

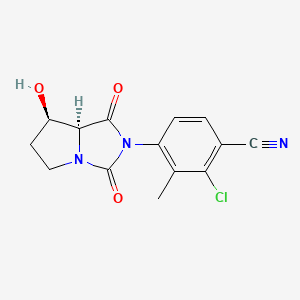
BMS-564929; BMS564929; 627530-84-1; 2-Chloro-4-[(7r,7as)-7-Hydroxy-1,3-Dioxotetrahydro-1h-Pyrrolo[1,2-C]imidazol-2(3h)-Yl]-3-Methylbenzonitrile; hydantoin,
CAS 627530-84-1, Squibb Bristol Myers Co
| Molecular Formula: |
C14H12ClN3O3 |
| Molecular Weight: |
305.71638 g/mol |
4-[(7R,7aS)-7-Hydroxy-1,3-dioxoperhydropyrrolo[1,2-c]imidazol-2-yl]-2-chloro-3-methylbenzonitrile
7-K,7aS)-2-Chloro-4-(7-hydroxy-l,3-dioxotetrahydropyrrolo[l,2- c]imidazol-2-yl)-3-methylbenzonitrile
4-[(7R,7aS)-7-hydroxy-1,3-dioxo-5,6,7,7a-tetrahydropyrrolo[1,2-c]imidazol-2-yl]-2-chloro-3-methylbenzonitrile
BMS-564929 is a highly potent, orally active and nonsteroidal tissue selective modulator of androgen receptor (AR) with Ki value of 2.11 nM.

BMS-564929 is a selective androgen receptor (AR) modulator with Ki value of 2.11 ± 0.16 nM [1].
The AR is a type of nuclear receptor that is activated by the androgenic hormones, testosterone, or dihydrotestosterone. The important function is regulating gene expression.
BMS-564929 is a muscle-tissue specific agonist for AR with a bicyclic hydantoin structure [2]. BMS-564929 is about 400-fold selective for AR vs. PR and more than 1000-fold selective for AR vs. GR, MR and ERα and β. In the C2C12 myoblast cell line, BMS-564929 shows a potency of 0.44 ± 0.03 nM compared with 2.81 ± 0.48 nM measured for testosterone
In castrated male rats, BMS-564929 is substantially more potent than testosterone (T) in promoting the growth of the levator ani muscle, and is highly selective for muscle vs. Prostate. Because of its potent oral activity and tissue selectivity, BMS-564929 is expected to yield beneficial clinical effects in muscle and other tissues with a more favorable safety way

