
Home » 2015 (Page 17)
Yearly Archives: 2015
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves Repatha to treat certain patients with high cholesterol
August 27, 2015
Release
The U.S. Food and Drug Administration today approved Repatha (evolocumab) injection for some patients who are unable to get their low-density lipoprotein (LDL) cholesterol under control with current treatment options.
Repatha, the second drug approved in a new class of drugs known as PCSK9 inhibitors, is approved for use in addition to diet and maximally-tolerated statin therapy in adult patients with heterozygous familial hypercholesterolemia (HeFH), homozygous familial hypercholesterolemia (HoFH), or clinical atherosclerotic cardiovascular disease, such as heart attacks or strokes, who require additional lowering of LDL cholesterol.
Familial hypercholesterolemia (encompassing both HeFH and HoFH) is an inherited condition that causes high levels of LDL cholesterol. A high level of LDL cholesterol in the blood is linked to cardiovascular or heart disease. Heart disease is the number one cause of death for Americans, both men and women. According to the Centers for Disease Control and Prevention, about 610,000 people die of heart disease in the United States every year– that equals one in every four deaths.
“Repatha provides another treatment option in this new class of drugs for patients with familial hypercholesterolemia or with known cardiovascular disease who have not been able to lower their LDL cholesterol enough with statins,” said John Jenkins, M.D., director of the Office of New Drugs, Center for Drug Evaluation and Research. “Cardiovascular disease is a serious threat to the health of Americans, and the FDA is committed to facilitating the development and approval of effective and safe drugs to address this important public health problem.”
Repatha is an antibody that targets a specific protein, called PCSK9. PCSK9 reduces the number of receptors on the liver that remove LDL cholesterol from the blood. By blocking PCSK9’s ability to work, more receptors are available to get rid of LDL cholesterol from the blood and, as a result, lower LDL cholesterol levels.
The efficacy and safety of Repatha were evaluated in one 52-week placebo-controlled trial and eight 12-week placebo-controlled trials in participants with primary hyperlipidemia, including two that specifically enrolled participants with HeFH and one that enrolled participants with HoFH. In one of the 12-week studies, 329 participants with HeFH, who required additional lowering of LDL cholesterol despite statins with or without other lipid-lowering therapies, were randomized to receive Repatha or placebo for 12 weeks. Participants taking Repatha had an average reduction in LDL cholesterol of approximately 60 percent, compared to placebo.
The most common side effects of Repatha include nasopharyngitis, upper respiratory tract infection, flu, back pain, and reactions such as redness, pain, or bruising where the injection is given. Allergic reactions, such as rash and hives, have been reported with the use of Repatha. Patients should stop using Repatha and get medical help if they experience symptoms of a serious allergic reaction.
Multiple clinical trials have demonstrated that statins lower the risk of having a heart attack or stroke. A trial evaluating the effect of adding Repatha to statins for reducing cardiovascular risk is ongoing.
Repatha is marketed by Amgen Inc., of Thousand Oaks, Calif.
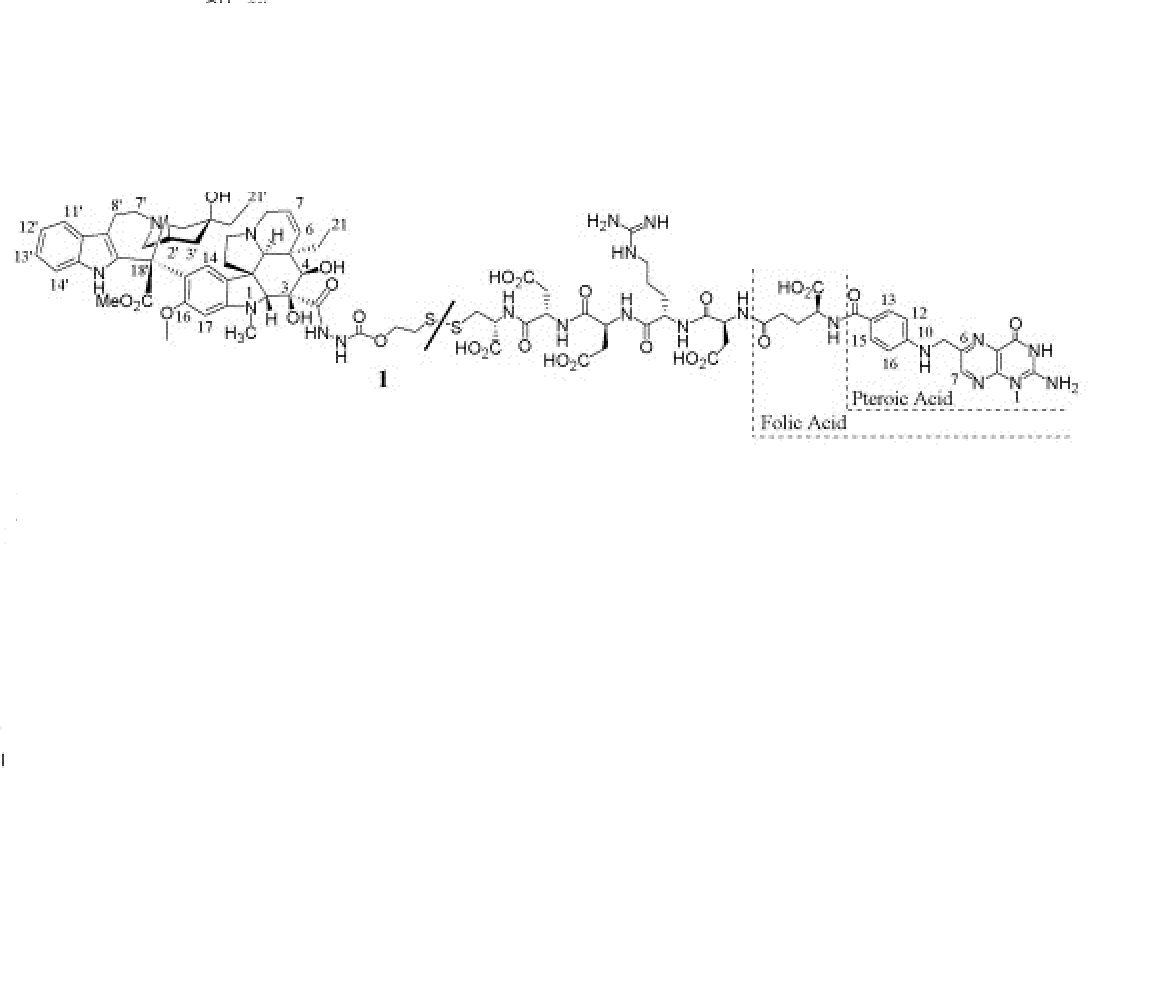
Vintafolide

