
Radezolid
869884-78-6 cas no
http://www.ama-assn.org/resources/doc/usan/radezolid.pdf
869884-78-6, RX-103, RX-1741, RX-O1_667, Radezolid (USAN/INN), UNII-53PC6LO35W
Molecular Formula: C22H23FN6O3
Molecular Weight: 438.454823
Rib-X Pharmaceuticals
Phase II completed
N-{[(5S)-3-(2-fluoro-4′-{[(1H-1,2,3-triazol-5-ylmethyl)amino]methyl}biphenyl-4-yl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide
Rib-X Pharmaceuticals has completed two Phase II clinical trials of radezolid for the treatment of pneumonia and uncomplicated skin infections. The trial completion dates were in 2008 and 2009, but to date the Phase III trials have not been initiated [1-6].
Radezolid (INN, codenamed RX-1741) is a novel oxazolidinone antibiotic being developed by Rib-X Pharmaceuticals, Inc. for the treatment of serious multi-drug–resistant infections. Radezolid has completed two phase-II clinical trials. One of these clinical trials was for uncomplicated skin and skin-structure infections (uSSSI) and the other clinical trial was for community acquired pneumonia (CAP).
Oxazolidinone antibiotics are a relatively new class of antibacterial agents with activity against a broad spectrum of gram-positive pathogens. The first member of this new class to be commercialized, linezolid, was approved in 2000. Since that time the development of linezolid resistant organisms has prompted efforts to discover more effective members of the oxazolidinone class.
A new family of biaryl oxazolidinone antibacterials with activity against both linezolid-susceptible and -resistant Gram-positive bacteria, as well as certain Gram-negative bacteria has been reported (see Bioorganic & Medicinal Chemistry Letters, 2008, 18, 6175-6178, and PCT Patent Publication WO 2005/019211).
Among the known biaryloxazolidinones is N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-bipheny- l-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide, more commonly known as radezolid (RX-1741), currently being developed for multi-drug-resistant infections.
Although a monohydrochloride salt of radezolid was disclosed in PCT Patent Publication WO 2006/133397, there is a continuing need for new salts and polymorphs thereof having improved properties such as solubility to optimize bioavailability on therapeutic administration.
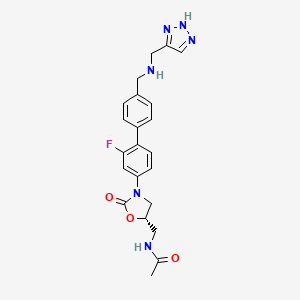 Radezolid
Radezolid
Synthesis 1
http://www.google.co.il/patents/WO2005019211A2?hl=iw&cl=en
Scheme A
Scheme B illustrates the synthesis of intermediates 7 and 8 of the present invention using Suzuki coupling chemistry between boronic acids and aryl triflates. Boronic ester 6 is treated with an appropriate aryl triflate to yield the BOC-protected biaryl 7. The BOC group of 7 is removed to provide amine 8, an intermediate useful in the synthesis of certain compounds of the present invention.
Scheme B
8, R = NH2-HCI Scheme C depicts the synthesis of intermediates 9-13, which are useful in producing certain methoxy-substituted biaryl derivatives of the present invention. Suzuki coupling of boronic ester 6 produces biaryl aldehyde 9, which can be reduced to alcohol 10. Mesylation of 10 yields 11 that can be converted to azide 12. Reduction of azide 12 yields amine 13.
Scheme C
Scheme D depicts the synthesis of pyridyl intermediates, which are useful for the synthesis of compounds of the present invention, via similar chemistry to that shown in Scheme C. Coupling of boronic ester 6 to a halopyridine aldehyde produces biaryl aldehyde 14. Aldehyde 14 serves as the precursor to intermediates 15-18 via chemistry described above.
Scheme D
Biaryl aldehyde 19 (Scheme E) can be synthesized from a Suzuki coupling of iodide 1 and 4-formylphenylboronic acid. Scheme E illustrates how intermediate aldehydes of type 19, 9, and 14 can be converted via reductive amination chemistry to other amines, such as amines 20-22, which are useful as intermediates for the synthesis of certain compounds of the invention.
