
Home » 2014 (Page 83)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
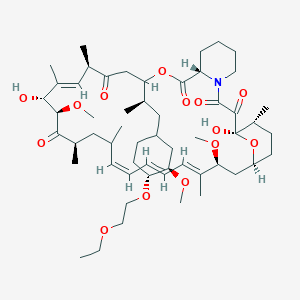
Umirolimus, Biolimus

Umirolimus, Biolimus
Biosensors (Originator)
40 -O-[(2′-ethoxy) ethyl]rapamycin
(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-12-{(2R)-1-[(1S,3R,4R)-4-(2-Ethoxyethoxy)-3-methoxycyclohexyl]-2-propanyl}-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36 -dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentone
Umirolimus inhibits T cell and smooth muscle cell proliferation, and was designed for use in drug eluting stents. This analog has a chemical modification at position 40 of the rapamycin ring. It has potent immunosuppressive properties that are similar to those of sirolimus, but the drug is more rapidly absorbed by the vessel wall, readily attaches and enters smooth muscle cell membranes causing cell cycle arrest at G0, and is comparable to sirolimus in terms of potency.
The key biologic event associated with the restenotic process is clearly the proliferation of smooth muscle cells in response to the expansion of a foreign body against the vessel wall. This proliferative response is initiated by the early expression of growth factors such as PDGF isoforms, bFGF, thrombin, which bind to cellular receptors.
However, the key to understanding the mechanism by which compounds like umirolimus inhibit cell proliferation is based on events which occur downstream of this growth factor binding. The signal transduction events which culminate in cell cycle arrest in the G1 phase are initiated as a result of ligand binding to an immunophilin known as FK binding protein-12. The FK designation was based on early studies conducted with tacrolimus, formerly known as FK-506, which binds this cytoplasmic protein with high affinity.
Subsequent investigations showed that rapamycin also binds to this intracellular target, forming an FKBP12–rapamycin complex which is not in itself inhibitory, but does have the capacity to block an integral protein kinase known as target of rapamycin (TOR). TOR was first discovered in yeast J.N. Heitman, N.R. Movva and M.N. Hall, Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast, Science 253 (1991), pp. 905–909. View Record in Scopus | Cited By in Scopus (434)and later identified in eukaryotic cells, where it was designated as mTOR, the mammalian target of rapamycin. The importance of mTOR is based on its ability to phosphorylate a number of key proteins, including those associated with protein synthesis (p70s6kinase) and initiation of translation (4E-BP1).
Of particular significance is the role that mTOR plays in the regulation of p27kip1, an inhibitor of cyclin-dependent kinases such as cdk2. The binding of agents like rapamycin and umirolimus to mTOR is thought to block mTOR’s crucial role in these cellular events, resulting in arrest of the cell cycle, and ultimately, cell proliferation.
Introduction
It is known that Biolimus A9, a rapamycin derivative, is an immunosuppressant, and is also proven to have anti-tumor and anti-fungal effect.
Several prior arts had disclosed the improvements of the product yield of rapamycin derivatives. U.S. Pat. No. 7,193,078 to Isozaki et al. disclosed a process for producing Biolimus A9, giving an example to obtain a yield of 46% by reacting rapamycin with 2-ethoxyethyl trifluoromethane sulfonate (or 2-ethoxyethyl triflate) in an organic solvent.
However, the Isozaki’s prior art still has the following drawbacks:
- 1. Even one example ever showed a 46% yield of Biolimus A9, it however just revealed a small-scale laboratory experiment with only one gram (1.09 mmol) of rapamycin and 1.95g (8.78 mmol) of 2-ethoxyethyl triflate. After amplifying or expanding the process to be larger scale, the yield will be remarkably reduced to thereby decrease the commercial or industrial value of this prior art (Note: The low yield after simulated process amplification will be hereinafter discussed in Examples 3, 4 of this application).
- 2. Even the reactant of 2-ethoxyethyl triflate is a compound with high activity, it is unstable and will be decomposed such as after being stored for one week at room temperature. Also, the triflate is not UV-absorbable and is therefore unsuitable for process tracking when proceeding the reaction. Such poor properties will affect the material storage, production scheduling and process tracking for commercially making the Biolimus A9.
sirolimus 42 – ether derivatives are a class of sirolimus derivative, is a new generation macrolide immunosuppressant and anticancer drugs. The compounds discovered by the Swiss company Sandoz, mainly applicable to organ transplant recipient’s immune suppression and cancer. The synthesis of such substances currently on the patent literature have W09409010, CN102127092A and CN102268015A.
Patent Document W09409010 on Synthesis of this type of structure are used sirolimus protected materials in acidic or neutral reaction conditions, and then removing the protective group to obtain the target product. Such as 42-0 – (4 – hydroxymethyl) benzyl – sirolimus, the first synthesis of the formula V, and then removal of silyl ether protecting groups have the formula VI.

This synthetic method has several drawbacks: 1, the reaction reagent relatively difficult to obtain; 2, the intermediate prepared in the reaction yield is low;
3, sirolimus, structural part to participate in a two-step reaction, reduction reaction yield, costs.
CN102127092A mention a synthetic sirolimus 42 – ether derivative everolimus one way. This synthetic route similar to the W09409010 (route of reaction formula 1), but with silane reagents and reaction conditions are different.First reaction toluene as solvent, 50 ° C _60 ° C between the reaction and after-treatment of the intermediate after the first column chromatography, yield 32%.The second step in tetrahydrofuran as a solvent, the reaction overnight at 0 ° C, after treatment by a column chromatography to give the product, the yield was 66%, with a total yield of 21.1%.
Reaction Scheme I:

This method has the defects include: 1, the reaction reagent relatively difficult to obtain. 2, the structure part of sirolimus to attend two-step reaction, reduction reaction yield, costs. 3, the use of highly toxic solvents, are not suitable for practical application. 4, the reaction temperature is relatively harsh, difficult to control.
CN102268015A discloses a method for synthesizing everolimus. The first step to sirolimus or sirolimus derivatives as raw materials in -20 ° C was added dropwise trifluoromethanesulfonic anhydride and incubated for 3 hours, was isolated intermediates 02, the yield was 87.4% or 95.32%. The second step of the intermediate 02 with ethylene glycol mono-protected in 50 ° C reaction intermediate 03 was isolated in a yield of 79.0% or 76.78%. The third step of dilute hydrochloric acid was added dropwise at room temperature intermediate 03 was deprotected product everolimus. The total yield was 48.4% or 52.5%. See Reaction Scheme 2 synthetic route.
Reaction Scheme 2:

The method of the defects as follows: 1, to protection and deprotection of ethylene glycol. 2, the reaction steps excessive structural part to participate in the sirolimus-step reaction. 3, the reaction yield improved, but also greatly increased operating costs.
………………..
.
Examples 5,42-0-(2_ ethoxy) ethyl – Synthesis of sirolimus
[0044] In the IOOml three-necked flask was added 2g of sirolimus, 2. 78g of 4_ dimethylaminopyridine, 5. 34g of chlorine acid glycol ester and 20ml of acetonitrile, 35 ° C The reaction was stirred 36 hours ended. The reaction solution was poured into an equal volume of saturated sodium bicarbonate solution and extracted with 5% potassium bisulfate solution was washed twice with a saturated sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated through the column. Silica gel column chromatography (EA: PE = I :20-2: 1), obtained by rotary evaporation 42-0 – (2 – ethoxy) ethyl – sirolimus I. 57g (yield: 73.3 %). HPLC analysis showed that: a purity of 88.2%.
…………


the Biolimus A9 of the present invention will be presented as below-mentioned:
Biolimus A9

Reaction Parameters
Quantity of Alkylbenzene Sulfonate: 1˜20 equivalents, preferably being 5˜10 equivalents, per equivalent of sirolimus.
Reaction Temperature: 40˜80° C., preferably being 55˜65° C. Reaction Time: 12˜72 hours, preferably being 16˜30 hours.
After the reaction is completed, the rough product is collected, washed, dried and purified to obtain the Biolimus A9 of the present invention with high yield of 45%.
Since the product Biolimus A9 is a polyene macrolide, which is easily oxidized and decomposed during the storage or material handling.
Accordingly, a proper antioxidant may be homogeneously mixed with the Biolimus A9 to enhance the stability when stored or handled.
The proper antioxidants may be selected from: Butylatd hydroxytoluene (BHT), DL-α-tocopherol, propyl gallate, ascorbyl palmitate, 3-tert-butyl-4-hydroxyanisole, 2-tert-butyl-4-hydroxyanisole, and fumaric acid.
The Butylated hydroxytoluene (BHT) is the most preferable antioxidant adapted for use in the present invention.
The process for making Biolimus A9 in accordance with the present invention will be described in detail in view of the following examples:
EXAMPLE 1
A. Synthesis of 2-ethoxyethyl pentafluorobenzene sulfonate
In a reaction flask, 25 grams (93.8 mmol) of pentafluorobenzene sulfonyl chloride (or pentafluorobenzene sulfochloride) and 86 ml of tetrahydrofuran were added and nitrogen gas was filled into the flask.
The flask is then cooled to 0° C. and is dripped therein with 2-ethoxyethanol (8.5g, 94.5 mmol) and triethyl amine (15 g, 148.5 mmol). After dripping, the reaction solution is stirred for 30 minutes, and then filtered, concentrated and the residue is separated from the solution and further purified by silica gel column chromatography to obtain a colorless oily product of 2-ethoxyethyl pentafluorobenzene sulfonate (26.6 g, 83.1 mmol) having a yield of 88.6%.
B. Synthesis of Biolimus A9
In a reaction flask, 1 g (1.1 mmol) of sirolimus, 7.8 g (60.3 mmol) of ethyl di-isopropyl amine, 3.5 ml of methylene chloride and 2.8 g (8.7 mmol) of 2-ethoxyethyl pentrafluorobenzene sulfonate as previously obtained were added therein.
The reaction mixture in the flask was heated to 60° C. and agitated for 23 hours. It is cooled, and further added therein with ethyl acetate (100 ml) and aqueous solution of hydrochloric acid (1N, 100 ml ) under agitation.
Then, it is settled for separating the organic and aqueous layers. The organic layer is collected, and washed with pure water (100 ml) and saturated saline (100 ml). The washed organic liquid is then dried and concentrated. The residue is then separated from the liquid and further purified by silica gel column chromatography to obtain white solid product of Biolimus A9(0.49 g, 0.5 mmol) with a yield of 45.4%.
EXAMPLE 2 Process Amplification of Example 1B
In a reaction flask, 10 g (10.9 mmol) of sirolimus, 78 g (603.5 mmol) of ethyl di-isopropyl amine, 35 ml of methylene chloride and 28 g (87.4 mmol) of 2-ethoxyethyl pentrafluorobenzene sulfonate were added therein.
The reaction mixture in the flask was heated to 60° C. and agitated for 24 fours. It is cooled, and further added therein with ethyl acetate (500 ml) and aqueous solution of hydrochloric acid (1N, 500 ml ) under agitation.
Then, it is settled for separating the organic and aqueous layers. The organic layer is collected, and washed with pure water (500 ml) and saturated saline (400 ml). The washed organic liquid is then dried and concentrated. The residue is then separated from the liquid and further purified by silica gel column chromatography to obtain white solid product of Biolimus A9(4.8 g, 4.9 mmol) with a yield of 44.5%.
This example is a process amplification of the previous Example 1, Step B, by amplifying or expanding the quantity of each reactant for about 10 times of that of the Example 1 (of small scale).
By the way, the production yield (44.5%) of this Example is still as high as that of the previous Example 1 of small scale. It indicates that the reproducibility of high yield can still be obtained in accordance with the present invention even after process amplification, proving that the present invention is suitable for commercialization or mass production. The product may be further purified to obtain a high-purity final product of Biolimus A9 such as by middle-performance liquid chromatography or the like. The Biolimus A9 thus obtained is identified by the X-ray powder diffractogramm as shown in the single drawing FIGURE as attached herewith.
EXAMPLE 3 Comparative Example for Simulating the Process of the Prior Art of U.S. Pat. No. 7,193,078
A. Synthesis of 2-ethoxyethyl trifluoromethane sulfonate
In a reaction flask, 2-ethoxyethanol (10 g, 111 mmol), methylene chloride (177 ml) and 2,6-dimethyl pyridine (23.8 g, 222.3 mmol) were added into the flask, which is filled therein with nitrogen gas. It is cooled to 0° C. and added dropwise with trifluoromethane sulfonic acid anhydride (37.6 g, 133.4 mmol). After completing the dripping of said sulfonic acid anhydride, the reaction mixture is agitated for one hour and a saturated aqueous solution of ammonium chloride (20 ml) is added and further agitated for 10 minutes.
It is then settled for separating the layers. The organic layer is collected, and is respectively washed with aqueous solution of hydrochloric acid (1N, 100 ml), pure water (100 ml), saturated aqueous solution of sodium bicarbonate (100 ml) and saturated saline (100 ml). The washed organic layer is dried, concentrated and the residue is then separated and further purified with silica gel column chromatography to obtain the oily product of 22.5 g (101.3 mmol) of 2-ethoxyethyl trifluoromethane sulfonate (or 2-ethoxyethyl triflate), with a yield of 91.3%.
B. Synthesis of Biolimus A9
In a reaction flask, sirolimus (1 g, 1.1 mmol), ethyl di-isopropyl amine (7.8 g, 60.3 mmol), methylene chloride (3.5 ml) and 2-ethoxyethyl triflate (2.0 g, 8.8 mmol) as previously made in Example 3A were added into the flask, which is filled with nitrogen gas. The reaction mixture is heated to 60° C. and is agitated for one hour and twenty minutes. Then, it is cooled, added with ethyl acetate (100 ml) and aqueous solution of hydrochloric acid (1N, 100 ml) and is further agitated. After agitation, it is settled for separating the layers. The organic layer is collected and respectively washed with pure water (100 ml), saturated saline (80 ml). The washed organic layer is dried and concentrated. The residue is then separated and purified by silica gel column chromatography to obtain white product of Biolimus A9 (0.48 g, 0.49 mmol), with a yield of 44.5%.
EXAMPLE 4 Comparative Example for Simulative Process Amplification of Example 3B
In a reaction flask, sirolimus (10 g, 10.9 mmol), ethyl di-isopropyl amine (78 g, 603.5 mmol), methylene chloride (35 ml) and 2-ethoxyethyl triflate (20 g, 88 mmol), each having a quantity about 10 times of that used in Example 3B, were added into the flask, which is filled with nitrogen gas. The reaction mixture is heated to 60° C. and is agitated for one hour and twenty minutes. Then, it is cooled, added with ethyl acetate (500 ml) and aqueous solution of hydrochloric acid (1N, 500 ml) and is further agitated. After agitation, it is settled for separating the layers. The organic layer is collected and respectively washed with pure water (500 ml), saturated saline (400 ml). The washed organic layer is dried and concentrated. The residue is then separated and purified by silica gel column chromatography to obtain white product of Biolimus A9 (2.9 g, 2.9 mmol), having a yield of 26.8% only.
Comparatively, via this process amplification, the yield of Biolimus A9 of the prior art is remarkably reduced in comparison with its small-scale production (Example 3B). Therefore, the prior art of U.S. Pat. No. 7,193,078 may be considered as a process especially suitable for small-scale production, such as a laboratory experiment, rather than a large-scale commercial or industrial production, which is thus inferior to this application, when compared with this application which has shown the high yields both in small-scale process (Example 1) and large-scale process (Example 2).
Accordingly, this application is more suitable for commercialization for mass production.
Moreover, the essential reactant of 2-ethoxyethyl triflate of the prior art (U.S. Pat. No. 7,193,078), even having high activity, is unstable because it will be decomposed into unknown compounds after one-week storage (by NMR spectrographic detection) as accompanied with physical change from its original colorless transparent liquid to a black viscous oily product, to thereby be inferior to this application because the 2-ethoxyethyl pentafluoro benzene sulfonate (which is obviously different from the 2-ethoxyethyl triflate as used in the prior art) of this application is still stable after one-week storage as aforementioned.
Furthermore, the 2-ethoxyethyl pentafluorobenzene sulfonate of this application may absorb ultra-violet rays to have a better tractability during the process proceeding than that of the 2-ethoxyethyl triflate (which is not UV-absorbable) of the prior art. So, this application is also beneficial for better production scheduling, reliable process tracking and efficient production management than the prior art.
So, this application is more suitable for commercial production even when considering the stability of product storage and improvement of process monitoring, control and management.
EXAMPLE 5
The Biolimus A9, as obtained from Example 2, is respectively added with anti-oxidant, namely Butylated Hydroxytoluene (or BHT), for 0.1%, 0.2, 0.5%, and 1% (w/w) based on 100% (wt) of Biolimus to enhance its stability at 40° C. by revealing a high yield of more than 99.4% even after six-week storage. Comparatively, a control test is provided by adding 0% of anti-oxidant (BHT) into Biolimus A9, resulting in a reduction of yield to be 69.7% after six-week storage. The yield data of different amounts of anti-oxidant as added into Biolimus A9 with respect to time lapse of weeks are summarized in Table 1 as below-mentioned.
………………………….
The O-(2-ethoxyethyl)-rapamycin can be produced by reaction between rapamycin and 2-ethoxyethyl triflate in the presence of N,N-diisopropylethylamine in methylene chloride.

