




| Patent | Submitted | Granted |
|---|---|---|
| NORMALIZATION OF CULTURE OF CORNEAL ENDOTHELIAL CELLS [US2015044178] | 2012-12-27 | 2015-02-12 |
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SEMAPIMOD
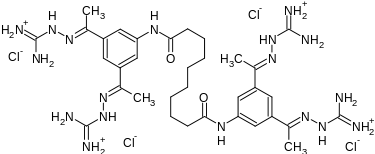

Semapimod Mesylate
CPSI-2364, AXD-455, CN-1493, CNI 1493
CAS No. 352513-83-8(Semapimod base)
Cas 164301-51-3 4x HCl
CAS 872830-80-3 (Semapimod mesylate)
MW 1129
CROHNS DISEASE, PHASE 1
N,N’-bis[3,5-bis[(E)-N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl]decanediamide
Decanediamide, N,N’-bis[3,5-bis[1-[(aminoiminomethyl)hydrazono]ethyl]phenyl]-, methanesulfonate
N,N’-Bis(3,5-bis(1-(carbamimidoylhydrazono)ethyl)phenyl)decanediamide
- N,N’-Bis(3-acetylphenyl)decane diamide tetrakis (amidinohydrazone)
- N,N’-bis(3,5-bis{(1E)-N-[amino(imino)methyl]ethanehydrazonoyl}phenyl)decanediamide
- N,N’-bis[3,5-bis[(E)-N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl]decanediamide
- N,N’-bis[3,5-bis[(E)-N-guanidino-C-methyl-carbonimidoyl]phenyl]decanediamide
A nitric oxide synthesis inhibitor and a p38 MAPK inhibitor potentially for the treatment of Crohn’s disease.

Crohn’s disease (CD) is a chronic inflammatory disease involving the upper and lower gastrointestinal tract and characterized by abdominal pain, weight loss, gastrointestinal bleeding and formation of fistulas between loops of bowel and from the bowel to the skin or other organs. Current therapy for active Crohn’s disease consists of symptomatic treatment, nutritional therapy, salicylates and immunosuppressants or surgical management.
Tumor necrosis factor a (TNF-a) plays a central role in the initiation and amplification of the granulomatous inflammatory reaction seen in CD (van Deventer, 1997). Increased TNF-a is present in gut mucosa as well as in stool of patients with active CD (Braegger et al, 1992). CNI-1493 is a synthetic guanylhydrazone compound that is an inhibitor of TNF-a synthesis. A monoclonal antibody to TNF, infliximab, is now approved for treatment of CD, but not all patients respond and many who do respond eventually become refractory to this treatment as well.
CNI-1493 is a synthetic compound which blocks the production of several inflammatory cytokines, including TNF. Because it blocks production of multiple inflammatory mediators, it may be more active than products targeted to a specific cytokine. In addition, as it is not a biologic, it should not cause hypersensitivity reactions or induce formation of antibodies.
The purpose of this trial is to determine if CNI-1493 is safe and effective in treating patients with moderate to severe Crohn’s Disease in a placebo controlled setting………https://clinicaltrials.gov/ct2/show/NCT00038766
Semapimod (INN), formerly known as CNI-1493, is an investigational new drug which has anti-inflammatory,[1] anti-cytokine,[2] immunomodulatory,[3] antiviral[4] and antimalarial[5] properties.
History
Semapimod was developed at the former Picower Institute for Medical Research, and is now licensed to Cytokine PharmaSciences. In 2000, Cytokine PharmaSciences licensed anti-infective applications of semapimod to Axxima Pharmaceuticals, but Axxima became insolvent in Dec. 2004 and its assets were acquired by GPC Biotech, which has recently merged into Agennix AG[1]. Although the disposition of Axxima’s partial rights to semapimod was not specified in these merger announcements, Cytokine PharmaSciences does not currently list any licensees for semapimod on its website.
Mechanism of action
Semapimod was first developed to inhibit nitric oxide synthesis by inflammatory macrophages, via inhibition of the uptake of arginine which macrophages require for nitric oxide synthesis.[1] Subsequently it was found that suppression of nitric oxide synthesis occurred even at semapimod concentrations 10-fold less than required for inhibition of arginine uptake, suggesting that this molecule was a more general inhibitor of inflammatory responses.[2] Further work revealed that semapimod suppressed the translation efficiency of tumor necrosis factor production.[6] Specifically, semapimod was found to be an inhibitor of p38 MAP kinase activation.[7] Surprisingly, however, the primary mode of action in vivo is now thought to be via stimulation of the vagus nerve, thereby down-regulating inflammatory pathways via the recently discovered cholinergic anti-inflammatory pathway.[8][9]
Pharmacology and clinical trials
In a preclinical study in rats, semapimod was found to suppress cytokine-storm induction by the anticancer cytokine interleukin-2 (IL-2) without decreasing its anticancer properties, allow larger doses of IL-2 to be administered.[10] A subsequent phase I trial in humans failed to show an increase in the tolerated dose of IL-2, although indications of pharmacological activity as an inhibitor of tumor necrosis factor production were observed.[11]
In a preliminary clinical trial of semapimod in patients with moderate to severe Crohn’s disease, positive clinical changes were observed, including endoscopic improvement, positive responses in some patients not responding to infliximab, healing of fistulae, and indications for tapering of steroids; no significant adverse effects were observed.[12]
In a small clinical trial against post-ERCP pancreatitis, significant suppression was not observed, although investigators observed a significant reduction of the incidence of hyperamylasemia and the levels of post-ERCP amylase.[13]
In the clinical trials above, semapimod tetrahydrochloride was administered by intravenous injection. This route has drawbacks such as dose-limiting phlebitis.[2] Recently Cytokine PharmaSciences has announced the development of novel salt forms of semapimod which are said to be orally absorbable; a phase I clinical trial of one of these salt forms, CPSI-2364, has been completed, and a phase II trial is planned for 2010.[3][4]
Chemistry
Semapimod is synthesized by reacting 3,5-diacetylaniline[14] with sebacoyl chloride in the presence of pyridine, followed by reaction of the resulting tetraketone with aminoguanidine hydrochloride.[1]
PATENT
-
N,N′-bis(3,5-diacetylphenyl) decanediamide tetrakis (amidinohydrazone) tetrahydrochloride (CNI-1493), which has the following structural formula:
-

SYNTHESIS
The reaction of decanedioyl dichloride (I) with 3,5-diacetylaniline (II) by means of pyridine in dichloromethane gives the corresponding diamide (III), which is condensed with aminoguanidine (IV) in refluxing aqueous ethanol to afford the target tetrakis amidinohydrazone. EP 0746312; EP 1160240; US 5599984; WO 9519767
http://www.google.com/patents/EP0746312A1?cl=en
References
- 1 Bianchi, M.; Ulrich, P.; Bloom, O.; Meistrell m, M. , I. I.; Zimmerman, G. A.; Schmidtmayerova, H.; Bukrinsky, M.; Donnelley, T.; Bucala, R.; Sherry, B.; Manogue, K. R.; Tortolani, A. J.; Cerami, A.; Tracey, K. J. (Mar 1995). “An inhibitor of macrophage arginine transport and nitric oxide production (CNI-1493) prevents acute inflammation and endotoxin lethality”. Molecular Medicine (Cambridge, Mass.) 1 (3): 254–266. ISSN 1076-1551. PMC 2229913. PMID 8529104.
- 2
- Bianchi, M.; Bloom, O.; Raabe, T.; Cohen, P. S.; Chesney, J.; Sherry, B.; Schmidtmayerova, H.; Calandra, T.; Zhang, X.; Bukrinsky, M.; Ulrich, P.; Cerami, A.; Tracey, K. J. (Mar 1996). “Suppression of proinflammatory cytokines in monocytes by a tetravalent guanylhydrazone”. The Journal of Experimental Medicine 183 (3): 927–936. doi:10.1084/jem.183.3.927. ISSN 0022-1007. PMC 2192362. PMID 8642296.
- 3
- Martiney, J.; Rajan, A. J.; Charles, P. C.; Cerami, A.; Ulrich, P. C.; MacPhail, S.; Tracey, K. J.; Brosnan, C. F. (Jun 1998). “Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage-deactivating agent” (Free full text). Journal of immunology (Baltimore, Md. : 1950) 160 (11): 5588–5595. ISSN 0022-1767. PMID 9605164.
- 4
- Hauber, I.; Bevec, D.; Heukeshoven, J.; Krätzer, F.; Horn, F.; Choidas, A.; Harrer, T.; Hauber, J. (Jan 2005). “Identification of cellular deoxyhypusine synthase as a novel target for antiretroviral therapy”. The Journal of Clinical Investigation (Free full text) 115 (1): 76–85. doi:10.1172/JCI21949. ISSN 0021-9738. PMC 539192. PMID 15630446.
- 5
- Specht, S.; Sarite, R.; Hauber, I.; Hauber, J.; Görbig, F.; Meier, C.; Bevec, D.; Hoerauf, A.; Kaiser, A. (May 2008). “The guanylhydrazone CNI-1493: an inhibitor with dual activity against malaria-inhibition of host cell pro-inflammatory cytokine release and parasitic deoxyhypusine synthase”. Parasitology research 102 (6): 1177–1184. doi:10.1007/s00436-008-0891-x. ISSN 0932-0113. PMID 18256853.
- 6
- Cohen, P. S.; Nakshatri, H.; Dennis, J.; Caragine, T.; Bianchi, M.; Cerami, A.; Tracey, K. J. (Apr 1996). “CNI-1493 inhibits monocyte/macrophage tumor necrosis factor by suppression of translation efficiency”. Proceedings of the National Academy of Sciences of the United States of America 93 (9): 3967–3971. Bibcode:1996PNAS…93.3967C. doi:10.1073/pnas.93.9.3967. ISSN 0027-8424. PMC 39469. PMID 8632999.
- 7
- Cohen, P. S.; Schmidtmayerova, H.; Dennis, J.; Dubrovsky, L.; Sherry, B.; Wang, H.; Bukrinsky, M.; Tracey, K. J. (May 1997). “The critical role of p38 MAP kinase in T cell HIV-1 replication”. Molecular Medicine (Cambridge, Mass.) 3 (5): 339–346. ISSN 1076-1551. PMC 2230081. PMID 9205949.
- 8
- Tracey, J. (Feb 2007). “Physiology and immunology of the cholinergic antiinflammatory pathway”. The Journal of Clinical Investigation (Free full text) 117 (2): 289–296. doi:10.1172/JCI30555. ISSN 0021-9738. PMC 1783813. PMID 17273548.
- 9
- Oke, L.; Tracey, J. (Mar 2008). “From CNI-1493 to the immunological homunculus: physiology of the inflammatory reflex” (Free full text). Journal of Leukocyte Biology 83 (3): 512–517. doi:10.1189/jlb.0607363. ISSN 0741-5400. PMID 18065685.
- 10
- Kemeny, M. M.; Botchkina, G. I.; Ochani, M.; Bianchi, M.; Urmacher, C.; Tracey, K. J. (1998). “The tetravalent guanylhydrazone CNI-1493 blocks the toxic effects of interleukin-2 without diminishing antitumor efficacy”. Proceedings of the National Academy of Sciences of the United States of America 95 (8): 4561–4566. Bibcode:1998PNAS…95.4561K. doi:10.1073/pnas.95.8.4561. PMC 22529. PMID 9539777.
- 11
- Atkins, M. B.; Redman, B.; Mier, J.; Gollob, J.; Weber, J.; Sosman, J.; MacPherson, B. L.; Plasse, T. (2001). “A phase I study of CNI-1493, an inhibitor of cytokine release, in combination with high-dose interleukin-2 in patients with renal cancer and melanoma”. Clinical Cancer Research 7 (3): 486–492. PMID 11297238.
- 12
- Hommes, D.; Van Den Blink, B.; Plasse, T.; Bartelsman, J.; Xu, C.; MacPherson, B.; Tytgat, G.; Peppelenbosch, M.; Van Deventer, S. (2002). “Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease”. Gastroenterology 122 (1): 7–14. doi:10.1053/gast.2002.30770. PMID 11781274.
- 13 Vanwesterloo, D.; Rauws, E.; Hommes, D.; De Vos, A.; Van Der Poll, T.; Powers, B.; Fockens, P.; Dijkgraaf, M.; Bruno, M. (2008). “Pre-ERCP infusion of semapimod, a mitogen-activated protein kinases inhibitor, lowers post-ERCP hyperamylasemia but not pancreatitis incidence”. Gastrointestinal Endoscopy 68 (2): 246–254. doi:10.1016/j.gie.2008.01.034. PMID 18455169.
- 14 Ulrich, P.; Cerami, A. (Jan 1984). “Trypanocidal 1,3-arylene diketone bis(guanylhydrazone)s. Structure-activity relationships among substituted and heterocyclic analogues”. Journal of Medical Chemistry 27 (1): 35–40. doi:10.1021/jm00367a007. ISSN 0022-2623. PMID 6690682.
| Patent | Submitted | Granted |
|---|---|---|
| Neural tourniquet [US2005282906] | 2005-12-22 | |
| Guanylhydrazone Salts, Compositions, Processes of Making, and Methods of Using [US2008262090] | 2008-10-23 | |
| Protective role of semapimod in necrotizing enterocolitis [US7795314] | 2007-12-06 | 2010-09-14 |
| METHOD OF TREATING ILEUS BY PHARMACOLOGICAL ACTIVATION OF CHOLINERGIC RECEPTORS [US2011112128] | 2011-05-12 | |
| Method of treating ileus by pharmacological activation of cholinergic receptors [US2007213350] | 2007-09-13 | |
| Pharmaceutically active aromatic guanylhydrazones [US2005171176] | 2005-08-04 | |
| Guanylhydrazone salts, compositions, processes of making and methods of using [US7244765] | 2006-01-19 | 2007-07-17 |
| GUANYLHYDRAZONE SALTS, COMPOSITIONS, PROCESSES OF MAKING, AND METHODS OF USING [US8034840] | 2008-06-19 | 2011-10-11 |
| METHOD FOR TREATING GLIOBLASTOMAS AND OTHER TUMORS [US2014323576] | 2014-03-14 | 2014-10-30 |
| Methods of treatment of fatty liver disease by pharmacological activation of cholinergic pathways [US8865641] | 2012-06-14 | 2014-10-21 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N,N’-bis[3,5-bis[N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl] decanediamide tetrahydrochloride
|
|
| Identifiers | |
| CAS Number | 164301-51-3 352513-83-8 (base) |
| ATC code | None |
| PubChem | CID: 5745214 |
| UNII | 9SGW2H1K8P |
| ChEMBL | CHEMBL2107779 |
| Chemical data | |
| Formula | C34H56Cl4N18O2 |
| Molecular mass | 890.73984 g/mol |
see………http://worlddrugtracker.blogspot.in/2015/12/semapimod.html
/////////Semapimod Mesylate, CPSI-2364, AXD-455, CN-149, PHASE 1, FERRING, CNI 1493
CC(=NN=C(N)N)C1=CC(=CC(=C1)NC(=O)CCCCCCCCC(=O)NC2=CC(=CC(=C2)C(=NN=C(N)N)C)C(=NN=C(N)N)C)C(=NN=C(N)N)C
Zucapsaicin for osteoarthritis

