PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).
The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.
14
Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N
2) DBU
P-1 P-2 P-3
a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
†Global API Chemistry, ‡MDR Chemical Science,§Analytical Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
A novel synthesis of GSK1265744, a potent HIV integrase inhibitor, is described. The synthesis is highlighted by an efficient construction of the densely functionalized pyridinone core as well as a highly diastereoselective formation of the acyl oxazolidine moiety. The latter exploits the target molecule’s ability to chelate to Mg2+, a key feature in the integrase inhibitor’s mechanism of action.
Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug
^“Adopted USANs”(PDF). American Medical Association. Retrieved 19 September 2014.
^World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).
SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).
Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).
Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).
MOLECULAR FORMULA C17H11ClF3N5O3
MOLECULAR WEIGHT 425.7
Merck Sharp & Dohme Corp
reverse transcriptase inhibitor
Doravirine (MK-1439) is a non-nucleoside reverse transcriptase inhibitor under development by Merck & Co. for use in the treatment of HIV infection. Doravirine demonstrated robust antiviral activity and good tolerability in a small clinical study of 7-day monotherapy reported at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013. Doravirine appeared safe and generally well tolerated with most adverse events being mild-to-moderate.[1][2]
investigational next-generation, non-nucleoside reverse transcriptase inhibitor (NNRTI), at the 21st Conference on Retroviruses and Opportunistic Infections (CROI). Interim data demonstrating potent antiretroviral (ARV) activity for four doses (25, 50, 100 and 200 mg) of once-daily, oral doravirine in combination with tenofovir/emtricitabine in treatment-naïve, HIV-1 infected adults after 24 weeks of treatment were presented during a late-breaker oral session. Based on these findings as well as other data from the doravirine clinical program, Merck plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
“Building on our long-standing commitment to the HIV community, Merck continues to evaluate new candidates we believe have the potential to make a meaningful difference in the lives of HIV patients,” said Daria Hazuda, Ph.D., vice president, Infectious Diseases, Merck Research Laboratories. “We look forward to advancing doravirine into Phase 3 clinical trials in the second half of 2014.”
Doravirine Clinical Data
This randomized, double-blind clinical trial examined the safety, tolerability and efficacy of once-daily doravirine (25, 50, 100 and 200 mg) in combination with once-daily tenofovir/emtricitabine versus efavirenz (600 mg), in treatment-naïve, HIV-1 infected patients. The primary efficacy analysis was percentage of patients achieving virologic response (< 40 copies/mL).
At 24 weeks, doravirine doses of 25, 50, 100, and 200 mg showed virologic response rates consistent with those observed for efavirenz at a dose of 600 mg. All treatment groups showed increased CD4 cell counts.
Proportion of Patients with Virologic
Response at 24 weeks (95% CI)
Mean CD4 Change
from Baseline (95% CI)
Treatment*
Dose (mg)
n/N
% <40
copies/mL
cells/μL
Doravirine
25
32/40
80.0 (64.6, 90.9)
158 (119, 197)
50
32/42
76.2 (60.5, 87.9)
116 (77, 155)
100
30/42
71.4 (55.4, 84.3)
134 (100, 167)
200
32/41
78.0 (62.4, 89.4)
141 (96, 186)
Efavirenz
600
27/42
64.3 (48.0, 78.4)
121 (73, 169)
Missing data approach:
Non-completer = Failure
Observed Failure
*In combination with tenofovir/emtricitabine
The incidence of drug-related adverse events was comparable among the doravirine-treated groups. The overall incidence of drug-related adverse events was lower in the doravirine-treated groups (n=166) than the efavirenz-treated group (n=42), 35 percent and 57 percent, respectively. The most common central nervous system (CNS) adverse events at week 8, the primary time point for evaluation of CNS adverse experiences, were dizziness [3.0% doravirine (overall) and 23.8% efavirenz], nightmare [1.2% doravirine (overall) and 9.5% efavirenz], abnormal dreams [9.0% doravirine (overall) and 7.1% efavirenz], and insomnia [5.4% doravirine (overall) and 7.1% efavirenz].
Based on the 24-week data from this dose-finding study, a single dose of 100 mg doravirine was chosen to be studied for the remainder of this study, up to 96 weeks.
About Doravirine
DORAVIRINE
Doravirine, also known as MK-1439, is an investigational next-generation, NNRTI being evaluated by Merck for the treatment of HIV-1 infection. In preclinical studies, doravirine demonstrated potent antiviral activity against HIV-1 with a characteristic profile of resistance mutations selected in vitro compared with currently available NNRTIs. In early clinical studies, doravirine demonstrated a pharmacokinetic profile supportive of once-daily dosing and did not show a significant food effect.
Merck’s Commitment to HIV
For more than 25 years, Merck has been at the forefront of the response to the HIV epidemic, and has helped to make a difference through our proud legacy of commitment to innovation, collaborating with the community, and expanding global access to medicines. Merck is dedicated to applying our scientific expertise, resources and global reach to deliver healthcare solutions that support people living with HIV worldwide.
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses
Bioorg Med Chem Lett 2014, 24(3): 917
The optimization of a novel series of non-nucleoside reverse transcriptase inhibitors (NNRTI) led to the identification of pyridone 36. In cell cultures, this new NNRTI shows a superior potency profile against a range of wild type and clinically relevant, resistant mutant HIV viruses. The overall favorable preclinical pharmacokinetic profile of 36 led to the prediction of a once daily low dose regimen in human. NNRTI 36, now known as MK-1439, is currently in clinical development for the treatment of HIV infection.
Scheme 1.
Reagents and conditions: (a) K2CO3, NMP, 120 °C; (b) KOH, tert-BuOH, 75 °C; (c) Zn(CN)2, Pd(PPh3)4, DMF, 100 °C.
Scheme 3.
Reagents and conditions: (a) K2CO3, DMF, −10 °C; (b) MeI or EtI, K2CO3, DMF.
Scheme I depicts a method for preparing compounds of Formula I in which hydroxypyridine 1-1 is alkylated with chlorotriazolinone 1-2 to provide 1-3 which can be selectively alkylated with an alkyl halide (e.g., methyl iodide, ethyl iodide, etc.) to afford the desired 1-4. Scheme I
Scheme II depicts an alternative route to compounds of the present invention, wherein fluorohydroxypyridine II-l can be alkylated with chlorotriazolinone II-2 to provide the alkylated product II-3 which can be converted to the desired II-5 via nucleophilic aromatic substitution (S] fAr) using a suitable hydroxyarene II-4.
Scheme II
Hydroxypyridines of formula I-l (Scheme 1) can be prepared in accordance with Scheme III, wherein a SNAr reaction between pyridine III-l (such as commercially available 2- chloro-3-fluoro-4-(trifluoromethyl)pyridine) and hydroxyarene H-4 can provide chloropyridine III-2, which can be hydrolyzed under basic conditions to the hydroxypyridine I-l. Scheme III
Another method for preparing hydroxypyridines of formula I-l is exemplified in Scheme IV, wherein S Ar coupling of commercially available 2-chloro-3-fluoro-4- nitropyridone-N-oxide IV-1 with a suitable hydroxyarene II-4 provides N-oxide IV-2, which can first be converted to dihalides IV-3 and then hydro lyzed to hydroxypyridine IV-4. Further derivatization of hydroxypyridine IV-4 is possible through transition metal-catalyzed coupling processes, such as Stille or boronic acid couplings using a PdLn catalyst (wherein L is a ligand such as triphenylphosphine, tri-tert-butylphosphine or xantphos) to form hydroxypyridines IV-5, or amination chemistry to form hydroxypyridines IV-6 in which R2 is N(RA)RB.
