
Home » 2016 (Page 51)
Yearly Archives: 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New patent, WO 2016001885, Dr Reddy’s Laboratories Ltd, Eliglustat hemitartarate
![]()
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Telangana, India Hyderabad 500034 (IN)
VELAGA, Dharma Jagannadha Rao; (IN).
PEDDY, Vishweshwar; (IN).
VYALA, Sunitha; (IN)
(WO2016001885) AMORPHOUS FORM OF ELIGLUSTAT HEMITARTARATE
Chemically Eliglustat is named N-[(1 R,2R)-2-(2,3-dihydro-1 ,4-benzodioxin-6-yl)-2-hydroxy-1 -(1 -pyrrolidinylmethyl)ethyl]-Octanamide(2R!3R)-2,3-dihydroxybutanedioate and the hemitartarate salt of eliglustat has the structural formula as shown in Formula I.

Formula I
Eliglustat hemitartrate (Genz-1 12638), currently under development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase, and is currently under development by Genzyme.
U.S. patent No. 7,196,205 discloses a process for the preparation of Eliglustat or a pharmaceutically acceptable salt thereof.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 discloses process for preparation of Eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of Eliglustat, (ii) a hemitartrate salt of Eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It has been disclosed earlier that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailablity patterns compared to crystalline forms [Konne T., Chem pharm Bull., 38, 2003(1990)]. For some therapeutic indications one bioavailabihty pattern may be favoured over another. An amorphous form of Cefuroxime axetil is a good example for exhibiting higher bioavailability than the crystalline form.
Solid amorphous dispersions of drugs are known generally to improve the stability and solubility of drug products. However, such dispersions are generally unstable over time. Amorphous dispersions of drugs tend to convert to crystalline forms over time, which can lead to improper dosing due to differences of the solubility of crystalline drug material compared to amorphous drug material. The present invention, however, provides stable amorphous dispersions of eliglustat hemitartrate. Moreover, the present invention provides solid dispersions of eliglustat hemitartrate which may be reproduced easily and is amenable for processing into a dosage form.
There remains a need to provide solid state forms of eliglustat hemitartarate which are advantageous in a cost effective and environment friendly manner.

EXAMPLES
Example 1 : Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 14 mL of dichloromethane at 26°C and stirred for 15 min. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 45°C. After distillation the solid was dried under vacuum at 45°C.
Example 2: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 70 mL of ethanol and stirred for 15 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid was dried under vacuum at 48°C.
Example 3: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 20 mL of methanol and stirred for 15 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid was dried under vacuum at 48°C.
Example 4: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and PVP-K30.
500mg of eliglustat hemitartarate and 500mg of PVP-K30 was dissolved in 20 mL of methanol and stirred for 10 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid is dried under vacuum at 48°C.
Example 5: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and hydroxy propyl cellulose.
500mg of eliglustat hemitartarate and 500 mg of hydroxy propyl cellulose was dissolved in 30 ml of methanol and stirred for 10 min at 25° – 30°C. The solution is distilled under reduced pressure at 49°C. After distillation the solid is dried under vacuum at 49°C.
Example 6: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and hydroxy propyl methyl cellulose.
500mg of eliglustat hemitartarate and 500 mg of hydroxy propyl methyl cellulose was dissolved in 30 mL of methanol and stirred for 10 min at 25° – 30°C. The solution is distilled under reduced pressure at 48°C. After distillation the solid is dried under vacuum at 48°C.
Example 7 Preparation of amorphous form of eliglustat hemitartarate.
3g of eliglustat hemitartarate was dissolved in 75 mL of methanol and stirred at 25°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected for spray drying at inlet temperature of 70°C and outlet temperature of 42°C to afford the title compound.
Example 8: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 30 mL of isopropanol and stirred at 56°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected to complete distillation under reduced pressure and drying at about 56°C to afford the title compound.
Example 9: Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 40 mL of ethyl acetate and stirred at about 63°C. Then methanol (5 mL) is added at the same temperature to obtain clear solution which was filtered to remove the undissolved particles. Then additional quantity of methanol (5mL) is added to the filtrate and the filtrate was again filtered to remove particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 10: Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 40 mL of acetone and stirred at about 55°C followed by addition of methanol (15 mL). The mixture is stirred at 55°C for clear solution and filtered to remove the undissolved particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 11 : Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 25 mL of isopropyl alcohol and 25 mL of ethanol. The mixture was stirred at about 58°C for dissolution and filtered to remove the undissolved particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 12 Preparation of amorphous form of eliglustat hemitartarate.
5g of eliglustat hemitartarate was provided in 300 mL of isopropyl alcohol and stirred at about 59°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected for spray drying at inlet temperature of 65°C and outlet temperature of 37°C to afford the title compound according to Fig. 6
Example 13: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Copovidone
500mg of eliglustat hemitartarate and 500mg of Copovidone were dissolved in 30 mL of methanol and stirred for clear solution, then filtered to make it particle free. The solvent from the filtrate was evaporated under reduced pressure at 45°C and obtained solid was subjected to drying at 45°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction.
Example 14: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Copovidone
2g of eliglustat hemitartarate and 2g of Copovidone were dissolved in 100 mL of methanol and stirred for clear solution, then filtered to make it particle free. The solvent from the filtrate was subjected to spray drying at inlet temperature of 70 at 45°C and outlet temperature of 42°C to afford the title compound. The resulting dispersion was found to be amorphous by X-ray powder diffraction.
Example 15: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate
2g of eliglustat hemitartarate was charged in 40 mL of methanol followed by addition of 2g of PVP K-30. The mixture was stirred for clear solution and filtered to make it particle free, the bed was washed with 20 mL of methanol. Then 2g of Syloid is added to the filtrate and filtrate is subjected to distillation under reduced pressure at about 57°C and obtained solid was subjected to drying at about 57°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 7a. The said dispersion is kept at 25°C under 40% relative humidity for 24 hours and PXRD was recorded and found to be amorphous according to Fig 7b.
Example 16: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate
2g of eliglustat hemitartarate was charged in 40 mL of methanol followed by addition of 2g of Copovidone. The mixture was stirred for clear solution and filtered to make it particle free, the bed was washed with 20 mL of methanol. Then 2g of Syloid is added to the filtrate and filtrate is subjected to distillation under reduced pressure at about 57°C and obtained solid was subjected to drying at about 57°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 8a. The said dispersion is kept at 25°C under 40% relative humidity for 24 hours and PXRD was recorded and found to be amorphous according to Fig. 8b and D90 of the resultant solid is about 437 microns.
Example 17: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Syloid
1 g of eliglustat hemitartarate was dissolved in 25 ml_ of methanol and filtered to make it particle free. Then 1 g of Syloid 244 FPNF was added to the filtrate and solvent from the filtrate was evaporated under reduced pressure at 56°C and obtained solid was subjected to drying at 56°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 9 and D90 of the resultant solid is about 4 microns.
Example 18: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Syloid
1 g of eliglustat hemitartarate was dissolved in 25 ml_ of methanol and filtered to make it particle free. Then 500mg of Syloid 244 FPNF was added to the filtrate and solvent from the filtrate was evaporated under reduced pressure at 56°C and obtained solid was subjected to drying at 56°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction.

PATENT
(WO2015059679) IMPROVED PROCESS FOR THE PREPARATION OF ELIGLUSTAT
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills Hyderabad 500034 (IN)
JAVED, Iqbal; (IN).
DAHANUKAR, Vilas Hareshwar; (IN).
ORUGANTI, Srinivas; (IN).
KANDAGATLA, Bhaskar; (IN)
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence: (i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate, (ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone, (iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine, (iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It is also an objective of the present application to provide an improved process for the preparation of eliglustat and a pharmaceutically acceptable salt thereof which is high yielding, simple, cost effective, environment friendly and commercially viable by avoiding repeated cumbersome and lengthy purification steps. It is a further objective of the present application to provide crystalline forms of eliglustat free base and its salts.
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %

G.V. Prasad, chairman, Dr Reddy’s Laboratories
//////////////New patent, WO 2016001885, Dr Reddy’s Laboratories Ltd, Eliglustat hemitartarate, WO 2015059679
Continuous ruthenium-catalyzed methoxycarbonylation with supercritical carbon dioxide
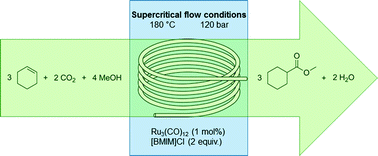
DOI: 10.1039/C5CY01883H, Paper
The methoxycarbonylation of cyclohexene with carbon dioxide over a ruthenium catalyst was realized in a micro flow system under supercritical conditions.

Department of Chemical Engineering and Chemistry
ir. S.C. (Stefan) Stouten –

P.O. Box 513
5600 MB EINDHOVEN
- Department:
- Department of Chemical Engineering and Chemistry
- Section:
- Micro Flow Chemistry and Process Technology
- Positioncategory:
- doctoral candidate (PhD) (PhD Stud.)
- Position:
- doctoral candidate
- Room:
- STW 0.
- Email:
- s.stouten@tue.nl

Volker Hessel
prof.dr. V. (Volker) Hessel

P.O. Box 513
5600 MB EINDHOVEN
- Department:
- Department of Chemical Engineering and Chemistry
- Section:
- Micro Flow Chemistry and Process Technology
- Positioncategory:
- Professor (HGL)
- Position:
- Full Professor
- Room:
- STW 1.45
- Tel:
- +31 40-247 2973
- Tel (internal):
- 2973
- Email:
- v.hessel@tue.nl
////////Continuous, ruthenium-catalyzed, methoxycarbonylation, supercritical carbon dioxide, flow reactor
NEW PATENT, WO2016001844, SUN PHARMACEUTICALS, AFATINIB DIMALEATE

AMORPHOUS FORM OF AFATINIB DIMALEATE
SUN PHARMACEUTICAL INDUSTRIES LIMITED
VERMA, Shyam Sunder; (IN).
SINGH, Shravan Kumar; (IN).
SINGH, Kaptan; (IN).
PRASAD, Mohan; (IN)
Afatinib dimaleate is a tyrosine kinase inhibitor, chemically designated as 2-butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(35)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2£)-, (2Z)-2-butenedioate (1:2) having the structure depicted by Formula I.

Formula I
U.S. Patent Nos. RE43,431 and 6,251,912 provide processes for the preparation of afatinib dimaleate.
U.S. Patent No. 8,426,586 and PCT Publication Nos. WO 2012/121764 and WO
2013/052157 provide processes for the preparation of crystalline forms of afatinib and their salts.
Example: Preparation of an amorphous form of afatinib dimaleate
In a round bottom flask, a mixture of afatinib (3 g) and ethyl acetate (30 mL) was heated to about 65°C to obtain a turbid solution. In another round bottom flask, a mixture of maleic acid (1.6 g) and ethyl acetate (30 mL) was heated to about 50°C to obtain a clear solution. The maleic acid solution was added to the afatinib solution, and then the reaction mixture was heated at about 75°C to about 80°C. The reaction mixture was stirred at about 75°C to about 80°C for about 1 hour. The reaction mixture was cooled to about
20°C to obtain a sticky material. The sticky material was scratched with a spatula, and then the reaction mixture was further stirred at about 20°C to about 25°C for about 1 hour. The material obtained was filtered, and then washed with ethyl acetate (20 mL). The solid obtained was dried under vacuum at about 45°C to about 50°C for about 15 hours to obtain the amorphous form of afatinib dimaleate.
Yield: 2.5 g (56%)


Sun Pharma chief Dilip Shanghvi
///////
NEW PATENT, TICAGRELOR, DR. REDDY’S LABORATORIES LIMITED, WO 2016001851
![]()
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara hills, Hyderabad, Telangana Hyderabad 500034 (IN)
DAHANUKAR, Vilas; (IN).
ELATI, Ravi Ram Chandrasekhar; (IN).
ORUGANTI, Srinivas; (IN).
RAPOLU, Rajesh Kumar; (IN).
KURELLA, Sreenivasulu; (IN)
![]()
The drug compound having the adopted name “ticagrelor” has chemical names: [1 S-(1 α,2α,3β(1 S*,2R*),5p)]-3-[7-[2-(3,4-difluorophenyl-cyclopropyl] amino]-5-(propylthio)-3H-1 ,2,3-triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)-cyclopentane-1 ,2-diol; or (1 S,2S,3fl,5S)-3-[7-{[(1 fl,2S)-2-(3,4-difluorophenyl) cyclopropyl]amino}-5-(propylthio)-3H 1 ,2,3]-triazolo[4,5-c/|pyrimidin-3-yl]-5-(2-hydroxyethoxy)cyclope ed by Formula I.