BMS-564,929 is an investigational selective androgen receptor modulator, which is being developed by Bristol-Myers Squibb for treatment of the symptoms of age-related decline in androgen levels in men (“andropause“). These symptoms may includedepression, loss of muscle mass and strength, reduction in libido and osteoporosis. Treatment with exogenous testosterone is effective in counteracting these symptoms but is associated with a range of side effects, the most serious of which is enlargement of the prostate gland, which can lead to benign prostatic hypertrophy and even prostate cancer. This means there is a clinical need for selective androgen receptor modulators, which produce anabolic effects in some tissues such as muscle and bone, but without stimulating androgen receptors in the prostate.[1]
BMS-564,929 is one such compound currently in early human clinical trials, which is an orally active, potent and selective agonist for androgen receptors (Ki 2.1nM, 20x functional selectivity for muscle tissue over prostate) and in studies on castrated rats it was shown to counteract decrease in muscle mass over time, and at higher doses even increased muscle mass, without significantly affecting prostate tissue.[2] It does however vastly reduce luteinizing hormone levels, it being an astonishing 33x more suppressive compound than testosterone,[3] which may be a problem in human clinical use.[4]
Selective androgen receptor modulators may also be used by athletes to assist in training and increase physical stamina and fitness, potentially producing effects similar to anabolic steroids but with significantly fewer side effects. For this reason, SARMs have already been banned by the World Anti-Doping Agency since January 2008 despite no drugs from this class yet being in clinical use, and blood tests for all known SARMs are currently being developed.[5][6]
| Patent |
Submitted |
Granted |
| Bicyclic modulators of androgen receptor function [US2004019063] |
2004-01-29 |
|
| BICYCLIC MODULATORS OF ANDROGEN RECEPTOR FUNCTION [US7772267] |
2008-05-08 |
2010-08-10 |
| Bicyclic modulators of androgen receptor function [US7405234] |
2004-09-16 |
2008-07-29 |
WO 2003096980
http://www.google.com/patents/WO2003096980A2?cl=en
Example 23
(7-K,7aS)-2-Chloro-4-(7-hydroxy-l,3-dioxotetrahydropyrrolo[l,2- c]imidazol-2-yl)-3-methylbenzonitrile
23 A. 3-Chloro-2-methylphenyIacetamide
To a solution of 3-chloro-2-methylaniline (3.00 g, 21.2 mmol) in 25 mL of EtOH at rt was added acetic anhydride (2.40 mL, 25.4 mmol), and the solution was stirred at rt for 2 h. The mixture was concentrated under reduced pressure to give 3.89 g (100%) of the desired acetamide. 1H NMR (DMSO- ) δ 2.05 (s, 3H), 2.20 (s, 3H), 7.16 (t, J = 1.1, 8.3, 1H), 7.25 (d, J = 8.3, 1H), 7.31 (d, J = 8.3, 1H), 9.55 (s, 1H); 13C NMR (DMSO- ) δ 15.1, 23.1, 124.4, 125.8, 126.7, 130.3, 133.7, 138.0, 168.3; HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA; 1 min hold, 4 mL/min UV detection at 220 nm, 2.32 min retention time; HPLC b) column: Shimadzu Shim-Pack VP-ODS CI 8 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold; 4 mL/min, UV detection at 220 nm, 2.20 min retention time (99%); MS (ES) m/z 184 [M+H]+.
no 23B. 4-Bromo-3-chloro-2-methylphenylacetamide
To a suspension of acetamide 23A (2.00 g, 10.9 mmol) in 15 mL of glacial AcOH cooled to approximately 15 °C was added bromine (1.67 mL, 32.7 mmol) over 20 min. The ice bath was removed and the solution was stirred for
2 h, poured into ice water with stirring, and the solid was then filtered and dried to give 2.75 g (96%) of the desired bromide. 1H NMR (DMSO-_i6) δ 2.05 (s,
3H), 2.28 (s, 3H), 7.29 (d, J = 8.3, 1H), 7.56 (d, J = 8.8, 1H), 9.60 (s, 1H); 13C NMR (DMSO–i6) δ 16.7, 23.1, 118.1, 125.5, 130.4, 132.7, 133.4, 137.1, 168.4;
HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10%
MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1 % TFA, 1 min hold,
4 mL/min, UV detection at 220 nm, 2.95 min retention time; HPLC b) column:
Shimadzu Shim-Pack VP-ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold,
4 mL/min, UV detection at 220 nm, 2.87 min retention time (98%); MS (ES) m/z 263 [M+H]+.
23C. 3-Chloro-4-cyano-2-methylphenylacetamide
A suspension of bromide 23B (2.70 g, 10.3 mmol) and copper cyanide (0.92 g,
10.3 mmol) in DMF (30 mL) was heated to 150 °C for 4 h. The suspension was cooled, poured into water with stirring, and the solid was filtered and dried to give 1.44 g (67%) of the desired nitrile. 1H NMR (DMSO-d6) δ 2.12 (s, 3H),
i n 2.29 (s, 3H), 7.72 (d, J = 8.8, 1H), 7.75 (d, J = 8.2, 1H), 9.73 (s, 1H); 13C NMR (DMSO- ) δ 15.3, 23.5, 107.7, 116.5, 123.0, 130.1, 131.5, 135.7, 142.3, 168.8; HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.23 min retention time; HPLC b) column: Shimadzu Shim-Pack VP-ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.13 min retention time (95%); MS (ES) m/z 209 [M+H]+.
23D. 3-Chloro-4-cyano-2-methylphenylaniline