Scheme IV
IV-1
– – Scheme V depicts the introduction of substitution at the five-position of the hydroxypyridines via bromination, and subsequent transition metal-catalyzed chemistries, such as Stille or boronic acid couplings using PdLn in which L is as defined in Scheme IV to form hydroxypyridines V-3, or amination chemistry to form hydroxypyridines V-4 in which R3 is N(RA)RB.
Scheme V
As shown in Scheme IV, fiuorohydroxypyridines II-l (Scheme II) are available from the commercially available 3-fluoroypridines VI- 1 through N-oxide formation and rearrangement as described in Konno et al., Heterocycles 1986, vol. 24, p. 2169.
Scheme VI
The following examples serve only to illustrate the invention and its practice. The examples are not to be construed as limitations on the scope or spirit of the invention.
The term “room temperature” in the examples refers to the ambient temperature which was typically in the range of about 20°C to about 26°C.
A mixture of the 3-bromo-5-chlorophenol (3.74 g; 18.0 mmol), 2-chloro-3-fluoro- 4-(trifluoromethyl)pyridine (3.00 g; 15.0 mmol) and 2CO3 (2.49 g; 18.0 mmol) in NMP (15 mL) was heated to 120°C for one hour, then cooled to room temperature. The mixture was then diluted with 250 mL EtOAc and washed with 3 x 250 mL 1 :1 H20:brine. The organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; load with toluene; 100:0 to 0:100 hexanes:CH2Cl2 over 40 minutes) provided title compound (1-2) as a white solid. Repurification of the mixed fractions provided additional title compound. lH NMR (400 MHz, CDCI3): δ 8.55 (d, J = 5.0 Hz, 1 H); 7.64 (d, J = 5.0 Hz, 1 H);
To a suspension of 3-(3-bromo-5-chlorophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1-2; 3.48 g; 8.99 mmol) in lBuOH (36 mL) was added KOH (1.51 g; 27.0 mmol) and the mixture was heated to 75°C overnight, at which point a yellow oily solid had precipitated from solution, and LCMS analysis indicated complete conversion. The mixture was cooled to room temperature, and neutralized by the addition of -50 mL saturated aqueous NH4CI. The mixture was diluted with 50 mL H2O, then extracted with 2 x 100 mL EtOAc. The combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90: 10 CH2Cl2:MeOH over 40 minutes) provided the title compound (1-3) as a fluffy white solid. lH NMR (400 MHz, DMSO): δ 12.69 (s, 1 H); 7.59 (d, J = 6.9 Hz, 1 H); 7.43 (t, J = 1.7 Hz, 1 H); 7.20 (t, J = 1.9 Hz, 1 H); 7.13 (t, J = 2.0 Hz, 1 H); 6.48 (d, J = 6.9 Hz, 1 H).
To a suspension of 3-(3-bromo-5-chlorophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1-3; 3.25 g; 8.82 mmol) in NMP (29 mL) was added CuCN (7.90 g; 88 mmol) and the mixture was heated to 175°C for 5 hours, then cooled to room temperature slowly. With increased fumehood ventilation, 100 mL glacial AcOH was added, then 100 mL EtOAc and the mixture was filtered through Celite (EtOAc rinse). The filtrate was washed with 3 x 200 mL 1 : 1 H20:brine, then the organic extracts were dried (Na2S04) and concentrated in vacuo.
Purification by ISCO CombiFlash (120 g column; dry load; 100:0 to 90:10 CH2Cl2:MeOH over 40 minutes), then trituration of the derived solid with Et20 (to remove residual NMP which had co-eluted with the product) provided the title compound (1-4). lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
The title compound was prepared as described in the literature: Cowden, C. J.; Wilson, R. D.; Bishop, B. C; Cottrell, I. F.; Davies, A. J.; Dolling, U.-H. Tetrahedron Lett. 2000, 47, 8661.
A suspension of the 3-chloro-5-{[2-hydroxy-4-(trifluoromethyl)pyridin-3- yl]oxy}benzonitrile (1-4; 2.00 g; 6.36 mmol), 5-(chloromethyl)-2,4-dihydro-3H-l,2,4-triazol-3- one (1-5; 0.849 g; 6.36 mmol) and K2CO3 (0.878 g; 6.36 mmol) in DMF (32 mL) was stirred for 2 hours at room temperature, at which point LCMS analysis indicated complete conversion. The mixture was diluted with 200 mL Me-THF and washed with 150 mL 1 : 1 : 1 H20:brine:saturated aqueous NH4CI, then further washed with 2 x 150 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 150 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. Purification by ISCO CombiFlash (80 g column; dry load; 100:0 to 90:10 EtOAc:EtOH over 25 minutes) provided the title compound (1-6) as a white solid. lH NMR (400 MHz, DMSO): δ 1 1.46 (s, 1 H); 1 1.39 (s, 1 H); 7.93 (d, J = 7.3 Hz, 1 H); 7.76 (s, 1 H); 7.58 (s, 1 H); 7.51 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.02 (s, 2 H).
A solution of 3-chloro-5-({2-oxo-l -[(5-oxo-4,5-dihydro-lH-l,2,4-triazol-3- yl)methyl]- 4-(trifluoromethyl)-l ,2-dihydropyridin-3-yl}oxy)benzonitrile (1-6; 2.37 g; 5.76 mmol) and K2CO3 (0.796 g; 5.76 mmol) in DMF (58 mL) was cooled to 0°C, then methyl iodide (0.360 mL; 5.76 mmol) was added. The mixture was allowed to warm to room
temperature, and stirred for 90 minutes, at which point LCMS analysis indicated >95%
conversion, and the desired product of -75% LCAP purity, with the remainder being unreacted starting material and 6/s-methylation products. The mixture was diluted with 200 mL Me-THF, and washed with 3 x 200 mL 1 : 1 H20:brine. The aqueous fractions were further extracted with 200 mL Me-THF, then the combined organic extracts were dried (Na2S04) and concentrated in vacuo. The resulting white solid was first triturated with 100 mL EtOAc, then with 50 mL THF, which provided (after drying) the title compound (1-1) of >95% LCAP. Purification to >99% LCAP is possible using Prep LCMS (Max-RP, 100 x 30 mm column; 30-60% CH3CN in 0.6% aqueous HCOOH over 8.3 min; 25 mL/min). lH NMR (400 MHz, DMSO): δ 1 1.69 (s, 1 H); 7.88 (d, J = 7.3 Hz, 1 H); 7.75 (s, 1 H); 7.62 (s, 1 H); 7.54 (s, 1 H); 6.67 (d, J = 7.3 Hz, 1 H); 5.17 (s, 2 H); 3.1 1 (s, 3 H). EXAMPLE 1A
A mixture of the 3-chloro-l-iodophenol (208 g; 816.0 mmol), 2-chloro-3-fluoro-
4-(trifluoromethyl)pyridine (155 g; 777.0 mmol) and K2CO3 (161 g; 1 165.0 mmol) in NMP (1.5 L) was held at 60°C for 2.5 hours, and then left at room temperature for 2 days. The mixture was then re-heated to 60°C for 3 hours, then cooled to room temperature. The mixture was then diluted with 4 L EtOAc and washed with 2 L water + 1 L brine. The combined organics were then washed 2x with 500 mL half brine then 500 mL brine, dried over MgS04 and concentrated to afford crude 1A-2. lH NMR (500 MHz, DMSO) δ 8.67 (d, J = 5.0 Hz, 1 H), 7.98 (d, J = 5.0 Hz, 1 H), 7.63-7.62 (m, 1 H), 7.42-7.40 (m, 1 H), 7.22 (t, J = 2.1 Hz, 1 H).
