
Home » 2014 (Page 74)
Yearly Archives: 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Buserelin a luteinizing hormone-releasing hormone (LHRH) agonist
 Buserelin
Buserelin
57982-77-1 cas no
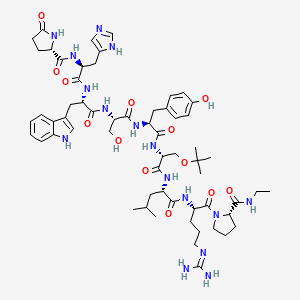
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-5-(diaminomethylideneamino)-1-[(2S)-2-(ethylcarbamoyl)pyrrolidin-1-yl]-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]-5-oxopyrrolidine-2-carboxamide
6-[O-(1,1-dimethylethyl)-D-serine]-9-(N-ethyl-L-prolinamide)-10-deglycinamideluteinizing hormone-releasing factor (pig)

Buserelin is a luteinizing hormone-releasing hormone (LHRH) agonist, a synthetic hormone which stimulates the pituitary gland’s gonadotrophin-releasing hormone receptor (GnRHR). It is used in prostate cancer treatment.
Buserelin stimulates the pituitary gland’s gonadotrophin-releasing hormone receptor (GnRHR). Buserelin desensitizes the GnRH receptor, reducing the amount of LH and testosterone. However, there is a concomitant surge in LH and testosterone levels with the decrease in androgens, so antiandrogens must administered.
Buserelin is a Gonadotropin-releasing hormone agonist (GnRH agonist). The drug’s effects are dependent on the frequency and time course of administration. GnRH is released in a pulsatile fashion in the postpubertal adult. Initial interaction of any GnRH agonist, such as buserelin, with the GnRH receptor induces release of FSH and LH by gonadotrophes. Long-term exposure to constant levels of buserelin, rather than endogenous pulses, leads to downregulation of the GnRH receptors and subsequent suppression of the pituitary release of LH and FSH.
Like other GnRH agonists, buserelin may be used in the treatment of hormone-responsive cancers such as prostate cancer or breast cancer, estrogen-dependent conditions (such as endometriosis or uterine fibroids), and in assisted reproduction.
It is normally delivered via a nasal spray, but is also available as an injection.
Buserelin acetate is marketed by Sanofi-Aventis under the brand name Suprefact and a generic form of Buserelin is now produced by CinnaGen under the brand name CinnaFact.
Buserelin is also marketed under the brand name Metrelef. Metrelef is approved to treat patients with endometriosis by suppression of ovarian hormone production. In ovulation induction Metrelef is used as a pituitary blockade as an adjunct togonadotrophin administration.

Buserelin, a synthetic gonadotropin-releasing hormone (GRH) agonist, specifically binds to GRH receptor presented at anter iorpituitary and increases or decreases the number of receptors in hypophysis through auto- regulation mechanism (G. Tolis et al., Tumor Growth Inhibition in Patients with Prostatic Carcinoma Treated with Luteinizing Hormone-Feleasing Hormone Agonists, Proc. Natl. Acad. Sci. , 79, pl658, 1982).
<5> The synthetic methods for preparing peptides are divided into two methods, i.e., liquid phase synthesis and solid phase synthesis. The liquid phase peptide synthesis of which all the reagents reacts together under the solution phase by being dissolved in the solution, has been reported to show rapid reaction rate however it has disadvantages such as the difficulty in separating and purification of the products. In a while, solid phase peptide synthesis which have been developed based on the theory of R. B. Merrifield, has been reported to have various advantages comparing with the former method for example, convenient to isolation and purification, the ‘applicability to automation (Bodanszky et al, In Peptide Synthesis, John Wiley & Sons, 1976). Lots of peptide synthetic resins have been developed to synthesize various peptides after the publication of the theory of R. B. Merrifield till now. For example, chloromethyl polystyrene resin had been developed by Merrifield and Wang resin having 4-alkoxybenzyl alcohol had been developed with modifying the former resin to overcome the disadvantages thereof at the early stage. Various resins to improve the disadvantages of conventional resins have been developed after then and the representative resins among those resins are trityl group introduced 2-chlorotrityl resin and rink amide resin which can provide amide group from the carboxyl terminal of peptide under mild cleavage condition, respectively.
<6> At the early stage, the simple structured type-peptides have been synthesized using by the resins however the complex structured type peptides showing various physiological activities have been synthesized mainly. The peptides comprising unnatural amino acids have been synthesized by chemical synthetic method since the peptides could not be prepared by enzymatic synthesis. Among them, the peptides comprising D-amino acid or aza-amino acid have been reported to have potent physiological activities and further to be developed as a medicine (USP Nos. 6,624,290; 6,069,163; 5,965,538; and 4,634,715). However, the novel method for preparing LH-RH such as goserelin or GnRH peptides using by solid phase synthesis has been still need till now since previously known methods, for example, the methods disclosed in USP No. 5,602,231; EP No. 0518655; USP No. 6,879,289; and USP No. 3,914,412, have been reported to have unsolved problems such as a limit to obtain pure product etc.
http://www.google.com/patents/WO2008044890A1?cl=en
Example 4: Preparation of buserelin
<98> Ig of 2-chlroro trityl chloride resin showing 0.9 mM/g of substitution rate was swollen with 10ml of DMF and the reaction mixture mixed with 768 mg of Fmoc-Arg (N02)-0H (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto to react together. The resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 615 mg of Fmoc-Leu-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 670 mg of Fmoc-D- SeKtBu)-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 859 mg of Fmoc-Tyr(OBzI)-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 726 mg of Fmoc-Ser(OBzI)-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above- described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 742 mg of Fmoc-Trp-0H (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above- described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 1.078g of Fmoc-His(Fmoc)-0H (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 244 mg of Pyr-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method.
<99> The resin was washed again and 2ml of 1% TFA (Trifluoroacetic acid)/DCM (dichloromethane) per 70mg of peptide resin was added to the resin, eluted to release the peptide from the resin and the elute was collected with 200 microliter of pyridine. The above-described step was repeated five times. The resin was washed with DCM (dichloromethane) and methanol and the elute was collected with the former elute. The elute was concentrated with evaporation and ether was added thereto to obtain the precipitated peptide. The precipitated peptide was performed to coupling reaction with 305 mg of Pro- NH-CH2CH3 (2.4mM) and 303mg of DIC (2.4 niM) in the presence of DCM
(dichloromethane) solvent. The solution was subjected to concentration with evaporator. The resulting concentrate was dissolved in EtOAc, washed with saturated NaHCOs solution, distilled water, 5% citrate solution and dried with anhydrous MgS(V The remaining MgS04 was discarded with filtration and the filtrate was concentrated with evaporation. The benzyl group and Cbz group among the side chain protecting group in the peptide were removed through catalytic hydrogen transfer reaction using by Pd/C and ammonium formate in the presence of methanol. The resulting peptide was purified with reverse phase column chromatography (Shimadzu H-kit, acetonitrile^water= 22:78 → 32:68, 1% increase/min) to isolate pure buserelin (Yield: 40%).
new patent
Solid state method for the preparation of buserelin, an LHRH analog useful for the treatment of sexual dysfunction, ovulation, puberty retardation and cancer. Method is under basic conditions and increases yield and purity. This appears to be the first PCT application from Hybio with this target, however several Chinese national filings have been published. Pan, Ma and Yuan are named on several previous solid phase synthesis PCT applications, most recently WO2013117135.
| US5212288 * | Feb 8, 1991 | May 18, 1993 | Syntex (U.S.A.) Inc. | Temporary minimal protection synthesis of serine-containing polypeptides |
| US5510460 * | May 26, 1995 | Apr 23, 1996 | Zeneca Limited | Peptide process |
| US5602231 * | May 26, 1995 | Feb 11, 1997 | Zeneca Limited | Process for making peptides |
| US6028172 * | Feb 10, 1998 | Feb 22, 2000 | Mallinckrodt Inc. | Reactor and method for solid phase peptide synthesis |
| US6897289 * | May 5, 2000 | May 24, 2005 | Lipotec, S.A. | Peptide synthesis procedure in solid phase |
Relugolix (TAK-385) in phase 2 By Takeda for the treatment of endometriosis and uterine fibroids
Relugolix (TAK-385)
1-[4-[1-(2,6-Difluorobenzyl)-5-(dimethylaminomethyl)-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl]-3-methoxyurea
N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
CAS NO 737789-87-6
-
C29-H27-F2-N7-O5-S
- 623.6383
Synonyms
-
N-(4-(1-((2,6-Difluorophenyl)methyl)-5-((dimethylamino)methyl)-1,2,3,4-tetrahydro-3-(6-methoxy-3-pyridazinyl)-2,4-dioxothieno(2,3-d)pyrimidin-6-yl)phenyl)-N’-methoxyurea
-
TAK-385
-
UNII-P76B05O5V6
Systematic Name
-
Urea, N-(4-(1-((2,6-difluorophenyl)methyl)-5-((dimethylamino)methyl)-1,2,3,4-tetrahydro-3-(6-methoxy-3-pyridazinyl)-2,4-dioxothieno(2,3-d)pyrimidin-6-yl)phenyl)-N’-methoxy-
TAK-385 is a luteinizing hormone-releasing hormone (LH-RH) receptor antagonist administered orally. By preventing LH-RH from binding with the LH-RH receptor in the anterior pituitary gland and suppressing the secretion of luteinizing hormone (LH) and follicle stimulation hormone (FSH) from the anterior pituitary gland, TAK-385 controls the effect of LH and FSH on the ovary, reduces the level of estrogen in blood, which is known to be associated with the development of endometriosis and uterine fibroids, and is expected to improve the symptoms of these disorders.
- *1 The hormone that controls the secretion of LH and FSH, gonadotropic hormones, secreted from the anterior pituitary gland.
- *2 A hormone that is secreted from the anterior pituitary gland by the action of LH-RH and encourages follicular maturation, ovulation and luteinization by acting on the ovaries.
- *3 A hormone that is secreted from the anterior pituitary gland by the action of LH-RH and encourages follicular maturation by stimulating the ovaries.
TAK-385, an oral antagonist of gonadotropin-releasing hormone (GnRH), was originated by Takeda. It is in phase II clinical trials for the treatment of endometriosis and for the treatment of uterine fibroids (myoma). Phase I clinical trials are also underway for the treatment of prostate cancer.
 Relugolix (TAK-385)
Relugolix (TAK-385)
…………….
http://www.google.co.in/patents/EP1591446A1?cl=en
(Production Method 1)

- (Production method 2)
-


- Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
-

-
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17 %) as colorless crystals.
1 H-NMR(CDCl3) δ: 2.15 (6H, s), 3.6-3.8 (2H, m), 3.82 (3H, s), 4.18 (3H, s), 5.35 (2H, s), 6.92 (2H, t, J = 8.2 Hz), 7.12 (1H, d, J = 8.8 Hz), 7.2-7.65 (7H, m), 7.69 (1H, s).

……………
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
1-{4-[1-(2,6-Difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (16b)
 tak 385
tak 385
Click to access jm200216q_si_001.pdf
…………………….
new patent
Method for the production of TAK-385 or its salt and crystals starting from 6-(4-aminophenyl)-1-(2,6-difluorobenzyl)-5-dimethylaminomethyl-3-(6-methoxypyridazin-3-yl) thieno[2,3-d] pyrimidine-2,4 (1H,3H)-dione or its salt. Takeda Pharmaceutical is developing relugolix (TAK-385), an oral LHRH receptor antagonist analog of sufugolix, for the treatment of endometriosis and uterine fibroids. As of April 2014, the drug is in Phase 2 trails. See WO2010026993 claiming method for improving the oral absorption and stability of tetrahydro-thieno[2,3-d]pyrimidin-6-yl]-phenyl)-N’-methoxy urea derivatives.
references
Discovery of TAK-385, a thieno[2,3-d]pyrimidine-2,4-dione derivative, as a potent and orally bioavailable nonpeptide antagonist of gonadotropin releasing hormone (GnRH) receptor
238th ACS Natl Meet (August 16-20, Washington) 2009, Abst MEDI 386
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
Ulipristal acetate for emergency contraception

Ulipristal acetate
17alpha-Acetoxy-11beta-[4-(dimethylamino)phenyl]-19-norpregna-4,9-diene-3,20-dione
(8S,11S,13S,14R,17R)-17-Acetoxy-11-[4-(dimethylamino)phenyl]-19-norpregna-4,9-diene-3,20-dione
REVIEW.http://www.fsrh.org/pdfs/ellaOneNewProductReview1009.pdf
126784-99-4 CAS
HRP-2000
PGL-4001
RTI-3021-012
UPA-UF
VA-2914
CHECK OUT NEW PATENTS BELOW NEW PATENTS IN 2014
WO-2014050105 Amorphous ulipristal acetate, ASKA Pharmaceutical Co Ltd
WO-2014050106 Crystalline polymorphic form of ulipristal acetate
WO-2014050107 Crystalline polymorphic form of ulipristal acetate
Ulipristal acetate (trade name EllaOne in the European Union, Ella in the U.S. for contraception,[1] and Esmya for uterine fibroid) is a selective progesterone receptor modulator (SPRM).

