
Home » 2014 (Page 90)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pamicogrel KB 3022 for Coagulation Disorders Therapy


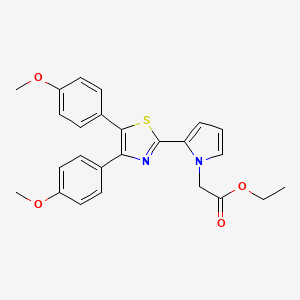 PAMICOGREL
PAMICOGRELPamicogrel (CAS NO.: 101001-34-7), with its systematic name of 1H-Pyrrole-1-acetic acid, 2-(4,5-bis(4-methoxyphenyl)-2-thiazolyl)-, ethyl ester, could be produced through many synthetic methods.
Following is one of the synthesis routes:
alpha-Bromo-4,4-dimethoxidesoxybenzoin (I) is cyclized with pyrrole-2-carbothioamide (II) in hot acetonitrile to produce 4,5-bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole (III), which is then condensed with ethyl bromoacetate (IV) in the prsence of NaOH and tetrabutylammonium bromide in refluxing dichloromethane – water.

- Reaction Scheme-I:
-


wherein R is as defined above, and Y is a halogen such as chlorine, bromine or iodine, or p-toluenesulfonyloxy group.
-
The process of the above reaction scheme-I can be carried out by reacting a compound (II) and an equimolar or excess amount of a compound (III) in the presence of a base or a phase transfer catalyst. In case of using a base such as metallic potassium, metallic sodium, potassium tert-butoxide etc.; the reaction is carried out in a solvent of tetrahydrofuran or dimethoxyethane at a temperature of from room temperature to a boiling point of the solvent for 1 to 24 hours. In case of using a phase transfer catalyst such as a quaternary ammonium salt (e.g. tetra-n-butylammonium bromide, methyltrioctylammonium chloride, etc.), the reaction is carried out in two phases of benzene or dichloromethane and 50 % aqueous sodium hydroxide or 60 % aqeuous potassium hydroxide at a temperature of from 0°C to a boiling point of the solvent for one minute to 24 hours.


- Reaction Scheme-II:
-

wherein X is a halogen such as bromine or chlorine.
-
The above process can be carried out by reacting a compound (IV) and an equimolar amount of a compound (V) in a solvent such as acetonitrile, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or an alcohol (e.g. ethanol) at a temperature of from 50°C to a boling point of the solvent for 10 minutes to 4 hours.

- Reaction Scheme-III:
-




- Reaction Scheme-IV:
-

wherein R1 is as defined above.
-
The process can be carried out by converting a compound (VII) into an oxime (VIII) by a conventional oxime forming reaction, heating the oxime (VIII) in acetic anhydride to obtain a compound (IX), and treating the compound (IX) with hydrogen sulfide, that is, by blowing hydrogen sulfide gas into a reaction system containing the compound (IX) in a solvent such as DMF, DMSO or pyridine in the presence of 0.5 to 5 equimolar amount of a base such as a tertiary amine (e.g. triethylamine) at a temperature of from 0° to 40°C for 3 to 24 hours

- Reaction Scheme-V:
-


- Reference Example 6
- 4,5-Bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole [compound of the formula (II)]:
-
Pyrrole-2-carbothioamide (cf. J. Org. Chem., 38, 667, 1973) (1.51 g, 12 mmole) and α-bromo-4,4′-dimethoxy- deoxybenzoin (cf. Aust. J. Chem., 8, 385. 1955) (4.02 g, 12 mmole) are dissolved in acetonitrile (120 ml). The mixture is stirred at 60°C for 50 minutes. After the reaction, the reaction mixture is distilled under reduced pressure to remove the solvent. To the resulting residue are added chloroform and aqueous solution of sodium carbonate, and the mixture is shaken. The chloroform layer is taken, and the aqueous layer is further extracted with chloroform. The chloroform layers are combined, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to remove the solvent. The residue is recrystallized from ligroin to give 4,5-bis(4-methoxyphenyl)-2-(pyrrol-2-yl)-thiazole (3.74 g, yield: 86 %).
-
M.p. 131.5 – 134.0°C
-
NMR (CDCl3, δ ppm): 3.7 (6H) , 6.1 (1H, dd) , 6.5-6.9 (6H), 7.1-7.5 (4H), 9.4-9.8 (lH).
-
Elementary analysis for C21H18N2O2S:


- Example 14
-
Ethyl 2-[4,5-bis(4-methoxyphenyl)thiazol-2-yl]-pyrrole-1-acetate (compound of the formula (I) wherein R1 = -CH2-COOC2H5):
- 4,5-Bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole obtained in the same manner as described in Reference Example 6 (3.62 g, 10 mmole), ethyl bromoacetate (1.67 g, 10 mmole), and tetra-n-butylammonium bromide (0.32 g, 1 mmole) are refluxed with vigorous stirring in two phases of dichloromethane (40 ml) and 50 % aqueous sodium hydroxide (40 ml) at room temperature for 2 minutes. To the mixture are added water and dichloromethane under ice-cooling, and the mixture is shaken. The dichloromethane layer is taken, and the aqueous layer is further extracted with dichloromethane. The dichloromethane layers are combined, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to remove the solvent. The residue is recrystallized from ligroin to give ethyl 2-[4,5-bis(4-methoxyphenyl)thiazol-2-yl]pyrrole-1-acetate (3.64 g, yield: 81 %).
-
M.p. 132.5 – 135.5°C
-
NMR (CDCl3, δ ppm): 1.2 (3H, t), 3.8 (6H), 4.15 (2H, q), 5.25 (2H, s), 6.25 (1H, dd), 6.7-6.95 (6H), 7.2-7.55 (4H).
-
Elementary analysis for C25H24N2O4S:


Determination of the antiplatelet agent. KB-3022, and its metabolite by high-performance liquid chromatography.Nakada Y, Ikuta Y, Kawashima T, Awata N.Chem Pharm Bull (Tokyo). 1990 Apr;38(4):1093-5.
 pamicogrel
pamicogrel| EP0037274A1 * | 30 Mar 1981 | 7 Oct 1981 | Eli Lilly And Company | Substituted triaryl thiazole compounds |
| EP0077024A2 * | 7 Oct 1982 | 20 Apr 1983 | Schering Aktiengesellschaft | Imidazole derivatives, process for their preparation and pharmaceutical products containing them |
| US4168315 * | 28 Sep 1977 | 18 Sep 1979 | The Upjohn Company | Dianisyl thiazole compound, compositions and method of antithrombotic treatment |
TAKEDA PHARMACEUTICALS 武田薬品工業株式会社 ON THE RISE
Tadataka Yamada, M.D., Chief Medical & Scientific Officer of Takeda

TAKEDA US CHICAGO OFFICE
TAKEDA PIPELINE SEE LINKS BELOW
1 https://www.takeda.com/investor-information/annual/files/ar2013_10_en.pdf
2. http://www.takeda.com/research/files/pipeline_20131031_en.pdf
3 http://www.takeda.com/research/pipeline/
- 2012 Download Entire File
 PDF 0.4MB 34P
PDF 0.4MB 34P
Takeda’s top executives had frequently pointed to TAK-875 as one of their best shots at coming up with an important new approach to treating diabetes. The drug is designed to spur insulin secretion in the pancreas and Takeda had confidently projected an approval in Japan in 2015 with a follow-up approval in the big U.S. market a year or two later.
The termination of the high-profile program caused some anxiety among investors. Takeda’s shares plunged 8% on the loss as analysts wondered how the pharma company could counter the loss of Actos, a $3.7 billion drug that accounted for about a quarter of its revenue in 2011.
Takeda won an approval on a trio of DPP-4 diabetes drugs–Nesina (alogliptin) and two combos with alogliptin, dubbed Oseni and Kazano–at the beginning of the year. But Takeda suffered some big delays in gaining acceptance, a common fate in this field, where regulators are particularly cautious about new drugs. And Merck had already solidified its lead in the DPP-4 market with Januvia whileOnglyza trailed closely behind it. Takeda had hoped that a combination of TAK-875 and Januvia could help regain some lost market territory–but that dream has clearly vanished as well.
| January 27, 2014 | |
| January 22, 2014 | |
| January 17, 2014 | |
| January 14, 2014 | |
| January 10, 2014 |
2013
| December 27, 2013 | ||
| December 25, 2013 | ||
| December 25, 2013 | ||
| December 24, 2013 | ||
| December 20, 2013 | ||
| December 20, 2013 | ||
| December 19, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 9, 2013 | ||
| December 5, 2013 | ||
| December 4, 2013 | ||
| November 30, 2013 | ||
| November 21, 2013 | ||
| November 19, 2013 | ||
| November 14, 2013 | ||
| November 12, 2013 | ||
| November 12, 2013 | ||
| October 21, 2013 | ||
| October 7, 2013 | ||
| October 2, 2013 | ||
| October 1, 2013 |
| September 26, 2013 | ||
| September 26, 2013 | ||
| September 24, 2013 | ||
| September 20, 2013 | ||
| September 20, 2013 | ||
| September 13, 2013 | ||
| September 13, 2013 | ||
| September 5, 2013 | ||
| September 2, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 22, 2013 | ||
| August 13, 2013 | ||
| August 1, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 30, 2013 | ||
| July 29, 2013 | ||
| July 26, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 18, 2013 | ||
| July 10, 2013 | ||
| July 1, 2013 |
CLIPPED
Takeda isn’t quite in the top 10 among global drugmakers, but the company boasts the 7th-largest pipeline in the industry, according to its presentation at the conference. Yamada noted that 31% of the pipeline assets are in late-stage trials. Millennium is leading development of three late-stage contenders, TAK-700 for prostate cancer, MLN9708 for multiple myeloma and MLN0002 for ulcerative colitis andCrohn’s disease.
In an effort to revive its diabetes franchise, Takeda is in the final stage of development for a first-of-a-kind GPR40 agonist called TAK-875, designed to provide glucose-dependent insulin secretion.
With a rich late-stage pipeline at Takeda, Yamada wants the company to focus on growing its ranks of earlier-stage drug candidates. To do this the company has landed a variety of deals, including the purchase of Intellikine for $310 million to acquire anti-cancer drugs and more recently the acquisition of Envoy Therapeutics last year for $140 million.
Takeda has formed a New Frontier Science group to scout out the hottest research in academia and elsewhere and form collaborations with scientists behind those innovations. At the J.P. Morgan conference, Yamada said, he was attending many meetings with members of the biotech community.