To a suspension of 3-(3-chloro-5-iodophenoxy)-2-chloro-4- (trifluoromethyl)pyridine (1A-2; 421 g, 970 mmol) in t-BuOH (1 L) was added KOH (272 g, 4850 mmol) and the mixture was heated to 75°C for 1 hour, at which point HPLC analysis indicated >95% conversion. The t-BuOH was evaporated and the mixture diluted with water (7mL/g, 2.4L) and then cooled to 0°C, after which 12N HC1 (~240mL) was added until pH 5. This mixture was then extracted with EtOAc (20mL/g, 6.5L), back extracted with EtOAc 1 x 5mL/g (1.5L), washed 1 x water:brine 1 : 1 (l OmL/g, 3.2L), 1 x brine (lOmL/g, 3.2L), dried over MgS04, filtered and concentrated to afford a crude proudct. The crude product was suspended in MTBE (2.25 L, 7mL/g), after which hexanes (1 L, 3 mL/g) was added to the suspension over ten minutes, and the mixturen was aged 30minutes at room temperature. The product was filtered on a Buchner, rinsed with MTBE hexanes 1 :2 (2 mL/g = 640 mL), then hexanes
A solution of 3-(3-chloro-5-iodophenoxy)-4-(trifluoromethyl)pyridin-2-ol (1A-3; 190 g; 457 mmol) in DMF (914 mL) was degassed for 20 minutes by bubbling N2, after which CuCN (73.7 g; 823 mmol) was added, and then the mixture was degassed an additional 5 minutes. The mixture was then heated to 120°C for 17 hours, then cooled to room temperature and partitioned between 6 L MeTHF and 2 L ammonium buffer (4:3: 1 = NH4CI
sat/water/NH-iOH 30%). The organic layer washed with 2 L buffer, 1 L buffer and 1 L brine then, dried over MgS04 and concentrated. The crude solid was then stirred in 2.2 L of refluxing
MeCN for 45 minutes, then cooled in a bath to room temperature over 1 hour, aged 30 minutes, then filtered and rinsed with cold MeCN (2 x 400mL). The solid was dried on frit under N2 atm for 60 hours to afford title compound 1-4. lH NMR (400 MHz, DMSO): δ 12.71 (s, 1 H); 7.75 (s, 1 H); 7.63-7.57 (m, 2 H); 7.54 (s, 1 H); 6.49 (d, J = 6.9 Hz, 1 H).
Steps lA(d) and lA(e)
The title compound 1-1 was then prepared from compound 1-4 using procedures similar to those described in Steps 1(d) and 1(e) set forth above in Example 1.
Crystalline anhydrous Form II of doravirine, useful for the treatment of HIV-1 and HIV-2 infections. The compound was originally claimed in WO2008076223. Also see WO2011120133. Merck & Co is developing doravirine (MK-1439), for the oral tablet treatment of HIV-1 infection. As of April 2014, the drug is in Phase 2 trials.
The next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) doravirine (formerly MK-1439) showed potent antiretroviral activity and good tolerability in combination with tenofovir/FTC (the drugs in Truvada) in a dose-finding study presented at the 21stConference on Retroviruses and Opportunistic Infections (CROI) last week in Boston.
NNRTIs are generally well tolerated and well suited for first-line HIV treatment, but as a class they are susceptible to resistance. Pre-clinical studies showed that Merck’s doravirine has a distinct resistance profile and remains active against HIV with common NNRTI resistance mutations including K103N and Y181C.
As reported at last year’s CROI, doravirine reduced HIV viral load by about 1.3 log in a seven-day monotherapy study. Doravirine is processed by the CYP3A4 enzyme, but it is neither a CYP3A4 inducer nor inhibitor, so it is not expected to have major drug interaction concerns.
Javier Morales-Ramirez from Clinical Research Puerto Rico reported late-breaking findings from a phase 2b study evaluating the safety and efficacy of various doses of doravirine versus efavirenz (Sustiva) for initial antiretroviral therapy.
This study included 208 treatment-naive people living with HIV from North America, Europe and Asia. More than 90% were men, 74% were white, 20% were black and the median age was 35 years. At baseline, the median CD4 cell count was approximately 375 cells/mm3 and 13% had received an AIDS diagnosis. Study participants were stratified by whether their viral load was above (about 30%) or below 100,000 copies/ml; median HIV RNA was approximately 4.5 log10.
Morales-Ramirez reported 24-week results from part 1 of the study, which will continue for a total of 96 weeks. In this part, participants were randomly allocated into five equal-sized arms receiving doravirine at doses of 25, 50, 100 or 200mg once daily, or else efavirenz once daily, all in combination with tenofovir/FTC.
At 24 weeks, 76.4% of participants taking doravirine had viral load below 40 copies/ml compared with 64.3% of people taking efavirenz. Response rates were similar across doravirine doses (25mg: 80.0%; 50mg: 76.2%; 100mg: 71.4%; 200mg: 78.0%). More than 80% of participants in all treatment arms reached the less stringent virological response threshold of <200 copies/ml.
Both doravirine and efavirenz worked better for people with lower pre-treatment viral load in an ad hoc analysis. For people with <100,000 copies/ml at baseline, response rates (<40 copies/ml) ranged from 83 to 89% with doravirine compared with 74% with efavirenz. For those with >100,000 copies/ml, response rates ranged from 50 to 91% with doravirine vs 54% with efavirenz.
Median CD4 cell gains were 137 cells/mm3 for all doravirine arms combined and 121 cells/mm3 for the efavirenz arm.
Doravirine was generally safe and well tolerated. People taking doravirine were less than half as likely as people taking efavirenz to experience serious adverse events (3.0% across all doravirine arms vs 7.1% with efavirenz) or to stop treatment for this reason (2.4 vs 4.8%). Four people taking doravirine and two people taking efavirenz discontinued due to adverse events considered to be drug-related.
The most common side-effects were dizziness (3.6% with doravirine vs 23.8% with efavirenz), abnormal dreams (9.0 vs 7.1%), diarrhoea (4.8 vs 9.5%), nausea (7.8 vs 2.4%) and fatigue (6.6 vs 4.8%). Other central nervous system (CNS) adverse events of interest included insomnia (5.4 vs 7.1%), nightmares (1.2 vs 9.5%) and hallucinations (0.6 vs 2.4%). Overall, 20.5% of people taking doravirine reported at least one CNS side-effect, compared with 33.3% of people taking efavirenz.
People taking doravirine had more favourable lipid profiles and less frequent liver enzyme (ALT and AST) elevations compared with people taking efavirenz.
The researchers concluded that doravirine demonstrated potent antiretroviral activity in treatment-naive patients, a favourable safety and tolerability profile, and fewer drug-related adverse events compared with efavirenz.
Based on these findings, the 100mg once-daily dose was selected for future development and will be used in part 2 of this study, a dose-confirmation analysis that will enrol an additional 120 participants.
In the discussion following the presentation, Daniel Kuritzkes from Harvard Medical School noted that sometimes it takes longer for viral load to go down in people who start with a high level, so with further follow-up past 24 weeks doravirine may no longer look less effective in such individuals.
Reference
Morales-Ramirez J et al. Safety and antiviral effect of MK-1439, a novel NNRTI (+FTC/TDF) in ART-naive HIV-infected patients. 21st Conference on Retroviruses and Opportunistic Infections, Boston, abstract 92LB, 2014.
Merck Moves Doravirine Into Phase 3 Clinical Trials
Wednesday Mar 19 | Posted by: roboblogger | Full story: EDGE
Earlier this month, at the 21st Conference on Retroviruses and Opportunistic Infections , Merck indicated plans to initiate a Phase 3 clinical trial program for doravirine in combination with ARV therapy in the second half of 2014.