Medical uses
Emergency contraception
For emergency contraception[2] a 30 mg tablet is used within 120 hours (5 days) after an unprotected intercourse or contraceptive failure.[3] It has been shown to prevent about 60% of expected pregnancies,[4] and prevents more pregnancies than emergency contraception with levonorgestrel.[5] Ulipristal acetate is available by prescription for emergency contraception in over 50 countries, with access through pharmacists without a prescription being tested in the United Kingdom.[6][7][8][9] Emergency contraception (EC) is a woman’s second chance for primary prevention of pregnancy.
A reproductive-age woman is a candidate for emergency contraception if she seeks care within 120 hours of unprotected intercourse (UPI), which is the window of pregnancy risk associated with a given act of intercourse based upon the estimated lifespan of sperm in the genital tract (Wilcox et al, 1995). Current hormonal methods of emergency contraception prevent at least half of expected pregnancies if taken within 72 hours of UPI (Von Hertzen et al, 1998).
Levonorgestrel at a total dose of 1.5 mg (taken in a single dose or two 0.75 mg doses 12 hours apart) is the current standard for hormonal emergency contraception and is licensed for use up to 72 hours after UPI. Clinical trials involving levonorgestrel used for emergency contraception more than 72 hours after intercourse do not conclusively establish efficacy rates because of insufficient sample size. Nevertheless, these studies reveal a trend towards markedly higher failure rates when levonorgestrel is taken 48 hours or more after unprotected intercourse (von Hertzen et al, 1998; Von Hertzen et al, 2002).
This trend may be explained by levonorgestrel mode of action for emergency contraception. Levonorgestrel acts by interfering with the LH peak but does not appear to interfere with the ovulatory process when taken close to ovulation, a time when intercourse is most likely to lead to fertilization (Croxatto et al, 2004; Marions et al, 2004; Wilcox et al, 2004). For a woman who presents for emergency contraception more than 72 hours after intercourse, the only currently available method proven to be highly effective is insertion of a copper contraceptive intra-uterine device (IUD). However, IUDs are not widely available in many countries and insertion can only be performed by a trained clinician. Furthermore, many women decline IUD insertion as a method of emergency contraception because the procedure is invasive, is relatively expensive and has a risk of complications including uterine perforation on insertion (Grimes et al, 2004). Additionally, many women seeking emergency contraception are not seeking a long acting contraceptive method.
There is, therefore, a need for a new hormonal emergency contraceptive that can be used and is highly effective up to 120 hours after UPI. Ulipristal acetate (also known as CDB-2914) is a selective progesterone receptor modulator that inhibits or delays ovulation in a dose-dependent fashion (Stratton et al, 2000). In a double-blind non-inferiority trial, ulipristal acetate was shown to be as efficacious as levonorgestrel for preventing pregnancy when used within 72 hours of UPI (Creinin et al, 2006). Moreover, study data suggest improved efficacy in preventing pregnancy from 48 to 72 hours when levonorgestrel efficacy markedly wanes. ulipristal acetate for use in providing post coital contraception in a female subject between about 3 to about 5 days, or between about 72 to about 120 hours, after unprotected intercourse.
A subject of the invention is thus a method for providing post coital contraception in a female subject, comprising providing the subject with a therapeutically effective amount of ulipristal acetate, between about 3 to about 5 days, or between about 72 to about 120 hours, after unprotected intercourse. It is further provided a kit comprising i) a dosage form comprising ulipristal acetate and ii) a printed matter stating that ulipristal acetate may be taken within 120 hours or 5 days after unprotected intercourse Any woman of reproductive age may need post-coital or emergency contraception at some point to avoid an unintended pregnancy. It is meant to be used in situations of unprotected intercourse, such as: when no contraceptive has been used;
when there is a contraceptive failure or incorrect use, including: – condom breakage, slippage, or incorrect use; – non-compliance with dosage regimen for combined oral contraceptive pills; – non-compliance with dosage regimen for progestogen-only pill (minipill); – more than two weeks late for a progestogen-only contraceptive injection (depot- medroxyprogesterone acetate or norethisterone enanthate); – more than seven days late for a combined estrogen-plus-progestogen monthly injection; – dislodgment, delay in placing, or early removal of a contraceptive hormonal skin patch or ring; – dislodgment, breakage, tearing, or early removal of a diaphragm or cervical cap; – failed coitus interruptus (e.g., ejaculation in vagina or on external genitalia); – failure of a spermicide tablet or film to melt before intercourse; – miscalculation of the periodic abstinence method or failure to abstain on fertile day of cycle; – IUD expulsion; or in cases of sexual assault when the woman was not protected by an effective contraceptive method.
Uliprisnil acetate, originally developed at the Research Triangle Institute, is a selective progesterone receptor modulator (SPRM) first launched in the E.U. in 2009 by HRA Pharma as emergency contraception within 120 hours (5 days) of unprotected sexual intercourse or contraceptive failure. The company filed for approval of this indication in the U.S. in 2009 and approval was obtained in 2010. In 2012, the product was approved in the E.U. for the pre-operative treatment of moderate to severe symptoms of uterine fibroids in adult women of reproductive age. First E.U. commercialization took place in Germany in March 2012 followed by the U.K. in April. The compound is being developed in phase II clinical trials at the National Institutes of Health (NIH) for the treatment of uterine fibroids and premenstrual syndrome (PMS). Two formulations of uliprisnil are in early clinical trials at the Population Council for the prevention of pregnancy: a vaginal ring and an intrauterine delivery system (IUS). Watson conducted phase III clinical studies for the treatment of women with anemia associated with uterine leiomyoma, however the development has been discontinued.
Uliprisnil acetate is a well-known steroid that possesses antiprogestational and antiglucocorticoid activity. In preclinical studies, the growth of lead follicles exposed to a midfollicular dose of the compound was delayed in a dose-related fashion, indicating that the compound may have an additional mechanism of action involving progesterone or estrogen antagonism.
In 2007, uliprisnil acetate was licensed to PregLem by HRA Pharma in Europe for the treatment of gynecological disorders excluding contraception. A license for North American was granted to HRA in 2010. In 2010, the compound was licensed to Watson (now Actavis) by HRA Pharma for the commercialization in the U.S. for use as emergency contraception. Also in 2010, Watson (now Actavis) obtained a license to uliprisnil for the treatment of uterine fibroids. In 2011, the product was licensed to Gedeon Richter by HRA Pharma for marketing and distribution in China, Russia and (Commonwealth of Independent States) CIS republics for the treatment of uterine myoma.
Treatment of uterine fibroids
Ulipristal acetate is used for pre-operative treatment of moderate to severe symptoms of uterine fibroids in adult women of reproductive age in a daily dose of a 5 mg tablet.[10] Treatment of uterine fibroids with ulipristal acetate for 13 weeks effectively controlled excessive bleeding due to uterine fibroids and reduced the size of the fibroids.[11][12][13] Two intermittent 3-month treatment courses of ulipristal acetate 10 mg resulted in amenorrhea at the end of the first treatment course in 79.5%, at the end of the second course in 88.5% of subjects. Mean myoma volume reduction observed during the first treatment course (−41.9%) was maintained during the second one (−43.7%).[10
Adverse effects
Common side effects include abdominal pain and temporary menstrual irregularity or disruption. Headache and nausea were observed under long-term administration (12 weeks), but not after a single dose.[3]
Interactions
Ulipristal acetate is metabolized by CYP3A4 in vitro. Ulipristal acetate is likely to interact with substrates of CYP3A4, like rifampicin, phenytoin, St John’s wort, carbamazepine or ritonavir, therefore concomitant use with these agens is not recommended.[10][14] It might also interact with hormonal contraceptives and progestogens such as levonorgestrel and other substrates of the progesterone receptor, as well as with glucocorticoids.[10]
Contraindications
Ulipristal acetate should not be taken by women with severe liver diseases[3] because of its CYP mediated metabolism. It has not been studied in women under the age of 18.[15]
Pregnancy
Unlike levonorgestrel, and like mifepristone, ulipristal acetate is embryotoxic in animal studies.[16] Before taking the drug, a pregnancy must be excluded.[3] The EMA proposed to avoid any allusion to a possible use as an abortifacient in the package insert to avert off-label use.[17] It is unlikely that ulipristal acetate could effectively be used as an abortifacient, since it is used in much lower doses (30 mg) than the roughly equipotent mifepristone (600 mg), and since mifepristone has to be combined with a prostaglandin for the induction of abortion.[18] However, data on embryotoxicity in humans are very limited, and it is not clear what the risk for an abortion or for teratogenicity (birth defects) is. Of the 29 women studied who became pregnant despite taking ulipristal acetate, 16 had induced abortions, six had spontaneous abortions, six continued the pregnancies, and one “was lost to follow-up“.[19]
Lactation
It is not recommended to breast feed within 36 hours of taking the drug since it is not known whether ulipristal acetate or its metabolites are excreted into the breast milk.[3][20]
Pharmacokinetics
In animal studies, the drug was quickly and nearly completely absorbed from the gut. Intake of food delays absorption, but it is not known whether this is clinically relevant.[21] Ulipristal acetate is metabolized in the liver, most likely by CYP3A4, and to a small extent by CYP1A2 and CYP2D6. The two main metabolites have been shown to be pharmacologically active, but less than the original drug. The main excretion route is via the faeces.[22]
Pharmacodynamics
As a SPRM, ulipristal acetate has partial agonistic as well as antagonistic effects on the progesterone receptor. It also binds to the glucocorticoid receptor, but has no relevant affinity to the estrogen, androgen and mineralocorticoid receptors.[23] Phase II clinical trials suggest that the mechanism might consist of blocking or delaying ovulation and of delaying the maturation of the endometrium.[24]
History
Ulipristal acetate was granted marketing authorization by the European Medicines Agency (EMA) in March 2009.[25] The U.S. Food and Drug Administration approved the drug for use in the United States on 13 August 2010,[26] following the FDA advisory committee’s recommendation.[27][28] Watson Pharmaceuticals announced the availability of ulipristal acetate in the United States on 1 December 2010, in retail pharmacies, clinics, and one on-line pharmacy, KwikMed.[29] Amorphous ulipristal acetate. ASKA is developing ulipristal acetate in Japan under license from HRA Pharma for the treatment of uterine fibroids and for emergency contraception. In March 2014, it was in phase II for both indications (in Japan). Also see the co-published WO2014050106 and WO2014050107. Crystalline polymorphic form C of ulipristal acetate.
Also claims its method of preparation. Appears to be the first filing from the assignee on this API, which was developed by HRA Pharma under license from the RTI, indicated in the US as an emergency contraceptive for prevention of pregnancy. In May 2011, ASKA signed an exclusive licensing agreement with HRA Pharma to develop and commercialize the API . In November 2013, ASKA had begun phase II development for emergency contraception and uterine fibroids [1339186] in Japan. Also see concurrently published WO2014050105 and WO2014050107. Crystalline polymorphic form B of ulipristal acetate. Also claims process for the preparation and composition comprising the same. Useful for the treatment of uterine leiomyoma.
Appears to be the first filing from the assignee on this API, see concurrently published WO2014050105 and WO2014050106. The drug was developed by HRA Pharma under license from the RTI, indicated in the US as an emergency contraceptive for prevention of pregnancy. In May 2011, ASKA signed an exclusive licensing agreement with HRA Pharma to develop and commercialize the API In November 2013, ASKA had begun phase II development in Japan for emergency contraception and uterine fibroids Buccal forms or devices are also useful, such as those described in U.S. patent application 20050208129 , herein incorporated by reference. U.S. patent application 20050208129 describes a prolonged release bioadhesive mucosal therapeutic system containing at least one active principle, with an active principle dissolution test of more than 70% over 8 hours and to a method for its preparation.
Said bioadhesive therapeutic system comprises quantities of natural proteins representing at least 50% by weight of active principle and at least 20% by weight of said tablet, between 10% and 20% of a hydrophilic polymer, and compression excipients, and comprising between 4% and 10% of an alkali metal alkylsulphate to reinforce the local availability of active principle and between 0.1 % and 1% of a monohydrate sugar.
Ulipristal acetate, formerly known as CDB-2914, designates within the context of this application 17α-acetoxy-11β-[4-N,N-dimethylamino-phenyl)-19-norpregna-4,9-diene-3,20-dione, represented by formula I:

Ulipristal acetate, and methods for its preparation, are described e.g., in U.S. Pat. Nos. 4,954,490; 5,073,548, and 5,929,262, as well as in international patent applications WO2004/065405 and WO2004/078709. Ulipristal acetate possesses antiprogestational and antiglucocorticoidal activity, and has been proposed for contraception, in particular for emergency contraception, and for the therapy of various hormonal diseases.
(Steroids, 2000,65, 395 ~ 400; US5929262A; CN1298409A; CN101466723A). Reaction is as follows:

Properties of this compound are further described in Blithe et al, Steroids. 2003 68(10-13):1013-7. So far, clinical trials have been conducted using oral capsules of ulipristal acetate (Creinin et al, Obstetrics & Gynecology 2006; 108:1089-1097; Levens et al, Obstet Gynecol. 2008, 111(5):1129-36). In order to increase the properties and clinical benefit of this molecule, there is a need for improved formulations thereof
-
Ulipristal acetate, formerly known as CDB-2914, is 17α-acetoxy-11β-[4-N, N-dimethylamino-phenyl)-19-norpregna- 4, 9-diene-3, 20-dione, represented by formula I:

-
It is a well-known steroid, more specifically a 19-norprogesterone, which possesses antiprogestational and antiglucocorticoidal activity. This compound, and methods for its preparation, are described in U. S. Patent Nos. 4,954, 490,5 , 073,548 , and 5,929, 262 , and international patent applications WO2004/065405 and WO2004/078709 . Properties of this compound are further described in Blithe et al, 2003.
-
Metabolites of CDB-2914, include those described in Attardi et al, 2004 , e.g. monodemethylated CDB-2914 (CDB-3877) ;didemethylated CDB-2914 (CDB-3963) ; 17alpha-hydroxy CDB-2914 (CDB-3236) ; aromatic A-ring derivative of CDB-2914 (CDB-4183).




-
It is now proposed to use ulipristal acetate or a metabolite thereof for treating uterine fibroids, more particularly for reducing or stopping bleeding in a patient afflicted with uterine fibroids, reducing the size of uterine fibroids and/or reducing uterine volume More particularly the inventors have shown in a randomized, placebo-controlled, double blinded, parallel trial, that ulipristal acetate significantly reduces fibroid volume after 3 months, and stops bleeding
-
Ulipristal acetate or a metabolite thereof alleviates symptoms of uterine fibroids, including bleeding, pelvic pain, pressure.
-
Ulipristal acetate or a metabolite thereof is useful for preventing or treating anemia in patients afflicted with uterine fibroids.
-
It is also useful for preventing or treating leiomyosarcomas and for preventing dissemination of uterine fibroids to other organs.





………………
synthesis
http://www.google.com.br/patents/US5929262
CA2216737A1, EP0817793A2, WO1996030390A2, WO1996030390A3
The United States Of America As Represented By The Department Of Health And Human Services 
 EXAMPLE 7 The Preparation of the Compound of Formula (I) (17α-Acetoxy-11β-(4-N,N-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione) From the Compound of Formula (VIII) 340 mL of acetic acid (5.92 mol) were added to a well stirred mixture containing 834 mL of trifluoroacetic anhydride (5.92 mol) in 2,300 mL of methylene chloride under argon. After stirring for 30 minutes at room temperature, 51.3 g of p-toluenesulfonic acid (0.26 mol) were added, and the mixture was chilled to 0 methylene chloride solution containing 128.3 g of the compound of formula (VIII) (0.30 mol) were added, and the reaction mixture was stirred at 0 cautious addition of a 4.5N potassium carbonate solution until the pH was in the range of 7.0-7.5. The reaction mixture was diluted with water and extracted with methylene chloride. The methylene chloride extracts were washed with water and brine, combined, and dried over sodium sulfate.
EXAMPLE 7 The Preparation of the Compound of Formula (I) (17α-Acetoxy-11β-(4-N,N-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione) From the Compound of Formula (VIII) 340 mL of acetic acid (5.92 mol) were added to a well stirred mixture containing 834 mL of trifluoroacetic anhydride (5.92 mol) in 2,300 mL of methylene chloride under argon. After stirring for 30 minutes at room temperature, 51.3 g of p-toluenesulfonic acid (0.26 mol) were added, and the mixture was chilled to 0 methylene chloride solution containing 128.3 g of the compound of formula (VIII) (0.30 mol) were added, and the reaction mixture was stirred at 0 cautious addition of a 4.5N potassium carbonate solution until the pH was in the range of 7.0-7.5. The reaction mixture was diluted with water and extracted with methylene chloride. The methylene chloride extracts were washed with water and brine, combined, and dried over sodium sulfate.
Evaporation of the solvent gave the acetate of formula (I) as a thick syrup. The above syrup was dissolved in 300 mL of isopropyl alcohol and evaporated. The dissolution and evaporation were repeated three times. Finally, the remaining solid, which retained isopropyl alcohol as solvent of recrystallization, was dissolved in ethyl acetate and evaporated to give a stable foam. The foam was quickly dissolved in ether, and this solution was set aside to crystallize. The solid that formed was collected by filtration, washed with ether, and dried in vacuo to yield 105.7 g of the compound of formula (I) as yellow crystals in 75% yield;
m.p. 183-185 1735 and 1714(–C═O), 1664 and 1661 (conjugated –C═O), 1563, 1518, 1441, 1351, 1305, 1252, 1203, 1171; NMR (CDCl.sub.3) δ0.38 (s, 18-CH.sub.3), 2.10 (s, 17-OAc), 2.14 (s, 21-CH.sub.3), 2.92 (s, –N(CH.sub.3).sub.2, 4.44 (d, C-11 H), 5.83 (br. s, C-4 H), 6.71 and 7.07 (d, aromatic H); MS(EI) m/z (relative intensity) 475(M.sup.+, 41), 134(18), 121 (100). Analysis calculated for C.sub.30 H.sub.37 NO.sub.4 : C, 75.76; H, 7.84; N, 2.94. Found. C, 75.80; H 7.96; N, 3.09.
……………..
SYNTHESIS http://www.google.com/patents/WO2004065405A1?cl=en
CA2514169A1, CA2514169C, CN1753905A, CN100354300C, EP1602662A1, EP2348033A2, EP2348033A3
EXAMPLE 1 Preparation of 17α-aceto-d-llβ-(4-N, N-dimetüaminofeniI)-19-norpregna-4 ,9-dien-3,20-dione [VA-2914] Raw Were charged 38.5 g of 3,3 – (l ,2-etanodioxi)-5α-hydroxy-llβ-(4-N, N-dimethylaminophenyl)-17α-acetoxy-19-norpregna-9-en-20-one [carbinol acetate] purified in a flask under nitrogen atmosphere at a temperature between 20 ° C and 22 ° C, and added 385 ml of deionized water and 17.91 g of HKSO. The resulting suspension was stirred until complete dissolution, for about 4 hours. The end of the reaction was determined by thin layer chromatography (TLC). Then added 3.85 g of neutral Al 2 O 3, stirred for 30 minutes, the suspension was filtered and the insolubles were washed with 38.5 ml of deionized water. To the filtrate were added 325 ml of ethyl acetate and the pH was adjusted to a constant value between 7.0 and 7.2 with sodium bicarbonate solution to 7% w / v. The phases were allowed to decant for 15 minutes and, after checking the absence of the final product therein by means of TLC, the phases were separated, discarding the aqueous phase. The resultant organic phase was added 192.5 ml of deionized water, stirred for 10 minutes and the phases were allowed to decant for 15 minutes.
After verifying the absence of aqueous phase final product by TLC, the phases were separated, discarding the aqueous phase. The resulting organic phase was concentrated under vacuum to a residue and obtained approximately 28 g of 17α-acetoxy-llβ-(4-N, N-dimethylaminophenyl)-19-norpregna-4,9-dien-3 ,20-dione [NA -2914] raw. EXAMPLE 2 Isopropanol hemisolvate obtaining 17α-acetoxy-llβ-(4-Ν, Ν-dimethylaminophenyl)-19-norpregna-4 ,9-dien-3 ,20-dione The crude 17α-acetoxy-l lβ-(4-Ν, Ν-dimethylaminophenyl)-19-norpregna-4 ,9-dien-3,20-dione obtained in Example 1 was added 2 x 38.5 ml isopropanol concentrating vacuum to a residue both times. The finally obtained solid was added 77 ml of isopropanol and heated until dissolved. Then allowed to cool to a temperature between 0 ° C and 5 ° C, and the temperature was maintained for 1 h. The resulting suspension was filtered and the cake washed with cold isopropanol.
The yield achieved was 96% molar (5.5% isopropanol content). Isopropanol hemisolvate obtemdo NA-2914 has been characterized by IR spectroscopy, DSC and XRD, as indicated in the description, and has the characteristics indicated therein and shown in Figures 1-3.
…………….
A new and efficient method for the synthesis of Ulipristal acetate
http://www.sciencedirect.com/science/article/pii/S0039128X14000634


In this study, we describe another new and efficient route for preparing Ulipristal acetate. The 1,4-addition compound 5 was greatly improved after the starting material ketone 1 was underwent epoxidation, cyanation, hydroxyl group protection and Grignard addition. The synthetic procedure is only 6 steps and the total yield is about 27.4%, which is much suitable for industrial process.
We have succeeded in finding another convenient and efficient synthetic route for the synthesis of Ulipristal acetate with a good yield.
•The yield of 11β-substituted isomer was greatly improved.
•The 17β-carbonitrile compound was obtained with high purity after the reaction.
•The yield of once Grignard addition dione was greatly improved.
•These synthetic procedures are much suitable for industrial process.
…………….
Volume 78, Issues 12–13, 11 December 2013, Pages 1293–1297
http://www.sciencedirect.com/science/article/pii/S0039128X13002122
We set out to describe a new and efficient route for preparing Ulipristal acetate with a good yield. The selected epoxidization conditions gave out 80% of 5α,10α-epoxide 2a in the two diastereoisomers which greatly improved the yield of 11β-substituted isomer 4a. And phenyl–sulfinyl compound 6 was synthesized from ketone 5 directly treated with phenylsulfenyl chloride in the presence of triethylamine. These synthetic procedures is only 8 steps, less than currently reported in the literature, but more suitable for industrial process.