Scheme E
Scheme F depicts the general synthesis of compounds of type la and lb from amines of type 5, 13, 18, and 20-22. Compounds of type la and lb are synthesized via acylation of amines 5, 13 and 18 and 20-22 with the appropriate acids using, for example, l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) as the coupling agent. Compounds 4001-4007 were specifically synthesized from amine 5 and the appropriate carboxylic acids. Scheme F
Scheme G highlights the synthesis of compounds of general structure II from amines of type 5 and 18. The amine can be acylated with carboxylic acids using EDCI (or other commonly employed peptide coupling reagents known in the art) to afford amides II.
Acid chlorides can be purchased or synthesized and allowed to react with amines 5 and 18, in the presence of bases such as triethylamine, to also produce amides II.
Alternatively, carboxylic acids can be pre-loaded onto a solid polymeric support, such as a tetrafluorophenol containing resin (TFP resin), and reacted with amines to yield amide products of general structure II (such as compounds 4008-4015).
Scheme G
Scheme H illustrates the synthesis of compounds of general structure Ilia from amines of type 5, 13, and 18 using reductive amination chemistry. For example, biaryl amine compounds 4016-4028 are synthesized in this manner. Scheme H
Scheme I depicts the synthesis of general structure Illb of the present invention from amine intermediate 8. For example, compounds 4029-4031 are synthesized using this reductive amination chemistry.
Scheme I
Scheme J shows the synthesis of compounds of general structure IVa and IVb. Amines 20, 21, and 22 can be converted to tertiary amines IVa, such as compounds 4032-4034 and 4036, using standard reductive amination chemistry employed earlier for other derivatives.
This reductive amination chemistry can be employed on biaryl aldehyde intermediates such as 19, 9, and 14 to yield optionally substituted amines of general structure IVb, illustrated by compound 4037.
Scheme J
producing compounds of the present invention. Known iodoaryl oxazolidinone intermediate 50 (see U.S. Patent Nos. 5,523,403 and 5,565,571) is coupled to a substituted aryl boronic acid (the Suzuki reaction) to produce biaryl alcohol 51. Mesylate 52, azide 53, and amine 54 are then synthesized using chemistry well known to those skilled in the art. Scheme 1
NaN3, DMF, 70 °C
……………….
NO 2
http://www.google.com/patents/US20100234615
| TABLE 1 |
|
| Compound |
|
| Number |
Structure |
|
|
| 1 |
|
Example 1 Synthesis of Compound 1
Compound 1 and its hydrochloride salt are synthesized according to the following Scheme:
4-Methoxybenzyl Azide
1001.
A solution of 4-methoxybenzyl chloride 1000 (51.8 g, 331.0 mmol) in anhydrous DMF (200 mL) was treated with solid sodium azide (21.5 g, 331.0 mmol, 1.0 equiv) at 25° C., and the resulting mixture was stirred at 25° C. for 24 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was quenched with H2O (400 mL) and ethyl acetate (EtOAc, 400 mL) at room temperature.
The two layers were separated, and the aqueous layer was extracted with EtOAc (200 mL). The combined organic extracts were washed with H2O (2×200 mL) and saturated NaCl aqueous solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude 4-methoxybenzyl azide (51.2 g, 53.95 g theoretical, 94.9% yield) was obtained as colorless oil, which by HPLC and 1H NMR was found to be essentially pure and was directly used in the subsequent reaction without further purifications. For 4-methoxybenzyl azide 1001:
1H NMR (300 MHz, CDCl3) δ 3.84 (s, 3H, ArOCH3), 4.29 (s, 2H, Ar—CH2), 6.96 (d, 2H, J=8.7 Hz), 7.28 (d, 2H, J=7.8 Hz).
C-[1-(4-Methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-Methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine
(1003 and 1004).
A solution of 4-methoxybenzyl azide 1001 (61.2 g, 375.5 mmol) in toluene (188 mL) was heated with propargylamine 1002 (commercially available, 30.97 g, 38.6 mL, 563.0 mmol, 1.5 equiv) at 25° C., and the resulting reaction mixture was warmed up to gentle reflux at 100-110° C. for 21 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was cooled down to room temperature before being concentrated in vacuo to remove the excess amount of propargylamine and solvent.
