
Home » 2014 (Page 89)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Why FDA Supports a Flexible Approach to Drug Development

We all know that just as every person is different, so too is every disease and every drug. And so we weren’t surprised by the results of a new study published in the Journal of … Continue reading →
FONDAPARINUX

FONDAPARINUX
Fondaparinux is a drug belonging to the group of the antithrombotic agents and are used to prevent deep vein thrombosis in patients undergoing orthopedic surgery. It is also used for the treatment of severe venous thrombosis and pulmonary
114870-03-0 ………..10x SODIUM SALT
| CAS number | 114870-03-0 FREE FORM |
|---|
| MF | C31H43N3Na10O49S8 10X SODIUM |
|---|---|
| MW | 1726.77 g/mol 10X SODIUM |
GSK-576428 Org-31540 SR-90107SR-90107A
launched 2002
Arixtra, Quixidar, Fondaparinux sodium, Fondaparin sodium, Arixtra (TN), Fondaparinux, Org-31540, AC1LCS4W, SR-90107A
Fondaparinux (Arixtra) is a synthetic pentasaccharide anticoagulant. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycan heparin and heparan sulfate (HS). This monomeric sequence in heparin and HS is thought to form the high affinity binding site for the natural anti-coagulant factor, antithrombin III (ATIII).
Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000-fold. Fondaparinux potentiates the neutralizing action ofATIII on activated Factor X 300-fold. Fondaparinux may be used: to prevent venous thromboembolism in patients who have undergone orthopedic surgery of the lower limbs (e.g. hip fracture, hip replacement and knee surgery); to prevent VTE in patients undergoing abdominal surgery who are are at high risk of thromboembolic complications; in the treatment of deep vein thrombosis (DVT) and pumonary embolism (PE); in the management of unstable angina (UA) and non-ST segment elevation myocardial infarction (NSTEMI); and in the management of ST segment elevation myocardial infarction (STEMI).

FONDAPARINUX
Fondaparinux (trade name Arixtra) is an anticoagulant medication chemically related to low molecular weight heparins. It is marketed byGlaxoSmithKline. A generic version developed by Alchemia is marketed within the US by Dr. Reddy’s Laboratories.
Fondaparinux is a synthetic pentasaccharide Factor Xa inhibitor. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycans heparin and heparan sulfate (HS). Within heparin and heparan sulfate this monomeric sequence is thought to form the high affinity binding site for the anti-coagulant factor antithrombin III (ATIII). Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000 fold. In contrast to heparin, fondaparinux does not inhibit thrombin.
Fondaparinux is given subcutaneously daily. Clinically, it is used for the prevention of deep vein thrombosis in patients who have had orthopedic surgery as well as for the treatment of deep vein thrombosis and pulmonary embolism.
One potential advantage of fondaparinux over LMWH or unfractionated heparin is that the risk for heparin-induced thrombocytopenia (HIT) is substantially lower. Furthermore, there have been case reports of fondaparinux being used to anticoagulate patients with established HIT as it has no affinity to PF-4. However, its renal excretion precludes its use in patients with renal dysfunction.
Unlike direct factor Xa inhibitors, it mediates its effects indirectly through antithrombin III, but unlike heparin, it is selective for factor Xa.[1]
Fondaparinux is similar to enoxaparin in reducing the risk of ischemic events at nine days, but it substantially reduces major bleeding and improves long term mortality and morbidity.[2]
It has been investigated for use in conjunction with streptokinase.[3]
Fondaparinux sodium, a selective coagulation factor Xa inhibitor, was first launched in the U.S. in 2002 by GlaxoSmithKline in a subcutaneous injection formulation for the prophylaxis of deep venous thrombosis (DVT) which may lead to pulmonary embolism in patients at risk for thromboembolic complications who are undergoing hip replacement, knee replacement, hip fracture surgery or abdominal surgery. The product is available in Japan for the treatment of acute deep venous thrombosis and acute pulmonary thromboembolism. In 2004, GlaxoSmithKline launched fondaparinux as an injection to be used in conjunction with warfarin sodium for the subcutaneous treatment of acute pulmonary embolism and DVT.
In 2007, GlaxoSmithKline received approval in the E.U. for the treatment of acute coronary syndrome (ACS), specifically unstable angina or non-ST segment elevation myocardial infarction (UA/NSTEMI) and ST-segment elevation myocardial infarction (STEMI), while in the U.S. an approvable letter was received for this indication. Currently, the drug is in clinical development at GlaxoSmithKline for the treatment of venous limb superficial thrombosis.

Fondaparinux Molecule
GlaxoSmithKline had filed a regulatory application in the E.U. seeking approval of fondaparinux sodium for the prevention of venous thromboembolic events (VTE), however; in 2008, the application was withdrawn for commercial reasons. Commercial launch in Japan for the product for the prevention of venous thromboembolism in high risk patients undergoing surgery in the abdomen took place in 2008.
In 2010, the EMA approved a regulatory application filed by GlaxoSmithKline seeking approval of a once-daily formulation of fondaparinux sodium for the treatment of adults with acute symptomatic spontaneous superficial-vein thrombosis (SVT) of the lower limbs without concomitant DVT. Product launch took place in the U.K. for this indication the same year.
The antithrombotic activity of fondaparinux is the result of antithrombin III (ATIII)-mediated selective inhibition of Factor Xa. By selectively binding to ATIII, the drug potentiates (about 300 times) the innate neutralization of Factor Xa by ATIII. Neutralization of Factor Xa, in turn, interrupts the blood coagulation cascade and thus inhibits thrombin formation and thrombus development. Fondaparinux does not inactivate thrombin (activated Factor II) and has no known effect on platelet function. At the recommended dose, no effects have been demonstrated on fibrinolytic activity or bleeding time.
Originally developed by Organon and Sanofi (formerly known as sanofi-aventis), fondaparinux sodium is currently available in approximately 30 countries. In 2004, Organon transferred its rights to the drug to Sanofi in exchange for revenues based on future sales from jointly developed antithrombotic products and in early 2005, GlaxoSmithKline also acquired the antithrombotic.
At the beginning of 2005, GlaxoSmithKline signed a two-year agreement with Adolor (acquired by Cubist in 2011) for the copromotion of fondaparinux sodium in the U.S. In Sepetember 2013, Aspen Pharmacare acquired Arixtra global rights (excluding China, India and Pakistan) from GlaxoSmithKline for the treatment of thrombosis with GlaxoSmithKline commercializing the product in Indonesia under licence from Aspen.
Chemical structure
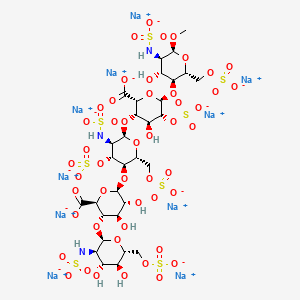
Abbreviations
- GlcNS6S = 2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
- GlcA = β-D-glucopyranuronoside
- GlcNS3,6S = 2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl
- IdoA2S = 2-O-sulfo-α-L-idopyranuronoside
- GlcNS6SOMe = methyl-O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
The sequence of monosaccharides is D-GlcNS6S-α-(1,4)-D-GlcA-β-(1,4)-D-GlcNS3,6S-α-(1,4)-L-IdoA2S-α-(1,4)-D-GlcNS6S-OMe, as shown in the following structure:

ARIXTRA (fondaparinux sodium) Injection is a sterile solution containing fondaparinux sodium. It is a synthetic and specific inhibitor of activatedFactor X (Xa). Fondaparinux sodium is methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-β-D-glucopyranuronosyl-( 1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-Osulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt.
The molecular formula of fondaparinux sodium is C31H43N3Na10O49S8 and its molecular weight is 1728. The structural formula is provided below:
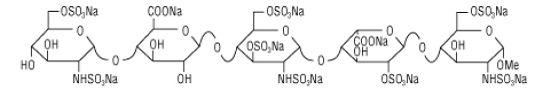
ARIXTRA is supplied as a sterile, preservative-free injectable solution for subcutaneous use.
Each single-dose, prefilled syringe of ARIXTRA, affixed with an automatic needle protection system, contains 2.5 mg of fondaparinux sodium in 0.5 mL, 5.0 mg of fondaparinux sodium in 0.4 mL, 7.5 mg of fondaparinux sodium in 0.6 mL, or 10.0 mg of fondaparinux sodium in 0.8 mL of an isotonic solutionof sodium chloride and water for injection. The final drug product is a clear and colorless to slightly yellow liquid with a pH between 5.0 and 8.0.
……………….
INTRODUCTION
In U.S. Patent No. 7,468,358, Fondaparinux sodium is described as the “only anticoagulant thought to be completely free of risk from HIT-2 induction.” The biochemical and pharmacologic rationale for the development of a heparin pentasaccharide in Thromb. Res., 86(1), 1-36, 1997 by Walenga et al. cited the recently approved synthetic pentasaccharide Factor Xa inhibitor Fondaparinux sodium. Fondaparinux has also been described in Walenga et al., Expert Opin. Investig. Drugs, Vol. 11, 397-407, 2002 and Bauer, Best Practice & Research Clinical Hematology, Vol. 17, No. 1, 89-104, 2004.
Fondaparinux sodium is a linear octasulfated pentasaccharide (oligosaccharide with five monosaccharide units ) molecule having five sulfate esters on oxygen (O-sulfated moieties) and three sulfates on a nitrogen (N- sulfated moieties). In addition, Fondaparinux contains five hydroxyl groups in the molecule that are not sulfated and two sodium carboxylates. Out of five saccharides, there are three glucosamine derivatives and one glucuronic and one L-iduronic acid. The five saccharides are connected to each other in alternate α and β glycosylated linkages (see Figure 1).
Figure 1 Fondaparinux Sodium

Monosaccharide E Monosaccharide D Monosaccharide C Monosaccharide B Monosaccharide A derived from derived from derived from derived from derived from
Monomer E Monomer D Monomer C Monomer B1 Monomer A2
Fondaparinux Sodium
Fondaparinux sodium is a chemically synthesized methoxy derivative of the natural pentasaccharide sequence, which is the active site of heparin that mediates the interaction with antithrombin (Casu et al., J. Biochem., 197, 59, 1981). It has a challenging pattern of O- and N- sulfates, specific glycosidic stereochemistry, and repeating units of glucosamines and uronic acids (Petitou et al, Progress in the Chemistry of Organic Natural Product, 60, 144-209, 1992).
The monosaccharide units comprising the Fondaparinux molecule are labeled as per the convention in Figure 1, with the glucosamine unit on the right referred to as monosaccharide A and the next, an uronic acid unit to its left as B and subsequent units, C, D and E respectively. The chemical synthesis of Fondaparinux starts with monosaccharides of defined structures that are themselves referred to as Monomers A2, Bl, C, D and E, for differentiation and convenience, and they become the corresponding monosaccharides in fondaparinux sodium.
Due to this complex mixture of free and sulfated hydroxyl groups, and the presence of N- sulfated moieties, the design of a synthetic route to Fondaparinux requires a careful strategy of protection and de-protection of reactive functional groups during synthesis of the molecule. Previously described syntheses of Fondaparinux all adopted a similar strategy to complete the synthesis of this molecule. This strategy can be envisioned as having four stages.
The strategy in the first stage requires selective de-protection of five out of ten hydroxyl groups. During the second stage these five hydroxyls are selectively sulfonated. The third stage of the process involves the de -protection of the remaining five hydroxyl groups. The fourth stage of the process is the selective sulfonation of the 3 amino groups, in the presence of five hydroxyl groups that are not sulfated in the final molecule. This strategy can be envisioned from the following fully protected pentasaccharide, also referred to as the late-stage intermediate.

In this strategy, all of the hydroxyl groups that are to be sulfated are protected with an acyl protective group, for example, as acetates (R = CH3) or benzoates (R = aryl) (Stages 1 and 2) All of the hydroxyl groups that are to remain as such are protected with benzyl group as benzyl ethers (Stage 3). The amino group, which is subsequently sulfonated, is masked as an azide (N3) moiety (Stage 4). R1 and R2 are typically sodium in the active pharmaceutical compound (e.g., Fondaparinux sodium).
This strategy allows the final product to be prepared by following the synthetic operations as outlined below: a) Treatment of the late- stage intermediate with base to hydrolyze (deprotect) the acyl ester groups to reveal the five hydroxyl groups. The two R1 and R2 ester groups are hydrolyzed in this step as well.

b) Sulfonation of the newly revealed hydroxyl groups.

c) Hydrogenation of the O-sulfated pentasaccharide to de-benzylate the five benzyl- protected hydroxyls, and at the same time, unmask the three azides to the corresponding amino groups.

d) On the last step of the operation, the amino groups are sulfated selectively at a high pH, in the presence of the five free hydroxyls to give Fondaparinux (Figure 1). While the above strategy has been shown to be viable, it is not without major drawbacks. One drawback lies in the procedure leading to the fully protected pentasaccharide (late stage intermediate), especially during the coupling of the D-glucuronic acid to the next adjacent glucose ring (the D-monomer to C-monomer in the EDCBA nomenclature shown in Figure 1). Sugar oligomers or oligosaccharides, such as Fondaparinux, are assembled using coupling reactions, also known as glycosylation reactions, to “link” sugar monomers together. The difficulty of this linking step arises because of the required stereochemical relationship between the D-sugar and the C-sugar, as shown below:

The stereochemical arrangement illustrated above in Figure 2 is described as having a β- configuration at the anomeric carbon of the D-sugar (denoted by the arrow). The linkage between the D and C units in Fondaparinux has this specific stereochemistry. There are, however, competing β- and α-glycosylation reactions.
The difficulties of the glycosylation reaction in the synthesis of Fondaparinux is well known. In 1991 Sanofi reported a preparation of a disaccharide intermediate in 51% yield having a 12/1 ratio of β/α stereochemistry at the anomeric position (Duchaussoy et al., Bioorg. & Med. Chem. Lett., 1(2), 99-102, 1991).
In another publication (Sinay et al, Carbohydrate Research, 132, C5-C9, 1984) yields on the order of 50% with coupling times on the order of 6- days are reported. U.S. Patent No. 4,818,816 {see e.g., column 31, lines 50-56) discloses a 50% yield for the β-glycosylation.
Alchemia’s U.S. Patent No. 7,541,445 is even less specific as to the details of the synthesis of this late-stage Fondaparinux synthetic intermediate. The ‘445 Patent discloses several strategies for the assembly of the pentasaccharide (1+4, 3+2 or 2+3) using a 2-acylated D-sugar (specifically 2-allyloxycarbonyl) for the glycosylation coupling reactions. However, Alchemia’s strategy involves late-stage pentasaccharides that all incorporate a 2-benzylated D- sugar.
The transformation of acyl to benzyl is performed either under acidic or basic conditions. Furthermore, these transformations, using benzyl bromide or benzyl trichloroacetimidate, typically result in extensive decomposition and the procedure suffers from poor yields. Thus, such transformations (at a disaccharide, trisaccharide, and pentasaccharide level) are typically not acceptable for industrial scale production.
Examples of fully protected pentasaccharides are described in Duchaussoy et al, Bioorg. Med. Chem. Lett., 1 (2), 99-102, 1991; Petitou et al, Carbohydr. Res., 167, 67-75, 1987; Sinay et al, Carbohydr. Res., 132, C5-C9, 1984; Petitou et al., Carbohydr. Res., 1147, 221-236, 1986; Lei et al., Bioorg. Med. Chem., 6, 1337-1346, 1998; Ichikawa et al., Tet. Lett., 27(5), 611-614, 1986; Kovensky et al, Bioorg. Med. Chem., 1999, 7, 1567-1580, 1999.
These fully protected pentasaccharides may be converted to the O- and N-sulfated pentasaccharides using the four steps (described earlier) of: a) saponification with LiOHZH2CVNaOH, b) O-sulfation by an Et3N- SO3 complex; c) de-benzylation and azide reduction via H2/Pd hydrogenation; and d) N-sulfation with a pyridine-SO3 complex.
Even though many diverse analogs of the fully protected pentasaccharide have been prepared, none use any protective group at the 2-position of the D unit other than a benzyl group. Furthermore, none of the fully protected pentasaccharide analogs offer a practical, scaleable and economical method for re-introduction of the benzyl moiety at the 2-position of the D unit after removal of any participating group that promotes β-glycosylation.
Furthermore, the coupling of benzyl protected sugars proves to be a sluggish, low yielding and problematic process, typically resulting in substantial decomposition of the pentasaccharide (prepared over 50 synthetic steps), thus making it unsuitable for a large [kilogram] scale production process.

Ref. 1. Sinay et al, Carbohydr. Res., 132, C5-C9, 1984.
Ref. 2. Petitou et al., Carbohydr. Res., 147, 221-236. 1986
It has been a general strategy for carbohydrate chemists to use base-labile ester-protecting group at 2-position of the D unit to build an efficient and stereoselective β-glycosidic linkage. To construct the β-linkage carbohydrate chemists have previously acetate and benzoate ester groups, as described, for example, in the review by Poletti et al., Eur. J. Chem., 2999-3024, 2003.
The ester group at the 2-position of D needs to be differentiated from the acetate and benzoates at other positions in the pentasaccharide. These ester groups are hydrolyzed and sulfated later in the process and, unlike these ester groups, the 2-hydroxyl group of the D unit needs to remain as the hydroxyl group in the final product, Fondaparinux sodium.
Some of the current ester choices for the synthetic chemists in the field include methyl chloro acetyl and chloro methyl acetate [MCA or CMA] . The mild procedures for the selective removal of theses groups in the presence of acetates and benzoates makes them ideal candidates. However, MCA/CMA groups have been shown to produce unwanted and serious side products during the glycosylation and therefore have not been favored in the synthesis of Fondaparinux sodium and its analogs. For by-product formation observed in acetate derivatives see Seeberger et al., J. Org. Chem., 2004, 69, 4081-93.
Similar by-product formation is also observed using chloroacetate derivatives. See Orgueira et al., Eur. J. Chem., 9(1), 140-169, 2003.
The levulinyl group can be rapidly and almost quantitatively removed by treatment with hydrazine hydrate as the deprotection reagent as illustrated in the example below. Under the same reaction conditions primary and secondary acetate and benzoate esters are hardly affected by hydrazine hydrate. See, e.g., Seeberger et al, J. Org. Chem., 69, 4081-4093, 2004.

The syntheses of Fondaparinux sodium described herein takes advantage of the levulinyl group in efficient construction of the trisaccharide EDC with improved β- selectivity for the coupling under milder conditions and increased yields.

Substitution of the benzyl protecting group with a THP moiety provides its enhanced ability to be incorporated quantitatively in position-2 of the unit D of the pentasaccharide. Also, the THP group behaves in a similar manner to a benzyl ether in terms of function and stability. In the processes described herein, the THP group is incorporated at the 2-position of the D unit at this late stage of the synthesis (i.e., after the D and C units have been coupled through a 1,2-trans glycosidic (β-) linkage). The THP protective group typically does not promote an efficient β- glycosylation and therefore is preferably incorporated in the molecule after the construction of the β-linkage.
Fondaparinux and sodium salt thereof can be prepared from pure compound of Formula II by following the teachings from Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991). A second aspect of the present invention provides a process for the preparation of 4-0- -D-glucopyranosyl-l,6-anhydro- -D-glucopyranose, represented by STR BELOW

……………………………..
SYNTHESIS
EP2464668A2 AND US8288515
The scheme below exemplifies some of the processes of the present invention disclosed herein.

The tetrahydropyranyl (THP) protective group and the benzyl ether protective group are suitable hydroxyl protective groups and can survive the last four synthetic steps (described above) in the synthesis of Fondaparinux sodium, even under harsh reaction conditions. Certain other protecting groups do not survive the last four synthetic steps in high yield.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


