
Home » 2014 (Page 86)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Vertex Pharmaceuticals: Another Step Forward For Kalydeco
.
On February 21st, Vertex Pharmaceuticals announces that the FDA approves a supplemental New Drug Application (sNDA) for orphan drug Kalydeco (Ivacaftor) for people with Cystic Fibrosis (CF), ages 6 and older, who have one of the 8 additional mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene :
• G178R
• S549N
• S549R
• G551S
• G1244E
• S1251N
• S1255P
• G1349D.
Kalydeco receives approval from the FDA in January 2012 for CF patients, ages 6 and older who have at least one copy of the G551D mutation. Thus, Kalydeco is currently approved for 9 mutations. The new approval affects approximately 150 in the United States.
The sNDA approval is based on previously announced data from a Phase III, 2-part, randomized, double-blind, placebo-controlled, cross-over study of 39 CF patients who have one of the above listed 8 mutations + the G970R mutation. Based on this…
View original post 262 more words
Immune cells regulate blood stem cells

Blood stem cell cultures: Blood stem cells from colonies (cell clusters) in vitro consisting of different blood cells. Nine blood stem cell colonies are illustrated in the image, which have developed into differentiated cell types, particularly into white blood cells (leukocytes).Credit: Department of Clinical Research of the University of Bern, Tumor-Immunology research group
Researchers in Bern, Switzerland have discovered that, during a viral infection, immune cells control the blood stem cells in the bone marrow and therefore also the body’s own defences. The findings could allow for new forms of therapy, such as for bone marrow diseases like leukaemia.
During a viral infection, the body needs various defence mechanisms – amongst other things, a large number of white blood cells (leukocytes) must be produced in the bone marrow within a short period of time. In the bone marrow, stem cells are responsible for this task: the blood stem cells. In…
View original post 390 more words
KW-4490 A PDE4 inhibitor from Kyowa Hakko Kirin
 KW 4490
KW 4490- cis-4-Cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxin-5-yl)cyclohexanecarboxylic Acid
- Cyclohexanecarboxylic acid, 4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxin-5-yl)-, cis–
- cis-4-Cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxin-5-yl)cyclohexane-1-carboxylic acid;
- cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylic acid
KF 66490; KW 4490;
MF C17 H19 N O5
A phosphodiesterase type 4 inhibitor, commonly referred to as a PDE4 inhibitor, is a drug used to block the degradative action ofphosphodiesterase 4 (PDE4) on cyclic adenosine monophosphate (cAMP). It is a member of the larger family of PDE inhibitors. The PDE4 family of enzymes are the most prevalent PDE in immune cells. They are predominantly responsible for hydrolyzing cAMP within both immune cells and cells in the central nervous system
PDE4 hydrolyzes cyclic adenosine monophosphate (cAMP) to inactive adenosine monophosphate (AMP). Inhibition of PDE4 blocks hydrolysis of cAMP, thereby increasing levels of cAMP within cells.

Practical synthesis of the PDE4 inhibitor, KW-4490
ORGN 699 |
|
Arata Yanagisawa, arata.yanagisawa@kyowa.co.jp1, Koichiro Nishimura2, Tetsuya Nezu2, Kyoji Ando2, Ayako Maki2, Eiichiro Imai2, and Shin-ichiro Mohri2. (1) Pharmaceutical Research Center, Medicinal Chemistry Research Laboratories, Kyowa Hakko Kogyo Co., Ltd, 1188 Shimotogari, Nagaizumi-cho, Sunto-gun, Shizuoka, Japan, (2) Pharmaceutical Research Center, Sakai Research Laboratories, Kyowa Hakko Kogyo Co., Ltd, 1-1-53 Takasu-cyo, Sakai-ku, Sakai, Osaka, Japan
|
A practical and scaleable synthesis of the PDE4 inhibitor, KW-4490 (1), was developed for the multi-kilogram preparation. This improved synthesis features construction of the 1-arylcyclohexene by Diels-Alder reaction, followed by a newly established acid-mediated hydrocyanation. The synthesis was achieved in 7 steps in 38% overall yield. Efforts toward increasing the regioselectivity in the Diels-Alder reaction, optimization of crystallization-induced dynamic resolution of the hydrocyanation product, and investigation of other synthetic routes will be presented. |
|
New Reactions and Methodology, Metal-Mediated Reactions, Physical Organic Chemistry, Molecular Recognition and Self-Assembly
7:00 PM-9:00 PM, Wednesday, August 20, 2008 Pennsylvania Convention Center — Blrm A/B, PosterDivision of Organic ChemistryThe 236th ACS National Meeting, Philadelphia, PA, August 17-21, 2008 |
A team at Kyowa Hakko Kirin in Japan has used a crystallisation-induced dynamic resolution in the synthesis of KW-4490, a PDE-4 inhibitor being developed for asthma and chronic obstructive pulmonary disease.6 Towards the end of the synthesis, they were faced with a mixture of cis and trans diastereomers of an intermediate derived from a hydrocyanation reaction, which was about 62:38 cis:trans; altering the conditions of the reaction did not give a selective process. The desired isomer was the cis, so they wanted to convert the unwanted trans isomer to cis to improve the yield (Scheme 2).
They first tried using a base-induced isomerisation using a base such as potassium t-butoxide, but although this worked to a degree the best ratio of products obtained was 75:25. The same result was obtained when they tested the system on both pure cis and trans isomers, indicating that this ratio represented the thermodynamic equilibrium. However, they realised that the cis isomer was less soluble in ethanol, so they thought the answer might lie in crystallisation-induced dynamic resolution.
They therefore suspended a crude mixture of the two isomers in ethanol and added a catalytic amount of potassium t-butoxide to effect the isomerisation. It was stirred and warmed, and hexane added portion-wise to crash the cis isomer out of solution. The group managed to increase the ratio of isomers to 99:1 by continuous isomerisation, with a 90% isolated yield.
Scheme 2: Kyowa Hakko Kirin found a way to improve the yield of the cis isomerA Practical Synthesis of the PDE4 Inhibitor, KW-4490
http://pubs.acs.org/doi/abs/10.1021/op1001287?prevSearch=KW%2B4490&searchHistoryKey=



Compound (XIII) is disclosed in WO00/14085 as being useful as a PDE-IV inhibitor. A method for the preparation of a typical compound among compounds (XIII) disclosed in WO00/14085 is as follows:


However, this method is not practically satisfactory as a industrially applicable preparation method, because of (1) requiring multiple steps, (2) low overall yield, (3) requiring purification by silica-gel column chromatography, and the like.
REFERENCE EXAMPLE 1
Synthesis of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid
(1) Synthesis of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid ethyl ester
Under a nitrogen atmosphere, trifluoromethanesulfonic acid (2.25 g) and trimethylsilylcyanide (1.57 mL) were dissolved in benzotrifluoride (10 mL), and a solution of 4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)-3-cyclohexenecarboxylic acid ethyl ester (0.79 g) prepared according to the method described in EXAMPLE 1 in benzotrifluoride (10 mL) was added dropwise at −25° C. After being stirred for for one hour at −20° C., an aqueous saturated sodium hydrogen carbonate was added and the mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was crystallized from ethanol (1 mL) to give a solid substance (0.64 g). The solid substance (0.030 g) was crystallized from a mixed solvent of diisopropyl ether and ethyl acetate (0.36 mL, diisopropyl ether/ethyl acetate=4/1) to give cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid ethyl ester (0.019 g, 47.3%) as a solid.
Melting point 131° C.
1H-NMR (CDCl3, δ ppm) 6.84 (d, J=8.9 Hz, 1H), 6.49 (d, J=8.9 Hz, 1H), 4.39−4.33 (m, 4H), 4.17 (q, J=7.1 Hz, 2H), 3.88 (s, 3H), 2.44 (brd, J=12.6 Hz, 2H), 2.32 (tt, J=11.8, 3.8 Hz, 1H), 2.18−1.95 (m, 4H), 1.86 (dt, J=3.6, 12.6 Hz, 2H), 1.28 (t, J=7.1 Hz, 3H).
[0184] IR (KBr, cm−1) 2953, 2228, 1722, 1607, 1504, 1460, 1381, 1325, 1281, 1117, 1043, 953, 787.
MS (m/z) 346(M+H)+.
(2) Synthesis of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid
To a suspension of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid ethyl ester (397 g) prepared according to the method described (1) of REFERENCE EXAMPLE 1 in ethanol (1.99 L) was added a 6 ml/L aqueous potassium hydroxide (377 mL), and the mixture was stirred for 4 hours at room temperature. After water (2.03 L) was added to the reaction mixture, a 6 mol/L aqueous hydrochloric acid (576 mL) was added to crystallize and to give cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid (366 g, 98.1%) as a solid.
Melting point 245° C.
1H-NMR (DMSO-d6, δ ppm) 12.26 (brs, 1H), 6.79 (d, J=8.9 Hz, 1H), 6.59 (d, J=8.9 Hz, 1H), 4.27 (dd, J=11.9, 5.0 Hz, 4H), 3.75 (s, 3H), 2.34−2.26 (m, 3H), 2.05−2.00 (m, 2H), 1.86−1.63 (m, 4H).
IR (KBr, cm−1) 3287, 2932,1728, 1609, 1508, 1454, 1285, 1119, 953, 802, 764.
MS (m/z) 318(M+H)+.

-
To a solution of 12 g (62 mmol) of 8-methoxy-1,4-benzodioxane-5-carbaldehyde in 140 ml of acetonitrile was added 12 g (110 mmol) of lithium bromide, and then 12 ml (95 mmol) of trimethylsilyl chloride was dropwise added thereto. After 15 minutes, the mixture was ice-cooled, and 19 ml (110 mmol) of 1,1,3,3-tetramethyldisiloxane was dropwise added thereto, followed by stirring at room temperature for 2 hours. The mixture was diluted with methylene chloride, and then was filtered through Celite. The solvent was evaporated in vacuo from the filtrate to give a pale yellow oily substance. To a solution of the obtained crude 5-bromomethyl-8-methoxy-1,4-benzodioxane in 180 ml of DMF was added 9.2 g (190 mmol) of sodium cyanide, followed by stirring at room temperature for 60 hours. To the mixture was added water under ice-cooling, and a solid separated out therefrom was collected by filtration to give 6.8 g (53%) of Compound 1a as an ash-colored solid.Melting Point: 121 – 125 °C
1H-NMR (CDCl3, δ, ppm) 3.60 (s, 2H), 3.88 (s, 3H), 4.33 (s, 4H), 6.50 (d, J = 8 Hz, 1H), 6.86 (d, J = 8 Hz, 1H).
MASS (m/z) 205 (M+).
- Example 1.
- 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl) cyclohexanone (Compound 1)(Step A)
- Synthesis of 2-(8-methoxy-1,4-benzodioxan-5-yl)acetonitrile (Compound 1a)
-
To a solution of 6.2 g (30 mmol) of Compound 1a obtained in Step A in 94 ml of acetonitrile were added 1.4 ml (3.0 mmol) of a 40% methanolic solution of Triton B and 27 ml (300 mmol) of methyl acrylate, followed by heating under reflux for 5 hours. The mixture was allowed to stand for cooling, and then poured into water, followed by extraction with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 2/1) to give 6.4 g (56%) of Compound 1b as a pale yellow oily substance.
1H-NMR (CDCl3, δ, ppm) 2.05-2.37 (m, 4H), 2.39-2.59 (m, 2H), 2.62-2.82 (m, 2H), 3.60 (s, 6H), 3.87 (s, 3H), 4.20-4.40 (m, 4H), 6.48 (d, J = 9 Hz, 1H), 7.01 (d, J = 9 Hz, 1H).
MASS (m/z) 377 (M+).
- (Step B) Synthesis of dimethyl 4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)pimelate (Compound 1b)
-
To a solution of 6.4 g (17 mmol) of Compound 1b obtained in Step B in 96 ml of 1,2-dimethoxyethane was added 2.0 g (50 mmol) of 60% sodium hydride. After heating under reflux for 3 hours, the mixture was allowed to stand for cooling, poured into ice water, acidified with a 6 mol/liter aqueous hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 2/1) to give 5.0 g (86%) of Compound 1c as a white solid.
Melting Point: 129 – 132 °C
1H-NMR (CDCl3, δ, ppm) 2.21-2.50 (m, 3H), 2.61-2.89 (m, 2H), 3.11(d, J = 15 Hz, 1H), 3.79 (s, 3H), 3.89 (s, 3H), 4.37 (s, 4H), 6.49 (d, J = 9 Hz, 1H), 6.84 (d, J = 9 Hz, 1H), 12.2 (s, 1H).
MASS (m/z) 345 (M+).
- (Step C) Synthesis of 4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-2-methoxycarbonylcyclohexanone (Compound 1c)
-
A mixture of 5.0 g (15 mmol) of Compound 1c obtained in Step C, 50 ml of DMSO, 5 ml of water, and 5.0 g of sodium chloride was stirred at 150°C for 5 hours. The mixture was allowed to stand for cooling, and water was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 3/1) to give 3.6 g (86%) of Compound 1 as a white solid.
Melting Point: 157 – 161 °C
1H-NMR (CDCl3, δ, ppm) 2.21-2.41 (m, 2H), 2.45-2.72 (m, 4H), 2.81-3.00 (m, 2H), 3.89 (s, 3H), 4.37 (s, 4H), 6.51 (d, J = 9 Hz, 1H), 6.88 (d, J = 9 Hz, 1H).
MASS (m/z) 287 (M+).
- (Step D) Synthesis of Compound 1
-
In 65 ml of THF was dissolved 10 g (41 mmol) of 5-bromo-8-methoxy-1,4-benzodioxane, and 28 ml (45 mmol) of a 1.59 mol/liter solution of n-butyl lithium in hexane was dropwise added thereto at -78°C. After 15 minutes, a solution of 9.6 g (61 mmol) of 1,4-cyclohexadione monoethyleneketal in 50 ml of THF was dropwise added thereto. The mixture was stirred for 1 hour, followed by stirring at room temperature for 20 minutes. Water was added thereto, the mixture was extracted with ethyl acetate, and the extract was washed with brine and dried over sodium sulfate. The solvent was evaporated therefrom, and the residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 1/1) to give 9.0 g (68%) of Compound 2a as a white solid.
Melting Point: 94 – 96 °C
1H-NMR (CDCl3, δ, ppm) 1.58-1.72 (m, 2H), 1.88-2.28 (m, 6H), 3.57 (s, 1H), 3.86 (s, 3H), 3.90-4.07 (m, 4H), 4.35 (s, 4H), 6.46 (d, J = 9 Hz, 1H), 6.82 (d, J = 9 Hz, 1H).
MASS (m/z) 322 (M+). -
- (Step B) Synthesis of Compound 2
-
In 4.9 ml of methylene chloride was dissolved 0.49 g (1.5 mmol) of Compound 2a obtained in Step A, 0.26 ml (1.9 mmol) of trimethylsilyl cyanide was added thereto at -78°C, then 0.20 ml (1.6 mmol) of a boron trifluoride-ethyl ether complex was dropwise added thereto, and the mixture was stirred for 10 minutes, followed by stirring at room temperature for 10 minutes. A saturated aqueous solution of sodium bicarbonate was added thereto and the mixture was extracted with ethyl acetate. The extract was washed with brine and dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 2/1) to give 0.30 g (61%) of Compound 2 as a colorless oily substance.
1H-NMR (CDCl3, δ, ppm) 1.79-1.95 (m, 2H), 2.06-2.20 (m, 4H), 2.30-2.46 (m, 2H), 3.87 (s, 3H), 3.90-4.07 (m, 4H), 4.36 (s, 4H), 6.48 (d, J = 9 Hz, 1H), 6.82 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
-
- Example 2. 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanone ethyleneketal (Compound 2)
- (Step A)Synthesis of 4-hydroxy-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanone ethyleneketal (Compound 2a)
-
In 2.9 ml of acetone was dissolved 0.29 g (0.87 mmol) of Compound 2 obtained in Example 2, 1.2 ml (7.2 mmol) of a 6 mol/liter aqueous hydrochloric acid was added thereto, and the mixture was heated under reflux for 3 hours. The mixture was allowed to stand for cooling and poured into a saturated aqueous solution of sodium bicarbonate, the mixture was extracted with ethyl acetate, and the extract was washed with brine. The mixture was dried over sodium sulfate, and the solvent was evaporated to give 0.23 g (92%) of Compound 1 as a white solid.
- Example 3. Compound 1
- Example 4. Methyl
cis-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylate (Compound 3) and methyltrans-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylate (Compound 4)(Step A) Synthesis of 2-[4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexylidene]-1,3-dithiane (Compound 3a)
-
To a solution of 5.0 ml (26 mmol) of 2-trimethylsilyl-1,3-dithiane in 50 ml of THF was added dropwise 17 ml (26 mmol) of a 1.54 mol/liter solution of n-butyl lithium in hexane under ice-cooling. After 10 minutes, the mixture was cooled to -78°C, and a solution of 3.6 g (13 mmol) of Compound 1 obtained in Example 1 in 40 ml of THF was dropwise added thereto. After 10 minutes, to the mixture was added brine, followed by addition of water at room temperature. The mixture was extracted with ethyl acetate, the extract was dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 4/1) to give 3.9 g (79%) of Compound 3a as a white solid.
Melting Point: 164 – 166 °C
1H-NMR (CDCl3, δ, ppm) 1.70-1.92 (m, 2H), 2.05-2.24 (m, 2H), 2.28-2.53 (m, 4H), 2.89 (t, J = 6 Hz, 4H), 3.18-3.38 (m, 2H), 3.87 (s, 3H), 4.36 (s, 4H), 6.47 (d, J = 9 Hz, 1H), 6.79 (d, J = 9 Hz, 1H).
MASS (m/z) 389 (M+).
-
In 120 ml of methanol was suspended 3.9 g (10 mmol) of Compound 3a obtained in Step A, 1.7 ml (20 mmol) of 70% perchloric acid, and 4.3 g (16 mmol) of mercury chloride (HgCl2) were added thereto, and the mixture was stirred for 4 hours. The mixture was diluted with methylene chloride and was filtered through Celite, the filtrate was poured into a saturated aqueous solution of sodium bicarbonate, and the mixture was extracted with methylene chloride. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 1/1) to give the crude Compound 3 as a white solid and also to give 0.18 g (5.5%) of Compound 4 as a colorless transparent oily substance. Compound 3 was further recrystallized from ethyl acetate to give 0.57 g (17%) of white crystals.
Compound 3
Melting Point: 123 – 124 °C
1H-NMR (CDCl3, δ, ppm) 1.75-2.22 (m, 6H), 2.27-2.51 (m, 3H), 3.71 (s, 3H), 3.88 (s, 3H), 4.36 (s, 4H), 6.48 (d, J = 9 Hz, 1H), 6.84 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
Compound 4
1H-NMR (CDCl3, δ, ppm) 1.92-2.38 (m, 8H), 2.70-2.88 (m, 1H), 3.69 (s, 3H), 3.87 (s, 3H), 4.36 (s, 4H), 6.48 (d, J = 9 Hz, 1H), 6.81 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
- (Step B) Synthesis of Compound 3 and Compound 4
Example 5.
cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylic acid (Compound 5)
-
To a mixture of 0.55 g (1.7 mmol) of Compound 3 obtained in Example 4 and 3.3 ml of methanol was added 3.3 ml of THF to dissolve them. To the mixture was dropwise added 2.6 ml of a 1.3 mol/liter aqueous solution of potassium hydroxide, followed by stirring at room temperature for 1 hour. The mixture was poured into water, ethyl acetate was added thereto, and an aqueous layer was extracted. The aqueous layer was acidified with a 1 mol/liter aqueous hydrochloric acid, and the precipitated solid was collected by filtration and re-slurried with ethanol to give 0.45 g (86%) of Compound 5 as a white solid.
Melting Point: 228 – 230 °C
1H-NMR (DMSO-d6 , δ, ppm) 1.59-1.90 (m, 4H), 1.94-2.10 (m, 2H), 2.20-2.45 (m, 3H), 3.75 (s, 3H), 4.27 (dd, J = 5, 12 Hz, 4H), 6.60 (d, J = 9 Hz, 1H), 6.79 (d, J = 9 Hz, 1H), 12.2 (br s, 1H).
MASS (m/z) 317 (M+).Elemental analysis: C17H19NO5 Found (%) C 64.09, H : 6.01, N : 4.51 Calcd. (%) C 64.34, H : 6.03, N : 4.41
-
Teixeira, M. M.; Gristwood, R. W.; Cooper, N.; Hellewell, P. G. Trends Pharmacol. Sci.1997, 18, 164– 170, [PubMed],
-
Dyke, H. J.; Montana, J. G. Exp. Opin. Investig. Drugs 2002, 11, 1– 13Lipworth, B. J. Lancet 2005, 365 ( 9454) 167– 175Kroegel, C.; Foerster, M. Exp. Opin. Investig. Drugs 2007, 16, 109– 124
-
Yanagawa, K. The 26th Medicinal Chemistry Symposium, 2007, 2P-29.Ohshima, E.;Yanagawa, K.; Manabe, H.; Miki, I.; Masuda, Y. PCT Int. Appl. WO0164666 A1, 2001.
-
Christensen, S. B.; Guider, A.; Forster, C. J.; Gleason, J. G.; Bender, P. E.; Karpinski, J. M.; DeWolf, W. E., Jr.; Barnette, M. S.; Underwood, D. C.; Griswold, D. E.; Cieslinski, L. B.; Burman, M.; Bochnowicz, S.; Osborn, R. R.; Manning, C. D.; Grous, M.; Hillegas, L. M.; O’Leary-Bartus, J.; Ryan, M. D.; Eggleston, D. S.; Haltiwanger, R. C.; Torphy, T. J. J. Med. Chem. 1998, 41, 821– 835
-
Caron, S.; Vazquez, E.; Wojcik, J. M. J. Am. Chem. Soc. 2000, 122, 712– 713Culkin, D. A.; Hartwig, J. F. Acc. Chem. Res. 2003, 36, 234– 245You, J.; Verkade, J. G. Angew. Chem., Int. Ed. 2003, 42, 5051–5053
-
Muratake, H.; Natsume, M. Tetrahedron 1990, 46, 6331– 6342Caron, S.; Vazquez, E. Org. Process Res. Dev. 2001, 5, 587– 592
-
North, M. In Comprehensive Organic Functional Group Transformations; Katritzky, A. R.;Meth-Cohn, O.; Rees, C. W., Eds.; Pergamon: Oxford, 1995; Vol. 3, p 614.
-
Lemaire, M.; Doussot, J.; Guy, A. Chem. Lett. 1988, 1581– 1584Guy, A.; Doussot, J.; Guette, J.-P.; Garreau, R.; Lemaire, M. Synlett 1992, 821– 822Kurti, L.; Czako, B.; Corey, E. J. Org. Lett. 2008, 10, 5247– 5250
-
Yanagisawa, A.; Nezu, T.; Mohri, S. Org. Lett. 2009, 11, 5286– 5289
-
Brown, H. C.; Cole, T. E. Organometallics 1983, 2, 1316– 1319
-
Kuivila, H. G.; Nahabedian, K. V. J. Am. Chem. Soc. 1961, 83, 2159– 2163Kuivila, H. G.; Nahabedian, K. V. J. Am. Chem. Soc. 1961, 83, 2164– 2166Nahabedian, K. V.; Kuivila, H. G. J. Am. Chem. Soc. 1961, 83, 2167–2174
-
Stang, P. J.; Fisk, T. E. Synthesis 1979, 438– 440Stang, P. J.; Treptow,W. Synthesis 1980, 283– 284Saulnier, M. G.; Kadow, J. F.; Tun, M. M.; Langley, D. R.; Vyas, D. M. J. Am. Chem. Soc. 1989, 111, 8320– 8321
-
Kato, S.; Chujo, I.; Suzuki, K. PCT Int. Appl. WO04000795 A1, 2004.
-
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457– 2483O’Keefe,D. F.; Dannock, M. C.; Marcuccio, S. M. Tetrahedron Lett. 1992, 33, 6679– 6680Segelstein, B. E.; Bulter, T. W.; Chenard, B. L. J. Org. Chem. 1995, 60, 12– 13Kong, K.-C.; Cheng, C.-H. J. Am. Chem. Soc. 1991, 113, 6313– 6315
-
Brands, K. M. J.; Davies, A. J. Chem. Rev. 2006, 106, 2711– 2733
-
Baker, W.; Jukes, E. H. T.; Subrahmanyam, C. A. J. Chem. Soc. 1934, 1681–1684Dallacker, F.; Van Wersh, J. Chem. Ber. 1972, 105, 3301–3305
- Yanagisawa, Arata; Organic Process Research & Development 2010, 14(5), P1182-1187
- Yanagisawa, Arata; Organic Letters 2009, 11(22), P5286-5289
- US 20010056117
- WO 2002059105
- WO 2000014085
| WO1998022455A1 * | Nov 19, 1997 | May 28, 1998 | Michio Ichimura | Oxygenic heterocyclic compounds |
| JP10147585A * | Title not available |
| WO2001064666A1 * | Mar 2, 2001 | Sep 7, 2001 | Kyowa Hakko Kogyo Kk | Oxygen-containing heterocyclic compounds |
| WO2002059105A1 * | Jan 25, 2002 | Aug 1, 2002 | Kyowa Hakko Kogyo Kk | Styrene derivatives and process for production thereof |
| WO2006041120A1 * | Oct 13, 2005 | Apr 20, 2006 | Daisuke Harada | Pharmaceutical composition |
| WO2006041121A1 * | Oct 13, 2005 | Apr 20, 2006 | Daisuke Harada | Remedies/preventives for chronic skin disease |
| WO2006123726A1 * | May 18, 2006 | Nov 23, 2006 | Daisuke Harada | Pharmaceutical composition |
| WO2011134468A1 | Apr 28, 2011 | Nov 3, 2011 | Leo Pharma A/S | Biaryl phosphodiesterase inhibitors |
| EP1362853A1 * | Jan 25, 2002 | Nov 19, 2003 | Kyowa Hakko Kogyo Co., Ltd | Styrene derivatives and process for production thereof |
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review