The oily residue was then treated with 30% ethyl acetate-hexane (v/v, 260 mL), and the resulting mixture was warmed up to reflux and stirred at reflux for 30 min before being cooled down to room temperature for 1 h. The pale-yellow solids were then collected by filtration, washed with 30% ethyl acetate-hexane (v/v, 2×100 mL), and dried in vacuo at 40° C. for overnight to afford the crude, cycloaddition product (78.8 g, 81.75 g theoretical, 96.4%) as a mixture of two regioisomers, C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004), in a ratio of 1.2 to 1 by 1H NMR.
The crude cycloaddition product was found to be essentially pure and the two regioisomers were not separated before being used directly in the subsequent reaction without further purification. For 1003 and 1004:
1H NMR (300 MHz, DMSO-d6) δ 1.82 (br. s, 2H, NH2), 3.72 and 3.73 (two s, 3H, Ar—OCH3), 5.47 and 5.53 (two s, 2H, ArCH2), 6.89 and 6.94 (two d, 2H, J=8.7 Hz, Ar—H), 7.17 and 7.29 (two d, 2H, J=8.7 Hz, Ar—H), 7.58 and 7.87 (two br. s, 1H, triazole-CH); C11H14N4O, LCMS (EI) m/e 219 (M++H) and 241 (M++Na).
4-({tert-Butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-Butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009).
Method A. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.0 g, 91.74 mmol) in 1,2-dichloroethane (DCE, 280 mL) was treated with 4-formylphenylboronic acid 1005 (commercially available, 12.39 g, 82.57 mmol, 0.9 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.2 g, 137.6 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture was concentrated in vacuo. The residue, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with tetrahydrofuran (THF, 100 mL) and water (H2O, 100 mL).
The resulting solution was subsequently treated with solid potassium carbonate (K2CO3, 37.98 g, 275.2 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.02 g, 91.74 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo.
The crude, regioisomeric 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 35.98 g, 37.32 g, 96.4%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo.
This crude material was directly used in the subsequent reaction without further purification. For 1008 and 1009:
1H NMR (300 MHz, DMSO-d6) δ 1.32 and 1.37 (two br. s, 9H, COOC(CH3)3), 3.70, 3.73 and 3.74 (three s, 3H, Ar—OCH3), 4.07-4.39 (m, 4H), 5.49 and 5.52 (two s, 2H), 6.70-8.04 (m, 9H, Ar—H and triazole-CH); C23H29BN4O5, LCMS (EI) m/e 453 (M++H) and 475 (M++Na).
Method B. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.06 g, 92.0 mmol) in tetrahydrofuran (THF, 300 mL) was treated with 4-formylphenylboronic acid (13.11 g, 87.4 mmol, 0.95 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.25 g, 138.0 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with water (H2O, 200 mL).
The resulting aqueous solution was subsequently heated with solid potassium carbonate (K2CO3, 38.0 g, 276 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.08 g, 92 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude, 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 38.45 g, 39.50 g, 97.3%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo. This crude material was found to be essentially identical in every comparable aspect as the material obtained from Method A and was directly used in the subsequent reaction without further purification.
(5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester
(1011 and 1012).
A suspension of the crude regioisomeric mixture of 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 37.62 g, 83.23 mmol) and N-[3-(3-fluoro-4-iodo-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1010, 28.32 g, 74.9 mmol, 0.90 equiv) in toluene (150 mL) was treated with powder K2CO3 (34.45 g, 249.7 mol, 3.0 equiv), EtOH (50 mL), and H2O (50 mL) at 25° C.,
and the resulting mixture was degassed three times under a steady stream of Argon at 25° C. Pd(PPh3)4 (866 mg, 0.749 mmol, 0.01 equiv) was subsequently added to the reaction mixture, and the resulting reaction mixture was degassed three times again under a stead stream of Argon at 25° C. before being warmed up to gentle reflux for 18 h. When TLC and HPLC/MS showed the coupling reaction was complete, the reaction mixture was cooled down to room temperature before being treated with H2O (100 mL) and ethyl acetate (100 mL). The two layers were then separated, and the aqueous layer was extracted with EtOAc (100 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×150 mL), H2O (100 mL), and the saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The residual oil was solidified upon standing at room temperature in vacuo to afford the crude, (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-y]methyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester (1011) and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1012) as a regioisomeric mixture.
