
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
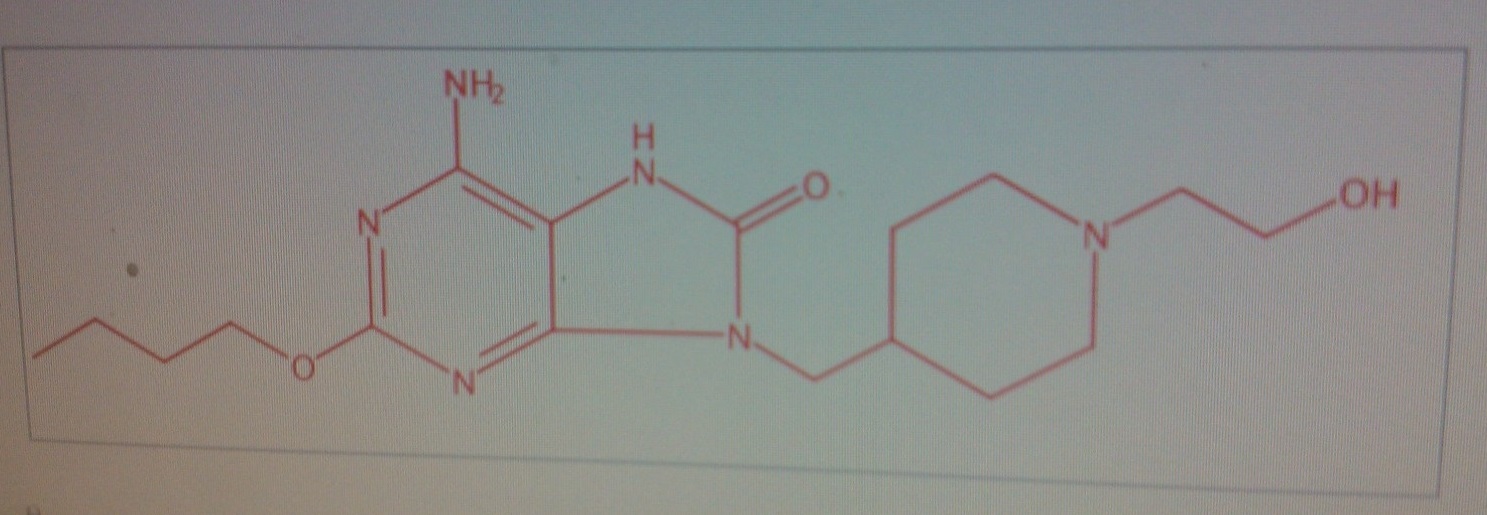
Probable GSK 2245035
 GSK 2245035 PROBABLE
GSK 2245035 PROBABLE8H-Purin-8-one, 6-amino-2-butoxy-7,9-dihydro-9-[[1-(2-hydroxyethyl)-4-piperidinyl]methyl]-
CAS NO 1264370-20-8
GSK 2245035
PHASE 2, Allergic asthma; Allergic rhinitis
Toll-like receptor 7 agonist
Immunomodulators; Interferon alfa 2a stimulants; Toll-like receptor 7 agonists
- 01 Aug 2014 GlaxoSmithKline completes a phase II trial in Allergic asthma and allergic rhinitis in Canada (NCT01788813)
- 31 Jul 2013 GlaxoSmithKline completes a phase II trial in Allergic asthma and allergic rhinitis in Canada (NCT01607372)
- 29 Mar 2013 GlaxoSmithKline initiates enrolment in a phase II trial for Allergic asthma and allergic rhinitis in Canada (NCT01788813)
Patent
https://www.google.com/patents/WO2011098451A1?cl=en
Example 2: 6-Amino-2-(butyloxy)-9-([1 -(2-hvdroxyethyl)-3-piperidinyllmethyl|-7,9-dihydro-8/-/-purin-8- one
2-(Butyloxy)-8-(methyloxy)-9-(-piperidinylmethyl)-9/-/-purin-6-amine (for example, as prepared for Intermediate 14) (33.4 mg, 0.1 mmol) was suspended in DMF (0.3 mL) was added to 2- bromoethanol (commercially available, for example, from Aldrich) (0.0071 mL, 0.100 mmol). DIPEA (0.040 mL, 0.23 mmol) was added. The reaction was shaken in a stoppered vial at ambient temperature overnight. The reaction mixture was diluted with DMSO (0.4 mL) and the resultant solution purified by MDAP (Method A). Appropriate fractions were combined and evaporated in vacuo. The residues was dissolved in 4M HCI in dioxane (0.4 mL) and allowed to stand at room temperature overnight. The solvent was dried under a stream of nitrogen in the Radleys blowdown apparatus. The residue was redissolved in methanol (0.5 mL) and applied to the top of a 0.5 g aminopropyl SPE (preconditioned with methanol, 2 CV). The cartridge was washed with methanol (2 mL). The solvent was dried under a stream of nitrogen in the Radleys blowdown apparatus to give the title compound (0.022 g).
LCMS (System A): tRET = 0.57min; MH+ 365
REF
pdf (892 KB), English, Pages 211
titis B vaccine both manufactured by GlaxoSmithKline. MPL is a nontoxic derivate … GSK2245035 compound that is a highly selective TLR7 agonist. Intranasal …
FDA approves Cholbam to treat rare bile acid synthesis disorders


March 17, 2015
Release
Today the U.S. Food and Drug Administration approved Cholbam (cholic acid) capsules, the first FDA approved treatment for pediatric and adult patients with bile acid synthesis disorders due to single enzyme defects, and for patients with peroxisomal disorders (including Zellweger spectrum disorders). Patients with these rare, genetic, metabolic conditions exhibit manifestations of liver disease, steatorrhea (presence of fat in the stool) and complications from decreased fat-soluble vitamin absorption.
Individuals with these rare disorders lack the enzymes needed to synthesize cholic acid, a primary bile acid normally produced in the liver from cholesterol. The absence of cholic acid in these patients leads to reduced bile flow, accumulation of potentially toxic bile acid intermediates in the liver (cholestasis), and malabsorption of fats and fat-soluble vitamins in the diet. If untreated, patients fail to grow and can develop life-threatening liver injury.
Cholbam is approved as an oral treatment for children aged three weeks and older, and adults. The manufacturer of Cholbam was granted a rare pediatric disease priority review voucher–a provision that encourages development of new drugs and biologics for the prevention and treatment of rare pediatric diseases.

“This approval underscores the agency’s commitment to making treatments available to patients with rare diseases,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research (CDER). “Prior to today’s approval, patients with these rare bile acid synthesis disorders had no approved treatment options.”
The efficacy of Cholbam for the treatment of patients with bile acid synthesis disorders due to single enzyme defects was assessed in an uncontrolled trial involving 50 patients treated over an 18 year period. An extension trial followed 21 of these patients and enrolled an additional 12 patients with interim efficacy data available for an additional 21 months. On average, patients were 4 years of age at the start of cholic acid treatment (range 3 weeks to 36 years). Response to treatment was evaluated by improvements in baseline liver function tests and weight. Responses were noted in 64 percent of patients with evaluable data. Two-thirds of patients survived greater than three years. Literature reports also supported the efficacy of Cholbam in this population.
The efficacy of Cholbam for the treatment of peroxisomal disorders, including Zellweger spectrum disorders, was assessed in an uncontrolled, treatment trial involving 29 patients treated over an 18 year period. An extension trial followed 10 of these patients and enrolled an additional two patients with interim efficacy data available for 21 additional months. The majority of patients were less than 2 years of age at the start of cholic acid treatment (range 3 weeks to 10 years). Response to treatment was evaluated by improvements in baseline liver function tests and weight. Responses were noted in 46 percent of patients with evaluable data. Forty-two percent of patients survived greater than 3 years.
Cholbam did not affect other manifestations of bile acid disorders due to single enzyme defects or peroxisomal disorders such as neurologic symptoms.
The most common side effect in patients treated with Cholbam was diarrhea. The use of Cholbam should be carefully monitored by an experienced hepatologist or pediatric gastroenterologist, and treatment discontinued in patients developing worsening liver function.
An observational study to assess the long-term safety of Cholbam will be required post-approval.
Cholbam is marketed by Baltimore, Maryland-based Asklepion Pharmaceuticals LLC.
GSK 2256294

GSK 2256294
GSK 2256294A
CAS 1142090-23-0
MF C21H24F3N7O
MW 447.46
Antiasthmatics, soluble epoxide hydrolase inhibitor
Chronic obstructive pulmonary disease COPD …PHASE 1
(1R,3S)- Cyclohexanecarboxamide, N-[[4-cyano-2-(trifluoromethyl)phenyl]methyl]-3-[[4-methyl-6-(methylamino)-1,3,5-triazin-2-yl]amino]-,
cis-N-[[4-Cyano-2-(trifluoromethyl)phenyl]methyl]-3-[[4-methyl-6-(methylamino)-1,3,5-triazin-2-yl]amino]cyclohexanecarboxamide
(1R,3S)-N-(4-cyano-2-(trifluoromethyl)benzyl)-3-(4-methyl-6-(methylamino)-1,3,5-triazin-2-ylamino)cyclohexanecarboxamide
cis-N-((4-Cyano-2-(trifluoromethyl)phenyl)methyl)-3-((4-methyl-6-(methylamino)-1,3,5-triazin-2-yl)amino)cyclohexanecarboxamide
Cyclohexanecarboxamide, N-((4-cyano-2-(trifluoromethyl)phenyl)methyl)-3-((4-methyl-6-(methylamino)-1,3,5-triazin-2-yl)amino)-, (1R,3S)-rel-
- Originator GlaxoSmithKline
- Class Antiasthmatics
- Mechanism of Action Epoxide hydrolase inhibitors
GSK 2256294 is a soluble epoxide hydrolase inhibitor in phase I clinical trials at GlaxoSmithKline for the oral treatment of patients with chronic obstructive pulmonary disease (COPD).
GSK2256294A is a potent, reversible, tight binding inhibitor of isolated recombinant human sEH (IC50 value 27 pM), and displays potent inhibition against the rat (IC50 = 61 pM) and murine (IC50 = 189 pM) orthologs of sEH. GSK2256294A also displays potent cellular inhibition (IC50 = 0.66 nM) of sEH in a cell line transfected with the human sEH enzyme.The selectivity of the compound has been demonstrated by testing against a large panel of enzymes, receptors and ion channels, including the phosphatase activity of EPHX2.
- 01 Jan 2015GlaxoSmithKline initiates enrolment in a phase I trial in Healthy volunteers in USA (NCT02262689)
- 09 Oct 2014GlaxoSmithKline plans a phase I trial in Healthy volunteers in USA (NCT02262689)
- 01 May 2014GlaxoSmithKline completes a phase I pharmacokinetics trial for Chronic obstructive pulmonary disease (in the elderly, in volunteers) in USA (NCT02006537)
PATENT
http://www.google.com/patents/WO2009049157A1?cl=en
Step 1:
4- (bromomethyl) -3- (trifluoromethyl) benzonitrile

A mixture of 4-methyl-3- (trifluoromethyl) benzonitrile (10g, 54mmOl) was dissolved in 200mL of carbon tetrachloride, and acid imide with N- desert shot glass (10.5g, 59mmol) and peroxybenzoate (benzoyl peroxide) (1.3g, 0.54mmol) processing. The reaction mixture was heated to reflux temperature and stirred for one week. SOmL water was then added, and the layers separated. The aqueous layer with methylene chloride (2X50mL) and extracted. The organic layers were washed with water (2X50mL), dried over magnesium sulfate, and concentrated to give 4- (bromomethyl) -3- (trifluoromethyl) benzonitrile (14g, 53mm0l), as a yellow oil which was used without further purification for the subsequent steps.
Step 2:
4- (aminomethyl) -3- (trifluoromethyl) benzonitrile

4- (bromomethyl) -3- (trifluoromethyl) benzonitrile (14g) was dissolved in 500mL of 5M methanol solution of ammonia, and the mixture was stirred at room temperature for 24 hours. The solvent was removed in vacuo to give a yellow solid, which was dissolved in IM HCl and extracted with diethyl ether (3X30mL). Then, with IM NaOH and the aqueous layer was adjusted to pH 9-10 and extracted with dichloromethane (3X80mL). Thus obtained 4- (aminomethyl) -3- (trifluoromethyl) benzonitrile (4.7g, 23mm0l, 43%), as a yellow solid. MS (ES) m / e 201 [M + H] + “1H NMR (400MHz, DMS0-D6) δ 8.2 (s, 1H), 8.15 (d, 1H), 8.0 (d, 1H), 3.9 (s, 2H).
Step 1:
4-chloro -N, 6- dimethyl-1,3,5-triazin-2-amine

Intermediate 13 (500mg, 3.07mmol) was added 25-30% methylamine (300uL, 3.07mmol) in aqueous CH3CN / H20 (15mL) in a solution. The mixture was cooled to (TC, with the pH adjusted to 9_10.pH IMNaOH maintained at 9-10 for 0.5 hours. The reaction progress was monitored by LCMS, the mixture was used in the subsequent step without any treatment.
Intermediate 19
3 – {[methyl (methyl-amino) triazin-2-yl 4 -1,3,5_ -6-] amino} cyclohexanecarboxylic acid was prepared