………..
http://www.google.com/patents/CN103145787A?cl=en
Reaction is as follows:

………..
WO2013063859A1
http://www.google.com/patents/WO2013063859A1?cl=en
Preparation of related reports Uli Division acetate compounds as follows:
1, U.S. Patent US4954490 methods, (see Reaction Scheme 1),
The method is based on the 3 – methoxy -19 – norpregn-1, 3,5 (10), 17 (20) – tetraene as a starting material, in turn by the addition, oxidation, reduction, hydrolysis, addition and elimination, oxidation of 17-hydroxy-19 – norpregn left the -4,9 – 31 women -3, 20 – dione (Compound V2), and then condensed by ethylene glycol, epoxidized-chloroperbenzoic acid, Bonus format, acid hydrolysis, acetylation of 10-step reaction by Uli acetate SECRETARY (Compound 1), and a melting point of 118-121 ° C the product was obtained by recrystallization with methanol ^. Due to the method, the step length, but difficult to obtain a starting material, the complexity of the reaction conditions, the required intermediate product was purified by column chromatography, the total yield is only 0.62%, Gao costs, the instability of the resulting product is not suitable pharmaceutically acceptable. And is not suitable for industrial production.
Reaction Formula I:


2, U.S. Patent No. US5929262 discloses his method Another method for preparing acetic acid Uli Division (see Reaction Scheme II), the reaction of formula II:

The method is based on 3,3 – ethylenedioxy-17) 8 – cyano -19 – norpregn -5 (10)-9 (11) – dien-17 alcohol (compound III) as a starting material, , first with dimethyl chloromethyl silane protected hydroxy, and then at the cryogenic-70Ό obtained by acid hydrolysis with the DBB / LI reagents After the reaction, a condensation reaction with ethylene glycol ketal, epoxy reaction, then the format of the reaction, The acid hydrolysis reaction and the acetylation reaction to obtain the target object and sequentially by treatment with isopropanol, ethyl acetate and crystallized from ether to obtain a yellow product with a melting point of 183-185Ό. The method expensive starting materials prices, harsh reaction conditions, need to be ultra-low temperature and water and oxygen reaction, high cost of low yield (total yield of about 14%), and therefore not suitable for industrial production.
3, World Patent WO2004078709 discloses a method for preparing (see Reaction Scheme III), the method the Πα hydroxy _19_ norpregn _ 4, 9 (10) _ diene-_ _ 3, 17-dione (Compound V2 ), followed by acetylation of 3 – bit carbonyl condensation, epoxy, Bonus format, acid hydrolyzed to give the target. Although the steps are shorter, but a starting material is from Compound VI was prepared by hydrolysis under acidic conditions to obtain a total yield of about 11.8% (starting from the compound VI operator), the actual reaction step is longer, lower yield, higher cost not suitable for industrial production.

In this method, 3,3 – ethylenedioxy -19 – norpregn -5 (10), 9 (11) – dien-17 – one (referred to as 3 – ketal compound II) as a starting material, by the addition of acetylene, benzene sub-sulfonyl chloride, and then by hydrolysis of sodium methoxide, acid hydrolysis, condensation of ethylene glycol, epoxy, Grignard reaction, acid hydrolysis and acetylation reaction of 9-step reaction to obtain a target object, isopropoxy alcohol crystallization with ethanol and water was heated at 70 ° C after 14h excluding solvate crystal. The method uses a greater risk of acetylene and odor of benzene times sulfonyl chloride, especially benzene times, unstable sulfonyl chloride, easy storage, decomposition of impurities involved in the reaction leads to a low yield, and benzene of times sulfonyl chloride of environmental pollution Further crystallization prolonged heating will produce new impurities, the total yield of 13.8% -15.8%, high cost, is not suitable for industrial production.
The existing methods, the methods 1, 2 and 4 are related to the preparation of compound VI, and also the starting materials in Method 3 Hydrolysis of compound VI is obtained. SUMMARY OF THE INVENTION
Technical problems to be solved by the present invention is to overcome these drawbacks,, study design Uli acetate Secretary industrialization production methods.
WO2013063859A1
http://www.google.com/patents/WO2013063859A1?cl=en


………
|
Ulipristal Acetate intermediates(7)
|
![19-Norpregn-9-ene-3,20-dione, 11-[4-(dimethylamino)phenyl]-5,17-dihydroxy-, cyclic 3,20-bis(1,2-ethanediyl acetal), (5α,11β)-](https://i0.wp.com/www.lanospharma.com/images/19-Norpregn-9-ene-3%2C20-dione%2C%2011-%5B4-%28dimethylamino%29phenyl%5D-5%2C17-dihydroxy-%2C%20cyclic%203%2C20-bis%281%2C2-ethanediyl%20acetal%29%2C%20%285%CE%B1%2C11%CE%B2%29-.gif)

![19-Norpregna-4,9-diene-3,20-dione, 17-(acetyloxy)-11-[4-(methylamino)phenyl]-, (11β)-](https://i0.wp.com/www.lanospharma.com/images/19-Norpregna-4%2C9-diene-3%2C20-dione%2C%2017-%28acetyloxy%29-11-%5B4-%28methylamino%29phenyl%5D-%2C%20%2811%CE%B2%29-.gif)




…………
Bioorganic & Medicinal Chemistry
Keywords: Synthesis. New drug molecules. New chemical entities. Medicine. Therapeutic agents. a b s t r a c t …. 1153. 22. Ulipristal acetate (ellaOne®).
…………. FORMULATION http://www.google.com/patents/WO2011091892A1?cl=en 
References
- “FDA approves ella™ tablets for prescription emergency contraception” (Press release). FDA. 13 August 2010. Retrieved 2013-06-12.
- Creinin, MD; Schlaff, W; Archer, DF; Wan, L; Frezieres, R; Thomas, M; Rosenberg, M; Higgins, J (2006). “Progesterone receptor modulator for emergency contraception: a randomized controlled trial”. Obstetrics and gynecology 108 (5): 1089–97. doi:10.1097/01.AOG.0000239440.02284.45. PMC 2853373. PMID 17077229.
- “Summary of Product Characteristics: ellaOne 30 mg tablet”. Retrieved 20 November 2010.
- “European Public Assessment Report for Ellaone. Summary for the public”. EMA. 2009. p. 2. Retrieved 22 November 2009.
- Glasier, A. F.; Cameron, S. T.; Fine, P. M.; Logan, S. J.; Casale, W.; Van Horn, J.; Sogor, L.; Blithe, D. L.; Scherrer, B.; Mathe, H.; Jaspart, A.; Ulmann, A.; Gainer, E. (2010). “Ulipristal acetate versus levonorgestrel for emergency contraception: A randomised non-inferiority trial and meta-analysis”. The Lancet 375 (9714): 555–562. doi:10.1016/S0140-6736(10)60101-8. PMID 20116841.
- Trussell, James; Cleland, Kelly (February 13, 2013). “Dedicated emergency contraceptive pills worldwide”. Princeton: Office of Population Research at Princeton University, Association of Reproductive Health Professionals. Retrieved March 25, 2014.
- ICEC (2014). “EC pill types and countries of availability, by brand”. New York: International Consortium for Emergency Contraception (ICEC). Retrieved March 25, 2014.
- HRA Pharma (March 2013). “Countries where ellaOne was launched”. Paris: HRA Pharma. Retrieved March 25, 2014.
- ECEC (2014). “Emergency contraception availability in Europe”. New York: European Consortium for Emergency Contraception (ECEC). Retrieved March 25, 2014. “Ulipristal acetate Emergency Contraception Pills (UPA ECPs), while available in most European countries since 2010, are not yet available in Albania, Estonia, Macedonia, Malta, Switzerland and Turkey. For now UPA ECPs are sold with a prescription in all countries, although provision without a prescription is currently being tested in the United Kingdom.”
- “Summary of Product Characteristics: Esmya 5mg tablet”. Retrieved 20 Febr 2014.
- Nieman, L. K.; Blocker, W.; Nansel, T.; Mahoney, S.; Reynolds, J.; Blithe, D.; Wesley, R.; Armstrong, A. (2011). “Efficacy and tolerability of CDB-2914 treatment for symptomatic uterine fibroids: A randomized, double-blind, placebo-controlled, phase IIb study”. Fertility and Sterility 95 (2): 767–772.e1–772. doi:10.1016/j.fertnstert.2010.09.059. PMID 21055739.
- Levens, E. D.; Potlog-Nahari, C.; Armstrong, A. Y.; Wesley, R.; Premkumar, A.; Blithe, D. L.; Blocker, W.; Nieman, L. K. (2008). “CDB-2914 for Uterine Leiomyomata Treatment”. Obstetrics & Gynecology 111 (5): 1129–1136. doi:10.1097/AOG.0b013e3181705d0e. PMC 2742990. PMID 18448745.
- Jacques Donnez; Tetyana F. Tatarchuk, Philippe Bouchard, Lucian Puscasiu, Nataliya F. Zakharenko, Tatiana Ivanova, Gyula Ugocsai, Michal Mara, Manju P. Jilla, Elke Bestel, Paul Terrill, Ian Osterloh, and Ernest Loumaye, for the PEARL I Study Group. “Ulipristal Acetate versus Placebo for Fibroid Treatment before Surgery”. New England Journal of Medicine. doi:10.1056/NEJMoa1103182. PMID 22296075.
- CHMP (2009:12, 14)
- CHMP (2009:33, 43)
- CHMP (2009:16)
- CHMP (2009:41)
- RCOG (2004). The Care of Women Requesting Induced Abortion : Evidence-based clinical guideline number 7 (PDF). London: RCOG Press. ISBN 1-904752-06-3. Archived from the original on 27 February 2008.
- CHMP (2009:37)
- CHMP (2009:43)
- CHMP (2009:12, 20)
- CHMP (2009:13–14, 21)
- Attardi, B.; Burgenson, J.; Hild, S.; Reel, J. (2004). “In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone”. The Journal of Steroid Biochemistry and Molecular Biology 88 (3): 277–288. doi:10.1016/j.jsbmb.2003.12.004. PMID 15120421.
- CHMP (2009:22–23)
- CHMP (2009). “Assessment Report for Ellaone”. EMA. Retrieved 22 November 2009.
- “FDA grants approval of ella for emergency contraception” (Press release). HRA Pharma. 13 August 2010. Retrieved 2010-08-15.
- Emma Hitt (18 June 2010). “FDA Panel Gives Ulipristal Acetate Unanimous Positive Vote for Emergency Contraception Indication”. Retrieved 2010-06-22.
- Harris, Gardiner (14 August 2010). “F.D.A. Approves 5-Day Emergency Contraceptive”. The New York Times. Retrieved 14 August 2010.
- Watson PR (1 December 2010). “Watson Launches ella(R)(ulipristal acetate)”. Retrieved 12 January 2010.\
| WO2004065405A1 | Jan 21, 2004 | Aug 5, 2004 | Crystal Pharma S A | Method of obtaining 17$g(a)-acetoxy-11$g(b)-(4-n,n-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione |
| WO2004078709A2 | Feb 13, 2004 | Sep 16, 2004 | Hyun K Kim | METHOD FOR PREPARING 17 α-ACETOXY-11β-(4-N,N-DIMETHYLAMINOPHENYL)-19-NORPREGNA-4,9-DIENE-3,20-DIONE, INTERMEDIATES THEREOF, AND METHODS FOR THE PREPARATION OF SUCH INTERMEDIATES |
| US4954490 | Jun 23, 1988 | Sep 4, 1990 | Research Triangle Institute | 11 β-substituted progesterone analogs |
| US5073548 | Apr 3, 1990 | Dec 17, 1991 | Research Triangle Institute | 11 β-substituted progesterone analogs |
| US5929262 | Mar 30, 1995 | Jul 27, 1999 | The United States Of America As Represented By The Department Of Health And Human Services | Method for preparing 17α-acetoxy-11β-(4-N, N-dimethylaminophyl)-19-Norpregna-4,9-diene-3, 20-dione, intermediates useful in the method, and methods for the preparation of such intermediates |
| US20050208129 | Apr 25, 2005 | Sep 22, 2005 | Bioalliance Pharma | Prolonged release bioadhesive therapeutic systems |
| WO2001074840A2 * | Mar 16, 2001 | Oct 11, 2001 | Carmie K Acosta | 17-alpha-substituted-11-beta-substituted-4-aryl and 21-substituted 19-norpregna 21-substituted 19-norpregnadienedione as antiprogestational agents |
| CN101466723A * | May 18, 2007 | Jun 24, 2009 | 吉瑞工厂 | Industrial process for the synthesis of 17a-acetoxy-11ss-[4-(n,n-dimethyl-amino)- phenyl]-19-norpregna-4,9-diene-3,20-dione and new intermediates of the process |
| CN102516345A * | Nov 1, 2011 | Jun 27, 2012 | 上海优拓医药科技有限公司 | Preparation method of ulipristal acetate and key intermediate thereof |
| US5929262 * | Mar 30, 1995 | Jul 27, 1999 | The United States Of America As Represented By The Department Of Health And Human Services | Method for preparing 17α-acetoxy-11β-(4-N, N-dimethylaminophyl)-19-Norpregna-4,9-diene-3, 20-dione, intermediates useful in the method, and methods for the preparation of such intermediates |

Biochips for better cancer therapy

Cancer is the second leading cause of disease-related death in the United States, and may overtake heart disease without aggressive new therapies. One promising area of cancer treatment is photodynamic therapy (PDT), which combines the agents of a photosensitive drug, light, and oxygen to attack cancerous tumors and lesions locally in the targeted region of the body by selective optical illumination.
Research being conducted by Prof. Euisik Yoon’s group aims to dramatically accelerate progress in PDT. And it is being accomplished through a lab-on-a-chip measuring about the size of a quarter. At the heart of this biochip is a 5x5mm testing area that will test the interaction of the drug, light, and oxygen simultaneously, generating results in a fraction of the time of current testing practices.
“In cancer research doctors are always looking for better drugs,” explained Dr. Xia Lou, a postdoctoral fellow in Prof. Yoon’s group. “But there has…
View original post 392 more words
Clinafloxacin from kyorin

Clinafloxacin
7-(3-Aminopyrrolidin-1-yl)-8-chloro-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid
7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid
(±)-7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid
105956-99-8 cas no
Clinafloxacin (INN) is a fluoroquinolone antibiotic. Its use is associated with phototoxicity and hypoglycaemia.[1]
Clinafloxacin is a novel quinolone with wide activity against the plethora of microorganisms encountered in intraabdominal infections.
Clinafloxacin is a chlorofluoroquinolone with excellent bioavailability and activity against gram-positive, gram-negative, and anaerobic pathogens . Typical MICs for α-streptococci are 0.06–0.12 µg/mL . MIC90 values for methicillin-resistant Staphylococcus aureus (MRSA) average 1.0 µg/mL. The MIC90 for enterococci is typically 0.5 µg/mL . Both intravenous and oral formulations have been developed . Several studies have demonstrated the efficacy of clinafloxacin monotherapy for serious infections Clinafloxacin was also active in animal models of endocarditis, including endocarditis due to ciprofloxacin-resistant S. aureus infection .



-
A mixture of 8-chloro-1-cyclopropyl-6,7-difluoro-1,4-di- hydro-4-oxo-3-quinolinecarboxylic acid (0.6 g), anhydrous acetonitrile (6 ml), 3-aminopyrrolidine (0.35 g) and DBU (0.31 g) was refluxed for an hour. Then, 3-aminopyrrolidine (0.2 g) was more added and further refluxed for 2 hours. After cooling, the resulting precipitate was collected by filtration, dissolved in water (9 ml) containing sodium hydroxide (0.12 g) and neutralized with acetic acid. The resulting precipitate was collected by filtration and washed with water and acetonitrile successively to give the title compound (0.52 g) as colorless powder, mp 237-238 °C (decompd.).
-
Analysis (%) for C17H17ClFN3O3·H2O, Calcd. (Found): C, 53.20 (52.97); H, 4.99 (4.62); N, 10.95 (10.83).
- Example 28 7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid
Example 29 7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
-
To a suspension of 7-(3-amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid (100 mg) in ethanol (2 ml) was added 0.2 ml of ethanol solution of hydrogen chloride (7.0 mmol HC1/ml) and then the mixture was concentrated. The resulting residue was recrystallized from methanol to give the title compound (79 mg) as light yellow prisms, mp 263-265 °C (decompd.).
-
Analysis (%) for C17H17ClFN3O3.HCl, Calcd. (Found): C, 50.76 (50.50); H, 4.51 (4.44); N, 10.45 (10.38).
…………………..
J. Med. Chem., 23, 1358 (1980)

-
structural formula D

may be readily prepared from the known starting material methyl 5-oxo-l-(phenylmethyl)-3-pyrrolidinecarboxylate, A, [J. Org. Chem., 26, 1519 (1961)] by the following reaction sequence.

-
The compound wherein R3 is hydrogen, namely 3-pyrrolidinemethanamine, has been reported in J. Org. Chem., 26, 4955 (1961).