Vintafolide, EC-145 , MK-8109
mw 1917.041, cas 742092-03-1, mf C86 H109 N21 O26 S2
(2S)-2-[(4-{[(2-amino-4-oxo-3H-pteridin-6-yl)methyl]amino}phenyl)formamido]-4-{[(1S)-1-{[(1S)-4-carbamimidamido-1-{[(1S)-2-carboxy-1-{[(1S)-2-carboxy-1-{[(1R)-1-carboxy-2-({2-[({[(1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-(methoxycarbonyl)-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10,11-dihydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraen-10-yl]formohydrazido}carbonyl)oxy]ethyl}disulfanyl)ethyl]carbamoyl}ethyl]carbamoyl}ethyl]carbamoyl}butyl]carbamoyl}-2-carboxyethyl]carbamoyl}butanoic acid
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-1,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Endocyte innovator
Vintafolide is an investigational targeted cancer therapeutic currently under development by Endocyte and Merck & Co.[1] It is a small molecule drug conjugate consisting of a small molecule targeting the folate receptor, which is overexpressed on certain cancers, such as ovarian cancer, and a potent chemotherapy drug, vinblastine.[2] It is being developed with a companion imaging agent, etarfolatide, that identifies patients that express the folate receptor and thus would likely respond to the treatment with vintafolide.[3] A Phase 3 study evaluating vintafolide for the treatment of platinum-resistant ovarian cancer (PROCEED trial) and a Phase 2b study(TARGET trial) in non-small-cell lung carcinoma (NSCLC) are ongoing.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
A Marketing Authorization Application (MAA) filing for vintafolide and etarfolatide for the treatment of patients withfolate receptor-positive platinum-resistant ovarian cancer in combination with doxorubicin, pegylated liposomal doxorubicin (PLD), has been accepted by the European Medicines Agency.[6] The drug received an orphan drug status in Europe in March 2012.[1] Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012.[1] The drug received orphan drug status in Europe in March 2012.[3]Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion imaging agent used to identify patients expressing the folate receptor that will likely respond to treatment with vintafolide.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
In 2014 Merck and Endocyte stopped a late-stage study of vintafolide in treating ovarian cancer on the recommendation of a data safety monitoring board, saying that the drug failed to improve progression-free survival.[7]
Vintafolide is folate-conjugated with DAVBLH, which is a derivative of the vinca alkaloid vinblastine.Vinblastine is a microtubule-destabilizing agent that binds tubulin and causes M phase-specific cell cycle arrest and apoptosis of mitotically active cells. Vinblastine is an extremely potent chemotherapeutic agent but has significant toxicities including bone marrow suppression, neurotoxicity, gastrointestinal toxicity and vesicant injury.
Endocyte’s desacetylvinblastinehydrazide/folate conjugate (EC-145) is a folate-targeted cytotoxic anticancer drug in early development for the treatment of non-small cell lung cancer (NSCLC) and breast cancer. The compound had been pre-registered in the E.U. by Merck for the treatment of ovarian cancer, but the application was withdrawn due to lack of efficacy.
In 2012, the product was licensed to Merck & Co. by Endocyte for worldwide exclusive development and commercialization. In 2014, however, this license agreement was terminated and Endocyte regained all rights.
Folates can serve as one-carbon donors in reactions that are critical in the de novo biosynthesis of purines and thymidylate, amino acid metabolism and methylation reactions. Folate can enter a cell by two routes: RFC or by membrane-bound FRs. RFC is a bidirectional anion transporter that is the normal entry method for reduced folates in most cells. By contrast, FRs are expressed in a limited distribution in normal tissues but are overexpressed in multiple cancers including ovarian, lung, breast and colorectal cancer. FRs bind folate derivatives with high affinity and mediate their internalization by endocytosis. Given that FRs are not typically expressed on the luminal surface of epithelial cells, making them inaccessible to normal circulation, they are attractive therapeutic targets with limited toxicity. In addition to the therapeutic agent vintafolide, a radiodiagnostic agent (99mTc-etarfolatide [EC20]) has been developed to allow single-photon emission computed tomography (SPECT) imaging to identify FR-expressing tissues (tumors).
In 2012, orphan drug designations were assigned in the E.U. for the treatment of ovarian cancer and to be used with folic acid for the diagnosis of positive folate-receptor status in ovarian cancer. In 2013, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer.
Vintafolide is a water-soluble derivative of folic acid and the vinca alkaloid DAVLBH. The molecules are connected through a hydrophilic L-peptide spacer and a disulfide linker (Figure 1). The disulfide linker serves as a cleavable bond that is necessary for drug release following receptor mediated endocytosis. The disulfide bond is reduced in the acidic environment of the endosome, leading to efficient release of vinblastine.

Vintafolide.
DAVBLH: Desacetylvinblastine hydrazide

Structure of vintafolide and mechanism of release of the payload in the endosome.

Mechanism of action
Folate is required for cell division, and rapidly dividing cancer cells often express folate receptors in order to capture enough folate to support rapid cell growth. Elevated expression of the folate receptor occurs in many diseases, including other aggressively growing cancers and inflammatory disorders.[8] Vintafolide binds to the folate receptor and is subsequently taken up by the cell through a natural internalization process called endocytosis. Once inside the cell, vintafolide’s linker releases the chemotherapy drug which kills the cell.[3]
……………
Bioorganic & Medicinal Chemistry Letters (2006), 16(19), 5093-5096
http://www.sciencedirect.com/science/article/pii/S0960894X06008079
An efficient synthesis of the folate receptor (FR) targeting conjugate EC145 is described. EC145 is a water soluble derivative of the vitamin folic acid and the potent cytotoxic agent, desacetylvinblastine monohydrazide. Both molecules are connected in regioselective manner via a hydrophilic peptide spacer and a reductively labile disulfide linker.

………approach for the design and regioselective synthesis of a FA-vinca alkaloid conjugate 1 (EC145,BELOW). As indicated in the retrosynthetic scheme, 1 can be assembled by tethering a FA-Spacer unit 2 to the highly potent cytotoxic molecule, desacetylvinblastine monohydrazide 3, via a linker containing a reducible disulfide bond. The latter is important for drug delivery applications since real-time imaging using a fluorescence resonance energy transfer technique has recently demonstrated that reduction-mediated release of the drug cargo from a disulfide linked FA-conjugate efficiently occurs within the endosomes of cancer cells.

-
Scheme 1.
Reagents and conditions: (i) a—Fmoc-Asp(OtBu)-OH, PyBOP, DIPEA, RT, 1 h; b—20% piperidine/DMF, rt, 10 min; (ii) a—Fmoc-Arg(Pbf)-OH, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iii) a—Fmoc-Glu-OtBu, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iv) N10-TFA-pteroic acid, PyBOP, DIPEA, rt, 1.5 h; (v) TFA/H2O/TIPS/EDT (92.5:2.5:2.5:2.5), rt, 1 h; (vi) aq NH4OH, pH 9.3, rt, 1 h.
- Selected 1H NMR data for 2 (D2O, 300 MHz): δ 8.68 (s, 1H, FA H-7), 7.57 (d, 2H,J = 8.4 Hz, FA H-12 & 16), 6.67 (d, 2H, J = 9 Hz, FA H-13 & 15), 4.40–4.75 (series of m, 5H), 4.35 (m, 2H), 4.16 (m, 1H), 3.02 (m, 2H), 2.55–2.95 (series of m, 8H), 2.42 (m, 2H), 2.00–2.30 (m, 2H), 1.55–1.90 (m, 2H), 1.48 (m, 2H).
- 1H NMR for compound 6 (DMSO-d6, 300 MHz): δ 8.38 (m, 1H), 8.16 (dt, 1H, J = 8 Hz, 1 Hz), 8.02 (dt, 1H, J = 8 Hz, 1 Hz), 7.88 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz), 7.7 (m, 2H), 7.63 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz,), 7.4–7.2 (br, 1H), 7.2 (m, 1H), 4.72 (t, 2H,J = 6 Hz), 3.36 (t, 2H, J = 6 Hz).
- Selected 1H NMR data for
EC145 (D2O, 300 MHz): δ 8.67 (s, 1H, FA H-7), 7.50 (br s, 1H, VLB H-11′), 7.30–7.40 (br s, 1H, VLB H-14′), 7.35 (d, 2H, J = 7.8 Hz, FA H-12 & 16), 7.25 (m, 1H, VLB H-13′), 7.05 (br s, 1H, VLB H-12′), 6.51 (d, 2H, J = 8.7 Hz, FA H-13 & 15), 6.4 (s, 2H, VLB H-14 & 17), 5.65 (m, 1H, VLB H-7), 5.5 (m, 1H, VLB H-6), 4.15 (m,1H, VLB H-8′), 3.82 (s, 3H, VLB C18′ –CO2CH3), 3.69 (s, 3H, VLB C16 –OCH3), 2.8 (s, 3H, VLB N–CH3), 1.35 (br s, 1H, VLB H-3′), 1.15 (m, 1H, VLB H-2′), 0.9 (t, 3H, J = 7 Hz, VLB H-21′), 0.55 (t, 3H, J = 6.9 Hz, VLB H-21).
 CLICK ON IMAGE FOR CLEAR VIEW
CLICK ON IMAGE FOR CLEAR VIEW
…………
WO 2004069159
http://www.google.com/patents/WO2004069159A2?cl=en
EXAMPLE 16b