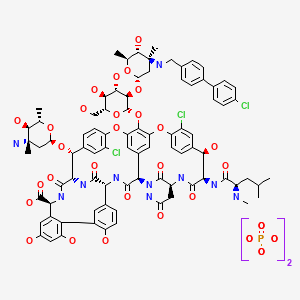
Oritavancin
(4R)-22-O-(3-Amino-2,3,6-trideoxy-3-C-methyl-alpha-L-arabinohexopyranosyl)-N3-(p-(p-chlorophenyl)benzyl)vancomycin
(3S, 6R, 7R, 22R, 23S, 26S, 36R, 38aR) -22 – (3-Amino-2 ,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyloxy) -3 – (carbamoylmethyl ) -10,19-dichloro-44-[2-O-[3 – (4′-chlorobiphenyl-4-ylmethylamino) -2,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyl] – beta-D-glucopyranosyloxy] –
| CAS No. | 171099-57-3 |
| CBNumber: | CB92451283 |
| Molecular Formula: | C86H97Cl3N10O26 |
| Formula Weight: | 1793.12 |
Also known as NDISACC-(4-(4-chlorophenyl)benzyl)A82846B and LY333328,N-(4-(4-chlorophenyl)benzyl)A82846B
Abbott (Supplier), Lilly (Originator), InterMune (Licensee)
The medicines company—
-
the Oritavancin Program Results.pdf
 phx.corporate-ir.net/External.File?item…t=1
phx.corporate-ir.net/External.File?item…t=1Jul 2, 2013 – Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review
PARSIPPANY, NJ — (Marketwired) — 02/19/14 — The Medicines Company (NASDAQ: MDCO) today announced that the U.S. Food and Drug Administration (FDA) has accepted the filing of a new drug application (NDA) for oritavancin, an investigational intravenous antibiotic, with priority review. The Medicines Company is seeking approval of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), administered as a single dose.
In December 2013, the FDA designated oritavancin as a Qualified Infectious Disease Product (QIDP). The QIDP designation provides oritavancin priority review, and an additional five years of exclusivity upon approval of the product for the treatment of ABSSSI. Priority review means the FDA’s goal is to take action on the application within six months, compared to 10 months under standard review. The FDA action date (PDUFA date) for oritavancin is August 6, 2014.
Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
In Dec 2008 the FDA declined to approve it, and an EU application was withdrawn.
In 2009 the development rights were acquired by The Medicine Co. who are running clinical trials for a possible new FDA application in 2013.[2]
Its structure is similar to vancomycin[3] It is a lipoglycopeptide
About Oritavancin
Oritavancin is an investigational intravenous antibiotic for which The Medicines Company is seeking approval in the treatment of ABSSSI caused by susceptible gram-positive bacteria, including MRSA. In clinical trials, the most frequently reported adverse events associated with oritavancin were nausea, headache, vomiting and diarrhea. Hypersensitivity reactions have been reported with the use of antibacterial agents including oritavancin.

Oritavancin shares certain properties with other members of the glycopeptide class of antibiotics, which includes vancomycin, the current standard of care for serious Gram-positive infections in the United States and Europe.[4] Data presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in September 2007 demonstrated that oritavancin possesses potent and rapid bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram positive bacteria, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterococci, and Streptococci.[5] Two posters presented at the meeting also demonstrated that oritavancin was more active than either metronidazole or vancomycin against strains of Clostridium difficile tested.[6]
Anthrax : Research presented at the American Society for Microbiology (ASM) 107th Annual General Meeting in May 2007, suggested oritavancin’s potential utility as a therapy for exposure to Bacillus anthracis, the gram-positive bacterium that causes anthrax, having demonstrated efficacy in a mouse model both pre- and post-exposure to the bacterium[7]
 oritavancin
oritavancin
The 4′-chlorobiphenylmethyl group disrupts the cell membrane of gram positive bacteria.[8] It also acts by inhibition of transglycosylation and inhibition of transpeptidation.[9]
Results have been presented (in 2003) but possibly not yet published from two pivotal Phase 3 clinical trials testing the efficacy of daily intravenous oritavancin for the treatment of complicated skin and skin-structure infections (cSSSI) caused by Gram-positive bacteria. The primary endpoints of both studies were successfully met, with oritavancin achieving efficacy with fewer days of therapy than the comparator agents (vancomycin followed by cephalexin). In addition, oritavancin showed a significantly improved safety profile with a 19.2 percent relative reduction in the overall incidence of adverse events versus vancomycin/cephalexin (p<0.001) in the second and larger pivotal trial.[10]
A Phase 2 clinical study was planned to run until May 2008 entitled “Single or Infrequent Doses for the Treatment of Complicated Skin and Skin Structure Infections (SIMPLIFI),” evaluating the efficacy and safety of either a single dose of oritavancin or an infrequent dose of oritavancin compared to the previously studied dosing regimen of 200 mg oritavancin given once daily for 3 to 7 days.[11] Results published May 2011.[12]
Regulatory submissions
USA
On February 11, 2008, Targanta submitted a New Drug Application (NDA) to the US FDA seeking approval of oritavancin;[13] in April 2008, the FDA accepted the NDA submission for standard review.[14] On 9 Dec 2008 the FDA said insufficient data for approval of oritavancin had been provided and they requested a further phase 3 clinical study to include more patients with MRSA.[15]
Europe
June 2008, Targanta’s Marketing Authorization Application (MAA) for oritavancin was submitted and accepted for review by the European Medicines Agency (EMEA),[16] but the company later withdrew the application in Aug 2009.[17]
About The Medicines Company
The Medicines Company’s purpose is to save lives, alleviate suffering, and contribute to the economics of healthcare by focusing on 3,000 leading acute/intensive care hospitals worldwide. Its vision is to be a leading provider of solutions in three areas: acute cardiovascular care, surgery and perioperative care, and serious infectious disease care. The company operates in the Americas, Europe and the Middle East, and Asia Pacific regions with global centers today in Parsippany, NJ, USA and Zurich, Switzerland.
“We look forward to working with the FDA during the review process, and sharing the knowledge we have gained in our studies of oritavancin,” said Matthew Wikler, MD, Vice President and Medical Director, Infectious Disease Care for The Medicines Company. “We believe that upon approval, oritavancin, administered as a single dose for the treatment of ABSSSI, will offer new options for both physicians and their patients for the treatment of these infections.”
The oritavancin NDA is based on data from two Phase 3 clinical trials, SOLO I and SOLO II, which were conducted under a Special Protocol Assessment (SPA) agreement with the FDA. These Phase 3 trials evaluated the efficacy and safety of a single 1200mg dose of oritavancin compared to 7 to 10 days of twice-daily vancomycin in adults with ABSSSI, including infections caused by MRSA. The combined SOLO studies were conducted in 1,959 patients (modified intent-to -treat population, or mITT), with 405 of the patients suffering from an ABSSSI with a documented MRSA infection.
 oritavancin
oritavancin
Drug substance
Oritavancin diphosphate
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=oritavancin
- LY 333328 diphosphate
- LY333328 diphosphate
- Oritavancin diphosphate
- UNII-VL1P93MKZN
- 192564-14-0 CAS NO
INTRODUCTION
Oritavancin
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.


vancomycin, desmethylvancomycin, eremomycin, teicoplanin (complex of five compounds), dalbavancin, oritavancin, telavancin, and A82846B (LY264826) having structures A, B, C, D, E, F, G and H:

R = B-2-Acetylamido-glucopyraπosyl- Attorney Docket No 33746-704 602


Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopeptides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglycopeptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, whileoritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci.
All three lipoglycopeptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clostridium spp. than dalbavancin, oritavancin or vancomycin. The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily administration. Dalbavancin and telavancin exhibit concentration-dependent activity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities.
Oritavancin’s activity is also considered concentration-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the pharmacodynamic parameter that best describes its activity. Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reactions, fever and diarrhoea were commonly observed withoritavancin therapy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Oritavancin diphosphate (oritavancin) is a semi-synthetic lipoglycopeptide derivative of a naturally occurring glycopeptide. Its structure confers potent antibacterial activity against gram-positive bacteria, including vancomycin-resistant enterococci (VRE), methicillin- and vancomycin-resistant staphylococci, and penicillin-resistant streptococci. The rapidity of its bactericidal activity against exponentially-growing S. aureus (≧3-log reduction within 15 minutes to 2 hours against MSSA, MRSA, and VRSA) is one of the features that distinguishes it from the prototypic glycopeptide vancomycin (McKay et al., J Antimicrob Chemother. 63(6):1191-9 (2009), Epub 2009 Apr. 15).
Oritavancin inhibits the synthesis of peptidoglycan, the major structural component of the bacterial cell wall by a mechanism that is shared with glycopeptides, such as vancomycin (Allen et al., Antimicrob Agents Chemother 41(1):66-71 (1997); Cegelski et al., J Mol Biol 357:1253-1262 (2006); Arhin et al., Poster C1-1471: Mechanisms of action of oritavancin in Staphylococcus aureus [poster]. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007; Chicago, Ill.). Oritavancin, like vancomycin, binds to the Acyl-D-Alanyl-D-Alanine terminus of the peptidoglycan precursor, lipid-bound N-acetyl-glucosamine-N-acetyl-muramic acid-pentapeptide (Reynolds, Eur J Clin Microbiol Infect Dis 8(11):943-950 (1989); Nicas and Allen, Resistance and mechanism of action.
In: Nagarajan R, editor. Glycopeptide antibiotics. New York: Marcel Dekker 195-215 (1994); Allen et al., Antimicrob Agents Chemother 40(10):2356-2362 (1996); Allen and Nicas, FEMS Microbiology Reviews 26:511-532 (2003); Kim et al., Biochemistry 45:5235-5250 (2006)). However, oritavancin inhibits cell wall biosynthesis even when the substrate is the altered peptidoglycan precursor that is present in VRE and vancomycin-resistant S. aureus (VRSA). Thus, the spectrum of oritavancin antibacterial activity extends beyond that of vancomycin to include glycopeptide-resistant enterococci and staphylococci (Ward et al., Expert Opin Investig Drugs 15:417-429 (2006); Scheinfeld, J Drugs Dermatol 6:97-103 (2007)). Oritavancin may inhibit resistant bacteria by interacting directly with bacterial proteins in the transglycosylation step of cell wall biosynthesis (Goldman and Gange, Curr Med Chem 7(8):801-820 (2000); Halliday et al., Biochem Pharmacol 71(7):957-967 (2006); Wang et al., Poster C1-1474: Probing the mechanism of inhibition of bacterial peptidoglycan glycotransferases by glycopeptide analogs. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007). Oritavancin also collapses transmembrane potential in gram positive bacteria, leading to rapid killing (McKay et al., Poster C1-682: Oritavancin disrupts transmembrane potential and membrane integrity concomitantly with cell killing in Staphylococcus aureus and vancomycin-resistant Enterococci. 46th Intersci Conf Antimicro Agents Chemo, San Francisco, Calif., Sep. 27-30, 2006). These multiple effects contribute to the rapid bactericidal activity of oritavancin.
Vancomycin (U.S. Patent 3,067,099); A82846A, A82846B, and A82846C (U.S. Patent 5,312,738, European Patent Publication 256,071 A1); PA-42867 factors A, C, and D (U.S. Patent4,946,941 and European Patent Publication 231,111 A2); A83850 (U.S. Patent No. 5,187,082); avoparcm (U.S. Patent 3,338,786 and U.S. Patent 4,322,343); actmoidin, also known as K288 (J. Antibiotics Series A 14:141 (1961); helevecardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 86/157,397); galacardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 89/221,320); and M47767 (European Patent Publication 339,982).
Oritavancin is in clinical development against serious gram-positive infections, where administration of the drug is via intravenous infusion using several dosages administered over a series of days. The development of alternative dosing regimens for the drug could expand treatment options available to physicians. The present invention is directed to novel dosing regimens.
Means for the preparation of the glycopeptide antibiotics, including oritavancin and analogs thereof, may be found, for example, in U.S. Pat. No. 5,840,684,
SYNTHESIS

LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
J Antibiot1996, 49, (6) :575-81
(3S,6R,7R,22R,23S,26S,36R,38aR)-3-(Carbamoylmethyl)-10,19-dichloro-7,28,30,32-tetrahydroxy-6-(N-methyl-D-leucylamido)-2,5,24,38,39-pentaoxo-22-(L-vancosaminyloxy)-44-[2-O-(L-vancosaminyl)-beta-D-glucopyranosyloxy]-2,3,4,5,6,7,23,24,25,26,36,37,38,38a-tetradecahydro-1H,22H-8,11:18,21-dietheno-23,36-(iminomethano)-13,16:31,36-dimetheno-[1,6,9]oxadiazacyclohexadecino[4,5-m][10,2,16]benzoxadiazacyclotetracosine-26-carboxylic acid; A82846B (I)
4′-chloro[1,1′-biphenyl]-4-carbaldehyde (II)
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
…………………..
EXAMPLE 4
Preparation of Compound 229
A three liter 3-necked flask was fitted with a
condenser, nitrogen inlet and overhead mechanical stirring apparatus. The flask was charged with pulverized A82846B acetate salt (20.0 g, 1.21 × 10-3 mol) and methanol (1000 mL) under a nitrogen atmosphere. 4′-chlorobiphenylcarboxaldehyde (2.88 g, 1.33 × 10-2 mol, 1.1 eq.) was added to this stirred mixture, followed by methanol (500 mL). Finally, sodium cyanoborohydride (0.84 g, 1.33 × 10-2 mol, 1.1 eq.) was added followed by methanol (500 mL). The resulting mixture was heated to reflux (about 65°C).
After 1 hour at reflux, the reaction mixture attained homogeneity. After 25 hours ac reflux, the heat source was removed and the clear reaction mixture was measured with a pH meter (6.97 at 58.0°C). 1 N NaOH (22.8 mL) was added
dropwise to adjust the pH to 9.0 (at 54.7°C). The flask was equipped with a distillation head and the mixture was concentrated under partial vacuum to a weight of 322.3 grams while maintaining the pot temperature between 40-45°C.
The distillation head was replaced with an addition funnel containing 500 mL of isopropanol (IPA). The IPA was added dropwise to the room temperature solution over 1 hour. After approximately 1/3 of the IPA was added, a granular precipitate formed. The remaining IPA was added at a faster rate after precipitation had commenced. The flask was weighed and found to hold 714.4 grams of the IPA/methanol slurry.
The flask was re-equipped with a still-head and
distilled under partial vacuum to remove the remaining methanol. The resulting slurry (377.8 g) was allowed to chill in the freezer overnight. The crude product was filtered through a polypropylene pad and rinsed twice with 25 mL of cold IPA. After pulling dry on the funnel for 5 minutes, the material was placed in the vacuum oven to dry at 40°C. A light pink solid (22.87 g (theory = 22.43 g) ) was recovered. HPLC analysis versus a standard indicated 68.0% weight percent of Compound 229 (4- [4-chlorophenyl] benzyl-A82846B] in the crude solid, which translated into a
corrected crude yield of 69.3%.
The products of the reaction were analyzed by reverse-phase HPLC utilizing a Zorbax SB-C18 column with ultraviolet light (UV; 230 nm) detection. A 20 minute gradient solvent system consisting of 95% aqueous buffer/5% CH3CN at time=0 minutes to 40% aqueous buffer/60% CH3CN at time=20 minutes was used, where the aqueous buffer was TEAP (5 ml CH3CN, 3 ml phosphoric acid in 1000 ml water).
………………….
Oritavancin (also termed N-(4-(4-chlorophenyl)benzyl)A82846B and LY333328) has the following Formula III:

References
- Targanta Revives Oritavancin: Next Weapon Against cSSSI? BioWorld Today, November 26, 2007
- “Biotechs pick up slack in antibiotics development”. 17 May 2011.
- http://www.farm.ucl.ac.be/Full-texts-FARM/Domenech-2009-1.pdf “Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization” 2009
- Scheinfeld, N (2007). “A comparison of available and investigational antibiotics for complicated skin infections and treatment-resistant Staphylococcus aureus and enterococcus“.J Drugs Dermatol. 6 (4): 97–103. PMID 17373167.
- 2007 ICAAC Posters: E-1612 “In Vitro Activity Profile of Oritavancin against a Broad Spectrum of Aerobic and Anaerobic Bacterial Pathogens”/E -1613 “In Vitro Activity Profile of Oritavancin (ORI) Against Organisms Demonstrating Key Resistance Profiles to Other Antimicrobial Agents”/E-1614 “In vitro Time Kill Studies of Oritavancin against Drug-resistant Isolates ofStaphylococcus aureus and Enterococci”/E-1615 “Anti-Enterococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1616 “Anti-Streptococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1617 “In Vitro Activity Profile of Oritavancin (ORI) Against Resistant Staphylococcal Populations From a Recent Surveillance Initiative”/E-1620 “Pharmacokinetic Concentrations of Oritavancin Kill Stationary-Phase and Biofilm Staphylococcus aureus In Vitro.” / Targanta Press Release September 19, 2007
- ICAAC 2007 Posters: “In Vitro Susceptibility of Genotypically Distinct Clostridium difficileStrains to Oritavancin” and “Activity of Metronidazole, Vancomycin and Oritavancin Against Epidemic Clostridium difficile Spores” / Targanta Press Release September 19, 2007
- ASM 2007 Poster: “Efficacy of Oritavancin in a Murine Model of Bacillus anthracis Spore Inhalation Anthrax” / Targanta Press Release May 24, 2007
- Belley; McKay, GA; Arhin, FF; Sarmiento, I; Beaulieu, S; Fadhil, I; Parr Jr, TR; Moeck, G (2010).“Oritavancin Disrupts Membrane Integrity of Staphylococcus aureus and Vancomycin-Resistant Enterococci To Effect Rapid Bacterial Killing”. Antimicrobial agents and chemotherapy 54(12): 5369–71. doi:10.1128/AAC.00760-10. PMC 2981232. PMID 20876372.
- Zhanel et al. (2012). “Oritavancin: Mechanism of Action”. Clin Infect Dis.doi:10.1093/cid/cir920.
- ICAAC 2003 Late-breaker poster: “Phase III Trial Comparing 3-7 days of Oritavancin vs. 10-14 days of Vancomycin/Cephalexin in the Treatment of Patients with Complicated Skin and Skin Structure Infections (cSSSI)” / InterMune Press Release September 15, 2003
- ClinicalTrials.gov NCT00514527
- Comparison of the Efficacy and Safety of Oritavancin Front-Loaded Dosing Regimens to Daily Dosing: An Analysis of the SIMPLIFI Trial. May 2011. doi:10.1128/AAC.00029-11.
- “Drugs.com, Targanta Submits Oritavancin New Drug Application”. Retrieved 2008-02-12.
- “FDA News, Targanta to Get FDA Decision by December”. Retrieved 2008-04-10.
- http://www.fiercebiotech.com/press-releases/fda-issues-complete-response-letter-oritavancin Dec 2008.
- “Pharmaceutical Business Review, EMEA accepts Targanta’s oritavancin MAA for review”. Retrieved 2008-06-26.
- http://www.nelm.nhs.uk/en/NeLM-Area/News/2009—August/24/European-application-for-investigational-antibiotic-oritavancin-withdrawn-/
- http://onlinelibrary.wiley.com/doi/10.1111/j.1574-6976.2003.tb00628.x/pdf
- http://www.pjps.pk/wp-content/uploads/pdfs/26/5/Paper-30.pdf
- Antimicrobial Agents and Chemotherapy, 2003 , vol. 47, 5 p. 1700 – 1706
- Antimicrobial Agents and Chemotherapy, 1999 , vol. 43, 1 p. 115 – 120
- Antimicrobial Agents and Chemotherapy, 1997 , vol. 41, 10 p. 2165 – 2172
- Tetrahedron, 2004 , vol. 60, 47 p. 10611 – 10618………… NMRhttp://www.sciencedirect.com/science/article/pii/S0040402004015108
Cooper, R.D.G.; Snyder, N.J.; Zweifel, M.J.; et al.; Reductive alkylation of glycopeptide antibiotics: Synthesis and antibacterial activity. J Antibiot 1996, 49, 6, 575-81.
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0667353; EP 1016670; EP 1031576 .
| EP0435503A1 * | Dec 11, 1990 | Jul 3, 1991 | Eli Lilly And Company | Improvements in or relating to gylcopeptide derivatives |
| US4639433 * | Aug 14, 1985 | Jan 27, 1987 | Eli Lilly And Company | Glycopeptide derivatives |
| US4698327 * | Apr 18, 1986 | Oct 6, 1987 | Eli Lilly And Company | Novel glycopeptide derivatives |
| US20040106590 * | Aug 29, 2003 | Jun 3, 2004 | Barry Eisenstein | Methods and reagents for treating infections of clostridium difficile and diseases associated therewith |
| US20050197333 | Dec 22, 2004 | Sep 8, 2005 | Van Duzer John H. | Rifamycin analogs and uses thereof |
| US20070014849 | Sep 20, 2006 | Jan 18, 2007 | Daniela Jabes | Use of ramoplanin to treat diseases associated with the use of antibiotics |
| US20030176327 * | Oct 18, 2002 | Sep 18, 2003 | Cassell Gail Houston | Antibiotics for treating biohazardous bacterial agents |
| US20040147441 | Aug 25, 2003 | Jul 29, 2004 | Leach Timothy S. | Methods and reagents for preventing bacteremias |
| WO1999010006A1 | Aug 18, 1998 | Mar 4, 1999 | Lilly Co Eli | Therapy for staphylococcus aureus |
| WO2000066144A2 | Apr 19, 2000 | Nov 9, 2000 | Lilly Co Eli | Monthly doses of glycopeptide antibiotics for treatment of streptococcus pneumoniae infections |
| WO2008097364A2 | Sep 24, 2007 | Aug 14, 2008 | Targanta Therapeutics Corp | Use of oritavancin for prevention and treatment of anthrax |
| WO1998052592A1 * | May 5, 1998 | Nov 26, 1998 | Lilly Co Eli | Urea and thiourea derivatives of glycopeptides |
| WO2002036612A1 * | Nov 2, 2001 | May 10, 2002 | Univ Cambridge Tech | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane-associating elements |
| WO2007138999A1 | May 25, 2007 | Dec 6, 2007 | Shionogi & Co | Glycopeptide antibiotic derivative |
| WO2009081958A1 | Dec 25, 2008 | Jul 2, 2009 | Shionogi & Co | Glycosylated glycopeptide antibiotic derivative |
| EP2314599A1 | Nov 24, 2005 | Apr 27, 2011 | National University Corporation Nagoya University | Glycopeptide antibiotic monomer derivatives |
| US5919756 * | May 1, 1997 | Jul 6, 1999 | Eli Lilly And Company | Amides |
| US5919771 * | Apr 30, 1998 | Jul 6, 1999 | Eli Lilly And Company | Urea and thiourea derivatives of glycopeptides |
| US7078380 | Nov 2, 2001 | Jul 18, 2006 | Cambridge University Technical Services Limited | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane associating elements |
| US8481696 | Dec 25, 2008 | Jul 9, 2013 | Shionogi & Co., Ltd. | Glycosylated glycopeptide antibiotic derivatives |