A solution of cyanoacetamide 23C (9.90 g, 47.4 mmol) in 100 mL of concentrated HCl EtOH (1 :1) was refluxed 30 min. The solution was then concentrated and dried under reduced pressure to give 9.41 g (98%) of the desired aniline as the hydrochloride salt. The free base of the aniline was obtained by suspending the salt in EtOAc and washing with saturated aqueous NaHC03 solution. The organic layer was then dried (MgS04), filtered and concentrated under reduced pressure. Η NMR (OMSO-dβ) δ 2.12 (s, 3H), 6.30 (s, 2H), 6.61 (d, J = 8.23, 1H), 7.36 (d, J = 8.23, 1H); 13C NMR DMSO-d6) δ 13.8, 96.9, 112.1, 118.3, 118.85, 132.2, 135.6, 152.5; HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.43 min retention time; HPLC b):column: Shimadzu Shim-Pack VP-ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 220 nm, 2.31 min retention time (99%); MS (ES) m/z 167 [M+H]+.
23E. 2-Chloro-4-isocyanato-3-methylbenzonitriIe
5
The title compound was prepared from compound 23D in a manner similar to that described in Experiments 2D to 2E.
l o 23F. (2S,3-R)-l-(3-Chloro-4-cyano-2-methylphenylcarbamoyl)-3-hydroxy- pyrrolidine-2-carboxylic acid methyl ester
To a solution of hydroxyproline compound IF (493 mg, 3.40 mmol) in CH2C12
15 (15 mL) was added 4 A molecular sieves (~ 3.0 g), followed by isocyanate 23E (725 mg, 3.22 mmol), and the resulting mixture was stirred at rt overnight, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 0.5% MeOH in EtOAc/hexane, 1: 1) to afford the title compound (736 mg) as an off-white solid. HPLC column: YMC S-5 0 C18 (4.6 x 50 mm), 0% to 100% B, 4 min gradient, 1 min hold (A = 90% H20 – 10% CH3CN – 0.1% TFA and B = 10% H20 – 90% CH3CN – 0.1% TFA), flow rate at 4 mL/min, UV detection at 220 nm, 1.57 min retention time (100%); MS (ES) m/z 338 [M+H]+. 23G. (7-R,7a5)-2-Chloro-4-(7-hydroxy-l,3-dioxotetrahydropyrroIo[l,2- c]imidazoI-2-yl)-3-methyIbenzonitrile.

To a suspension of cz‘s-3-hydroxyproline methyl ester, HCl salt (4.91 g, 27 mmol) in CH2C12 (100 mL) cooled to 0 °C was added -Pr2NEt (4.79 mL, 27.5 mmol). After stirring at rt for 15 min, isocyanate 23E was added as a solid in one portion through a powder addition funnel, rinsing with 50 mL CH2C12. The resulting light brown solution was stirred at rt until urea formation was complete (~ 2 h). To the mixture was then added DBU (4.6 mL, 30 mmol), and the resulting brown colored solution was stirred at rt until hydantoin formation was complete (~ 15 h). The product (4.72 g, 62%) was collected by filtration and washing with CH2C12 (2x). The mother liquor was then diluted with CH2C12 and washed with H20 (2x), 1 N HCl (2x) and brine. After removal of most of the solvent under reduced pressure, further product (1.2 g, 16%) was collected by filtration and washing with CH2C12 (2x). Recrystallization of the 4.72 g of crude product from hot THF and hexane gave 4.5 g of analytically pure product.
1H NMR (DMSO- ) δ 2.05-2.11 (m, 1H), 2.15-2.22 (m, 1H), 2.20, 2.24 (s, 3H), 3.29-3.33 (m, 1H), 3.59-3.68 (m, 1H), 4.42-4.50 (m, 2H), 5.64, 5.72 (d, J = 3.9, 3.3, 1H), 7.22, 7.51 (d, J = 8.3, 1H), 7.96 (d, I = 8.2,1H);
13C NMR (OMSO-d6) δ 15.4, 15.6, 35.5, 35.6, 43.3, 43.4, 68.8, 69.3, 69.8, 112.9, 113.1, 115.8, 128.1, 128.7, 132.1, 136.3, 136.4, 136.9, 137.1, 158.6, 169.1, 169.6;

