
Home » 2015 (Page 6)
Yearly Archives: 2015
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zydus gets USFDA nod for clinical trials of Saroglitazar

November 19, 2015
New Delhi: Zydus Cadila has received US health regulator’s nod to initiate phase II clinical trials of Saroglitazar, its new drug for treating high fat levels in body due to diabetes, obesity, and sedentary habits.
“United States Food and Drug Administration (USFDA) has endorsed company’s plan to initiate a phase II clinical trial of Saroglitazar in patients with severe hypertriglyceridemia,” Zydus Cadila said in a statement.
http://www.medicaldialogues.in/zydus-gets-usfda-nod-for-clinical-trials-of-sarolitazar/

//////////////
Mirogabalin


| Originator |
Daiichi Sankyo
|
|---|---|
| Therapeutic Claim |
Treatment of fibromyalgia
|
Phase III clinical trials at Daiichi Sankyo for the treatment of pain associated with fibromyalgia
| Class |
Analgesic drugs (small molecules)
|
|---|---|
| Mechanism of action |
CACNA2D1 protein modulators
|
SYNTHESIS
 SEE
SEE
[(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid benzenesulfonatee

DESIRED
[(1S,5R,6R)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid , optical isomer of the compound
 UNDESIRED
UNDESIRED
Mirogabalin (DS-5565) is a drug developed by Daiichi Sankyo and related to drugs such as gabapentin and pregabalin. Similarly to these drugs, mirogabalin binds to the α2δ calcium channels (1 and 2), but with significantly higher potency than pregabalin. It has shown promising results in Phase II clinical trials for the treatment of diabetic peripheral neuropathic pain,[1][2] and is currently in Phase III trials.
Mirogabalin, a voltage-dependent calcium channel subunit alpha-2/delta-1 ligand, is in phase III clinical trials at Daiichi Sankyo for the treatment of pain associated with fibromyalgia. The company is also conducting phase III clinical studies for the treatment of chronic pain and pain associated with diabetic peripheral neuropathy.
Mirogabalin besylate
cas 1138245-21-2
UNII: 01F4FRP8YL
C12-H19-N-O2.C6-H6-O3-S, 367.4635
Bicyclo(3.2.0)hept-3-ene-6-acetic acid, 6-(aminomethyl)-3-ethyl-, (1R,5S,6S)-, benzenesulfonate (1:1)
SEE
Tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate…..http://www.google.com/patents/US20140094623?cl=zh
PATENT
WO 2009041453
https://www.google.co.in/patents/EP2192109A1
(Example 21) [(1S,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid (exemplary compound No: 8, optically active form of the compound of Example 8)
(21-a) Resolution of tert-butyl (±)-[(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate
Tert-butyl (±)-[(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (230 g, 778 mmol) was resolved using Chiralpak IC (N-Hex:EtOH=98:2, 1.0 mL/min, 40°C) manufactured by Daicel Chemical Industries, Ltd. to respectively obtain 115 g of a peak 1 (retention time: 5.2 min) and 93.7 g of a peak 2 (retention time: 6.3 min).
(21-b) Tert-butyl ([(1R,5S,6S)-6-(tert-butoxycarbonylamino)methyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate
Tert-butyl [(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (peak 1, 7.0 g, 23.7 mmol) was dissolved in ethanol (60 mL) and water (21 mL). To the solution, iron powder (13.27 g, 237 mmol) and ammonium chloride (628.1 mg, 11.9 mmol) were added, and the mixture was stirred for 5.5 hours under heating to reflux. The mixture was allowed to cool, then diluted with saturated saline, a saturated aqueous solution of sodium bicarbonate, and ethyl acetate, and filtered through Celite to remove insoluble matter. The filtrate was separated into organic and aqueous layers. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure to obtain a pale yellow oil substance (7.02 g). This substance was dissolved in dichloromethane (200 mL). To the solution, (Boc)2O (5.25 g, 25 mmol) and triethylamine (5.01 g, 50 mmol) were added, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and the residue was then purified by silica gel chromatography to obtain the title compound of interest as a pale yellow oil substance (8.82 g, <100%). (21-c) [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
A 4 N hydrochloric acid-ethyl acetate solution (100 mL) was added to tert-butyl (1R,5S,6S)-[6-(tert-butoxycarbonylaminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (9.82 g, 23.7 mmol), and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure. The residue was dissolved in dichloromethane. To the solution, triethylamine was added dropwise, and the resulting powder was collected by filtration, then washed with dichloromethane, and then dried to obtain 4.02 g of a white powder. This powder was washed with ethanol and ethyl acetate to obtain the title compound of interest as a white powder (2.14 g, 43%).
(Example 31) [(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid benzenesulfonate (exemplary compound No: 8, optically active benzenesulfonate)
(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid (4.50 g, 20.6 mmol) was dissolved by heating in a 1 M aqueous solution (22.7 mL) of benzenesulfonic acid monohydrate, and the solution was then allowed to cool to room temperature. The resulting solid was collected by filtration. The solid was washed with water (15 mL) and then dried using a vacuum pump to obtain the compound of interest as a colorless solid (6.45 g, 77%).
PATENT
JP 2010241796
PATENT
WO 2012169475
-
 Reference Example 1[6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
Reference Example 1[6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid(1-a) Ethyl 4-ethyl-3-hydroxyhept-6-enoateSodium hydride (>63% oil, 2.09 g, 55 mmol) was added to a solution of ethyl 3-oxohexanoate (7.91 g, 50 mmol) in tetrahydrofuran (50 mL) under ice cooling, and the mixture was stirred in this state for 10 minutes. To the reaction solution, n-butyllithium (1.58 M solution in hexane, 34.8 mL, 55 mmol) was added dropwise, and the mixture was further stirred for 10 minutes under ice cooling. Then, allyl bromide (4.7 mL, 55 mmol) was added thereto, and the mixture was stirred in this state for 1 hour and then further stirred at room temperature for 4 hours. To the reaction solution, 1 N hydrochloric acid and a saturated aqueous solution of ammonium chloride were added, followed by extraction with n-pentane. The organic layer was washed with saturated saline and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was dissolved in ethanol (80 mL). To the solution, sodium borohydride (1.51 g, 40 mmol) was added under ice cooling, and the mixture was stirred in this state for 2 hours. 1 N hydrochloric acid (50 mL) was added thereto, and the mixture was stirred for 30 minutes. Then, saturated saline was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography to obtain the compound of interest as a pale yellow oil substance (3.64 g, 37%, mixture of diastereomers).
(1-b) 4-Ethyl-3-hydroxyhept-6-enoic acid
(1-c) Tert-butyl 3-ethylbicyclo[3.2.0]hept-3-en-6-ylideneacetate
-
1H-NMR (400 MHz, CDCl3): δ ppm:
-
Major isomer: 1.06 (3H, t, J=7.4 Hz), 1.45 (9H, s), 2.07-2.22 (3H, m), 2.59-2.70 (2H, m), 2.87-2.96 (1H, m), 3.30 (1H, ddt, J=8.6, 18.4, 2.7 Hz), 3.86-3.88 (1H, m), 5.22-5.23 (1H, m), 5.45-5.47 (1H, m).
-
Minor isomer: 1.08 (3H, t, J=7.3 Hz), 1.49 (9H, s), 2.07-2.21 (3H, m), 2.43-2.47 (1H, m), 2.59-2.70 (1H, m), 2.75-2.85 (1H, m), 2.87-2.96 (1H, m), 4.28-4.31 (1H, m), 5.35-5.38 (1H, m), 5.45-5.47 (1H, m).
(1-d) Tert-butyl [3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate
(1-e) [6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
1H-NMR (400 MHz, CDCl3): δ ppm: 0.91 (3H, t, J=7.5 Hz), 1.28 (3H, t, J=7.2 Hz), 1.43-1.55 (2H, m), 1.98-2.28 (2H, m), 2.45-2.48 (2H, m), 2.88-2.93 (1H, m), 4.07-4.10 (1H, m), 4.10-4.20 (2H, m), 5.01-5.09 (2H, m), 5.75-5.86 (1H, m).Ethyl 4-ethyl-3-hydroxyhept-6-enoate (3.64 g, 18.2 mmol) was dissolved in a 2 N solution of potassium hydroxide in methanol (120 mL), and the solution was stirred overnight at room temperature. From the reaction solution, the solvent was distilled off under reduced pressure. To the residue, a 1 N aqueous sodium hydroxide solution (200 mL) was then added, followed by extraction with diethyl ether. The aqueous layer was made acidic by the addition of concentrated hydrochloric acid under ice cooling, followed by extraction with diethyl ether again. The organic layer was washed with saturated saline and dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure to obtain the compound of interest as a pale yellow oil substance (3.14 g, <100%, mixture of diastereomers).1H-NMR (400 MHz, CDCl3): δ ppm: 0.91-0.96 (3H, m), 1.39-1.52 (3H, m), 2.01-2.28 (2H, m), 2.52-2.55 (2H, m), 4.05-4.15 (2H, m), 5.03-5.10 (2H, m), 5.74-5.86 (1H, m).4-Ethyl-3-hydroxyhept-6-enoic acid (3.13 g, 18.2 mmol) was dissolved in acetic anhydride (15 mL). To the solution, potassium acetate (4.27 g, 43.6 mmol) was added, and the mixture was stirred at room temperature for 100 minutes. The reaction solution was heated to reflux and stirred for 3.5 hours to form “3-ethylbicyclo[3.2.0]hept-6-en-6-one” in the reaction solution. To the reaction solution, ice water and toluene were then added, and this mixture was stirred overnight at room temperature. The mixture was separated into aqueous and organic layers by the addition of saturated saline (50 mL) and toluene (20 mL). Then, the organic layer was washed with a 1 N aqueous sodium hydroxide solution and saturated saline in this order, then dried over anhydrous magnesium sulfate, and filtered. The filtrate was added to a reaction solution prepared by adding sodium hydride (>65% oil, 761.9 mg, 20 mmol) to a solution of tert-butyl dimethoxyphosphorylacetate (4.48 g, 20 mmol) in tetrahydrofuran (50 mL) under ice cooling, and the mixture was further stirred for 1 hour. The reaction solution was separated into aqueous and organic layers by the addition of a saturated aqueous solution of ammonium chloride and saturated saline. The aqueous layer was subjected to extraction with ethyl acetate. The organic layers were combined, then washed with saturated saline, and then dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography to obtain the compound of interest as a pale yellow oil substance (1.32 g, 31%, E/Z mixture).Tert-butyl [3-ethylbicyclo[3.2.0]hept-3-en-6-ylideneacetate (1.32 g, 5.63 mmol) was dissolved in nitromethane (7 mL). To the solution, 1,8-diazabicyclo[5.4.0]undec-7-ene (1.2 mL, 7.3 mmol) was added, and the mixture was heated with stirring at 50 to 60° C. for 7 hours. The mixture was allowed to cool, and a saturated aqueous solution of potassium dihydrogen phosphate was then added thereto, followed by extraction with ethyl acetate. Then, the organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography to obtain the compound of interest as a colorless oil substance (1.39 g, 84%).1H-NMR (400 MHz, CDCl3): δ ppm: 1.09 (3H, t, J=7.4 Hz), 1.46 (9H, s), 1.52 (1H, dd, J=7.6, 13.2 Hz), 2.06 (1H,d, 16.6 Hz), 2.14 (2H, q, J=7.4 Hz), 2.30 (1H, ddd, J=2.4, 7.6, 13.2 Hz), 2.47 (2H, s), 2.49 (1H, dd, J=7.6,16.6 Hz), 2.86 (1H, quint, J=7.6 Hz), 3.21-3.22 (1H, m), 4.75 (1H, d, J=11.7 Hz), 4.84 (1H, d, J=11.7 Hz), 5.27 (1H, s).Tert-butyl [3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (1.09 g, 4.71 mmol) was dissolved in ethanol (10 mL) and water (5 mL). To the solution, iron powder (1.32 g, 23.5 mmol) and ammonium chloride (249.6 mg, 4.71 mmol) were added, and the mixture was stirred for 2 hours under heating to reflux. The mixture was allowed to cool, then diluted with saturated saline, a saturated aqueous solution of sodium bicarbonate, and ethyl acetate, and filtered through Celite to remove insoluble matter. The filtrate was separated into organic and aqueous layers. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate, and the solvent was then distilled off under reduced pressure. To the residue, a 4 N solution of hydrochloric acid in ethyl acetate (20 mL) was added, and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure. The residue was suspended in dichloromethane. To the suspension, triethylamine was added dropwise, and the resulting powder was collected by filtration, then washed with dichloromethane, and then dried to obtain the compound of interest as a white powder (425.1 mg, 43%).1H-NMR (400 MHz, CD3OD): δ ppm: 1.10 (3H, t, J=7.4 Hz), 1.48 (1H, dd, J=7.5, 12.5 Hz), 2.03-2.08 (2H, m), 2.14 (2H, q, J=7.4 Hz), 2.46 (1H, d, J=16.2 Hz), 2.46-2.53 (1H, m), 2.51 (1H, d, J=16.2 Hz), 2.85 (1H, quint, J=7.5 Hz), 3.09-3.10 (1H, m), 3.14 (1H, d, J=13.0 Hz), 3.18 (1H, d, J=13.0 Hz), 5.38 (1H, dd, J=1.7, 3.7 Hz).(Step of Performing Optical Resolution from Diastereomeric Mixture)- Reference Example 2Tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate
-
 Acetonitrile (4.7 L, 8.6 v/w) was added to tert-butyl [6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (627.0 g, net: 543.6 g, 2.05 mol, 85:15 diastereomeric mixture), and the mixture was stirred at 40° C. To the reaction solution, D-mandelic acid (116.3 g, 0.76 mmol, 0.37 eq.) was added, and the mixture was stirred at 40° C. for 1 hour and then allowed to cool slowly to 3° C. After stirring at 3° C. for 1 hour, the resulting crystal was collected by filtration. Then, the crystal was dried under reduced pressure under the condition of 40° C. to obtain tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate as a white powder (251.2 g, yield: 29.4%, 97.6% ee, 99.6% de).
Acetonitrile (4.7 L, 8.6 v/w) was added to tert-butyl [6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (627.0 g, net: 543.6 g, 2.05 mol, 85:15 diastereomeric mixture), and the mixture was stirred at 40° C. To the reaction solution, D-mandelic acid (116.3 g, 0.76 mmol, 0.37 eq.) was added, and the mixture was stirred at 40° C. for 1 hour and then allowed to cool slowly to 3° C. After stirring at 3° C. for 1 hour, the resulting crystal was collected by filtration. Then, the crystal was dried under reduced pressure under the condition of 40° C. to obtain tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate as a white powder (251.2 g, yield: 29.4%, 97.6% ee, 99.6% de). -
1H-NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (3H, t, J=7.6 Hz), 1.28-1.35 (1H, m), 1.39 (9H, s), 1.96-2.11 (4H, m), 2.28 (1H, d, J=15.6 Hz), 2.33 (1H, d, J=15.6 Hz), 2.36-2.40 (1H, m), 2.72 (1H, quint, J=7.6 Hz), 3.00 (1H, d, J=13.2 Hz), 3.03 (1H, d, J=13.2 Hz), 3.31 (1H, br s), 4.54 (1H, s), 5.21-5.23 (1H, m), 7.13-7.25 (3H, m), 7.35-7.37 (2H, m).
-
[α]20 D −104.4° (C=0.108, MeOH).
-
Anal. calcd for C24H35NO5: C, 69.04; H, 8.45; N, 3.35; Found C, 69.15; H, 8.46; N, 3.46.
-