Cinnamon cuts blood glucose levels in diabetes patients
Consumption of cinnamon is associated with favorable reductions in plasma glucose and lipid levels, according to research published in the September/October issue of the Annals of Family Medicine.
Robert W. Allen, Pharm.D., of the Western University of Health Sciences in Pomona, Calif., and colleagues used data from 10 randomized, controlled trials involving 543 patients with type 2 diabetes to conduct an update of a previous systematic review and meta-analysis examining the effect of cinnamon consumption on glucose and lipid levels.
The researchers found that cinnamon, in daily doses of 120 mg/d to 6 g/d for four to 18 weeks, was associated with a significant reduction in levels of fasting plasma glucose (?24.59 mg/dL), but no significant effect on glycosylated hemoglobin. Cinnamon intake also was linked to significant changes in lipid levels, including decreases in levels of total cholesterol (?15.60 mg/dL), low-density lipoprotein cholesterol (LDL-C) (?9.42 mg/dL), and triglycerides…
View original post 89 more words
Sonidegib/Erismodegib..Novartis Cancer Drug LDE225 Meets Primary Endpoint in Phase 2


Sonidegib/Erismodegib
CODE DESIGNATION ..LDE225, NVP-LDE-225
Treatment of medulloblastoma PHASE3 2014 FDA FILING
Treatment of advanced basal cell carcinoma PHASE3 2014 FDA FILING
Treatment of SOLID TUMORS..PHASE1 2017 FDA FILING
READMalignant Solid Tumors of Childhood
THERAPEUTIC CLAIM Oncology, Antineoplastics & Adjunctive Therapies
CHEMICAL NAMES
1. [1,1′-Biphenyl]-3-carboxamide, N-[6-[(2R,6S)-2,6-dimethyl-4-morpholinyl]-3-pyridinyl]-2-
methyl-4′-(trifluoromethoxy)-, rel-
2. N-{6-[(2R,6S)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl}-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide
N-[6-[(2S,6R)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl]-2-methyl-3-[4-(trifluoromethoxy)phenyl]benzamide
N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide
MOLECULAR FORMULA C26H26F3N3O3
MOLECULAR WEIGHT 485.5
SPONSOR Novartis Pharma AG
CAS REGISTRY NUMBER 956697-53-3 free form
NOTE… DIPHOSPHATE SALT IS THE DRUG WITH CAS 1218778-77-8
sonidegib – European Medicines Agency READ THIS..
Summary EudraCT Number: 2012-004022-21 Sponsor’s Protocol … READ THIS
About the Study
The Phase II, randomized, double-blind BOLT (Basal cell carcinoma Outcomes in LDE225 Trial) study was designed to assess the safety and efficacy of two oral dose levels of LDE225 (200 mg and 800 mg) in patients with locally advanced or metastatic basal cell carcinoma[4], which are subtypes of advanced basal cell carcinoma.
The primary endpoint was the proportion of patients achieving an objective response rate, defined as a confirmed complete response and partial response as their best overall response per modified RECIST criteria, within six months of starting treatment with LDE225. Key secondary endpoints of the study included assessing the duration of tumor responseand the rate of complete response. Other secondary endpoints included progression-free survival, time to tumor response and overall surviva
Sonidegib (INN) or Erismodegib (USAN), also known as LDE225 is a Hedgehog signalling pathway inhibitor (via smoothened antagonism) being developed as an anticancer agent by Novartis.[1][2] It has been investigated as a potential treatment for:
- Pancreatic cancer[3][4][5][6]
- Breast cancer[7][8]
- Basal cell carcinoma of the skin[9][10][11]
- Small cell lung cancer[12]
- Medulloblastoma[13][14]
- Advanced solid tumours (including ovarian, breast, pancreatic, stomach, oesophageal cancers and glioblastoma multiforme)[15][16][17]
- Acute leukaemia[18]
- Chronic myeloid leukaemia[19]
- Myelofibrosis and Essential thrombocythaemia[20]
NVP-LDE-225, a product candidate developed by Novartis, is in phase III clinical trials for the treatment of medulloblastoma and basal cell carcinoma. Phase II trials are in progress for the treatment of adult patients with relapsed or refractory or untreated elderly patients with acute leukemia.
Early clinical trials are ongoing for the oral treatment of advanced solid tumors, for the treatment of myelofibrosis in combination with ruxolitinib and for the treatment of small cell lung cancer. A phase II clinical trial for the treatment of basal cell carcinomas in Gorlin’s syndrome patients with a cream formulation of NVP-LDE-225 was discontinued in 2011 since the formulation did not demonstrate tumor clearance rate sufficient to support further development.
Dana-Farber Cancer Institute and the Massachusetts General Hospital are conducting phase I clinical trials for the treatment of locally advanced or metastatic pancreatic cancer in combination with chemotherapy. In 2009, orphan drug designation was assigned in the E.U. for the treatment of Gorlin syndrome.
It has demonstrated significant efficacy against melanoma in vitro and in vivo.[21] It also demonstrated efficacy in a mouse model of pancreatic cancer.[22]
NVP-LDE225 Diphosphate salt (Erismodegib, Sonidegib)

- Synonym:Erismodegib, Sonidegib
- CAS Number:1218778-77-8
- Mol. Formula:C26H26F3N3O3 ∙ 2H3PO4
- MW:681.5
- nmr.http://www.chemietek.com/Files/Line2/Chemietek,%20NVP-LDE225%20[02],%20NMR.pdf
- hplc–http://www.chemietek.com/Files/Line3/Chemietek,%20NVP-LDE225%20[02],%20HPLC.pdf
Brief Description:
About LDE225
LDE225 (sonidegib) is an oral, investigational, selective smoothened inhibitor being studied in a variety of cancers. Smoothened (SMO) is a molecule that regulates the hedgehog (Hh) signaling pathway, which plays a critical role in stem cell maintenance and tissue repair. LDE225 is currently in clinical development for a variety of diseases including myelofibrosis, leukemia and solid tumors.
Given that LDE225 is an investigational compound, the safety and efficacy profile has not yet been fully established. Access to this investigational compound is available only through carefully controlled and monitored clinical trials. These trials are designed to better understand the potential benefits and risks of the compound. Given the uncertainty of clinical trials, there is no guarantee that LDE225 will ever be commercially available anywhere in the world.
Possibility (LDE225) is effective in medulloblastoma relapsed or refractory hedgehog pathway inhibitor sonidegib has been revealed. That the anti-tumor effect was observed in some patients and tolerability in 1/2 test phase.
4th Quadrennial Meeting of the World Federation of Neuro-Oncology in conjunction with the 18th Annual Meeting of the Society for Neuro-Oncology, which was held in San Francisco November 21 to 24 in (WFNO-SNO2013), rice Dana-Farber It was announced by Mark Kieran Mr. Children’s Hospital Cancer Center.
The research group, announced the final results of the Phase 1 trial that target advanced solid cancer in children of sonidegib. 1 dose increased multi-test phase, was initiated from 372mg/m2 once-daily dosing to target children under the age of 18 more than 12 months. (233mg/m2 group 11 people, 16 people 372mg/m2 group, 11 people group 425mg/m2, 680mg/m2 group 21 women) who participated 59 people, including medulloblastoma 38 patients. 12 median age was (2-17).
Creatine phosphokinase elevation of grade 4 only were seen at 372mg/m2 as dose-limiting toxicity only, and became two recommended dose phase and 680mg/m2. Nausea muscle pain creatine kinase rise malaise (22.0%) (15.3%) (15.3%), (13.6%), vomiting side effects were many, was (13.6%). Hypersensitivity vomiting creatine kinase increased (3.4%) (1.7%) (1.7%), rhabdomyolysis side effects of grade 3/4 was (1.7%). (One group 372mg/m2, 425mg/m2 group one) complete response was obtained in two people, a strong correlation was found between the activation of the hedgehog pathway and effect.
Phase III clinical trials that target medulloblastoma the activated hedgehog pathway currently are underway.
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and diagnostic tools, over-the-counter and animal health products. Novartis is the only global company with leading positions in these areas. In 2013, the Group achieved net sales of USD 57.9 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 136,000 full-time-equivalent associates and operate in more than 140 countries around the world.

The following Examples serve to illustrate the invention without limiting the scope thereof, it is understood that the invention is not limited to the embodiments set forth herein, but embraces ali such forms thereof as come within the scope of the disclosure,

Step 1:
To a solution of 2-chloro-5-nitro-pyridine 1 (5.58 g, 35.2 mmoL) and c/s-2,6- dimethylmorpholine (4.05 g, 35.2 mmoL) in anhydrous DMF (30 mi.) was added K2CO3 (9.71 g, 70.4 mnrtoL). The mixture was heated at 50ºC overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 3 as a yellow solid, after purification by silica gel chromatography, obtained pure product (7.80 g, 93.2%). LC-MS m/z: 238.2 (M+ 1).
Step 2:
The above material 3 (7.3Og. 30.8 mmoL) was hydrogenated in the presence of 10% Pd-C (1.0 g) in MeOH (120 ml) under hydrogen overnight. The suspension was filtered through celite and the filtrate was concentrated to give the crude product 4 (5.92 g) as a dark brown oil which was used directly in the next step without further purification. LC-MS m/z. 208.2 (M+1).
Step 3:
To a solution of 3-bromo-2-methyl benzoic acid (2.71 g, 12.6 mmoL), 6-((2S,6R)-2,6- dimethylmorpholino)pyridin-3-arnine 4 (2.61 g, 12.6 mmoL), and HATU (4.80 g, 12.6 mmoL) in anhydrous DMF (30 mL) was added diisopropylethylamine (6.58 mL, 37.8 mmoL) dropwise. The resulting mixture was stirred overnight at room temperature. The reaction mixture was diluted with water (50 mL), and then extracted with EtOAc (3×120 mL). The organic layer was dried and concentrated to give the crude product. This crude product was then purified by flash column chromatography using 30% EtOAc in hexane as eiuent to give 5 as a white solid (4.23 g, 83.0%). LC-MS m/z: 404.1 (M+1).
Step 4:
A mixture of 4-(trif!uoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo- N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-ylJ-4-methyl-benzamide 5 (250 mg, 0.62mmol), Pd(PPh3)4 (36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL) in a sealed tube was heated at 130ºC overnight. The reaction mixture was diluted with EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and concentrated to give the crude product which was then purified by preparative mass triggered HPLC (C18 column, etuted with CH3CN-H2O containing 0.05% TFA) to give N-(6-((2S,6R)-2,6-dimethyfmorpholino)pyridin-3-yl)-2-rnethyl- 4′-(trifluoromethoxy)biphenyi-3-carboxamide (183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
The resultant crystalline product (Form A) was converted to the amorphous form by dissolving in 3% w/w aqueous ethanol, and the resultant solution spray dried at about 150ºC.
Form B was prepared by heating the amorphous form in an oven at 110ºC for 2 hours. In a further embodiment, the invention relates to a process step or steps, or an intermediate as described herein.

…………………………..
SYNTHESIS

| LC-MS m/z 486.2 (M + 1) |

Step 1: To a solution of 3-iodo-4-methyl-benzoic acid (10.0 g, 38.2 mmol) in methanol (70 ml) is added concentrated sulfuric acid (0.5 ml). The reaction mixture is heated at 70° C. for 48 hours, cooled to room ambient temperature and then concentrated. After that, ethyl acetate (100 ml) and aqueous NaHCO3 (saturated, 100 ml) solution are added to the residue. The organic layer is separated and washed again with aqueous NaHCO3 (saturated, 100 ml) solution. The organic layer is separated, dried over anhydrous Na2SO4 and concentrated to yield 3-iodo-4-methyl-benzoic acid methyl ester 1. It is used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.87 (d, 1H, J=8.4 Hz), 7.48 (d, 1H, J=8.4 Hz), 3.85 (s, 3H), 3.35 (s, 3H); LC-MS m/z: 277.0 (M+1).
Step 2: To a round-bottom flask containing 3-iodo-4-methyl-benzoic acid methyl ester (1.38 g, 5.00 mmol), 4-cyanophenylboronic acid (1.10 g, 7.48 mmol), palladium acetate (168 mg, 0.748 mmol), 2-(dicyclohexylphosphino)biphenyl (0.526 g, 1.50 mmol) and potassium fluoride (0.870 g, 15.0 mmol) is added anhydrous 1,4-dioxane (15 ml). The flask is purged with argon and sealed. The mixture is stirred at 130° C. for 18 hours, cooled to ambient temperature and then water (20 ml) and ethyl acetate (20 ml) are added. Solid is removed under vacuum filtration. The filtrate is extracted with EtOAc (20 ml×2). The organic layers are combined, washed with aqueous HCl (5%, 20 ml) and saturated NaHCO3 (20 ml). It is dried over MgSO4, and concentrated. The residue is purified by silica gel column chromatography (EtOAc/Hexane, gradient) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2; LC-MS m/z: 252.1 (M+1).
Step 3: To a solution of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2 (2.56 g, 10.3 mmol) in 1,4-dioxane-H2O (1:1 mixture, 20 ml) is added NaOH (1.22 g, 30.2 mmol)). The reaction is stirred at ambient temperature for 24 hours. To this mixture is added aqueous HCl (1 N, 36 ml) and it is then extracted with ethyl acetate (40 ml×3). The organic layers are combined, dried over anhydrous Na2SO4. The solver is removed. The solid obtained is washed with small amount of acetonitrile and air dried to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3: 1H NMR (DMSO-d6) δ 7.94 (d, 2H, J=8.0 Hz), 7.84 (dd, 1H, J1=8.4 Hz, J2=1.2 Hz), 7.75 (d, 1H, J=1.2 Hz), 7.61 (d, 2H, J=8.0 Hz), 7.48 (d, 1H, J=8.4 Hz), 2.29 (s, 3 H); LC-MS m/z 238.1 (M+1).
Step 4: To a suspension of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3 (40 mg, 0.17 mmol) in anhydrous methylene chloride (5 ml) is added 2 drops of DMF. Then oxalyl chloride (32 mg, 22 μl, 0.25 mmol) is added. The mixture is stirred at ambient temperature until it turns clear. After that, it is concentrated, re-dissolved in anhydrous methylene chloride (3 ml), and added to a solution of 4-(morpholine-4-sulfonyl)-phenylamine (61 mg, 0.25 mmol) and triethylamine (34 mg, 47 μl, 0.33 mmol) in methylene chloride (2 ml). The mixture is stirred for 2 hours, concentrated and the residue is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [4-(morpholine-4-sulfonyl)-phenyl]-amide: 1H NMR (DMSO-d6) δ 10.64 (s, 1H), 8.07 (d, 2H, J=8.8 Hz), 7.97 (d, 2H, J=8.4 Hz), 7.95 (d, 1H, J=8.8 Hz), 7.89 (s, 1H), 7.43 (d, 2H, J=8.4 Hz), 7.67 (d, 2H, J=8.8 Hz), 7.53 (d, 2H, J=8.8 Hz), 3.63 (m, 4H), 2.84 (m, 4H) 2.32 (s, 3H); LC-MS m/z: 462.1 (M+1).
Example 2 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide

Step 1: To a solution of 2-chloro-5-nitro-pyridine 4 (2.38 g, 15 mmol.) and cis-2,6-dimethylmorpholine (1.73 g, 15 mmol.) is added K2CO3 (4.14 g, 30 mmol.). The mixture was heated at 50° C. overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 6 as a yellow solid. The crude product is used directly in next step without further purification. LC-MS m/z: 238.1 (M+1).
Step 2: The above crude material 6 is hydrogenated in the presence of Pd—C (0.2 g) in MeOH (100 mL) under hydrogen over 10 h. The suspension is filtered through celite and the filtrate is concentrated to give the crude product 7 as a dark brown oil which is used directly in the next step without further purification. LC-MS m/z: 208.1 (M+1).
Step 3: To a solution of 3-bromo-4-methyl benzoic acid (108 mg, 0.5 mmol.), 6-(2,6-Dimethyl-morpholin-4-yl)-pyridin-3-ylamine 7 (104 mg, 0.5 mmol.), amd HATU (190 mg, 0.5 mmol.) in dry DMF (5 mL) is added triethylamine (139 uL, 1.0 mmol.) dropwise. The resulting mixture is stirred at room temperature for 2 h. After concentration, the residue is partitioned between EtOAc and water. The organic layer is dried and concentrated to give the crude product. The final compound is purified by flash column chromatography using 50% EtOAc in hexane as eluent to give 8 as a white solid. LC-MS m/z: 404.1 (M+1).
Step 4: A mixture of 4-cyanophenyl boronic acid (18 mg, 0.12 mmol), 3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide 8 (40 mg, 0.1 mmol), Pd(PPh3)4 (11 mg, 0.01 mmol), and Na2CO3 (42 mg, 0.4 mmol) in a combined solvent system of toluene (0.2 mL) and water (0.2 mL) and ethanol (0.05 mL) is heated at 140° C. under microwave irradiation for 30 min. The reaction mixture is diluted with EtOAc and water. The aqueous layer is extracted with EtOAc. The combined organic layer is washed with brine and concentrated to give the crude product which is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide. LC-MS m/z: 427.2 (M+1).

4-(Trifluoromethoxy)phenylboronic acid
- CAS Number 139301-27-2
- Linear Formula CF3OC6H4B(OH)2
- Molecular Weight 205.93
CONDENSE WITH …3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamideACS Medicinal Chemistry Letters, 2010 , vol. 1, 3 p. 130 – 134