Takeda Pharmaceutical Company Limited (武田薬品工業株式会社 Takeda Yakuhin Kōgyō Kabushiki-gaisha?) is the largest pharmaceutical company in Japan and Asia and a top 15 pharmaceutical company. The company has over 30,000 employees worldwide and achieved $16.2 billion USD in revenue during the 2012 fiscal year.[1] The company is focused on metabolic disorders, gastroenterology, neurology, inflammation, as well asoncology through its independent subsidiary, Millennium: The Takeda Oncology Company.[2] Its headquarters is located in Chuo-ku, Osaka, and it has an office in Nihonbashi, Chuo, Tokyo.[3][4] In January 2012, Fortune Magazine ranked the Takeda Oncology Company as one the 100 best companies to work for in the United States.
Takeda Pharmaceuticals was founded on June 12, 1781 and was incorporated on January 29, 1925.
Takeda’s Japanese logo
In 1977, Takeda first entered the U.S. pharmaceutical market by developing a joint venture with Abbott Laboratories called TAP Pharmaceuticals.[5]Through TAP Pharmaceuticals, Takeda and Abbott launched the blockbusters Lupron (leuprolide) in 1985 and Prevacid (lansoprazole) in 1995.
One of the firm’s mainstay drugs is Actos, a compound in the thiazolidinedione class of drugs used in the treatment of type 2 diabetes. Launched in 1999, Actos has become the best-selling diabetes drug in the world with $4 billion USD in sales during the 2008 fiscal year.[6]
In February 2005, Takeda announced its acquisition of San Diego, California-based Syrrx, a company specializing in high-throughput X-ray crystallography, for $270 million.[7]
In February 2008, Takeda acquired the Japanese operations of Amgen and rights to a dozen of the California biotechnology company’s pipeline candidates for the Japanese market.[8]
In March 2008, Takeda and Abbott Laboratories announced plans to conclude their 30-year old joint venture, TAP Pharmaceuticals, that had over $3 billion in sales in its final year. The split resulted in Abbott acquiring U.S. rights to Lupron and the drug’s support staff. On the other hand, Takeda received rights to Prevacid and TAP’s pipeline candidates. The move also increased Takeda’s headcount by 3,000 employees.[9]
In April 2008, Takeda announced that it was acquiring Millennium Pharmaceuticals of Cambridge, Massachusetts, a company specializing in cancerdrug research, for $8.8 billion. The acquisition brought in Velcade, a drug indicated for hematological malignancies, as well as a portfolio of pipeline candidates in the oncology, inflammation, and cardiovascular therapeutic areas. Millennium now operates as an independent subsidiary, serving as the global center of excellence in oncology under its new name: “Millennium: The Takeda Oncology Company.” [10]
In May 2008, the company licensed non-exclusively the RNAi technology platform developed by Alnylam Pharmaceuticals, creating a potentially long-term partnership between the companies.[11]
On May 19, 2011, Takeda Pharmaceutical and Nycomed announced that Takeda will acquire Nycomed for € 9.6 billion. The acquisition was completed by September 30, 2011.[12]
On April 11, 2012, Takeda Pharmaceutical and URL Pharma announced that Takeda will acquire URL Pharma for $800 million. The acquisition is expected to be completed within 60 days.
On 25 May 2012, Takeda announced the purchase of Brazilian pharmaceutical company Multilab by R$ 540 million.[13]

Takeda Midosuji Building, headquarters of Takeda Pharmaceutical Company, inChuo-ku, Osaka, Japan
Takeda operates two primary bases in Japan in Osaka and Tokyo. Its United States subsidiary is based in Deerfield, Illinois, and all Global Operations outside of Japan and U.S. are based in Opfikon (Zurich), Switzerland. The company maintains research & development sites in Osaka and Tsukuba, Japan; San Diego andSan Francisco, United States; Cambridge, United Kingdom; and Singapore.[14]
The company has manufacturing facilities in Japan, China, Indonesia, Italy, and Ireland.[15] Following the Nycomed acquisition, the Takeda manufacturing sites have been extended with facilities in Argentina,Austria,Belgium,Brazil,Denmark, Estonia,Germany,Mexico,Norway and Poland. Takeda has overseas marketing presences in the U.S., UK, France, Italy, Germany, Austria, Switzerland, Spain, China, Taiwan, Philippines, Thailand, Indonesia, and Singapore. It has recently[when?] announced its first foray into Canada, Portugal, Spain, Mexico, and Ireland.[15]

AT INDONESIA
Products
Some of the key products that Takeda produces on behalf of partners include:[16]
- Actos (pioglitazone) – Type 2 Diabetes
- Amitiza (lubiprostone) – Chronic idiopathic constipation
- Basen (voglibose) – Type 2 Diabetes
- Benet (risedronic acid) – Osteoporosis (Japan)
- Blopress (candesartan) – Hypertension
- Enbrel (etanercept) – Inflammatory diseases (Japan)
- Dexilant (dexlansoprazole) – Gastroesophageal reflux disease – name changed to Dexilant in U.S.
- Lupron/Leuplin (leuprorelin) – GnRH agonist for prostate cancer and endometriosis
- Prevacid/Takepron (lansoprazole) – Gastroesophageal reflux disease
- Rozerem (ramelteon) – Insomnia
- Uloric (febuxostat) – Gout
- Velcade (bortezomib) – Multiple myeloma and mantle cell lymphoma (Millennium Pharmaceuticals)
![]() AT UK
AT UK
References
- “Financial Results for Fiscal 2012” (PDF). Takeda Pharmaceutical Company Limited. May 9, 2013. Retrieved June 13, 2013.
- “Takeda Initiates Cardiovascular Outcomes Trial for Alogliptin, An Investigational Treatment for Type 2 Diabetes”. Newsblaze.com. 2009-08-28. Retrieved 2010-09-18.
- “FAQ.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Q : Where is Takeda located? A : The Head Office is located in Osaka, Japan, and the Tokyo Head Office is located in Tokyo, Japan.”
- “Overview.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Headquarters Head Office 1-1, Doshomachi 4-chome, Chuo-ku, Osaka 540-8645” and “Tokyo Head Office 12-10, Nihonbashi 2-chome, Chuo-ku, Tokyo 103-8668”
- “TAP Pharmaceutical Products, Inc.: Private Company Information – BusinessWeek”. Investing.businessweek.com. 2008-04-30. Retrieved 2010-09-18.
- Decker, Susan (2009-07-06). “Takeda Sues Torrent to Stop Generic Copy of Actos Diabetes Pill”. Bloomberg. Retrieved 2010-09-18.
- Somers, Terri (2005-02-08). “Japanese drug giant taking over Syrrx here | The San Diego Union-Tribune”. Signonsandiego.com. Retrieved 2010-09-18.
- “Takeda, Amgen in exclusive tie-up for Japanese market”. MarketWatch. 2008-02-04. Retrieved 2010-09-18.
- Marrazzo, Amanda (2008-05-15). “Featured Articles From The Chicago Tribune”. Archives.chicagotribune.com. Retrieved 2010-09-18.
- “MILLENNIUM: The Takeda Oncology Company | About Millennium | Our History”. Mlnm.com. Retrieved 2010-09-18.
- staff (2008-06-15). “Takeda Signs On as Alnylam’s Asian Partner for $150M Upfront”. Genetic Engineering & Biotechnology News (print) (Mary Ann Liebert, Inc.). p. 14.
- http://www.takeda.com/press/article_43116.html
- Hirschler, Ben (May 25, 2012). “Farmacêutica Takeda comprará Multilab por até R$ 540 mi”. Grupo Abril (in portuguese). Exame. Retrieved January 27, 2013.
- “Locations | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “By Business | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “Annual Reports | Investor Information | Takeda Pharmaceutical Company Limited”. Takeda.com. Retrieved 2010-09-18.
 |
|
| Native name | 武田薬品工業株式会社 |
|---|---|
| Type | Public KK |
| Traded as | |
| Industry | Pharmaceuticals |
| Founded | Doshomachi, Osaka, Japan (June 12, 1781) |
| Headquarters | 1-1, Doshomachi Yonchome,Chuo-ku, Osaka, Japan |
| Key people | Yasuchika Hasegawa (President & CEO) |
| Revenue | |
| Operating income | |
| Net income | |
| Total assets | |
| Total equity | |
| Employees | 30,481 (2012) |
| Website | takeda.com (Global website) |
References:
|
|