HPLC a) column: Phenominex ODS C18 4.6 x 50 mm, 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH 10% H2O/0.1% TFA; 1 min hold; 4 mL/min, UV detection at 254 nm, 2.07 and 2.32 min retention time; HPLC b) column: Shimadzu Shim-Pack VP-ODS C18 (4.6 x 50 mm), 4 min gradient, 10% MeOH/90% H2O/0.1% TFA to 90% MeOH/10% H2O/0.1% TFA, 1 min hold, 4 mL/min, UV detection at 254 nm, 1.93 and 2.23 min retention time; Chiral HPLC column: Daicel Chiralcel OD 4.6 x 250 mm, isocratic, 30 min, 25% isopropanol/hexanes, 1 mL/min, UV detection at 254 nm; Shimadzu HPLC: 17.99 min retention time (>99%): Column: Hypercarb 5μ, 4.6 x 100 mm, 25 °C, isocratic, 30 min ACN/Η20 (35:65); 1 mL/min,
10.99 min retention time; MS (ES) m/z 306 [M+H]+. Alternatively, compound 23G can also be prepared by the following procedure: A solution of 22C (0.10 g, 0.28 mmol) and copper cyanide (0.03 g, 0.34 mmol) in DMF (1 mL) was refluxed for 3 h, cooled to rt, and diluted with water. The resulting solid was filtered, washed with water, dried and purified using preparative HPLC to afford the title compound (27 mg).
Alternatively, compound 23G can also be prepared by the following procedures: A solution of 22C (0.10 g, 0.278 mmol) and copper cyanide (0.03g, 0.334 mmol) in DMF (1 mL) was refluxed for 3 h, cooled to rt and diluted with water. The resulting solid was filtered, washed with water, dried and purified using preparative HPLC to afford the title compound (27 mg). HPLC: 99% at 2.06, 2.34 min (retention time) (Conditions: Phenom. Lura C18 (4.6 x 50 mm); Eluted with 0% to 100% B, 4 min gradient (A = 90% H20 – 10% MeOH – 0.1% TFA and B = 10% H20 – 90% MeOH – 0.1% TFA); Flow rate at 4.0 mL/min. UV detection at 220 nm). Chiral HPLC: retention time = 11.04 min (99%); Conditions: OD (4.6 x 250 mm); Eluted with 25% isopropanol in hexane for 30 min at 1 mL/min. MS (ES) m/z 306 [M+l]+.
References
- Gao, W; Dalton, JT (2007). “Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs)”. Drug Discovery Today 12 (5–6): 241–8. doi:10.1016/j.drudis.2007.01.003. PMC 2072879. PMID 17331889.
- Ostrowski, J; Kuhns, JE; Lupisella, JA; Manfredi, MC; Beehler, BC; Krystek Jr, SR; Bi, Y; Sun, C et al. (2007). “Pharmacological and x-ray structural characterization of a novel selective androgen receptor modulator: potent hyperanabolic stimulation of skeletal muscle with hypostimulation of prostate in rats”. Endocrinology 148 (1): 4–12. doi:10.1210/en.2006-0843. PMID 17008401.
- http://antaeuslabs.blogspot.com/2011/01/hydantoin-derivative-sarms-bms-564929.html
- Gao, W; Dalton, JT (2007). “Ockham’s Razor and Selective Androgen Receptor Modulators (SARMs): Are We Overlooking the Role of 5α-Reductase?”. Molecular interventions 7 (1): 10–3.doi:10.1124/mi.7.1.3. PMC 2040232. PMID 17339601.
- Thevis, M; Kohler, M; Schlörer, N; Kamber, M; Kühn, A; Linscheid, MW; Schänzer, W (2008). “Mass spectrometry of hydantoin-derived selective androgen receptor modulators”. Journal of mass spectrometry : JMS 43 (5): 639–50. doi:10.1002/jms.1364. PMID 18095383.
- Thevis, M; Kohler, M; Thomas, A; Maurer, J; Schlörer, N; Kamber, M; Schänzer, W (2008). “Determination of benzimidazole- and bicyclic hydantoin-derived selective androgen receptor antagonists and agonists in human urine using LC-MS/MS”. Analytical and Bioanalytical Chemistry 391 (1): 251–61. doi:10.1007/s00216-008-1882-6. PMID 18270691.
External links
//////////
TAKE A TOUR
TAKE A TOUR
TAKE A TOUR
Jejuri, Pune district, Maharashtra, INDIA
-
en.wikipedia.org/wiki/Jejuri
Jejuri is a city and a municipal council in Pune district in the Western Indian state of Maharashtra. It is famous for the main temple of Lord Khandoba.






