An example of the O-alkylrapamycin derivative (with R=hydroxyalkyl) is O-(2-hydroxyethyl)-rapamycin represented by the general formula 3 below.
The O-(2-hydroxyethyl)-rapamycin can be produced by reaction between rapamycin and t-butyldimethylsilyloxyethyl triflate in the presence of N,N-diisopropylethylamine in methylene chloride, followed by deprotecting of t-butyldimethylsilyl group.

EXAMPLES
The invention will be described with reference to the following examples, which demonstrate the efficient production of O-alkylrapamycin derivatives by the process of the present invention.
Example 1
- (1) Synthesis of 2-ethoxyethyl Triflate
In a round bottom flask containing a stirring bar was placed 9.0 g (100 mmol) of ethoxyethanol. The atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. The flask was given 160 mL of methylene chloride and 23.3 mL (120 mmol) of 2,6-lutidine. The flask cooled with ice was given dropwise 20.2 mL (120 mmol) of trifluoromethanesulfonic acid anhydride over 20 minutes. After stirring for 1 hour, the reaction liquid was mixed with 20 mL of saturated solution of ammonium chloride. The resulting mixture was washed sequentially with 1N hydrochloric acid (100 mL), deionized water (100 mL), saturated solution of sodium hydrogen carbonate (100 mL), and saturated aqueous solution of sodium chloride (100 mL). The organic layer was separated and dried with anhydrous sodium sulfate. With the sodium sulfate filtered off, the solution was concentrated under reduced pressure. The residue underwent silica gel chromatography. Thus there was obtained 15.03 g (67.6% yields) of 2-ethoxyethyl triflate from the fraction in eluate of 20% ethyl acetate-hexane.
- (2) Synthesis of 40-O-[(2′-ethoxy)ethyl]rapamycin
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of methylene chloride for dissolution. To the flask was further added 10 mL (57.5 mmol) of N,N-diisopropylethylamine and 1.95 g (8.78 mmol) of the previously synthesized 2-ethoxyethyl triflate with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated by using a rotary evaporator. The concentrated solution was purified using a column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1 v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 494 mg (0.501 mmol) of the desired product (46% yields).
Example 2
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of chloroform for dissolution. To the flask was further added 10 mL (57.5 mmol) of N,N-diisopropylethylamine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 451 mg (0.458 mmol) of the desired product (42% yields).
Example 3
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of methylene chloride for dissolution. To the flask was further added 8 mL (57.4 mmol) of triethylamine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica-gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 344 mg (0.349 mmol) of the desired product (32% yields).
Example 4
In 2 mL of methanol was dissolved 500 mg of the 40 -O-[(2′-ethoxy)ethyl]rapamycin which had been obtained in Example 1. The resulting solution was added dropwise to 20 mL of deionized water with stirring. The solids which had precipitated out were filtered off and washed with a small amount of water and finally dried under reduced pressure at 40° C. for more than 10 hours. Thus there was obtained 483 mg of white powder.
This product gave an NMR chart as shown in FIG. 1. This NMR chart indicates the structure of 40 -O-[(2′-ethoxy) ethyl]rapamycin represented by the general formula 4.

Comparative Example
A sample of 40-O-[(2′-ethoxy)ethyl]rapamycin was synthesized by the process disclosed in WO94/09010 official gazette so as to evaluate yields.
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of toluene for dissolution. To the flask was further added 467 mg (4.36 mmol) of 2,6-lutidine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 247 mg (0.251 mmol) of the desired product (23% yields).
NMR

http://www.google.com/patents/US20050192311
This product gave an NMR chart as shown in FIG. 1. This NMR chart indicates the structure of 40-O-[(2′-ethoxy)ethyl]rapamycin represented by the general formula 4.

| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050131008 | Nov 12, 2004 | Jun 16, 2005 | Sun Biomedical, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US7193078 * | Mar 1, 2005 | Mar 20, 2007 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| WO2012017449A1 | Aug 2, 2011 | Feb 9, 2012 | Meril Life Sciences Pvt. Ltd | Process for preparation of novel 42-0-(heteroalkoxyalkyl) rapamycin compounds with anti-proliferative properties” |
| US7872122 | May 8, 2009 | Jan 18, 2011 | Chunghwa Chemical Synthesis & Biotech Co., Ltd. | Process for making Biolimus A9 |
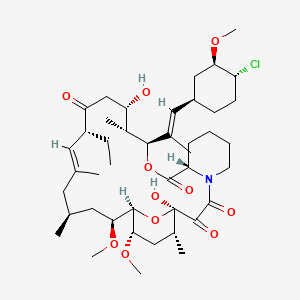
Pimecrolimus Пимекролимус…For treatment of mild to moderate atopic dermatitis.

Pimecrolimus
137071-32-0 cas
(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)- 3-{(E)-2-[(1R,3R,4S)-4-Chloro-3-methoxycyclohexyl]- 1-methylvinyl}-8-ethyl-5,6,8,11,12,13,14,15,16,17,
18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy- 14,16-dimethoxy-4,10,12, 18-tetramethyl-15,19-epoxy- 3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1, 7,20,21(4H,23H)-tetrone
The systematic name of pimecrolimus is (lR,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(lE)-2- {(1 R,3R,4S)-4-chloro-3-methoxycyclohexyl} – 1 -methylvinyl] – 17-ethyl- 1,14- dihydroxy-23,25-dimethoxy-13,19,21,27-tetramethyl-ll,28-dioxa-4-aza- tricyclo[22.3.1.04‘9]octacos-18-ene-2,3,10,16-tetraone.
Pimecrolimus is the 32 epichloro derivative of ascomycin.
|
4-11-2008
|
Pharmaceutical Composition
|
| Canada | 2200966 | 2006-12-19 | expiry 2015-10-26 |
| United States | 6423722 | 1998-12-26 | 2018-12-26 |
PATENT AND EXPIRY DATE
| 5912238 | Jun 15, 2016 | |
| 5912238*PED | Dec 15, 2016 | |
| 6352998 | Oct 26, 2015 | |
| 6352998*PED | Apr 26, 2016 | |
| 6423722 | Jun 26, 2018 | |
| 6423722*PED | Dec 26, 2018 |
Viktor Gyollai, Csaba Szabo, “Methods of preparing pimecrolimus.” U.S. Patent US20060142564, issued June 29, 2006.

NDA..021302, 13 DEC 2001… VALEANT BERMUDA..ELIDEL1% TOPICAL CREAM
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is currently available as a topical cream, once marketed by Novartis, (however Galderma will be promoting the molecule in Canada in early 2007) under the trade name Elidel.
NMR…http://file.selleckchem.com/downloads/nmr/S500401-Pimecrolimus-NMR-Selleck.pdf
HPLC…….http://file.selleckchem.com/downloads/hplc/S500401-Pimecrolimus-HPLC-Selleck.pdf
http://file.selleckchem.com/downloads/hplc/S500401-Pimecrolimus-HPLC-Selleck.pdf
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is available as a topical cream, once marketed by Novartis (however, Galderma has been promoting the compound in Canada since early 2007) under the trade name Elidel.