CMC CENTRE
The Chemistry, Manufacturing and Controls (CMC) Center is a global organization responsible for overall R&D activities ranging from chemical information on development candidates to the processes leading to “manufacturing” of pharmaceutical products.
The main sites are located in Osaka and consist of the following laboratories: the Chemical Development Laboratories in charge of R&D for developing the manufacturing methods of active pharmaceutical ingredients and the manufacturing of drug substances for clinical samples; the Pharmaceutical Technology R&D Laboratories in charge of R&D for dosage forms, manufacturing and packaging, as well as manufacturing of clinical samples; and the Analytical Development Laboratories in charge of R&D for the development of analytical methods and stability studies of clinical samples. In addition, Hikari Bio-Manufacturing Technology Laboratories is located in Hikari (Yamaguchi) and this is where antibody drug substances are manufactured.
As for overseas sites, the Cambridge Biologics CMC Group (Massachusetts) and the Chicago Pharmaceutical Science Group (Illinois) are located in the USA, while the CMC Center Europe is mainly located in Roskilde, Denmark. All research and development activities at Takeda are promoted with the cooperation of these sites.
List of Publications of Takeda Research Laboratories
Trelagliptin succinate (SYR-472) for the treatment of type 2 diabetes.

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)

trelagliptin succinate
Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE NOF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I



Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
US20080227798 AND US20120197018
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
13 NMR TRELAGLIPTIN SUCCINATE

1H NMR TRELAGLIPTIN SUCCINATE

Zanamivir, Relenza…For the prevention and treatment of influenza A and B.

Zanamivir
139110-80-8
APPROVED 26-7-96……. GSK NDA 021036
A guanido-neuraminic acid that is used to inhibit neuraminidase.
Zanamivir INN /zəˈnæmɨvɪər/ is a neuraminidase inhibitor used in the treatment and prophylaxis of influenza caused by influenza A virus andinfluenza B virus. Zanamivir was the first neuraminidase inhibitor commercially developed. It is currently marketed by GlaxoSmithKline under the trade name Relenza as a powder for oral inhalation.
The drug is approved for use for the prevention and treatment of influenza in those over the age of 7 in the United States, Canada, European Union, and many other countries. It is not recommended for people with respiratory problems and ailments.
| United States | 6294572 | APPROVED 1994-12-15 | EXPIRY 2014-12-15 |
| United States | 5360817 | 1993-07-26 | 2013-07-26 |
| Canada | 2291994 | 2003-10-14 | 2011-04-24 |
| Canada | 2081356 | 2000-02-22 | 2011-04-24 |
| Patent No | PatentExpiry | use code |
|---|---|---|
| 5360817 | Jul 26, 2013 | |
| 5648379 | Jul 15, 2014 | U-274 |
| 5648379 | Jul 15, 2014 | U-721 |
| 5648379 | Jul 15, 2014 | U-722 |
| 6294572 | Dec 15, 2014 |
Zanamivir was discovered in 1989 by scientists led by Peter Malcolm Colman and Joseph Varghese at the CSIRO, in collaboration with theVictorian College of Pharmacy, Monash University, and scientists at Glaxo, UK. Zanamivir was the first of the neuraminidase inhibitors. The discovery was initially funded by the Australian biotechnology company Biota and was part of Biota’s ongoing program to develop antiviral agents throughrational drug design. Its strategy relied on the availability of the structure of influenza neuraminidase, by X-ray crystallography. It was also known, as far back as 1974, that 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA), a sialic acid analogue, is an inhibitor of neuraminidase. Sialic acid (N-acetyl neuraminic acid, NANA), the substrate of neuraminidase, is itself a mild inhibitor of the enzyme, but the dehydrated derivative DANA, a transition-state analogue, is a better inhibitor.
Computational chemistry techniques were used to probe the active site of the enzyme, in an attempt to design derivatives of DANA that would bind tightly to the amino acid residues of the catalytic site, and so would be potent and specific inhibitors of the enzyme. The GRID software by Molecular Discovery was used to determine energetically favourable interactions between various functional groups and residues in the catalytic site canyon. This investigation showed that there is a negatively charged zone in the neuraminidase active site that aligns with the C4hydroxyl group of DANA. This hydroxyl is, therefore, replaced with a positively charged amino group; the 4-amino DANA was shown to be 100 times better as an inhibitor than DANA, owing to the formation of a salt bridge with a conserved glutamic acid (119) in the active site. It was also noticed that Glu 119 is at the bottom of a conserved pocket in the active site, just big enough to accommodate a more basic functional positively charged group, such as a guanidino group, which was also larger than the amino group. Zanamivir, a transition-state analogue inhibitor of neuraminidase, was the result.
As Biota was a small company, it did not have the resources to bring zanamivir to market by itself. In 1990, zanamivir patent rights were licensed to Glaxo, now GlaxoSmithKline (GSK). In 1999, the product was approved for marketing in the US and subsequently has been registered by GSK in a total of 70 countries (GlaxoSmithKline News release, 2006). Zanamivir is delivered via Glaxo’s proprietary Diskhaler inhalation device. The license agreement entitled Biota to receive a 7% royalty on Glaxo’s sales of zanamivir.
|
Chemical name: |
5- Acetamido- 2, 6- anhydro- 3, 4, 5- trideoxy- 4- guanidino- D- glycero- D- galacto- non- 2- enonic acid |
| Synonyms: | Zanamivir, GG167, 4-guanidino-Neu5Ac2en and 2,3- Didehydro- 2, 4- dideoxy- 4- guanidino- N- acetyl- D- neuraminic acid(2R,3R,4S)-4-guanidino-3-(prop-1-en-2-ylamino)-2-((1R,2R)-1,2,3-trihydroxypropyl)-3,4-dihydro-2H-pyran-6-carboxylic acid |
| Empirical formula: |
C12H20N4O7 |
| Structural formula: | |
| Molecular weight: | 332.31g |
| Beilstein number: | 7083099 |
| Normal State: | Powder |
| Colour: | White to ‘off white’ |
| Melting point: | 325oC |
| Optical rotary power: | Type [�]Conc: 0.9g/100mlSolvent: H2OOptical rotary power: 41 degWavelength: 589nmTemp: 20oC |
| CAS number: | 139110-80-8 |
| Solubility: | 18mg/mL in water at 20oC |
Zanamivir is used for the treatment of infections caused by influenza A virus and influenza B virus. There is low to moderate evidence that it decreases the risk of one’s getting influenza by 1% to 12% in those exposed. In otherwise-healthy individuals, benefits overall appear to be small.It is unclear whether it affects the risk of one’s need to be hospitalized or the risk of death. An independent analysis of its effects by the Cochrane collaboration was awaiting release of trial data as of 2012. The evidence for a benefit in preventing influenza is weak in children with concerns of publication bias in the literature. As of 2009 no influenza has shown any signs of resistance. Since then genes expressing resistance to were found in patients infected with Influenza A H7N9 and who were treated with corticosteroids.
 ZANAMIVIR
ZANAMIVIR
Mass

| 1H NMR |
| Hydrogen | Chemical shift /ppm |
| (1H, d, 3-H) | 5.53 |
| (2H, 2dd, 4- and 6-H) | 4.50 – 4.38 |
| (1H, dd, 5-H) | 4.21 |
| (2H, dd+ddd, 9-Ha and 8-H) | 4.00-3.88 |
| (2H, 2dd, 9-Hb and 7-H) | 3.70-3.62 |
| (3H, s, Ac) | 2.05 |
|
|
| 13C NMR |
| Carbon | Shift /ppm |
| (C=O, Ac) | 177.3 |
| (C-1) | 172.1 |
| (guanidino) | 159.9 |
| (C-2) | 152.1 |
| (C-3) | 106.8 |
| (C-6) | 78.3 |
| (C-8) | 72.6 |
| (C-7) | 71.0 |
| (C-9) | 65.9 |
| (C-4) | 54.0 |
| (C-5) | 50.6 |
| (Me) | 24.8 |
ref 12
IR spectra:
| The following peaks are present in the IR spectra of Relenza: 3332cm-1, 1676cm-1, 1600cm-1, 1560cm-1, 1394cm-1, 1322cm-1 and 1281cm-1. |
UV spectra
| The maximum peak is 235nm giving E = 199 dm-3 mol-1cm-1 |
ref 13for above