A mixture of 4-(trifluoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo-N-[6-(2,6-
dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide E (250 mg, 0.62mmol), Pd(PPh3)4
(36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL)
in a sealed tube was heated at 1300C overnight. The reaction mixture was diluted with EtOAc
and water. The aqueous layer was extracted with EtOAc. The combined organic layer was
washed with brine and concentrated to give the crude product which was then purified by
preparative mass triggered HPLC (C18 column, eluted with CH3CN-H2O containing 0.05% TFA)
to give N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide (5m, 183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
HRMS (m/z): [M+H]+
calcd for C26H27N3O3F3 486.2005; found 486.1986,
1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.15 (s, 1H), 8.43 (d, 1H), 7.94 (dd, 1H), 7.52-7.43
(m, 5H), 7.38 (m, 1H), 7.33 (m, 1H), 6.86 (d, 1H), 4.06 (d, 2H), 3.62 (m, 2H), 2,34 (m, 2H), 2.22
(s, 3H), 1.16 (d, 6H).
http://pubs.acs.org/doi/suppl/10.1021/ml1000307/suppl_file/ml1000307_si_001.pdf
Reference
- “LDE225 – PubChem”. PubChem. National Institutes of Health. Retrieved 16 February 2014.
- Pan, S; Wu, X; Jiang, J; Gao, W; Wan, Y; Cheng, D; Han, D; Liu, J; Englund, NP; Wang, Y; Peukert, S; Miller-Moslin, K; Yuan, J; Guo, R; Matsumoto, M; Vattay, A; Jiang, Y; Tsao, J; Sun, F; Pferdekamper, AC; Dodd, S; Tuntland, T; Maniara, W; Kelleher, JF; Yao, Y; Warmuth, M; Williams, J; Dorsch, M (10 June 2010). “Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist”. ACS Medicinal Chemistry Letters 1 (3): 130–134. doi:10.1021/ml1000307.
- “A Biomarker Study to Identify Predictive Signatures of Response to LDE225 (Hedgehog Inhibitor) In Patients With Resectable Pancreatic Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Gemcitabine + Nab-paclitaxel With LDE-225 (Hedgehog Inhibitor) as Neoadjuvant Therapy for Pancreatic Adenocarcinoma”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose-escalation, and Safety Study of LDE225 and Gemcitabine in Locally Advanced or Metastatic Pancreatic Cancer Patients”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Pilot Study of a Hedgehog Pathway Inhibitor (LDE-225) in Surgically Resectable Pancreas Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study With LDE225 in Combination With Docetaxel in Triple Negative (TN) Advanced Breast Cancer (ABC) Patients (EDALINE)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014.
- “LDE225 in Treating Patients With Stage II-III Estrogen Receptor- and HER2-Negative Breast Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “To Evaluate the Safety, Local Tolerability, PK and PD of LDE225 on Sporadic Superficial and Nodular Skin Basal Cell Carcinomas(sBCC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Trial to Evaluate the Safety, Local Tolerability, Pharmacokinetics and Pharmacodynamics of LDE225 on Skin Basal Cell Carcinomas in Gorlin Syndrome Patients”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Combination of the Hedgehog Inhibitor, LDE225, With Etoposide and Cisplatin in the First-Line Treatment of Patients With Extensive Stage Small Cell Lung Cancer (ES-SCLC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase III Study of Oral LDE225 Versus (vs) Temozolomide (TMZ) in Patients With Hedge-Hog (Hh)-Pathway Activated Relapsed Medulloblastoma (MB)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase I Dose Finding and Safety Study of Oral LDE225 in Children and a Phase II Portion to Assess Preliminary Efficacy in Recurrent or Refractory MB”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Phase Ib, Dose Escalation Study of Oral LDE225 in Combination With BKM120 in Patients With Advanced Solid Tumors”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose Finding and Safety of Oral LDE225 in Patients With Advanced Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “LDE225 and Paclitaxel in Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study of Efficacy and Safety of LDE225 in Adult Patients With Relapsed/Refractory Acute Leukemia”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Nilotinib and LDE225 in the Treatment of Chronic or Accelerated Phase Myeloid Leukemia in Patients Who Developed Resistance to Prior Therapy”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase Ib/II Dose-finding Study to Assess the Safety and Efficacy of LDE225 + INC424 in Patients With MF”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- Jalili, A; Mertz, KD; Romanov, J; Wagner, C; Kalthoff, F; Stuetz, A; Pathria, G; Gschaider, M; Stingl, G; Wagner, SN (30 July 2013). “NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo.” (PDF). PloS one 8 (7): e69064. doi:10.1371/journal.pone.0069064. PMC 3728309.PMID 23935925.
- Fendrich, V; Wiese, D; Waldmann, J; Lauth, M; Heverhagen, AE; Rehm, J; Bartsch, DK (November 2011). “Hedgehog inhibition with the orally bioavailable Smo antagonist LDE225 represses tumor growth and prolongs survival in a transgenic mouse model of islet cell neoplasms.”. Annals of Surgery 254 (5): 818–23.doi:10.1097/SLA.0b013e318236bc0f. PMID 22042473.
- ChemMedChem, 2013 , vol. 8, 8 p. 1261 – 1265
- ACS Med. Chem. Lett., 2010, 1 (3), pp 130–134.
- MORE REF
sonidegib
Skin Cancer Foundation. “Skin Cancer Facts.” Available at:http://www.skincancer.org/skin-cancer-information/skin-cancer-facts . Accessed on February 14, 2014.
Rubin AI, Chen EH, Ratner D (2005). Current Concepts: Basal-Cell Carcinoma. N Engl J Med; 353:2262-9.
ClinicalTrials.gov. “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)” Available at:http://clinicaltrials.gov/ct2/show/NCT01327053?term=%22LDE225%22+and+%22BOLT%22&rank=1. Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Complete Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45652 . Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Partial Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45819 . Accessed on February 14, 2014.
Wong C S M, Strange R C, Lear J T (2003). Basal cell carcinoma. BMJ; 327:794-798.
Copcu E, Aktas A. Simultaneous two organ metastases of the giant basal cell carcinoma of the skin. Int Semin Surg Oncol. 2005;2:1-6. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC544837/ . Accessed on February 14, 2014.
Skin Cancer Foundation. “Basal Cell Carcinoma Treatment Options.” Available athttp://www.skincancer.org/skin-cancer-information/basal-cell-carcinoma/bcc-treatment-options . Accessed on February 14, 2014.
Stuetz A, et al. LDE225, a specific smoothened inhibitor, for the topical treatment of nevoid basal cell carcinoma syndrome (Gorlin’s syndrome). Melanoma Research. 2010; 20:e40. Available at:http://journals.lww.com/melanomaresearch/Fulltext/2010/06001/FC24_LDE225,_a_specific_smoothened_inhibitor,_for.87.aspx#FC24_LDE225%2C_a_specific_smoothened_inhibitor%2C_for.87.aspx?s=2&_suid=139234380607909969110518506816.
Novartis.com. “The Pipeline of Novartis Oncology: LDE225.” Available at:http://www.novartisoncology.com/research-innovation/pipeline.jsp #. Accessed on February 14, 2014.
Children’s Medical Research Center, Children’s Memorial Hospital/Northwestern University Feinberg School of Medicine. “The Sonic hedgehog/patched/gli signal transduction pathway.” Available at http://www.childrensmrc.org/iannaccone/gli/ . Accessed on February 14, 2014.
Gupta S, Takebe N, LoRusso P. Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol. 2010 July; 2(4): 237-250. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3126020/ . Accessed on February 14, 2014.
| WO2004078163A2 | Feb 26, 2004 | Sep 16, 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007113120A1 | Mar 22, 2007 | Oct 11, 2007 | Frank Hoffmann | Stamping apparatus with feed device |
| WO2007131201A2 * | May 4, 2007 | Nov 15, 2007 | Irm Llc | Compounds and compositions as hedgehog pathway modulators |
| WO2008154259A1 | Jun 4, 2008 | Dec 18, 2008 | Irm Llc | Biphenylcarboxamide derivatives as hedgehog pathway modulators |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ



FDA Breakthrough Therapy Designation: Fourth Drug Receives FDA Approval
.
The FDA approves on February 12, the fourth drug to have the coveted Breakthrough Therapy Designation (BTD). The approval is for orphan drug Imbruvica (Ibrutinib) as a single agent for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who have received at least one prior therapy. Imbruvica, a once-daily, oral kinase inhibitor, is developed and commercialized by Pharmacyclics and Janssen Biotech. Imbruvica is the :
• 1st FDA BTD drug to receive approval in 2014
• 1st FDA BTD drug to receive approval for a 2nd indication – Mantle Cell Lymphoma (MCL) on 11.13.13
• 2nd FDA BTD drug to receive approval for the same indication, Chronic Lymphocytic Leukemia (CLL) – Genentech’s Gazyva (Obinutuzumab) receives approval for CLL on 11.01.13
• 3rd FDA BTD drug to receive approval for an oncology indication
• 4th FDA BTD drug to receive approval.
The media, investors, patients, regulatory agencies, and the pharmaceutical industry…
View original post 194 more words
MOXIFLOXACIN, Bay-12-8039
 MOXIFLOXACIN Bay-12-8039 US FDA:link CAS 354812-41-2 186826-86-8 HYDROCHLORIDE
MOXIFLOXACIN Bay-12-8039 US FDA:link CAS 354812-41-2 186826-86-8 HYDROCHLORIDE
| 1-cyclopropyl-7-[(1S,6S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo- quinoline-3-carboxylic acid |

Moxifloxacin is a fourth-generation synthetic fluoroquinolone antibacterial agent developed by Bayer AG (initially called BAY 12-8039). It is marketed worldwide (as the hydrochloride) under the brand names Avelox, Avalox, and Avelon for oral treatment. In most countries, the drug is also available in parenteral form for intravenous infusion. Moxifloxacin is also sold in an ophthalmic solution (eye drops) under the brand names Vigamox, Moxezafor the treatment of conjunctivitis (pink eye). A United States patent application was submitted on 30 June 1989, for Avelox (moxifloxacin hydrochloride).[1] In 1999 Avelox was approved by theU.S. Food and Drug Administration (FDA) for use in the United States.[2] In the United States, moxifloxacin is licensed for the treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, community acquired pneumonia, complicated and uncomplicated skin and skin structure infections, and complicated intra-abdominal infections.[3] In the European Union, it is licensed for acute bacterial exacerbations of chronic bronchitis, non-severe community-acquired pneumonia, and acute bacterial sinusitis. Based on its investigation into reports of rare but severe cases of liver toxicity and skin reactions, the European Medicines Agency recommended in 2008 that the use of the oral (but not the IV) form of moxifloxacin be restricted to infections in which other antibacterial agents cannot be used or have failed.[4] In the US, the marketing approval does not contain these restrictions, though the label contains prominent warnings against skin reactions. 
- Acute Exacerbations of Chronic Bronchitis (AECB)
- Acute Bacterial Sinusitis (ABS)
- Community Acquired Pneumonia (CAP)
Additional indications were approved by the FDA as follows:
- April 2001: Uncomplicated Skin and Skin Structure Infections (uSSSI)[8]
- May 2004: Community Acquired Pneumonia caused by multi-drug resistant Streptococcus pneumoniae.[9]
- June 2005: Complicated Skin and Skin Structure Infections (cSSSI)[10]
- November 2005: Complicated Intra-Abdominal Infections (cIAI).[11]
The European Union requires that moxifloxacin only be prescribed when other antibiotics that have been initially recommended for treatment cannot be used or have failed.[12][13] At the current time,[when?] there are no approved uses within the pediatric population for Oral and I.V. moxifloxacin. A significant number of drugs found within this class, including moxifloxacin, are not licensed by the FDA for use in children due to the risk of permanent injury to the musculoskeletal system.[14][15][16] In ophthalmology, moxifloxacin is approved for the treatment of conjunctival infections caused by susceptible bacteria.[17] Note: Moxifloxacin may be licensed for other uses, or restricted, by the various regulatory agencies worldwide Marketing authorisations for the tablet and injectable forms of Moxifloxacin are held by Bayer, while Alcon (now a subsidiary of Novartis) produces ophthalmic solutions for treating conjunctivitis under the brand names of Moxeza, Vigamox, and Moxivig. Avelox generated sales of USD320 million in the first 9 months of 2013. Moxifloxacin is available in three distinct administration forms. Formulated as a salt, Moxifloxacin hydrochloride is sold as an oral 400 mg film-coated tablet and as an injectable solution for infusion by Bayer; although in the US it is distributed by Merck Sharp and Dohme under license from Bayer. Alcon has formulated Moxifloxacin hydrochloride as a 0.5% ophthalmic solution under license from Bayer. Moxifloxacin was first discovered in 1988 and received the first market authorisation eleven years later in 1999 in the US. Moxifloxacin hydrochloride is a synthetic broad-spectrum antibacterial agent. The active moiety, moxifloxacin has been shown to be clinically active against most strains of microorganisms such as aerobic gram-positive microorganisms including staphylococcus aureus, streptococcus pneumonia (penicillin-susceptible strains) and streptococcus pyogenes, aerobic gram-negative microorganisms including haemophilus influenza hemophilus parainfluenzae, klebisiella pneumonia. Moxifloxacin is commercially available under the brand name of AVELOX® marketed by Bayer pharms. VIGAMOX® (moxifloxacin hydrochloride ophthalmic solution) 0.5% is a sterile solution for topical ophthalmic use. Moxifloxacin hydrochloride is an 8-methoxy fluoroquinolone anti-infective, with a diazabicyclononyl ring at the C7 position.
 |
C21H24FN304•HC1 Mol Wt 437.9 Chemical Name: l-Cyclopropyl-6-fluoro-l,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolol[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid, monohydrochloride. Moxifloxacin hydrochloride is a slightly yellow to yellow crystalline powder. Each mL of VIGAMOX® solution contains 5.45 mg moxifloxacin hydrochloride, equivalent to 5 mg moxifloxacin base. Contains: Active: Moxifloxacin 0.5% (5 mg/mL); Inactives: Boric acid, sodium chloride, and purified water. May also contain hydrochloric acid/sodium hydroxide to adjust pH to approximately 6.8. VIGAMOX® solution is an isotonic solution with an osmolality of approximately 290 mOsm/kg. syn…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html Market Considerations Amongst the US approvals, Dr. Reddy’s, Teva, Torrent, and Aurobindo have received tentative approvals for the 400 mg oral tablet formulation. Akorn, Teva and Apotex have received tentative approvals for a Moxifloxacin hydrochloride ophthalmic solution. No 180 day period of exclusivity has been awarded since all patents were found to be valid. In the UK, Teva, Rivopharm, and Double-E Pharma have received marketing authorisations for the 400 mg Moxifloxacin tablets, while Noridem has received a market authorisation for the equivalent 400mg/250ml solution for infusion. A similar trend of generic competition, for tablets and infusions, following molecule patent expiry is expected throughout Europe. Currently no generic market authorisations for ophthalmic formulations have been granted in major European countries. However, Sandoz and Hexal have gained market authorisations in some European markets for the ophthalmic dosage form following Novartis’ acquisition of Alcon. In Canada the only generic manufacturer holding a marketing authorisation is Sandoz, however, this was granted as a New Drug Submission rather than as an ANDS. Following patent expiries from mid-2014, the European and North American markets are likely to have significant competition if the numbers of companies filing litigation, ANDS, ANDAs and the like in the northern hemisphere is anything to go by.  MOXIFLOXACIN
MOXIFLOXACIN
History
Moxifloxacin was first patented (United States patent) in 1991 by Bayer A.G., and again in 1997.[47] Avelox was subsequently approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999 to treat specific bacterial infections.[2] Ranking 140th within the top 200 prescribed drugs in the United States for 2007[48] moxifloxacin, in the same manner asciprofloxacin, has proven to be a blockbuster drug for Bayer A. G., generating billions of dollars in additional revenue. In 2007 alone, Avelox generated sales of $697.3 million dollars worldwide.[26] Moxifloxacin is also manufactured by Alcon as Vigamox. syn………http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html
Patent
A United States patent application was made on 30 June 1989, for Avelox (moxifloxacin hydrochloride),(Bayer A.G. being the assignee), which was subsequently approved on 5 February 1991. This patent was scheduled to expire on 30 June 2009. However, this patent was extended for an additional two and one half years on 16 September 2004, and as such is not expected to expire until 2012.[49] Moxifloxacin was subsequently (ten years later) approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999. There have been at least four additional United States patents filed regarding moxifloxacin hydrochloride since the 1989 United States application,[47][50] as well as patents outside of the USA.
Additional regulatory history
6/12/2002 Changes made to minimize the impact of warnings concerning adverse reactions.[51] 26 June 2003 New Zealand Pharmacovigilance warns of moxifloxacin induced respiratory insufficiency.[52] 10/6/2003 Changes made to minimize the impact of post marketing reports as well as the risk of tendon injuries.[53] 29 December 2008 Addition of numerous adverse reactions associated with the use of moxifloxacin.[54] 27 April 2009 Issuance of a Medication Guide and revisions to include new safety information including the addition of the Black Box Warning to the Medication Guide. The FDA had determined that Moxifloxacin poses a serious and significant public health concern, requiring the distribution of a Medication Guide.[55] 24 June 2009 Updating of the carton and container labels to include a statement to let dispensers know that a Medication Guide must be dispensed with the product.(emphasis added)[56] Patent related
|
As indicated by the Key Patent Indicator (Fig. 2), patents in the families with priority DE3824072A, 15/07/1988 (‘072), and DE4200414A, 10/01/1992, (‘414) provide protection for the Moxifloxacin molecule and are considered to be the main constraint to generic entry. As patents in the ‘414 family have expired or were never granted, the only remaining constraint to generic entry is the ‘072 family. The term of the Australian patent of this family have been extended to 19 June 2014 while the Canadian member will enjoy the longer term of 17 years from grant, expiring in November 2015. Supplementary protection certificates (SPCs) have been granted in France, Germany, Spain and the UK, and will expire in June 2014. Given that there are less than 2 years until these SPCs expire, and that as yet no applications for paediatric extension of the SPCs have been published in Europe, they are unlikely to be extended by 6 months on the basis of the approved Paediatric Investigation Plans. The US member, 4,990,517 (‘517), protecting the general structure of the Moxifloxacin molecule, expired in June 2012, after enjoying 6 month paediatric extension on top of a 901 day s156 extension. However, the absence of generics on the US market is due to Bayer securing a divisional patent, 5,607,942 (‘942). This patent claims the Moxifloxacin molecule specifically and is due to expire in September 2014, after being awarded a 6 month paediatric extension. Members of the ‘072 family from both Canada and the US have been the subject of litigation after generic manufacturers identified these patents in paragraph IV filings, and the equivalent in Canada, as early as 2006. After filing an infringement suit in the US against Teva in relation to US ‘517, US ‘942, Bayer enjoyed a satisfying validification of their patents when Teva agreed that it would be infringing two of the patents, while the third was decided by the court to be equally valid. In Canada, Novopharm, Cobalt, Apotex, Mylan and Apotex have also all tested the litigation waters relating to the equivalent patent with no success noted so far. A third patent family that promises to be a constraint for generic ophthalmic formulations is Alcon’s 1998 patent, US10250498P (Fig. 2), which identifies an ophthalmic formulation of Moxifloxacin and its use in the treatment and prevention of eye infections. Patents in this family are set to expire in August 2019. US members 6,716,830 (‘830) and 7,671,070 (‘070) have been awarded a 6 month paediatric extension, extending their expiration until March 2020. The validity of US ‘830 was upheld following Teva filing paragraph IV certifications to manufacture generic Vigamox. Teva has since appealed this ruling. In addition, Alcon has filed patent infringement suits against Watson, Lupin and Apotex in relation to US ‘070 after they submitted Abbreviated New Drug Applications (ANDAs)with paragraph IV filings in preparation for commercialisation of a Moxeza/Vigamox generic equivalent. There has been no outcome from these suits to date. In addition, applications for Orders of Prohibition against Cobalt, Apotex and Teva have also been noted for the equivalent Canadian patent following the filing of Abbreviated New Drug Submissions (ANDS) by these companies. The equivalent European patent 1,117,401 was revoked following opposition by Teva filed in the European patent office. Its divisional patents, 1,384,478 (granted) and 2,301,541 (accepted) have restricted claims to the use of Moxifloxacin in the topical treatment of ophthalmic infections caused by P. aeruginosa and H. influenza, respectively. This may provide a prepared generic competitor an opportunity to launch their Moxifloxacin ophthalmic equivalent in Europe soon after the expiry of patents protecting the molecule, subject to legal review of the remaining claims of the patent. Alcon have secured additional protection for their Moxeza ophthalmic formulation by way of patents in the family with priority US5987708P (09/06/2008). Patent claims specify ratios of Moxifloxacin to inactive ingredients and additional inactive ingredients and therefore generic competitors are likely to circumvent the patent by reformulation. Lupin has filed paragraph IV certifications to US8450311, which is currently subject of a patent infringement suit. Families with priorities DE19546249A (12/12/1995), DE19751948A (24/11/1997), DE19855758A (10/11/1998) and US36433499A (30/07/1999) are not considered to be a constraint to generic entry because the protected technologies are likely to be circumvented. Generic equivalents In 2007, the U.S. District Court for the District of Delaware held that two Bayer patents on Avelox (moxifloxacin hydrochloride) are valid and enforceable, and infringed by Dr. Reddy’s ANDA for a generic version of Avelox.[70][71] The district court sided with Bayer, citing the Federal Circuit’s prior decision in Takeda v. Alphapharm[72] as “affirming the district court’s finding that defendant failed to prove a prima facie case of obviousness where the prior art disclosed a broad selection of compounds, any one of which could have been selected as a lead compound for further investigation, and defendant did not prove that the prior art would have led to the selection of the particular compound singled out by defendant.” According to Bayer’s press release[70] announcing the court’s decision, it was noted that Teva had also challenged the validity of the same Bayer patents at issue in the Dr. Reddy’s case. Within Bayer’s first quarter 2008 stockholder’s newsletter[73] Bayer stated that they had reached an agreement with Teva Pharmaceuticals USA, Inc., the adverse party, to settle their patent litigation with regard to the two Bayer patents. Under the settlement terms agreed upon, Teva would obtain a license to sell its generic moxifloxacin tablet product in the U.S. shortly before the second of the two Bayer patents expires in March 2014.
- Economic impact: adverse reactions:
The advocacy group Public Citizen has lobbied for increasing safety warnings and for the removal of some fluoroquinolone drugs from clinical practice.[74][75][76][77][78][79][80][81]
|
3-5-1997
|
7-(1-pyrrolidinyl)-3-quinolone- and – naphthyridone-carboxylic acid derivatives as antibacterial agents and feed additives
|
|
6-9-2000
|
INHIBITORS OF MULTIDRUG TRANSPORTERS INHIBITORS OF MULTIDRUG TRANSPORTERS
|
|
|
5-19-2000
|
PHARMACEUTICAL MOXIFLOXACIN PREPARATION
|
|
|
5-12-2000
|
AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
1-20-2000
|
COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS
|
|
|
12-17-1999
|
NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES
|
|
|
4-2-1999
|
MEDICAMENT FORMULATION WITH A CONTROLLED RELEASE OF AN ACTIVE AGENT
|
|
|
8-28-1998
|
COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS
|
|
11-10-2006
|
Amorphous moxifloxacin hydrochloride
|
|
|
10-4-2006
|
Method for producing 8-methoxy-quinolinecarboxylic acids
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
7-13-2005
|
Aqueous pharmaceutical composition containing moxifloxacin or salts thereof
|
|
|
5-25-2005
|
Method for producing 8-methoxy-quinolinecarboxylic acids
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
5-21-2003
|
Method for the enantiomer separation of cis-8-benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane
|
|
|
11-31-2000
|
OPTICALLY ACTIVE QUINOLINE CARBOXYLIC ACID DERIVATIVES HAVING 7-PYRROLIDINE SUBSTITUTES CAUSING OPTICAL ACTIVITY AND A PROCESS FOR PREPARING THEREOF
|
|
|
11-17-2000
|
ANTIBACTERIAL OPTICALLY PURE BENZOQUINOLIZINE CARBOXYLIC ACIDS, PROCESSES, COMPOSITIONS AND METHODS OF TREATMENT (S)-BENZOQUINOLIZINE CARBOXYLIC ACIDS AND THEIR USE AS ANTIBACTERIAL AGENTS
|
|
|
8-32-2000
|
COMPOSITIONS AND METHODS FOR IMPROVED DELIVERY OF HYDROPHOBIC THERAPEUTIC AGENTS
|
|
6-18-2010
|
MULTI-ARM POLYMER PRODRUGS
|
|
|
4-9-2010
|
COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID
|
|
|
7-22-2009
|
Tri-, tetra-substituted-3-aminopyrrolidine derivative
|
|
|
7-3-2009
|
NOVEL HYDRATE FORM
|
|
|
3-20-2009
|
Multi-Arm Polymer Prodrugs
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
|
|
11-21-2008
|
Phosphonated Fluoroquinolones, Antibacterial Analogs Thereof, and Methods for the Prevention and Treatment of Bone and Joint Infections
|
|
|
8-15-2008
|
Multi-arm polymer prodrugs
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
7-4-2012
|
TRI-, TETRA-SUBSTITUTED-3-AMINOPYRROLIDINE DERIVATIVE
|
|
|
6-13-2012
|
Process for the Synthesis of Moxifloxacin Hydrochloride
|
|
|
12-2-2011
|
COMPACTED MOXIFLOXACIN
|
|
|
10-12-2011
|
Treatment of bacterial diseases of the respiratory organs
|
|
|
9-16-2011
|
Novel Hydrate Form
|
|
|
9-2-2011
|
NOVEL POLYMORPH OF MOXIFLOXACIN HYDROCHLORIDE
|
|
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-10-2011
|
SYNTHESIS OF (4aS,7aS)-OCTAHYDRO-1H-PYRROLO[3,4-b]PYRIDINE
|
|
|
7-30-2010
|
MULTI-ARM POLYMER PRODRUGS
|
|
|
6-30-2010
|
Multi-arm polymer prodrugs
|
DESCRIPTION
-
Moxifloxacin is a therapeutic agent that shows a broad spectrum antibacterial action. Moxifloxacin is the international non-proprietary name (INN) for 1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid.
-
The structure of moxifloxacin corresponds to formula (I):

-
Racemic moxifloxacin was firstly described in EP-A-350733 and, particularly, moxifloxacin having a (S,S)-configuration is described inEP-A-550903 .
-
In experimental example 19 of EP-A-550903 and example Z19 of EP-A-591808 , a method for preparing and isolating moxifloxacin base is described. The same method is described in patent document EP-A-592868 ). These published patent applications neither describe nor suggest the possible existence of a crystalline form of moxifloxacin base. In WO-A-2008059521 it is disclosed that by performing the mentioned examples an acetonitrile solvated form of moxifloxacin with low purity is obtained, and so the obtained solvate form can not be used as such in pharmaceutical formulations. In the above mentioned examples, the obtained moxifloxacin base crude is purified and isolated by chromatography using methylene chloride/methanol/17% aqueous ammonia as the solvent system. The purification process disclosed in said documents has been reproduced by the authors of the present invention, but only an amorphous form of moxifloxacin was obtained. This process for the purification and isolation of moxifloxacin base is complex and difficult to perform on industrial scale due to the need for purifying the product by column chromatography.
-
WO-A-9926940 discloses a process for the preparation of moxifloxacin from a difluoro precursor comprising the step of adjusting the pH to 6.8- 7.0. However, the reproduction of this example shows that at this pH moxifloxacin hydrochloride or a mixture of moxifloxacin hydrochloride and moxifloxacin base is obtained, since the X-ray diffractogram of the isolated compound corresponds with the hydrochloride moxifloxacin. This fact has also been confirmed by the reproduction of the subsequent crystallization described in the international patent application of the previous compound in ethanol/water. The behaviour of this solid was very different regarding its solubility in respect what was described in the international patent application.
-
WO-A-2004091619 and WO-A-2007010555 disclose the preparation of moxifloxacin base as a solid. Nevertheless, they do not describe nor suggest the preparation of a crystalline form of moxifloxacin base. The authors of the present invention have proved that the X-ray diffractogram of the product obtained by reproducing the reference example disclosed in WO-A-2004091619 , based on a pH adjustment to 7.0-7.2, corresponds to the X-ray diffractogram of the monohydrate of moxifloxacin hydrochloride disclosed in US 5849752 . Similarly, by reproducing the reference example of WO-A-2007010555 , also for obtaining moxifloxacin base but based on a pH adjustment to 5.0-6.0, it is neither expected to obtain moxifloxacin base due to the adjustment of the solution within a range of acidic pH values.
-
[0008]WO-A-2008059521 discloses a process for the preparation of a crystalline form of moxifloxacin base, designated as Form I, by recrystallization in a ketosolvent, such as acetone. This form is characterized by its X-ray diffraction pattern corresponding to a hemihydrate. The best yield obtained is a 78.2% starting from moxifloxacin hydrochloride in example 18. This crystalline form has a tendency to occlude solvent molecules within the crystalline network in amounts very superiors to the allowed ones by for Guidelines Residual Solvents (CPMP/ICH/283/95) and can be difficult to impossible to remove by drying, what forces to carry out laborious treatments, either physical, or chemical to reach allowed solvent levels. The presence of any non-aqueous solvents in amounts over the allowed ones would not make this crystalline form suitable for the preparation of pharmaceutical formulations.
-
The existence of polymorphs is unpredictable and there is no a priori established procedure to prepare an unknown polymorph. The difference in the physical properties of different morphological forms results from the orientation and intermolecular interactions of adjacent molecules are complexes in the bulk solid. Furthermore, the different solid forms of a pharmaceutically active ingredient can have different characteristics, and offer certain advantages in methods of manufacture and also in pharmacology. Thus, the discovery of new solid forms can contribute to clear improvements in the efficiency of methods of production and/or improvements in the characteristics of the pharmaceutical formulations of the active ingredients, since some forms are more adequate for one type of formulation, and other forms for other different formulations.