Formula I
Ticagrelor is the active ingredient in the commercially available BRILINTA® tablets for oral administration.
Ticagrelor and related compounds are disclosed in International Patent Application Publication Nos. WO 00/34283 and WO 99/05143 as pharmaceutically active Ρ2τ (which are now usually referred to as P2Y12) receptor antagonists. Such antagonists can be used, inter alia, as inhibitors of platelet activation, aggregation, or degranulation. International Patent Application Publication Nos. WO 01 /92263 and WO 2010/030224 A1 , WO 2012085665 A2, WO 2012138981 A2 and WO 2013037942 A1 disclose processes for preparing ticagrelor.
The processes for the preparation of traizolo [4,5-d] pyrimidine derivatives preferably Ticagrelor and related compounds, described in the above mentioned prior art suffer from disadvantages since the processes involve tedious and cumbersome procedures such as lengthy and multiple synthesis steps, reactions under pressure and high temperature, longer reaction times, tedious work up procedures and multiple crystallizations or isolation steps, column chromatographic purifications and thus resulting in low overall yields of the product. Ticagrelor obtained by the processes described in the prior art does not have satisfactory purity and unacceptable amounts of impurities are formed along with Ticagrelor at various stages of the processes that are difficult to purify and thus get carried forward in the subsequent steps thus affecting the purity of final compound. Thus, there remains a need to prepare compounds of Formula I of high purity and in good yield while overcoming the drawbacks presented by the previously described processes.

Formula V Formula V”
In a preferred embodiment, present application provide compounds of Formula IV with specific groups i.e. compounds of Formula IV and Formula IV”,

Formula IV Formula IV”
In a preferred embodiment, present application provides a compound of Formula II with specific groups i.e. compounds of Formula ΙΓ and Formula II”,

Formula ΙΓ Formula II”
In a preferred embodiment, present application provides a compound of Formula l la with specific grou

Formula lla

Formula VII’ Formula VII”
In a preferred embodiment, present application provides compounds of Formula Vila with specific groups i.e. compounds of Formula Vila’ and Formula “,

Formula Vila’ Formula Vila”
G.V. Prasad, chairman, Dr Reddy’s Laboratories
EXAMPLES
EXAMPLE 1 : Preparation of 2-bromo-N,N-diphenylacetamide (FORMULA Vile).
A flask is charged with Ν,Ν-diphenyl amine (25 g) and dichloromethane (350 mL) under nitrogen atmosphere. The reaction mixture is cooled to 0°C followed by addition of solution of triethyl amine (20.7 mL) and bromoacetyl chloride (38.72 mL) in dichloromethane (181 mL). The mixture is cooled to room temperature and then stirred for about 16 hours. The completion of the reaction is monitored by TLC. The reaction mixture is diluted with dichloromethane (250 mL) and then washed with 0.5N aqueous hydrochloric acid solution (3×150 mL), brine (100 mL). The organic layer is separated and subjected to distillation under vacuum at 45°C. The obtained compound is recrystallized from hexane (250 mL) and methanol (100 mL) to afford the title compound.
EXAMPLE 2: Preparation of benzyl ((3aS,4R,6S,6aR)-6-(2-(diphenylamino)-2-oxoethoxy)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (FORMULA Vila).
A flask is charged with sodium hydride (2.85 mL, 60% dispersion in oil) and dimethyl formamide (10 mL) under nitrogen atmosphere. The reaction mixture is then cooled to -30°C followed by addition of a solution of benzyl
((3aS,4R,6S,6aR)-6-hydroxy-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (20 g) in dimethyl formamide (40 mL). The mixture is stirred at -30°C for about 45 minutes, then a solution of 2-bromo-N,N-diphenylacetamide (22.65 g) in dimethyl formamide (60 mL) is added at the same temperature. The reaction mixture is allowed to attain room temperature and stirred at the same for 3 hours and completion of the reaction is monitored by TLC. The reaction mixture is quenched with ice-cold water (200 mL) and extracted with ethyl acetate (3×150 mL). The organic layer is combined and washed with water (3×100 mL), brine (100 mL) and then organic layer is then subjected to complete distillation under vacuum at 45°C. The crude so obtained is treated with MTBE (150 mL) and stirred at room temperature for overnight followed by filtration of obtained solid to afford the title compound.
EXAMPLE 3: Preparation of 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyl tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula VII)
A flask is charged with benzyl ((3aS,4R,6S,6aR)-6-(2-(diphenylamino)-2-oxoethoxy)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (15 g), ethanol (300 mL), 10% Pd/C (1.5 g) and ammonium formate (5.49 g). The reaction mixture is stirred at 80°C for 8 hours and completion of the reaction is monitored by TLC. Then reaction mixture is cooled to room temperature and filtered through celite bed, bed is washed with ethyl acetate (100 mL). The filtrate is subjected to complete distillation under vacuum at 45°C. Then ethanol (120 mL), L-tartaric acid (4.88 g) is added to the crude compound and mixture is stirred for 4 hours at room temperature. To the mixture, MTBE (300 mL) is added at the same temperature. The solvent is distilled under vacuum at 35°C to afford the gummy solid. Then MTBE (100 mL) is added to the gummy solid and mixture is stirred for 10-12 hours. The solid obtained is filtered and washed with MTBE (50 mL). The solid obtained is dissolved in water and sodium bicarbonate solution (200 mL) is added, desired compound is extracted in ethyl acetate (100 mL). The solvent is subjected to distillation (upto 40%) followed by addition of hexane (150 mL) and ethyl acetate (20 mL). The mixture is stirred at -10°C, then solid is recovered followed by drying under vacuum at 40°C. The crude compound is purified by column chromatography using methanol and dichloromethane (5:95) to afford the title compound.
EXAMPLE 4: Preparation of 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2- (propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula V)
A flask is charged with 4,6-dichloro-2-(propylthio)pyrimidin-5-amine (6.5 g),
2- (((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (10.39 g), sodium bicarbonate (9.13 g) and water. The mixture is stirred at 95-100°C for 15-20 hours till completion of the reaction (as monitored by TLC). Then water (20 mL) and ethyl acetate (25 mL) are added at room temperature. The layers are separated and aqueous layer is extracted with ethyl acetate (20 mL). The organic layers are combined and washed with brine solution (2×25 mL). The organic layer is subjected to complete distillation under vacuum at 40-45°C. The obtained crude compound is purified by column chromatography using ethyl acetate and hexane (30:70) to afford the title compound.
EXAMPLE 5: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula IV)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (6 g), acetic acid (30 mL) and water (6 mL). The mixture is cooled to 0 to -5°C followed by addition of addition of sodium nitrite solution (768 mg in 6 mL of water). The mixture is stirred for 1 hour at the same temperature and then mixture is allowed to attain room temperature, and further stirred for 1 hour. The completion of the reaction is monitored by TLC and then toluene (60 mL) is added. The layers are separated, organic layer is washed with saturated solution of potassium carbonate and subsequently organic layer is dried with sodium sulphate and used for next reaction.
EXAMPLE 6: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin- 3- yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula II)
A flask is charged with (1 R,2S)-2-(3,4-difluorophenyl)cyclopropan-1 -amine mandelate (3.25 g), diisopropylethyl amine (6.1 mL), toluene (60 mL) and stirred for 30 minutes at room temperature. Then slowly, toluene layer containing 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (60 mL) is added over a period of 10 minutes. The reaction mixture is stirred at room temperature for overnight and completion of the reaction is monitored by TLC. The reaction mixture is diluted with water (60 mL), layers are separated and aqueous layer is extracted with toluene (2×30 mL). The combined organic layers are washed with brine (60 mL) and then subjected to complete distillation under vacuum at 45°C to afford the crude compound. The crude compound is purified by column chromatography using ethyl acetate and hexane (80:20).
EXAMPLE 7: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol (Formula lib)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (3 g), THF (90 mL) and mixture is cooled to 0°C. Then portion wise, Lithium aluminium hydride (940 mg) is added over a period of 10 minutes and mixture is stirred at 0°C for 1 hour. The reaction mixture is then stirred at room temperature for 5 hours and progress of the reaction is monitored by TLC. Then mixture is cooled to 0-5°C and quenched with ice cold water (100 mL) and then diluted with ethyl acetate (30 mL). The layers are separated and organic layer after drying is used for next step.
EXAMPLE 8: Preparation of Ticagrelor (Formula I)
A flask is charged with organic layer containing 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3] triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol (1 10 mL) and 2% hydrochloric acid solution (75 mL). The reaction mixture is stirred at room temperature for 48 hours and progress of the reaction is monitored by TLC. Then the reaction mixture is diluted with ethyl acetate (50 mL), layers are separated. The organic layer is sequentially washed with water (50 mL), brine solution (50 mL) followed by complete distillation under vacuum at 45°C. The crude compound is dissolved in ethyl acetate (12 mL) and then hexane (50 mL) is added. The mixture is stirred for 2 hours followed by isolation of solid by filtration. The obtained solid is dissolved in ethyl acetate (12 mL) and treated with charcoal followed by filtration. The filtrate is subjected to complete distillation and obtained solid is purified by column chromatography using ethyl acetate:hexane (1 :1 ) and methanohdichloromethane (5:95) to afford the title compound.
EXAMPLE 9: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol (Formula lib)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (100 mg), THF (3 mL) and cooled to 0-5°C followed by addition of Vitride (0.04 mL) at 0-5°C over a period of 5 minutes. The mixture is stirred at same temperature for 1 hour and progress of the reaction is monitored by TLC. Additional amount of Vitride (0.13 mL) is added to the mixture and stirred for additional 6 hours. After completion of reaction, reaction mixture is cooled to 0-5°C and quenched with saturated sodium potassium tartrate solution (10 mL) and extracted with ethyl acetate (20 mL). The organic layer is subjected to complete distillation under reduced pressure and obtained material is purified by column chromatography using ethyl acetate: hexane (1 :1 ) and methanohdichloromethane (5:95) to afford the title compound.
EXAMPLE 10: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)acetaldehyde (Formula lib’)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (100 mg), THF (5 mL) and mixture is cooled to 0°C. Then portion wise, Lithium aluminium hydride (15 mg) is added over a period of 1 0
minutes and mixture is stirred at 0°C for 1 hour. The progress of the reaction is monitored by TLC. After completion of the reaction, mixture is quenched with ice cold water (5 mL) and diluted with ethyl acetate (10 mL). The layers are separated and organic layer after drying is subjected to complete distillation followed by purification using preparative TLC using 40% ethyl acetate in hexane to afford the title compound.
EXAMPLE 11 : Preparation of 2-bromo-1 -morpholinoethan-1 -one
A flask is charged with bromoacetyl bromide (25 mL), dichloromethane (500 mL) and mixture is stirred under nitrogen atmosphere. The reaction mixture is cooled to -25°C followed by slow addition of morpholine (72.7 mL in 500 mL of DCM) at the same temperature over a period of 30 minutes. The reaction mixture is stirred at -25°C for 15 minutes, then allowed to attain room temperature at which it is further stirred for 4 hours. The completion of the reaction is monitored by TLC and reaction mixture is sequentially washed with water (2×250 mL) and brine solution (2×100 mL). The organic solvent is subjected to distillation to afford the title compound.
EXAMPLE 12: Preparation of benzyl ((3aS,4R,6S,6aR)-2,2-dimethyl-6-(2-morpholino-2-oxoethoxy)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (Formula Vila”)
A flask is charged with sodium hydride (60%, 4.29 g), DMF (90 mL) and cooled to -30°C. Then, benzyl ((3aS,4R,6S,6aR)-6-hydroxy-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (30 g) is added to the reaction mixture at the same temperature over a period of 25 minutes and mixture is stirred at -30°C for 1 hour. Then 2-bromo-1 -morpholinoethan-1 -one (24.36 g) is added to the reaction mixture at -30°C over a period of 20 minutes and temperature is raised to room temperature. The mixture is stirred at RT for 1 hour. The progress of the reaction is monitored by TLC and after completion, the reaction mixture is quenched with ice cold water followed by extraction with ethyl acetate. The organic layer is separated and subjected to distillation to afford the title compound.
EXAMPLE 13: Preparation of 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula VII”)
A flask is charged with benzyl ((3aS,4R,6S,6aR)-2,2-dimethyl-6-(2-morpholino-2-oxoethoxy)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate, ethanol (1 0 g), ammonium formate (4.35 g) and 10% Pd/C (1 g). The reaction mixture is heated to 80°C and then stirred for 2 hours. The progress of the reaction is monitored by TLC and after completion of the reaction, mixture is cooled to room temperature, filtered and washed with ethyl acetate (100 mL). The filtrate is distilled under reduced pressure and obtained compound is purified by column chromatography using methanol-DCM (5:95) to afford the title compound.
EXAMPLE 14: Preparation of 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2- (propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula V”)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (8 g), water (24 mL) and stirred for 10 minutes. Then sodium bicarbonate (8.9 g), 4,6-dichloro-2-(propylthio)pyrimidin-5-amine (6.3 g) and water (24 mL) is added and mixture is heated to 95-100°C at which point it is stirred for 15 hours. The progress of the reaction is monitored by TLC and on completion reaction mixture is cooled to room temperature followed by addition of water (24 mL) and ethyl acetate (40 mL). The layers are separated and aqueous layer is extracted with ethyl acetate (20 mL). The organic layers are combined, washed with brine solution (2×40 mL) and subjected to distillation under vacuum at 45°C to afford the title compound.
EXAMPLE 15: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3] dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula IV”)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (4 g), acetic acid (20 mL) and stirred under nitrogen atmosphere for 10 minutes. Then water (8 mL) is added and mixture is cooled to -5 to 0°C followed by slow addition of sodium nitrite (650 mg). The mixture is stirred at 0°C for 1 hour and progress of the reaction is monitored by TLC. After completion of the reaction, mixture is extracted with toluene (40 mL and 20 mL). The combined toluene layer is sequentially washed with potassium
carbonate solution (40 mL) and brine solution (2×20 mL) followed by distillation under vacuum to afford the desired compound.
EXAMPLE 16: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula II”)
A flask is charged with (1 R,2S)-2-(3,4-difluorophenyl)cyclopropan-1 -amine mandelate (2.5 g) and toluene (20 mL) followed by drop-wise addition of diisopropylethylamine (4.7 mL), then mixture is stirred for 10 minutes at RT. Then toluene layer containing 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3] dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (4 g in 55 mL) is added to the above mixture and reaction mass is stirred for 15 hours at room temperature. The progress of the reaction is monitored by TLC followed by addition of water (20 mL) on completion of reaction. The layers are separated, aqueous layer is extracted with toluene (20 mL). The organic layers are combined, washed with brine solution (2×20 mL) and then subjected to distillation under vacuum at 45°C to afford the crude compound. The crude compound is purified by column chromatography using hexane to afford the title compound.
EXAMPLE 17: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (1 g) in tetrahydrofuran (20 mL) and stirred under nitrogen atmosphere followed by addition of vitride (1 .53 mL) over a period of 10 minutes. The reaction mixture is stirred for 1 hour at room temperature and progress of the reaction is monitored by TLC. On completion, the mixture is quenched with sodium potassium tartrate (5 mL). The mixture is extracted with ethyl acetate (10 mL), then layers are separated and organic layer is subjected to distillation under vacuum at 45°C. The obtained material is dissolved in THF (20 mL) and slowly lithium aluminiumhydride (0.1 17 g) is added to the mixture at 0-5°C. Then mixture is stirred at room temperature for 1 hour and progress of the reaction is monitored by TLC. On completion of reaction, it is quenched with ice-cold water (20 mL) and extracted with ethyl acetate (15 mL). The layers are separated and organic layer is used for next step.
EXAMPLE 18: Preparation of Ticagrelor
A flask is charged with organic layer containing 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3] triazolo [4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy) ethan-1 -ol (30 mL) and 2% hydrochloric acid solution (30 mL). The reaction mixture is stirred at room temperature for overnight and progress of the reaction is monitored by TLC. Then the reaction mixture is diluted with ethyl acetate (20 mL), layers are separated. The organic layer is washed with brine solution (20 mL) followed by complete distillation under vacuum at 45°C. The crude compound is purified by column chromatography using ethyl acetate:hexane (7:10) and methanol :d ic h I oro methane (5:95) to afford the title compound.
EXAMPLE 19: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula IV)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (5 g) and acetonitrile (50 mL) for clear solution. To this, isoamyl nitrite (1 .5 g) is added over a period of 5 minutes. The reaction mixture is maintained at room temperature for 5 hours and completion of the reaction is monitored by TLC. Then water (50 mL) and toluene (50 mL) are added and layers are separated. The aqueous layer is extracted with toluene (50 mL) and total organic layers are combined, subjected to distillation under vacuum to afford the title compound.
Anji Reddy