Pimecrolimus is an ascomycin macrolactam derivative. It has been shown in vitro that pimecrolimus binds to macrophilin-12(also referred to as FKBP-12) and inhibits calcineurin. Thus pimecrolimus inhibits T-cell activation by inhibiting the synthesis and release of cytokines from T-cells. Pimecrolimus also prevents the release of inflammatory cytokines and mediators from mast cells.
Pimecrolimus is a chemical that is used to treat atopic dermatitis (eczema). Atopic dermatitis is a skin condition characterized by redness, itching, scaling and inflammation of the skin. The cause of atopic dermatitis is not known; however, scientists believe that it may be due to activation of the immune system by various environmental or emotional triggers. Scientists do not know exactly how pimecrolimus reduces the manifestations of atopic dermatitis, but pimecrolimus reduces the action of T-cells and mast cells which are part of the immune system and contribute to responses of the immune system. Pimecrolimus prevents the activation of T-cells by blocking the effects of chemicals (cytokines) released by the body that stimulate T-cells. Pimecrolimus also reduces the ability of mast cells to release chemicals that promote inflammation.
Pimecrolimus, like tacrolimus, belongs to the ascomycin class of macrolactam immunosuppressives, acting by the inhibition of T-cell activation by the calcineurin pathway and inhibition of the release of numerous inflammatory cytokines, thereby preventing the cascade of immune and inflammatory signals.[1] Pimecrolimus has a similar mode of action to that of tacrolimus but is more selective, with no effect on dendritic (Langerhans) cells.[2] It has lower permeation through the skin than topical steroids or topical tacrolimus[3] although they have not been compared with each other for their permeation ability through mucosa. In addition, in contrast with topical steroids, pimecrolimus does not produce skin atrophy.[4] It has been proven to be effective in various inflammatory skin diseases, e.g., seborrheic dermatitis,[5] cutaneous lupus erythematosus,[6]oral lichen planus,[7] vitiligo,[8] and psoriasis.[9][10] Tacrolimus and pimecrolimus are both calcineurin inhibitors and function as immunosuppressants.[11]
Ascomycin macrolactams belong to a new group of immunosuppressive, immunomodulatory and anti-inflammatory agents and include, e.g., ascomycin (FK520), tacrolimus (FK506) and pimecrolimus (ASM 981). The main biological effect of ascomycin macrolactams appears to be the inhibition of the synthesis of both Th1 and Th2-type cytokines in target cells.
As used herein, the term “ascomycin macrolactam” means ascomycin, a derivative of ascomycin, such as, e.g., tacrolimus and pimecrolimus, or a prodrug or metabolite of ascomycin or a derivative thereof.
Ascomycin, also called immunomycin, is a structurally complex macrolide produced by Streptomyces hygroscopicus. Ascomycin acts by binding to immunophilins, especially macrophilin-12. It appears that ascomycin inhibits the production of Th1 (interferon- and IL-2) and Th2 (IL-4 and IL-10) cytokines. Additionally, ascomycin preferentially inhibits the activation of mast cells, an important cellular component of the atopic response. Ascomycin produces a more selective immunomodulatory effect in that it inhibits the elicitation phase of allergic contact dermatitis but does not impair the primary immune response when administered systemically. The chemical structure of ascomycin is depicted below.

Tacrolimus (FK506) is a synthetic derivatives of ascomycin. As a calcineurin inhibitor, it works through the FK-binding protein and inhibits the dephosphorylation of nuclear factor of activated T cells (NFAT), thereby preventing the transport of the cytoplasmic component of NFAT to the cell nucleus. This leads to transcriptional inhibition of proinflammatory cytokine genes such as, e.g., interleukin 2, which are dependent on the nuclear factor of activated NFAT. The chemical structure of tacrolimus is depicted below.

Pimecrolimus, an ascomycin derivative, is a calcineurin inhibitor that binds with high affinity to the cytosolic receptor macrophilin-12, inhibiting the calcium-dependent phosphatase calcineurin, an enzyme required for the dephosphorylation of the cytosolic form of the nuclear factor of the activated T cell (NF-AT). It thus targets T cell activation and proliferation by blocking the release of both TH1 and TH2 cytokines such as IF-g, IL-2, -4, -5, and -10.3 It also prevents the production of TNF-a and the release of proinflammatory mediators such as histamine, hexosaminidase, and tryptase from activated mast cells.3 It does not have general antiproliferative activity on keratinocytes, endothelial cells, and fibroblasts, and in contrast to corticosteroids, it does not affect the differentiation, maturation, functions, and viability of human dendritic cells. The chemical structure of pimecrolimus is depicted below.

Pimecrolimus is an anti-inflammatory compound derived from the macrolactam natural product ascomycin, produced by certain strains of Streptomyces.

In January 2006, the United States Food and Drug Administration (FDA) announced that Elidel packaging would be required to carry a black box warning regarding the potential increased risk of lymph node or skin cancer, as for the similar drug tacrolimus. Whereas current practice by UKdermatologists is not to consider this a significant real concern and they are increasingly recommending the use of such new drugs.[12]
Importantly, although the FDA has approved updated black-box warning for tacrolimus and pimecrolimus, the recent report of the American Academy of Dermatology Association Task Force finds that there is no causal proof that topical immunomodulators cause lymphoma or nonmelanoma skin cancer, and systemic immunosuppression after short-term or intermittent long-term topical application seems an unlikely mechanism.[13] Another recent review of evidence concluded that postmarketing surveillance shows no evidence for this systemic immunosuppression or increased risk for any malignancy.[14] However, there are still some strong debates and controversies regarding the exact indications of immunomodulators and their duration of use in the absence of active controlled trials.[15] Dermatologists’ and Allergists’ professional societies, the American Academy of Dermatology[1], and the American Academy of Allergy, Asthma, and Immunology, have protested the inclusion of the black box warning. The AAAAI states “None of the information provided for the cases of lymphoma associated with the use of topical pimecrolimus or tacrolimus in AD indicate or suggest a causal relationship.”[2].

ELIDEL® (pimecrolimus) Cream 1% contains the compound pimecrolimus, the immunosuppressant 33-epi-chloro-derivative of the macrolactam ascomycin.
Chemically, pimecrolimus is (1R,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(1E)-2{(1R,3R,4S)-4-chloro-3-methoxycyclohexyl}-1-methylvinyl]-17-ethyl-1,14-dihydroxy-23,25 dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-aza-tricyclo[22.3.1.0 4,9]octacos-18-ene2,3,10,16-tetraone.
The compound has the empirical formula C43H68CINO11 and the molecular weight of 810.47. The structural formula is
 |
Pimecrolimus is a white to off-white fine crystalline powder. It is soluble in methanol and ethanol and insoluble in water.
Each gram of ELIDEL Cream 1% contains 10 mg of pimecrolimus in a whitish cream base of benzyl alcohol, cetyl alcohol, citric acid, mono- and di-glycerides, oleyl alcohol, propylene glycol, sodium cetostearyl sulphate, sodium hydroxide, stearyl alcohol, triglycerides, and water.
The second representative of the immunosuppressive macrolides for topical application – after tacrolimus (Protopic ®) – has 21 October in the trade. Pimecrolimus is approved for short-term and intermittent long-term treatment for patients aged two years who suffer from mild to moderate atopic dermatitis.
Pimecrolimus is a lipophilic derivative of macrolactam Ascomycin. The macrolides inhibit the production and release of pro-inflammatory cytokines by blocking the phosphatase calcineurin.The anti-inflammatory effect unfolds the drug in the skin. Since he is only minimally absorbed to not measurable, it hardly affects the local or systemic immune response. Therefore, the authorization neither restricts nor a maximum daily dose treatable area or duration of therapy.The cream can also be applied on the face, head and neck, and in skin folds, but not simultaneously with other anti-inflammatory topical agents such as glucocorticoids.
In studies in phases II and III patients aged three months and treated a maximum of one year.In two six-week trials involving 186 infants and young children as well as 403 children and adolescents, the verum symptoms and itching decreased significantly better than the cream base. Already in the first week of itching in 44 percent of children and 70 percent of the infants improved significantly. In adults, pimecrolimus was less effective than 0.1 percent betamethasone 17-valerate.
In the long-term treatment the verum significantly reduced the incidence of flares, revealed two studies with 713 and 251 patients. About a half and one year each about twice as many of the small patients were free of acute disease exacerbations than with the cream base (example: 61 versus 34 per cent of children, 70 versus 33 percent of infants older than six months). Moreover, the use of topical corticosteroids decreased significantly.
In a study of 192 adults with moderate to severe eczema half suffered six months no relapses more (24 percent with placebo). In the long-term therapy pimecrolimus was less effective than 0.1 percent triamcinolone acetonide cream and 1 percent hydrocortisone cream in adults.
The new topicum is-apart from burning and irritation at the application site – relatively well tolerated. It is neither kontaktsensibilisierend still phototoxic or sensitizing and does not cause skin atrophy. As in atopic Ekzen but usually a long-term therapy is necessary studies can reveal long-term adverse effects of the immunosuppressant on the skin only beyond one year.Also available from direct comparative studies between tacrolimus and pimecrolimus. They could help to delineate the importance of the two immunosuppressants.
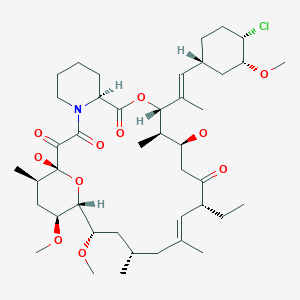

Pimecrolimus (registry number 137071-32-0; Figure 1) is a macro lide having anti-inflammatory, antiproliferative and immunosuppressive properties. This substance is present as an active ingredient in the Elidel ® drug recently approved in Europe and in the USA for topical treatment of inflammatory conditions of the skin such as atopic dermatitis.

Figure 1: structural formula of pimecrolimus
19th Ed., vol. π, pg. 1627, spray-drying consists of bringing together a highly dispersed liquid and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. Spray-drying however is often limited to aqueous solutions unless special expensive safety measures are taken. Also, in spite of the short contact time, certain undesirable physical and chemical characteristics of the emerging solids are in particular cases unavoidable. The turbulence present in a spray-drier as a result of the moving air may alter the product in an undesirable manner. Modifications to the spray-drying technique are disclosed in WO 03/063821 and WO 03/063822. [00012] European Patent EP 427 680 Bl discloses a method of synthesizing amorphous pimecrolimus (Example 66a). The method yields amorphous pimecrolimus as a colorless foamy resin.
U.S. Patent No. US 6,423,722 discloses crystalline forms of pimecrolimus, such as form A, form B, etc. US 722 also contend that by performing example 66a from the European Patent EP 427 680 Bl, amorphous pimecrolimus is obtained.
The preparation of pimecrolimus was described for the first time in the patent application EP427680 on behalf of Sandoz. Used as raw material in such document is ascomycin (compound identified by registry number 11011-38-4), a natural product obtained through fermentation from Streptomyces strains (such as for example Streptomyces hygroscopicus var ascomyceticus, or Streptomyces hygroscopicus tsukubaensis N°9993). Pimecrolimus is obtained from the ascomycin through a sequence of four steps of synthesis (scheme 1)

Scheme 1 : synthesis process described in EP427680
From a structural point of view, pimecrolimus is the 33-epi-chloro derivative of ascomycin. As described in EP427680, the simultaneous presence – in the structure of ascomycin – of two secondary hydroxyl groups in position 24 and in position 33, requires the protection of the hydroxyl in position 24 before substituting the second hydroxyl in position 33 with an atom of chlorine.
In order to obtain the monoprotection of the hydroxyl in position 24 of ascomycin, such synthesis process provides for the preparation of 24,33-disilyl derivative and the subsequent selective removal of the silyl ester in position 33.
The high ratio between the silylating agent and the substrate and the non-complete selectivity of the subsequent step of deprotection requires carrying out two chromatographic purifications on the column of silica gel (Baumann K., Bacher M., Damont A., Hogenauer K., Steck A. Tetrahedron, (2003), 59, 1075-1087). The general yields of such synthesis process are not indicated in literature; an experiment by the applicant revealed that such yields amount to about 16% molar starting from ascomycin.
Other synthesis processes were recently proposed as alternatives to the synthesis of EP427680.
In particular, the International patent application WO2006040111 on behalf of Novartis provides for the direct substitution of the hydroxyl in position 33 of ascomycin with an atom of chlorine and a second alternative, described in the international patent application WO2006060614 on behalf of Teva, uses – as a synthetic intermediate – a sulfonate derivative in position 33 of ascomycin. Both the proposed synthetic alternatives are not entirely satisfactory in that in WO2006040111 the proposed halogenating agents (chlorophosphorane and N- chlorosuccinimide) are not capable, according to the same authors, of regioselectively substituting the hydroxyl function in position 33, while in WO2006060614 the quality characteristics of the obtained product are, even after chromatographic purification and/or crystallisation, low for a product to be used for pharmaceutical purposes (i.e. purity of 96% as described in the experimental part).
Generally, purified enzymatic systems may be used for the organic synthesis of polyfunctional molecules (Wang Y-F, Wong C-H. J Org Chem (1988) 53, 3127- 3129; Santaniello E., Ferraboschi P., Grisenti P., Manzocchi A. Chem. Rev. (1992), 92(5), 1071-140; Ferraboschi P., Casati S., De Grandi S., Grisenti P., Santaniello E. Biocatalysis (1994), 10(1-4), 279-88); WO2006024582). WO2007103348 and WO2005105811 describe the acylation of rapamycin in position 42 in the presence of lipase from Candida antartica.
…………………….

Scheme 2: synthesis of pimecrolimus for enzymatic transesterification of ascomycin.