Zucapsaicin (珠卡赛辛)
cis-Capsaicin; (Z)-Capsaicin
Zucapsaicin; Civamide; Cis-Capsaicin; 25775-90-0; (Z)-Capsaicin; (Z)-N-(4-Hydroxy-3-methoxybenzyl)-8-methylnon-6-enamide;
(Z)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide
CAS No. 25775-90-0
| MF C18H27NO3 | |
| Molecular Weight: | 305.41188 g/mol |
|---|
WINSTON INNOVATOR
SANOFI
(Zuacta®/Civanex®
A medication used to treat osteoarthritis of the knee and other neuropathic pain.TRPV1 CHANNEL AGONIST



Zucapsaicin (Civanex) is a medication used to treat osteoarthritis of the knee and other neuropathic pain. It is applied three times daily for a maximum of three months. It reduces pain, and improves articular functions. It is the cis-isomer of capsaicin. Civamide, manufactured by Winston Pharmaceuticals, is produced in formulations for oral, nasal, and topical use (patch and cream).[1]
Zucapsaicin has been tested for treatment of a variety of conditions associated with ongoing nerve pain. This includes herpes simplex infections; cluster headaches and migraine; and knee osteoarthritis.[2]
Civanex (zucapsaicin) cream is a TRPV-1 modulator in development for the treatment of signs and symptoms of osteoarthritis of the knee.
Zucapsaicin, the cis-isomer of the natural product capsaicin, is a
topical analgesic that was initially developed by Winston Pharmaceuticals
and approved in Canada in July 2010 for the treatment of
severe pain in adults with osteoarthritis of the knee.
Bronson, J.; Dhar, M.; Ewing, W.; Lonberg, N. In Annual Reports in MedicinalChemistry; John, E. M., Ed.; Academic Press, 2011; Vol. 46, p 433.
The advantagesof zucapsaicin compared with naturally-occurring capsaicin, are reported to be a lesser degree of local irritation (stinging, burning,
erythema) in patients and a greater degree of efficacy in preclinical
animal models of pain.
Bernstein, J. E. U.S. 5063060, 1991.
Bernstein, J. E. U.S. 20050084520 A1, 2005.
The analgesic action of both
zucapsaicin and capsaicin is mediated through the transient receptor
potential vanilloid type 1 (TRPV1) channel, a ligand-gated ion
channel expressed in the spinal cord, brain, and localized on neurons
in sensory projections to the skin, muscles, joints, and
gut.
Westaway, S. M. J. Med. Chem. 2007, 50, 2589.
The scale preparation of zucapsaicin likely parallels the original
approach described by Gannett and co-workers involving the
coupling of vanillylamine with (Z)-8-methylnon-6-enoyl chloride.
Gannett, P. M.; Nagel, D. L.; Reilly, P. J.; Lawson, T.; Sharpe, J.; Toth, B. J. Org.Chem. 1988, 53, 1064.
Orito and co-workers elaborated this original approach in
an effort to prepare both capsaicin and zucapsaicin on gram-scale,
Kaga, H.; Miura, M.; Orito, K. J. Org. Chem. 1989, 54, 3477.
References
- 1 Winston Pharmaceuticals website http://www.winstonlabs.com/productdevelopment/civamide.asp
- 2 Zucapsaicin information from the National Library of Medicine http://druginfo.nlm.nih.gov/drugportal
Janusz, John M.; Buckwalter, Brian L.; Young, Patricia A.; LaHann, Thomas R.; Farmer, Ralph W.; et al. Journal of Medicinal Chemistry, 1993 , vol. 36, # 18 p. 2595 – 2604
Journal of Organic Chemistry, , vol. 53, # 5 p. 1064 – 1071
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(Z)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide
|
|
| Clinical data | |
| Trade names | Civanex |
| Routes of administration |
Topical |
| Identifiers | |
| CAS Number | 25775-90-0 |
| ATC code | M02AB02 |
| PubChem | CID: 1548942 |
| ChemSpider | 1265956 |
| UNII | 15OX67P384 |
| Synonyms | Civamide; (Z)-Capsaicin; cis-Capsaicin |
| Chemical data | |
| Formula | C18H27NO3 |
| Molecular mass | 305.41188 g/mol |
////Zucapsaicin
Oc1ccc(cc1OC)CNC(CCCC\C=C/C(C)C)=O
see………..http://apisynthesisint.blogspot.in/2015/12/zucapsaicin-for-osteoarthritis.html
Iptakalim Hydrochloride 盐酸埃他卡林

Iptakalim Hydrochloride 盐酸埃他卡林
NDA Filed china
A K(ir) 6.1/SUR2B activator potentially for the treatment of pulmonary arterial hypertension.

179.7, C9H21N.HCl
CAS No. 642407-44-1(Iptakalim)
642407-63-4(Iptakalim Hydrochloride)
N-(1-methylethyl)-2,3-dimethyl-2-butylamine
| Catholic Healthcare West (D/B/A/ St. Joseph’s Hospital And Medical Center) |


