin-4-yl)-phenyl} -propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %. To anhydrous ethanol (400 ml) was added SOCl2 (22 ml, 306 mmol) dropwise at 0-
5°C. Crude acid (26.8 g ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-450C for 6-12h. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2CO3 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2- amino-6- (R- 1 -[4-chloro-2-(3-methyl-pyrazol- 1 -yl)-phenyl]-2,2,2-trifluoro-ethoxy} – pyrimidin-4-yl)-phenyl} -propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. 1H-NMR (400 MHz, CD3OD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
PATENT
WO 2011056916
https://www.google.com/patents/WO2011056916A1?cl=en
PATENT
WO 2010065333
CLIP,……..PL CHECK ERROR



REFERENCES
Kulke, M.H.; Hoersch, D.; Caplin, M.E.; et al.
Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome
J Clin Oncol 2017, 35(1): 14
| WO2010056992A1 * | Nov 13, 2009 | May 20, 2010 | The Trustees Of Columbia University In The City Of New York | Methods of preventing and treating low bone mass diseases |
| US7709493 | May 20, 2009 | May 4, 2010 | Lexicon Pharmaceuticals, Inc. | 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds and methods of their use |
| US20090088447 * | Sep 25, 2008 | Apr 2, 2009 | Bednarz Mark S | Solid forms of (s)-ethyl 2-amino-3-(4-(2-amino-6-((r)-1-(4-chloro-2-(3-methyl-1h-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)-pyrimidin-4-yl)phenyl)propanoate and methods of their use |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9199994 | Sep 5, 2014 | Dec 1, 2015 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9512122 | Sep 1, 2015 | Dec 6, 2016 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
///////////telotristat ethyl, fast track designation,priority review,orphan drug designation, Xermelo , Woodlands, Texas-based, Lexicon Pharmaceuticals, Inc, fda 2017, LX 1606, LX 1032
O=C(OCC)[C@@H](N)Cc1ccc(cc1)c2cc(nc(N)n2)O[C@H](c3ccc(Cl)cc3n4ccc(C)n4)C(F)(F)F
O=C(OCC)[C@@H](N)CC1=CC=C(C2=NC(N)=NC(O[C@H](C3=CC=C(Cl)C=C3N4N=C(C)C=C4)C(F)(F)F)=C2)C=C1.O=C(O)CNC(C5=CC=CC=C5)=O