PATENT
WO 2012169474
PATENT
a compound having the formula (Va) (and its enantiomers), and to carry out optical resolution by chloride with optically active organic amine, and is a process for preparing a compound having the general formula (VIa) .
[Formula 19] The solvent used in this step, MTBE, CPME, ethers such as THF; aromatic hydrocarbons such as toluene; esters such as ethyl acetate; EtOH, alcohols such as diisopropyl alcohol CH; s three nitriles such as CN; ketones such as acetone; or is a mixed solvent of these solvents and water, preferably toluene, ethyl acetate, CH 3 CN, are MTBE, More preferably, toluene, MTBE. Optically active organic amine used in this step, preferably, (1R, 2R) -trans-1- amino-2-indanol, (S) -2- phenylglycinol, (R) -1- ( p- tolyl) ethylamine, (1R, 2S) -2- amino-1,2-diphenyl ethanol, (S) -1- (2- naphthyl) ethylamine, (R) -1- (4- bromophenyl) ethylamine, (1S, 2R) – (+) – 1- amino-2-indanol is a L- phenylalaninol, etc., more preferably, (1R, 2R) -trans-1- amino-2-indanol, (S ) -2-phenylglycinol. Equivalent of the optically active organic amine to be used have the general formula (Va) compound having a relative (and its enantiomers) are 0.5-1.1 equivalents. The reaction temperature of this step is such as about 0-50 ℃, preferably, after aging the crystals at about 10-30 ℃, is obtained by filtering the compound of formula (VIa). The time required to chloride present step is not particularly limited, but is usually 4 to about 48 hours. In this step, (1) with respect to (Va) compound with (and its enantiomers), directly to a compound of formula (VIa) with the desired configuration by the action of the above-mentioned optically active amine How to get, or, with respect to (2) compounds having formula (Va) (or its enantiomer), first, quinine, (1S, 2S) -trans-1- amino-2-indanol, (R) -2- by the action of an optically active amine such as phenylglycinol, it allowed to temporarily deposit the enantiomer having the unnecessary configuration, after removing the precipitate by filtration, against followed by compound obtained from the filtrate, (1R, 2R ) -trans-1- amino-2-indanol, by the action of optically active amines such as (S) -2- phenylglycinol, to precipitate the salt of the compound of formula (VIa) with the desired configuration How to get Te, one of the methods is used.
 Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).
Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).[Formula 20] specific production method according to the present method will be described later as a reference example.

Formula (V) or a compound having the general formula (VI) from (and / or its enantiomer) is a process for preparing a compound of formula (VII) (and / or its enantiomer), the general formula (V) is a compound having (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst, reduction with a solvent, or a compound having the general formula (VI) (and / or its enantiomer) solution compounds having the general formula (V) obtained by salt (and / or its enantiomer) solution, under a hydrogen atmosphere to carry out a reduction reaction in the presence of a metal catalyst, by a compound of formula (VII) This is a method of manufacturing a.
Formula 21] (1) Kaishio step formula compound with a (VI) (and / or its enantiomer) is suspended in an organic solvent, washed with an aqueous solution obtained by adding an acid, by liquid separation and the organic layer , compounds having general formula (V) (and / or its enantiomer) solution containing it can get. The solvent used in this step include aromatic hydrocarbons such as toluene, ethers such as MTBE, an ester such as ethyl acetate, and the like, preferably toluene, or is MTBE. Acid used in this step is not particularly limited, hydrochloric acid, sulfuric acid, phosphoric acid, citric acid, malonic acid can be used.

compounds having the general formula (V) (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst was reduced in a solvent, a cyano group (or a nitro group) and an amino group It is converted into, and is a step for preparing a compound of formula (VII). This reaction is usually carried out in a neutral or basic conditions.
The solvent used in this step include aromatic hydrocarbons such as toluene, MTBE, ethers such as THF, alcohols of C1-C4, or is water, preferably toluene, MTBE, or water , and the Particularly preferred is water.
Metal catalyst used in this step, vinegar Sanskrit nickel, sponge cobalt, or palladium – is carbon, preferably, sponge nickel (eg, Kawaken Fine Chemicals Co., Ltd. of PL-9T, NDT-65, NDT- 90, NDHT-90M, NDHT-M3, and the like, or, Nikko Rika Co., Ltd. R-100, R-200, such as R-205, R-211, R-2311), or, sponge cobalt (for example, the river Research ODHT-60 manufactured by Fine Chemical Co., Ltd., OFT-55, or the like, or is a Nikko Rika Co., Ltd. R-400, R-400K, such as R-401, R-455, such as A-8B46 manufactured by Johnson Matthey) .
In this step, when carrying water as a solvent is usually added to the base. As the base used, preferably an inorganic base, particularly preferred are lithium hydroxide, sodium hydroxide, alkali metal hydroxides such as potassium hydroxide.
In this step, by the addition of aqueous ammonia, it is possible to improve the yield, it is not necessarily added aqueous ammonia.
In this step, by the addition of dimethyl polysiloxane, it is possible to suppress the generation of bubbles from the reaction liquid, it is not necessarily added dimethylpolysiloxane.
The reaction temperature in this step is about 20-60 ℃, preferably, is about 30-50 ℃.
The reaction time of this step, the raw material is not particularly limited as long as it is a time that is substantially consumed, it is usually 2 to about 12 hours.
In this step, after the completion of the reaction, the catalyst was removed by filtration, by adding an acid to the filtrate, by then crystallizing the compound of formula (VII), and filtering and washing the precipitate, pure products a you can get.
Chemical Formula 22] The solvent used in this step include water, anisole, aqueous acetone, water CH 3 CN, MTBE water – acetone, anisole – acetate, anisole – acetone, anisole – acetate – acetone, acetone – water -CH 3 CN single like, or it is a mixed solvent, preferably, water, anisole. The organic acid used in this step is an organic acid that is pharmacologically today preferably a benzenesulfonic acid. Equivalent of the organic acid used in this step is preferably a compound having the formula (VII) with respect to (and / or its enantiomer) is about 1.00-1.10 equivalents. This step is carried out in the range of usually about -15-50 ℃, preferably, after aging the crystals at a temperature of about -10-30 ℃, filtration, by washing, the general formula (VIII) a compound having a (and / or its enantiomers) get. The time required for chloride in this step is not particularly limited, but is usually 1 to about 24 hours.
 In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.
In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.content of the enantiomers represented by the formula (XI): 1.0% less than
the formula (XII) and the double bond represented by the formula (XIII) The total content of regioisomers: less than 0.5%
(Note that each content is calculated from the area percentage of the free form of formula (IX) (VII) in the by test High Performance Liquid Chromatography)
[formula 23] [of 24]


The internal standard substance in a magnetic resonance spectra (NMR), and using tetramethylsilane and abbreviations indicate the multiplicity, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and brs = It shows a broad singlet.
In the name of the compound, “R” and “S” indicate the absolute configuration at the asymmetric carbon. Furthermore, “RS” and “SR” indicates that the asymmetric carbon atom is racemic. In addition, “(1RS, and 5SR) -” if such a can shows the relative arrangement of the 1-position and the 5-position, as well shows only one of the diastereomers, its diastereomers are racemic We show that.
In the name of the compound, “E” and “Z” indicates the arrangement of positional isomers in the structure of the compound having a position isomerism.


(Example 7) [(1R, 5S)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] propane two acid diethyl Diethyl [(1R, 5S) -3-ethylbicyclo [3.2.0 ] hept-3-en-6-Ylidene] Propanedioate [of 31] to CPME (159 mL), 0 ° C with Ti (Oi-Pr) 4 (16.0 mL, 54.6 mmol) After addition of, TiCl 4 and stirred for 1 hour at (18.0 mL, 164 mmol) and over 8 minutes was added dropwise 0 ° C. Then diethyl malonate (25.72 g, 161 mmol), was added (1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (19.87 g, 146 mmol), 30-40 ° it was stirred for 4 hours at C. The reaction was quenched with water (100 mL), and extracted with toluene (40 mL). After the organic layer is concentrated under reduced pressure, to obtain a crude product of the title compound as a yellow oil (43.61 g).

(Example 8) [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] propane diacid di -tert- butyl (racemic) Di-tert-butyl [( RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] Propanedioate (Racemate) [of 32] with respect to THF (30 mL), and TiCl at 0 ° C 4 and (1.6 mL, and the mixture was stirred for 30 minutes was added 14.7 mmol). Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (1.00 g, 7.34 mmol), malonic acid di -tert- butyl (1.91 g, 8.81 mmol) was added After stirring for 1.5 hours, it was added pyridine (2.2 mL, 29.4 mmol). 0 ° 3.5 hours after stirring at C, and subjected to stirring overnight with warming to room temperature, quenched with water (10 mL), and extracted two times with toluene (10 mL). After washed with saturated brine (10 mL), the solvent was distilled off under reduced pressure, silica gel column chromatography (hexane: ethyl acetate = 20: 1) and subjected to purification to give the title compound (2.26 g, 92% ). 1 H NMR (CDCl 3 ) (500 MHz): delta = 1.07 (3H, t, J = 7.5 Hz), 1.47 (9H, s), 1.52 (9H, s), 2.06-2.14 (2H, M), 2.16 -2.24 (1H, m), 2.60-2.69 (2H, m), 2.90 (1H, quint, J = 7.0 Hz), 3.25 (1H, ddd, J = 18.6, 8.5, 3.5 Hz), 4.12-4.23 (1H , m), 5.36 (1H, s).