To 2,4-dichloro-6-methyl-1,3,5-triazine (2.291g, 13.97mmol) and methylamine (6.98ml, 13.97mmol) was added dropwise IN NaOH, to maintain the pH of 10. The reaction mixture was stirred for 30 minutes. Subsequently, a solution of 3-amino-cyclohexane – carboxylic acid (2.0g, 13.97mmol), was added dropwise to maintain a pH of 10 INNaOH. The reaction mixture was heated to 70 ° C overnight. Cooling the reaction mixture was directly purified by preparative HPLC. MS (ES +): m / e266.2 [M + H] +. 1H NMR (400MHz, DMS0-D6) δ 9.0_8.5 (bm, 2Η), 3.9 (bs, 1Η), 2.9 (m, 2Η), 2.3 (s, 3Η), 2.2 (s, 3Η), 1.9- 1.7 (bm, 4Η), 1.4-1.1 (bm, 4Η).
Step 2:
3 – {[4_-methyl-6- (methylamino) _1,3,5_ triazine _2_ yl] amino} cyclohexanecarboxylic acid

in (TC, 4-chloro -N, 6- dimethyl-1,3,5-triazin-2-amine mixture (485mg, 3.07mmol) was added 3-amino-cyclohexyl burning acid (527mg, 3.68mmol). The mixture was allowed to warm to room temperature .pH maintained between 9 to 10 for 3 hours. The mixture was concentrated and the product was purified by HPLC to afford 0.6g (2.26mmol, 74% yield) of the desired product, as a white solid .MS (ES +): m / e 266.2 [M + H] + “
Example 74
(cis)-N-{[4-cyano-2-(trifluoromethyl)phenyl]methyl}-3-{[4-methyl-6-(methylamino)-1 ,3,5- triazin-2-yl]amino}cyclohexanecarboxamide
To a solution of 3-{[4-methyl-6-(methylamino)-1 ,3,5-triazin-2- yl]amino}cyclohexanecarboxylic acid (0.100 g, 0.264 mmol) in N,N-Dimethylformamide (DMF) (4 ml) was added 4-(aminomethyl)-3-(trifluoromethyl)benzonitrile (0.053 g, 0.264 mmol) followed by diisopropylethylamine (0.101 ml, 0.580 mmol) and 1 H-1 ,2,3- benzotriazol-1-yloxy-tris(dirnethylamino)-phosphonium hexafluorophosphate (BOP reagent, 0.128 g, 0.290 mmol). The reaction was stirred at room temperature for 4 hours and then purified by preparative HPLC to provide (cis)-N-{[4-cyano-2- (trifluoromethyl)phenyl]methyl}-3-{[4-methyl-6-(methylamino)-1 ,3,5-triazin-2- yl]amino}cyclohexanecarboxamide (83 mg, 0.148 mmol, 56 %). MS (ES) m/e 448
[M+H]+. 1H NMR (400 MHz, DMSO-D6) D 7.8 (bs, 1 H), 7.3 (bs, 1 H), 7.2 (m, 1 H), 6.9 (m, 1 H), 3.8 (bs, 2H), 3.3 (bm, 1 H), 2.2 (bm, 4H), 1.8 – 1.5 (bm, 4H), 1.3 – 1.1 (bm, 4H), 0.8 – 0.5 (bm, 4H)
PATENT
http://www.google.com/patents/CN101896065B?cl=en
(cis) -N – {[4- cyano-2- (trifluoromethyl) phenyl] methyl} -3 – {[4_-methyl-6- (methylamino) _1,3, .5- triazin-2-yl] amino} cyclohexanecarboxamide

Example 74
(cis) -N- {[4- cyano-2- (trifluoromethyl) phenyl] methyl} -3- {[4_ methyl _6_ (methylamino) -1,3 , 5-triazin-2-yl] amino} cyclohexanecarboxamide

To 3 – {[4_-methyl-6- (methylamino) -l, 3,5- triazin-2-yl] amino} cyclohexanecarboxylic acid (0.1OOg,
0.264mmol) in N, N- dimethylformamide (DMF) (4ml) was added 4- (aminomethyl) -3- (trifluoromethyl) benzonitrile (0.053g, 0.264mmol), followed by the addition of diisopropylethylamine (0.1Olml, 0.580mmol) and 1H-1,2,
3- benzotriazol-1-yloxy – tris (dimethylamino) _ scale hexafluorophosphate (Β0Ρ reagent, 0.128g, 0.290mmol). The reaction mixture was stirred at room temperature for 4 hours, and then purified by preparative HPLC to afford (cis) -N- {[4- cyano-2- (trifluoromethyl) phenyl] methyl} -3 – {[4_ methyl-6- (methylamino) -1,3,5_ triazin-2-yl] amino} cyclohexane carboxamide (83mg, 0.148mmol, 56%) “MS (ES) m / e 448 [ M + H] +. 1H NMR (400MHz, DMS0-D6) δ 7.8 (bs, 1H), 7.3 (bs, 1H), 7.2 (m, 1H), 6.9 (m, 1H), 3.8 (bs, 2H) , 3.3 (bm, 1H), 2.2 (bm, 4H),
1.8-1.5 (bm, 4H), 1.3-1.1 (bm, 4H), 0.8-0.5 (bm, 4H).
SMILES Cc1nc(nc(n1)N[C@H]2CCC[C@H](C2)C(=O)NCc3ccc(cc3C(F)(F)F)C#N)NC
GSK 2126458, Omipalisib, PI3K/mTOR inhibitor

GSK 2126458
CAS 1086062-66-9
OMipalisib;GSK2126458;GSK-2126458;GSK2126458 (GSK458);GSK212;
2,4-Difluoro-N-[2-methoxy-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl]benzenesulfonamide;
2,4-Difluoro-N-[2-Methoxy-5-[4-(pyridazin-4-yl)quinolin-6-yl]pyridin-3-yl]benzenesulfonaMide
2,4-Difluoro-N-[2-methoxy-5-[4-(4-pyridazinyl)quinolin-6-yl]pyridin-3-yl]benzenesulfonamide
phosphoinositide 3 kinase inhibitor
idiopathic pulmonary fibrosis
PHASE 1
MW 505.49598
MF C25H17F2N5O3S
GSK…….http://www.gsk.com/media/280387/product-pipeline-2014.pdf
![]()
Omipalisib (GSK2126458): Omipalisib, also known as GSK2126458, is a small-molecule pyridylsulfonamide inhibitor of phosphatidylinositol 3-kinase (PI3K) with potential antineoplastic activity. PI3K inhibitor GSK2126458 binds to and inhibits PI3K in the PI3K/mTOR signaling pathway, which may trigger the translocation of cytosolic Bax to the mitochondrial outer membrane, increasing mitochondrial membrane permeability and inducing apoptotic cell death. Bax is a member of the proapoptotic Bcl2 family of proteins. PI3K, often overexpressed in cancer cells, plays a crucial role in tumor cell regulation and survival.
GlaxoSmithKline (GSK) is developing omipalisib (GSK-2126458), a phosphoinositide 3-kinase/mammalian target of rapamycin (PI3K/mTOR) inhibitor as well as mTOR complex 1 and 2 inhibitor, for the potential oral treatment of cancer and idiopathic pulmonary fibrosis
MEDKOO

![]()
![]()
![]()
![]()
|
Certificate of Analysis: |
|
|
QC data: |
GSK2126458 is a highly potent PI3K and mTOR inhibitor. In vivo, GSK2126458 showed anti-tumor activity in both pharmacodynamic and tumor growth efficacy models. GSK2126458 reduced the phosphorylated AKT, p70S6K contents in a dose and time dependent way. The IC50 of GSK2126458 is 2 nM for pAKT in the HCC1954 breast carcinoma cell line. In various human tumor cells, GSK2126458 had a width of inhibitory activity for potent cell growth and induced cell death. Notably, GSK2126458 acted mainly by not induction of apoptosis but cell cycle arrest, particularly in G1-phase
GlaxoSmithKline (GSK) is developing omipalisib (GSK-2126458), a phosphoinositide 3-kinase/mammalian target of rapamycin (PI3K/mTOR) inhibitor as well as mTOR complex 1 and 2 inhibitor, for the potential oral treatment of cancer and idiopathic pulmonary fibrosis
GSK-2126458 is a phosphatidylinositol 3-Kinase (PI3K) inhibitor in early clinical development for the oral treatment of solid tumors and for the oral treatment of lymphoma. Early clinical studies are ongoing for the treatment of idiopathic pulmonary fibrosis. The compound is being developed b GlaxoSmithKline.
In August 2009, a phase I trial began for solid tumors and lymphoma . In April 2012, phase Ib co-clinical trials in advanced prostate cancer (PC) were underway . In March 2013, a phase I trial was initiated in the UK in patients with idiopathic pulmonary fibrosis
In April 2014, a phase I, open-label, multicenter, dose-escalation study (study number P3K113794) and safety data were presented at the 105th AACR meeting in San Diego, CA. Advanced solid tumor patients (n = 69) received oral continuous GSK-2126458 or intermittent GSK-2126458 bid + trametinib. For GSK-2126458 and trametinib, the MTD in QD cohort was 2 and 1 mg, respectively, and also 1 and 1.5 mg, respectively
PAPER ![]()
![]()
![]()
![]()
![]()
![]()
![]()
Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rampamycin
ACS Med Chem Lett 2010, 1(1): 39

Phosphoinositide 3-kinase α (PI3Kα) is a critical regulator of cell growth and transformation, and its signaling pathway is the most commonly mutated pathway in human cancers. The mammalian target of rapamycin (mTOR), a class IV PI3K protein kinase, is also a central regulator of cell growth, and mTOR inhibitors are believed to augment the antiproliferative efficacy of PI3K/AKT pathway inhibition. 2,4-Difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl}benzenesulfonamide (GSK2126458, 1) has been identified as a highly potent, orally bioavailable inhibitor of PI3Kα and mTOR with in vivo activity in both pharmacodynamic and tumor growth efficacy models. Compound 1 is currently being evaluated in human clinical trials for the treatment of cancer.

synthesis![]()
![]()
![]()
![]()
![]()


………………..
PATENT
WO 2008144463
http://www.google.co.in/patents/WO2008144463A1?cl=en
Example 345
2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3- pyridinyl } benzenesulf onamide
a) 6-bromo-4-(4-pyridazinyl)quinoline
Dissolved 6-bromo-4-iodoquinoline (17.43 g, 52.2 mmol), 4- (tributylstannanyl)pyridazine (19.27 g, 52.2 mmol), and PdC12(dppf)-CH2C12 (2.132 g, 2.61 mmol) in 1,4-dioxane (200 mL) and heated to 105 °C. After 3 h, added more palladium catalyst and heated for 6 h. Concentrated and dissolved in methylene chloride/methanol. Purified by column chromatography (combiflash) with 2% MeOH/EtOAc to 5% MeOH/EtOAc to give the crude title compound. Trituration with EtOAc furnished 6-bromo-4-(4-pyridazinyl)quinoline (5.8 g, 20.27 mmol, 38.8 % yield). MS(ES)+ m/e 285.9, 287.9 [M+H]+.
b) 2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3- pyridinyl } benzenesulf onamide A slurry of 6-bromo-4-(4-pyridazinyl)quinoline (4.8 g, 16.78 mmol), bis(pinacolato)diboron (4.69 g, 18.45 mmol) , PdC12(dppf)-CH2C12 (530 mg, 0.649 mmol) and potassium acetate (3.29 g, 33.6 mmol) in anhydrous 1,4-dioxane (120 ml) was heated at 100 °C for 3 h. The complete disappearance of the starting bromide was observed by LCMS. The reaction was then treated with N-[5-bromo-2- (methyloxy)-3-pyridinyl]-2,4-difluorobenzenesulfonamide (6.68 g, 17.61 mmol) and another portion of PdC12(dppf)-CH2C12 (550 mg, 0.673 mmol), then heated at 110 °C for 16 h. The reaction was allowed to cool to room temperature, filtered, and concentrated. Purification of the residue by chromatography (Analogix; 5% MeOH / 5% CH2C12 / 90% EtOAC) gave 6.5 g (76%) desired product. MS(ES)+ m/e 505.9 [M+H]+.
INTERMEDIATES:
Intermediate 1 Similar but not same
Scheme A:

Conditions: a) Tributyl(vinyl)tin, Pd(PPh3)4, dioxane, reflux; b) OsO4, NaIO4, 2,6- lutidine, r-BuOH, dioxane, H2O, rt; c) (4-pyridyl)boronic acid, Pd(PPh3)4, 2 M K2CO35 DMF, 100 DC.
4-(4-pyridinyl)-6-quinolinecarbaldehydeSimilar but not same

a) 4-chloro-6-ethenylquinoline
A mixture of 6-bromo-4-chloroquinoline (6.52 g, 26.88 mmol; see J. Med. Chem., H 268 (1978) ), tributyl(vinyl)tin (8.95 g, 28.22 mmol), and tetrakistriphenylphospbine palladium (0) (0.62 g, 0.54 mmol) in 1,4-dioxane (150 mL) was refluxed for 2.0 h, cooled to room temperature, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (0-4% MeOH:CH2Cl2) to give the title compound (5.1 g) as a pale yellow solid. MS (ES)+ m/e 190 [M+H]+. This material was used directly in the next step.
b) 4-chloro-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-ethenylquinoline (5.1 g, 26.88 mmol), 2,6-lutidine
(5.76 g, 53.75 mmol), sodium (meta) periodate (22.99 g, 107.51 mmol), and osmium tetroxide (5.48 g of a 2.5% solution in tert-butanol, 0.538 mmol) in l,4-dioxane:H2θ (350 mL of 3: 1 mixture) was stirred for 3.5 h at room temperature and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2Cb) to give the title compound (4.26 g, 83% for 2 steps) as a pale yellow solid. MS (ES)+ m/e 192 [M+H]+.
c) 4-(4-pyridmyl)-6-qumolinecarbaldehyde
A mixture of 4-chloro-6-quinolinecarbaldehyde (3.24 g, 16.92 mmol), A- pyridylboronic acid (3.12 g, 25.38 mmol), tetrakistriphenylphosphine palladium (0) (0.978 g, 0.846 mmol), and 2M aqueous K2CO3 (7.02 g, 50.76 mmol, 25.4 mis of 2M solution) in DMF (100 mL) was heated at 100 °C for 3.0 h and cooled to room temperature. The mixture was filtered through Celite and the Celite was washed with EtOAc. The filtrate was transferred to a separatory funnel, washed with water and saturated NaCl, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (5% MeOH:CH2Cl2) to give the title compound (2.03 g, 51%) as a tan solid. MS (ES)+ m/e 235 [M+H]+.
Intermediate 2
Preparation of 2-amino-5 -bromo-N,N-dimethyl-3 -pyridinesulfonamideSimilar but not same

a) 2-ammo-5-bromo-3-pyridinesulfonyl chloride
To a cooled (0 °C) solution of chlorosulfonic acid (58 mL) under vigorous stirring was added 5-bromo-2-pyridinamine (86.7 mmol) portionwise. The reaction mixture was then heated at reflux for 3 hrs. Upon cooling to room temperature, the reaction mixture was poured over ice (-100 g) with vigorous stirring. The resulting yellow precipitate was collected by suction filtration, washing with cold water and petroleum ether to provide the title compound as an orange-yellow solid (18.1 g, 77% yield). MS(ES)+ m/e 272.8 [M+H]+.
* Other sulfonyl chlorides can be prepared using this procedure by varying the choice of substituted aryl or heteroaryl.
b) 2-amino-5-bromo-N,N-dimethyl-3-pyridinesulfonamide
To a cold (0 DC) suspension of 2-amino-5-bromo-3-pyridinesulfonyl chloride (92.1 mmol) in dry 1,4-dioxane (92 mL) was added pyridine (101.3 mmol) followed by a 2M solution of dimethylamine in THF (101.3 mmol). The reaction was allowed to warm to rt for 2 h, heated to 50 DC for 1 h, then cooled to rt. After standing for 2 h, the precipitate was collected by filtration and rinsed with a minimal amount of cold water. Drying the precipitate to constant weight under high vacuum provided 14.1 g (55%) of the title compound as a white solid. MS(ES)+ m/e 279.8, 282.0 [M+H]+.
Intermediate 3
Preparation of 2-amino-N,N-dimethyl-5-(4,4,5,5-tetramethyl-l,3.2-dioxaborolan-2- yl)-3 -pyridinesulfonamideSimilar but not same

c) To a solution of 2-amino-5-bromo-N,N-dimethyl-3 -pyridinesulfonamide (7.14 mmol) in 1,4-dioxane (35 mL) was added 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi-l,3,2- dioxaborolane (7.86 mmol), potassium acetate (28.56 mmol) and [1,1 ‘- bis(diphenylphosphmo)-ferrocene] dichloropalladium(II) dichloromethane complex (1 :1) (0.571 mmol). The reaction mixture was stirred at 100 °C for 18 h. The reaction was concentrated in vacuo, re-dissolved in ethyl acetate (50 mL) and purified on silica using 60% ethyl acetate/hexanes to yield the title compound as a tan solid (86 %). IH ΝMR (400 MHz, DMSOd6) δ ppm 8.41 (d, 1 H, J =1.52), 7.92 (d, 1 H, J = 1.77), 2.68 (s, 6 H), 1.28 (s, 12 H).
* Other boronate or boronic acids can be prepared using this procedure by varying the choice of aryl or heteroaryl bromide. Scheme 17:

Conditions: a) NaO(Rl), (Rl)OH, O 0C to room temperature; b) SnCl2-2H2O, ethyl acetate, reflux; c) (R2)SO2C1, pyridine, O 0C to room temperature.
Intermediate 4
Preparation of N-r5-bromo-2-(methyloxy)-3-pyridinyll-2,4- difluorobenzenesulfonamide
N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4- difluorobenzenesulfonamide a) 5-bromo-2-(methyloxy)-3-nitropyridine

To a cooled (0 °C) solution of 5-bromo-2-chloro-3-nitropyridine (50 g, 211 mmol) in methanol (200 mL) was added dropwise over 10 minutes 20% sodium methoxide (50 mL, 211 mmol) solution. The reaction, which quickly became heterogeneous, was allowed to warm to ambient temperature and stirred for 16 h. The reaction was filtered and the precipitate diluted with water (200 mL) and stirred for 1 h. The solids were filtered, washed with water (3 x 100 mL) and dried in a vac oven (40 °C) to give 5-bromo-2-(methyloxy)-3-nitropyridine (36 g, 154 mmol, 73.4 % yield) as a pale yellow powder. The original filtrate was concentrated in vacuo and diluted with water (150 mL). Saturated ammonium chloride (25 mL) was added and the mixture stirred for 1 h. The solids were filtered, washed with water, and dried in a vac oven (40 °C) to give a second crop of 5-bromo-2-(methyloxy)-3- nitropyridine (9 g, 38.6 mmol, 18.34 % yield). Total yield = 90%. MS(ES)+ m/e 232.8, 234.7 [M+H]+.
b) 5-bromo-2-(methyloxy)-3-pyridinamine

To a solution of 5-bromo-2-(methyloxy)-3-nitropyridine (45 g, 193 mmol) in ethyl acetate (1 L) was added tin(II) chloride dihydrate (174 g, 772 mmol). The reaction mixture was heated at reflux for 4 h. LC/MS indicated some starting material remained, so added 20 mol% tin (II) chloride dihydrate and continued to heat at reflux. After 2 h, the reaction was allowed to cool to ambient temperature and concentrated in vacuo. The residue was treated with 2 N sodium hydroxide and the mixture stirred for 1 h. The mixture was then with methylene chloride (1 L), filtered through Celite, and washed with methylene chloride (500 mL). The layers were separated and the organics dried over magnesium sulfate and concentrated to give 5-bromo-2-(methyloxy)-3-pyridinamine (23 g, 113 mmol, 58.7 % yield). The product was used crude in subsequent reactions. MS(ES)+ m/e 201.9, 203.9 [M+H]+.
c) N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4-difluorobenzenesulfonamide
To a cooled (0 °C) solution of 5-bromo-2-(methyloxy)-3-pyridinamine (20.3 g, 100 mmol) in pyridine (200 mL) was added slowly 2,4-difluorobenzenesulfonyl chloride (21.3 g, 100 mmol) over 15 min (reaction became heterogeneous). The ice bath was removed and the reaction was stirred at ambient temperature for 16 h, at which time the reaction was diluted with water (500 mL) and the solids filtered off and washed with copious amounts of water. The precipitate was dried in a vacuum oven at 50 °C to give N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4- difluorobenzenesulfonamide (12 g, 31.6 mmol, 31.7 % yield) MS(ES)+ m/e 379.0, 380.9 [M+H]+.
References
|
1: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
2: Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, Gilmer TM, Martin AM, Marmorstein R, Schultz DC, Speicher DW, Karakousis GC, Xu W, Amaravadi RK, Xu X, Schuchter LM, Herlyn M, Nathanson KL. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013 Sep 26;4(6):1090-9. doi: 10.1016/j.celrep.2013.08.023. Epub 2013 Sep 19. PubMed PMID: 24055054; PubMed Central PMCID: PMC3956616.
3: Kim HG, Tan L, Weisberg EL, Liu F, Canning P, Choi HG, Ezell SA, Wu H, Zhao Z, Wang J, Mandinova A, Griffin JD, Bullock AN, Liu Q, Lee SW, Gray NS. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem Biol. 2013 Oct 18;8(10):2145-50. doi: 10.1021/cb400430t. Epub 2013 Aug 13. PubMed PMID: 23899692; PubMed Central PMCID: PMC3800496.
4: Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, Esmaeli B, Woodman SE. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. doi: 10.1158/1078-0432.CCR-11-3227. Epub 2012 Jun 25. PubMed PMID: 22733540; PubMed Central PMCID: PMC3935730.
5: Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012 Apr;11(4):909-20. doi: 10.1158/1535-7163.MCT-11-0989. Epub 2012 Mar 2. PubMed PMID: 22389471.
6: Wang M, Gao M, Miller KD, Sledge GW, Zheng QH. [11C]GSK2126458 and [18F]GSK2126458, the first radiosynthesis of new potential PET agents for imaging of PI3K and mTOR in cancers. Bioorg Med Chem Lett. 2012 Feb 15;22(4):1569-74. doi: 10.1016/j.bmcl.2011.12.136. Epub 2012 Jan 10. PubMed PMID: 22297110.
7: Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18(20):2995-3014. Review. PubMed PMID: 21651476.
8: Leung E, Kim JE, Rewcastle GW, Finlay GJ, Baguley BC. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther. 2011 Jun 1;11(11):938-46. Epub 2011 Jun 1. PubMed PMID: 21464613; PubMed Central PMCID: PMC3127046.
GSK 2269557 In Phase 1….Asthma , COPD, is it COMPD A OR B?
 COMPD A
COMPD A
Compd A OR B IS GSK 2269557
DATA FOR COMPD A
6-(1H-indol-4-yl)-4-[5-[[4-(1-methylethyl)-1-piperazinyl]methyl]-2-oxazolyl]-1H-Indazole,
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole
EMAIL ME amcrasto@gmail.com
Phosphoinositide 3ΌΗ kinases (hereinafter PI3Ks) are a family of signal transducer enzymes which are involved in various cellular functions including cell growth, proliferation and differentiation. A wide variety of retroviruses and DNA-based viruses activate the PI3K pathway as a way of preventing host cell death during viral infection and ultimately exploiting the host cell synthesis machinery for its replication (Virology 344(1) p. 131-8 (2006) by Vogt et al.; and Nat. Rev. Microbiol. 6(4) p. 265-75 (2008) by Buchkovich et al). It has therefore been postulated that PI3K inhibitors may have potential therapeutic benefit in the treatment of viral infections such as influenza virus infection, in addition to the more established treatment of cancer and inflammatory diseases.
The Influenza NS1 protein activates Class la PI3Ks by binding to their regulatory subunit p85beta but not to other Class la regulatory subunits such as p85alpha. The recent crystal structure of the NS1-p85beta complex (Hale et al. Proc. Natl. Acad. Sci. U S A. 107(5) p.1954-1959 (2010)) is also suggestive of an interaction with the p110 kinase subunit providing a mechanism for catalytic activation of the kinase domain. This observation provides a rationale for isoform specificity not only with the p85 regulatory subunit but also potentially with the p110 catalytic subunit too. The function of PI3K during influenza virus infection has also been investigated by, for example, Ehrhardt et al. (Cell. Microbiol. 8(8) p. 1336-1348 (2006)), and the role of PI3K5 signalling in morbidity and lung pathology induced by influenza virus infection has been reported in WO 2010/083163.
There remains a need to provide compounds which are inhibitors of the activity or function of PI3K5 which may be useful in the treatment or prevention of influenza virus infection.
GSK 2269557 is an inhaled phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor in early clinical trials at GlaxoSmithKline for the treatment of patients with asthma and also for the treatment of chronic obstructive pulmonary disease (COPD) in patients who smoke cigarettes.
- 18 Nov 2014GlaxoSmithKline plans a phase II trial in Chronic obstructive pulmonary disease in Belgium, Denmark, the Netherlands and Russia (NCT02294734)
- 01 Jun 2014Phase-II clinical trials in Chronic obstructive pulmonary disease in Germany (Inhalation)
- 01 May 2014GlaxoSmithKline plans a phase II trial for Chronic obstructive pulmonary disease in Germany (NCT02130635)
EMAIL ME amcrasto@gmail.com
CLICK ON IMAGES TO VIEW SIMILAR ROUTES FOR COMPD A AND B
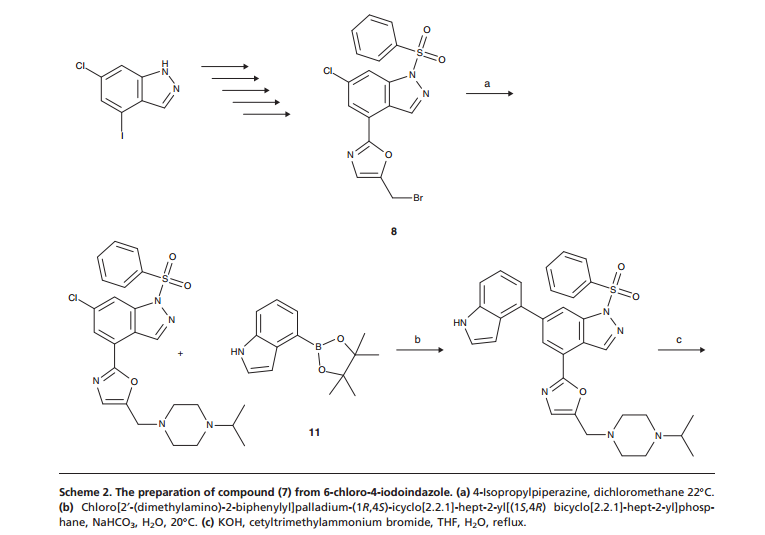
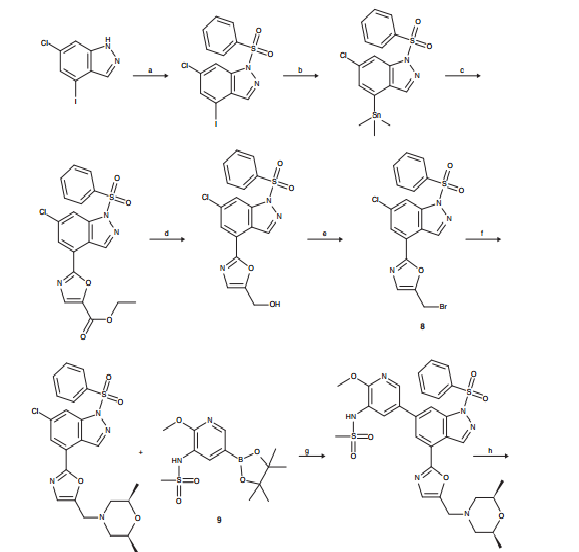
CLICK ON IMAGE TO VIEW
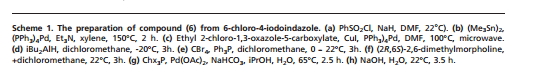
…………………………………………………………………….
COMPD A
WO 2012032065
http://www.google.com/patents/WO2012032065A1?cl=en
Example 68
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole

Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1/-/-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 100°C for 30 min. Additional 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 10°C for 30 min, then 140°C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 : 1 , v/v) and purified by MDAP (method H). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml_) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 120°C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 45°C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 : 1). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 : 1). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane: DCM (1 : 1) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 50°C overnight. This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 : 1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441.
1 H NMR (400MHz ,DMSO-d6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
Method C
Potassium hydroxide (145.6 g) was added to a suspension of 6-(1 H-indol-4-yl)-4-(5-{[4-(1- methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (300.7 g) and cetyltrimethylammonium bromide (9.3 g) in tetrahydrofuran (6.0 L) and water (30 ml) stirring under nitrogen at ambient temperature. The mixture was heated at reflux for 17 hours and was then cooled to 20-25°C. Ethyl acetate (3.0 L) and water (3.0 L) were added, stirred for 10 minutes and then separated. The organic layer was extracted with hydrochloric acid (1 M, 1 x 3.0 L, 2 x 1.5L) and the acidic extracts combined and basified to ~pH 8 by the addition of saturated sodium carbonate solution (2.1 L). After ageing for 30 minutes the resultant suspension was filtered, washed with water (300 ml) and the solid dried under vacuum at 65°C to give the title compound as a pale yellow solid (127.9 g).
LCMS (Method B): Rt 2.44 min, MH+ 441.
…………………………………………………………………………
WO 2010125082
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 6
6-(1 H-lndol-4-yl)-4-(5-{[4-(1 -methylethyl)-1 -piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1 H- indazole

Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1H-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 1000C for 30 min. Additional 4-(4, 4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 1O0C for 30 min, then 14O0C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and purified by MDAP (method A). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml.) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 1200C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 450C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 :1 ). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 :1 ). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane:DCM (1 :1 ) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 500C overnight.
This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 :1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441. 1H NMR (400MHz ,DMSOd6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
EMAIL ME amcrasto@gmail.com
COMPD B
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 1
Λ/-[5-[4-(5-{[(2/?,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-
6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide

Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1 ,3-oxazol-2- yl)-1-(phenylsulfonyl)-1 H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1 ,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium- 1 (1 /?,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 12O0C under microwave irradiation for 1 h. Additional chloroP’^dimethylamino^-biphenylyOpalladium-^I R^S^bicycloP^.ilhept^- yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 12O0C under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2x 2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3ml, 1 :1 , v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2x 25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 4O0C for 3 h to give the title compound as a white solid (26 mg). LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4- morpholinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1 ,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 800C. Chloro[2′-(dimethylamino)-2- biphenylyl]palladium-1 (1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)-bicyclo[2.2.1]hept-2- yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 800C.
The reaction mixture was cooled to 450C, sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45 0C for 4 hours. The mixture was cooled to RT and diluted with water (610 ml_). Dichloromethane (920 ml.) was added, and the mixture was filtered twice through Celite (washed with 200 ml. 1 ,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1 ,4-dioxane/DCM 2:1 (500 ml_). The aqueous phase was neutralised with hydrochloric acid to pH -7 and extracted with 1 ,4- dioxane/DCM 2:1 (1 L), then 1 ,4 dioxane/DCM 1 :1 (2×500 ml_). The organics were washed with brine (500 ml_), and filtered through Celite (washed with 200 ml. 1 ,4 dioxane/DCM 2:1 ), and evaporated to yield a dark black solid, which was purified in 4 batches:
Batch 1 : 28g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
Batch 2: 3Og was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
Batch 4: 29g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1 100 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 600C for 5hrs to give the title compound as an off-white solid (45.51 g). LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield -23 g of a solid residue that was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 ml_). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65°C overnight to give the title compound as an off-white solid (11.9O g). LCMS (Method A): Rt 0.62 mins, MH+ 513.
………………………………………..
http://www.google.co.in/patents/US8735390
Example 1N-[5-[4-(5-{[(2R,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide

Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 120° C. under microwave irradiation for 1 h. Additional chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 120° C. under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2×2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3 ml, 1:1, v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1:1, v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2×25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 40° C. for 3 h to give the title compound as a white solid (26 mg).
LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B
N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 80° C. Chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 80° C.
The reaction mixture was cooled to 45° C., sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45° C. for 4 hours. The mixture was cooled to RT and diluted with water (610 mL). Dichloromethane (920 mL) was added, and the mixture was filtered twice through Celite (washed with 200 mL 1,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1,4-dioxane/DCM 2:1 (500 mL). The aqueous phase was neutralised with hydrochloric acid to pH ˜7 and extracted with 1,4-dioxane/DCM 2:1 (1 L), then 1,4 dioxane/DCM 1:1 (2×500 mL). The organics were washed with brine (500 mL), and filtered through Celite (washed with 200 mL 1,4 dioxane/DCM 2:1), and evaporated to yield a dark black solid, which was purified in 4 batches:
- Batch 1: 28 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
- Batch 2: 30 g was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
- Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
- Batch 4: 29 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1100 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 60° C. for 5 hrs to give the title compound as an off-white solid (45.51 g).
LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield ˜23 g of a solid residue that was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 mL). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65° C. overnight to give the title compound as an off-white solid (11.90 g).
LCMS (Method A): Rt 0.62 mins, MH+ 513.
Method C
10M Sodium hydroxide solution (0.70 ml) was added to a stirred suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (1.17 g) in water (5.8 ml). The resulting mixture was stirred at room temperature for 3.75 hours and was then washed with ethyl acetate (2×6 ml). The layers were separated and the aqueous phase was acidified to pH 6 with 2M hydrochloric acid (0.8 ml). The acidified aqueous layer was extracted twice with ethyl acetate (11 ml then 5 ml). The combined ethyl acetate extracts were dried by azeotropic distillation and diluted with further ethyl acetate (11 ml). The misture was stirred at room temperature for 112 hours. The slurry was seeded and then stirred at room temperature for 48 hours. The resultant suspension was filtered, washed with ethyl acetate (2×2 ml) and the solid dried under vacuum at 40° C. to give the title compound as a pale yellow solid (0.58 g).
LCMS (Method B): Rt 1.86 min, MH+ 513.
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
Preparation of Polymorphs of Compound A
Form (II)
Ethyl acetate (15 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (2.1 g) and was stirred at ambient conditions overnight. The resultant slurry was filtered and dried under vacuum at 50° C. to give a new solid state form (91 ckw/w).
1H NMR (400 MHz, DMSO d6) d=13.49 (br s, 1H), 9.39 (s, 1H), 8.58 (s, 1H), 8.42 (d, J=2.2 Hz, 1H), 7.99 (d, J=2.2 Hz, 1H), 7.93 (d, J=1.2 Hz, 1H), 7.88 (s, 1H), 7.35 (s, 1H), 4.00 (s, 3H), 3.74 (s, 2H), 3.58 (m, 2H), 3.11 (s, 3H), 2.80 (d, J=10.3 Hz, 2H), 1.78 (t, J=10.3 Hz, 2H), 1.05 (d, J=6.4 Hz, 6H)
SODIUM SALT OF COMPD B
http://www.google.com/patents/US20140256721
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
http://www.google.com/patents/US20140256721
Preparation of Salts of Compound ASodium Salt
Methanol (2 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (0.3 g) followed by aqueous sodium hydroxide (0.129 ml) to give a solution. Tert-butylmethylether (4 ml) was added to the solution followed by seed crystals of the sodium salt and this suspension was stirred overnight at ambient conditions. The suspension was filtered, washed with tert-butylmethylether (2 ml) and air dried to give the sodium salt (0.2312 g) as a hydrate.
NMR: Consistent with salt formation
1H NMR (400 MHz, DMSO d6) d=13.35 (br s, 1H), 8.53 (s, 1H), 7.90 (d, J=1.2 Hz, 1H), 7.73 (s, 1H), 7.65 (d, J=2.5 Hz, 1H), 7.62 (d, J=2.2 Hz, 1H), 7.33 (s, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.59 (m, 2H). 2.83 (d, J=10.3, 2H), 2.61 (s, 3H), 1.78 (t, J=10.5 Hz, 2H), 1.05 (d, J=6.1 Hz, 6H)
EMAIL ME amcrasto@gmail.com
EMAIL ME amcrasto@gmail.com
| US20100280029 * | 28 Apr 2010 | 4 Nov 2010 | Julie Nicole Hamblin | Novel compounds |
| WO2010125082A1 | 28 Apr 2010 | 4 Nov 2010 | Glaxo Group Limited | Oxazole substituted indazoles as pi3-kinase inhibitors |
| US20140256721 * | 14 Apr 2014 | 11 Sep 2014 | Glaxosmithkline Intellectual Property Development Limited | Novel Polymorphs and Salts |
| WO2012032065A1 | 6 Sep 2011 | 15 Mar 2012 | Glaxo Group Limited | Indazole derivatives for use in the treatment of influenza virus infection |
| WO2012032067A1 | 6 Sep 2011 | 15 Mar 2012 | Glaxo Group Limited | Polymorphs and salts of n- [5- [4- (5- { [(2r,6s) -2, 6 – dimethyl – 4 -morpholinyl] methyl} – 1, 3 – oxazol – 2 – yl) – 1h- inda zol-6-yl] -2- (methyloxy) – 3 – pyridinyl] methanesulfonamide |
| WO2012055846A1 | 25 Oct 2011 | 3 May 2012 | Glaxo Group Limited | Polymorphs and salts of 6-(1h-indol-4-yl)-4-(5- { [4-(1-methylethyl)-1-pi perazinyl] methyl} -1,3-oxazol-2-yl)-1h-indazole as pi3k inhibitors for use in the treatment of e.g. respiratory disorders |
| WO2012064744A2 * | 8 Nov 2011 | 18 May 2012 | Lycera Corporation | Tetrahydroquinoline and related bicyclic compounds for inhibition of rorϒ activity and the treatment of disease |
| WO2013088404A1 | 14 Dec 2012 | 20 Jun 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2014068070A1 | 31 Oct 2013 | 8 May 2014 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for preventing antiphospholipid syndrome (aps) |
| US8524751 | 5 Mar 2010 | 3 Sep 2013 | GlaxoSmithKline Intellecutual Property Development | 4-oxadiazol-2-YL-indazoles as inhibitors of P13 kinases |
| US8536169 | 3 Jun 2009 | 17 Sep 2013 | Glaxo Group Limited | Compounds |
| US8575162 | 28 Apr 2010 | 5 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8580797 | 28 Apr 2010 | 12 Nov 2013 | Glaxo Smith Kline Intellectual Property Development Limited | Compounds |
| US8586583 | 2 Oct 2012 | 19 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8586590 | 2 Oct 2012 | 19 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8609657 | 2 Oct 2012 | 17 Dec 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8658635 | 3 Jun 2009 | 25 Feb 2014 | Glaxosmithkline Intellectual Property Development Limited | Benzpyrazol derivatives as inhibitors of PI3 kinases |
| US8735390 | 6 Sep 2011 | 27 May 2014 | Glaxosmithkline Intellectual Property Development Limited | Polymorphs and salts |
| US8765743 | 3 Jun 2009 | 1 Jul 2014 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
…..

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA

.




 .
.