Nigella Sativa Kills 89% of Lung Cancer Cells in Vitro: Researchers have just shown that nigella sativa (also known as black seed or black cumin) seed oil killsup to 89% of human lung cancer cells (A-549) after just 24 hours, while a non-oil extract from the seeds killed up to 77% of the cancer cells.
The extracts were prepared from seeds obtained at a local market. Nigella sativa is a powerful medicinal herb which has been used for thousands of years in traditional Chinese, Ayurvedic, Unani and Arabic medicine. It is best known for its potent anti-inflammatory and antioxidant properties, and has been used to suppress coughs, treat kidney stones, diarrhea and stomach pain. But modern science has now also uncovered nigella’s powerful anti-diabetes and anti-cancer effects.
This super herb has already shown potent activity against cancer of the breast, prostate, kidney, pancreas, liver, colon and cervix in previous lab studies, and this new study has shown new activity against lung cancer. Good health and cancer prevention should always start with a well-balanced diet focused on organic vegetables, fruit and whole foods (consuming at least half in the raw state). But nigella sativa may offer sizeable benefits for those wanting an extra measure of protection.
Nigella sativa is an annualflowering plant, native to south and southwest Asia. It grows to 20–30 cm (7.9–11.8 in) tall, with finely divided, linear (but not thread-like) leaves. The flowers are delicate, and usually coloured pale blue and white, with five to ten petals. The fruit is a large and inflated capsule composed of three to seven united follicles, each containing numerous seeds. The seed is used as a spice.
Etymology
Nigella sativa seed
The scientific name is a derivative of Latin niger (black).[2]
Common names
In English, Nigella sativa seed is variously called fennel flower,[3]nutmeg flower,[3]black caraway,[3]Roman coriander,[3] and also called black cumin.[3] Other names used, sometimes misleadingly, are onion seed and black sesame, both of which are similar-looking, but unrelated.Blackseed and black caraway may also refer to Bunium persicum.[4]
The seeds are frequently referred to as black cumin (as in Assamese: kaljeera or kolajeera or Bengalikalo jeeray), But black cumin (kala Jeera)[clarification needed] is different than Nigella sativa (Kali Jeeri).[citation needed] In south Indian language Kannada it is called [ಕೃಷ್ಣ ಜೀರಿಗೆ] “Krishna Jeerige”, but this is also used for a different spice, Bunium persicum.
In English-speaking countries with large immigrant populations, it is also variously known as kaljeera (Assamese কালজীৰা kalzira or ক’লাজীৰাkolazira), kalo jira (Bengali: কালোজিরাkalojira, black cumin), karum cheerakam, habbat al-barakah (Arabic حبة البركة) Kurdish “reşke” (rashkeh) (Tamil கருஞ்சீரகம்), kalonji (Hindi कलौंजी kalauṃjī or कलोंजी kaloṃjī, Urdu كلونجى kaloṃjī) or mangrail (Hindi मंगरैल maṃgarail), “Kala Jira in Marathi” ketzakh (Hebrew קצח), chernushka (Russian), çörek otu (Turkish), garacocco (Cypriot Turkish), ḥebbit al-barakah, seed of blessing (Arabic), siyah daneh (Persian سیاهدانه siyâh dâne), jintan hitam (Indonesian), karim jeerakam (കരിംജീരകം) in Malayalamor කළු දුරු in Sinhala, Karto Jeera in Beary.
It is used as part of the spice mixture paanch phoran or panch phoron (meaning a mixture of five spices) and by itself in a great many recipes in Bengali cookery and most recognizably in naan bread.[5]
The Turkish name çörek otu literally means “bun’s herb” from its use in flavouring the çörek buns. Such braided-dough buns are widespread in the cuisines of Turkey and its neighbours (see Tsoureki τσουρέκι). In Bosnian, the Turkish name for Nigella sativa is respelled as čurekot. The seed is used in Bosnia, and particularly its capital Sarajevo, to flavour pastries (Bosnian: somun) often baked on Muslim religious holidays.
The Arabic approbation about Bunium bulbocastanum (Kaala Jeera) Hebbit il barakah, meaning the “seed of blessing” is also applied toNigella sativa (Kali Jeeri).
Characteristics
Nigella sativa has a pungent bitter taste and smell. It is used primarily in confectionery and liquors. Peshawarinaan is, as a rule, topped with kalonji seeds. Nigella is also used in Armenian string cheese, a braided string cheese called Majdouleh or Majdouli in the Middle East.
History
According to Zohary and Hopf, archaeological evidence about the earliest cultivation of N. sativa “is still scanty”, but they report supposed N. sativa seeds have been found in several sites from ancient Egypt, including Tutankhamun‘s tomb.[6] Although its exact role in Egyptian culture is unknown, it is known that items entombed with a pharaoh were carefully selected to assist him in the afterlife.
The earliest written reference to N. sativa is thought to be in the book of Isaiah in the Old Testament, where the reaping of nigella and wheat is contrasted (Isaiah 28: 25, 27). Easton’s Bible dictionary states the Hebrew word ketsah refers to N. sativa without doubt (although not all translations are in agreement). According to Zohary and Hopf, N. sativawas another traditional condiment of the Old World during classical times; and its black seeds were extensively used to flavour food.[6]
Found in Hittite flask in Turkey from 2nd millennium BCE.[7]
History of medicineIn the Unani Tibb system of medicine, black cumin (Bunium bulbocastanum) is regarded as a valuable remedy for a number of diseases. Sayings of the Islamic prophet Muhammadunderline the significance of black cumin. According to a hadith narrated by Abu Hurairah, he says, “I heard Allah’s Apostle saying, ‘There is healing in black seed (haba sowda) for all diseases except death.'” [8][9]
The black cumin (Bunium bulbocastanum) seeds have been traditionally used in the Middle East and Southeast Asian countries for a variety of ailments. Nigella seeds are sold as black cumin in small bundles to be rubbed until warm, when they emit an aroma similar to black cumin which opens clogged sinuses in the way that do eucalyptus or Vicks.
Nestlé has purportedly filed a patent application covering use of Nigella sativa as a food allergy treatment.[10] Yet the firm denies the claim of patenting the plant, stating that the patent would only cover “the specific way that thymoquinone – a compound that can be extracted from the seed of the fennel flower – interacts with opioid receptors in the body and helps to reduce allergic reactions to food”.[11]
Medical studies
Thymoquinone, found in the seed oil extract of N. sativa, has been shown to have anti-neoplastic effects in rats and mice and in cultured human cells from several types of cancer, including pancreatic ductal adenocarcinoma.[12] It has protective antioxidant and anti-inflammatory effects, and promotes apoptosis (cell death) of the cancer cells.[12]
Black cumin
Nigella sativa oil
Original black cumin (Bunium bulbocastanum) is rarely available, so N. sativa is widely used instead; in India, Carum carvi is the substitute. Cumins are from the Apiaceae (Umbelliferae) family, but N. sativa is from Ranunculaceae family. Black cumin (not N. sativa) seeds come as paired or separate carpels, and are 3–4 mm long. They have a striped pattern of nine ridges and oil canals, and are fragrant (Ayurveda says, “Kaala jaaji sugandhaa cha” (black cumin seed is fragrant itself)), blackish in colour, boat-shaped, and tapering at each extremity, with tiny stalks attached; it has been used for medicinal purposes for centuries, both as a herb and pressed into oil, in Asia, the Middle East, and Africa.
Ali BH, Blunden G (April 2003). “Pharmacological and toxicological properties of Nigella sativa”. Phytother Res17 (4): 299–305.doi:10.1002/ptr.1309. PMID12722128.