The compounds of Examples 16a and 16b were prepared from the peptidyl fragment Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH , prepared according to the general procedure described in Scheme 12. The Michael addition of this peptidyl fragment to the maleimido derivative of seco-CBI-bis-indole resulted in the folate conjugates Example 16a. The peptidyl fragment also reacted with either the thiosulfonate or pyridyldithio-activated vinblastine to form Example 16b. The maleimido derivative of seco-CBI-bis-indole, and the thiosulfonate and pyridyldithio- activated vinblastine intermediates were prepared using the procedures described herein for other examples.
……………..
https://www.google.com/patents/WO2012142281A1?cl=en
Folate-targeted drugs have been developed and are being tested in clinical trials as cancer therapeutics. EC145, also known as vintafolide, comprises a highly potent vinca alkaloid cytotoxic compound, desacetylvinblastine hydrazide (DAVLBH), conjugated to folate. The EC 145 molecule targets the folate receptor found at high levels on the surface of epithelial tumors, including non-small cell lung carcinomas (NSCLC), ovarian, endometrial and renal cancers, and others, including fallopian tube and primary peritoneal carcinomas. It is believed that EC 145 binds to tumors that express the folate receptor delivering the vinca moiety directly to cancer cells while avoiding normal tissue. Thus, upon binding, EC 145 enters the cancer cell via endocytosis, releases DAVLBH and causes cell death or inhibits cell function. EC 145 has the following formula

EC145
and has been accorded the Chemical Abstracts Registry Number 742092-03-1. As used herein, according to the context, the term EC 145 means the compound, or a pharmaceutically acceptable salt thereof; and the compound may be present in a solid, solution or suspension in an ionized form, including a protonated form. EC145 is disclosed in U.S. Patent No. 7,601,332; and particular uses and an aqueous liquid pH 7.4, phosphate-buffered formulation for intravenous administration are disclosed in WO 2011/014821. As described in WO 2011/014821, it is necessary to store the aqueous liquid formulation in the frozen state to ensure its stability. To avoid this necessity, a formulation is needed which has adequate stability at ambient temperature.
As one aspect of the invention described herein, there is provided a pharmaceutical composition of EC145 which is a lyophilized solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
In another aspect of the invention, there is provided a pharmaceutical composition of EC 145 which is an X-ray amorphous solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-{[(2-Amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)-5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5-({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro-1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7-methanoazacycloundecino[5,4-b]indol-9-carboxylate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 742092-03-1 |
| ATC code | L01CA06 |
| ChemSpider | 27444385 |
| Synonyms | EC-145 |
| Chemical data | |
| Formula | C86H109N21O26S2 |
| Molecular mass | 1917 g/mol |
References
- Sridharan, Balaji (Apr 16, 2012). “Endocyte soars on cancer drug deal with Merck”. Reuters.
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Kuo, Phillip H. (February 2013). “Companion Imaging Diagnostics for Targeted Therapies”. Radiology Today 14 (2): 32.
- “Merck, Endocyte in Development Deal”. Drug Development & Discovery magazine. 2012-04-25.
- Dosio, F.; Milla, P.; Cattel, L. (2010). “EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers”. Current opinion in investigational drugs (London, England : 2000) 11 (12): 1424–1433. PMID 21154124.
- “EMA Accepts For Review MAA Filings For Vintafolide And Etarfolatide”. rttnews.com. 2012-11-27.
- Garde, Damian (2014-05-02). “Merck halts study of the billion-dollar cancer drug vintafolide”. Fierce Biotech. Retrieved 21 April 2015.
- “Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay” 338 (2). March 2005. pp. 284–93. doi:10.1016/j.ab.2004.12.026.PMID 15745749.
-
WO2008098970A1 * Feb 13, 2008 Aug 21, 2008 Pf Medicament Anhydrous crystalline vinflunine salts, method of preparation and use thereof as a drug and means of vinflunine purification WO2010150100A1 * Jun 23, 2010 Dec 29, 2010 Entarco Sa The use of spinosyns and spinosyn compositions against diseases caused by protozoans, viral infections and cancer WO2011014821A1 * Jul 30, 2010 Feb 3, 2011 Endocyte, Inc. Folate-targeted diagnostics and treatment US20100247669 * Sep 30, 2010 Cerulean Pharma Inc. Polymer-agent conjugates, particles, compositions, and related methods of use
////////Vintafolide, BMS-753493, DAVBLH, Desacetylvinblastine hydrazide, EC-145 , MK-8109 , phase 2
Sandoz, Pfizer closing Mumbai API plant

Sandoz closing Mumbai API plant but says it remains committed to India

Sandoz will shutter an Indian API facility in 2016 as part of a manufacturing refocus in the region.
From online sources
Drug major Sandoz will discontinue operations at its Turbhe site (Maharashtra) by end December 2016, as part of global plans to optimise its manufacturing network.

The Turbhe sites employs 170 people and manufactures antibiotics and active pharmaceutical ingredients (API), a note from the company said. Sandoz is the generic drugs arm of pharmaceutical company Novartis.
“Sandoz will refocus its manufacturing set up in India as part of its strategy to optimise its global manufacturing network, while continuing to serve patients in India,” the company said. As part of the plan, Sandoz will focus its manufacturing at other sites which employ over 1,300 employees and produce over three billion tablets and 180 tonnes of API annually, it added. The company has two manufacturing facilities at Kalwe and Mahad.
“We made the announcement today to ensure our associates are informed as soon as possible about our decisions and to ensure a transparent process,” Vivek Devaraj, Sandoz Country Head in India, said in the statement. “We are committed to managing the process with care, sensitivity and respect for all impacted associates at Turbhe, to supporting our customers through the transition and to meeting patient needs for access to important medicines,” he added.
In 2012, the company had shut down its formulations and API development centres, respectively. Drug companies have in the past shut down plants in India as a fallout of global strategies, mergers and acquisitions. At present, Pfizer’s plant in Thane faces an uncertain future.

……………………
ABOUT PFIZER
 There has been practically no production at Pfizer’s Thane plant (pic above) since 2013.
There has been practically no production at Pfizer’s Thane plant (pic above) since 2013.