.
 MANUDEVI
MANUDEVI

Suven Life gets patent for neuro-degenerative drug
March 16, 2015
Drug firm Suven Life Sciences has been granted a patent each by the US and New Zealand for a drug used in the treatment of neuro-degenerative diseases.
The patents are valid until 2030 and 2031, respectively, Suven Life Sciences said in a filing to the BSE.
Commenting on the development, Suven Life CEO Venkat Jasti said: “We are very pleased by the grant of these patents to Suven for our pipeline of molecules in CNS arena that are being developed for cognitive disorders with high unmet medical need with huge market potential globally.”
SUVEN, Chief executive and chairman Venkat Jasti

The company has “secured patents in USA and New Zealand to one of their new chemical entity (NCE) for CNS therapy through new mechanism of action – H3 Inverse agonist…,” Suven Life Sciences said.
With these new patents, Suven has a total of 20 granted patents from US and 23 granted patents from New Zealand.
“These granted patents are exclusive intellectual property of Suven and are achieved through the internal discovery research efforts.
“Products out of these inventions may be out-licensed at various phases of clinical development like at Phase-I or Phase-II,” Suven said.
http://www.bseindia.com/xml-data/corpfiling/AttachLive/suven_life_sciences_ltd_160315.pdf

Suven Life Sciences secures 2 (two) Product Patents for their NCE’s through New mechanism of action – H3 Inverse Agonist in USA & New Zealand HYDERABAD, INDIA (March 16, 2015) – Suven Life Sciences Ltd (Suven) announced today that they secured patents in USA (us 8912179) and New Zealand (614567) to one of their New Chemical Entity (NCE) for CNS therapy through new mechanism of action – H3 Inverse agonist and these patents are valid until 2030 and 2031 respectively. The granted claims of the patent include the class of selective H3 ligands discovered by Suven and are being developed as therapeutic agents and are useful in the treatment of cognitive impairment associated with neurodegenerative disorders



Suven Life Sciences Ltd.
6th Floor, SDE Serene Chambers,
Avenue – 7, Road No. 5, Banjara Hills,
Hyderabad-500 034, Telangana, INDIA
Phone : +91-40-2354-1142, 2354-3311
Fax : +91~40~2354-1152
Email id: info@suven.com
INDIAN PATENT
- Nirogi, Ramakrishna; Shinde, Anil Karbhari; Kambhampati, Ramasastri; Namala, Rambabu; Dwarampudi, Adi Reddy; Kota, Laxman; Gampa, Murlimohan; Kodru, Padmavathi; Tiriveedhi, Taraka Naga Vinaykumar; Kandikere, Vishwottam Nagaraj; et al
- From Indian Pat. Appl. (2012), IN 2010CH02551
PATENT
http://www.google.com/patents/US8912179
The present invention relates to heterocyclyl compounds of formula (I) and their pharmaceutically acceptable salts, its process of preparation and compositions containing them, for the treatment of various disorders that are related to Histamine H3 receptors.

1-Cyclobutyl-piperidin-4-ol (1.6 grams, 10 mmol) in tetrahydrofuran (20 mL) was treated with cooled and stirred suspension of sodium hydride (0.9 grams, 18 mmol) in tetrahydrofuran (20 mL) slowly over a period of 30 minutes; the reaction mixture was stirred for 1 hour. A solution of 2-Bromo-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester (3 grams, 9 mmol, obtained in preparation 1) in tetrahydrofuran (30 mL) was added drop wise over a period of 15 minutes and refluxed the reaction for 6 hours. Reaction mass was quenched with ice cold water and the product was extracted with ethyl acetate (3×50 mL). Combined organics were washed with water followed by brine and dried over anhydrous sodium sulphate. Organic volatiles were evaporated under vacuum. The residue was purified by flash chromatography (ethylacetate/n-hexane, 1/1) to obtain the title compound (2.0 grams).
1H-NMR (δ ppm): 1.48 (9H, s), 1.65-1.72 (2H, m), 1.85-1.92 (4H, m), 2.01-2.07 (4H, m), 2.18-2.19 (2H, m), 2.57 (2H, m), 2.62-2.66 (2H, m), 2.71-2.75 (1H, m), 3.70 (2H, m), 4.43 (2H, m), 4.93 (1H, m);
Mass (m/z): 394.2 (M+H)+.
Step (ii): Preparation of 2-(1-Cyclobutyl-piperidin-4-yloxy)-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridineA solution of 2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester (2.0 grams, 5 mmol, obtained in above step) in dichloromethane (30 mL) was treated with trifluroacetic acid (5.0 mL, 50 mmol) at 0° C. Reaction mass was stirred for 4 hours. After completion of reaction, the reaction mass was quenched into ice cold water and adjust pH to 10, by using 40% aqueous sodium hydroxide solution. The product was extracted with dichloromethane (3×50 mL), combined organics were washed with water followed by brine and dried over anhydrous sodium sulphate. Organic volatiles were evaporated under vacuum to obtain the title compound (1.3 grams).
1H-NMR (δ ppm): 1.68-1.74 (2H, m), 1.85-1.93 (4H, m), 2.06 (4H, m), 2.19 (2H, m), 2.60-2.61 (4H, m), 2.73-2.80 (1H, m), 2.90-3.10 (1H, m), 3.13-3.16 (2H, m), 3.85 (2H, s), 4.90-4.93 (1H, m);
Mass (m/z): 294.2 (M+H)+.
Step (iii): Preparation of 1-[2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-propan-1-oneA solution of 2-(1-Cyclobutyl-piperidin-4-yloxy)-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridine (1.3 grams, 4 mmol, obtained in above step) and triethylamine (1.9 mL, 13 mmol) in dichloromethane (30 mL) was cooled to 0° C. Propionylchloride (0.4 mL, 5 mmol) in dichloromethane (5 mL) was added drop wise over a period of 15 minutes and stirred the reaction for 30 minutes. Reaction mass was poured onto ice cold water and the product was extracted with ethyl acetate (3×50 mL). Combined organics were washed with water followed by brine and dried over anhydrous sodium sulphate. Organic volatiles were evaporated under vacuum. The residue was purified by flash chromatography (methanol/chloroform, 2/98) to obtain the title compound (1.0 gram).
1H-NMR (δ ppm): 1.17-1.21 (3H, m), 1.65-1.72 (5H, m), 1.87-1.91 (4H, m), 2.01-2.07 (4H, m), 2.22 (1H, m), 2.38-2.45 (2H, m), 2.45 (1H, m), 2.68-2.76 (3H, m), 3.72-3.74 (1H, m), 4.47-4.62 (2H, m), 4.92-4.94 (1H, m).
Mass (m/z): 350.4 (M+H)+.
Step (iv): Preparation of 1-[2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-propan-1-one tartrateA solution of 1-[2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-propan-1-one (0.8 grams, 2.3 mmol, obtained in above step) in methanol (10 mL) was treated with L(+)-Tartaric acid (0.34 grams, 2.3 mmol) at 0° C. Stirred the reaction mass for about 1 hour and the solvent was evaporated under vacuum to dryness. The solids were washed with diethyl ether and dried under vacuum to obtain the title compound (1.1 grams).
1H-NMR (δ ppm): 1.12-1.20 (3H, m), 1.82-1.87 (2H, m), 2.16-2.32 (7H, m), 2.45-2.55 (2H, m), 2.63-2.66 (3H, m), 2.72 (1H, m), 3.20 (2H, m), 3.47-3.50 (1H, m), 3.66-3.70 (1H, m), 3.81-3.88 (2H, m), 4.45 (2H, s), 4.60 (2H, s), 5.18 (5H, m);
Mass (m/z): 350.4 (M+H)+.
| Publication number | US8912179 B2 |
| Publication type | Grant |
| Application number | US 13/818,152 |
| PCT number | PCT/IN2010/000740 |
| Publication date | Dec 16, 2014 |
| Filing date | Nov 15, 2010 |
| Priority date | Sep 2, 2010 |
| Also published as | CA2812970A1, 4 More » |
| Inventors | Ramakrishna Nirogi, Anil Karbhari Shinde,Ramasastri Kambhampati, Rambabu Namala,Adi Reddy Dwarampudi, Laxman Kota,Murlimohan Gampa, Padmavathi Kodru,Taraka Naga Vinaykumar Tiriveedhi,Vishwottam Nagaraj Kandikere, Nageshwara Rao Muddana, Ramanatha Shrikantha Saralaya, Pradeep Jayarajan, Dhanalakshmi Shanmuganathan, Ishtiyaque Ahmad,Venkateswarlu Jasti, Less « |
| Original Assignee | Suven Life Sciences Limited |
| Export Citation | BiBTeX, EndNote, RefMan |
| Patent Citations (12), Non-Patent Citations (10), Classifications (16),Legal Events (1) | |
| External Links: USPTO, USPTO Assignment, Espacenet | |
……………….
Banjara Hills,Hyderabad
 TAJ KRISHNA
TAJ KRISHNA