Colorectal cancer stem cells thrive in conditions of inflammation. A University of Colorado Cancer Center study presented today at the American Association for Cancer Research (AACR) Annual Meeting 2014 shows that the chemical silibinin, purified from milk thistle extract, affects cell signaling associated with inflammation and thus also the formation and survival of colorectal cancer stem cells.
“We have been deeply involved in this line of research that extends from silibinin to its chemopreventive properties in colorectal cancer, and the current study takes another important step: we see both a likely chemopreventive mechanism and the result of this mechanism in animal models,” says Sushil Kumar, PhD, postdoctoral fellow in the lab of Rajesh Agarwal, PhD, co-program leader of Cancer Prevention and Control at the CU Cancer Center and professor at the Skaggs School of Pharmacy and Pharmaceutical Sciences.
The group compared mice chemically treated to develop inflammation-dependent colorectal cancer…
A new nonsurgical approach to treating chronic pain and stiffness associated with knee osteoarthritis has demonstrated significant, lasting improvement in knee pain, function, and stiffness. This safe, two-solution treatment delivered in a series of injections into and around the knee joint is called prolotherapy.
David Rabago, MD, and a team of researchers from the University of Wisconsin School of Medicine and Public Health, and Meriter Health Services, Madison, WI, report substantial improvement among participants in the one-year study who received at least three of the two-solution injections. Symptom improvement ranged from 19.5-42.9% compared to baseline status.
Apigenin, which abounds in particular parsley, have protective effects against cancer. Indeed, a U.S. study showed that apigenin alters the process of gene regulation in cancer cells, which has the effect of making them sensitive to the new process of cell death. Credits: H. Zell
Apigenin, a very natural chemical compound present in the Mediterranean diet, breaks immortality of cancer cells. A result obtained by researchers at the Ohio State University (USA).
Process chemistry is the arm of pharmaceutical chemistry concerned with the development and optimization of a synthetic scheme and pilot plant procedure to manufacture compounds for the drug development phase. Process chemistry is distinguished from medicinal chemistry, which is the arm of pharmaceutical chemistry tasked with designing and synthesizing molecules on small scale in the early drug discovery phase.
Medicinal chemists are largely concerned with synthesizing a large number of compounds as quickly as possible from easily tunable chemical building blocks (usually for SAR studies). In general, the repertoire of reactions utilized in discovery chemistry is somewhat narrow (for example, the Buchwald-Hartwig amination, Suzuki coupling and reductive amination are commonplace reactions).[1] In contrast, process chemists are tasked with identifying a chemical process that is safe, cost and labor efficient, “green,” and reproducible, among other considerations.
Oftentimes, in searching for the shortest, most efficient synthetic route, process chemists must devise creative synthetic solutions that eliminate costly functional group manipulations and oxidation/reduction steps.
This article will focus exclusively on the chemical and manufacturing processes associated with the production of small molecule drugs. Biological medical products (more commonly called “biologics”) represent a growing proportion of approved therapies, but the manufacturing processes of these products are beyond the scope of this article.
Additionally, the many complex factors associated with chemical plant engineering (for example, heat transfer and reactor design) and drug formulation will be treated cursorily.
Process Chemistry Considerations
Cost efficiency is of paramount importance in process chemistry and, consequently, is a focus in the consideration of pilot plant synthetic routes. The drug substance that is manufactured, prior to formulation, is commonly referred to as the active pharmaceutical ingradient (API) and will be referred to as such herein.
API production cost can be broken into two components: the “material cost” and the “conversion cost.”[2] The ecological and environmental impact of a synthetic process should also be evaluated by an appropriate metric (e.g. the EcoScale).
An ideal process chemical route will score well in each of these metrics, but inevitably tradeoffs are to be expected. Most large pharmaceutical process chemistry and manufacturing divisions have devised weighted quantitative schemes to measure the overall attractiveness of a given synthetic route over another. As cost is a major driver, material cost and volume-time output are typically weighted heavily.
The chemical and processing industries (CPI) provide the building blocks for many products. By using large amounts of heat and energy to physically or chemically transform materials, these industries help meet the world’s most fundamental needs for food, shelter and health, as well as products that are vital to such advanced technologies as computing, telecommunications and biotechnology.
These industries face major challenges to meet the needs of the present without compromising the needs of the future generations in the face of increasing industrial competitiveness. This translates into the need to make processes much more energy efficient, safer and more flexible, and to reduce emissions to meet the many competitive challenges within a global economy.
The chemical and processing industries refer to processes where materials undergo chemical conversion during their production into finished products, as well as – or instead of – the physical conversions common to industry in general.
In the chemical process industry the products differ chemically from the raw materials as a result of undergoing one or more chemical reactions during the manufacturing process.
The chemical process industries broadly include the traditional chemical industries, both organic and inorganic; the petroleum industry; the petrochemical industry, which produces the majority of plastics, synthetic fibers, and synthetic rubber from petroleum and natural-gas raw materials; and a series of allied industries in which chemical processing plays a substantial part.
While the chemical process industries are primarily the realm of the chemical engineer and the chemist, they also involve a wide range of other scientific, engineering, and economic specialists.
Material Cost
The material cost of a chemical process is the sum of the costs of all raw materials, intermediates, reagents, solvents and catalysts procured from external vendors. Material costs may influence the selection of one synthetic route over another or the decision to outsource production of an intermediate.
Conversion Cost
The conversion cost of a chemical process is a factor of that procedure’s overall efficiency, both in materials and time, and its reproducibility. The efficiency of a chemical process can be quantified by its atom economy, yield, volume-time output, and environmental factor (E-factor), and its reproducibility can be evaluated by the Quality Service Level (QSL) and Process Excellence Index (PEI) metrics.
An illustrative example of atom economy using the Claisen rearrangement and Wittig reaction.
Atom Economy
The atom economy of a reaction is defined as the number of atoms from the starting materials that are incorporated into the final product. Atom economy can be viewed as an indicator of the “efficiency” of a given synthetic route.[3]
For example, the Claisen rearrangement and the Diels-Alder cycloaddition are examples of reaction that are 100 percent atom economical. On the other hand, a prototypical Wittig reaction has especially poor atom economy (merely 20 percent in the example shown).
Process synthetic routes should be designed such that atom economy is maximized for the entire synthetic scheme. Consequently, “costly” reagents such as protecting groups and high molecular weight leaving groups should be avoided where possible. An atom economy value in the range of 70 to 90 percent for an API synthesis is ideal, but it may be impractical or impossible to access certain complex targets within this range. Nevertheless, atom economy is a good metric to compare two routes to the same molecule.
Yield
Yield is defined as the amount of product obtained in a chemical reaction. According to Vogel’s Textbook of Practical Organic Chemistry, yields around 100% are called quantitative, yields above 90% are excellent, yields above 80% are very good, yields above 70% are good, yields above 50% are fair, and yields below 40% are poor. The yield that has practical significance in a process chemistry setting is the isolated yield, referring to the yield of the isolated product after all extraction and purification steps. In a final API synthesis, isolated yields of 80 percent or above for each synthetic step are expected.
An illustrative example of convergent synthesis.
There are several strategies that are employed in the design of a process route to ensure adequate overall yield of the pharmaceutical product. The first is the concept of convergent synthesis. Assuming a very good to excellent yield in each synthetic step, the overall yield of a multistep reaction can be maximized by combining several key intermediates at a late stage that are prepared independently from each other.
Another strategy to maximize isolated yield (as well as time efficiency) is the concept of telescoping synthesis (also called one-pot synthesis). This approach describes the process of eliminating workup and purification steps from a reaction sequence, typically by simply adding reagents sequentially to a reactor. In this way, unnecessary losses from these steps can be avoided.