Moxifloxacin Hydrochloride namely (4aS-Cis) -l-cyclopropyl-7- (2, 8- diazabicyclo [4.3.0] non-8-yl) -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylic acidhydrochloride has the formula

Moxifloxacin Hydrochloride Moxifloxacin is a fluoroquinolone broad spectrum antibacterial particularly against Gram-positive bacteria significantly better than those of Sparfloxacin and Ciprofloxacin that was disclosed in EP No 350,733 and EP No 550,903. Moxifloxacin has activity against Gram- negative and Gram-positive organisms, including Streptococcus pneumonia, Staphylococcus aureus, Pseudomonas aeruginosa, particularly against the respiratory disease-causing pathogens like Mycoplasma pneumonia, Mycobacterium tuberculosis, Chlamydia pneumoniae and the activity shown to be unaffected by B-lactamases . US Patent No 5,157,117 discloses (l-cyclopropyl-6, 7-difluoro-8-methoxy- 4-oxo-l, 4-dihydro-3-quinoline carboxylic acid-O3, 04)bis (acyloxy-O) borate and process for its preparation by reacting ethyl-1- cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylate with Boric acid and acetic anhydride in presence of zinc chloride and its conversion to Gatifloxacin hydrochloride. WO 2005/012285 discloses the process for the preparation of moxifloxacin hydrochloride using a novel intermediate namely (4aS-Cis)-(1-cyclopropyl-7-(2,8-diazabicyclo[4,3,0]non-8-yl)-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic acid-O3,O4)bis(acycloxy-O)borate. Hydrates of Moxifloxacin hydrochloride known are the anhydrous and monohydrate. US Patent No. 5,849,752 discloses the monohydrate of Moxifloxacin hydrochloride and its preparation by treating the anhydrous crystalline form with ethanol/ water mixtures. The prior art disclosed in European Patent No’s EP 350,733, EP 550,903 and EP 657,448 discloses the preparation of Moxifloxacin hydrochloride involving the condensation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid or its esters with (S,S) 2,8-Diaza bicyclo [4.3.0] nonane in presence of a base and its conversion to hydrochloride at higher temperatures leading to the desired Moxifloxacin along with its positional isomer namely (4aS-Cis)-l- cyclopropyl-6- (2, 8-diazabicyclo [4.3.0] non-8-yl) -7-fluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid as a major impurity. As the impurity and the Moxifloxacin are positional isomers they are difficult to separate. Purification of Moxifloxacin to remove this isomer results in lower yields thereby increasing the product cost. Similarly methods described in the prior art involves the preparation of Moxifloxacin and then its conversion to its hydrochloride thereby incorporating an additional step in the manufacturing process also leading to lowering of yields. Moxifloxacin and its pharmacologically acceptable salts are disclosed in European patents EP 350733, EP 550903 and EP 657,448. The disclosed process for the preparation of moxifloxacin hydrochloride comprises of condensing l-cyclopropyl-6,7- difluoro-8-methoxy-4-oxo-l,4-dihydro-3-quinoline carboxylic acid or its esters with (S,S)2,8-diazobicyclo[4.3.0]nonane, in presence of a base at high temperature followed by conversion into hydrochloride salt . This process not only produces desired moxifloxacin hydrochloride but also its positional isomer namely l-cyclopropyl-7-fluoro- 1,4- dihydro -8- methoxy -6- (4aS,7aS)- octahydro- 6H -pyrrolo [3,4-b] pyridine-6-yl] -4- oxo-quinolinecarboxylic acid as a major impurity which is difficult to separate. The purification of moxifloaxcin to remove this isomer results in lower yields thereby increasing the product cost. The International publication WO 2005/012285 discloses an improved process for the preparation of moxifloxacin hydrochloride incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin hydrochloride from the ethyl 1 -cyclopropyl-6,7-difluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3-quinolme carboxylate through a novel intermediate (4aS-cis)-l-cyclopropyl-7-(2,8-diazabicyclo[4.3.0]non-8- yl)-6-fluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3 -quinolinecarboxylicacid-03,04)bis(acyloxy -0)-borate. US patent application 6897315 discloses a process for the preparation of 8-methoxy-3-quinoline carboxylic acid especially moxifloxacin incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin from 8-halo moxifloxacin derivative using methanol and potassium tertiary butoxide. US patent 5639886 discloses one-pot process for the preparation of 3-quinoline carboxylic acid derivatives including moxifloxacin. WO 2004 091619 claims anhydrous crystalline form-Ill of moxifloxacin hydrochloride and WO 2004/039804 claims amorphous form of moxifloxacin hydrochloride. US Pat.No.5, 849,752 discloses specific crystalline forms of anhydrous moxifloxacin mono hydrochloride and monohydrated moxifloxacin mono hydrochloride. Anhydrous moxifloxacin mono hydrochloride disclosed in US Pat. No.5, 849,752 has been designated as “Form-I” and the hydrated form as “Form-II” in US Pat. No.7,230,006. It also discloses a novel crystalline Form-Ill of anhydrous moxifloxacin mono hydrochloride. US patent US 5,480,879 discloses the melting range of moxifloxacin in example part as 203-208°C and does not speaks about polymorphism of moxifloxacin. Experiment executed as per the procedure given in example Zl 9 of US 5,480,879 and resulted in acetonitrile solvated form of moxifloxacin with low purity and the obtained solvated form can not used for formulations U.S. Pat. No. 5,849, 752 (“the ‘752 patent”), incorporated by reference, described two crystalline forms of moxifloxacin hydrochloride namely, anhydrous moxifloxacin hydrochloride and monohydrated moxifloxacin hydrochloride. For convenience, the anhydrous crystalline form described in the 752 patent is designated as “Form I”, and the hydrated form as “Form II”. According to U.S. Pat. No. ‘752’, moxifloxacin hydrochloride monohydrate Form II was obtained by stirring a suspension of the anhydrous moxifloxacin hydrochloride in aqueous media until hydration. Moxifloxacin hydrochloride monohydrate of ‘752’ was also prepared by crystallizing moxifloxacin hydrochloride from a media having a water content which is stoichiometrically sufficient but limited to 10%. WO patent application publication No. 04/091619 disclosed anhydrous Form III of moxifloxacin hydrochloride. WO patent application publication No. 04/039804 disclosed amorphous form of moxifloxacin hydrochloride. WO 2005/054240 disclosed two novel crystalline forms which were designated as Form A and Form B of moxifloxacin hydrochloride. WO patent application publication No. 07/010555 disclosed two crystalline forms which were Form X and Form Y of moxifloxacin hydrochloride. According to WO Publication No. 2007/010555, Form Y was obtained by crystallization of moxifloxacin hydrochloride from the mixture of methanol and water in the ratio of about 8:1 by volume. WO patent application publication No. 07/148137 disclosed hydrate form of moxifloxacin hydrochloride. According to WO Publication No. 2007/148137, moxifloxacin hydrochloride monohydrate was obtained by crystallization moxifloxacin hydrochloride by humidification of moxifloxacin hydrochloride at 50-90% relative humidity at 25-60° C. for 8 to 24 hours. WO patent application publication No. 08/028959 disclosed crystalline form of moxifloxacin hydrochloride. According to WO Publication No. 2008/028959, moxifloxacin hydrochloride was obtained by dissolving moxifloxacin hydrochloride in a mixture of methanol and water and adding acetone and recovering moxifloxacin hydrochloride crystalline form. WO patent application publication No. 08/059521 disclosed process for the preparation of anhydrous crystalline form I of moxifloxacin hydrochloride. WO patent application publication No. 08/095964 disclosed crystalline form of moxifloxacin base.  …………………… SYNTHESIS Drugs Fut 1997,22(2),109 36th Intersci Conf Antimicrob Agents Chemother (Sept 15-18, New Orleans) 1996,Abst. F1.
…………………… SYNTHESIS Drugs Fut 1997,22(2),109 36th Intersci Conf Antimicrob Agents Chemother (Sept 15-18, New Orleans) 1996,Abst. F1.  The anhydrization of pyridine-2,3-dicarboxylic acid (I) with acetic anhydride gives the corresponding anhydride (II), which by treatment with benzylamine (III) is converted into the benzylimide (IV). The hydrogenation of (IV) with H2 over Pd/C yields 8-benzyl-2,8-diazabicyclo[4.3.0]nonane-7,9-dione (V), which is further hydrogenated with LiAlH4, affording (?-cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VI) (1). The optical resolution of (VI) by separation of the cis-(R,R)-isomer as crystalline L-(+)-tartrate and further purification of the cis-(S,S)-isomer (VII) as the D-(-)-tartrate affords enantiomerically pure (S,S)-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VII). The debenzylation of (VII) by hydrogenolysis with H2 over Pd/C gives (S,S)-2,8-diazabicyclo[4.3.0]nonane (VIII), which is condensed with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) in basic medium and finally salified with HCl. The 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) has been obtained as follows: The reaction of 2,4,5-trifluoro-3-methoxybenzoyl chloride (X) with malonic acid monoethyl ester monopotassium salt (XI) by means of triethylamine gives 2-(2,4,5-trifluoro-3-methoxybenzoyl)acetic acid ethyl ester (XII), which is condensed with triethyl orthoformate yielding the corresponding ethoxymethylene derivative (XIII). The reaction of (XIII) with cyclopropylamine affords the cyclopropylaminomethylene derivative (XIV), which is finally cyclized to (IX) by means of NaF in DMF. ……………………….. J Label Compd Radiopharm 2000,43(8),795
The anhydrization of pyridine-2,3-dicarboxylic acid (I) with acetic anhydride gives the corresponding anhydride (II), which by treatment with benzylamine (III) is converted into the benzylimide (IV). The hydrogenation of (IV) with H2 over Pd/C yields 8-benzyl-2,8-diazabicyclo[4.3.0]nonane-7,9-dione (V), which is further hydrogenated with LiAlH4, affording (?-cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VI) (1). The optical resolution of (VI) by separation of the cis-(R,R)-isomer as crystalline L-(+)-tartrate and further purification of the cis-(S,S)-isomer (VII) as the D-(-)-tartrate affords enantiomerically pure (S,S)-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VII). The debenzylation of (VII) by hydrogenolysis with H2 over Pd/C gives (S,S)-2,8-diazabicyclo[4.3.0]nonane (VIII), which is condensed with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) in basic medium and finally salified with HCl. The 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) has been obtained as follows: The reaction of 2,4,5-trifluoro-3-methoxybenzoyl chloride (X) with malonic acid monoethyl ester monopotassium salt (XI) by means of triethylamine gives 2-(2,4,5-trifluoro-3-methoxybenzoyl)acetic acid ethyl ester (XII), which is condensed with triethyl orthoformate yielding the corresponding ethoxymethylene derivative (XIII). The reaction of (XIII) with cyclopropylamine affords the cyclopropylaminomethylene derivative (XIV), which is finally cyclized to (IX) by means of NaF in DMF. ……………………….. J Label Compd Radiopharm 2000,43(8),795  The condensation of 2,4,5-trifluoro-3-methoxybenzoyl chloride (I) with 14C-labeled diethyl malonate (II) by means of MgCl2 and TEA gives the benzoylmalonate (III), which is monodecarboxylated with TsOH in refluxing water, yielding the benzoylacetate (IV). The reaction of (IV) with triethyl orthoformate and Ac2O at 140 C affords the benzoylacrylate (V), which is treated with cyclopropylamine (VI) in cyclohexane to provide ethyl 3-(cyclopropylamino)-2-(2,4,5-trifluoro-3-methoxybenzoyl)acrylate (VII). The cyclization of (VII) by means of K2CO3 in hot N-methylpyrrolidone gives the quinolone carboxylate (VIII), which is hydrolyzed with NaOH in hot methanol, affording the carboxylic acid (IX). Finally, this compound is condensed with (S,S)-2,8-diazabicyclo[4.3.0]octane (X) by means of 1,4-diazabicyclo[2.2.2]octane (DABCO) in refluxing acetonitrile. …………………….
The condensation of 2,4,5-trifluoro-3-methoxybenzoyl chloride (I) with 14C-labeled diethyl malonate (II) by means of MgCl2 and TEA gives the benzoylmalonate (III), which is monodecarboxylated with TsOH in refluxing water, yielding the benzoylacetate (IV). The reaction of (IV) with triethyl orthoformate and Ac2O at 140 C affords the benzoylacrylate (V), which is treated with cyclopropylamine (VI) in cyclohexane to provide ethyl 3-(cyclopropylamino)-2-(2,4,5-trifluoro-3-methoxybenzoyl)acrylate (VII). The cyclization of (VII) by means of K2CO3 in hot N-methylpyrrolidone gives the quinolone carboxylate (VIII), which is hydrolyzed with NaOH in hot methanol, affording the carboxylic acid (IX). Finally, this compound is condensed with (S,S)-2,8-diazabicyclo[4.3.0]octane (X) by means of 1,4-diazabicyclo[2.2.2]octane (DABCO) in refluxing acetonitrile. ……………………. 
 …………………… EP1651630A1 The reaction scheme is given below: Stage-I Acetic anhydride
…………………… EP1651630A1 The reaction scheme is given below: Stage-I Acetic anhydride


Ethyl-l-cyclopropyl-6, 7- ( l-cyclopropyl-6, 7-difluoro-l, 4- difluoro-1, 4-dihydro-8- dihydro-8- methoxy-4-oxo-3- methoxy-4-oxo-3-quinoline quinoline carboxylic acid-03, 04) carboxylate . Bis ( acetate-O) -borate (Borate complex) Stage-II Triet yl amine Acetonitrile


l-cyclo propyl-6, 7-difluoro- [S, S] -2, 8-diazabicyclo- (1- cyclo propyl-6, fluoro-7 (2, 8- 1, 4-dihydro-8- methoxy-4-oxo [4 ,3.0]nonane Diazabicyclo-nonane) 1,4- -3-quinoline carboxylic acid- dihydro-8-methoxy-4-oxo-3 03,04)Bis ( acetate-O) -borate- quinoline carboxylic acid- (Borate complex) (03,04) bis (acetate-O) -borate itage-III

(1- cyclo propyl-6, fluoro- (2, 8- Moxifloxacin HCI pseudohydrate Diazabicyclo-nonane) 1,4- dihydro-8-methoxy-4-oxo-3 -quinolinecarboxylicacid- (O^O4) bis (acetate-O) -borate Stage-TV

Moxifloxacin HCI pseudohydrate Moxifloxacin HCI monohydrate EXAMPLE – I Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo- l,4-dihydro-3-quinoline carboxylic acid-O3,O*)bis (acyloxy-O)borate Acetic anhydride (175 g) is heated to 70°C and boric acid (30 g) is slowly added lot wise in a temperature range of 70°C to 90°C. The temperature is then raised, maintained under reflux for 1 hr followed by cooling to about 70°C. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylate (100 g) is added under stirring. The temperature is then raised and maintained for 1 hr in the range of 100°C to 105°C. The reaction mass is cooled to 0°C, chilled water (400 ml) is added slowly followed by cold water (600 ml) at temperature 0°C to 5°C and maintained for 2 hrs at 0°C to 5°C. The product which is a boron acetate complex is filtered, washed with water (500 ml) and dried at 55°C to 60°C under vacuum to constant weight. The dry wt is 130.0 g corresponding to yield of 95.2%. Stage-2: Preparation of (4aS-Cis) -l-Cyclopropyl-7- (2, 8-diazabicyclo [4.3.0]non-8-yl) -6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3-quinoline carboxylicacid-03,0*)bis (acyloxy-O)borate The boron acetate complex (130 g) prepared in stage 1 is suspended in acetonitrile (650 ml), and [S, S] -2, 8-diazabicyclo [4.3.0] nonane (47 g) and triethyl amine (72.9 g) are added. The temperature is raised to reflux and maintained for 1 hr. at reflux, followed by cooling to about 40°C. The solvent is removed under vacuum at temperature below 40°C, and n-hexane (200 ml) is added. After maintaining the reaction mass for 1 hr at room temperature the product is isolated by filtration followed by washing of the wet cake with n-hexane . The product is dried at about 45°C to about 50°C to constant weight. Dry wt of the novel intermediate is 117.0 g corresponding to yield of 71.5%. Elemental analysis: C: 56.42%, H: 5.62%, N: 7.76% and the calculated values for the intermediate, formula C25H29BFN308C: 56.6%, H: 5.47%, N: 7.92% IR Spectrum (KBr, cm-1) : 3415, 3332, 2936, 1718, 1630, 1573, 1526, 1445, 1273, 1042, 935, 860, 798, 682 ^ NMR (200 MHz, CDC13, ppm) : 9.00 (1H), 7.82 (1H), 4.12 (4H), 3.57 (3H), 3.43 (4H), 3.07 (2H) , 2.75 (2H), 2.4 (1H),’ 2.1 (6H), 1.84 (2H) , 1.6 (1H), 1.31 (2H) Mass Spectrum (MJ : 530.3 [M+H] , 470.2 [M+ – CH3COOH] , 428.2 [M+– (CH3CO)20, 100%], 402.2, 388.2 Stage -3: Preparation of Moxifloxacin Hydrochloride pseudohydrate The intermediate (117 g) prepared stage-2 is dissolved in ethanol (600 ml) by stirring for about 30 min. at room temperature and the insolubles if any are filtered off. pH of the filtrate is adjusted to about 0.5 by addition of hydrochloric acid at room temperature and maintained for 2 hrs. The reaction mass is cooled, and maintained for two hrs, at about 0°C to about 5°C. The product is filtered, washed with chilled ethanol (50 ml) and dried at about 50°C to about 55°C till constant weight. The dry weight of the Moxifloxacin hydrochloride pseudohydrate is 87.5g corresponding to yield of 91.0%. Water content of the product by KF is 0.64% w/w. X-ray diffraction pattern data are given in Table-1 EXAMPLE – II Stage- 2 : Preparation of Moxifloxacin pseudohydrate with out isolating (4aS-Cis) -l-Cyclopropyl-7- (2 , 8-diazabicyclo [4.3.0] on-8-yl) -6-fluoro- 8-methoxy-4-oxo-l,4-dihydro-3-quinolinecarboxylicacid-03,04)bis (acyloxy-O) borate The boron acetate complex (130 g) prepared in stage-1 of Example-1 is suspended in acetonitrile (650ml) and [S, S] -2, 8-Diazabicyclo [4.3.0]nonane (47 g) & triethyl amine (72.9 g) are added. Temperature of the reaction mass is raised to reflux, maintained for 1 hr. at reflux and cooled to room temperature. Methanol (600 ml) is added and maintained for 30 min at room temperature to obtain a clear solution. The solution is filtered to remove insolubles if any and pH of the filtrate is adjusted to about 0.5 with hydrochloric acid (57.5 g) . The reaction mass is maintained for 2 hrs at temperature in the range of about 20°C to about 25°C, cooled to 0°C followed by maintaining the reaction mass at about 0°C to about 5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at about 50°C to 55°C until constant weight. Dry wt of the Moxifloxacin hydrochloride pseudohydrate is 88g corresponding to yield of 68.7%. EXAMPLE – III : Preparation of Moxifloxacin Hydrochloride monohydrate Moxifloxacin hydrochloride (50 g) prepared as above is suspended in a mixture of ethanol (250 ml) and hydrochloric acid (25 ml) . Raised the temperature, maintained for two hrs at 40°C to 45°C followed by cooling to about 25°C. The product is filtered and dried under vacuum at 50-55°C until become constant weight. Dry wt of Moxifloxacin hydrochloride monohydrate is 46 g corresponding to yield of 90.5%. The IR spectral data and XRD pattern are identical with available Moxifloxacin hydrochloride monohydrate.