GSK 2793660, Trying to crack the structure
 COMPD A
COMPD A
 COMPD B
COMPD B
 COMPD C
COMPD C
 COMPD D
COMPD D
 A
A B
B C
C D
DGSK 2793660
DATA FOR A
HCL SALT CAS 1613458-78-8
BASE CAS 1613458-70-0
C20 H27 N3 O3 . Cl H
MW OF BASE…..357.45
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
2H-Pyran-4-carboxamide, 4-amino-N-[(1S,2E)-4-(2,3-dihydro-1H-indol-1-yl)-1-ethyl-4-oxo-2-buten-1-yl]tetrahydro-, hydrochloride (1:1)
DATA FOR B
1613458-79-9 HCL SALT
1613458-71-1 BASE
C22 H31 N3 O3 . Cl H
MW 385.50 OF BASE
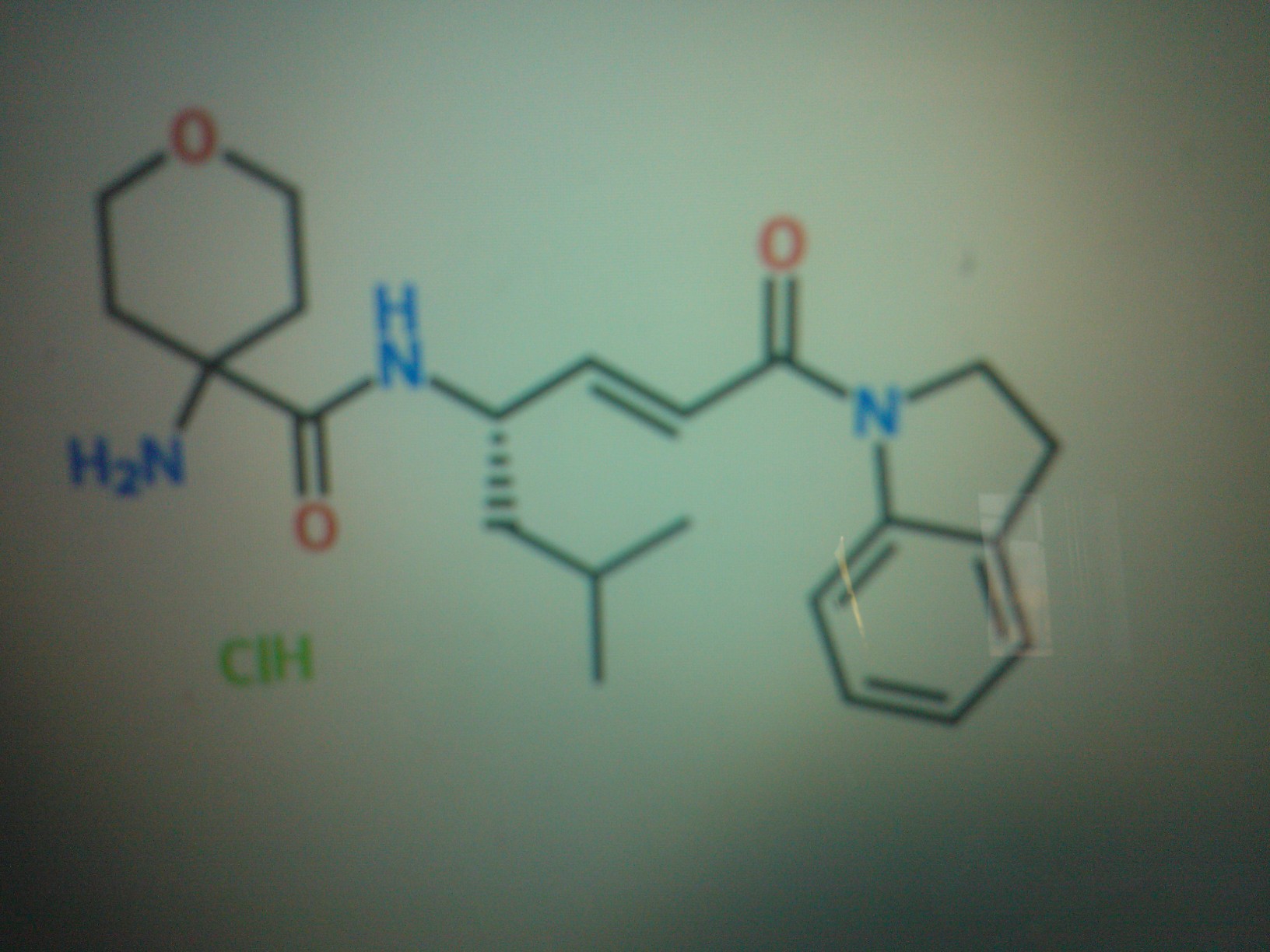
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
DATA FOR C
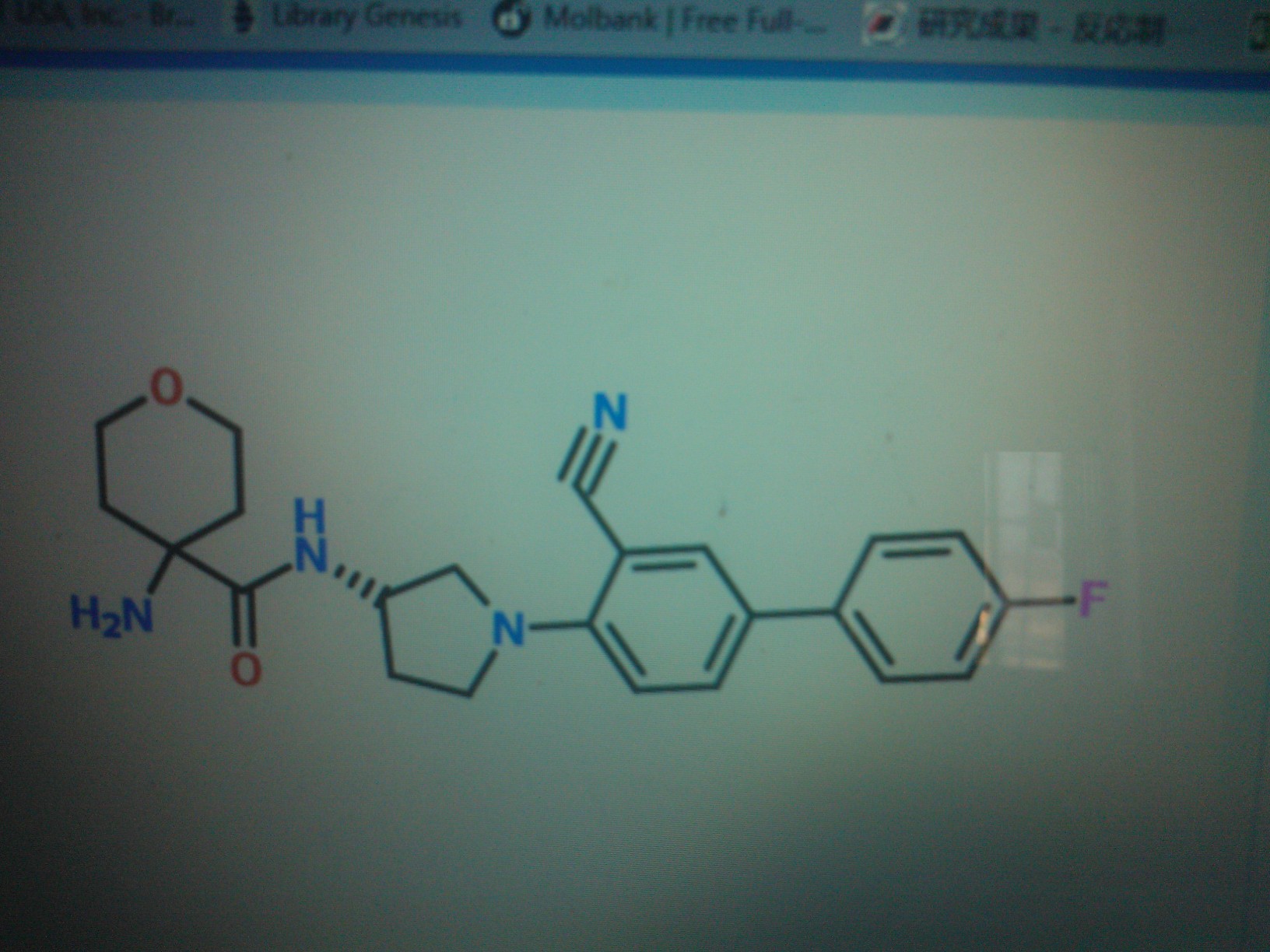
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
C24 H27 F N4 O . Cl H, MW 442.957
CAS OF BASE 1394001-73-0
CAS OF HCL 1394001-71-8
DATA FOR D
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
CAS OF BASE 1394001-74-1
CAS OF HCL 1394001-72-9
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Bronchiectasis
Dipeptidyl peptidase I inhibitor
http://www.gsk.com/media/280387/product-pipeline-2014.pdf
This study is the first administration of GSK2793660 to humans and will evaluate the safety, tolerability, PK and PD of single oral ascending doses of GSK2793660, and of repeat oral doses of GSK2793660 in healthy subjects. The study will comprise two parts (Part A and Part B). Part A will consist of two cohorts of subjects, each taking part in a three-way cross over study, with ascending doses of GSK2793660 and placebo. Available safety, PK and PD data will be reviewed before each dose escalation. This will be followed by a food-effect arm in the cohort that received what is deemed to be the target clinical dose. Part B is planned to consist of up to two cohorts of subjects, each taking part in one 14 day repeat dose study period. Subjects will be dosed on Day 1 and then on Days 3-15. It is planned that two doses will be evaluated. The dose(s) to be tested will be selected based on safety, PK, and PD from Part A. The study is intended to provide sufficient confidence in the safety profile of the molecule and information on target engagement to allow progression to further studies………..https://clinicaltrials.gov/ct2/show/NCT02058407
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Cathepsins are a family of enzymes included in the papain superfamily of cysteine proteases. Cathepsins B, C, F, H, K, L, S, V, and X have been described in the scientific literature. Cathepsin C is also known in the literature as Dipeptidyl Peptidase I or “DPPI.”
A number of recently published studies have begun to describe the role cathepsin C plays in certain inflammatory processes. See e.g. Adkison et al., The Journal of Clinical Investigation 109:363-371 (2002); Tran et al., Archives of Biochemistry and Biophysics 403 : 160-170 (2002); Thiele et al., The Journal of Immunology 158: 5200-5210 (1997);
Bidere et al., The Journal of Biological Chemistry 277: 32339-32347 (2002); Mabee et al., The Journal of Immunology 160: 5880-5885 (1998); McGuire et al., The Journal of
Biological Chemistry, 268: 2458-2467 (1993); and Paris et al., FEBS Letters 369: 326-330 (1995). From these studies, it appears that cathepsin C is co-expressed in granules of neutrophils and other leukocytes with certain serine proteases and cathepsin C functions to process the pro-forms of the serine proteases to active forms. Serine proteases are released from the granules of leukocytes recruited to sites of inflammation. Once activated, these proteases have a number of functions including degradation of various extracellular matrix components, which together can propagate tissue damage and chronic inflammation.
Studies in both cathepsin C deficient mice, and the human cathepsin C deficiency
Papillon-Lefevre syndrome clearly demonstrate that cathepsin C is required for the
activation of the neutrophil serine proteases in azurophilic granules such as neutrophil elastase (NE), cathepsin G, and proteinase 3. See Pham, C. T. et al., J. Immunol. 173 :
7277-7281 (2004).
A number of respiratory diseases are associated with an overabundant
acculumation of neutrophils and the presence of increased levels of at least some
neutrophil serine proteases. These enzymes are believed to play a role in the pathology of several respiratory diseases, such as Chronic Obstructive Pulmonary Disease (“COPD”), cystic fibrosis (CF), and non-cystic fibrosis (non-CF) bronchiectasis. Each of these diseases is associated with increased levels of E in particular, and E at least is considered to play a role in the progression of disease. See Ranes, J. and Stoller, J. K., Semin. Respir. Crit. Care Med 26: 154-166 (2005); Saget, S. D. et al., Am. J. Resp. Crit. Care Med. 186: 857-865 (2012); Tsang, K. W. et al., Chest 117: 420-426 (2000).
Additional roles of the other proteases is emerging. See Hartl, D. et al., Nature Med. 13 : 1423-1430 (2007); Korkmaz, B. et al., Pharm. Rev. 62: 726-759 (2010).
Cigarette smoking is a significant risk factor for developing COPD. Exposure to cigarette smoke and other noxious particles and gases may result in chronic inflammation of the lung. In response to such exposure, inflammatory cells such as CD8+ T cells, macrophages, and neutrophils are recruited to the area. These recruited inflammatory cells release proteases, which are believed to play a major role in the disease etiology by a number of mechanisms. Proteases released from recruited cells include the serine proteases NE as above; granzymes A and B, released from cytotoxic T cells or natural killer cells; and chymases, released from mast cells. Cathepsin C appears to be involved in activating all of these enzymes to some extent.
A number of studies with cathepsin C deficient mice have suggested roles for cathepsin C in disease models. Cathepsin C knockout mice are resistant to lung airspace enlargement and inflammatory cell infiltration in both cigarette smoke and ozone exposure models of COPD. See Guay et al., Current Topics in Medicinal Chemistry, 2010, 10, 708- 716; See also Podolin et al. (2008), Inflammation Research, 57(Suppl 2) S104.
In a model of rheumatoid arthritis (“RA”), another chronic inflammatory disease where cathepsin C may play a role, neutrophils are recruited to the site of joint
inflammation and release cathepsin G, NE, and proteinase 3, which are believed to be responsible in part for cartilage destruction associated with RA (Hu, Y. and Pham, C. T. Arthritis Rheum. 52: 2553-2558 (2005); Zen, K. et al, Blood 117:4885-4894 (2011)). Other models where cathepsin C may play a role include osteoarthritis, asthma, Multiple Sclerosis, and Anti-Neutrophil Cytoplasmic Autoantibody (ANCA)-related diseases (e.g. ANCA-associated vasculitis). See e.g. Matsui, K., Yuyama, N., Akaiwa, M., Yoshida, N. L., Maeda, M., Sugita, Y., Izuhara, K., Gene 293(1-2): 1-7 (2002); Wolters, P. J., Laig- Webster, M., Caughey, G. H., American Journal of Respiratory Cell & Molecular Biology 22(2): 183-90 (2000); Schreiber et al., J. Am. Soc. Nephrol. 23 :470-482 (2012). Cathepsin C has been demonstrated to have a role in neutrophil migration in the development of aortic aneurysms by a mechanism which has not been clearly elucidated (Pagano, M. B. et al., PNAS 104: 2855-2860 (2007)).
One approach to treating these conditions is to inhibit the activity of the serine proteases involved in the inflammatory process, especially NE activity. See e.g.,
Ohbayashi, Expert Opin. Investig. Drugs 11(7): 965-980 (2002); Shapiro, Am. J. Respir. Cell Mol. Biol. 26: 266-268 (2002). Indeed, a potent and selective inhibitor of NE was found to improve lung function in patients with bronchiectasis (Stockley, R. et al. Respir. Med. 107, 524-533 (2013)). In light of the role cathepsin C plays in activating certain serine proteases, especially NE, it is desirable to prepare compounds that inhibit its activity, which thereby inhibit serine protease activity. Thus, there is a need to identify compounds that inhibit cathepsin C, which can be used in the treatment of a variety of conditions mediated by cathepsin C.
There are additional activities of cathepsin C that may also be related to disease etiology. Cathepsin C is highly expressed in the lung epithelium where it may play a role in the processing of other enzymes not yet identified. Cathepsin C has also been reported to cleave kallikrein-4, which is believed to play a role in dental enamel maturation (Tye, C. E. et al. J. Dental Res. 88: 323-327 (2009)). Finally, cathepsin C is itself released from cells and may play a direct role in the degradation of matrix proteins.
DATA FOR A
WO 2014091443
http://www.google.com/patents/WO2014091443A1?cl=en

