
Home » 2014 (Page 75)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Dandelion, Burdock, and Cancer
 burdock
burdock dandelion
dandelionDandelion root and burdock root are my two most commonly prescribed herbs when chronic conditions require anti-inflammatory, blood purifying alteratives for gentle detoxification. This includes conditions such as arthritis and cancer. I’ve studied literally hundreds of herbs from around the world, and considering cost, availability, palatability (no small matter, as people with chronic disease like cancer need to be able to take their herbs at least three times a day for months) – there are probably no two more simple and powerful anticancer herbs on the planet than dandelion and burdock.*
After prescribing both of these in strong dose clinically for years with great results (patients feel better, or experience slowing or even complete remission of some cancers), I learned that many professional British medical herbalists also use the same two-herb combination for conditions requiring blood, lymphatic and liver detoxification.
http://www.planetherbs.com/michaels-blog/dandelion-burdock-and-cancer.html
Glenmark’s novel molecule ‘GRC 27864’ for chronic inflammatory diseases including pain entering human trials

Glenmark’s novel molecule ‘GRC 27864’ for chronic inflammatory diseases including pain entering human trials
- GRC 27864 is a potent, selective, orally bioavailable inhibitor of mPGES-1
- The molecule has successfully completed pre-clinical and Phase 1 enabling studies. Regulatory submission has been filed for Phase 1 trial (first-in-human)with MHRA, UK
- mPGES-1 inhibitors selectively block the production of PGE2 while sparing other prostanoids of physiological importance
- With this announcement, Glenmark has reaffirmed its position globally in the development of novel pain therapies
Mumbai, India: April 3, 2014: Glenmark Pharmaceuticals today announced that its Novel Chemical Entity (NCE) ‘GRC 27864’ is entering human trials. This NCE program targets Microsomal Prostaglandin E synthase-1 (mPGES-1) as a novel therapeutic target in pain management. Selective mPGES-1 inhibitors are expected to inhibit increased prostaglandin E2 (PGE2) production in the disease state without affecting other prostanoid metabolites and, consequently, may be devoid of the GI(gastrointestinal) and cardiovascular side effects seen with NSAIDs and COX-2 inhibitors, respectively.
Glenmark has completed preclinical studies and Phase 1 enabling GLP studies for its selected lead molecule, GRC 27864 and has filed a Phase 1 application forfirst-in-human trial with the MHRA, UK. The Phase 1 studies are to be initiated soon and are likely to get completed by January 2015. Following this, Glenmark will also be initiating a proof of concept study in patients with acute pain.
ANTHONY CRASTO’S NEW DRUG APPROVALS TOUCHES 2 LAKH VIEWS IN 179 COUNTRIES
ANTHONY CRASTO’S NEW DRUG APPROVALS TOUCHES 2 LAKH VIEWS IN 179 COUNTRIES

DR ANTHONY MELVIN CRASTO Ph.D
WORLDDRUGTRACKER,
OTHERS
- Eurekamoments in Organic Chemistry
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- Drug Scaleup and Manufacturing International
SEE ALSO
- Organic Chemistry by Dr Anthony
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India, in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now sSanofi, Searle India ltd, now Rpg lifesciences, etc. he is now helping millions, has million hits on google on all organic chemistry websites. His New Drug Approvals, Green Chemistry International, Eurekamoments in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide . He gas good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, polymorphism etc He suffered a paralytic stroke in dec 2007 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
Personal Links
- my sites on the net
- DR ANTHONY MELVIN CRASTO
- GOOGLE GROUP ORGANIC PROCESS DEVELOPMENT
- mixxt
- epernicus
- scipeople
- jimdo
- yolasite
- my cv
- slidestaxx
- wordpress blog
- ABOUT ME
- BRANDSITE
- SKILLPAGES
- Academia.edu
- RESEARCHGATE
- DIIGO
- SLIDESHATE
- WIX
- WIX BLOG
- ISSUU
- SCRIBD
- BIZ
- GOOGLE BLOG
- APNACIRCLE
- Eurekamoments in Organic Chemistry
- Organic Chemistry by Dr Anthony
- Green Chemistry International
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
- DR ANTHONY MELVIN CRASTO Ph.D
- Pharmaceuticals
- Medicinal chemistry
- Organic chemistry literature
- Patent related site
- Green chemistry
- Reagents
- R & D
- Molecules
- Heterocyclic chem
- Sourcing
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- GOOGLE PLUS
- Drug Scaleup and Manufacturing International


amcrasto@gmail.com
email me if u like my posts
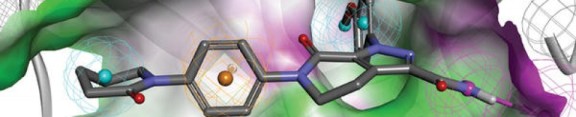
Cenicriviroc in Phase 2 for HIV by Takeda/Tobira

Cenicriviroc
TAK-652; TBR-652
(-)-(S)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-[4-(1-propyl-1H-imidazol-5-ylmethylsulfinyl)phenyl]-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate
497223-25-3 , Molecular Formula: C41H52N4O4S Molecular Weight: 696.94098
497223-28-6 (mesylate) C41 H52 N4 O4 S . C H4 O3 S, 793.047
Cenicriviroc (TAK-652, TBR-652) is an experimental drug candidate for the treatment of HIV infection.[1] It is being developed by Takeda Pharmaceutical and Tobira Therapeutics.
TBR-652 (formerly TAK-652) is a highly potent and orally active CCR5 antagonist in phase II clinical trials at Takeda for the treatment of HIV infection. Tobira Therapeutics is evaluating the compound in preclinical studies for the treatment of rheumatoid arthritis.
TBR-652 binds CCR5 receptors to interfere with the entry of the HIV-1 virus into macrophages and activated T-cells by inhibiting fusion between viral and cellular membranes. This mechanism of action differs from currently used HIV treatments such as nucleoside reverse transcriptase inhibitors and protease inhibitors.
In 2007, Takeda entered into an agreement with Tobira pursuant to which Tobira obtained exclusive worldwide rights to develop, manufacture and commercialize TBR-652 for the treatment of HIV infection.
Cenicriviroc is an inhibitor of CCR2 and CCR5 receptors,[2] allowing it to function as an entry inhibitor which prevents the virus from entering into a human cell. Inhibition of CCR2 may have an anti-inflammatory effect.
A double-blind, randomized, placebo-controlled clinical study to assess the antiviral activity, safety, and tolerability of cenicriviroc was conducted in 2010. HIV-infected patients taking cenicriviroc had significant reductions in viral load, with the effect persisting up to two weeks after discontinuation of treatment.[3] Additional Phase II clinical trials are underway.[4]
Phase IIb data presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in March 2013 showed similar viral suppression rates of 76% for patients taking 100 mg cenicriviroc, 73% with 200 mg cenicriviroc, and 71% with efavirenz. Non-response rates were higher with cenicriviroc, however, largely due to greater drop-out of patients. A new tablet formulation with lower pill burden may improve adherence. Looking at immune and inflammatory biomarkers, levels of MCP-1 increased and soluble CD14 decreased in the cenicriviroc arms.[5]
Although HIV has been largely rendered a chronic infection, there remains a need for new drugs because of the virus’s propensity to develop resistance to the drugs used to keep it at bay.
Pfizer’s maraviroc was the first drug that acted on the cells to prevent viral entry by antagonising the CCR5 co-receptor. Several others have been investigated and have failed; another that is undergoing clinical trials is Takeda’s cenicriviroc, which has been licensed to Tobira Therapeutics. Unlike maraviroc, the new agent also acts at the CCR2 co-receptor, which is implicated in cardiovascular and metabolic diseases.
In a Phase I double blind, placebo controlled trial designed to study safety, efficacy and pharmacokinetics, treatment-experienced but CCR5 antagonist-naïve patients with HIV-1 were given doses of 25, 50, 75, 100 or 150mg of the drug, or placebo once a day for 10 days.2 The maximum median reductions in HIV-1 RNA values were 0.7, 1.6, 1.8 and 1.7 log10 copies/ml for the respective doses, with a median time to nadir of 10 to 11 days. The effect on CD-4 cell counts was negligible. There was also a significant reduction in levels of monocyte chemotactic protein 1, suggesting that CCR2 was also being blocked. The drug was both generally safe and well tolerated, and no patients withdrew from the trial due to adverse events.
In another Phase I trial, designed to look at pharmacokinetics and pharmacodynamics and carried out in a similar patient population, subjects were given the drug as oral monotherapy for 10 days, again in doses of 25, 50, 75, 100 and 150mg, or placebo.3 The drug was well absorbed into the systemic circulation, and the concentration levels declined slowly, with meant elimination half-lives of one to two days. Potent, dose-dependent reductions in viral load were seen, and again it was generally safe and well tolerated across all levels.
In June 2011, Tobira initiated a multi-centre, double blind, double dummy, 48-week comparative Phase IIb trial in 150 patients with HIV-1 infection. Subjects are being given 100 or 200mg once-daily doses of the drug to evaluate its efficacy, safety and tolerability.
PATENTS
WO 2003014105
WO 2003076411
WO 2005116013
WO 2007144720
WO 2011163389
US 20130079233
WO 2013167743
See also
ancriviroc (formerly known as SCH-C), vicroviroc which has the chemical name (4,6-dimethylprymidine-5-yl){4- [(3S)-4-{(1 R)-2-methoxy-1 -[4-(trifluoromethyl)phenyl]ethyl}-3-methylpiperazin-1 -yl]-4-methylpiperidin-1 – yljmethanone, PRO-140, apliviroc (formerly known as GW-873140, Ono-4128, AK-602), AMD-887, INC- B9471 , CMPD-167 which has the chemical name N-methyl-N-((1R,3S,4S)-3-[4-(3-benzyl-1-ethyl-1H- pyrazol-δ-yOpiperidin-i-ylmethylH-IS-fluorophenyllcyclopent-i-yll-D-valine), methyl1-endo-{8-[(3S)-3- (acetylamino)-3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7-tetrahydro-1 H- imidazo[4,5-c]pyridine-5-carboxylate, methyl 3-endo-{8-[(3S)-3-(acetamido)-3-(3-fluorophenyl)propyl]-8- azabicyclo[3.2.1]oct-3-yi}-2-methyl-4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridine-5-carboxylate, ethyl 1- endo-{8-[(3S)-3-(acetylamino)-3-(3-fiuorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7- tetrahydro-1 H-imidazo[4,5-c]pyridine-5-carboxylate and N-{(1S)-3-[3-endo-(5-lsobutyryl-2-methyl-4,5,6,7- tetrahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1-(3-fluorophenyl)propyl}acetamide) and pharmaceutically acceptable salts, solvates or derivatives of the above. The last four compounds are disclosed in WO 03/084954 and WO 05/033107.


http://pubs.acs.org/doi/full/10.1021/jm0509703

Compound (S)-(−)-5b (TAK-652) also inhibited the replication of six macrophage-tropic (CCR5-using or R5) HIV-1 clinical isolates in peripheral blood mononuclear cells (PBMCs) (mean IC90 = 0.25 nM).
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide ((S)–(−)-5a). The 1 N HCl (160 mL) was added to 1931 (35.68 g, 53.4 mmol), and the mixture was extracted with EtOAc. To the aqueous layer was added 25% aqueous K2CO3 (160 mL), and the mixture was extracted with a mixture of EtOAc and i-PrOH (4:1). The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo to give (S)-18. To a solution of 16a (18.0 g, 41.1 mmol) and DMF (0.5 mL) in THF (180 mL) was added thionyl chloride (SOCl2) (4.50 mL, 61.7 mmol) at room temperature. After being stirred at room temperature for 1.5 h, the reaction mixture was concentrated in vacuo. A solution of the residue in THF (200 mL) was added dropwise to a mixture of (S)-18 and triethylamine (Et3N) (35.0 mL, 251 mmol) in THF (150 mL) under ice cooling. After being stirred at room temperature for 4 h, water was added to the reaction mixture. The mixture was washed with 10% aqueous AcOH, saturated aqueous NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on a NH silica gel (hexane/EtOAc = 1:5 → 1:8 → 1:9) to give 21.14 g (75%) of (S)-(−)-5a as a yellow amorphous powder, [α]D = −132.5° (C = 0.507%, EtOH). 1H NMR (300 MHz, CDCl3) δ 0.87−1.03 (9H, m), 1.34−1.49 (2H, m), 1.50−1.85 (8H, m), 2.55−2.65 (2H, m), 3.15−3.25 (2H, m), 3.52−3.58 (4H, m), 3.75−3.83 (4H. m), 4.02 (1H, d, J = 13.8 Hz), 4.08−4.17 (3H, m), 6.56 (1H, d, J = 1.0 Hz), 6.80 (1H, d, J = 8.8 Hz), 6.96 (2H, d, J = 8.8 Hz), 7.31−7.46 (7H, m), 7.55 (1H, s), 7.76 (2H, d, J = 8.8 Hz), 7.98 (1H, s). Anal. (C40H50N4O4S·0.25H2O) C, H, N.
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate ((S)–(−)-5b). The free base of (S)-(−)-5b was prepared in 80% yield from 16band 19 by a method similar to that described for (S)-(−)-5a. To a solution of the free base of (S)-(−)-5b (64.91 g, 93.1 mmol) in EtOAc (600 mL) was added dropwise a solution of methanesulfonic acid (8.95 g, 93.1 mmol) in EtOAc (160 mL) at room temperature. After being stirred at room temperature for 4 h, the crystals were collected by filtration and washed with EtOAc to give 69.09 g (94%) of (S)-(−)-5b as yellow crystals. The crystals (68.0 g) were purified by recrystallization from 2-butanone to give 58.9 g (85%) of (S)-(−)-5b as yellow crystals, mp 145.5−147.5 °C, [α]D = −191.2° (c = 0.508%, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 0.82−0.97 (12H, m), 1.29−1.39 (2H, m), 1.40−1.55 (4H, m), 1.65−1.85 (2H, m), 2.00−2.25 (1H, m), 2.29 (3H,s), 2.38−2.60 (2H, m), 3.10 (2H, d, J = 7.8 Hz), 3.30−3.60 (4H, m), 3.70 (2H, t, J = 4.8 Hz), 3.98 (2H, t,J = 6.6 Hz), 4.10 (2H, t, J = 4.8 Hz), 4.34 (1H, d, J = 15.0 Hz), 4.68 (1H, d, J = 15.0 Hz), 6.87 (1H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.7 Hz), 7.16 (1H, s), 7.42−7.60 (8H, m), 7.93 (2H, d, J = 8.7 Hz), 9.05 (1H, s), 10.18 (1H, s). Anal. (C42H56N4O7S2) C, H, N.
…………………
WO 2003014105 OR US20090030032
http://www.google.st/patents/US20090030032?hl=pt-PT&cl=un
EXAMPLE 7 Preparation of Compounds 9 and 10
8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propyl-1H-imidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzazocin-5-carboxamide (317 mg) was resolved by using CHIRAKCEL OJ 50 mm ID×500 mL (hexane/ethanol) to give (−)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (142 mg) (Compound 9) and (+)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (143 mg) (Compound 10).
Compound 9
[α]D=−127.4° (C=0.533% in ethanol).
Compound 10
[α]D=+121.0° (C=0.437% in ethanol).
………………………….
WO 2003076411
http://www.google.st/patents/WO2003076411A1?cl=en
http://www.google.st/patents/US20050107606?hl=pt-PT&cl=en