This crude product (43.36 g, 49.28 g theoretical, 88%) was used directly in the subsequent reaction without further purification. For the mixture of 1011 and 1012: 1H NMR (300 MHz, DMSO-d6) δ 1.35 and 1.38 (two br. s, 9H, COO(CH3)3), 1.85 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.73 and 3.76 (two s, 3H, Ar—OCH3), 3.79 (dd, 1H, J=6.6, 9.1 Hz), 4.18 (t, 1H, J=9.1 Hz), 4.35-4.43 (m, 4H), 4.73-4.81 (m, 1H), 5.50 (br. s, 2H), 6.90 and 6.98 (two d, 2H, J=8.7 Hz), 7.28 and 7.32 (two d, 2H, J=8.7 Hz), 7.35 (dd, 2H, J=2.2, 8.6 Hz), 7.42 (dd, 1H, J=2.2, 8.6 Hz), 7.49-7.63 (m, 4H, aromatic-H), 7.90 and 7.99 (two br. s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3); C35H39FN6O6, LCMS (EI) m/e 659 (M++H) and 681 (M++Na).
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1013)
and
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H–[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1014).
A solution of a regioisomeric mixture of (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1011 and 1012, 37.28 g, 56.65 mmol) in ethyl acetate (EtOAc, 150 mL) and methanol (MeOH, 30 mL) was treated with a solution of 4 N hydrogen chloride in 1,4-dioxane (113.3 mL, 453.2 mmol, 8.0 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 12 h. When TLC and HPLC/MS showed that the N-BOC deprotection reaction was complete,
the solvents were removed in vacuo. The residue was then suspended in 250 mL of 5% methanol (MeOH) in acetonitrile (CH3CN), and the resulting slurry was stirred at room temperature for 1 h. The solids were then collected by filtration, washed with toluene (2×100 mL) and 5% methanol in acetonitrile (2×50 mL), and dried in vacuo to afford a regioisomeric mixture of the crude, (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 30.0 g, 33.68 g theoretical, 89.1% yield) as off-white crystals in a ratio of 1.2 to 1.
This material was found by 1H NMR and HPLC/MS to be essentially pure and was directly used in the subsequent reactions without further purification. For the regioisomeric mixture of 1013 and 1014:
1H NMR (300 MHz, DMSO-d6) δ 1.84 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.71 and 3.74 (two s, 3H, Ar—OCH3), 3.80 (dd, 1H, J=6.6, 9.1 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.23-4.30 (m, 4H), 4.73-4.80 (m, 1H), 5.58 and 5.70 (two s, 2H), 6.88 and 6.93 (two d, 2H, J=8.7 Hz), 7.15 and 7.32 (two d, 2H, J=8.7 Hz), 7.43 (dd, 2H, J=2.2, 8.6 Hz), 7.52-7.62 (m, 6H, aromatic-H), 8.28 (s, 1H, triazole-CH), 8.32 (t, 1H, J=5.8 Hz, NHCOCH3), 9.91 and 10.32 (two br. s, 2H, ArCH2N+H2); C30H31FN6O4, LCMS (EI) m/e 559 (M++H) and 581 (M++Na).
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide hydrochloride (1 hydrochloride salt).
A solution of the crude regioisomeric mixture of (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-1H-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 29.17 g, 49.07 mmol) in trifluoroacetic acid(TFA, 150 mL) was warmed up to 65-70° C., and the resulting reaction mixture was stirred at 65-70° C. for 12 h. When TLC and HPLC/MS showed that the deprotection reaction was complete, the solvents were removed in vacuo.
The residual solids were then treated with ethyl acetate (EtOAc, 100 mL) and H2O (150 mL) before being treated with a saturated aqueous solution of sodium carbonate (30 mL) at room temperature. The resulting mixture was then stirred at room temperature for 1 h before the solids were collected by filtration, washed with EtOAc (2×50 mL) and H2O (2×50 mL), and dried in vacuo at 40-45° C. to afford the crude, (5S)-N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl)-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1 as the free base, 18.9 g, 21.49 g theoretical, 87.9%) as off-white powders, which by HPLC/MS and 1H NMR was found to be one pure regioisomer and this regioisomer was found to be identical as the material obtained from deprotection of 1013 alone by the same method.