The ester moieties in EDCBA Pentamer were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 16 hours to give the pentasaccharide intermediate API1. The five hydroxyl moieties in API1 were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60° C. for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2. The intermediate API2 was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the five benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3. This transformation occurs by reacting API2 with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3 were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours. The acidic work-up procedure of the reaction removes the THP group to provide crude fondaparinux which is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final API, fondaparinux sodium
Experimental Procedures Preparation of IntD1 Bromination of Glucose Pentaacetate
To a 500 ml flask was added 50 g of glucose pentaacetate (C6H22O11) and 80 ml of methylene chloride. The mixture was stirred at ice-water bath for 20 min HBr in HOAc (33%, 50 ml) was added to the reaction mixture. After stirring for 2.5 hr another 5 ml of HBr was added to the mixture. After another 30 min, the mixture was added 600 ml of methylene chloride. The organic mixture was washed with cold water (200 ml×2), Saturated NaHCO3(200 ml×2), water (200 ml) and brine (200 ml×2). The organic layer was dried over Na2SO4 and the mixture was evaporated at RT to give white solid as final product, bromide derivative, IntD1 (˜95% yield). C14H19BrO9, TLC Rf=0.49, SiO2, 40% ethyl acetate/60% hexanes; Exact Mass 410.02.
Preparation of IntD2 by Reductive Cyclization
To a stirring mixture of bromide IntD1 (105 g), tetrabutylammonium iodide (60 g, 162 mmol) and activated 3 Å molecular sieves in anhydrous acetonitrile (2 L), solid NaBH4 (30 g, 793 mmol) was added. The reaction was heated at 40° C. overnight. The mixture was then diluted with dichloromethane (2 L) and filtered through Celite®. After evaporation, the residue was dissolved in 500 ml ethyl acetate. The white solid (Bu4NI or Bu4NBr) was filtered. The ethyl acetate solution was evaporated and purified by chromatography on silica gel using ethyl acetate and hexane as eluent to give the acetal-triacetate IntD2 (˜60-70% yield). TLC Rf=0.36, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD3 by De-Acetylation
To a 1000 ml flask was added triacetate IntD2 (55 g) and 500 ml of methanol. After stirring 30 min, the reagent NaOMe (2.7 g, 0.3 eq) was added and the reaction was stirred overnight. Additional NaOMe (0.9 g) was added to the reaction mixture and heated to 50° C. for 3 hr. The mixture was neutralized with Dowex 50Wx8 cation resin, filtered and evaporated. The residue was purified by silica gel column to give 24 g of trihydroxy-acetal IntD3. TLC Rf=0.36 in SiO2, 10% methanol/90% ethyl acetate.
Preparation of IntD4 by Benzylidene Formation
To a 1000 ml flask was added trihydroxy compound IntD3 (76 g) and benzaldehyde dimethyl acetate (73 g, 1.3 eq). The mixture was stirred for 10 min, after which D(+)-camphorsulfonic acid (8.5 g, CSA) was added. The mixture was heated at 50° C. for two hours. The reaction mixture was then transferred to separatory funnel containing ethyl acetate (1.8 L) and sodium bicarbonate solution (600 ml). After separation, the organic layer was washed with a second sodium bicarbonate solution (300 ml) and brine (800 ml). The two sodium carbonate solutions were combined and extracted with ethyl acetate (600 ml×2). The organic mixture was evaporated and purified by silica gel column to give the benzylidene product IntD4 (77 g, 71% yield). TLC Rf=0.47, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD5 by Benzylation
To a 500 ml flask was added benzylidene acetal compound IntD4 (21 g,) in 70 ml THF. To another flask (1000 ml) was added NaH (2 eq). The solution of IntD4 was then transferred to the NaH solution at 0° C. The reaction mixture was stirred for 30 min, then benzyl bromide (16.1 ml, 1.9 eq) in 30 ml THF was added. After stirring for 30 min, DMF (90 ml) was added to the reaction mixture. Excess NaH was neutralized by careful addition of acetic acid (8 ml). The mixture was evaporated and purified by silica gel column to give the benzyl derivative IntD5. (23 g) TLC Rf=0.69, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD6 by Deprotection of Benzylidene
To a 500 ml flask was added the benzylidene-acetal compound IntD5 (20 g) and 250 ml of dichloromethane, the reaction mixture was cooled to 0° C. using an ice-water-salt bath. Aqueous TFA (80%, 34 ml) was added to the mixture and stirred over night. The mixture was evaporated and purified by silica gel column to give the dihydroxy derivative IntD6. (8 g, 52%). TLC Rf=0.79, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of IntD7 by Oxidation of 6-Hydroxyl
To a 5 L flask was added dihydroxy compound IntD6 (60 g), TEMPO (1.08 g), sodium bromide (4.2 g), tetrabutylammonium chloride (5.35 g), saturated NaHCO3 (794 ml) and EtOAc (1338 ml). The mixture was stirred over an ice-water bath for 30 min To another flask was added a solution of NaOCl (677 ml), saturated NaHCO3 (485 ml) and brine (794 ml). The second mixture was added slowly to the first mixture (over about two hrs). The resulting mixture was then stirred overnight. The mixture was separated, and the inorganic layer was extracted with EtOAc (800 ml×2). The combined organic layers were washed with brine (800 ml). Evaporation of the organic layer gave 64 g crude carboxylic acid product IntD7 which was used in the next step use without purification. TLC Rf=0.04, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of Monomer D by Benzylation of the Carboxylic Acid
To a solution of carboxylic acid derivative IntD7 (64 g) in 600 ml of dichloromethane, was added benzyl alcohol (30 g) and N-hydroxybenzotriazole (80 g, HOBt). After stirring for 10 min triethylamine (60.2 g) was added slowly. After stirring another 10 min, dicyclohexylcarbodiimide, (60.8 g, DCC) was added slowly and the mixture was stirred overnight. The reaction mixture was filtered and the solvent was removed under reduced pressure followed by chromatography on silica gel to provide 60.8 g (75%, over two steps) of product, Monomer D. TLC Rf=0.51, SiO2 in 40% ethyl acetate/60% hexanes.
Synthesis of the BA Dimer
Step 1. Preparation of BMod1, Levulination of Monomer B1
A 100 L reactor was charged with 7.207 Kg of Monomer B1 (21.3 moles, 1 equiv), 20 L of dry tetrahydrofuran (THF) and agitated to dissolve. When clear, it was purged with nitrogen and 260 g of dimethylamino pyridine (DMAP, 2.13 moles, 0.1 equiv) and 11.05 L of diisopropylethylamine (DIPEA, 8.275 kg, 63.9 moles, 3 equiv) was charged into the reactor. The reactor was chilled to 10-15° C. and 13.7 kg levulinic anhydride (63.9 mol, 3 equiv) was transferred into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. Completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. When the reaction was complete, 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. The mixture was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight. The yield of the isolated syrup of BMod1 was 100%.
Synthesis of the BA Dimer
Step 2. Preparation of BMod2, TFA Hydrolysis of BMod1
A 100 L reactor was charged with 9296 Kg of 4-Lev Monomer B1 (BMod1) (21.3 mol, 1 equiv). The reactor chiller was turned to <5° C. and stirring was begun, after which 17.6 L of 90% TFA solution (TFA, 213 mole, 10 equiv) was transferred into the reactor. When the addition was complete, the reaction was monitored by TLC and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, the reactor was chilled and 26.72 L of triethylamine (TEA, 19.4 Kg, 191.7 mole, 0.9 equiv) was transferred into the reactor. An additional 20 L of water and 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer was extracted (EtOAc layer) with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 50:50, 80:20 (EtOAc/heptane), 100% EtOAc, 5:95, 10:90 (MeOH/EtOAc). The pure fractions were pooled and evaporated to a syrup. The yield of the isolated syrup, BMod2 was 90%.
Synthesis of the BA Dimer
Step 3. Preparation of BMod3, Silylation of BMod2
A 100 L reactor was charged with 6.755 Kg 4-Lev-1,2-DiOH Monomer B1 (BMod2) (17.04 mol, 1 equiv), 2328 g of imidazole (34.2 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., then 5.22 L tert-butyldiphenylchloro-silane (TBDPS-Cl, 5.607 Kg, 20.4 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The yield of BMod3 was about 80%.
Synthesis of the BA Dimer
Step 4. Preparation of BMod4, Benzoylation
A 100 L reactor was charged with 8.113 Kg of 4-Lev-1-Si-2-OH Monomer B1 (BMod3) (12.78 mol, 1 equiv), 9 L of pyridine and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 1.78 L of benzoyl chloride (2155 g, 15.34 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The DCM solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Isolated syrup BMod4 was obtained in 91% yield.
Synthesis of the BA Dimer
Step 5. Preparation of BMod5, Desilylation
A 100 L reactor was charged with 8.601 Kg of 4-Lev-1-Si-2-Bz Monomer B1 (BMod4) (11.64 mol, 1 equiv) in 30 L terahydrofuran. The reactor was purged with nitrogen and chilled to 0° C., after which 5.49 Kg of tetrabutylammonium fluoride (TBAF, 17.4 mol, 1.5 equiv) and 996 mL (1045 g, 17.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred at ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction took approximately 6 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. Pure fractions were pooled and evaporated to a syrup. The intermediate BMod5 was isolated as a syrup in 91% yield.
Synthesis of the BA Dimer
Step 6: Preparation of BMod6, TCA Formation
A 100 L reactor was charged with 5.238 Kg of 4-Lev-1-OH-2-Bz Monomer B1 (BMod5) (10.44 mol, 1 equiv) in 30 L of DCM. The reactor was purged with nitrogen and chilled to 10-15° C., after which 780 mL of diazabicyclo undecene (DBU, 795 g, 5.22 mol, 0.5 equiv) and 10.47 L of trichloroacetonitrile (TCA, 15.08 Kg, 104.4 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under a nitrogen atmosphere. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of dichloromethane were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/Heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of BMod6 was 73%.
Synthesis of the BA Dimer
Step 7. Preparation of AMod1, Acetylation of Monomer A2
A 100 L reactor was charged with 6.772 Kg of Monomer A2 (17.04 mole, 1 eq.), 32.2 L (34.8 Kg, 340.8 moles, 20 eq.) of acetic anhydride and 32 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C. When the temperature reached −20° C., 3.24 L (3.63 Kg, 25.68 mol, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) was transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 3-5 hours for completion. When the reaction was complete, extraction was performed with 3×15 L of 10% sodium bicarbonate and 20 L of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The syrup was purified in a 200 L silica column using 140 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of AMod1 was 83%.
Synthesis of the BA Dimer
Step 8. Preparation of AMod3,1-Methylation of AMod1
A 100 L reactor was charged with 5891 g of acetyl Monomer A2 (AMod1) (13.98 mole, 1 eq.) in 32 L of dichloromethane. The reactor was purged with nitrogen and was chilled to 0° C., after which 2598 mL of trimethylsilyl iodide (TMSI, 3636 g, 18 mol, 1.3 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 2-4 hours to reach completion. When the reaction was complete, the mixture was diluted with 20 L of toluene. The solution was evaporated to a syrup and was co-evaporated with 3×6 L of toluene. The reactor was charged with 36 L of dichloromethane (DCM), 3.2 Kg of dry 4 Å Molecular Sieves, 15505 g (42 mol, 3 equiv) of tetrabutyl ammonium iodide (TBAI) and 9 L of dry methanol. This was stirred until the TBAI was completely dissolved, after which 3630 mL of diisopropyl-ethylamine (DIPEA, 2712 g, 21 moles, 1.5 equiv) was transferred into the reactor in one portion. The completion of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 16 hours for completion. When the reaction was complete, the molecular sieves were removed by filtration. Added were 20 L EtOAc and extracted with 4×20 L of 25% sodium thiosulfate and 20 L 10% NaCl solutions. The organic layer was separated and dried with 8-12 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70 and 40:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to give intermediate AMod3 as a syrup. The isolated yield was 75%.
Synthesis of the BA Dimer
Step 9. Preparation of AMod4, DeAcetylation of AMod3
A 100 L reactor was charged with 4128 g of 1-Methyl 4,6-Diacetyl Monomer A2 (AMod3) (10.5 mol, 1 equiv) and 18 L of dry methanol and dissolved, after which 113.4 g (2.1 mol, 0.2 equiv) of sodium methoxide was transferred into the reactor. The reaction was stirred at room temperature and monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was complete, Dowex 50Wx8 cation resin was added in small portions until the pH reached 6-8. The Dowex 50Wx8 resin was filtered and the solution was evaporated to a syrup (bath temp. 40° C.). The syrup was diluted with 10 L of ethyl acetate and extracted with 20 L brine and 20 L water. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The isolated yield of the syrup AMod4 was about 88%.
Synthesis of the BA Dimer
Step 10. Preparation of AMod5,6-Benzoylation
A 100 L reactor was charged with 2858 g of Methyl 4,6-diOH Monomer A2 (AMod4) (9.24 mol, 1 equiv) and co-evaporated with 3×10 L of pyridine. When evaporation was complete, 15 L of dichloromethane, 6 L of pyridine were transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −40° C. The reactor was charged with 1044 mL (1299 g, 9.24 mol, 1 equiv) of benzoyl chloride. When the addition was complete, the reaction was allowed to warm to −10° C. over a period of 2 hours. The reaction was monitored by TLC (60% ethyl acetate/hexane). When the reaction was completed, the solution was evaporated to a syrup (bath temp. 40° C.). This was co-evaporated with 3×15 L of toluene. The syrup was diluted with 40 L ethyl acetate. Extraction was carried out with 20 L of water and 20 L of brine solution. The Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 25:70 and 30:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. The isolated yield of the intermediate AMod5 was 84%.
Synthesis of the BA Dimer
Step 11. Preparation of BA1, Coupling of Amod5 with BMod6
A 100 L reactor was charged with 3054 g of methyl 4-Hydroxy-Monomer A2 (AMod5) from Step 10 (7.38 mol, 1 equiv) and 4764 g of 4-Lev-1-TCA-Monomer B1 (BMod6) from Step 6 (7.38 mol, 1 equiv). The combined monomers were dissolved in 20 L of toluene and co-evaporated at 40° C. Co evaporation was repeated with an additional 2×20 L of toluene, after which 30 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to below −20° C. When the temperature was between −20° C. and −40° C., 1572 g (1404 mL, 11.12 moles, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) were transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to 0° C. and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (40:70 ethyl acetate/heptane). The reaction required 3-4 hours to reach completion. When the reaction was complete, 926 mL (672 g, 6.64 mol, 0.9 equiv) of triethylamine (TEA) was transferred into the mixture and stirred for an additional 30 minutes, after which 20 L of water and 10 L of dichloromethane were transferred into the reactor. The solution was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 2×20 L water and 20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and used in the next step. The isolated yield of the disaccharide BA1 was about 72%.
Synthesis of the BA Dimer
Step 12, Removal of Levulinate Methyl [(methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl]-2-deoxy-α-D-glucopyranoside
A 100 L reactor was charged with 4.104 Kg of 4-Lev BA Dimer (BA1) (4.56 mol, 1 equiv) in 20 L of THF. The reactor was purged with nitrogen and chilled to −20 to −25° C., after which 896 mL of hydrazine hydrate (923 g, 18.24 mol, 4 equiv) was transferred into the reactor. Stirring was continued and the reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 2-3 hour for the completion, after which 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (ETOAc layer) was extracted with 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to dryness. The isolated yield of the BA Dimer was 82%. Formula: C42H43N3O13; Mol. Wt. 797.80.
Synthesis of the EDC Trimer
Step 1. Preparation of EMod1, Acetylation
A 100 L reactor was charged with 16533 g of Monomer E (45 mole, 1 eq.), 21.25 L acetic anhydride (225 mole, 5 eq.) and 60 L of dichloromethane. The reactor was purged with nitrogen and was chilled to −10° C. When the temperature was at −10° C., 1.14 L (1277 g) of boron trifluoride etherate (BF3.Et2O, 9.0 moles, 0.2 eq) were transferred into the reactor. After the complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by TLC (30:70 ethyl acetate/heptane) and HPLC. The reaction took approximately 3-6 hours to reach completion. When the reaction was completed, the mixture was extracted with 3×50 L of 10% sodium bicarbonate and SOL of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The isolated yield of EMod1 was 97%.
Synthesis of the EDC Trimer
Step 2. Preparation of EMod2, De-Acetylation of Azidoglucose
A 100 L reactor was charged with 21016 g of 1,6-Diacetyl Monomer E (EMod1) (45 mole, 1 eq.), 5434 g of hydrazine acetate (NH2NH2.HOAc, 24.75 mole, 0.55 eq.) and 50 L of DMF (dimethyl formamide). The solution was stirred at room temperature and the reaction was monitored by TLC (30% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was completed, 50 L of dichloromethane and 40 L of water were transferred into the reactor. This was stirred for 30 minutes and the layers were separated. This was extracted with an additional 40 L of water and the organic phase was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of intermediate EMod2 was 100%.
Synthesis of the EDC Trimer
Step 3. Preparation of EMod3, Formation of 1-TCA
A 100 L reactor was charged with 12752 g of 1-Hydroxy Monomer E (EMod2) (30 mole, 1 eq.) in 40 L of dichloromethane. The reactor was purged with nitrogen and stirring was started, after which 2.25 L of DBU (15 moles, 0.5 eq.) and 15.13 L of trichloroacetonitrile (150.9 moles, 5.03 eq) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After the reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (30:70 ethyl acetate/Heptane) and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, 40 L of water and 20 L of DCM were charged into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 40 L water and the DCM solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90 (DCM/EtOAc/heptane), 20:5:75 (DCM/EtOAc/heptane) and 20:10:70 DCM/EtOAc/heptane). Pure fractions were pooled and evaporated to give Intermediate EMod3 as a syrup. Isolated yield was 53%.
Synthesis of the EDC Trimer
Step 4. Preparation of ED Dimer, Coupling of E-TCA with Monomer D
A 100 L reactor was charged with 10471 g of 6-Acetyl-1-TCA Monomer E (EMod3) (18.3 mole, 1 eq., FW: 571.8) and 6594 g of Monomer D (16.47 mole, 0.9 eq, FW: 400.4). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. This was repeated with co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) were transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to −40° C. When the temperature was between −30° C. and −40° C., 2423 g (2071 mL, 9.17 moles, 0.5 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 2040 mL of triethylamine (TEA, 1481 g, 0.8 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The ED Dimer was obtained in 81% isolated yield.
Synthesis of the EDC Trimer
Step 5. Preparation of ED1 TFA, Hydrolysis of ED Dimer
A 100 L reactor was charged with 7.5 Kg of ED Dimer (9.26 mol, 1 equiv). The reactor was chilled to <5° C. and 30.66 L of 90% TFA solution (TFA, 370.4 mol, 40 equiv) were transferred into the reactor. When the addition was completed the reaction was allowed to warm to room temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexanes) and HPLC. The reaction took approximately 3-4 hours to reach completion. When the reaction was completed, was chilled and 51.6 L of triethylamine (TEA, 37.5 Kg, 370.4 mole) were transferred into the reactor, after which 20 L of water & 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60, 50:50, 60:40 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. Isolated yield of ED1 was about 70%.
Synthesis of the EDC Trimer
Step 6. Preparation of ED2, Silylation of ED1
A 100 L reactor was charged with 11000 g of 1,2-diOH ED Dimer (ED1) (14.03 mol, 1 equiv), 1910.5 g of imidazole (28.06 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 3.53 L butyldiphenylchloro-silane (TBDPS-Cl, 4.628 Kg, 16.835 mol, 1.2 equiv) was charged into the reactor. When the addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (50% ethyl acetate/hexane) and HPLC. The reaction required 4-6 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Intermediate ED2 was obtained in 75% isolated yield.
Synthesis of the EDC Trimer
Step 7. Preparation of ED3, D-Levulination
A 100 L reactor was charged with 19800 g of 1-Silyl ED Dimer (ED2) (19.37 moles, 1 equiv) and 40 L of dry tetrahydrofuran (THF) and agitated to dissolve. The reactor was purged with nitrogen and 237 g of dimethylaminopyridine (DMAP, 1.937 moles, 0.1 equiv) and 10.05 L of diisopropylethylamine (DIPEA, 63.9 moles, 3 equiv) were transferred into the reactor. The reactor was chilled to 10-15° C. and kept under a nitrogen atmosphere, after which 12.46 Kg of levulinic anhydride (58.11 moles, 3 eq) was charged into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. The completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and by HPLC. 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The ED3 yield was 95%.
Synthesis of the EDC Trimer
Step 8. Preparation of ED4, Desilylation of ED3
A 100 L reactor was charged with 19720 g of 1-Silyl-2-Lev ED Dimer (ED3) (17.6 mol, 1 equiv) in 40 L of THF. The reactor was chilled to 0° C., after which 6903 g of tetrabutylammonium fluoride trihydrate (TBAF, 26.4 mol, 1.5 equiv) and 1511 mL (26.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred and allowed to warm to ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction required 3 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of ED4 was about 92%.
Synthesis of the EDC Trimer
Step 9. Preparation of ED5, TCA Formation
A 100 L reactor was charged with 14420 g of 1-OH-2-Lev ED Dimer (ED4) (16.35 mol, 1 equiv) in 30 L of dichloromethane. The reactor was purged with nitrogen and stirring was begun, after which 1222 mL of diazabicycloundecene (DBU, 8.175 mol, 0.5 equiv) and 23.61 Kg of trichloroacetonitrile (TCA, 163.5 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours for reaction completion. When the reaction was completed, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of intermediate ED5 was about 70%.
Synthesis of the EDC Trimer
Step 10.
Preparation of EDC Trimer, Coupling of ED5 with Monomer C 6-O-acetyl-2-azido-3,4-di-O-benzyl-2-deoxy-α-D-glucopyranosyl-(1→4)-benzyl (3-O-benzyl-2-O-levulinoyl)-β-D-glucopyranosyluronate-(1→4)-(3-O-acetyl-1,6-anhydro-2-azido)-2-deoxy-β-D-glucopyranose
A 100 L reactor was charged with 12780 g of 2-Lev 1-TCA ED Dimer (ED5) (7.38 mole, 1 eq., FW) and 4764 g of Monomer C (7.38 mole, 1 eq). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. Repeated was co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −20° C. When the temperature was between −20 and −10° C., 2962 g (11.2 moles, 0.9 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm to 5° C. and stirring was continued. Completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 1133 g of triethylamine (TEA, 0.9 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. Dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of EDC Trimer was 48%. Formula: C55H60N6O18; Mol. Wt. 1093.09. The 1H NMR spectrum (d6-acetone) of the EDC trimer is shown in FIG. 3.

Preparation of EDC1
Step 1:
Anhydro Ring Opening & Acetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-1,3,6-tri-O-acetyl-β-D-glucopyranose
7.0 Kg (6.44 mol) of EDC Trimer was dissolved in 18 L anhydrous Dichloromethane. 6.57 Kg (64.4 mol, 10 eq) of Acetic anhydride was added. The solution was cooled to −45 to −35° C. and 1.82 Kg (12.9 mol, 2 eq) of Boron Trifluoride etherate was added slowly. Upon completion of addition, the mixture was warmed to 0-10° C. and kept at this temperature for 3 hours until reaction was complete by TLC and HPLC. The reaction was cooled to −20° C. and cautiously quenched and extracted with saturated solution of sodium bicarbonate (3×20 L) while maintaining the mixture temperature below 5° C. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. The resulting syrup of EDC1 (6.74 Kg) was used for step 2 without further purification. The 1H NMR spectrum (d6-acetone) of the EDC-1 trimer is shown in FIG. 4.

Preparation of EDC2
Step 2:
Deacetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-β-D-glucopyranose
The crude EDC1 product obtained from step 1 was dissolved in 27 L of Tetrahydrofuran and chilled to 15-20° C., after which 6 Kg (55.8 mol) of benzylamine was added slowly while maintaining the reaction temperature below 25° C. The reaction mixture was stirred for 5-6 hours at 10-15° C. Upon completion, the mixture was diluted with ethyl acetate and extracted and quenched with 10% citric acid solution (2×20 L) while maintaining the temperature below 25° C. The organic layer was extracted with 10% NaCl/1% sodium bicarbonate (1×20 L). The extraction was repeated with water (1×10 L), dried over anhydrous sodium sulfate and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel yielded 4.21 Kg (57% yield over 2 steps) of EDC2[ also referred to as 1-Hydroxy-6-Acetyl EDC Trimer]. The 1H NMR spectrum (d6-acetone) of the EDC-2 trimer is shown in FIG. 5.

Preparation of EDC3
Step 3:
Formation of TCA Derivative 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-1-O-trichloroacetimidoyl-β-D-glucopyranose
4.54 Kg (3.94 mol) of EDC2 was dissolved in 20 L of Dichloromethane. 11.4 Kg (78.8 mol, 20 eq) of Trichloroacetonitrile was added. The solution was cooled to −15 to −20° C. and 300 g (1.97 mol, 0.5 eq) of Diazabicycloundecene was added. The reaction was allowed to warm to 0-10° C. and stirred for 2 hours or until reaction was complete. Upon completion, water (20 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. Column chromatographic separation using silica gel and 20-60% ethyl acetate/heptane gradient yielded 3.67 Kg (72% yield) of 1-TCA derivative, EDC3. The 1H NMR spectrum (d6-acetone) of the EDC-3 trimer is shown in FIG. 6.

Preparation of EDCBA1
Step 4:
Coupling of EDC3 with BA Dimer Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
3.67 Kg (2.83 mol) of EDC3 and 3.16 Kg (3.96 mol, 1.4 eq) of BA Dimer was dissolved in 7-10 L of Toluene and evaporated to dryness. The resulting syrup was coevaporated with Toluene (2×15 L) to remove water. The dried syrup was dissolved in 20 L of anhydrous Dichloromethane, transferred to the reaction flask, and cooled to −15 to −20° C. 898 g (3.4 mol, 1.2 eq) of triethylsilyl triflate was added while maintaining the temperature below −5° C. When the addition was complete, the reaction was immediately warmed to −5 to 0° C. and stirred for 3 hours. The reaction was quenched by slowly adding 344 g (3.4 mol, 1.2 eq) of Triethylamine. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. The resulting syrup of the pentasaccharide, EDCBA1 was used for step 5 without further purification. The 1H NMR spectrum (d6-acetone) of the EDCBA-1 pentamer is shown in FIG. 7.

Preparation of EDCBA2
Step 5:
Hydrolysis of Levulinyl moiety Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)—O-[benzyl 3-O-benzyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl)-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
The crude EDCBA1 from step 4 was dissolved in 15 L of Tetrahydrofuran and chilled to −20 to −25° C. A solution containing 679 g (13.6 mol) of Hydrazine monohydrate and 171 g (1.94 mol) of Hydrazine Acetate in 7 L of Methanol was added slowly while maintaining the temperature below −20° C. When the addition was complete, the reaction mixture was allowed to warm to 0-10° C. and stirred for several hours until the reaction is complete, after which 20 L of Ethyl acetate was added and the reaction was extracted with 10% citric acid (2×12 L). The organic layer was washed with water (1×12 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-45% ethyl acetate/heptane gradient yielded 2.47 Kg (47.5% yield over 2 steps) of EDCBA2. The 1H NMR spectrum (d6-acetone) of the EDCBA-2 pentamer is shown in FIG. 8.

Preparation of EDCBA Pentamer
Step 6:
THP Formation Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
2.47 Kg (1.35 mol) of EDCBA2 was dissolved in 23 L Dichloroethane and chilled to 10-15° C., after which 1.13 Kg (13.5 mol, 10 eq) of Dihydropyran and 31.3 g (0.135 mol, 0.1 eq) of Camphorsulfonic acid were added. The reaction was allowed warm to 20-25° C. and stirred for 4-6 hours until reaction was complete. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCE. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-35% ethyl acetate/heptane gradient yielded 2.28 Kg (88.5% yield) of fully protected EDCBA Pentamer. The 1H NMR spectrum (d6-acetone) of the EDCBA pentamer is shown in FIG. 9.

Preparation of API1
Step 1:
Saponification Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-α-L-Idopyranosyluronosyl-(1→4)-2-azido-3-O-benzyl-2-deoxy-α-D-glucopyranoside disodium salt
To a solution of 2.28 Kg (1.19 mol) of EDCBA Pentamer in 27 L of Dioxane and 41 L of Tetrahydrofuran was added 45.5 L of 0.7 M (31.88 mol, 27 eq) Lithium hydroxide solution followed by 5.33 L of 30% Hydrogen peroxide. The reaction mixture was stirred for 10-20 hours to remove the acetyl groups. Then, 10 L of 4 N (40 mol, 34 eq) sodium hydroxide solution was added. The reaction was allowed to stir for an additional 24-48 hours to hydrolyze the benzyl and methyl esters completely. The reaction was then extracted with ethyl acetate. The organic layer was extracted with brine solution and dried with anhydrous sodium sulfate. Evaporation of the solvent under vacuum gave a syrup of API1 [also referred to as EDCBA(OH)5] which was used for the next step without further purification.
Preparation of API2
Step 2:
O-Sulfonation Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-azido-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
The crude product of API1 [aka EDCBA(OH)5] obtained in step 1 was dissolved in 10 L Dimethylformamide. To this was added a previously prepared solution containing 10.5 Kg (66 moles) of sulfur trioxide-pyridine complex in 10 L of Pyridine and 25 L of Dimethylformamide. The reaction mixture was heated to 50° C. over a period of 45 min. After stiffing at 1.5 hours at 50° C., the reaction was cooled to 20° C. and was quenched into 60 L of 8% sodium bicarbonate solution that was kept at 10° C. The pH of the quench mixture was maintained at pH 7-9 by addition of sodium bicarbonate solution. When all the reaction mixture has been transferred, the quench mixture was stirred for an additional 2 hours and pH was maintained at pH 7 or greater. When the pH of quench has stabilized, it was diluted with water and the resulting mixture was purified using a preparative HPLC column packed with Amberchrom CG161-M and eluted with 90%-10% Sodium Bicarbonate (5%) solution/Methanol over 180 min. The pure fractions were concentrated under vacuum and was then desalted using a size exclusion resin or gel filtration (Biorad) G25 to give 1581 g (65.5% yield over 2 steps) of API2 [also referred to as EDCBA(OSO3)5]. The 1H NMR spectrum (d6-acetone) of API-2 pentamer is shown in FIG. 10.

Preparation of API3
Step 3:
Hydrogenation Methyl O-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-amino-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
A solution of 1581 g (0.78 mol) of O-Sulfated pentasaccharide API2 in 38 L of Methanol and 32 L of water was treated with 30 wt % of Palladium in Activated carbon under 100 psi of Hydrogen pressure at 60-65° C. for 60 hours or until completion of reaction. The mixture was then filtered through 1.0μ and 0.2μ filter cartridges and the solvent evaporated under vacuum to give 942 g (80% yield) of API3 [also referred to as EDCBA(OSO3)5(NH2)3]. The 1H NMR spectrum (d6-acetone) of API-3 pentamer is shown in FIG. 11.

Preparation of Fondaparinux Sodium
Step 4:
N-Sulfation & Removal of THP Methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)—O-β-D-glucopyranuronosyl-(1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N-sulfated EDCBA(OSO3)5(NHSO3)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux Sodium form.
Analysis of the Fondaparinux sodium identified the presence of P1, P2, P3, and P4 in the fondaparinux. P1, P2, P3, and P4 were identified by standard analytical methods.
INTERMEDIATES
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.

The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.

Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:

Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:


The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:


Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, havinga free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:

The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.


In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
…………………………
A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodium, Org Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c

An improved method for the simultaneous removal of O-benzyl and N-carboxybenzyl groups as well as reducing azide groups to amines in protected heparin-like pentasaccharides, a key process in fondaparinux sodium synthesis, is reported. Under catalytic transfer hydrogenation conditions, using readily available and inexpensive ammonium formate, the hydrogenolysis is done in less than an hour in good yield and purity. This procedure represents a major advantage over the previously published procedures, the latter of which involve several hours/days of hydrogenation reaction under catalytic reduction using gaseous hydrogen.

Synthesis of Compound 1 (FONDAPARINUX)
………………
SYNTHESIS
In the synthesis of Fondaparinux sodium, the monomers XII, XVIII, XXVII, XXXVIII, XXXXI and dimers XIX, XX, XL described herein may be made either by processes described in the art or, by a process as described herein. The XII and XVIII monomers may then linked to form a disaccharide XX, XXXIX and XXVII monomers may then linked to form a disaccharide XL, XLIII and XX dimers may then linked to form a tetrasaccharide, XLVII tetramer and XLV monomer may be linked to form a pentasaccharide (XLVIII) pentamer. The XLVIII pentamer is an intermediate that may be converted through a series of reactions to fondaparinux sodium. This strategy described herein provides an efficient method for multi-kilogram preparation of fondaparinux in high yields and highly stereoselective purity.
Fondaparinux sodium (LIII) was prepared in 3 synthetic steps from O – S pentasaccharide (L) using the following procedure:

Fondaparinux Sodium (LIII)
Preparation of Fondaparinux sodium (LIII)—
N- sulfonation of Deprotected Pentasaccharide (LI) methyl 0-2-deoxy-3,6-di-0- sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l— >4)-0-2-0-sulfo-a-L- idopyranurosyl-( 1— >4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranoside,decasodium salt
A solution of deprotected pentasaccharide (LI) (145 gm) in water (2.54 V) was adjusted to a pH of 9.5 – 10.5 with 1 N NaOH solution. S03-pyridine complex (156 gm) was added into 3 lots every 15 min, the pH being maintained at 9.5-10.5 by automatic addition of 1 N NaOH. The mixture was stirred for 2 hrs at RT, during this aqueous NaOH (IN solution) was added to maintain pH at 9.5 – 10.5. After neutralization to pH 7 – 7.5 by addition of HC1 solution, the mixture was evaporated using vacuum. The residue was dissolved in water (1.6 L) at RT, to this solution was added acetone (1.6 L) at RT. The reaction mass was cooled to 5°C – 1 0 °C and stirred for 1 hr. The solid was filtered and washed with cold acetone: water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (1.6 L) at RT, and to this solution was added acetone(1.6 L) at RT. The mixture was cooled to 5 to 10°C and stirred for 1 hr. The solid was filtered and washed with cold acetone/water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (0.7 L) and charcoal (40 gm) was added at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. The pH of the clear filtrate was adjusted to 8.0 – 8.5 with IN NaOH solution and distilled off completely under vacuum below 55 °C. The residue was dissolved in 0.5 M NaCl solution and layered onto a column of Dowex® 1×2 -400 resins using a gradient of NaCl solution (0.5 to 10M). The product fractions were combined and distilled off under vacuum below 55 °C up to 1 – 2 L volume. The solid was filtered off and the clear filtrate was distilled off under vacuum below 55 °C up to slurry stage and subjected to azeotropic distillation with methanol two times. The solid residue was stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The above solid was dissolved in water and the pH adjusted to 4 – 4.5 with IN HC1 solution and charcoalized three times with 26 gm of charcoal at RT for 15-30 minutes and filtered off. To the clear filtrate was added 0.39 kg of NaCl, then methanol was added (35 volume) at RT and the mixture was stirred for 15-30 minutes. The solution was decanted and the sticky mass was stirred with methanol (0.65 L) at RT for 15-30 minutes. The solid was filtered off and dissolved in water, and the pH adjusted to 8 – 8.5 with IN NaOH solution. The solution was filtered through 0.45 micron paper & distilled off completely under vacuum below 55°C. The solution was subjected to azeotropic distillation with methanol to give highly pure fondaparinux sodium (97.17 gm) (HPLC purity 99.7%).
SOR Results
Three batches of product made in accordance with the present processes provided the following stereoisomeric optical rotation results:
Specification: Between +50.0° and +60.0°.
Batch- 1 = +55.1°
Batch-2 = +55.7° Batch-3 = +55.4°.
INTERMEDIATES
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; MS is molecular sieve; DMF is dimethyl formamide; Bn is benzyl; MDC is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; MeOH is methanol; RT is room temperature; Ac2O is acetic anhydride; HBr is hydrogen bromide; EtOAc is ethyl acetate; Cbz is benzyloxycarbonyl; CADS is chloro acetyl disaccharide; HDS is hydroxy disaccharide; NMP is N-methylpyrrolidone.
Methyl 3-O-benzyl-4-O-monochloro acetyl-β-L-idopyranuronate

Route of Synthesis for α-Methyl-6-o-acetyl-3-o-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-α-D-glucopyranoside

Methyl 6-O-acetyl-3-O-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-4-O-(methyl-2-O acetyl-3-O-benzyl-α-L-idopyranosyluronate)-glucopyranoside

Route of Synthesis for 1,6-Anhydro-2-azido-3-O-acetyl-2-deoxy-beta-D-glucopyranose

Route of synthesis for Methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranuronate

Route of synthesis for 3-O-Acetyl-1,6-anhydro-2-azido-4-O-2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyl methyluronate-beta-D-glucopyranose
(or)
3-O-Acetyl-1,6-anhydro-2-azido-2-deoxy-4-O-(methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyluronate)-beta-D-glucopyranose

Route of Synthesis for 1,6-Anhydro-2-azido-3,4-di-O-benzyl-2-deoxy-beta-D-glucopyranose

Synthesis of Disaccharide XLIII
Disaccharide XLIII was prepared in 2 synthetic steps from CADS sugar (XL) using the following procedure:

CADS sugar XL was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative XLII. This step was carried out using the reactants CADS, AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 20 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl CADS (XLII) was brominated at the anomeric carbon using titanium tetra bromide in MDC andethylacetate and stirring at 20° C.-50° C. for 6-16 hours, preferably 6 hours, to give the bromo derivative, (XLIII) after work-up and recrystallization from solvent/alcohol.
Synthesis of the Monosaccharide (XLV)
The monosaccharide (XLV) was prepared in 2 synthetic steps from monomer (XLI) using the following procedure:

Mono sugar (XLI) was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative (XLIV). This step was carried out using the reactants Mono sugar (XLI), AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 24 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl Mono sugar (XLIV) was brominated at the anomeric carbon using titanium tetra bromide in MDC and ethyl acetate and stirring at 20° C.-50° C. for 6-20 hours, preferably 16 hours, to give the bromo derivative, (XLV) after work-up and recrystallization from ether.
Synthesis of the Hydroxy Tetrasaccharide (XLVII)
The hydroxy tetrasaccharide (XLVII) was prepared in 2 synthetic steps from disaccharide (XLIII) and HDS (XX) using the following procedure:

Disaccharide (XLIII), was coupled with disaccharide (XX) in the presence of silver carbonate, silver per chlorate and 4 A° MS in MDC and stirred at ambient temperature for 5-12 hrs, preferably 4-6 hours, in the dark followed by work-up and purification in water/methanol to give the tetrasaccharide (XLVI). The d echloroacetylation of tetrasaccharide (XLVI) was carried out in THF, ethanol and pyridine in the presence of thiourea at reflux for 6 to 20 hrs, preferably 12 hours, to give the hydroxy tetrasaccharide (XLVIII).
Synthesis of the Pentasaccharide (XLVIII)
The pentasaccharide (XLVIII) was prepared in 2 synthetic steps from monosaccharide (XLV) and tetrasaccharide (XLVII) using the following procedure:

Monosaccharide (XLV), was coupled with tetrasaccharide (XLVII) in the presence of 2,4,6-collidine, silver triflate and 4 A° MS in MDC and stirred at −10° C. to −20° C. for 1 hr in the dark followed by work-up and purification by column chromatography to give the pentasaccharide (XLVIII).
Synthesis of OS Pentasaccharide (L)
The OS pentasaccharide (L) was prepared in 2 synthetic steps from pentasaccharide (XLVIII) using the following procedure:

Pentasaccharide (XLVIII) was deacetylated in the presence of NaOH in mixture of solvents of MDC, methanol and water at 0° C. to 35° C., for 1-2 hrs followed by work-up and distillation to obtain deacetylated pentasaccharide (XLIX) which was subjected to O-sulfonation in DMF in the presence of SO3-trimethylamine (TMA) at 50° C. to 100° C., preferably 50° C.-55° C., for 6-24 hrs, preferably 12 hours, followed by salt removal through Sephadex® resin and column chromatography purification, then pH adjustment by dilute NaOH to give OS pentasaccharide (L).
…………………………
INTERMEDIATE
highly pure 4-Ο-β-ϋ- glucopyranosyl- 1 ,6-anhydro- -D-glucopyranose