References
- Rubinstein, E. (2001). “History of quinolones and their side effects.”. Chemotherapy. 47 Suppl 3: 3–8; discussion 44–8.doi:10.1159/000057838. PMID 11549783.
| EP0106489A2 * | Sep 6, 1983 | Apr 25, 1984 | Warner-Lambert Company | Antibacterial agents |
| EP0153163A2 * | Feb 15, 1985 | Aug 28, 1985 | Warner-Lambert Company | 7-Substituted-1-cyclopropyl-6,8-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids; 7-substituted-1-cyclopropyl-1,4-dihydro-6-fluoro-4-oxo-1,8-naphthyridine-3-carboxylic acids; their derivatives; and a process for preparing the compounds |
| BE899399A1 * | Title not available | |||
| GB2057440A * | Title not available |
| Examples of | ||
| reported trade | ||
| names for products | ||
| containing the 6- | ||
| 6-Fluoroquinolin- | fluoroquinolin- | |
| 4(1H)-one | 4(1H)-one | Structure |
| amifloxacin |
 |
|
| balofloxacin |
 |
|
| ciprofloxacin | Cipro®, Ciprobay, & Ciproxin |
 |
| clinafloxacin |
 |
|
| danofloxacin | Advocin & Advocid |
 |
| difloxacin | Dicural® & Vetequinon |
 |
| enrofloxacin | Baytril® |
 |
| fleroxacin | Megalone |
 |
| flumequine | Flubactin |
 |
| garenoxacin |
 |
|
| gatifloxacin | Tequin® & Zymar® |
 |
| grepafloxacin | Raxar |
 |
| ibafloxacin |
 |
|
| levofloxacin | Levaquin®, Gatigol, Tavanic, Lebact, Levox, & Cravit |
 |
| lomefloxacin | Maxaquin® |
 |
| marbofloxacin | Marbocyl® & Zenequin |
 |
| moxifloxacin | Avelox® & Vigamox® |
 |
| nadifloxacin | Acuatin, Nadoxia, & Nadixa |
 |
| norfloxacin | Noroxin®, Lexinor, Quinabic, & Janacin |
 |
| ofloxacin | Floxin®, Oxaldin, & Tarivid |
 |
| orbifloxacin | Orbax® & Victas |
 |
| pazufloxacin |
 |
|
| pefloxacin |
 |
|
| pradofloxacin |
 |
|
| prulifloxacin |
 |
|
| rufloxacin | Uroflox |
 |
| sarafloxacin | Floxasol, Saraflox, Sarafin |
 |
| sitafloxacin |
 |
|
| sparfloxacin | Zagam |
 |
| temalioxacin | Omniflox |
 |
| enoxacin | Penetrex & Enroxil |
 |
| gemifloxacin | Factive |
 |
| tosufloxacin |
 |
|
| trovafloxacin | Trovan |
 |
PRULIFLOXACIN by Nippon Shinyaku Co.

PRULIFLOXACIN
(RS)-6-Fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl]-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid
6-Fluoro-1-methyl-7-(4-(5-methyl-2-oxo-1,3-dioxelen-4-yl)methyl-1-piperazinyl)-4-oxo-4H-(1,3)thiazeto(3,2-a)quinoline-3-carboxylic acid
123447-62-1 CAS NO
OPT-99
Launched – 2002 BY NIPPON SHINYAKU
SYNTHESIS…….http://www.drugfuture.com/synth/syndata.aspx?ID=151640
Prulifloxacin is an older synthetic chemotherapeutic antibiotic of the fluoroquinolone drug class[1][2] undergoing clinical trials prior to a possible NDA (New Drug Application) submission to the U.S. Food and Drug Administration (FDA). It is a prodrug which is metabolized in the body to the active compound ulifloxacin.[3][4] It was developed over two decades ago by Nippon Shinyaku Co. and was patented in Japan in 1987 and in the United States in 1989.[5][6]
It has been approved for the treatment of uncomplicated and complicated urinary tract infections, community-acquired respiratory tract infections in Italy and gastroenteritis, including infectious diarrheas, in Japan.[3][7] Prulifloxacin has not been approved for use in the United States.
Prulifloxacin is a novel fluoroquinolone antibiotic that was launched pursuant to a collaboration between Meiji Seika and Nippon Shinyaku in 2002 for the oral treatment of systemic bacterial infections, including acute upper respiratory tract infection, bacterial pneumonia, prostatitis, cholecystitis, bacterial enteritis, internal genital infections, otitis media, sinusitis and others. It is currently marketed in a tablet formulation. A once-daily formulation to be taken over a three-day period is in phase III clinical trials at Optimer Pharmaceuticals to be used in the treatment of bacterial gastroenteritis, including traveler’s diarrhea. The formulation had been in phase II trials at the company for the treatment of urinary tract infections, however, no recent development for this indication have been reported. The drug has also been studied at Optimer for the treatment of community-acquired respiratory tract infections, but recent progress reports for this indication have not been made available.
Prulifloxacin has in vitro activity against a wide range of gram-negative and gram-positive microorganisms. Its antibacterial action results from inhibition of DNA gyrase and topoisomerase IV, both Type II isomerases. DNA gyrase is an essential enzyme that is involved in the replication, transcription, and repair of bacterial DNA. Topoisomerase IV is an enzyme known to play a key role in the partitioning of the chromosomal DNA during bacterial cell division. Together, the Type II topoisomerases remove the positive supercoils that accumulate ahead of a translocating DNA polymerase, allowing DNA replication to continue unhindered by topological strain. Fluoroquinolones may be active against pathogens that are resistant to penicillins, cephalosporins, aminoglycosides, macrolides and tetracyclines, as they possess a distinct mechanism of action from these antibiotics.
Prulifloxacin was discovered by Nippon Shinyaku and codeveloped with Meiji Seika in Japan. Nippon Shinyaku granted Angelini a manufacturing and marketing license for Italy in 1993. Exclusive Korean manufacturing and commercialization rights were acquired by Yuhan from Nippon Shinyaku in March 2003. In June 2004, Optimer was granted exclusive development and commercialization rights to prulifloxacin in the U.S. from Nippon Shinyaku. Finally, Recordati signed a nonexclusive licensing agreement with Angelini for the marketing and sale of prulifloxacin in Spain in October 2004. In March 2009, the product was licensed to Lee’s Pharmaceuticals by Nippon Shinyaku for marketing in China as an oral treatment of bacterial infection. In 2010, prulifloxacin was licensed to Algorithm by Nippon Shinyaku in North Africa and the Middle East for the development and marketing for the treatment of bacterial infections.
History
In 1987 a European Patent (EP 315828) for prulifloxacin (Quisnon ) was issued to the Japanese based pharmaceutical company, Nippon Shinyaku Co., Ltd (Nippon). Ten years after the issuance of the European patent, marketing approval was applied for and granted in Japan (March 1997). Subsequent to being approved by the Japanese authorities in 1997 prulifloxacin (Quisnon) was co-marketed and jointly developed in Japan with Meiji Seika as licensee (Sword).[6]
In more recent times, Angelini ACRAF SpA, under license from Nippon Shinyaku, has fully developed prulifloxacin, for the European market.[8] Angelini is the licensee for the product in Italy. Following its launch in Italy, Angelini launched prulifloxacin in Portugal (January 2007) and it has been stated that further approvals will be sought in other European countries.[9][10]
Prulifloxacin is marketed in Japan and Italy as Quisnon (Nippon Shinyaku); Sword (Meiji); Unidrox (Angelini) and generic as Pruquin.
In 1989 and 1992 United States patents (US 5086049) were issued to Nippon Shinyaku for prulifloxacin. It was not until June 2004, when Optimer Pharmaceuticals acquired exclusive rights to discover, develop and commercialize prulifloxacin (Pruvel) in the U.S. from Nippon Shinyaku Co., Ltd., that there were any attempts to seek FDA approval to market the drug in the United States. Optimer Pharmaceuticals expects to file an NDA (new drug application) for prulifloxacin some time in 2010. As the patent for prulifloxacin has already expired, Optimer Pharmaceuticals has stated that this may have an effect on the commercial prospects of prulifloxacin within the United States market.[11]
Licensed uses
Prulifloxacin has been approved in Italy ,Japan,China,India and Greece (as indicated), for treatment of infections caused by susceptible bacteria, in the following conditions:
- Italy
- Acute uncomplicated lower urinary tract infections (simple cystitis)
- Complicated lower urinary tract infections
- Acute exacerbation of chronic bronchitis
- Japan
- Gastroenteritis, including infectious diarrheas
- Other countries
- Prulifloxacin has not been approved for use in the United States, but may have been approved in other Countries, other than that which is indicated above.
Availability
Prulifloxacin is available as:
- Tablets (250 mg, 450 mg or 600 mg)
In most countries, all formulations require a prescription.

Prulifloxacin is chemically known as 6-fluoro-1-methyl-7-{4-[(5-methyl-2-oxo-1 ,3-dioxol- 4-yl)methyl]piperazin-1-yl}-4-oxo-4H-[1 ,3]-thiazeto-[3,2-a]-quinoline-3-carboxylic acid, and it has the structure as shown below as formula I:

FORMULA I
Prulifloxacin has significant antibacterial activity and has been marketed as a synthetic antibacterial agent.
Prulifloxacin was first disclosed in US 5,086,049. The patent discloses a process for the preparation of prulifloxacin by the condensation of ulifloxacin with a 4-halomethyl-5- methyl-1 ,3-dioxolen-2-one of formula III

wherein X is halo selected form chloro, bromo or iodo, in the presence or absence of an aprotic solvent and a base to obtain prulifloxacin free base which is recrytallised with chloroform-methanol. In an exemplified process, ethyl 6,7-difluoro-1-methyl-4-oxo-4H- (1 ,3)-thiazeto-(3,2-a)-quinoline-3-carboxylate is condensed with piperazine in the presence of dimethyl formamide and purified by column chromatography followed by basic hydrolysis to give ulifloxacin, which is then converted to prulifloxacin.
The above process involves column chromatography. Prulifloxacin prepared by this method has a purity of 60-65% containing impurities in unacceptable levels. Removal of these impurities by usual purification procedures, such as recrystallisation, distillation and washing, is difficult and requires extensive and expensive multiple purification processes. This further decreases the overall yield. A method involving column chromatographic purifications and multiple purifications cannot be used for large-scale operations, thereby making the process commercially non-viable.
European Patent No. 315828 disclosed a variety of quinoline carboxylic acid derivatives and pharmaceutically acceptable salts thereof. These compounds are exhibiting antibacterial activity and useful as remedies for various infectious diseases. Among them prulifloxacin, chemically (+)-6-Fluoro- 1 -methyl-7-[4-(5-methyl-2-oxo-1 ,3-dioxolen-4-ylmethyl)-1 -piperazinyl]-4-oxo-4H- [1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid is a fluoroquinolone antibacterial prodrug which shows potent and broad-spectrum antibacterial activity both in vitro and in vivo. Prulifloxacin also showed superior activity against strains of Enterobacteriaceae and Pseudomonas aeruginosa. Prulifloxacin is represented by the following structure:

Processes for the preparation of prulifloxacin and related compounds were disclosed in European Patent No. 315828 and UK Patent Application No. GB 2190376.
In – the preparation of prulifloxacin, 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid of formula I:

is a key intermediate. According to the UK Patent Application No. GB 2190376, the compound of the formula I was prepared by the reaction of 3,4-difluroaniline with carbon disulfide and triethylamine to give triethylammonium N-(3,4- difluorophenyl)dithio carbamate, which by reaction with ethyl chloroformate and triethylamine in chloroform is converted into 3,4-difluorophenyl isothiocyanate, followed by reaction with diethyl malonate and KOH in dioxane affords the potassium salt, which is then treated with methoxymethyl chloride in dimethylformamide to give diethyl 1-(3,4-difluorophenylamino)-1- (methoxymethylthio)-rnethylene-rnalonate. The cyclization of the thio compound at 2400C in diphenyl ether affords ethyl 6,7-difluoro-4-hydroxy-2- methoxymethylthioquinoline-3-carboxylate, which by treatment with HCI in ethanol gives ethyl δy-difluoro^-hydroxy^-mercaptoquinoline-S-carboxylate. The cyclization of the mercapto compound with 1,1-dibromoethane by means of potassium carbonate and potassium iodide in hot dimethylformamide yields ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate, which is condensed with piperazine in dimethylformamide to afford ethyl 6- fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate, which is then subjected to hydrolysis with potassium hydroxide in hot tert-butanol to give the compound of formula I.
The compound of formula I obtained by the process described in the UK Patent Application No. GB 2190376 is not satisfactory from purity point of view, the reaction between ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2- a]quinoline-3-carboxylate and piperazine requires longer time about 48 hours to complete, the yield obtained is not satisfactory, and the process also involves column chromatographic purifications. Methods involving column chromatographic purifications cannot be used for large-scale operations, thereby making the process commercially not viable. According to the European Patent No. 315828, prulifloxacin is prepared by reacting 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a] quinoline-3-carboxylic acid with 4-bromomethyl-5-methyl-1 ,3-dioxolen-2-one in presence of potassium bicarbonate in dimethylformamide. However, a need still remains for an improved and commercially viable process of preparing pure prulifloxacin that will solve the aforesaid problems associated with process described in the prior art and will be suitable for large- scale preparation, in terms of simplicity, purity and yield of the product.
Prulifloxacin is chemically 6-fluoro-l-methyl-7-{4-[(5-methyl-2-oxo-l,3-dioxol-4- yl)methyl]piperazin-l-yl}-4-oxo-4H-[l,3]thiazeto[3,2-α]quinoline-3-carboxylic acid of Formula I having the structure as depicted below:

FORMULA I
Prulifloxacin has significant antibacterial activity and has been marketed as a synthetic antibacterial agent. U.S. Patent No. 5,086,049 provides a process for the preparation of prulifloxacin by reacting 6-fluoro-l-methyl-4-oxo-7-piperazin-l-yl-4H- [l,3]thiazeto[3,2-α]quinoline-3-carboxylic acid of Formula II,

FORMULA II and 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III,

FORMULA III using N,N-dimethylformamide as a solvent. 4-(Bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III is used in excess to one mole of the compound of Formula II. The process provided in U.S. Patent No. 5,086,049 further involves concentrating the reaction mixture, pouring the residue into water and isolating prulifloxacin by filtration. The resulting prulifloxacin is recrystallized from chloroform-methanol.
However, U.S. Patent No. 5,086,049 does not provide any method to remove the unreacted or the excess of 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III used as a starting material. The present inventors have observed that it is difficult to obtain prulifloxacin with pharmaceutically acceptable purity by following the process provided in U.S. Patent No. 5,086,049, which is typically contaminated by process related impurities including 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one
A need still remains for an improved and commercially-viable process for preparing pure prulifloxacin that will solve the aforesaid problems associated with the process described in the prior art and that will be suitable for large-scale preparation, in terms of simplicity, purity and yield of the product.
EP1626051 A1 mentions that Type I, Type II and Type III crystals of prulifloxacin are obtained by crystallization from acetonitrile as reported in lyakuhin Kenkyu, Vol. 28 (1), (1997), 1-11. However, the conditions of crystallization from acetonitrile for preparing Type I, Type II and Type III crystals are not disclosed in lyakuhin Kenkyu, Vol. 28 (1), (1997), 1-11. EP1626051A1 further mentions that Type III crystals have been marketed by considering the solubility, absorbability, therapeutic effect and the like of the respective crystal forms.
US 2007/0149540 discloses a crystal of prulifloxacin acetonitrile solvate (Compound B) which is an intermediate for producing preferentially the type III crystal of prulifloxacin. A crystal of Compound B can be preferentially precipitated by controlling the supersaturation concentration in crystallization using acetonitrile as a solvent, subsequently; the type III crystal of Compound A can be produced by performing desolvation of the crystal.
WO 2008/111018 discloses processes for the preparation of Type I, Type II and Type III crystals of prulifloxacin. There is disclosed a process for preparing Type I crystals by controlled cooling over a period of 7 to 9 hours and prolonged drying over 24 hours. The inventors of the present invention have found that Type I and Type III crystals prepared according to the WO 2008/111018 process are unstable and the process is non-reproducible.
WO 2010/0084508 discloses processes for the preparation of Type I, Type II and Type III crystals of prulifloxacin.
WO 2008/059512 discloses a process for the preparation of prulifloxacin using novel intermediates.
WO 2008/111016 discloses a process for the preparation of prulifloxacin having purity of about 99% or above. It would be a significant contribution to the art to provide a crystalline form of prulifloxacin, which is consistent and to provide industrially viable methods of preparation, pharmaceutical formulations, and methods of use thereof.

…………………
SYNTHESIS
http://www.google.com/patents/WO2012001357A1?cl=en
Scheme 1.