Mr G.V. Prasad, CEO, Dr. Reddy’s Labs

G V Prasad and Mr K. Satish Reddy
///////////
NEW PATENT, TICAGRELOR, DR. REDDY’S LABORATORIES LIMITED, WO 2016001851
New Patent from Zydus Cadila, Canagliflozin, US 20160002275
CADILA HEALTHCARE LIMITED [IN]

(2S,3R,4R,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is also known as Canagliflozin, is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2) which is chemically represented as compound of Formula (I).
U.S. Pat. No. 7,943,788 B2 discloses canagliflozin and a process for its preparation.
U.S. Pat. No. 7,943,582 B2 (the ‘582 patent) discloses crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate and process for preparation thereof.
U.S. PG-Pub. No. 2011/0212905 discloses crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate and process for preparation thereof.
U.S. PG-Pub. Nos. 2009/0233874, 2010/099883 and 2008/0146515 discloses similar process for the preparation of canagliflozin substantially as same as shown in scheme-1 below.
International (PCT) Publication No. WO 2011/079772 discloses a process for the preparation of canagliflozin by reduction of keto group of acetyl protected compound followed by hydrolysis.
U.S. PG-Publication No. 2014/0128595 discloses a process for the preparation of canagliflozin from anhydroglucopyranose derivative substantially as same as shown in scheme-2 below.
The prior-art processes requires sequence of protection/deprotection of canagliflozin obtained in the course of the reactions and further purification or crystallization to obtain canagliflozin in reasonably pure form. This sequences of processes results in high amount of yield loss.
In view of the above prior art, there is provided a novel, efficient and convenient process for preparation of canagliflozin which is at least a useful alternative to the prior art as well as an efficient and convenient method for purification of canagliflozin without sequence of protection and deprotection.
Scheme-3.

Ahmedabad-based pharma giant Cadila Healthcare’s chairman and managing director, Pankaj Patel,
EXAMPLES
Example-1Preparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Va) (5 g) and 150 mL toluene at 25° C. 1.5 mL (1.6M) n-butyl lithium in hexane was added dropwise at room temperature and the solution was stirred for 30 minutes. This solution was cooled to −78° C. and added dropwise to a solution of 3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV) (6.4 g) in 100 mL toluene and the mixture was stirred for 3 hours. The reaction mixture was treated with 2.5 g methanesulfonic acid in 100 mL methanol and stirred for 1 hour. The reaction mass was warmed to 25° C. and then added to pre-cool saturated sodium bicarbonate solution and resulting mass was extracted with ethyl acetate. The extract was washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain compound of Formula (III).
Example-1APreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Va) (5 g) and 150 mL toluene at 25° C. 1.5 mL (1.6M) n-butyl lithium in hexane was added dropwise at room temperature and the solution was stirred for 30 minutes. This solution was cooled to −78° C. and added dropwise to a solution of 3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV) (6.4 g) in 100 mL toluene and the mixture was stirred for 3 hours. The reaction mixture was treated with 2.5 g methanesulfonic acid in 100 mL methanol and stirred for 1 hour. The reaction mixture warmed to room temperature and stirred for 8 hours. Saturated sodium bicarbonate solution was added to the reaction mixture and the separated aqueous layer was extracted with toluene. The organic layer was distilled to remove toluene and the residue was dissolved in 50 mL methylene dichloride, washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain residue. The residue was treated with 150 mL diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed withdiisopropyl ether to obtain compound of Formula (III).
Example-1BPreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 5 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 100 g 2-(5-iodo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Vb), 114.35 g 3,4,5-tris((trimethylsilyl)oxy)-6-(((tri-methylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV), 2 L toluene and 1 Ltetrahydrofuran at 30° C. The reaction mixture was cooled to −78° C. and 171.45 mL n-butyl lithium in hexane (1.6M) was added and the solution was stirred for 3 hours. The reaction mixture was treated with 94.16 g methanesulfonic acid in 1500 mL methanol and stirred for 1 hour. The reaction mixture warmed to 25° C. and stirred for 8 hours. The reaction mixture was cooled to 5° C. and saturated sodium bicarbonate solution was added to the reaction mixture and stirred for 30 min. The separated aqueous layer was extracted with toluene. The organic layer was distilled to remove toluene and the residue was dissolved in 300 mL methylene dichloride and 200 g silica gel of 60-120 mesh was added. The reaction mixture was stirred for 30 min at 30° C., washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain residue. The residue was treated with 1 L diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed with diisopropyl ether to obtain compound of Formula (III).
Example-2APreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (IIa1)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 10 g compound of Formula (III), 80 mL methylene dichloride and 4.3 g N-methylmorpholine at −5 to 5° C. 2.7 g trimethylsilyl chloride was added slowly and stirred for 1 hour. After confirming the reaction completion TLC, 30 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The organic layer was evaporated under reduced pressure to obtain compound of Formula (IIa).
Example-2BPreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (IIa1)
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 100 g compound of Formula (III) and 900 mL methanol at 30° C. and stirred for 1 hour. The reaction mixture was filtered to remove silica gel and washed with methanol. The filtrate was distilled under vacuum to remove methanol completely, 350 mL methylene dichloride and 42.63 g N-methylmorpholine were added to the residue and cooled to at −5 to 5° C., 34.34 g trimethylsilyl chloride was lot-wise added and stirred for 45 min. After confirming the reaction completion TLC, 300 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The separated organic layer was dried over sodium sulfate and filtered to obtain compound of Formula (IIa1).
Example-3APreparation of Canagliflozin of Formula (I)
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added solution of compound (IIa) prepared in example-2B and cooled to −70° C. 8 mL triethylsilane and 5.5 mL boron trifluoridediethyl etherate were added dropwise within 1 hour maintaining the reaction temperature between −70° C. The reaction was warmed to −30° C. and stirred for 30 min. The reaction mixture was then added to freshly preparedsodium bicarbonate solution at 5° C. and then allowed to warm to room temperature and stirred for 20 mints to adjust the pH of 7-8. The reaction mass was then slowly added to cold water. The resulting mass was extracted with ethyl acetate. The combined organic layers were washed with saturated bicarbonatesolution, dried over Na2SO4 and evaporated under reduced pressure to obtain canagliflozin having purity 86% by HPLC.
Example-3BPreparation of Canagliflozin of Formula (I)
In 2 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added the solution of compound (IIa) prepared in example-2B and cooled to −70° C. 67.38 g triethylsilane and 83.08 g boron trifluoridediethyl etherate were added dropwise within 1 hour maintaining the reaction temperature between −70° C. The reaction was warmed to −30° C. and stirred for 3 hours. The reaction mixture was then added to freshly prepared sodium bicarbonate solution at 5° C. and then allowed to warm to room temperature and stirred for 20 mints to adjust the pH of 7-8. The reaction mixture was then slowly added to cold water. The separated aqueous layer was extracted with 200 mL methylene dichloride. The combined organic layer was washed with 300 mL water and distilled completely to remove methylene dichloride. The resulting residue extracted with 500 mL ethyl acetate and stirred to obtain clear solution. The reaction mixture was treated with brine and saturated bicarbonate solution to separate the layers. The separated organic layer was dried over sodium sulfate, charcoalized and filtered. The filtrate is distilled to remove ethyl acetate completely under vacuum. The residue was dissolved in 300 mL methylene dichloride and 200 g silica gel of 60-120 mesh was added. The reaction mixture was stirred for 30 min at 30° C. and distilled completely under reduced pressure to obtain residue. The residue was treated with 500 L diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed with diisopropyl ether to obtain canagliflozin (I) having purity 87% by HPLC.
Example-4Preparation of (3R,4S,5R,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl)tris(oxy)tris(trimethylsilane) (IIb1)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 10 g compound of Formula (III), 100 mL methylene dichloride and 15 g N-methylmorpholine at 0 to 5° C. 12.7 g trimethylsilyl chloride was added slowly and stirred for 1 hour. After confirming the reaction completion by TLC, 300 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The organic layer was evaporated under reduced pressure to obtain compound of Formula (IIb1).
Example-5Preparation of Canagliflozin
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added 20 g compound (IIb1) prepared in example-4 and 100 mL methylene dichloride at −25° C. to −30° C. 11 mL triethylsilane and 7.8 mL boron trifluoridediethyl etherate was added drop wise within 1-2 hours maintaining the reaction temperature between −25° C. to −30° C. The reaction was stirred for 30 min and then allowed to warn to room temperature and stirred for 1.5-2 hours. The reaction mixture was then slowly added to cold water. The reaction mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated bicarbonate solution, dried over sodium sulfate and evaporated under reduced pressure to obtain canagliflozin having purity 86% by HPLC.
Example-6Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 85%) and 100 mLtoluene were stirred to obtain a clear solution. 10 g Polyvinylpyrrolidone was added to the solution and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in ethyl acetate and water mixture for 30 min. The separated ethyl acetate layer was evaporated to dryness to obtain pure canagliflozin. (7.1 g. Purity 96.55% by HPLC).
Example-7Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 87%) and 100 mLtoluene were stirred in in a round bottom flask to obtain a clear solution. 10 g β-cyclodextrin was added to the solution and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in ethyl acetate and water mixture for 30 min. The separated ethyl acetate layer was treated with activated carbon, filtered and evaporated to dryness to obtain pure canagliflozin. (7.9 g, Purity 98.93% by HPLC).
Example-8Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 87%) and 0.25 g activated carbon were stirred in 100 mL toluene for 15-20 min and filtered. 10 g β-cyclodextrin was added to the filtrate and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in isopropyl acetate and water mixture for 30 min. The separated isopropyl acetate layer was evaporated to dryness to obtain pure canagliflozin. (7.7 g, Purity 99.12% by HPLC).
Example-9Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g canagliflozin (purity 87%) and 100 mLtoluene were stirred to obtain a clear solution. 10 g hydroxy propyl methyl cellulose was added to the solution and stirred for 2-3 hour. The reaction mixture was filtered, washed with toluene. The solid was stirred in isopropyl acetate and water mixture for 30 min and dried to obtain pure canagliflozin. (Purity 97-98% by HPLC).
Example-10Purification of Canagliflozin
In 2 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 100 g canagliflozin (purity 87%) obtained in example-3B and 900 mL methanol were stirred for 45 min at 30° C. The reaction mixture was filtered to remove silica gel. The filtrate was distilled under vacuum completely below 45° C. 400 mL toluene was added and heated to 55° C. to obtain a clear solution. The reaction mixture was filtered and the filtrate was added 100 g β-cyclodextrine. The reaction mixture was heated at 75° C. for 30 min and cooled to 30° C. and further stirred for 30 min. 5 g canagliflozin β-cyclodextrin complex was added to the solution and further cooled to 5° C. The reaction mixture was stirred for 3 hours and filtered. The wet-cake was treated with 300 mL isopropyl acetate and heated at 75° C. for 30 min. The reaction mixture was cooled to 30° C. and stirred for 6 hours and further cooled to 5° C. and stirred for 3 hours. The reaction mixture was filtered and washed with isopropyl acetate and dried at 30° C. to obtain crystalline canagliflozin β-cyclodextrine complex having 40 g pure canagliflozin with 99% purity by HPLC.
Example-11Preparation of Amorphous Canagliflozin
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 100 g canagliflozin β-cyclodextrine (purity 98%) obtained in example-10 and 400 mL acetone were stirred for 30 min at 30° C. The reaction mixture was filtered to remove β-cyclodextrine. The filtrate was distilled under vacuum completely below 45° C. 400 mL acetone was added to the residue to get clear solution at 30° C. 5 g activated charcoal was added and stirred for 20 min. The reaction mixture was filtered and the filtrate was spray dried using JISL Mini spray drier LSD-48 keeping feed pump at 30 rpm, inlet temperature at 60° C., outlet temperature at 40° C. and 2 Kg/cm2 hot air supply. The product was collected from cyclone and is further dried at 40° C.±5° C. under vacuum for 12 hours to get 80 g of amorphous canagliflozin having 99.6% purity by HPLC.
WO2014195966
https://www.google.co.in/patents/WO2014195966A2?cl=un
Canagliflozin is inhibitor of sodium dependent glucose transporter inhibitor (SGLT) which is chemically represented as (25′,3i?,4/?,55,,6 ?)-2-{3-[5-[4-Fluoro-phenyl]-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol having (I).