Jejuri is situated 48 km from Pune in Maharashtra State. Jejuri can be reached is by Road or Rail from Pune. Number of State Transport buses ply from Pune. It can be reached by Express trains from Pune Railway Station. GKP LTT Express Train no.15018 departure 0450 hrs from Pune PN arrival Jejuri JJR 0548 hrs, Maharshtra Express Train no.11040 departure 0450 hrs from Pune PN arrival Jejuri JJR 0549 hrs Koyana Express Train no.11029 departure 0045 hrs from Pune PN arrival Jejuri JJR 0148 hrs Sahyadri Express Train no.11023 departure 2205 hrs from Pune PN arrival Jejuri JJR 2308 hrs.These trains runs all days.
Jejuri is one of the most famous religious places in Maharashtra. The Village Jejuri is popularly known as Khanderayachi Jejuri.
Jejuri’s Khandoba Temple is built on a hill, which is approximately 51 kilometers away from Pune Railway Station. As the Temple is on the hill, one has to ascend more than 200 steps. But the ascending is not so tough and the wonderful view of Jejuri village is superb. If weather permits, One can easily see the spectacular view of Dive and Saswad Ghat. One can enjoy number of `Deep Mala’ (lamp post) while climbing the hill. Jejuri is really popular for its old Deep Malas.
The Jejuri temple was constructed in 1608. The Sabhamandap (Audience Hall) and other parts of the structure were completed subsequently. In 1742, Holkars constructed pillars and completed battlements and tank. The devotees added gateways, stairways, lamp pillars, cloisters etc.
The Idol of Lord khandoba in the Temple is beautiful.
The shepherd community considers Khandoba as their family deity.
One must visit Jejuri to look the Crystal Stands. Jejuri is one of the important temples in Maharashtra with historical significance.
Khandobacha Yelkot, Yelkot Yelkot Jay Malhar, Sadanandacha Yelkot, Kadepathar Maharaj Ki Jay are some of the popular terms here.
One can find many idols in and nearby the Jejuri Temple.
Temple Festivals :-
Chaitra Pournima, Dussehra, Champa Shashthi, Paush Pournima, Magh Pournima, Mahashivratri, Somvati Amavasya, Guru Pournima
Pandit/Brahmin for Pooja in Jejuri :-
Upadhye Guruji-9850150797, 02115-253152
Accommodation :-
Shree Siddhi Lodge and Hotel # 02115-253090
Best time to visit Jejuri :-
Throughout the year
Nearest Railway Station :-
Jejuri Railway Station
Distance :-
Mumbai – Jejuri – 206 Kilometers (By Road) Via Mumbai Pune Express Way
Nearby Attractions / Holy Places :-
Shree Mayureshwar Temple, Morgaon (Ashta Vinayak)
Pandeshwar
Bhuleshwar
Saswad
Kanifnath Temple
Balaji Temple


A painting depicts Khandoba riding a white horse with Mhalsa, accompanied with a dog and attendants including a Waghya dancing before him.
Khandoba and Mhalsa killing demons Mani-Malla — a popularoleograph, c.1880.
| Khandoba |

Khandoba (center) in his four armed form, the two metal images depict him with his wives. The sanctum of the newer Jejuri temple. |