(Example 9) 5 – [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -2,2-dimethyl-1,3-dioxane -4-6- dione (racemic) 5 – [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] 2,2-dimethyl-1,3-dioxane-4-6-dione (Racemate) [of 33] THF for (80 mL), TiCl at 0 ° C 4 was stirred for 10 minutes was added (4.5 mL, 41 mmol). Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (2.81 g, 20.6 mmol), Meldrum’s acid (3.57 g, 24.8 mmol) was added and after stirring for 50 minutes , pyridine (6.53 g, 82.6 mmol) it was added. After 1.5 h stirring at 0 ° C, and subjected to stirring overnight with warming to room temperature, quenched with water (80 mL), and extracted three times with toluene (50 mL). The organic layers with saturated brine (50 mL), washed with 1 M HCl (10 mL), after distilling off the solvent, silica gel column chromatography (hexane: ethyl acetate = 9: 1-6: 1) to perform purification, as a white solid to give the title compound (4.51 g, 83.2%). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.05 (3H, t, J = 7.6 Hz), 1.69 (3H, s), 1.71 (3H, s), 2.11 (2H, Q, J = 7.6 Hz ), 2.20-2.35 (1H, m), 2.65-2.85 (1H, m), 2.92-3.13 (2H, m), 3.47-3.63 (1H, m), 4.45-4.59 (1H, m), 5.43 (1H , s). 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.1, 24.3, 27.59, 27.64, 34.1, 42.3, 42.8, 60.7, 104.4, 108.5, 119.4, 150.3, 160.1, 160.7.

(Example 10) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid dimethyl Dimethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate [of 34] Dimethyl [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en- 6-ylidene] propanedioate (517 mg, 1.66 mmol) was dissolved in MeOH (5.2 mL), was added sodium cyanide (90 mg, 1.84 mmol) at room temperature and stirred for 2 hours at room temperature. After quenching with 10% aqueous acetic acid (5 mL), and extracted three times with ethyl acetate (5 mL), the solvent was distilled off under reduced pressure to give the title compound as an oil (667 mg). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.08 (3H, t, J = 7.6 Hz), 1.80 (1H, dd, J = 12.4, 8.0 Hz), 2.01-2.22 (3H, M), 2.54 (1H, dd, J = 16.8, 7.6 Hz), 2.73 (1H, ddd, J = 12.8, 8.8, 2.8 Hz), 3.18 (1H, quint, J = 7.6 Hz), 3.67 (1H, s), 3.78 ( . 3H, s), 3.82 (3H, s), 5.16-5.28 (1H, M) 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.2, 24.4, 32.1, 37.5, 39.2, 42.5, 52.9, 53.0 , 54.6, 55.0, 118.8, 123.2, 153.9, 166.62, 166.63.

(Example 11) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid diethyl Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate [of 35] Diethyl obtained by the method shown in Example 7 [(1R, 5S) -3-ethylbicyclo [3.2 .0] hept-3-en-6-Ylidene] Propanedioate crude product (43.61 g, 146 mmol) was dissolved in EtOH (262 mL) and was added sodium cyanide (7.15 g, 146 mmol) at room temperature , it was stirred for 4 hours at room temperature. Acetate (8.76 g), after the reaction quenched with water (180 mL), the solvent it was concentrated to approximately 340 mL under reduced pressure. Water was added (80 mL), then extracted three times with ethyl acetate (150 mL), the solvent was distilled off under reduced pressure to give the title compound as an oil (HPLC quantitative value: 44.29 g, 96.3% (( 1R, 5S) -3-Ethylbicyclo [3.2.0] total yield from hept-3-en-6-one)). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.07 (3H, t, J = 7.6 Hz), 1.28 (3H, t, J = 7.2 Hz), 1.31 (3H, t, J = 7.2 Hz), 1.80 (1H, dd, J = 12.6, 7.6 Hz), 2.01-2.19 (3H, m), 2.53 (1H, dd, J = 16.8, 7.6 Hz), 2.72 (1H, ddd, J = 12.6, 9.2, 2.8 Hz), 3.16 (1H, quint, J = 7.6 Hz), 3.61 (1H, s), 3.67-3.82 (1H, M), 4.15-4.33 (4H, M), 5.21-5.26 (1H, M). 13 C NMR (CDCl 3 ) (100 MHz):. delta = 12.2, 14.0, 24.4, 32.2, 37.7, 39.3, 42.5, 55.0, 55.2, 62.00, 62.02, 119.0, 123.3, 153.7, 166.21, 166.23 (HPLC analysis conditions) Diethyl [(1R, 5S, 6R) -6-cyano-3-ethylbicyclo [3.2.0] hept-3-en-6-yl] propanedioate quantification method column: Cadenza CW-C18 (Imtakt, 3 μm, 4.6 mm × 150 mm), 40 ° Cdetection wavelength: UV 205 nm mobile phase: MeCN: 0.1% AcOH aqueous solution = 10: 90-80: 20 (gradient) (0-2 min: MeCN 10%, 2-17 min: MeCN 10 → 80%, 17-25 min: MeCN 80%, 25-30 min: MeCN 80 → 10%, 40 min: STOP) measurement time: 40 min flow rate: 1.0 mL / min retention time: Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate: 18.6 min, Diethyl [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3 en-6-ylidene] propanedioate: 19.7 min







Column: CHIRALPAK AD-RH 4.6 × 250 mm
mobile phase: 10 mM pH 2.0 phosphate buffer / MeCN = 25/75 (isocratic)
flow rate: 1.0 mL / min
Column temperature: 40 ° C
Detection wavelength: UV 210 nm
analysis time: 80 minutes
retention time: (1S, 5R, 6R) – Body: 35.2 min, (1R, 5S, 6S) – Body: 42.1 min

(Example 28) [(1R, 5S, 6S)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid benzyl amine salt Benzylammonium [(1R, 5S, 6S) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetate [of 52] Diethyl obtained by the method of Example 12 [(1RS, 5SR, 6RS) -6-cyano -3-Ethylbicyclo [3.2.0] Hept- 3-en-6-YL] After the addition of EtOH (390 mL) to CPME solution of propanedioate, heating under reflux, 8 N aqueous solution of potassium hydroxide (6.9 mL, 55.07 mmol ) after adding a total of 5 times every 1 hour, refluxed for 5 hours and returned to room temperature. The addition of water (60 mL) and 8N aqueous potassium hydroxide (24 mL) to the above EtOH solution, and after stirring for 2 h at 26-27 ° C, under reduced pressure at an external temperature of 40-45 ° C until 150 mL It was concentrated. To remove the organic layer by water (180 mL) and toluene (90 mL) was added for liquid separation. The resulting aqueous solution Toluene (150 mL) added, cooled to, was added concentrated hydrochloric acid 42.5 mL at 2-9 ° C, the pH was adjusted to 1.4. By separation to remove the aqueous layer was added toluene (300 mL) benzylamine (23.6 g, 220.28 mmol) and. After stirring for 30 minutes at 44-46 ° C make the inoculation, and concentrated under reduced pressure until 300 mL at 44-46 ° C. After stirring overnight at 22-23 ° C, and crystals were filtered off. And vacuum dried at 40 ° C, was obtained as a white crystalline title compound 54.4 g (79.2% from (1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one) a.

(Example 33) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) – 6 (aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid [of 57] Benzylammonium [(1R, 5S, 6S) -6-cyano-3-Ethylbicyclo [3.2. 0] hept-3-en-6-yl] acetate (40.0 g) in toluene (200 mL), was added 2 mol / L hydrochloric acid (100 mL) at room temperature and dissolved. And allowed to stand the solution to drain the aqueous layer to obtain an organic layer. To the stirred addition of 10% aqueous sodium chloride solution (about 100 mL), and the aqueous layer was removed after standing. The solution of water (100 mL) was added to, was adjusted to 10.0 to pH added 8 mol / L aqueous potassium hydroxide solution (about 15.7 mL), the organic layer was removed to standing. The solution to the sponge cobalt (10 g), 28% aqueous ammonia (13 mL), 2% dimethylpolysiloxane / toluene solution (2 mL) was added and warmed to 40 ° C in a hydrogen gas pressure (0.45 MPa) It was stirred for 8 hours.After cooling to room temperature, filtering the reaction mixture to remove the sponge cobalt. The sponge cobalt on the filter it was washed with water (80 mL). The resulting solution was stirred for 0.5 hours added the activated carbon (4 g), to remove the charcoal by filtration. The activated carbon on the filter it was washed with water (60 mL). The solution I was adjusted to about pH 6.0 with concentrated hydrochloric acid (about 32.7g) a. Then, after stirring for 0.5 hours was added potassium chloride (55.0 g), and cooled to 0 ° C. The resulting was filtered and crystals were washed with 20% brine cooled to about 0 ° C (80 mL), and dried overnight in vacuum at 50 ° C to give the title compound as white crystals (26.9 g, content 88.3 %, 88.7% content in terms of yield).

(Example 34) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) – 6 (aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid [of 58] (R) -Phenylethanaminium [(1R, 5S, 6S) -6-cyano-3 ethylbicyclo [3.2.0] hept-3-en-6-yl] acetate (35.9g, 99.2 mmol, 95.7% de, ee 99.2%) in toluene (120 mL) and 1 mol / L hydrochloric acid (150 mL) was added , it was stirred. After removing the aqueous layer, the organic layer was washed twice with water (120 mL), and concentrated. The obtained residue in MTBE to (150 mL) and sponge nickel (10.1 g) was added, under hydrogen pressure (approximately 4 atm) and stirred for 3 hours at room temperature. The reaction of 2 mol / L aqueous potassium hydroxide solution (72 mL) was added, After stirring for 30 minutes, a sponge nickel was filtered off. It was washed with a filtration sponge nickel 2 mol / L potassium hydroxide solution (12 mL). After combining the filtrate and washings, the organic layer was removed to obtain an aqueous layer. The organic layer was re-extracted with 2M aqueous potassium hydroxide solution. The matched aqueous layer was cooled, after adjusting the pH adding concentrated hydrochloric acid (about 12 mL) to 7.5, and the mixture was stirred at 0 ° C for about 3 hours. Filtered the precipitated crystals were washed with ice-cold water (24 mL), and dried under reduced pressure at 50 ° C, to give the title compound (18.3g, 88%, 99.8% de) and.

(Example 35) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid one benzenesulfonate [(1R, 5S, 6S)-6-(aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid Monobenzenesulfonate [of 59] MTBE (83 mL), acetone (4.0 mL), water ( with respect to a mixture of 0.98 mL), at 0 ° C [(1R, 5S, 6S) -6- (Aminomethyl) -3-ethylbicyclo [3.2.0] hept-3-en-6-yl] acetic acid ( 4.07 g, 19.5 mmol) was added and stirred to form a slurry solution. This BsOH (3.08 g, 19.5 mmol) it was added acetone (10.1 mL) solution of. 0 ° After stirring for 1 hour at C, and stirred for 2 hours and allowed to warm to room temperature. Over 1 hour and gradually cooled to -10 ° C, and stirred for 2.5 hours. The resulting was filtered crystals, after washing with acetone and cooled to 0 ° C (12 mL), and by vacuum-dried at 40 ° C, as white crystals of the title compound was obtained (6.44 g, 90.1% ). Various spectrum data of the obtained title compound was almost (extent the structure can be identified) coincides with (described in Patent Documents 5 and 6) the known information. (Purity measurement method -1) column: Cadenza CW-C18 (Imtakt, 3 μm, 4.6 mm × 150 mm), 40 ° C detection wavelength: UV 205 nm mobile phase: MeCN: 5 mM ammonium hydrogen carbonate aqueous solution = ten ninety -80: 20 (gradient) (0-12 min: MeCN 10%, 12-27 min: MeCN 10 → 80%, 27-45 min: MeCN 80%, 45-50 min: MeCN 80 → 10%, 50- 60 min: MeCN 10%, 60 min: STOP) measurement time: 60 min flow rate: 1.0 mL / min infusion sample concentration: 5mg / mL sample injection volume: 2μL retention time: the title compound (as free form): 12.5 min diastereoisomers Marr (Compound X): 13.5 min double bond position isomer (compound XII or XIII): 9.4 min, 9.6 min, 11.4 min