Example 1 : Preparation and purification of 4-0- -D-grucopyranosyl-L6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2′,3′,4′,6′-hepta-(9-acetyl- -D-ceilobioside represented by Formula I;

(400 g) in isopropyl alcohol (4 L) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (355 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was then heated to 50°C to 55°C and stirred for 30 minutes. The solid obtained was filtered and washed with isopropyl alcohol (400 mL). The solid was stirred with isopropyl alcohol (2.8 L) at 50°C for 30 minutes followed by filtering and washing with isopropyl alcohol (400 mL). The resultant solid was suspended into methanol (800 mL to 1600 mL) followed by cooling to 2°C to 5°C. The pH of the suspension was adjusted to 2 to 3 using 15% methanolic hydrochloride. The solid so obtained was filtered and washed with methanol (400 mL). Solvent was recovered from the filtrate to dryness under vacuum to obtain the pure compound of Formula II as foamy solid.
Yield: 142 g
Example 2: Preparation and purification of 4-Q- -D-grucopyranosyl-l,6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2 ,3 ^ ^‘-hepta-O-acetyl- -D-cellobioside of Formula I (100 g) in methanol (300 mL) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (88.6 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was cooled to 2°C to 5°C and 15% methanolic hydrogen chloride was added to it until the pH of the mixture reached 2 to 3. At this pH, the reaction mixture was filtered and the residual solid was washed with methanol (100 mL). The solvent was recovered from the filtrate under vacuum. The solid material so obtained was stirred with dichloromethane (500 mL) followed by removal of solvent through decantation/filtration. The resultant solid was stirred with isopropyl alcohol (500 mL), filtered and dried to obtain the pure compound of Formula II.
Yield: 29 g
………………………
SYNTHESIS
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:
Conversion of FPP (also referred to a Fully Protected Pentamer) to FondaparinuxSodium:

Reagents: 1. NaOH, H202, LiOH, Dioxane, RT, 24-48 h; 2. Py.S03, DMF, 60°C, 2h, CG-161 purification; 3. 10% Pd/C, H2, 72h; 4. (a) Py.S03, NaOH, NH4OAc, 12h, (b) HiQ NH4OAc/ NaCl ion-exchange, Sephadex Desalt and (c) HiQ NaCl ion-exchange, Sephadex Desalt. The ester moieties in EDCBA Pentamer-CB were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 24- 48 hours to give the pentasaccharide intermediate API1-CB. The five hydroxyl moieties in API1-CB were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60°C for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2-CB. The intermediate API2-CB was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the six benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3-CB. This transformation occurs by reacting API2-CB with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3-CB were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours . The crude fondaparinux is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final product, fondaparinux sodium.
Preparation of Fondaparinux Sodium – Step 4: N-Sulfation of API-3-CB:
Methyl 0-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l→4)-0^-D- glucopyranuronosyl-(l→4)-0-2-deoxy-3,6-di-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl- (l→4)-0-2-0-sulfo-a-L-idopyranuronosyl-(l→4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D- glucopyranoside, decasodium salt
To a solution of 25.4 gram (16.80 mmol, leq) of API-3-CB in 847 mL of water was slowly added 66.85 gram (446.88 mmol, 25eq) of sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2N sodium hydroxide solution. The reaction was allowed to stir for 4 hours at pH 9.0 – 9.5. When reaction was completed, the pH was adjusted 7.0 by using 70 mL of 50 mmol Ammonium acetate solution pH -3.5. The resulting N-Sulfated Cellobiose mixture was purified using Ion-Exchange
Chromatographic Column followed by desalting using size exclusion resin to gave gram ( %) of the purified Fondaparinux Sodium form.
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N- sulfated EDCBA(OS03)5(NHS03)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux sodium form.
INT
SCHEME 1 – Synthesis of Monomer A-2 & AMod5 fBuildinq Block Al

Reagents: 1. NaOMe, MeOH, RT, 2hr, 50wx resin; 2. (Bu3Sn)20 (0.8equiv), ACN, MS, reflux, 3h; 3.l2 (1.5 equiv), 5°C to RT, 2h; 4. NaH (2 equiv), DMF, p-MeOC6H4CH2Br (PMB-Br, 2.5 equiv), -20°C to RT, 2h; 5. NaN3, DMF, 120°C, 12h; 6. NaH, DMF, BnBr, 0°C to RT, 3h.; 7. BF3.Et20, Ac20, DCM, -20°C to RT, 3h; 8. (a) TMS-I, TBAI, RT, 2h; (b) DIPEA, MeOH, 16h, RT; 9. NaOMe, Dowex 50WX8-100 resin H+ form, RT, 3h; 10. Pyridine, Bz-CI, -40°C to -10°C, 2h;
Scheme 2 – Synthesis of Monomer B-1 and BMod6 fBuildinq Block B1

Reagents: 1. NaH, BnBr, THF, DMF, 0° to 65°C, 3h; 2. 66% Acetic Acid/H20, 40 °C, 16h; 3. Nal04, (Bu)4NBr, DCM, H20, Dark, 3h; 4. (PhS)3CH, n-BuLi, THF, -78 °C, 3h; 5. CuCI2/CuO, MeOH, H20, 3h; 6. 90% TFA/H20, DCM, RT, 2h; 7. DMF, CSA 2-methoxypropene, 0° to RT, 16hrs; MeOH, TEA. 8. Lev20, DIPEA, RT, 16h; 9. 90% TFA, RT, 4h; 10. Imidazole, TBDPSi-CI, RT, 3h; 11. Pyridine, BzCI, RT, 3h; 12. TBAF, RT, 3h; 13. TCA, DBU, RT, 2h; Also see, e.g., Bonnaffe et al., Tetrahedron Lett., 41, 307-311, 2000; Bonnaffe et al., Carbohydr. Res., 2003, 338, 681-686, 2003; and Seeberger et al., J. Org. Chem., 2003, 68, 7559- 7561, 2003.
……………………..
Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
1H NMR (D2O) δ: 5.68 (d, J = 3.8 Hz, 1H, H-1D), 5.56 (d, J = 3.4 Hz, 1H, H-1F), 5.24 (d, J = 3.8 Hz, 1H, H-1G), 5.07 (d, J = 3.5 Hz, 1H, H-1H), 4.68 (d, J = 7.9 Hz, 1H, H-5G), 4.54 (dd, J = 11.4, 2.2 Hz, 1H, H-1E), 4.48-4.34 (m, 6H, H-6F, 6H, 6’H, 6D, 3E, 2G), 4.33-4.30 (m, 1H, H-6’F), 4.25-4.17 (m, 4H, H-4G, 3G, 6’D, 5F), 4.06-3.98 (m, 2H, H-4F, 5H), 3.94 (dd, J = 9.7, 2.2 Hz, 1H, H-5D), 3.92-3.86 (m, 2H,H-3E, H-4E), 3.85-3.80 (m, 2H, H-5E, 4H), 3.73-3.60 (m, 3H, H-3H, 3D, 4D), 3.53-3.44 (m, 2H, H-2F, H-2E), 3.47 (s, 3H, OMe), 3.34 (dd, J = 10.2, 3.7 Hz, 1H, H-2H), 3.31(dd, J = 10.2, 3.7 Hz, 1H, H-2D)

FONDAPARINUX
……………………………………..
Synthesis of intermediates
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.
The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.
Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:
Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:
The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:
Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, having a free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:
The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.
In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
………………………………
Intermediates listed on the internet
Fondaparinux sodium Intermediates
Fondaparinux sodium N-4

……………………………….
Fondaparinux sodium N-3
114903-05-8

a-D-Glucopyranoside, Methyl O-2-azido-2-deoxy-3,4-bis-O-(phenylMethyl)-a-D-glucopyranosyl-(14) -O-2,3-bis-O-(phenylMethyl)-b-D-glucopyranuronosyl-(14)-O-2-azido- 2-deoxy-a-D-glucopyranosyl-(14)-O-3-O-(phenylMethyl)-a-L-idopyranu ronosyl-(14)-2-deoxy-2
FSC

114903-05-8

|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||
|
|||||||
| Description | |||||||
|
|||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
References
- “Medscape.com”. Retrieved 2009-01-23.
- “NEJM — Comparison of Fondaparinux and Enoxaparin in Acute Coronary Syndromes”. Retrieved 2009-01-23.
- Peters RJ, Joyner C, Bassand JP, et al. (February 2008). “The role of fondaparinux as an adjunct to thrombolytic therapy in acute myocardial infarction: a subgroup analysis of the OASIS-6 trial”.Eur. Heart J. 29 (3): 324–31. doi:10.1093/eurheartj/ehm616. PMID 18245119.
- WO 2013003001
- Synthesis of heparin fragments: A methyl alpha-pentaoside with high affinity for antithrombin III
Carbohydr Res 1987, 167: 67 - A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodiumOrg Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c
- WO 2012047174
- US 2012116066
- WO 2013011460 RANBAXY
- WO 2013115817
- The unique antithrombin III binding domain of heparin: A lead to new synthetic antithrombotics
Angew Chem Int Ed Engl 1993, 32(12): 1671 - Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991).
- Carbohydrate Research, 101, p. 148-151 (1982),
- Chemistry – A European Journal, 2012 , vol. 18, 34 pg. 10643 – 10652
- Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
- Analytical Chemistry, 2006 , vol. 78, 6 pg. 1774 – 1779
PATENTS
| US4818816 * | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof |
| US6376663 * | Nov 29, 1996 | Apr 23, 2002 | Macquarie Research Ltd. | Desalting and purification of oligosaccharides and their derivatives |
| US7541445 * | Sep 6, 2002 | Jun 2, 2009 | Alchemia Limited | Synthetic heparin pentasaccharides |
| US20040048785 * | Jun 18, 2003 | Mar 11, 2004 | Societe L’oreal S.A. | C-glycoside compounds for stimulating the synthesis of glycosaminoglycans |
| US20040149200 * | Jun 11, 2002 | Aug 5, 2004 | Tsuyoshi Shimose | Crystals of an oligosaccharides and process for preparation thereof |
| US20110105418 * | Jul 30, 2010 | May 5, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof |
| WO2011014793A2 * | Jul 30, 2010 | Feb 3, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof | |
| AU2008200616A1 | Title not available | ||||
| JPS63218691A * | Title not available | ||||
| US4818816 | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof | |
| US7468358 | Oct 27, 2004 | Dec 23, 2008 | Paringenix, Inc. | Method and medicament for sulfated polysaccharide treatment of heparin-induced thrombocytopenia (HIT) syndrome | |
| US84771910 | Title not available | ||||
| USPP23055709 | Title not available |
FONDAPARINUX



The three specialties available in the United States – dalteparin (Fragmin, Pfizer), enoxaparin (Lovenox, Sanofi-Aventis) and tinzaparin (Innohep, Bristol-Myers Squibb) – the first two are found in Brazil, enoxaparin under the names Lovenox, Cutenox and Dripanina.

Eluxadoline …Diarrhea-predominant irritable bowel syndrome
Eluxadoline
5 JAN 2014
Furiex Pharmaceuticals Inc. more than doubled in its best day of trading after its experimental drug alleviated diarrhea and abdominal pain caused by irritable bowel syndrome in two studies.
The drug eluxadoline met targets for improvements in stool consistency and abdominal pain that were developed in conjunction with U.S. and European regulators, the company said today. Furiex will apply for approval in June, Chairman Fred Eshelman said in an investor call today. He estimated annual sales of $750 million to $1 billion.
“By our math, it looks like a pretty doggone good market,” Eshelman said on the call, noting that there is only one currently approved drug available in the U.S. for the condition.
Diarrhea-predominant irritable bowel syndrome is a chronic disorder that affects about 28 million patients in the U.S. and Europe, Furiex said in the statement.Furiex said it would apply by mid-year for U.S. approval of the drug, eluxadoline, to treat diarrhea-predominant irritable bowel syndrome (IBS-d), a debilitating bowel disorder that affects about 28 million people in the United States and major European markets.
Furiex said it expected to seek European approval in early 2015.
“We believe that there are a lot of patients out there who need this drug. There is a huge unmet need,” Furiex Chief Medical Officer June Almenoff said in a telephone interview.
Currently approved drugs for IBS address constipation associated with the disorder, but there are few options for diarrhea predominant IBS.
Furiex founder and chairman Fred Eshelman said he believes the drug has the potential for blockbuster sales, which he defined as annual sales of between $750 million and $1 billion.
Eluxadoline was tested at two doses against a placebo over the course of 12 weeks to meet requirements by the U.S. Food and Drug Administration, and for 26 weeks for European health regulators, in Phase III studies involving 2,428 patients, Furiex said.
For the combined goal of improvement in abdominal pain and stool consistency for at least half the days in the study, eluxadoline achieved a statistically significant improvement at the 100 milligram and 75 mg doses through 12 weeks in both studies.
On the 26-week measure, the higher dose succeeded in both studies but the lower dose missed statistical significance in one of the two trials, according to initial results released by the company.
The success appeared to be driven by the percentage of patients reporting improvements in diarrhea, which ranged from 30 percent to 37 percent versus 22 percent and 20.9 percent for the placebo groups.
When the composite goal was broken into its two components, researchers found a numerical improvement in pain response rates that did not achieve statistical significance.
The drug appeared to be safe and well-tolerated in both studies, Furiex said. The most commonly reported side effects were constipation and nausea.
The company plans to present a far more detailed analysis of the late stage studies at an upcoming medical meeting.
“We’re very excited about the path ahead and about how this can transform patients’ lives,” Almenoff said.
Eluxadoline
5-({[(2S)-2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)propanoyl][(1S)-1-(4-phenyl-1H-imidazol-2-yl)ethyl]amino}methyl)-2-methoxybenzoic acid
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
864821-90-9 CAS
JNJ-27018966
Molecular Formula: C32H35N5O5
Molecular Weight: 569.6508
Agents for Irritable Bowel Syndrome, mu-Opioid Agonists, delta-Opioid Antagonists
Mu Delta is a locally active mu opioid receptor agonist and delta opioid receptor antagonist in phase III clinical evaluation at Furiex Pharmaceuticals for the oral treatment of diarrheal predominant irritable bowel syndrome (d-IBS).
The product candidate holds an advantage over currently marketed products for this indication because it acts locally on the enteric nervous system, possibly decreasing adverse effects on the central nervous system. In 2011, fast track designation was assigned in the U.S. for the treatment of d-IBS. In 2011, Mu Delta was licensed to Furiex Pharmaceuticals by Janssen for the treatment of d-IBS, granting an option to Furiex to continue development and commercialization following phase II proof of concept studies.
The opioid receptors were identified in the mid-1970’s, and were quickly categorized into three sub-sets of receptors (mu, delta and kappa). More recently the original three types of receptors have been further divided into sub-types. Also known is that the family of opioid receptors are members of the G-protein coupled receptor (GPCR) super-family. More physiologically pertinent are the well established facts that opioid receptors are found throughout the central and peripheral nervous system of many mammalian species, including humans, and that modulation of the respective receptors can elicit numerous, albeit different, biological effects, both desirable and undesirable (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye, T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269; J. V. Aldrich, “Analgesics”, Burger’s Medicinal Chemistry and Drug Discovery, 5thEdition, Volume 3: Therapeutic Agents, John Wiley & Sons, Inc., 1996, pp. 321-441). In the most current literature, the likelihood of heterodimerization of the sub-classes of opioid receptors has been reported, with respective physiological responses yet undetermined (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development”, Drug Development 2000, pp. 203-238).
A couple biological effects identified for opioid modulators have led to many useful medicinal agents. Most significant are the many centrally acting mu opioid agonist modulators marketed as analgesic agents to attenuate pain (e.g., morphine), as well as peripherally acting mu agonists to regulate motility (e.g., loperamide). Currently, clinical studies are continuing to evaluate medicinal utility of selective delta, mu, and kappa modulators, as well as compounds possessing combined sub-type modulation. It is envisioned such explorations may lead to agents with new utilities, or agents with minimized adverse side effects relative to currently available agents (examples of side effects for morphine includes constipation, respiratory depression, and addiction potential). Some new GI areas where selective or mixed opioid modulators are currently being evaluated includes potential treatment for various diarrheic syndromes, motility disorders (post-operative ileus, constipation), and visceral pain (post operative pain, irritable bowel syndrome, and inflammatory bowel disorders) (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development” Drug Development, 2000, pp. 203-238).
Around the same time the opioid receptors were identified, the enkephalins were identified as a set of endogenous opioid ligands (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye; T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269). Schiller discovered that truncating the original pentapeptide enkephalins to simplified dipeptides yielded a series of compounds that maintained opioid activity (Schiller, P. WO 96/06855). However one potential drawback cited for such compounds is the likelihood of their inherent instability (P. W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), pp. 313-316).
More recently, a series of opioid pseudopeptides containing heteroaromatic or heteroaliphatic nuclei were disclosed, however this series is reported showing a different functional profile than that described in the Schiller works. (L. H. Lazarus et al., Peptides 2000, 21, pp. 1663-1671).
Most recently, works around morphine related structures were reported by Wentland, et al, where carboxamido morphine derivatives and it’s analogs were prepared (M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 1717-1721; M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 623-626). Wentland found that substitution for the phenol moiety of the morphine related structures with a primary carboxamide led anywhere from equal activities up to 40 fold reduced activities, depending on the opioid receptor and the carboxamide. It was also revealed that any additional N-substitutions on the carboxamide significantly diminished the desired binding activity.
Compounds of the present invention have not been previously disclosed and are believed to provide advantages over related compounds by providing improved pharmacological profiles.
Opioid receptor modulators, agonists or antagonists are useful in the treatment and prevention of various mammalian disease states, for example pain and gastrointestinal disorders such as diarrheic syndromes, motility disorders including post-operative ileus and constipation, and visceral pain including post-operative pain, irritable bowel syndrome and inflammatory bowel disorders.
It is an object of the present invention to provide opioid receptor modulators. It is a further object of the invention to provide opioid receptor agonists and opioid receptor antagonists. It is an object of the present invention to provide opioid receptor ligands that are selective for each type of opioid receptor, mu, delta and kappa. It is a further object of the present invention to provide opioid receptor ligands that modulate two or three opioid receptor types, mu, delta and kappa, simultaneously.
It is an object of the invention to provide certain instant compounds that are also useful as intermediates in preparing new opioid receptor modulators. It is also an object of the invention to provide a method of treating or ameliorating a condition mediated by an opioid receptor. And, it is an object of the invention to provide a useful pharmaceutical composition comprising a compound of the present invention useful as an opioid receptor modulator.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1 h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid is an opoid receptor modulator (mu receptor agonist and delta receptor antagonist) and may be useful for treating irritable bowel syndrome, pain or other opioid receptor disorders.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and methods of making this molecule are disclosed in
US application 2005/02033143. Example 9 of US application 2005/02033143 makes the hydrochloride salt of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
Applicants have discovered a process of making the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and two novel crystals of this zwitterion. In Applicant’s hands, these novel crystals provide improved properties and can be purified at higher purity. Applicant’s new process results in improved and less costly process manufacturing conditions than the procedure disclosed in US application 2005/02033143.
………………..
FIG. 6 is the molecular structure of the zwitterion 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.

…………………..
SYNTHESIS OF 5-formyl-2- methoxy-benzoic acid methyl ester
Example 8: 2-Methoxy-5-formylbenzoic acid

Lithium hydroxide (1.04g, 0.043mol, 3eq) in water (lOmL) was added to a stirred solution of methyl 2-methoxy-5-formylbenzoate (2.8g, 0.014mol, leq) in a mixture of tetrahydrofuran (30mL) and methanol (20mL). The solution was stirred overnight, acidified to pH 1 with 10% HCl and the organic solvents removed in vacuo. The aqueous solution was extracted with ethyl acetate (lOOmL) and the organic solution washed with brine (lOOmL), then extracted with saturated aqueous sodium bicarbonate (3 x lOOmL). The basic solution was washed with ethyl acetate (lOOmL), then acidified to pH 1 with 10% HCl and back extracted with dichloromethane (3 x lOOmL). The organic solution was dried over sodium sulfate and evaporated in vacuo to give a cream coloured powder (2.01g, 77%). 1H NMR (CDC13) δ 9.99 (s, IH, O=C- H), 4.14 (s, 3H, CH3).
………………
ANALOGOUS METHOD TO PREPARE..2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester

USE 5-formyl-2- methoxy-benzoic acid methyl ester for 3,4- dimethoxybenzaldehyde, TO GET 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester
Example 4
(3,4-Dimethoxy-benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine

A solution of 1-(4-phenyl-1 W-imidazol-2-yl)-ethylamine (0.061 g, 0.33 mmol) of Example 3, and 0.55 g (0.33 mmol) of 3,4-dimethoxybenzaldehyde in 5 ml_ of anhydrous methanol was stirred at room temperature for 1 h and then cooled to about 0-100C in an ice bath for 1 h. The reaction was treated carefully with 0.019 g (0.49 mmol) of sodium borohydride in one portion and maintained at about 0-100C for 21 h. Cold 2M aqueous HCI was added dropwise (30 drops), the mixture was stirred for 5 min, and then partially concentrated in vacuo unheated. The residual material was taken up in EtOAc to yield a suspension that was treated with 5 ml_ of cold 3M aqueous NaOH and stirred vigorously until clear. The phases were separated and the aqueous layer was extracted three times additional with EtOAc. The combined extracts were dried over MgSO4, filtered, and concentrated to yield (3,4-dimethoxy- benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine as a light yellow oil (HPLC: 87% @ 254nm and 66% @ 214 nm).
MS (ES+) (relative intensity): 338.1 (100) (M+1)
This sample was of sufficient quality to use in the next reaction without further purification.
…………………..
SYNTHESIS
In an embodiment, the present invention is directed to processes for the preparation of the compound of formula (IV)

also known as, 5-({[2-amino-3-(4-carbamoyl-2,5-dimethyl-phenyl)- propionyl]-[1 -(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid
Example 1
(S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2.6-dimethyl-phenyl)- propionic acid


STEP A: Trifluoromethanesulfonic acid 4-bromo-3,5-dimethyl-phenyl ester
To a cooled (0°C) solution of 4-bromo-3,5-dimethylphenol (3.05 g, 15.2 mmol) in pyridine (8 ml_) was added trifluoromethanesulfonic anhydride (5.0 g, 17.7 mmol) dropwise. After completion of addition, the resulting mixture was stirred at 0°C for 15 min, and then at room temperature overnight. The reaction was quenched by addition of water, and then extracted with EtOAc. The organic extracts were washed sequentially with water, 2N HCI (2x ), brine, and then dried over MgSO4. Filtration and evaporation to dryness yielded compound 1 b as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 2.45 (6H, s), 7.00 (2H, s).
Step B: 4-Bromo-3,5-dimethylbenzoic acid
Into a solution of compound 1 b (6.57 g, 19.7 mmol) in DMF (65 ml_) were added K2CO3 (13.1 g, 94.7 mmol), Pd(OAc)2 (0.44 g, 1.97 mmol) and 1 ,1′-bis(diphenylphosphino)ferrocene (2.29 g, 4.14 mmol). The resulting mixture was bubbled in gaseous CO for 10 min and was heated to 60°C for 7.5h with a CO(9) balloon. The cooled mixture was partitioned between aqueous NaHCO3 and EtOAc, and filtered. The aqueous phase was separated, acidified with aqueous 6N HCI, extracted with EtOAc, and then dried over Na2SO4. Filtration and concentration of the filtrate yielded crude compound 1c as a brown residue, which was used in the next step without further purification. STEP C: Method A: 4-Bromo-3,5-dimethyl-benzamide
Into a suspension of compound 1c in DCM (40 ml_) was added SOCI2 (3.1 rnL, 42 mmol) and the mixture was heated at reflux for 2 h. Upon removal of the solvent by evaporation, the residue was dissolved in DCM (40 ml_) and then ammonium hydroxide (28% NH3 in water, 2.8 ml_) was added. The reaction mixture was heated at 5O0C for 2 h and concentrated. The residue was diluted with H2O, extracted with EtOAc, and the organic portion was dried over Na2SO4. After filtration and evaporation, the residue was purified by flash column chramotagraphy (eluent: EtOAc) to yield compound 1 d as an off-white solid.
1H NMR (300 MHz, CD3CN): δ 2.45 (6H, s), 5.94 (1 H, br s), 6.71 (1 H, br s), 7.57 (2H, s)
MS(ES+)(relative intensity): 228.0 (100%) (M+1).
Step C: Method B: 4-Bromo-3,5-dimethyl-benzamide
A mixture of compound 1 b (3.33 g, 10 mmol), PdCI2 (0.053 g, 0.3 mmol), hexamethyldisilazane (HMDS, 8.4 ml_, 40 mmol), and DPPP (0.12 g, 0.3 mmol) was bubbled with a gaseous CO for 5 min and then stirred in a CO balloon at 80°C for 4 h. To the reaction mixture was added MeOH (5 ml_). The reaction mixture was stirred for 10 min, diluted with 2N H2SO4 (200 ml_), and then extracted with EtOAc. The EtOAc extract was washed with saturated aqueous NaHCO3, brine, and then dried over Na2SO4. Filtration and evaporation of the resultant filtrate yielded a residue, which was purified by flash column chromatography (eluent: EtOAc) to yield compound 1d as a white solid.
Step D: 2-terf-Butoxycarbonylaminoacrylic acid methyl ester
To a suspension of /V-Boc-serine methyl ester (Compound 1e, 2.19 g, 10 mmol) and EDCI (2.01 g, 10.5 mmol) in DCM (70 ml_) was added CuCI (1.04 g, 10.5 mmol). The reaction mixture was stirred at room temperature for 72 h. Upon removal of the solvent, the residue was diluted with EtOAc, washed sequentially with water and brine and then dried over MgSO4. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :4) to yield compound 1f as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 1.49 (9H, s), 3.83 (3H, s), 5.73 (1 H, d, J = 1.5 Hz), 6.16 (1 H1 S), 7.02 (1 H, s).
STEP E: (2)-2-fert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)acrylic acid methyl ester
A flask charged with compound 1d (0.46 g, 2.0 mmol), compound 1f (0.80 g, 4.0 mmol), tri-o-tolylphosphine (0.098 g, 0.32 mmol) and DMF (8 ml_) was purged with N2(g) 3 times. After the addition of tris(dibenzylideneacetone)dipalladium (0) (0.074 g, 0.08 mmol) and TEA (0.31 ml_, 2.2 mol), the reaction mixture was heated at 110°C for 24 h. At that time, the reaction was quenched by addition of water, and then extracted with EtOAc. The organic phase was washed with 1 N HCI, saturated aqueous NaHCO3, brine, and dried over MgSO4. The mixture was concentrated to a residue, which was purified by flash column chromatography (eluent: EtOAc:hexane~1 :1 to EtOAc only) to yield compound 1g as a white solid.
1H NMR (300 MHz, CD3OD): δ 1.36 (9H, s), 2.26 (6H, s), 3.83 (3H, s), 7.10 (1 H, s), 7.56 (2H, s); 13C NMR (75 MHz, DMSO-d6): δ 17.6, 25.7, 50.2, 78.7, 124.9, 126.4,
128.3, 131.2, 135.2, 135.5, 152.8, 164.3, 169.6;
MS (ES+) (relative intensity): 349.1 (38%)(M+1).
STEP F: (S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid methyl ester
Into a reactor charged with a solution of compound 1g (0.56 g, 1.6 mmol) in degassed MeOH (80 mL) was added [Rh(COd)(H1R-DIPAMP)J+BF4 “ under a stream of argon. The reactor was sealed and flushed with H2, stirred at 6O0C under 1000 psi of H2 for 14 days. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :1) to yield compound 1 h as a white solid. ee: >99%; 1H NMR (300 MHz, CDCI3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J = 7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1 H, m), 5.12 (1 H, d, J = 8.7 Hz), 5.65 (1 H, br s), 6.09 (1 H, br s), 7.46 (2H, s);
MS(ES+) (relative intensity): 250.9 (100) (M-BoC)+.
STEP G: (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid
Into an ice-cooled solution of compound “I h (0.22 g, 0.63 mmol) in THF (3.5 ml_) was added an aqueous LiOH solution (1 N, 3.5 ml_) and the reaction mixture stirred at 0°C. Upon completion of the reaction, the reaction mixture was concentrated and the aqueous phase was neutralized with cooled aqueous 1 N HCI at 0°C, and then extracted with EtOAc. The combined extracts were dried over Na2SO4 overnight. Filtration and evaporation of the filtrate to dryness yielded compound 1j as a white solid. 1H NMR (300 MHz, DMSO-cfe): δ 1.30 (9H, s), 2.32 (6H, s), 2.95(1 H, dd,
J= 8.8, 13.9 Hz), 3.10 (1 H, dd, J= 6.2, 14.0 Hz), 4.02-4.12 (1 H, m), 7.18-7.23 (2H, m), 7.48 (2H1 s), 7.80 (1 H, s);
MS(ES+) (relative intensity): 236.9 (6) (M-BoC)+.
Example 5
5-((r2-Amino-3-(4-carbamoyl-2.6-dimethyl-phenyl)-propionvn-n-(4-phenyl- 1 H-imidazol-2-yl)-ethvπ-aminol-methyl)-2-methoxy-benzoic acid


STEP A. 2-Methoxy-5-{[1-(4-phenyl-1 W-imidazol-2-yl)-ethylamino]-methyl}- benzoic acid methyl ester
Using the procedures described for Example 4, substituting 5-formyl-2- methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4- dimethoxybenzaldehyde, 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester was prepared.
STEP B. 5-({[2-ferf-ButoxycarbonylmethyI-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 H-imidazoI-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid methyl ester
Using the procedure of Example 3 for the conversion of Cpd 3d to Cpd 3e, substituting 2-methoxy-5-{[1-(4-phenyl-1 /-/-imidazol-2-yl)-ethylamino]- methylj-benzoic acid methyl ester for Cpd 3d and substituting 2-tert- Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionic acid for 2- tø/t-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 5a was prepared.
STEP C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid
5-({[2-tørf-Butoxycarbonylmethyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)- propionyl]-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid methyl ester was dissolved in an ice-chilled (0-10°C), mixed solvent system of THF (10 ml_) and MeOH (5 ml_). A LiOH H2O/water suspension (2.48 M; 3.77 ml_) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3 x 26 ml_). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to yield a pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2CI2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOHZCH2CI2 system as follows: Initial 100% CH2CI2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 5-({[2-terf- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4- phenyl-1 /-/-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 5b, as a white solid.
STEP D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1 – (4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A portion of Cpd 5b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 ml_)/THF (5 ml_), filtered, and subsequently treated with gaseous HCI for 15 min. After completion of the HCI addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield Cpd 5c as a white solid di-HCI salt.
Example 2
Racemic 2-terf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethvl- phenvD-propionic acid