Formula-2a Formula-2b

Formula-3a Formula-3b Formula-3c Formula-3d

Formula-4b

Formula-4c

SCHEME-5:


![[1860-5397-9-265-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Due to the elaborate substitution pattern of the parent quinolone ring systems these compounds are usually prepared via a linear consecutive sequence. In the case of moxifloxacin, an intramolecular base catalysed nucleophilic aromatic substitution is used to prepare the bicyclic ring system of the highly substituted aromatic1.104 (Scheme 19). A SNAr reaction is then used to introduce the saturated piperidinopyrrolidine appendage 1.105to furnish the desired structure [57-60]. In order to obtain a high yield for the substitution reaction a one-pot procedure was developed, initial masking of the acid (1.104) is achieved by silylation with subsequent borane chelate formation. Addition of the amine nucleophile 1.105 under basic conditions then renders the desired product in high yield. The available patent literature however does not comment on regioselectivity issues of the SNAr reaction due to the presence of the second fluoride substituent in the substrate, although not necessarily as electronically favourable for displacement it is certainly more accessible.
![[1860-5397-9-265-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i19.png?scale=2.0&max-width=1024&background=FFFFFF)
The saturated (S,S)-2,8-diazabicyclo[4.3.0]nonane (1.105) used in the final step can be prepared by a double nucleophilic substitution between tosylamine and 2,3-bis-chloromethylpyridine (1.112) followed by catalytic reduction of the resulting bicycle using palladium on carbon in acetic acid (Scheme 20). As the corresponding sulfonamide 1.113 was found to be a crystalline solid a resolution using (D)-(+)-O,O-dibenzoyltartaric acid was reported to separate the enantiomers [Petersen, U.; Schenke, T.; Krebs, A.; Schenke, T.; Philipps, T.; Grohe, K.; Bremm, K.-D.; Endermann, R.; Metzger, K. G. New Quinoline and Naphthyridinonecarboxylic Acid Derivatives. Ger. Patent DE 4 208 792 A1, March 23, 1993.].
![[1860-5397-9-265-i20]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i20.png?scale=2.0&max-width=1024&background=FFFFFF)
| First way of enantioselective synthesis of moxifloxacin intermediate LI GuangXun, WU Lei, FU QingQuan, TANG Zhuo, ZHANG XiaoMei
A new method of enantioselective synthesis of (S,S)-2,8-diazobicyclo [4.3.0] nonane was found by using (R)-2-amino-2-phenyl-ethanol as chiral induction reagent. The entire synthetic process included 8 steps which were easy to operate with high yield. The purification method was only simple recrystallization or even used directly in the next step without further purification. The total yield was 29%.
|
|||
2013 Vol. 56 (3): 307-311 [Abstract] ( 22 ) [ PDF (518 KB) ] ( 92 ) [Supporting Information] DOI: 10.1007/s11426-012-4803-7
|


- Drugwatch. “Avelox Patent Family”. Retrieved 16 September 2012.
- “Details for NDA:021085”. DrugPatentWatch. Retrieved 17 July 2009.
- “www.accessdata.fda.gov”.
- “www.emea.europa.eu”.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021085s047,021277s041lbl.pdf
- “Avelox”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1999/21085ltr.pdf
- http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21-085S010_Avelox_Approv.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2004/21085se1-022,21277se1-017ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s026,021277s022ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s027,029,021277s024,025ltr.pdf.
- “Moxifloxacin: restricted use : MHRA”.
- European Medicines Agency (24 July 2008). “European Medicines Agency recommends restricting the use of oral moxifloxacin-containing medicines”. Retrieved 20 July 2009.
- “SYNOPSIS”. Retrieved 29 January 2009.
- Karande SC, Kshirsagar NA (February 1992). “Adverse drug reaction monitoring of ciprofloxacin in pediatric practice”. Indian Pediatr 29 (2): 181–8. PMID 1592498.
- Dolui SK, Das M, Hazra A (2007). “Ofloxacin-induced reversible arthropathy in a child”. Journal of Postgraduate Medicine 53 (2): 144–5. doi:10.4103/0022-3859.32220 .PMID 17495385.
- “Center for drug evaluation and research Application number 21-598”. Food and Drug Administration (FDA). 15 April 2005. Retrieved 21 July 2009.
- Jump up^ Babar, S. (October 2013). “SIADH Associated With Ciprofloxacin.”. The Annals of Pharmacotherapy (Sage Publiahing) 47 (10): 1359–1363.doi:10.1177/1060028013502457 . ISSN 1060-0280.PMID 24259701. Retrieved November 139,2013.
- Renata Albrecht (28 July 2004). “NDA 21-085/S-024, NDA 21-277/S-019”. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (31 May 2007). “NDA 21-085/S-036, NDA 21-277/S-030”. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (15 February 2008). “NDA 21-085/S-038, NDA 21-277/S-031”. Division of Special Pathogen and Transplant Products. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- DEPARTMENT OF HEALTH & HUMAN SERVICES (28 February 2008). “NDA 21-085/S-014, S-015, S-017”. Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Nea Zealand Government (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 30 January 2009.
- http://www.mhra.gov.uk/Safetyinformation/DrugSafetyUpdate/CON087781, retrieved 2012-11-10 Missing or empty
|title=(help) - http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title=(help) - “EU agency recommends restricting moxifloxacin use”. Reuters. 24 July 2008. Retrieved 21 July 2009.
- Renata Albrecht (3 October 2008). “NDA 21-085/S-040, NDA 21-277/S-034”. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Updated Labeling for Antibiotic Avelox (Moxifloxacin) Regarding Risk of Severe Liver Injury / Information Update: 2010-42 For immediate release 22 March 2010http://www.hc-sc.gc.ca/ahc-asc/media/advisories-avis/_2010/2010_42-eng.php
- Saint F, Gueguen G, Biserte J, Fontaine C, Mazeman E (September 2000). “[Rupture of the patellar ligament one month after treatment with fluoroquinolone]”. Rev Chir Orthop Reparatrice Appar Mot (in French) 86 (5): 495–7.PMID 10970974.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title=(help) - Bayer (December 2008). “AVELOX (moxifloxacin hydrochloride) Tablets AVELOX I.V. (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). p. 19. Retrieved 2 November 2010. Unknown parameter
|line=ignored (help) - “Moxifloxacin”. University of Maryland Medical Center. 2009. Retrieved 22 July 2009.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf
- Shin HC, Kim JC, Chung MK (September 2003). “Fetal and maternal tissue distribution of the new fluoroquinolone DW-116 in pregnant rats”. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 136 (1): 95–102.doi:10.1016/j.cca.2003.08.004 . PMID 14522602.
- Dan M, Weidekamm E, Sagiv R, Portmann R, Zakut H (February 1993). “Penetration of fleroxacin into breast milk and pharmacokinetics in lactating women” (PDF).Antimicrob. Agents Chemother. 37 (2): 293–6.doi:10.1128/AAC.37.2.293 . PMC 187655.PMID 8452360.
- Noel GJ, Bradley JS, Kauffman RE (October 2007). “Comparative safety profile of levofloxacin in 2523 children with a focus on four specific musculoskeletal disorders”.Pediatr. Infect. Dis. J. 26 (10): 879–91.doi:10.1097/INF.0b013e3180cbd382 . PMID 17901792.
- Joette Meyer; Renata Albrecht (16 March 2004).“Division of Special Pathogen and Immunologic Drug Products Summary of Clinical Review of Studies Submitted in Response to a Pediatric Written Request”. Food and Drug Administration (FDA). Retrieved 22 July 2009.
- Elbe DH, Chang SW (February 2005). “Moxifloxacin-warfarin interaction: a series of five case reports”. Ann Pharmacother 39 (2): 361–4. doi:10.1345/aph.1E179 .PMID 15632222.
- Karen E. Brown. “Top Ten Dangerous Drug Interactions in Long-Term Care”. Medication Management Project. Retrieved 1 August 2009.
- Stass HH (29 June – 3 July 1997). “Bay 12-8039 (BA) does not interact with theophylline (TH)”. Presented at the 20th International Congress of Chemotherapy.
- Medscape: Medscape Access
- http://69.20.19.211/cder/warn/dec99/wl121599.pdf
- Renata Albrecht (16 May 2002). “NDA 21-085/S-012”.Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Drlica K, Zhao X (1 September 1997). “DNA gyrase, topoisomerase IV, and the 4-quinolones”. Microbiol Mol Biol Rev. 61 (3): 377–92. PMC 232616. PMID 9293187.
- “Drug card for Moxifloxacin (DB00218)”. Canada: DrugBank. 19 February 2009. Retrieved 3 August 2009.
- Alffenaar J. W. C., van Altena R., Bökkerink H. J (2009). “Pharmacokinetics of moxifloxacin in cerebrospinal fluid and plasma in patients with tuberculous meningitis”. Clinical Infectious Diseases 49 (7): 1080–2. doi:10.1086/605576 .PMID 19712035.
- “Inventors/Applicants”. patentlens.net. 3 October 2006. Retrieved 17 July 2009.
- Ed Lamb (1 May 2008). “Top 200 Prescription Drugs of 2007”. Pharmacy Times. Retrieved 21 July 2009.
- http://www.uspto.gov/web/offices/pac/dapp/opla/term/certs/4990517.pdf
- US 4990517 A (Feb 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+4990517#list US 5051509 A (Sep 1991) Nagano et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5051509#list US 5059597 A (Oct 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5059597#list US 5395944 A (Mar 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5395944#list US 5416096 A (May 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5416096#list
- Renata Albrecht (12 June 2002). “NDA 21-085/S-006, S-007 – NDA 21-277/S-002”. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- The 114Th Medicines Adverse Reactions Committee Meeting; Professor P. Ellis, Professor D.C.G. Skegg, Dr M. Rademaker, Dr F. McClure, Dr N. Rafter, Dr M. Tatley (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 12 August 2009.
- Renata Albrecht, (6 October 2003). “NDA 21-085/S-019 – NDA 21-277/S-011”. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Renata Albrecht (6 October 2003). “NDA 21-085/S-039 NDA 21-277/S-033”. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Bayer HealthCare Pharmaceuticals Inc. (August 2008).“MEDICATION GUIDE AVELOX (AV-eh-locks) (moxifloxacin hydrochloride) Tablets AVELOX I.V. (AV-eh-locks) (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Ozlem Belen (24 June 2009). “NDA 21-085/S-041 NDA 21-277/S-035”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Bailey RR, Natale R, Linton AL (October 1972). “Nalidixic acid arthralgia”. Can Med Assoc J 107 (7): 604 passim.PMC 1940945. PMID 4541768.
- Bailey RR, Kirk JA, Peddie BA (July 1983). “Norfloxacin-induced rheumatic disease”. N Z Med J 96 (736): 590.PMID 6223241.
- Szarfman A, Chen M, Blum MD (January 1995). “More on fluoroquinolone antibiotics and tendon rupture” (PDF). N Engl J Med 332 (3): 193.doi:10.1056/NEJM199501193320319 . PMID 7800023.
- “Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399)”. Public Citizen. 1 August 1996. Retrieved on 27 December 2008.
- “Reports of adverse events with fluoroquinolones”. FDA Medical Bulletin 26 (3). October 1996.[dead link] Retrieved on 27 December 2008.
- “Madigan, Public Citizen, petition FDA for “black box” warning regarding potential adverse effects of certain popular antibiotics” (Press release). Office of the Illinois Attorney General. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen Petitions the FDA to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781)”. Public Citizen. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen v. Food and Drug Administration (FDA) (Fluoroquinolone)”. Public Citizen. 3 January 2008. Retrieved 27 December 2008.
- Ravn, Karen (18 August 2008). “Behind the FDA’s ‘black box’ warnings”. Los Angeles Times. Retrieved 27 December 2008.
- “FDA Requests Boxed Warnings on Fluoroquinolone Antimicrobial Drugs” (Press release). U.S. Food and Drug Administration. 8 July 2008. Retrieved 11 October 2008.
- “Drugs@FDA”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- “MedWatch: The FDA Safety Information and Adverse Event Reporting Program”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- MacCarthy, Paul (22 October 2008). “Important Change in the Avelox (moxifloxacin hydrochloride) and Cipro (ciprofloxacin) Complete Prescribing Information – Addition of Boxed Warning and Medication Guide Regarding Tendinitis and Tendon Rupture”. Bayer HealthCare Pharmaceuticals. Retrieved 27 December 2008.
- Bayer AG (6 November 2007). “Ruling in Bayer’s favor over Avelox patents”. Bayer. Retrieved 29 August 2009.
- http://www.orangebookblog.com/Bayer_20v._20Dr._20Reddy_27s.pdf
- “United States Court of Appeals for the Federal Circuit”. uscourts.gov. 28 June 2007. Retrieved 29 August 2009.
- Bayer AG (24 April 2008). “Risk Report”. Bayer. Retrieved 29 August 2009.
- In The United States District Court For The District Of Columbia Public Citizen, Inc. VS. Food And Drug Administration 3 January 2008
- Office of the Attorney General State of Illinois Lisa Madigan Citizen Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics 18 May 2005
- Public Citizen’s Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781) 29 August 2006
- Public Citizen’s Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399) 1 August 1996
- Public Citizen’s Petition to Ban the Antibiotic Gatifloxacin (Tequin) (HRG Publication #1768)
- Public Citizen’s Petition to immediately ban the antibiotic Trovafloxacin (Trovan). (HRG Publication #1485) Date: 3 June 1999
- Public Citizen’s Petition to immediately stop the distribution of dangerous, misleading prescription drug information to the public. HRG Publication #1442 Date: 9 June 1998
- June 2004, A petition To the United States Congress to immediately take action to protect consumers from the reckless and negligent abuses of the FDA and the following Pharmaceutical Companies: Bayer, Ortho-McNeill, Pfizer, Merck, Bristol-Myers Squibb, Sanofi Winthrop, Bertek Pharmaceuticals – Rhone-Poulenc Rorer and Barr. These companies manufacture and distribute fluoroquinolone antibiotics in the United States in a manner that fails to warn of serious adverse event risks, and downplays and fails to warn physicians of the serious risks associated with fluoroquinolone therapy.
| EP0350733A2 | Jun 30, 1989 | Jan 17, 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0591808A1 | Sep 27, 1993 | Apr 13, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| EP0592868A1 | Sep 29, 1993 | Apr 20, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| US5849752 | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO1999026940A2 | Nov 12, 1998 | Jun 3, 1999 | Bayer Ag | Method for producing 8-methoxy-quinoline carboxylic acids |
| WO2004091619A1 | Apr 9, 2004 | Oct 28, 2004 | Reddys Lab Ltd Dr | A crystalline form iii of anhydrous moxifloxacin hydrochloride and a process for preparation thereof |
| WO2007010555A2 | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| CN102030751B | Dec 1, 2010 | Nov 21, 2012 | 上虞京新药业有限公司 | Process for crystallizing moxifloxacin hydrochloride |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
| WO2005012285A1 * | Aug 5, 2004 | Feb 10, 2005 | Satyanarayana Chava | An improved process for the preparation of moxifloxacin hydrochloride |
| EP0443498A1 * | Feb 18, 1991 | Aug 28, 1991 | Kyorin Pharmaceutical Co., Ltd. | Isoindoline derivatives |
| EP0550903A1 * | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| US6323213 * | Feb 12, 1997 | Nov 27, 2001 | Bayer Aktiengesellschaft | Possibly substituted 8-cyano-1-cyclopropyl-7-(2,8-diazabicyclo-[4.3.0]-nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinolin carboxylic acids and their derivatives |
| US5849752 * | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO2007010555A2 * | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 * | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| WO2008138759A1 * | Apr 30, 2008 | Nov 20, 2008 | Sandoz Ag | Process for the preparation of moxifloxacin hydrochloride |
| WO2007148137A1 * | Jun 22, 2007 | Dec 27, 2007 | Generics Uk Ltd | Novel hydrate form of moxifloxacin monohydrochloride |
| WO2008028959A1 * | Sep 7, 2007 | Mar 13, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin hydrochloride |
| WO2008059223A2 * | Nov 13, 2007 | May 22, 2008 | Cipla Ltd | Process for the synthesis of moxifloxacin hydrochloride |
| WO2008095964A1 * | Feb 6, 2008 | Aug 14, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin base |
| WO2009087151A1 * | Jan 7, 2009 | Jul 16, 2009 | Chemo Iberica Sa | Polymorphic forms of moxifloxacin hydrochloride and processes for preparation thereof |
| CN102603738B | Feb 24, 2012 | Dec 11, 2013 | 天津市汉康医药生物技术有限公司 | 一种稳定的盐酸莫西沙星化合物 |
| EP2083010A1 | Jan 8, 2008 | Jul 29, 2009 | Chemo Ibérica, S.A. | Polymorphic Forms of Moxifloxacin hydrochloride and processes for preparation thereof |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US8198451 | Nov 13, 2007 | Jun 12, 2012 | Cipla Limited | Process for the synthesis of moxifloxacin hydrochloride |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
 syn…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html spectroscopy…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html
syn…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html spectroscopy…………. http://orgspectroscopyint.blogspot.in/2015/06/moxifloxacin.html
The secret of fertile sperm

Progesterone and other fatty signaling molecules are critical for sperm fertility. Credit: C. Cain
To better understand the causes of male infertility, a team of Bay Area researchers is exploring the factors, both physiological and biochemical, that differentiate fertile sperm from infertile sperm. At the 58th Annual Biophysical Society Meeting, which takes place Feb. 15-19, 2014, in San Francisco, Calif., the team will present its work to identify and characterize proteins known as ion channels, which are crucial for sperm fertility and expressed within a sperm cell’s plasma membrane.
“Any knowledge gained in this area may help create much-needed diagnostic testing and treatments for male infertility, which is in essence an idiopathic disease, because at this time 80 percent of male infertility cases can’t be diagnosed or treated,” said Melissa Miller, a postdoctoral fellow who will present the team’s findings at the meeting. Miller works in the labs of both…
View original post 328 more words
The US FDA has issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate)高三尖杉酯碱 for chronic myeloid leukaemia (CML).


Omacetaxine mepesuccinate 高三尖杉酯碱
Alkaloid from Cephalotaxus harringtonia; FDA approved orphan drug status for Ceflatonin in the treatment of chronic myeloid leukemia due to being an inducer of apoptosis in myeloid cells and inhibitor of angiogenesis.
26833-87-4 CAS NO
1-((1S,3aR,14bS)-2-Methoxy-1,5,6,8,9,14b-hexahydro-4H-cyclopenta(a)(1,3)dioxolo(4,5-h)pyrrolo(2,1-b)(3)benzazepin-1-yl) 4-methyl (2R)-2-hydroxy-2-(4-hydroxy-4-methylpentyl)butanedioate
1-((11bS,12S,14aR)-13-methoxy-2,3,5,6,11b,12-hexahydro-1H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-d]cyclopenta[b]pyrrolo[1,2-a]azepin-12-yl) 4-methyl 2-hydroxy-2-(4-hydroxy-4-methylpentyl)succinate
Also known as: NSC-141633,
- BRN 5687925
- Ceflatonin
- CGX-635
- Homoharringtonine
- Myelostat
- NSC 141633
- Omacetaxine mepesuccinate
- Omapro
- Synribo
- UNII-6FG8041S5B
- 高三尖杉酯碱
CGX-635-14 (formulation), CGX-635, HHT, ZJ-C, Myelostat, Ceflatonin
USFDA on 26th October 2012 APPROVED
| Formula | C29H39NO9 |
|---|---|
| Mol. mass | 545.62 g/mol |
The US Food and Drug Administration has now issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate) for chronic myeloid leukaemia (CML).
Synribo is indicated for adult patients with chronic phase (CP) or accelerated phase (AP) CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).
Read more at: http://www.pharmatimes.com/Article/14-02-17/US_green_light_for_Teva_s_CML_drug_Synribo.aspx#ixzz2tdkbGFcw
Homoharringtonine is an angiogenesis-inhibiting and apoptosis-inducing alkaloid which was approved in October 2012 by the FDA for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKI). In November 2012, the product was commercialized as Synribo(R) on the U.S. market by Teva.
The original developer, ChemGenex, selected homoharringtonine for the combination trials due to its complementary mechanism of action that can reduce Bcr-Abl protein expression associated with resistance to imatinib mesylate.
In 2004, the compound received orphan drug designation from the EMEA for the treatment of AML and CML. Orphan drug designation was granted by the FDA for the treatment of CML in 2006 and for the treatment of myelodysplasia in 2009. Fast track designation was assigned to homoharringtonine for CML in 2006. In 2009, the product was licensed to Hospira by ChemGenex Pharmaceuticals for development and marketing in Europe, the Middle East and parts of Africa.
Homoharringtonine, AKA HHT or omacetaxine mepesuccinate, is a cephalotaxine ester and protein synthesis inhibitor with established clinical activity as a single agent in hematological malignancies. Homoharringtonine is synthesized from cephalotaxine, which is an extract from the leaves of the plant, Cephalotaxus species. In October 2005, homoharringtonine received Orphan Drug designation from the EMEA for the treatment of chronic myeloid leukemia (CML). Then in March 2006, homoharringtonine received Orphan Drug status from the FDA for the treatment of CML. In November 2006, homoharringtonine, for the treatment of CML, was granted Fast Track designation by the FDA. Most recently, in October 2012, homoharringtonine was marketed under the brand name Synribo” and FDA approved for patients who are intolerant and/or resistant to two or more tyrosine kinase inhibitors used to treat accelerated or chronic phase CML
Omacetaxine mepesuccinate is administered subcutaneously and acts differently from TKIs. It may have a therapeutic advantage for patients who have failed TKIs. Omacetaxine is currently in global phase 2/3 clinical trials for CML and has been granted Orphan Drug designations by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMEA) as well as Fast Track status by the FDA. In vitro and animal model trails are promising and recent results showed that omacetaxine has potential to treat resistant leukemia mainly CML and ALL.
| PATENT | ||
|---|---|---|
|
3-25-2011
|
CEPHALOTAXUS ESTERS, METHODS OF SYNTHESIS, AND USES THEREOF
|
Tetrahedron Letters,Vo1.23,No.34,pp 3431-3434 … – Brock University
Omacetaxine mepesuccinate (INN, trade name Synribo) is a semi-synthetic analogue of an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of chronic myelogenous leukemia (CML). It was approved by the US FDA in October 2012 for the treatment of adult patients with CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).[1]
Omacetaxine mepesuccinate is a semisynthetic derivative of the cytotoxic plant alkaloid homoharringtonine isolated from the evergreen tree Cephalotaxus with potential antineoplastic activity. Omacetaxine mepesuccinate binds to the 80S ribosome in eukaryotic cells and inhibits protein synthesis by interfering with chain elongation. This agent also induces differentiation and apoptosis in some cancer cell types. Omacetaxine mepesuccinate (INN, or homoharringtonine, trade name Synribo) is an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of Chronic Myelogenous Leukemia. It was approved by the USFDA on 26th October 2012 for the treatment of adult patients with chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs)
Omacetaxine is indicated for use as a treatment for patients with chronic myeloid leukaemia who are intolerant of tyrosine kinase inhibitors.[2][3]
In June 2009, results of a long-term open label Phase II study were published, which investigated the use of omacetaxine infusions in CML patients. After twelve months of treatment, about one third of patients showed a cytogenetic response.[4] A study in patients who had failed imatinib and who had the drug resistant T315I mutation achieved cytogenetic response in 28% of patients and haematological response in 80% of patients, according to preliminary data.[5]
Phase I studies including a small number of patients have shown benefit in treating myelodysplastic syndrome (MDS, 25 patients)[6] and acute myelogenous leukaemia (AML, 76 patients).[7] Patients with solid tumors did not benefit from omacetaxine.[8]
Omacetaxine is a protein translation inhibitor. It inhibits protein translation by preventing the initial elongation step of protein synthesis. It interacts with the ribosomal A-site and prevents the correct positioning of amino acid side chains of incoming aminoacyl-tRNAs. Omacetaxine acts only on the initial step of protein translation and does not inhibit protein synthesis from mRNAs that have already commenced translation.[9]
Omacetaxine mepesuccinate
SYNRIBO contains the active ingredient omacetaxine mepesuccinate, a cephalotaxine ester. It is a protein synthesis inhibitor. Omacetaxine mepesuccinate is prepared by a semi-synthetic process from cephalotaxine, an extract from the leaves of Cephalotaxus sp. The chemical name of omacetaxine mepesuccinate is cephalotaxine, 4-methyl (2R)-hydroxyl-2-(4-hydroxyl-4-methylpentyl) butanedioate (ester).
Omacetaxine mepesuccinate has the following chemical structure:
 |
The molecular formula is C29H39NO9 with a molecular weight of 545.6 g/mol. SYNRIBO for injection is a sterile, preservative-free, white to off-white, lyophilized powder in a single-use vial. Each vial contains 3.5 mg omacetaxine mepesuccinate and mannitol.
SYNRIBO is intended for subcutaneous administration after reconstitution with 1.0 mL of 0.9% Sodium Chloride Injection, USP. The pH of the reconstituted solution is between 5.5 and 7.0.
…………………………………..
INTRODUCTION
Harringtonines 3 are particular cephalotaxanes formed by attachement of a branched hydroxyacyloxy side-chain at the 3-position of various cephalotaxines moieties. Harringtoriines are natural esters of cephalotaxines exhibiting generally a strong cytotoxic activity. However the lost only one atom of this minimal structure lead to a dramatic lost of activity (see below). Some example of harringtonines are harringtonine
3a, homoharringtonine 3b, drupangtonine 3c, anhydroharringtonine 3d and neoharringtonine 3e.
SCHEME 1 DEFINITION NOMENCLATURE AND NUMBERING OF CEPHALOTAXANES