| Patent | Submitted | Granted |
|---|---|---|
| Bicyclic [gamma]-amino acid derivative [US7947738] | 2010-09-30 | 2011-05-24 |
| Optical Resolution Methods for Bicyclic Compounds Using Enzymes [US2015038738] | 2014-10-10 | 2015-02-05 |
| WO2015005298A1 * | Jul 8, 2014 | Jan 15, 2015 | Daiichi Sankyo Company,Limited | METHOD FOR PRODUCING OPTICALLY ACTIVE BICYCLIC γ-AMINO ACID DERIVATIVE |
CONSTRUCTION
References
- Vinik A, Rosenstock J, Sharma U, Feins K, Hsu C, Merante D, et al. Efficacy and safety of mirogabalin (DS-5565) for the treatment of diabetic peripheral neuropathic pain: a randomized, double-blind, placebo- and active comparator-controlled, adaptive proof-of-concept phase 2 study. Diabetes Care. 2014 Dec;37(12):3253-61. doi: 10.2337/dc14-1044. PMID 25231896
- Vinik A, Sharma U, Feins K, Hsu C, Merante D. DS-5565 for the Treatment Of Diabetic Peripheral Neuropathic Pain: Randomized, Double-Blind, Placebo- And Active Comparator-Controlled Phase II Study (S20.004) Neurology April 8, 2014; 82(10): Supplement S20.004
Tokyo, Japan – (February 4, 2015) – Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo) today announced enrollment of the first patients in large-scale, multi-national clinical programs evaluating the safety and efficacy of investigational mirogabalin (DS-5565), the first preferentially selective alpha-2 delta ligand. The phase 3 clinical program across Asia includes the REDUCER (An Asian, phase 3, multicenter, RandomizEd, Double-blind, placebo-controlled 14-week stUdy of DS-5565 in patients with diabetiC pEripheral neuRopathic pain followed by a 52-week open-label extension) study and the NEUCOURSE (An AsiaN, phasE 3, mUltiCenter, randomized, dOUble-blind, placebo-contRolled 14-week study of DS-5565 in patientS with postherpetic neuralgia followed by a 52-week open-label Extension) study which will evaluate investigational mirogabalin for the treatment of diabetic peripheral neuropathic pain (DPNP) and postherpetic neuralgia (PHN), respectively. The phase 3 global ALDAY (A Randomized, Double-Blind, Placebo- and Active-Controlled Study of DS-5565 in Patients with Pain Associated with Fibromyalgia) clinical program is ongoing and will evaluate mirogabalin for the treatment of pain associated with fibromyalgia in three identical studies.
“Pain associated with the neurologic conditions of diabetic peripheral neuropathic pain, postherpetic neuralgia and fibromyalgia can be debilitating,” said Lesley Arnold, MD, Professor of Psychiatry and Behavioral Neuroscience and Director of the Women’s Health Research Program, University of Cincinnati and lead investigator of the ALDAY program. “New treatment options are needed to help people living with these neurologic conditions relieve and manage their chronic pain and hopefully, improve their function and quality of life.”
“We are pleased that our global clinical development program evaluating the efficacy and safety of mirogabalin continues to move forward and has progressed into phase 3,” said Mahmoud Ghazzi, MD, PhD, Executive Vice President and Global Head of Development for Daiichi Sankyo. “Daiichi Sankyo is committed to identifying and studying new medicines that could help improve the management of chronic pain for people with diabetic peripheral neuropathy, postherpetic neuralgia and pain associated with fibromyalgia.”
About the REDUCER and NEUCOURSE Phase 3 Clinical Studies
The REDUCER study will last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan and Korea. The NEUCOURSE study will also last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan, Korea, Singapore, Malaysia and Thailand. The studies will include about 750 patients each with either diabetic peripheral neuropathic pain or postherpetic neuralgia, respectively. The objectives of the double-blind studies are to evaluate safety and efficacy of mirogabalin by comparing change in the average daily pain score (ADPS) from baseline to Week 14 in patients receiving a total daily dose of either 15 mg, 20 mg or 30 mg of mirogabalin versus placebo. Both studies will be followed by one-year open-label extension studies to assess long-term safety and efficacy of mirogabalin. For more information on the REDUCER study in patients with diabetic peripheral neuropathic pain, please visit
https://www.clinicaltrials.gov/ct2/show/NCT02318706?term=Mirogabalin&rank=3.
For more information on the NEUCOURSE study in patients with postherpetic neuralgia, please visithttps://www.clinicaltrials.gov/ct2/show/NCT02318719?term=Mirogabalin&rank=1.
About the ALDAY Phase 3 Clinical Program
The ALDAY program is a large clinical phase 3 program evaluating mirogabalin for the treatment of pain associated with fibromyalgia, and includes three, randomized, double-blind, placebo- and active-controlled studies, and an open label safety study that will be carried out over the next three years. Approximately 4,000 patients with pain associated with fibromyalgia will be enrolled at approximately 800 clinical centers at more than 40 countries worldwide. The primary objective of the studies in the ALDAY program is to compare change in weekly ADPS from baseline to Week 13 in patients receiving a total daily dose of either 15 mg or 30 mg of mirogabalin versus placebo. Weekly ADPS is based on daily pain scores reported by the patient that best describes his or her worst pain over the previous 24 hours. The primary objective of the phase 3 open-label extension study is to assess the long-term safety of a total daily dose of mirogabalin 15 mg or mirogabalin 30 mg in patients with pain associated with fibromyalgia. For more information on the studies in the ALDAY program, please visit
https://clinicaltrials.gov/ct2/show/NCT02187471?term=DS5565&rank=1
https://clinicaltrials.gov/ct2/show/NCT02187471?term=ds-5565&rank=2
https://clinicaltrials.gov/ct2/show/NCT02146430?term=ds-5565&rank=3
For more information on the open-label extension study, please visithttps://clinicaltrials.gov/ct2/show/NCT02234583?term=ds-5565&rank=4
For patient recruitment or additional clinical study information, please visit http://www.aldaystudy.com/.
About Diabetic Peripheral Neuropathic Pain
Diabetic peripheral neuropathy is a disorder that causes nerve damage to the extremities and is one of the most common long-term complications of diabetes.1 Symptoms include sharp pains or increased sensitivity, numbness, loss of balance and coordination, tingling, burning, or prickling sensations, which typically worsen at night.1 Up to 50 percent of people with diabetes have peripheral neuropathy2 and it is estimated that between 11 and 26 percent of people with diabetes experience diabetic peripheral neuropathic pain (DPNP).3-6 However, DPNP is often undertreated and underreported.2
About Postherpetic Neuralgia
Postherpetic neuralgia is pain that occurs after recovering from shingles, an infection that is caused by the herpes zoster (chickenpox) virus. Pain from postherpetic neuralgia can range in severity, and is typically described as burning, sharp, or stabbing.7 Other symptoms include sensitivity to touch, itching, numbness, and in rare cases, muscle weakness or paralysis can occur.7 The risk of developing postherpetic neuralgia increases with age and it mainly affects people older than 60.7 Studies have shown that only half of all patients affected with the condition will be relieved from pain within a year.8 Most people will require more than one treatment to help ease the pain.7
About Fibromyalgia
Fibromyalgia is a chronic disorder that causes widespread muscle pain, generalized tender points and fatigue.9 Other common symptoms include sleep disturbances, morning stiffness, memory and thinking problems (sometimes called fibro fog), tingling in the hands and feet and headaches.9 Fibromyalgia is often misdiagnosed and suboptimally treated.10-17 The overall estimated prevalence of fibromyalgia is approximately two to three percent in the general population, with a higher prevalence in women.18-22 Pain that occurs with fibromyalgia has a substantial impact on the patient, and can be associated with societal and economic burdens.23-29
About Mirogabalin
Mirogabalin is an investigational drug that is currently being studied for the treatment of DPNP, PHN and pain associated with fibromyalgia. Mirogabalin is preferentially selective in regards to how it binds to α2δ-1 subunit, a protein that may help to regulate how the brain processes pain signals. It has a unique binding profile and long duration of action.30*,31
About Daiichi Sankyo
Daiichi Sankyo Group is dedicated to the creation and supply of innovative pharmaceutical products to address the diversified, unmet medical needs of patients in both mature and emerging markets. While maintaining its portfolio of marketed pharmaceuticals for hypertension, dyslipidemia and bacterial infections used by patients around the world, the Group has also launched treatments for thrombotic disorders and is building new product franchises. Furthermore, Daiichi Sankyo research and development is focused on bringing forth novel therapies in oncology and cardiovascular-metabolic diseases, including biologics. The Daiichi Sankyo Group has created a “Hybrid Business Model,” to respond to market and customer diversity and optimize growth opportunities across the value chain. For more information, please visit: www.daiichisankyo.com.
| trial(s) |
|
|---|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1R,5S,6S)-6-(aminomethyl)-3-ethyl-bicyclo(3.2.0)hept-3-ene-6-acetic acid
|
|
| Identifiers | |
| CAS Registry Number | 1138245-21-2 |
| PubChem | CID: 49802951 |
| ChemSpider | 32701007 |
| Chemical data | |
| Formula | C12H19NO2 |
| Molecular mass | 209.285 g/mol |
USP revises Chapter on Pharmaceutical Water
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg)
Changes to the fundamental monograph on pharmaceutical water <1231> Water for Pharmaceutical Purposes from the US-American Pharmacopeia have been published for comments in the Pharmacopeial Forum 41(5). The revision presented in the current draft mainly has a structural nature. The content of the monograph has been reorganised in 9 new chapters which aim at improving readibility and searchability of the content searched:
1. INTRODUCTION
2. SOURCE WATER CONSIDERATIONS
3. WATERS USED FOR PHARMACEUTICAL MANUFACTURING AND TESTING PURPOSES
4. VALIDATION AND QUALIFICATION OF WATER PURIFICATION, STORAGE, AND DISTRIBUTION SYSTEMS
5. DESIGN AND OPERATION OF PURIFIED WATER AND WATER FOR INJECTION SYSTEMS
6. SAMPLING
7. CHEMICAL EVALUATIONS
8. MICROBIAL EVALUATIONS
9. ALERT AND ACTION LEVELS AND SPECIFICATIONS
The draft document is available for free on the website of the USP Pharmacopeial Forum. You only need to register for free. The deadline for comments is 20 November 2015.
http://www.gmp-compliance.org/enews_5070_USP-revises-Chapter–1231–on-Pharmaceutical-Water_n.html
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide
Cas 1820758-44-8
C24 H18 F N3 O4 S
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide


Glycogen synthase kinase-3 (GSK-3) is a constitutively active, ubiquitous serine/threonine kinase that takes part in a number of physiological processes ranging from glycogen metabolism to apoptosis. GSK-3 is a key mediator of various signaling pathways, such as the Wnt and the insulin/AKT signaling pathways.
Therefore, dysregulation of GSK-3 has been linked to various human diseases, such as cancer, diabetes, and neurodegenerative diseases.Two related isoforms of GSK-3 exist in mammals, GSK-3α and -β, which share a sequence identity within their catalytic domains of 98%.
Beyond the catalytic domains they show significant differences. Although these isoforms are structurally related, they are not functionally equivalent, and one cannot compensate for loss of the other.
The debate on the respective contributions of the isoforms GSK-3α and GSK-3β on the pathogenesis of different diseases is ongoing.
Various studies indicate that the therapies of certain diseases benefit from specific targeting of GSK-3α and GSK-3β. GSK-3α was recently identified as a differentiation target in acute myeloid leukemia (AML). AML is a hematopoietic malignancy defined by uncontrolled proliferation and disrupted myeloid differentiation. AML is the second most common form of leukemia in adults.
The current treatment of AML with conventional chemotherapy is very aggressive yet ineffective for the majority of patients with the disease.Thus, alternative targeted treatment approaches for AML are highly desirable. GSK-3α recently emerged as a potential target in this disease.
PAPER