Although zanamivir was the first neuraminidase inhibitor to the market, it had only a few months lead over the second entrant, oseltamivir (Tamiflu), with an oral tablet formulation.
According to the CDC, Tamiflu, zanamivir’s main competitor, is not as effective at treating the influenza viruses as zanamivir, especially in H1N1 seasonal flu. In fact, tests showed 99.6% of the tested strains of seasonal H1N1 flu and 0.5% of 2009 pandemic flu were resistant to Tamiflu, while no flu samples, seasonal or pandemic, showed any resistance to zanamivir.
When first marketed in the US in 1999/2000, zanamivir captured only 25% of the influenza antiviral market, despite a huge promotional campaign. By the end of that season, Tamiflu was outselling zanamivir 3:1. During that season, zanamivir experienced worldwide safety warnings involving the risk of bronchospasm and death. Glaxo then reduced the marketing of zanamivir, and Tamiflu’s dominance increased. More than US$20 million worth of zanamivir sold by Glaxo in the first US season was returned to the company in the next two seasons because zanamivir’s sales to patients were far less than expected.
Biota commenced legal proceedings in 2004 alleging Glaxo’s reduced marketing of zanamivir to be a breach of contract. Biota claimed approximately A$700m from Glaxo. After Biota spent four years trying to progress its case, and incurring A$50m in legal costs, the company abandoned the claim in July 2008, recovering only A$20 million, including legal costs following settlement at mediation. Biota had refused an earlier tactical offer from Glaxo of A$75 million plus legal costs.
In August 2006, Germany announced it would buy 1.7 million doses of zanamivir, as part of its preparation strategy against bird flu. “Germany’s purchase shows that countries are starting to take a balanced view of influenza preparedness,” says Simon Tucker, head of research at Melbourne-based Biota, where zanamivir was originally developed.
In April 2009, many cases of swine flu (H1N1-type virus) were reported in US and Mexico. Zanamivir is one of only two drugs prescribed to treat it. A study published in June 2009 emphasized the urgent need for augmentation of oseltamivir (Tamiflu) stockpiles, with additional antiviral drugs including zanamivir, based on an evaluation of the performance of these drugs in the scenario that the 2009 H1N1 swine flu neuraminidase (NA) were to acquire the Tamiflu-resistance (His274Tyr) mutation, which is currently widespread in 99.6% of all tested seasonal H1N1 strains.n January 2011, GSK announced that it would commence phase III trials for intravenous zanamivir in a study that will span 20 countries in the Northern and Southern Hemispheres.
Recently, the reported oseltamivir-resistance H5N1 virus neuraminidase still retaining susceptibility to zanamivir indicates that the structure of zanamivir has some advantages over oseltamivir in binding to the active pocket of H5N1 neuraminidase.
As a proven anti-influenza drug target, neuraminidase continues to be attractive for the development of new inhibitors. The crystal structure of H5N1 avian influenza neuraminidase (PDB code: 2HTY) provides the three-dimensional structural information and opportunity for finding new inhibitors in this regard, because the existing inhibitors, such as oseltamivir and zanamivir, were developed based on different structures of neuraminidase, such as subtypes N9 and N2, and type B genus of influenza virus.
ZANAMIVIR
Chemistry

- Scheigetz, J.; Zamboni, R.; Bernstein, M. A.;Roy, B. (December 1995). “A syntheses of 4-a-guanidino-2-deoxy-2,3-didehydro n-acetylneuraminic acid”. Organic Letters 27 (6): 637–644.doi:10.1021/ol901511x. Retrieved 2010-11-14.
Zanamivir synthetic process in the world
Together with oseltamivir, zanamivir is the only medicine which can prevent influenza on humans caused by H5N1 and H1N1 virus. Vietnam prepared oseltamivir (Tamiflu) medicine. But there was no zanamivir – the first influenza medicine belonging N1 kind, discovered and commercialized before oseltamivir. The scientific name of zanamivir is acid 5-acetamido-4-guanidino-6-(1,2,3-trihydroxy-propyl)-5,6-dihydro-4H-pyran-2-carboxylic. The discovery of zanamivir opens research possibilities for new medicines which have the same effect on enzyme neuraminidase inhibitor to prevent and treat influenza.
Acid sialic is an input to synthetize zanamivir. The name acid sialic (Neu5Ac2en) is used to indicate derivation at O- and N- positions of acid neuraminic, just for acid N-axetylneuraminic. Acid sialic of carbohydrate groups is on animal cells and microorganism, especially in glycoprotein and gangliosid. The commercial acid sialic is extracted from whey of the cheese and milk process as well as egg yolk, and costs about 5,000 USD per kilo.
In 1994, zanamivir was first synthesized and made public by Von Itzstein and other scientists from the Department of Pharmaceutical Chemistry under Monash University (Australia). Then, Chandler and co-workers of Glaxo company (GSK, Britain) acquired results, improved reaction steps and made them public in 1995. Accordingly, this method produced 8.3% of general output. The synthetic process is described in Figure 1.

Figure 1: Zanamivir synthetic process according to Chandler
Up to now, the research of Chandler has been the only publication about zanamivir synthetic method, the output of which is greater than milligrams, and it reproduces details about reaction conditions and physiochemical properties of the requisite substances.
Recently, a research group of Yao (China) proposed a new approach to synthetize into intermediate compound 5. Researchers started from another material – D-glucono-δ-lactone, which is cheaper than acid sialic. However, the synthetic process is longer and much complicated, including 24 steps, with lower productivity (0.2%).
Researching on synthesizing Zanamivir from Acid sialic by Institute of Chemistry
Synthetizing methyl N-acetylneuraminate (2) and O-pentaacetoxy (3) from acid sialic
Scientists from the Institute of Chemistry used acid sialic (axit N-acetylneuraminic) 98% from China as the input for the zanamivir synthetic process. They decided to use the method of Warner, using ion exchange resin Dowex-H, with the role of catalyst. Reaction was performed in the room in 10 hours. The output was metyl (2) este product of acid N-acetylneuraminic with a productivity of 99%.
Then, to synthetize O-pentaacetoxy (3), scientists applied axetyl effective chemistry method recently published, using BF3.OEt2catalysis at 00C. Productivity in this case exceeded 95%.

Figure 2: The diagram of O-pentaacetoxy 3 derivative making
The use of catalysts which were ion exchange resin Dowex-H (for este chemical reaction) and BF3.OEt2 (for axetyl chemical reaction) had more advantages than the method by scientists from Glaxo.
Synthesizing intermediate compound – oxazoline (4) key from O-pentaacetoxy (3)

Figure 3: Diagram to synthesize oxazoline (4) from O-pentaacetoxy (3) according to a and b methods
Firstly, scientists conducted a survey on oxazoline (4) synthetic process according to Chandler’s process. O-pentaacetoxy (3) compound was separated from two types of OAc and formed oxazoline round thanks to the effect of strong acid Lewis, which was TMSOTf at 520C in 2.5 hour. The productivity of this reaction achieved 40%. The pilot instead of TMSOTf by BF3.OEt2 catalysis in dichloromethane at room temperature at night, the productivity of the reaction to form oxazoline round from penta-acetoxy (5) was similar to the method using TMSOTf (42%). To increase productivity, scientists made a survey on one-pot method, directly from metyl este (2) to oxazoline (4), without passing O-pentaacetoxy (3), gave the highest productivity (73,3%) and was the most economic effectiveness.
Synthesizing zanamivir from oxazoline (4) intermediate compound
The next, scientists successfully conducted reactions from oxazoline (4) intermediate compound to Zanamivir (9) final product (Figure 1). Zanamivir product had IR and NMR data which were compatible with their structure.
Therefore, scientists from the Institute of Chemistry under Vietnam Academy of Science and Technology built a stable process, including seven major steps, synthesizing from acid sialic with the general productivity of 6.6% (the productivity made public in the world was 8.3%). Especially, in the first period, from acid sialic to oxazolin (4) was optimized and gave a general productivity of 74%, higher than the productivity made public by (61.7%). However, the productivity gained in the later period is still low. Now, synthesizing zanamivir influenza medicine still continues to be researched.
……………………
Beau and coworkers assembled the core dihydropyran framework of zanamivir congeners via a combination of PBM reaction and Iron(III)-promoted deprotection-cyclization sequence. A stereochemically-defined α-hydroxyaldehyde 2, diallylamine and a dimethylketal-protected boronic acid 1 is coupled to form the acyclic, stereochemically-defined amino-alcohol 3, which then undergoes an Iron(III)-promoted cyclization to form a bicyclic dihydropyran 4. Selective opening of the oxazoline portion of the dihydropyran intermediate 4 with water or timethylsilyl azide then furnish downstream products that have structures resembling the Zanamivir family members.