Finally, to minimize overall cost, synthetic steps involving expensive reagents, solvents or catalysts should be designed into the process route as late stage as possible, to minimize the amount of reagent used.
In a pilot plant or manufacturing plant setting, yield can have a profound effect on the material cost of an API synthesis, so the careful planning of a robust route and the fine-tuning of reaction conditions are crucially important. After a synthetic route has been selected, process chemists will subject each step to exhaustive optimization in order to maximize overall yield. Low yields are typically indicative of unwanted side product formation, which can raise red flags in the regulatory process as well as pose challenges for reactor cleaning operations.
Volume-Time Output
The volume-time output (VTO) of a chemical process represents the cost of occupancy of a chemical reactor for a particular process or API synthesis. For example, a high VTO indicates that a particular synthetic step is costly in terms of “reactor hours” used for a given output. Mathematically, the VTO for a particular process is calculated by the total volume of all reactors (m3) that are occupied times the hours per batch divided by the output for that batch of API or intermediate (measured in kg).
The process chemistry group at Boehringer-Ingelheim, for example, targets a VTO of less than 1 for any given synthetic step or chemical process.
Additionally, the raw conversion cost of an API synthesis (in dollars per batch) can be calculated from the VTO, given the operating cost and usable capacity of a particular reactor. Oftentimes, for large-volume APIs, it is economical to build a dedicated production plant rather than to use space in general pilot plants or manufacturing plants.
Environmental Factor (E-factor) and Process Mass Intensity (PMI)
Both of these measures, which capture the environmental impact of a synthetic reaction, intend to capture the significant and rising cost of waste disposal in the manufacturing process. The E-factor for an entire API process is computed by the ratio of the total mass of waste generated in the synthetic scheme to the mass of product isolated.
A similar measure, the process mass intensity (PMI) calculates the ratio of the total mass of materials to the mass of the isolated product.
For both metrics, all materials used in all synthetic steps, including reaction and workup solvents, reagents and catalysts, are counted, even if solvents or catalysts are recycled in practice. Inconsistencies in E-factor or PMI computations may arise when choosing to consider the waste associated with the synthesis of outsourced intermediates or common reagents. Additionally, the environmental impact of the generated waste is ignored in this calculation; therefore, the environmental quotient (EQ) metric was devised, which multiplies the E-factor by an “unfriendliness quotient” associated with various waste streams. A reasonable target for the E-factor or PMI of a single synthetic step is any value between 10 and 40.
Quality Service Level (QSL)
The final two “conversion cost” considerations involve the reproducibility of a given reaction or API synthesis route. The quality service level (QSL) is a measure of the reproducibility of the quality of the isolated intermediate or final API. While the details of computing this value are slightly nuanced and unimportant for the purposes of this article, in essence, the calculation involves the ratio of satisfactory quality batches to the total number of batches. A reasonable QSL target is 98 to 100 percent.
Process Excellence Index (PEI)
Like the QSL, the process excellence index (PEI) is a measure of process reproducibility. Here, however, the robustness of the procedure is evaluated in terms of yield and cycle time of various operations. The PEI yield is defined as follows:
In practice, if a process is high-yielding and has a narrow distribution of yield outcomes, then the PEI should be very high. Processes that are not easily reproducible may have a higher aspiration level yield and a lower average yield, lowering the PEI yield.
Similarly, a PEI cycle time may be defined as follows:
For this expression, the terms are inverted to reflect the desirability of shorter cycle times (as opposed to higher yields). The reproducibility of cycle times for critical processes such as reaction, centrifugation or drying may be critical if these operations are rate-limiting in the manufacturing plant setting. For example, if an isolation step is particularly difficult or slow, it could become the bottleneck for an API synthesis, in which case the reproducibility and optimization of that operation become critical.
For an API manufacturing process, all PEI metrics (yield and cycle times) should be targeted at 98 to 100 percent.
EcoScale
In 2006, Van Aken, et al.[4] developed a quantitative framework to evaluate the safety and ecological impact of a chemical process, as well as minor weighting of practical and economical considerations. Others have modified this EcoScale by adding, subtracting and adjusting the weighting of various metrics. Among other factors, the EcoScale takes into account the toxicity, flammability and explosive stability of reagents used, any nonstandard or potentially hazardous reaction conditions (for example, elevated pressure or inert atmosphere), and reaction temperature. Some EcoScale criteria are redundant with previously considered criteria (e.g. E-factor).
Synthetic Case Studies
Boehringer Ingelheim HCV Protease Inhibitor (BI 201302)
Macrocyclization is a recurrent challenge for process chemists, and large pharmaceutical companies have necessarily developed creative strategies to overcome these inherent limitations. An interesting case study in this area involves the development of novel NS3 protease inhibitors to treat Hepatitis C patients by scientists at Boehringer-Ingelheim.[5] The process chemistry team at BI was tasked with developing a cheaper and more efficient route to the active NS3 inhibitor BI 201302, a close analog of BILN 2061. Two significant shortcomings were immediately identified with the initial scale-up route to BILN 2061, depicted in the scheme below.[6] The macrocyclization step posed four challenges inherent to the cross-metathesis reaction.
High dilution is typically necessary to prevent unwanted dimerization and oligomerization of the diene starting material. In a pilot plant setting, however, a high dilution factor translates into lower throughput, higher solvent costs and higher waste costs.
High catalyst loading was found to be necessary to drive the RCM reaction to completion. Because of high licencing costs of the ruthenium catalyst that was used (1st generation Hoveyda catalyst), a high catalyst loading was financially prohibitive. Recycling of the catalyst was explored, but proved impractical.
Long reaction times were necessary for reaction completion, due to slow kinetics of the reaction using the selected catalyst. It was hypothesized that this limitation could be overcome using a more active catalyst. However, while the second-generation Hoveyda and Grubbs catalysts were kinetically more active than the first-generation catalyst, reactions using these catalysts formed large amounts of dimeric and oligomeric products.
An epimerization risk under the cross-methathesis reaction conditions. The process chemistry group at Boehringer-Ingelheim performed extensive mecahnistic studies showing that epimerization most likely occurs through a ruthenacyclopentene intermediate.[7] Furthermore, the Hoveyda catalyst employed in this scheme minimizes epimerization risk compared with the alalogous Grubbs catalyst.
Additionally, the final double SN2 sequence to install the quinoline heterocycle was identified as a secondary inefficiency in the synthetic route.
Analysis of the cross-methathesis reaction revealed that the conformation of the acyclic precursor had a profound impact on the formation of dimers and oligomers in the reaction mixture. By installing a Boc protecting group at the C-4 amide nitrogen, the Boehringer-Ingelheim chemists were able to shift the site of initiation from the vinylcyclopropane moiety to the nonenoic acid moiety, improving the rate of the intramolecular reaction and decreasing the risk of epimerization. Additionally, the catalyst employed was switched from the expensive 1st generation Hoveyda catalyst to the more reactiveless expensive Grela catalyst.[8] These modifications allowed the process chemists to run the reaction at a standard reaction dilution of 0.1-0.2 M, given that the rates of competing dimerization and oligomerization reactions was so dramatically reduced.