Example 21 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
To a solution of 8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (45 g) in tetrahydrofuran (135 ml) was added N,N-dimethylformamide (230 mg) and added dropwise thionyl chloride (12.45 g) at 10 to 15° C., and the resulting solution was stirred at the same temperature for 40 minutes to prepare an acid chloride.
Separately, to a solution of (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine in tetrahydrofuran (270 ml) was added pyridine (27.59 g), the resulting mixture was adjusted to 5° C. or lower, and then thereto was added dropwise the acid chloride solution at 5° C. or less, and the resulting mixture was stirred at the same temperature for 2 hours. To the mixture were added water (270 ml) and 20% aqueous citric acid solution (180 ml), tetrahydrofuran was distilled off under reduced pressure and the residue was extracted with ethyl acetate. The extract was sequentially washed with water, saturated sodium bicarbonate solution and water, and then the solvent was distilled off. To the residue was added ethyl acetate (360 ml), added heptane (360 ml) at 40° C. and added seed crystals of (−)-8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide (10 mg), and the mixture was stirred at 25° C. for 2 hours and stirred at 5° C. for 1 hour. The precipitated crystals were collected by filtration to obtain 63.97 g (yield: 92.1%) of the title compound. Melting point: 120-122° C.
Elemental analysis value: in terms of C41H52N4O4S
Calcd. value: C, 70.66; H, 7.52; N, 8.04.
Analytical value: C, 70.42; H, 7.52; N, 8.01
Industrial Applicability
According to the present invention, an optically active sulfoxide derivative having CCR5 antagonism or an intermediate compound thereof can be prepared without causing side reactions such as racemization and Pummerer rearrangement. In particular, Process 7 is industrially advantageous since it is possible to prepare an optically active Compound (II) by asymmetric oxidization in the presence of an optically active acid.
Example 20 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
According to the same method as that described in Example 15, the title compound was produced from 8-[4-(2-butoxyethoxy)phenyl]-1-propyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid and (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine.
1H-NMR (CDCl3, δ, 300 MHz) 0.88-1.01 (9H, m), 1.37-1.42 (2H, m), 1.57-1.80 (8H, m), 2.63 (2H, br), 2.77 (3H, s), 3.27 (2H, br), 3.51-3.57 (4H, m), 3.77-3.86 (4H, m), 3.90-4.05 (1H, m), 4.14 (2H, t, J=4.6 Hz), 4.25 (1H, d, J=14.6 Hz), 6.73 (1H, s), 6.84 (1H, d, J=8.7 Hz), 6.93 (2H, d, J=8.8 Hz), 7.21 (2H, d, J=8.7 Hz), 7.40-7.48 (4H, m), 7.61 (1H, s), 7.89 (2H, d, J=8.7 Hz), 8.65 (1H, s), 9.27 (1H, br)
Elemental analysis value: in terms of C41H54N4O7S2
Calcd. value: C, 63.21; H, 6.99; N, 7.19; S, 8.23.
Analytical value: C, 63.00; H, 7.09; N, 7.41; S, 8.25
Example 15 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (986 mg) was dissolved in tetrahydrofuran (3 ml) and thereto was added N,N-dimethylformamide (one drop). Subsequently, to the resulting solution was added dropwise oxalyl chloride (0.2 ml, 2.29 mmol) under ice-cooling and the mixture was stirred for 80 minutes under ice-cooling to prepare an acid chloride.
Separately, (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine (689 mg) was added to tetrahydrofuran (7 ml) and the resulting solution was cooled to 5° C. To the solution was added dropwise pyridine (0.62 ml) and added dropwise the acid chloride solution at 3 to 5° C., and the mixture was stirred for 2 hours under ice-cooling. To the mixture was added water (20 ml) at 10° C. or lower and the mixture was extracted with ethyl acetate. The organic layer was sequentially washed with water, saturated sodium bicarbonate solution and water, and concentrated under reduced pressure. Thereto was added toluene and the mixture was concentrated under reduced pressure. Thereto was added acetonitrile and the mixture was concentrated under reduced pressure. The residue was dissolved in acetonitrile (7 ml) and acetone (7 ml), thereto was added dropwise methanesulfonic acid (209 mg), and added seed crystals and the mixture was stirred at room temperature for 100 minutes. Subsequently, to the mixture was added acetone-acetonitrile (1:1, 5 ml). After stirring at room temperature overnight, the mixture was stirred for 2.5 hours under ice-cooling. The precipitated crystals were collected by filtration and washed with the ice-cooled acetone (9 ml). The crystals were dried at 40° C. under reduced pressure to obtain 1.51 g (yield: 87%) of the title compound as yellow crystals.
1H-NMR (300 MHz, DMSO-d6, δ): 0.78-0.96 (12H, m), 1.25-1.40 (2H, m), 1.41-1.51 (4H, m), 1.65-1.85 (2H, m), 2.05-2.15 (1H, m), 2.30 (3H, s), 2.35-2.50 (2H, m), 3.05-3.15 (2H, m), 3.30-3.55 (4H, m), 3.65-3.70 (2H, m), 3.90-4.05 (2H, m), 4.05-4.10 (2H, m), 4.30 (1H, d, J=14.73 Hz), 4.65 (1H, d, J=14.73 Hz), 6.85 (1H, d, J=8.97 Hz), 6.97 (1H, d, J=8.79 Hz), 7.17 (1H, s), 7.35-7.75 (6H, m), 7.92 (2H, d, J=8.79 Hz), 9.08 (1H, s), 10.15 (1H, s).
Elemental analysis value: in terms of C41H52N4O4S.CH4SO3
Calcd. value: C, 63.61; H, 7.12; N, 7.06; S, 8.09.
Found value: C, 63.65; H, 7.23; N, 7.05; S, 8.08.
………………………….
References
- Klibanov, Olga M.; Williams, Shannon H.; Iler, Cameron A (2010). “Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection”. Current Opinion in Investigational Drugs 11 (8): 940–950. PMID 20721836.
- Baba, Masanori; Takashima, Katsunori; Miyake, Hiroshi; Kanzaki, Naoyuki; Teshima, Koichiro; Wang, Xin; Shiraishi, Mitsuru; Iizawa, Yuji (2005). “TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans”. Antimicrobial Agents and Chemotherapy 49 (11): 4584–4591. doi:10.1128/AAC.49.11.4584-4591.2005. PMC 1280155. PMID 16251299.
- C. Reviriego (2011). Drugs of the Future 36 (7): 511–517. doi:10.1358/dof.2011.36.7.1622066.
- “Tobira Therapeutics Initiates Phase 2b Trial of Cenicriviroc”. The Body. July 5, 2011.
- CROI 2013: CCR5/CCR2 Inhibitor Cenicriviroc Has Both Anti-HIV and Anti-inflammatory Effects. Highleyman, Liz. HIVandHepatitis.com. 7 March 2013.
|
11-26-2012
|
Chemokine receptor antagonists.
|
Journal of medicinal chemistry
|
|
|
6-1-2011
|
Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects.
|
Journal of acquired immune deficiency syndromes (1999)
|
|
|
8-1-2010
|
Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection.
|
Current opinion in investigational drugs (London, England : 2000)
|
|
|
3-1-2009
|
The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property?
|
Molecular pharmacology
|
|
|
2-1-2007
|
Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652.
|
Antimicrobial agents and chemotherapy
|
|
|
9-10-2006
|
[Progress in AIDS therapy].
|
Nihon Naika Gakkai zasshi. The Journal of the Japanese Society of Internal Medicine
|
|
|
3-23-2006
|
Highly potent and orally active CCR5 antagonists as anti-HIV-1 agents: synthesis and biological activities of 1-benzazocine derivatives containing a sulfoxide moiety.
|
Journal of medicinal chemistry
|
|
|
11-1-2005
|
TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans.
|
Antimicrobial agents and chemotherapy
|
|
|
1-27-2005
|
Stereoselective synthesis of [L-Arg-L/D-3-(2-naphthyl)alanine]-type (E)-alkene dipeptide isosteres and its application to the synthesis and biological evaluation of pseudopeptide analogues of the CXCR4 antagonist FC131.
|
Journal of medicinal chemistry
|
|
|
1-1-2005
|
TAK-652, a novel CCR5 inhibitor, has favourable drug interactions with other antiretrovirals in vitro.
|
Antiviral therapy
|
……………….
Chemical structures of selected small molecule CCR5 inhibitors. A. Maraviroc (MVC, Selzentry), B. Vicriviroc (VCV), C. Cenicriviroc (TBR-652), D. PF-232798.

ACH-702 the isothiazoloquinolone in preclinical from Achillion Pharmaceuticals (USA)

ACH-702
7-[3(R)-(2-Aminopropan-2-yl)pyrrolidin-1-yl]-9-cyclopropyl-6-fluoro-8-methoxy-2,3,4,9-tetrahydroisothiazolo[5,4-b]quinoline-3,4-dione
(7^-7-[3-(1-AMrNO-I-METHYLETHYL)PYRROLiDiN-I-YL]-P-CYCLOPROPYL-6-FLUORO-8-METHOXY-PH-ISOTHIAZOLO[5,4-B]QUINOLINE-3,4-DIONE
(R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-l-yl]-9-cyclopropyl-6-fluoro-8-methoxy- 9H-isothiazolo[5, 4-b] -quinoline-3, 4-dione
922491-46-1 free base
922491-09-6 (hydrochloride)468.973, C21 H25 F N4 O3 S . Cl H
ACH-0139586
ACH-702
Achillion Pharmaceuticals (USA)

pre clinical
Achillion Pharmaceuticals is working on the discovery of compounds in a new subclass of quinolones, the isothiazoloquinolones. The most advanced compound is ACH-702, which is at the pre-clinical stage of development [1-3].
 ACH 702
ACH 702
The utility of isothiazoloquinolines as pharmaceutical agents has been discussed in the literature. For example, Pinol, et al discussed the use of isothiazoloquinolines as medical bactericides in US Patent 5,087,621, including

The Proctor & Gamble Company discussed antimicrobial quinolones including the following compound:

in published application no. US 2003008894.
The use of isothiazoloquinoline compounds as TNF production inhibitors has also been discussed, for example by Sankyo Co., Ltd. in JPl 010149, which includes the following compound

Bayer Aktiengesellschaft has discussed bicycle[3.3.0]oct-7-yl containing compounds useful for treating H. pylori infections in WO 98/26768, including isothiazoloquinolines, having the general structure shown below in which Y may be sulfur joined to the carboxamide group to form a 5-membered ring

Otsuka Pharmaceutical Co., Ltd. has discussed the use of isothiazoloquinolines as antibacterial agents in JP 01193275, including the following carbamate-containing compound

Abbott Laboratories has discussed the use of isothiazoloquinolines as antineoplastic agents in US Patent No. 5,071,848 and has discussed the use of tricyclic quinolones as antibacterial agents in US 4,767,762. The Abbott compounds have hydrogen, halogen, or lower alkyl as substituents at the 6- and 8- positions of the isothiazoloquinoline core.
………………
Synthesis
WO2008021491A2
http://www.google.com/patents/WO2008021491A2?cl=en
EXAMPLE 1. SYNTHESIS OF (7^-7-[3-(1-AMrNO-I-METHYLETHYL)PYRROLiDiN-I-YL]-P-
CYCLOPROPYL-6-FLUORO-8-METHOXY-PH-ISOTHIAZOLO[5,4-B]QUINOLINE-3,4-DIONE (5). Step 1. Ethyl l-cyclopropyl-6, 7-difluoro-2-methanesulfonyl-8-methoxy-4-oxo-l,4-dihydro- quinoline-3-carboxylate (6)
Oxonβ®
MeOHZH2O


6
Water (180 mL), followed by Oxone® (Dupont Specialty Chemicals) (170 g, 277 mmol), is added to a suspension of 1 in MeOH (510 mL). The reaction mixture is heated with stirring at 55-60 0C for 3 h. The reaction mixture is cooled to room temperature, diluted with water (40 mL), and stirred at 5 0C (ice bath) for 30 min. The resulting crystals are collected by filtration, washed with water (2 x 100 mL), and dried to afford 6 (13.8 g). This material was used in the next step without further purification, mp 177-178 0C. 1H NMR (DMF-^7): J0.62 (m, IH), 1.11 (m, 2H), 1.29 (m, IH), 1.32 (t, JH-H = 7.0 Hz, 3H), 3.76 (s, 3H), 4.18 (m, IH), 4.21 (d, JH-F = 2.0 Hz, 3H), 4.33 (q, JH–H = 7.0 Hz, 2H), 7.64 (dd, JH-F = 10.0 Hz, 8.5 Hz, IH). Step 2 (R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-yl]-l-cyclopropyl-6-fluoro-2- methanesulfonyl-8-methoxy-4-oxo-l , 4-dihydro-quinoline-3-carboxylic acid ethyl ester (7)