STEP A: Racemic 2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid methyl ester
To a reactor charged with a solution of compound 1g (0.68 g, 1.95 mmol) in MeOH (80 mL) was added 10% Pd-C (0.5 g). The reactor was connected to a hydrogenator and shaken under 51 psi of H2 overnight. The mixture was filtered through a pad of Celite and the filtrate was concentrated to dryness to yield compound 2a as a white solid.
The 1H NMR spectrum was identical to that of (S)-2-tert- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)propionic acid methyl ester, compound 1 h.
STEP B: Racemic 2-terf-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid
Following the procedure described for Example 1 , STEP G (preparation of (S)-2-teAt-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid), compound 2b – racemic 2-te/?-butoxycarbonylamino-3- (4-carbamoyl-2,6-dimethyl-phenyl)propionic acid – was prepared.
…………….
POLYMORPHS
Example 1 Preparation of the zwitterion of 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A 1 L three-necked round-bottomed flask equipped with a mechanical stirrer, addition funnel and a thermocouple was charged without agitation. 34.2 g of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid (see Example 9 of US 2005/0203143), 340 ml of acetone, and 17 ml of 204 mmolar concentrated HCl were combined in the flask. The stirring was started and the resulting slurry formed a clear solution. This solution was heated to 45° C. under vigorous stirring and aged at this temperature for a period of two hours. After the completion, the reaction mass was cooled to ambient temperature and the supernatant was removed by suction. The vessel along with the residue was rinsed with 20 ml of acetone and then removed as previously. 170 ml of water was added and the reaction mass and was aged under stirring until a homogeneus solution resulted. This solution was then added over a period of ˜½ hr to a solution of 90 ml of 1N NaOH and water. The pH was adjusted to 6.5-7.0 accordingly. The resulting slurry was aged for about 2 hrs at ambient temperature, cooled to 10-15° C., aged at that temperature for about 1 hr, and then filtered. The solid was washed with 10 ml water, air-dried for a period of 4 to 5 hrs, and then placed in a vacuum oven at 50-55° C. until the water content was less than 3%.
Example 2 Preparation of the Form α Crystal
The Form α crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at 0-25% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1-3 respectively.
Example 3 Preparation of the Form β crystal
The Form β crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at greater than 60% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1, 4, and 5 respectively.
…………….
SYNTHESIS
Example 9 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid

A. 2-Methoxy-5{[1-(4-phenyl-1 H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester.
Using the procedures described for Example 3, substituting 5-formyl-2-methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4-dimethoxybenzaldehyde, 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester was prepared.
B. 5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester.
Using the procedure of Example 1 for the conversion of Cpd 1d to Cpd 1e, substituting 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester for Cpd 1 d and substituting 2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl-propionic acid of Example 8 for 2-tert-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 9a was prepared.
C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[11-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester was dissolved in an ice-chilled (0-10° C.), mixed solvent system of THF (10 mL) and MeOH (5 mL). A LiOH.H2O/water suspension (2.48 M; 3.77 mL) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3×26 mL). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to give 2.26 g (146% of theory) of pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2Cl2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOH/CH2Cl2 system as follows: Initial 100% CH2Cl2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 1.74 g (113% of theory) of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 9b, as a white solid.
D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
A portion of Cpd 9b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 mL)/THF (5 mL), filtered, and subsequently treated with gaseous HCl for 15 min. After completion of the HCl addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214 nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield 0.19 g (71%) of desired Cpd 9c as a white solid di-HCl salt.
Example 8 (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester

A. (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester. Into a cool solution of Boc-L-(2,6-diMe)Tyr-OMe (7.0 g, 21.6 mmol; Sources: Chiramer or RSP AminoAcidAnalogues) and N-phenyltrifluoromethanesulfonimide (7.9 g, 22.0 mmol) in dichloromethane (60 mL) was added triethylamine (3.25 mL, 23.3 mmol). The resulting solution was stirred at 0° C. for 1 h and slowly warmed to rt. Upon completion, the reaction was quenched by addition of water. The separated organic phase was washed with 1 N NaOH aqueous solution, water and dried over Na2SO4 overnight. After filtration and evaporation, the residue was purified by flash column chromatography (eluent: EtOAc-hexane: 3:7) to give the desired product (9.74 g, 99%) as a clear oil; 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.06 (2H, d, J=7.7 Hz), 3.64 (3H, s), 4.51-4.59 (1H, m), 5.12 (1H, d, J=8.5 Hz), 6.92 (2H, s); MS (ES+) (relative intensity): 355.8 (100) (M−Boc)+.
B. (S)4-(2-tert-Butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethylbenzoic acid. To a suspension of (S)-2-tert-butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester (9.68 g, 21.3 mmol), K2CO3 (14.1 g, 0.102 mol), Pd(OAc)2 (0.48 g, 2.13 mmol) and 1,1′-bis(diphenylphosphino)ferrocene (2.56 g, 4.47 mmol) in DMF (48 mL) was bubbled in gaseous CO for 15 min. The mixture was heated to 60° C. for 8 h with a CO balloon. The cool mixture was partitioned between NaHCO3 and EtOAc, and filtered. The aqueous layer was separated, acidified with 10% citric acid aqueous solution, extracted with EtOAc, and finally dried over Na2SO4. Filtration and concentration of the filtrate resulted in a residue. The residue was recrystallized from EtOAc-hexanes to afford the desired product (7.05 g, 94%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.42 (6H, s), 3.14 (2H, J=7.4 Hz), 3.65 (3H, s), 4.57-4.59 (1H, m), 5.14 (1H, d, J=8.6 Hz), 7.75 (2H, s); MS(ES+) (relative intensity): 251.9 (100) (M−Boc)+.
C. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid methyl ester. Into a stirring solution of (S)-4-(2-tert-butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethyl benzoic acid (3.00 g, 8.54 mmol), PyBOP (6.68 g, 12.8 mmol) and HOBt (1.74 g, 12.8 mmol) in DMF (36 mL) was added DIPEA (5.96 mL, 34.2 mmol) and NH4Cl (0.92 g, 17.1 mmol). The resulting mixture was stirred at rt for 40 min before being partitioned between aqueous NH4Cl solution and EtOAc. The separated organic phase was washed sequentially with 2N citric acid aqueous solution, saturated aqueous NaHCO3 solution, and brine, then dried over Na2SO4 overnight. After filtration and concentration, the residue was purified by flash column chromatography (eluent: EtOAc) to give the product. (3.00 g, 100%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J=7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1H, m), 5.12 (1H, d, J=8.7 Hz), 5.65 (1H, brs), 6.09 (1H, br s), 7.46 (2H, s); MS(ES+) (relative intensity): 250.9 (100) (M−Boc)+.
D. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid. Into an ice-cooled solution of methyl ester from Step C (2.99 g, 8.54 mmol) in THF (50 mL) was added an aqueous LiOH solution (1N, 50 mL) and stirred at 0° C. Upon consumption of the starting materials, the organic solvents were removed and the aqueous phase was neutralized with cooled 1N HCl at 0° C., and extracted with EtOAc, and dried over Na2SO4 overnight. Filtration and evaporation to dryness led to the title acid (S)-2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid (2.51 g, 87%); 1H NMR (300 MHz, DMSO-d6): δ 1.30 (9H, s), 2.32 (6H, s), 2.95 (1H, dd, J=8.8, 13.9 Hz), 3.10 (1H, dd, J=6.2, 14.0 Hz), 4.02-4.12 (1H, m), 7.18-7.23 (2H, m), 7.48 (2H, s), 7.80 (1H, s); MS(ES+) (relative intensity): 236.9 (6) (M−Boc)+.
…………………..
PATENTS
1.WO 2005090315
2..WO 2006099060
3.WO 2009009480
4. WO 2010062590
5.US 2011263868 *
|
12-24-2010
|
NOVEL COMPOUNDS AS OPIOID RECEPTOR MODULATORS
|
|
|
8-32-2010
|
Compounds as opioid receptor modulators
|
|
|
6-23-2010
|
Compounds as opioid receptor modulators
|
|
|
2-12-2010
|
PROCESS FOR THE PREPARATION OF OPIOD MODULATORS
|
|
|
12-9-2009
|
Process for the preparation of opioid modulators
|
| US7629488 * | Mar 6, 2006 | Dec 8, 2009 | Janssen Pharmaceutica N.V. | Process for the preparation of opioid modulators |
| US7741356 * | Mar 14, 2005 | Jun 22, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7786158 * | Oct 24, 2007 | Aug 31, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7994206 | Jul 7, 2008 | Aug 9, 2011 | Janssen Pharmaceutica, N.V. | Crystals and process of making 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid |
| CN1950342A | Mar 14, 2005 | Apr 18, 2007 | 詹森药业有限公司 | Novel compounds as opioid receptor modulators |
Update july 2015
Eluxadoline
Trade Name: Viberzi®
Research Code: JNJ-27018966, JNJ27018966, JNJ 27018966
Chemical Name: 5 – [[[(2S) -2-amino-3- [4- (aminocarbonyl) -2,6-dimethylphenyl ] -1- oxopropyl] [(1S) -1- (4-phenyl-1H-imidazol-2-yl) ethyl] amino] methyl] -2-methoxybenzoic acid
CAS No: 864821-90-9
MOA: mu opioid receptor agonist
Indication: Irritable bowel syndrome with diarrhea (IBS-D)
Approval Date: May 27, 2015 (US)
Originator: Furiex Pharmaceuticals Inc ( Furiex acquired Eluxadoline from Janssen in 2011 )
Developer: Forest Laboratories Inc. (acquired by Actavis PLC in 2014 )

FDA grants breakthrough therapy designation to Promacta (EU trade name: Revolade)
![]()
Earlier this week, Ligand Pharmaceuticals Inc. ( LGND ) announced that the U.S. Food and Drug Administration (FDA) granted breakthrough therapy designation to Promacta (EU trade name: Revolade). Ligand and its partner GlaxoSmithKline ( GSK ) are looking to get Promacta approved for the treatment of cytopenias in patients suffering from severe aplastic anemia (SAA), who are unresponsive to immunosuppressive therapy.
The FDA granted breakthrough therapy designation to Promacta based on data from an open-label phase II National Institute of Health (NIH) study (n = 43) evaluating Promacta in treatment experienced SAA patients, who showed insufficient response to immunosuppressive therapy.
The designation, which was enacted as part of the 2012 Food and Drug Administration Safety and Innovation Act, is granted to potential new treatments for serious or life-threatening diseases or conditions where the initial clinical data shows that the treatment has the potential to demonstrate substantial improvement on one or more clinically significant endpoints compared to existing treatments. The designation should help fasten the development and review process for the candidate.
We note that Promacta is already approved for the treatment of thrombocytopenia (reduced platelet count) in patients with chronic hepatitis C virus (HCV) infection to enable the initiation and maintenance of interferon-based therapy. Promacta is also approved for thrombocytopenia in patients with chronic idiopathic thrombocytopenia (ITP).
PROMACTA (eltrombopag) Tablets contain eltrombopag olamine, a small molecule thrombopoietin (TPO) receptor agonist for oral administration. Eltrombopag interacts with the transmembrane domain of the TPO receptor (also known as cMpl) leading to increased platelet production. Each tablet contains eltrombopag olamine in the amount equivalent to 12.5 mg, 25 mg, 50 mg, 75 mg, or 100 mg of eltrombopag free acid.
Eltrombopag olamine is a biphenyl hydrazone. The chemical name for eltrombopag olamine is 3′-{ (2Z)-2-[1 -(3,4-dimethylphenyl)-3-methyl-5-oxo- 1,5-dihydro-4H-pyrazol-4- ylidene]hydrazino}-2′-hydroxy-3-biphenylcarboxylic acid – 2-aminoethanol (1:2). It has the molecular formula C25H22N4O4•2(C2H7NO). The molecular weight is 564.65 for eltrombopag olamine and 442.5 for eltrombopag free acid. Eltrombopag olamine has the following structural formula:
 |
Eltrombopag olamine is practically insoluble in aqueous buffer across a pH range of 1 to 7.4, and is sparingly soluble in water.
The inactive ingredients of PROMACTA are: Tablet Core: magnesium stearate, mannitol, microcrystalline cellulose, povidone, and sodium starch glycolate. Coating: hypromellose, polyethylene glycol 400, titanium dioxide, polysorbate 80 (12.5 mg tablet), FD&C Yellow No. 6 aluminum lake (25 mg tablet), FD&C Blue No. 2 aluminum lake (50 mg tablet), Iron Oxide Red and Iron Oxide Black (75 mg tablet), or Iron Oxide Yellow and Iron Oxide Black (100 mg tablet).
Biogen’s multiple sclerosis drug Tecfidera obtains EU approval
US-based Biogen Idec has received approval from the European Commission (EC) for its Tecfidera (dimethyl fumarate) as a first-line oral treatment for people with relapsing-remitting multiple sclerosis (RRMS), the most common form of multiple sclerosis (MS).
Biogen’s multiple sclerosis drug Tecfidera obtains EU approval click here
Lysosomal Storage Disorders: Advocacy Group Receives FDA Orphan Designations
This is the second Blog Post in a series over the next week that will examine Lysosomal Storage Disorders (LSDs) in the rare disease and orphan drug space. This Blog Post presents an advocacy group that receives two FDA Orphan Drug Designations (ODDs) in 2013 for treatment of rare diseases. The chart below identifies the gene therapy that receives FDA ODD where the sponsor is a rare disease advocacy group.
Row Num | Generic Name | Designation Date | Orphan Designation |
1 | recombinant adeno- associated virus vector AAV2/rh8 expressing human B-hexosaminidase A and B subunits | 03-25-2013 | Sandhoff Disease |
2 | recombinant adenovirus vector AAV2/rh8 expressing human B-hexosaminidase A & B subunits | 03-25-2013 | Tay-Sachs Disease |
.
** “Generic Name” Column Link = Is the FDA Orphan Drug Product Designation Database Record.
The National Tay-Sachs and Allied Diseases Association (NTSAD) announces in June 2013 that the FDA…
View original post 211 more words
How to Handle Drug Polymorphs… Case Study of Trelagliptin Succinate

PART 1………..http://drugsynthesisint.blogspot.in/p/gliptin-series.html
PART 2 ……http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html


If you’re involved in late drug discovery, API manufacture, drug product formulation, clinical material production, or manufacture of final dosage form, a basic understanding and awareness of solid form issues could save you a great deal of difficulty, time, and money during drug development.


Investigational new drug, writing an application for clinical trial authorization, permission marketing …The control of polymorphism in drug candidates is now ubiquitous.
READ………….An Overview of Solid Form Screening During Drug Development, http://www.icdd.com/ppxrd/10/presentations/PPXRD-10_Ann_Newman.pdf
ANN NEWMAN
When addressing the subject of polymorphism, the first reference that comes to mind is that of the occurred during the manufacture of ritonavir incident. Abbott molecule inhibitor of HIV protease marketed as Norvir, is a cautionary example of the challenges of polymorphism.
Indeed, during the production of ritonavir in 1997, a new polymorph unmarked emerged. Its precipitation and unexpected outbreak led to the cessation of the production of Norvir and seriously compromise the process. The incident has deeply marked the pharmaceutical industry.
It is ironic that the process used to discover pharmaceutical drug targets is the same one that decreases the actual efficacy of those drugs once ingested. If you remember from basic chemistry, there are compounds that exist in highly ordered crystalline states and those that remain in amorphous form.
The discovery of drug targets has often been accomplished through X-ray crystallography, which requires a sample (for example, of a defective enzyme linked to cancer or high cholesterol) to be crystallized so that the diffraction patterns can be made sense of. Scientists may spend years trying to crystallize one molecule or compound so that they can identify regions that, for example, may be blocked by pharmaceuticals.
However, when it comes to the molecular arrangement of those pharmaceuticals, crystallization actually decreases their bioavailability and solubility. Thus, it may be better for these drugs to be in amorphous form. Pierric Marchand, general manager of the company Holodiag, dedicated to the study and characterization of solid state, summarizes that ” today, it is not reasonable to not worry about the problem of polymorphism ” .
” In recent years, manufacturers have realized the essential side of expertise , “says Jean-Rémi David, commercial director Calytherm. The services company specializing in the field of physico-chemical analysis, based in Herault, has just relocated last year in supporting pharmaceutical development to meet demand. ” This is a concern for all deal with potential impacts on the effectiveness or the formulation , “says Stéphane Suchet, quality manager in the group of fine and specialty chemicals Axyntis.
Polymorphic forms are the amorphous and crystalline forms such as hydrate or solvate forms. When a molecule of interest exists in polymorphic forms, it is called polymorphism, according to the definition of the FDA (Food and Drug Administration).
Polymorphism is present at all stages of development of a drug from research to marketing. ” Keep in mind that organic molecule loves to polymorphism , “says Marchand Pierric. However, for a marketing authorization for example, must learn the criteria for the polymorphism of the molecule. ” In terms of the formulation, for example we can check whether the selected polymorphism is unchanged , “explains Pierric Marchand.

A significant influence on several levels Because the consequences of polymorphism are multiple. ” They are at three levels: bioequivalence, manufacturability and stability “lists Fabienne Lacoulonche, founder and scientific director of Calytherm.In terms of bioequivalence, different polymorphic forms may have different properties of solubility and dissolution rate … ” For poorly soluble active ingredients, you can have much more bioavailable than other crystalline forms , “Fabienne Lacoulonche information.
In terms of manufacturability, some parameters such as temperature, moisture can lead to changes in the crystalline form. ” The complexity is to anticipate changes polymorphism, both at laboratory scale, pilot and industrial , “adds the founder of Calytherm. Finally, polymorphism plays on stability. Active ingredient or finished product, are subjected to stability studies in this direction. ” When the molecule is identified, we try to highlight the existence of several forms of polymorphism, explains Stéphane Suchet (Axyntis) .
When developing a new substance, the assessment is systematic . ” Isolation of crystals from a screening is carried out in different solvents by various analytical techniques. Ideally, it will be concluded the absence of polymorphism. ” But if different polymorphic forms are present, we rework the terms of our crystallization process to control the formation of the same polymorph reproducibly ideally form the thermodynamically more stable , “says Stéphane Suchet. X-ray diffraction and other thermal analysis ”
The ICH guidelines provide decision trees to guide the industry in controlling polymorphism says Fabienne Lacoulonche (Calytherm.) We use it for writing the CTD (Common Technical Document) .
“Polymorphism is a phenomenon” complex and difficult to control, because the crystallization is dependent on many parameters , she develops. must understand the maximum . ” For this, several analytical methods are available to industry. The main technique is the X-ray diffraction ” It is a robust, rapid, which allows to characterize the different polymorphs , “summarizes Pierric Marchand (Holodiag).Non-destructive, it can work both on small quantities on large samples. Temperature and atmosphere are controlled, and analytical capabilities are broad.

But if this technique indispensable allows for routine and development, it is not sufficient in itself. Just to add a battery of additional tests, thermal analysis. ” It takes coupling methods “ confirms Fabienne Lacoulonche (Calytherm). The X-ray diffraction is a method of choice, but sometimes it is not sufficient.
The coupling with a thermal analysis method (technical ATG, or DSC thermal analysis, differential scanning calorimetry or thermomicroscopique) allows to distinguish between two polymorphic whose RX diffractograms obtained are comparable.
TGA can be coupled with IR or mass spectrometry, DSC with RX. Raman spectroscopy is also part of complementary methods. ” The difficulty increases when we want to characterize the shape of the active ingredient in the finished product , says Fabienne Lacoulonche. example, by X-ray diffraction, the peaks related to the active ingredient in the diffractogram of the finished product may be masked by those excipients: it is then necessary to use other methods, such as Raman microscopy. “In general, a single method of analysis is not sufficient to characterize the polymorphism of an active substance in the active substance or finished product: the complementarity of different methods that will conclude precisely on the polymorphism of a crystalline substance.
In addition, ” the diffractometer remains an expensive device, which requires installation in an air-conditioned and a cooling room , “says Marchand Pierric (Holodiag). To this is added the need to have expertise and qualified personnel to carry out the analyzes. ” We must master these techniques and the ability to interpret the results , “says Jean-Rémi David (Calytherm). However, polymorphism is a “problem well under control , “said Stéphane Suchet (Axyntis),” systematically evaluated although it is however not always immune to miss a polymorphic form, knowing that the screening performed in the development can never be completely comprehensive … ”

FDA
FDA may refuse to approve an ANDA referencing a listed drug if the application contains insufficient information to show that the drug substance is the “same” as that of the reference listed drug. A drug substance in a generic drug product is generally considered to be the same as the drug substance in the reference listed drug if it meets the same standards for identity.
In most cases, the standards for identity are described in the USPalthough FDA may prescribe additional standards when necessary. Because drug product performance depends on the product formulation, the drug substance in a proposed generic drug product need not have the same physical form (particle size, shape, or polymorph form) as the drug substance in the reference listed drug. An ANDA applicant is required to demonstrate that the proposed product meets the standards for identity, exhibits sufficient stability and is bioequivalent to the reference listed drug.
FDA PRESENTATION……polymorphs and co-crystals – ICDD Regulatory Considerations on Pharmaceutical Solids: Polymorphs/Salts and Co-Crystals.. THIS IS A MUST READ ITEM
Over the years FDA has approved many generic drug products based upon a drug substance with different physical form from that of the drug substance in the respective reference listed drug (e.g., warfarin sodium, famotidine, and ranitidine). Also many ANDAs have been approved in which the drug substances differed from those in the corresponding reference listed drugs with respect to solvation or hydration state (e.g., terazosin hydrochloride, ampicillin, and cefadroxil). Several regulatory documents and literature reports (67-69) address issues relevant to the regulation of polymorphism.
The concepts and principles outlined in these are applicable for an ANDA. However, certain additional considerations may be applicable in case of ANDAs. Often at the time FDA receives an ANDA a monograph defining certain key attributes of the drug substance and drug product may be available in the Unites States Pharmacopoeia (USP). These public standards play a significant role in the ANDA regulatory review process and in case of polymorphism, when some differences are noted, lead to additional requirements and considerations.
This commentary is intended to provide a perspective on polymorphism in pharmaceutical solid in the context of ANDAs. It highlights major considerations for monitoring and controlling drug substance polymorphs and describes a framework for regulatory decisions regarding drug substance “sameness” considering the role and impact of polymorphism in pharmaceutical solids.

Since polymorphs exhibit certain differences in physical (e.g., powder flow and compactability, apparent solubility and dissolution rate) and solid state chemistry (reactivity) attributes that relate to stability and bioavailability it is essential that the product development and the FDA review process pay close attention to this issue.
This scrutiny is essential to ensure that polymorphic differences (when present) are addressed via design and control of formulation and process conditions to physical and chemical stability of the product over the intended shelf-life, and bioavailability/bioequivalence.
Most pharmaceuticals are distributed as solid doseages. In order to take action, they must dissolve in the gut and be absorbed into the blood stream. In many cases, the rate at which the drug dissolves can limit its effectiveness. Pharmaceutical compounds can be packed into more than one arrangement in the solid states known as polymorphs. Rapid and efficient methods of polymorph formation can be used to increase drug efficacy and shelf life.
Regulatory agencies worldwide require that, as part of any significant filing, a company has to demonstrate that it has made a reasonable effort to identify the polymorphs of their drug substance and has checked for polymorph interconversions. Due to the unpredictable behaviour of polymorphs and their respective differences in physicochemical properties, companies also have to demonstrate consistency in manufacturing between batches of the same product. Proper understanding of the polymorph landscape and nature of the polymorphs will contribute to manufacturing consistency.
POLYMORPHISM AND PATENTS http://www.collegio.unibo.it/uploads/ideas/joelbernstein.pdf A MUST CLICK FOR PHARMA CHEMISTS

READ………..High-throughput crystallization: polymorphs, salts, co-crystalsand solvates of pharmaceutical solidshttp://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.85.5397&rep=rep1&type=pdf

Definitions
“Crystalline”, as the term is used herein, refers to a material, which may be hydrated and/or solvated, and has sufficient ordering of the chemical moiety to exhibit a discernable diffraction pattern by XRPD or other diffraction techniques. Often, a crystalline material that is obtained by direct crystallization of a compound dissolved in a solution or by interconversion of crystals obtained under different crystallization conditions, will have crystals that contain the solvent used in the crystallization, termed a crystalline solvate. Also, the specific solvent system and physical embodiment in which the crystallization is performed, collectively termed crystallization conditions, may result in the crystalline material having physical and chemical properties that are unique to the crystallization conditions, generally due to the orientation of the chemical moieties of the compound with respect to each other within the crystal and/or the predominance of a specific polymorphic form of the compound in the crystalline material.
Depending upon the polymorphic form(s) of the compound that are present in a composition, various amounts of the compound in an amorphous solid state may also be present, either as a side product of the initial crystallization, and/or a product of degradation of the crystals comprising the crystalline material. Thus, crystalline, as the term is used herein, contemplates that the composition may include amorphous content; the presence of the crystalline material among the amorphous material being detectably among other methods by the composition having a discernable diffraction pattern.
The amorphous content of a crystalline material may be increased by grinding or pulverizing the material, which is evidenced by broadening of diffraction and other spectral lines relative to the crystalline material prior to grinding. Sufficient grinding and/or pulverizing may broaden the lines relative to the crystalline material prior to grinding to the extent that the XRPD or other crystal specific spectrum may become undiscernable, making the material substantially amorphous or quasi-amorphous. Continued grinding would be expected to increase the amorphous content and further broaden the XRPD pattern with the limit of the XRPD pattern being so broadened that it can no longer be discerned above noise. When the XRPD pattern is broadened to the limit of being indiscernible, the material may be considered no longer a crystalline material, but instead be wholly amorphous. For material having increased amorphous content and wholly amorphous material, no peaks should be observed that would indicate grinding produces another form.
“Amorphous“, as the term is used herein, refers to a composition comprising a compound that contains too little crystalline content of the compound to yield a discernable pattern by XRPD or other diffraction techniques. Glassy materials are a type of amorphous material. Glassy materials do not have a true crystal lattice, and technically resembling very viscous non-crystalline liquids. Rather than being true solids, glasses may better be described as quasi-solid amorphous material. “Broad” or “broadened”, as the term is used herein to describe spectral lines, including XRPD, NMR and IR spectroscopy, and Raman spectroscopy lines, is a relative term that relates to the line width of a baseline spectrum. The baseline spectrum is often that of an unmanipulated crystalline form of a specific compound as obtained directly from a given set of physical and chemical conditions, including solvent composition and properties such as temperature and pressure.
For example, broadened can be used to describe the spectral lines of a XRPD spectrum of ground or pulverized material comprising a crystalline compound relative to the material prior to grinding. In materials where the constituent molecules, ions or atoms, as solvated or hydrated, are not tumbling rapidly, line broadening is indicative of increased randomness in the orientation of the chemical moieties of the compound, thus indicative of an increased amorphous content. When comparisons are made between crystalline materials obtained via different crystallization conditions, broader spectral lines indicate that the material producing the relatively broader spectral lines has a higher level of amorphous material.
“About” as the term is used herein, refers to an estimate that the actual value falls within ±5% of the value cited. “Forked” as the term is used herein to describe DSC endotherms and exotherms, refers to overlapping endotherms or exotherms having distinguishable peak positions
. 
Classes of multicomponent pharmaceutical materials. (a) Schematic of crystalline materials showing neutral and charged species. The red box indicates polymorphs are possible for all the multicomponent crystals contained within the box (adapted from Reference 7). (b) Schematic of amorphous solid dispersions showing binary, ternary, and quaternary possibilities for polymers and surfactants. Other solubilization techniques using cyclodextrins and phospholipids are included for completeness but have a different mechanism for solubilization when compared to polymer and surfactant systems.
The red box indicates that properties can change with water or solvent content. General methods for precipitating and crystallizing a compound may be applied to prepare the various polymorphs described herein. These general methods are known to those skilled in the art of synthetic organic chemistry and pharmaceutical formulation, and are described, for example, by J. March, “Advanced Organic Chemistry: Reactions, Mechanisms and Structure ” 4th Ed. (New York: Wiley-Interscience, 1992).
In general, a given polymorph of a compound may be obtained by direct crystallization of the compound or by crystallization of the compound followed by inter-conversion from another polymorphic form or from an amorphous form. Depending on the method by which a compound is crystallized, the resulting composition may contain different amounts of the compound in crystalline form as opposed to as an amorphous material.
Also, the resulting composition may contain differing mixtures of different polymorphic forms of the compound. Compositions comprising a higher percentage of crystalline content {e.g., forming crystals having fewer lattice defects and proportionately less glassy material) are generally prepared when using conditions that favor slower crystal formation, including slow solvent evaporation and those affecting kinetics.
Crystallization conditions may be appropriately adjusted to obtain higher quality crystalline material as necessary. Thus, for example, if poor crystals are formed under an initial set of crystallization conditions, the solvent temperature may be reduced and ambient pressure above the solution may be increased relative to the initial set of crystallization conditions in order to slow down crystallization.  Precipitation of a compound from solution, often affected by rapid evaporation of solvent, is known to favor the compound forming an amorphous solid as opposed to crystals. A compound in an amorphous state may be produced by rapidly evaporating solvent from a solvated compound, or by grinding, pulverizing or otherwise physically pressurizing or abrading the compound while in a crystalline state.
Precipitation of a compound from solution, often affected by rapid evaporation of solvent, is known to favor the compound forming an amorphous solid as opposed to crystals. A compound in an amorphous state may be produced by rapidly evaporating solvent from a solvated compound, or by grinding, pulverizing or otherwise physically pressurizing or abrading the compound while in a crystalline state.
Seven crystalline forms and one amorphous solid were identified by conducting a polymorph screen (Example 3). Described herein are Form A, Form B, Form C, Form D, Form E, Form F, Form G, and Amorphous Form of Compound I. Where possible, the results of each test for each different polymorph are provided. Forms A, C, D and E were prepared as pure forms. Forms B, F, and G were prepared as mixtures with Form A.
Various tests were performed in order to physically characterize the polymorphs of Compound I including X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), hot stage microscopy, Fourier transform infrared spectroscopy (FT-IR), Fourier transform Raman spectrometry, linked thermogravimetric-infrared spectroscopy (TG-IR), solution proton nuclear magnetic resonance (1H-NMR), solid state 13carbon nuclear magnetic resonance (13C-NMR), and moisture sorption and desorption analysis (M S/Des).