Scheme 3. Synthesis of pimecrolimus for enzyme-catalyzed alcoholysis from 33,24- diacetate of ascomycin
Example 1
Preparation of the 33-acetyl derivative of ascomvcin (compound I of scheme II)
Lipase from Candida antarctica (CAL B, Novozym 435) [0.140 g (2 U/mg)
FLUKA] was added to a solution of ascomycin (100 mg; 0.126 mmol) in toluene (8 ml) and vinyl acetate (4.5 eq; 0.473 g). The reaction is kept under stirring at the temperature of 30° C for 80 hrs then the enzyme is taken away for filtration and the filtrate is concentrated at low pressure to obtain 105 mg of 33-acetyl ascomycin.
A sample of such intermediate was purified for analytical purposes by chromatography on silica gel (n-hexane/acetone = 8/2 v/v as eluents) and thus crystallised by acetone/water.
The following analysis were carried out on such sample: 1H-NMR (500MHz) δ:
2.10 (CH3CO), 3.92 and 4.70 (24CH and 33CH); IR (cm-1): 3484.245, 2935.287,
1735.331, 1649.741, 1450.039,
1372.278; DSC: endotherm at 134.25° C; [α]D=-74,0° (c=0.5 CHCl3).
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H7iNO13: C 64.80%; H, 8.58%; N, 1.68%;
O, 24.94%
Elementary analysis found: C 64.78%; H, 8.54%; N, 1.59%; O, 24.89%
Preparation of the 24-tgrt-butyldimethylsilylether-33 -acetyl derivative of ascomvcin (intermediate 24-silyl-33-Oac; compound II of scheme 2)
2,6-lutidine (0.29Og; 2.7 mmolels) and tert-butyldimethylsilyl triflate (0.238g; 0.9 mmoles) are added to a solution of 33-acetyl derivative of ascomycin (150 mg;
0.18 mmoles) in dichloromethane (5ml). The reaction is left under stirring at ambient temperature for 30 minutes. After this period the reaction mixture is washed with a solution saturated with sodium bicarbonate (5 ml) and organic phase obtained is washed in sequence with HCl 0.1N (5 ml 3 times) and with a solution at 30% of NaCl (5ml). The organic phase is anhydrified on sodium sulphate, filtered and concentrated to residue under vacuum to obtain 128 mg of product.
Spectrum of MS (ESI +): m/z: 970.5 (M+23; 100.0%)
1H-NMR (500 MHz) δ: 0.05 and 0.06 ((CHs)2Si), 0.90 ((CH3)3C-Si), 2.10
(CH3CO), 4.70 (33CH)
IR (cm-‘): 3462.948, 2934.450, 1739.236, 1649.937
Elementary analysis calculated for C51H85NOi3Si: C 64.59%; H, 9.03%; N, 1.48%; O, 21.93%
Elementary analysis found: C 64.50%; H, 9.05%; N, 1.41%; O, 21.88%
DSC= endoderma a 236,43° C. [α]D=-81,4° (c=0.5 CHCl3).
Preparation of 24-tert-butyldimethylsilylether of ascomycin (intermediate 24- silyl-33-OH; compound III of scheme 2) n-octan-1-ol (0.035g; 0.265 mmoles) and CAL B (Novozym 435) [0.100 g (2
U/mg) FLUKA] are added to a solution of 24-tert-butyldimethylsilylether-33- acetyl derivative of ascomycin (50 mg; 0.053 mmoles) in tert-butylmethylether (4 ml). The reaction is kept under stirring at the temperature of 40° C for 120 hours.
After this period the reaction mixture is filtered and the filtrate is evaporated to residue under vacuum to obtain a reaction raw product which is purified by chromatography on silica gel: 44 mg of product (0.048 mmoles) are recovered through elution with petroleum ether/acetone 7/3.
The chemical/physical properties of the obtained product match those of a reference sample obtained according to patent EP427680.
Preparation of 24-tert-butyldimethylsilylether-33-epi-chloro ascomycin
(intermediate 24-silyl-33-chloro; compound IV of scheme 2)
A solution of 24-silyl FR520, i.e. 24-silyl ascomycin (165 g; 0.18 moles) in anhydrous toluene (1.4 litres) and pyridine (50 ml) is added to a suspension of dichlorotriphenylphosphorane (99.95g) in anhydrous toluene (1.1 litres), under stirring at ambient temperature (20-25 °C) in inert atmosphere.
After adding, the reaction mixture is heated at the temperature of 60° C for 1 hour.
After this period the temperature of the reaction mixture is taken to 25° C and thus the organic phase is washed in sequence with water (1 time with 1 L) and with an aqueous solution of NaCl at 10% (4 times with 1 L each time), then it is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain about 250 g of a moist solid of toluene. Such residue product is retaken with n- hexane (500 ml) and then evaporated to dryness (in order to remove the toluene present). The residue product is diluted in n-hexane (500 ml) under stirring at ambient temperature for about 45 minutes and then the undissolved solid taken away for filtration on buckner (it is the sub-product of dichlorophosphorane).
The filtrate is concentrated at low pressure to obtain 148.6 g of a solid which is subsequently purified by chromatography on silica gel (elution with n- heptane/acetone = 9/1) to obtain 123 g (0.13 moles) of product.
The chemical/physical properties of the obtained product match those described in literature (EP427680).
Preparation of the pimecrolimus from 24-fert-butyldimethylsilylether-33-epi- chloro ascomycin
The intermediate 24-silyl-33 chloro (123g; 0.13 Moles; compound IV of scheme
2) is dissolved under stirring at ambient temperature in a dichloromethane/methanol mixture=l/l=v/v (1.1 litres) then p-toluenesulfonic acid monohydrate (10.11 g) is added.
The reaction is kept under stirring at the temperature of 20-25° C for 72 hours, thus a solution of water (600 ml) and sodium bicarbonate (4.46 g) is added to the reaction mixture. The reaction mixture is kept under stirring at ambient temperature for 10 minutes, the organic phase is then prepared and washed with an aqueous solution at 10% of sodium chloride (600 ml).
The organic phase is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain 119 g of raw pimecrolimus. Such raw product is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 66 g (81.5 mmoles) of purified pimecrolimus.
The chemical/physical data obtained matches the data indicated in literature.
Example 2
Preparation of ascomvcin 24.33-diacetate (intermediate 24, 33-diacetate; compound V of scheme 3)
DMAP (4.5 eq; 0.136 g) and acetic anhydride (4.5 eq; 0.114 g) are added to a solution of ascomycin (200 mg; 0.25 mmoles) in pyridine (2.5 ml), under stirring at the temperature of 0° C.
The reaction is kept under stirring for 1.5 hours at the temperature of 0° C then it is diluted with water and it is extracted with ethyl acetate (3 times with 5 ml). The organic extracts are washed with HCl 0.5 N (5 times with 10 ml), anhydrified on
Na2SO4 concentrated under vacuum.
The residue product was purified by chromatography on silica gel (n- hexane/acetone 8/2 v/v as eluent) to obtain ascomycin 24,32-diacetate (210 mg;
0.24 mmoles).
We carried out the following analysis on such purified sample:
1H-NMR (500 MHz) δ: 2.02 and 2.06 (2 CH3CO), 5.20 and 4.70 (24CH and
33CH);
IR (Cm-1): 3462.749, 2935.824, 1734.403, 1650.739, 1449.091, 1371.079.
DSC: endothermic peak at 234.10° C ; [α]D=- 100.0° (C=0.5 CHCl3).
Spectrum of MS (ESI+): m/z: 898.4 (100.0%; m+23).
Elementary analysis calculated for C47H73NO14: C 64.44%; H 8.40%; N 1.60%; O
25.57%
Elementary analysis found: C 64.55%; H 8.44%; N 1.61%; O 25.40%
Preparation of the 24-acetyl ascomycin (intermediate 24-acetate-33-OH; compound VI of scheme 3)
Lipase from Candida antartica (CAL B Novozym 435) [1.1 g (2 U/mg) FLUKA] is added to a solution of ascomycin 33,24-diacetate (500 mg; 0.57 mmol) in
TBDME (25 ml) and n-octan-1-ol (4.5 eq; 0.371 g). The reaction is kept under stirring at 30° C for 100 hours, then the enzyme is taken away for filtration and the obtained filtrate is concentrated under low pressure to obtain 425 mg (0.51 mmoles) of product.
A sample was purified for analytical purposes by chromatography on silica gel (n- hexane/acetone = 7:3 v/v as eluents) and thus crystallised by acetone/water.
We carried out the following analysis on such purified sample: 1H-NMR
(500MHz) δ: 2.05 (CH3CO); IR (an 1): 3491.528, 2935.860, 1744.728, 1710.227,
1652.310, 1448.662, 1371.335. DSC: endothermic peak at 134.68° C; [α]D=-
102.7° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H71NO13: C 64.80%; H, 8.58%; N, 1.68%;
0, 24.94%
Elementary analysis found: C 64.71%; H, 8.49%; N, 1.60%; O, 24.97%
Preparation of the 24-acetyl-33epi-chloro ascomycin (intermediate 24-Acetate-33- chloro; compound VII of scheme 3) Supported triphenylphosphine (0.335 g; 1.1 mmoles) is added to a solution of 24- acetyl ascomycin (400 mg; 0.48 mmoles) in carbon tetrachloride (5 ml). The reaction mixture is kept under reflux for 3 hours then it is cooled at ambient temperature. The obtained suspension is filtered and the filtrate is concentrated to residue under vacuum to obtain 0.45g of reaction raw product which is purified by chromatography on silica gel: 163mg (0.19 mmoles) of product are obtained by elution with petroleum ether/acetone = 90/10.
1H-NMR δ: 2.08 (CH3CO); 4.60 (33CH); IR (Cm“1)= 3464.941, 2934.360,
1738.993, 1650.366, 1450.424, 1371.557; DSC: endothermic peak at 231.67° C
[α]D=-75.2° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 874.3 (M+23; 100.0%)
Elementary analysis calculated for C45H70ClNO12: C 63.40%; H, 8.28%; Cl,
4.16%; N, 1.64%; O, 22.52%
Elementary analysis found: C 63.31%; H, 8.30%; Cl, 4.05%; N, 1.58%; O,
22.42%.
Preparation of pimecrolimus from 24-acetyl-33-epi-chloro ascomycin
A solution of 24-acetyl-33-epi-chloro ascomycin (200 mg; 0.23 mmoles; compound VII) in methanol (2 ml) and HCl 3N (1 ml) is stirred at ambient temperature for 40 hours. After this period, the reaction is neutralised with an aqueous bicarbonate solution, the methanol evaporated under vacuum. The mixture is extracted with dichloromethane (3 times with 5 ml), anhydrified on sodium sulphate, filtered and concentrated to residue to obtain a residue product which is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 78 mg of purified pimecrolimus (0.096 mmoles).
The chemical/physical characteristics of the obtained product matches the data indicated in literature for pimecrolimus.
Example 4 (comparative*)
Verification of the method of synthesis of pimecrolimus described in EP427680 Imidazole (508 mg) and tert-Butyldimethylsilylchloride (1.125 g) are added in portions to a solution of 2g (2.53 mmoles) of ascomycin in anhydrous N,N- dimethylformamide (40 ml). The reaction mixture is kept under stirring at ambient temperature for 4.5 days. The reaction is thus processed diluting it with ethyl acetate (200 ml) and processing it using water (5 x 100 ml). The organic phase is separated, anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy raw product which is subsequently purified by chromatography on silica gel (1:30 p/p): 2.1 g (2.05 mmoles; yields 81% molars) of ascomycin 24,33 disilyl intermediate are obtained by elution with n- hexane/ethyl acetate 3/1. The chemical/physical data of such intermediate matches that indicated in EP427680.
2.1 g (2.05 mmoles) of ascomycin 24,33 disilyl intermediate are dissolved in a solution under stirring at the temperature of 0°C composed of acetonitrile (42 ml) and aqueous HF 40% (23.1 ml). The reaction mixture is kept under stirring at the temperature of 0°C for 2 hours then it is diluted with dichloromethane (30 ml). Then the reaction is washed in sequence with a saturated aqueous solution using sodium bicarbonate (30 ml) and water (30 ml). The separated organic phase is anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy residue which is subsequently purified by chromatography on silica gel (1:30 p/p): 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate are obtained by elution with dichloromethane/methanol 9/1. The chemical/physical data of such intermediate matches that obtained on the compound III scheme 2 and matches the data of literature indicated in EP427680. A mixture of 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate, triphenylphosphine (337 mg) in carbon tetrachloride (36.4 ml) is heated under stirring under reflux for 15 hours. After this period the reaction mixture is evaporated to residue under vacuum to obtain a solid product purified by chromatography on silica gel (1:30 p/p): 535 mg (0.57 mmoles; yields 63% molars) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are obtained by elution with n-hexane/ethyl acetate 2/1. The chemical/physical data of such intermediate matches those we obtained on compound IV scheme 2 and matches the data of literature indicated in EP427680.
535 mg (0.57 mmoles) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are dissolved under stirring at ambient temperature in acetonitrile (16.4 ml) and aqueous HF 40% (0.44 ml). The reaction mixture is kept under stirring at ambient temperature for 45′ and then it is diluted with ethyl acetate (100 ml). The organic phase is thus washed in sequence with an aqueous solution of sodium bicarbonate (70 ml) with water (2 x 70 ml) and thus it is anhydrified on sodium sulphate, filtered and evaporated under vacuum to obtain a solid which is subsequently purified by chromatography on silica gel (1 :30 p/p): 323 mg (0.399 mmoles; yields 70% molars) of pimecrolimus is obtained by elution with n- hexane/ethyl acetate 2/3. The chemical/physical characteristics of the obtained product matches the data indicated in literature regarding pimecrolimus; the overall yield of the process is 16%.
………………………..
POLYMORPHS…….WO2006060615A1
Example 7: Preparation of amorphous pimecrolimus by precipitation [00094] 19,5 g purified pimecrolimus (colorless resin) was dissolved in 217 ml acetone at 4O0C and concentrated. Residue: 38,76 g. The residue was diluted with 6 ml distilled water with stirring. Finally 1 ml acetone was added. This solution was added slowly to 2 L chilled distilled water that was stirred efficiently. After the addition had been completed, the suspension was stirred 20 min at O0C. Then the solid was filtered and dried at 450C in vacuum oven overnight. Product: 15,65 g yellowish solid. Amorphous (XRD, DSC).
Example 8: Preparation of amorphous pimecrolimus by grinding
[00095] Procedure of grinding: 200 mg of Pimecrolimus sample was ground gently in an agate mortar using a pestle for half a minute. ,
References
- Allen BR, Lakhanpaul M, Morris A, Lateo S, Davies T, Scott G, Cardno M, Ebelin ME, Burtin P, Stephenson TJ (2003). “Systemic exposure, tolerability, and efficacy of pimecrolimus cream 1% in atopic dermatitis patients”. Arch Dis Child 88 (11): 969–973. doi:10.1136/adc.88.11.969.PMC 1719352. PMID 14612358.
- Meingassner JG, Kowalsky E, Schwendinger H, Elbe-Bürger A, Stütz A (2003). “Pimecrolimus does not affect Langerhans cells in murine epidermis”. Br J Dermatol 149 (4): 853–857.doi:10.1046/j.1365-2133.2003.05559.x. PMID 14616380.
- Billich A, Aschauer H, Aszódi A, Stuetz A (2004). “Percutaneous absorption of drugs used in atopic eczema: pimecrolimus permeates less through skin than corticosteroids and tacrolimus”. Int J Pharm 269 (1): 29–35. doi:10.1016/j.ijpharm.2003.07.013.PMID 14698574.
- Firooz A, Solhpour A, Gorouhi F, Daneshpazhooh M, Balighi K, Farsinejad K, Rashighi-Firoozabadi M, Dowlati Y (2006). “Pimecrolimus cream, 1%, vs hydrocortisone acetate cream, 1%, in the treatment of facial seborrheic dermatitis: a randomized, investigator-blind, clinical trial”. Archives of Dermatology 142 (8): 1066–1067. doi:10.1001/archderm.142.8.1066.PMID 16924062.
- Firooz A, Solhpour A, Gorouhi F, Daneshpazhooh M, Balighi K, Farsinejad K, Rashighi-Firoozabadi M, Dowlati Y (2006). “Pimecrolimus cream, 1%, vs hydrocortisone acetate cream, 1%, in the treatment of facial seborrheic dermatitis: a randomized, investigator-blind, clinical trial”. Archives of Dermatology 142 (8): 1066–1067. doi:10.1001/archderm.142.8.1066.PMID 16924062.
- Kreuter A, Gambichler T, Breuckmann F, Pawlak FM, Stücker M, Bader A, Altmeyer P, Freitag M (2004). “Pimecrolimus 1% cream for cutaneous lupus erythematosus”. J Am Acad Dermatol 51(3): 407–410. doi:10.1016/j.jaad.2004.01.044. PMID 15337984.
- Gorouhi F, Solhpour A, Beitollahi JM, Afshar S, Davari P, Hashemi P, Nassiri Kashani M, Firooz A (2007). “Randomized trial of pimecrolimus cream versus triamcinolone acetonide paste in the treatment of oral lichen planus”. J Am Acad Dermatol 57 (5): 806–813.doi:10.1016/j.jaad.2007.06.022. PMID 17658663.
- Boone B, Ongenae K, Van Geel N, Vernijns S, De Keyser S, Naeyaert JM (2007). “Topical pimecrolimus in the treatment of vitiligo”. Eur J Dermatol 17 (1): 55–61. doi:10.1111/j.1610-0387.2006.06124.x. PMID 17081269.
- Kreuter A, Sommer A, Hyun J, Bräutigam M, Brockmeyer NH, Altmeyer P, Gambichler T (2006). “1% pimecrolimus, 0.005% calcipotriol, and 0.1% betamethasone in the treatment of intertriginous psoriasis: a double-blind, randomized controlled study”. Arch Dermatol 142 (9): 1138–1143. doi:10.1001/archderm.142.9.1138. PMID 16983001.
- Jacobi A, Braeutigam M, Mahler V, Schultz E, Hertl M (2008). “Pimecrolimus 1% cream in the treatment of facial psoriasis: a 16-week open-label study”. Dermatology 216 (2): 133–136.doi:10.1159/000111510. PMID 18216475.
- Scheinfeld N (2004). “The use of topical tacrolimus and pimecrolimus to treat psoriasis: a review”. Dermatol. Online J. 10 (1): 3. PMID 15347485.
- N H Cox and Catherine H Smith (December 2002). “Advice to dermatologists re topical tacrolimus” (DOC). Therapy Guidelines Committee. British Association of Dermatologists.
- Berger TG, Duvic M, Van Voorhees AS, VanBeek MJ, Frieden IJ; American Academy of Dermatology Association Task Force (2006). “The use of topical calcineurin inhibitors in dermatology: safety concerns Report of the American Academy of Dermatology Association Task Force”. J Am Acad Dermatol 54 (5): 818–823. doi:10.1016/j.jaad.2006.01.054.PMID 16635663.
- Spergel JM, Leung DY (2006). “Safety of topical calcineurin inhibitors in atopic dermatitis: evaluation of the evidence”. Curr Allergy Asthma Rep 6 (4): 270–274. doi:10.1007/s11882-006-0059-7. PMID 16822378.
- Stern RS (2006). “Topical calcineurin inhibitors labeling: putting the “box” in perspective”.Archives of Dermatology 142 (9): 1233–1235. doi:10.1001/archderm.142.9.1233.PMID 16983018.
| WO2005105811A1 | Apr 12, 2005 | Nov 10, 2005 | Ping Cai | Regiospecific synthesis of rapamycin 42-ester derivatives |
| WO2006024582A1 | Jul 26, 2005 | Mar 9, 2006 | Poli Ind Chimica Spa | A method for the preparation of mycophenolate mofetil by enzimatic transesterification |
| WO2006040111A2 | Oct 10, 2005 | Apr 20, 2006 | Novartis Ag | Heteroatoms-containing tricyclic compounds |
| WO2006060614A1 | Dec 1, 2005 | Jun 8, 2006 | Teva Gyogyszergyar Zartkoeruen | Methods for preparing pimecrolimus |
| WO2007103348A2 | Mar 5, 2007 | Sep 13, 2007 | Wyeth Corp | Process for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants |
| EP0427680A1 | Nov 7, 1990 | May 15, 1991 | Sandoz Ltd. | Heteroatoms-containing tricyclic compounds |
- Elidel official homepage
- FDA News
- NPS RADAR
- Article about American Academy of Dermatology speaking out against black box warning
- Report of the Calcineurin Task Force of the ACAAI and AAAAI