10 6 7
A mixture containing compound (6) (3.88 g, 9.67 mmol), compound 10 (1.64 g, 12.8 mmol), anhydrous DIEA (5.05 g, 39.1 mmol, dried over 4A sieves), and anhydrous DMF (40 mL) is heated at 70 0C under an atmosphere of argon gas. After heating for 4.5 h (LC-MS analysis shows ~7% compound (6) remained), the reaction mixture is cooled to room temperature, diluted with EtOAc (200 mL), and washed with water (100 mL). The aqueous layer is extracted with EtOAc (100 mL), and the combined organic layers are washed with a saturated aqueous solution of sodium bicarbonate (100 mL). The organic layer is diluted with water (100 mL) and treated with an aqueous solution of HCl (4 N) until the aqueous layer is acidic (pH 2—3 after shaking the mixture vigorously). The organic layer is separated, and this process is repeated. The combined aqueous layers are diluted with EtOAc (100 mL) and treated with an aqueous solution of sodium hydroxide (6 N) until the aqueous layer is basic (pH ~8 after shaking the mixture vigorously). The aqueous layer is separated, and this process is repeated. The combined organic layers are dried over magnesium sulfate, filtered, and concentrated under reduced pressure giving an orange solid (3.27 g of an~80:20 mixture of compound (7) and impurity B). This solid is recrystallized from hot EtOAc (~60 mL) furnishing 2.18 g (44% yield) of pure compound 7 as a bright yellow solid. LC-MS mlz calcd for C24H32FN3O6S 509 ([M+]); found 510 ([M + H]+).
This reaction should not be allowed to proceed for more than a few hours (not overnight) as prolonged reaction time can lead to the formation of more side products. The product should be —95% pure (based on HPLC), with only a trace amount of impurity B. Step 3. (R)-7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-yl]-l-cyclopropyl-6-fluoro-2-mercapto-8- methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid ethyl ester (8)

7 8
Compound 7 (1.04 g, 2.04 mmol) is partially dissolved in DME (40 mL) under an atmosphere of argon. Sodium hydrosulfide hydrate (Aldrich, 72.6% by titration, 465 mg, 6.02 mmol) in water (3.0 mL) is added to this solution. The resulting mixture is sparged slowly with argon for 30 min.
The progress of the reaction is monitored by HPLC-MS, and judged to be complete (<2% of 7 remains) after 11.5 h. Excess sodium hydrosulfide is quenched upon addition of aq HCl (4.5 mL, 4 N).
The resulting orange solution (pH ~2) is sparged with argon (30 min) to remove the generated hydrogen sulfide. Step. 4 (R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-l-yl]-9-cyclopropyl-6-fluoro-8-methoxy- 9H-isothiazolo[5, 4-b] -quinoline-3, 4-dione (5)

A solution of potassium carbonate (4.26 g, 30.8 mmol) in water (25 mL) is next added to this solution to give a clear yellow solution (pH 9-10). The clear yellow solution is then sparged with argon for ~5 min. Finally, hydroxylamine-0-sulfonic acid (0.93 g, 8.2 mmol) is added portionwise as a solid, with immediate evolution of gas and formation of the product as a yellow precipitate. After stirring for 16 h, the reaction mixture (pH 10.2) is acidified with aq HCl to pH 8.3 (the approximate isoelectric point of 5) causing additional product to precipitate from solution. The reaction mixture is concentrated under reduced pressure (final volume -40 mL). The yellow precipitate is collected by centrifugation, washed with water (3 x 40 mL, with sonication), and lyophilized to give 0.80 g of 5.
……………………..
WO2007014308A1
http://www.google.com/patents/WO2007014308A1?cl=en
EXAMPLE 5. SYNTHESIS OF I-METHYL-I-PYRROLIDIN-3-YL ETHYLAMINE (5)
1 -Methyl- l-pyrrolidin-3-yl-ethylamine is prepared in accordance with the synthetic scheme below.
N O

5 P
Step 1. Synthesis of (S)-l-benzylpyrrolidin-3-yl methanesulfonate (N).
Methanesulfonyl chloride (15 mL, 0.19 mol) is added to a cooled (0 0C) solution of toluene (300 mL) containing (5)-l-benzylpyrrolidin-3-ol (24.5 g, 0.14 mol) and triethylamine (80 mL, 0.57 mol). The resulting mixture is stirred at 0 °C for 15 min, and allowed to warm to room temperature with stirring for 2h. The mixture is quenched with a 5% aqueous solution of sodium bicarbonate (250 mL). The organic layer is washed with a 5% aqueous solution of sodium bicarbonate (2 x 250 mL), washed with water (I x 250 mL), dried over magnesium sulfate, and concentrated under reduced pressure to give N (35.1 g, 99 %) as an orange oil. 1H NMR (300 MHz, CDCl3): £2.07 (m, IH), 2.30 (m, IH), 2.49 (m, IH), 2.75-2.90 (m, 3H), 2.98 (s, 3H), 3.61 (d, J= 13.0 Hz, IH), 3.68 (d, J= 13.0 Hz, IH), 5.18 (m, IH), 7.15-7.30 (m, 5H). LCMS mlz calcd for C12H17NO3S 255 ([M+]); found 256 ([M + H]+, 100%), 160 (40%). Steps 2 and 3. Syntheses of(R)-l-benzylpyrrolidine-3~carbonitrile (O) and 2-((R)-I- benzylpyrrolidin-3-yl)propan-2-amine (P).
The syntheses of O and P are described previously by Fedij et al. (Fedij, V.; Lenoir, E. A., Ill; Suto, M. J.; Zeller, J. R.; Wemple, J. Tetrahedron: Asymmetry 1994, J, 1131- 1134). Step 4. Synthesis ofl-((R)-Methyl~l-pyrrolidin-3-yl)-ethylamine (5).
A mixture containing P (7.4 g), 20% palladium hydroxide on carbon (7.5 g), and ethanol (75 niL) is stirred under an atmosphere of hydrogen gas (50 psi) at 45 °C for 24 h. The mixture is filtered and the filtrate is concentrated under reduced pressure to give 5 (4.1 g, 95 %) as a yellow oil. This material is stored under an atmosphere of argon gas. 1H NMR (300 MHz, CDCl3): J1.09 (s, 6H), 1.51 (m, IH), 1.64 (br s, 3H), 1.81 (m, IH), 2.06 (apparent pentet, J= 8.5 Hz, IH), 2.69 (dd, J= 11.0 Hz, J= 8.5 Hz, IH), 2.94 (m, 2H), 3.00 (dd, J= 11.0 Hz, J= 8.5 Hz, IH). LCMS mlz calcd for C7H16N2 128 ([M+]); found 129 ([M + H]+, 60%), 112 (100%).
EXAMPLE 6. GENERAL METHOD FOR THE FINAL AMINE-COUPLING STEP: SYNTHESIS OF 7-((R)-3-
(2-AMINOPROPAN-2-YL)PYRROLIDIN- 1 -YL)-9-CYCLOPROPYL-6-FLUORO-8- METHOXYISOTHIAZOLO[5,4-B]QUINOLINE-3 ,4(2H,9H)-DIONE HYDROCHLORIDE
[0261 ] 7-((R)-3-(2-Aminopropan-2-yl)pyrrolidin- 1 -yl)-9-cyclopropyl-6-fluoro-8- methoxyisothiazolo[5,4-b]quinoline-3,4(2H,9H)-dione hydrochloride is prepared in accordance with the synthetic scheme below.

Synthesis ofJ-ffRJS-^-aminopropan^-ylJpyrrolidin-l-ylJ-P-cyclopropyl-o-fluoro-S- methoxyisothiazolofS, 4-bJguinoline-3,4(2H, 9H)-dione hydrochloride (6).
Under an atmosphere of argon, a reaction vessel is charged with 5 (206.0 mg, 1.6 mmol), 3 (328.6 mg, 1.0 mmol), dimethyl sulfoxide (4.5 mL), and ΛζN-diisopropylethylamine (750 μL, 4.3 mmol). The resulting mixture is irradiated with microwaves (CEM Discover) at 125 0C for 1 h (conventional heating may also be used — 115 °C in an oil bath for 14 h), allowed to cool, and evaporated to dryness under reduced pressure (-70 °C/2-3 mm Hg). The oily residue is triturated with ethyl acetate (15 mL) and the resulting powder is collected by centrifugation. This solid is purified using preparative HPLC to give the desired product. Preparative HPLC is performed using a YMC Pack Pro C18 150 x 30.0 mm 5//m column coupled to a YMC Pack Pro 50 x 20 mm 5/an column with an isocratic elution of 0.37 min at 95:5 H2OiCHsCN containing 0.1% TFA followed by a 15.94 min linear gradient elution from 95:5 to 25:75, followed by a 0.69 min linear gradient from 25:75 to 5:95 at a flow rate of 30.0 mL/min with UV detection at 254. The crude material is loaded as a solution containing acetic acid (~2 mL), methanol (~1 mL), and water (~1 mL). The purified product is isolated as the TFA salt and is converted to the corresponding hydrochloride salt by addition of a solution of hydrogen chloride (~1.25 M in methanol) followed by evaporation; this process is repeated twice to give a yellow solid. Purity by HPLC: >99%; tR = 10.08 min. 1H NMR (300 MHz, TFA-d): δ 1.28 (m, 2H), 1.53 (m, 2H), 1.66 (s, 6H), 2.43 (m, IH), 2.57 (m, IH), 3.35 (m, IH), 3.97 (s, 3H), 4.01-4.38 (m, 5H), 8.17 (d, J= 12.0 Hz, IH, aromatic). 19F(1H) (282 MHz, TFA-J): δ-\ 18.0 (s). 13C(1H) (75 MHz, TFA-d): £13.5, 13.9, 25.0, 25.1, 29.1, 39.7, 49.6, 59.4 (br, W1/2 « 14 Hz), 59.8 (br, W1/2 « 14 Hz), 60.0, 66.8, 106.0, 112.1 (dJc_F = 23.0 Hz), 137.5 (br m, W1/2 « 24 Hz), 138.4, 144.8 (br, W1/2 » 10 Hz), 155.3 (dJc_F = 255.0 Hz), 169.8, 170.1, 171.5 (br, W1/2 « 9 Hz). LCMS mlz calcd for C21H25FN4O3S 432 ([M+]); found 433 ([M + H]+). Anal. Calcd for C21H25FN4O3S-l.5HCM.5H2O: C, 49.05; H, 5.78; N, 10.90; Cl, 10.34. Found: C, 49.30; H, 5.60; N, 10.83; Cl, 10.00.
EXAMPLE 3. SYNTHESIS OF 9-CYCLθPRθPYL-6,7-DiFLUθRθ-8-METHθχγ-9H-isoτHiAzθLθ[5,4- 5]QUlNOLlNE-3,4-DIONE (Compound 3).
9-Cyclopropyl-6,7-difluoro-8-methoxy-9H-isothiazolo[5,4-&]quinoline-3,4-dione (3) is prepared in accordance with the synthetic scheme below.