Sandoz India’s Turbhe plant to down shutters by December 2016
Less than a week after Sandoz, the generic division of Novartis, announced that it would discontinue operations at its Turbhe site by end December 2016, another MNC, Pfizer India announced the closure of its manufacturing facility at Thane, two months from today, from September 16, 2015.
According to Pfizer India spokesperson, the Thane plant was commissioned in the 1960s, manufacturing medicines for both domestic and international markets but there has been ‘practically been no production activity at this plant since 2013′. Hence closure of the site would not impact supply of Pfizer India’s medicines.
Both plant closures are a consolidation of manufacturing facilities, with the shutting down of older facilities and re-direction to more modern facilities, with Pfizer India’s statement attributing the decision to ‘an assessment of its long term viability and its ability to achieve the needed production.’
132 of the 212 Pfizer India workmen at the Thane plant had already taken up the voluntary retirement scheme (VRS) offered by the company and the statement indicated that the remaining 80 workmen who continued to receive full wages despite plant inactivity, would also receive requisite compensation as mandated by law.
While the close down process is in the final stages at Pfizer India’s Thane facility, Sandoz’ July 10 announcement is the beginning of the process at its Turbhe plant, which employs 170 associates and manufactures antibiotics and APIs.
“We made the announcement (on July 10) to ensure our associates are informed as soon as possible about our decisions and to ensure a transparent process,” said Vivek Devaraj, Sandoz Country Head in India. He said the company was “committed to managing the process with the utmost care, sensitivity and respect for all impacted associates at Turbhe, to supporting our customers through the transition and to meeting patient needs for access to important medicines.” Manufacturing would now focus at its other sites which employ over 1,300 associates and produce over three billion tablets and 180 tonnes of API annually.”
/////////Sandoz, shutter, Indian API facility, Pfizer
Mahendra Chemicals gets FDA Warning Letter with Focus on “Data Integrity”
DRUG REGULATORY AFFAIRS INTERNATIONAL

A Warning Letter issued by the US Food & Drug Administration (FDA) to an Indian API manufacturer on 13 July 2015 shows again a clear focus on the missing integrity of data. Specifically, the following issues are addressed:
1. Activities were not recorded at the time they were carried out and original data were deleted:
Entries in the manufacturing protocols were made only days after the relevant activities had been conducted. Further, batches were released before all results were available.
In particular the use of “rough notes” was criticised as these original data were completely destroyed after transfer in the batch records.
2. Due to unauthorised access to data systems, data could be modified or deleted:
Specifically HPLC, GC, and Karl Fischer Titrators were concerned. For instance, for the GC instrument multiple copies of raw data were found in the waste. And there was no password regulation for the data systems…
View original post 100 more words
FDA approves Praluent for the treatment of high LDL cholesterol
26 August 2015
Sanofi and Regeneron have announced that the US Food and Drug Administration (FDA) has approved Praluent® (alirocumab) Injection.

Praluent is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease (ASCVD), who require additional lowering of low-density lipoprotein (LDL) cholesterol. The effect of Praluent on cardiovascular morbidity and mortality has not been determined.
////////Sanofi, Regeneron, US Food and Drug Administration, FDA, approved, Praluent® , alirocumab
Non compliance at Parabolic drugs
DRUG REGULATORY AFFAIRS INTERNATIONAL

Statement “non compliance GMP”. Officina Farmaceutica: Parabolic Drugs Limited – INDIA (30/07/2015)
Following the inspection, conducted by the inspectorate Italian, under the program of inspections of the EDQM, at the Indian site in question, the same was not “in compliance” with the GMP.
It calls on companies to verify, with urgency, if the medicines containing the following active substances / intermediate production Dicloxacillin SODIUM, amoxicillin trihydrate, PIVAMPICILLIN, Flucloxacillin SODIUM, SODIUM cloxacillin, AMPICILLIN trihydrate, AMPICILLIN ANHYDROUS, Bacampicillin HYDROCHLORIDE authorized for the Italian market and / or products for export, showing this as a possible supplier of active / intermediate Officina Farmaceutica: PARABOLIC DRUGS LIMITED, PDL-2 – Plot No. 45, Industrial Area, Phase II, Panchkula District of Haryana, 134113 , INDIA .
The communication must be sent only by all companies Holders of marketing authorizations or Officine pharmaceutical manufacturers of medicines containing these materials pharmacologically active / production intermediates produced at…
View original post 194 more words
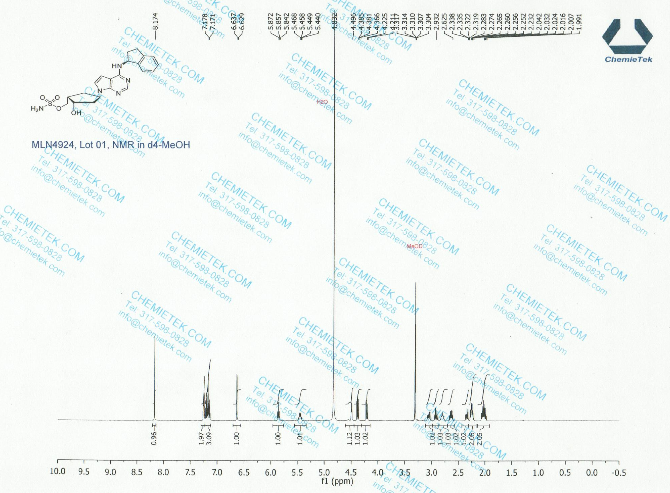
Pevonedistat

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE



| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation
AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR

 Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
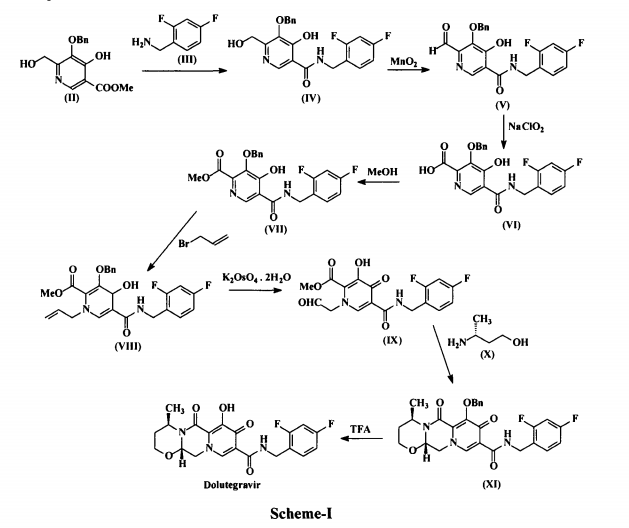
Dolutegravir (I) is chemically known as (4/?,12aS)-N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2//-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide. Dolutegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. Dolutegravir is being marketed under the trade name Tivicay®. US 8,129,385 disclosed Dolutegravir or its pharmaceutically acceptable salts thereof. US ‘385 also discloses a process for the preparation of Dolutegravir (I). The process involves the condensation of 5-benzyloxy-4-hydroxy-6-hydroxymethyl nicotinic acid (II) with 2,4-difluorobenzylamine (III) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-4-hydroxy-6-hydroxymethyl nicotinic acid amide (IV), which is further under goes oxidation using manganese dioxide (Mn02) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-6-formyl-4-hydroxy-nicotinic acid amide (V). This amide compound (V) is reacted with sodium chlorite (NaClCh) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4- hydroxy-pyridine-2-carboxylic acid (VI), which is further treated with methanol (MeOH) to produce 3-benzyloxy-5-(2,4-difluorobenzyl)-4-hydroxy-pyridine-2-carboxylic acid methyl ester (VII).