| WO2005117837A1 * | Jun 1, 2005 | Dec 15, 2005 | Lorant Gyuricza | Process for preparation of amorphous form of a drug |
| EP0427680A1 * | Nov 7, 1990 | May 15, 1991 | Sandoz Ltd. | Heteroatoms-containing tricyclic compounds |
| EP0480623A1 * | Oct 2, 1991 | Apr 15, 1992 | Merck & Co., Inc. | New halomacrolides and derivatives having immunosuppressive activity |
| US6423722 * | Oct 17, 2000 | Jul 23, 2002 | Novartis Ag | Crystalline macrolides and process for their preparation |
Cancer-fighting compound in figs and celery targets aggressive breast tumors

It is rare for a natural molecule to garner the attention of medical researchers for two completely different cancer-fighting properties, but the compound psoralen has done just that. Found in figs, celery and other fruits and vegetables, psoralen is already used to treat lymphoma—as well as skin conditions such as psoriasis—based on its ability to stop DNA from copying itself and triggering cell death when combined with UV light.
Now researchers at Duke University have found that UV light activation of psoralen also has the ability to kill breast cancer cells that overproduce the protein HER2. About one-third of breast tumors are HER2-positive, along with stomach, ovarian, and other types of cancer. HER2-positive breast cancer is considered one of the most aggressive forms of the disease, because the HER2 protein encourages cancer cells’ unchecked growth. The most promising drugs for HER2-positive cancer, such as lapatinib and trastuzumab, can block the…
View original post 52 more words
PIRODAVIR



A mixture of 10.4 parts of 3-chloro-6-methylpyridazine, 22.4 parts of ethyl 4-[2-(4-piperidinyl)ethoxy]benzoate butanedioate (1:1), 8.6 parts of sodium carbonate and 0.9 parts of N,N-dimethylformamide was stirred for 3 hours in an oil bath at .+-.150.degree. C. After cooling, water and dichloromethane were added and the layers were separated. The organic layer was dried, filtered and evaporated. The residue was purified by column chromatography over silica gel using a mixture of trichloromethane and ethanol (99:1 by volume) as eluent. The pure fractions were collected and the eluent was evaporated. The residue was crystallized from a mixture of 2,2′-oxybispropane and 2-propanone (75:25 by volume). The precipitated product was filtered off and dried, yielding 17 parts (56.8%) of ethyl 4-[2-[1-(6-methyl-3-pyridazinyl)-4-piperidinyl]-ethoxy]benzoate; mp. 130.1.degree. C. (comp. 1).

Scheme 1. Synthesis of Pirodavir (3) and Related Compounds
| US2985657 * | Oct 12, 1959 | May 23, 1961 | Paul A J Janssen | 1-(aroylalkyl)-4-heterocyclylpiperazines |
| US4068383 * | Sep 30, 1976 | Jan 17, 1978 | Hoechstmass Balzer Gmbh & Co. | Tape measure reel |
| US4451476 * | Oct 17, 1983 | May 29, 1984 | Sterling Drug Inc. | Isoxazoles as antiviral agents |
| US4604127 * | May 15, 1985 | Aug 5, 1986 | Eli Lilly And Company | Herbicidal pyridazinylimidazolidinone compounds |
| EP0137242A2 * | Aug 20, 1984 | Apr 17, 1985 | Sterling Winthrop Inc. | (Substituted) Phenyl-aliphatic-isoxazoles useful as antiviral agents and preparation thereof |
| EP0156433A2 * | Mar 15, 1985 | Oct 2, 1985 | Janssen Pharmaceutica N.V. | Anti-virally active pyridazinamines |
| EP0211457A2 * | Jul 9, 1986 | Feb 25, 1987 | Janssen Pharmaceutica N.V. | Novel (4-substituted-piperazinyl)pyridazines |
| JPS5877866A * | Title not available |
FDA Secure Supply Chain Pilot Program: 13 companies prequalified

FDA Secure Supply Chain Pilot Program: 13 companies prequalified
In August 2013, the FDA initiated the so called Secure Supply Chain Pilot Program (SSCPP) to enhance the security of imported drugs. Now, the first companies have been listed. Read more.
In August 2013, the U.S. Food and Drug Administration (FDA) initiated the so called Secure Supply Chain Pilot Program (SSCPP) to enhance the security of imported drugs.
The goal was to enable qualified firms to expedite the importation of active pharmaceutical ingredients and finished drug products into the United States.