Examples of harringtonines

Examples of cephalotaxines

Harringtonine 3a (n = 2) Anhydroharringtonine 3d Homoharringtonine 3b (n = 3)

(-)-Cephalotaxine 2a


Drupacine 2b Drupangtonine 3c Neoharringtonine 3e (n = 2)
…………………………………
The term “cephalotaxanes” refers to compounds or salts thereof which have a basic skeleton of formula

where p is equal to 1 or 2 (it being possible for the two units to be identical or different and linked via a single bond or an oxygen atom), which can contain various oxygenated substituents (aliphatic or aromatic ethers, free or esterified alcohols, substituted or free enols and/or phenols, bridged ethers, and more generally any substituent usually encountered in the natural state on compounds of this type).
Harringtonines are alkaloids which are of high interest in anticancer chemotherapy, in particular on certain haematosarcomas which are multi-resistant to the existing therapies. The selectivity of harringtonines, which is based on a novel mechanism of action relating to protein synthesis, is such that this series is favoured with a great future in anticancer therapy.
Several literature compilations give a seemingly exhaustive review of all of the knowledge relating to cephalotaxanes, these compilations being, chronologically: [C. R. Smith, Jr, R. G. Powell and K. L. Mikolajczack, Cancer Treat. Rep., Vol. 60, 1157 (1976); C. R. Smith, Jr, L. Kenneth, K. L. Mikolajczack and R. G. Powell in “Anticancer Agent Based on Natural Product Model”, 391 (1980); Liang Huang and Zhi Xue in “The Alkaloids”, Vol. XXIII (A. Brossi Ed.), 157 (1984); M. Suffness and G. A. Cordell in “The Alkaloids, Chemistry and Pharmacology” (A. Brossi Ed.), Vol. 25, 57-69, 295-298 (1’987); P. J. O’Dwyer, S. A. King, D. F. Hoth, M. Suffness and B. Leyland-Jones, Journal of Clinical Oncology, 1563 (1986); T. Hudlicky, L. D. Kwart and J. W. Reed, in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, 199 (1998)].
Antiparasitic activities, in particular on the haematozoon of malaria, have also been recognized [J. M. Whaun and N. D. Brown, Ann Trop. Med. Par., Vol. 84, 229 (1990)].
Homo-harringtonine (HHT), the most active member of the series, is active at and above daily doses of 2.5 mg/m2 of body area per 24 hours, i.e., as a guide, at doses twenty times lower than that for Taxol. HHT has already undergone fourteen phase I and II clinical trials and it is the only known product capable of a 70% reinduction of full haematological remissions in patients suffering from chronic myeloid leukaemias that have become resistant to alpha-interferon [S. O’Brien, H. Kantarjian, M. Keating, M. Beran, C. Koler, L. E. Robertson, J. Hester, M. Rios, M. Andreeff and M. Talpaz, Blood, 332 (1995); Leukemia Insights, Vol. 3, No. 1 (1998)].
Harringtonines were extracted over 35 years ago from an exclusively Asiatic cephalotaxacea known as Cephalotaxus harringtonia, following the programme of research into novel anticancer agents in the plant kingdom developed by the National Cancer Institute. In fact, the Cephalotaxus alkaloids consist essentially (at least 50%) of cephalotaxine, a biosynthetic precursor of the harringtonines, the latter individually representing only a few percent of the total alkaloids.
Besides their low concentration in the natural state in plant starting material, harringtonines are mixed with many congeners which have very similar chemical structures. Thus, in a high resolution high performance liquid chromatography (HPLC) chromatogram of a semi-purified alkaloid extract, no less than several tens of cephalotaxine esters are counted.
Numerous antileukemia drugs have been investigated but so far, there is no single drug that is effective and safe. As discussed in U.S. 3,497,593, an alkaloid from Tylophora plant is said to have antitumor activity against mouse leukemia (L-1210). U.S. 3,928,584 discloses an organic composition derived from tree sap and is said to have activity against mouse leukemia P-388. Also U.S. 4,431,639 discloses that an extract of Rhisoma Stractylis promotes the production of lymphocytes in the circulating blood, consequently eliminating cancer growth
-
Harringtonine or Homoharringtonine, hereinafter referred to as HH, has been known to be effective against acute chronic granulocytic and monocytic leukemia (Journal of Chinese Internal Medicine 3:162-164, 1978). However, it is highly toxic and causes damage to heart and hematopoietic organs. The results of experiments in animals, such as mice, rabbits and dogs, indicate that most of them die from cardiotoxicity after receiving the drug. Therefore, there is a need to improve the HH drug for safe use against leukemia. This drug is of special importance in that all known antileukemia drugs are effective against lymphatic leukemia and there are no effective drugs for treating nonlymphatic leukemia
All the literature from 1972 to the present date [Mikolajczack et al., Tetrahedron, 1995 (1972); T. Hudlicky, L. D. Kwart and J. W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxane 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3 totally preformed to give a harringtonine 4b, i.e. the conversion 2a+3e(4b as described in the example featured in the scheme below

- ……………………………………………………..
SYNTHESIS
Tetrahedron Lett 1982,23(34),3431, J Org Chem 1983,48(26),5321

The oxidation of 2-methyl-1-cyclopentene-1-carbaldehyde (I) with O3 and Ag2O gives 2,6-dioxoheptanoic acid (II), which is esterified with cephalotaxine (III) by means of (COCl)2, yielding the ester (IV). Reformatsky reaction of (IV) with methyl bromoacetate (V) and Zn affords the adduct (VI), which is treated with an excess of methylmagnesium iodide to provide the target homoharringtonine (as a single diastereomer), along with some starting cephalotaxine that is separated by chromatography.
………………………………
SYNTHESIS
EP 1064285; FR 2776292; WO 9948894, Tetrahedron Lett 1999,402931

The intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII) has been obtained by several related methods: 1. The Grignard condensation of 4-methyl-3-pentenyl bromide (I) with diethyl oxalate (II) in HF gives the 2-oxoheptenoate (III), which is condensed with methyl acetate (IV) by means of LiHMDS in THF to yield 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V).
The cyclization of (V) by means of Ts-OH in hot toluene or by means of hot aqueous formic acid affords 2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid ethyl ester (VI), which is hydrolyzed with KOH in boiling water to provide the corresponding dicarboxylic acid (VII). Finally, this compound is regioselectively monoesterified by means of BF3/MeOH in methanol to furnish the intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII). 2.
The reaction of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with HCl in hot methanol gives 3-(ethoxycarbonyl)-3,7-dihydroxy-7-methyloctanoic acid methyl ester (IX), which is then cyclized by means of ZnCl2 in hot dichloroethane to yield the previously described intermediate (VIII). 3. The hydrolysis of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with KOH in refluxing methanol/water gives the corresponding diacid (X), which is regioselectively monoesterified by means of BF3/MeOH in methanol to yield 3-carboxy-3-hydroxy-7-methyl-6-octenoic acid methyl ester (XI).
Finally, this compound is cyclized by means of Ts-OH in hot toluene to afford the previously described carboxylic intermediate (VIII). The racemic acid (VIII) is submitted to optical resolution by esterification with quinine (XII) by means of 2,4,6-trichlorobenzoyl chloride and TEA or DCC to give a diastereomeric mixture of esters (XIII) that is separated by preparative HPLC to obtain the desired diastereomer (XIV).
The hydrolysis of (XIV) with KOH in refluxing ethanol/water gives the corresponding chiral dicarboxylic acid (XV), which is regioselectively monoesterified with BF3/MeOH in methanol to yield the chiral (R)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (XVI).
The esterification of (XVI) with cephalotaxine (XVII) by means of 2,4,6-trichlorobenzoyl chloride and TEA in toluene affords the corresponding ester (XVIII), which is treated with HBr in dichloromethane/HOAc, providing the bromoester (XIX). Finally, this compound is treated with NaHCO3, CaCO3 or BaCO3 in acetone/water to give the target hydroxyester.
………………………………………….
EXTRACTION
-
Throughout the specification, the concentration of the solvent is the same as first given unless stated otherwise. Redeuced pressure means about 2,27 kPa (17 mm Hg. abs), l is liter, kg is kilogram. ml is milliliter. Yield in weight %.
Example 1. HH is extracted from the skins, stems, leaves and seeds of Cephalotaxus fortunel Hook and other related species, such as Cephalotaxus sinensis Li, C. hainanensis, and C. wilsoniana, including C.oliveri mast and C.harringtonia. -
1 kg of finely ground Cephalotaxus fortunel Hook is extracted with 8 l of 90% ethanol at room temperature for 24 hrs. The solution is filtered to yield a filtrate A and filtercake. The filtercake is percolated with ethanol and filtered again to yield filtrate B. A and B are combined and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, 2% HCl is added to adjust the pH to 2.5. The solids are separated from the solution by filtration to yield a filtrate C. The solids are washed once with 2% HCl and filtered to yield a filtrate D. C and D are combined and the pH adjusted to 9.5 by adding saturated sodium carbonate solution. The alkaline filtrate is extracted with chloroform and the chloroform layer separated from the aqueous layer. This extration process is repeated five times. All the chloroform extracts are combined and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue respectively.
-
The solid alkaloid is then dissolved in 20 ml. of 6% citric acid in water. The solution is divided into three equal portions. These are adjusted to pH 7,8 and 9 by adding saturated sodium carbonate solution.
-
The portions having pH 8 and 9 are combined and extracted with chloroform. The chloroform extracts are distilled under reduced pressure, whereby chloroform is removed and recovered and a solid residue of crude Harringtonine is obtained.
-
The crude Harringtonine is dissolved in pure ethanol i.e. alkaloid : anhydrous ethanol 1:10 , and crystallized. The crystals are refined by recrystalliation in diethyl ether. Overall yield of Harringtonine is about 0.1% including yield from mixed HH from the subsequent process.
Harringtonine has the following chemical structure:
wherein R is

- melting point:
- 135° – 137°C
- crystal:
- colorless
- infrared spectrum:
- 3750, 1660, 1505, 1490, 1050, and 945 cm⁻¹.

-
The portion having a pH of 7 and the mother liquors from the foregoing crystallization of Harringtonine are combined and passed through a liquid chromatographic column of diameter to height ratio 1:50 packed with alumina. The column is finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids are mixture of HH. The mixed HH is then separated from each other by countercurrent distribution employing chloroform and pH 5 buffer. The first fraction of the countercurrent distribution is Homoharringtonine and the last fraction of the countercurrent distribution is Harringtonine. Homoharringtonine is purified by crystallization in methyl alcohol.
Homoharringtonine has the following chemical structure:
wherein R is

- yield:
- 0.02%
- melting point:
- 144° – 146°C
- infrared spectrum:
- 3500∼3400, 1750, 1665, 1030 and 940 cm⁻¹.







…………………………………………………………………………..
EXTRACTION
All the literature from 1972 to the present date [Mikolajczack et al.,Tetrahedron, 1995 (1972); T. Hudlicky, L.D. Kwart and J.W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S.W. Pelletier Ed.), Vol. 5, 639 (1987); M.A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxine 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3e totally preformed to give a harringtonine 4b , i.e. the conversion 2a + 3e ( 4b as described in the example featured in the scheme below

Example 46
Preparation of purified (-) cephalotaxine from total alkaloidic extract of Cephalotaxus sp
-
[0319]

-
Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica,; 23; 7; 835 (1980)]
-
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1 éq) showed the following results:
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).
Presence of 11 ± 5 % de (+)-cephalotaxine.
[α]22 = -134,0° (c = 0,214; CHCl3) : calculated rate 25 ± 5 % - Batch B: slightly racemized (1%)
[α]19 = -173,3° (c = 0,208; CHCl3)
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).

Enantiomeric enrichment of the natural cephalotaxine:
-
Crude chromatographied cephalotaxine (20g) was dissolved at 55°C in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity.
[α]20 D =-130° (C1, CHD3) corresponding to 10 % of racemization. The crystallized product thus obtained (20g) was dissolved again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-)-cephalotaxine [α]20 D= -185°(C1,CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20 = 0,5° (C1 ; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (-)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (-)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical evaluation of the enantiomeric purity of natural cephalotaxine:
-
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19.
The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11% ± 3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
Example 47Preparation of homoharringtonine, from anhydro-homoharringtonine:

1)° Method A
-
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at -10°C. After stirring at -10°C for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3 × 230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
-
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (214 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at -10°C a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at -10°C for 3 hours, was added water (13 ml) and then the temperature was raised to 20°C. After stirring at 20°C for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3 × 20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.




……………………
SEMISYNTHESIS
EXAMPLE 27 Preparation of homoharringtonine as a pharmaceutical use from crude semi-synthetic homoharringtonine resulting from example 25 by preparative high-performance liquid chromatography

1°) Method A
Crude homoharringtonine (35 g) is dissolved in buffer (triethylamine (1.55/1000) in deionised water and orthophosphoric acid to adjust pH to 3. The solution was filtered then injected on a preparative high-performance liquid chromatograph equipped with axial compression and high pressure pump (stationary phase: n-octadecylsilane, 15 μm, porosity 100, 1 kg; mobile phase; buffer/tetrahydrofurane 85/15). Elution was performed at a flow rate of 0.2 l/min. Fractions contain was monitored by U.V. detector and TLC. Retained fraction were finally checked by HPLC then combined, alkalinised with 2.5% aqueous ammonia and extracted with dichloromethane (4×400 ml). After concentration under reduced pressure homoharringtonine is obtained as a pale yellow resin which on trituration in a 8/2 water-methanol mixture gave pure homoharringtonine as a white crystalline solid (mp=127° C.), HPLC purity was higher than 99.8%.
2°) Method B
Same procedure of purification as method A was performed but mobile phase buffer/methanol (68/32) was used instead buffer/tetrahydrofurane.
3°) Method C
Same procedure of purification as method A was performed but mobile phase buffer/acetonitrile (85/15) was used instead buffer/tetrahydrofurane.
EXAMPLE 28 Preparation of homoharringtonine as a pharmaceutical use from semi-purified natural cephalotaxine
Crude homoharringtonine, prepared according to Example 25 from a partially racemized natural cephalotaxine and purified by chromatography and crystallisation according to the method A of Example 27, gave an homoharringtonine showing a non natural enantiomeric epi-homoharringtonine content less than 0.05%.
EXAMPLE 46 Preparation of purified (−) cephalotaxine from total alkaloidic extract of cephatotaxus sp

Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica; 23; 7; 835 (1980)]
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1éq) showed the following results:
Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine); 5.99 (1H, OCH2O (−)-cephalotaxine) and 5.76 (1H, OCH2O (−)-cephalotaxine). Presence of 11±5% de (+)-cephalotaxine. [α]22=−134,0°(c=0,214; CHCl3): calculated rate 25±5%
Batch B: slightly racemized (1%) [α]19=−173,3°(c=0,208; CHCl3)
Enantiomeric Enrichment of the Natural Cephalotaxine:
Crude chromatographied cephalotaxine (20 g) was dissolved at 55° C. in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity, [α]20 D=−130°(C1, CHD3) corresponding to 10% of racemization. The crystallized product thus obtained (20 g) was dissolyed again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-−)-cephalotaxine [α]20 D=−185°(C1, CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20=0,5°(C1; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (−)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (−)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical Evaluation of the Enantiomeric Purity of Natural Cephalotaxine:
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19. The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11%±3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
EXAMPLE 47
Preparation of homoharringtonine, from anhydro-homoharringtonine