For 1 as the free base: 1H NMR (300 MHz, DMSO-d6) δ 1.85 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.74 (s, 2H), 3.77 (s, 2H), 3.79 (dd, 1H, J=6.4, 9.2 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.72-4.81 (m, 1H), 7.39-7.62 (m, 7H, aromatic-H), 7.73 (s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH); C22H23FN6O3, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
A suspension of 1 free base (18.0 g, 41.1 mmol) in ethyl acetate (EtOAc, 80 mL), and methanol (MeOH, 20 mL) was treated with a solution of 4.0 N hydrogen chloride in 1,4-dioxane (41.1 mL, 164.4 mmol, 4.0 equiv) at room temperature, and the resulting mixture was stirred at room temperature for 8 h. The solvents were then removed in vacuo, and the residue was further dried in vacuo before being treated with a mixture of 10% methanol in acetonitrile (80 mL). The solids were collected by filtration, washed with 10% MeOH/acetonitrile (2×40 mL), and dried in vacuo to afford 1 hydrochloride salt (18.13 g, 19.50 g theoretical, 93% yield) as off-white crystals.
The crude 1 hydrochloride salt can be recrystallized from acetonitrile and water, if necessary, according to the following procedure: A suspension of the crude 1 hydrochloride salt (50.0 g) in acetonitrile (1250 mL) was warmed up to reflux before the distilled water (H2O, 280 mL) was gradually introduced to the mixture. The resulting clear yellow to light brown solution was then stirred at reflux for 10 min before being cooled down to 45-55° C. The solution was then filtered through a Celite bed at 45-55° C., and the filtrates were gradually cooled down to room temperature before being further cooled down to 0-5° C. in an ice bath for 1 h. The solids were then collected by filtration, washed with acetonitrile (2×50 mL), and dried in vacuo at 40° C. for 24 h to afford the recrystallized 1 hydrochloride salt (42.5 g, 50.0 g theoretical, 85% recovery) as off-white crystals.
For 1: 1H NMR (300 MHz, DMSO-d6) δ 1.86 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.84 (dd, 1H, J=6.4, 9.2 Hz), 4.19 (t, 1H, J=9.1 Hz), 4.24 (br. s, 2H), 4.31 (br. s, 2H), 4.74-4.79 (m, 1H), 7.44 (dd, 1H, J=2.2, 8.6 Hz), 7.57-7.66 (m, 6H, aromatic-H), 8.17 (s, 1H, triazole-CH), 8.30 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH);
13C NMR (75 MHz, DMSO-d6) δ 22.57, 40.69, 41.50, 47.36, 49.23, 71.85, 105.70 (d, J=28.5 Hz), 114.14 (d, J=2.9 Hz), 122.29 (d, J=13.3 Hz), 128.82 (d, J=3.0 Hz), 130.70, 130.94, 131.0, 131.22, 135.30, 137.92 (br. s), 139.66 (d, J=11.2 Hz), 154.11, 159.13 (d, J=243.5 Hz), 170.19;
C22H23FN6O3—HCl, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
……………………………..
http://www.sciencedirect.com/science/article/pii/S0960894X0801192X

References
- Zhou J, Bhattacharjee A, Chen S, et al. (December 2008). “Design at the atomic level: design of biaryloxazolidinones as potent orally active antibiotics”.Bioorg. Med. Chem. Lett. 18 (23): 6175–8. doi:10.1016/j.bmcl.2008.10.011. PMID 18947996.
- Skripkin E, McConnell TS, DeVito J, et al. (October 2008). “Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance”.Antimicrob. Agents Chemother. 52 (10): 3550–7. doi:10.1128/AAC.01193-07. PMC 2565890. PMID 18663023.
- Lawrence L, Danese P, DeVito J, Franceschi F, Sutcliffe J (May 2008). “In vitro activities of the Rχ-01 oxazolidinones against hospital and community pathogens”. Antimicrob. Agents Chemother. 52 (5): 1653–62. doi:10.1128/AAC.01383-07. PMC 2346622. PMID 18316525.