Hypertension is a multifactorial disorder, and effective blood pressure control is not achieved in most individuals. According to the most recent report of the American Heart Association, for 2010, the estimated direct and indirect financial burden for managing hypertension is estimated to be $76.6 billion. Overall, almost 75% of adults with cardiovascular diseases/comorbidities have hypertension, which is associated with a shorter overall life expectancy. Alarmingly, rates of prehypertension and hypertension are increasing among children and adolescents due, in part, to the obesity epidemic we currently face. There is also the problem of an aging population and the growing rates of diabetes and obesity in adults, all factors that are associated with high blood pressure.Thus, the need is great for novel drugs that target the various contributing causes of hypertension and the processes leading to end organ damage.
Iptakalim (IPT), chemically 2, 3–dimethyl-N-(1-methylethyl)-2-butanamine hydrochloride, is novel adenosine triphosphate–sensitive potassium (KATP) channel opener. KATP channels are composed of discrete pore-forming inward rectifier subunits (Kir6.1s) and regulatory sulphonylurea subunits (SUR).IPT shows high selectivity for cardiac KATP (SUR2A/Kir6.2) and vascular KATP (SUR2B/Kir6.1 or SUR6B/Kir6.2). Because of this high selectivity, IPT does not exhibit the adverse side effects associated with the older nonspecific K+ channel openers, which limit their use to the treatment of severe or refractory hypertension. IPT produces arteriolar and small artery vasodilatation, with no significant effect on capacitance vessels or large arteries. Vasodilatation is induced by causing cellular hyperpolarization via the opening of K+ channels, which in turn decreases the opening probability of L-type Ca2+ channels. Of particular note, IPT is very effective in lowering the blood pressure of hypertensive humans but not of those with normal blood pressure.
-
The present compd relates generally to a novel method for decreasing a human’s cravings for cigarettes and reducing instances of relapse during detoxification once smoking abstinence has been achieved, and more specifically, to a method for decreasing nicotine use by treating a human with a novel type of nicotinic acetylcholine receptor antagonist, iptakalim hydrochloride (IPT).
-
Cigarette smoking is a prevalent, modifiable risk factor for increased morbidity and mortality in the United States, and perhaps in the world. Smokers incur medical risks attributable to direct inhalation. Bystanders, termed passive smokers, also incur medical risks from second-hand smoke. Society, as a whole, also bears the economic costs associated with death and disease attributable to smoking. Although the majority of smokers have tried repeatedly to quit smoking, eighty percent of smokers return to tobacco in less than two years after quitting. Therefore, tobacco dependence is a health hazard for millions of Americans.
-
Nicotine is the biologically active substance that is thought to promote the use of tobacco products by approximately one-quarter of the world populations. Tobacco-related disease is personally and economically costly to the any nation. Unfortunately, once use of tobacco has begun, it is hard for a smoker to quit because of nicotinic dependence and addiction.
-
The initiation and maintenance of tobacco dependence in a human is due to certain bio-behavioral and neuromolecular mechanisms. Nicotinic acetylcholine receptors (nAChRs) in humans are the initial binding sites for nicotine. The binding of nicotine to nAChRs is thought to modulate the brain’s “reward” function by triggering dopamine release in the human brain. The nAChRs exist as a diverse family of molecules composed of different combinations of subunits derived from at least sixteen genes. nAChRs are prototypical members of the ligand-gated ion channel superfamily of neurotransmitter receptors. nAChRs represent both classical and contemporary models for the establishment of concepts pertaining to mechanisms of drug action, synaptic transmission, and structure and function of transmembrane signaling molecules.
-
Basic cellular mechanisms of nicotinic dependence also involve the functional state changes during repeated nicotinic agonists exposure and receptor changes in the number of receptors during chronic nicotinic exposure. nAChRs can exist in many different functional states, such as resting, activated, desensitized or inactivated The activation and/or desensitization of nAChRs plays an important role in initiating nicotinic tolerance and dependence. Recovery from receptor activation and/or desensitization contributes to nicotinic withdrawal symptoms.
-
The most abundant form of nAChRs in the brain contains α4 and β2 subunits. α4β2-nAChRs bind nicotine with high affinity and respond to levels of nicotine found in the plasma of smokers. α4β2-nAChR also have been implicated in nicotine self-administration, reward, and dependence. Therefore, selective drug action at nAChRs, especially at those containing α4 subunits, is thought to be an ideal way for nicotine cessation and reducing nicotine withdrawal syndrome. Unfortunately, thus far, no optimal compound can meet this purpose. The brain-blood-barrier permeable nAChR antagonist, mecamylamine is popularly used systemically but exhibits much less nAChR subtype selectivity.
-
Although a variety of psychopharmacological effects contribute to drug reinforcement, actions on the mesolimbic dopaminergic pathway is the predominant hypothesis for mechanisms of nicotinic reward. The mesolimbic dopaminergic pathway originates in the ventral tegmental area (VTA) of the midbrain and projects to forebrain structures including the prefrontal cortex and to limbic areas such as the olfactory tubercle, the amygdala, the septal region, and the nucleus accumbens. Many studies have indicated that dopamine release in the nucleus accumbens of the human brain is “rewarding” or signals an encounter with a “reward” from the environment. Other substances, such as alcohol, cocaine, and opiates, operate in the same manner, resulting in a cycle of substance or alcohol abuse.
-
Therefore, a considerable need exists for a novel compound that can selectively block α4 subtypes of nAChRs to prevent smoking-induced “reward”, to limit increasing nicotine-induced dopamine release, and/or to diminish nicotinic withdrawal symptoms.
Patent
https://www.google.com/patents/US20040266822
Example 1
-
Production of N-(1-methylethyl)-2,3-dimethyl-2-butylamine (Compound 1): Method 1. The solution of 7.6 g (0.0745 mole) 2,3-dimethyl-2-butanol in 3.24 mL glacial acetic acid was cooled and maintained at −5 to −8 degree of centigrade (° C.), then was added 7.3 g (0.49 mole) of powdered potassium cyanide in several times under stirring. 32.4 mL concentrated sulfuric acid was added dropwise while keeping the temperatue below 20° C., after which, the reaction mixture was stirred for 3.5 hours below 20° C. and another 6 hours at room temperature, then stood overnight. After poured into ice colded water, the mixture was adjusted to pH10 with 20% aqueous sodium hydroxide solution, and extracted with ether (×4). The extract was dried over anhydrous sodium sulfate. After filtration on the next day, the dessicator was removed, and the filtrate was evaporated off the ether, then distilled in vacuum to give 8.8 g (yield 91.6%) N-[2-(2,3-dimethylbutyl)]-fomide; bp 105-108° C./5 mmHg.
-
To the mixture of 7.7 g (0.0597 mole) N-[2-(2,3-dimethylbutyl)]-formide, 6.2 mL ethanol and 51.6 mL wate, 17.4 mL concentrated hydrochloric acid was added. The reaction mixture was refluxed for 4 hours in the oil bath, then distilled off ethanol in vacuum. The residue was adjusted to above pH12 with 40% aqueous sodium hydroxide solution, and extracted with ether. The extract was dried over anhydrous potassiun carbonate. After recovering the ether, The residue was distilled at atmosphere to give 3.75 g (yield 62.2%) 2,3-dimethyl-2-butylamine, bp 97-104° C.
-
The mixture of 10.6 g (0.15 mole) 2,3-dimethyl-2-butylamine, 6.45 g (0.0524 mole) 2-bromopropane, 3.0 mL glycol and 22.0 mL toluene was added into an autoclave, and heated with stirring for 17 hours at temperature of 170° C., after which, the organic layer was separated and extracted with 6N hydrochloric acid (15 mL×4). The extract was combined and washed once with toluene, then adjusted to pH 12-13 with 4% aqueous sodium hydroxide in the ice bath. The mixture was extracted with ether and then dried over anhydrous potassium carbonate. After recovering the ether, The filtrate was distilled to yield the fraction of bp 135-145° C. (yield 68.8%). The hydrochloride’s Mp is 228-230° C. (1-PrOH-Et2O). Elemental analysis for C9H22ClN(%): Calculated C, 60.14; H, 12.34; N, 7.79, Cl 19.73; Found C, 60.14; H, 12.48; N, 7.31, Cl 19.67.
-
1H-NMR(D2O, ppm) 0.98(d, J=6.75H, 6H), 1.33(s, 6H), 1.37(d, J=6.46, 6H), 2.10(m, 1H), 3.70(m, 1H). MS(m/z) 143 (M+), 100(B).
-
Method 2. To the mixture of 288 mL glacial acetic acid, 412 g (6.86 mole) urea and 288 g (3.43 mole) 2,3-dimethyl-2-butene, the solution of 412 mL concentrated sulfuric acid and 412 mL of glacial acetic acid was added dropwise under stirring, while maintaining the reaction temperature at the range of 45° C. to 50° C., then stirred for 5 hours at the temperature of 50-55° C. The mixture stood overnight. Next day, the mixture was reacted for another 7 hours at the temperature of 50-55° C., then poured into the solution of 1200 g (30 mole) sodium hydroxide in 8L glacial water. The resulting solid was filtered, washed with water (200 mL×5) and dried to give 404 g (yield 81.8%) N-(2,3-dimethyl-2-butyl)urea as white solid, mp 175-176° C. Elemental analysis for C7H16N2O(%): Calculated C 58.30, H 11.18, N 19.42; Found C, 58.70; H, 11.54; N, 19.25, 1H-NMR(CDCl3, ppm) 0.88-0.91(d, 6H, 2×CH3), 1.26(s, 6H, 2×CH3), 2.20-2.26(m, 1H, CH), 4,45(br, 2H), 4.65(br, 1H). MS(m/z) 145.0, 144.0(M+), 143.0, 129.1, 101.0, 86.1, 69.1, 58.0(B).
-
To the mixture of 196 g (1.36 mole) N-(2,3-dimethyl-2-butyl)urea and 392 mL glycol or tri-(ethanol)amine, a solution of 118 g (2.95 mole) sodium hydroxide in 118 mL water was added. The reaction mixture was heated for 8 hours in an oil bath at temperature of 120° C., then distilled at atmosphere to collect the fraction of bp 95-102° C. To the fraction, 75 g anhydrous potassium carbonate and. 40 g sodium hydroxide were added. The resulting mixture was distilled to give 88.5 g (yield 64.3%) 2,3-dimethyl-2-butylamine as colorless liquid, bp 99-101° C.
-
1H-NMR(CDCl3, ppm) 0.88-0.91(d, 6H, 2×CH3), 1.04 (s, 6H, 2×CH3), 1.53(m, 1H, CH).
-
To a 50.0 ml autoclave, 10.6 g (0.15 mole) 2,3-dimethyl-2-butylamine, 6.45 g (0.0524 mol) 2-bromopropane, 3.0 ml glycol and 22.0 ml toluene were added, and heated with stirring for 17 hours at 170° C., after which the organic layer was seperated and extracted with 6N hydrochloric acid (15 ml×4). The extract was combined and washed once with toluene, then adjusted to pH 12-13 with 4% aqueous sodium hydroxide in the ice bath. The mixture was extracted with ether and then dried over anhydrous potassium carbonate the ether was recovered, and distilled to give the fraction of bp 135-145° C. (yield 68.8%). mp of the hydrochloride is 228-230° C., (i-PrOH: Et2O). Elemental analysis for C9H22ClN(%): Calculated C, 60.14; H, 12.34; N, 7.79, Cl 19.73; Found C 60.14, H 12.48, N 7.31, Cl 19.67. 1H-NMR(D2O, ppm) 0.98(d, J=6.75H, 6H), 1.33(s, 6H), 1.37(d, J=6.46, 6H), 2.10(m, 1H), 3.70(m, 1H). MS(m/z) 143 (M+), 100(B).
-
Method 3. a solution of 0.10 mole enamine (prepared from the condensation of methyl iso-propyl ketone and iso-propylamine) in 20 mL hexane was filled with N2 and added dropwise to a solution containing 0.10 mole lithium methide with stirring in ice bath. After the reaction is complete, the mixture was poured into 500 g glacial water, and stirred. The aqueous layer was extracted with ether (×2). The resulting organic layer was concentrated. 3N hydrochloric acid was added to acified the organic layer to pH<1. The mixture was kept minutes and adjusted to pH>11 with 10% aqueous sodium hydroxide, then extracted with ether (×3). The extract was dried over anhydrous potassium carbonate and filtered. The filtrate was distilled at atmosphere to give a fraction of bp 140-145° C. with a yield of 80%.
REF
http://www.google.com/patents/US20060293393
//////Iptakalim Hydrochloride, 盐酸埃他卡林 , K(ir) 6.1/SUR2B activator, pulmonary arterial hypertension, nda
see……….http://apisynthesisint.blogspot.in/2015/12/iptakalim-hydrochloride.html
Tesmilifene , Antagonist of intracellular histamine

Tesmilifene
BMS-217380; BMY-33419; DPPE
CAS No. 98774-23-3(Tesmilifene), 92981-78-7(Tesmilifene hydrochloride)
Tesmilifene
CAS 98774-23-3
N,N-Diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine
DPPE
MFC19H25NO
MW 283.41
Percent Composition: C 80.52%, H 8.89%, N 4.94%, O 5.65%

Hydrochloride
CAS 92981-78-7
BMS-217380-01; BMY-33419
MF C19H25NO.HCl
MF 319.87
Percent Composition: C 71.34%, H 8.19%, N 4.38%, O 5.00%, Cl 11.08%
Properties: White crystals from isopropanol + acetone (3:1), mp 156-158°. pKa 10.9.
Melting point: mp 156-158°
pKa: pKa 10.9
Therap-Cat: Antineoplastic adjunct (chemosensitizer).
Tesmilifene is a novel potentiator of chemotherapy which, when added to doxorubicin, achieved an unexpected and very large survival advantage over doxorubicin alone in a randomized trial in advanced breast cancer.
PHASE 23 FOR An estrogen receptor antagonist potentially for the treatment of advanced breast cancer, gastric cancer
Tesmilifene is a novel agent that augments cytotoxicity of various chemotherapeutic agents both in vitro and in vivo. It binds selectively to the high-affinity microsomal antiestrogen binding site (Ki=50nm) but has no affinity for estrogen receptors. Inhibits concanavalin-A-induced histamine release in mast cells and acts as a novel antagonist of intracellular histamine.
US 4803227