1°) Method A
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at −10° C. After stirring at −10° C. for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3×230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03 g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (21.4 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at −10° C. a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at −10° C. for 3 hours, was added water (13 ml) and then the temperature was raised to 20° C. After stirring at 20° C. for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3×20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.
…………………………………
EXTRACTION
The remarkable clinical efficacy of Homoharringtonine (HHT) resulting in lot of observations of complete remission of leukemia and other solid cancer in human being since 1988. Recently, research articles reported that the HHT efficacy in glaucoma, inhibition of Hepatities B virus replication and using in bone marrow transplantation. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” [Susan O’Brien, at al.; Sequential homoharringtonine and interferon-α in the treatment of early chronic phase chronic myelogenous leukemia; Blood, Vol 93, No 12 (June 15), 1999: pp 4149-4153]. Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.” (Susan O’Brien, at al.; Simultaneous homoharringtonine and interferon-α in the treatment of patients with chronic-phase chronic myelogenous leukemia; American Cancer Society; Apr. 1, 2002, Vol 94, No. 7).
On Nov. 8, 1988, U.S. Pat. No. 4,783,454 titled Process for producing harringtonine and homoharringtonine disclosed the technique of isolation of a purified HHT from bark of Cephalotaxus. However, the natural source ofCephalotaxus is very limited. Trees of Cephalotaxus grow slowly. Bark ofCephalotaxus has very low content of HHT. Extracting HHT from bark ofCephalotaxus the yield was about 0.02% only. More important to harvest bark ofCephalotaxus will kill and destroy trees. Supply of HHT is very short now. Therefore, it is necessary to find a new manufacturing method.
DETAILED DESCRIPTION
Great progress has been made in research on Homoharringtonine (HHT) production and on future generation HHT drug since 1988. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.”
The good clinical results of HHT in treating cancer brought to the major problem, which is the supply of HHT both short term and long term. It is apparent that a huge amount of bark of Cephalotaxus is needed for collection, extraction and purification of HHT. It is clear that due to the slow growth of the trees ofCephalotaxus, which is a nature source of HHT, and the killing of trees by harvesting bark is not a sustainable resource for HHT production.
Present invention disclosed new methods for producing HHT. The new methods of producing HHT are shown as follows.
1. Tissue Culture (Plant Cell Culture):
Culture manipulation to promote secretion of HHT is a new way for an extracellular product HHT. The biosynthetic methods can yield more HHT through precursor of HHT feeding. The production of HHT increased significantly after the addition of the precursors and special biochemical agents. Content of precursor of HHT abounds in tree and it is very cheap. The present methods include several significant developments in technique of culture plant tissues that are
-
- (a) yields of HHT selected from rapid growth, resistance to infections organisms; and
- (b) HHT can excrete into media.
Traditional method of plant culture is very difficult to overcome the problem of high cost. Therefore, traditional method appears too long to have commercial value. HHT is secondary metabolite of Cephalotaxus. Secondary compound acts in defense against the harmful effects of toxins, carcinogens or mutagens found in the plant. In fact, traditional method is very difficult to increase HHT contenting in plant tissues. The present new method uses a special biochemical agent for increasing content of HHT and more easily to purify HHT from other metabolites.
More important is that the key of the present new technique for producing high content of HHT in plant cell culture is to increase production of HHT by directed fermentation through precursor of HHT feeding. The present new methods are used special metabolite of Cephalotaxus for markedly enhance production of HHT. Therefore, the present invention disclosed a new source for the long term of producing HHT.
2. Using Precursor of HHT:
Recent research’s results have established that direct production of HHT from its precursor and advances in biosynthetic understanding for HHT metabolism. Biosynthesis or semisynthesis of HHT from major nonactivity ingredients is well established through great advances in special biochemistry reactions. Using precursor of HHT for semisynthesis and increase of production in plant cell culture are new developing methods for producing HHT.
3. Using Leaves:
Our new method use leaves of tree of Cephalotaxus not use the bark. So far, the extraction of HHT is used bark. The leaves are harvested from the trees ofCephalotaxus, which grow in mountains of South China. The natural source of leaves is very abundance. The new methods do not use bark. Therefore, it can avoid destroy trees. The natural source of Cephalotaxus tree is very limited and slow growing. In fact, bark of Cephalotaxus has very low yield of HHT. The yield of HHT from Cephalotaxus bark is about 50-100 mg/kg of dried bark. The present new method, therefore, has a great economic and environmental value.
4. Semisynthesis:
HHT has received important chemical studies particularly in regard to structure and anticancer activity relationship and semisynthesis.
A great progress in biochemistry allows semisynthesis to use precursor of HHT from leaves of Cephalotaxus and to produce HHT. The total chemical synthesis of HHT appears too long to have commercial value too. Semisynthesis method can yield a high efficient conversion of precursor to HHT. It is other better biological source for manufacturing HHT. This new method uses closing chemical analogues to convert to HHT. This analogue is produced from leaves or other organ of Cephalotaxus. The present invention disclosed that new methods and techniques of manufacturing HHT could avoid chopping down Cephalotaxus trees which governmental environmentalists are trying to have declared a threatened species.
5. Using Taxol Residual
The anticancer drug Taxol is the most promising new chemotherapeutic agents that developed for cancer treatment in the past twenty years. Taxol has a unique mechanism of action. It has been shown to promote tubulin polymerization and stabilize microtubules against depolymerization. The FDA approved the clinical use of Taxol for several types of cancer. So far, annual sales of Taxol are more than $2 billion in market. Taxol is extracted from bark or leaves of an evergreen tree named Taxus species including Taxus brevifolia (or called Pacific yew). After Taxol has been extracted from bark or leaves, all residual materials of Taxus brecifolia named Taxus residual, which are waste.
Both taxol and HHT can be extracted from yew tree. The content of taxol is less than 0.01% in yew tree. The content of HHT in yew tree is about 0.01% -0.22%. The content of HHT is much higher than content of Taxol. Taxol extracted from bark of yew is difficult and expensive. One reason is that the presences of closely related congeners are similar to Taxol. A major congener is Cephalomannine (CPM), which is a waster of process in manufacturing of Taxol.
The chemical and physical characters are very close between Taxol and Cephalomannine (CPM).
CPM characterized by the same ring structure as Taxol and distinguishes from them only in C-13 ester structure. The present invention disclosed that CPM and related derivative are used to produce HHT.
The following specific examples will provide detailed illustrations of methods of producing relative drugs, according to the present invention and pharmaceutical dosage units containing demonstrates its effectiveness in treatment of cancer cells. These examples are not intended, however, to limit or restrict the scope of the invention in any way, and should not be construed as providing conditions, parameters, reagents, or
EXAMPLE 1
Production of HHT by Culture Cells
So far, HHT is extracted from bark and skins of Cephalotaxus species. However, growth of Cephalotaxus species is very slow and concentration of HHT in plant is extremely low. Furthermore, it is difficult to harvest the plants because of their low propagation rate and the danger of drastic reduced in plant availability. Also, cost of total chemical synthesis of HHT is very expensive and is not available for commerce now. For the reasons given above it is more difficult to obtainCephalotaxus on a large scale for long time. Therefore, Cephalotaxus cell cultures are one of best methods for obtaining HHT. In this present invention, special elicitation is disclosed and it will significantly increase production of HHT.
The methods of cell and tissue culture are disclosed as below.
Parts of bark, stems, leaves, or roots of Cephalotaxus species were surface disinfected by treatment in 70% ethanol for 10 minutes and followed by 0.1 HgCl2for 3 minutes. Plant materials were washed five times for 10 minutes each by sterilized water. Parts of plant were cut into small pieces (0.5-1 mm) and put pieces to Murashige and Skoog’s (MS) medium and supplemented with derivative of new active ingredient of phylum mycota (IPM), precursor of HHT which is a derivative of Cephalotaxus (CEP), tyrosine (TYR) naphthaleneacetic acid (NAA), Kinetin (3 mg/L), and 3% sucrose (w/v). PH of medium was adjusted to 5.7˜5.8. Agar (10 g/L) added to medium. Callus tissues are collected from agar media and suspension cultured cells were harvested by filtration and cultured in MS medium.
The cultures were kept in a culture room at 26° C.±1° C. Friable callus tissues were obtained. The callu was inoculated into 4 L of MS liquid medium containing sucrose, derivative of CEP, PHE, TYR, NAA and Kinetin. Then callus tissues were cultivated 26° C. for 35 days on rotary shaker operated at 120 rpm in the dark. Cells were subcultured into fresh medium of same composition every 2 weeks and maintained at 120 rpm at 26°±1° C. Packed cell volume (PCV), fresh weight (FW), dry weight (DW), concentration of HHT and concentration of sugar were determined every 5th day. The cells were harvested and dried.
In general, callus and suspension cultures of cephalotaxus species grow very slow and no production of free or esterified HHT. However, according to the present invention, addition of IPM to cultures cause a drastic increasing in HHT after 30 days of incubation. For example, in control group (no IPM), HHT in cultured cells is 0.020 mg/g dry weight, but in treatment group (addition of IPM) HHT is about 0.050 mg/g dry weight. Therefore, IPM can increase 250% of content of HHT. It has resulted in plant cell culture systems that producing HHT at concentration higher than those produced by the mother plant. The production of HHT increases significantly after the addition of precursors (CEP). Addition of CEP can increase HHT. Obviously, the present invention provided a new commercial and economic method for producing HHT. The IPM and precursors (CEP) play key role in cultured cells.
EXAMPLE 2
Semi-Synthesis of HHT
HHT shows a significant inhibitory activity against leukemia and other cancer. Concentration of HHT, however, has only 0.01% in natural sources. Cephalotazine (CEP) is major alkaloids present in plant extracts and the concentration ofCephalotaxus has about 1%. Therefore, concentration of CEP is about 100 times higher then HHT in nature plant sources. But CEP is inactive. For the reason given above, semisynthesis of HHT from CEP will increase huge natural sources of HHT.
-
- (1) Extraction of CEP
10 kg of dried stems or leaves or roots of Cephalotaxus species were milled, placed in a percolator, along 80 L of 95% of ethanol, and allowed to stand 24 hours. The ethanol was recovered under reduced pressure (below 40° C.). 20 L of 5% tartaric acid was added to concentrated ethanol solution. The ammonia water was added to the acidic solution and adjusted pH to 9. The solution of pH 9 was filtered and yielded a filtrate. The filtrate was extracted with CHCl3. CHCl3 was recovered under reduced pressure and residue was obtained. The residue was chromatographed packed with alumna and eluted by CHCl3-MeOH (9:1). Eluate was concentrated under reduced pressure. Residue was dried under vacuum. The product is CEP.
-
- (2) Semisynthesized HHT from CEP
Materials and Methods
Melting points were determined on a Fisher-Johns apparatus. Infrared spectra were obtained on a Perkin-Elmer 567 infrared spectrophotometer or on a Beckman 4230 IR spectrophotometer. Peak positions were given in cm−1. The IR spectra of solid samples were measured as potassium bromide dispersions, and the spectra of liquids were determined in chloroform or carbon tetrachloride solutions. NMR spectra were measured on a Varian A-60, Perkin-Elmer R-32, Varian EM-390, or Brüker WH-90 NMR spectrometer. Chemical-shift values were given in parts per million downfield from Me4Si as an internal standard. Mass spectra were run on an AE1 MS-12 Finnigan 3300, or CEC21-110B mass spectrometer.
Preparative thin-layer chromatography was accomplished using 750-μm layers of aluminum oxide HF-254 (type E), aluminum oxide 60 PF-254 (type E), silica gel HF-254 (type 60 PF-254), or silica gel GF-254. Visualization was by short-wave ultraviolet light. Grace silica gel, Grade 923, and Woelm neutral aluminum oxide, activity III, were used for column chromatography. Analytical thin-layer chromatography was run on plastic sheets precoated with aluminum oxide F-254 neutral (type T), 200-μm thick, and on Polygram Sil G/UV254 (silica gel), 250 μm on plastic sheets. Visualization was usually by short-wave ultraviolet light, phosphomolybdic acid, or iodoplatinate.
Preparation of α-Ketoester-Harringtonine
1 g of Benzene-α-acetone Na was put into 10 L of benzene. Mixture was stirred at room temperature then was dissolved in 10 L of pyridine and stirred at 0° C. Oxalic chloride was added from a dropping funnel to solution of pyridine. Stirring was continued while the solution warmed to room temperature and stand overnight. Excess reagent was removed. This solution was dissolved in CH2Cl2and cooled to near 0° C. in an ice water bath. 5 g of CEP, 2.5 L of CH2Cl2 and 2.5 L of pyridine were added to cold CH2Cl2 solution. Manipulations were done in a dry N2 atmosphere and all glassware heat-dried just before use. The suspension was stirred at room temperature and overnight. The mixture was washed with 10% Na2CO3 and saturated aqueous NaCl, then dried with auhydrous magenesium sulfate, and filtered and the solvents were removed in vacuo. Evaporation provided as an amorphous solid α-ketoester-harringtonine (mp 143˜145° C.).
Semi-Synthesis of HHT
10 L of CH3CHBrCOOEt and activated zin dust and THF were added to the α-ketoester-harringtonine (at −78° C.) for 6 hours followed by slow warming to room temperature with stirred. The reaction mixture was diluted with 10 L CHCl3 and 10 L H2O and solid Na2CO3 was added. CHCl3 was evaporated under reduced pressure and residue was obtained.
The residue was purified by chromatography on alumina. The column was flushed with chloroform and followed by chloroform-methanol (9:1). The solvents were recovered under reduced pressure to provide as a solid. Solid was dissolved in pure ethanol and crystallized. The crystals were refined by recrystalization in diethyl ether. The crystals dried under vacuum. The product is HHT, which has the following characters:
[α]D −119° (C=0.96),
MSm/e (%): 689 (M+, 3), 314 (3), 299 (20), 298 (100), 282 (3), 266 (4), 20 (3), 150 (8), 131 (12), 73 (18)
EXAMPLE 3
HHT Extracted from Plant Tissue
Extraction of HHT has several major methods which including extraction by organic solvent, chromatograph and adjust pH.
HHT was extracted from plant tissue culture, plant cells or leaves of Cephalotaxusspecies.
1 kg of ground Cephalotaxus fortunei Hook was extracted with 10 liters of water at room temperature for 24 hrs. To filtered the solution to yield a filtrate. Ten liters of 90% ethanol added to filtrate. The mixture was Centrifugalized to yield a sediment. Percolated the sediment with ethanol and filter again to yield filtrate, combined filtrates, and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, added 10% of HCl to adjust the pH to 2.5. To separated the solids from the solution by filtration to yield a filtrate (1). Washed the solids once with 2% HCl and filtered to yield a filtrate (2). Combined (1) and (2) and adjusted the pH to 9.5 by adding saturated sodium carbonate solution. Extracted the alkaline filtrate with chloroform and separated the chloroform layer from the aqueous layer. To repeated this extraction process five times. Combined all the chloroform extracts and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue obtained. The solid alkaloid was then dissolved in 6% citric acid in water. The solution was divided into three equal portions. These were adjusted to pH 7, 8 and 9 by adding saturated sodium carbonate solution. The portions having pH 8 and 9 were combined and extracted with chloroform. The chloroform extracts were distilled under reduced pressure, whereby chloroform was removed and recovered and crude HHT was obtained. The crude HHT was dissolved in pure ethanol and crystallized. The crystals were refined by recrystallization in diethyl ether. The crude HHT obtained.
The portion having a pH of 7 passed through a liquid chromatographic column packed with alumina of diameter to height 1:50. The column was finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids were mixture crude of HHT. Combined crude HHT and then separated from each other by countercurrent distribution employing chloroform and pH 5 buffers. The first fraction of the countercurrent distribution was HHT. HHT was purified by crystallization in methyl alcohol. The crystallization was purified by recrystallization in methyl alcohol and dried under vacuum.
…………………….
Example 1 : Preparation of harringtonine drug substance by purification of commercial natural harringtonine
A. Analytical profile of starting product
By combination of HPLC analysis with UV detection (see Figure 6) and mass spectrometry detection (see figure 7 and 8) a total of 6.5% of related compound (identified as b,c: position isomer of harringtonine = 3.4%; d: homoharringtonine = 3%; e: 4′-demethyl harringtonine = 0.01%; f: drupacine derivative: 0.05%) are found in the starting product.
B. Chromatography of natural harringtonine
Natural harringtonine (5 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 80 mm; length: 1000 mm) containing 1000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi). Unwanted fractions are discarded based upon in-line UV spectrophotometric detection. Kept fractions are collected in 16 separate containers which each are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase). During the development phase, a dual in-line UV-MS detection is used. After discarding of the fractions representing more than 0.5 % of the total content of harringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure. Then crude concentrated solution of harringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 1.5 %. C. Crystallization of raw harringtonine
Under a laminar flow hood, the above raw harringtonine (4.1 grams) is dissolved in methanol (5ml), at 30°C. The resulting alcoholic solution was filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized rotary flask. Then, desionized water (50mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of harringtonine is kept under vacuum and rotation is continued during appearance of white crystals of pure harringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (10 mL x 2). The white translucent crystals are then dried using high vacuum at 40°C for 24 hours. Overall yield is 76%. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 400 therapeutic units dosed at 10mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 7 and 9 compare HPLC chromatogram before and after purification in using this process. Table II shows the comparison of the corresponding related compound content.

For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC) thermogravimetry, 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 5 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline harringtonine obtained by this process. A series of sharp absorption bands are noted at 615, 654, 674, 689, 709, 722, 750, 761 805, 850, 928, 989, 1022, 1033, 1062, 1083, 1112, 1162, 1205, 1224, 1262, 1277, 1308, 1340, 1364, 1382, 1438 1486, 1508, 1625, 1656, 1725, 1745, 2883, 2936, 2972, 3079, 3353, 3552 and 3647 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG) Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 12 mg of harringtonine drug substance were accurately weighed (12.4471 mg) into a DSC pan. The sample was heated from 25°C to 200°C at a rate of 10°C/min. The DSC data were obtained following a standard method in the art. The DSC curve of crystalline harringtonine drug substance ((Figure 4), exhibits a melting endotherm at 79.5 °C . No subsequent decomposition occurred under the upper tested temperature 200°C. Simultaneous TG measurement, indicated a loss on drying of 1.3 % which did not correspond to a lost of structural molecule of solvent or water.
Example 2: Preparation of homoharringtonine drug substance by purification of raw semi- synthetic (hemi-synthetic) homoharringtonine
A. Analytical profile of starting product
Crude reaction mixture of raw homoharringtonine contains a potential of 250 grams of homoharringtonine DS together with process impurities such as catalyst, unchanged starting product (anhydro-homo-harringtonine), and some related side product. HPLC analysis with UV detection (see left-side chromatogram on Figure 10) indicated a total of 9 % of related impurities. B. Chromatography of semi-synthetic homoharringtonine
Raw semi-synthetic homoharringtonine (550 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 450 mm; length: 1000 mm) containing 48,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 540 L/hour). Unwanted fractions are discarded based upon by- passed in-line UV spectrophotometric detector. Kept fractions are collected in 30 separate stainless steel containers (20 or 50 L each) which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 % (see rigth-side chromatogram on Figure 10)
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above raw homoharringtonine DS (210 grams) is dissolved in methanol (240 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (2400mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (450 mL x 2). The white cryitals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 88% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 40,000 therapeutic units dosed at 5mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 11 shows HPLC chromatogram before and after crystallization. Total of related impurities of homoharringtonine DS is 0.03%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 3 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline homoharringtonine obtained by this process. A series of sharp absorption bands are noted at 612, 703, 771 , 804, 826, 855, 879, 932, 1029, 1082, 1119,
1135, 1161 , 1191 , 1229, 1274, 1344, 1367, 1436, 1457, 1488, 1505, 1653, 1743, 2814, 2911 ,
2958, 3420, and 3552 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG)
Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 11 mg of homoharringtonine drug substance were accurately weighed (10.6251 mg) into a DSC pan. The sample was heated from 25°C to 250°C at a rate of 5°C/min. The
DSC data were obtained following a standard method in the art. The DSC curve of crystalline homoharringtonine drug substance (Figure 1), exhibits a melting endotherm at 145.6 °C.
Melting range performed by the capillary method (Bucchi Apparatus) gave 143-145°C. Literature indicated 144-146°C [Anonymous, Acta Bot. Sin. 22, 156 (1980) cited by L. Huang and Z. Xue, Cephalotaxus Alkaloids, in “The Alkaloids”, vol. XXIII, pp157, (1988).
Crystallization medium was not published. This is the only literature reference regarding melting point of a crystalline form of HHT] X-Ray Powder Diffraction
X-ray powder diffraction pattern was collected on a INEL microdiffractomer, model
DIFFRACTINEL. Powdered homoharringtonine DS was packed in a glass capillary tube and was analyzed according to a standard method in the art. The X-ray generator was opered at 45 kV and 40 mA, using the copper Kalpha line as the radiation source. The sample was rotated along the chi axis and data was collected between 0 and 120 deg 2-theta. A collection time of 1200 sec was used. As showed on Figure 2, the x-ray powder diffraction for this crystalline form of homoharringtonine shows a typical pattern including major reflection peaks at approximately 7.9, 9.2, 10.9, 14.9 16.0, 17.7, 19.5, 19.7, 21.78, 23.1 , 25.3, 25.4 and 25.7 deg 2-theta.
Example 3: Preparation of homoharringtonine drug substance by purification of a commercial sample of impure homoharringtonine from Chinese source
A. Analytical profile of starting product
Analytical HPLC chromatogram of natural homoharringtonine (China National Pharmaceutical) is displayed on Figure 12 (bottom left).
B. Chromatography of Natural Homoharringtonine
Natural homoharringtonine (25 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 200 mm; length: 1000 mm) containing 12,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 120 IJhour). Unwanted fractions are discarded based upon bypassed in-line UV spectrophotometric detector. Kept fractions are collected in 22 separate stainless steel containers which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 %.
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above chromatographied homoharringtonine DS (18 grams) is dissolved in methanol (35 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (300 mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs.
Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water
(50 mL x 2). The white crystals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 84% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current
Good Manufacturing Practices were applied.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 12 (bottom right) shows HPLC chromatogram after crystallization. Total of related impurities of homoharringtonine DS is 0.05%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry. Infrared Spectra, Differential Scanning Calorimetry (DSC) and X-Ray Powder Diffraction gave patterns strictly superimposable to the one of example 2 obtained from semi-synthetic homoharringtonine (Figure 3, 1 , and 2, respectively).
………………………………….
KOREAN PAPER.. LINK
Title: 한국산 개비자(Cephalotaxus koreans)에서의 Harringtonine과 Homoharringtonine의 확인 및 함량 분석
Author: 박호일 ; 이연 (한국생물공학회)
Source: 한국생물공학회지 = Korean journal of biotechnology and bioengineering; ISSN:1225-7117 @ 1225-7117 @ ; VOL.11; NO.6; PAGE.689-695; (1996)
Pub.Country: Korea
Language: Korean
Abstract: Harringtonine and homoharringtonine known as anti-cancer agents were isolated from Korean native plumyew(Cephalotaxus koreana) using column chromatography(CHCl3:MeOH=19:1, Rf=0.28). The structure of the mixture of two compounds was characterized by 1H-NMR. Comparison of our spectra of harringtonine and homoharringtonine with previously reported ones indicated that the two are identical. The contents of harringtonine and homoharringtonine in the needles, stems, and roots of Korean native plumyew were determined by high performance liquid chromatography(HPLC). The contents of both compounds varied with the site of location and the part of plant. The content of harringtonine was higher in needles and roots than in stems, whereas the content of homoharringtonlne was lower than harringtonine. Homoharringtonine contents in needles at Mt. Palgong, Mt. Dukyu, Mt. Baekyang, Mt. Jiri, and Namhae were higher than in stems and roots. But homoharringtonine contents in needles al Mt. Jokye and Jindo were lower than in stems and roots.
http://img.kisti.re.kr/originalView/originalView.jsp
……………………………………………………………………………….
SYNTHESIS OF HOMOHARRINGTONINE AND SEPARATION OF ITS STEREOMERS
-
[PDF]
Chapter 1 Drug Discovery from Plants – Springer
LC-NMR-MS and LC-SPE-NMR to accelerate their future discovery. Keywords …..Ceflatonine (34), a synthetic version of homoharringtonine produced by.
…………………………………………………………………………….
References
- “Synribo (omacetaxine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 18 February 2014.
- “SYNRIBO (omacetaxine mepesuccinate) injection, powder, lyophilized, for solution [Cephalon, Inc.]”. DailyMed. Cephalon, Inc. October 2012. Retrieved 18 February 2014.
- Sweetman, S, ed. (14 November 2012). Omacetaxine Mepesuccinate. “Martindale: The Complete Drug Reference”. Medicines Complete(Pharmaceutical Press).
- Li, Y. F.; Deng, Z. K.; Xuan, H. B.; Zhu, J. B.; Ding, B. H.; Liu, X. N.; Chen, B. A. (2009). “Prolonged chronic phase in chronic myelogenous leukemia after homoharringtonine therapy”. Chinese medical journal122 (12): 1413–1417. PMID 19567163. edit
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. (2009). “Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009”. Cancer 115 (23): 5382–5393.doi:10.1002/cncr.24601. PMID 19739234. edit
- Wu, L.; Li, X.; Su, J.; Chang, C.; He, Q.; Zhang, X.; Xu, L.; Song, L.; Pu, Q. (2009). “Effect of low-dose cytarabine, homoharringtonine and granulocyte colony-stimulating factor priming regimen on patients with advanced myelodysplastic syndrome or acute myeloid leukemia transformed from myelodysplastic syndrome”. Leukemia & Lymphoma50 (9): 1461. doi:10.1080/10428190903096719. edit
- Gu, L. F.; Zhang, W. G.; Wang, F. X.; Cao, X. M.; Chen, Y. X.; He, A. L.; Liu, J.; Ma, X. R. (2010). “Low dose of homoharringtonine and cytarabine combined with granulocyte colony-stimulating factor priming on the outcome of relapsed or refractory acute myeloid leukemia”.Journal of Cancer Research and Clinical Oncology 137 (6): 997–1003.doi:10.1007/s00432-010-0947-z. PMID 21152934. edit
- Kantarjian, H. M.; Talpaz, M.; Santini, V.; Murgo, A.; Cheson, B.; O’Brien, S. M. (2001). “Homoharringtonine”. Cancer 92 (6): 1591–1605.doi:10.1002/1097-0142(20010915)92:6<1591::AID-CNCR1485>3.0.CO;2-U. PMID 11745238. edit
- Wetzler M, Segal D. Omacetaxine as an Anticancer Therapeutic: What is Old is New Again. Current Pharmaceutical Design 2011;17:59-64
- Concise total synthesis of (±)-cephalotaxine via a transannulation strategy: Development of a facile reductive oxy-nazarov cyclization
Org Lett 2011, 13(13): 3538 - The first semi-synthesis of enantiopure homoharringtonine via anhydrohomoharringtonine from a preformed chiral acyl moiety
Tetrahedron Lett 1999, 40: 2931 - Synthesis of homoharringtonine and its derivative by partial esterification of cephalotaxine
Tetrahedron Lett 1982, 23(34): 3431 - Construction of chiral tertiary alcohol stereocenters via the (2,3)-Meisenheimer rearrangement: Enantioselective synthesis of the side-chain acids of homoharringtonine and harringtonine
J Org Chem 2013, 78(2): 339 - Studies in Cephalotaxus alkaloids. Stereospecific total synthesis of homoharringtonine
J Org Chem 1983, 48(26): 5321 - Chemistry – A European Journal, 2008 , vol. 14, 14 pg. 4293 – 4306
| WO2000040269A2 * | Jan 5, 2000 | Jul 13, 2000 | Clarence C Lee | Pharmaceutical compositions for treatment of diseased tissues |
| WO2002032904A1 * | Oct 17, 2000 | Apr 25, 2002 | Oncopharm Corp | New cephalotaxanes, their method of preparation and their use in treatment of cancers, leukemias, parasites including thus resistant to usual chemotherapeutic agents and as reversal agents |
| EP0393575A1 * | Apr 17, 1990 | Oct 24, 1990 | G.D. Searle & Co. | Neoplasia treatment compositions containing antineoplastic agent and side-effect reducing protective agent |
| USH271 * | Dec 18, 1985 | May 5, 1987 | The United States Of America As Represented By The Secretary Of The Army | Treatment of malaria with esters of cephalotaxine |
| US7169774 | Jun 25, 2004 | Jan 30, 2007 | Stragen Pharma S.A. | Cephalotaxane derivatives and their processes of preparation and purification |
| US7842687 | May 25, 2006 | Nov 30, 2010 | Chemgenex Pharmaceuticals, Inc. | Cephalotaxane derivatives and their processes of preparation and purification |
| US8466142 | Mar 3, 2009 | Jun 18, 2013 | Sloan-Kettering Institute For Cancer Research | Cephalotaxus esters, methods of synthesis, and uses thereof |
| Reference | ||
|---|---|---|
| 1 | * | KANTARJIAN H.M. ET AL: “Chronic myelogenous leukemia – Progress at the M. D. Anderson Cancer Center over the past two decades and future directions: First Emil J Freireich Award Lecture.” CLINICAL CANCER RESEARCH, (1997) 3/12 II (2723-2733). , XP001095529 |
| 2 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 1. Pharmacokinetic study in dogs and HHT determination in blood in using LC-MS method.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 179B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095449 |
| 3 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 2. Tolerance in humans and case report of a refractory patient with AML treated by very small dose of SQ HHT.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 202B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095450 |
| 4 | * | WHAUN J M ET AL: “TREATMENT OF CHLOROQUINE -RESISTANT MALARIA WITH ESTERS OF CEPHALOTAXINE HOMOHARRINGTONINE.” ANN TROP MED PARASITOL(1990) 84(3), 229-237, XP008006193 |
1H NMR

13 CNMR

HPLC