With this program, FDA wants to better focus its imports surveillance resources on preventing the entry of high-risk drugs that are the most likely to compromise the quality and safety of the U.S. drug supply.
The SSCPP is a voluntary program. Each firm accepted to participate in the program will be allowed to have up to five drugs subject to expedited import entry review. The SSCPP will be jointly administered by FDA’s Center for Drug Evaluation and Research (CDER) and Office of Regulatory Affairs (ORA).
Currently, the following companies have been accepted into the program:
- AbbVie Inc.
- Allergan, Inc.
- Astellas U.S. Technologies, Inc.
- Bristol-Myers Squibb Company
- Celgene Corporation
- GE Healthcare Inc.
- GlaxoSmithKline LLC
- Merck Sharp & Dohme Corporation
- Mylan Pharmaceuticals Inc.
- Novartis Pharmaceuticals Corporation
- Pfizer, Inc.
- Teva Pharmaceutcials USA, Inc.
- Watson Laboratories, Inc.
Source: FDA press release

Japanese Pharmacopoeia and Japanese GMP Regulations available online
Japanese Pharmacopoeia and Japanese GMP Regulations available online
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can download documents on GMP as well as on marketing authorisations for medicinal products. An English version of the Japanese Pharmacopoeia (JP) is also available. You will find the direct links in the News.
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can find in the section “Regulations and Procedures” under the heading “GMP” requirements regarding the inspection of manufacturers of medicinal products and APIs who want to introduce their products into Japan.
Now, a document was supplemented in January 2014 which describes which documents have to be submitted to the Japanese Agency within a pre-approval inspection and/ or a periodical post-approval inspection.
Go to the PMDA webpage to get more information.
There, you can also access the current Japanese Pharmacopoeia Sixteenth Edition in English.
Source: PMDA, Japan
Indian Regulators promote two levels of GMP
GMP deviations and even data falsification have been identified in a number of companies in India. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? Read more in our GMP News
GMP deviations and even data falsification have been identified in a number of companies in India. The FDA has issued numerous Warning Letters, the EU has published GMP Non Compliance Reports in its EudraGMDP database and EDQM has withdrawn various CEPs because of GMP inspection findings.
In an article published by Regulatory Focus on 28 January 2014 the question has been raised whether Indian companies have a chronic data falsification problem. The article lists 7 companies in India which have received a Warning Letter in the past months – all of them because of GMP deviations and because of “actually or potentially tampering with their data”. In addition to the 7 companies the Ranbaxy case is a story of its own. Not only one facility was found to manipulate data but several sites of the company are involved. For this reason the US FDA has issued a consent decree of permanent injunction against Ranbaxy. All manufactured products in the facilities concerned are now subject to an FDA import alert. In a press release the FDA states: “Because this company continued to violate current good manufacturing practice regulations and falsify information on drug applications, the FDA took these actions in an effort to protect consumers.” Dara Corrigan, FDA associate commissioner for regulatory affairs goes on: “The FDA continues to be committed to protecting consumers from potentially unsafe products that may be offered on the market.” On January 23, 2014 the FDA added an additional facility of Ranbaxy to the existing consent decree.
So far, the Indian Authority did not initiate the same measures like US and European counterparts. This also questions the supervision system in India. If inspections have been performed by Indian Inspectors at the concerned facilities why did they fail to make the same findings? The Drug Controller General of India, Mr. G.N. Singh, gave an interesting interpretation: According to an interview published by live mint & Wall Street Journal he said: “…it must be stated that every country has different measures and we cannot judge Ranbaxy by standards set up by the American drug regulator“. When Mr Singh was asked about the problems identified at three Ranbaxy plants he stated: “Some of those were found to be true and my office had told Ranbaxy to take corrective measures. Similar procedures will be followed in this case as well. But I do not think this is a situation which will warrant withdrawal of drugs from the domestic market. Our biggest objective is to maintain good quality of medicines and we are doing that. There are no drugs in the Indian market that are not up to the standards stated under the Drugs and Cosmetics Act.” In a final statement in the interview he also mentioned that he is “not worried about issues of quality.” In another interview with the Business Standard Press Mr Singh made an alarming statement for all customers of medicinal products and APIs in Europe and the US. “If I follow US standards, I will have to shut almost all drug facilities“. If this is the truth EU and US customers are in big trouble because products not complying to EU/US GMP standard (e.g. ICH Q7 GMP for APIs) would need to be taken from the market immediately.
This all looks like it will not fit together. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? It can only mean that dual standards exist. This would result in two quality levels, an international and a domestic quality level. Such a policy possibly causes questions by Indian patients who have to accept a different and probably lower quality standard.
It does not look like the Indian Regulators will re-think the GMP inspection approach and the quality standard in their country. Instead of acting in his own country the Drug Controller General of India announced inspections in the US and the EU.
But what are the international implications of this strategy? European Regulators need to react as they require from the Indian Authority to issue Written Confirmations of GMP compliance. Without a Written Confirmation APIs can not enter EU market. Currently more than 200 Written Confirmations have been issued by Indian Authority. If the inspections which have been performed as a prerequisite for issuing a Written confirmation were not based on the international standard ICH Q7 (GMP for APIs) the Written Confirmations are no longer valid documents. This issue might be raised by an EU court if a substandard API in a medicinal product will cause a health risk to patients in Europe.

Lysosomal Storage Disorders: Orphan Drugs For Niemann-Pick Disease
This is the sixth Blog Post in a series that will examine Lysosomal Storage Disorders (LSDs) in the rare disease and orphan drug space. This Blog Post reviews orphan drugs for the treatment of Niemann-Pick Disease (NPD).
Introduction
Niemann-Pick Diseases (NPDs) are a subgroup of LSDs that affect metabolism and are caused by genetic mutations. NPD is named after the two doctors who described the symptoms in the early part of the 20th century – Dr. Albert Niemann and Dr. Luddwick Pick. NPDs are characterized by the harmful accumulation of quantities of fatty substances, or lipids, in the cells of the spleen, lungs, bone marrow, liver, and brain. The three most commonly recognized forms of NPD are:
• Niemann-Pick Types A & B (ASMD or Acid Sphingomyelinase Deficiency)
• Niemann-Pick Type C (NPC).
Niemann-Pick types A and B are caused by a deficiency of an enzyme called acid sphingomyelinase. The enzyme deficiency leads to enlargement…
View original post 278 more words
Cidofovirסידופוביר سيدوفوفير


CIDOFOVIR
(S)-1-(3-Hydroxy-2-phosphonylmethoxypropyl)cytosine
[(S)-2-(4-Amino-2-oxo-1,2-dihydropyrimidin-2-yl)-1-(hydroxymethyl)ethoxymethyl]phosphonic acid
113852-37-2 CAS
120362-37-0 (Na salt)
149394-66-1 (dihydrate)
launched 1996 Gilead
SYNTHESIS.. CHEMDRUG
Rega Instituut (Originator)
For the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS)
US5142051 PATENT
| Canada | 1340856 | 1999-12-21 | EXPIRY 2016-12-21 |
| United States | 5142051 | 1993-06-26 | 2010-06-26 |
Cidofovir is a DNA polymerase inhibitor that was launched in 1996 by Gilead for the intravenous treatment of cytomegaloviral (CMV) retinitis in AIDS patients. Early clinical trials are underway at the National Institute for Allergy & Infectious Disease (NIAID) for the treatment of BK virus nephropathy (BKVN) in patients who have undergone kidney transplants.
Cidofovir suppresses CMV replication by selective inhibition of viral DNA synthesis. Biochemical data support selective inhibition of CMV DNA polymerase by cidofovir diphosphate, the active intracellular metabolite of cidofovir. Cidofovir diphosphate inhibits herpesvirus polymerases at concentrations that are 8- to 600-fold lower than those needed to inhibit human cellular DNA polymerases alpha, beta, and gamma1, 2, 3. Incorporation of cidofovir into the growing viral DNA chain results in reductions in the rate of viral DNA synthesis.
Cidofovir was originally developed under a collaboration between the Academy of Sciences of the Czech Republic and the Rega Institute for Medical Research. In 1991 and 1992, Gilead entered into license agreements with the Rega Institute that covered a large number of nucleotide analogue compounds and structures, including cidofovir. The drug became the subject of a marketing collaboration between Gilead and Pfizer (formerly Pharmacia & Upjohn) in August 1996 that covers all countries outside the U.S.
Cidofovir (brand name Vistide) is an injectable antiviral medication primarily used as a the treatment for cytomegalovirus (CMV) retinitis (an infection of the retina of the eye) in patients with AIDS.[1][2]
Its only indication that has received regulatory approval worldwide is cytomegalovirus retinitis.[1][2] Cidofovir has also shown efficacy in the treatment ofaciclovir-resistant HSV infections.[3] Cidofovir has also been investigated as a treatment for progressive multifocal leukoencephalopathy with successful case reports of its use.[4] Despite this meta-analyses have failed to demonstrate any efficacy in AIDS patients,[5] and the limited data in non-AIDS patients fail to demonstrate any efficacy either.[6] Cidofovir might have anti-smallpox efficacy and might be used on a limited basis in the event of a bioterror incident involving smallpox cases.[7] A cidofovir derivative with much higher activity against smallpox that can be taken orally has been developed.[8] It has inhibitory effects on varicella-zoster virus replication in vitro although no clinical trials have been done to date, likely due to the abundance of safer alternatives such as aciclovir.[9] Cidofovir shows anti-BK virus activity in a subgroup of transplant patients.[10] Cidofovir is being investigated as a complementary intralesional therapy against papillomatosis caused by HPV.[11][12]
It first received FDA approval on the 26th of June 1996,[13] TGA approval on the 30th of April 1998[2] and EMA approval on the 23rd of April 1997.[14]
Other
It has been suggested as an antitumour agent, due to its suppression of FGF2.[15][16]
Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, by Antonín Holý, and developed by Gilead Sciences[20] and is marketed with the brand name Vistide by Gilead in the USA, and by Pfizerelsewhere.
The chemical name of cidofovir is 1-[(S)-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC), with the molecular formula of C8H14N3O6P•2H2O and a molecular weight of 315.22 (279.19 for anhydrous). The chemical structure is:

Cidofovir is a white crystalline powder with an aqueous solubility of ≥ 170 mg/mL at pH 6 to 8 and a log P (octanol/aqueous buffer, pH 7.1) value of -3.3.
Cidofovir Injection is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL.
The formulation is pH-adjusted to 7.4 (range 7.1 to 7.7) with sodium hydroxide and/or hydrochloric acid and contains no preservatives. The appropriate volume of Cidofovir Injection must be removed from the single-use vial and diluted prior to administration
INTRODUCTION
Cidofovir’s chemical formula is C8H14N3O6P and its IUPAC name is ({[(S)-1-(4-amino-2-oxo-1,2-dihydropyrimidin-1-yl)-3-hydroxypropan-2-yl]oxy}methyl)phosphonic acid. Cidofovir has also been described as (S)-(1-(4-amino-2-oxopyrimidin-1(2H)-yl)-3-hydroxypropan-2-yloxy)methylphosphonic acid as well as possibly by other chemical names. Its chemical structure is:

Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, and developed by Gilead Sciences. Today, cidofovir is an injectable antiviral medication for the treatment of cytomegalovirus (CMV) retinitis in patients with AIDS. It suppresses CMV replication by selective inhibition of viral DNA polymerase and therefore prevention of viral replication and transcription. It is an acyclic nucleoside phosphonate, and is therefore independent of phosphorylation by viral enzyme, in contrast to, for instance, acyclovir.
Cidofovir is marketed with the brand name Vistide® by Gilead in the United States and by Pfizer in other parts of the world. Vistide® is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL. The formulation is pH-adjusted to 7.4 with sodium hydroxide and/or hydrochloric acid and contains no preservatives. Renal impairment is the major toxicity of Vistide®.
Presently, there are no Orange Book patents listed as having claims which cover Vistide®, although previously U.S. Pat. No. 5,142,051 was listed in the Orange Book for Vistide®. The ‘051 patent is not directed specifically to cidofovir or its crystalline forms. Instead, it broadly discloses N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases.
Cytomegalovirus (Cytomegaoviyns, CMV) is one of the biggest dangers of the herpes virus, the body’s infection rates as high as 50% to 80% of the current adult prevalence rate of more than 95%, generally showed a latent infection, most infections had no clinical symptoms, but under certain conditions, the invasion of organs and systems to produce more severe disease. The virus can invade the lung, liver, kidney, salivary gland, mammary gland and other polymorphonuclear leukocytes and lymphocytes, and, since the long-term or intermittent saliva, milk sweat, blood, urine, semen, exclude uterine secretions of the virus. Spread through a variety of ways in the mouth, genital tract, placenta, blood transfusion or organ transplantation.
When the body’s immune dysfunction, such as infected with HIV, cancer patients undergoing radiotherapy, chemotherapy, organ or bone marrow transplantation immunosuppressive anti-rejection etc will stimulate active infection, can cause acute retinitis, interstitial pneumonia, gastroenteritis and encephalitis, blindness or death without treatment rate of over 70%. With the rise in HIV infection rates and organ transplants extensively for anti-CMV drugs is also increasing demand.
cidofovir (cidofovir, HPMPC) are novel ether derivatives of cytidine phosphono chemical name
[5]-NL [(3 – hydroxy-2 – methoxy-phosphonic acid) glycerol]-N4-cytosine, Molecular structure of the formula (I):