synthesis







Intermediate 1
1,1-dimethylethyl ((l -l-{[methyl(methyloxy)amino]carbonyl}propyl)carbamate
To a solution of (2,S)-2-({[(l,l-dimethylethyl)oxy]carbonyl}amino)butanoic acid (2.50 g, 12.3 mmol) in THF (15.0 mL) was added Ι,Γ-carbonyldiimidazole (2.39 g, 14.8 mmol) portionwise over about 10 min. After stirring 30 min at RT, a solution of Ν,Ο- dimethylhydroxylamine hydrochloride (1.32 g, 13.5 mmol) and DIPEA (2.36 mL, 13.5 mmol) in DMF (4.0 mL) was added. The reaction mixture was stirred for 2 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HC1 (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.60 g, 88%) as a clear, colorless oil. LC-MS m/z 247 (M+H)+, 0.94 min (ret time).
Intermediate 2
1,1-dimethylethyl [(lS -l-formylpropyl] carbamate
To a solution of L1AIH4 (0.453 g, 11.9 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution of 1, 1-dimethylethyl ((l,S)-l-{[methyl(methyloxy)amino]carbonyl}- propyl)carbamate (2.67 g, 10.8 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6.5 mL) followed by 5% aq. potassium bisulfate (6.5 mL). The reaction mixture was washed with 1 M aq. HC1 (3 x 10 mL), saturated aq. NaHC03 (3 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil.
Intermediate 3
methyl (2E V)-4-({ [(1 , l-dimethylethyl)oxy] car bonyl} amino)-2-hexenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (4.35 g, 13.0 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 2 in Et20 (15 mL). The reaction mixture was stirred at RT overnight. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.44 g, 55% over two steps) as a clear, colorless oil. LC-MS m/z 244 (M+H)+, 0.98 min (ret time). Intermediate 4
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-2-hexenoic acid
Li OH (2.95 g, 123 mmol) was added to a solution of methyl (2£, S 4-({[(1, 1- dimethylethyl)oxy]carbonyl}amino)-2-hexenoate (6 g, 24.66 mmol) in THF (50 mL), MeOH (10.00 mL), and water (50.0 mL). The reaction was stirred overnight at RT. After 18.5 h, the reaction mixture was concentrated under reduced pressure to remove the THF and MeOH. Water (40 mL) was added, and aqueous mixture was adjusted to pH = 3 with 6 M aq. HC1, as measured by pH paper. EtOAc (80 mL) was added, the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were dried over Na2S04, concentrated under reduced pressure, and dried under high vacuum, giving 6.09 g of the title compound. LC-MS m/z 230 (M+H)+, 0.77 min (ret time).
Intermediate 5
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl] carbamate
A solution of 50 wt% *T3P in EtOAc (22.00 mL, 37.0 mmol) was added dropwise via addition funnel to a solution of (2£,,4,S)-4-({[(l, l-dimethylethyl)oxy]carbonyl}- amino)-2-hexenoic acid (5.65 g, 24.64 mmol), 2,3-dihydro-lH-indole (2.76 mL, 24.64 mmol), and Et3N (11 mL, 79 mmol) in CH2C12 (90 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 30 min, the reaction was quenched by dropwise addition of saturated aq. NaHC03 (50 mL). The layers were separated, and the reaction was washed with 10% citric acid (1 x 50 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 7.21 g (89%) of the title compound. LC-MS m/z 331 (M+H)+, 1.05 (ret time). Intermediate 6
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine
trifluoroacetate
TFA (25 mL, 324 mmol) was added to a solution of 1, 1-dimethylethyl [(1^,2£)-4- (2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]carbamate (7.21 g, 21.82 mmol) in CH2C12 (25 mL). The reaction was stirred at RT. After 3.5 h, CH2C12 (200 mL) was added, and the reaction was concentrated under reduced pressure and dried under high vacuum. LC-MS m/z 231 (M+H)+, 0.69 (ret time).
Intermediate 7
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino carbonyl)tetrahydro-2H-pyran-4-yl]carbamate
A solution of 50 wt% UT3P in EtOAc (1.3 mL, 2.184 mmol) was added dropwise to a solution of [(l,S’,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine trifluoroacetate (500 mg, 1.452 mmol), 4-((tert-butoxycarbonyl)amino)tetrahydro-2H- pyran-4-carboxylic acid (356 mg, 1.452 mmol), and Et3N (1 mL, 7.21 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction mixture was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 251 mg (38%) of the title compound. LC-MS m/z 458 (M+H)+, 0.96 (ret time).
Example 1
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.23 mL, 2.76 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.549 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp) for 1 h 45 min. The solvent was evaporated under reduced pressure. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 193.3 mg (89%) of the title compound. LC-MS m/z 358 (M+H)+, 0.68 (ret time).
1H MR (400 MHz, METHANOL-^) δ ppm 8.14 (br. s., 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.53 Hz, 1 H); 7.02 – 7.09 (m, 1 H); 6.83 (dd, J=15.18, 6.65 Hz, 1 H); 6.49 (d, 7=14.8 Hz, 1 H); 4.56 (d, 7=7.28 Hz, 1 H); 4.22 (br. s., 2 H); 3.95 (d, 7=7.53 Hz, 1 H); 3.88 – 3.94 (m, 1 H); 3.71 – 3.78 (m, 2 H); 3.23 (br. s., 2 H); 2.39 – 2.46 (m, 2 H); 1.79 – 1.86 (m, 2 H); 1.75 (s, 1 H); 1.72 (d, 7=8.28 Hz, 1 H); 1.00 (t, 7=7.40 Hz, 3 H)
DATA FOR B
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride







http://www.google.com/patents/WO2014091443A1?cl=en
Intermediate 8
N -{[(l,l-dimethylet leucinamide
To a solution ofN-(tert-butoxycarbonyl)-L-leucine (3.00 g, 13.0 mmol) in THF (25.0 mL) was added Ι,Γ-carbonyldiimidazole (2.52 g, 15.6 mmol) portionwise over about 10 min. After stirring 1 h at RT, a solution of N,O-dimethylhydroxylamine hydrochloride (1.39 g, 14.3 mmol) and DIPEA (2.49 mL, 14.3 mmol) in DMF (6.0 mL) was added. The reaction mixture was stirred for 2.5 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HCl (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.34 g, 66%) as a clear, colorless oil. LC-MS m/z 275 (M+H)+, 1.17 min (ret time).
Intermediate 9
1,1-dimethylethyl [(lS -l-formyl-3-methylbutyl]carbamate
To a solution of L1AIH4 (0.356 g, 9.38 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution ofN2-{[(l, l-dimethylethyl)oxy]carbonyl}-N1-methyl-N1-(methyloxy)-L- leucinamide (2.34 g, 8.53 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6 mL) followed by 5% aq. potassium bisulfate (6 mL). The reaction mixture was washed with 1 M aq. HCl (2 x 10 mL), saturated aq. NaHC03 (2 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil. Intermediate 10
methyl (2E 4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (3.42 g, 10.2 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 9 in Et20 (15 mL). The reaction mixture was stirred for 15 h at RT. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.74 g, 75% over two steps) as a clear, colorless oil. LC-MS m/z 272 (M+H)+, 1.22 min (ret time).
Intermediate 11
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoic acid
To a solution of methyl (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6- methyl-2-heptenoate (5.00 g, 18.43 mmol) in THF (15 mL), MeOH (15.0 mL), and water (15 mL) was added Li OH (2.206 g, 92.00 mmol). After stirring for 2 h at RT, the reaction mixture was concentrated in vacuo. The reaction mixture was acidified with 6 M aq. HC1 to pH = 5 and then extracted with EtOAc. The organic layer was washed with water, dried over Na2SC”4, filtered, and concentrated in vacuo to afford the title compound (4.7 g, 99%) as a white semi-solid. LC-MS m/z 158 (M+H-Boc)+, 0.94 min (ret time).
Intermediate 12
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]carbamate
To a solution of (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2- heptenoic acid (4.70 g, 18.26 mmol) in DMF (30.0 mL) were added BOP reagent (8.08 g, 18.26 mmol) and DIPEA (6.38 mL, 36.5 mmol). After stirring at RT for 5 min, 2,3-dihydro- lH-indole (2.053 mL, 18.26 mmol) was added and stirring continued overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04, filtered, concentrated in vacuo and purified by flash column chromatography (0-20% EtOAc/hexanes) to afford the title compound (4.83 g, 74%) as a white solid. LC-MS m/z 359 (M+H)+, 1.18 min (ret time).
Intermediate 13
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten-l-yl]amine trifluoroacetate
To a solution of 1, 1-dimethylethyl [(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2- methylpropyl)-4-oxo-2-buten-l-yl]carbamate (3.21 g, 8.95 mmol) in CH2C12 (10.0 mL) was added TFA (10 mL, 130 mmol). The reaction mixture was stirred for 17.5 h at RT and then concentrated under reduced pressure and dried under high vacuum to afford the title compound. LC-MS m/z 259 (M+H)+, 0.76 min (ret time).
Intermediate 14
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l- l]amino}carbonyl)tetrahydro-2H- ran-4-yl]carbamate
A solution of 50 wt% ¾P in EtOAc (1.2 mL, 2.016 mmol) was added dropwise to a solution of [(15′,2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]amine trifluoroacetate (500 mg, 1.343 mmol), 4-((tert- butoxycarbonyl)amino)tetrahydro-2H-pyran-4-carboxylic acid (329 mg, 1.343 mmol), and Et3N (0.93 mL, 6.71 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 204 mg (31%) of the title compound. LC-MS m/z 486 (M+H)+, 1.07 min (ret time).
Example 2
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.22 mL, 2.64 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(1^2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.517 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp). After 1 h 45 min, the solvent was evaporated under reduced pressure at 60 °C. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 130.6 mg (60%) of the title compound. LC-MS m/z 386 (M+H)+, 0.79 (ret time). 1H MR (400 MHz, METHANOL- d4) δ ppm 8.15 (d, J=7.03 Hz, 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.65 Hz, 1 H); 7.06 (t, J=7.91 Hz, 1 H); 6.81 (dd, J=15.18, 6.40 Hz, 1 H); 6.49 (br. s., 1 H); 4.73 – 4.85 (m, 2 H); 4.21 (t, J=8.28 Hz, 2 H); 3.91 – 3.97 (m, 2 H); 3.70 – 3.77 (m, 2 H); 3.25 – 3.21 (m, 2 H); 2.35 – 2.48 (m, 2 H); 1.82 (d, J=14.31 Hz, 2 H); 1.63 – 1.71 (m, 2 H); 1.50 – 1.57 (m, 1 H); 0.98 (dd, J=11.92, 6.40 Hz, 6 H).
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
http://www.google.im/patents/WO2012112733A1?cl=en
Example 1
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride

HCI salt
A solution of 1,1-dimethylethyl [l-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl]amino}carbonyl)cyclohexyl]carbamate (44 mg, 0.087 mmol) in HCI (4 M solution in 1,4-dioxane, 1.0 mL, 4.00 mmol) was stirred at RT for 1 h. The reaction mixture was diluted with Et20 (5 mL), and the mixture was filtered and washed with Et20 (2 x 2 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (33.5 mg, 87%). LC-MS m/z 407 (M+H)+, 0.94 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.65 – 7.72 (m, 2 H), 7.52 – 7.59 (m, 2 H), 7.10 – 7.17 (m, 2 H), 6.89 (d, J=8.53 Hz, 1 H), 4.50 – 4.58 (m, 1 H), 3.94 (dd, J=10.29, 6.53 Hz, 1 H), 3.80 (dt, J=9.41, 7.09 Hz, 1 H), 3.67-3.71 (m, 1 H), 3.64 (dd, J=10.29, 4.52 Hz, 1 H), 2.29 – 2.37 (m, 1 H), 2.04 – 2.16 (m, 3 H), 1.78 – 1.88 (m, 5 H), 1.45 – 1.62 (m, 3 H).
DATA FOR D
http://www.google.im/patents/WO2012112733A1?cl=en
Example 2
4-amino- V-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3-pyrrolidinyl]tetrahydro-2H- pyr -4-carboxamide hydrochloride
HCI salt
A solution of 1,1-dimethylethyl [4-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl] amino }carbonyl)tetrahydro-2H-pyran-4-yl] carbamate (183 mg, 0.360 mmol) in HC1 (4 M solution in 1,4-dioxane, 2.0 mL, 8.00 mmol) was stirred at RT for 0.5 h. The reaction mixture was diluted with Et20 (10 mL), and the mixture was filtered and washed with Et20 (2 x 5 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (122.8 mg, 77%). LC-MS m/z 409 (M+H)+, 0.87 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.66 – 7.72 (m, 2 H), 7.53 – 7.60 (m, 2 H), 7.11 – 7.18 (m, 2 H), 6.89 (d, J=8.78 Hz, 1 H), 4.53 – 4.60 (m, 1 H), 3.87 – 3.97 (m, 3 H), 3.78 – 3.84 (m, 1 H), 3.64 – 3.76 (m, 4 H), 2.30 – 2.44 (m, 3 H), 2.11 – 2.19 (m, 1 H), 1.77 – 1.84 (m, 2 H).
| WO2004002491A1 * | 25 Jun 2003 | 8 Jan 2004 | David J Aldous | Morpholine and tetrahydropyran drivatives and their use as cathepsin inhibitors |
| WO2008121065A1 * | 28 Mar 2008 | 9 Oct 2008 | Astrazeneca Ab | Novel pyrrolidine derivatives as antagonists of the chemokine receptor |
| US20070032484 * | 25 Jul 2006 | 8 Feb 2007 | Roche Palo Alto Llc | Cathepsin K inhibitors |
| US20020107266 * | Dec 11, 2001 | Aug 8, 2002 | Marguerita Lim-Wilby | Amides used particularly in the treatment, prevention or amelioration of one or more symptoms of malaria or Chagas’ disease; inhibiting the activity of falcipain or cruzain |
| US20100286118 * | May 6, 2010 | Nov 11, 2010 | Rhonan Ford | Substituted 1-cyanoethylheterocyclylcarboxamide compounds 750 |
| WO2012109415A1 | Feb 9, 2012 | Aug 16, 2012 | Glaxosmithkline Llc | Cathepsin c inhibitors |
KHK 7580 structure cracked……Evocalcet
KHK 7580 …..example
| 3.008 |
 |
|
 |
2HCl | MS · APCI: 375[M + H]+ |

in EP1757582
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid
4-[(3S)-3-[[(1R)-1-(1-naphthalenyl)ethyl]amino]-1-pyrrolidinyl]-Benzeneacetic acid,
cas will be updated
BASE ….870964-67-3
DI HCL SALT …….870856-31-8
MF C24 H26 N2 O2 BASE
MW 374.48 BASE
KHK-7580
KHK-7580; MT-4580
Mitsubishi Tanabe Pharma Corp… innovator
Kyowa Hakko Kirin Co Ltd.. licencee
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1-yl)phenylacetic acid,
useful as calcium-sensitive receptor (CaSR) agonists for treating hyperparathyroidism. a CaSR agonist, being developed by Kyowa Hakko Kirin, under license from Mitsubishi Tanabe, for treating secondary hyperparathyroidism (phase 2 clinical, as of March 2015).
WILL BE UPDATED
WO2005115975,/EP1757582
http://www.google.co.in/patents/EP1757582A1?cl=en
Example no
| 3.008 |
|
|
|
2HCl | MS · APCI: 375[M + H]+ |
Mitsubishi Tanabe Pharma Corporation
The present invention provides a novel crystal form of an arylalkylamine
compound. Specifically, a novel crystal form of
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid has
excellent stability, and is therefore useful as an active ingredient for
a medicine. The present invention also provides an industrially
advantageous method for producing an arylalkylamine compound.