Step 1. Synthesis of 2,4, 5-trifluoro-3-methoxybenzoyl chloride (A)
A mixture of 2,4,5-trifluoro-3-methoxybenzoic acid (154 mg, 0.75 mmol) and thionyl chloride (8 mL) is refluxed for 4 h. Excess thionyl chloride is removed in vacuo, and the remaining residue is used directly in the next synthetic step. Step 2. Synthesis of (Z)-ethyl 3-hydroxy-3-(2,4,5-trifluoro-3-methoxyphenyl)aaγlate (B).
Compound B is prepared using the general method of Wierenga and Skulnick (Wierenga, W.; Skulnick, H. I. J. Org. Chein. (1979) 44: 310-311). H-Butyllithium (1.6 M in hexanes) is added to a cooled (-78 °C) solution of tetrahydrofliran (10 mL) containing ethyl hydrogen malonate (180 juL, 1.50 mmol) and 2,2′-bipyridyl (~1 mg as indicator). The temperature of the reaction mixture is allowed to rise to ca. -5 0C during the addition of n- butyllithium. Sufficient n-butyllithium (2.8 mL, 4.48 mmol) is added until a pink color persists at -5 0C for 5-10 min. A solution of 2,4,5-trifluoro-3-methoxybenzoyl chloride (0.75 mmol, vide supra) in tetrahydrofuran (~3 mL) is added in one portion to the reaction mixture that had been recooled to -78 0C. The resulting mixture is allowed to warm to room temperature, diluted with ethyl acetate (50 mL), and quenched with a 1 M aqueous solution of hydrochloric acid. The organic layer is washed with a 5% aqueous solution of sodium bicarbonate (2 x 30 mL), followed by brine (2 x 50 mL), dried over sodium sulfate, and evaporated under reduced pressure to give the crude product. This material is purified by flash column chromatography (eluting with 20% v/v ethyl acetate in hexanes) to give pure B as a white solid. 1H NMR (300 MHz, CDCl3): (enol, predominant tautomer, >90%) δ 1.32 (t, JH–H = 7.0 Hz, 3H, CO2CH2CH3), 4.02 (apparent t, JH–F = 1.0 Hz, 3H, OCH3), 4.25 (q, JH–H = 7.0 Hz, 2H, CO2CH2CH3), 5.79 (s, IH, CH3C(OH)=CH- CO2CH2CH3), 7.39 (ddd, JH_F= 11.0 Hz, 8.5 Hz, 6.5 Hz, IH, aromatic), 12.68 (s, IH, OH). 19F(1H) NMR (282 MHz, CDCl3): <5-146.8 (dd, JF_F = 21.5 Hz, 10.5 Hz, IF), -140.2 (dd, JF_F = 21.5 Hz, 13.5 Hz, IF), -131.3 (dd, JF_F = 13.5 Hz, 10.5 Hz, IF).
Step 3. Synthesis ofζEyethy^-^ZJ-N-cyclopropy^methylthioJcarbonoimidoylJS-hydroxyS- (2, 4, 5-trifluoro-3-methoxyphenyl)acrylate (C)
Sodium hydride (60% in mineral oil, 31 mg, 0.78 mmol) is added portionwise to a cooled (0 °C) solution containing B (200 mg, 0.73 mmol), cyclopropyl isothiocyanate (120 /JL, 1.2 mmol), and dimethylformamide (2 mL). The resulting mixture is allowed to warm to room temperature with stirring overnight (18 h). Methyl iodide (80 juL, 1.2 mmol) is added to the resulting solution and stirred for an additional 4 h (until TLC indicated the complete consumption of B). The reaction mixture is diluted with ethyl acetate (100 mL) and quenched by addition of a saturated aqueous solution of ammonium chloride (30 mL). The organic layer is washed with brine (4 x 30 mL), dried over sodium sulfate, and evaporated under reduced pressure to give the crude product. This material is purified by flash column chromatography (eluting with 40% v/v ethyl acetate in hexanes) to give C as a yellow oil. 1H NMR (300 MHz, CDCl3): (50.86 (m, 2H, cyclopropyl CH2), 0.97 (m, 5H), 2.52 (s, 3H, SCH3), 3.00 (m, IH, cyclopropyl CH), 3.96 (q, JH–H = 7.0 Hz, 2H, CO2CH2CH3), 4.02 (apparent t, JH–F = 1.0 Hz, 3H, OCH3), 6.96 (m, IH, aromatic), 11.71 (s, IH). 19F(1H) NMR (282 MHz, CDCl3): £-149.9 (br, IF), -141.4 (br, IF), -135.7 (br, IF).
Step 4. Synthesis of ethyl l-cyclopropyl-6,7-difluoro-8-methoxy-2-(methylthio)-4-oxo-l,4- dihydroquinoline-3-carboxylate (D)
Sodium hydride (60% in mineral oil, 82 mg, 2.1 mmol) is added portionwise to a solution of C (760 mg, 1.95 mmol) in dimethylformamide (15 mL) at room temperature. The reaction mixture is heated at 80 0C for 3 d (until TLC indicates the complete consumption of B), cooled to room temperature, and quenched by addition of a saturated aqueous solution of ammonium chloride (10 mL). The mixture is extracted with ethyl acetate (3 x 50 mL). The combined organic extracts are washed with brine (4 x 30 mL), dried over sodium sulfate, and evaporated under reduced pressure to give crude D. This material is purified by flash column chromatography (eluting with 30% v/v ethyl acetate in hexanes) to D as a pale yellow oil.1H NMR (300 MHz, CDCl3): £0.73 (m, 2H, cyclopropyl CH2), 1.19 (m, 2H, cyclopropyl CH2), 1.38 (t, JH–H = 7.0 Hz, 3H, CO2CH2CH3), 2.66 (s, 3Η, SCH3), 3.74 (m, IH, cyclopropyl CH), 4.08 (d, JH–F = 2.5 Hz 3H, OCH3), 4.40 (q, JH_H = 7.0 Hz, 2H, CO2CH2CH3), 7.76 (dd, JH_F = 10.5 Hz, 8.5 Hz IH, aromatic). 19F(1H) NMR (282 MHz, CDCl3): £-146.8 (d, JF_F = 21.0 Hz, IF), – 137.7 (d, JF–F = 21.0 Hz, IF). LCMS mlz calcd for C17H17F2NO4S 369 ([M+]); found 370 ([M + H]+).
Step 5. Synthesis of ethyl l-cyclopropyl-6,7-difluoro-8-methoxy-2-(methylsulfinyl)-4-oxo-l,4- dihydroquinoline-3-carboxylate (E)
m-Chloroperoxybenzoic acid (<77%, 34 mg, 0.15 mmol) is added in one portion to a solution of D (50 mg, 0.14 mmol) in methylene chloride (3 mL) at room temperature. The reaction mixture is stirred for 1 h, diluted with ethyl acetate (20 mL), and washed with a 5% aqueous solution of sodium bicarbonate (2 x 10 mL). The organic layer is dried over sodium sulfate and evaporated under reduced pressure to give the crude product. This material is purified by preparative thin-layer chromatography (eluting with 10% v/v hexanes in ethyl acetate) to give pure E as a white solid. 1H NMR (300 MHz, CDCl3): £0.62 (m, IH, cyclopropyl CH2), 1.00 (m, IH, cyclopropyl CH2), 1.13 (m, IH, cyclopropyl CH2), 1.29 (m, IH, cyclopropyl CH2), 1.36 (t, JH_H = 7.5 Hz, 3H, CO2CH2CH3), 3.22 (s, 3Η, S(O)CH3), 3.85 (m, IH, cyclopropyl CH), 4.09 (d, JH-F = 2.5 Hz, 3H, OCH3), 4.37 (q, JH–H = 7.5 Hz, 2H, CO2CH2CH3), 7.75 (dd, JH–F = 10.0, 8.0 Hz, IH, aromatic). 19F(1H) NMR (282 MHz, CDCl3): £-145.2 (d, JF_F = 21.0 Hz, IF), -136.2 (d, JF_F = 21.0 Hz, IF). LCMS mlz calcd for C17H17F2NO5S 385 ([M+]); found 386 ([M + H]+).
Step 6. Synthesis of ethyl l-cyclopropyl-βJ-difluoro-l-mercaptoS-methoxy-^oxo-lA- dihydroquinoline-3-carboxylate (F).
Anhydrous sodium hydrogen sulfide (Alfa Aesar, 20 mg, 0.36 mmol) is added in one portion to a solution of DMF (6 mL) containing E (93 mg, 0.24 mmol) at room temperature. The resulting solution is heated at 40 0C for 2-3 h (until TLC indicated complete consumption of E) and allowed to cool to room temperature. The reaction mixture is quenched by addition of a 5% aqueous solution of hydrochloric acid (20 mL) and extracted with ethyl acetate (2 x 25 mL). The combined organic extracts are washed with brine (4 x 25 mL), dried over sodium sulfate, and evaporated to dryness under reduced pressure to give crude F in quantitative yield. This material is used directly in the next synthetic step to prevent its oxidative degradation. LCMS mlz calcd for C16H15F2NO4S 355 ([M+]); found 356 ([M + H]+) Step 7. Synthesis of9-cyclopropyl-6,7-difluoro-8-methoxyisothiazolo[5,4-b]quinoline- 3,4(2H,9H)-dione (3).
A solution of sodium bicarbonate (820 mg, 9.8 mmol) in water (14 mL) is added to a solution of F (348 mg, 0.98 mmol) in tetrahydrofuran (10 mL) at room temperature. Hydroxylamine-O-sulfonic acid (465 mg, 4.1 mmol) is added in one portion to this mixture. The reaction mixture is stirred at room temperature for ~3 h and quenched by addition of an aqueous solution of 5% hydrochloric acid (100 mL). The precipitate that formed is collected by filtration, washed with water (3 x 5 mL), and dried in vacuo to give 3 as a white solid. This product is of sufficient purity (>95% by 1H NMR spectroscopy) to use directly in the final amine-coupling step. 1HNMR (300 MHz, DMSO-J6): Jl.12 (m, 4H, cyclopropyl CH2), 3.85 (m, IH, cyclopropyl CH), 4.01 (d, JH–F= 1.5 Hz, 3H, OCH3), 7.85 (dd, JH_F = 11.0 Hz, 9.0 Hz, IH, aromatic). 19F(1H) NMR (282 MHz, DMSO-J6): £-146.4 (d, JF_F = 23.0 Hz, IF), -140.2 (d, JF_ F = 23.0 Hz, IF). LCMS mlz calcd for C14H10F2N2O3S 324 ([M*]); found 325 ([M + H]+).
REFERENCES
- Achillion Pharmaceuticals. About ACH-702. Available online: http://www.achillion.com/PL/pdf/04_ach_702_bg.pdf (accessed on 2 May 2013).
- Pucci, M.J.; Podos, S.D.; Thanassi, J.A.; Leggio, M.J.; Bradbury, B.J.; Deshpande, M. In vitro and in vivoprofiles of ACH-702, an isothiazoloquinolone, against bacterial pathogens. Antimicrob. Agents Chemother. 2011, 55, 2860–2871, doi:10.1128/AAC.01666-10.
- Achillion Pharmaceuticals, Inc. SEC filling form 10-Q quarterly report filed August 7, 2013. Available online: http://ir.achillion.com/secfiling.cfm?filingID=1193125–13–324297 (accessed on 28 September 2013).
- An efficient method for the synthesis of (R)-3-(1-amino-1-methylethyl)pyrrolidines for the antiinfective agent, PD 138312
Tetrahedron Asymmetry 1994, 5(7): 1131 - WO 2007014308
- WO 2008021491
-
WO2011031745A1 Sep 8, 2010 Mar 17, 2011 Achaogen, Inc. Antibacterial fluoroquinolone analogs -
HASHIMOTO, A. ET AL.: “Practical synthesis and molecular structure of a potent broad-spectrum antibacterial isothiazoloquinolone” ORG. PROCESS RESEARCH & DEVELOPMENT, vol. 11, 16 March 2007 (2007-03-16), pages 389-398, XP002465315 2 * WANG, Q. ET AL.: “Isothiazoloquinolones with Enhanced Antistaphylococcal Activities against Multidrug-Resistant Strains: Effects of Structural Modifications at the 6-, 7-, and 8-Positions” J. MED. CHEM., vol. 50, 2007, pages 199-210, XP002465316 -
WO2005019228A1 * Aug 4, 2004 Mar 3, 2005 Achillion Pharmaceuticals Inc Isothiazoloquinolones and related compounds as anti-infective agents WO2006118605A2 * Nov 10, 2005 Nov 9, 2006 Achillion Pharmaceuticals Inc 8a, 9-dihydro-4a-h-isothiazolo[5,4-b] quinoline-3, 4-diones and related compounds as anti-infective agents WO2007014308A1 * Jul 27, 2006 Feb 1, 2007 Achillion Pharmaceuticals Inc 8-methoxy-9h-isothiazolo[5,4-b]quinoline-3,4-diones and related compounds as anti-infective agents -
Citing Patent Filing date Publication date Applicant Title WO2008021491A2 * Aug 16, 2007 Feb 21, 2008 Achillion Pharmaceuticals Inc Method for synthesis of 8-alkoxy-9h-isothiazolo[5,4-b]quinoline-3,4-diones WO2011031745A1 Sep 8, 2010 Mar 17, 2011 Achaogen, Inc. Antibacterial fluoroquinolone analogs EP2488532A2 * Oct 15, 2010 Aug 22, 2012 Rib-X Pharmaceuticals, Inc. Antimicrobial compounds and methods of making and using the same US7902365 Aug 16, 2007 Mar 8, 2011 Achillion Pharmaceuticals, Inc. Method for synthesis of 8-alkoxy-9H-isothiazolo[5,4-B]quinoline-3,4-diones US8138346 Mar 4, 2011 Mar 20, 2012 Achillion Pharmaceuticals, Inc. Method for synthesis of 8-alkoxy-9H-isothiazolo[5,4-B]quinoline-3,4-diones
MG 96077 in Pre-Clinical for Gram-negative bacteria