The challenge for glycogen synthase kinase-3 (GSK-3) inhibitor design lies in achieving high selectivity for one isoform over the other. The therapy of certain diseases, such as acute myeloid leukemia (AML), may require α-isoform specific targeting. The scorpion shaped GSK-3 inhibitors developed by our group achieved the highest GSK-3α selectivity reported so far but suffered from insufficient aqueous solubility. This work presents the solubility-driven optimization of our isoform-selective inhibitors using a scorpion shaped lead. Among 15 novel compounds, compound 27 showed high activity against GSK-3α/β with the highest GSK-3α selectivity reported to date. Compound 27 was profiled for bioavailability and toxicity in a zebrafish embryo phenotype assay. Selective GSK-3α targeting in AML cell lines was achieved with compound 27, resulting in a strong differentiation phenotype and colony formation impairment, confirming the potential of GSK-3α inhibition in AML therapy
Evaluation of Improved Glycogen Synthase Kinase-3α Inhibitors in Models of Acute Myeloid Leukemia
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01200
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01200/suppl_file/jm5b01200_si_001.pdf
compound 27 as a colorless solid. HPLC: 96%, tR = 6.93 min.
1H NMR (DMSO-d6, 500 MHz, 300 K): δ (ppm) = 4.32 (td, J = 5.2 Hz, J = 3.7 Hz, 4H), 4.60 (s, 2H), 7.05 (d, J = 8.4 Hz, 1H), 7.25 (dd, J = 9.1 Hz, J = 2.7 Hz, 1H), 7.31 (td, J = 8.6 Hz, J = 2.8 Hz, 1H), 7.38 (m, 3H), 7.41 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.73 (s, 1H).
13C NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 114.3 (d, JC–F = 21 Hz), 115.0, 115.9 (d, JC–F = 21 Hz), 115.9, 118.1, 120.0, 128.6 (2C), 128.8 (2C), 132.0 (d, JC–F = 8 Hz), 134.8, 135.5, 138.9, 139.0 (d, JC–F = 7 Hz), 143.8, 146.7, 160.9 (d, JC–F = 247 Hz), 162.7, 164.9, 169.5.
EI-MS: m/z = 463 (100, [M+]), 464 (26, [M+ + H]), 465 (7, [M+ + 2H].
ABOUT Boris Schmidt
Boris Schmidt
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany*Phone: +49 6151 163075. Fax: +49 6151 163278.E-mail: Schmidt_boris@t-online.de.
………………………………………….
ABOUT Theresa Neumann
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany
////////FC(C=C1C(N)=O)=CC=C1C(C=C2)=CC=C2CSC3=NN=C(O3)C4=CC5=C(OCCO5)C=C4
AMG-319

AMG-319
N-((1S)-1-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine, WO2008118468
(S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine
CAS 1608125-21-8
Chemical Formula: C21H16FN7
Exact Mass: 385.14512
Phosphoinositide-3 kinase delta inhibitor
AMGEN, PHASE 2

PI3K delta isoform selective inhibitor that is being investigated in human clinical trials for the treatment of PI3K-mediated conditions or disorders, such as cancers and/or proliferative diseases
Useful for treating PI3K-mediated disorders such as acute myeloid leukemia, myelo-dysplastic syndrome, myelo-proliferative diseases, chronic myeloid leukemia, T-cell acute lymphoblastic leukemia, B-cell acute lymphoblastic leukemia, non-Hodgkins lymphoma, B-cell lymphoma, or breast cancer.
Amgen is developing AMG-319, a small molecule PI3K-δ inhibitor, for treating lymphoid malignancies and solid tumors including, head and neck squamous cell carcinoma.
AMG-319 is a highly selective, potent, and orally bioavailable small molecule inhibitor of the delta isoform of the 110 kDa catalytic subunit of class IA phosphoinositide-3 kinases (PI3K) with potential immunomodulating and antineoplastic activities. PI3K-delta inhibitor AMG 319 prevents the activation of the PI3K signaling pathway through inhibition of the production of the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3), thus decreasing proliferation and inducing cell death. Unlike other isoforms of PI3K, PI3K-delta is expressed primarily in hematopoietic lineages. The targeted inhibition of PI3K-delta is designed to preserve PI3K signaling in normal, non-neoplastic cells.
PATENT
http://www.google.com/patents/WO2008118468A1?cl=en
PATENT
WO2013152150
http://www.google.com/patents/WO2013152150A1?cl=en
PATENT
Example 4: Method of making N-((lSM-(7-fluoro-2-(2-pyridinyl)- 3-quinolinyl)ethyl)-9H-purin-6-amine
N-((l S)- 1 -(7-Fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine (4) is synthesized in four steps beginning with (S)-l-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethanamine hydrochloride (1). A nucleophilic aromatic substitution between coupling partners 1 and purine 5 affords the penultimate intermediate 2. Cleavage of the p-methoxybenzyl (PMB) group leads to the isolation of the desired butyl acetate solvate 3. A crystalline form change is induced through an aqueous-acetone recrystallization to afford the target hydrate 4.
Synthetic Scheme
Step 1. Preparation of PMB protected pyridylpurinamine tosylate (2)
(S)- 1 -(7-Fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethanamine is prepared similar to that described in US20130267524. The (S)-l-(7-fluoro-2-(pyridin-2-
yl)quinolin-3-yl)ethanamine hydrochloride (1) is coupled to PMB-chloropurine (5, prepared similar to that described in J. Med. Chem. 1988,31, 606-612) in the presence of K2CO3 in IPA. Upon reaction completion the K2CO3 is removed via filtration and the product is crystallized by the addition of /?-toluenesulfonic acid (pTSA). Isolation of the PMB-protected pyridylpurinamine tosylate (2) is conducted via filtration.

Dry 100 L reactor under nitrogen. Set the temperature to 20 ± 5 °C. Charge (l S)-N-chloro-l-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethanamine HCl salt (1) to the reactor. Then 9-(4-methoxybenzyl)-6-chloro-9H-purine (5) is added. Potassium carbonate is added to the reactor. Isopropyl alcohol is added to the reactor and the mixture is heated to 80 °C and stirred for 24 hours. Additional isopropyl alcohol is added to the reactor and the mixture is cooled to 20 °C. The mixture is filtered through Celite and the solid is washed with isopropyl alcohol and the isopropyl alcohol solutions containing 9-(4-methoxybenzyl)-N-((S)- 1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine are collected.
The 9-(4-methoxybenzyl)-N-((S)- 1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine isopropyl alcohol solution is heated to 50 °C. /^-Toluene sulfonic acid monohydrate is dissolved in isopropyl alcohol and added to the 9-(4-methoxybenzyl)-N-((S)-l-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine in portions. The mixture is slowly cooled to 20 ± 5 °C over 6 ± 2 hrs. The crystalline 9-(4-methoxybenzyl)-N-((S)- 1 -(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)- 9H-purin-6-amine toluene sulfonic acid salt is collected, rinsed with isopropyl alcohol and dried with vacuum.
Example 5: Method of Making the Crystalline Hydrate Form of N-((1S)-1- (7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine Step 1: Isolation of a Butyl Acetate (BuOAc) Solvate of N-((lS)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine (3)
To a 2 L jacketed reactor equipped with a condenser, a mechanical stirrer, and a bubbler, under an atmosphere of N2, was added N-((l S)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9-(4-methoxybenzyl)-9H-purin-6-amine (2, 100.0 g, 0.148 mol), followed by acetic acid (AcOH; 240 mL) and 1 -dodecanethiol (71.1 mL, 0.295 mol). The vessel was evacuated and back-filled with nitrogen three times. Methanesulfonic acid (MSA; 28.7 mL, 0.443 mol) was added to the vessel over 10 minutes. Then, the reaction was heated to 80 °C and stirred for 20 hrs. The reaction was then cooled to ambient temperature, after which toluene (1000 mL) and water (700 mL) were sequentially added. The solution was then stirred for 30 minutes. The phases were separated by removing the organic phase, adding another charge of toluene (1000 mL) to the aqueous phase, and the mixture was stirred for another 30 minutes. After removing the organic phase again, the aqueous phase was charged to a jacketed 5 L reactor equipped with a mechanical stirrer followed by n-butyl acetate (1500 mL,) and heated to 50 °C. The aqueous phase was neutralized to pH 6.3 with 10 N NaOH (350 mL). The organic (BuOAc) phase was azeotropically dried to 600 ppm water, while keeping a constant volume. The dried organic phase was polish filtered at 50 °C to remove salts, which were subsequently washed with hot BuOAc (285 mL). The BuOAc was charged back into the 2 L jacketed reactor equipped with a mechanical stirred and distillation apparatus, and then concentrated to 54 mg/g of N-((l S)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine in solution. The solution was then seeded with 1 wt% seed of the BuOAc solvate of N-((l S)- 1 -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. The slurry was further concentrated to 300 mL total volume and cooled to ambient temperature over 1 hour. Heptane (460 mL) was added dropwise to the solution, and the solution was aged overnight. The supernatant concentration was checked, and determined to be 5.3 mg/g of N-((l S)-l-(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. The supernatant was filtered and the resulting solid cake was washed with 1 : 1 BuOAc:heptane (280 mL), followed by heptane (280 mL). The washed cake was then
allowed to dry on the filter. The BuOAc solvate of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine was obtained as a white solid (59.5 g, 99.6 LCAP, 86.3 wt%, 90 % corrected yield). !H NMR (400 MHz, CDC13) δ 13.72 (s, 1H), 8.80 (s, 1H), 8.37 (s, 1H), 8.31 (s, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 18.8 Hz, 2H), 7.76 (t, J = 1 1.6 Hz, 2H), 7.39 (s, 1H), 7.31 (td, J = 8.7, 2.5 Hz, 1H), 6.15 (s, 1H), 4.06 (t, J = 6.7 Hz, 1H), 2.04 (s, 1H), 1.65 – 1.44 (m, 3H), 1.39 (dt, J = 14.9, 7.4 Hz, 1H), 1.33 – 1.20 (m, 2H), 0.93 (t, J = 7.4 Hz, 1H), 0.88 (t, J = 6.8 Hz, 1H); 13C NMR (101 MHz, CDC13) δ 152.28 (s), 148.46 (s), 138.10 (s), 137.22 (s), 135.58 (s), 129.47 (s), 124.80 (s), 123.53 (s), 1 13.24 – 1 13.09 (m), 1 12.89 (d, J = 20.3 Hz), 64.40 (s), 48.60 (s), 31.91 (s), 30.67 (s), 29.05 (s), 22.72 (s), 19.15 (s), 14.15 (s); IR: 3193, 3087, 2967, 2848, 1738, 1609, 1493, 1267, 1242, 1 143, 933, 874, 763, 677, 646, 627, 606, 581 , 559, 474 cm“1; exact mass m/z calcd for C2iH16FN7, (M + H)+386.1451 , found 386.1529; MP = 144 °C.
Step 2: Isolation of the Crystalline Hydrate of N-((lS)-l-(7-fluoro-2-(2-pyridinvn-3-quinolinyl)ethyl)-9H-purin-6-amine 4
To a 100 L reactor with its jacket set to 20 °C, 1.206 kg butyl acetate solvate of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine 3 was charged, followed by 6.8 L of acetone and 6.8 L of water. The resulting mixture was stirred at 90 rpm under nitrogen for 13 minutes to ensure complete dissolution of all solids. During these charges, the reactor contents increased in temperature that maximized at 26 °C. The solution was then transferred to another clean 100 L reactor through a 5 μιη filter, and stirred at 85 rpm under nitrogen. The solution was heated to 45 °C, and water (14.8 L) was added to reach a water content (by Karl Fischer, KF) of 75 wt%. The reactor solution was assayed by HPLC and shown to contain 42 mg/g N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. The solution was seeded with a slurry of 1 13 g of the crystalline hydrate of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine in 1 L water, and the seed slurry was rinsed into the reactor with an additional 1 L water. The reactor contents were cooled to 0 °C over 16 h and held at that temperature for 1 h. The supernatant was then assayed, and found to contain 7.6 mg/g of N-((l S)- l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine. Next, 10 L of water was added to the reactor over 38 min and aged for 1 h. The supernatant was assayed at 4.9 mg/g, and the solids were isolated by filtration. The solids were washed with an acetone/water solution (140 mL acetone in 2.7 L water), then 4 L water, and dried under nitrogen on the filter for 68 h. The crystalline hydrate of N-((l S)-l -(7-fluoro-2-(2-pyridinyl)-3-quinolinyl)ethyl)-9H-purin-6-amine was isolated as an off-
white solid (1.12 kg, 616 ppm acetone, 3.73 wt% water, 99.56 LCAP, 95.88 wt%). This material was co-milled at 3900 rpm using a 0.024″ screen to yield an off-white powder (1.09 kg, 99.7 LCAP, 95.4 wt%, 75% yield). Calculated losses were 212 g (18%) to liquors, 5.5g (0.5%) to washes, and 23 g (2%) to fouling. ¾ NMR (400 MHz, DMSO) δ 12.86 (s, 1H), 8.69 (s, 1H), 8.64 (s, 1H), 8.27 (s, 1H), 8.10 (s, 1H), 8.06 – 7.91 (m, 4H), 7.76 (dd, J = 10.4, 2.4 Hz, 1H), 7.50 (ddd, J = 19.2, 9.5, 3.6 Hz, 2H), 6.03 (s, 1H), 3.38 (s, 2H), 1.63 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 163.58, 161.12, 158.36, 157.94, 151.99, 147.98, 146.49, 146.36, 136.82, 134.07, 130.24, 130.14, 124.69, 124.65, 123.30, 1 17.36, 1 17.1 1, 112.10, 1 1 1.90, 46.02, 22.01. HRMS m/z Calcd. for C2iH17FN7 (M + H): 386.15295. Found: 386.15161.
PAPER
1: Cushing TD, Hao X, Shin Y, Andrews K, Brown M, Cardozo M, Chen Y, Duquette J, Fisher B, Gonzalez-Lopez de Turiso F, He X, Henne KR, Hu YL, Hungate R, Johnson MG, Kelly RC, Lucas B, McCarter JD, McGee LR, Medina JC, San Miguel T, Mohn D, Pattaropong V, Pettus LH, Reichelt A, Rzasa RM, Seganish J, Tasker AS, Wahl RC, Wannberg S, Whittington DA, Whoriskey J, Yu G, Zalameda L, Zhang D, Metz DP. Discovery and in vivo evaluation of (S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and related PI3Kδ inhibitors for inflammation and autoimmune disease. J Med Chem. 2015 Jan 8;58(1):480-511. doi: 10.1021/jm501624r. Epub 2014 Dec 3. PubMed PMID: 25469863.
http://pubs.acs.org/doi/abs/10.1021/jm501624r