http://worldwide.espacenet.com/publicationDetails/biblio?DB=worldwide.espacenet.com&II=0&ND=3&adjacent=true&locale=en_EP&FT=D&date=20150312&CC=WO&NR=2015034031A1&KC=A1
Mitsubishi Tanabe Pharma Corporation
The present invention provides a novel crystal form of an arylalkylamine compound. Specifically, a novel crystal form of 4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid has excellent stability, and is therefore useful as an active ingredient for a medicine. The present invention also provides an industrially advantageous method for producing an arylalkylamine compound.
………………….
Reference Example 3.001

(1) To a mixed solution containing 33.5 g of 3-hydroxypiperidine and 62.7 ml of triethylamine dissolved in 250 ml of methylene chloride was added dropwise a solution of 55.7 ml of benzyloxycarbonyl chloride in 150 ml of methylene chloride, and the mixture was stirred at room temperature for 16 hours. To the reaction mixture were added a saturated aqueous citric acid and chloroform, the mixture was stirred and the liquids were separated. The organic layer was dried, the solvent was evaporated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 75.5 g of benzyl 3-hydroxypiperidine-1-carboxylate.
MS•APCI (m/z): 236 [M+H]+
(2) 800 ml of a solution of 52.4 ml of oxalyl chloride in methylene chloride was cooled to −78° C., 53.2 ml of DMSO was added dropwise to the solution, and the mixture was stirred at −78° C. for 0.5 hour. A solution of 75.5 g of benzyl 3-hydroxypiperidine-1-carboxylate dissolved in 200 ml of methylene chloride was added dropwise to the mixture, and further 293 ml of triethylamine was added dropwise to the same, and the mixture was stirred for 16 hours while a temperature thereof was gradually raised to room temperature. To the reaction mixture were added a saturated aqueous sodium bicarbonate solution and chloroform, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated to obtain 83.7 g of 1-benzyloxycarbonyl-3-piperidone. MS•APCI (m/z): 234 [M+H]+
(3) To a solution of 83.7 g of 1-benzyloxycarbonyl-3-piperidone dissolved in 1.2 liters of methylene chloride was added 55.0 g of (R)-(+)-1-(1-naphthyl)ethylamine, and after the mixture was stirred at room temperature for 2 hours, 69 ml of acetic acid and 160 g of sodium triacetoxy borohydride were added to the mixture, and the mixture was stirred at room temperature for 15 hours. To the reaction mixture was added an aqueous sodium hydroxide to make the mixture basic, and then, chloroform was added to the mixture, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 98.7 g of benzyl 3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
(4) To a solution of 40.95 g of triphosgene dissolved in 800 ml of methylene chloride was added dropwise a mixed solution containing 80.6 g of benzyl 3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate and 86.6 ml of triethylamine dissolved in 200 ml of methylene chloride at 0° C., and the mixture was stirred at room temperature for 16 hours. To the reaction mixture was added water, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was washed with 200 ml of diethyl ether, and the crystal collected by filtration was recrystallized from chloroform and diethyl ether to obtain 48.9 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate.
Further, the filtrate was purified by silica gel column chromatography (hexane:ethyl acetate=8:1→0:1) to obtain 5.82 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate and 14.5 g of benzyl (S)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate.
(5) To a solution containing 54.6 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate dissolved in 700 ml of tetrahydrofuran was added 350 ml of water, and the mixture was stirred under reflux for 15 hours. After tetrahydrofuran was evaporated, a saturated aqueous sodium bicarbonate solution and chloroform were added thereto, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 24.3 g of benzyl (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
(6) To a solution containing 24.2 g of benzyl (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate dissolved in 250 ml of methanol was added 2.5 g of palladium carbon (10% wet), and the mixture was shaked under hydrogen atmosphere at 3 atm at room temperature for 40 hours. Palladium carbon was removed, and the solvent was evaporated, the residue was washed with ethyl acetate-chloroform (10:1), and collected by filtration to obtain 15.3 g of (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine (the following Reference example Table, Reference example 3.001(a)). MS•APCI (m/z): 255 [M+H]+
(7) By using 14.5 g of benzyl (S)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate, the same treatment was carried out as in the above-mentioned (5) to obtain 4.74 g of benzyl (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
Moreover, by using 4.7 g of benzyl (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate, the same treatment was carried out as in the above-mentioned (6) to obtain 2.89 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine. MS•APCI (m/z): 255 [M+H]+
(8) To a solution of 3.46 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine dissolved in 15 ml of methanol was added dropwise 20 ml of a solution of 4M hydrochloric acid in ethyl acetate, and the mixture was stirred. The reaction mixture was concentrated under reduced pressure, diethyl ether was added to the residue, washed and dried to obtain 3.33 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine dihydrochloride
| 3.008 |
|
|
|
2HCl | MS · APCI: 375[M + H]+ |
| TABLE A3 | |||||
 |
|||||
| Example No. | R1—X— |
|
—Ar | Salt | Physical properties, etc. |
…………………..
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
do not miss out on above click
http://www.kyowa-kirin.com/research_and_development/pipeline/
KHK7580 -Secondary Hyperparathyroidism
![]()
![]()
![]()
![]()
JP
| Company | Mitsubishi Tanabe Pharma Corp. |
| Description | Calcium receptor agonist |
| Molecular Target | |
| Mechanism of Action | Calcium-sensing receptor (CaSR) agonist |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Thyroid disease |
| Indication Details | Treat hyperparathyroidism in patients receiving hemodialysis; Treat secondary hyperparathyroidism (SHPT) |
| Regulatory Designation | |
| Partner |
August 29, 2014
Kyowa Hakko Kirin Announces Commencement of Phase 2b Clinical Study of KHK7580 in Patients with Secondary Hyperparathyroidism in Japan
Tokyo, Japan, August 29, 2014 — Kyowa Hakko Kirin Co., Ltd. (Tokyo: 4151, President and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) today announced the initiation of a phase 2b clinical study evaluating KHK7580 for secondary hyperparathyroidism patients receiving hemodialysis in Japan.
This randomized, placebo-controlled, double-blind, parallel-group, multi-center study is designed to evaluate efficacy and safety in cohorts comprising KHK7580, its placebo and cinacalcet and initial dose of KHK7580 for secondary hyperparathyroidism patients receiving hemodialysis.
KHK7580 is a small molecular compound produced by Mitsubishi Tanabe Pharma Corporation (President & Representative Director, CEO: Masayuki Mitsuka, “Mitsubishi Tanabe Pharma”). Kyowa Hakko Kirin signed a license agreement of KHK7580 with Mitsubishi Tanabe Pharma for the rights to cooperative research, develop, market and manufacture the product in Japan and some part of Asia on March 2008.
The Kyowa Hakko Kirin Group is contributing to the health and prosperity of the world’s people by pursuing advances in life sciences and technology and creating new value.
Outline of this study
| ClinicalTrials.gov Identifier | |
|---|---|
| Target Population | Secondary hyperparathyroidism patients receiving hemodialysis |
| Trial Design | Randomized, placebo-controlled, double-blind (included open arm of cinacalcet), parallel-group, multi-center study |
| Administration Group | KHK7580, Placebo, cinacalcet |
| Target Number of Subjects | 150 |
| Primary Objective | Efficacy |
| Trial Location | Japan |
| Trial Duration | Jul. 2014 to Jun. 2015 |
Contact:
Kyowa Hakko Kirin
Media Contact:
+81-3-3282-1903
or
Investors:
+81-3-3282-0009
Update on march 2016
New comment waiting approval on New Drug Approvals |
|

http://www.medkoo.com/products/6729

Name: Evocalcet
CAS#: 870964-67-3
Chemical Formula: C24H26N2O2
Exact Mass: 374.19943
Evocalcet is a calcium-sensing receptor agonist. The calcium-sensing receptor (CaSR) is a Class C G-protein coupled receptor which senses extracellular levels of calcium ion. The calcium-sensing receptor controls calcium homeostasis by regulating the release of parathyroid hormone (PTH). CaSR is expressed in all of the organs of the digestive system. CaSR plays a key role in gastrointestinal physiological function and in the occurrence of digestive disease. High dietary Ca2+ may stimulate CaSR activation and could both inhibit tumor development and increase the chemotherapeutic sensitivity of cancer cells in colon cancer tissues. (Last update: 12/15/2015).
Synonym: MT-4580; MT 4580; MT4580; KHK-7580; KHK7580; KHK 7580; Evocalcet
IUPAC/Chemical Name: 2-(4-((S)-3-(((R)-1-(naphthalen-1-yl)ethyl)amino)pyrrolidin-1-yl)phenyl)acetic acid
2
https://tripod.nih.gov/ginas/app/substance/f580b9fd

http://www.drugspider.com/drug/evocalcet
| INN name |
Evocalcet
|
||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lab Code(s) |
MT-4580
KHK-7580
|
||||||||||||||||||||||||||||||||||||||||
| Chemical name |
{4-[(3S)-3-{[(1R)-1-(Naphthalen-1-yl)ethyl]amino}pyrrolidin-1-yl]phenyl}acetic acid
|
||||||||||||||||||||||||||||||||||||||||
| Chemical structure |
 |
||||||||||||||||||||||||||||||||||||||||
| Molecular formula |
C24H26N2O2
|
||||||||||||||||||||||||||||||||||||||||
| SMILES |
O=C(O)CC1=CC=C(N2C[C@@H](N[C@@H](C3=C4C=CC=CC4=CC=C3)C)CC2)C=C1
|
||||||||||||||||||||||||||||||||||||||||
| CAS registry number |
870964-67-3
|
||||||||||||||||||||||||||||||||||||||||
| Orphan Drug Status |
No
|
||||||||||||||||||||||||||||||||||||||||
| On Fast track |
No
|
||||||||||||||||||||||||||||||||||||||||
| New Molecular Entity |
Yes
|
||||||||||||||||||||||||||||||||||||||||
| Originator | |||||||||||||||||||||||||||||||||||||||||
| Developer(s) | |||||||||||||||||||||||||||||||||||||||||
| Class | |||||||||||||||||||||||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||||||||||||||||||||||
| Clinical trial(s) |
|
||||||||||||||||||||||||||||||||||||||||
| Updated on |
11 Oct 2015
|
///////////////
SMILES Code: O=C(O)CC1=CC=C(N2C[C@@H](N[C@@H](C3=C4C=CC=CC4=CC=C3)C)CC2)C=C1
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease
CS-3150, (XL550)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
CAS 1632006-28-0 for S conf
MF C22 H21 F3 N2 O4 S
MW 466.47
(S)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
CAS 1632006-28-0 for S configuration
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide, CAS 880780-76-7
(+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-82-4
(-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-83-5
WO2008 / 126831 (US Publication US2010-0093826)http://www.google.co.in/patents/EP2133330A1?cl=en
WO 2006012642..compound A;..http://www.google.com/patents/WO2006012642A2?cl=en
WO2006 / 012642 (US Publication US2008-0234270)
WO 2015030010…http://www.google.com/patents/WO2015030010A1?cl=en
- Originator Exelixis
- Developer Daiichi Sankyo Company..
Daiichi Sankyo Company,Limited, 第一三共株式会社 - Class Antihypertensives; Small molecules
- Mechanism of Action Mineralocorticoid receptor antagonists

JAPAN PHASE 2……….Phase 2 Study to Evaluate Efficacy and Safety of CS-3150 in Patients with Essential Hypertension
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-121921
Phase II Diabetic nephropathies; Hypertension
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
- 01 May 2013 Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
-
Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.


Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
| Description | Small molecule antagonist of the mineralocorticoid receptor (MR) |
| Molecular Target | Mineralocorticoid receptor |
| Mechanism of Action | Mineralocorticoid receptor antagonist |
| Therapeutic Modality | Small molecule |
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
Several patents WO-2014168103,
WO-2015012205 and WO-2015030010
XL-550, claimed in WO-2006012642,
………………………………………………………………….
http://www.google.co.in/patents/EP2133330A1?cl=en
(Example 3)(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
-
After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
-
Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid.
1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s). -
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained.
1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
-
Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
-
Isomer C: (+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: +7.1° (c = 1.0, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.91 (s, 1H), 7.87-7.79 (m, 5H), 7.67-7.58 (m, 2H), 7.51 (s, 1H), 7.35 (d, 1H, J = 7.0 Hz), 3.78-3.65 (m, 4H), 3.05 (s, 3H), 2.07 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1260.
Retention time: 4.0 min. -
Isomer D: (-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: -7.2° (c = 1.1, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.88-7.79 (m, 6H), 7.67-7.58 (m, 2H), 7.50 (s, 1H), 7.36 (d, 1H, J = 7.5 Hz), 3.79-3.65 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1257.
Retention time: 4.5 min.
……………………………………………….
WO 2014168103
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014168103
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
………………………………………………
Patent literature
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Angiotensin II receptor 桔抗 agent
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
 OLMESARTAN
OLMESARTAN

{kind=link}
{kind=link}