The methyl ester compound (VII) is reacted with 3-bromopropene to produce l-allyl-3-benzyloxy-5-(2,4-difluorobenzyl)-4-oxo-l,4-dihydro-pyridine-2- carboxylic acid methyl ester (VIII), which is further reacted with potassium osmate dihydrate (K2OSO4.2H2O) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4-oxo-l-(2-oxo-ethyl)-l,4-dihydropyridine-2-carboxylic acid methyl ester (IX). The compound (IX) is reacted with (R)-3-amino-l-butanol (X) to produce benzyloxy Dolutegravir (XI), which is deprotected by treating with TFA to produce Dolutegravir (I). The process is as shown in scheme-I below:
The major disadvantage with the above prior-art process is that it involves large no of steps and tedious work-up procedures to isolate the required product. This results a longer period of time cycle is required to produce Dolutegravir (I), which in turn renders the process more costly and less eco friendly. Further the above processes are low yielding and with less purity. US 8,217,034 discloses variant process for the preparation of Dolutegravir.
This process involves the reaction of methyl l-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-l,4-dihydro-2-pyridine carboxylate (XII) with (R)-3-amino-l-butanol (X) to produce (4R, 12o5)-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2//-pyrido[ 1 \2′,4,5] pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIII), which is further undergoes bromination using NBS to produce (4R,12aS)-9-bromo-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIV). The bromo Compound (XIV) is condensed with 2,4-difluorobenzylamine (III) in the presence of Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) to produce benzyloxy Dolutegravir (XI), which is hydrogenated in the presence of Pd/C to produce Dolutegravir (I). The process is as shown in Scheme-II below:

The major disadvantage with the above prior art process of preparing Dolutegravir is the use of expensive reagent tetrakis(triphenylphosphine)palladium (Pd(PPh3)4> in coupling step. Use of this reagent on industrial scale is not preferred, which makes the process more expensive. WO 2011/119566 discloses another variant process for the preparation of Dolutegravir.
This process involves the reaction of l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid (XV) with acetic acid in presence of methane sulfonic acid to produce 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), which is further condensed with (R)-3-amino-l-butanol (X) to produce (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[ 1 ‘,2’:4,5]pyrazino[2,1 -b] [ 1,3]-oxazine-9-carboxylic acid (XVII). This acid Compound XVII is acylated with 2,4-difluorobenzylamine (III) in the presence of carbonyldiimidazole (CDI) to produce methoxy Dolutegravir (XVIII), which is demethylated in the presence of lithium bromide (LiBr) to produce Dolutegravir (I).
The process is as shown in Scheme-3 below:
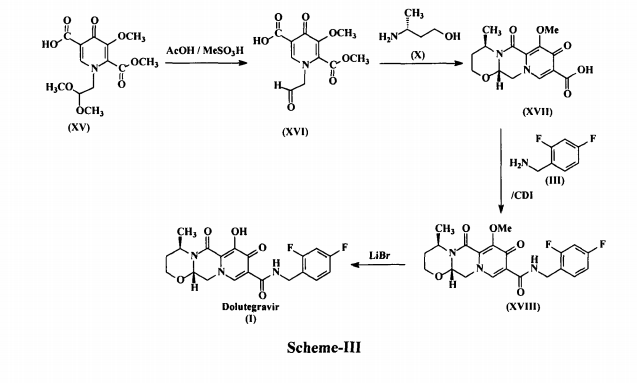
The major disadvantage of the above prior art process of preparing Dolutegravir is the use of expensive and highly moisture sensitive reagent, 1,1-carbonyldiimidazole (CDI), during acylation. Use of this reagent on industrial scale is not preferred due to anhydrous conditions required in the process. However, there is always a need for alternative preparative routes, which for example, involve fewer steps, use reagents that are less expensive and/or easier to handle, consume smaller amounts of reagents, provide a higher yield of product, have smaller and/or more eco-friendly waste products, and/or provide a product of higher purity. Hence, there is a need to develop cost effective and commercially viable process for the preparation of Dolutegravir of formula (I). The present invention is related to a process for the preparation of pure Dolutegravir of formula (I), wherein optically active acid addition salt of (R)-3-amino-l-butanol (X) is directly condensed with 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI) instead of condensing with free base of (R)-3-amino-1-butanol (X). The present invention is also related to a process for the preparation of pure Dolutegravir of formula (I), wherein, inexpensive and easily handling condensing reagents in the condensation of (4R, 12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[l’,2′:4,5]pyrazino [2,l-b][l,3]oxazine-9-carboxylic acid (XVII) with 2,4-difluorobenzylamine (III).


AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR
| APPLICATION NUMBER | 1361/CHE/2013 |
| APPLICANT NAME | AUROBINDO PHARMA LTD |
| DATE OF FILING | 27/03/2013 |
| PUBLICATION DATE (U/S 11A) | 16/01/2015 |
In another embodiment, 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4- dihydropyridine-3-carboxylic acid (XVI) used in the present invention is prepared by reacting 4-methoxyacetoacetate (XIX) with N,N-dimethyl-l,l- bis(methyloxy)methanamine (DMF-DMA) (XX) to produce methyl-2- (dimethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-(dimethylamino)-2 [(methyloxy)acetyl]-2-propenoate) (XXI), which is reacted with aminoacetaldehyde dimethyl acetal (XXII) to produce methyl-2-(2,2-dimethoxyethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-{[2,2-bis(methyloxy)ethyl]amino}-2-[(methyloxy) acetyl]-2-propenoate) (XXIII).
The compound (XXIII) is contacted with dimethyl ethanedioate in presence of alkali metal alkoxide to produce dimethyl-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-l ,4-dihydropyridine-2,5-dicarboxylate (XXIV), which is selectively hydrolyzed with a base to produce l-[2,2-bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l ,4-dihydro-3-pyridinecarboxylic acid (XV). The compound (XV) is treated with a catalytic amount of a strong protic acid in the presence of acetic acid in an organic solvent to produce a reaction mixture containing 5- methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), The process is as shown in Scheme-IV below:

The following examples illustrate the nature of the invention and are provided for illustrative purposes only and should not be construed to limit the scope of the invention.
Example-1:
EXAMPLES: Example-1: Process for the preparation of Dolutegravir
Step-i: Preparation of (/?)-3-amino-l-butanol tartarate salt: D-(+) Tartaric acid (12.7 g, 0.085 mol) was added in to a solution of (i?,5)-3-amino-l-butnaol (7.5 g, 0.084 mol) in methanol (100 ml) at 40 °C. The reaction mixture was stirred for about 1 hour at 35-40 °C and the reaction mass was cooled to 0-5°C and maintained for 30-40 minutes. The obtained solid was filtered and washed with chilled methanol (10 ml) at 0-5 °C. The solid was dried to get (i?)-3-amino-l-butanol tartarate salt (8.0 g, 40%).
Step-ii: Preparation of (4rt,12a£)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[l’,2′;4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxylic acid (XVII): l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l,4-dihydro-3-pyridinecarboxylic acid (XV) (lOOg; 0.3175 moles) was suspended in acetonitrile (800 ml) and heated to 80-82°C. A mixture of acetic acid (95.25 g), methanesulfonic acid (9.14 g; 0.09525 moles) and acetonitrile (200 ml) were added to the slurry at 80-82°C. The reaction mass was continued at 80-82°C to complete the reaction. After completion of the reaction, anhydrous sodium acetate (65 g) and (/?)-3-amino-l-butanol tartrate salt (79.68g; 0.3334 moles) were added at 20-25°C and stirred at 60-65°C to complete the reaction. The reaction mass was concentrated and acidified with IN aqueous hydrochloric acid (750 ml) and extracted with methylene chloride (1500 ml) at ice cold temperature. The organic layer was separated, concentrated, treated with hot methanol (350 ml) for 2 h, filtered, washed with methanol and dried to yield (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxylic acid (XVII) (72 g; HPLC purity: 99.07%).
Step-iii: Process for the preparation of Dolutegravir (I). Method A: Triethylamine (3.61 g; 0.0357 moles) was added to the suspension of (4R,12aS)-7- methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 – b][l,3]oxazine-9-carboxylic acid (XVII) (10 g; 0.0325 moles) in methylene chloride (50 ml), and cooled to 10-15°C. Pivaloyl chloride (4.3 g; 0.0357 moles) was added to the reaction mass, and stirred at 10-15°C for 1 h. Thereafter, 2,4-difiuorobenzylamine (5.58 g; 0.0389 moles) was added at 10-15°C and then warmed to 20-25°C to complete the reaction. After completion of the reaction, IN aqueous hydrochloric acid (20 ml) was added, organic layer was separated, washed with 5% w/w aqueous sodium bicarbonate solution (10 ml) followed by 15% w/w aqueous sodium chloride solution (10 ml) and concentrated. To the concentrated mass, acetonitrile (100 ml) and Lithium bromide (5.08 g; 0.0584 moles) were added and heated to 65-70°C for 3 h to complete the reaction. After completion of the reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml), concentrated to about 50 ml and DM water was added to crystallize the product at 20-25°C. The slurry was stirred for 2 h, filtered, washed with DM water and dried to yield (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxamide (I) (11.5 g, HPLC purity: 99.63%).
Method B: Isobutyl chloroformate (4.65 gm, 0.03404 moles) in methylene chloride (10 ml) was added to the solution of N-methylmorpholine (3.45 gm, 0.03410 moles) and (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino-[2,1 -b][l,3]oxazine-9-carboxy!ic acid (XVII) (10.0 gm, 0.03245 moles) in methylene chloride (60 ml) at -10 to 0°C in about 1 h. 2,4-Difloro benzyl amine (4.88 gm, 0.03409 moles) in methylene chloride (10 ml) was added to the cold reaction mass, and stirred at 20-30°C for completion of reaction. After completion of reaction, the reaction mass was washed with 5%w/w aqueous sodium bicarbonate solution (20 ml), IN hydrochloric acid (20 ml), DM water (20 ml) and concentrated. Acetonitrile (120 ml) and lithium bromide (4.8 gm, 0.05516 moles) were added to the concentrated mass, and stirred at 70-80°C for 3 h to complete the reaction. After completion of reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml) and concentrated to about 50 ml. DM Water (100 ml) was added to the concentrated reaction mass and stirred for 2 h at 25-30°C to crystallize the product. The product was filtered, washed with DM Water (50 ml) and dried to yield Dolutegravir (I) (10.7 gm, HPLC purity: 99.60%).
Example-2: Process for the preparation of Dolutegravir (I) (4R, 12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide (XVIII) (2 g, 0.0046 moles) was suspended in isopropyl alcohol (20 ml) and lithium bromide (0.8 g, 0.00924 moles) was added and stirred at 70-80°C for 15 h to complete the reaction. After completion of reaction the reaction mass was acidified with 5N aqueous hydrochloric acid (5 ml) and concentrated. DM Water (20 ml) was added to the concentrated mass and stirred at 25-30°C to crystallize the product. The product was filtered, washed with DM Water and dried to yield Dolutegravir (I) (1.5 g, HPLC purity: 97.93%).


/////
FDA warns Mylan about cGMP violations at its Indian facilities
DRUG REGULATORY AFFAIRS INTERNATIONAL

The US FDA has warned Mylan about manufacturing concerns at three of its plants in India.
In a warning letter to the generic drug manufacturer, the FDA said it had found ‘significant violations of current good manufacturing practice’ during inspections at the plants in August and September last year and in February this year.
The inspections relate to Mylan’s Agila Specialty Formulation Facility (SFF), Sterile Product Division (SPD), and Onco Therapies Limited (OTL) sites in Bangalore.

Some of the violations cited were failure to establish and follow appropriate written procedures designed to prevent microbiological contamination of drug products, such as the use of gloves with tears and pinholes, as well as deficiencies in environmental monitoring and poor monitoring of staff……..http://www.manufacturingchemist.com/news/article_page/FDA_warns_Mylan_about_cGMP_violations_at_its_Indian_facilities/111318/cn48579?dm_i=8EU,3MBVR,9ETTTY,D0ENC,1

Recently the Food and Drug Administration (FDA) began ramping up inspections of offshore manufacturing facilities and the results are shocking. Although cGMP violations have been found worldwide, experts are…
View original post 508 more words
Bempedoic Acid
Bempedoic Acid
ETC-1002, ESP-55016
CAS 738606-46-7
- C19H36O5
- MW 344.486 Da
8-Hydroxy-2,2,14,14-tetramethylpentadecanedioic acid
8-Hydroxy-2.2.14,14-tetramethylpentadecanedioic acid
ATP Citrate Lyase Inhibitor and AMP-activated Protein kinase (AMPK) activator
Indication: Hypercholesterolemia
Development Stage: Phase II
Developer: Esperion Therapeutics
Esperion Therapeutics was founded in April 2008 by former executives of, and investors in, the original Esperion Therapeutics which was founded in July 1998 and was bought by Pfizer for $ 1.3 billion in 2004 and then spun out in 2008. ETC-1002 was first discovered at the original Esperion, and Esperion subsequently acquired the rights to it from Pfizer in 2008. Esperion own the exclusive worldwide rights to ETC-1002.
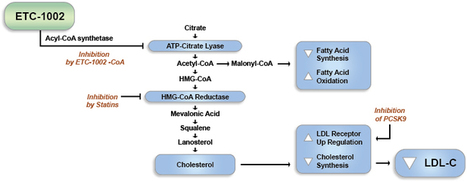

Bempedoic Acid ( ETC-1002) has a UNIQUE Dual mechanism of Action That has the Potential to Regulate Both lipid and Carbohydrate Metabolism. ETC-1002 Appears to Work by inhibitin ATP citrate lyase (ACL), a Key Enzyme in the Cholesterol biosynthetic pathway, and activating a Complementary Enzyme, 5′-adenosine monophosphate-activated Protein kinase (AMPK). Both Enzymes are Known to Play Significant roles in the synthesis of Cholesterol and glucose in the liver. By inhibitin Cholesterol synthesis in the liver, Causes ETC-1002 the liver to take up LDL particles from the blood, which reduces LDL-C levels.