The target product can be prepared by reacting para-benzylphenol (I) with 2-diethylaminoethylchloride hydrochloride (II) either by means of NaOH in H2O or with K2CO3 in DMF/acetone (at 60 C in both cases), followed by treatment with HCl to obtain the corresponding hydrochloride salt.
| EP 0153160; JP 1985190742; US 4803227 |
US 4803227
http://www.google.com/patents/US4803227
Tesmilifene is a small molecule chemopotentiator under development by YM BioSciences, a Candian pharmaceutical company that specialises in the development of cancer treatments. It is indicated for use in combination with standard cytotoxic drugs, such as taxanes and anthracyclines, which are widely used in the treatment of metastatic disease – when cancers spread to distant sites in the body.
Tesmilifene, the company’s lead investigational compound, is currently in phase III development for patients with metastatic breast cancer. At the end of January 2007, an independent safety monitoring board advised the company that its ongoing registration trial should be stopped; it was considered unlikely that significant differences in overall survival (primary endpoint) between treatment arms would emerge over time. The company had hoped that the addition of tesmilifene to standard epirubicin/cyclophosphamide therapy would confer a survival benefit similar to that seen in its earlier phase III trial.
In light of these disappointing results, YM BioSciences plans a detailed analysis of its phase III data in advanced breast cancer to see if it can identify why tesmilifene failed to add clinical benefit in this trial.
DRUG RESISTANCE LIMITS EFFECTIVENESS OF CHEMOTHERAPY
Cytotoxic drugs have proved potent weapons in the fight against malignant tumours and are considered first-line therapy for the treatment of many cancers. However, while patients often respond well to a first course of chemotherapy over time the response to drug treatment diminishes and the tumour may eventually become drug resistant. In some cases resistance can develop across several classes of anti-cancer drugs, leading to multidrug resistance. The development of drug resistance limits the effectiveness of many anti-cancer agents and is an important contributor to cancer deaths.
The development of agents that can overcome drug resistance is seen as one of the most important areas of cancer research and for which there is significant unmet need. Various approaches are being explored to boost the use of cytotoxic agents including chemopotentiators, chemoprotectants and liposomal formulations.
Clearly any agent that can prevent or reverse drug resistance would have a major impact on treatment strategies, enhancing the benefits of standard cytotoxic drugs.
TESMILIFENE MAY BOOST CYTOTOXIC EFFECTS OF ANTHRACYCLINES
Anthracyclines are a class of cytotoxic agents with proven efficacy in the treatment of breast cancer. They include agents such as doxorubicin and epirubicin among others. Because patients with metastatic breast cancer may have received anthracycline therapy for earlier stage breast cancer (adjuvant therapy) or following disease recurrence, there is a risk that they will fail to respond to continued treatment.
A phase III trial in 305 patients with advanced breast cancer has shown that when tesmilifene is combined with doxorubicin it appears to improve survival over treatment with doxorubicin alone. In this trial approximately half the patients were treated with both tesmilifene and doxorubicin, while the other half received doxorubicin alone. Although there were no significant differences in tumour response rates, progression-free survival, or average duration of response between treatment arms at endpoint, overall survival was significantly improved in the combination arm. Among patients treated with tesmilifene and doxorubicin overall survival was 23.6 months compared with 15.6 months for those treated with doxorubicin alone.
Researchers have suggested that tesmilifene may enhance the anti-tumour effects of anthracyclines in several ways:
- Reducing the cancer cell’s ability to become resistant
- Decreasing the metabolism or “break-down” of doxorubicin
- Disrupting the cancer cell’s energy source
TESMILIFENE REGISTRATION TRIAL
In March 2004 YM BioSciences began its pivotal international phase III trial of tesmilifene in metastatic breast cancer. By September 2005, 723 patients had been enrolled in the trial, which was designed once again to compare the efficacy and safety of tesmilifene and an antrhacycline (epirubicin) with epirubicin alone.
“At the end of January 2007, an independent safety monitoring board advised the company that its ongoing registration trial should be stopped.”
Given the survival benefit seen in the earlier trial, which was carried out by the Canadian National Cancer Institute, the company was optimistic about outcome in its pivotal registration trial. However, an interim analysis of 351 events suggested that significant differences in overall survival were unlikely to be seen between the two treatment arms as the data matured and the trial was brought to a premature end.
In addition to its work on anthracyclines, YM BioSciences has also been exploring the potential of tesmilifene to enhance the efficacy of taxanes, also standard chemotherapy for metastatic breast cancer. Other potential applications include:
- Adjuvant therapy for breast cancer, i.e. immediately post-surgery and before the cancer has recurred or metastasised
- Hormone-refractory prostate cancer
- Lung cancer
- Non-Hodgkin’s lymphoma
MARKETING COMMENTARY
Although there have been major advances in the treatment of breast cancer in the last 10 to 15 years, it remains a disease for which improved treatments are still urgently needed. Estimates from the WHO suggest that metastatic breast cancer will claim the lives of over 40,000 patients a year.
Current treatments for metastatic breast cancer are rarely curative but can nonetheless do much to improve patients’ quality of life or duration of survival. . By boosting the cytotoxic effects of standard chemotherapy agents such as anthracyclines, while protecting healthy cells, tesmilifene was thought to have potential to extend the benefits of cytotoxic therapy to more patients. This is now in doubt following premature ending of its pivotal registration trial in advanced breast cancer.
Literature References: Intracellular histamine antagonist with chemopotentiating and cytoprotective activity. Structurally similar to tamoxifen, q.v., although binds anti-estrogen binding site (AEBS) with no affinity for the estrogen receptor.
Prepn: L. J. Brandes, M. W. Hermonat, Biochem. Biophys. Res. Commun. 123, 724 (1984); and use as antineoplastic: eidem, US 4803227 (1989 to Univ. Manitoba); and study of binding affinity: M. Poirot et al., Bioorg. Med. Chem. 8, 2007 (2000). Spectral analysis of interaction with P450 isozymes: L. J. Brandes et al., Cancer Chemother. Pharmacol. 45, 298 (2000).
Clinical evaluation in combination with cyclophosphamide in prostate cancer: L. J. Brandes et al., J. Clin. Oncol. 13, 1398 (1995); in combination with doxorubicin in breast cancer: L. Reyno et al., J. Clin. Oncol. 22, 269 (2004).
Bioorg Med Chem 2000,8(8),2007
Product Literature References
Enhancement of cytotoxicity of natural product drugs against multidrug resistant variant cell lines of human head and neck squamous cell carcinoma and breast carcinoma by tesmilifene.: P. J. Ferguson, et al.; Cancer Lett. 274, 279 (2009), Abstract;
Phase III study of N,N-diethyl-2-[4-(phenylmethyl) phenoxy]ethanamine (BMS-217380-01) combined with doxorubicin versus doxorubicin alone in metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group St: L. Reyno, et al.; J. Clin. Oncol. 22, 269 (2004), Abstract;
Synergy between tamoxifen and cisplatin in human melanoma cells is dependent on the presence of antiestrogen-binding sites.: J.A. Jones, et al.; Cancer Res. 57, 2657 (1997), Abstract;
Influence of DPPE on histamine release from isolated rat mast cells.: N. Grosman; Agents Actions 41, 1 (1994), Abstract;
Histamine is an intracellular messenger mediating platelet aggregation.: S.P: Saxena, et al.; Science 243, 1596 (1989), Abstract;
///////Tesmilifene, Antineoplastic Adjunct, Chemosensitizer, PHASE 3, Tesmilifene hydrochloride, BMY-33419, BMS-217380, DPPE, N,N-DPPE, Antagonist of intracellular histamine
CCN(CC)CCOC1=CC=C(C=C1)CC2=CC=CC=C2
see……….http://apisynthesisint.blogspot.in/2015/12/tesmilifene-antagonist-of-intracellular.html
RQ 00000010 for the treatment of GERD, functional dyspepsia and chronic constipation.


RQ 00000010
CAS 907607-22-1
| Molecular Formula: | C22H27F3N2O6 |
|---|---|
| Molecular Weight: | 472.45479 g/mol |
HSMMHNBGQLGCBY-UHFFFAOYSA-N;
RaQualia Pharma Inc
PFIZER INNOVATOR
RQ-00000010; RQ-10
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
4-[[4-[[4-(2,2,2-trifluoroethoxy)-1,2-benzoxazol-3-yl]oxymethyl]piperidin-1-yl]methyl]oxane-4-carboxylic acid
PHASE 1 for the treatment of GERD, functional dyspepsia and chronic constipation.
Useful for treating diseases mediated by 5-HT4 receptor activity eg such as gastroesophageal reflux disease (GERD), gastric motility disorder, dyspepsia, constipation, esophagitis, diabetes, CNS and cardiovascular diseases.
RaQualia, following its spin-out from Pfizer, is developing RQ-00000010, a 5-HT4 receptor partial agonist, for the treatment of gastric motility disorders, including gastroparesis associated with Parkinson’s disease.
In November 2015, the drug was reported to be in phase 1 clinical development. RaQualia and licensee CJ CheilJedang are investigating the drug for the treatment of GERD, functional dyspepsia and chronic constipation.
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid is disclosed in PL1 as a 5-HT4 receptor agonist, which is useful in the treatment or alleviation of disease conditions mediated by 5-HT4 receptor activity; in particular 5-HT4 receptor agonistic activity, such as gastroesophageal reflux disease (GERD), gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia (FD), irritable bowel syndrome (IBS), constipation, dyspepsia, esophagitis, gastroesophageal disease, gastritis, nausea, central nervous system disease, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disorders, cardiac failure, heart arrhythmia, diabetes, and apnea syndrome (See NPL 1 to 13 and PL 2 to 7).
Simply an white solid has been produced in the previously known methods of preparing 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid, described in PL 1. A generic disclosure of pharmaceutically-acceptable salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid of the instant application is disclosed, and the free base of the compound of the instant invention is disclosed and claimed, in PL 1 having an international filing date of December 6, 2006, assigned to the assignee hereof. Thus any salts of the compound have been neither pacifically described nor synthesized in prior art.
It has been found that HCl-salt, HBr-salt, pTSA-salt and EDSA-salt of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid shown below, can be isolated as a crystalline form which has advantageous properties such as ease of making a formulation, high solubility, and good stability. In addition the salts of the present invention are more easily purified than a non-crystalline form disclosed in PL 1 (WO2006/090224) and crystalline form disclosed in PL 3 (WO2012/157288).
Patent Literature
{PL 1} WO2006/090224.
{PL 2} US Patent No. 6,106,864.
{PL 3} WO2012/157288
{PL 4} WO00/35298.
{PL 5} WO91/11172.
{PL 6} WO94/02518.
{PL 7} WO98/55148.
Non Patent Literature
{NPL 1} Bockaert J. et al., TiPs 13; 141-145, 1992.
{NPL 2} Ford A. P et al., Med. Res. Rev. 13: 633-662, 1993.
{NPL 3} Gullikson G. W. et al., Drug Dev. Res. 26; 405-417, 1992.
{NPL 4} Richard M. Eglen et al., TiPs 16; 391-398, 1995.
{NPL 5} Bockaert J. et al., CNS Drugs 1; 6-15, 1994.
{NPL 6} Romanelli M. N. et al., Arzheim Forsch./Drug Res., 43; 913-918, 1993.
{NPL 7} Kaumann A. J. et al., Naunyn-Schmiedebergs Arch Pharmacol., 344; 150-159, 1991.
{NPL 8} Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
{NPL 9} Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
{NPL 10} Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
{NPL 11} Pharmaceutical Technology On-line, 25(2), 1-14, by Verma et al. (2001).
{NPL 12} J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
{NPL 13} Evrard, B., et al., Journal of Controlled Release 96 (3), pp. 403-410, 2004.
{NPL 14} Byrn S. R. et al., Solid-State Chemistry of Drugs 2nd ed., pp 3-43 and 461-503, 1999, SSCI, Inc.