Formula I
[PRULIFLOXACIN]
Example 1
Preparation of ethyl-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-(1 ,3)-thiazeto-[3,2-a]- quinoline-3-carboxylate (formula III)
5,6-difluoro-1-methyl-4-oxo-4H-[1 ,3]-thiazeto-[3,2-a]-quinoline-3-carboxylic acid ethyl ester of formula (II) (100 gms, 0.321 moles) was stirred in 500 ml of DMF at room temperature. Piperazine (76 gms, 0.882 moles) was added at room temperature and stirred for 10 minutes. The temperature was slowly raised to 50-55°C and the reaction mass was stirred at 50-55°C for 5 hours. After completion of the reaction, the reaction mass was cooled to 25-30°C and stirred for 2 hours. The reaction mass was further chilled to 10-15°C and stirred for 2 hours. The precipitated solid was filtered, washed of chilled DMF (2 x 50 ml). The solid was slurry washed with water (300 ml), filtered, washed with water ( 2 x 100 ml) and dried under vacuum at 70-75°C to yield the title compound [90 gms, 74 % yield, 95% HPLC purity].
Example 2
Preparation of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid (formula IV)
Ethyl-6-fluoro-1 -methyl-4-oxo-7-(1 -piperazinyl)-4H-(1 ,3)-thiazeto-[3,2-a]-quinoline-3- carboxylate (100 gms, 0.265 moles) was stirred in water (600 ml) at 25-30°C. To this potassium hydroxide solution (50 gms of potassium hydroxide flakes is dissolved in 200 ml of water) was added and the reaction mass was heated to 80-85°C. The contents were stirred for 1 hour and after completion of reaction, the reaction mass was cooled to 25-30°C. The pH of the reaction mass was adjusted to 6.5-7.0 using 1:1 aqueous acetic acid solution. The contents were stirred at room temperature for 1 hour. The precipitated solid was filtered, washed with water (2 x 100 ml). The solid was slurried in methanol (300 ml) for 1 hour at 25-30°C, filtered, washed with methanol (2 x 50 ml) and dried under vacuum at 70-75°C to yield the title compound [90 gms, 97% yield, 96% HPLC purity]. Example 3
Preparation of prulifloxacin
To a solution of 4-(chloromethyl)-5-methyl-1,3-dioxol-2-one (55 gms, 0.371 moles) in 50 ml of DMF at 25-30°C, sodium bromide (77 gms, 0.748 moles) was added and the reaction mass was slowly heated to 40-45°C. The contents were stirred at 40-45°C for 2 hours, acetone ( 500 ml) was added at 40-45°C and stirred for 3 hours. The reaction mass was filtered over hyflo, and the bed washed with acetone (100 ml). The solvent was completely distilled off under vacuum below 45°C to yield 4-(bromomethyl)-5- methyl-1 ,3-dioxol-2-one (formula V).
To a solution of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid of formula IV (100 gms, 0.286 moles) in 4.0 It of acetonitrile, DIPEA (70 ml , 0.402 moles)) was added at room temperature, stirred for 10 minutes. The reaction mass was cooled to 10-15°C and a solution of 4-(bromomethyl)-5-methyl- 1 ,3-dioxol-2-one (formula V) in 500 ml of acetonitrile was slowly added at 10-15°C over a period of 1 hour. The contents were stirred at 25-30°C for 20 hour, filtered over hyflo, and the bed washed with 200 ml of acetonitrile. The solvent was distilled off completely under vacuum below 50°C. Acetonitrile (100 ml) was added at 50°C and the contents were stirred for 30-60 minutes. The reaction mass was slowly chilled to 0-5°C and the precipitated solid was filtered, washed with acetonitrile (25 ml) and dried to yield 65 gms of prulifloxacin. Example 4
Preparation of Type I crystals of prulifloxacin
Prulifloxacin (65 gms) was added to 200 ml of DMF at 25-30°C and heated to 80-85°C for 1 hour. The mixture was then slowly cooled to 25-30°C, stirred for 2 hours, chilled to 0-5°C for 2 hours. The precipitated solid was filtered and dried under vacuum at 70- 75°C to yield Type I crystals of prulifloxacin (55 gms, 99.5 % HPLC purity).
Example 5
Preparation of prulifloxacin
(55 gms, 0.371 moles) of 4-(chloromethyl)-5-methyl-1 ,3-dioxol-2-one is taken in 5.0 ml of DMF at 25-30°C. (77 gms, 0.748 moles) of sodium bromide is added and slowly heated the reaction mass to 40-45°C. The contents are stirred at 40-45°C for 2 hours, 500 ml of acetone is added at 40-45°C and stirred for 3 hours. The reaction mass is clarified over hyflo, and the bed washed with 100 ml of acetone to yield a solution of 4- (bromomethyl)-5-methyl-1 ,3-dioxol-2-one (formula V).
To a solution of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid of formula IV (100 gms, 0.286 moles) in 3.5 Its of acetone was at room temperature DIPEA (70 ml, 0.402 moles) and stirred for 10 minutes. The reaction mass was cooled to 10-15°C and a solution of 4-(bromomethyl)-5-methyl-1 ,3- dioxol-2-one (formula V) in acetone was slowly added to the reaction mass at 10-15°C over a period of 1 hour. The contents were further stirred at 25-30°C for 20 hour, filtered over hyflo and the bed washed with 200 ml of acetone. The solvent was distilled off completely under vacuum below 50°C. Acetonitrile (100 ml) was added at 50°C and the contents were stirred for 30-60 minutes. The reaction mass was further chilled to 0- 5°C and stirred for 2 hours. The precipitated solid was filteredand dried to yield prulifloxacin.
………………….
http://www.google.com/patents/WO2008059512A1?cl=en
novel process for preparing 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid of formula I:

which comprises: a) reacting the difluoro-quinoline compound of formula

wherein R represents hydrogen atom or alkyl containing 1 to 4 carbon atoms; with boric acid of formula III:

in presence of acetic anhydride and acetic acid to give borane compound of formula IV:

b) reacting the borane compound of formula IV with piperazine of formula V:
HN NH V
to give piperazine compound of formula Vl:

c) treating the compound of formula Vl with an alkaline metal hydroxide, carbonate or bicarbonate to obtain the compound of formula I.
Prulifloxacin and pharmaceutically acceptable acid addition salts of prulifloxacin can be prepared by using the compound of formula I by known methods for example as described in the European Patent No. 315828. Borane compound of the formula IV and Vl are novel and forms part of the invention. Preferably the reaction in step (a) is carried out at about 300C to reflux temperature more preferably at about 800C to reflux temperature and still more preferably at reflux temperature.
Example 1 Step-I:
Acetic anhydride (24 ml) and acetic acid (11 ml) are added to boric acid (3.5 gm) under stirring at 25 – 300C, the contents are heated to reflux and then stirred for 3 hours at reflux. The reaction mass is cooled to 1000C, ethyl 6,7- difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate (20 gm) is added at 1000C, the contents are heated to reflux and then refluxed for 2 hours. The reaction mass is cooled to 25 – 350C, toluene (200 ml) is added under stirring, the reaction mass is cooled to 50C and then stirred for 1 hour at 5 – 100C. Filtered the solid, washed with 20 ml of toluene and then dried to give 25.5 gm of 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate -O3,04/bis/acetato-0/-borone. Step-I I: Acetonitrile (125 ml), dimethylsulfoxide (125 ml) and piperazine (13.8 gm) are added to 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate-03,04/bis/acetato-0/-borone (25.5 gm, obtained in step-l) under stirring at 25 – 350C, the contents are heated to 850C and then stirred for 3 hours at 80 – 850C to form a clear solution. The solution is cooled to 100C and then stirred for 1 hour at 10 – 150C. Filtered the solid, washed with 25 ml of acetonitrile and then dried to give 26 gm of 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/acetato-0/- borone. Step-Ill: Water (155 ml), potassium hydroxide (17 gm) are added to 6-fluoro-1- methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate- O3,O4/bis/ acetato-0/-borone (26 gm, obtained in step-ll) under stirring at 25 – 350C, the contents are heated to 650C and then stirred for 4 hours at 60 – 650C. The reaction mass is cooled to 250C, filtered the undesired solid on hi-flow bed and then pH of the resulting filtrate is adjusted to 7 – 7.5 with 50% HCI solution at 25 – 300C. The separated solid is stirred for 1 hour at 25 – 300C, filtered the solid, washed with 35 ml of water and then dried to give 17 gm of 6-fluoro-1- methyl-4-oxo-7-(1 -piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid (HPLC Purity: 98.5%). Example 2 Step-I:
Acetic anhydride (12 ml) and acetic acid (5.5 ml) are added to boric acid (1.25 gm) under stirring at 25 – 300C, the contents are heated to reflux and then stirred for 3 hours at reflux. The reaction mass is cooled to 1000C, 6,7-difluoro-1- methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid (10 gm) is added at 1000C, the contents are heated to reflux and then refluxed for 3 hours. The reaction mass is cooled to 500C, toluene (100 ml) is added under stirring at 500C, the resulting mass is cooled to 100C and then stirred for 1 hour at 10 – 150C. Filtered the solid, washed with 20 ml of toluene and then dried to give 10 gm of 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate -O3 , 04/bis/acetato-0/-borone . Step-I I:
Acetonitrile (50 ml), dimethylsulfoxide (50 ml) and piperazine (5.5 gm) are added to 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate-03,04/bis/acetato-0/-borone (10 gm, obtained in step-l) under stirring at 25 – 350C, the contents are heated to 850C and then stirred for 3 hours at 80 – 850C to form a clear solution. The solution is cooled to 100C and then stirred for 1 hour at 10 – 150C. Filtered the solid, washed with 10 ml of acetonitrile and then dried to give 10.4 gm of 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/acetato-0/- borone. Step-Ill :
Water (62 ml), potassium hydroxide (7 gm) are added to 6-fluoro-1-methyl-4- oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/ acetato-OAborone (10.4 gm, obtained in step-ll) under stirring at 25 – 350C, the contents are heated to 650C and then stirred for 4 hours at 60 – 650C. The reaction mass is cooled to 250C, filtered the undesired solid on hi-flow bed and then pH of the resulting filtrate is adjusted to 7 – 7.5 with 50% HCI solution at 25 – 300C. The separated solid is stirred for 30 minutes at 25 – 300C, filtered the solid, washed with 20 ml of water and then dried to give 68 gm of 6-fluoro-1- methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid (HPLC Purity: 98.6%). Example 3
Acetonitrile (560 ml) and potassium bicarbonate (8 gm) are added to 6- fluoro-i-methyM-oxo-y-CI-piperazinyO^H-CI .SKhiazetofS^-alquinoline-S- carboxylic acid (14 gm, obtained as per the processes described in examples 1 and 2) under stirring at 25 – 300C, the contents are cooled to 150C and then the solution of 4-bromomethyl-5-methyl-1 ,3-dioxolen-2-one (10 gm) in acetonitrile (140 ml) is added at 15 – 200C for 30 to 45 minutes. The contents are stirred for 25 hours at 25 to 300C, filtered and the resulting filtrate is distilled under vacuum. To the residue added acetonitrile (70 ml), cooled the mass to 200C and then stirred for 1 hour to 1 hour 30 minutes at 20 – 250C. Filtered the solid, washed the solid with 15 ml of chilled acetonitrile and then dried to give 16 gm of prulifloxacin crude (HPLC Purity: 98.8%).
To the prulifloxacin crude (obtained above) added acetonitrile (200 ml) at 25 – 300C, the contents are heated to reflux and then refluxed for 30 minutes. To the reaction mass added activated carbon (5 gm) and refluxed for 15 minutes. The reaction mass is filtered on hi-flo bed, the resulting filtrate is cooled to 200C and then stirred for 3 – 4 hours at 20 – 250C. Filtered the solid, washed with 20 ml of acetonitrile and then dried to give 14 gm of prulifloxacin (HPLC Purity: 99.9%).
…………………
http://www.google.com/patents/WO2008111016A1?cl=en
In a first aspect, a process for the preparation of prulifloxacin is provided, the process comprising: a) reacting a compound of Formula II with a compound of Formula III to obtain prulifloxacin;


FORMULA III
FORMULA II
b) contacting the prulifloxacin obtained in step a) with an acid in a biphasic solvent system, wherein the biphasic solvent system comprises water and a water- immiscible organic solvent; c) separating the aqueous layer from the reaction mixture obtained in step b); d) treating the aqueous layer with a base; and e) isolating prulifloxacin.
The process described in steps b – e above may be carried out with prulifloxacin made from any process however.
The compounds of Formula II and Formula III may be prepared according to the methods provided in U.S. Patent No. 5,086,049.
Example 1: Process for the Preparation of Prulifloxacin:
Step A): A solution of 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one (35.5 g, 0.184 mole) in N,N-dimethylformamide (200 ml) was added dropwise at 0 to 5° C to a stirred solution of 6-fluoro-l-methyl-4-oxo-7-piperazin-l-yl-4H-[l,3]thiazeto[3,2-α]quinoline-3- carboxylic acid (50 g, 0.143 mole and potassium bicarbonate (15.8 g, 0.1578 mole) in N,N-dimethylformamide (200 ml). The resulting mixture was stirred at 25° to 28°C for 3 to 4 hours. After the completion of the reaction, the reaction mixture was poured into water (1250 ml). The solid obtained was filtered, washed with water (100 ml), and subsequently dissolved in a mixture of chloroform: methanol (7:3; 1250 ml). The lower organic layer was separated and water (500 ml) was added to the organic layer. A dilute aqueous solution of hydrochloric acid was added to the biphasic reaction mixture to adjust pΗ to 0.8 to 1.0. The reaction mixture was stirred for 15 minutes, allowed to settle and the upper aqueous layer was separated. The process was repeated twice and the aqueous layers were combined. Activated charcoal (10%) was added to the combined aqueous layer and stirred for 30 minutes, filtered and cooled to 20° to 25° C. The pΗ of the reaction mixture was adjusted to 6.5 to 7.0 by adding an aqueous solution of sodium bicarbonate. The solid obtained was extracted with chloroform (375 ml), stirred for 15 minutes and the organic layer was separated. The aqueous layer was further extracted with a mixture of chloroform: methanol (7:3 ratio; 50 ml). The combined organic layer was distilled under vacuum at 35° to 40° C to recover the solvent up to 125 ml. The reaction mass so obtained was stirred for 3 to 4 hours at 28° to 30° C, filtered and washed with chilled chloroform (50 ml). The wet cake obtained was dried at 45° C for 12 hours to obtain the title compound. Step B): The prulifloxacin (30 g) obtained in Step A) was suspended in a mixture of chloroform: ethanol (10:1, v/v, 585 ml: 58.5 ml) and heated to reflux temperature. Activated carbon (3.9 gm) was added to the partially cleared solution and refluxed for 30 minutes, followed by filtration through Celite bed. The bed was further washed with chloroform: ethanol (10:1, v/v, 585 ml: 58.5 ml). The filtrate so obtained was distilled at atmospheric pressure till to partially remove the solvent. The concentrate so obtained was stirred at about 25° C for 1 hour, and filtered. The solid obtained was washed with chloroform: ethanol (39 ml X 2), dried under vacuum at 45° C for 12 hours to obtain the title compound. Yield: 22 g
HPLC Purity: 99%
………………………….
SEE
Studies on pyridonecarboxylic acids. 1. Synthesis and antibacterial evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto[3, 2-a]quinoline-3-carboxylic acids
J Med Chem 1992, 35(25): 4727
http://pubs.acs.org/doi/pdf/10.1021/jm00103a011

The reaction of 3,4-difluoroaniline (I) with carbon disulfide and triethylamine gives triethylammonium N-(3,4-difluorophenyl)dithiocarbamate (II), which by reaction with ethyl chloroformate and triethylamine in chloroform is converted into 3,4-difluorophenyl isothiocyanate (III). The reaction of (III) with diethyl malonate and KOH in dioxane affords the potassium salt (IV), which is treated with chloromethyl methyl ether in DMF to give the corresponding methoxymethylsulfanyl compound (V). The cyclization of (V) at 240 C in diphenyl ether affords 6,7-difluoro-4-hydroxy-2-(methoxymethylsulfanyl)quinoline-3-carboxylic acid ethyl ester (VI), which by treatment with HCl in ethanol gives the corresponding mercapto compound (VII). The cyclization of (VII) with 1,1-dibromoethane by means of K2CO3 and KI in hot DMF yields 5,6-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid ethyl ester (VIII), which is condensed with piperazine (IX) in DMF to afford the corresponding piperazino-derivative (X). The hydrolysis of (X) with KOH in hot tert-butanol gives the corresponding free acid (XI) , which is finally condensed with 4-(bromomethyl)-5-methyl-1,3-dioxol-2-one (XII) by means of KHCO3 in DMF.
………………….
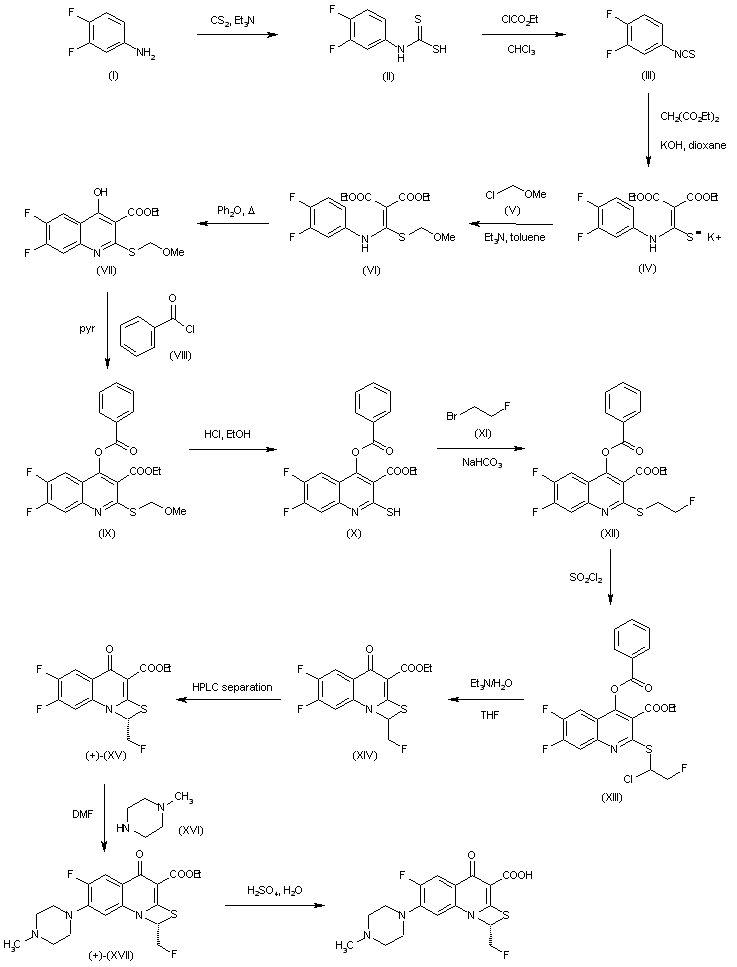
Treatment of 3,4-difluoroaniline (I) with CS2 and Et3N gives triethylammonium dithiocarbamate (II), which reacts with ethyl chloroformate in chloroform to yield (III). Isothiocyanate (III) is converted into the potassium salt (IV) by reaction with diethyl malonate and KOH in dioxane and then transformed into methoxymethyl thioether (VI) by means of reagent (V) and Et3N in toluene. Cyclization of (VI) by heating in diphenyl ether affords quinoline (VII), which then reacts with benzoyl chloride (VIII) in pyridine to furnish (IX). Benzoyloxy derivative (IX) is converted into (X) by means of HCl in EtOH, and its reaction with 1-bromo-2-fluoroethane (XI) and NaHCO3 yields compound (XII). Chlorination of (XII) with SO2Cl2 in hexane provides (XIII), which by simultaneous hydrolysis and intramolecular cyclization by means of Et3N /H2O in THF provides the mixture of isomers (XIV). (+)-(XV) is obtained by HPLC chromatography of (XIV) on a chiral stationary phase. Treatment of (+)-(XV) with 1-methylpiperazine (XVI) in DMF provides ethyl ester (+)-(XVII), which is finally hydrolyzed by means of H2SO4 in H2O.
INTERMEDIATES