Salt screening
Physicochemical properties of drug substances, such as solubility, dissolution rate, and physicochemical stability can be altered significantly by salt formation. Consequently, important properties of the drug product such as bioavailability or shelve life can be radically influenced. Crystallics’ technology platform for crystallization screening accommodates salt screening studies using only minimal amounts of drug substance while still performing a large number of experiments. High-throughput salt screening is used for both early phase salt selection studies and broad patent protection.
Salt selection – A powerful strategy for crystal form optimization
Pharmaceutical developers have focused efforts on finding and formulating a thermodynamically stable crystalline form with acceptable physical properties for a given compound. This is reasonable, given the need to avoid cascading from a meta-stable form to a more stable one in unpredictable fashion.
Occasionally certain physical properties, such as low aqueous solubility, are limiting to performance of the compound, leading to poor oral bioavailability or insufficient solubility for an injection formulation. One of the main strategies used to affect physical performance of a compound and one that is often employed by pharmaceutical scientists is the practice of salt selection (23). At least half of compounds in marketed products are in the form of a salt for one reason or another.
This fact alone speaks to the versatility of the salt selection approach. Salt forms of a pharmaceutical can have many benefits, such as improved stability characteristics, optimal bioavailability and aqueous solubility for an injectable formulation. Salts, like all other crystalline forms, are subject to polymorphism and solvate formation, thus requiring the same form identification studies as are needed for a neutral compound.
A remarkable example of co-optimization of properties is indinavir (HIV protease inhibitor), which is marketed as the sulfate salt ethanol solvate (24,25) The crystalline free base has variable oral bioavailability in dogs (26,27) and humans (28). While acidic solutions of the base compound showed good oral pharmacokinetics, the stability of the drug in acidic solution is not consistent with a product (26). Therefore, the discovery of the salt form ensured both shelf stability and robust bioavailability performance. The salt selection strategy is limited in two ways.
First, salt formation relies on the presence of one or more ionizable functional groups in the molecule; many drugs and development compounds lack this feature.
Second, our ability to predict a priori whether a given compound will form a crystalline salt (or salts) is non-existent. The ability to actively identify crystalline salt forms has been confined to manual empirical evaluation using multiple salt formers for a given acid or base. Recently advances have been made in the area of high-throughput salt selection and crystal engineering strategies associated with salt formation (14,29-32).
In one case, we have advocated the simultaneous assessment of polymorphism as a way to help rank the developability of different crystalline salts (14). While salt forms will continue to have a prominent place in pharmaceutical science, the need for enhanced productivity dictates that every advantage must be sought to aid the design of an appropriate crystalline form of an active molecule.
Specifically, the ability to design scaffolds into crystalline forms will enhance our capacity to convert interesting molecules into effective drugs. Crystal engineering offers some additional tools in this regard. 
CASE STUDY FORM A ONLY US8084605
TRELAGLIPTIN SUCCINATE
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared.
For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days.
The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE). Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR. Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1. Table 1. Characteristic XRPD Peaks (CuKa) of Form A


The above set of XRPD peak positions or a subset thereof can be used to identify Form A. One subset comprises peaks at about 11.31, 11.91, 12.86, 14.54, 15.81, 16.83, 17.59, 19.26, 19.52, 21.04, 22.32, 26.63, and 29.87 °2Θ. Another subset comprises peaks at about 11.31, 11.91, 19.26, 21.04, and 22.32 °2Θ; the peaks of this subset show no shoulder peaks or peak split greater than 0.2 °2Θ. Another subset comprises peaks at about 11.31, 11.91 and 22.32 °2Θ.  Figure 2 is a TGA thermogram of Form A. TGA analysis showed that Form A exhibited insignificant weight loss when heated from 25 0C to 165 0C; this result is indicative that Form A was an anhydrous solid.
Figure 2 is a TGA thermogram of Form A. TGA analysis showed that Form A exhibited insignificant weight loss when heated from 25 0C to 165 0C; this result is indicative that Form A was an anhydrous solid.  Figure 3 shows a characteristic DSC thermogram of Form A. DSC analysis showed a single endothermic event occurred at approximately 195 0C (peak maximum). This endothermic event was confirmed by hot stage microscopy which showed the melting of Form A, which onset around 177 0C and the melting point estimated to be at approximately 184 0C.
Figure 3 shows a characteristic DSC thermogram of Form A. DSC analysis showed a single endothermic event occurred at approximately 195 0C (peak maximum). This endothermic event was confirmed by hot stage microscopy which showed the melting of Form A, which onset around 177 0C and the melting point estimated to be at approximately 184 0C. 
Figure 4 (A-D) shows a characteristic FT-IR spectrum of Form A. The major bands expressed in reciprocal wavelengths (wavenumber in cm”1) are positioned at about 3815, 3736, 3675, 3460, 3402, 3141, 3098, 3068, 3049, 2953, 2934, 2854, 2760, 2625, 2536, 2481, 2266, 2225, 2176, 1990, 1890, 1699, 1657, 1638, 1626, 1609, 1586, 1553, 1517, 1492, 1478, 1450, 1419, 1409, 1380, 1351, 1327, 1289, 1271, 1236, 1206, 1180, 1158, 1115, 1087, 1085, 1064, 1037, 1027, 971, 960, 951, 926, 902, 886, 870, 831, 820, 806, 780, 760, 740, 728, 701, 685, 668, 637, 608, 594, 567, 558, and 516 cm”1 (values rounded to the nearest whole number). This unique set of IR absorption bands or a subset thereof can be used to identify Form A.
One such subset comprises absorption bands at about 3141, 3098, 3068, 3049, 2953, 2934, 2854, 2266, 2225, 1699, 1657, 1609, 1586, 1553, 1517, 1492, 1478, 1450, 1380, 1351, 1327, 1236, 1206, 1115, 1063, 902, 886, 870, 820, 780, 760, 685, 608, 594, and 516 cm 1. Another subset comprises absorption bands at about 3141, 2953, 2934, 2854, 2266, 2225, 1699, 1657, 1450, 1206, 886, 760, 685, 594, and 516 cm 1. Yet another subset comprises absorption bands at about 3141, 2953, 2934, 2266, 1699, 1657, 1450, and 1206 cm 1.
Aprepitant case study FTIR.. READING MATERIAL http://alpha.chem.umb.edu/chemistry/ch361/spring%2005/ftir%20polymorph.pdf
Figure 5 (A-D) shows a characteristic Raman spectrum of Form A. The major Raman bands expressed in reciprocal wavelengths (wavenumber in cm”1) are positioned at about 3100, 3068, 3049, 2977, 2954, 2935, 2875, 2855, 2787, 2263, 2225, 2174, 1698, 1659, 1626, 1607, 1586, 1492, 1478, 1451, 1439, 1409, 1400, 1382, 1351, 1328, 1290, 1281, 1271, 1237, 1223, 1213, 1180, 1155, 1134, 1115, 1084, 1063, 1035, 971, 952, 926901, 868, 805, 780, 759, 740, 727, 701, 686, 669, 609, 594, 566, 558, 516, 487, 479, 433, 418, 409, 294, 274, 241, 218, 191 and 138 cm”1 (values rounded to the nearest whole number). This unique set of Raman bands or a subset thereof may be used to identify Form A.
One such subset comprises Raman bands at about 2954, 2935, 2225, 1698, 1659, and 1607 cm”1. Another subset comprises Raman bands at about 3068, 2954, 2935, 2225, 1698, 1659, 1607, 1586, 1223, 1180, 901, 780, 759, 669, and 516 cm”1. Yet another subset comprises Raman bands at about 3100, 3068, 2225, 1698, 1659, 1607, 1586, 1351, 1237, 1223, 1180, 1155, 1134, 1115, 1063, 952, 926, 901, 868, 805, 780, 759, 740, 669, 609, and 516 cm”1.
Form A was further characterized by solution 1H NMR and solid-state 13carbon NMR. The spectra are reported in Figures 6 and 7, respectively. Chemical assignments were not performed; however, the spectra are consistent with the known chemical structure of Compound I. US8084605

 Example 11. Characterization of Form A Material prepared by the procedure of Example 1 was designated as Form A. The material was characterized by XRPD, TGA, DSC, hot stage microscopy, FT-IR, FT- Raman, 1H NMR, and 13C NMR. The analyses were conducted according to the procedures outlined in Section B of Example 3.
Example 11. Characterization of Form A Material prepared by the procedure of Example 1 was designated as Form A. The material was characterized by XRPD, TGA, DSC, hot stage microscopy, FT-IR, FT- Raman, 1H NMR, and 13C NMR. The analyses were conducted according to the procedures outlined in Section B of Example 3.
The characteristic spectra and thermograms for Form A are reported in Figures 1-7. The characterization data are summarized in Table D. Table D. Characterization Data of Form A of Compound I US8084605

Amorphous solid dispersion screening
Using the amorphous form of a drug substance offers several advantages with respect to dissolution rate and solubility of the substance. However, reduced chemical stability, increased hygroscopicity and, most important, physical instability are the major drawbacks of using the amorphous phase in the final drug product. These drawbacks can be overcome by stabilizing the amorphous phase of the API in a polymer matrix, e.q. an amorphous solid dispersion. Amorphous phases dissolve more rapidly than crystalline forms, and can significantly increase bioavailability of poorly water soluble drugs substances. However, the use of amorphous materials requires confidence that crystallization will not occur during the product lifespan. For a material that has never been obtained in a crystalline form, focus should be put on attempting to crystallize it. Crystallics has extensive experience of obtaining crystalline phases from amorphous materials.
Dispersions of a drug substance onto a polymeric matrix has received increased attention in recent years. A successful dispersion results in an amorphous solid material and will show improved dissolution rates and higher apparent solubility characteristics, as well as, sufficient resistance to chemical degradation and should be physically stable e.q. sufficient high glass transition temperature avoiding crystallization of the API.
A variety of factors contribute to the formation of a suitable Amorphous Solid Dispersion (ASD), including the nature of the polymer, the drug polymer ratio, the impact of surfactants and the solvent used in the process. Crystallics has developed high-throughput solid dispersion screening technology in order to find the optimal combination of these factors.
Example 10. Preparation of Amorphous Form US8084605
A sample of Compound I (40 mg) was dissolved in 1000 μl of water. The solution was filtered through a 0.2 μm nylon filter into a clean vial then frozen in a dry ice/acetone bath. The vials were covered with a Kimwipe then placed on a lyophilizer overnight. The resulting solids yielded Amorphous Form. 8. Amorphous Form The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis. 
TGA analysis (Figure 27) showed a 1.8% weight loss from 25 0C to 95 0C, which was likely due to loss of residual solvent. 
DSC analysis (Figure 28) showed a slightly concave baseline up to an exotherm at 130 0C (recrystallization), followed by an endotherm at 194 0C, which results from the melting of Form A. Hot stage microscopy confirmed these recrystallization and melting events (micrographs not included). An approximate glass transition was observed (Figure29) at an onset temperature of 82 0
C. 
Moisture sorption/desorption data (Figure 30 and Example 25) showed a 1.0% weight loss on equilibration at 5% relative humidity. Approximately 8% of weight was gained up to 65% relative humidity. Approximately 7% of weight was lost at 75% relative humidity. This is likely due to the recrystallization of the amorphous material to a crystalline solid. A 4.4% weight gain was observed on sorption from 75% to 95% relative humidity. Approximately 4.7% weight was lost on desorption from 95% to 5% relative humidity. 
The solid material remaining after the moisture sorption analysis was determined to be Form A by XRPD (Figure 31). Table H. Characterization Data of Amorphous Form US8084605

T=temperature, RH=relative humidity, MB = moisture sorption/desorption analysis Example 19: Relative Humidity Stressing Experiments

Table B. Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool; CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation Table C. Crystallization Experiments of Compound I in Various Solvent/Antisolvent

a precipitated by evaporation of solvent Table A. Approximate Solubilities of Compound I US8084605


a) Approximate solubilities are calculated based on the total solvent used to give a solution; actual solubilities may be greater because of the volume of the solvent portions utilized or a slow rate of dissolution. Solubilities are reported to the nearest mg/mL.
Example 3.
Polymorph Screen Compound I as prepared by the method described in Example 1 was used as the starting material for the polymorph screen. Solvents and other reagents were of ACS or HPLC grade and were used as received. A. Sample Generation. Solids for form identification were prepared via the following methods from Compound I.
1. Fast Evaporation (FE) A solution of Compound I was prepared in test solvents. The sample was placed in the hood, uncovered, to evaporate under ambient conditions. The solids were analyzed by XRPD for form identification.
2. Slow Evaporation (SE) A solution of Compound I was prepared in test solvents. The sample was placed in the hood, covered with foil rendered with pinholes, to evaporate under ambient conditions. The solids were analyzed by XRPD for form identification.
3. Room Temperature (RT) Slurries An excess amount of Compound I was slurried in test solvent on a rotating wheel for approximately 5 or 7 days. The solids were typically collected by vacuum filtration, air dried in the hood, and analyzed by XRPD for form identification.
4. Elevated Temperature Slurries Excess Compound I was slurried in test solvents at 47 0C on a shaker block for approximately 5 days. The solids were collected by vacuum filtering, dried in the hood, and then analyzed by XRPD for form identification.
5. Slow Cooling Crystallization (SC)
A saturated or near saturated solution of Compound I was prepared at elevated temperature. The samples were filtered through warmed 0.2 μm filters into warmed vials. The heat source was turned off and the samples slowly cooled to ambient temperature. If precipitation did not occur within a day the samples were placed in the refrigerator. The samples were transferred to a freezer if precipitation did not occur within several days. The solids were collected by decanting the solvent or vacuum filtration, dried in the hood and analyzed by XRPD for form identification.
6. Crash Cooling Crystallization (CC) A saturated or near saturated solution of Compound I was prepared at elevated temperature. The samples were filtered through warmed 0.2 μm filters into warmed vials then rapidly cooled in an acetone/dry ice or ice bath. If precipitation did not occur after several minutes the samples were placed in the refrigerator or freezer. Solids were collected by decanting solvent or vacuum filtration, dried in the hood, and then analyzed by XRPD. Samples that did not precipitate under subambient conditions after several days were evaporated in the hood and analyzed by XRPD for form identification.
7. Solvent/Antisolvent Crystallization (S/AS) A solution of Compound I was prepared in test solvent. A miscible antisolvent was added with a disposable pipette. Precipitate was collected by vacuum filtration or decanting solvent. The samples were stored under subambient conditions if precipitation did not occur. If solids were not observed after several days the samples were evaporated in the hood. Collected solids were analyzed by XRPD for form identification.
8. Relative Humidity (RH) Stressing Experiments Samples of Compound I were placed uncovered in approximately 58%, 88%, and 97% relative humidity jars. The samples were stored in the jars for approximately 8 days. The solids were collected and analyzed by XRPD for form identification.
9. Lyophilization Compound I was dissolved in water in a glass vial. The solution was frozen by swirling the vial in an acetone/dry ice bath. The frozen sample was placed on the lyophilizer until all of the frozen solvent was removed. The solids were collected and analyzed by XRPD for form identification.
10. Grinding Experiments Aliquots of Compound I were ground manually with a mortar and pestle as a dry solid and a wet paste in water. The samples were ground for approximately three minutes. The solids were collected and analyzed by XRPD for form identification.
11. Dehydration Experiments Hydrated samples of Compound I were dehydrated at ambient conditions (2 days) and in an ambient temperature vacuum oven (1 day). The solids were collected and analyzed by XRPD for form identification.
12. Vapor Stress Experiments Amorphous Compound I was placed in acetone, ethanol, and water vapor chambers for up to eight days. The solids were collected and analyzed by XRPD for form identification.
STABILITY STUDY Stability studies are commonly performed for new drug entities with chemical stability and impurity formation being investigated. It is also important to monitor the physical stability under these same conditions to anticipate any form changes that may occur. As an example, many hydrates will dehydrate to a lower hydrate or anhydrous form at elevated temperatures. Anhydrous materials can also undergo form transformations to other anhydrous forms upon heating.
These types of changes can be monitored using heating studies in an oven with subsequent XRPD analysis or in-situ variable temperature XRPD can be used to look for changes. In other cases, anhydrates will convert to hydrates or the API in an amorphous solid dispersion may crystallize under elevated relative humidity (RH) conditions.
Equilibration in RH chambers with subsequent analysis by XRPD or in-situ variable RH XRPD experiments can be used to readily identify these form changes. Once the effect of temperature and RH on form changes is understood, this can be factored into other processes such as drying, formulation, storage, and packaging B. Sample Characterization. The following analytical techniques and combination thereof were used determine the physical properties of the solid phases prepared.
1. X-Ray Powder Diffraction (XRPD)
XRPD is commonly used as the initial method of analysis for form screens. For polymorph, salt, and co-crystal screens XRPD is used to determine if a new form has been produced by comparing the powder pattern to all known forms of the API and the counterion/guest. If a new form is found by XRPD, additional characterization by other methods is in order. For amorphous solid dispersion screens, XRPD is used to confirm a lack of crystallinity indicated by an amorphous halo in the powder pattern.
The halos will move depending on the concentration and interactions of the API and polymer. Computational methods have also been used with XRPD data to establish miscibility of amorphous solid dispersions X-ray powder diffraction is a front line technique in solid form screening and selection based on its ability to give a fingerprint of the solid-state structure of a pharmaceutical material. Understanding the solid forms of a pharmaceutical compound provides a road map to help direct a variety of development activities ranging from crystallization, formulation, packaging, storage, and performance.
Different screening and selection strategies are warranted in early and late development because different information is needed at the various stages. Solid form selection and formulation approaches need to be investigated together and tailored to the situation. It is important to include solid form selection and possible changes in form as part of the risk management strategy throughout the drug development process.
X-ray powder diffraction (XRPD) analyses were performed using an Inel XRG- 3000 diffractometer equipped with a CPS (Curved Position Sensitive) detector with a 2Θ (2Θ) range of 120°. Real time data were collected using Cu-Ka radiation starting at approximately 4 °2Θ at a resolution of 0.03 °2Θ. The tube voltage and amperage were set to 40 kV and 30 mA, respectively. The pattern is displayed from 2.5 to 40 °2Θ. Samples were prepared for analysis by packing them into thin- walled glass capillaries. Each capillary was mounted onto a goniometer head that is motorized to permit spinning of the capillary during data acquisition. The samples were analyzed for approximately 5 minutes.
Instrument calibration was performed using a silicon reference standard. Peak picking was performed using the automatic peak picking in the Shimadzu XRD-6000 Basic Process version 2.6. The files were converted to Shimadzu format before performing the peak picking analysis. Default parameters were used to select the peaks.
2. Thermogravimetric Analysis (TGA)
Thermogravimetric (TG) analyses were performed using a TA Instruments 2950 thermogravimetric analyzer. Each sample was placed in an aluminum sample pan and inserted into the TG furnace. The furnace was first equilibrated at 25 0C, then heated under nitrogen at a rate of 10 °C/min, up to a final temperature of 350 0C. Nickel and Alumel™ were used as the calibration standards.
3. Differential Scanning Calorimetry (DSC)
Differential scanning calorimetry (DSC) was performed using a TA Instruments differential scanning calorimeter 2920. The sample was placed into an aluminum DSC pan, and the weight accurately recorded. The pan was covered with a lid and then crimped. The sample cell was equilibrated at 25 0C and heated under a nitrogen purge at a rate of 10 °C/min, up to a final temperature of 350 0C. Indium metal was used as the calibration standard. Reported temperatures are at the transition maxima. For studies of the glass transition temperature (Tg) of the amorphous material, the sample cell was equilibrated at ambient temperature, then heated under nitrogen at a rate of 20 °C/min, up to 100 0C. The sample cell was then allowed to cool and equilibrate at -20 0C. It was again heated at a rate of 20 °C/min up to 100 0C and then cooled and equilibrated at -20 0C. The sample cell was then heated at 20 °C/min up to a final temperature of 350 0C. The Tg is reported from the onset point of the transition.
4. Hot Stage Microscopy.
Hot stage microscopy was performed using a Linkam hot stage (model FTIR 600) mounted on a Leica DM LP microscope. The samples were prepared between two cover glasses and observed using a 20χ objective with crossed polarizers and first order compensator. Each sample was visually observed as the stage was heated. Images were captured using a SPOT Insight™ color digital camera with SPOT Software v. 3.5.8. The hot stage was calibrated using USP melting point standards.
5. Thermogravimetric-Infrared (TG-IR)
Thermogravimetric infrared (TG-IR) analyses were acquired on a TA Instruments thermogravimetric (TG) analyzer model 2050 interfaced to a Magna 560® Fourier transform infrared (FT-IR) spectrophotometer (Thermo Nicolet) equipped with an Ever-Glo mid/far IR source, a potassium bromide (KBr) beamsplitter, and a deuterated triglycine sulfate (DTGS) detector. The TG instrument was operated under a flow of helium at 90 and 10 cc/min for the purge and balance, respectively. Each sample was placed in a platinum sample pan, inserted into the TG furnace, accurately weighed by the instrument, and the furnace was heated from ambient temperature to 250 0C at a rate of 20 °C/min.
The TG instrument was started first, immediately followed by the FT-IR instrument. Each IR spectrum represents 32 co-added scans collected at a spectral resolution of 4 cm“1. A background scan was collected before the beginning of the experiment. Wavelength calibration was performed using polystyrene. The TG calibration standards were nickel and Alumel™. Volatiles were identified from a search of the High Resolution Nicolet TGA Vapor Phase spectral library.
6. Fourier Transform Infrared Spectroscopy (FT-IR)
Infrared spectra were acquired on a Magna-IR 560® or 860® Fourier transform infrared (FT-IR) spectrophotometer (Thermo Nicolet) equipped with an Ever-Glo mid/far IR source, an extended range potassium bromide (KBr) beamsplitter, and a deuterated triglycine sulfate (DTGS) detector. A diffuse reflectance accessory (the Collector™, Thermo Spectra-Tech) was used for sampling. Each spectrum represents 256 co-added scans collected at a spectral resolution of 4 cm“1. Sample preparation consisted of physically mixing the sample with KBr and placing the sample into a 13 -mm diameter cup. A background data set was acquired on a sample of KBr. A Log 1/R (R = reflectance) spectrum was acquired by taking a ratio of these two data sets against each other. Wavelength calibration was performed using polystyrene. Automatic peak picking was performed using Omnic version 7.2.
7. Fourier Transform Raman Spectroscopy (FT-Raman)
FT-Raman spectra were acquired on a Raman accessory module interfaced to a Magna 860® Fourier transform infrared (FT-IR) spectrophotometer (Thermo Nicolet). This module uses an excitation wavelength of 1064 nm and an indium gallium arsenide (InGaAs) detector. Approximately 0.5 W of Nd)YVO4 laser power was used to irradiate the sample. The samples were prepared for analysis by placing the material in a glass tube and positioning the tube in a gold-coated tube holder in the accessory. A total of 256 sample scans were collected from at a spectral resolution of 4 cm“1, using Happ-Genzel apodization. Wavelength calibration was performed using sulfur and cyclohexane. Automatic peak picking was performed using Omnic version 7.2.
8. Solid State Nuclear Magnetic Resonance Spectroscopy (13C-NMR)
The solid-state 13C cross polarization magic angle spinning (CP/MAS) NMR spectrum was acquired at ambient temperature on a Varian UN1TYINOVA-400 spectrometer (Larmor frequencies: 13C = 100.542 MHz, 1H = 399.799 MHz). The sample was packed into a 4 mm PENCIL type zirconia rotor and rotated at 12 kHz at the magic angle. The spectrum was acquired with phase modulated (SPINAL-64) high power 1H decoupling during the acquisition time using a 1H pulse width of 2.2 μs (90°), a ramped amplitude cross polarization contact time of 5 ms, a 30 ms acquisition time, a 10 second delay between scans, a spectral width of 45 kHz with 2700 data points, and 100 co-added scans. The free induction decay (FID) was processed using Varian VNMR 6.1C software with 32768 points and an exponential line broadening factor of 10 Hz to improve the signal-to- noise ratio. The first three data points of the FID were back predicted using the VNMR linear prediction algorithm to produce a flat baseline. The chemical shifts of the spectral peaks were externally referenced to the carbonyl carbon resonance of glycine at 176.5 ppm. 9. Solution Nuclear Magnetic Resonance Spectroscopy (1H-NMR) The solution 1H NMR spectrum was acquired at ambient temperature with a
spectrometer at a 1H Larmor frequency of 399.803 MHz. The sample was dissolved in methanol. The spectrum was acquired with a 1H pulse width of 8.4 μs, a 2.50 second acquisition time, a 5 second delay between scans, a spectral width of 6400 Hz with 32000 data points, and 40 co-added scans. The free induction decay (FID) was processed using Varian VNMR 6.1C software with 65536 points and an exponential line broadening factor of 0.2 Hz to improve the signal-to-noise ratio. The spectrum was referenced to internal tetramethylsilane (TMS) at 0.0 ppm. 10.
Moisture Sorption/Desorption Analysis Moisture sorption/desorption data were collected on a VTI SGA-100 Vapor Sorption Analyzer. Sorption and desorption data were collected over a range of 5% to 95% relative humidity (RH) at 10% RH intervals under a nitrogen purge. Samples were not dried prior to analysis. Equilibrium criteria used for analysis were less than 0.0100% weight change in 5 minutes, with a maximum equilibration time of 3 hours if the weight criterion was not met. Data were not corrected for the initial moisture content of the samples. NaCl and PVP were used as calibration standards
Does solid form matter?
Sometimes the properties of two solid forms of a drug are quite similar. In other cases, the physical and chemical properties can vary dramatically and have great impact on pharmacokinetics, ease of manufacturing, and dosage form stability. Properties that can differ among solid forms of a substance include color, solubility, crystal shape, water sorption and desorption properties, particle size, hardness, drying characteristics, flow and filterability, compressibility, and density.
Different solid forms can have different melting points, spectral properties, and thermodynamic stability. In a drug substance, these variations in properties can lead to differences in dissolution rate, oral absorption, bioavailability, levels of gastric irritation, toxicology results, and clinical trial results. Ultimately both safety and efficacy are impacted by properties that vary among different solid forms. Stability presents a special concern, since it’s easy to inadvertently generate the wrong form at any point in the development process.
Because energy differences between forms are usually relatively small, form interconversion is common and can occur during routine API manufacturing operations and during drug product formulation, storage, and use. The stakes are high. Encountering a new solid form during late stages of development can delay filing. A new form appearing in drug product can cause product withdrawal.
When should a search for solid forms begin?
The key to speed in the drug development process is to do it right the first time. For solid pharmaceuticals, that means:
- identify the optimum solid form early in drug development
- make the same form for clinical material and commercial API
- develop a crystallization process that assures control of solid form
- produce a drug product with solid form stability through expiration
scientists strongly recommend that investigation of possible solid forms of a new chemical entity be carried out as early in the development process as drug supply will allow. The best approach has three stages. The first stage, more relevant to some development processes than to others, is a milligram-scale abbreviated screen on efficacious compounds prior to final IND candidate selection. This early information can be used to guide selection of salts and solid forms for scale-up and toxicology studies. The second stage is full polymorph screening and selection of optimum solid form. This stage is important to all development processes and should certainly occur before the first GMP material is produced. In the case of ionic drugs, various salts should be prepared and screened for polymorphs and hydrates. The third stage, an exhaustive screen carried out before drug launch, is an effort to find and patent all of the forms of a high-potential drug. Staging the screening in this way optimizes the balance among the factors of early knowledge of options, probability of commercial success, and judicious investment of R&D money.
 Delay in understanding solid form issues results in problems like different batches of clinical material having different solid forms. Another common and preventable dilemma arises when clinical trials are carried out with one form while commercial production generates another. In this case, bridging studies are required to demonstrate to regulatory agencies that the clinicals are relevant. ICH guidelines require a search for solid forms, comparison of properties that might affect product efficacy, and, if appropriate, setting of solid form specifications.
Delay in understanding solid form issues results in problems like different batches of clinical material having different solid forms. Another common and preventable dilemma arises when clinical trials are carried out with one form while commercial production generates another. In this case, bridging studies are required to demonstrate to regulatory agencies that the clinicals are relevant. ICH guidelines require a search for solid forms, comparison of properties that might affect product efficacy, and, if appropriate, setting of solid form specifications.
How is solid form controlled in API manufacture?
It is important to control solid form during API synthesis in order to demonstrate complete process control to regulatory agencies. Different solid forms can have different solubilities and can affect recovery of API. Purification efficiencies can vary due to differential exclusion of impurities. Filtration and transfer characteristics often differ between forms. Ease of drying can vary due to different abilities to bind solvent and water in the crystal lattice. A prevalent but incorrect belief is that solid form is determined primarily by choice of crystallization solvent. In fact, it is well established that parameters like temperature, supersaturation level, rate of concentration or cooling, seeding, and ripening can have an overriding effect. These variables must be controlled to ensure consistency of solid form in API.
Can solid form problems arise in drug products, too?
The potential for solid form variation does not end at API production. Solid form issues remain through formulation, manufacture, storage, and use of drug product. It is common to observe form transformation during standard manufacturing operations like wet granulation and milling. Excipient interactions and compaction can induce form changes. Changes can occur in the final dosage form over time. Suspensions, including those in transdermal patches, are particularly vulnerable because they provide a low-energy pathway (dissolution/recrystallization) for form interconversion. Lyophile cakes are normally amorphous, but can crystallize on storage leading to difficulty in reconstitution. Even products containing drug in solution, such as filled gel caps, can be affected if the solution is or becomes supersaturated with respect to one of the possible solid forms of the drug.
How can you tell when you have a solid form problem?
Whenever there is a specification failure in drug product or drug substance, solid form changes should be considered in the search for causes. Particularly symptomatic is failure to meet melting point or dissolution specifications. Changes in humidity, crystallization conditions, or crystallization solvent can produce unwanted forms. Solvents known to readily produce solvates include water, alcohols, chlorinated hydrocarbons, cyclic ethers, ketones, nitriles, and amides. Changes in the appearance of gel caps or cracking of tablet coatings can indicate solid form problems. Various solid-state analytical techniques can be used to identify solid form in API. Some techniques can even determine solid form of API in intact final dosage form. Among the most useful techniques for solid-state characterization are melting point, DSC, TGA, hot stage and optical microscopy, solid-state NMR, IR and Raman spectroscopy, and X-ray powder diffraction.
Is there any good news about polymorphism?
Polymorphism presents opportunities as well as challenges. Investigation of the properties of different forms of a commercial drug can lead to new products with improved onset time, greater bioavailability, sustained release properties, or other therapeutic enhancements. New forms can bring improvements in manufacturing costs or API purity. These improvements are patentable and can provide a competitive advantage. An underutilized potential of polymorphism is to solve formulation problems that cause the abandonment of potentially useful drugs in which much investment has already been made. 
SOLUBILITY
Solubility is an important parameter for new molecules especially with the emergence of many poorly soluble compounds in the drug discovery and development pipeline. Polymorphic forms can exhibit solubility differences that vary within a factor of 1-5, amorphous solid dispersions show an improvement one or two orders of magnitude higher, and salts and co-crystals fall between these extremes . A comparison of solubility values of pure forms will provide important information when deciding on a solid form or dosage form. X-ray powder diffraction will allow identification of pure forms for these types of measurements.
However, form changes during solubility and dissolution experiments are also possible and need to be investigated. Solids remaining at the end of solubility and dissolution experiments should always be analyzed initially by XRPD to determine if a form transformation has occurred under these conditions. If a form change has occurred, XRPD patterns can be compared to known forms (polymorphs, hydrates, salts, free acid/base) in order to identify the solids remaining. If a pattern is obtained that does not correspond to known forms, complementary methods will be needed to determine properties such as hydration state or a change in stoichiometry as would be observed from breaking a salt and forming the free acid/base or the formation of salts in buffered solutions.
FORMULATION
Formulators are charged with the responsibility to formulate a product which is physically and chemically stable, manufacturable, and bioavailable. Most drugs exhibit structural polymorphism, and it is preferable to develop the most thermodynamically stable polymorph of the drug to assure reproducible bioavailability of the product over its shelf life under a variety of real-world storage conditions. There are occasional situations in which the development of a metastable crystalline or amorphous form is justified because a medical benefit is achieved. Such situations include those in which a faster dissolution rate or higher concentration are desired, in order to achieve rapid absorption and efficacy, or to achieve acceptable systemic exposure for a low-solubility drug.
Another such situation is one in which the drug remains amorphous despite extensive efforts to crystallize it. If there is no particular medical benefit, there is less justification for accepting the risks of intentional development of a metastable crystalline or amorphous form. Whether or not there is medical benefit, the risks associated with development of a metastable form must be mitigated by laboratory work which provides assurance that (a) the largest possible form change will have no substantive effect on product quality or bioavailability, and/or (b) a change will not occur under all reasonable real-world storage conditions, and/or (c) analytical methodology and sampling procedures are in place which assure that a problem will be detected before dosage forms which have compromised quality or bioavailability can reach patients.
Crystal engineering and co-crystals
Crystal engineering is generally considered to be the design and growth of crystalline molecular solids with the aim of impacting material properties. A principal tool is the hydrogen bond, which is responsible for the majority of directed intermolecular interactions in molecular solids. Co-crystals are multi-component crystals based on hydrogen bonding interactions without the transfer of hydrogen ions to form salts – this is an important feature, since Brønsted acid-base chemistry is not a requirement for the formation of a co-crystal.
Co-crystallization is a manifestation of directed self-assembly of different components. Co-crystals have been described of various organic substances over the years (33,34) and given various names, such as addition compounds (35,36) molecular complexes (37,38) and heteromolecular co-crystals (39). Regardless of naming convention, the essential meaning is that of a multi-component crystal where no covalent chemical modification of the constituents occurs as a result of the crystal formation. Pharmaceuticals co-crystals have only recently been discussed as useful materials for drug products.
Pharmaceutical co-crystals
Pharmaceutical co-crystals can be defined as crystalline materials comprised of an active pharmaceutical ingredient (API) and one or more unique co-crystal formers, which are solids at room temperature. Co-crystals can be constructed through several types of interaction, including hydrogen bonding, p-stacking, and van der Waals forces. Solvates and hydrates of the API are not considered to be co-crystals by this definition. However, co-crystals may include one or more solvent/water molecules in the crystal lattice. An example of putative design, a construction and preparation process is shown in Figure 2 for the 5-fluororuracil:urea 1:1 co-crystal(40).
This real example neatly illustrates the opportunity and challenge that exists currently with designing pharmaceutical co-crystals. Firstly, the ‘design’ is challenging because we have no ability to predict the exact crystal structure that may result from a crystallization attempt. By analogy to the challenge of deriving protein structure from first principles, the primary sequence (chemical structure in our case) is known and elements of secondary structure (the 2-D tape construction in Figure 2) are somewhat discernible from primary information. Prediction of the actual 3-D folded conformation (tertiary structure or obtained by self-assembly) is not possible. In other words, while we currently have the ability to project which things associate in what approximate manner on the secondary level, crystal structure prediction is essentially an intractable proposition.
By extension, and just as the exact function of a protein and quantitative parameters of activity are not predictable from primary and secondary structure, the prediction of crystal properties is not possible in the absence of structural information and measurements. There is early evidence that practitioners were aware that apparent co-crystallization of drugs could lead to useful preparations (41). In fact, a ‘chemical compound’ composed of sulfathiazole and proflavin dubbed flavazole was used to treat bacterial infection during the Second World War (42).
The case of flavazole reveals insight into how two different molecules might interact in a putative co-crystal:“… flavazole is definitely a chemical compound containing equimolar proportions of sulphathiazole and proflavin base. It is believed that combination occurs through the acidic sulphonamide group (SO2NH) of the sulphathiazole and the basic centres of the proflavin. Perhaps the most realistic expression of the formula would be to place proflavin and sulphathiazole side by side with a comma between them.” (42) In the second half of the 20th century, interest in co-crystals evolved into the directed study of intermolecular interactions in crystalline solids (43-45). The technical development of routine single-crystal structure determination led to a watershed of data, now largely accessible through the Cambridge Structural Database (CSD) (46,47).
The structural data have become useful for understanding the intermolecular interactions in co-crystals in atomic level detail (48). Using insight gained from analysis of the CSD and directed experimentation, scientists attempt design of co-crystals with specific properties, such as color or non-linear optical response, by selecting starting components with appropriate molecular properties likely to exhibit specific intermolecular interactions in a crystal (49-52).
However, even when chemically compatible functional groups are present it is not possible to accurately predict if a co-crystal, a eutectic mixture or simply a physical mixture will result from any given experiment. As a result of these complexities, attention has been directed at the identification and characterization of intermolecular packing motifs with the goal of developing principles for co-crystal materials (53).