PATENT
WO2006090224
| PFIZER JAPAN INC. |
EXAMPLE 1 :
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
Step 1. Methyl 2-hvdroxy-6-(2,2,2-trifluoroethoxy)benzoate
A mixture of 5-hydroxy-2,2-dimethyl-4tø-1 ,3-benzodioxin-4-one (123 g, 633 mmol, Synth. Commun.
1994, 24t 1025), potassium carbonate (262 g, 1.9 mol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (95.8 mL, 665 mmol) in Λ/,Λ/-dimethylformamide (600 mL) was stirred at 50 0C for 30 min. Then methanol (300 ml_) was added to the mixture, and stirring was continued for 5 h at that temperature. After cooling to room temperature, the mixture was diluted with water (500 ml_) and neutralized with 2Λ/ hydrochloric acid. Product was extracted with a mixture of ethyl acetate-hexane (5:1 , 500 mL x 3). Combined organic layers were washed with water (500 mL), dried over magnesium sulfate and concentrated under reduced pressure. The residual solid was recrystallized from methanol-water to afford 125 g (79%) of the desired product as colorless crystals.
1H-NMR (CDCI3) δ: 11.47 (1 H, s), 7.36 (1 H, t, J = 8.4 Hz), 6.72 (1 H, dd, J = 1.1 , 8.4 Hz), 6.38 (1 H, q, J = 8.1 Hz), 4.36 (2 H, q, J= 8.0 Hz), 3.96 (3 H, s).
MS (ESI) m/z: 251 (M+H) +, 249 (M-H) \
Step 2. 4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-ol
To a solution of hydroxylamine sulfate (120 g, 732 mmol) in water (360 mL) was added potassium carbonate (121 g, 875 mmol) at 0 0C. After 30 min of stirring, sodium sulfite (3.74 g, 29.7 mmol) and a methanolic solution of methyl 2-hydroxyl-6-(2,2,2-trifluoroethoxy)benzoate (36.4 g, 146 mmol, EXAMPLE 1 , step 1 , in 360 mL of methanol) were added to the mixture. Then the mixture was warmed to 50 °C and stirred for 30 h. After cooling to room temperature, reaction mixture was partially concentrated to approx. 2/3 volume and acidified with 2Λ/ hydrochloric acid. Product was extracted three times with ethyl acetate. Combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford the desired product as a crystalline solid. Crude product (36.3 g) was used for the next step without further purification.
The described above crude product (5.56 g, 22.14 mmol) was suspended in tetrahydrofuran (22.0 mL) and heated at 50 °C. 1 ,1 ‘-carbonyldiimidazole (7.54 g, 46.48 mmol) was added to the suspension at 50 °C. After addition, the mixture was stirred at 50 0C for 14 h, the mixture was cooled to room temperature. 2Λ/ hydrochloric acid was added to the mixture and extracted with ethyl acetate. The organic layer was extracted with 10% aq. potassium carbonate (100 mL x 5). The water layers were acidified with 2Λ/ hydrochloric acid and extracted with ethyl acetate (200 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give brown solid. The residual solid was recrystallized from ethyl acetate/hexane to give 3.21 g (61 %) of the title compound as colorless needles.
1H-NMR (CDCl3) δ: 7.53 (1 H1 1, J = 8.5 Hz), 7.14 (1 H, d, J= 8.5 Hz), 6.73 (1 H, d, J = 7.9 Hz), 4.63 (2 H, q, J= 8.0 Hz), 3.83 (1 H, br).
MS (ESI) m/z: 234 (M+H) +, 232 (M-H) “.
Step 3. rMethoxy(tetrahydro-4H-pyran-4-ylidene)methoxyKtrimethyl)silane
To a stirred solution of diisopropylamine (5.2 mL, 37 mmol) in tetrahydrofuran (15 mL) was added dropwise n-butyllithium (1.6 M in hexane, 21 mL, 34 mmol) at 0 0C and stirred for 20 min. A mixture of methyl tetrahydro-2W-pyran-4-carboxylate (4.5 g, 31 mmol) and trimethylsilyl chloride (4.3 mL, 34 mmol) was added to the mixture at -40 0C, then trimethylsilyl chloride (0.4 mL, 0.3 mmol) was added to the mixture. The mixture was stirred at room temperature for 2 h. The volatile components were removed by evaporation and the residual mixture was filtered through a pad of celite washing with hexane. The filtrate was evaporated to give 6.9 g (quant.) of the title compound as a clear yellow oil.
1H-NMR (CDCI3) δ: 3.64-3.59 (4 H, m), 3.52 (3 H, s), 2.24 (2 H, t, J = 5.6 Hz), 2.15 (2 H, t, J = 5.4 Hz), 0.22 (9 H, s).
Step 4. Methyl 4-{f4-(hvdroxymeth’vDpiperidin-1 -yllmethylltetrahvdro^rt-pyran^-carboxylate
To a stirred mixture of piperidin-4-ylmethanol (5.0 g, 43.4 mmol), f-butyldimethylsilylchloride (7.2 g, 47.8 mmol), and triethylamine (7.3 ml_, 52.1 mmol) in dichloromethane (50 mL) was added 4-dimethylaminopyridine (530 mg, 4.3 mmol) at 0 0C. After being stirred at 0 0C for 2 h, 50 mL of water was added to the mixture. The mixture was extracted with dichloromethane (50 mL x 3) and the extracts were combined, dried over sodium sulfate, and concentrated in vacuo to give 10.2 g of a crude oil. The residual oil was dissolved with 86 mL of ethanol, and potassium carbonate (7.2 g, 52.1 mmol) and paraformaldehyde (1.56 g, 52.1 mmol) were added to the solution. After being stirred at room temperature for 2 days, the mixture was filtered and the filtrate was concentrated in vacuo to give a yellow oil. The residual oil was dissolved with 45 mL of acetonitrile and magnesium chloride (414 mg, 4.3 mmol) was added to the solution. [methoxy(tetrahydro-4H-pyran-4-ylidene)methoxy](trimethyl)silane (11.3 g, 52.1 mmol, EXAMPLE 1 , step 3) was added to the mixture at 0 0C. After being stirred at 0 0C for 20 h, 100 mL of 2Λ/ hydrochloric acid was added to the mixture. The mixture was stirred for 30 min and washed with diethyl ether (100 mL x 2). The water layer was neutralized with aq. ammonia and extracted with ethyl acetate (100 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give a yellow oil. The residual oil was purified by silica gel column chromatography (dichloromethane/methanol/aq. ammonia 400: 10: 1 ) to give 6.8 g (41%) of the title compound as a colorless waxy solid.
1H-NMR (CDCI3) δ: 3.75-3.90 (2 H, m), 3.71 (3 H, s), 3.40-3.55 (4 H, m), 2.73 (2 H, m), 2.49 (2 H, m), 2.10-2.25 (2 H, m), 1.95-2.10 (2 H, m), 1.50-1.70 (4 H, m), 1.30-1.50 (2 H, m), 1.10-1.30 (2 H, m).
MS (ESI) m/z: 272 (M+H) +.
Step 5. Methyl 4-{r4-((r4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -yllmethyll-tetrahydro-2H-pyran-4-carboxylate
A mixture of 4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-ol (230 mg, 1 mmol, EXAMPLE 1 , step
2), methyl 4-{[4-(hydroxymethyl)piperidin-1 -yl]methyl}tetrahydro-2/-/-pyran-4-carboxylate (270 mg, 1 mmol, EXAMPLE 1 , step 4), and cyanomethyltributylphosphorane (400 mg, 1.5 mmol) in toluene (1.0 mL) was stirred at 100 0C for 16 h. After cooling, the mixture was concentrated in vacuo to give a dark brown oil. The residual oil was purified by silica gel column chromatography (hexane/ethyl acetate 2 : 1 ) to give 250 mg (51 %) of the title compound as a white solid.
1H-NMR (CDCl3) δ: 7.44 (1 H, dd, J= 7.9, 8.4 Hz), 7.12 (1 H, d, J= 8.4 Hz), 6.61 (1 H, d, J= 7.9 Hz), 4.49 (2 H, q, J= 8.1 Hz), 4.24 (2 H, d, J= 6.4 Hz), 3.88-3.78 (2 H, m), 3.72 (3 H, s), 3.54-3.41 (2 H, m), 2.83-2.71 (2 H, m), 2.52 (2 H, s), 2.35-1.29 (11 H, m).
MS (ESI) m/z: 487 (M+H) +.
Step 6. 4-(r4-(ir4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -ylimethylltetrahydro-2H-pyran-4-carboxylic acid
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2/+pyran-4-carboxylate (89 mg, 0.18 mmol, EXAMPLE 1 , Step 5) in tetrahydrofuran (1 mL), methanol (1 ml_) and 2 Λ/ aq. sodium hydroxide (1 ml_) was stirred at 70 °C for 17 h. The mixture was neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate was filtered.
The precipitate was triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) δ: 7.59 (1 H1 dd, J= 8.1 , 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J= 8.7 Hz), 4.19 (2 H, d, J= 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H was not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) “.
m.p.: 171.7 °C.
IR (KBr) v: 2950, 1617, 1527, 1188, 1113 cm”1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
PATENT
WO2015174098
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015174098
PATENT
WO2014080633
http://www.google.com/patents/WO2014080633A1?cl=en
PATENT
WO 2015178020
The present invention relates to novel salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid. More particularly, the invention relates to salt forms (HCl-salt, HBr-salt, p-toluenesulfonate salt and ethanedisulfonate salt), and to processes for the preparation of, compositions containing and to uses of, such salt forms.
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A slurry of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-methyl}tetrahydro-2H-pyran-4-carboxylic acid (1.326 kg, 2.807 mol, a white solid) in ethyl acetate (18.564 L) is dissolved at 70 oC. The solution is cooled to 64 oC during 35 min and 200 mg of seed crystal (0.423 mmol) is seeded to the mixture. The mixture is cooled to 40 oC over 5 h period and stirred at this temperature for 14.5 h. The slurry is gradually cooled to 19 oC during 6 h period and the mixture is stirred at this temperature for 46 h. The formed precipitate is collected by filtration and the filter cake is washed with 2.0 L of ethyl acetate. The filter cake is dried under reduced pressure at 50 oC to afford 1.140 kg of the desired crystalline form of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-
methyl}tetrahydro-2H-pyran-4-carboxylic acid (86%).
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
m.p. (DSC onset): 169 oC.
The temperature has a margin of error of +/- 1 oC.
Crystallinity by PXRD: Crystal (Figure 1): Main peaks at 2-Theta: 5.9, 9.3, 9.8, 11.9, 13.7, 14.3, 15.0, 17.8, 18.2-19.3, 19.7, 22.6, 23.4-24.5 and 24.9 (o ). Each peak has a margin of error of +/- 0.2.
IR nu (diffuse reflection) (Figure 6): 4389-4383, 3426, 2943-2937, 2120, 1904, 1724, 1614, 1535, 1508, 1437, 1420, 1287, 1261, 1221, 1180, 1121, 1094, 1059, 1022, 991, 974, 957, 934, 918, 868, 827, 783, 746, 731, 654, 638, 615, 588, 554, 542 and 507 cm-1. Each peak has a margin of error of +/- 2 cm-1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.76; H, 5.74; N, 5.85.
PATENT
WO2012/157288
http://www.google.co.in/patents/WO2012157288A1?cl=pt-PT
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-Trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylate (89 mg, 0.18 mmol, PCT WO2006090224 EXAMPLE 1, Step 5) in tetrahydrofuran (1 mL), methanol (1 mL) and 2 N aq. sodium hydroxide (1 mL) is stirred at 70 oC for 17 h. The mixture is neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate is filtered. The precipitate is triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) –.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
| Patent | Submitted | Granted |
|---|---|---|
| Benzisoxazole Derivatives [US2008207690] | 2008-08-28 | |
| 5-HT4 Receptor Agonist as a Prokinetic Agent [US2014051726] | 2012-03-23 | 2014-02-20 |
| Polymorph Form of 4-methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid [US2014187583] | 2012-05-18 | 2014-07-03 |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
WO-2015178020-A1 |
2015-11-26 |
EN
|
|
||
|
2.
WO-2015174098-A1 |
2015-11-19 |
EN
|
|
||
|
3.
US-9187463-B2 |
2015-11-17 |
|
|||
|
4.
US-20150322055-A1 |
2015-11-12 |
|
|||
|
5.
EP-2922849-A1 |
2015-09-30 |
EN
|
|
||
|
6.
EP-2710002-A4 |
2014-10-01 |
EN
|
|
||
|
7.
US-8816090-B2 |
2014-08-26 |
|
|||
|
8.
EP-1856114-B1 |
2014-08-20 |
EN
|
|
||
|
9.
US-20140187583-A1 |
2014-07-03 |
|
|||
|
10.
WO-2014080633-A1 |
2014-05-30 |
EN
|
|
||
|
11.
EP-2710002-A1 |
2014-03-26 |
EN
|
|
||
|
12.
US-20140051726-A1 |
2014-02-20 |
|
|||
|
13.
EP-2688648-A1 |
2014-01-29 |
EN
|
|
||
|
14.
WO-2012157288-A1 |
2012-11-22 |
EN
|
|
||
|
15.
WO-2012127878-A1 |
2012-09-27 |
EN
|
|
||
|
16.
US-20080207690-A1 |
2008-08-28 |
|
|||
|
17.
EP-1856114-A1 |
2007-11-21 |
EN
|
|
||
|
18.
WO-2006090224-A1 |
2006-08-31 |
EN
|
|
see……….http://apisynthesisint.blogspot.in/2015/12/rq-00000010-for-treatment-of-gerd.html
/////c12c(cccc1onc2OCC3CCN(CC3)CC4(CCOCC4)C(=O)O)OCC(F)(F)F
C1CN(CCC1COC2=NOC3=C2C(=CC=C3)OCC(F)(F)F)CC4(CCOCC4)C(=O)O
Lefucoxib (乐福昔布)