| Reaction scheme part 1: |
| The commercially available N-acetyl-neuraminic acid 1 is the starting reagent for the most direct approach to the synthesis of 4-guanidino-Neu5Ac2en (Relenza). In reaction scheme 1 the steps for the conversion of N-acetyl-neuraminic acid 1 to its 4-amino analogue is shown. Step 1 is the addition of methanolic HCl (MeOH and HCl gas), which produces the methyl ester of 1, followed by acetic anhydride in pyridine with 4-(dimethylamino)pyridine catalysis, which produces the penta-acetoxy compound, 2. In step 2, 2 is converted into the oxazoline 3 at high yield using trimethylsilyl trifluoromethanesulfonate (TMSOTf) in ethyl acetate at 52oC. In step 3, the azido compound, 4, is produced by the reaction of 3 with trimethylsilyl azide in tert-butyl alcohol at 80oC. In step 4 catalytic sodium methoxide in methanol was used to remove the acetate protecting groups from 4 to give triol 5. The 4-amino analogue, 6 was made in step 5, by hydrolysis using triethylamine in water, hydrogenolysis with a Lindlar catalyst and finally the addition of Dowex 2 * 8 resin. The triethylamine salt of the 6 was made during hydrogenolysis and the purpose of the Dowex 2 * 8 resin was to desalt this intermediate. The chemical names of the compounds are: |
| 1: N-acetyl-neuraminic acid |
| 2: 5- Acetamido- 3,5- dideoxy- D- glycero- �- D- galacto- 2- nonulo- pyranosonic acid methyl ester |
| 3: Methyl (3aR, 4R, 7aR)- 2- Methyl- 4- [(1’S, 2’R)- 1′, 2′, 3′ – triacet- oxypropyl]- 3a, 7a- dihydro- 4H- pyrano [3, 4-d] oxazole- 6- carboxlate. |
| 4: 5- Acetamido- 7, 8, 9- tri- O- acetyl- 2, 6- anhydro- 4- azido- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid methyl ester. |
| 5: 5- Acetamido- 2, 6- anhydro- 4- azido- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid methyl ester. |
| 6: 5- Acetamido- 4- amino- 2, 6- anhydro- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid. |

Part one of reaction scheme
| Synthesis of reactant necessary for part 2 of reaction: |
| Aminoiminomethane-sulfonic acid (AIMSA), 7, which is necessary for the conversion of compound 6 into Relenza, 9, is synthesised in Reaction scheme 2. The oxidizing solution necessary for the reaction is prepared by the addition of peracetic acid to 30% hydrogen peroxide and then conc. sulfuric acid. This is followed by acetic anhydride and, once the reaction has completed, methanol. Thiourea is dissolved in methanol and added slowly to the oxidizing solution.to produce compound 7. Note that any crystals that form are removed and that the reaction needs to be carried out under cooled conditions. See the reference source for more experimental details. |

Synthesis of AIMSA
| Reaction scheme part 2: |
| Reaction scheme 3 shows the conversion of compound 6 into Relenza For route A, 3 mol equivalent of AIMSA, 7, and 3 mol equivalent of potassium carbonate are added in a portionwise manner to compound 6 over an eight hour period. A yield of about 48% of the crystalline product 8 should be obtained for this method. An alternative route is to treat compound 6 with 1.1 mol equivalent of cyanogen bromide in the presence of sodium acetate in methanol. Route B step 1 gives compound 9, which can be converted into the final product 8 by treating it with ammonium hydroxide and ammonium formate at 85oC. A 36% yield of the purified product can be obtained after purification with ion-exchange chromatography and crystallisation. The chemical names of the compounds in this scheme are: |
| 8. 5- Acetamido- 2, 6- anhydro- 3, 4, 5- trideoxy- 4- guanidino- D- glycero- D- galacto- non- 2- enonic acid. (Relenza) |
| 9. 5- Acetylamino- 2, 6- anhydro- 4- cyanoamino- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid |

Part 2 of reaction scheme
ref are 13 and 14
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
SYNTHESIS FROM PATENT EP2276479A2
ZANAMIVIR AND BOC PROTECTED ZANAMIVIR
The synthesis of zanamivir is shown in Scheme 1. The starting material used for zanamivir synthesis is sialic acid 1, which was converted to the methyl ester 2, in presence of Dowex H+ as described in detail in reference 104. The hydroxyl groups of 2 are protected with acetyl groups to give compound 3, which was then converted to the oxazoline derivative 4 in the presence of trimethyltrifluoromethanesulfonate as described in detail in reference 105. Azide 5 was synthesized from 4 in presence of azidotrimethylsilane as described in detail in reference 105. The azide is reduced to the corresponding amine 6 by using Lindlar’s catalyst, and the amine is in turn converted to the guanidine derivative 7 as described in detail in reference 106. The final step involves the deprotection of the methyl ester and acetyl groups in the presence of methanolic sodium hydroxide to give Boc-protected zanamivir 8 as described in detail in reference 106. 8, 1H NMR (CD3OD) δ (ppm) 5.6 (d, J = 2.0 Hz, IH), 5.01 (dd, J = 9.6, 2.1 Hz, IH), 4.25 (dd, J = 10.8, 1.1 Hz, IH), 4.18 (dd, J = 10.6, 9.6 Hz, IH), 3.89 (ddd, J = 9.4, 6.2, 2.7 Hz, IH), 3.84 (dd, J = 11.3, 2.8 Hz, IH), 3.67 (dd, J = 11.3, 5.8 Hz, IH), 3.57(d, J = 9.3 Hz, IH), 1.9 (s, 3H), 1.55 (s, 9H), 1.50 (s, 9H); ESI-MS: 533 (M+H)+.
Scheme 1



a) Dowex H Methanol b) Aceticanhydride DMAP pyridine c) trimethylsilyl tπfluorαmethane sulfonate ethylacetate d) azidotrimethylsilane butanol e) Lindlar’s catalyst ethanol f) N N’-bis-tert-butoxycarbonyMH-pyrazole-i carboxamidine tetrahydrofuran g) sodium hydroxide methanol
104. Martin, R., K.L. Witte, and C-H. Wong, The synthesis and enzymatic incorporation of sialic acid derivatives for use as tools to study the structure, activity, and inhibition of glycoproteins and other glycoconjugates. Bioorganic & Medicinal Chemistry, 1998. 6(8): p. 1283-1292.
105. Malcolm Chandler, M.J.B., Richard Conroy, Brian Lamount, Bina Patel, Vipulkumar K. Patel, Ian P. Steeples, Richard Storer, Naill G. Weir, Michael
Wrightm Christopher Williamson, Synthesis of the potent influenza neuraminidase inhibitor 4-guanidino Neu5Ac2en. X-Ray molecular structure of S-acetamido^-amino^^-anhydro-S^^-trideoxy-D-erythro-L-gluco- nononic acid. J. Chem. Soc, Perkin Trans. 1, 1995: p. 1173 – 1180.
106. Masuda, T., et al., Synthesis and anti-influenza evaluation of polyvalent sialidase inhibitors bearing 4-guanidino-Neu5Ac2en derivatives. Chem Pharm Bull (Tokyo), 2003. 51(12): p. 1386-98
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
The active component of RELENZA is zanamivir. The chemical name of zanamivir is 5- (acetylamino)-4-[(aminoiminomethyl)-amino]-2,6-anhydro-3,4,5-trideoxy-D-glycero-D-galacto non-2-enonic acid. It has a molecular formula of C12H20N4O7 and a molecular weight of 332.3. It has the following structural formula:
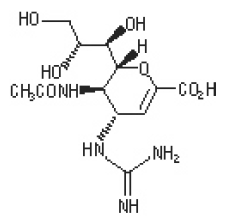 |
Zanamivir is a white to off-white powder for oral inhalation with a solubility of approximately 18 mg/mL in water at 20°C.
RELENZA is for administration to the respiratory tract by oral inhalation only. Each RELENZA ROTADISK contains 4 regularly spaced double-foil blisters with each blister containing a powder mixture of 5 mg of zanamivir and 20 mg of lactose (which contains milk proteins). The contents of each blister are inhaled using a specially designed breath-activated plastic device for inhaling powder called the DISKHALER. After a RELENZA ROTADISK is loaded into the DISKHALER, a blister that contains medication is pierced and the zanamivir is dispersed into the air stream created when the patient inhales through the mouthpiece. The amount of drug delivered to the respiratory tract will depend on patient factors such as inspiratory flow. Under standardized in vitro testing, RELENZA ROTADISK delivers 4 mg of zanamivir from the DISKHALER device when tested at a pressure drop of 3 kPa (corresponding to a flow rate of about 62 to 65 L/min) for 3 seconds.
CLIP
On Zanamivir
Total Synthesis of Anti-Influenza Agents Zanamivir and Zanaphosphor via Asymmetric Aza-Henry Reaction