Figure 2. Steps involved in crystal engineering of a pharmaceutical phase, exemplified by the real example of co-crystallization of 5-fluorouracil and urea. Scientists in India have reported a rare example of synthon polymorphism in co-crystals of 4,4′-bipyridine and 4-hydroxybenzoic acid.

Polymorphism is defined as the ability of a material to exist in more than one form or crystal structure. It has important implications for the properties of such materials; for example in pharmaceuticals, the dissolution rate of a drug can be dependent on the polymorphic form. While this is a common phenomenon in single crystals it is much less common in co-crystals, systems where the structure has at least two distinct components. Gautam Desiraju from the Indian Institute of Science, found that when 4,4′-bipyridine and 4-hydroxybenzoic acid were dissolved together in a solvent such as methanol they would co-crystallise to form two different polymorphs. They noticed that a third form, a pseudopolymorph, was also present.
PROSPECTS FOR CRYSTAL ENGINEERING AND PHARMACEUTICAL CO-CRYSTALS
At the beginning of the 21st century, the field of crystal engineering has experienced significant development. Importantly, crystal engineering principles are now being actively considered for application to pharmaceuticals to modulate the properties of these valuable materials (54).
Because the physical properties that influence the performance of pharmaceutical solids are reasonably well appreciated, there is a unique opportunity to apply crystal engineering techniques and the appropriate follow-up studies to solve real world problems, such as poor physical and chemical stability or inadequate dissolution for appropriate biopharmaceutical performance of an oral drug. As structures and series of pharmaceutical co-crystals have begun to appear, we again find that properties cannot be predicted from the structures. Nevertheless, occasional trends have been suggested.
For example, insoluble drug compounds co-crystallized with highly water soluble complements tend to achieve kinetic solubilities in aqueous media several times greater than the pure form (55,56).
There are also more possible phases for each given active compound to consider, thus there will arguably be a greater opportunities for property enhancement. In terms of stability enhancement and solubilization, the example of the series of itraconazole co-crystals with pharmaceutically acceptable 1,4-diacids (55) suggests a strategy alternative to amorphous drug formulation. The co-crystal options presented retain the stability inherent in a crystalline state, while allowing for solubilization that significantly exceeds that of crystalline itraconazole base and rivals the performance of the engineered amorphous bead formulation (Sporanox®).
Where are we now? From recent literature it appears that knowledge gained over the past century and increasingly sophisticated screening techniques developed within the last decade are paving the way towards design of co-crystals with potentially improved pharmaceutical properties (55-58) In terms of the application to pharmaceutical systems, the field of crystal engineering is developing the retro-synthetic understanding of crystal structure using reasoning that is analogous to that applied by organic chemists. For example, the retro-synthetic approach in covalent synthesis operates on the level of a single molecule, while the analogous effort in crystal engineering focuses on the “supermolecule”:
 piracetam
piracetam
The assemblies that define the crystalline arrangement of the molecules as they self-organize into the solid-state. The parallels between the development of crystal engineering and synthetic organic chemistry run still deeper. Methodologies for carrying out these crystallizations are being developed alongside the development of new robust motifs (6,53,55,57,60). The importance of the solubility and dissolution relationships of the components of a putative co-crystal is becoming a matter of significant investigation (56,60). The same can be said for the roles of additives in templating novel forms.
Mechanical milling of materials has also been documented as a means to make co-crystals, and a recent example of polymorphic forms of caffeine:glutaric acid illustrates the opportunities of this type of processing to influence crystal form (61). With an increase in the understanding of the modes of self-assembly, one can start to address the design aspect towards making pharmaceutical co-crystals.
There remain several limitations to the application of what is currently known to the design of useful materials. As mentioned earlier, it remains intractable to reliably predict crystal structure. Multi-component crystals are well out of reach for prediction due in part to complex energetic landscapes, lack of appropriate charge density models and a large number of degrees of freedom, making computation unfeasible. Moreover, there is only a qualitative understanding of the interplay between intermolecular interactions and materials performance, especially for properties relevant to pharmaceuticals such as solubility, dissolution profile, hygroscopicity and melting point.
But the saving grace of the co-crystal approach comes in two guises: Complementarity and diversity. On the topic of complementarity, it is possible, by way of CSD database mining for instance, to identify trends of hetero-synthon occurrence in model systems. As for the diversity aspect, the space of possible co-crystal formers is large, limited only by pharmaceutical acceptability. Coupled with parameters such as stoichiometry variation and increase in the number of components (binary systems can be expanded into ternary ones, etc.), the opportunities appear vast.
THE FUTURE OF CRYSTAL ENGINEERING IN PHARMACEUTICAL SCIENCE
READ
Novel Challenges in Crystal Engineering: Polymorphs and New …
Where are we going? At this point, we have only just scratched the surface of materials science-driven pharmaceutical product design. In the 21st century, practitioners of pharmaceutical chemistry need to enumerate and exploit the opportunities of crystal form design that nature affords us, and thus gain increasing ability to design the materials we need from the molecules that we seek to convert into pharmaceuticals.
Learning will be facilitated by advances in crystallization automation (6,62), microscopy-spectroscopy techniques (Raman and IR microscopy) and new techniques such as terahertz spectroscopy and AFM, along with increasingly sophisticated X-ray diffraction lab instrumentation. In addition, further enhancements in the data mining tools associated with the CSD operating on an ever increasing number of high-quality crystal structures will undoubtedly lead to new knowledge and principles of interaction. 
The challenge placed before pharmaceutical scientists, now and in the future, is the following: (i) to understand the requirement of a particular compound in terms of materials structure and properties, and (ii) to creatively integrate crystal engineering within the limits of pharmaceutical acceptability of components to obtain new forms of active ingredients with desirable properties for formulation and delivery. It should become the collective mantra of medicinal chemists, process engineers and pharmaceutical scientists to “design and make the material we need.” This mantra can form the common aspiration for an industry that is in significant need of innovation and productivity enhancement.
Applications and Advantages
Applications
- Drug companies can use this technology to protect themselves against others generating and patenting polymorphs
Advantages
- Having more than one solid form of a drug allows optimization of drug dissolution behavior and shelf life

IP AND POLYMORPHS


REFERENCES
| [1] | E. L Paul, H.-H. Tung, and M. Midler. Organic crystallization processes. Powder Technology, 150:133-143, 2005. |
| [2] | C. R.Gardner,, C. T. Walsh and Ö. Almarsson. Drugs as materials: Valuing physical form in drug discovery, Nature Reviews Drug Disc, 926-934, 2004. |
| [3] | W. Ostwald. Studien uber die Bildung und Umwandlung fester Korper. Z Phys Chem, 22:289, 1897. |
| [4] | L. Kofler and A. Kofler. Thermo-mikro-methoden zur Kenneichnung organischer Stoffe und stoffgemische. Innsbruck, Wagner, 1954. |
| [5] | Bernstein J., Polymorphism in Molecular Crystals. Oxford University Press: New York, New York, 2002. |
| [6] | S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. Remenar, M. Read, A. Lemmo, S. Ellis, M. J. Cima and C. R .Gardner. High-throughput Crystallization: Polymorphs, Salts, Co-crystals and Solvates of Pharmaceutical Solids. Adv Drug Deliv RevB, 275-300, 2004. |
| [7] | D. Sharmistha and D. J.Grant. Crystal Structures of Drugs: Advances in Determination, Prediction and Engineering. Nature Reviews Drug Discovery,3: 42-57, 2004. |
| [8] | D. Singhal and W. Curatolo. Drug Polymorphism and dosage form design: a practical perspective. Adv Drug Deliv Rev, 56: 335-347, 2004. |
| [9] | N. Lewis. Shedding Some Light on Crystallization Issues: Lecture Transcript for the First international Symposium on Aspects of Polymorphism and Crystallization – Chemical Development Issues. Org Proc Res Dev, 4: 407-412, 2000. |
| [10] | J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm Res,18: 859-866, 2001. |
| [11] | A. T. Hulme, S. L. Price and D. A. Tocher. A New Polymorph of 5-Fluorouracil Found Following Computational Crystal Structure Predictions. J Am Chem Soc, 127:1116-1117, 2005. |
| [12] | J. Lucas and P. Burgess. When Form Equals Substance: The Value of Form Screening in Product Life-Cycle Management. Pharma Voice, 2004. |
| [13] | J. Lucas and P. Burgess. The Paxil Patent: Four Simple Words, One Complex Claim Construction Case. Pharmaceutical Law & Industry, 2:2004. |
| [14] | J. F. Remenar, J. M. MacPhee, B. K. Larson, V. A. Tyagi, J. Ho, D. A. McIlroy, M. B. Hickey, P. B. Shaw and Ö. Almarsson.. Salt selection and simultaneous polymorphism assessment via high-throughput crystallization: the case of sertraline, Org Proc Res Dev, 7:990-996, 2003. |
| [15] | S. R. Byrn, R. R. Pfeiffer, M. Ganey, C. Hoiberg, and G. Poochikian. Pharmaceutical Solids: A strategic approach to regulatory considerations. Pharmaceutical Research, 12:945, 1995. |
| [16] | ICH Steering Committee. 11-10-2000. Good Manufacturing Practice Guide For Active Pharmaceutical Ingredients Q7a, ICH Harmonized Tripartite Guideline. |
| [17] | Ö. Almarsson and C. R. Gardner. Novel approaches the issues of developability. Current Drug Discovery, January:21-26, 2003. |
| [18] | B. C. Hancock and G.Zografi. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci, 86:1-12, 1997. |
| [19] | A. R. M. Serajuddin. Solid Dispersion of Poorly Water-Soluble Drugs: Early Promises, Subsequent Problems, and Recent Breakthroughs. J Pharm Sci, 88:1058- 1066, 1999. |
| [20] | L.Yu. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev, 48:27-42, 2001. |
| [21] | See www.tricor.com |
| [22] | Y. Wu, A. Loper, E. Landis, I. Hettrick, L. Novak, K. Lynn, C. Chen, K. Thompson, R. Higgins, S. Shelukar, G. Kwei and D. Storey. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm, 285:135-46, 2004. |
| [23] | P. H. Stahl and M. Nakano Pharmaceutical Aspects of the Drug Salt Form. Handbook of Pharmaceutical Salts: Properties, Selection, and Use. New York: Wiley-VCH/VCHA, 2002. |
| [24] | J. H. Lin, D. Ostovic and J. P. Vacca, The Integration of Medicinal Chemistry, Drug Metabolism, and Pharmaceutical Research and Development in Drug Discovery and Development, chapter 11: The Story of Crixivan®, and HIV Protease Inhibitor, in Integration of Pharmaceutical Discovery and Development (ed. Borchardt et al.), Plenum Press, New York 1998. |
| [25] | B. D. Johnson, A. Howard, R. Varsolona, J. McCauley and D. K. Ellison. Indinavir Sulfate, in Harry G. Brittain (ed),Analytical Profiles of Drug Substances and Excipients, Academic Press: San Diego, 1999; V26, pp. 319-357. |
| [26] | G.Y. Kwei, L. B. Novak, L. A. Hettrick, E. R. Reiss, D. Ostovic, A. E. Loper, C. Y. Lui, R. J. Higgins, I. W. Chen, J. H. and Lin. , Rediospecific Intestinal Absorption of HIV protease inhibitor L-735,524 in beagle dogs. Pharm Res, 12:884, 1995. |
| [27] | J. H. Lin, I.-W. Chen, K. J. Vastag, and D. Ostovic. pH-dependent oral absorption of L-735,524, a potent HIV protease inhibitor, in rats and dogs. Drug Metab Disp 23:730-735, 1995. |
| [28] | K. C. Yeh, P. J. Deutsch, H. Haddix, M. Hesney, V. Hoagland, W. D. Ju, S. J. Justice, B. Osborne, A. T. Sterrett, J. A. Stone, E. Woolf and S. Waldman. Single-Dose Pharmacokinetics of Indinavir and the Effect of Food. Antimicrob. Agents Chemother, 42:332, 1998. |
| [29] | P J. Desrosiers. The potential of preform. Modern Drug Discovery, 7:40-43, 2004. |
| [30] | E A. Collier. A crystallization / crystal engineering approach to aid salt selection – anions. UMIST – Institute of Science and Technology, Dept. of Chem. Eng. 2004. |
| [31] | O. Félix, M. W. Hosseini, A. De Cian and J. Fischer. Crystal engineering of 2-D hydrogen bonded molecular networks based on the self-assembly of anionic and cationic modules. Chem Commun, 281-282, 2000. |
| [32] | C. B. Aakeroy and M. Niewenhuzen. Hydrogen-bonded layers of hydrogen malate anions – a framework for crystal engineering. J Am Chem Soc, 116:10983-10991, 1994. |
| [33] | For example, d-glucose:sodium chloride monohydrate is described in F. v. Kobell and J. F. Prakt Chemie, 28:489, 1843. |
| [34] | Quinhydrone was described in F. Wöhler. Untersuchungen über das Chinon. Annalen, 51:153, 1844. |
| [35] | For example, A. Buguet. Cryoscopy of Organic Mixtures and Addition Compounds. Compt Rend, 149:857-8, 1910. |
| [36] | H. Grossmann. Thiourea. Chemiker-Zeitung,31:1195-6, 1908. |
| [37] | For example see, A. Damiani, P. De Santis, E. Giglio, A. M. Liquori, R. Puliti and A. Ripamonti. The crystal structure of the 1:1 molecular complex between 1,3,7,9-tetramethyluric acid and pyrene. Acta Crystallogr,19:340-8, 1965. |
| [38] | J. N. Van Niekerk and D. H. Saunder. The crystal structure of the molecular complex of 4,4′-dinitrobiphenyl with biphenyl. Acta Crystallogr,1:44, 1948. |
| [39] | S. Pekker, E. Kovats, G. Oszlanyi, G. Benyei, G. Klupp, G. Bortel, I. Jalsovszky, E. Jakab, F. Borondics, K. Kamaras, M. Bokor, G. Kriza, K. Tompa and G. Faigel. Rotor-stator molecular crystals of fullerenes with cubane. Nature Materials, 4:764-767, 2005. |
| [40] | 5-Fluorouracil:urea. C4H3N2O2F.CH4N2O; C2/c; a = 9.461(3) Å, b = 10.487(3) Å, c = 15.808(4) Å, β = 99.89(7)º; Z = 4; T = 100(2) K; GOF = 1.023, R2 = 0.0663; wR2 = 0.1753. |
| [41] | C. G. Santesson. Addition Combinations. Archiv fuer Experimentelle Pathologie und Pharmakologie, 118:313-24, 1926. |
| [42] | J. McIntosh, R. H. M. Robinson, F. R. Selbie, J. P. Reidy, H. Elliot Blake and L. Guttmann. Lancet, 249:97-99, 1945. |
| [43] | K. Hoogsteen. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Crystallogr, 16:907-16, 1963. |
| [44] | F. S. Mathews,and A. Rich. The molecular structure of a hydrogen-bonded complex of N-ethyladenine and N-methyluracil. J Mol Bio, 8(1):89-95, 1964. |
| [45] | H. M. Sobell, K. Tomita and A. Rich. The crystal structure of an intermolecular complex containing a guanine and a cytosine derivative. Proc Natl Acad Sci USA, 49:885-92, 1963. |
| [46] | F. H. Allen. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr, B58:380-388, 2002. |
| [47] | I. J. Bruno,; J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr, B58:389-397, 2002. |
| [48] | F. H. Allen, O. Kennard and R. Taylor. Systematic analysis of structural data as a research technique in organic chemistry Acc Chem Res, 16:146-153, 1983. |
| [49] | M. C. Etter. A new role for hydrogen-bond acceptors in influencing packing patterns of carboxylic acids and amides. J Am Chem Soc, 104:1095-6, 1982. |
| [50] | M. C. Etter and G. M. Frankenbach. Hydrogen-bond directed cocrystallization as a tool for designing acentric organic solids. Chem Mater, 1:10-12, 1989. |
| [51] | Desiraju, G. R., Crystal Engineering. The Design of Organic Solids, Materials Science Monographs 54, Elsevier, Amsterdam, 1989. |
| [52] | J. A. R. P. Sarma and G. R. Desiraju. Crystal engineering via donor-acceptor interactions. X-ray crystal structure and solid state reactivity of the 1:1 complex, 3,4-dimethoxycinnamic acid-2,4-dinitrocinnamic acid. J Chem Soc, Chem Commun, :45-46, 1983. |
| [53] | For example, C. B. Åakeroy, J. Desper and B. A. Helfrich. CrystEngComm, 6:19-24, 2004. |
| [54] | Ö. Almarsson and M. J. Zaworotko. Crystal Engineering of the Composition of Pharmaceutical Phases. Do Pharmaceutical Co-crystals Represent a New Path to Improved Medicines? Chem Commun, :1889 – 1896, 2004. |
| [55] | J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee,H. Guzmán and Ö. Almarsson. Crystal engineering of novel cocrystals of a triazole drug with 1,4-dicarboxylic acids. J Am Chem Soc, 125:8456-8457, 2003. |
| [56] | S. L. Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly and G. P. Stahly. Crystal Engineering Approach To Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids. J Am Chem Soc,126:13335-13342, 2004. |
| [57] | S. Fleischman, L. Morales; B. Moulton, N. Rodríguez-Hornedo, R. Bailey Walsh and M. J. Zaworotko. Crystal engineering of the composition of pharmaceutical phases.Chem Commun,186, 2003. |
| [58] | A. V. Trask, W. D. S. Motherwell and W. Jones. Solvent-drop Grinding: Green Polymorph Control of Cocrystallisation. Chem Commun, 890-891, 2004. |
| [59] | N. Variankaval, R. Wenslow, J. Murry, R. Hartman, R. Helmy, E. Kwong, S.-D. Clas, C. Dalton and I. Santos. Preparation and Solid-State Characterization of Nonstoichiometric Cocrystals of a Phosphodiesterase-IV Inhibitor and L-Tartaric Acid. Cryst Growth Des, 6:690-700 2006. |
| [60] | S. J. Hehm, B. Rodriguez-Spong and N Rodriguez-Hornedo. Phase Solubility Diagrams of Cocrystal Are Explained by Solubility Product and Solution Complexation: Cryst Growth Des, 6:592-600, 2006. |
| [61] | A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter. Selective Polymorph Transformation via Solvent-drop Grinding. Chem Commun,, 880-882, 2005. |
| [62] | A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter. Selective Polymorph Transformation via Solvent-drop Grinding. Chem Commun,, 880-882, 2005. |
63……..POLYMORPHISM AND PATENTS http://www.collegio.unibo.it/uploads/ideas/joelbernstein.pdf 64…Aprepitant case study FTIR.. READING MATERIALhttp://alpha.chem.umb.edu/chemistry/ch361/spring%2005/ftir%20polymorph.pdf 65…..READ………….An Overview of Solid Form Screening During Drug Development, http://www.icdd.com/ppxrd/10/presentations/PPXRD-10_Ann_Newman.pdf 66…..CRYSTALLIZATION..http://www.intechopen.com/books/advanced-topics-on-crystal-growth/crystallization-from-the-conformer-to-the-crystal
67International Conference on Harmonization Q6A Guideline: Specifications for New Drug Substances and Products: Chemical Substances, October 1999.
68Center for Drug Evaluation and Research Guidance: Submitting Supporting Documentation in Drug Applications for the Manufacture of Drug Substances, February 1987.
69S. Byrn, R. Pfeiffer, M. Ganey, C. Hoiberg, and G. Poochikian. Pharmaceutical solids: A strategic approach to regulatory considerations. Pharm. Res. 12:945-954 (1995). 70
- H. Brittain. Methods for the characterization of polymorphs and solvates. In H. G. Brittain (ed.) Polymorphism in Pharmaceutical Solids. Marcel Dekker, Inc., New York, 1999, pp. 227-278.
- L. X. Yu and G. L. Amidon GL. Analytical Solutions to Mass Transfer. In: G. L. Amidon, P. I. Lee, and E. M. Topp (eds.) Transport Processes in Pharmaceutical Systems. Marcel Dekker, Inc., 1999, p. 23-54.
- G. L. Amidon, H. Lennernas, V. P. Shah, and J. R. Crison. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12:413-420 (1995).
- L. X. Yu, G. L. Amidon, J. E. Polli, H. Zhao, M. Mehta, D. P. Conner, V. P. Shah, L. J. Lesko, M.-L. Chen, V. H. L. Lee, and A. S. Hussain. Biopharmaceutics Classification System: The scientific basis for biowaiver extension. Pharm. Res. 19:921-925 (2002).
- S. R. Byrn, R. R. Pfeiffer, and J. G. Stowell. Solid-State Chemistry of Drugs. 2nd Edition, SSCI, Inc., West Lafayette, Indiana, pp. 259-366.
- H. G. Brittain and E. F. Fiese. Effect of pharmaceutical processing on drug polymorphs and solvates. In H. G. Brittain (ed.) Polymorphism in Pharmaceutical Solids. Marcel Dekker, Inc., New York, 1999, pp. 331-362.
71 CRYSTALS POLYMORPHS IN PHARMAhttp://www.fcfar.unesp.br/arquivos/475753.pdf
72 API………….POLYMORPHISM pharmaceutical ingredients (APIs).http://www.ncbi.nlm.nih.gov/pubmed/19275600
73 polymorphs and co-crystals – ICDD POWER POINT PRESENTATION
74Thermodynamic stability and transformation of pharmaceutical … http://pac.iupac.org/publications/pac/pdf/2005/pdf/7703×0581.pdf
75http://www.imc.cas.cz/nmr/projekt/ws/springer.pdf
76 High-throughput crystallization: polymorphs, salts, co-crystalsand solvates of pharmaceutical solidshttp://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.85.5397&rep=rep1&type=pdf
77 Crystalline Solid – University of Utah College of Pharmacy Homepharmacy.utah.edu/pharmaceutics/pdf/Crystalline.pdfForm – a term encompassing all solids – polymorphs, solvates, amorphous … inPolymorphism in Pharmaceutical Solids
78..related to xrd
- Bish, DL and Post, JE, editors. 1989. Modern Powder Diffraction. Reviews in Mineralogy, Vol. 20. Mineralogical Society of America
- Cullity, B. D. 1978. Elements of X-ray diffraction. 2nd ed. Addison-Wesley, Reading, Mass
- I. Ivanisevic, R. B. McClurg and P. J. Schields: Uses of X-Ray Powder Diffraction in the Pharmaceutical Industry, Pharmaceutical Sciences Encyclopedia: Drug Discovery, Development and Manufacturing, Ed. by Shayne C. Gad., p. 1-42 (2010).
- S. R. Byrn, S. Bates and I. Ivanisevic: Regulatory Aspects of X-ray Powder Diffraction, Part 1, American Pharmaceutical Review, p.55-59 (2005)
- D. Beckers: The power of X-ray analysis, Pharmaceutical Technology Europe (2010), p.29,30.
- M. Sakata, S. Aoyagi, T. Ogura & E. Nishibori (2007): Advanced Structural Analyses by Third Generation Synchrotron Radiation Powder Diffraction, AIP Conference Proceedings, Vol. 879, pp. 1829-1832 (2007).
- Bergamaschi, A.; Cervellino, A.; Dinapoli, R.; Gozzo, F.; Henrich, B.; Johnson, I.; Kraft, P.; Mozzanica, A.; Schmitt, B.; Shi, X.: The MYTHEN detector for X-ray powder diffraction experiments at the Swiss Light Source. J. Synchrotron Rad. 17 (2010) 653–668.
- R.B. Von Dreele, P.W. Stephens, G.D. Smith, and R.H. Blessing: The First Protein Crystal Structure Determined from X-ray Powder Diffraction Data: a Variant of T3R3 Human Insulin Zinc Complex Produced by Grinding,Acta Cryst. D 56, 1549-53 (2000).
- Margiolaki, I., Wright, J. P., Fitch, A. N., Fox, G. C. & Von Dreele, R. B.: Synchrotron X-ray powder diffraction study of hexagonal turkey egg-white lysozyme, Acta Cryst. D61, 423–432 (2005). See also: Margiolaki, I. & Wright, J. P.: Powder crystallography on macromolecules, Acta Cryst. A64, 169–180 (2008).
- Fundamentals of Crystallography,C. Giacovazzo Ed., International Union of Crystallography, Oxford Science Publications, Third Edition (2011).
- The basics of Crystallography and Diffraction (Third edition, 2010), Christopher Hammond, IUCr, Oxford Science Publications; ISBN 978-0-19-954645-9. See also: X-Ray Structure Determination, A practical Guide, George H. Stout and Lyle H. Jensen, Wiley Interscience.
- Bruni G, Gozzo F, Capsoni D, Bini M, Macchi P, Simoncic P, Berbenni V., Milanese C., Girella A., Ferrari S. and Marini A., Thermal, Spectroscopic, and Ab Initio Structural Characterization of Carprofen Polymorphs, J. Pharm. Sciences 100(6), 2321 (2011).
- Brunelli, M., Wright, J. P., Vaughan, G. B. M., Mora, A. J. & Fitch, A. N.: Solving Larger Molecular Crystal Structures from Powder Diffraction Data by Exploiting Anisotropic Thermal Expansion. Angew. Chem. (2003) 115, 2075–2078.
- T. Wessels, Ch. Baerlocher and L.B. McCusker: Single-crystal-like diffraction data from polycrystalline materials, Science (1999), 284, 477-479.
- Shankland, K., David, W. I. F., Csoka, T. & McBride, L.: Structure solution of Ibuprofen from powder diffraction data by the application of a genetic algorithm combined with prior conformational analysis, Intl. J. Pharmaceut. (1998) 165, 117–126.
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni and R. Rizzi: The dual-space resolution bias correction algorithm: applications to powder data, J. Appl. Cryst. (2010). 43, 798-804.
- G.Oszlanyi and A. Suto: The charge flipping algorithm, Acta Cryst. (2008). A64, 123–134 and references herein.
- Boccaleri, E., Carniato, F., Croce, G., Viterbo, D., van Beek, W., Emerich H. and Milanesio, M.,In-situ simultaneous Raman/high-resolution X-ray powder diffraction study of transformations occurring in materials at non-ambient conditions, J. Appl. Cryst., 2007, 40, 684-693.
- Scarlett N.V.Y. and Madsen I. C., Quantification of phases with partial or no known crystal structures, Powder Diffraction (2006) 21, 278-284
- Giannini C., Guagliardi A. and Millini R., Quantitative phase analysis by combining the Rietveld and the whole-pattern decomposition methods, J. Appl. Cryst., 2002, 35, 481-490.
- Scardi, P.; Leoni, M.: Line profile analysis: pattern modeling versus profile fitting, J. Appl. Cryst. 39 (2006) 24–31. Scardi, P.; Leoni, M.: Whole Powder Pattern Modelling, Acta Crystall. A58 (2002) 190–200
- Local structure from total scattering and atomic pair distribution function (PDF) analysis, In Powder diffraction: theory and practice, (Royal Society of Chemistry, London England, 2008), Robert E. Dinnebier and Simon J. L. Billinge, Eds., pp. 464 – 493.
- Neder R. B. And Korsunskiy V. I., Structure of nanoparticles from powder diffraction data using the pair distribution function, 2005 J. Phys.: Condens. Matter 17 S125
- A. Cervellino, C. Giannini, A. Guagliardi, Determination of nanoparticle, size distribution and lattice parameter from x-ray data for monoatomic materials with f.c.c. cubic unit cell, J. Appl. Cryst. 36, 1148-1158 (2003).
- P.W. Stephens, D.E. Cox, and A.N. Fitch, Synchrotron Radiation Powder Diffraction in Structure Determination by Powder Diffraction, pp. 49-87, edited by W.I.F. David, K. Shankland, L.B. McCusker, and C. Baerlocher, (Oxford University Press, 2002)
- Joel Bernstein: Polymorphism in Molecular Crystals, IUCr Monographs on Crystallography (2002), Oxford Science Publications.
- Polymorphism in Pharmaceutical Solids, Ed. By Harry G. Brittain, Drugs and The Pharmaceutical Sciences, Vol. 192
- Tremayne, M.: The impact of powder diffraction on the structural study of organic materials., Philosophical Transactions of the Royal Society of London Series A: Chemistry and Life Sciences Triennial Issue, 362: 2691 (2004).
- Law D, Schmitt EA, Marsh KC, Everitt EA,Wang W, Fort JJ, Krill SL, Qiu Y. Ritonavir-PEG 8000 amorphous solid dispersions: in vitro and in vivo evaluations. J. Pharm. Sci. 2004; 93 (3): 563–567.
- Bruni G., Berbenni V., Milanese C., Girella A., Cardini A., Lanfranconi S. and Marini A.,Determination of the nateglinide polymorphic purity through DSC, J. Pharm. and Biomed. Anal. 54 (2011) 1196-1199.
- Aaltonen J., Alleso M., Mirza S., Koradia V., Gordon K.C and Rantanen J., Solid form screening – A review. European Journal of Pharmaceutics and Biopharmaceutics 71 (2009), 23-37.
- Srivastava D., The Food and Drug Administration and Patent Law at a Crossroads: The Listing of Polymorph Patents as a Barrier to Generic Drug Entry. Food and Drug Law Journal, Vol. 59, No.2 (2004), 339-354.
- Grabowski H. G., Kyle M., Mortimer R., Long G. & Kirson N., Evolving Brand-name And Generic Drug Competition May Warrant A Revision Of The Hatch-Waxman Act. Health Affairs, 30, no.11 (2011):2157-2166.
- Rakowski W. A and Mazzochi D. M., The case of disappearing polymorph: ‘Inherent anticipation’ and the impact of Smithkline Beecham Corp. v Apotex Corp. (Paxil®) on patent validity and infringement by inevitable conversion’. Journal of Generic Medicine, Vol.1, No.2 (2006):131-139.
- Cabri W., Ghetti P., Pozzi G. and Alpegiani M., Polymorphisms and Patent, Market, and Legal Battles: Cefdinir Case Study. Organic Process Research & Development 2007, 11, 64-72.
- Bauer J, Spanton S, Henry R, Quick J, Dziki W, Porter W, Morris J., Ritonavir: an extraordinary example of conformational polymorphism, Pharm Res. 2001 Jun;18(6):859-66.
79………..related to estimation
- McCrone, W.C. in Physics and Chemistry of the Organic Solid State, Vol. 2, (Eds.: D. Fox, M.M. Labes, A. Weissberger), Interscience, New York, 1965, pp. 725-767.
- Bernstein, J., Davey, R.L., and Henck, JO, Concomitant Polymorphism, Angew.Chem.Int. Ed. 1999,38, 3440-3461.
- Polymorphism in Pharmaceutical Solids, Second Edition (Drugs and the Pharmaceutical Sciences) Harry G. Brittain (Editor) Informa HealthCare; 2nd edition (July 27, 2009)
- Otsuka, M., Kato, F. and Matsuda, Y., Determination of indomethacin polymorphic contents by chemometric near-infrared spectroscopy and conventional powder X-ray diffractometry, Analyst, 2001, 126, 1578 – 1582.
- Patel, A.D., Luner, P. E. and Kemper, M. S., Low-level Determination of Polymorph composition in Physical Mixtures by Near-Infrared Reflectance Spectroscopy, J Pharm Sci 2001, 90, 360-370.
- Agatonovic-Kustrin S, Wu V, Rades, T, Saville, D, Tucker, I G, Powder diffractometric assay of two polymorphic forms of ranitidine hydrochloride Int J Pharm 1999, 184, 107.
- Hurst, V.J., Schroeder, P.A., and Styron, R.W. Accurate quantification of quartz and other phases by powder X-ray diffractometry. Analytica Chimica. Acta, 1997, 337, 233-252
- Campbell Roberts, S.N., Williams A.C., Grimsey, I.M., and Booth S.W., Quantitative analysis of mannitol polymorphs. X-ray powder diffractometry—exploring preferred orientation effects J. Pharm. Biomed. Anal., 2002, 28, 1149-1159.
SEE
PART 1………..http://drugsynthesisint.blogspot.in/p/gliptin-series.html
PART 2 ……http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html
FDA Breakthrough Therapy Designation: 32 And Counting
On February 3rd, GlaxoSmithKline (GSK) announces that Promacta (US)/Revolade (Europe) (Eltrombopag) receives the coveted FDA Breakthrough Therapy Designation (BTD) for cytopenias in patients with Severe Aplastic Anemia (SAA), who have had insufficient response to Immunosuppressive Therapy (IST). The drug is not approved or licensed anywhere in the world for this indication.
SAA is a rare disorder where the bone marrow fails to make enough new blood cells. There are currently no therapies approved for this indication. About forty percent (40%) of patients who do not respond to initial IST die within 5 years of diagnosis.
Regulatory Actions
• Receives FDA ODD in November 2013 for Aplastic Anemia
• Receives FDA BTD in February 2014 for Aplastic Anemia
• Receives FDA ODD in May 2008 & FDA approval in November 2008 for Idiopathic Thrombocytopenia Purpura.
This is the 32nd BTD that is announced by a sponsor company since…
View original post 150 more words
SUMATRIPTAN …Avanir files new drug application for migraine drug