154330-67-3
Ethyl 6,7-difluoro-2-ethylmercapto-4-hydroxyquinoline-3-carboxylate

154330-68-4
Ethyl 4-acetoxy-6,7-difluoro-2-(ethylthio)quinoline-3-carboxylate

113046-72-3
Ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate

113028-17-4
Ethyl 6-fluoro-1-methyl-4-oxo-7-(1-piprazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate

112984-60-8
6-Fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid
REFERENCES
- Nelson, Jennifer M.; Chiller, Tom M.; Powers, John H.; Angulo, Frederick J. (2007). “Food Safety: Fluoroquinolone‐Resistant Campylobacter Species and the Withdrawal of Fluoroquinolones from Use in Poultry: A Public Health Success Story”. Clinical Infectious Diseases 44 (7): 977–80. doi:10.1086/512369. PMID 17342653.
- Kawahara S (1998). “[Chemotherapeutic agents under study]”. Nippon Rinsho (in Japanese) 56 (12): 3096–9. PMID 9883617.
- Fritsche, T. R.; Biedenbach, D. J.; Jones, R. N. (2008). “Antimicrobial Activity of Prulifloxacin Tested against a Worldwide Collection of Gastroenteritis-Producing Pathogens, Including Those Causing Traveler’s Diarrhea”. Antimicrobial Agents and Chemotherapy 53 (3): 1221–4. doi:10.1128/AAC.01260-08. PMC 2650572.PMID 19114678.
- Giannarini, Gianluca; Tascini, Carlo; Selli, Cesare (2009). “Prulifloxacin: clinical studies of a broad-spectrum quinolone agent”. Future Microbiology 4 (1): 13–24.doi:10.2217/17460913.4.1.13. PMID 19207096.
- JP patent 1294680, Kise Masahiro; Kitano Masahiko; Ozaki Masakuni; Kazuno Kenji; Matsuda Masato; Shirahase Ichiro; Segawa Jun, “Quinolinecarboxylic Acid Derivative”, issued November 28, 1989
- Prulifloxacin. Drugfuture.com. Retrieved on 2010-11-03.
- Anonymous (2002). “Prulifloxacin [‘Quisnon’; Nippon Shinyaku] has been approved in Japan”. Inpharma 1 (1362): 22.
- Research and Development Department of Angelini. Angelinipharma.com. Retrieved on 2010-11-03.
- Nippon Shinyaku, Annual Report 2007
- “Prulifloxacin. NAD-441A, NM 441, Quisnon”. Drugs in R&D 3 (6): 426–30. 2002.PMID 12516950.
- Annual Report 2008, p. 34
Segawa,J,Mashiko kitano, Kenji Kazuno et al, Studies on Pyridonecarboxylic acids,1.Sythesis and antibacterial Evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto [3,2-]quionoline- 3-caroboxylic acids[J].J Med Chem. 1992,35(25):4727-4738.
Masato Matsuoka, Jun Segawa, Yoshihiko.et al, Studies on Pyridone Carb oxylic acids. V.A Practial synthesis of Ethyl 6,7–Difuoro-1-methyl-4-oxo-[1,3] Thiazeto [3,2-a]quinoline-3- Caroboxylate a key intermediate for the new tricyclic quinolone, prulifloxacin (NM441) and Versatile new syntheses of the 2-thioquinoline Skeleton[J].J Heterocyclic Chem.1997,34,1773-1779.
|
3-13-1996
|
Sustained release capsule
|
|
|
10-11-1995
|
Method of manufacturing solid dispersion
|
|
|
2-5-1992
|
7(4-(5 METHYL-2-OXO-1,3-DIOXALEN-4-YL)METHYL 1-PIPERZINYL)-4-OXO-4H-(1,3)THIAZETO(3,2-A)QUINOLINE-3-CARBOXYLIC ACIDS
|
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
|
|
|
2-11-2011
|
PROCESS FOR THE PREPARATION OF PURE PRULIFLOXACIN
|
|
|
8-6-2010
|
PROCESS FOR PREPARATION OF PRULIFLOXACIN USING NOVEL INTERMEDIATES
|
|
|
5-7-2010
|
PROCESS FOR THE PREPARATION OF CRYSTALS OF PRULIFLOXACIN
|
|
|
4-9-2010
|
COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID
|
|
|
12-11-2009
|
Compounds and Methods for modulating the Silencing of a Polynucleotide of Interest
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
2-6-2004
|
Medicinal composition
|
| WO2008059512A1 | Nov 17, 2006 | May 22, 2008 | Hetero Drugs Ltd | Process for preparation of prulifloxacin using novel intermediates |
| WO2008111016A1 | Mar 14, 2008 | Sep 18, 2008 | Ranbaxy Lab Ltd | Process for the preparation of pure prulifloxacin |
| WO2008111018A2 | Mar 14, 2008 | Sep 18, 2008 | Ranbaxy Lab Ltd | Process for the preparation of crystals of prulifloxacin |
| WO2010084508A2 | Dec 10, 2009 | Jul 29, 2010 | Elder Pharmaceuticals Ltd. | Process for the preparation of type i, type ii and type iii crystalline prulifloxacin |
| EP0315828A1 * | Oct 26, 1988 | May 17, 1989 | Nippon Shinyaku Company, Limited | Quinolinecarboxylic acid derivatives |
| EP1626051A1 | Apr 28, 2004 | Feb 15, 2006 | Nippon Shinyaku Co., Ltd. | Crystals of quinolinecarboxylic acid derivative solvate |
| US5086049 | Apr 8, 1991 | Feb 4, 1992 | Nipponshinyaku Co., Ltd. | 7[4-(5 methyl-2-oxo-1,3-dioxalen-4-yl)methyl 1-piperzinyl]-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acids |
| US20070149540 | Apr 28, 2004 | Jun 28, 2007 | Nippon Shinyaky Co., Ltd. | Crystals of quinolinecarboxylic acid derivative solvate |
EXTRA INFO
http://www.google.com/patents/EP2524922A1?cl=en
-
formula 1 is S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl]-4-oxo-4H-[1,3]thiazet o[3,2-α]quinoline-3-carboxylic acid (levo-prulifloxacin for short); its stereo configuration is S configuration; it has optical property of levorotatory polarized light:
-
 S-(-) ulifloxacin (as shown in formula 2 below) as raw material and the compound as shown in the following formula 3 are reacted in organic solvent in the presence of alkaline material. The reaction formula is shown below:
S-(-) ulifloxacin (as shown in formula 2 below) as raw material and the compound as shown in the following formula 3 are reacted in organic solvent in the presence of alkaline material. The reaction formula is shown below:
-
S-(-)-ulifloxacin and R-(+)-ulifloxacin are prepared according to the method disclosed in CN101550142A .
-
Japanese scholars Masato Matsuoka et al. have proved the absolute configuration of optically pure prulifloxacin. The study (see the publication Chem. Pharm. Bull. 43(7) 1238-1240 (1995)) verifies that (-)-ulifloxacin is S configuration while (+)-ulifloxacin is the enantiomer of R configuration by applying chemical methods together with single-crystal X-ray diffraction.
-
Accordingly, R-prulifloxacin can be prepared from R-(+)-ulifloxacin and the compound of formula 3 by the method described hereinbefore.
-
[0022]The reaction formula is depicted below:

-
S-prulifloxacin prepared in accordance with the present invention is determined to be laevorotatory by optical rotation measurement, so it is S-(-)-prulifloxacin. R-prulifloxacin prepared in accordance with the present invention is determined to be dextrorotatory by optical rotation measurement, so it is R-(+)-prulifloxacin.
-
The present invention studied the absorption features of S-(-)-prulifloxacin and R-(+)-prulifloxacin on circular polarized light by circular dichroism spectroscopy. The two spectrograms are mirror images of each other, which proves that S-(-)-prulifloxacin and R-(+)-prulifloxacin are enantiomer of each other.
-
Comparing the circular dichroism spectrogram as depicted in figure 4 with the circular dichroism spectrogram of analogue of the similar structure with known absolute configuration as disclosed in the publication Chem. Pharm. Bull. 47(12) 1765-1773 (1999), it is found that (-)-prulifloxacin has similar Cotton effect to the two analogues reported in the publication, ethyl S-(-)-6,7-difluoro-1-methyl-4-oxo-4H-[1,3] thiazeto[3,2-α]quinoline-3-carboxylate and ethyl S-(-)-6, 7-difluoro-1-fluoromethyl-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylate; so does (+)-prulifloxacin. The results also verify on the other hand that the absolute configuration of levo-prulifloxacin of the present invention is S type while the absolute configuration of dextro-prulifloxacin is R type.
-
The compound of the present invention and physiologically acceptable acid can be prepared to salts: dissolving or suspending S-(-)-prulifloxacin in solvent such as chloroform, DMF and the like; adding into acid or acid solution (for example, hydrochloric acid or hydrogen chloride-methanol solution and the like) while stirring; precipitating and filtering to obtain solid salt from the solvent solution, or alternatively removing solvent from the salt solution directly by concentration, spray drying and the like to obtain the salt of S-(-)-prulifloxacin. The obtained solid may be further recrystallized.
Example 1 Preparation of (S)-(-)-uliflourxacin
-
105 g of racemic uliflourxacin was dissolved in 1,500 mL of dimethyl sulfoxide. 27 g of D-tartaric acid was dissolved in 405 mL of dimethyl sulfoxide dropwise while stirring. After stirring at room temperature for 20 hours, the precipitate was filtrated. The collected solid was dried under vacuum to obtain 86 g solid, which was recrystallized in dimethyl sulfoxide to obtain 37 g of levoulifloxacin-D-tartrate, with C49.08%, H5.06%, N9.50%, S7.44% shown by elemental analysis (molecular formula: C16H16FN3O3S·1/2C4H6O6·H2O, calculated values: C48.86%, H4.78, N9.50%, S7.25%). Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After precipitation, filtration, and drying, 24.5 g of (S)-uliflourxacin was obtained, having a chemical name (S)-(-)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto [3,2-α]quinoline-3-carboxylic acid.
-
Specific rotation [α]20 D= -133° (c=0.5, 0.1 mol/L methanesulfonic acid); 1H-NMR (DMSO-d6) δ2.11 (3H, d, j=6.2 Hz), 2.87 (4H, m), 3.19 (4H, m), 6.40 (1H, q, j=6.2 Hz), 6.89 (1H, d, j=7.4Hz), 7.79 (1H, d, j=13.9Hz), optical purity e.e. 96%.
Example 2 Preparation of (R)-(+)-uliflourxacin
-
105 g of racemic uliflourxacin was dissolved in 1,500 mL of DMSO. 27 g of L-tartaric acid was dissolved in 405 mL dimethyl sulfoxide dropwise while stirring to allow that the solution became turbid and the precipitation occurred. The solution was stirred at room temperature for 20 hours and then filtered. The collected solid was dried under vacuum to obtain 82 g solid which was recrystallized in dimethyl sulfoxide to obtain 34 g of dextrouliflourxacin-L-tartarte. Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After filtration and drying, 22 g of (R)-uliflourxacin was obtained, having a chemical name (R)-(+)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline -3-carboxylic acid.
-
Specific rotation [α]20 D= +132.4° (c=0.5, 0.1 mol/L methanesulfonic acid), optical purity e.e. 96%.
Example 3 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin prepared in Example 1, 2.02 g (0.02 mol) of triethylamine and 20 ml of dimethylformamide (hereinafter referred to as DMF) were mixed and stirred. After the solution was cooled to -5∼5 °C, 0.012 mol of 4-bromomethyl-5-methyl-1,3-dioxolen-2-one (hereinafter referred to as DMDO-Br) in DMF (5 ml) solution was added thereinto, followed by stirring at -5∼5 °C for 3 hours. The reaction solution was poured into 100 ml of ice water, stirred for 30 minutes, and then filtered. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.9 g of S-(-)-prulifloxacin was obtained, having a chemical name: S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl ]-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylic acid, with a purity of 98% and a yield rate of 63%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
Example 4 Preparation of R-(+)-prulifloxacin
-
R-(+)-prulifloxacin prepared in Example 2 was used as raw material to prepare 2.7 g of target product R-(+)-prulifloxacin in accordance with the method as described in Example 3, with a yield rate of 60.7% and a purity of 98%. Specific rotation [α]20 D= +108° (c=0.5, 0.1 mol/L methanesulfonic acid).
-
Comparing the circular dichroism spectrogram as depicted in Figure 4 with the circular dichroism spectrogram of analogue of the similar structure with known absolute configuration as disclosed in the publication Chem. Pharm. Bull. 47(12) 1765-1773 (1999), it was found that (-)-prulifloxacin has similar Cotton effect with the two analogues reported in the publication, ethyl S-(-)-6,7-difluoro-1-methyl-4-oxo-4H-[1,3] thiazeto[3,2-α]quinoline-3-carboxylate and ethyl S-(-)-6, 7-difluoro-1-fluoromethyl-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylate; so does (+)-prulifloxacin. The results also verify on the other hand that the absolute configuration of levo-prulifloxacin of the present invention is S type while the absolute configuration of dextro-prulifloxacin is R type.
-
Conclusion: The absolute configuration of the sample prepared in Example 3 is S configuration, as shown in the formula below:

Example 5 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 1.2 g (0.012 mol) of anhydrous potassium bicarbonate and 20 ml of dimethylsulfoxide were mixed and stirred. 0.012 mol of DMDO-Br in DMSO (5 mL) solution was added dropwise at -20 °C. Stirring proceeded at -20 °C for 3 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.5 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 54%.
Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
Example 6 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 1.04 g (0.008 mol) of N,N-diisopropylethylamine and 20 mL of N,N-dimethylformamide (DMF) was mixed and stirred, 0.008 mol of DMDO-Br in DMF (5 mL) solution was added thereinto. The solution was heating to 60 °C and reacted for 15 minutes. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.0 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 43%.
Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
Example 7 Preparation of S-(-)-prulifloxacin
-
10 g (0.029 mol) of S-(-)-uliflourxacin, 30 ml of N,N-dimethylacetylamide and 14.7 g (0.145mol) of triethylamine was mixed and cooled to 5~10 °C. 8.5 g (0.03 mol) 4-(p-toluenesulfonic acid-1-methyl ester)-5-methyl-1,3-dioxolen-2-one in 25 ml of N,N-dimethylacetylamide solution was added dropwise while stirring. After addition, the solution was reacted at room temperature for 10 hours. The reaction solution was poured into 200 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 7.46 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 57%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 8 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 0.79 g (0.05 mol) of potassium carbonate and 20 ml of dimethylformamide (DMF) was mixed and stirred. 0.012 mol of DMDO-Br in DMF (5ml) solution was added at -10 °C. At the same temperature, the solution was reacted for 2 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.2 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 48%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 9 Preparation of S-(-)-prulifloxacin
-
3.49 g (0.01 mol) of S-(-)-uliflourxacin, 0.79 g (0.02 mol) of diisopropylamine and 20 ml of dimethylformamide (DMF) was mixed and stirred. 0.02 mol of DMDO-Br in DMF (5ml) solution was added at 0 °C. At the same temperature, the solution was reacted for 2 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.5 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 54%. Specific rotation [α]20D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 10 Preparation of R-(+)-prulifloxacin
-
In accordance with the method as described in Example 5, the raw material R-(+)-prulifloxacin was prepared to 2.5 g of the target product R-(+)-prulifloxacin with a purity of 98% and a yield rate of 54%. Specific rotation [α]20 D= +108° (c=0.5, 0.1 mol/L methanesulfonic acid).
Example 11 Preparation of levo-prulifloxacin hydrochloride
-
0.5 g of S-(-)-prulifloxacin was dissolved in 15 mL of chloroform and then 0.5 mL of 33% (v/v) hydrochloric acid- methanol solution was added while stirring. The solution was filtered and the filtration residue was washed with methanol. The collected solid was dried to obtain 450 mg said compound with a yield rate of 83%. The melting point of the product is higher than 220 °C (the sample became darker during the test).
- S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl] -4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid hydrochloride
Example 12 Preparation of levo-prulifloxacin mesylate
-
0.5 g of S-(-)-prulifloxacin was dissolved in 15 mL of chloroform and then 0.5 mL of 50% methanesulfonic acid- methanol solution was added while stirring. The solution was filtered and the filtration residue was washed with methanol. The collected yellow solid was dried with calcium chloride under vacuum for 24 hours and further dried with calcium chloride at 80 °C under vacuum for 5 hours to obtain 470 mg said compound with a yield rate of 78%. The melting point of the product is higher than 220 °C (the sample became darker during the test).
- S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl] -4- oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylic acid mesylate
Example 13 Preparation of levo-prulifloxacin hydrochloride
-
0.5 g of S-(-)-prulifloxacin was dissolved in 15 mL of chloroform and then 0.5 mL of 33% (v/v) hydrochloric acid- methanol solution was added while stirring. The solution was dried by evaporation. Methanol was added to the residue and stirred for 10 minutes. The solution was filtered and the filtration residue was washed with methanol. The collected solid was dried to obtain 460 mg said compound with a yield rate of 85%.
-