Additionally, the process chemistry team envisioned a SNAr strategy to install the quinoline heterocycle, instead of the SN2 strategy that they had employed for the synthesis of BILN 2061. This modification prevented the need for inefficient double inversion by proceeding through retention of stereochemistry at the C-4 position of the hydroxyproline moiety.[9]
It is interesting to examine this case study from a VTO perspective. For the unoptimized cross-metathesis reaction using the Grela catalyst at 0.01 M diene, the reaction yield was determined to be 82 percent after a reaction and workup time of 48 hours. A 6-cubic meter reactor filled to 80% capacity afforded 35 kg of desired product. For the unoptimized reaction:
This VTO value was considered prohibitively high and a steep investment in a dedicated plant would have been necessary even before launching Phase III trials with this API, given its large projected annual demand. But after reaction development and optimization, the process team was able to improve the reaction yield to 93 percent after just 1 hour (plus 12 hours for workup and reactor cleaning time) at a diene concentration of 0.2 M. With these modifications, a 6-cubic meter reactor filled to 80% capacity afforded 799 kg of desired product. For this optimized reaction:
Thus, after optimization, this synthetic step became less costly in terms of equipment and time and more practical to perform in a standard manufacturing facility, eliminating the need for a costly investment in a new dedicated plant.
Simvastatin, originally developed by Merck, is the most frequently prescribed statin today, with more nearly 100 million prescriptions filled in 2010, according to IMS Health. The traditional synthesis of the drug entailed a multi-step chemical process starting from Lovastatin. The chemical process was using large amounts of hazardous reagents as well as large quantities of solvents.
Professor Yi Tang, at UCLA conceived an initial synthesis that used an engineered enzyme. Codexis Inc. licensed the intellectual property from UCLA, optimized the initial enzyme and developed the new process for commercial use as shown in Figure 1. Following the quantitative hydrolysis of lovastatin to monacolin J acid, Codexis developed a novel, non-natural acyl donor enzyme to regioselectively acylate the C8 position and effect cyclization to simvastatin. This mild bioenzymatic process reduces the 4 steps chemical synthesis to only two steps. The Codexis process is significantly more efficient, cost effective and environmentally friendly.
This is the reaction scheme for producing the drug Simvastatin. The process was an award-winner at last month’s Green Chemistry Challenge Awards held by the Environmental Protection Agency
“We started working on Simvastatin in 2008 and completed the planning process in 2010,” Huisman said. “Then, we started the commercialization process, which takes time because you need regulatory approval of the new process we were working on. We licensed some technology from Yi Tang and UCLA and were then able to continue.”
Codexis took the three-step process used to make Simvastatin and cut out two of the steps, Huisman said.
“From the starting material, it (Simvastatin) has three reactive groups, or hydroxy groups, and what we need to do is convert two of the three groups,” Huisman explained. “We took out a protective step and a de-protective step. We took out two of the steps, and it was intense chemical processing. We then were able to accomplish everything in one step. We also circumvented the use of several nasty chemicals, as well.”
By cutting out two steps, “the overall yield goes up tremendously, about 35 percent,” Huisman added. “And we’re generating 25 times less waste than we did in the old process.”
Huisman said the new process doesn’t change the drug’s effects at all, and that scientists have been trying to do this type of work on commercial drugs for decades.
“In order for this to be a commercial process, the enzyme needs to be improved,” he said. “We needed to speed up the enzyme 1,000-fold to make this process workable; it took a team of scientists about nine months to optimize the enzymes and speed it up.”
Codexis 2 step enzymatic process versus the 4 step chemical synthesis
AZIDES
A popular procedure for making 5-substituted tetrazoles is the reaction of sodium azide with a nitrile, often in the presence of an ammonium salt. The example shown below is from Organic Syntheses (Novartis Process R&D and Ley’s group at Cambridge), providing the useful enantiocatalyst shown on an 80 mmol scale. The excess sodium azide was destroyed with sodium nitrite and sulfuric acid, which converts hydrazoic acid into nitrogen and nitrous oxide gases.
While the above procedure may be popular, any time you use sodium azide you should be thinking, “hydrazoic acid can be generated, it’s explosive and toxic, and I need to take the appropriate safety precautions.” That’s precisely what happened during some recent process R&D work at Merck Frosst on the steroyl-CoA desaturase inhibitor MK-8245. The discovery chemistry route used NaN3/pyridinium chloride as shown below, but the process group felt that the potential for significant amounts of hydrazoic acid generation was too high.
Armed with the ability to detect hydrazoic acid in the headspace above the reaction mixture using online IR, the Merck Frosst researchers surveyed alternatives. Sharpless’s zinc bromide procedure, proposed to minimize hydrazoic acid formation by control of the pH, led to a reading of 2000 ppm of HN3 in the headspace, which is below the detonation threshold of 15,000 ppm but was still felt to be undesirable. In their own survey of conditions, the Merck Frosst scientists found something quite new and significant: Reaction with sodium azide in the presence of a catalytic amount of zinc oxide in aqueous THF (pH 8) proceeded efficiently, and most notably, with only 2 ppm of HN3 in the headspace! They were able to make 7 kg of the tetrazole in one run in nearly quantitative yield. Nice!
I’d be remiss if I didn’t mention Bu3SnN3 and Me3SiN3/Cu(I) as sodium azide surrogates, sometimes used on large scale. Shown below is an application to valsartan (see here and here) with recycling of the tin by-products. The intermediate stannyl tetrazole and leftover Bu3SnN3 were converted with HCl to Bu3SnCl, which was then converted to the fluoride, which was removed by filtration and recycled to Bu3SnCl.
Recently, large pharmaceutical process chemists have relied heavily on the development of enzymatic reactions to produce important chiral building blocks for API synthesis. Many varied classes of naturally occurring enzymes have been co-opted and engineered for process pharmaceutical chemistry applications. The widest range of applications come from ketoreductases and transaminases, but there are isolated examples from hydrolases, aldolases, oxidative enzymes, esterases and dehalogenases, among others.[10]
One of the most prominent uses of biocatalysis in process chemistry today is in the synthesis of Januvia®, a DPP-4 inhibitor developed by Merck for the management of type II diabetes. The traditional process synthetic route involved a late-stage enamine formation followed by rhodium-catalyzed asymmetric hydrogenation to afford the API sitagliptin. This process suffered from a number of limitations, including the need to run the reaction under a high-pressure hydrogen environment, the high cost of a transition-metal catalyst, the difficult process of carbon treatment to remove trace amounts of catalyst and insufficient stereoselectivity, requiring a subsequent recrystallization step before final salt formation.[11][12]
Merck’s process chemistry department contracted Codexis, a medium-sized biocatalysis firm, to develop a large-scale biocatalytic reductive amination for the final step of its sitagliptin synthesis. Codexis engineered a transaminase enzyme from the bacteria Arthrobacter through 11 rounds of directed evolution. The engineered transaminase contained 27 individual point mutations and displayed activity four orders of magnitude greater than the parent enzyme. Additionally, the enzyme was engineered to handle high substrate concentrations (100 g/L) and to tolerate the organic solvents, reagents and byproducts of the transamination reaction. This biocatalytic route successfully avoided the limitations of the chemocatalyzed hydrogenation route: the requirements to run the reaction under high pressure, to remove excess catalyst by carbon treatment and to recrystallize the product due to insufficient enantioselectivity were obviated by the use of a biocatalyst. Merck and Codexis were awarded the Presidential Green Chemistry Challenge Award in 2010 for the development of this biocatalytic route toward Januvia®.[13]
ATORVASTATIN
Biocatalytic process development firm Codexis was recognized with the award in the greener reaction conditions category for developing a “green-by-design” enzymatic process to replace a chemical process for making ethyl (R)-4-cyano-3-hydroxybutyrate. This chemical, also known as hydroxynitrile, is the key chiral building block used to make atorvastatin, the active ingredient in Pfizer‘s cholesterol-lowering drug Lipitor.
The new process is helping to lower atorvastatin’s long-term production costs, according to John H. Grate, senior vice president of R&D and chief technology officer at Codexis. The savings could be financially significant for Pfizer and future generics manufacturers given that Lipitor is the world’s top pharmaceutical, with annual sales of about $13 billion.