- Hanselmann R, Job G, Johnson G, Lou R, Martynow JG, Reeve MM (2009). “Synthesis of an antibacterial compound containing a 1,4-substituted 1H-1,2,3-triazole- a scaleable alternative to the “click” reaction””. Organic Process Research and Development 14: 152–158. doi:10.1021/op900252a.
- Franceschi F, Duffy EM (March 2006). “Structure-based drug design meets the ribosome”. Biochem. Pharmacol. 71 (7): 1016–25.doi:10.1016/j.bcp.2005.12.026. PMID 16443192.
- Ohlsen K (November 2009). “Novel antibiotics for the treatment of Staphylococcus aureus“. Expert Rev. Clin. Pharmacol. 2 (6): 661–72.doi:10.1586/ecp.09.26.
- Sutcliffe, J.A. Antibiotics in development targeting protein synthesis. Ann. NY Acad. Sci. 2011, 1241, 122–152, doi:10.1111/j.1749-6632.2011.06323.x.
- Rib-X. Radezolid. Available online: http://www.rib-x.com/pipeline/radezolid.php#development (accessed on 14 April 2013).
- Rib-X Pharmaceuticals, Inc. Safety and efficacy study of oxazolidinone to treat pneumonia. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00640926 (accessed on 14 April 2013).
- Rib-X Pharmaceuticals, Inc. Safety and efficacy study of oxazolidinones to treat uncomplicated skin infections. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00646958 (accessed on 14 April 2013).
- Shaw, K.J.; Barbachyn, M.R. The oxazolidinones: Past, present, and future. Ann. NY Acad. Sci. 2011, 1241, 48–70, doi:10.1111/j.1749-6632.2011.06330.x.
- Skripkin, E.; McConnell, T.S.; DeVito, J.; Lawrence, L.; Ippolito, J.A.; Duffy, E.M.; Sutcliffe, J.; Franceschi, F. Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance.Antimicrob. Agents Chemother. 2008, 52, 3550–3557, doi:10.1128/AAC.01193-07.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| US6969726 * |
Jun 2, 2004 |
Nov 29, 2005 |
Rib X Pharmaceuticals Inc |
Biaryl heterocyclic compounds and methods of making and using the same |
| US20050043317 * |
Jun 2, 2004 |
Feb 24, 2005 |
Jiacheng Zhou |
Biaryl heterocyclic compounds and methods of making and using the same |
|
|
9-17-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
9-17-2010
|
Process for the synthesis of triazoles
|
|
|
4-28-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
11-26-2008
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
10-26-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
12-13-2006
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
11-30-2005
|
Biaryl heterocyclic compounds and methods of making and using the same
|

October 10, 2012
QIDP Designation for Radezolid for Acute Bacterial Skin and Skin Structure Infections, Community-acquired Bacterial Pneumonia
Rib-X Pharmaceuticals announced that the FDA designated radezolid as a Qualified Infectious Disease Product (QIDP) for the indications of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CABP).
The QIDP designation will enable Rib-X to benefit from certain incentives for the development of new antibiotics, including an additional five years of market exclusivity, priority review and eligibility for fast-track status, provided under the new Generating Antibiotic Incentives Now (GAIN) program. GAIN was included in the FDA Safety and Innovation Act (FDASIA), formerly known as PDUFA V, which received bipartisan Congressional support and was signed into law by President Obama in July 2012.
Radezolid has completed two Phase 2 clinical trials with an oral formulation in uncomplicated skin and skin structure infections (uSSSI) and in CABP. A Phase 1 study with an IV formulation was recently completed in healthy subjects. Rib-X recently announced data from a positive Phase 1 IV dosing study conducted in healthy subjects and an in vivo long-term safety study vs. linezolid (Zyvox; Pfizer).
Radezolid is a next-generation oxazolidinone with a safety profile permitting long-term treatment of resistant infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
For more information call (203) 624-5606 or visit www.rib-x.com



 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






















































 MRX I
MRX I

































 valnemulin
valnemulin retapamulin
retapamulin