WO 2004067489


6.13
7-Bromo-2,2-dimethylheptanoic acid ethyl ester
 7-Bromo-2,2-dimethylheptanoic acid ethyl ester
7-Bromo-2,2-dimethylheptanoic acid ethyl ester
Under argon atmosphere and cooling with an ice-bath, a solution of lithium diisopropylamide in THF (1.7 L, 2.0 M, 3.4 mol) was slowly dropped into a solution of 1 ,5- dibromopentane (950 g, 4.0 mol) and ethyl isobutyrate (396 g, 3.4 mol) in THF (5 L) while keeping the temperature below +5 DC. The reaction mixture was stiπed at room temperature for 20 h and quenched by slow addition of saturated ammonium chloride solution (3 L). The resulting solution was divided into three 4-L portions. Each portion was diluted with saturated ammonium chloride solution (5 L) and extracted with ethyl acetate (2 ‘ 2 L). Each 4-L portion of ethyl acetate was washed with saturated sodium chloride solution (2 L), 1 N hydrochloric acid (2 L), saturated sodium chloride solution (2 L), saturated sodium bicarbonate solution (2 L), and saturated sodium chloride solution (2 L). The three separate ethyl acetate layers were combined into a single 12-L portion, dried over magnesium sulfate, and concenfrated in vacuo to give the crude material (1.7 L) which was purified by vacuum distillation. Two fractions were obtained: the first boiling at 88 – 104 °C / 0.6 ton (184.2 g), the second at 105 – 120 °C / 1.4 ton (409.6 g) for atotal yield of 60 %. 1H NMR (300 MHz, CDC13/TMS): δ (ppm): 4.11 (q, 2 H, J = 7.2 Hz), 3.39 (t, 2 H, J = 6.8 Hz), 1.85 (m, 2 H), 1.56 – 1.35 (m, 4 H), 1.24 (t, 3 H, J = 7.2 Hz), 1.31 – 1.19 (m, 2 H), 1.16 (s, 6 H). 13C NMR (75 MHz, CDCI3/TMS): δ (ppm): 177.9, 60.2, 42.1, 40.5, 33.8, 32.6, 28.6, 25.2, 24.2, 14.3. HRMS (El, pos): Calcd. for CπH22Brθ2 (MH+): 265.0803, found: 265.0810.
6.18
2,2,14.14-Tetramethyl-8-oxo-pentadecanedioic acid diethyl ester
 p- toluenesulfonyl methyl isocyanide
p- toluenesulfonyl methyl isocyanide
 8-isocyano-2,2,14,14-teframethyl-8-(toluene-4-sulfonyl)-pentadecanedioic acid diethyl ester
8-isocyano-2,2,14,14-teframethyl-8-(toluene-4-sulfonyl)-pentadecanedioic acid diethyl ester
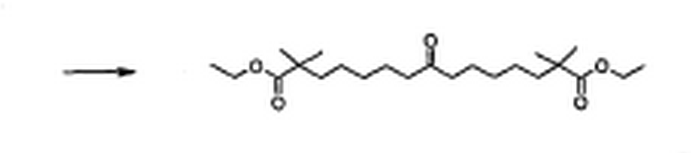 2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester
2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester
Under Ar atmosphere, to a solution of 7-bromo-2,2-dimethylheptanoic acid ethyl ester (26.50 g, 100 mmol), tetra-n-butylammonium iodide (3.69 g, 10 mmol) and p- toluenesulfonyl methyl isocyanide (9.80 g, 50 mmol) in anhydrous DMSO (300 mL) was added sodium hydride (4.80 g, 20.5 mmol, 60 % dispersion in mineral oil) at 5 – 10 oC. The reaction mixture was stiπed at room temperature for 20 h and quenched with ice-water (300 mL). The product was extracted with dichloromethane (3 D 100 mL). The combined organic layers were washed with water (200 mL), half-saturated NaCl solution (2 ‘ 200 ■– ■ ■• •• ■• .. <i„ ‘ir ι., – ib,
mL), and saturated NaCl solution (200 mL), dried over MgS04, and concentrated in vacuo to get the crude 8-isocyano-2,2,14,14-teframethyl-8-(toluene-4-sulfonyl)-pentadecanedioic acid diethyl ester (36.8 g) as an orange oil, which was used in the next step without purification. To a solution of this crude product (36.8 g) in dichloromethane (450 mL) was added concentrated hydrochloric acid (110 mL) and the mixture was stiπed at room temperature for 1 h. The solution was diluted with water (400 mL) and the aqueous layer was extracted with dichloromethane (200 mL). The combined organic layers were washed with saturated NaHC0 solution (2 x 150 mL) and saturated NaCl solution (150 mL). The organic solution was dried over Na2S04 and concenfrated in vacuo. The residue was subjected to column chromatography (silica gel, hexanes : ethyl acetate = 11 : 1) to give 2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester (12.20 g, 66 % over two steps) as a colorless oil. lH NMR (300 MHz, CDC13/TMS): δ (ppm): 4.11 (q, 4 H, J – 6.9 Hz), 2.37 (t, 4 H, J – 7.5 Hz), 1.58 – 1.47 (m, 8 H), 1.35 – 1.10 (m, 8 H), 1.24 (t, 6 H, J = 7.2 Hz), 1.15 (s, 12 H). 13C NMR (75 MHz, CDC13/TMS): δ (ppm): 211.6, 178.3, 60.5, 43.1, 42.5, 40.9, 30.1, 25.5, 25.1, 24.1, 14.7. HRMS (LSIMS, nba): Calcd. for C23IL3O5 (MH+): 399.3110, found: 399.3129.
6.19
8-Oxo-2,2,14,14-tetramethylpentadecanedioic acid
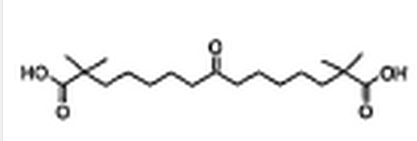
A solution of KOH (25 g) in water (50 mL) was added to a solution of 2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester (10.69 g, 155 mmol) in ethanol (400 mL), then heated at reflux for 4 h. After cooling, the solution was evaporated to a volume of ca. 50 mL and diluted with water (800 mL). The organic impurities were removed by extracting with dichloromethane (2 x 200 mL). The aqueous layer was acidified to pH 2 with concentrated hydrochloric acid (50 mL) and extracted with methyl tert.-butyl ether (MTBE, 3 x 200 mL). The combined organic layers were dried over magnesium sulfate and concenfrated in vacuo to give the crude product (9.51 g) as an oil. Crystallization from hexanes / MTBE (50 mL : 25 mL) afforded 8-oxo-2,2,14,14- teframethylpentadecanedioic acid (6.92 g, 79 %) as waxy, white crystals. M.p.: 83 – 84 °C. 1H NMR (300 MHz, CDCI3/TMS): δ (ppm): 12.03 (s, 2 H), 2.37 (t, 4 H, J = 7.3 Hz), 1.52 – 1.34 (m, 8 H), 1.28 – 1.10 (m, 8 H), 1.06 (s, 12 H). 13C NMR (75 MHz, CDCI3/TMS): δ (ppm): 210.5, 178.8, 41.7, 41.2, 29.1, 25.0, 24.4, 23.1. HRMS (LSIMS, gly): Calcd. for C19H3505 (MH+): 343.2484, found: 343.2485.
6.20
8-Hydroxy-2.2.14,14-tetramethylpentadecanedioic acid
Under nitrogen atmosphere, sodium borohydride (0.06 g, 1.6 mmol) was added to a stiπed solution of 8-oxo-2,2,14,14-tetramethylpentadecanedioic acid (1.18 g, 3.4 mmol) in methanol (50 mL) at 0 °C. The reaction progress was momtored by thin layer chromatography (silica; hexanes : ethyl acetate = 50 : 50). Additional sodium borohydride was added after 1 h (0.48 g, 13 mmol). After 8 h, the reaction mixture was hydrolyzed with water (50 mL) and acidified with concenfrated hydrochloric acid (3 mL) to pH 1. The solution was diluted with water (50 mL) and exfracted with dichloromethane (4 x 25 mL). The combined organic layers were washed with saturated sodium chloride solution (2 x 30 mL), dried over magnesium sulfate, concentrated in vacuo, and dried in high vacuo to give 8-hydroxy-2,2,14,14-tetramethylpentadecanedioic acid (0.7 g, 60 %) as a very viscous oil.
!H NMR (300 MHz, CDC13/TMS): δ (ppm): 7.42 (br. s, 3 H), 3.59 (br. s, 1 H), 1.65 – 1.00 (m, 20 H), 1.18 (s, 12 H). 13C NMR (75 MHz, CDC13/TMS): δ (ppm): 184.5, 71.8, 42.1, 40.5, 37.0, 29.8, 25.2, 25.1, 24.9, 24.8.
HRMS (FAB): Calcd. for Cι9H3705 (MH+): 345.2635, found: 345.2646. HPLC: 83.8 % purity.
………………..
PAPER
Journal of Medicinal Chemistry, 47 (24), 6082-6099;. 2004
http://pubs.acs.org/doi/abs/10.1021/jm040006p