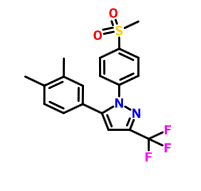
Lefucoxib (乐福昔布)
5-(3,4-dimethyl-phenyl)-1-methanesulfonyl-3-trifluoromethol-pyrazole
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole
CAS 849048-84-6
![]()
| Molecular Formula: | C19H17F3N2O2S |
|---|---|
| Molecular Weight: | 394.41069 g/mol |
IND FILED
Prostaglandin G/H Synthase 2 (PTGS2; COX-2) Inhibitors
A COX-2 inhibitor potentially for the treatment of rheumatoid arthritis.
cyclooxygenase-2 (COX-2) inhibitor
National Center of Biomedical Analysis
![]()
 CHINA FLAG
CHINA FLAG

PATENT
CN 1468854
http://www.google.com/patents/CN1468854A?cl=en
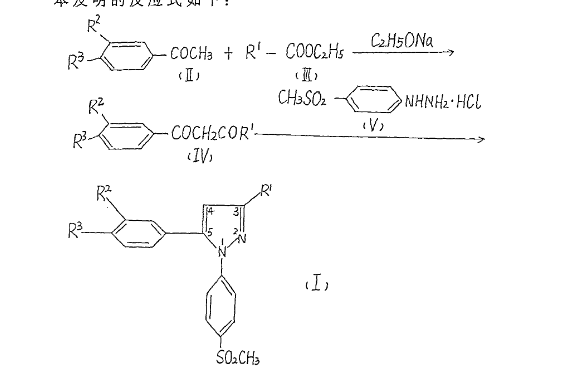
Example 1
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1)
1- (3,4- two toluene-yl) -4,4,4-trifluoro-methyl – D-1,3-dione (IV1) of sodium metal was weighed 2.3g (0.1mol) was added 50ml of anhydrous toluene to prepare a sodium sand. After cooling, ethanol was added dropwise 12ml, and then heated at 60 ℃, complete reaction of sodium metal. After cooling to room temperature, was added 3,4-dimethylphenyl ethanone 23.8g (0.1mol) and trifluoroacetic ethyl acetate 20ml (0.2mol), reacted at 100 ℃ 5 hours. Toluene was distilled off under reduced pressure, a 10% aqueous hydrochloric acid was added, the pH was adjusted to 2-3, extracted with ethyl acetate, washed with water, dried over anhydrous MgSO4, ethyl acetate was distilled off under reduced pressure. Then under reduced pressure, distillation, collecting fractions 105-107 ℃ / 0.7mmHg, was 14.6g, 60% yield.
1- [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1) take the above-prepared substituted (IV1) 2.38g (0.01mol ), 15ml of ethanol, then added p-methanesulfonyl phenyl hydrazine salt alkoxide 2.3g (0.01ml). Was refluxed for 15 hours. Place the refrigerator overnight, the crystals were collected by filtration, recrystallized from ethanol, mp 129-31 ℃, to give 3.1 g.
Elemental analysis: C19H17F3N2O2S Calculated: C, 57.86; H, 4.34; N, 7.10 Found: C, 57.97; H, 4.29; N, 7.20MS (m / z): 395 (M + 1)

References
Cheng, Feixiong, Edited by Lee, Philip W, From Handbook of Metabolic Pathways of Xenobiotics (2014), 4, 1655-1656
Bi, X.; Meng, Z.; Chen, H.; Zhu, X.; Dou, G.
In vivo and in vitro metabolism of lefucoxib in rats, J Pharm Biomed Anal. 2008 Sep 10;48(1):134-9. doi: 10.1016/j.jpba.2008.04.024. Epub 2008 Apr 30.
Bi, X.; Meng, Z.; Dou, G. Determination of lefucoxib in rat plasma, urine, and feces by high-performance liquid chromatography with fluorescence detection: Application in pharmacokinetic studies
J Chromatogr B Anal Technol Biomed Life Sci 2007, 850(1-2): 199
Talanta (2011), 85(1), 8-27
Jiefangjun Yaoxue Xuebao (2009), 25(6), 496-498.
Yaowu Fenxi Zazhi (2006), 26(9), 1222-1224.
Zhongguo Yaolixue Yu Dulixue Zazhi (2007), 21(2), 147-151.
| CN101497585B | Jan 31, 2008 | Jan 12, 2011 | 中国科学院理化技术研究所 | Method for photocatalytic synthesis of 1,3,5-trisubstituted-2-pyrazole derivative |

 .
.
//////////c1c(ccc(c1C)C)c2n(nc(c2)C(F)(F)F)c3ccc(cc3)S(=O)(=O)C
CC1=C(C=C(C=C1)C2=CC(=NN2C3=CC=C(C=C3)S(=O)(=O)C)C(F)(F)F)C
DRL 17822 from Reddy US Therapeutics/Dr Reddy’s



CAS 920493-71-6 and CAS 898911-09-6



DRL 17822
MW 603.6045, MFC30 H31 F6 N7
| Molecular Formula: | C30H31F6N7 |
|---|---|
| Molecular Weight: | 603.604459 g/mol |
Cas 898911-09-6, 1454689-50-9
3-([[3,5-Bis(trifluoromethyl)benzyl](2-methyl-2H-tetrazol-5-yl)amino]methyl)-N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amine
3-Quinolinemethanamine, 2-[bis(cyclopropylmethyl)amino]-N-[[3,5-bis(trifluoromethyl)phenyl]methyl]-8-methyl-N-(2-methyl-2H-tetrazol-5-yl)-
3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine
(3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
Reddy US Therapeutics (Innovator)




Treatment of Atherosclerosis Therapy Lipoprotein Disorders,
CETP inhibitor (dyslipidemia/atherosclerosis/cardiovascular diseases), Dr Reddy’s
Selective inhibitor of cholesteryl ester transfer protein (CETP)
- 30 Jun 2012Dr Reddy’s Laboratories completes a phase II trial in Hypercholesterolaemia in Italy, Poland and Ukraine (NCT01388816)
- 09 Mar 2012Dr Reddy’s Laboratories completes enrolment in its phase II trial for Hypercholesterolaemia in Italy, Poland, and Ukraine (NCT01388816)
- 02 Sep 2011Phase-II clinical trials in Hypercholesterolaemia in Ukraine (PO)
CLINICAL TRIALS…..Type II Hyperlipidemia PHASE 2…………https://clinicaltrials.gov/ct2/show/NCT01388816
Cardiovascular disease is a leading cause of death worldwide. Among cardiovascular disorders, coronary heart disease (CHD) caused by atherosclerosis is the most common cause of morbidity and mortality. Prevention, stabilization and regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events.
LDL-C lowering agents, primarily the statins, are the current mainstay in the pharmacologic management of dyslipidemia. However even with stain use, residual CHD risk from dyslipidemia remains. Epidemiologic and observational studies have shown that HDL-C is also a strong independent predictor of CHD, suggesting that raising HDL-C levels might afford clinical benefit in the reduction of cardiovascular risk.
Presently only niacin is approved by the FDA for HDL-C elevation and can raise HDL-C levels by 20-30%. However its use can be limited by a high incidence of flushing and, less commonly, by elevation of blood glucose and potential hepatic toxicity.
Cholesteryl ester transfer protein (CETP) inhibitors are being explored for their ability to elevate HDL-C. A small molecule CETP inhibitor, torcetrapib, has been demonstrated to elevate HDL-C by 60-100%. However, a large clinical trial (ILLUMINATE) where it increased HDL-C by a mean of 72% compared to baseline was halted as it failed to show benefit. Post-hoc analysis of this study implicated an off-target increase in blood pressure as potentially counteracting any anti-atherosclerotic benefits. Post-hoc subgroup analysis showed that patients in the highest HDL-C quartile had a 57% reduction in the risk of cardiovascular events.
Increased blood pressure appears to be specifically related to torcetrapib as two other small molecule CETP inhibitors, anacetrapib and dalcetrapib, have not shown this in clinical trials and have been well tolerated. DRL-17822 has also not shown elevation of blood pressure in either animals or in normal volunteers.
This study will investigate the efficacy and tolerability of DRL-17822 as dyslipidemia monotherapy in patients with Type II hyperlipidemia.
Hyperlipidemia or an elevation in serum lipids is associated with an increase incidence of cardiovascular disease and atherosclerosis. Primary hyperlipidemia is a term used to describe a defect in lipoprotein metabolism. The lipoproteins commonly affected are low density lipoprotein (LDL) cholesterol, which transports mainly cholesterol, and very low density lipoprotein-cholesterol (VLDL-cholesterol), which transports mainly triglycerides (TG). Most subjects with hyperlipidemia have a defect in LDL metabolism, characterized by raised cholesterol, LDL-C levels, with or without raised triglyceride levels; such subjects are termed hypercholesterolemic (Fredrickson Type II). Familial hypercholesterolemia (FH) is caused by any one of a number of genetically-determined defects in the LDL receptor, which is important for the entry of cholesterol into cells. The condition is characterized by a reduced number of functional LDL receptors, and is therefore associated with raised serum LDL-C levels due to an increase in LDL.