The development and optimization of a series of quinolinylpurines as potent and selective PI3Kδ kinase inhibitors with excellent physicochemical properties are described. This medicinal chemistry effort led to the identification of 1 (AMG319), a compound with an IC50 of 16 nM in a human whole blood assay (HWB), excellent selectivity over a large panel of protein kinases, and a high level of in vivo efficacy as measured by two rodent disease models of inflammation.
(S)-N-(1-(7-Fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (1)
//////////
C[C@H](NC1=C2N=CNC2=NC=N1)C3=CC4=CC=C(F)C=C4N=C3C5=NC=CC=C5
New 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors in Model of Mood Disorders
CAS 1452582-16-9, 428.47, C23 H26 F2 N4 O2
1H-Indazole-3-carboxamide, 5-(2,3-difluorophenyl)-N-[[1-(2-methoxyethyl)-4-piperidinyl]methyl]-
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
![]()
1 H-indazole-3-carboxamide compounds acting as glycogen synthase kinase 3 beta (GSK-33) inhibitors and to their use in the treatment of GSK-33-related disorders such as (i) insulin-resistance disorders; (ii) neurodegenerative diseases; (iii) mood disorders; (iv) schizophrenic disorders; (v) cancerous disorders; (vi) inflammation, (vii) substance abuse disorders; (viii) epilepsies; and (ix) neuropathic pain.
Protein kinases constitute a large family of structurally related enzymes, which transfer phosphate groups from high-energy donor molecules (such as adenosine triphosphate, ATP) to specific substrates, usually proteins. After phosphorylation, the substrate undergoes to a functional change, by which kinases can modulate various biological functions.
In general, protein kinases can be divided in several groups, according to the substrate that is phosphorylated. For example, serine/threonine kinase phosphorylates the hydroxyl group on the side chain of serine or threonine aminoacid.
Glycogen synthase kinases 3 (GSK-3) are constitutively active multifunctional enzymes, quite recently discovered, belonging to the serine/threonine kinases group.
Human GSK-3 are encoded by two different and independent genes, which leads to GSK-3a and GSK-33 proteins, with molecular weights of about 51 and 47 kDa, respectively. The two isoforms share nearly identical sequences in their kinase domains, while outside of the kinase domain, their sequences differ substantially (Benedetti et al., Neuroscience Letters, 2004, 368, 123-126). GSK-3a is a multifunctional protein serine kinase and GSK-33 is a serine-threonine kinase.
It has been found that GSK-33 is widely expressed in all tissues, with widespread expression in the adult brain, suggesting a fundamental role in neuronal signaling pathways (Grimes and Jope, Progress in Neurobiology, 2001, 65, 391-426). Interest in glycogen synthase kinases 3 arises from its role in various physiological pathways, such as, for example, metabolism, cell cycle, gene expression, embryonic development oncogenesis and neuroprotection (Geetha et al., British Journal Pharmacology, 2009, 156, 885-898).
GSK-33 was originally identified for its role in the regulation of glycogen synthase for the conversion of glucose to glycogen (Embi et al., Eur J Biochem, 1980, 107, 519-527). GSK-33 showed a high degree of specificity for glycogen synthase.
Type 2 diabetes was the first disease condition implicated with GSK- 3β, due to its negative regulation of several aspects of insulin signaling pathway. In this pathway 3-phosphoinositide-dependent protein kinase 1 (PDK-1 ) activates PKB, which in turn inactivates GSK-33. This inactivation of GSK-33 leads to the dephosphorylation and activation of glycogen synthase, which helps glycogen synthesis (Cohen et al., FEBS Lett, 1997, 410, 3-10). Moreover, selective inhibitors of GSK-33 are expected to enhances insulin signaling in prediabetic insulin- resistant rat skeletal muscle, thus making GSK-33 an attractive target for the treatment of skeletal muscle insulin resistance in the pre-diabetic state (Dokken et al., Am J. Physiol. Endocrinol. Metab., 2005, 288, E1 188-E1 194).
GSK-33 was also found to be a potential drug target in others pathological conditions due to insulin-resistance disorders, such as syndrome X, obesity and polycystic ovary syndrome (Ring DB et al., Diabetes, 2003, 52: 588-595).
It has been found that GSK-33 is involved in the abnormal phosphorylation of pathological tau in Alzheimer’s disease (Hanger et al., Neurosci. Lett, 1992, 147, 58-62; Mazanetz and Fischer, Nat Rev Drug Discov., 2007, 6, 464-479; Hong and Lee, J. Biol. Chem., 1997, 272, 19547- 19553). Moreover, it was proved that early activation of GSK-33, induced by apolipoprotein ApoE4 and β-amyloid, could lead to apoptosis and tau hyperphosphorylation (Cedazo-Minguez et al., Journal of Neurochemistry, 2003, 87, 1 152- 1 164). Among other aspect of Alzheimer’s disease, it was also reported the relevance of activation of GSK-33 at molecular level (Hernandez and Avila, FEBS Letters, 2008, 582, 3848-3854).
Moreover, it was demonstrated that GSK-33 is involved in the genesis and maintenance of neurodegenerative changes associated with Parkinson’s disease (Duka T. et al., The FASEB Journal, 2009; 23, 2820- 2830).
Accordingly to these experimental observations, inhibitors of GSK-33 may find applications in the treatment of the neuropathological consequences and the cognitive and attention deficits associated with tauopathies; Alzheimer’s disease; Parkinson’s disease; Huntington’s disease (the involvement of GSK-33 in such deficits and diseases is disclosed in Meijer L. et al., TRENDS Pharm Sci, 2004; 25, 471 -480); dementia, such as, but not limited to, vascular dementia, post-traumatic dementia, dementia caused by meningitis and the like; acute stroke; traumatic injuries; cerebrovascular accidents; brain and spinal cord trauma; peripheral neuropathies; retinopathies and glaucoma (the involvement of GSK-33 in such conditions is disclosed in WO 2010/109005).
The treatment of spinal neurodegenerative disorders, like amyotrophic lateral sclerosis, multiple sclerosis, spinal muscular atrophy and neurodegeneration due to spinal cord injury has been also suggested in several studies related to GSK-33 inhibition, such as, for example in Caldero J. et al., “Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord”, Neuroscience. 2010 Feb 17;165(4):1353-69, Leger B. et al., “Atrogin-1 , MuRF1 , and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients”, Muscle Nerve, 2009 Jul; 40(1 ):69-78, and Galimberti D. et al., “GSK33 genetic variability in patients with Multiple Sclerosis”, Neurosci Lett. 201 1 Jun 1 5;497(1 ):46- 8. Furthermore, GSK-33 has been linked to the mood disorders, such as bipolar disorders, depression, and schizophrenia.
Inhibition of GSK-33 may be an important therapeutic target of mood stabilizers, and regulation of GSK-33 may be involved in the therapeutic effects of other drugs used in psychiatry. Dysregulated GSK-33 in mood disorder, bipolar disorder, depression and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression and the ability of neurons to respond to stressful, potentially lethal conditions (Jope and Ron, Curr. Drug Targets, 2006, 7, 1421- 1434).
The role of GSK-33 in mood disorder was highlighted by the study of lithium and valproate (Chen et al., J. Neurochem., 1999, 72, 1327- 1330; Klein and Melton, Proc. Natl. Acad. Sci. USA, 1996, 93, 8455-8459), both of which are GSK-33 inhibitors and are used to treat mood disorders. There are also existing reports from the genetic perspective supporting the role of GSK-33 in the disease physiology of bipolar disorder (Gould, Expert. Opin. Ther. Targets, 2006, 10, 377-392).
It was reported a decrease in AKT1 protein levels and its phosphorylation of GSK-33 at Serine-9 in the peripheral lymphocytes and brains of individuals with schizophrenia. Accordingly, this finding supports the proposal that alterations in AKT1 -GSK-33 signaling contribute to schizophrenia pathogenesis (Emamian et al., Nat Genet, 2004, 36, 131- 137).
Additionally, the role of GSK-33 in cancer is a well-accepted phenomenon.
The potential of small molecules that inhibit GSK-33 has been evidenced for some specific cancer treatments (Jia Luo, Cancer Letters, 2009, 273, 194-200). GSK-33 expression and activation are associated with prostate cancer progression (Rinnab et al., Neoplasia, 2008, 10, 624-633) and the inhibition of GSK3b was also proposed as specific target for pancreatic cancer (Garcea et al., Current Cancer Drug Targets, 2007, 7, 209-215) and ovarian cancer (Qi Cao et al., Cell Research, 2006, 16 671 -677). Acute inhibition of GSK-33 in colon-rectal cancer cells activates p53-dependent apoptosis and antagonizes tumor growth (Ghosh et al., Clin Cancer Res 2005, 1 1 , 4580-4588).
The identification of a functional role for GSK-33 in MLL-associated leukaemia suggests that GSK-33 inhibition may be a promising therapy that is selective for transformed cells that are dependent on HOX overexpression (Birch et al., Cancer Cell, 2010, 1 7, 529-531 ).
GSK-33 is involved in numerous inflammatory signalling pathways, for example, among others GSK-33 inhibition has been shown to induce secretion of the anti-inflammatory cytokine IL-1 0. According to this finding, GSK-33 inhibitors could be useful to regulate suppression of inflammation (G. Klamer et al., Current Medicinal Chemistry, 2010, 17(26), 2873-2281, Wang et al., Cytokine, 2010, 53, 130-140).
GSK-33 inhibition has been also shown to attenuate cocaine-induced behaviors in mice. The administration of cocaine in mice pretreated with a GSK-33 inhibitor demonstrated that pharmacological inhibition of GSK3 reduced both the acute behavioral responses to cocaine and the long- term neuroadaptations produced by repeated cocaine (Cocaine-induced hyperactivity and sensitization are dependent on GSK3, Miller JS et al. Neuropharmacology. 2009 Jun; 56(8):1 1 16-23, Epub 2009 Mar 27).
The role of GSK-33 in the development of several forms of epilepsies has been demonstrated in several studies, which suggest that inhibition of GSK-33 could be a pathway for the treatment of epilepsy (Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy, Lohi H et al., Hum Mol Genet. 2005 Sep 15;14(18):2727-36 and Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease, Purl R et al., J Biol Chem. 2009 Aug 21 ;284(34) 22657-63). The relationship between GSK-33 inhibition and treatment of neuropathic pain has been demonstrated in Mazzardo-Martins L. et al., “Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreases neuropathic pain in mice: evidence for the mechanisms of action”, Neuroscience. 2012 Dec 13;226, and Xiaoping Gu et al., “The Role of Akt/GSK33 Signaling Pathway in Neuropathic Pain in Mice”, Poster A525, Anesthesiology 2012 October 13-17, 2012 Washington.
A review on GSK-33, its function, its therapeutic potential and its possible inhibitors is given in “GSK-33: role in therapeutic landscape and development of modulators” (S. Phukan et al., British Journal of Pharmacology (2010), 160, 1- 19).
WO 2004/014864 discloses 1 H-indazole-3-carboxamide compounds as selective cyclin-dependant kinases (CDK) inhibitors. Such compounds are assumed to be useful in the treatment of cancer, through a mechanism mediated by CDK2, and neurodegenerative diseases, in particular Alzheimer’s disease, through a mechanism mediated by CDK5, and as anti-viral and anti-fungine, through a mechanism mediated by CDK7, CDK8 and CDK9.
Cyclin-dependant kinases (CDKs) are serine/threonine kinases, first discovered for their role in regulating the cell cycle. CDKs are also involved in regulating transcription, mRNA processing, and the differentiation of nerve cells. Such kinases activate only after their interaction and binding with regulatory subunits, namely cyclins.
Moreover, 1 H-indazole-3-carboxamide compounds were also described as analgesics in the treatment of chronic and neuropathic pain (see, for example, WO 2004/074275 and WO 2004/101 548) and as 5-HT4 receptor antagonists, useful in the treatment of gastrointestinal disorders, central nervous system disorders and cardiovascular disorders (see, for example, WO 1994/101 74).
Patent
WO 2013124158
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
SEE ENTRY 8