The potent anti-influenza agents, zanamivir and its phosphonate congener, are synthesized by using a nitro group as the latent amino group at C4 for asymmetric aza-Henry reaction with a chiral sulfinylimine, which is derived from inexpensive d-glucono-δ-lactone to establish the essential nitrogen-containing substituent at C5. This method provides an efficient way to construct the densely substituted dihydropyran core of zanamivir and zanaphosphor without using the hazardous azide reagent.
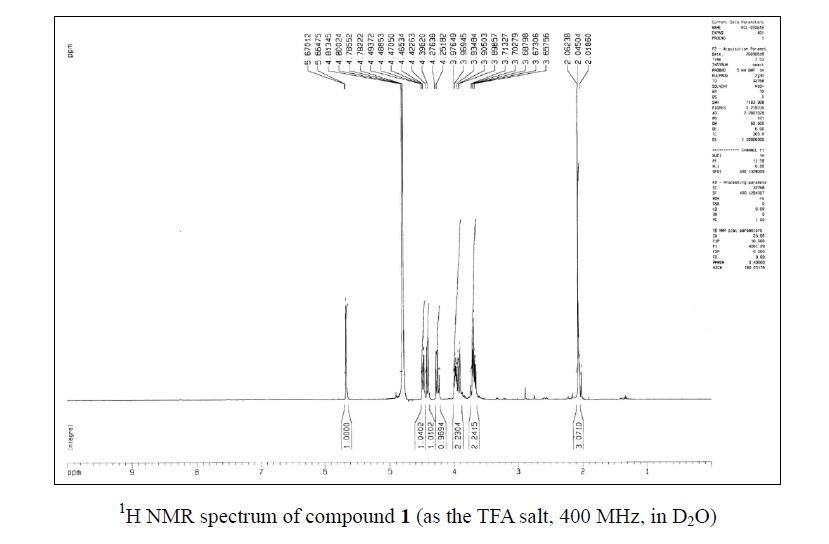
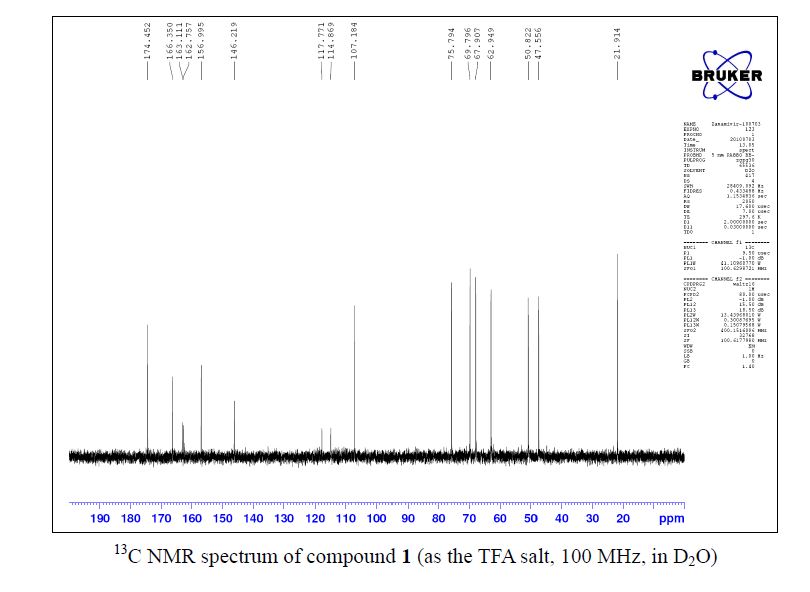
Zanamivir as the TFA salt (40 mg, 90 %). C14H21F3N4O9; colorless solid, mp 260262 oC;
1H NMR (400 MHz, D2O) δ 5.67 (1 H, d, J = 2.1 Hz), 4.48 (1 H, dd, J = 9.3, 2.1 Hz), 4.41 (1H, d, J = 10.6 Hz), 4.26 (1 H, dd, J = 10.6, 9.3 Hz), 3.98–3.90 (2 H, m), 3.71–3.66 (2 H, m),2.06 (3 H, s);
13C NMR (100 MHz, D2O) δ 174.5, 166.4, 162.9 (CO2 of TFA, q, J = 35.4 Hz ),157.0, 146.2, 116.3 (CF3 of TFA, q, J = 290.2 Hz ), 107.2, 75.8, 69.8, 67.9, 62.9, 50.8, 47.6,21.9;
ESI–HRMS calcd for C12H20N4O7Na: 355.1230, found: m/z 355.1288 [M + Na]+.
Introduction
Relenza (Zanamivir for oral inhalation) is the first in a new generation of influenza virus-specific drugs known as neuraminidase inhibitors, which work by interferring with the life cycles of influenza viruses A and B. It prevents the virus spreading infection to other cells by blocking the neuraminidase enzyme present on the surface of the virus. Relenza is available as a powder that is administered by inhalation of 2 blisters from the rotadisk inside the diskhaler (Fig. 1) twice daily for five days. This means that 20mg of Relenza is delivered to the principal site of viral replication each day.The main method for preventing influenza since the 1960s is by vaccination and although this and anti-viral drugs such as amantadine and its analogue rimantadine have long been available (since 1976 and 1993 respectively), they are only of limited use because of the constant mutation of the virus. This chameleon-like nature also means that the virus can become unrecognizable to the human immune system and thus repeatedly infect millions of people year after year.