Formula (I)
U.S. Patent No, 7,943,788 B2 (the ‘788 patent) discloses canagliflozin or salts thereof and the process for its preparation.
U.S. Patent Nos. 7,943,582 B2 and 8,513,202 B2 discloses crystalline form of 1 -(P-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate and process for preparation thereof. The US ‘582 B2 and US ‘202 B2 further discloses that preparation of the crystalline form of hemi-hydrate canagliflozin typically involves dissolving in a good solvent (e.g. ketones or esters) crude or amorphous compound prepared in accordance with the procedures described in WO 2005/012326 pamphlet, and adding water and a poor solvent (e.g. alkanes or ethers) to the resulting solution, followed by filtration.
U.S. PG-Pub. No. 2013/0237487 Al (the US ‘487 Al) discloses amorphous dapagliflozin and amorphous canagliflozin. The US ‘487 Al also discloses 1:1 crystalline complex of canagliflozin with L-proline (Form CS1), ethanol solvate of a 1: 1 crystalline complex of canagliflozin with D-proline (Form CS2), 1 :1 crystalline complex of canagliflozin with L-phenylalanine (Form CS3), 1:1 crystalline complex of canagliflozin with D-proline (Form CS4).
The US ‘487 Al discloses preparation of amorphous canagliflozin by adding its heated toluene solution into n-heptane. After drying in vacuo the product was obtained as a white solid of with melting point of 54.7°C to 72.0°C. However, upon repetition of the said experiment, the obtained amorphous canagliflozin was having higher amount of residual solvents. Therefore, the amorphous canagliflozin obtained by process as disclosed in US ‘487 Al is not suitable for pharmaceutical preparations.
The US ‘487 Al further discloses that amorphous canagliflozin obtained by the above process is hygroscopic in nature which was confirmed by Dynamic vapor sorption (DVS) analysis. Further, it was observed that the amorphous form underwent a physical change between the sorption/desorption cycle, making the sorption/desorption behavior different between the two cycles. The physical change that occurred was determined to be a conversion or partial conversion from the amorphous state to a crystalline state. This change was supported by a change in the overall appearance of the sample as the humidity increased from 70% to 90% RH.
The canagliflozin assessment report EMA/718531/2013 published by EMEA discloses that Canagliflozin hemihydrate is a white to off-white powder^ practically insoluble in water and freely soluble in ethanol and non-hygroscopic. Polymorphism has been observed for canagliflozin and the manufactured Form I is a hemihydrate, and an unstable amorphous Form II. Form I is consistently produced by the proposed commercial synthesis process.
Therefore, it is evident from the prior art that the reported amorphous form of canagliflozin is unstable and hygroscopic as well as not suitable for pharmaceutical preparations due to higher amount of residual solvents above the ICH acceptable limits.
Hence, there is a need to provide a stable amorphous form of canagliflozin which is suitable for pharmaceutical preparations.
Crystalline solids normally require a significant amount of energy for dissolution due to their highly organized, lattice like structures. For example, the energy required for a drug molecule to escape from a crystal is more than from an amorphous or a non-crystalline form. It is known that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form (Econno T., Chem. Pharm. Bull., 1990; 38: 2003-2007). For some therapeutic indications, one bioavailability pattern may be favoured over another.
An amorphous form of some of the drugs exhibit much higher bioavailability than the crystalline forms, which leads to the selection of the amorphous form as the final drug substance for pharmaceutical dosage from development. Additionally, the aqueous solubility of crystalline form is lower than its amorphous form in some of the drugs, which may resulted in the difference in their in vivo bioavailability. Therefore, it is desirable to have amorphous forms of drugs with high purity to meet the needs of regulatory agencies and also highly reproducible processes for their preparation.
In view of the above, it is therefore, desirable to provide canagliflozin amorphous form as well as an efficient, economical and eco-friendly process for the preparation of highly pure canagliflozin amorphous form.
Example-l:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 25.0 g of canagliflozin was dissolved in 250.0 mL of methanol mixture at 25°C to 30°C. The content was stirred for 30 minutes at 25°C to 30°C. To this, 1.0 g charcoal was added and stirred for 30 minutes at 25°C to 30°C. The content was filtered through Hyflo-supercel, and the Hyflo-supercel pad was washed with 50.0 mL methanol. The filtrate was concentrated under vacuum below 45°C followed by spray drying in JISL Mini spray drier LSD-48 under the below conditions. The product was collected from cyclone and is further dried at 55°C±5°C under vacuum for 16 hours to get 19.0 g of amorphous canagliflozin.

The spray-dried canagliflozin is amorphous in nature. The obtained product contains residual solvent well within ICH limit.
The obtained solid was amorphous canagliflozin as is shown by the X-ray diffraction pattern shown in FIG.1.
Example-2:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 25.0 g of canagliflozin was dissolved in 250.0 mL of acetone mixture at 25°C to 3O°C. The content was stirred for 30 minutes at 25°C to 30°C. To this, 1.0 g charcoal was added and stirred for 30 minutes at 25°C to 30°C. The content was filtered through Hyflo-supercel, and the Hyflo-supercel pad was washed with 50.0 mL acetone. The filtrate was concentrated under vacuum below 45°C followed by spray drying in JISL Mini spray drier LSD-48 under the below conditions. The product was collected from cyclone and is further dried at 55°C±5°C under vacuum for 16 hours to get 20.0 g of amorphous canagliflozin.

The spray-dried canagliflozin is amorphous in nature. The compound is having residual acetone less than 0.5% by GC.
The obtained solid was amorphous canagliflozin as is shown by the X-ray diffraction pattern shown in FIG.2.
Example-3:
Preparation of amorphous form of canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g of canagliflozin was dissolved in 125 mL methanol and heated to obtain clear solution at 65°C. The solution was distilled to remove methanol completely. The compound thus obtained was amorphous canagliflozin.
Example-4:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g of canagiiflozin was dissolved in 125 mL acetone and heated to obtain clear solution at 65°C. The solution was distilled to remove acetone completely. The compound thus obtained was amorphous canagiiflozin. The compound is having residual acetone less than 0.5% by GC.
Example 5:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottom flask equipped with mechanical stirrer, thermometer and an addition funnel, canagiiflozin (0.5 gm, 1.02 mmol), PVP K-30 (4 gm, 8 times) and 88% methanol in water (12.5ml, 25V) were heated to 65-70°C to get clear solution. The reaction mixture was stirred for 1 hour, concentrated under vacuum (1.5 mbar) at 65-70°C and degassed under vacuum (1.5 mbar) for 1 hour at 70°C to obtain the title compound in amorphous form.
Example 6:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottom flask equipped with mechanical stirrer, thermometer and an addition funnel, canagiiflozin (0.5 gm, 1.02 mmol), HPMC-AS (1 gm, 2 times) in 88% methanol in water (12.5 ml, 25V) were heated at 65 to 70°C to get clear solution. The reaction mixture was stirred for 2 hours, concentrated under vacuum (1.5 mbar) at 70°C and degassed under vacuum (1.5 mbar) for lhr at 70°C to obtain the title compound in amorphous form.
Example-7:
Preparation of canagliflozin-L-Proline crystalline complex
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel; 25.0 g of canagiiflozin, 6.06 g L-proline and 250 mL ethanol were heated to 75-80°C, stirred for 15 min and then cooled down to 25-30°C. The mass was filtered and washed with ethanol to obtain canagliflozin-L-proline crystalline complex.
Example-8:
Preparation of amorphous canagliflozin from canagliflozin-L-proline crystaUine complex
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of Canagliflozin-L-Proline Crystalline Complex and 250 mL of ethyl acetate were stirred to get a clear solution, washed with 2×150 mL of water and the organic layer was distilled. To the residue 100 mL of isopropyl acetate and 2.5 mL of water was added and heated to 75-80°C, stirred for 15 min and cooled down to 25-30°C. The mass filtered and washed with isopropyl acetate to obtain canagliflozin. The obtained canagliflozin was subjected to spray dyring under conditions of example-2 using acetone solvent to obtain amorphous canagliflozin. Purity > 99.5% by HPLC. The compound is having residual acetone less than 0.5% by GC.
The obtained solid was amorphous canagliflozin as shown by the X-ray diffraction pattern shown in FIG.2.
HPLC Purity of amorphous canagliflozin was measured by using following chromatographic conditions:
Equipment: Shimadzu LC2010C HPLC system equipped with a dual
wavelength UV-VIS detector or equivalent
Column: romasil C-8 (250mmx4.6 mm, 5 μπι) or equivalent
Flow rate: 1.5 mL/minute
Column oven temp.: 30°C
Wavelength: 210 nm
Injection Volume: 10 μΐ, .
Diluent: Mobile Phase A: Mobile Phase B (30:70)
Mobile Phase A: Buffer:Acetonitrile:Methanol (60:30: 10)
Mobile Phase B: Acetonitrile: Methanol (80:20)
Example-9:
Preparation of amorphous form of Canagliflozin as per Example-2 of US ‘487 Al In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to get clear solution. 100 mL of n-heptane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 65°C under vacuum for 72 hours. The residual n-heptane was 44000 ppm by GC after 72 hours drying.
Example-10:
Replacing toluene with ethyl acetate in above example-9
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to obtain clear solution. 100 mL of n-heptane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 65°C under vacuum for 72 hours. The residual n-heptane was -44000 ppm by GC after 72 hours drying.
Example-11:
Replacing n-heptane with cyclohexane in above example-9
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to obtain clear solution. 100 mL of cyclohexane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 55°C under vacuum for 72 hours. The residual cyclohexane was >5000 ppm by GC after 72 hours drying.
Example-12:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel; 25.0 g of canagliflozin and 250 mL of ethyl acetate were stirred to get clear solution and then ethyl acetate was removed under reduced pressure to obtain 20.0 g of amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 55°C under vacuum for 72 hours. The residual ethyl acetate was -8450 ppm by GC after 72 hours drying.
///////////////New Patent, Zydus Cadila, Canagliflozin, US 20160002275
Shanghai Hengrui’s potent inhibitors of Human Uric Acid Transporter 1 (hURAT1)