MG 96077
poster
MG96077 – MethylGene
………..http://methylgene.solocom.biz/files/2011/10/poster102.pdf ……………..lot of data presented
Mirati Therapeutics (USA)
Pre-Clinical for Gram-negative bacteria
Beta-Lactamase Inhibitors—Non-beta-Lactam Phosphonate-Based
Mirati Therapeutics is seeking partners to continue the development of the compound MG96077, a non-beta-lactam phosphonate-based beta-lactamase inhibitor that has shown an inhibitory profile for both class A and class C beta-lactamase enzymes [1,2].
Potent, irreversible inhibitor of serine β-lactamases that efficiently protects βlactams from hydrolysis in a variety of class
A- and class C-producing organisms-
 |
September 14, 2009 13:23 ET
MethylGene Presents Preclinical Data for Its Beta-Lactamase Inhibitor, MG96077, at the 49th Annual ICAAC Meeting
MONTREAL, QUEBEC–(Marketwire – Sept. 14, 2009) – MethylGene Inc. (TSX:MYG) today disclosed preclinical data for MG96077, a novel, broad spectrum, non-beta-lactam beta-lactamase inhibitor (BLI). MG96077 possesses a broad-spectrum inhibitory profile for both class A and class C beta-lactamase enzymes, including extended spectrum beta-lactamases (ESBLs). In addition, the compound overcomes resistance in beta-lactam-resistant organisms such as Pseudomonas aeruginosa. The data were presented in a poster session at the 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) Annual Meeting in San Francisco, California.
Poster C1-1373: Novel Beta-Lactamase Inhibitor Potentiates and Extends the Antibacterial Activity of Imipenem against Beta-Lactam-Resistant P. aeruginosa and K. pneumoniae
MG96077 was tested in combination with imipenem, a commonly-used antibiotic agent for a variety of serious infections.
A series of in vitro and in vivo preclinical studies focused on comparing the combination of MG96077 and imipenem to imipenem alone, or imipenem plus currently approved BLIs, were performed. Greater than 90 percent of imipenem-resistant clinical isolates of Pseudomonas aeruginosa and Klebsiella pneumoniae were rendered susceptible with the addition of MG96077 to imipenem. The combination of imipenem and any of the three currently approved BLIs did not achieve greater than 61 percent coverage.
Furthermore, the combination of imipenem and MG96077 in vivo demonstrated 3-6 log reduction in colony forming units (CFU) and a 100 percent survival rate in combating imipenem-resistant P. aeruginosa infections of mouse spleen and lung. The pharmacokinetic properties of MG96077 were similar to imipenem in preclinical studies with no observable drug-drug interactions.
Thus, MG96077 is a novel beta-lactamase inhibitor that restores efficacy to imipenem against a high percentage of imipenem-resistant Pseudomonas and Klebsiella strains and, therefore, may address the clinical need for antibacterial therapies with more potent coverage of resistant gram-negative organisms.
MethylGene retains exclusive rights to MG96077 and a series of related molecules. Additional data has been developed regarding MG96077 compared to other beta-lactam antibiotics, as well as other compounds in the series paired with various beta-lactam antibiotics.
“Antibiotic resistance rates are increasing among several problematic gram-negative pathogens, including P. aeruginosa, K. pneumoniae, Acinetobacter spp. and Enterobacteriaceae that are often responsible for serious hospital-acquired infections. In these studies, MG96077 appears to demonstrate activity in a variety of organisms and we look forward to further evaluation of this compound in what is a growing antibiotic market in need of novel treatments,” said Donald F. Corcoran, President and Chief Executive Officer of MethylGene.
About MethylGene
MethylGene Inc. (TSX:MYG) is a publicly-traded, clinical stage, biopharmaceutical company focused on the discovery, development and commercialization of novel therapeutics with a focus on cancer. The Company’s product candidates include: MGCD265, an oral, multi-targeted kinase inhibitor targeting the c-Met, VEGF, Ron and Tie-2 receptor tyrosine kinases that is in Phase I and Phase II clinical trials for cancers; MGCD290, a fungal Hos2 inhibitor being developed for use in combination with fluconazole for serious fungal infections that is in Phase I clinical studies; and MGCD0103, an oral, isoform-selective HDAC inhibitor which has been in multiple clinical trials for solid tumors and hematological malignancies and is licensed to Taiho Pharmaceutical Co. Ltd. A fourth compound discovered using MethylGene’s HDAC platform, EVP-0334 – a potential cognition enhancing agent, is in a Phase I study sponsored by EnVivo Pharmaceuticals Inc. MethylGene also has a funded collaboration with Otsuka Pharmaceutical Co. Ltd. for applications in ocular diseases using the Company’s proprietary kinase inhibitor chemistry. Please visit our website at www.methylgene.com.
- Martell, L.A.; Rahil, G.; Vaisburg, A.; Young, K.; Hickey, E.; Hermes, J.; Dininno, F.; Besterman, J.M. A Novel Beta-Lactamase Inhibitor Potentiates and Extends the Antibacterial Activity of Imipenem against β-Lactam-Resistant P. aeruginosa and K. pneumoniae. In Proceedings of 49th ICAAC Annual Meeting, San Francisco, CA, USA, 14 September 2009.
- Mirati Therapeutics. MG96077. Available online: http://mirati.com/other-pipeline-assets/mg96077(accessed on 9 July 2013).
- 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) Annual Meeting in San Francisco, California.
Poster C1-1373: Novel Beta-Lactamase Inhibitor Potentiates and Extends the Antibacterial Activity of Imipenem against Beta-Lactam-Resistant P. aeruginosa and K. pneumoniae
ATL1102 for MS – Toxicology Study Main Findings
| Sequence Type: DNA fragment | |
| CTGAGTCTGTTTTCCATTCT |
ATL 1102
The antisense oligonucleotide is complementary to a region in the 3’UTR of human ITGA4 (integrin alpha 4) cDNA whose sequence is 5′-CTGAGTCTGTTTTCCATTCT-3′
Phosphorothioate antisense oligonucleotide consisting of a 9-nucleotide central region of deoxynucleotides flanked by 3 2′-O-methoxyethyl (2′-MOE) nucleotides on the 5′ end and 8 2′-MOE nucleotides on the 3′ end.
TOORAK, Australia, April 1, 2014 /PRNewswire/ — Antisense Therapeutics Limited (“ANP” or the “Company”) is pleased to advise that results from a chronic toxicity study in monkeys indicate that ATL1102, an antisense oligonucleotide currently under development for the treatment of multiple sclerosis (MS), was well-tolerated when given subcutaneously for a 6-month dosing period at the 2 dose levels tested (1.5 and 3mg/kg/dose). The Company believes that the preclinical and clinical experience to date with ATL1102 should allow dosing in future trials at or above the 1.5 mg/kg/dose level.
read at
http://www.sys-con.com/node/3037721
ATL-1102
ISIS-107248
TV-1102
ITGA4 Expression Inhibitors
Signal Transduction Modulators
PHASE 2
Antisense Therapeutics
Isis Pharmaceuticals
Antisense Therapeutics Limited (ASX: ANP) is an Australian publicly listed biopharmaceutical drug discovery and development company. Its mission is to create, develop and commercialise second generation antisense pharmaceuticals for large unmet markets. ANP has 4 products in its development pipeline that it has in-licensed from Isis Pharmaceuticals Inc., world leaders in antisense drug development and commercialisation – ATL1102 (injection) which has successfully completed a Phase II efficacy and safety trial, significantly reducing the number of brain lesions in patients with multiple sclerosis, ATL1103 a second-generation antisense drug designed to block GHr production and thereby lower blood IGF-I levels and is in clinical development as a potential treatment for growth and other GH-IGF-I disorders, ATL1102 (inhaled) which is at the pre-clinical research stage as a potential treatment for asthma and ATL1101 a second-generation antisense drug at the pre-clinical stage being investigated as a potential treatment for cancer.
ATL1102 is a second generation antisense inhibitor of CD49d, a subunit of VLA-4 (Very Late Antigen-4). In inflammation, white blood cells (leukocytes) move out of the bloodstream into the inflamed tissue, for example, the Central Nervous System (CNS) in MS, and the lung airways in asthma. The inhibition of VLA-4 may prevent white blood cells from entering sites of inflammation, thereby slowing progression of the disease. VLA-4 is a clinically validated target in the treatment of MS. Antisense inhibition of VLA-4 has demonstrated positive effects in a number of animal models of inflammatory disease including MS with the MS animal data having been published in a peer reviewed scientific journal. ATL1102 was previously shown by Antisense Therapeutics to be highly effective in reducing MS lesions in a Phase IIa clinical trial in MS patients.
ATL-1102 is an antisense oligonucleotide in phase II clinical trials at Isis Pharmaceuticals and Antisense Therapeutics for the treatment of relapsing-remitting multiple sclerosis (MS) in a subcutaneous injection formulation. Phase I clinical trials in a subcutaneous injections for stem cell mobilization and preclinical studies of an inhalation formulation of the drug candidate for the treatment of asthma are also being conducted at Antisense Therapeutics.
ATL-1102 is complementary to nt 4288-4207 (3’UTR) of human integrin alpha 4 (ITGA4) cDNA, and thus inhibits ITGA4 expression, blocking the synthesis of CD49d, a subunit of very late antigen-4 (VLA-4). VLA-4 is known to play a part in both the onset and progression of MS, and its inhibition may prevent white blood cells from entering the central nervous system.
ATL-1102 was originally developed at Isis Pharmaceuticals. In December 2001, Isis and Circadian Technologies formed Antisense Therapeutics, established to focus on the discovery and development of antisense therapeutics. As part of the company’s formation, Antisense Therapeutics received a license to ATL-1102 and entered into a five-year antisense drug discovery and development program with Isis. In 2008, Antisense licensed ATL-1102 to Teva. In 2010, Teva terminated its licensee agreement with Antisense for the development of ATL-1102 for the treatment of relapsing-remitting multiple sclerosis. The company stated that the compound was not on line with its preferred product pipeline. In 2001, ATL-1102 was licensed to Antisense Therapeutics by Isis Pharmaceuticals. In 2012, development and commercialization rights to the product were licensed to Tianjin International Joint Academy of Biotechnology and
Contact Information:
Website: www.antisense.com.au
Managing Director: Mark Diamond +61 (3) 9827 8999
USA Investor/Media: Joshua Drumm +(1) 212 375 2664;jdrumm@tiberend.com
Australia Investor/Media: Simon Watkin +61 (0)413 153 272;simon@marketconnect.com.au
SOURCE Antisense Therapeutics Limited
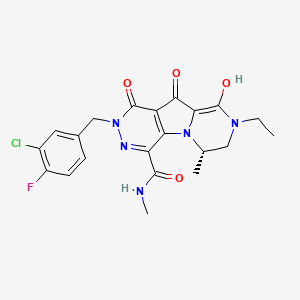
MK 2048 an HIV integrase inhibitor from Merck

MK 2048
Molecular Formula: C21H21ClFN5O4 Molecular Weight: 461.873943
869901-69-9, 3oyl, 3oyn
| (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide |
6(S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
|
5-27-2009
|
Hiv Integrase Inhibitors
|
MK-2048 is a second generation integrase inhibitor, intended to be used against HIV infection. It is superior to the first available integrase inhibitor,raltegravir, in that it inhibits the HIV enzyme integrase 4 times longer. It is being investigated for use as part of pre-exposure prophylaxis (PrEP). [1]
It is being developed by Merck & Co.[2]
![]()
MK-2048 is a second generation integrase inhibitor for HIV-1 integrase. MK-2048 inhibits subtype B and subtype C integrase activities. MK-2048 inhibits R263K mutants slightly more effectively than G118R mutants.
MK-2048 inhibits S217H intasome and, by contrast, MK2048 remains fully active against the N224H intasome. MK2048 displays substantially lower dissociation rates compared with raltegravir, another integrase inhibitor.
MK-2048 is active against viruses resistant to RAL and EVG. MK-2048 exposure leads to the selection of G118R as a possible novel resistance mutation after 19 weeks. MK-2048, with continued pressure, subsequently leads to an additional substitution, at position E138K, after 29 weeks, within the IN gene.
Although the G118R mutation alone confers only slight resistance to MK-2048 but not to RAL or EVG, its presence arouses a dramatic reduction in viral replication capacity compared to wild-type NL4-3. E138K both partially restores viral replication capacity and also contributes to increased levels of resistance against MK-2048.

Structure of MK-2048 with important pharmacophore highlighted

…………………..
Synthesis
WO2005110415A1
http://www.google.as/patents/WO2005110415A1?cl=en
EXAMPLE 62 6(S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide

Step 1: te rt-Butyl[( 1 S)-2-(ethylamino)- 1 -methylethyl] carbamate To a cold (0 °C) solution of N-(tø/ -butoxycarbonyl)-L-alanine N’-methoxy-N’- methylamide (15.6 g, 67.2 mmol) in anhydrous THF (150 mL) and diethyl ether (400 mL), solid lithium aluminum hydride (5.1 g, 134.3 mmol) was added portionwise over a period of 30 minutes. The mixture was stirred at room temperature for 3 hours and cooled back to 0 °C. The reaction was treated carefully with an aqueous solution of potassium hydrogen sulfate (250 mL, 1M). The resultant mixture was diluted with diethyl ether.
The organic extract was washed successively with dilute hydrochloric acid, and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide the corresponding aldehyde as colorless solid. Without further purification, a cold (0 °C), stirred solution of the intermediate aldehyde (10.7 g, 61.8 mmol) and ethylamine hydrogen chloride (10.1 g, 123.5 mmol) in methanol (72 mL) was treated with sodium triacetoxyborohydride (17.2 g, 80.9 mmol) in one portion. The mixture was allowed to warm up to room temperature.
After stirring at room temperature overnight, the solution was concentrated under vacuum. The residue was partitioned between diethyl ether and cold aqueous sodium hydroxide (1.5 M). The ethereal extract was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide the titled compound. lH NMR (400 MHz, CDCI3) δ 4.68 (br s, IH), 3.75 (br t, IH), 2.62 (m, 5 H), 1.13 (d, J = 6.7 Hz, 3H),
1.09 (t, J = 7.0 Hz, 3H). ES MS M+l = 203
Step 2: ført-Butyl { ( 1 S)-2-[(bromoacetyl)ethylamino] – 1 -methylethyl } carbamate To a cold (0 °C) stirred solution of ?ert-butyl[(lS)-2-(ethylamino)-l- methylethyl]carbamate (11.0 g, 54.6 mmol) in a mixture of ethyl acetate (107 mL) and saturated aqueous sodium bicarbonate (65 mL), bromoacetyl bromide (12.1 g, 60.0 mmol) was added portionwise under an atmosphere of nitrogen. The mixture was allowed to warm up to room temperature over a period of 3.5 hours. The organic phase was separated, washed successively with saturated aqueous sodium bicarbonate, and brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was concentrated as a solution in toluene under vacuum to afford the title compound. ES MS M+l = 323, 325.
Step 3: fe7 -Butyl (2S)-4-ethyl-2-methyl-5-oxopiperazine-l-carboxylate To a stirred slurry of sodium hydride (1.7 g, 69.8 mmol) in anhydrous THF (800 mL), a solution of tert-butyl{(lS)-2-[(bromoacetyl)ethylamino]-l-methylethyl}carbamate (17.4 g, 53.7 mmol) in anhydrous THF (100 mL) was added dropwise over a period of 1 hour under an atmosphere of nitrogen. The reaction mixture was stirred at room temperature for two hours, cooled in an ice-water bath, and quenched with dropwise addition of aqueous citric acid (80 mL, 1M). The mixture was concentrated under vacuum. The residue was partitioned between chloroform and saturated aqueous sodium bicarbonate. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a gradient of 0-15% acetonitrile in chloroform. Collection and concentration of appropriate fractions provided the title compound. lH NMR (400 MHz, CDCI3) δ 4.46 (br s, IH), 4.24 (d, J = 18.4 Hz, 1 H), 3.78 (d, J = 18.4 Hz, 1 H),
3.64 (dd, J = 12.3, 4.2 Hz, 1 H), 3.54 (heptet, J = 7.1 Hz, 1 H), 3.38 (heptet, J = 7.1 Hz, 1 H), 2.99 (dd, J = 12.3, 1.8 Hz, 1 H), 1.47 (s, 9H), 1.21 (d, J = 6.8 Hz, 3H), 1.14 (t, J = 7.1 Hz, 3H). ES MS M+l = 243.
Step 4: (5S)-l-Ethyl-5-methylpiperazin-2-one hydrochloride Anhydrous hydrogen chloride gas was bubbled into a cold (-20 °C) solution of tert-butyl (2S)-4-ethyl-2-methyl-5-oxopiperazine-l-carboxylate (10.5 g, 43.4 mmol) in ethyl acetate (250 mL) under nitrogen. After the solution was saturated with hydrogen chloride, the reaction mixture was stirred in an ice-water bath for 30 minutes. The product mixture was purged with nitrogen, concentrated under vacuum to provide the title hydrogen chloride salt as pale yellow solid. lH NMR (400 MHz, DMSO-d6) δ 10.00 (br d, 2H), 3.72 (d, J = 16.6 Hz, 1 H), 3.62(d, J = 16.6 Hz, 1 H),
3.49-3.35 (m, 5 H), 3.29 (heptet, /= 7.3 Hz, 1 H), 1.31 (d, / = 6.6 Hz, 3H), 1.05 (t, J = 7.1 Hz, 3H).
Step 5: Ethyl (4S)-2-ethyl-8-hydroxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7- carboxy late Anhydrous ammonia gas was bubbled into a cold (0 °C) solution of (5S)-l-Ethyl-5- methylpiperazin-2-one hydrochloride (5.8 g, 32.3 mmol) in chloroform for 30 minutes. The resultant slurry was filtered and concentrated under vacuum. The residual oil was concentrated as a solution in toluene under vacuum, redissolved in toluene (120 mL) and treated with diethyl ethoxymethylenemalonate (7.0 g, 32.3 mmol) and heated in a sealed flask in an oil bath at 100 °C overnight. The resultant solution was concentrated under vacuum. The residual oil was concentrated as a solution in toluene under vacuum to provide the corresponding diethyl { [(2S)-4-ethyl-2-methyl-5- oxopiperazin-l-yl]methylene}malonate. Without further purification, to a solution of the malonate (10.5 g, 33.5 mmol) in anhydrous THF (330 mL) warmed with an external oil bath at 65 °C under an atmosphere of nitrogen, a solution of lithium bis(trimethylsilyl)amide (35.1 mL, 1 M, 35.1 mmol) was added. The solution was heated at the same temperature for one hour and concentrated under vacuum. The residue was partitioned between dichloromethane and hydrochloric acid (1M). The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with diethyl ether. The solid precipitated was filtered, washed with diethyl ether to provide the title compound as pale brown solid. lH NMR (400 MHz, CDCI3) δ 8.43 (s, IH), 7.11 (s, IH), 4.32 (q, J = 7.1 Hz, 2H), 4.24 (m, IH), 3.65-
3.35 (m, 4H), 1.51 (d, J = 6.4 Hz, 3H), 1.36 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H). ES MS M+l = 267
Step 6: Ethyl (4S)-2-ethyl-8-methoxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7- carboxylate A mixture of ethyl (4S)-2-ethyl-8-hydroxy-4-methyl-l -oxo- 1,2,3, 4-tetrahydropyrrolo[ 1,2- a]pyrazin-7-carboxylate (6.6 g, 24.8 mmol), anhydrous potassium carbonate (13.7 g, 99.1 mmol, 325 mesh), and iodomethane (4.2 g, 29.7 mmol) in anhydrous DMF (123 mL) was stirred at room temperature overnight. The mixture was filtered and concentrated under vacuum. The residue was partitioned between chloroform and dilute hydrochloric acid. The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a gradient of 0-3% methanol in chloroform. Collection and concentration of appropriate fractions provided the title compound. Residual methanol was removed by concentrating from its solution in toluene under vacuum. lH NMR (400 MHz, CDCI3) δ 7.19 (s, IH), 4.29 (q, J = 7.1 Hz, 2 H), 4.24 (m, IH), 4.03 (s, 3H), 3.70-
3.32 (m, 4 H), 1.52 (d, J = 6.6 Hz, 3H), 1.35 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.2 Hz, 3H). ES MS M+l = 281
Step 7: Ethyl (4S)-6-bromo-2-ethyl-8-methoxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2- a]pyrazin-7-carboxylate To a mixture of ethyl (4S)-2-ethyl-8-(methoxy)-4-methyl-l-oxo-l,2,3,4- tetrahydropyrrolo[l,2- ]pyrazine-7-carboxylate (6.2 g, 22.1 mmol) and sodium bicarbonate (20.0 g, 238.0 mmol) in dichloromethane (500 mL) at 0 °C, a solution of bromine in dichloromethane (24.2 mmol, 0.5 M) was added over a period of 60 minutes. The reaction mixture was stirred at room temperature for 2 h, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluted with ethyl acetate. Collection and concentration of appropriate fractions provided the corresponding bromide. Residual ethyl acetate was removed by concentrating from its solution in benzene under vacuum. lH NMR (400 MHz, CDCI3) δ 4.58 (br m, IH), 4.34 (m, IH), 3.99 (s, 3H), 3.92 (dd, J = 13.0, 4.0 Hz,
IH), 3.67 (heptet, J = 7.1 Hz, 1 H), 3.49 (heptet, J = 7.1 Hz, 1 H), 3.23 (d, J = 13.0 Hz, IH), 1.40 (d, J = 7.1 Hz, 3H), 1.38 (t, 7 = 7.0 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). ES MS M+l = 359, 361.
Step 8: Ethyl (4S)-2-ethyl-8-(methoxy)-6-[methoxy(oxo)acetyl]-4-methyl-l-oxo-l,2,3,4- tetrahydropyrrolo[ 1 ,2- ]pyrazine-7-carboxylate To a cold (-78 °C) solution of ethyl (4S)-6-bromo-2-ethyl-8-methoxy-4-methyl-l-oxo- l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7-carboxylate (8.51 g, 23.7 mmol) in anhydrous THF (800 mL) under an atmosphere of dry nitrogen, a solution of n-BuLi in hexane (10.5 mL, 26.3 mmol, 2.5 M) was added. The resultant mixture was stirred at -78 °C for 20 minutes. A solution of dimethyl oxalate (6.4 g, 53.8 mmol; dried from concentration from benzene under vac) in anhydrous THF (30 mL) was added. The reaction mixture was stirred at -78 °C for 1 hour and cannulated into a mixture of aqueous sulfuric acid (240 mL, 2M) and THF (200 mL) maintained between at -5 to -35 °C. The mixture was extracted with ethyl acetate (3 times). The organic extracts were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluted with 40 to 100% ethyl acetate- hexane gradient. Collection and concentration of appropriate fractions provided the titled compound. lH NMR (400 MHz, CDCI3) δ 5.07 (m, IH), 4.29 (q, J = 7.2 Hz, 2H), 4.00 (s, 3H), 3.99-3.93 (m, IH), 3.89 (s, 3H), 3.74-3.66 (m, IH), 3.53-3.48 (m, IH), 3.23 (dd, J = 1.3, 13.2 Hz, IH), 1.46 (d, J = 6.6 Hz, 3H), 1.36 (t, J = 7.2 Hz, 3H), 1.22 (t, 7= 7.1 Hz, 3H). ES MS M+l = 367
Step 9: (6S)-8-Ethyl-10-methoxy-6-methyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carbohydrazide A mixture of ethyl (4S)-2-ethyl-8-(methoxy)-6-[methoxy(oxo)acetyl]-4-methyl-l-oxo- l,2,3,4-tetrahydropyrrolo[l,2-α]pyrazine-7-carboxylate (3.3 g, 8.9 mmol) and anhydrous hydrazine (1.7 mL, 53.7 mmol) in methanol (400 mL) was stirred at room temperature for one hour. The reaction mixture was concentrated under vacuum. The residue was concentrated from toluene. The resultant gummy solid was treated with methanol (20 mL). Diethyl ether was added to the resultant slurry which was filtered to provide the title compound as white solid. lH NMR (400 MHz, CDCI3) δ 8.99 (br s, 2H), 5.54 (br m, IH), 4.12 (m, IH), 4.10 (s, 3H), 3.81 (m, IH),
3.39 (m, IH), 3.21 (d, 7 = 12.6 Hz, IH), 1.44 (d, 7 = 6.4 Hz, 3H), 1.23 (t, 7 = 7.3 Hz, 3H). ES MS M+l =
335
Step 10: (6S)-8-Ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide To a solution of (6S)-8-ethyl-10-methoxy-6-methyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carbohydrazide (0.39 g, 1.2 mmol) and methylamine (5.9 mL, 11.8 mmol; 2 M in THF) in anhydrous dichloromethane (25 mL) in a water bath at room temperature, a solution of iodine (0.60 g, 2.4 mmol) in dichloromethane was added dropwise.
After the addition was completed, an aqueous solution of sodium sulfite was added and the mixture was stirred vigorously for 10 minutes. The organic phase was separated, diluted with chloroform, and washed with brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was triturated with a mixture of ethanol (7 mL) and diethyl ether (25 mL). The white solid precipitated was obtained by filtration and dried from its solution in toluene under vacuum. 1H NMR (400 MHz, CDCI3) δ 11.57 (s, IH), 7.38 (m, IH), 5.95 (br m, IH), 4.17 (s, 3H), 4.03 (dd, 7 =
13.4, 3.8 Hz, 1 H), 3.76 (heptet, 7 = 7.1 Hz, 1 H), 3.50 (heptet, 7 = 7.1 Hz, 1 H), 2.99 (dd, 7 = 12.9, 1.0 Hz, 1 H), 3.03 (d, 7 = 5.0 Hz, 3H), 1.44 (d, 7 = 6.6 Hz, 3H), 1.23 (t, 7 = 7.2 Hz, 3H). ES MS M+l = 334 Step 11: (6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[r,2′: l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide To a cold (0 °C) solution of (6S)-8-ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[l’,2′: l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.58 g, 4.73 mmol) in anhydrous DMF (50 mL), a solution of lithium bis(trimethylsilyl)amide (4.97 mL, 4.97 mmol, 1 M in THF) was added. After stirring at the same temperature for 25 minutes, 3-chloro-4-fluorobenzyl bromide (1.27 g, 5.68 mmol) was added. The reaction mixture was stirred at room temperature for 10 minutes and concentrated under vacuum. The residue was partitioned between chloroform and brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a 1-5% methanol in ethyl acetate gradient. Collection and concentration of appropriate fractions provided the title compound. lH NMR (400 MHz, CDCI3) δ 7.46 (dd, 7 = 6.9, 2.2 Hz, IH), 7.32 (m, IH), 7.09 (t, 7 = 7.6 Hz, IH), 7.03
(br signal, IH), 5.92 (m, IH), 5.32 (d, 7 = 14.1 Hz, IH), 5.26 (d, 7= 14.1 Hz, IH), 4.14 (s, 3H), 3.97 (dd, 7 = 13.2, 3.7 Hz, IH), 3.73 (heptet, 7 = 7.2 Hz, 1 H), 3.51 (heptet, 7 = 7.1 Hz, IH), 3.21 (dd, 7= 13.2, 1.7 Hz, IH), 3.03 (d, 7 = 5.0 Hz, 3H), 1.42 (d, 7 = 6.6 Hz, 3H), 1.23 (t, 7 = 7.1 Hz, 3H). ES MS M+l = 476
Step 12:
(6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d3pyridazine-4-carboxamide
To a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-l,9- dioxo-l,2,6,7,8,9-hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.15 g, 2.41 mmol) in anhydrous dichloromethane (800 mL), a solution of boron tribromide in dichloromethane (3.14 mL, 3.14 mmol; 1 M) was added. After stirring at room temperature for 5 minutes, the reaction mixture was treated with anhydrous methanol, stirred for 30 minutes, and concentrated under vacuum. The procedure was repeated twice. The residue was dissolved in a mixture of methanol and acetonitrile and treated with aqueous sodium hydroxide. The mixture was subjected to purification on preparative reverse phase high pressure column chromatography. Collection and lyophilization of appropriate fractions provided the title compound as white amorphous solid.
MK 2048
lH NMR (400 MHz, CDCI3) δ 7.48 (dd, 7 = 7.0, 2.2 Hz, IH), 7.33 (m, IH), 7.09 (t, 7 = 8.7 Hz, IH), 6.01 (m, IH), 5.33 (d, 7= 14.1 Hz, IH), 5.27 (d, 7 = 14.1 Hz, IH), 3.99 (dd, 7= 12.8, 4.0 Hz, 1 H), 3.71(heptet, 7 = 7.1 Hz, 1 H), 3.49 (heptet, 7 = 7.1 Hz, 1 H), 3.24 (dd, 7 = 13.2, 1.5 Hz, 1 H), 3.03 (d, 7 = 5.1 Hz, 3H), 1.42 (d, 7 = 6.6 Hz, 3H), 1.24 (t, 7 = 7.3 Hz, 3H). ES MS M+l = 462
The amorphous product was dissolved in boiling methanol (1.4 g/200 mL). Upon cooling in an ice-water bath, a precipitate formed which was separated by obtained by filtration to afford a white crystalline solid.
MK 2048sodium salt
The corresponding sodium salt was prepared by treatment of a solution of (6S)-2-(3- chloro-4-fluorobenzyl)-8-ethyl- 10-hydroxy-N,6-dimethyl-l ,9-dioxo- 1 ,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (920 mg, 1.99 mmol) in aqueous acetonitrile with aqueous sodium hydroxide (1.03 equivalent), followed by lyophilization of the resultant solution.
(5S)-1-ethyl-5-methylpiperazin-2-one
 1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2
1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2