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


























 AIRPORT
AIRPORT





 FLAG
FLAG









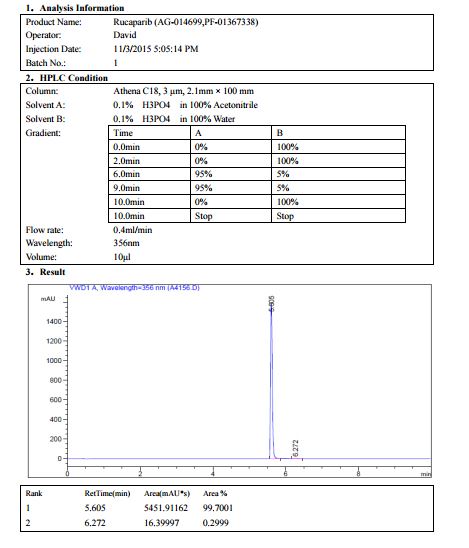
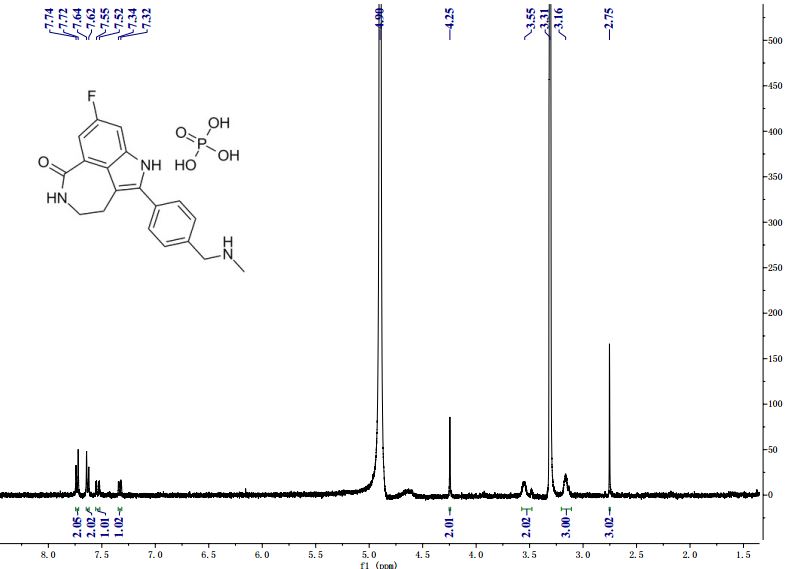
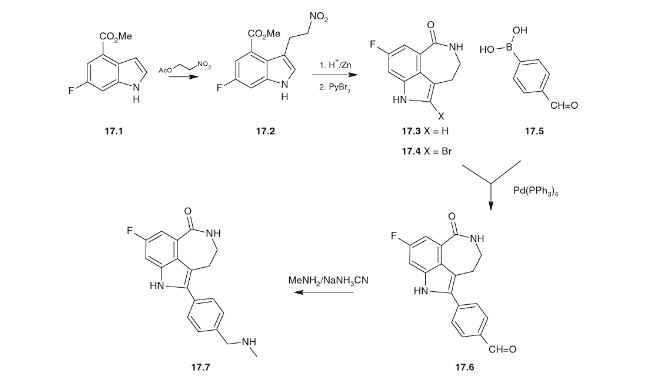
 Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.




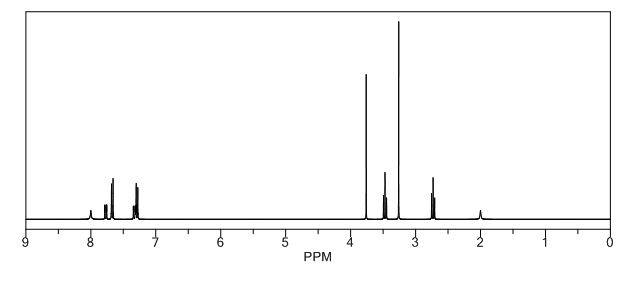

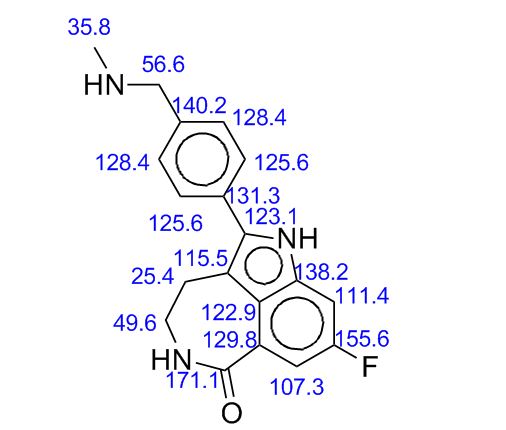
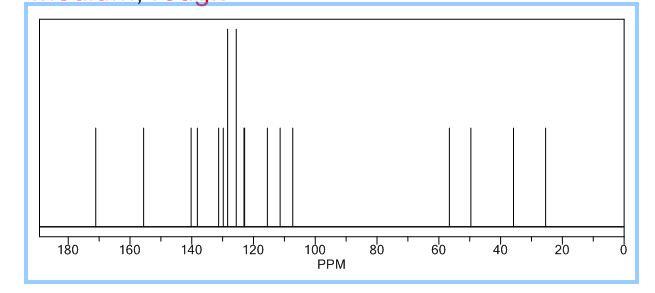



























 .
. .
.
 .
.
 .
.



 .
.









 .
.












 .
. .
.
 .
. .
. .
. .
. .
.
 .
. .
.


















 Pirarubicin Hydrochloride
Pirarubicin Hydrochloride 


 .
.")

 .
.






 GOLCONDA
GOLCONDA