| MF | C 1 4 H 1 2 BrNO 2 S |
|---|---|
| MW | 338.21958 g / mol |
1- (6-bromoquinolin-4-yl) sulfanylcyclobutane-1-carboxylic acid
CAS…….1638327-48-6
Cyclobutanecarboxylic acid, 1-[(6-bromo-4-quinolinyl)thio]-
COMING ………….
MS m / z (ESI): 338.0 [M + l]
1H NMR (400 MHz, DMSO) δ 13.17 (s, 1H), 8.75-8.79 (m, 1H), 8.24 (s, 1H), 7.87-7.98 (m, 2H), 7.21-7.25 (m, 1H), 2.83-2.95 (m, 2H), 2.30-2.41 (m, 2H), 2.16-2.27 (m, 1H), 1.97-2.08 (m, 1H)
WO-2014183555-A1 / 2014-11-20
http://www.google.co.in/patents/WO2014183555A1?cl=en
PROCEDURE
6-bromo-quinoline-4-thiol
A mixture of 6-bromo-4-chloro-quinoline 3a (260 mg, 1.1 mmol, using known methods “Bioorganic &
Medicinal Chemistry Letters, 2012, 22 (4), 1569-1574 “prepared to give) and sodium sulfide (100 mg, 1.3 mmol) was added to 4 mL of N, N- dimethyl formamide, plus complete, heated 80 ° C, the reaction was stirred for 2 hours. To the reaction mixture was added 50 mL of water, 1 M hydrochloric acid was added dropwise to the reaction solution to pH 5-6, extracted with ethyl acetate (50 mL X 3), the combined organic phases, with no over anhydrous sodium sulfate, filtered, and the filtrate concentrated under reduced pressure to give the title product 6-bromo-quinolin-4-thiol 3b (257 mg, yellow oil), it was used directly in the next reaction.
The second step
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl carboxylate
Under an argon atmosphere, 6-bromo-quinolin-4-thiol 3b (257 mg, 1.1 mmol), 1- bromo-cyclobutyloxy embankment carboxylate (266 mg, 1.3 mmol) and cesium carbonate (371 mg, 1.1 mmol) were sequentially added to 5 mL of N, N- dimethylformamide and heated to 60 ° C, the reaction was stirred for 2 hours. The reaction solution was filtered, the filter cake washed with ethyl acetate (10 mL X 3) and the filtrate was concentrated under reduced pressure to give the title product l – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c ( 300 mg, brown oil). Yield: 77%.
MS m / z (ESI): 368.2 [M + l]
1H MR (400 MHz, CDCl 3 ) [delta] 8.67 (d, = 4.77 Hz, IH), 8.31 (d, = 2.13 Hz, IH), 7.94 (d, = 8.91Hz, IH), 7.78 (dd, = 9.03, 2.13Hz, IH), 7.15 (d, = 4.89Hz, IH), 4.16 (q, = 7.15Hz, 2H), 2.86-3.04 (m, 2H), 2.39-2.51 (m, 2H), 2.25-2.37 ( m, IH), 2.00-2.15 (m, IH), 1.16 (t, = 7.09Hz, 3H)
third step
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid
L – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c (100 mg, 0.27 mmol) and lithium hydroxide monohydrate (23 mg, 0.55 mmol) was dissolved in 6 mL of tetrahydrofuran, ethanol and water (^ = 4: 1: 1) mixed solvent, the reaction was stirred for 3 hours. 1M hydrochloric acid was added dropwise to the reaction solution pH of 5 to 6, liquid separation, the aqueous phase was extracted (10 mL X 3) with dichloromethane, the combined organic phases, the organic phase was washed with a saturated sodium chloride solution (10 mL XI), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure, the resulting A by thin layer chromatography in a developing solvent system, and the residue was purified to give the title product l – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid 3 (20 mg, white solid), yield: 22%.
MS m / z (ESI): 338.0 [M + l]
1H NMR (400 MHz, DMSO) δ 13.17 (s, 1H), 8.75-8.79 (m, 1H), 8.24 (s, 1H), 7.87-7.98 (m, 2H), 7.21-7.25 (m, 1H), 2.83-2.95 (m, 2H), 2.30-2.41 (m, 2H), 2.16-2.27 (m, 1H), 1.97-2.08 (m, 1H)
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid
First step
6-bromo-quinoline-4-thiol
A mixture of 6-bromo-4-chloro-quinoline 3a (260 mg, 1.1 mmol, a known method of “Bioorganic &
Medicinal Chemistry Letters, 2012, 22 (4), 1569-1574 “prepared to give) and sodium sulfide (100 mg, 1.3 mmol) was added to 4 mL of N, N- dimethyl formamide, plus complete, heated 80 ° C, the reaction was stirred for 2 hours. To the reaction mixture was added 50 mL of water, 1 M hydrochloric acid was added dropwise to the reaction solution to pH 5-6, extracted with ethyl acetate (50 mL X 3), the combined organic phases, with no over anhydrous sodium sulfate, filtered, and the filtrate concentrated under reduced pressure to give the title product 6-bromo-quinolin-4-thiol 3b (257 mg, yellow oil), it was used directly in the next reaction.
The second step
L – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl
Under an argon atmosphere, 6-bromo-quinolin-4-thiol 3b (257 mg, 1.1 mmol), 1- bromo-cyclobutyloxy embankment carboxylate (266 mg, 1.3 mmol) and cesium carbonate (371 mg, 1.1 mmol) were added to 5 mL of N, N- dimethylformamide and heated to 60 ° C, the reaction was stirred for 2 hours. The reaction mixture was filtered, the filter cake washed with ethyl acetate (10 mL X 3) and the filtrate was concentrated under reduced pressure to give the title product l – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c ( 300 mg, brown oil). Yield: 77%.
MS m / z (ESI): 368.2 [M + l]
1H MR (400 MHz, CDC1 3) δ 8.67 (d, = 4.77Hz, IH), 8.31 (d, = 2.13Hz, IH), 7.94 (d, = 8.91Hz, IH), 7.78 (dd, = 9.03, 2.13Hz, IH), 7.15 (d, = 4.89Hz, IH), 4.16 (q, = 7.15Hz, 2H), 2.86-3.04 (m, 2H), 2.39-2.51 (m, 2H), 2.25-2.37 ( m, IH), 2.00-2.15 (m, IH), 1.16 (t, = 7.09Hz, 3H) Step
L – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid
L – ((6-bromo-quinolin-4-yl) thio) ethyl cyclobutyl 3c (100 mg, 0.27 mmol) and lithium hydroxide monohydrate (23 mg, 0.55 mmol) was dissolved in 6 mL of tetrahydrofuran, ethanol and water (^ = 4: 1: 1) mixed solvent, the reaction was stirred for 3 hours. 1M hydrochloric acid was added dropwise to the reaction solution pH of 5 to 6, liquid separation, the aqueous phase was extracted (10 mL X 3) with dichloromethane, the combined organic phases, the organic phase was washed with a saturated sodium chloride solution (10 mL XI), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure, to the resulting thin layer chromatography using a developing solvent system A and the residue was purified to give the title product l – ((6-bromo-quinolin-4-yl) thio) cyclobutyl acid 3 (20 mg, white solid), yield: 22%. MS m / z (ESI): 338.0 [M + l]
1H NMR (400 MHz, DMSO) δ 13.17 (s, 1H), 8.75-8.79 (m, 1H), 8.24 (s, 1H), 7.87-7.98 (m, 2H), 7.21-7.25 (m, 1H), 2.83-2.95 (m, 2H), 2.30-2.41 (m, 2H), 2.16-2.27 (m, 1H), 1.97-2.08 (m, 1H)
Discovery of potent and orally bioavailable inhibitors of Human Uric Acid Transporter 1 (hURAT1) and binding mode prediction using homology model
- Shanghai Hengrui Pharmaceutical Co. Ltd, 279 Wenjing Rd., Shanghai 200245, China
This Letter describes the Discovery of a series of potent inhibitors of Human Uric Acid Transporter 1 (hURATl). Lead generation via 3D pharmacophore Analysis and Optimization resulted in compound 41 . With an IC 50 of 33.7 nM, 41 Also Demonstrated good Oral Bioavailability in RAT (74.8%) and displayed a consistent PK profile across all species tested (rat, dog and monkey).

http://www.sciencedirect.com/science/article/pii/S0960894X1530353X
Volume 26, Issue 2 , 15 January 2016, Pages 277-282

//////// Shanghai Hengrui, inhibitors of Human Uric Acid Transporter 1 (hURAT1), 1- (6-bromoquinolin-4-yl) sulfanylcyclobutane-1-carboxylic acid
c13cc (ccc3nccc1SC2 (C (= O) O) CCC2) Br
BMS 955829

(4R,5R)-5-(2,5-difluorophenyl)-4-(5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one
cas 1375751-08-8
Chemical Formula: C22H14F2N2O2
Exact Mass: 376.1023
Bristol-Myers Squibb Company INNOVATOR
BMS-955829 is a Positive allosteric modulators (PAMs). BMS-955829 shows high functional PAM potency, excellent mGluR5 binding affinity, low glutamate fold shift, and high selectivity for the mGluR5 subtype. BMS-955829 is a potent mGluR5 PAM (EC50 = 2.6 ± 1.0 nM; n = 6), devoid of inherent mGluR5 agonist activity (EC50 > 30μM). The measured binding Ki of BMS-955829 was found to be 1.6 nM, which was in good agreement with its functional potency.