(S)-1-[(R)-2-Di-(4-methoxy-3,5-dimethylphenyl-phosphino)ferrocenyl]-ethyl-dicyclohexylphosphine
SL-J006-2
(5S)-l-Ethyl-5-methylpiperazin-2-one was alternatively prepared as follows:
Step 1: N^rf-Butoxycarbonyl-N^ethylglycinamide Ethylamine (37 g, 0.82 mol) was condensed into a pressure vessel at 0 °C. N-(tert- butoxycarbonyl)glycine methyl ester (50 mL, 0.34 mol) was added. The vessel was sealed and the mixture was stirred at room temperature overnight. The product mixture was concentrated under vacuum and the residue was passed through a pad of silica gel eluting with ethyl acetate. The solution was concentrated under vacuum to provide the title compound as a clear oil. lH NMR (400 MHz, CDCI3) δ 6.11 (br s, IH), 5.18 (br s, IH), 3.77 (d, 7 = 5.7 Hz, 2H), 3.31 (q, 7 = 7.1
Hz, 2H), 1.15 (t, 7 = 7.1 Hz, 3H).
Step 2: l-Ethyl-5-methylpyrazin-2(lH)-one A cold (0 °C) solution of N^tø^butoxycarbonyl-N^ethylglycinamide (68.0 g, 0.33 mol) in anhydrous dichloromethane (500 mL) was saturated with anhydrous hydrogen chloride gas. After stirring at the same temperature for 1.5 hours, the solution was recharged with more hydrogen chloride gas and stirred for additional 15 minutes. The reaction mixture was concentrated under vacuum. The residue was dissolved in methanol, diluted with toluene, and concentrated under vacuum to afford the intermediate N-ethylglycinamide HCI salt.
This was stored under vacuum overnight and used without further purification. A solution of N-ethylglycinamide HCI salt (44.2 g, 0.32 mol), aqueous sodium hydroxide (640 mL, 1M), water (350 mL), pyruvic aldehyde (20.9 mL, 40% solution in water) was heated in an oil bath at 120 °C for one hour. The reaction mixture was cooled and saturated with solid sodium chloride. The mixture was extracted with chloroform (4×250 mL).
The combined organic extract was dried over anhydrous sodium sulfate, filtered, and passed through a plug of silica gel. The silica gel was rinsed successively with ethyl acetate and then 2% methanol in ethyl acetate. The eluted fractions were combined and concentrated under vacuum. The residual solid was recrystallized from diethyl ether to afford the title compound as pale yellow solid. lH NMR (400 MHz, CDCI3) δ 8.11 (s, IH), 6.92 (s, IH), 3.92 (q, 7 = 7.2 Hz, 2H), 2.28 (s, 3H), 1.37 (t, 7 = 7.2 Hz, 3H).
Step 3: (5 S)- 1 -Ethyl-5-methylpiperazin-2-one
A mixture of chloro-l,5-cyclooctadiene iridium (I) dimer (34 mg, 51 μmol) and (S)-l-[(R)-2-di-(3,5-bis(trifluoromethyl)phenyl)phosphino)ferrocenyl]ethyldicyclohexylphosphine (44 mg, 51 μmol; Solvias AG, SL-J006-2) in a mixture of 1:2 toluene and methanol (100 mL; purged with nitrogen for 15 minutes) was sonicated under an atmosphere of nitrogen for 15 minutes. To the resultant mixture, iodine (0.39 g, 1.52 mmol) and l-ethyl-5-methylpyrazin-2(lH)-one (7.0 g, 50.66 mmol) was added. The resultant mixture was heated in an oil bath at 50 °C under an atmosphere of hydrogen gas at 800 psi for 48 hours. The product mixture was filtered through a pad of Celite. The filtrate was concentrated under vacuum. The residue was treated with chloroform saturated with ammonia gas (100 mL). The resultant suspension was filtered through a pad of Celite, which was the rinsed with chloroform saturated with ammonia gas. The combined filtrate was concentrated under vacuum. The residue was concentrated as a solution in toluene for subsequent reaction. lH NMR (400 MHz, CDCI3) δ 3.58 (d, 7 = 17.2 Hz, IH), 3.53(d, 7 = 17.2 Hz, IH), 3.49-3.35 (m, 2H),
1.19 (d, 7 = 5.9 Hz, 3H), 1.14 (t, 7 = 7.2 Hz, 3H).
……………..
US 7538112
http://www.google.com/patents/US7538112
Step 12: (6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
To a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.15 g, 2.41 mmol) in anhydrous dichloromethane (800 mL), a solution of boron tribromide in dichloromethane (3.14 mL, 3.14 mmol; 1 M) was added. After stirring at room temperature for 5 minutes, the reaction mixture was treated with anhydrous methanol, stirred for 30 minutes, and concentrated under vacuum. The procedure was repeated twice. The residue was dissolved in a mixture of methanol and acetonitrile and treated with aqueous sodium hydroxide. The mixture was subjected to purification on preparative reverse phase high pressure column chromatography. Collection and lyophilization of appropriate fractions provided the title compound as white amorphous solid.
1H NMR (400 MHz, CDCl3) δ 7.48 (dd, J=7.0, 2.2 Hz, 1H), 7.33 (m, 1H), 7.09 (t, J=8.7 Hz, 1H), 6.01 (m, 1H), 5.33 (d, J=14.1 Hz, 1H), 5.27 (d, J=14.1 Hz, 1H), 3.99 (dd, J=12.8, 4.0 Hz, 1 H), 3.71 (heptet, J=7.1 Hz, 1 H), 3.49 (heptet, J=7.1 Hz, 1 H), 3.24 (dd, J=13.2, 1.5 Hz, 1 H), 3.03 (d, J=5.1 Hz, 3H), 1.42 (d, J=6.6 Hz, 3H), 1.24 (t, J=7.3 Hz, 3H). ES MS M+1=462
The amorphous product was dissolved in boiling methanol (1.4 g/200 mL). Upon cooling in an ice-water bath, a precipitate formed which was separated by obtained by filtration to afford a white crystalline solid.
The corresponding sodium salt was prepared by treatment of a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (920 mg, 1.99 mmol) in aqueous acetonitrile with aqueous sodium hydroxide (1.03 equivalent), followed by lyophilization of the resultant solution.
References
- Keith Alcorn. Ralvetgravir shows potential for use as PrEP drug AIDSmap.com. 28 April 2009. Accessed 8 Nov 2009.
- Mark Mascolini. Merck Offers Unique Perspective on Second-Generation Integrase Inhibitor. 10th International Workshop on Clinical Pharmacology of HIV Therapy, April 15–17, 2009, Amsterdam. Accessed 8 Nov 2009.
| WO2011121105A1 | 1 Apr 2011 | 6 Oct 2011 | Tibotec Pharmaceuticals | Macrocyclic integrase inhibitors |
| EP1756114A2 * | 3 May 2005 | 28 Feb 2007 | Merck and Co., Inc. | Hiv integrase inhibitors |
| US7517929 | 3 Dec 2004 | 14 Apr 2009 | Momentive Performance Materials Inc. | Star-branched silicone polymers as anti-mist additives for coating applications |
| US5693640 * | 6 Jun 1995 | 2 Dec 1997 | Merck, Sharp & Dohme, Ltd. | Pyridazino-indole derivatives |
| US5756501 * | 3 Dec 1996 | 26 May 1998 | American Home Products Corporation | Saturated and unsaturated pyridazino 4,5-B! indolizines useful as antidementia agents |
Valnemulin

Valnemulin
101312-92-9,
| (3aS,4R,5S,6S,8R,9R,9aR,10R)-6-ethenyl- 5-hydroxy-4,6,9,10-tetramethyl-1-oxodecahydro- 3a,9-propano-3aH-cyclopenta[8]annulen-8-yl- [(R)-2-(2-amino-3-methylbutanoylamino)-1,1-dimethtylethyl sulfanyl]acetate [[2-[(R)-2-Amino-3-methylbutyramido]-1,1-dimethylethyl]thio]acetic acid (3aS,4R,5S,6S,8R,9R,9aR,10R)-decahydro-5-hydroxy-1-oxo-4,6,9,10-tetramethyl-6-vinyl-3a,9-propano-3aH-cyclopentacycloocten-8-yl ester |
launched 1999 novartis for bacterial infection


Valnemulin (trade name Econor) is a pleuromutilin antibiotic used to treat swine dysentery, ileitis, colitis and pneumonia. It is also used for the prevention of intestinal infections of swine.[1] Valnemulin has been observed to induce a rapid reduction of clinical symptoms ofMycoplasma bovis infection, and eliminate M. bovis from the lungs of calves.[2]
Pleuromutilin, a compound of formula

is a naturally occurring antibiotic, e.g. produced by the basidomycetes Pleurotus mutilus and P. passeckerianus, see e.g. The Merck Index, 12th edition, item 7694.
A number of further pleuromutilins having the principle ring structure of pleuromutilin and having e.g. antibacterial activity, have been developed.
A pleuromutilin of the present invention includes a pleuromutilin having the basic structural elements as set out in formula

wherein R is vinyl or ethyl and the dotted line is a bond or is no bond.
The following numbering system is used in the present application:

The dotted line between positions 19 and 20 (and between positions 1 and 2) is a bond or is no bond. In a compound of formula A or of formula PLEU a hydrogen atom in positions 4, 7 and/or 8 of the ring system may be replaced by deuterium, and if the dotted line between positions 1 and 2 is no bond (single bond between positions 1 and 2) the ring system may be further substituted in positions 1 and/or 2, e.g. by halogen, deuterium or hydroxy. The group —O— in position 14 is further substituted, preferably by a substituted carbonyl group.
Examples of pleuromutilins according to the present invention includes e.g.
-
- A compound as disclosed in U.S. Pat. No. 3,716,579, e.g. of formula

- wherein R is CH3—(CH2)7—CH═CH—(CH2)7—COO—, CH3—(CH2)4—CH═CH—CH2—CH═CH—(CH2)7—COO—, CH3—(CH2)9—CH═CH—(CH2)7—COO— or hydrogen;
- A compound as disclosed in GB1312148, e.g. of formula


-
- A compound as disclosed in U.S. Pat. No. 4,130,709, e.g. of formula

-
- A compound as disclosed in U.S. Pat. No. 4,129,721; e.g. of formula

and the 19,20-dihydro derivative thereof and the tetra(C2-6)alkanoyl derivatives thereof;
-
- A compound as disclosed in EP0013768, e.g. of formula

-
- A compound as disclosed in EP0153277, e.g. an N-acyl-14-O-[(1-amino-2-methylpropan-2-yl)thioacetyl]-mutilin or 19,20-dihydromutilin, such as of formula

wherein R1 is vinyl or ethyl positions 19 and 20), and R2 is optionally hydroxy-substituted aminoalkyl or a 5-membered saturated heterocycle, e.g. including Valnemulin (Econor®) of formula

-
- A compound as disclosed in U.S. Pat. No. 516,526, e.g. of formula

wherein R1 and R2 independently of each other are H, alkyl, alkenyl, cycloalkyl, aryl or aralkyl;
-
- A compound as disclosed in WO9322288, e.g. of formula

wherein R1 and R2 are independently of each other H, alkyl, or, R1 and R2 together with the carbon atom to which they are attached are cycloalkyl; and R3 and R4 independently of each other are H, alkyl or substituted alkyl;
-
- A compound as disclosed in WO9725309, e.g. of formula

wherein Y is carbamoyloxy, wherein the N-atom is unsubstituted or mono- or disubstituted, such as a compound of formula

see more at……..
-
- A compound as disclosed in EP0153277, e.g. an N-acyl-14-O-[(1-amino-2-methylpropan-2-yl)thioacetyl]-mutilin or 19,20-dihydromutilin, such as of formula
wherein R1 is vinyl or ethyl positions 19 and 20), and R2 is optionally hydroxy-substituted aminoalkyl or a 5-membered saturated heterocycle, e.g. including Valnemulin (Econor®) of formula
-
- A compound as disclosed in U.S. Pat. No. 516,526, e.g. of formula
wherein R1 and R2 independently of each other are H, alkyl, alkenyl, cycloalkyl, aryl or aralkyl;
-
- A compound as disclosed in WO9322288, e.g. of formula
wherein R1 and R2 are independently of each other H, alkyl, or, R1 and R2 together with the carbon atom to which they are attached are cycloalkyl; and R3 and R4 independently of each other are H, alkyl or substituted alkyl;
-
- A compound as disclosed in WO9725309, e.g. of formula
wherein Y is carbamoyloxy, wherein the N-atom is unsubstituted or mono- or disubstituted, such as a compound of formula
see more at…………….http://www.google.com/patents/US8088823
-
Valnemulin is known from EP-0.153.277 and is described specifically therein in example 12.
Valnemulin is also known by the commercial name Econor®. -
As is generally known, this compound has antibacterial properties, e.g. following oral or parenteral administration, and is used for the prevention or cure of a series of bacterial infections in the field of animal health. The broad spectrum of activity includes Streptococcus aronson, Staphylococcus aureus, Mycoplasma arthritidis, Mycoplasma bovigenitalium, Mycoplasma bovimastitidis, Mycoplasma bovirhinis,Mycoplasma sp., Mycoplasma canis, Mycoplasma felis, Mycoplasma fermentans, Mycoplasma gallinarum, Mycoplasma gallisepticum, A. granularum, Mycoplasma hominis, Mycoplasma hyorhinis,Actinobacillus laidlawii, Mycoplasma meleagridis, Mycoplasma neurolyticum, Mycoplasma pneumonia und Mycoplasma hyopneumoniae.
-
WO 98/01127 describes its excellent activity against an illness complex that can arise whenever animals are kept in a very restricted space (increased stocking density) e.g. for transport purposes, and are thus exposed to great stress. The most frequent pathogens that play a decisive role in this instance are Mycoplasma hyopneumoniae, Serpulina(formerly Treponema) hyodysenteriae, Serpulina pilosicoli, Lawsonia intracellularis, Mycoplasma gallisepticum, Pasteurella multocida, Actinobacillus (Haemophilus) pleuropneumoniae and Haemophilus parasuis, whereby diseases of the respiratory tract and other infections often occur together and lead to a complex clinical picture. All herd animals are affected, e.g. cattle, sheep and pigs, but also poultry.
-
In its free form (valnemulin base), valnemulin is relatively unstable and is therefore primarily used in the form of its salts, particularly as the hydrochloride. A current method of administering antibiotics in the field of animal health is the injection, since it is suitable for administering a controlled single dose and thus a quantity tailored to individual needs. This is often crucial to successful control of many infectious diseases in the field of animal medicines. In contrast, oral administration cannot be controlled nearly so well, and is more customary in human medicine.
-
However, it has been shown that aqueous injection solutions and even oily injection suspensions of the salts of valnemulin are poorly tolerated by most domestic animals and in particular by pigs. Damage ranging from mild skin irritation to poorly healing necroses, has been observed. This is also one of the reasons that valnemulin has mainly been used orally until now. In addition, aqueous solutions usually do not show the desired depot action. A further problem is that valnemulin cannot be produced in technical quantities in the free form, as the so-called valnemulin base, but occurs as the salt, and has therefore been used for therapy as the salt.
………………………………….
syn
http://www.google.com/patents/CN102675173B?cl=en
valnemulin hydrochloride (Valnemulin hydrochloride) chemical name: [[2_ [[(2R) _2_ amino-3 – methyl-1 – oxobutyl] amino] -1,1 – dimethyl- ethyl] thio] acetic acid (3aS, 4R, 5S, 6S, 8R, 9R, 9aR, I OR) -6 – vinyl-decahydro-5 – hydroxy -4,6,9,10 – tetramethyl-1 – oxo _3a, 9 – propanol _3aH_ cyclopenta cyclooctene en-8 – yl ester hydrochloride, the chemical structure of formula I as shown. A molecular weight of 601.28, as a white amorphous powder characters have strong hygroscopicity, soluble in water, ethanol, methyl tert-butyl ether in almost insoluble, mp 174-177Ό.