Gilead developed by the United States, in May 1996 the FDA approved injectable celecoxib Duofu Wei listed, France and Canada also continued with the approval of the use of the trade name Vistide. Its CAS number is 113852-37-2, formula C8H14N3O6P, the structure of formula (I). Cidofovir for CMV is highly inhibitory activity of certain ganciclovir or foscarnet resistant strains of the virus are also active. And herpes simplex virus (HSV), herpes zoster virus (VZV), human papillomavirus (HPV), also has a strong activity.
Its mechanism of action: cidofovir having a phosphoric acid group, a ring-opening mechanism of the antiviral nucleoside phosphonate compound (ANP) and the consistent cyclic nucleoside analogues are nucleosides or virus in vivo kinase activation into triphosphate metabolite, thereby inhibiting viral replication by DNA polymerase and reverse transcriptase. Unlike the three-step cyclic nucleoside analogues must phosphorylation reaction, ring opening nucleoside phosphonate group containing phosphorus compound itself, eliminating the first step of the phosphorylation reaction speed, and thus a higher activity. Cidofovir is absorbed when the cells in the cell pyrimidine nucleoside phosphorylase kinase (P bandit kinase and NDP kinase) to effect conversion of the active metabolite monophosphate (HPMPCp), diphosphate (HPMPCpp) and a bile acid base adducts. Cidofovir diphosphate inhibits viral DNA polymerase or reverse transcriptase activity, and its corresponding natural dNTP incorporated into the viral DNA chain competition, since no 3 – hydroxy end, continue to extend the DNA chain termination. Can slow the synthesis of DNA, viral DNA and to the loss of stability, thereby inhibiting viral replication, transcription of the ability to reduce viral DNA to exert antiviral activity. Compared with other anti-CMV drugs, cidofovir characteristics: significant and lasting effect, started the first two weeks administered once a week, then only administered once every two weeks, easy to use, and to reduce its toxicity side effects.
Several major techniques are based on the synthesis of cidofovir cytosine as starting material, mainly carried out to improve the synthesis of the side chain.
(I) J. Med Chem, 1989,32,1457 ~ 1463 discloses a synthetic process:

The route to cytosine as the raw material, with a chiral side chain by condensation, deprotection and reduction can be obtained in three steps cidofovir.However, chiral side chain subject to a six-step reaction system. The total yield is low, adverse side. And using Me3SiBr, so that the costs and the risk of surge, is not conducive to industrial production.
(2) US 5591852,1995-1-7; US 2005/023833 & WO 2006/014429 and US 2009/0270618, Tetrahedron Lett 1994,35,3243-3246 and “Chinese Journal of New Drugs”, 2007,16. , 1272-1274 for the synthesis of a lot of improvements:

Benzoyl cytosine with a chiral starting material and trityloxymethyl ethylene oxide condensation, deprotection and hydrolysis was then prepared by deprotection cidofovir group. The synthetic steps to make some shorter, but still use expensive Me3SiBr, adverse ones, the low yield of the security at the cost of industrial production is still unfavorable. (Several different patent protection only in the order of the amino cytosine different!)
(3) Patent Publication No. CN1690065A, CN1690066A, CN1690067A (2005 年 11 月 2 Publication Date) and the “Chinese Journal of Medicinal Chemistry” 2007,17,41-46, reported a new synthetic route:

The route of process steps is too long, the total yield is low, side effects side. But not conducive to industrial production.
(4) Patent No. CN 101205215A (25 June 2008 publicly) announced a halogen epoxy propane as a starting material for the synthesis route:
Use of the route (R) – epihalohydrin reaction with cytosine, cytosine ring because alkaline easily cause epoxy ring-opening reaction of the ring, but side reactions, the purified product is not, nor is suitable for industrial production.
Subsequently, the patent number CN 101525352A (2009 年 9 月 9 Publication Date) discloses (4) based on the modified route through epoxypropionate alkane ether in the form of a direct reaction with cytosine, after a series of similar steps obtain the final product cidofovir.
In view of the clinical application of cidofovir more favorable therapeutic effect in, looking for a high yield and because of economic and practical, easy to control, the risk of small synthetic methods and technology is now more urgent needs.
Synthesis

Brodfuehrer, P; Howell, Henry G.; Sapino, Chester; Vemishetti, Purushotham (1994). “A practical synthesis of (S)-HPMPC”. Tetrahedron Letters 35 (20): 3243. doi:10.1016/S0040-4039(00)76875-4.
………………………………………
CN 102268040

, Example 1:
1 Synthesis of 4,4 ‘- dimethoxytrityl methyl – (R) – glycidol (Compound III): The 5 04 g (15 mmoDDMT-Cl grain port 0 20 g (1 52 mmol… ) 4_ dimethylaminopyridine (DMAP) was dissolved in 100 mL CH2C12 cooled to 0 ° C, was added dropwise 10 mL TEA was slowly added 2. 00 g (27mmol) hydroxymethyl chiral oxirane (Compound II ) addition was completed, the reaction warmed to room temperature naturally. fly 4 h, until TLC until the disappearance of the detection DMT-Cl, the reaction was stopped by filtration, the filtrate was washed with saturated NaHC03 solution (50mLX2), saturated NaCl solution (50 mLX2), anhydrous Na2S04 dried, filtered, and concentrated to a viscous colorless directly, i.e., 5 08 g of 4,4 ‘-dimethoxy-triphenylmethyl _ -.. (R) – glycidol (Compound III), yield 90 %, HPLC purity 99%.
2, Synthesis (S)-N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl] cytosine (Compound IV):. Under nitrogen to 3 56 g (32 mmol) of cytosine was added 150 mL of anhydrous N, N-dimethylformamide (DMF), and at room temperature, was added portionwise 1. 24 g (31 mmol, molar concentration of 60%) NaH, 0. 5 h after adding 11 92 g (31 mmol) 4,4 ‘-. dimethoxytrityl methyl – (R) – glycidol (Compound III), plus finished warming up to 10 (Tll (TC reaction . 6-8 h and then filtered, and the filtrate evaporated under reduced pressure DMF, the remaining solid phase was added 500 mL of ethyl acetate and 50 mL of water, separated and the organic layer was washed with saturated NaHC03 solution (50 mL X 2), saturated NaCl solution (50 mL X 2), dried over anhydrous Na2S04 filtered and dried, and concentrated to give 13 90 g of a white solid, S Jie (S)-Nl-[(2 -.. hydroxy-3 – (methoxy-dimethoxytrityl ) propyl] cytosine (Compound IV), yield 92%, HPLC purity 98%.
3 Synthesis ⑶-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V):
75 ~ 80 ° C under the conditions, 48 76 g (0 100 mol.) (S) _N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl]. Cytosine (Compound IV) was added to 150 mL anhydrous DMF, and then inputs 8. 5g (0. 050 mol) tert-butoxide, magnesium reaction 0.5-1 h, tosyloxy added diethyl 32 methylsulfinyl . 2 g (0. 100 mol), the reaction epileptic 8 h, p-toluenesulfonic acid was added to neutralize the excess alkali to neutral distilled DMF, ethyl acetate (300 mLX 3) washing the combined ethyl acetate phase was concentrated to give a solid, i.e., synthetic 58 18 g (S)-Nl-. {[2 – (diethoxy-phosphono-methoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V), yield 89%, HPLC purity greater than 95%.
4 Synthesis of (S)-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – hydroxy] propyl} cytosine (Compound VI): The 10 g (S)-Nl- {[2 – (phosphono-methoxy ethoxy) -3 – (methoxy-dimethoxytrityl)] propyl}-cell
Pyrimidine (compound V) was dissolved in a concentration of 70 mL of 80% acetic acid solution, 90 ° C reaction. After 5 h, cooled to room temperature, 50 mL of water and 30 mL of dichloromethane, and the organic phase washed with water (30 mL X2) and the combined aqueous phase was concentrated to give crude 9. 5 g, can be performed directly in the next reaction.
can also be separated by flash column chromatography (CH2C12 = MeOH = 10: 1), 4.6 g obtained as a pale yellow oil, i.e. (S)-Nl-{[2 – (methoxy diethoxy phosphono ) -3 – hydroxy] propyl} cytosine (Compound VI), yield 90%.
5 was synthesized ⑶-Nl-{[2_ (diphosphonic acid methoxy) -3 – hydroxy] propyl} cytosine (Compound I):
The 9.5g (S)-Nl-{[2 – (methoxy diethoxy phosphonomethyl) -3 – hydroxy] propyl} cytosine (Compound VI) into a crude product containing 5 76 g (0.. 045 mol) solution of hydrogen iodide, hydroiodic acid, and after reflux for 4-5 h. (50 mLX 2) wash solution was separated with ethyl acetate. The aqueous phase was added sodium hydroxide to adjust pH between 3 Γ3 6, filtered, recrystallized from methanol to give 3.81 g of white crystalline solid, S Jie (S)-Ni-{[2 -.. (Diphosphonic acid methoxy yl) -3 – hydroxy] propyl} cytosine (Compound I), yield 88% (containing two crystal water), HPLC purity greater than 99%.
…………………………………………
POLYMORPHS
Example 7 Amorphous Cidofovir
Intermediate 5 (FIG. 7; 0.5 g, 0.054 mol) was heated with a solution of sodium methoxide in methanol (0.5 M, 15 mL, 7.5 mmol) at 72° C. for 14.5 h then at 90° C. for 5.5 h. The reaction mixture was quenched with water (10 mL) and filtered through a bed of ion exchange resin Dowex® 50WX8 100-200 (H). The filtrate was cycled through the ion exchange bed (2 times) then washed successively with 1:1 methanol:water (40 mL), methanol (40 mL) and 4% triethylamine:methanol (50 mL). This ion-exchange bed was further washed with 48:48:4 methanol:water:triethylamine (100 mL) until no UV absorbance was detected in the filtrate. This reaction produced intermediate 7 (FIG. 7) together with cyclic cidofovir impurity. This mixture was then dissolved in 6 N HCl and heated to 65° C. After cooling the reaction mixture to room temperature, ethyl acetate was charged and stirred and the aqueous layer separated. The aqueous was stirred with ethanol (50 mL). The precipitated material was filtered and the solid was washed with ethanol. The ethanol filtrate was concentrated. The concentrated material was taken up in acetonitrile and stirred with trimethylsilyl bromide (19 mL) at room temperature for 18 h. The reaction mixture was filtered and the filtrate concentrated. The residue was taken up in toluene (30 mL) and ammonium hydroxide (28%, 50 mL) was charged and stirred at room temperature. The organic phase was separated and the aqueous phase was concentrated to dryness. Water (20 mL) and ethanol (15 mL) were added to the residue. The mixture pH was 6 and was adjusted to pH 3 with concentrated HCl (2 mL) then adjusted to pH 4 to 4.5 with 28% NH4OH. After stirring for 0.5 h, the mixture was cooled, filtered and the solids washed with 2:1 EtOH:H2O and dried under vacuum for 18 h. The isolated solid was taken up in water (10 mL) and 28% NH4OH added to give a solution. Concentrated HCl was added to the solution until pH 4 was reached. Ethanol (13 mL) was charged and the mixture stirred at −17° C. for 18 h, filtered and the solids washed with 2:1 EtOH:water (2×8 mL), dried under vacuum at 35° C. The cidofovir isolated in this manner was determined to be in the amorphous form by XRPD.