SYNTHESIS AND INTERMEDIATES…….https://www.google.co.in/patents/WO2012064603A1?cl=en
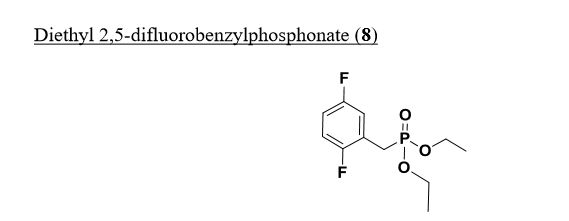
Intermediate 73

Diethyl 2,5-difluorobenzylphosphonate. A mixture of 2-(bromomethyl)-l,4- difluorobenzene (3 g, 14.49 mmol) and triethyl phosphite (7.72 ml, 43.5 mmol) was heated to 160 °C with stirring for 4 hours, cooled to ambient temperature and concentrated under high vacuum to remove most triethyl phosphite. The resulting residue was purified by column chromatography (20% to 30 % EtO Ac/Toluene) providing diethyl 2,5-difluorobenzylphosphonate (3.76 g, 13.52 mmol, 93 % yield) as colorless oil. ¾ NMR (500MHz, DMSO-d6) δ 7.30 – 7.10 (m, 3H), 4.05 – 3.91 (m, 4H), 3.31 – 3.20 (m, 2H), 1.18 (t, J=7.0 Hz, 6H). MS Anal. Calcd. for [M+H]+ CiiHieFzOsP: 265.2; found 265.3.

Intermediate 74

(E)-3-Bromo-5-(2,5-difluorostyryl)pyridine. To a stirred solution of diethyl 2,5-difluorobenzylphosphonate (63.5 g, 240 mmol) and 5-bromonicotinaldehyde (50.7 g, 264 mmol) in tetrahydrofuran (1923 ml) was added potassium tert-butoxide in tetrahydrofuran (312 ml, 312 mmol) at -10 °C. After three hours, the reaction mixture was allowed to warm to ambient temperature and stirring was continued for another 16 hours at which time the reaction mixture was diluted with ether (800 mL) and washed with H2O. The organic layer was dried over anhydrous magnesium sulfate, filered and concentrated to provide a yellow wax to which was added 300 mL of hexane and after sonication filtered to provide (is)-3-bromo-5-(2,5- difluorostyryl)pyridine (54 g, 173 mmol, 72.1%) as a white solid. XH NMR
(500MHz, DMSO-d6) δ 8.78 (d, J=1.8 Hz, IH), 8.63 (d, J=2.1 Hz, IH), 8.44 (t, J=2.0 Hz, IH), 7.67 (ddd, J=9.4, 6.0, 3.2 Hz, IH), 7.56 – 7.48 (m, IH), 7.46 – 7.40 (m, IH), 7.34 (td, J=9.6, 4.6 Hz, IH), 7.24 (tt, J=8.3, 3.6 Hz, IH). MS Anal. Calcd. for [M+H]+ Ci3H9BrF2N: 296.0; found 298.1


Intermediate 75

Tert-butyl (lR,2R)-l-(5-bromopyridin-3-yl)-2-(2,5-difluorophenyl)-2- hydroxyethylcarbamate. A solution of tert-butyl carbamate (4.18 g, 35.0 mmol) in propanol (39 ml) was sequentially treated with sodium hydroxide (1.376 g, 34.4 mmol) in water (72 ml) and tert-butyl hypochlorite (3.88 ml, 34.4 mmol). After 5 min of stirring, the reaction mixture was cooled to 0 °C. A solution of
(DHQD)2PHAL (0.555 g, 0.677 mmol) in propanol (39 ml), a solution of (E)-3- bromo-5-(2,5-difluorostyryl)pyridine (3.34 g, 11.28 mmol) in propanol (68 ml) , and potassium osmate dihydrate (0.166 g, 0.451 mmol) were sequentially added. The reaction mixture was stirred for three additional hours at 0 °C, warmed to ambient temperature and after an additional 16 hours the light yellow homogenous solution was quenched with saturated aqueous sodium sulfite (100 mL). The aqueous phase was extracted with ethyl acetate( 2 X 50 mL), the combined organic phases were washed with brine (100 mL), dried over anhydrous magnesium sulfate and concentrated to afford a residue which was purified via column chromatography (25% to 40 % EtO Ac/Hex) to provide tert-butyl (7R,2R)-l-(5-bromopyridin-3-yl)-2- (2,5-difluorophenyl)-2-hydroxyethylcarbamate (2.2991 g, 5.09 mmol, 45.1 % yield) as an optically enriched mixture of enantiomers. XH NMR (500MHz, DIVISOR) δ 8.56 (d, J=1.8 Hz, IH), 8.40 (s, IH), 8.03 (s, IH), 7.52 (d, J=9.5 Hz, IH), 7.25 (br. s., IH), 7.10 (t, J=5.6 Hz, 2H), 5.89 (d, J=4.9 Hz, IH), 5.03 (t, J=5.0 Hz, IH), 4.83 (dd, J=8.9, 5.2 Hz, IH), 1.40 – 1.34 (m, 9H), MS Anal. Calcd. for [M+H]+
Ci8H2oBrF2 203: 429.1; found 431.3.
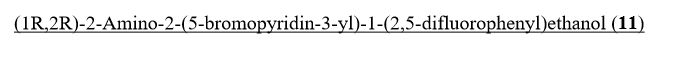
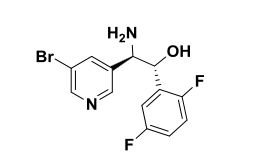
Intermediate 77

(lR,2R)-2-Amino-2-(5-bromopyridin-3-yl)-l-(2,5-difluorophenyl)ethanol To a stirred solution of tert-butyl tert-butyl (7R,2R,)-l-(5-bromopyridin-3-yl)-2-(2,5- difluorophenyl)-2-hydroxyethylcarbamate (2.30 g, 5.09 mmol) in methylene chloride (30 mL) was added HC1 in dioxane (30 ml, 120 mmol). The reaction mixture was placed in an oil bath set to 50 °C. After three hours, the reaction mixture was concentrated providing (7R,2R^-2-amino-2-(5-bromopyridin-3-yl)-l-(2,5- difluorophenyl)ethanol 2HC1 salt (2.10 g, 4.97 mmol, 98 % yield) as an optically enriched yellow wax. XH NMR (500MHz, DMSO-d6) δ 8.95 (d, J=3.7 Hz, 2H), 8.64 (d, J=2.4 Hz, 1H), 8.45 (d, J=1.5 Hz, 1H), 8.31 (t, J=2.0 Hz, 1H), 7.47 – 7.09 (m, 3H), 7.04 (td, J=9.2, 4.4 Hz, 1H), 5.29 (d, J=9.2 Hz, 1H), 4.57 (dd, J=9.0, 5.3 Hz, 1H). Anal. Calcd. for [M+H]+ Ci3H12BrF2N20: 329.0; found 331.2.

Intermediate 78

(4R,5R)-4-(5-Bromopyridin-3-yl)-5-(2,5-difluorophenyl)oxazotidin-2-one. To optically enriched (7R,2R)-2-amino-2-(5-bromopyridin-3-yl)-l-(2,5- difluorophenyl)ethanol, 2 HC1 (2.019 g, 4.82 mmol) in tetrahydrofuran (98 ml) was added diisopropylethylamine (2.95 ml, 16.87 mmol) and the resultant solution was stirred for ten mintues at ambient temperature, cooled to 0 °C and
carbonyldiimidazole (1.094 g, 6.75 mmol) was added. After an additional three hours at 0 °C the reaction mixture was warmed to ambient temperature and allowed to stir for another 16 hours. 2M ¾ in methanol (5ml) was added and after ten mintues the suspension was filtered and concentrated to a pink oil which was purified by column chromatography (25% to 40 % EtO Ac/Hex) providing (4R,5R)-4-(5- bromopyridin-3-yl)-5-(2,5-difluorophenyl)oxazolidin-2-one (1.353 g, 3.62 mmol, 75 % yield) as an optically enriched white solid. ¾ NMR (500MHz, DMSO-d6) δ 8.80 – 8.68 (m, 1H), 8.55 (d, J=2.1 Hz, 2H), 8.16 (t, J=2.1 Hz, 1H), 7.46 – 7.28 (m, 3H), 5.71 – 5.58 (m, 1H), 5.02 (d, J=6.7 Hz, 1H). MS Anal. Calcd. for [M+H]+ Ci4H10BrF2 2O2: 355.0; found 357.2.
Intermediate 79

(4R,5R)-4-(5-Bromopyridin-3-yl)-5-(2,5-difluorophenyl)oxazotidin-2-one. Method – 2 A mixture of tert-butyl ((lR,2R)-l-(54oromopyridin-3-yl)-2-(2,5- difluorophenyl)-2-hydroxyethyl)carbamate and tert-butyl ((lR,2R)-2-(5- bromopyridin-3-yl)-l-(2,5-difluorophenyl)-2-hydroxyethyl)carbamate (about 6: 1 ratio) (101 g, 236 mmol) in tetrahydrofuran (590 mL) was cooled to -7 °C with a methanol/ice bath. To this mixture was added a solution of 1 M potassium tert- butoxide in tetrahydrofuran (590 mL, 590 mmol) via an addition funnel while maintaining the internal temperature < 3 °C. The reaction mixture was stirred with a cooling bath for 30 min and then allowed to warm up to room temperature. After 20 h, the reaction was deemed complete by LC/MS. The reaction mixture was concentrated to dryness to give crude product. Another identical scale reaction was performed. The crude products of the two batches were combined to work up together. They were treated with ethyl acetate (1.75 L) and water (1.75 L). The layers were separated. The organic layer was washed with brine (1.75 L), dried (sodium sulfate), and evaporated to give 161.5 g of crude product as a brown solid. This was purified by ISCO to give 67.1 g (42% yield). LC/MS (ES+) 355/357 (M+H, 100; Br isotope pattern); XH NMR (400MHz, CDCl3) δ 8.75 (d, J=2.2 Hz, 1H), 8.53 (d, J=1.8 Hz, 1H), 7.97 (t, J=2.0 Hz, 1H), 7.29 – 7.23 (m, 1H), 7.18 – 7.09 (m, 2H), 6.40 (s, 1H), 5.56 (d, J=5.7 Hz, 1H), 4.84 (d, J=5.5 Hz, 1H); Calcd for
Ci4H9N2BrF202: C, 47.34; H, 2.55; N, 7.86; Br, 22.50; F, 10.69. Found: C, 47.29; H, 2.61; N, 7.87; Br, 22.40; F, 10.37. Note: Chiral HPLC of the above sample showed 4.7% of the enantiomer. The (4S, 55) enantiomer can be purged by recrystallization from methanol to give > 99.9 ee with 67% recovery.
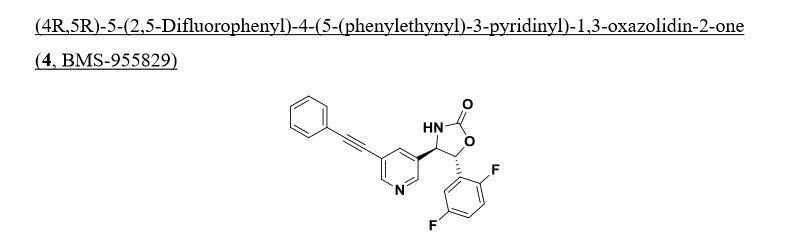

Scheme 1.

Pd(0)/Cu(l)/ TBAF Scheme 2.

cheme 4.

R’ = H, alkyl
Scheme 8.


cheme 11.

Scheme 12.


Scheme 14.

Scheme 15.

R” = H, alkyl R” = alkyl
cheme 16.

R’ = alky I
R” = alkyl

Scheme 17.