Keto-substituted hydrocarbons with 11−19 methylene and bis-terminal hydroxyl and carboxyl groups have been synthesized and evaluated in both in vivo and in vitro assays for their potential to favorably alter lipid disorders including metabolic syndrome. Compounds were assessed for their effects on the de novo incorporation of radiolabeled acetate into lipids in primary cultures of rat hepatocytes as well as for their effects on lipid and glycemic variables in obese female Zucker fatty rats [Crl:(ZUC)-faBR] following 1 and 2 weeks of oral administration. The most active compounds were found to be symmetrical with four to five methylene groups separating the central ketone functionality and the gem dimethyl or methyl/aryl substituents. Furthermore, biological activity was found to be greatest in both in vivo and in vitro assays for the tetramethyl-substituted keto diacids and diols (e.g., 10c, 10g,14c), and the least active were shown to be the bis(arylmethyl) derivatives (e.g., 10e, 10f,14f). Compound 14c dose-dependently elevated HDL-cholesterol, reduced triglycerides, and reduced NEFA, with a minimum effective dose of 30 mg/kg/day. Compound 10g dose-dependently modified non-HDL-cholesterol, triglycerides, and nonesterified fatty acids, with a minimum effective dose of 10 mg/kg/day. At this dose, compound 10g elevated HDL-cholesterol levels 2−3 times higher than pretreatment levels, and a dose-dependent reduction of fasting insulin and glucose levels was observed.
ONLY KETO COMPD DESCRIBED
2,2,14,14-Tetramethyl-8-oxopentadecanedioic Acid (10g). According to the procedure given for 10f, 9g (8.54 g, 21.4 mmol) was saponified with KOH (85%, 4.53 g, 68.6 mmol) in EtOH (13 mL) and water (5 mL) at reflux for 4 h. The solid product obtained after usual workup was recrystallized from Et2O/hexanes (50 mL/50 mL), affording 10g (4.16 g, 57%) as colorless needles.
Mp: 82−83 °C.
1H NMR (CDCl3): δ 11.53 (br, 2H), 2.39 (t, 4H, J = 7.3), 1.60−1.50 (m, 8 H), 1.30−1.20 (m, 8 H), 1.18 (s, 12 H).
13C NMR (CDCl3): δ 211.7, 185.0, 42.8, 42.3, 40.4, 29.7, 25.1, 24.8, 23.8.
HRMS (LSIMS, gly): calcd for C19H35O5 (MH+) 343.2484, found 343.2444.
HPLC: Alltima C-8 column, 250 × 4.6 mm, 5 μm; 60% acetonitrile/40% 0.05 M KH2PO4, flow rate 1.0 mL/min; RI, tR 6.50 min, 92.6% pure.
Anal. (C19H34O5): C, H.
A PRECURSOR OF Bempedoic Acid
PATENTs/PAPERS
WO2005068412,
WO2004067489,
Journal of Medicinal Chemistry, 47 (24), 6082-6099;. 2004
US20040198814
Esperion Therapeutics


Ringing the bell were Roger Newton, Esperion’s founder and chief science officer, and Tim Mayleben, the CEO and president.
Esperion raised about $73 million in its offering on June 26. It hopes to use the funds to conduct two Phase 3 U.S. Food and Drug Administration trials next year on a cholesterol-lowering drug with the working name of ETC-1002.
ETC-1002 has shown good results in preliminary human trials in lowering LDL, the so-called bad cholesterol, in patients who are either intolerant or resistant to such statin drugs as Lipitor and Torvast. More results are expected this summer.
This was the second IPO for a drug company called Esperion. The first Esperion was founded in 1998 to create a drug to raise HDL, the so-called good cholesterol. It went public in 2000 and was sold to Pfizer Inc. for $1.3 billion in 2004.
In 2008, as part of closing its Michigan operations, Pfizer sold the name and rights to some small molecules back to Newton.
 Esperion Therapeutics founder and chief scientific officer Roger Newton, left, and CEO and President Tim Mayleben celebrate the company’s initial public …
Esperion Therapeutics founder and chief scientific officer Roger Newton, left, and CEO and President Tim Mayleben celebrate the company’s initial public …


 Esperion cofounder Roger Newton was one of the Key players in the Development of LDL-Cholesterol Lowering Pfizer’s statin atorvastatin (Lipitor), the Biggest Selling Drug of All time with Annual Sales of Almost $ 13 Billion Dollars in 2006 at ITS Peak.
Esperion cofounder Roger Newton was one of the Key players in the Development of LDL-Cholesterol Lowering Pfizer’s statin atorvastatin (Lipitor), the Biggest Selling Drug of All time with Annual Sales of Almost $ 13 Billion Dollars in 2006 at ITS Peak.

Esperion President and CEO Tim Mayleben (left) and Chief Science Officer Roger Newton in the company’s labs at the Michigan Life Science and Innovation Center.Mayleben previously told AnnArbor.com that the drug being developed by the company, which is housed at the Michigan Life Science and Innovation Center in Plymouth Township, is undergoing the second round of “phase two” clinical tests. Most drugs go through three phases of testing before the results are submitted to the Food and Drug Administration. Mayleben said he does not expect the company to submit ETC-1002 to the FDA for approval for at least another three years.,Newton, a co-inventor of Lipitor and founder of the first Esperion, raised more than $22 million to buy the intellectual property from the original company back from Pfizer when the company closed its Ann Arbor offices in 2007…….http://www.annarbor.com/business-review/ann-arbor-pharmaceutical-company-esperion-therapeutics-to-ring-nasdaq-opening-bell-wednesday/
……………………
Michigan Life Science and Innovation Center in Plymouth Township
 SRI International’s Helen Parish (from left), David Sahner and Elizabeth Wood in November 2013 at the site of the nonprofit’s new clinical laboratory at the Michigan Life Science and Innovation Center in Plymouth Township.
SRI International’s Helen Parish (from left), David Sahner and Elizabeth Wood in November 2013 at the site of the nonprofit’s new clinical laboratory at the Michigan Life Science and Innovation Center in Plymouth Township.
 Michigan Life Science and Innovation Center
Michigan Life Science and Innovation Center
 Esperion Therapeutics CEO Roger Newton in his laboratory at the Michigan Life Science Innovation Center in Plymouth Township.
Esperion Therapeutics CEO Roger Newton in his laboratory at the Michigan Life Science Innovation Center in Plymouth Township.
 Pfizer Inc. announced Jan. 22, 2007 that it would close its Ann Arbor research campus on Plymouth Road and Huron Parkway. In the photo at left, then-Ann Arbor SPARK CEO Michael Finney, then Gov. Jennifer Granholm and Ann Arbor Mayor John Hieftje speak at a press conference addressing Pfizer’s announcement.
Pfizer Inc. announced Jan. 22, 2007 that it would close its Ann Arbor research campus on Plymouth Road and Huron Parkway. In the photo at left, then-Ann Arbor SPARK CEO Michael Finney, then Gov. Jennifer Granholm and Ann Arbor Mayor John Hieftje speak at a press conference addressing Pfizer’s announcement.
///////Bempedoic Acid, PHASE 2, Esperion Therapeutics, Roger Newton, Tim Mayleben, ETC 1002, ESP 55016