Valnemulin hydrochloride is a new generation of pleuromutilins semi-synthetic antibiotics, are two terpenes, and tiamulin belong to the same class of drugs that are animal-specific antibiotics. Mainly used for prevention and treatment of pigs, cattle, sheep and poultry mycoplasma disease and Gram-positive bacterial infections. 1999 by the European Commission approved for the prevention and treatment of swine dysentery swine dysentery infection caused by the spirochete short and swine enzootic pneumonia caused by the Mycoplasma pneumoniae infection is the first Europe-wide approval of veterinary drugs premixes, is as a veterinary prescription drugs. September 9, 2010, approved by the Ministry of Agriculture Bulletin No. 1457 of Valnemulin hydrochloride developed materials and premixes for the state Department of Agriculture approved new animal drug II. Therefore, in our veterinary clinic has broad application prospects.
Chemical synthesis of hydrochloric Valnemulin has a lot of literature. Most of which are to pleuromutilins as raw material, p-toluenesulfonyl chloride sulfonation reaction with dimethyl cysteamine hydrochloride, and then protected with an amino group, a carboxyl group is activated D-valine reaction, the final hydrolysis Valnemulin hydrochloride.
Chinese patent discloses a method for preparing CN102225905A Valnemulin hydrochloride method, which is after the Pleuromutilin reacted with tosyl chloride, after 1 – amino-2 – methyl-2 – thiolate substituted acid to give (2 – amino-1 ,1 – dimethylethyl) thio] acetic acid (3aS, 4R, 5S, 6S, 8R, 9R, 9aR, 10R) -6 – vinyl decahydro _5_ hydroxy -4,6,9,10 – tetramethyl-1 – oxo-3a, 9 – propanol _3aH_ cyclopenta cyclooctene – (4H) -8 spare ester; other with D- valine methyl acetoacetate and the reaction with chloroacetic acid anhydride as isobutyl, and then with (2 – amino-1 ,1 – dimethylethyl) thio] acetic acid (3aS, 4R, 5S, 6S, 8R , 9R, 9aR, I OR) -6 – vinyl decahydro-5 – hydroxy -4,6,9,10 – tetramethyl _1_ oxo _3a, 9 – propanol _3aH_ garrison diene ring and cyclooctene – (4H) -8 ester amide hydrochloride later deprotected valnemulin hydrochloride was obtained, with ether as the solvent for this process, morpholine catalyst. In this line, the spent acid to activate Dioxide valine carboxy, increasing the difficulty of activation, and in the last reaction step using ether as the solvent, low-boiling inflammable volatile ether, in the operation increased risk. [0007] Chinese patent CN101318921A also opened a method for preparing Valnemulin, comprising the following steps: (1) preparation of chlorinated Pleuromutilin: to Pleuromutilin as raw materials, the reaction with HCl, convert it to Chloride pleuromutilin; (2) Preparation of N-allyloxycarbonyl group of valine: valine as starting material, which was reacted with allyl chloroformate to obtain N-protected amino group is allyloxycarbonyl group valine; (3) 1,1 – dimethyl-2 – Preparation of (N-allyloxycarbonyl group valinamido) ethanethiol: N-allyloxycarbonyl group of valine obtained acid chloride with oxalyl chloride and _ chloride with 1,1-dimethyl-2 – aminoethanethiol intermediate obtained by reacting 1,1 – dimethyl -2 – (N-allyloxycarbonyl group valinamido) ethanethiol; (4) Preparation of valnemulin of: 1,1 – dimethyl -2 – (N-allyloxycarbonyl group valinamido) reaction with ethanethiol pleuromutilins chloride to obtain an amino protected valnemulin In palladium catalyzed hydrogenation of carbon was going to protect Valnemulin. The disadvantage of this method is the use of a pleuromutilin in the chloride solution of HCl in methanol, increased corrosion of the equipment; allyl protecting group removal in the spent catalyst is palladium on carbon, palladium on carbon is too high and difficult recovery rates , thereby increasing the cost of the final product.
Starting materials Pleuromutilin by higher fungi Basidiomycetes Pleurotus a strain produced by submerged culture mainly against Gram-positive bacteria and mycoplasma active antimicrobial substances. As a semi-synthetic precursor substance, its content and impurities have a significant impact on the synthesis of hydrochloric acid Valnemulin quality standards. Recrystallization can effectively remove impurities and improve the content, thus effectively simplify the purification process to remove impurities and hydrochloric La Vergne wonderful forest to provide a guarantee from the source.

Example 1:
A Valnemulin hydrochloride chemical synthesis method, as follows:
(I) purified pleuromutilin
[0021] 250g of pleuromutilin first dissolved in methyl tert-butyl ether 2000mL stirred solution clear. Activated carbon was then added 50g, stirred for 3 h at room temperature decolorization, activated carbon was filtered off and the filtrate was concentrated to about 500mL heating when a large number of crystal precipitation, heating was stopped, cooled to room temperature and stirring was continued for 4h. After the crystals were centrifuged and dried to obtain fine pleuromutilin 220g (yield 88%).
References
- Econor: Product Profile
- Stipkovits, L.; Ripley, P.; Tenk, M.; Glávits, R.; Molnár, T.; Fodor, L. (2005). “The efficacy of valnemulin (Econor) in the control of disease caused by experimental infection of calves with”. Research in Veterinary Science 78 (3): 207–215. doi:10.1016/j.rvsc.2004.09.005.PMID 15766939.
| US4278674 | Nov 29, 1979 | Jul 14, 1981 | Sandoz Ltd. | Substituted 14-desoxy-mutilin compositions |
| US4517178 * | Jun 13, 1983 | May 14, 1985 | Rikagaku Kenkyusho | Novel antibiotic 76-11, process for the production thereof, anticoccidiosis agent and domestic animals growth accelerator comprising the same as an effective ingredient |
| US7534814 * | Jun 14, 2004 | May 19, 2009 | Nabriva Therapeutics Ag | Mutilin derivatives and their use as antibacterials |
| US20050159377 | Jun 16, 2004 | Jul 21, 2005 | Smithkline Beecham Corporation | Methods of modulating activity of prokaryotic ribosomes |
| EP0013768A1 | Dec 31, 1979 | Aug 6, 1980 | Sandoz Ag | New pleuromutilin derivatives, their production and pharmaceutical compositions containing them |
| WO2000071560A1 | May 4, 2000 | Nov 30, 2000 | Lisa Anne Hegg | Methods of modulating activity of prokaryotic ribosomes |
| WO2002012199A1 | Aug 2, 2001 | Feb 14, 2002 | Steven Aitken | Heterocyclic mutilin esters and their use as antibacterials |
| WO2002022580A1 | Sep 11, 2001 | Mar 21, 2002 | Gerd Ascher | Antibacterials mutilins |
BC-7013 a Topical pleuromutilin antibiotic agent from Nabriva

BC-7013 (topical)
[14-O-[(3-Hydroxymethyl-phenylsulfanyl)-acetyl]-mutilin]
Pleuromutilins
Nabriva
Gram-positive
poster……….https://jmilabs.com/data/posters/ICAAC2009/F1-1521.pdf
BC–7013 [14-O-[(3-Hydroxymethyl-phenylsulfanyl)-acetyl]-mutilin] is a novel semi-synthetic pleuromutilin derivative that inhibits prokaryotic protein synthesis.
Pleuromutilins were discovered as natural-product antibiotics in 1950. Tiamulin was the first pleuromutilin compound to be approved for veterinary use in 1979, followed by valnemulin in 1999. It was not until 2007 that retapamulin became the first pleuromutilin approved for use in humans. However, retapamulin is limited to topical application. Recent advances in lead optimization have led to the synthesis of pleuromutilins that combine potent antibacterial activity with favorable pharmaceutical properties, making these compounds suitable for oral and intravenous delivery. Most pleuromutilins have an antibacterial spectrum that spans the common pathogens involved in both skin and respiratory tract infections. Two new pleuromutilins, BC-3205 and BC-7013 (both Nabriva Therapeutics AG), have entered clinical trials. In this review, the key properties of pleuromutilin derivatives, designed primarily through modifications at the C(14) side chain, are presented, and the potential of these compounds in systemic therapy in humans is discussed.
Discovered in 1959, pleuromutilins have the potential to be developed as a new class of antibiotics for systemic use in humans. Although in 2007 retapamulin became the first pleuromutilin approved for topical use in humans, it was not until 2011 that a pleuromutilin antibiotic, BC‑3781, was tested successfully in a Phase 2 clinical trial for systemic use in patients.
BC‑7013 belongs to a series of proprietary Nabriva pleuromutilins, which have been designed by Nabriva’s medicinal chemists to fulfill the specific requirements of a topical antibacterial agent. The clinical study is designed to evaluate safety and tolerability of BC‑7013.
In recent years, bacterial infections resistant to most forms of current antibiotics have appeared throughout the world and are currently the third leading cause of death in the US and Western Europe. Pleuromutilins represent a new class of antibiotics, inhibiting bacterial protein synthesis by binding to unique sites on the 50S subunit of the ribosome. These new antibiotics have two distinct advantages: they have a very low potential for cross-resistance with other established antibacterial classes and display a very low potential for resistance development.
“This is the second pleuromutilin antibiotic Nabriva has moved into clinical trials since our inception 18 months ago, emphasising our view of the potential of this new antibiotic class”, said Rodger Novak, Chief Operating Officer of Nabriva.
Nabriva’s first pleuromutilin program to enter the clinic, BC‑3205, is an oral agent with activity against gram positive and gram negative bacteria and atypicals. BC‑3205 is currently in a multi-dose Phase 1 trial.
Pleuromutilin antibiotics are a novel clinically validated class of antibiotics that specifically inhibit bacterial protein synthesis. Their antibacterial profile covers resistant pathogens, including MRSA, that cause diseases such as respiratory tract and skin infections.
Nabriva Therapeutics’ pleuromutilins are unique antimicrobial compounds that interfere with bacterial protein synthesis via a specific interaction with the 23S rRNA of the 50S bacterial ribosome subunit. These antibacterials have a distinct anti-bacterial profile. Their unique mechanism of action implies a very low probability of cross resistance with other antibacterials. In an industry first, Nabriva’s world class medicinal chemistry expertise achieved the development of intravenous and orally available pleuromutilins clearing the way for i.v. and oral therapy with this antibiotic class. This achievement constitutes a significant milestone in providing appropriate medication for the treatment of life-threatening bacterial infections offering a distinctly different class of antibiotics for the treatment of bacterial diseases.
- New class of antibiotics on the human market
- High target specificity plus a unique mode of action ensures differentiation from existing antibiotic classes and compounds in development
- Very low propensity for resistance development
- Accessibility to both Gram-positive & Gram-negative ribosomes translates into broad spectrum activity against a wide range of infections
- Excellent safety profile
- Oral, intraveneous and topical delivery
Nabriva is the first company with proof of concept achieved for the systemic use of pleuromutilins in patients. Two new pleuromutilins, BC‑3781 and BC‑7013, from Nabriva have shown excellent results in clinical studies. BC‑3781 is about to enter Phase 3. Since Nabriva´s pleuromutilin antibiotics have an ideal anti-bacterial spectrum for both skin and respiratory infections and are available as both oral and i.v. formulations, they address a significant medical need and constitute an excellent commercial opportunity.
mutilins

Mutilin is minor metabolite of the pleuromutilin family, originally isolated from Pleurotus mutilus. Mutilin is formed by hydrolysis of the hydroxyacetyl ester of pleuromutilin, and is a degradation product and in vivo metabolite of
pleuromutilin. Interest in mutilin has focused on its potential as a substrate for generating unique metabolites via
biosynthesis to provide a broader range of targets for semi-synthetic modification.
pleuromutilin
Nabriva. Pleuromutilins. Available online: http://www.nabriva.com/programs/pleuromutilins/ (accessed on 7 December 2012).
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. NY Acad. Sci. 2011, 1241, 71–81, doi:10.1111/j.1749-6632.2011.06219.x.