Fandofloxacin In phase 2 by Dong Wha Pharmaceutical Co Ltd

DW-116; fandofloxacin
164150-99-6 FREE BASE ,
164151-00-2., 164150-85-0
6-fluoro-1-(5-fluoropyridin-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxoquinoline-3-carboxylic acid
6-Fluoro-1-(5-fluoropyridin-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Dong Wha Pharmaceutical Co Ltd
Molecular Formula: C20H18F2N4O3
Molecular Weight: 400.378726
synthesis………….http://www.drugfuture.com/synth/syndata.aspx?ID=226498
http://www.google.com.mx/patents/WO1995005373A1
synthesis 1

Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
synthesis 2
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
Synthesis, pharmacokinetics, and biological activity of a series of new pyridonecarboxylic acid antibacterial agents bearing a 5-fluoro-2-pyridyl group or a 3-fluoro-4-pyridyl group at N-1
J Heterocycl Chem 1997, 34(3): 1021
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
10-28-2005
|
Identification and use of effectors and allosteric molecules for the alteration of gene expression
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
4-20-2000
|
NOVEL QUINOLONE CARBOXYLIC ACID DERIVATIVES
|
|
|
3-6-1996
|
Quinolone carboxylic acid derivatives
|
|
|
2-24-1995
|
NOVEL QUINOLONE CARBOXYLIC ACID DERIVATIVES
|
ELLAGIC ACID A CANCER FIGHTING WONDER

ELLAGIC ACID
2,3,7,8-Tetrahydroxy-chromeno[5,4,3-cde]chromene-5,10-dione
as a very potent CK2 inhibitor

Ellagic acid is a natural phenol antioxidant found in numerous fruits and vegetables. The antiproliferative and antioxidant properties of ellagic acid have spurred preliminary research into the potential health benefits of ellagic acid consumption.
Ellagic acid is the dilactone of hexahydroxydiphenic acid.
Ellagic acid is an antioxidant and an anti-proliferative compound present in fruits, nuts and vegetables. In spite of evidences for anticancer activity in various cancer cell-lines, human cancer cells, the mechanistic role of ellagic acid is not conclusive enough to be recommended for a clinical use. The present review provides information about the chemopreventive role of ellagic acid in oral cancer and proposes molecular basis for ellagic acid’s inhibitory activity against oral cancer. We show that ellagic acid modulates growth of tumor cells through regulation of multiple cell signaling pathways including cell proliferation pathway (cyclin dependent kinase 2, cyclin A2, cyclin B1, cyclin D1, c-myc, PKCα), cell survival/apoptosis pathway (Bcl-XL, Bax, Caspase 9/3, Akt), tumor suppressor pathway (p53, p21), inflaming Metastasis pathways (IL-1 beta, TNF-α, matrix metalloproteinases 9/3, COX-2), angiogenesis pathways (VEGF), cell immortalization (TERT), NF-κβ.

Chinese medicine…Cordyceps ( dong chong xia cao ) 冬蟲草 དབྱར་རྩྭ་དགུན་འབུ་ ………..to treat many diseases related to lungs, kidney, and also used as a natural Viagra.

Ophiocordyceps sinensis (left) growing out of the head of a dead caterpillar
Ophiocordyceps sinensis is a fungus that parasitizes larvae of ghost moths and produces a fruiting body valued as an herbal remedy. The fungus germinates in the living larva, kills and mummifies it, and then the stalk-like fruiting body emerges from the corpse. It is known in English colloquially as caterpillar fungus, or by its more prominent foreign names (see below): yartsa gunbu or yatsa gunbu (Tibetan), or Dōng chóng xià cǎo (Chinese: 冬虫夏草; literally “winter worm, summer grass”). Of the various entomopathogenic fungi, Ophiocordyceps sinensis is one that has been used for at least 2000 years[2] to treat many diseases related to lungs, kidney, and also used as a natural Viagra. This fungus is not yet cultivated commercially,[3] despite the fact that several fermentable strains of Ophiocordyceps sinensis are isolated by Chinese Scientists.[4] Overharvesting and over exploitation have led to the classification of O. sinensis as an endangered species in China.[5] Additional research needs to be carried out in order to understand its morphology and growth habit for conservation and optimum utilization.

The moths in which O. sinensis grows are ambiguously referred to as “ghost moth”, which identifies either a single species or the genus Thitarodes, and the species parasitized by O. sinensis may be one of several Thitarodes that live on the Tibetan Plateau (Tibet, Qinghai, West-Sichuan, SW-Gansu & NW Yunnan), and the Himalayas (India, Nepal, Bhutan).
O. sinensis is known in the West as a medicinal mushroom, and its use has a long history in Traditional Chinese medicine as well as Traditional Tibetan medicine.[6] The hand-collected fungus-caterpillar combination is valued by herbalists and as a status symbol;[7] it is used as an aphrodisiac and treatment for ailments such as fatigue and cancer, although such use is mainly based on traditional Chinese medicine and anecdote. Recent research however seems to indicate a variety of beneficial effects in animal testing, including increased physical endurance through heightened ATP production in rats.[8]

| Cordyceps Sinensis |
| Cordyceps sinensis (Berk.) Sacc. and the usually the larvae are the remains of Hepialus varians |
| tonifies lung yin and kidney yang. For impotence, chronic lower back pain, afraid of cold, over abundance of mucus and tears, chronic cough and wheezing from deficiency, blood in phlegm from consumption due tokidney yang deficiency (shenyangxu). |
Cordyceps ( Dong Chong Cao ) 冬蟲草 Chinese Herbal Articles also known as chong cao, dong chong cao, yarsa gumba (Nepalese name of Tibetan origin), yartsa gunbu (dbyar rtswa dgun ‘bu) Tibetan name 蟲草, 冬蟲草. It belong to the “Ascomycetes or Clavicipitaceae” family.
Cordyceps ( Dong Chong Cao ) 冬蟲草 has a sweet, warm properties. It is use for treating the lung and kidney.

|
1. Improves auto-immune system.
2. Protects kidneys from toxins.
3. Protects kidneys from exhaustion.
4. Protects liver from toxins and treats and prevents cirrhosis of liver.
5. Protect the heart from the damaging effect of ouabain (C29H44O12.8H2O).
6. Anti-arrhythmia.
7. Anti-rejection effect in cornea transplant.
8. Antibiotic effect.
9. Inhibits contraction of smooth muscles.
|

Cordyceps ( Dong Chong Cao ) 冬蟲草 use in large dosages and/or long term usage can be toxic to kidneys.
According to the classics Medical Material, “Ben Cao Bei Yao” 本草備要, the best dong chong xia cao 冬蟲夏草, are produced in Sichuan. Today, most of them are produced in Xizang (Tibet) and Qinghai. Because the sizes the larvae are larger, they fetch higher prices.
According to the classics Medical Material, “Ben Cao Bei Yao” 本草備要, the best dong chong xia cao 冬蟲夏草, are produced in Sichuan. Today, most of them are produced in Xizang (Tibet) and Qinghai. Because the sizes the larvae are larger, they fetch higher prices.
Taxonomic History/ Systematics

Caterpillars with emergingOphiocordyceps sinensis
Morphological Features
Similar to other Cordyceps]] species, O. sinensis consists of two parts, a fungal endosclerotium (caterpillar) and stroma.[2] The stroma is the upper fungal part and is dark brown or black, but can be a yellow color when fresh and, longer than the caterpillar itself, usually 4–10 cm. It grows singly from the larval head, and is clavate, sublanceolate or fusiform and distinct from the stipe.[9] The stipe is slender, glabrous, and longitudinally furrowed or ridged. The fertile part of the stroma is the head. The head is granular due to the ostioles of the embedded perithecia.[2] The perithecia are ordinally arranged and ovoid [9] The asci are cylindrical or slightly tapering at both ends, and may be straight or curved, with a capitate and hemispheroid apex and may be two to four spored.[2] Similarly, ascospores are hyaline, filiform, multiseptate at a length of 5-12 um and subattenuated on both sides.[9] Perithecial, ascus and ascospore characters in the fruiting bodies are the key identification characteristics of O. sinensis. Ophiocordyceps (Petch) Kobayasi species produce whole ascospores and do not separate into part spores which is different from other Cordyceps species, which produce either immersed or superficial perithecia perpendicular to stromal surface and the ascospores at maturity are disarticulated into part spores.[10] Generally Cordyceps species possess brightly colored and fleshy stromata, but O. sinensis had dark pigments and tough to pliant stromata, a typical characteristic feature of most of the Ophiocordyceps species.[3]
Important developments in Classification
The species was first described scientifically by Miles Berkeley in 1843 as Sphaeria sinensis;[11] Pier Andrea Saccardo transferred the species to the genus Cordyceps in 1878.[12]The scientific name‘s etymology is from the Latin cord “club”, ceps “head”, and sinensis “from China“. The fungus was known as Cordyceps sinensis until 2007, when molecularanalysis was used to emend the classification of the Cordycipitaceae and the Clavicipitaceae, resulting in the naming of a new family Ophiocordycipitaceae and the transfer of several Cordyceps species to Ophiocordyceps.[10] Based on a molecular phylogenetic study, Sung et al. (2007) separated the megagenus Cordyceps into four genera as it was polyphyletic, viz. Cordyceps (40 spp.), Ophiocordyceps (146 spp.), Metacordyceps (6 spp.) and Elaphocordyceps (21 spp.), while the remaining 175 spp. were left in Cordyceps. As a result, C. sinensis was transferred to Ophiocordyceps, hence renamed as O. sinensis.[2]
Common Names[edit]
In Tibetan it is known as དབྱར་རྩྭ་དགུན་འབུ་ (ZYPY: yartsa gunbu, Wylie: dbyar rtswa dgun ‘bu, “summer grass winter bug”), which is the source of the Nepali यार्शागुम्बा, yarshagumba,yarchagumba or yarsagumba. The transliteration in Bhutan is Yartsa Guenboob. It is known as keera jhar, keeda jadi, keeda ghas or ‘ghaas fafoond in Hindi. Its name in Chinese Dōng chóng xià cǎo (冬蟲夏草) means “winter worm, summer grass” (i.e., “worm in the winter, [turns to] plant in the summer”). The Chinese name is a literal translation of the original Tibetan name, which was first recorded in the 15th Century by the Tibetan doctor Zurkhar Namnyi Dorje. In colloquial Tibetan Yartsa gunbu is often shortened to simply “bu” or “yartsa”.
In traditional Chinese medicine, its name is often abbreviated as chong cao (蟲草 “insect plant”), a name that also applies to other Cordyceps species, such as C. militaris. InJapanese, it is known by the Japanese reading of the characters for the Chinese name, tōchūkasō (冬虫夏草).
Strangely, sometimes in Chinese English language texts Cordyceps sinensis is referred to as aweto [Hill H. Art. XXXVI: The Vegetable Caterpillar (Cordiceps robertsii). Transactions and Proceedings of the Royal Society of New Zealand 1868-1961. Vol 34, 1901;396-401], which is the Māori name for Cordyceps robertsii, a species from New Zealand.
The English term “vegetable caterpillar” is a misnomer, as no plant is involved. “Caterpillar fungus” is a preferable term.
Nomenclature of the anamorph
Since the 1980s, 22 species in 13 genera have been attributed to the anamorph of O. sinensis. Of the 22 species, Cephalosporium acreomonium is the zygomycetous species ofUmbelopsis, Chrysosporium sinense has very low similarity in RAPD polymorphism, hence it is not the anamorph. Likewise, Cephalosporium dongchongxiacae, C. sp. sensu,Hirsutella sinensis and H. hepiali and Synnematium sinnense are synonymous and only H. sinensis is only validly published in articles. Cephalosporium sinensis possibly might be synonymous to H. sinensis but there is lack of valid information. Isaria farinose is combined to Paecilomyces farinosus and is not the anamorph. Several species like Isaria sp. Verticella sp. Scydalium sp. Stachybotrys sp. were identified only up to generic level, and thus it is dubious that they are anamorph. Mortierella hepiali is discarded as anamorph as it belongs to Zygomycota. Paecilomyces sinensis and Sporothrix insectorum are discarded based on the molecular evidence. P. lingi appeared only in one article and thus is discarded due to incomplete information. Tolypocladium sinense, P. hepiali, and Scydalium hepiali, have no valid information and thus are not considered as anamorph toOphiocordyceps sinensis. V. sinensis is not considered anamorph as there is no valid published information. Similarly, Metarhizium anisopliae is not considered anamorph as it has widely distributed host range, and is not restricted only in high altitude.[13] Thus Hirsutella sinensis is considered the validly published anamorph of O. sinensis. Cordyceps nepalensis and C. multiaxialis which had similar morphological characteristics to C. sinensis, also had almost identical or identical ITS sequences and its presumed anamorph, H. sinensis. This also confirms H. sinensis to be anamorph of O. sinensis and suggests C. nepalensis and C. multiaxialis are synonyms.[14] Evidence based on microcyclic conidiation from ascospores and molecular studies [2] support H. sinensis as the anamorph of the caterpillar fungus, O. sinensis.
Ecology
The caterpillars prone to infection by O. sinensis generally live 6 inches underground [4] in alpine grass and shrub-lands on the Tibetan Plateau and the Himalayas at an altitude between 3,000 and 5,000 m (9,800 and 16,400 ft). The fungus is reported from the northern range of Nepal, Bhutan, and also from the northern states of India, apart from northern Yunnan, eastern Qinghai, eastern Tibet, western Sichuan, southwestern Gansu provinces.[4] The fungus consumes its host from inside out as they hibernate in alpine meadows. Usually the larvae are more vulnerable after shedding their skin, during late summer. The fungal fruiting body disperses spores which infect the caterpillar. The infected larvae tend to remain vertical to the soil surface with their heads up. The fungus then germinates in the living larva, kills and mummifies it, and then the stalk-like fruiting body emerges from the head and the fungus finally emerges from the soil by early spring.[15] Fifty-seven taxa from seven genera (1 Bipectilus, 1 Endoclita, 1 Gazoryctra, 12 Hepialus, 2Magnificus, 3 Pharmacis, and 37 Thitarodes [3]) are recognized as potential hosts of O. sinensis.
Reproduction Biology
Ophiocordyceps sinensis has both teleomorphic and anamorphic phases. Spending up to five years underground before pupating, the Thitarodes caterpillar is attacked while feeding on roots. It is not certain how the fungus infects the caterpillar; possibly by ingestion of a fungal spore or by the fungus mycelium invading the insect through one of the insect’s breathing pores. The dark brown to black fruiting body (or mushroom) emerges from the ground in spring or early summer, the long, usually columnar fruiting body reaches 5–15 cm above the surface and releases spores.
In late autumn, chemicals on the skin of the caterpillar interact with the fungal spores and release the fungal mycelia, which then infects the caterpillar.[4] After invading a host larva, the fungus ramifies throughout the host and eventually kills it. Gradually the host larvae become rigid due to the production of fungal sclerotia. Fungal sclerotia are multihyphal structures that can remain dormant and then germinate to produce spores. After over-wintering, the fungus ruptures the host body, forming a sexual sporulating structure (a perithecial stroma) from the larval head in summer that is connected to the sclerotia (dead larva) below ground and grows upward to emerge from the soil.[16] The slow growing O. sinensis grows at a comparatively low temperature, i.e., below 21oC. Temperature requirements and growth rates are crucial factors that identify O. sinensis from other similar fungi.[3]
Use in medicine
It is used as a curative to many diseases, anti- aging,[17] hypoglycemic,[18] aphrodisiac and also treatment against cancer. Ophiocordyceps sinensis serves against kidney and lung problems and stimulates the immune system; it is used for treatment of fatigue, night sweating, respiratory disease, hyperglycemia, hyperlipidemia, asthenia after severe illness, arrhythmias and other heart diseases and liver disease.[4]
Traditional Asian medicines