……………………………..
Journal of the American Chemical Society, 2011 , vol. 133, 7 p. 2264 – 2274
http://pubs.acs.org/doi/abs/10.1021/ja109823e

………………………………………………..
READ ALSO
Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457
http://pubs.acs.org/doi/abs/10.1021/jm00127a010
References
- “Vistide (cidofovir) dosing, indications, interactions, adverse effects, and more”.Medscape Reference. WebMD. Retrieved 4 February 2014.
- “Product Information VISTIDE®”. TGA eBusiness Services. Gilead Sciences Pty Ltd. 3 September 2013. Retrieved 5 February 2014.
- Chilukuri, S; Rosen, T (2003 Apr). “Management of acyclovir-resistant herpes simplex virus.”.Dermatologic clinics 21 (2): 311–20. doi:10.1016/S0733-8635(02)00093-1.PMID 12757254.
- Segarra-Newnham M, Vodolo KM (June 2001). “Use of cidofovir in progressive multifocal leukoencephalopathy”. Ann Pharmacother 35 (6): 741–4. doi:10.1345/aph.10338.PMID 11408993.
- De Luca, A; Ammassari, A; Pezzotti, P; Cinque, P; Gasnault, J; Berenguer, J; Di Giambenedetto, S; Cingolani, A; Taoufik, Y; Miralles, P; Marra, CM; Antinori, A; Gesida 9/99, IRINA, ACTG 363 Study, Groups (September 2008). “Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis.”. AIDS (London, England) 22 (14): 1759–67.doi:10.1097/QAD.0b013e32830a5043. PMID 18753934.
- Langer-Gould, A; Atlas, SW; Green, AJ; Bollen, AW; Pelletier, D (28 July 2005). “Progressive Multifocal Leukoencephalopathy in a Patient Treated with Natalizumab”. New England Journal of Medicine 353 (4): 375–381. doi:10.1056/NEJMoa051847. PMID 15947078.
- De Clercq E (July 2002). “Cidofovir in the treatment of poxvirus infections”. Antiviral Res. 55(1): 1–13. doi:10.1016/S0166-3542(02)00008-6. PMID 12076747.
- Bradbury, J (March 2002). “Orally available cidofovir derivative active against smallpox.”.Lancet 359 (9311): 1041. doi:10.1016/S0140-6736(02)08115-1. PMID 11937193.
- Magee, WC; Hostetler, KY; Evans, DH (August 2005). “Mechanism of Inhibition of Vaccinia Virus DNA Polymerase by Cidofovir Diphosphate”. Antimicrobial Agents and Chemotherapy49 (8): 3153–3162. doi:10.1128/AAC.49.8.3153-3162.2005. PMC 1196213.PMID 16048917.
- Araya CE, Lew JF, Fennell RS, Neiberger RE, Dharnidharka VR (February 2006).“Intermediate-dose cidofovir without probenecid in the treatment of BK virus allograft nephropathy”. Pediatr Transplant 10 (1): 32–7. doi:10.1111/j.1399-3046.2005.00391.x.PMID 16499584.
- Broekema FI, Dikkers FG (August 2008). “Side-effects of cidofovir in the treatment of recurrent respiratory papillomatosis”. Eur Arch Otorhinolaryngol 265 (8): 871–9. doi:10.1007/s00405-008-0658-0. PMC 2441494. PMID 18458927.
- Soma MA, Albert DM (February 2008). “Cidofovir: to use or not to use?”. Curr Opin Otolaryngol Head Neck Surg 16 (1): 86–90. doi:10.1097/MOO.0b013e3282f43408.PMID 18197029.
- “Cidofovir Monograph for Professionals – Drugs.com”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 5 February 2014.
- “Vistide : EPAR -Product Information” (PDF). European Medicines Agency. Gilead Sciences International Ltd. 7 November 2013. Retrieved 5 February 2014.
- Liekens S, Gijsbers S, Vanstreels E, Daelemans D, De Clercq E, Hatse S (March 2007). “The nucleotide analog cidofovir suppresses basic fibroblast growth factor (FGF2) expression and signaling and induces apoptosis in FGF2-overexpressing endothelial cells”. Mol. Pharmacol.71 (3): 695–703. doi:10.1124/mol.106.026559. PMID 17158200.
- Liekens S (2008). “Regulation of cancer progression by inhibition of angiogenesis and induction of apoptosis”. Verh. K. Acad. Geneeskd. Belg. 70 (3): 175–91. PMID 18669159.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3. edit
- “Vistide (cidofovir)” (package insert). Gilead Sciences. September 2010. DOSAGE AND ADMINISTRATION: Dosage.
- Safrin, S; Cherrington, J; Jaffe, HS (September 1997). “Clinical uses of cidofovir”. Reviews in Medical Virology 7 (3): 145–156. doi:10.1002/(SICI)1099-1654(199709)7:3<145::AID-RMV196>3.0.CO;2-0. PMID 10398479.
- “Press Releases: Gilead”. Retrieved 2007-12-05.
- Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457 - Synthesis and antiherpesvirus activity of (S)-1-((3-hydroxy-2-phosphonylmethoxy)propyl)cytosine (HPMPC) and related nucleotide analoguesNucleosides Nucleotides 1989, 8(5-6): 923
- Journal of Pharmaceutical Sciences, 2012 , vol. 101, 9 p. 3249 – 326
- BRODFUEHRER P R ET AL: “A Practical Synthesis of (S)-HPMPC“, 19940101, vol. 35, no. 20, 1 January 1994 (1994-01-01), pages 3243-3246, XP002012084
- http://www.chemdrug.com/databases/8_0_nhnpseknoegbdisx.html CHEMDRUG SYNTHESIS
| 2 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of nucleotide analogs bearing the (S)-(3-hydroxy-2-phosphonylmethoxy)propyl moiety attached to adenine, guanine, and cytosine“, ACS SYMPOSIUM SERIES , 401(NUCLEOTIDE ANALOGUES ANTIVIRAL AGENTS), 88-102 CODEN: ACSMC8; ISSN: 0097-6156, 1989, XP002677024, |
| 3 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of the nucleotide analog (S)-1-[3-hydroxy-2-(phosphonylmethoxy)prop yl]cystosine“, JOURNAL OF MEDICINAL CHEMISTRY , 32(7), 1457-63 CODEN: JMCMAR; ISSN: 0022-2623, 1989, XP002677026, |
| 4 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; JIANG, XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir”, XP002677148, retrieved from STN Database accession no. 2009:1568897 -& JIANG XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir“, JIEFANGJUN YAOXUE XUEBAO – PHARMACEUTICAL JOURNAL OF CHINESE PEOPLE’S LIBERATION ARMY, ZHONGGUO RENMIN JIEFANGJUN, ZONGHOUQINBU, WEISHENGBU, YAOPIN YIQI JIANYANSUO, CN, vol. 25, no. 5, 1 January 2009 (2009-01-01), pages 395-397, XP008152443, ISSN: 1008-9926 |
| 5 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JIANFENG ET AL: “Improved synthesis of cidofovir”, XP002677149, retrieved from STN Database accession no. 2008:1052727 -& LIU JIANFENG ET AL: “Improved synthesis of cidofovir“, ZHONGGUO YAOWU HUAXUE ZAZHI – CHINESE JOURNAL OF MEDICINAL CHEMISTRY, GAI-KAI BIANJIBU, SHENYANG, CN, vol. 17, no. 1, 1 February 2007 (2007-02-01), pages 41-43, XP008152444, ISSN: 1005-0108 |
| 6 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LU, NING ET AL: “Synthesis of cidofovir”, XP002677151, retrieved from STN Database accession no. 2007:1124844 -& LU NING ET AL: “Synthesis of cidofovir“, ZHONGGUO XIN YAO ZAZHI – CHINESE NEW DRUGS JOURNAL, GAI-KAN BIANJIBU, BEIJING, CN, vol. 16, no. 16, 1 January 2007 (2007-01-01), pages 1272-1274, XP008152436, ISSN: 1003-3734 |
| 7 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; YI, HONG ET AL: “Synthesis of antiviral agent cidofovir”, XP002677150, retrieved from STN Database accession no. 2007:1249882 -& YI HONG ET AL: “Synthesis of antiviral agent cidofovir“, ZHONGGUO KANGSHENGSU ZAZHI/ CHINESE JOURNAL OF ANTIBIOTICS, SICHUAN, CN, vol. 31, no. 7, 1 January 2006 (2006-01-01), pages 412-413, XP008152413, ISSN: 1001-8689 |
| 8 | * | FROMTLING R A ET AL: “Cidofovir. HPMPC. GS-504. GS-0504. Viside“, DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 21, no. 10, 1 January 1996 (1996-01-01), pages 1003-1013, XP008152410, ISSN: 0377-8282 |
| 9 | * | HOLY A: “SYNTHESES OF ENANTIOMERIC N-(3-HYDROXY-2-PHOSPHONOMETHOXYPROPYL) DERIVATIVES OF PURINE AND PYRIMIDINE BASES“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 3, 1 January 1993 (1993-01-01), pages 649-674, XP009042514, ISSN: 0010-0765, DOI: 10.1135/CCCC19930649 |
| 10 | * | JOANNE J BRONSONA ET AL: “A New Synthesis of the Potent and Selective Anti-Herpesvirus Agent (S)-1-[3-Hydroxy-2-(Phosphonylmethoxy)Prop yl]Cytosine“, NUCLEOSIDES, NUCLEOTIDES AND NUCLEIC ACIDS, TAYLOR & FRANCIS, PHILADELPHIA, PA , vol. 9, no. 6 1 January 1990 (1990-01-01), pages 745-769, XP008152412, ISSN: 1525-7770, DOI: 10.1080/15257779008043142 Retrieved from the Internet: URL:http://www.tandfonline.com/doi/abs/10.1080/15257779008043142 [retrieved on 2006-10-04] |
| 11 | * | PETR ALEXANDER AND ANTONÍN HOL: “General Method of Preparation of N-[(S)-(3-Hydroxy-2-phosphonomethoxypropyl )] Derivatives of Heterocyclic Bases“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 5, 1 May 1993 (1993-05-01), pages 1151-1163, XP008152404, ISSN: 0010-0765, DOI: 10.1135/CCCC19931151 |
| 12 | * | SNOECK, ROBERT ET AL: “(S)-1-(3-Hydroxy-2-phosphonylmethoxypropy l)cytosine, a potent and selective inhibitor of human cytomegalovirus replication“, ANTIMICROBIAL AGENTS AND CHEMOTHERAPY , 32(12), 1839-44 CODEN: AMACCQ; ISSN: 0066-4804, 1988, XP002677023, |
| 13 | * | SYMERSK AND A HOL J: “Structure of 1-(S)-(3-hydroxy-2-phosphonylmethoxypropyl )cytosine; an antiviral agent“, ACTA CRYSTALLOGRAPHICA SECTION C. CRYSTAL STRUCTURE COMMUNICATIONS, MUNKSGAARD, COPENHAGEN, DK, vol. 47, no. 10, 15 October 1991 (1991-10-15), pages 2104-2107, XP008152403, ISSN: 0108-2701, DOI: 10.1107/S0108270190013257 |
| 14 | * | ULRIKA ERIKSSON: “Synthesis and biological evaluation of novel cidofovir prodrugs targeting HPepT1“, SYNTHESIS AND BIOLOGICAL EVALUATION OF NOVEL CIDOFOVIR PRODRUGS TARGETING HPEPT1- A DISSERTATION PRESENTED TO THE FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA IN PARTIAL FULFILLMEN , 1 January 2005 (2005-01-01), pages 1-287, XP008152406, Retrieved from the Internet: URL:http://search.proquest.com/pqdtscieng/docview/305421788/fulltextPDF/1371C222DB27F96BE2D/2?accountid=29404 |
| 15 | * | WEBB, ROBERT R., II ET AL: “Synthesis of (S)-N1-(3-hydroxy-2-phosphonylmethoxy)prop ylcytosine, (S)-HPMPC“, TETRAHEDRON LETTERS , 29(43), 5475-8 CODEN: TELEAY; ISSN: 0040-4039, 1988, XP002677025 |
| CN1559429A * | Feb 18, 2004 | Jan 5, 2005 | 肖广常 | 注射用西多福韦冻干粉针剂 |
| CN1690066B * | Apr 19, 2004 | Apr 28, 2010 | 横店集团成都分子实验室有限公 | 抗病毒剂西多福韦新衍生物 |
| CN101525352A * | Feb 16, 2009 | Sep 9, 2009 | 苏州凯达生物医药技术有限公司 | 西多福韦及其中间体的制备方法 |
| CN102268040A * | Sep 5, 2011 | Dec 7, 2011 | 扬州三友合成化工有限公司 | 抗病毒药物西多福韦的一种合成方法 |
| US5142051 | Jul 17, 1987 | Aug 25, 1992 | Ceskoslovenska Akademie Ved | N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases and a therapeutical composition therefrom with antiviral activity |
Trial to test laser acupuncture treatment for osteoarthritis
The potential for laser acupuncture to provide painless and effective treatment for osteoarthritis knee pain is being put to the test in a clinical trial beginning in Sydney. Traditional Chinese medicine practitioner Meikin Li Rees is recruiting 60 participants for the trial, being undertaken as part of her PhD research at UTS.
“Osteoarthritis (OA) is the most common form of arthritis and the major cause of musculoskeletal pain and immobility in elderly people worldwide,” Ms. Rees said. “In Australia, arthritis affects some 3.4 million people – nearly 17 per cent of the population.
“Of the proportion of Australians affected, 60 per cent are women and 60 per cent of all people living with arthritis are of working age. “If the current trend continues, one in five people, or around 4.6 million Australians, are forecast to be living with arthritis by 2020.”
Ms. Rees said the Osteoarthritis Research Society International already recommends needle acupuncture
View original post 151 more words