R’ = H, alkyl
R” = H, alkyl
Scheme 18.
R’ = H, alkyl R’ = H, alkyl

P T/US2011/059339

COMPD IS 185

Example 185

(4R, 5R)-5-(2, 5-difluorophenyl)-4-(5-(phenylethynyl)-3-pyridinyl)-l, 3-oxazolidin-2- one.
To a stirred solution of optically enriched (4R,5R)-4-(5-bromopyridin-3-yl)-5- (2,5-difluorophenyl)oxazolidin-2-one (1.25 g, 3.25 mmol) in triethylamine (70 mL) was added ethynylbenzene (0.592 mL, 5.28 mmol), copper(I) iodide (67 mg, 0.352 mmol), and triphenylphosphine (653 mg, 2.464 mmol). Nitrogen was bubbled through the mixture for 10 mintues before adding dichlorobis(triphenylphosphine)- palladium(II) (202 mg, 0.282 mmol) with continued nitrogen gas bubbling. After an additional 10 mintues the reaction mixtrue was heated to reflux for 16 hours, cooled to ambient temperature, diluted with EtOAc, washed with water (3X), brine, dried over magnesium sulfate, and concentrated in vacuo. Column chromatography (25% – -> 40% EtO Ac/Hex) provided optically enriched (4R,5R)-5-(2,5-difluorophenyl)-4- (5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one which was separated by chiral SFC chromatography (Chiralcel OJ-H preparative column, 30 x 250mm, 5μιη, Mobile Phase: 40% MeOH (0.1%DEA) in C02 @ 150Bar, Temp: 35°C, Flow rate: 70.0 mL/min. for 16 min, UV monitored @ 280 nM . tR = 9.23 min) to provide (1.38 g, 2.99 mmol, 85 % yield) of pure single enantiomer (4R,5R)-5-(2,5-difluorophenyl)- 4-(5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one.
‘H NMR (500 MHz, DMSO-i¾) δ ppm 8.77 (d, J=2.21 Hz, 1 H) 8.57 (s, 1 H) 8.56 (d, J=2.20 Hz, 1 H) 8.07 (t, J=2.05 Hz, 1 H) 7.58 – 7.66 (m, 2 H) 7.44 – 7.52 (m, 3 H) 7.39 – 7.45 (m, 1 H) 7.28 – 7.39 (m, 2 H) 5.67 (d, J=6.62 Hz, 1 H) 5.04 (d, J=6.62 Hz, 1 H). 13C NMR (126 MHz,
DMSO-i¾) δ ppm 157.28; 157.24 (d, J=240.70 Hz) 155.92 (d, J=245.20 Hz) 151.63; 147.70; 136.78; 135.02; 131.57; 129.43; 128.89; 126.63 (dd, J=14.99, 7.72 Hz) 121.51; 119.47; 117.83 (dd, J=23.60, 9.10 Hz) 117.50 (dd, J=24.50, 8.20 Hz); 114.60 (dd, J=26.34, 4.54 Hz); 92.86; 85.76; 78.12; 59.43;
LCMS (ESI) m/z calcd for C22H15F2N202: 377.11, found 377.20[M+H]+;
HRMS (ESI) m/z calcd for
C22H15F2N202: 377.1096, found 377.1096 [M+H]+.
SEE
WO2015054103, OXAZOLIDINONES AS MODULATORS OF MGLUR5
PAPER

Positive allosteric modulators (PAMs) of the metabotropic glutamate receptor subtype 5 (mGluR5) are of interest due to their potential therapeutic utility in schizophrenia and other cognitive disorders. Herein we describe the discovery and optimization of a novel oxazolidinone-based chemotype to identify BMS-955829 (4), a compound with high functional PAM potency, excellent mGluR5 binding affinity, low glutamate fold shift, and high selectivity for the mGluR5 subtype. The low fold shift and absence of agonist activity proved critical in the identification of a molecule with an acceptable preclinical safety profile. Despite its low fold shift, 4 retained efficacy in set shifting and novel object recognition models in rodents.
Discovery and Preclinical Evaluation of BMS-955829, a Potent Positive Allosteric Modulator of mGluR5
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00450
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00450/suppl_file/ml5b00450_si_001.pdf
SEE…………http://orgspectroscopyint.blogspot.in/2016/01/bms-955829.html
///////BMS 955829, mGluR5, positive allosteric modulator, schizophrenia, cognition, neurotoxicity, Bristol-Myers Squibb
FC1=CC=C(C=C1[C@H]([C@@H](C2=CC(C#CC3=CC=CC=C3)=CN=C2)N4)OC4=O)F
AZD 2716

AZD2716
- Antiplaque candidate drug
AstraZeneca INNOVATOR
(R)-7(AZD2716) a novel, potent secreted phospholipase A2 (sPLA2) inhibitor with excellent preclinical pharmacokinetic properties across species, clear in vivo efficacy, and minimized safety risk. Based on accumulated profiling data, (R)-7 was selected as a clinical candidate for the treatment of coronary artery disease.
Chiral HPLC using a Chiralcel OJ 5 μm 20×250 mm
column with heptane/EtOH/formic acid ((10:90:0.1; 15 ml/min, 40 °C, 260 nm) as mobile
phase to yield (S)-7 and (R)-7
(R)-7:tR=5.8 min [α]D20 15.4 (c 0.5, ACN), 99.7 %ee. desired
(S)-7: tR=9.2 min. 99.0 % ee. undesired
LINK
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.6b00188
![]()
SYNTHESIS
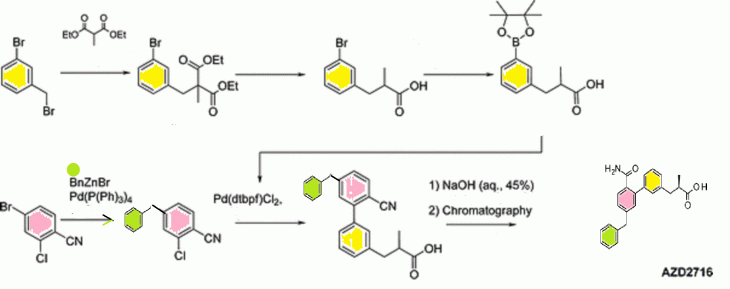
1H NMR (400 MHz, DMSO-d6): δ 1.04 (d, J = 6.6 Hz, 3H), 2.55–2.68 (m, 2H), 2.95 (dd, J = 6.1, 12.8 Hz, 1H), 4.00 (s, 2H), 7.13–7.37 (m, 13H), 7.49–7.54 (m, 1H), 12.2 (s, br, 1H).
13C NMR (151 MHz, DMSO): δ 16.7, 39.1, 40.7, 41.0, 126.3, 126.4, 127.3, 127.8, 128.0, 128.2, 128.7, 128.9, 129.2, 130.3, 135.3, 139.2, 139.5, 140.5, 141.2, 142.7, 171.3, 177.1.
HRMS (ESI): [M + H]+ m/z calcd for C24H24NO3 374.1751, found 374.1748.
1H NMR


13C NMR
An Enantioselective Hydrogenation of an Alkenoic Acid as a Key Step in the Synthesis of AZD2716
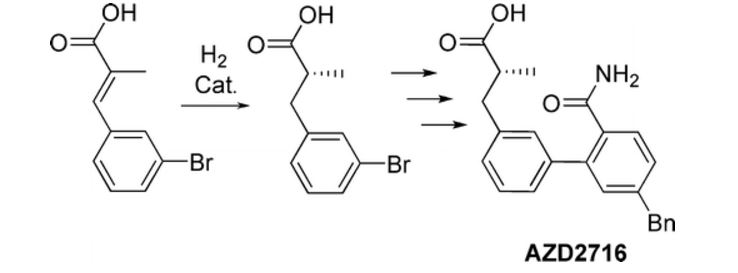
A classical resolution of a racemic carboxylic acid through salt formation and an asymmetric hydrogenation of an α,β-unsaturated carboxylic acid were investigated in parallel to prepare an enantiomerically pure alkanoic acid used as a key intermediate in the synthesis of an antiplaque candidate drug. After an extensive screening of rhodium- and ruthenium-based catalysts, we developed a rhodium-catalyzed hydrogenation that gave the alkanoic acid with 90% ee, and after a subsequent crystallization with (R)-1-phenylethanamine, the ee was enriched to 97%. The chiral acid was then used in sequential Negishi and Suzuki couplings followed by basic hydrolysis of a nitrile to an amide to give the active pharmaceutical ingredient in 22% overall yield.
Paper

Expedited structure-based optimization of the initial fragment hit 1 led to the design of (R)-7(AZD2716) a novel, potent secreted phospholipase A2 (sPLA2) inhibitor with excellent preclinical pharmacokinetic properties across species, clear in vivo efficacy, and minimized safety risk. Based on accumulated profiling data, (R)-7 was selected as a clinical candidate for the treatment of coronary artery disease.
Discovery of AZD2716: A Novel Secreted Phospholipase A2 (sPLA2) Inhibitor for the Treatment of Coronary Artery Disease
http://pubs.acs.org/doi/full/10.1021/acsmedchemlett.6b00188

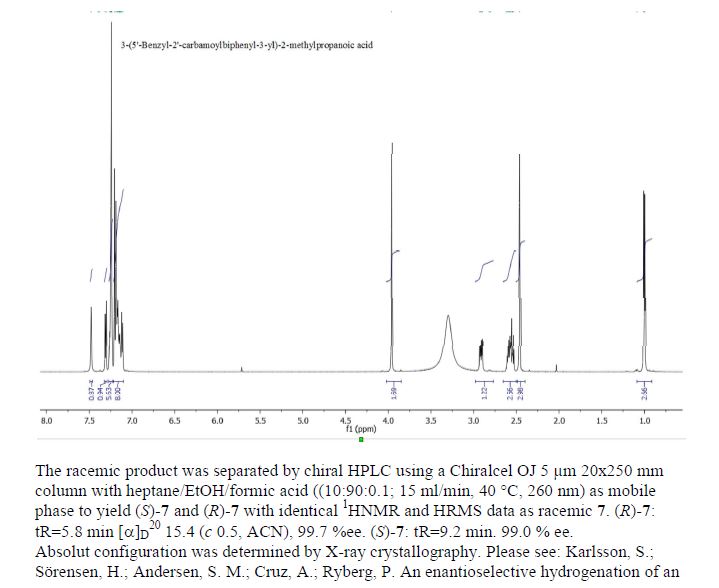
akenoic acid as a key step in the sysnthesis of AZD2716. Org. Proc. Res. Dev. 2016, 20(2),
262-269).
/////////atherosclerosis, coronary artery disease, fragment screening, fragment-based drug discovery, Secreted phospholipase A2, sPLA2, AZD2716, AZD-2716, AZD 2716, PRECLINICAL
c1c(cc(c(c1)C(=O)N)c2cccc(c2)CC(C(=O)O)C)Cc3ccccc3
Dofequidar fumarate

Dofequidar fumarate
Phase III
A P-glycoprotein inhibitor potentially for the treatment of breast cancer and non-small lung cancer (NSCLC).
![]()
MS-209; Dofequidar fumarate
CAS No. 129716-58-1 (Dofequidar FREE )
CAS No 153653-30-6 (Dofequidar fumarate 1;1)…..C34H35N3O7, 597.66
5-[3-[4-(2,2-Diphenylacetyl)piperazin-1-yl]-2-hydroxypropoxy]quinoline sesquifumarate
1-[4-(2,2-Diphenylacetyl)piperazin-1-yl]-3-(quinoliln-5-yloxy)-2-propanol sesquifumarate
1-(Diphenylacetyl)-4-[(2RS)-2-hydroxy-3-(5-quinolyloxy)propyl]piperazine sesquifumarate
CAS Number 158681-49-3, C30H31N3O3 · 1.5 C4H4O4, Molecular Weight 655.69
4-(Diphenylacetyl)-a-[(5-quinolinyloxy)methyl]-1-Piperazineethanol (E)-2-butenedioate fumarate (1:1.5), C30 H31 N3 O3 . 3/2 C4 H4 O4

Dofequidar fumarate(MS-209 fumarate), an orally active quinoline compound, has been reported to overcome MDR by inhibiting ABCB1/P-gp, ABCC1/MDR-associated protein 1, or both.
Dofequidar fumarate(MS-209 fumarate), an orally active quinoline compound, has been reported to overcome MDR by inhibitingABCB1/P-gp, ABCC1/MDR-associated protein 1, or both.
IC50 value:
Target: P-gp
in vitro: MS-209 at 3 microM effectively overcame docetaxel resistance in MDR cancer cells, and this concentration was achieved in blood plasma for > 7 h without serious toxicity [1]. MS-209 restored chemosensitivity of SBC-3 / ADM cells to VP-16, ADM, and VCR in a dose-dependent manner in vitro [2]. dofequidar inhibits the efflux of chemotherapeutic drugs and increases the sensitivity to anticancer drugs in CSC-like side population (SP) cells isolated from various cancer cell lines. Dofequidar treatment greatly reduced the cell number in the SP fraction [3]. In 4-1St cells, which are extremely resistant to ADM and VCR, MS-209 at a concentration of 3 microM enhanced the cytotoxicity of ADM and VCR, 88- and 350-fold, respectively [4].
in vivo: Treatment with docetaxel alone at the maximal tolerated dose (MTD) showed an apparent antitumor activity to an intrinsically resistant HCT-15 tumor xenograft, and MS-209 additionally potentiated the antitumor activity of docetaxel. Against a MCF-7/ADM tumor xenograft expressing larger amounts of P-gp, docetaxel alone at the MTD showed no antitumor activity, whereas the MTD of docetaxel combined with MS-209 greatly reduced MCF-7/ADM tumor growth [1]. Intravenous injection with SBC-3 or SBC-3 / ADM cells produced metastatic colonies in the liver, kidneys and lymph nodes in natural killer (NK) cell-depleted severe combined immunodeficiency (SCID) mice, though SBC-3 / ADM cells more rapidly produced metastases than did SBC-3 cells. Treatment with VP-16 and ADM reduced metastasis formation by SBC-3 cells, whereas the same treatment did not affect metastasis by SBC-3 / ADM cells. Although MS-209 alone had no effect on metastasis by SBC-3 or SBC-3 / ADM cells, combined use of MS-209 with VP-16 or ADM resulted in marked inhibition of metastasis formation by SBC-3 / ADM cells to multiple organs [2].
Dofequidar fumarate is a multidrug resistance (MDR)-reversing quinoline derivative that interacts directly with P-glycoprotein and inhibits the efflux of antitumor agents. The agent had been in phase III clinical development by Nihon Schering (now Bayer) for the treatment of advanced and recurrent breast cancer and non-small lung cancer (NSCLC) and at the National Cancer Institute in combination with docetaxel for the treatment of solid tumors. In 2000, Schering AG obtained dofequidar fumarate when Nihon Schering acquired Mitsui Pharmaceuticals, originator of the compound.