Medicinal use of the caterpillar fungus apparently originated in Tibet and Nepal. So far the oldest known text documenting its use was written in the late fourteen hundreds by the Tibetan doctor Zurkhar Nyamnyi Dorje (Wylie: Zur mkhar mnyam nyid rdo rje)[1439-1475]) in his text: Man ngag bye ba ring bsrel (“Instructions on a Myriad of Medicines”). A translation is available at Winkler.[19]
The first mention of Ophiocordyceps sinensis in traditional Chinese Medicine was in Wang Ang’s 1694 compendium of materia medica, Ben Cao Bei Yao.[20] In the 18th Century it was listed in Wu Yiluo‘s Ben cao cong xin (“New compilation of materia medica”).[21] No sources have been published to uphold widespread claims of “thousands of years of use in Chinese medicine” or use of “chong cao since the 7th Century Tang Dynasty in China”. The ethno-mycological knowledge on caterpillar fungus among the Nepalese people is documented byDevkota(2006) The entire fungus-caterpillar combination is hand-collected for medicinal use.
The fungus is a medicinal mushroom which is highly prized by practitioners of Tibetan medicine, Chinese medicine and traditional Folk medicines, in which it is used as an aphrodisiac and as a treatment for a variety of ailments from fatigue to cancer. In Chinese medicine it is regarded as having an excellent balance of yin and yang as it is apparently both animal and vegetable. Assays have found thatOphiocordyceps species produce many pharmacologically active substances. They are now cultivated on an industrial scale for their medicinal value. However, no one has succeeded so far in growing the larva cum mushroom artificially. The biological process that forms the Ophiocordyceps is still unknown and true cultivation has yet to be realized.[3] All artificial products are derived from mycelia grown on grains or in liquids.
According to Bensky et al. (2004), laboratory-grown C. sinensis mycelia have similar clinical efficacy and less associated toxicity. He notes a toxicity case of constipation, abdominal distension, and decreased peristalsis, two cases of irregular menstruation, and one case report ofamenorrhea following ingestion of tablets or capsules containing C. sinensis. In Chinese medicine C. sinensis is considered sweet and warm, entering the lung and kidney channels; the typical dosage is 3–9 grams.[22]
Research

Cordycepin, a compound isolated from the “Caterpillar fungus”.
Some work has been published in which Ophiocordyceps sinensis has been used to protect the bone marrow and digestive systems ofmice from whole body irradiation.[23] An experiment noted Ophiocordyceps sinensis may protect the liver from damage.[24] An experiment conducted with mice noted the mushroom may have an anti-depressant effect.[25] Researchers have noted that the caterpillar fungus has ahypoglycemic effect and may be beneficial for people with insulin resistance.[26][27][28][29][30] There is also experimental evidence of the supposed energizing effect of the fungus, as it has been shown to increase endurance through heightened ATP production in rats.[8]
A March 2013 study on Cordyceps Sinensis documented the medicinal fungus’ anti-inflammatory properties.[31] Scientists were able to show Cordyceps Sinensis’ ability to suppress interleukin-1b and interleukin-18 secretion by inhibiting both canonical and non-canonical inflammasomes. Inflammasomes have long been associated with auto-inflammatory diseases, such as gout. The study used a specific anamorphic mycelial form of Cordyceps Sinensis known as Hirsutella Sinensis.
Introduction to the Western world
| ) |

Ophiocordyceps sinensis
The Western world was largely unaware of Ophiocordyceps prior to 1993. The fungus dramatically caught the world’s eye due to the performance of three female Chinese athletes, Wang Junxia, Qu Yunxia, and Zhang Linli. These athletes broke five world records for 1,500, 3,000 and 10,000 meter dashes at the National Games in Beijing, China. The number of new world records set at a single track event attracted much attention and suspicion. Following the races, the women were expected by some to fail drug tests for anabolic steroids. However, the athletes’ tests revealed no illegal substances, and coach Ma Junren told the reporters that the runners were takingOphiocordyceps sinensis and turtle blood at his request. However for the 2000 Sydney Olympics, Ma Junren withdrew some of his athletes at the last minute. It was speculated that a new doping test would have revealed illegal substances, thus half a dozen Chinese field and track athletes were left at home.
Economics and impact

Many shops in downtown Lanzhouadvertise Dōng chóng xià cǎo (冬虫夏草) among other local specialties.
In rural Tibet, yartsa gunbu has become the most important source of cash income. The fungi contributed 40% of the annual cash income to local households and 8.5% to the GDP in 2004. Prices have increased continuously, especially since the late 1990s. In 2008, one kilogram traded for US$3,000 (lowest quality) to over US$18,000 (best quality, largest larvae). The annual production on the Tibetan Plateau was estimated in 2009 at 80–175 tons.[32] The Himalayan Ophiocordyceps production might not exceed a few tons.
In 2004 the value of a kilogram of caterpillars was estimated at about 30,000 to 60,000 Nepali rupees in Nepal, and about Rs 100,000 in India.[33] In 2011 the value of a kilogram of caterpillars was estimated at about 350,000 to 450,000 Nepali rupees in Nepal. A 2012 BBC article indicated that in north Indian villages a single fungus was worth Rs 150 (about £2 or $3), which is more than the daily wage of a manual laborer.[34]
According to Daniel Winkler, the price of Ophiocordyceps sinensis has risen dramatically on the Tibetan Plateau, basically 900% between 1998 and 2008, an annual average of over 20% (after inflation). However, the value of big sized caterpillar fungus has increased more dramatically than smaller size Cordyceps, regarded as lower quality.[20]
| Year | % Price Increase | Price/kg (Yuan) |
|---|---|---|
| 1980s | 1,800 | |
| 1997 | 467% (incl. inflation) | 8,400 |
| 2004 | 429% (incl. inflation) | 36,000 |
| 2005 | 10,000–60,000 | |
| 2013 | 125,000–500,000 |
Because of its high value, inter-village conflicts over access to its grassland habitats has become a headache for the local governing bodies and in several cases people were killed. In November 2011, a court in Nepal convicted 19 villagers over the murder of a group of farmers during a fight over the prized aphrodisiac fungus. Seven farmers were killed in the remote northern district of Manang in June 2009 after going to forage for Yarchagumba. [35]
Its value gave it a role in the Nepalese Civil War, as the Nepalese Maoists and government forces fought for control of the lucrative export trade during the June–July harvest season.[36] Collecting yarchagumba in Nepal had only been legalised in 2001, and now demand is highest in countries such as China, Thailand, Vietnam, Korea and Japan. By 2002, the herb was valued at R 105,000 ($1,435) per kilogram, allowing the government to charge a royalty of R 20,000 ($280) per kilogram.
The search for Ophiocordyceps sinensis is often perceived to pose a threat to the environment of the Tibetan Plateau where it grows. While it has been collected for centuries and is still common in such areas, current collection rates are much higher than in historical times.
Ophiocordyceps producers like to perpetuate the story that unscrupulous harvesters insert twigs into the ascocarps of wild C. sinensis to increase their weight and therefore the price paid. A tiny twig is only used when the ascocarp is broken from the caterpillar, and has nothing to do with artificially increasing weight. Supposedly, at some point in the past, someone inserted lead wires with which to increase weight; however, each year hundreds of millions of specimens are harvested and this appears to have been a one-time occurrence.
Cultivated C. sinensis mycelium is an alternative to wild-harvested C. sinensis, and producers claim it may offer improved consistency. Artificial culture of C. sinensis is typically by growth of pure mycelia in liquid culture (in China) or on grains (in the West). The first time in Vietnam, Prof. Aca. Dr. Dai Duy Ban together with scientists and DAIBIO Company and DAIBIO Great Traditional Medicine Family Clinic discovered the Cordyceps sinensis as Isaria cerambycidae N.SP. to develop Fermentation DAIBIO Cordyceps Sinensis.[37]Ascocarps are not produced through in vitro cultivation.
References
- “Ophiocordyceps sinensis (Berk.) G.H. Sung, J.M. Sung, Hywel-Jones & Spatafora 2007″. MycoBank. International Mycological Association. Retrieved 2011-07-19.
- Shrestha, B., Weimin, Z., Yongjie, Z., & Xingzhong, L. (2010). What is the Chinese caterpillar fungus Ophiocordyceps sinensis (Ophiocordycipitaceae)?. Mycology: An International Journal On Fungal Biology, 1(4), 228-236. doi:10.1080/21501203.2010.536791.
- Hsieh, C., et al., A Systematic Review of the Mysterious Caterpillar Fungus Ophiocordyceps sinensis in Dong-ChongXiaCao and Related Bioactive Ingredients. Vol. 3. 2013. 16-32.
- Zhu JS, Halpem GM, Jones K. 1998. The scientific rediscovery of an ancient Chinese herbal medicince: Cordyceps sinensis. I. J Alt Complem Med 4:289-303.
- Xiao-Liang, W., & Yi-Jian, Y. (2011). Host insect species of Ophiocordyceps sinensis: a review. Zookeys, 12743-59. doi:10.3897/zookeys.127.802
- Halpern, Miller (2002). Medicinal Mushrooms. New York, New York: M. Evans and Company, Inc. pp. 64–65. ISBN 0-87131-981-0
- http://www.npr.org/2011/10/09/141164173/caterpillar-fungus-the-viagra-of-the-himalayas
- ^ Jump up to:a b Rajesh Kumar, P.S. Negi, Bhagwat Singh, Govindasamy Ilavazhagan, Kalpana Bhargava, Niroj Kumar Sethy (2011). “Cordyceps sinensis promotes exercise endurance capacity of rats by activating skeletal muscle metabolic regulators”. Journal of Ethnopharmacology 136: 260–266.
- Sung, G. H., et al. (2007). “A multi-gene phylogeny of Clavicipitaceae (Ascomycota, Fungi): identification of localized incongruence using a combinational bootstrap approach.” Molecular Phylogenetics and Evolution 44(3): 1204-1223.
- Sung GH, Hywel-Jones NL, Sung JM, Luangsa-Ard JJ, Shrestha B, Spatafora JW. (2007). “Phylogenetic classification of Cordyceps and the clavicipitaceous fungi”.Studies in Mycology 57: 5–59. doi:10.3114/sim.2007.57.01. PMC 2104736.PMID 18490993.
- Berkeley MJ. (1843). “On some entomogenous Sphaeriae”. London Journal of Botany 2: 205–11.
- Saccardo PA. (1878). “Enumeratio Pyrenomycetum Hypocreaceorum hucusque cognitorum systemate carpologico dispositorum” (PDF). Michelia (in Latin) 1 (3): 277–325.
- Jiang, Y. Y., & Yao, Y. J. (n.d). Names related to Cordyceps sinensis anamorph. Mycotaxon, 84245-254.
- Liu, Z., Liang, Z., Liu, A., Yao, Y., Hyde, K. D., & Yu, Z. (n.d). Molecular evidence for teleomorph-anamorph connections in Cordyceps based on ITS-5.8S rDNA sequences. Mycological Research, 106(9), 1100-1108.
- Stone, R. (2008). Last Stand for the Body Snatcher of the Himalayas?. Science, (5905), 1182. doi:10.2307/20145300
- Xing, X. K., & Guo, S. X. (2008). The Structure and Histochemistry of Sclerotia of Ophiocordyceps sinensis. Mycologia, (4), 616. doi:10.2307/20444986.
- Ji DB, Ye J, Li CL, Wang YH, Zhao J, Cai SQ (2009) Antiaging effect of Cordyceps sinensis extract. Phytotherapy Research 23 (1): 116-122. Doi: 10.1002/ptr.2576
- Zhang GQ, Huang YD, Bian Y, Wong JH, Ng TB, Wang HX (2006) Hypoglycemic activity of the fungus Cordyceps militaris, Cordyceps sinensis, Tricholoma mongolicum and Omphalia lapidescens in streptozotocin-induced diabetic rats. Applied Microbiology and Biotechnology 72 (6): 1152-1156. Doi: 10.1007/s00253-006-0411-9.
- Winkler D. (2008). “The mushrooming fungi market in Tibet exemplified by Cordyceps sinensis and Tricholoma matsutake“. Journal of the International Association of Tibetan Studies. In: In the Shadow of the Leaping Dragon: Demography, Development, and the Environment in Tibetan Areas (4).
- Winkler D. (2008). “Yartsa Gunbu (Cordyceps sinensis) and the fungal commodification of the rural economy in Tibet AR”. Economic Botany 62 (3): 291–305.doi:10.1007/s12231-008-9038-3.
- Wu Y (1757). “Ben cao cong xin” – “New compilation of materia medica” (in Chinese).
- Jump up^ Bensky D, Gamble A, Clavey S, Stöger E, Bensky L. Lai (2004). Materia Medica: Chinese Herbal Medicine (3rd ed.). Seattle, Washington: Eastland Press. ISBN 978-0-939616-42-8.
- Liu W-C, Wang S-C, Tsai M-L, Chen, M-C, Wang Y-C, Hong J-H, McBride WH, Chiang C-S. (2006). “Protection against radiation-induced bone marrow and intestinal injuries byCordyceps sinensis, a Chinese herbal medicine”. Radiation Research 166 (6): 900–907.doi:10.1667/RR0670.1. PMID 17149981.
- WS, Hsu SL, Chyau CC, Chen KC, Peng RY. (July 2009). “Compound Cordyceps TCM-700C exhibits potent hepatoprotective capability in animal model”. Fitoterapia 81(1): 1–7. doi:10.1016/j.fitote.2009.06.018. PMID 19596425.
- Nishizawa K, Torii K, Kawasaki A, et al. (2007). “Antidepressant-like effect ofCordyceps sinensis in the mouse tail suspension test”. Biological and Pharmaceutical Bulletin 30 (9): 1758–62. doi:10.1248/bpb.30.1758. PMID 17827735.
- Kiho T, Hui J, Yamane A, Ukai S. (1993). “Polysaccharides in fungi. XXXII. Hypoglycemic activity and chemical properties of a polysaccharide from the cultural mycelium of Cordyceps sinensis“. Biological and Pharmaceutical Bulletin 16 (12): 1291–3. doi:10.1248/bpb.16.1291. PMID 8130781.
- Kiho T, Yamane A, Hui J, Usui S, Ukai S. (1996). “Polysaccharides in fungi. XXXVI. Hypoglycemic activity of a polysaccharide (CS-F30) from the cultural mycelium of Cordyceps sinensis and its effect on glucose metabolism in mouse liver”. Biological and Pharmaceutical Bulletin 19 (2): 294–6. doi:10.1248/bpb.19.294. PMID 8850325.
- Zhao CS, Yin WT, Wang JY, et al. (2002). “CordyMax Cs-4 improves glucose metabolism and increases insulin sensitivity in normal rats”. Journal of Alternative and Complementary Medicine 8 (3): 309–14. doi:10.1089/10755530260127998.PMID 12165188.
- Lo HC, Tu ST, Lin KC, Lin SC. (2004). “The anti-hyperglycemic activity of the fruiting body of Cordyceps in diabetic rats induced by nicotinamide and streptozotocin”. Life Sciences 74 (23): 2897–908. doi:10.1016/j.lfs.2003.11.003. PMID 15050427.
- Li SP, Zhang GH, Zeng Q, et al. (2006). “Hypoglycemic activity of polysaccharide, with antioxidation, isolated from cultured Cordyceps mycelia”. Phytomedicine 13 (6): 428–33.doi:10.1016/j.phymed.2005.02.002. PMID 16716913.
- Huang, T. et al. (March 2013). “Hirsutella sinensis mycelium suppresses interleukin-1b and interleukin-18 secretion by inhibiting both canonical and non-canonical inflammasomes.” (PDF). Scientific Report. 3, 1374;.
- Winkler, D. (2009). “Caterpillar Fungus (Ophiocordyceps sinensis) Production and Sustainability on the Tibetan Plateau and in the Himalayas”. Asian Medicine 5 (2): 291. doi:10.1163/157342109X568829.
- Sharma S. (2004). “Trade of Cordyceps sinensis from high altitudes of the Indian Himalaya: Conservation and biotechnological priorities” (PDF). Current Science 86(12): 1614–9.
- Jeffrey, Craig (2012-07-07). “The ‘Viagra’ transforming local economies in India”. BBC News. Retrieved July 9, 2012.
- Staff (14 November 2011) ‘Himalayan viagra’: Six men get life for Nepal murders BBC News Asia, Retrieved 9 July 2012
- Baral N, Heinen JT. (2005). “The Maoist people’s war and conservation in Nepal”.Politics and the Life Sciences 24 (1): 2–11. doi:10.2990/1471-5457(2005)24[2:TMPWAC]2.0.CO;2.
- DAIBIO Cordyceps Sinensis in Vietnam
- Winkler, D. 2005. Yartsa Gunbu – Cordyceps sinensis. Economy, Ecology & Ethno-mycology of a Fungus Endemic to the Tibetan Plateau. In: A.BOESI & F. CARDI (eds.). Wildlife and plants in traditional and modern Tibet: Conceptions, Exploitation and Conservation. Memorie della Società Italiana di Scienze Naturali e del Museo Civico di Storia Naturale di Milano, Vol. 33.1:69–85.
- Zhang Y., Zhang S., Wang M., Bai F. & Liu X. (2010). “High Diversity of the Fungal Community Structure in Naturally-Occurring Ophiocordyceps sinensis“. PLoS ONE 5(12): e15570. doi:10.1371/journal.pone.0015570.
External links
Yartsa Gunbu (Cordyceps sinensis) in Tibet
- Daniel Winkler’s Cordyceps blog
- Nepal’s Nature – The Himalayan Viagra
- Page at Everything2.com
- Image gallery of Cordyceps sinensis
- The first time in Vietnam, Prof.Aca.D.Sc Dai Duy Ban with his scientists discovered Cordyceps Sinensis as Isaria cerambycidae N.SP. and Fermentation Daibio Cordyceps Sinensis by Daibio Great Family Traditional Medicine Clinic Company
- Daibio Cordyceps Sinensis in Vietnam
- An Electronic Monograph of Cordyceps and Related Fungi
- Cordyceps information from Drugs.com
- Cordyceps sinensis (Berk.) Sacc. Medicinal Plant Images Database (School of Chinese Medicine, Hong Kong Baptist University) (English) (traditional Chinese)
- Chinese Caterpiller Fungus Chinese Medicine Specimen Database (School of Chinese Medicine, Hong Kong Baptist University) (English) (traditional Chinese)
- Tibet’s Golden “Worm” August 2012 National Geographic (magazine)