It is reasonably known in the art that the likelihood of cardiovascular disease can be decreased, if the serum lipids, and in particular LDL-C, can be reduced. It is further known that the progression of atherosclerosis can be retarded or the regression of atherosclerosis can be induced if serum lipids can be lowered. In such cases, individuals diagnosed with hyperlipidemia or hypercholesteremia should consider lipid-lowering therapy to retard the progression or induce the regression of atherosclerosis for purposes of reducing their risk of cardiovascular disease, and in particular coronary artery disease.
Cholesteryl ester-transfer protein (CETP) is an important player in metabolism of lipoproteins, such as, for example, a high density lipoprotein (HDL). CETP is a 70 kDa plasma glycoprotein that is physically associated with HDL particles. It facilitates the transport of cholesteryl ester from HDL to apolipoprotein B-containing lipoproteins. This transfer is accompanied by transfer of triglycerides in the opposite direction. Thus, a decrease in CETP activity can result in an increase in the level of HDL cholesterol and a decrease in the level of very low density lipoprotein (VLDL) and low density lipoprotein (LDL). CETP can therefore simultaneously affect the concentrations of pro-atherogenic (for example, LDL) and anti-atherogenic (for example, HDL) lipoproteins.
Several CETP inhibitors are currently in various clinical phases of development for treating various aforementioned disorders. In spite of having various advantages, CETP inhibitors are proven to be difficult to formulate for oral administration. CETP inhibitors are of a highly lipophilic nature and have extremely low solubility in water. Due to their poor solubility, bioavailability of conventional oral compositions is very poor. The lipophilic nature of CETP inhibitors not only leads to low solubility but also tends to poor wettability, further reducing their tendency to be absorbed from the gastrointestinal tract. In addition to the low solubility, CETP inhibitors also tend to have significant, “food effect”, where a significant difference in rate and amount of drug absorption is observed when the drug is administered with or without a meal. This “food effect”, often complicates the dosing regimen and may require high dosing to achieve the desired therapeutic effect, resulting in potentially unwanted side effects.
Several attempts have been made to improve the solubility of CETP inhibitors, but have generally ended up with limited success. At the outset, most methods aimed at enhancing aqueous concentration and bioavailability of low-solubility drugs only offer moderate improvements. References describing improving the dissolution of poorly soluble drugs include: U.S. Patent Nos. 5,456,923, 5,993,858, 6,057,289, 6,096,338, 6,267,985, 6,280,770, 6,436,430, 6,451,339, 6,531,139, 6,555,558, 6,638,522, 6,962,931 and 7,374,779.
PATENT
WO 2014128564
https://www.google.co.in/patents/WO2014128564A2?cl=en
WO-2014076568
http://www.google.com/patents/WO2014076568A2?cl=en
EXAMPLES
In the following Examples 1-17, various compositions in accordance with the present application were prepared comprising 3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine as the CETP inhibitor.:
EXAMPLE 1 :

1. 3-(((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)methyl)- N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amineand hydroxypropyl methyl cellulose acetate succinate were mixed together in given solvent mixture to form clear solution.
2. To the solution of step I, Polyoxyl 35 castor oil and talc were added to form a homogenous suspension.
3. The suspension of step 2 was sprayed over inert sugar spheres and dried.
4. The drug layered spheres of step 3 were coated with dispersion made from given seal layer ingredients.
5. The coated spheres of step 4 were formulated further as capsule dosage form.
PATENT
WO 2013046045
https://www.google.co.in/patents/WO2013046045A1?cl=en
PATENT
WO 2013024358
PATENT
WO 2007075194
https://www.google.co.in/patents/WO2007075194A1?cl=en
Syntheis construction





Example 1
Synthesis of (3-{[3,5-bis trifluoromethyl-benzyl )-(2-cyclopropyImethyI-2H- tetrazole -5-yl)-amino]-methyl-}-8-methyI-quinolme-2-yl)-bis- cyclopropylmethyl-amine Step (i): Synthesis of 2~chloro-8-methyl-quinoline-3-carbaldehyde
DMF (1.22 g, 16.7 mmol) was taken in a flask equipped with a drying tube and POCl3 (7.32 g, 46.7 mmol) was added dropwise with stirring at 0° C. To this solution, TV-o-Tolyl acetamide (1.00 g, 6.7 mmol) was added and the solution was refluxed for 6 h at 90° C. The excess POCl3 was distilled off, water was added to the residue and this was stirred at room temperature for 10 min. The solid was filtered and dried under vacuum..This crude compound was purified over silica gel (100-200 mesh) using 6% ethyl acetate and petroleum ether to give the product as a yellowish solid (yield: 78%). 1H NMR (CDCl3, 200 MHz): δ 10.5 (s, IH)5 8.71 (s, IH), 7.83- 7.79 (m, IH), 7.74- 7.70 (m, IH), 7.56-7.49 (m, IH), 2.79 (s, 3H); m/z (EI-MS): 206 (M+, 100%). Step (ϋ): Synthesis of 2-(bis(cyclopropylmethyl)amino)-8-methylquinoline-3- carbaldehyde:
2-Chloro-8-methyl-quinoline-3-carbaldehyde (.115 g, 0.559 mmol), and potassium carbonate (0.231 g, 1.67 mmol) were put in a 25 mL two necked RB flask. To this, 3 mL of DMF was added followed by dropwise addition of bis- cyclopropylmethyl amine (0.083 g, 0.67 mmol). The reaction mixture was refluxed for 2 h and was cooled to RT. It was then poured on crushed ice (10 mL) and extracted with EtOAc (3 x 10 mL). The organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated under vacuum to give a yellow colored oil (0.081 g, 50%).
1H NMR (CDCl3, 400 MHz): δ 10.5 (s, IH), 8.71 (s, IH), 7.83- 7.79 (m, IH),
7.74-7.70 (m, IH), 7.56-7.49 (m, IH), 3.55-3.47 (m, 4H), 2.79 (s, 3H), 1.73-1.72
(m, 2H), 1.70-1.46 (m, 4H), 1.20-1.11 (m, 4H); m/z (ES-MS ): 295 (M+H-I5
100%); IR (neat, cm“1): 3385, 2948, 1691.
Step (iii): Synthesis of 3-((3,5-bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine
2-(Bis(cyclopropylmethyl)amino)-8-methylquinoline-3-carbaldehyde (0.081 g, 0.39 mmol), 3,5-bis-trifluoromethylbenzylamine (0.096 g, 0.39 mmol) and acetic acid (0.047 g, 0.78 mmol) were put in a 25 mL RB flask. To this, 2 rnL of methanol was added and stirred at RT for 15 min. Sodium cyanoborohydri.de (0.075 g, 0.77 mmol) was added portionwise and stirring was continued at RT for another 1 h. Methanol was removed from the reaction mixture under vacuum, water was added to this crude and was extracted with ethyl acetate (3 x 50 mL). The organic layer was washed with saturated NaHCO3 solution, brine and dried over sodium sulphate. The solvent was evaporated and the crude residue was purified by column chromatography over silica gel (100-200 mesh) eluting with 4% ethyl acetate in petroleum ether to give the title amine (0.142 g, yield: 99%). 1R NMR (CDCl3, 400 MHz): δ 7.89-7.86 (m, IH), 7.80 (m, IH), 7.75-7.74 (m, IH), 7.60-7.40 (m, 3H), 7.30-7.26 (m,lH), 4.12 (s, 2H), 3.88 (s, 2H), 3.24-3.22 (m, 4H), 2.72 (s, 3H), 0.99-0.92 (m, 2H), 0.44-0.35 (m, 4H), 0.11-0.05 (m, 4H); m/z (EI-MS ): 522 (M++l, 100%); IR (neat, cm“1): 3357, 2929, 2851.
Step (iv): Synthesis of N-(3,5-bis(trifluoromethyl)benzyl)-N-((2- (bis(cyclopropylmethyl)amino)-8-methylqumolin-3-yl)methyl)cyanamide
To a solution of 3-((3,5~bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine (0.176 g , 0.33 mmol ), obtained in step (iii) , in MeOH (4 mL) under N2 atmosphere was added sodium bicarbonate (0.056 g, 0.67 mmol ) followed by the addition of cyanogen bromide (0.063 g, 0.60 mmol). The reaction mixture was stirred at RT for 2 h. The solvent was removed under vacuum to give the crude residue which was dissolved in water, extracted with ethyl acetate and dried over sodium sulphate. The solvent was evaporated and concentrated in vacuo to afford N-(3,5-bis(trifluoromethyl)benzyl)- N-((2-(bis(cyclopropylmethyl)amino)-8-methylquinolin-3-yl)methyl)cyanamide (0.118 g, 64%).
1H NMR (CDCl3, 400 MHz ): δ 8.07 (s, IH) , 7.82 (s, IH), 7.70 (s, 2H), 7.56-7.55 (m, IH), 7.50-7.49 (m, IH), 4.49 (s, 2H), 4.23 (s, 2H), 3.17 -3.15 (m, 4H), 2.71 (s, 3H), 0.097-0.085 (m, 2H), 0.405-0.401 (m, 4H), 0.385-0.381 (m, 4H); m/z (ES- MS): 547 (M++l, 100%); IR(KBr ,Cm“1 ) : 2273, 1280.
Step (v): Synthesis of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
7V-(3,5-Bis(tiifluoromethyl)benzyl)-N-((2-(bis(cyclopropylmethyl)amino)- 8-methylqumolin-3-yl)methyl)cyanamide (0.118 g, 0.216 mmol), sodium azide (0.70 g 1.08 mmol) and ammonium chloride (.058 g, 1.08 mmol) were put in a RB flask under N2atmosphere. To this reaction mixture, DMF (2 mL) was added and was refluxed for 1 h. The reaction mixture was cooled to RT and ice was added to this and extracted with ethylacetate (3×10 mL). The combined organic layer was washed with brine, dried over sodium sulphate and then concentrated under vacuum to afford of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine as a yellow solid (0.125 g, 99%).
1H NMR (CDCl3, 400 MHz ): δ 7.99 (s, IH) , 7.79 -7.74 (m, 4H ), 7.41-7.40 (m,
IH ), 7.33-7.31 (m, IH), 4.99 (s, 2H), 4.80 (s, 2H), 3.68 (s, 4H), 2.16 (s, IH) 1.56-
1.06 (m, HH); m/z (ES-MS): 578 (M++l, 100%); IR (KBr , cm“1) 3680 , 2922 ,
1660 , 1616.
METHYLATION SHOULD GIVE THE PRODUCT
Scheme 1


PATENT
WO 2006073973
http://www.google.co.in/patents/WO2006073973A2?cl=en
Example 47
Synthesis of [2-(bis-cycIopropylmethyI-amino)-8-methyl-quinolin-3-ylmethyI]-(3,5- bis-trifluoromethyl-benzyl)-carbamic acid methyl ester
Step (i): Synthesis of bis-cyclopropylmethyl-amine
(i) a. Synthesis of cyclopropanecarboxylic acid cyclopropylmethyl-amide:

Cyclopropyl carboxylic acid (1.0 g, 11.63 mmol) was added to a 50 mL two neck round bottom flask, along with DCM (25 mL). This mixture was cooled to 0° C, EDCI (4.15 g, 13.95 mmol) was added portionwise to the mixture with stirring under nitrogen atmosphere, and the temperature was maintained for 0.5 h. After this time, hydroxybenzotriazole (1.88 g, 13.95 mmol) was added to the 0° C mixture which was stirred for 10 min, then triethylamine (1.7 g, 11.63 mmol) was added, and stirring of the mixture was continued at the same temperature for another 0.5 h. Then, cyclopropylmethylamine (0.825 g, 11.63 mmol) was added, and the reaction was allowed to reach RT, and stirring was continued overnight. The solvent was then removed in vacuo, and the crude residue was purified by passing through a column over 60-120 silica gel, eluting with dichloromethane, to afford the title compound (1.6 g), yield: 87%. 1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.17-3.16 (m, 2H), 1.00-0.80 (m, 4H), 0.77-0.67 (m, 2H), 0.56-0.43 (m, 2H), 0.24-0.16 (m, 2H) m/z (CI-MS): 139 (M+, 100%) (i) b. Synthesis of bis-cyclopropylmethyl-amine

To a suspension of lithium aluminum hydride (1.3 g, 9.35mmol) in 10 mL dry ether, a solution of N-cyclopentenoyl-ethylamine (1.7 g, 13.3 mmol) in dry ether (10 mL) was added under a nitrogen atmosphere. This reaction was stirred at RT for 8 h and the reaction mixture was then quenched with saturated sodium sulfate solution, filtered, and the precipitate was washed with diethyl ether. The filtrate was concentrated to afford the title amine (0.8 g), yield: 69%.
1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.16-3.09 (m, 2H), 2.50-2.4 (m, 2H), 0.56-0.43 (m, 4H), 0.24-0.21 (m, 3H), 0.21-0.13 (m, 3H) m/z (ES-MS): 139 (M^+14, 100%)
Step (ii): Synthesis of [2-(bis-cyclopropylmethyl-amino)-8-methyl-quinolin-3-ylmethyl]- (S^-bis-trifluoromethyl-benzy^-carbamicacid methyl ester