DMSO-de; δ 13.09 (s, 1 H), 8.23-8.42 (m, 2H), 7.72 (dd, J=0.82, 8.69 Hz, 1 H), 7.55 (td, J=1.76, 8.74 Hz, 1 H), 7.24-7.49 (m, 3H), 3.40 (t, J=6.04 Hz, 2H), 3.22 (s, 3H), 3.18 (d, J=6.40 Hz, 2H), 2.84 (d, J=11.53 Hz, 2H), 2.42 (t, J=5.95 Hz, 2H), 1.82- 2.02 (m, 2H), 1.41 -1.71 (m, 3H), 1.06-1.31 (m, 2H)
PAPER

Hit Optimization of 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors with in Vivo Activity in Model of Mood Disorders
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208

http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
Angelini S.p.A., Angelini Research Center,
![]()
/////
COCCN1CCC(CNC(=O)c2n[nH]c3ccc(cc23)c4cccc(F)c4F)CC1
AZD 1080
 .
.
AZD 1080
2-Hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]-1H-indole-5-carbonitrile
2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1H-indole-5-carbonitrile
AZD1080 is a selective, orally active, brain permeable GSK3 inhibitor, inhibits human GSK3α and GSK3β with Ki of 6.9 nM and 31 nM, respectively, shows >14-fold selectivity against CDK2, CDK5, CDK1 and Erk2.
Cas 612487-72-6, AZD1080,
AZD-1080, a glycogen synthase kinase 3 (GSK-3) inhibitor, had been in early clinical trials for the treatment of Alzheimer’s type dementia by AstraZeneca
| Astrazeneca Ab |
PATENTS
WO 2003082853
http://www.google.com/patents/WO2003082853A1?cl=en
PAPER
Organic Process Research & Development (2008), 12(3), 540-543.
http://pubs.acs.org/doi/abs/10.1021/op800020r

A mild and robust method for the large-scale palladium-catalysed cyanation of aryl bromides has been developed. The reaction is sensitive to cyanide poisoning of the catalyst, and it was found that the order of adding the reagents had a strong impact on the performance of the reaction. Addition of the cyanide source to a preheated mixture of the other reagents was critical for achieving a robust and scaleable process. This improved protocol allowed the reaction to be run to full conversion within 3 h at 50 °C on a 6.7 kg scale. Furthermore, it led to the identification of several new efficient catalysts for the reaction.
2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1H-indole-5-carbonitrile (2) (5.2 kg, 15.6 mol), 90% yield with a purity of >90% by HPLC. 1H NMR (d6-DMSO, 400 MHz) δ 14.79 (broad s, 1H), 10.86 (broad s, 1H), 8.08 (s, 1H), 7.95 (s, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.27 (dd,J = 8.0, 0.9 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 3.57 (t, J = 4.4 Hz, 4H), 3.36 (s, 2H), 2.36 (broad s, 4H); 13C NMR (d6-DMSO, 100 MHz) δ 168.8, 148.6, 141.8, 137.0, 136.1, 125.4, 123.9, 122.3, 121.1, 118.8, 118.3, 108.7, 101.3, 84.6, 66.1, 58.4, 52.8. MS (ES) m/z [M + 1] 335.
PAPER
Topics in Organometallic Chemistry (2012), 42(Organometallics as Catalysts in the Fine Chemical Industry), 125-134.
http://link.springer.com/chapter/10.1007%2F3418_2011_25
PATENT
https://www.google.co.in/patents/WO2007089193A1?cl=en

In the above scheme, preferably Rl is bromo and X is chloro.
Synthesis of 2-Hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl] lH-indole-5-carbonitrile citrate
Example 14
2-Hydroxy-3-r5-(moφholin-4-ylmethyl)pyridin-2-yl1 lH-indole-5-carbonitrile citrate salt 2-Hydroxy-3-[5-(moφholin-4-ylmethyl)pyridin-2-yl] lH-indole-5-carbonitrile (5.14 kg, 15.4 mol) was suspended in ethanol (54 L) at room temperature. The suspension was heated to an inner temperature of 700C and a solution of citric acid (3.424 kg, 17.82 mol, 1.300 eq)) in water (103 L) was added keeping the inner temperature above 650C. The mixture was heated to reflux. After this the resulting solution was mixed with activated charcoal (0.412 kg) and reflux continued for 3.5 h after which the reaction mixture was clear filtered at 830C followed by cooling to room temperature over 20 h. After filtration the precipitate was washed twice with a cold mixture of ethanol/water (6.9 L/13.7 L). Drying under vacuum at 5O0C gave 6.648 kg, 82.2% yield of 2-hydroxy-3-[5-(morpholin- 4-ylmethyl)pyridin-2-yl]lH-indole-5-carbonitrile citrate having a purity of at least 98%. The palladium content was less than 1 ppm and the zinc content was lower than 10 ppm. 1H NMR (Jd-DMSO3 400 MHz) δ 14.7 (br s, 1 H), 11.55 (s, 1 H), 10.98 (s, IH), 8.31 (s, 1 H), 8.08 (br d, J= 1.84Hz, IH), 8.02 (s, IH), 7.90 (br d, J = 8.92Hz, 1 H), 7.31 (d, J = 8.0 Hz, 1 H), 7.02 (d, J= 8.0Hz), 4.28 (s, 2 H), 3.97 (m, 2 H), 3.94 (m, 2H), 3.35 (m, 9H), 3.32 (m, 2H) ppm; 13C NMR (d6-DMSO, 400MHz) δ 168.9, 148.5, 142.7, 139.8, 137.5,126.4, 124.9, 124.8, 120.9, 119.4, 118.4, 113.3, 109.0, 101.6, 85.7, 63.1, 55.5, 50.3, 40.1, 39.9, 39.7, 39.2, 39.0, 38.8ppm; MS (ES) m/z [M++l] 335.
LIK 066, Licogliflozin diprolinate
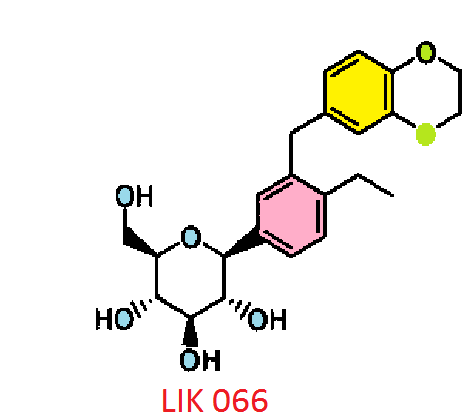
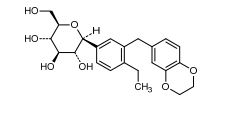
Licogliflozin
LIK 066
Licogliflozin diprolinate

LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
Licogliflozin, a SGLT-1/2 inhibitor, is in phase II clinical development at Novartis for the treatment of metabolic disorders, for the treatment of heart failure in patients with type 2 diabetes, for the treatment of obesity and for the treatment of polycystic ovary syndrome (PCOS) in overweight and obese women. Phase II trials for the treatment of type 2 diabetes had been discontinued.
EMA/415156/2014 European Medicines Agency decision P/0183/2014 of 24 July 2014 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3- dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran3,4,5-triol (2:1) (LIK066) (EMEA-001527-PIP01-13) in accordance with Regulation (EC) No 1901/2006 of the European Parliament and of the Council
1. Opinion of the Paediatric Committee on the agreement of a Paediatric Investigation Plan and a deferral and a waiver. 2014, EMEA-001527-PIP01-13 (here) [ Novartis revealed the IUPAC name here].
Where name is given
http://www.who.int/medicines/publications/druginformation/issues/DrugInformation2017_Vol31-4/en/


http://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf?ua=1
SEE ALSO


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS
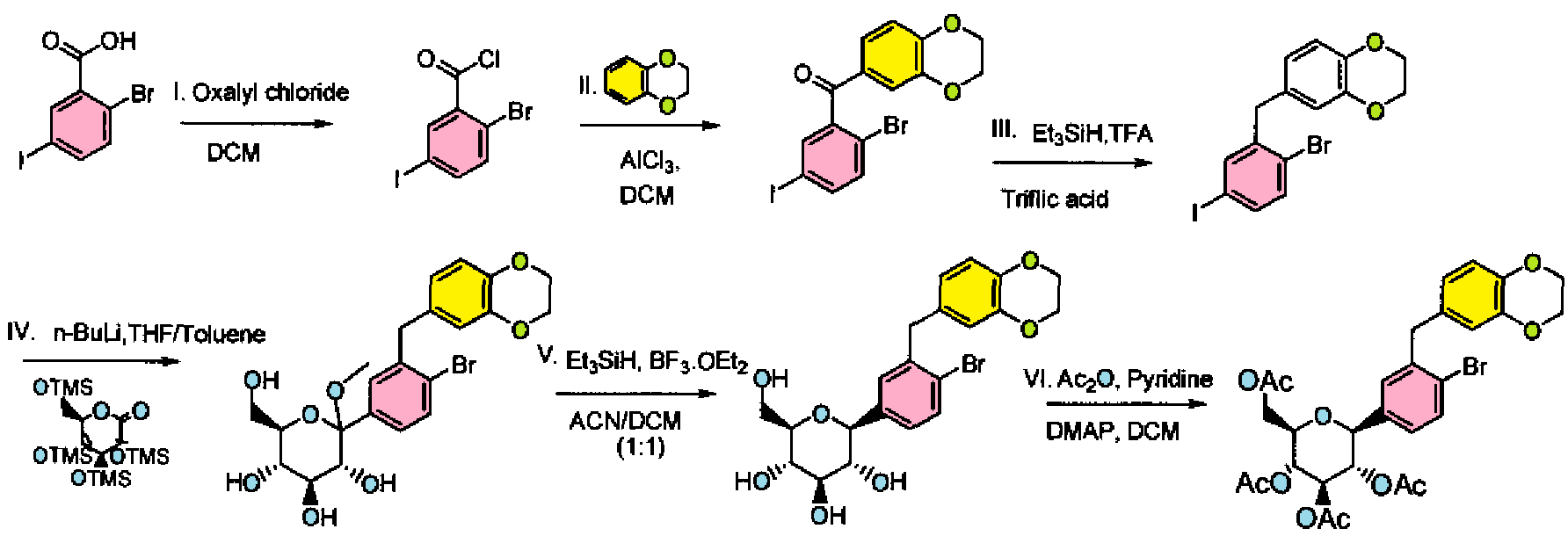
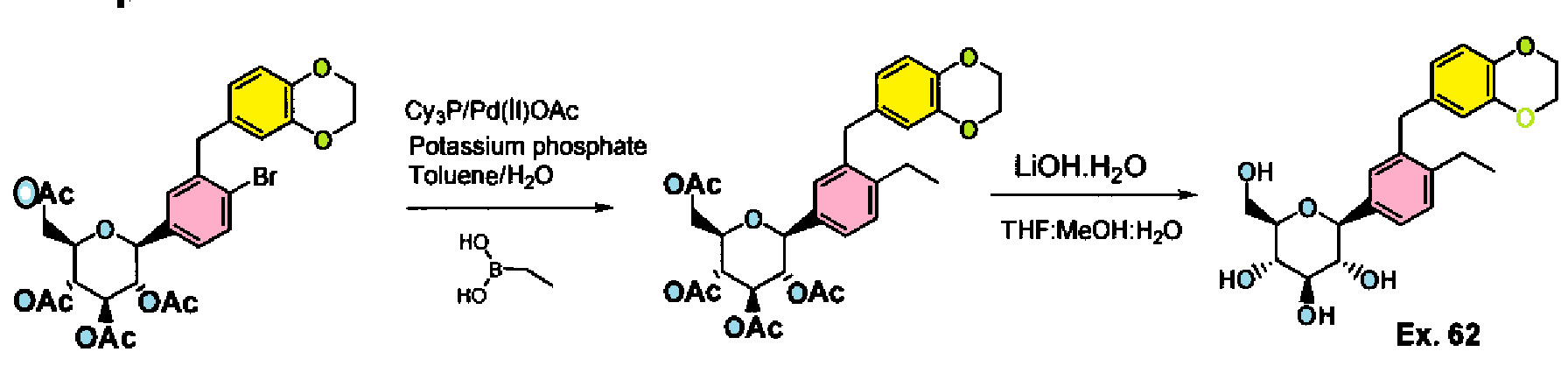
PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Ex. 57 Ex. 58
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE

Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////Licogliflozin diprolinate
see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
EV 077

EV-077
SER 150 (formerly EV-077)
Also known as: formerly EV-077-3201
EV-077-3201-2TBS
CAS 1384128-29-3
Evolva INNOVATOR
Oral thromboxane receptor antagonist and thromboxane synthase inhibitor
EV-077 is a small compound being developed for the treatment of complications of diabetes. In Phase 2. Outlicensed to Serodus in 2013.
In 2013, Serodus licensed the product candidate for the treatment of diabetic nephropathy and it is conducting phase II clinical trials on this research.
EV-077 is an oral, small molecule compound, belonging to a new structural class. Preclinical and early clinical studies indicate EV-077 has potential in reducing vascular inflammation by inhibiting the activity of prostanoids and isoprostanes – in particular in diabetes. Towards the end of 2011, the Russian Patent Office granted patent protection for EV-077 in the treatment of complications of diabetes for a term extending to 2026. Evolva has outlicenced EV-077 to Serodus in 2013. Serodus aims to bring EV-077 further through clinical development and at a future time point decide whether Serodus or a partner will conduct the final clinical trials.
EV-077 is in development as a potential pharmaceutical for the treatment of diabetic nephropathy and other diabetic complications. It is in Phase II clinical studies.
In 2013, Evolva out-licensed EV-077 to Serodus (Oslo, Norway). Serodus aims to bring EV-077 through Phase II and then decide whether or not to partner for the final clinical trials and commercialisation. Evolva is entitled to clinical and regulatory milestones as well as a single-digit royalty on sales. If Serodus sublicenses EV-077 then Evolva will receive up to 30% of Serodus’ total licensing income.
As of Q2 2015 Serodus continues active development of EV-077.
– See more at: http://www.evolva.com/ev-077/#sthash.4mgJ3E0f.dpuf
Patients with diabetes mellitus (DM) have increased propensity to generate thromboxane A2 (TXA2) and other eicosanoids which can contribute to their heightened platelet reactivity. EV-077 is a potent thromboxane receptor antagonist and thromboxane synthase inhibitor and thus represents an attractive therapy in patients with DM. However, the effects of EV-077 on pharmacodynamic (PD) profiles in patients with DM and coronary artery disease (CAD) while on antiplatelet therapy is poorly explored and represented the aim of this in vitro pilot investigation. Patients with DM and stable CAD (n = 10) on low-dose aspirin (81 mg/day) were enrolled and then switched to clopidogrel (75 mg/day) monotherapy for 7-10 days. PD assessments were conducted while on aspirin and on clopidogrel using light transmittance aggregometry following stimuli with U-46619 [TXA2 stable analogue (7 μM)], arachidonic acid [AA (1 mM)], collagen (3 μg/mL) and adenosine diphosphate [ADP (5 μM and 20 μM)] with and without in vitro EV-077. EV-077 completely inhibited U-46619-stimulated platelet aggregation (p = 0.005 for both aspirin and clopidogrel) and also showed a significant reduction of collagen-induced aggregation (aspirin p = 0.008; clopidogrel p = 0.005). EV-077 significantly reduced AA-induced platelet aggregation in clopidogrel (p = 0.009), but not aspirin (p = 0.667) treated patients. Ultimately, EV-077 significantly reduced ADP-mediated platelet aggregation in both aspirin (ADP 5 μM p = 0.012; ADP 20 μM p = 0.032) and clopidogrel (ADP 5 μM p = 0.007; ADP 20 μM p = 0.008) treated patients. In conclusion, in DM patients with CAD on aspirin or clopidogrel monotherapy, in vitro EV-077 exerts potent platelet inhibitory effects on multiple platelet signaling pathways. These data support that EV-077 has only additive platelet inhibiting effects on top of standard antiplatelet therapies. These findings warrant further investigation in ex vivo settings.
Description
EV-077 is a small compound being developed for the treatment of complications of diabetes. In Phase 2. Outlicensed to Serodus in 2013.
Situation Overview
Diabetes and its complications are major global health care problems. Based on estimates by the International Diabetes Federation (IDF), there were 366 million diabetics worldwide in 2011, a number which is expected to increase to 552 million by 2030. IDF estimates the number of deaths in 2011 at 4.6 million and total spending on diabetic health care at USD 465 billion.
EV-077 is an oral, small molecule compound, belonging to a new structural class. EV-077 is being developed for the reduction of vascular inflammation by inhibiting the activity of prostanoids and isoprostanes ��� in particular in diabetes. Towards the end of 2011, the Russian Patent Office granted patent protection for EV-077 in the treatment of complications of diabetes for a term extending to 2026. Additional patent applications are pending in all major territories. Evolva has outlicenced EV-077 to Serodus in 2013.
Mechanism of Action
Preclinical and early clinical studies indicate EV-077 has potential in reducing vascular inflammation by inhibiting the activity of prostanoids and isoprostanes in particular in diabetes. The mechanism of action of EV-077 means that it can potentially ameliorate or prevent a range of diabetic complications (including loss of kidney function, reduced peripheral blood flow and increased risk of thrombosis) that derive from the following chain of events:
- Diabetic patients have a reduced sensitivity to insulin which increases overall glucose levels in the body;
- This increase in glucose increases oxidative stress;
- The oxidative stress generates a high level of isoprostanes and prostanoids;
- The isoprostanes and prostanoids chronically activate thromboxane prostanoid receptors, that are located on the walls of blood vessels (endothelial cells and smooth muscle cells) and the surface of platelets;
- Activation of the thromboxane prostanoid receptors causes vascular inflammation and increased platelet reactivity;
- An increased number of vascular events and a progressive deterioration of circulatory and renal function.
Clinical Trials
In November 2011, Evolva received regulatory clearance to progress EV-077 into Phase IIa clinical studies for the treatment of complications of diabetes. It is a single-centre study, conducted in Germany. The study was a randomized, double-blind, and placebo-controlled, and investigated the efficacy and safety of EV-077 in type 2 diabetics with a heightened risk of diabetic vascular complications. Measurements included blood flow and platelet reactivity, biomarkers for oxidative stress and vascular inflammation as well as markers of the function of organs that are often impaired in diabetes (e.g. kidney).
In May 2012, the study was terminated. Interim results for the first 32 patients enrolled in the Phase IIa study show promising efficacy data, indicating that 300mg EV-077 given orally twice daily to patients with type 2 diabetes provided anti-platelet activity, reduced exercise-induced proteinuria and increased forearm blood flow. This was achieved with only a slight increase in bleeding time. The analysis also indicated that EV-077 was generally well tolerated, with adverse events mostly limited to increases in liver enzymes, which were transient or resolved after discontinuation.
In parallel with the Phase IIa study, Evolva is conducting epidemiological studies to identify high risk diabetic patient subgroups that can potentially derive particular benefit from the administration of EV-077. Given success, this is expected to expedite both further clinical development (by reducing the size and duration of late stage clinical trials) and the eventual approval process.
Partners by Region
Evolva has outlicensed EV-077 to Serodus in 2013. Serodus aims to bring EV-077 further through clinical development and at a future time point decide whether Serodus or a partner will conduct the final clinical trials.
WO 2014011273
http://www.google.com/patents/WO2014011273A2?cl=en
Journal of Thrombosis and Haemostasis (2011), 9(10), 2109-2111
Thrombosis Research (2012), 130(5), 746-752
European Journal of Clinical Pharmacology (2013), 69(3), 459-465
Biochemical and Biophysical Research Communications (2013), 441(2), 393-398
Journal of Thrombosis and Thrombolysis (2014), 37(2), 131-138
http://www.google.co.in/patents/WO2008089461A1?cl=en
(Z)-6-((2S,4S,5R)-2-(2-chlorophenyl)-4-(2-hydroxyphenyl)-1 ,3-dioxan-5-yl)hex-4-enoic acid has the 3 groups all up, which has a dramatic effect on its biological activities:

see
WO 2011057262
/////////////// SEE……..http://drugsynthesisint.blogspot.in/2015/11/ev-077.html
Zydus Cadila’s new 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds in pipeline for diabetes type 2
List of compounds as DPP-IV inhibitors


Watch out on this post as I get to correct structure………..
![]()

2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds

One Example of 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds
CAS 1601479-87-1
(2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[5-(methylsulfonyl)-3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]tetrahydro-2H-pyran-3-amine
MW 399.45, C18 H23 F2 N3 O3 S
INTRODUCTION
Dipeptidyl peptidase IV , CD26; DPP-IV; DP-IV inhibitors acting as glucose lowering agents reported to be useful for the treatment of type 2 diabetes. compound inhibited human DPP-IV enzyme activity (IC50 < 10 nM) in fluorescence based assays.
It lowered glucose levels (with -49.10% glucose change) when administered to C57BL/6J mice at 0.3 mg/kg p.o. in oral glucose tolerance test (OGTT).
Compound displayed the following pharmacokinetic parameters in Wistar rats at 2 mg/kg p.o.: Cmax = 459.04 ng/ml, t1/2 = 59.48 h and AUC = 4751.59 h·ng/ml.
Dipeptidyl peptidase 4 (DPP-IV) inhibitor that inhibited human DPP-IV enzyme activity with an IC50 of < 10 nM in a fluorescence based assay.
Watch out on this post as I get to correct structure………..![]()

PATENT
http://www.google.com/patents/WO2014061031A1?cl=en
Compound 8: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz): 7.32-7.28 (m, IH), 7.26-7.23 (m, 2H), 4.77 (d, IH, J= 10Hz), 4.32(dd, IH, J,= 2.0Hz, J2= 10.8Hz), 4.19 (s, 4H), 3.89-3.83 (m, 4H), 3.70- 3.65 (m, IH), 3.61 (t, IH, J= 11.6Hz), 3.53-3.46 (m, IH), 3.04 (s, 3H), 2.65-2.62 (dd, IH, Ji= 1.2Hz, J2= 12Hz), 1.84 (q, IH, J = 12 Hz); ESI-MS: (+ve mode) 400.0 (M+H)+ (100 %); HPLC: 99.4 %.
Compound 4: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(hexahydropyrrolo[3, 4-c Jpyrrol- 2(lH)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz):

.23-7.20 (m, 2H), 4.64 (d, IH, J= 10.4 Hz), 4.38-4.35 (dd, IH, J,= 2.4Hz, J2= 10.4Hz), 3.69 (t, IH, J= 11Hz), 3.57-3.53 (m, 4H), 3.34-3.30 (m, 8H), 2.68-2.65 (m, IH), 2.04 (q, IH, J = 1 1.6 Hz); ESI-MS: (+ve mode) 323.9 (M+H)+ (100 %), 345.9 (M+Na)+ (20%); HPLC: 98.6 %

PATENT
IN 2012MU03030
“NOVEL DPP-IV INHIBITORS”
3030/MUM/2012
Abstract:
The present invention relates to novel compounds of the general formula (I) their tautomeric forms, their enantiomers, their diastereoisomers, their pharmaceutically accepted salts, or pro-drugs thereof, which are useful for the treatment or prevention of diabetes mellitus (DM), obesity and other metabolic disorders. The invention also relates to process for the manufacture of said compounds, and pharmaceutical compositions containing them and their use.

Pankaj R. Patel (right), Chairman and Managing Director,
////////////2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds, DPP-IV inhibitors