Fig 1: The diskhaler used to administer Relenza. Each blister in the Rotadisk contains 5mg of the drug
Why there is a need for a more effective influenza treatment: At present influenza is basically an uncontrolled disease and an effective method is needed for both the prevention and treatment of it. In the 20th century there were some major pandemics such as the 1918-1919 Spanish ‘flu which killed 20 million people world wide, the 1957 Asian ‘flu, the 1968 Hong Kong ‘flu and the 1977 Russian ‘flu12 These viruses also affect different animals, especially domesticated chickens and turkeys and in Hong Kong in 1997 a virulent bird flu virus, started infecting and killing people for the first time ever. Of the 18 people affected 6 died, although there was no evidence that the virus was able to spread between people. Given the antigenic properties of the influenza virus, in the future the virus may be passed from person to person, and because human immune systems are not prepared for avian viruses the effects on the population could be grave. It would not be possible to prepare vaccines in time and anti-viral drugs are not always adequate.
Advantages of Relenza over previous treatments:
Relenza has a number of advantages over the existing treatments for influenza. It does not cause significant side effects and the development of zanamivir-resistant viruses is not expected to occur readily in patients. This is because selection of drug-resistant mutants characterized by changes in neuraminidase requires prolonged passage in tissue culture and may be a biological cripple. If started within two days of the onset of influenza symptoms and if a fever is present, the duration of illness is decreased by an average of 1.5 days. It appears to decrease the severity of flu symptoms for the remainder of the illness, as well as decreasing the number of complications from the flu. It is also possible that Relenza could be used as a method of ‘flu prevention although it has not yet been approved for this use.
Comparison of the symptoms of the ‘flu with that of a common cold:
| People infected by an influenza virus suffer a lot more than those with a cold. As you can see from the table below, some of the symptoms are similar, but with a cold they are less severe.Influenza also becomes more serious when it leads to secondary bacterial pneumonia or primary influenza viral pneumonia or when it exacerbates underlying medical conditions such as pulmonary or cardiac disease. In children, the symptoms are similar to those observed in adults, however children often have higher fevers and younger ones may develop gastrointestinal manifestations. It should be noted that Relenza is not effective on people with colds or other viral illnesses. |
| Influenza | Cold |
| Sore throat | Mild sore throat |
| High fever and chills | Low-grade fever |
| Non-productive cough | Cough |
| Severe muscle aches | Congestion |
| Headache | |
| Intense fatigue. |
The effect of Relenza on patients with respiratory diseases:Relenza is not generally recommended for the treatment of patients with respiratory dieseases such as asthma or chronic obstructive pulmonary disease (COPD) and has carried an approval since its approval in July 1999. Some patients with underlying airway diseases have experienced serious adverse events following treatment, with some fatal outcomes although causality has been difficult to establish. It has been recommended that patients with asthma have a fast-acting bronchodilator inhaler available and use it about 15 minutes before taking Relenza
Successfulness of Relenza:The sialidase inhibitory activities (determined by methods described in reference 7) of Relenza compared to the more recent neuraminidase inhibitor Oseltamivir are shown in the table below9.IC50 is the concentration that reduces enzyme activity by 50%.
| Compound | Influenza A IC50 (�M) | Influenza B IC50 (�M) |
| Relenza | 0.005 | 0.004 |
| Oseltamivir | 0.002 | 0.032 |
The results demonstrate that both compounds are good inhibitors of influenza A and B, with Oseltamivir being more selective towards Influenza A and Relenza showing a better overall performance. In phase I and II tests reported by the Lancet5, no important adverse effects were found in healthy patients or those reported to have mild to moderate asthma following an inhaled administration of 40mg/day of Relenza. There was a significant improvement of the symptoms of people taking Relenza compared to those taking the placebo.
1940s: Discovery that the influenza virus’s enzyme was destroying receptors on red blood cellsF.This was discovered by George Hirst, who noticed that when red blood cells were mixed with fluids from influenza infected chicken embryos in cold conditions the cells were very heavily agglutinated by the virus. These red cells dispersed when warmed up and could not be re-agglutinated in the cold with fresh virus. This led him to the conclusion that the influenza virus’s enzyme was destroying receptors on red blood cells.
The finding of sialidase (also known as neuraminidase):Alfred Gottschalk heard of Hirst’s experiment and interpretation of results, and this led him to believe that there was a “split product”. He discovered sialic or neuraminic acid (Fig 2), a type of sugar, and the enzyme on the virus was called neuraminidase (or sialidase). At this time it was thought that it was the neuraminidase which was responsible for the observations made by Hirst, but it was later shown by Robin Valentine, W. Graeme Laver, Norbert Bischofberger and Robert G. Webster that the hemagglutinin (receptor-binding) and neuraminidase (receptor-destroying) activities of the virus resided in two quite different spikes on the surface of the virus.

Fig 2: Sialic Acid
Discovery of how new pandemic strains of ‘flu A occured.
Ed Kilbourne, W. Graeme Laver, Norbert Bischofberger and Robert G. Webster realised that hybrid viruses could be formed by infecting cells simultaneously with two different Type A flu viruses. This was because the RNA pieces coding the various virus proteins reassorted, some of the viruses contained the hemagglutinin from one parent and the neuraminidase from the other. This “mating” of two parent viruses to give a hybrid virus explained how new pandemic strains of ‘flu A occurred, and led to a very good way of producing influenza viruses with any desired combination of hemagglutinin and neuraminidase spikes. This helped towards finding a way of producing pure neuraminidase which was later essential for crystal growth and drug design experiments.
The crystallization of neuraminidase:
Laver, Bischofberger and Webster isolated one type of influenza virus by sucking off the allantoic fluid surrounding the embryo of infected chicken eggs and purifying this. The virus particles were incubated with an enzyme capable of digesting proteins. This enzyme was selected to split the “heads” of the neuraminidase spikes off the virus particle without destroying them and to leave behind or destroy the hemagglutinin spike. The neuraminidase “heads” obtained were concentrated using high-speed centrifugation. The tiny pellet of neuraminidase heads examined had a crystalline appearance, and X-ray diffraction analysis of larger crystals showed that they were made of protein.
Neu5Ac2en (DANA) was shown to inhibit influenza neuraminidase:
Different variants of ‘flu neuraminidase were known to exist, each containing an amino acid sequence that varies between types of neuraminidase apart from one small sequence.It was seen that the conserved amino acids came together when the neuraminidase polypeptide folded up to form the active enzyme. This formed a well conserved cavity which was the active catalytic site of the neuraminidase enzyme. It became apparent that a plug-drug could be made to exactly fit into the active site and inhibit the neuraminidase activity from other influenza viruses. A synthetic analog of sialic acid called Neu5Ac2en (DANA) (Fig 3) was shown to inhibit the influenza virus neuraminidase, but not sufficiently enough to be used treatment for the ‘flu in humans.
Fig 3: Neu5Ac2en (DANA)

Fig 3: Neu5Ac2en (DANA)
The plug drug.Mark von Itzstein and colleagues discovered that replacing the OH at the 4 position of sialic acid with a positively charged amino group made a better inhibitor than sialic acid or its analogue, DANA. Replacing the OH at the 4 position of sialic acid with a guanidino group led to a potent inhibitor of ‘flu neuraminidase. This compound was given the names GG167 and Zanamivir and is now more commonly known as Relenza. Peter Colman soaked the substrate for sialic acid in neuraminidase crystals and used X-ray crystallography to determine the three-dimensional structure of the crystals. The strong binding of Relenza by ‘flu neuraminidase which was seen is due to the positively charged guanidino group being anchored by the negatively charged glutamic acids. More details about this are provided in the immunology section.
Immunology

|
Fig 4: The influenza viruses as seen under the electron microscope. Neuraminidase and haemagglutin spikes are visible. |
||
Structure of the flu virus:Influenza (Fig 4) is an RNA virus which may exist as any shape from round balls to long, spaghetti-like filaments. The genome of this virus is associated with five different viral proteins and is surrounded by a lipid membrane, which means that influenza belongs to the “enveloped” group of viruses. Eight separate pieces of ribonucleic acid (RNA) make up the influenza virus genome and each piece of RNA specifies the amino acid sequence of one and sometimes two of the virus’s proteins. The segmented nature of the RNA allows differenet flu viruses to easily “mate” with each other to form hybrid progeny viruses with bits of RNA from each parent virus.Two glycoprotein molecules, known as hemagglutinin (HA) and neuraminidase (NA) (Fig 5) are stuck onto the lipid envelope of the virus and both play a crucial role in the infection of the epithelial cells of the upper respiratory tract. HA is a rod-shaped triangular molecule.and NA exists as a mushroom shaped spike with a box-like head on top of a long stalk, containing a hydrophobic region by which it is embedded in the viral membrane.. |
||
|
Fig 5: The Neuraminidase enzyme |
||
| The enzyme Neuraminidase, also known as sialidase, is a tetramer with C-4 symmetry and an approximate molecular weight of 250 000. It contains a symmetrical folding pattern of six four-stranded antiparallel �-sheets arranged like propeller blades. Nine types of neuraminidase have been identified for influenza A and only one subtype for influenza B, and only 30% of the overall amino acid sequence is conserved between all known types of neuraminidase8 – these are the amino acids which line and surround the walls of the binding pocket. If they mutate, the enzyme is inactivated, so the virus could not mutate to escape from a drug which interfered with this site. So neuraminidase offers an attractive site for therapeutic intervention in influenza infections. | ||
|
|
||
How the influenza virus works:The influenza virus (like all viruses) can only replicate after invading selected living cells and growing inside them. It makes thousands of new virus particles from the cellular machinery and then goes on to infect other cells.. Hemagglutinin allows the virus to infect the epithelial cells of the upper respiratory tract by attaching it to cells through receptors on the cell containing sialic acid, it fuses the cell membrane with the membrane of the virus, allowing the RNA of the virus to get inside the cell and thus instruct the cell to make thousands of new virus particles. After this viral replication, the progeny virions must be released from the cell to repeat the cell cycle of infection.Neuraminidase removes the sialic acid receptors from the host cell and other newly made virus particles by cleavage of -glycosidic bonds. This enables the virus to escape from the cell in which it grew and spread in the body to infect other cells. The action of NA may also facilitate viral mobility through the mucus of the respiratory tract. 
|
| Fig. 6: The life cycle of the influenza virus. Click once on this image to see a larger version | ||
|
The life cycle of the influenza virusG begins with the individual virus entering the cell lining of the respiratory tract (letter a in Fig. 6), and the cell being induced to take up the virus because hemagglutinin on the virus binds to the sialic acid (b and c in Fig 6). The virus then dispatches its genetic material (made up of RNA) and its internal proteins to the nucleus of the cell (e and f). Messenger RNA is produced when some of the internal proteins duplicate the RNA (f). This messenger RNA is used by the cell as a template for making viral proteins (g and h) and genes which become new viral particles and leave the cell covered in sialic acid. This sialic acid needs to be removed so that the hemagglutinin molecules on one particle don’t attach to the sialic acid on other ones, thus causing the new viruses to clump together and stick to the cell. The sialic acid is removed from the surface of the new viral particle by neuraminidase (j) and the new viral particles are able to travel and invade other cells (k). |
||
| How Relenza works:
Relenza adopts a position within the active site of the enzyme and copies the geometry of the sialoside hydrolysis transition state9. It can achieve very good binding through appropriate presentation of its four pendent substituents and contains a hydrogen bonding glycerol sidechain. The guanidino group in Relenza is believed to form salt bridges with Glu 119 in the neuraminidase active site and add a strong charge interaction with Glu 2278. Two hydroxyl groups of the 6-glycerol side chain are hydrogen bonded to Glu276 and the 4-hydroxyl is oriented towards Glu119. The NH group of the 5-N acetyl side chain interacts with a bound water molecule on the floor of the active site. The carbonyl oxygen of the same side chain is hydrogen bonded to Arg152 and the methyl group enters a hydrophobic pocket lined by Ile222 and Trp178. The glycosidic oxygen projects into bulk solvent.
|