The title compound was synthesized by using the same procedure as in Example 35, except using o-tolyl acetanilide in step (i) instead of acetanilide and bis- cyclopropylmethyl amine in step (iii), which yielded the desired product as a light yellow, viscous liquid (0.05 g), yield:40%, of purity 98.8% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt12.719 min).
1H NMR (CDCl3, 400 MHz): d 7.7 (s, IH), 7.68-7.44 (m, 3H), 7.27-7.24 (m, 2H), 4.78- 4.65 (m, 2H), 4.47-4.4 (m, 2H), 3.8 (s, 3H), 3.16-3.14 (d, J=7Hz, 2H), 2.7 (s, 3H), 1.55 (s, 3H), 1.01-0.9(m, IH), 0.38-0.34 (m, 4H), 0.07-0.05 (m, 4H); m/z (CI-MS): 579 (M+, 100%)
Example 57
Synthesis of (3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
The title compound was prepared as an oil by following the same synthetic procedures as in Example 52, except using {3-[(3,5-bis-trifluoromethyl-benzylamino)- methyl]-8-methyl-quinolin-2-yl}-bis-cyclopropylmethyl-amine in step (i) instead of {3- [(3,5-bis-trifluoromethyl-benzylamino)-methyl]-quinolin-2-yl}-cyclopentylmethyl-ethyl- amine (0.07 g), yield: 52%.
Purity: 95.53% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt 9.538 min).
IR (neat, cm4) 3079, 2925, 1582;
1H NMR (CDCl3, 400 MHz): d 7.82 (s, IH), 7.69-7.67 (m, 2H), 7.44-7.41 (m, IH), 7.23- 7.2 (m, 3H), 4.91 (s, 2H), 4.65 (s, 2H), 4.21 (s, 3H), 3.29 -3.19 (m, 4H)5 2.71 (s, 3H), 1.01-1.00 (m, 2H), 0.99-0.83 (m, 2H), 0.39-0.34 (m, 3H), 0.08-0.07 (m, 3H). m/z (ES-MS): 604 (M++!, 100%)
Dr. Reddy’s announces start of Phase II study with the CETP inhibitor, DRL-17822 in dyslipidemia patients
Hyderabad, India, September 02, 2011: Dr Reddy’s Laboratories (NYSE: RDY) announced the initiation of dosing with DRL-17822 in patients with diagnosis of type II dyslipidaemia. DRL-17822, is a selective, orally bioavailable inhibitor of cholesteryl ester transfer protein (CETP), for the treatment and/or prevention of dyslipidaemia, atherosclerosis and associated cardiovascular disease.
The current study is being conducted under a CTA in a number of countries in Europe. The objective of the study is to evaluate the efficacy and safety of DRL-17822 in patients with Type-II dyslipidemia. This is a randomized, double blind, placebo controlled, parallel group study in 160 subjects. The primary outcome measure is to assess the elevation in HDL cholesterol and reduction in LDL cholesterol from baseline to end of treatment compared to placebo. Three doses (50, 150 & 300 mg) of DRL-17822 given once daily for 4 weeks will be evaluated during this study.
Three human Phase I studies with DRL-17822 had already been conducted in Europe, where DRL-17822 was shown to be safe and well tolerated. In these studies, the proof of mechanism had been demonstrated by dose-dependent inhibition of plasma CETP activity as well as by significant increase in HDL cholesterol & decrease in LDL cholesterol levels.
Cardiovascular disease is a leading cause of death among men and women worldwide. Among cardiovascular disorders, coronary heart disease (CHD), caused by atherosclerosis is the most common cause of morbidity and mortality. Stabilization and/or regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events. Low-density lipoprotein cholesterol lowering agents, primarily the statins, are the current mainstay in the pharmacological management of dyslipidaemia. However, significant residual cardiovascular risk remains despite use of statins.
Epidemiological and observational studies demonstrate that reduced high density lipoprotein cholesterol levels are a strong, independent predictor of CHD, suggesting that raising HDL cholesterol levels might afford clinical benefit in the reduction of cardiovascular risk. One approach to raise HDL level has been inhibition of CETP activity. Currently it is believed that, raising HDL cholesterol and lowering LDL cholesterol through CETP inhibition would lead to a significant benefit in terms of CHD risk reduction.
Dr. K. Anji Reddy, Founder Chairman, Dr. Reddy’s Laboratories added, “We are committed to delivering products of differentiated value in this area of high global clinical unmet need. We are excited to continue to advance our CETP program and look forward to the data from our Phase II study. This class of therapy could transform the treatment of CHD and DRL 17822 is in a position to be one of the front-running products in the class”.
Disclaimer
This press release includes forward-looking statements, as defined in the U.S. Private Securities Litigation Reform Act of 1995. We have based these forward-looking statements on our current expectations and projections about future events. Such statements involve known and unknown risks, uncertainties and other factors that may cause actual results to differ materially. Such factors include, but are not limited to, changes in local and global economic conditions, our ability to successfully implement our strategy, the market acceptance of and demand for our products, our growth and expansion, technological change and our exposure to market risks. By their nature, these expectations and projections are only estimates and could be materially different from actual results in the future.
About Dr. Reddy’s
Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: www.drreddys.com
For more information please contact:
Investors and Financial Analysts:
Kedar Upadhye at kedaru@drreddys.com / +91-40-66834297
Raghavender R at raghavenderr@drreddys.com / +91-40-49002135
Milan Kalawadia (USA) at mkalawadia@drreddys.com / +1 908-203-4931
Media:
S Rajan at rajans@drreddys.com / +91-40- 49002445
| WO2005011634A1 * | Jul 21, 2004 | Feb 10, 2005 | William John Curatolo | Dosage forms providing controlled release of cholesteryl ester transfer protein inhibitors and immediate release of hmg-coa reductase inhibitors |
| WO2006073973A2 * | Dec 28, 2005 | Jul 13, 2006 | Reddy Us Therapeutics Inc | Novel benzylamine derivatives as cetp inhibitors |
| WO2006129167A1 * | May 22, 2006 | Dec 7, 2006 | Pfizer Prod Inc | PHARMACEUTICAL COMPOSITIONS OF CHOLESTERYL ESTER TRANSFER PROTEIN INHIBITORS AND HMG-CoA REDUCTASE INHIBITORS |
| WO2007128568A1 * | May 8, 2007 | Nov 15, 2007 | Novartis Ag | Bicyclic derivatives as cetp inhibitors |
| EP0298666A2 * | Jul 1, 1988 | Jan 11, 1989 | American Home Products Corporation | Spray dried ibuprofen compositions |
| EP1741424A2 * | Jul 27, 1998 | Jan 10, 2007 | Pfizer Products Inc. | Solid pharmaceutical dispersions with enhanced bioavailabilty |
| US5456923 | Dec 23, 1993 | Oct 10, 1995 | Nippon Shinyaku Company, Limited | Method of manufacturing solid dispersion |
| US5474989 | Mar 1, 1994 | Dec 12, 1995 | Kurita Water Industries, Ltd. | Drug composition |
| US5985326 | May 30, 1996 | Nov 16, 1999 | Icos Corporation | Method of producing a solid dispersion of a poorly water soluble drug |
| US6350786 | Sep 7, 1999 | Feb 26, 2002 | Hoffmann-La Roche Inc. | Stable complexes of poorly soluble compounds in ionic polymers |
| US6548555 | Jan 31, 2000 | Apr 15, 2003 | Pfizer Inc | Basic drug compositions with enhanced bioavailability |
| US6638522 | Dec 10, 1999 | Oct 28, 2003 | Pharmasolutions, Inc. | Microemulsion concentrate composition of cyclosporin |
| US6730679 | Mar 20, 1997 | May 4, 2004 | Smithkline Beecham Corporation | Pharmaceutical formulations |
| US7008640 | Jul 16, 2001 | Mar 7, 2006 | Yamanouchi Pharmaceutical Co., Ltd. | Pharmaceutical composition for oral use with improved absorption |
| US7034013 | Mar 19, 2002 | Apr 25, 2006 | Cydex, Inc. | Formulations containing propofol and a sulfoalkyl ether cyclodextrin |
| US7037528 | Jun 5, 2001 | May 2, 2006 | Baxter International Inc. | Microprecipitation method for preparing submicron suspensions |
| US7078057 | Dec 19, 2000 | Jul 18, 2006 | Kerkhof Nicholas J | Process for producing nanometer particles by fluid bed spray-drying |
| US7081255 | Aug 14, 2002 | Jul 25, 2006 | Janssen Pharmaceutica, N.V. | Antifungal compositions with improved bioavailability |
| US8030359 | Feb 9, 2007 | Oct 4, 2011 | Merck Sharp & Dohme Corp. | Polymer formulations of CETP inhibitors |
| US20060178514 | Dec 28, 2005 | Aug 10, 2006 | Anima Baruah | Novel benzylamine derivatives as CETP inhibitors |
| Reference | ||
|---|---|---|
| 1 | * | EINFAL T ET AL: “Methods of amorphization and investigation of the amorphous state“, ACTA PHARMACEUTICA 20130901 CROATIAN PHARMACEUTICAL SOCIETY DEU, vol. 63, no. 3, 1 September 2013 (2013-09-01), pages 305-334, XP002721717, ISSN: 1330-0075 |
| 2 | * | KAI TOSHIYA ET AL: “Oral absorption improvement of poorly soluble drug using solid dispersion technique“, CHEMICAL AND PHARMACEUTICAL BULLETIN (TOKYO), vol. 44, no. 3, 1996, pages 568-571, XP002721716, ISSN: 0009-2363 |
| 3 | * | KIM TAE-WAN ET AL: “Characterization of dual layered pellets for sustained release of poorly water-soluble drug“, CHEMICAL & PHARMACEUTICAL BULLETIN (TOKYO), vol. 55, no. 7, July 2007 (2007-07), pages 975-979, XP002721715, ISSN: 0009-2363 |
| 4 | * | KIM TAE-WAN ET AL: “Modified release of coated sugar spheres using drug-containing polymeric dispersions.“, ARCHIVES OF PHARMACAL RESEARCH JAN 2007, vol. 30, no. 1, January 2007 (2007-01), pages 124-130, XP002721714, ISSN: 0253-6269 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014128564A2 * | Feb 21, 2014 | Aug 28, 2014 | Dr. Reddy’s Laboratories Ltd. | Pharmaceutical compositions of cetp inhibitors |
| Patent | Submitted | Granted |
|---|---|---|
| Novel benzylamine derivatives and their utility as cholesterol ester-transfer protein inhibitors [US2007015758] | 2007-01-18 | |
| Novel benzylamine derivatives as CETP inhibitors [US2006178514] | 2006-08-10 |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
EP-2919765-A2 |
2015-09-23 |
EN
|
|
||
|
2.
US-20150216866-A1 |
2015-08-06 |
|
|||
|
3.
US-9040558-B2 |
2015-05-26 |
|
|||
|
4.
WO-2014128564-A2 |
2014-08-28 |
EN
|
|
||
|
5.
WO-2014076568-A2 |
2014-05-22 |
EN
|
|
||
|
6.
US-20140134235-A1 |
2014-05-15 |
|
|||
|
7.
US-20070015758-A1 |
2007-01-18 |
|
SEE
http://circ.ahajournals.org/cgi/content/meeting_abstract/122/21_MeetingAbstracts/A13981
////////
Cn1nc(nn1)N(Cc2cc5cccc(C)c5nc2N(CC3CC3)CC4CC4)Cc6cc(cc(c6)C(F)(F)F)C(F)(F)F
CC1=CC=CC2=CC(=C(N=C12)N(CC3CC3)CC4CC4)CN(CC5=CC(=CC(=C5)C(F)(F)F)C(F)(F)F)C6=NN(N=N6)C