PAPER
Structure-activity relationship of newly synthesized quinoline derivatives for reversal of multidrug resistance in cancer
J Med Chem 1997, 40(13): 2047
5-[3-{4-(2,2-Diphenylacetyl)piperazin-1-yl}-2-hydroxypropoxy]quinoline 1.5Fumarate (16, MS-209)
free form of 16 (7.37 g, 70%): mp 161−162 °C; 1H-NMR (CDCl3) δ 2.2−2.8 (m, 6 H), 3.5−3.6 (m, 2H), 3.7−3.9 (m, 2H), 4.1−4.3 (m, 3H), 5.20 (s, 1H), 6.86 (d, 1H, J = 7.3 Hz), 7.2−7.4 (m, 11H), 7.59 (t, 1H, J = 8.1 Hz), 7.71 (d, 1H, J = 8.1 Hz), 8.54 (d, 1H, J = 7.3 Hz), 8.91 (dd, 1H, J = 2, 4 Hz); IR (KBr) 2954, 1630, 1587, 1268, 1091, 802, 748, 703 cm-1.
16 1.5Fumarate(1.0 g, 60%): mp 210 °C dec; 1H-NMR (DMSO-d6) δ 2.2−2.6 (m, 6H), 3.4−3.6 (m, 4H), 4.0−4.2 (m, 3H), 5.53 (s, 1H), 6.63 (s, 3H), 7.03 (d, 1H, J = 8.1 Hz), 7.2−7.4 (m, 10H), 7.5−7.7 (m, 3H), 8.61 (d, 1H, J = 8.1 Hz), 8.89 (dd, 1H, J = 1.5, 4.4 Hz); IR (KBr) 3424, 1644, 1592, 1277, 1180, 1110, 799 cm-1.
Patent
WO 2004099151
https://www.google.co.in/patents/WO2004099151A1?cl=en
A method for producing the purest rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyI] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate and the purest rac-1 – {4- [2-hydroxy-3- (5-quinoly loxy) propylene l] piperazin-1-yl} -2,2-diphenylethan-1 -one fumarate
The invention relates to a method for producing the purest rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate as well as rac -1- {4- [2- hydroxy-3- (5-quinolyloxy) propyl] piperazin-1-yl} -2,2-diphenylethan-1-one fumarate with a purity of at least 99.55%
The multidrug resistance modulator rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] – piperazin-1-yl} -2,2-diphenylethan-1 -one fumarate, its preparation and use as carcinostatic drug is described as well as other derivatives of this compound in EP 575,890.
According to the process described in EP 575 890 A process for the preparation of pure rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-dϊphenylethan-1-one fumarate is first by coupling the two modules epoxiline (B) (5- (2,3-epoxypropoxy) – quinoline) and Diphenpiperazid (C) (N- (2,2-Diphenylacetyl) piperazine), the free base 5- [3- {4- (2,2-diphenylacetyl) piperazin-1-yl} -2-hydroxypropoxy] quinoline isolated as a crude product. This implementation includes two sub-stages. First, the Epoxylat with hydroxyquinoline (A) is reacted. In the second step the epoxiline (B) (5- (2,3-epoxypropoxy) -quinolin) by Diphenpiperazid (C) (N- (2,2-Diphenylacetyl) piperazine) is opened, it gives the secondary alcohol (D). This reaction takes place in ethanol, water catalyzes the conversion. The workup / isolation is then carried out by precipitation from acetone / water and drying under vacuum at 60 ° C.
The overall reaction results from the following scheme:

On the isolation of the free base, the many impurities (purity of the crude product is typically about 80%), joins in the next step a very expensive cleaning procedures. After charcoal treatment of the free base and the formation of the fumarate in methanol, the free base is again prepared by treatment with dilute sodium hydroxide solution for purification. Subsequently, as the last step, repeated fumarate formation. The two fumarate formations are procedurally identical and differ only in the batch size (T. Suzuki et al., J. Med. Chem. (1997) 40, 2047) (JP 2000281653). Starting from the crude free base, the typical yield for this laboratory cleaning sequence 45% of theory.
A disadvantage of this method is not only the low yield (about 50% loss in the final stage), but also the complex technical implementation, which binds many operational capacities and thus caused increased costs. A particular disadvantage is the extremely poor filterability of the free base, the filter must be dried partially over several weeks.
Despite the high procedural expenses according to this known method, the extremely high purity requirements of rac-1 – {4- [2-hydroxy-3- (5- quinolyloxy) propyl] piperazine-1-yl} -2,2-diphenylethane-1 -one fumarate not always be achieved completely satisfactory.
. Furthermore provides the method described in EP 575 890 any reasonable results during scale-up an overview of the individual reactions are the following scheme:
It has now been found that these known disadvantages can be overcome with the process of this invention. In the process of this invention also the epoxiline (B) and Diphenpiperazid (C) is first coupled by opening of the epoxide. But is not the free base (D) but after the addition of solid fumaric acid directly the fumarate salt (E) is then isolated as a crude product.
The present application thus provides a process for the preparation of pure rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1 -one fumarate , which is characterized in that firstly
a) a Epoxytosylat of structure I
OTs
(0 with

b) 5-hydroxyquinoline (II)
(II) and cesium carbonate in a suitable solvent and at a suitable temperature to 5- (2,3-epoxypropoxy) -quinolin of formula III

allowed to react, and then the 5- (2,3-epoxypropoxy) -quinolin of formula III
c) with N- (2,2-Diphenylacefyl) piperazine of the formula IV

in a suitable solvent and at a suitable temperature followed by the addition of solid fumaric acid to the crude rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] – piperazin-1-yl} -2,2-diphenylethane 1-one fumarate of the formula V
And subsequently reacting (V)
d) the thus formed crude rac-1 – fumarate {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1 -one (V) is isolated and is dissolved in a solvent mixture of methanol and methylene chloride, is treated with activated carbon and subsequently filtered through a pressure filter having silica gel as column material, and the thus obtained pure rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate (V) is crystallized from a suitable alcohol.
Preparation Example
Preparation of rac-1 – 4- [2-Hy droxy-3- (5-quinolyloxy) propylene l] -piperazin-1 -yl> -2,2-diphenylethan-1-one fumarate
A) Under nitrogen, 44.2 g of 5-hydroxy-quinoline and 151.9 g of cesium carbonate with 560 ml acetone will give at room temperature together and stirred for 30 minutes at 60 ° C bath temperature. At 50 ° C internal temperature 73.0 g of 5- (2,3-epoxypropoxy) -quinolin dissolved in
153.3 g of dichloromethane, admit. The mixture is stirred at 50 ° C for two hours. The mixture is filtered at 50 ° C. The filter residue (inorganic salts) is washed with 560 ml of 50 ° C warmed acetone. 85.4 g are then N- (2,2-diphenyl-acetyl) piperazine admit and concentrated at a bath temperature of 40 ° C under vacuum to 374 g final weight. It will then add 374 g of demineralized water and 2
Stirred at 40 ° C hours. Then 255 g of acetone and 201 g of demineralized water will admit. The mixture is cooled to room temperature and 89.1 g of fumaric acid are in solid form to Gege-ben. It is stirred for 60 minutes at 60 ° C bath temperature and then stirred at 0 ° C for 2 hours. The solid is suction filtered and washed with 150 ml of ice-cold methanol. The filter residue is dried at 60 ° C under vacuum.
Yield: 65 – 85% of theory
B) 56.0 g of the thus prepared rac-1 – {4- [2-hydroxy-3- (5-quinolyIoxy) propyl] -piperazin-1-yl} – 2,2-diphenylethan-1-one fumarate were nitrogen and treated at room temperature with 5.6 g of activated carbon, Norit SX plus, 672 ml of methanol and 1008 ml of dichloromethane. The resulting suspension is stirred at a bath temperature of 75 ° C to warm to reflux temperature and refluxed for 30 min. At an internal temperature of 40 ° C is rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate in solution. The mixture is then filtered hot through 300% silica gel and the silica gel with 560 ml of a mixture of 168 ml of methanol and 392 ml of dichloromethane at room temperature RT. The solution is concentrated at a bath temperature of 40 ° C and an initial vacuum of 400 mbar to a final volume of 517 ml. The ultimate vacuum of 350 mbar. The distilled volume is about the difference in volume (about 1, 7 I). There are 404 ml of methanol was added so that a final volume of 921 ml is achieved. The solution is cooled to 0 ° C, whereupon the product precipitates. The resulting suspension is stirred for 2 hours at 0 ° C and then filtered through a paper filter. The filter residue is washed with 56.0 ml of ice-cold methanol. The filter residue is dried at 60 ° C and under vacuum at 100 mbar for 10 hours.
Yield (. Uncorr): 47.29 g (84.45% FS)
Purity: 99.65% (HPLC, 100% method)
References on Dofequidar fumarate
http://jco.ascopubs.org/content/25/4/411.full.pdf
SEE………http://apisynthesisint.blogspot.in/2016/01/ms-209-dofequidar-fumarate.html
///////////MS-209, Dofequidar fumarate, PHASE 3
What are “complex manufacturing processes”? A recent reply from the EMA
The Variations Regulation (EC) no. 1234/2008 of the European Commission defines the procedure for variations of existing marketing authorisations. The “detailed guidelines for the various categories of variations“, which were published in the consolidated version in August 2013 in the European Official Journal, explain the interpretation and application of this Variations Regulation.
Although the “detailed guidelines” describe a number of scenarios of possible variations in some detail, there are formulations in the Guideline text which require clarification due to their blur. The EMA adopted such a case in a recent update of itsquestions and answers collection “Quality of Medicines Questions and Answers: Part 1” to concretise the case through a statement.
It is about the term “complex manufacturing processes”, which is used in two scenarios associated with type II variations (found in the “detailed guidelines” p 40ff):
- Replacement or addition of a manufacturing site for part or all of the manufacturing process of the finished product (Guideline change code B.II.b.1)
…
c) Site where any manufacturing operation(s) take place, except batch release, batch control, and secondary packaging, for biological/immunological medicinal products, or for pharmaceutical forms manufactured by complex manufacturing processes. - Change in the batch size (including batch size ranges) of the finished product (Guideline change code B.II.b.4)
…
d) The change relates to all other pharmaceutical forms manufactured by complex manufacturing processes .
The EMA now clarified this term as follows:
- Guideline Change Code B.II.b.1: Complex manufacturing processes are given when the understanding of the relation between quality characteristics of the product and its in vivo efficacy is lacking. This is often the case in innovative medicines such as products of nanomedicine.
- Guideline Change Code B.II.b.4: Complex manufacturing processes are those which contain one or more sub-steps, where a scale-up can lead to problems.
In both scenarios, the approving authority will decide on a case by case basis. If the applicant submits the variation as a Type IB, he must provide a valid justification that the production process is not “complex”. However, in doubt the authority may upgrade the variation to a Type II. Therefore, the EMA recommends that the applicant clarifies the situation with the authority before submitting the variation.
What are “complex manufacturing processes”? A recent reply from the EMA………..http://www.gmp-compliance.org/enews_05072_What-are-%22complex-manufacturing-processes%22-A-recent-reply-from-the-EMA.html