|
Fig 7. Relenza bound to neuraminidase |
||
|
The binding involved in Fig 7 is shown more clearly in Fig 8 below. Neuraminidase can no longer remove the sialic acid receptors from the host cell and newly made virus particles because of this binding. Therefore the virsuse ‘clump’ together or to the host cell and cannot go on to effect new cells. |
||
 |
||
|
Fig 8: Depiction of interaction of Relenza (GG 167) in the neuraminidase binding site6 |
||
References
| 1): K. J. Lui and A. P. Kendal, Am. J. Public Health, 1987, 77, 712 |
| 2): Scheiget, Zambonis, Bernstein and Roy, Org. Prep. Proced. Int., 1995, 27, 637- 644 |
| 3): Glaxo Wellcome Inc. Relenza� (zanamivir for inhalation) [package insert]. Research Triangle Park, NC: Glaxo Wellcome, Inc., 1999 |
| 4): N Seppa, Scientific American, July 10th 1999, Volume 156 |
| 5): L. Gubareva, Lancet, March 4th 2000, 355: 827-35 |
| 6): J. Medicinal Chemistry. 1999, 42, 2332-2343 |
| 7):P Smith, S Sollis, P Howes, P Cherry, I Starkey, K Cobley, H Weston, J Scicinski, A Merritt, A Whittington, P Wyatt, N Taylor, D Green, R Bethall, S Madar, R Fenton, P Morley, T Pateman, A Beresford. A. J. Med. Chem, 41, 1998, 787-797 |
| 8): C Kim, W Lew, M Williams, H Liu, L Zhang, S Swaminathan, N Bischofberger, M Chen, D Mendel, C Tai, G Laver, R Stevens, J Am Chem Soc, 1997, 119, 681-690 |
| 9): P Smith, J Robinson, D Evans, S Sollis, P Howes, N Trivedi and R Bethell, Bioorganic and Medicinal Chemistry Letters 9, 1999, 601-604 |
| 10): A. J. Hay, A. J. Wolstenholme, J. J. Skehel and M. H. Smith. EMBO J,. 1985, 4, 3021: L. J. Holsinger and R. A. Lamb, Cell, 1992, 69, 517 |
| 11): J. C. Stoof, J. Booij, B. Drukarch and E. C. Wolters, Eur. J. Pharmacol., 1992, 213, 439 |
| 12): W. Graeme Laver, Norbert Bischofberger, and Robert G. Webster, Perspectives in Biology and Medicine 43.2 (2000) 173-192. This can be seen by visitinghttp://www.press.jhu.edu/journals/perspectives_in_biology_and_medicine/v043/43.2laver.html nmr |
| 13): M. Chandler, M. J. Bamford, R. Conroy, B. Lamont, B. Patel, V. K. Patel, I. P. Steeples, R. Storer, N. G. Weir, M. Wright, C. Williamson, J. Chem. Soc. Perkin Trans. 1, 1995, 1173- 1180 nmr synth |
| 14): A. E. Miller, J. J. Bischoff, Synthesis, 1986, 777- 779 |
| 15): G. D. Allena, S. T. Brookesa, A. Barrow, b, J. A. Dunnc and C. M. Grossec, Journal of Chromatography B: 1999, 732, 383-393 |
Zanamivir
139110-80-8
APPROVED 26-7-96……. GSK NDA 021036
A guanido-neuraminic acid that is used to inhibit neuraminidase.
READ AT
TASIMELTION…FDA Approves Hetlioz: First Treatment for Non-24 Hour Sleep-Wake Disorder in Blind Individuals

TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
VEC-162, BMS-214778, 609799-22-6, Hetlioz, UNII-SHS4PU80D9,

January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
Tasimelteon
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
References
- ‘Time-bending drug’ for jet lag. BBC News. 2 December 2008
- Vachharajani, Nimish N., Yeleswaram, Krishnaswamy, Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Shantha MW Rajaratnam, Mihael H Polymeropoulos, Dennis M Fisher, Thomas Roth, Christin Scott, Gunther Birznieks, Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet 373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Audio interview with Joseph Hull of Harvard, spring 2011
- Vanda Pharmaceuticals seeks FDA approval
- Recent progress in the development of agonists and antagonists for melatonin receptors.Zlotos DP.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
 TASIMELTION
TASIMELTION
PATENTS
|
10-15-2010
|
PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE
|
|
|
8-21-2009
|
TREATMENT FOR DEPRESSIVE DISORDERS
|
|
|
5-10-2000
|
Benzopyran derivatives as melatonergic agents
|
|
|
11-10-1999
|
Benzodioxa alkylene ethers as melatonergic agents
|
|
|
6-19-1998
|
BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS
|
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc. 1938, 60, 2794.
- Oppolzer, W.; Chapuis, C.; Bernardinelli, G. Helv. Chim. Acta 1984, 67, 1397.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, G.; Carroll,, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Oppolzer, W.; Mills, R. J.; Pachinger, W.; Stevenson, T. Helv. Chim. Acta 1986, 69, 1542; Oppolzer, W.; Schneider, P. Helv. Chim. Acta 1986, 69, 1817; Oppolzer, W.; Mills, R. J.; Réglier, M. Tetrahedron Lett. 1986, 27, 183; Oppolzer, W.; Poli. G.Tetrahedron Lett. 1986, 27, 4717; Oppolzer, W.; Poli, G.; Starkemann, C.; Bernardinelli, G. Tetrahedron Lett. 1988, 29, 3559.
- Oppolzer, W.; Barras, J-P. Helv. Chim. Acta 1987, 70, 1666.
- Curran, D. P.; Kim, B. H.; Daugherty, J.; Heffner, T. A. Tetrahedron Lett. 1988, 29, 3555.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
Org. Synth. 1990, 69, 158 |
- Org. Syn. Coll. Vol. 8, 110
- Org. Syn. Coll. Vol. 9, 212
-
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Reychler, M. A. Bull. Soc. Chim. III 1889, 19, 120.
- Armstrong, H. E.; Lowry, T. M. J. Chem. Soc., Trans. 1902, 81, 1441.
- Dauphin, G.; Kergomard, A.; Scarset, A. Bull. Soc. Chim. Fr. 1976, 862.
- Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc. 1982, 104, 5412.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I 1988, 1753.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
- For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
- Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
- Davis, F. A.; Sheppard, A. C. Tetrahedron 1989, 45, 5703.
- Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc. 1987, 109, 3370.
- Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron 1985, 41, 4747.
- Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett. 1986, 27, 5079.
- Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc. 1983, 105, 3123.
- Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett. 1987, 28, 5115.
- Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett. 1988, 29, 4269.
- Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem. 1986, 51, 2402.
- Davis, F. A.; Haque, M. S. J. Org. Chem. 1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem. 1989, 54, 2021.
- Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem. 1987, 52, 5288.
- Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett. 1989, 30, 779.
- Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc. 1990, 112, 6679.

dedicated to lionel my son



my daughter Aishal

THEY KEEP ME GOING
India’s GVK BIO goes international with Aragen deal

GVK Biosciences (GVK BIO), the discovery research and development organisation based in Hyderabad, India, has reached a definitive agreement to acquire Aragen Bioscience, a preclinical contract research organisation operating out of Morgan Hill, US and specialising in high-value biologics services.
No financial terms were disclosed for the deal, in which GVK BIO is acquiring the capital stock of Aragen Bioscience. It is the Indian company’s first international acquisition.
According to a report in India’s Business Standard, GVK is taking a 65% stake in Aragen Biosciences and will acquire the remaining 35% over a period of two years, leaving Aragen to function as a separate entity.
Read more at: http://www.pharmatimes.com/Article/14-01-30/India_s_GVK_BIO_goes_international_with_Aragen_deal.aspx#ixzz2s3enmHuZ
Follow us: @PharmaTimes on Twitter