SUMATRIPTAN, GR-43175
1-[3-(2-dimethylaminoethyl)-1H-indol-5-yl]- N-methyl-methanesulfonamide
3-[2-(Dimethylamino)ethyl]-N-methyl-1H-indole-5-methanesulfonamide
| Formula | C14H21N3O2S |
|---|---|
| Mol. mass | 295.402 g/mol |
| CAS number | 103628-46-2 |
|---|
Avanir Pharmaceuticals has filed a new drug application (NDA) with the US Food and Drug Administration (FDA) for approval of its new breath-powered investigational drug-device combination product, ‘AVP-825’, for the acute treatment of migraines. click on title Avanir files new drug application for migraine drug
 SUMATRIPTAN
SUMATRIPTAN
SUMATRIPTAN SUCCINATE

AVP-825 is an investigational drug-device combination product consisting of low-dose sumatriptan powder delivered intranasally utilizing a novel Breath Powered delivery technology. If approved, AVP-825 would be the first and only fast-acting, dry-powder intranasal form of sumatriptan for the treatment of migraine.
The Breath Powered delivery technology is activated by user’s breath to propel medications deep into the nasal cavity where absorption is more efficient and consistent than through most other routes. A user exhales into the device, automatically closing the soft palate and sealing off the nasal cavity completely. Through a sealing nosepiece placed into the nostril, the exhaled breath carries medication from the device directly into one side of the nose. Narrow nasal passages are gently expanded and medication is dispersed deep into the nasal cavity reaching areas where it can be rapidly absorbed. As the medication is delivered, the air flows around to the opposite side of the nasal cavity and exits through the other nostril. Closure of the soft palate helps prevent swallowing or inhalation of sumatriptan powder into the lungs.
| Canada | 2469019 | APPROVED 2005-09-13 | EXP 2022-12-04 |
| United States | 6135979 | 1997-03-21 | 2017-03-21 |
| United States | 5705520 | 1994-12-10 | 2011-12-10 |
| Canada | 2098302 | 2001-10-16 | 2011-12-10 |
| Patent No | PatentExpiry | use code |
|---|---|---|
| 5307953 | Dec 2, 2012 | |
| 5307953*PED | Jun 2, 2013 | |
| 5554639 | Sep 10, 2013 | U-232…METHOD OF TREATING MIGRAINE |
| 5554639*PED | Mar 10, 2014 |
Sumatriptan is a synthetic drug belonging to the triptan class, used for the treatment of migraine headaches. Structurally, it is an analog of the naturally occurring neuro-active alkaloids dimethyltryptamine (DMT), bufotenine, and 5-methoxy-dimethyltryptamine, with an N-methyl sulfonamidomethyl- group at position C-5 on the indole ring.[1]
Sumatriptan is produced and marketed by various drug manufacturers with many different trade names such as Sumatriptan, Imitrex, Treximet, Imigran, Imigran recovery.
Large doses of sumatriptan can cause sulfhemoglobinemia, a rare condition in which the blood changes from red to greenish-black, due to the integration of sulfur into the hemoglobin molecule.[2] If sumatriptan is discontinued, the condition reverses within a few weeks.
Serious cardiac events, including some that have been fatal, have occurred following the use of sumatriptan injection or tablets. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation.
The most common side-effects[3] reported by at least 2% of patients in controlled trials of sumatriptan (25, 50, and 100 mg tablets) for migraine are atypical sensations (paresthesias and warm/cold sensations) reported by 4% in the placebo group and 5–6% in the sumatriptan groups, pain and other pressure sensations (including chest pain) reported by 4% in the placebo group and 6–8% in the sumatriptan groups, neurological events (vertigo) reported by less than 1% in the placebo group and less than 1% to 2% in the sumatriptan groups. Malaise/fatigue occurred in less than 1% of the placebo group and 2–3% of the sumatriptan groups. Sleep disturbance occurred in less than 1% in the placebo group to 2% in the sumatriptan group.
 SUMATRIPTAN
SUMATRIPTAN
Sumatriptan is structurally similar to serotonin (5HT), and is a 5-HT (types 5-HT1D and 5-HT1B[4]) agonist. The specific receptor subtypes it activates are present on the cranial arteries and veins. Acting as an agonist at these receptors, sumatriptan reduces the vascular inflammation associated with migraines.
The specific receptor subtype it activates is present in the cranial and basilar arteries. Activation of these receptors causes vasoconstriction of those dilated arteries. Sumatriptan is also shown to decrease the activity of the trigeminal nerve, which, it is presumed, accounts for sumatriptan’s efficacy in treating cluster headaches. The injectable form of the drug has been shown to abort a cluster headache within fifteen minutes in 96% of cases.[5]
Sumatriptan is administered in several forms; tablets, subcutaneous injection, and nasal spray. Oral administration (as succinate) suffers from poorbioavailability, partly due to presystemic metabolism—some of it gets broken down in the stomach and bloodstream before it reaches the target arteries. A new rapid-release tablet formulation has the same bioavailability, but the maximum concentration is achieved on average 10–15 minutes earlier. When injected, sumatriptan is faster-acting (usually within 10 minutes), but the effect lasts for a shorter time. Sumatriptan is metabolised primarily by monoamine oxidase A into an indole acetic acid analogue, part of which is further conjugated with glucuronic acid. These metabolites are excreted in the urine and bile. Only about 3% of the active drug may be recovered unchanged.
There is no simple, direct relationship between sumatriptan concentration (pharmacokinetics) per se in the blood and its anti-migraine effect (pharmacodynamics). This paradox has, to some extent, been resolved by comparing the rates of absorption of the various sumatriptan formulations, rather than the absolute amounts of drug that they deliver.[6][7]
Sumatriptan was the first clinically available triptan (in 1991). In the United States, it is available only by medical prescription. However, it can be bought over the counter in the UK and Sweden in 50 mg dosage. Several dosage forms for sumatriptan have been approved, including tablets, solution for injection, and nasal inhalers.
On April 15, 2008, the US FDA approved Treximet, a combination of sumatriptan and naproxen, an NSAID.[8] This combination has shown a benefit over either medicine used separately.[9]
In July 2009, the US FDA approved a single-use jet injector formulation of sumatriptan. The device delivers a subcutaneous injection of 6 mg sumatriptan, without the use of a needle.Autoinjectors with needles have been previously available in Europe and North America for several years.[10]
Phase III studies with a iontophoretic transdermal patch (Zelrix/Zecuity) started in July 2008.[11] This patch uses low voltage controlled by a pre-programmed microchip to deliver a single dose of sumatriptan through the skin within 30 minutes.[12][13]Zecuity was approved by the US FDA in January 2013.[14]

Sumatriptan vials 100 5509
On November 6, 2008, Par Pharmaceutical announced that it would begin shipping generic versions of sumatriptan injection (sumatriptan succinate injection) 4 mg and 6 mg starter kits and 4 mg and 6 mg pre-filled syringe cartridges to the trade immediately. In addition, Par anticipates launching the 6 mg vials early in 2009.[15]
Mylan Laboratories Inc., Ranbaxy, Sandoz, Dr. Reddy’s Pharmaceuticals and other companies have received FDA approval for generic versions of Imitrex tablets in 25-, 50-, and 100-milligram doses since 2009. The drug is available in U.S. and European markets, since Glaxo’s patent protections have expired in those jurisdictions. However, sales of a generic delivered via nasal spray are still restricted in the United States.
See also Sumavel DosePro (above).[10]
Chemistry
hydrogenation of nitrile with pd/c in presence of dimethyl amine
…………………


The diazotation of 4-amino-N-methylbenzenemethanesulfonamide (I) with NaNO2-HCl followed by reduction with SnCl2 gives the 4-hydrazino compound (II), which is condensed with (phenylthio)acetaldehyde (III) in ethanol yielding the ethylideneamino compound (IV). The cyclization of (IV) with HCl in ethanol affords N-methyl-3-(phenylthio)-1H-indole-5-methansulfonamide (V), which is desulfurized with RaNi in refluxing ethanol-water to give N-methyl-1H-indole-5-methanesulfonamide (VI). The reaction of (VI) with oxalyl chloride and dimethylamine yields the oxalyl derivative (VII), which is finally reduced with LiAlH4 in refluxing THF.

The condensation of hydrazine (II) with 4,4-dimethoxy-N,N-dimethylbutylamine (VIII) by means of HCl in water gives the butylidenehydrazino compound (IX), which is cyclized with polyphosphate ester (PPE) in CHCl3.

……………………
Beilstein J. Org. Chem. 2011, 7, 442–495.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S9
ref are below article
Indoles
The neuroamine transmitter serotonin contains an indole ring, so it is not surprising that indoles are a recurring theme in many drugs affecting central nervous system (CNS) function including antidepressants, antipsychotics, anxiolytics and antimigraine drugs, as well as psychedelic agents. Indole is also one of the best represented heterocyclic motifs present in the top selling pharmaceuticals, being found in eight of the top 200 drugs, with five of these belonging to the triptan family of antimigraine treatments. The classical Fischer indole synthesis is usually reported as one of the first choice routes to prepare these scaffolds. Drugs such as GSK’s serotonin receptor modulators sumatriptan (49, Imitrex) and zolmitriptan (50, Zomig) use the Fischer indole synthesis at a late stage in order to form the desired compound albeit in only low to moderate yields (Scheme 9).
![[1860-5397-7-57-i9]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i9.png)
However, in sumatriptan the indole product resulting from the Fischer synthesis can still react further which leads to the formation of by-products and significantly reduced yields. One way to minimise this was to protect the nitrogen of the sulfonamide group prior to indole formation [11]. This leads not only to an increased yield in the indole forming step (to 50%) but also facilitates chromatographic purification. The dimethylamino group can be present from the beginning of the synthesis or can be introduced via displacement of chloride or reduction of a cyano moiety. Alternatively, the dimethyl ethylene amine side chain can be introduced in position 3 via a Friedel–Crafts-type acylation. The resulting acid chloride is transformed in situ to the corresponding amide which on reduction with lithium aluminium hydride affords sumatriptan (Scheme 10) [12].
![[1860-5397-7-57-i10]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i10.png)
In the standard Fischer indole synthesis a hydrazine, which is most commonly derived from the corresponding diazonium salt, is reacted with a suitable carbonyl compound. Alternatively, the Japp–Klingemann reaction can be used to directly couple the diazonium salt with a β-ketoester to obtain a hydrazone which can then undergo indole ring formation (Scheme 11) [13].
![[1860-5397-7-57-i11]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i11.png)
As can be seen from Scheme 11 the indole 59 prepared via the Japp–Klingemann reaction is substituted at position 2 by an ester group which prevents reaction with electrophiles, thereby reducing the amount of undesired by-products. A simple sequence of hydrolysis and decarboxylation then affords sumatriptan [14].
All the reported methods for the synthesis of sumatriptan begin with the sulfonamide group already present on the aromatic ring and several routes are possible to introduce this functional group. The scalable route to the sulfonamides inevitably involves the preparation of the sulfonyl chloride intermediate which is then trapped with the desired amine. The sulfonyl chloride can also be prepared from the corresponding hemithioacetal 61 by treatment with NCS in wet acetic acid (Scheme 12). This efficient oxidation produces only methanol and formaldehyde as by-products [15].
![[1860-5397-7-57-i12]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i12.png)
- 11. Pete, B.; Bitter, I.; Szántay, C., Jr.; Schön, I.; Töke, L. Heterocycles 1998, 48, 1139–1149. doi:10.3987/COM-97-8087
- 12…Oxford, A. W. Indole Derivative. U.S. Patent 5,037,845, Aug 6, 1991.
- 13…Japp, F. R.; Klingemann, F. Chem. Ber. 1887, 20, 2942–2944. doi:10.1002/cber.188702002165
- Pete, B.; Bitter, I.; Harsányi, K.; Töke, L. Heterocycles 2000, 53, 665–673. doi:10.3987/COM-99-8815
- Kim, D.-W.; Ko, Y. K.; Kim, S. H. Synthesis 1992, 12, 1203–1204. doi:10.1055/s-1992-26333
[15
References for full article
- The presence of the sulfonamide group in the molecule does not make sumatriptan a “sulfa drug”, since it does not have any anti-microbial properties.
- “Patient bleeds dark green blood”. BBC News. 8 June 2007. Retrieved 6 March 2010.
- Tablets
- Razzaque Z, Heald MA, Pickard JD, et al. (1999). “Vasoconstriction in human isolated middle meningeal arteries: determining the contribution of 5-HT1B- and 5-HT1F-receptor activation”.Br J Clin Pharmacol 47 (1): 75–82. doi:10.1046/j.1365-2125.1999.00851.x. PMC 2014192.PMID 10073743.
- Treatment of acute cluster headache with sumatriptan. The Sumatriptan Cluster Headache Study Group. N Engl J Med 1991;325:322-6.
- Fox, A. W. (2004). “Onset of effect of 5-HT1B/1D agonists: a model with pharmacokinetic validation”. Headache 44 (2): 142–147. doi:10.1111/j.1526-4610.2004.04030.x.PMID 14756852. edit
- Freidank-Mueschenborn, E.; Fox, A. (2005). “Resolution of concentration-response differences in onset of effect between subcutaneous and oral sumatriptan”. Headache 45 (6): 632–637. doi:10.1111/j.1526-4610.2005.05129a.x. PMID 15953294. edit
- GSK press release – Treximet (sumatriptan and naproxen sodium) tablets approved by FDA for acute treatment of migraine
- Brandes JL, Kudrow D, Stark SR, et al. (April 2007). “Sumatriptan-naproxen for acute treatment of migraine: a randomized trial”. JAMA 297 (13): 1443–54.doi:10.1001/jama.297.13.1443. PMID 17405970.
- Brandes, J.; Cady, R.; Freitag, F.; Smith, T.; Chandler, P.; Fox, A.; Linn, L.; Farr, S. (2009). “Needle-free subcutaneous sumatriptan (Sumavel DosePro): bioequivalence and ease of use.”. Headache 49 (10): 1435–1444. doi:10.1111/j.1526-4610.2009.01530.x.PMID 19849720. edit
- ClinicalTrials.gov NCT00724815 The Efficacy and Tolerability of NP101 Patch in the Treatment of Acute Migraine (NP101-007)
- SmartRelief -electronically assisted drug delivery (iontophoresis)
- Pierce, M; Marbury, T; O’Neill, C; Siegel, S; Du, W; Sebree, T (2009). “Zelrix: a novel transdermal formulation of sumatriptan”. Headache 49 (6): 817–25. doi:10.1111/j.1526-4610.2009.01437.x. PMID 19438727.
- Zecuity Approved by the FDA for the Acute Treatment of Migraine
- “PAR PHARMACEUTICAL BEGINS SHIPMENT OF SUMATRIPTAN INJECTION”. Par Pharmaceutical. 2008-11-06. Retrieved 2008-11-25.
- Serotonin 5HT1-receptor agonist. Prepn: M. D. Dowle, I. H. Coates, DE 3320521; eidem, US 4816470; A. W. Oxford, GB 2162522 (1983, 1989, 1986 all to Glaxo).
- Receptor binding studies: P. P. A. Humphrey et al., Br. J. Pharmacol.94, 1123 (1988); P. Schoeffter, D. Hoyer, Arch. Pharmacol. 340, 135 (1989).
- LC-MS determn in plasma: J. Oxford, M. S. Lant, J. Chromatogr. 496, 137 (1989).
- Clinical evaluations in migraine: A. Doenicke et al., Lancet 1, 1309 (1988);
- Subcutaneous Sumatriptan International Study Group, N. Engl. J. Med. 325, 316 (1991); in acute cluster headache: Sumatriptan Cluster Headache Study Group, ibid. 322.
- Review of pharmacology and clinical experience: S. J. Peroutka, Headache 30 (Suppl. 2), 554-560 (1990).
- Drugs Fut 1989,14(1),35
|
1-4-2012
|
Noncardiotoxic pharmaceutical compounds
|
|
|
7-9-2010
|
NON-MUCOADHESIVE FILM DOSAGE FORMS
|
|
|
1-22-2010
|
Fixed Combination Dosage Forms for the Treatment of Migraine
|
|
|
12-11-2009
|
ACTIVE AGENT DELIVERY SYSTEMS AND METHODS FOR PROTECTING AND ADMINISTERING ACTIVE AGENTS
|
|
|
10-9-2009
|
PHARMACEUTICAL COMPOSITIONS COMPRISING A TRIPTAN AND A NONSTEROIDAL ANTI-INFLAMMATORY DRUG
|
|
|
10-9-2009
|
ACTIVE AGENT DELIVERY SYSTEMS AND METHODS FOR PROTECTING AND ADMINISTERING ACTIVE AGENTS
|
|
|
5-7-2009
|
Patient controlled drug delivery device
|
|
|
3-20-2009
|
DEUTERIUM-ENRICHED SUMATRIPTAN
|
|
|
3-13-2009
|
Rapid dissolution of combination products
|
|
|
2-19-2009
|
A METHOD OF IDENTIFYING MODULATORS OF CELL SURFACE MEMBRANE RECEPTORS USEFUL IN THE TREATMENT OF DISEASE
|
|
4-8-1992
|
PREPARATION OF INDOLE DERIVATIVES
|
|
|
1-10-1992
|
PHARMACEUTICAL PREPARATIONS
|
|
|
10-32-1991
|
SYSTEM AND METHOD FOR DETERMINING THREE-DIMENSIONAL STRUCTURES OF PROTEINS
|
|
|
8-7-1991
|
Indole derivative
|
|
|
7-4-1990
|
Pharmaceutical formulations
|
|
|
8-8-1984
|
Fuel and water homogenizer
|
Avanir Pharmaceuticals, Inc. is a biopharmaceutical company focused on bringing innovative medicines to patients with central nervous system disorders of high unmet medical need. As part of our commitment, we have extensively invested in our pipeline and are dedicated to advancing medicines that can substantially improve the lives of patients and their loved ones. For more information about Avanir, please visit http://www.avanir.com.
AVANIR® is a trademark or registered trademark of Avanir Pharmaceuticals, Inc. in the United States and other countries. All other trademarks are the property of their respective owners.
Avanir Pharmaceuticals, Inc. licensed exclusive rights for the development and commercialization of AVP-825, a novel Breath Powered intranasal system containing a low-dose sumatriptan powder from OptiNose Inc. of Yardley, PA.
IMITREX Tablets contain sumatriptan succinate, a selective 5-HT1B/1D receptor agonist. Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole- 5-methanesulfonamide succinate (1:1), and it has the following structure:
IMITREX Tablets contain sumatriptan succinate, a selective 5-HT1B/1Dreceptor agonist. Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole- 5-methanesulfonamide succinate (1:1), and it has the following structure:
 |
The empirical formula is C14H21N3O2S•C4H6O4, representing a molecular weight of 413.5. Sumatriptan succinate is a white to off-white powder that is readily soluble in water and in saline.
Each IMITREX Tablet for oral administration contains 35, 70, or 140 mg of sumatriptan succinate equivalent to 25, 50, or 100 mg of sumatriptan, respectively. Each tablet also contains the inactive ingredients croscarmellose sodium, dibasic calcium phosphate, magnesium stearate, microcrystalline cellulose, and sodium bicarbonate. Each 100-mg tablet also contains hypromellose, iron oxide, titanium dioxide, and triacetin.
Topiroxostat 托匹司他 for gout and hyperuricemia


Topiroxostat
托匹司他
FUJI YAKUHIN ……..INNOVATOR
Approved in japan PMDA JUNE 28 2013
Xanthine oxidase inhibitor
FOR GOUT AND HYPERURICEMIA
Launched – 2013, Fuji YakuhinSanwa, Topiloric Uriadec
IUPAC Name: 4-(5-pyridin-4-yl-1H-1,2,4-triazol-3-yl)pyridine-2-carbonitrile
CAS Registry Number: 577778-58-6
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1)
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole
Synonyms: 4-(5-PYRIDIN-4-YL-1H-1,2,4-TRIAZOL-3-YL)PYRIDINE-2-CARBONITRILE,
AC1NRB9T, Topiroxostat (JAN/INN), DB01685, D09786, FYX-051
SK-0910
4-[5-PYRIDIN-4-YL-1H-[1,2,4]TRIAZOL-3-YL]-PYRIDINE-2-CARBONITRILE,
C13H8N6 MF,248.2482 MW
 TOPIROXOSTAT
TOPIROXOSTAT
托匹司他
A xanthine oxidase inhibitor used to treat gout and hyperuricemia.

PATENT EXP 3/12/22, US /EU/CN

FYX-051, TOPIROXOSTAT is a xanthine oxidase inhibitor. This agent was approved in Japan by Fuji Yakuhin and Sanwa for the treatment of gout and hyperuricemia in 2013 and launched at the same year. In 2009, the compound was licensed to Sanwa by Fuji Yakuhin in Japan for the codevelopment and commercialization of gout.
The number of patients with hyperuricemia in Japan is reported to be 1.25 million and the number suffering from asymptomatic hyperuricemia is estimated to reach several millions. Hyperuricemia is becoming a popular disease.
Presently, hyperuricemia and gout due to hyperuricemia are treated by improving the living environment and administering various drug therapies for each period when an attack of gout is predicted to occur (presymptomatic period), when an attack of gout occurs, or when an attack of gout subsides. That is, preventive therapy is conducted in the presymptomatic period by administering colchicines as well as controlling the daily living environment. When an attack occurs, drug therapy using non-steroidal or steroidal anti-inflammatory agents is mainly conducted. After the attack subsides, patients are given guidance to improve their lifestyle. When improvement is judged insufficient, an assessment is made as to whether hyperuricemia is caused by reduced excretion of uric acid or by increased production of uric acid followed by treatment with drugs, which exhibit a uricosuric effect, such as probenecid and benzbromarone, those which inhibit resorption of uric acid, such as sulfinpyrazone, those which improve acidurea conditions, such as citrates, and xanthine oxidase inhibitors which inhibit production of uric acid, such as allopurinol. Colchicine is said to be able to prevent about 90% of attacks through inhibiting chemotaxis and phagocytosis of leukocytes, such as neutrophils, if administration thereof has been completed within a few hours before the attack. Since colchicine has various adverse effects, however, the use thereof is limited to the minimum and it is therefore difficult to timely administer it.
Accordingly, drug therapies are mainly adopted, but only allopurinol is available for the treatment of a disease caused by increased production of uric acid. However, a metabolite of allopurinol, oxypurinol, tends to accumulate and may cause calculi formation. Furthermore, this drug has been reported to induce adverse events such as rash, a decreased renal function and hepatitis, and it is not easy to administer.
Examples of compounds having xanthine oxidase inhibiting activity that can be used for treating gout caused by increased production of uric acid and that are effective for hyperuricemia and gout due to hyperuricemia have been described in J. Medicinal Chemistry, 1975, Vol. 18, No. 9, pp. 895–900, Japanese Patent Publication No. 49-46622 and Japanese Patent Publication No. 50-24315, which disclose some 1,3,5-substituted or 3,5-substituted 1,2,4-triazole compounds.
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1) has a xanthine oxidase inhibitory activity and serum uric acid level known as the agent that reduces (Patent Document 1).

The method for producing the compound (1), for example, 2 by Reissert Henze reaction isonicotinic acid methyl N-oxide – is a cyano isonicotinate, and the hydrazide which is then, 4 – this condensed cyanopyridine After obtaining a hydrazide of isonicotinic acid N-oxide (Patent Document 1, Example 12) and method, a cyano group after introduction, 4 by Reissert Henze reaction – method of condensing a cyano pyridine is known (Patent Document 1, Example 39).Further, 4 – as a starting material cyano-N-oxide, a triazole ring after construction (Patent Document 3), Reissert Henze unprotected or (Patent Document 2) to protect the ring condensed with isonicotinic acid hydrazide method of obtaining the compound (1) by introducing a cyano group by the reaction have also been reported.
The crystalline polymorph, yet the same molecule with the same chemical composition, the molecular arrangement in the crystal are different, and are different crystalline states. The pharmaceutical compounds having crystal polymorphism such the differences in physicochemical properties, affect pharmacological activity, solubility, bioavailability, stability and the like are known.Therefore, when the crystal polymorphism is present in a pharmaceutically useful compound, producing compounds of the crystalline form highly useful from polymorphs thereof is desirable.
WO 2003/064410 discloses WO 2005/009991 discloses Japanese Patent Publication No. 2005-41802
However, 4 of the above Patent Document – no description about the presence of crystalline polymorph on carbonitrile – pyridine-2-[yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol] It has not been, to these manufacturing methods, it is disclosed a method for the purpose of improving the chemical purity and yield, there is no description of the crystallographic plane.
Method of producing topiroxostat, useful for preventing or treating gout; and its intermediates. Picks up from WO2012060308, claiming the use of this topiroxostat for treating renal dysfunction. Along with the concurrently published WO2014017515, claiming crystalline Forms I and II of this compound, which, Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017516
Crystalline Forms I and II of topiroxostat, useful for preventing or treating gout. Along with the concurrently published WO2014017516, claiming a method of producing this compound. Picks up from WO2012060308, claiming a method of treating renal dysfunction using topiroxostat, which Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017515
novel 1,2,4-triazole compounds having an optionally substituted 2-cyanopyridin-4-yl group at 3-position and an optionally substituted aromatic group at 5-position inhibit a xanthine oxidase and are useful for treatment of gout and hyperuricemia, and have previously filed a patent application (Patent Document 1). The compounds can be prepared according to a method shown by the following reaction scheme:
-
 wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
Patent Document 1: JP-A-2002-017825 -
-
A compound represented by formula (1) which is a starting material may be prepared by a method described in, for example, JP-A-47-7120, JP-A-61-152661A, JP-A-62-149673, JP-A-2002-528447, or European Patent Application No. 559363 specification. However, it is preferable to prepare compound (1) according to the following reaction scheme:
-

-


SYNTHESIS



PATENT
- Example 2
-
To the toluene solution obtained in Example 1 (2) was added 2-propanol (700 mL), and the mixture was stirred. To the resulting solution was added p-toluenesulfonic acid monohydrate (151.16 g) and the resulting mixture was stirred for 8 hours at an internal temperature of 80°C. The mixture was brought to room temperature, and the precipitated crystals were taken out and washed with 2-propanol (210 mL×2). The white crystals were dried under reduced pressure at 60°C for 15 hours to give 106.0 g of the captioned compound as white crystals. Subsequently, 90.0 g of the crystals was suspended in a mixture of 2-butanol (49 mL) and water (491 mL) and heated to an internal temperature of 80°C for 1 hour. The internal temperature was brought to room temperature, and the crystals were filtered and washed with a mixture of 2-butanol and water (1:10) (270 mL×3). The resulting crystals were dried under reduced pressure at 60°C for 15 hours to give 75.7 g of the captioned compound in a high purity.
-
1H―NMR(DMSO-d6)δppm:2.29(s,3H), 7.11 (m,2H), 7.48 (dd, 2H, J=6.48, 1.62Hz) , 8.32-8.35(m, 3H) , 8.57(dd, 1H, J=1.62, 0.81Hz) , 8.94-8.98(m, 3H)
- Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonate
Example 3
Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
-
To the white crystals (50.5g) obtained in Example 2 was added 2-propanol (937.5 mL) and water (312.5 mL), and the resulting mixture was heated and dissolved at an internal temperature of 80°C. Immediately thereafter, the solution was filtered and the filtrate was cooled to an internal temperature of 20°C. To the resulting suspension was added dropwise 0.52 mol/l of an aqueous sodium hydrogen carbonate solution (250 mL), and the mixture was stirred at room temperature for 2 hours. Then the crystals were filtered and washed with water (150 mL×3) and 2-butanol (150 mL×2). The crystals were dried under reduced pressure at 80°C for 15 hours to give 29.4 g of the captioned compound as pale yellow crystals.
-
1H―NMR(DMSO-d6)δppm:8.02(dd, 2H, J=4.59, 1.62Hz),8.32(dd, 1H, J=5.13, 1.62Hz), 8.55(dd, 1H, J=1.62, 1.08Hz), 8.80(dd, 2H, J=4.59, 1.62Hz), 8.93 (dd, 1H, J=5.13, 1.08Hz)
SYNTHESIS
Example 12
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of methyl isonicotinate N-oxide
13.9 g of isonicotinic acid N-oxide was added to 209 ml of methylene chloride, 29.7 g of 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline was further added thereto, and the mixture was stirred under argon atmosphere at room temperature for one hour. 32.1 g of methanol was added to this mixture, which was stirred at room temperature for 17 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (3:1) was used as an eluent to yield 11.1 g of a white powder.
1H-NMR (CDCl3) δppm: 3.95 (3H, s), 7.88 (2H, d, J=7.25 Hz), 8.22 (2H, J=7.25 Hz)
2) Production of Methyl 2-cyanoisonicotinate
11.1 g of the crystal obtained in 1) was dissolved in 170 ml of acetonitrile, 14.6 g of triethylamine and 21.5 g of trimethylsilylnitrile were added thereto, and the mixture was refluxed under argon atmosphere for 16 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (95:5) was used as an eluent to yield 8.44 g of a pale yellow powder.
1H-NMR (CDCl3) δppm: 4.01 (3H, s), 8.08 (1H, d, J=5.45 Hz), 8.24 (1H, s), 8.90 (1H, d, J=5.45 Hz)
3) Production of 2-cyanoisonicotinic acid hydrazide
8.44 g of the crystal obtained in 2) was added to 85 ml of methanol, 1.84 g of hydrazine was further added thereto, and the mixture was stirred under argon temperature for 2 hours. After the solvent was evaporated under reduced pressure, chloroform was added to the residue, which was stirred at room temperature for one hour. The precipitated crystal was filtered, washed with chloroform and dried with a vacuum pump to yield 4.15 g of a pale yellow powder.
1H-NMR (DMSO-d6) δppm: 4.72 (2H, s), 8.05 (1H, d, J=5.12 Hz), 8.31 (1H, s),8.90 (1H, d, J=5.12 Hz), 10.23 (1H, s)
4) Production of the Object Compound
2.67 g of 4-cyanopyridine was dissolved in 40 ml of methanol, 0.83 g of sodium methoxide was added thereto, and the mixture was stirred at room temperature for one hour. Then 4.15 g of the crystal obtained in 3) was added and the mixture was refluxed for 37 hours. After the reaction completed, the precipitated solid was filtered, washed with methanol and dried with a vacuum pump to yield 3.66 g of the object compound as a yellow powder.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54, 1.57 Hz), 8.31 (1H, dd, J=5.11, 1.65 Hz), 8.53 (1H, dd, J=1.65, 0.50 Hz), 8.80 (2H, dd, J=4.54, 1.57 Hz), 8.93 (1H, dd, J=5.11, 0.50 Hz)
Example 39
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of isonicotinic acid (N-2-tert-butoxycarbonyl)hydrazide-1-oxide
585 ml of methylene chloride was added to 39.0 g of isonicotinic acid N-oxide, and after 34.0 g of triethylamine was further added thereto, the mixture was cooled under argon atmosphere to −15° C. 33.5 g of ethyl chlorocarbonate in 117 ml of methylene chloride was added dropwise to this mixture, which was stirred at a temperature from −5 to −10° C. for one hour. Then 44.4 g of tert-butyl ester of carbamic acid in 117 ml of methylene chloride was added dropwise to this mixture and it was allowed to slowly rise to room temperature while it was stirred. The precipitated solid was filtered after 15 hours, washed with methylene chloride, and dried with a vacuum pump to yield 49.7 g of white crystal.
1H-NMR (DMSO-d6) δppm: 1.42 (9H, s), 7.82 (2H, d, J=7.09 Hz), 8.33 (2H, d, J=7.09 Hz), 9.02 (1H, s), 10.44 (1H, s)
Production of 2-cyanoisonicotinic acid hydrazine 1½ P-Toluenesulfonic acid salt
228 ml of dioxane was added to 30.4 g of the crystal obtained in 1), and after 13.1 g of trimethylsilyl cyanide and 38.8 g of N,N-dimethylcarbamoyl chloride were further added thereto, the mixture was stirred under argon atmosphere at 60° C. for 5 hours. After the solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and subsequently washed with 1.5 M sodium carbonate aqueous solution and a saturated saline solution and dried over magnesium sulfate. After the magnesium sulfate was filtered off, the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue, 68.5 g of p-toluenesulfonic acid monohydrate was added thereto, and the mixture was stirred at room temperature for 22 hours. The precipitated crystal was filtered, washed with ethyl acetate, and dried with a vacuum pump to yield 40.3 g of white crystal 2).
1H-NMR (DMSO-d6) δppm: 2.28 (4.5H, s), 7.12 (3H, dd, J=7.92 & 0.66 Hz), 7.48 (3H, dd, J=7.92 & 0.66 Hz), 8.10 (1H, dd, J=5.11 & 1.81 Hz), 8.39 (1H, dd, J=1.81 & 0.33 Hz), 8.99 (1H, dd, J=5.11 & 0.33 Hz)
3) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
9.98 g of 4-cyanopyridine was dissolved in 250 ml of methanol, and after 7.77 g of sodium methoxide was added thereto, the mixture was stirred at room temperature for one hour. Then 40.3 g of the crystal obtained in 2) was added and the mixture was refluxed for 24 hours. After the reaction completed, the precipitated crystal was filtered, washed with methanol, and dried with a vacuum pump to yield 16.3 g of yellow crystal.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54 & 1.57 Hz), 8.31 (1H, dd, J=5.11 & 1.65 Hz), 8.53 (1H, dd, J=1.65 & 0.50 Hz), 8.80 (2H, dd, J=4.54 & 1.57 Hz), 8.93 (1H, dd, J=5.11 & 0.50 Hz)
4) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
45 ml of ethanol and 15 ml of 1-methyl-2-pyrrolidone were added to 3.0 g of the crystal obtained in 3), and the mixture was heated and stirred at 80° C. for 19 hours. The crystal was filtered, subsequently washed with a mixture of ethanol and 1-methyl-2-pyrrolidone (3:1) and ethanol, and dried with a vacuum pump to yield 2.71 g of yellow crystal.
5) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonic acid salt
5 ml of ethanol and 30 ml of water were added to 2.48 g of the crystal obtained in 4), and after 3.8 g of p-toluenesulfonic acid monohydrate was further added thereto, the mixture was stirred at room temperature for 5 hours. The precipitated crystal was filtered, subsequently washed with a mixture of ethanol and water (1:6), water and then ethanol, and dried with a vacuum pump to yield 3.5 g of white crystal.
1H-NMR (DMSO-d6) δppm: 2.28 (3H, s), 7.12 (2H, dd, J=7.75 & 0.50 Hz), 7.48 (2H, dd, J=7.75 & 0.50 Hz), 8.33 (1H, dd, J=5.12 & 1.65 Hz), 8.45 (2H, d, J=6.11 Hz), 8.57 (1H, dd, J=1.65 & 0.66 Hz), 8.96˜9.02 (3H, m)
6) Production of the object compound
17 ml of ethanol and 17 ml of water were added to 3.36 g of the crystal obtained in 5), and the mixture was stirred at room temperature for 30 minutes. A solution of sodium carbonate (0.74 g of sodium carbonate in 17 ml of water) was further added, and the mixture was stirred at room temperature for 2 hours. The precipitated crystal was filtered, subsequently washed with water and ethanol, and dried with a vacuum pump to yield 1.89 g of the object compound as a pale yellow crystal.
TOPIROXOSTAT
SYNTHESIS

(First step)
The first step, 4 – is a step of obtaining a compound (3) is reacted in the presence of an alkali metal alkoxide, cyano-N-oxide and (2), and isonicotinic acid hydrazide.
4 used in this reaction – isonicotinic acid hydrazide and (2) a cyano-N-oxide is a known compound both, I can be prepared by known means.
The alkali metal alkoxide is used, 6 alkoxide alkali metal C 1-C are preferred, sodium methylate, sodium ethylate and the like can be given as specific examples. The reaction is preferably carried out in a solvent, as the solvent, alcohol solvents such as methanol, ethanol and the like are preferable.
The reaction is preferably first in a solvent, is treated with an alkali metal alkoxide compound (2) and then to react the isonicotinic acid hydrazide. First, heated to reflux under cooling, at 80 ℃ from 15 ℃ preferably, 30 minutes and 12 hours in general, the reaction temperature in the reaction with an alkali metal alkoxide (2) with the compound is reacted 1-4 hours, preferably about. Under the temperature conditions, using an excess amount or one equivalent of 30 minutes to 12 hours usually, reaction with isonicotinic acid hydrazide Subsequent to reaction for 1 to 5 hours, preferably.
Example 1:
Synthesis 4 oxide (3) – – – (4 – pyridin-carbonyl) -4 – N “pyridine hydrazide imide -1 was suspended in 40mL of methanol cyanopyridine-N-oxide and (2) 5.00g, sodium was added to methylate 22.4mg, and the mixture was stirred for 2 hours under 40 ℃ nitrogen atmosphere. was cooled to room temperature. reaction solution was stirred for 4 hours at 40 ℃ was added isonicotinic acid hydrazide 5.71g at the same temperature, precipitated The filtrated crystals were, washed with methanol 15mL, and dried 15 hours at 80 ℃, N “- to give (3) 9.60g oxide – (4 – pyridin) -4 – pyridine-hydrazide imide -1.
1 H-NMR (DMSO-d 6) δ (ppm): 6.98 (br, 2H), 7.81 (d, 2H, J = 5.77Hz), 7.85 (d, 2H, J = 7 .09 Hz), 8.29 (d, 2H, J = 7.09Hz), 8.73 (d, 2H, J = 5.77Hz), 10.37 (br, 1H)
MS m / z: 256 [M-H] –
(Second step)
The second step is a step of obtaining compound (4) by cyanation agent cyano compound (3).
As the cyanation agent used, trialkyl cyanide alkali metal cyanide, sodium cyanide, potassium cyanide and the like, zinc cyanide, trimethylsilyl cyanide and the like.
The cyanation reaction is preferably, for example, be carried out (Heterocycles, Vol.22, No.5, 1994) by Reissert Henze reaction. This reaction, for example, to give compound (4) by an organic solvent in the compound (3), and after activation with carbamoyl halide, and reacting the cyano agent. The alkylcarbamoyl halide used in the carbamoylation is a first step in Reissert Henze reaction, 6 alkylcarbamoyl halide di C 1-C dimethylcarbamoyl chloride, and di-propyl carbamoyl chloride can be used, preferably, dimethylcarbamoyl is chloride. The solvent used in this reaction, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran and acetonitrile can be used, however, N, N-dimethylformamide is preferred. Further, 15 ~ 60 ℃, more preferably 30 ~ 50 ℃ reaction temperature. The reaction time is preferably 1 to 24 hours, more preferably 1 to 3 hours. As the cyanation agent used in the cyanation reaction followed, cyano agents above can be used, sodium cyanide, potassium cyanide, zinc cyanide, and trimethylsilyl cyanide, and more preferably, it is sodium cyanide . -20 ~ 60 ℃ is preferred, more preferably -10 ~ 40 ℃, reaction temperature is 1-4 hours.
Is a novel compound (4) The compound obtained in this second step, it is useful as an intermediate for the production of compound (1). If through Compound (4) can be synthesized in good yield and easily without the need for purification in the second step is also possible, and can be produced (1) Compound industrially efficiently compound (4).
Synthetic N “hydrazide (4) – (4 – pyridine carbonyl) -4 – pyridine carboxylic acid N’-(carboxylic imidoyloxy – 2 – – cyano-4)
Example 2
4 pyridine hydrazide imide -1 – oxide ( was suspended in N, N-dimethylformamide 48mL and 3) 10.0g, under nitrogen atmosphere, followed by stirring for 1 hour was added dimethylcarbamoyl chloride 9.20g at 40 ℃. was added sodium cyanide 2.48g at the same temperature, After cooling to 5 ℃ below. reaction mixture was stirred for 1 hour, the crystals were collected by filtration. precipitate was successively added dropwise a 5% aqueous sodium bicarbonate solution 100mL, and 100mL water, and washed with water 100mL, at 80 ℃ for 15 h and dried under reduced pressure to give 4 – hydrazide (4) 9.28g of pyridine-carboxylic acid N’-(carboxylic imide yl – 2 – cyano-4).
1 H-NMR (DMSO-d 6) δ (ppm): 7.15 (br, 2H), 7.82 (d, 2H, J = 5.61Hz), 8.14 (d, 1H, J = 5 .11 Hz), 8.37 (s, 1H), 8.75 (d, 2H, J = 5.61Hz), 8.86 (d, 1H, J = 5.11Hz), 10.47 (br, 1H )
MS m / z: 265 [M-H] –

(Third step)
The third step is a step of obtaining a compound (1) by the presence of an acid catalyst, the cyclization reaction of the compound (4).
As the acid, organic phosphoric acid, p-toluenesulfonic acid, such as hydrochloric acid, inorganic acids can be used, inorganic acids, phosphoric acid is particularly preferable. As the reaction solvent, water, 2 – butanol, 2 – mixed solvent of alcohol and water or alcohol, propanol, ethanol and the like can be used, but water and 2 – I was mixed 5:1 to 10:1 butanol solvent. The reaction temperature and time, 60 ~ 100 ℃, preferably 2 to 12 hours at 70 ~ 90 ℃, I want to 8-10 hours, preferably.
Intermediates and compounds of the present invention the method (1) can be isolated and purified from the washed reaction mixture, recrystallization, by means of various conventional chromatography.
Example 3:
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile 4 Synthesis of (1) – pyridine-carboxylic acid N’- (2 – cyano-4 – carboxylic imide yl) water 82mL, 2 hydrazide (4) 9.25g – butanol was added 8.2mL, phosphate 4.00g, was stirred for 8 h at 80 ℃. After cooling to room temperature, the reaction mixture was precipitated crystals were collected by filtration, water: 2 – were washed with a mixed solution of 92.5mL butanol = 10:1. The 13 h and dried under reduced pressure at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl) – 1 H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1 I got a) 7.89g.
Topiroxostat
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
MS m / z: 247 [M-H] –
PATENT
Synthetic water-carbonitrile p-toluenesulfonate – pyridine Example 1: 4 – [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol]: 2 – butanol = was added monohydrate 6.62g p-toluenesulfonic acid in a mixed solution of 55mL of 10:1, 4 at 80 ℃ – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl] pyridine-2 – – triazol-3 was added carbonitrile 7.85g, and the mixture was stirred at the same temperature for 1 hour. After cooling to room temperature, the reaction mixture, and the precipitated crystals were collected by filtration, and water: 2 – were washed with a mixed solution of 40mL of butanol = 10:1. The dried under reduced pressure for 10 hours at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile p-toluene I got a sulfonate 12.6g.
1 H-NMR (DMSO-d 6) δ (ppm): 2.29 (s, 3H), 7.11 (m, 2H), 7.48 (dd, 2H, J = 6.48,1.62 Hz ) ,8.32-8 .35 (m, 3H), 8.57 (dd, 1H, J = 1.62,0.81 Hz) ,8.94-8 .98 (m, 3H)
– [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazole and potassium carbonate 8.22g, 4 in a mixed solution of 80mL of ethanol = 9:1: preparation water of crystal form I: Example 2 I was dissolved carbonitrile p-toluenesulfonate 10.0g – -3 – yl] pyridine-2. After stirring for 5 hours plus 15mL 6M hydrochloric acid at 20 ℃, was the precipitated crystals were collected by filtration, and washed with water 100mL. The 23 h and dried under reduced pressure at 80 ℃, 4 – to obtain carbonitrile 5.78g – pyridin-2 [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazole. Having a DSC as shown in FIG 4 and the powder X-ray diffraction pattern shown in FIG 1, the resulting crystals were type-I crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
N, N carbonitrile 40.0g – preparation of 4 Form II – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl – triazol-3]-2: Example 3 – dimethylformamide was added 300mL, and stirred for 25 min at 150 ℃. After cooling to room temperature the solution, and the precipitated crystals were collected by filtration, and washed twice with water 200mL, 4 and dried under reduced pressure overnight at 80 ℃ the crystal – [5 – (pyridin-4 – yl)-1H-1 , 2,4 – I got carbonitrile 30.4g – yl] pyridine-2 – triazole-3. Having a DSC as shown in FIG 5 and powder X-ray diffraction pattern shown in FIG 2, the resulting crystals were type II crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
The 25 ℃, about 2g carbonitrile, – preparation of the hydrate 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2: Example 4 I was stored for 14 days under conditions of relative humidity 97%. Having a DSC as shown in FIG 7 and the powder X-ray diffraction pattern shown in FIG 3, the obtained crystal was a hydrate.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
Test Example: solubility test Type I crystal by crystal form, II-type crystal, and water solubility of the hydrate was calculated by absorbance measurement method, a saturated solution concentration of each sample. I Figure 8 shows the results.Whereas the 6.2μg/mL water solubility of crystalline Form I, II type crystal 4.2μg/mL, hydrate was 1.9μg/mL.
From Figure 8, the water solubility of Form II and Form I crystals is good, water-soluble type I crystal is particularly good.
NMR
BMCL Volume 19, Issue 21, 1 November 2009, Pages 6225–6229
http://www.sciencedirect.com/science/article/pii/S0960894X09012372?np=y
view compd 39 and ignore rest
 TOPIROXOSTAT, FYX O51
TOPIROXOSTAT, FYX O51
view compd 39 and ignore rest
| 1 | * | Baldwin, J.J., J. Med. Chem.; 1975; 18(9); 895-900, especially p. 898, lines 3-5. |
| 2 | * | Geldard, J.F. et al., J. Org. Chem.; 1965; 30(1); 318-319, especially p. 319, starting line 33. |
| 3 | * | Lever, A.B.P., Inorg. Chem; 1990; 29; 1271-1285, especially p. 1275, line 18 and 19. |
Nucleosides, Nucleotides and Nucleic Acids, 2008 , vol. 27, 6-7 pg. 888 – 893
Inoue, Tsutomu; Sato, Takahiro; Ashizawa, Naoki; Iwanaga, Takashi; Matsumoto, Koji; Nagata, Osamu; Nakamura, Hiroshi
Bioorganic and Medicinal Chemistry Letters, 2009 , vol. 19, 21 pg. 6225 – 6229
WO 2012060308
WO 2007148835
WO 2005009991
| WO2003064410A1 * | Dec 3, 2002 | Aug 7, 2003 | Naoki Ashizawa | Novel 1,2,4-triazole compound |
| US3882134 * | May 21, 1973 | May 6, 1975 | Merck & Co Inc | 1-Substituted-3,5-dipyridyl-1,2,4-triazoles |
| US3947577 * | Jan 8, 1975 | Mar 30, 1976 | Merck & Co., Inc. | Anti-hyperuricemia composition |
| US3984558 * | Nov 29, 1974 | Oct 5, 1976 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds used as bronchodilators |
| US4011218 * | Dec 3, 1974 | Mar 8, 1977 | Merck & Co., Inc. | 1,2,4-triazoles |
| US4104393 * | Sep 2, 1977 | Aug 1, 1978 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds |
| US5571897 * | Dec 5, 1991 | Nov 5, 1996 | Wallac Oy | Luminescent lanthanide chelates |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-9199970-B2 |
2015-12-01 |
|
|||
|
2.
US-20150322006-A1 |
2015-11-12 |
|
|||
|
3.
US-20150309021-A1 |
2015-10-29 |
|
|||
|
4.
US-20150291543-A1 |
2015-10-15 |
|
|||
|
5.
EP-2927219-A1 |
2015-10-07 |
EN
|
|
||
|
6.
US-20150274680-A1 |
2015-10-01 |
|
|||
|
7.
EP-2913053-A1 |
2015-09-02 |
EN
|
|
||
|
8.
EP-2511844-B1 |
2015-08-12 |
EN
|
|
||
|
9.
EP-2712861-B1 |
2015-07-29 |
EN
|
|
||
|
10.
US-20150203490-A1 |
2015-07-23 |
|
|||
|
11.
US-20150191463-A1 |
2015-07-09 |
|
|||
|
12.
US-20150166510-A1 |
2015-06-18 |
|
|||
|
13.
EP-2878594-A1 |
2015-06-03 |
EN
|
|
||
|
14.
EP-2878598-A1 |
2015-06-03 |
E
|
|
||
|
15.
EP-2878595-A1 |
2015-06-03 |
EN
|
|
||
|
16.
US-20150126558-A1 |
2015-05-07 |
|
|||
|
17.
US-8987473-B2 |
2015-03-24 |
|
|||
|
18.
EP-2842948-A1 |
2015-03-04 |
EN
|
|
||
|
19.
EP-2776028-A1 |
2014-09-17 |
EN
|
|
||
|
20.
US-20140256748-A1 |
2014-09-11 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2-pyridinecarbonitrile
|
|
| Clinical data | |
| Trade names | Topiloric, Uriadec |
| Legal status |
|
| Identifiers | |
| CAS Number | 577778-58-6 |
| ATC code | None |
| PubChem | CID: 5288320 |
| ChemSpider | 4450517 |
| Chemical data | |
| Formula | C13H8N6 |
| Molecular mass | 248.24 g/mol |
/////////////
C1=CN=CC=C1C2=NC(=NN2)C3=CC(=NC=C3)C#N