Hydroxynitrile is used in the early stages of atorvastatin synthesis to build the chiral dihydroxy acid side chain that’s essential to the drug’s activity, Grate told C&EN. Demand for the intermediate is about 200 metric tons per year, and it’s currently being made by several fine chemicals producers. The competition to supply the intermediate to Pfizer has spurred several firms to chase after a better way to prepare hydroxynitrile (Angew. Chem. Int. Ed.2005,44, 362).
Chemical engineering professor Galen J. Suppes of the University of Missouri, Columbia, was honored with the academic award for his group’s work to create a low-cost catalytic process to convert the glycerol by-product from biodiesel production into propylene glycol–turning 1,2,3-propanetriol into 1,2-propanediol. At first glance, this achievement may not sound that exciting. But the repercussions of Suppes’s accomplishment are expected to have a major impact on the future use of biodiesel fuel, the world glycerol market, and the environmental health and safety of antifreeze and deicing chemicals.
Photo by Rob Hill/MU Publications
GREEN SOLUTION Suppes and his group uncovered ideal reaction conditions for the catalytic conversion of by-product glycerol to useful propylene glycol.
Biodiesel is a mixture of fatty acid methyl esters made by esterifying soybean oil or other vegetable oil or animal fat. The triglycerides in the oil consist of three long fatty acid chains connected to a propyl headgroup. Sodium hydroxide is used to cleave the chains, which in turn are reacted with methanol to form methyl esters, leaving the residual glycerol headgroup as a by-product. About 1 kg of crude glycerol is formed for every 9 kg of biodiesel produced.
Millions of gallons of glycerol are flooding the world market as biodiesel production is ramping up in the U.S. and Europe, Suppes explained. The fallout from this glycerol glut is that chemical companies have shuttered some glycerol production plants and are considering glycerol as a starting material to make a host of feedstock chemicals (C&EN, Feb. 6, page 7).
Suppes entered the picture about four years ago when he realized that an inexpensive method to convert glycerol to propylene glycol could be valuable, he said. Utilizing the glycerol not only would help offset the cost of biodiesel production, but the inexpensive propylene glycol could be used as a low-toxicity replacement for ethylene glycol in automotive antifreeze.
Suppes’s system involves low-pressure hydrogenolysis of glycerol using a copper chromite catalyst, CuO•Cr2O3 (Appl. Catal. A2005,281, 225). In the two-step process, glycerol is first dehydrated to form acetol (1-hydroxy-2-propanone), which is then hydrogenated to form propylene glycol.
GREEN LEFTOVERS Glycerol by-product from biodiesel production can be used as a feedstock in Suppes’ process to produce acetol or propylene glycol from renewable resources.
Copper chromite hydrogenolysis catalysts aren’t new, but the success of the Missouri process is in achieving high selectivity for propylene glycol by controlling the temperature and hydrogen pressure of the reaction, Suppes noted. In the past, researchers tended to use reaction temperatures that were too high, leading to a higher percentage of by-products. Thus, they “missed the window of opportunity to achieve high selectivity,” Suppes said. Tinkering with temperature, pressure, and several different catalysts, Suppes and his colleagues optimized the system to operate at about 220 °C and less than 10 bar versus about 260 °C and more than 150 bar for other systems.
Another key part of the synthesis is the ability to isolate the acetol intermediate, Suppes added. Acetol is a synthetic starting material used to make polyols. But when made from petroleum, it costs about $5.00 per lb, discouraging its widespread use. Suppes envisions that producing acetol from biomass-based glycerol using his process could lower the cost to 50 cents per lb, “opening up even more potential applications and markets for products made from glycerol.”
Suppes’s propylene glycol process has been patented and is being licensed through the Missouri Soybean Merchandising Council, which provided partial funding for the research. The first commercial facility, with an annual capacity of 11.5 million gal, is being built in an undisclosed location in the U.S. by Senergy Chemical. It’s expected to be in operation by the end of this year.
E7398, INN eribulin mesylate
The most awe-inspiring example of a positive tangible outcome from the combination of basic research into the synthesis of a system, and a correctly weighted assessment of ‘scalability’, is Halaven® (2, E7398, INN eribulin mesylate). Most chemists in industry and academia alike would have considered using total synthesis to support clinical development and commercialization of this compound a ‘fool’s errand,’ but the Kishi group and Eisai Inc. did not. The fact is that this compound solves a major clinical problem, so taking on the issues (length of synthesis, stability limitations, stereochemical problems, etc.) had a big payoff (reducing the relative weighting or importance of these factors in assessing the viability of a commercial chemical synthesis). As depicted below, a highly convergent approach, combined with powerful methodology for stitching together key fragments 5 and 6 (Nozaki–Hiyama–Kishi (NHK) coupling) and a strategy of targeting crystalline intermediates were all key elements that culminated in this landmark accomplishment
The commercial synthesis of Halaven® (2), a landmark achievement in process chemistry
Artemisinin (Cook, 2012).
(+)-Artemisinin (41) is currently the most effective drug against Plasmodium falciparum malaria as part of an artemisinin-based combination therapy (ACT). Although it can be isolated on an industrial scale from Artemisia annua, the market price of artemisinin (41) has fluctuated widely and traditional extraction does not provide enough material to meet the worldwide demand. Interestingly, recent efforts towards a cheaper and more efficient production of artemisinin (41) have mainly taken place in the areas of synthetic biology, semisynthesis and plant engineering, while there has been a lack of practical approaches using a straightforward total synthesis. Despite the fact that all the total syntheses of artemisinin, until 2010, were impressive from a feasibility point of view, none of them provided a solution for the low-cost synthesis of 41. This changed when Cook’s group recently published a scalable synthesis of artemisinin (41), which provides a blueprint for the cost-effective production of 41 and its derivatives below Key to their successful strategy was the use of reaction cascades that rapidly built complexity, starting from the cheap feedstock chemical, cyclohexenone (42). The latter was first subjected to a one-pot conjugate addition/alkylation sequence, to give ketone 43. A three-step sequence consisting of formylation, cycloaddition and a Wacker-type oxidation, yielded 9.4 g of methyl ketone 44. The challenging formation of the unusual peroxide bridge was initially met with failure, but was eventually realized by a reaction with singlet oxygen to give 41 amongst other oxidized intermediates. The entire synthetic sequence was conducted on a gram scale, required only three chromatographic purifications and was carried out in only five flasks. Considering the low cost of the commodity chemicals used and the conciseness of Cook’s synthesis, it is certainly worth being further investigated.
Cook’s scalable route to (+)-artemisinin (41).
Continuous/Flow Manufacturing
In recent years, much progress has been made in the development and optimization of flow reactors for small-scale chemical synthesis (the Jamison Group at MIT and Ley Group at Cambridge University, among others, have pioneered efforts in this field). The pharmaceutical industry, however, has been slow to adopt this technology for large-scale synthetic operations. For certain reactions, however, continuous processing may possess distinct advantages over batch processing in terms of safety, quality and throughput.
A case study of particular interest involves the development of a fully continuous process by the process chemistry group at Eli Lilly and Company for an asymmetric hydrogenation to access a key intermediate in the synthesis of LY500307,[14] a potent ERβ agonist that is entering clinical trials for the treatment of patients with schizophrenia, in addition to a regimen of standard antipsychotic medications. In this key synthetic step, a chiral rhodium-catalyst is used for the enantioselective reduction of a tetrasubstituted olefin. After extensive optimization, it was found that in order to reduce the catalyst loading to a commercially practical level, the reaction required hydrogen pressure up to 70 atm. The pressure limit of a standard chemical reactor is about 10 atm, although high-pressure batch reactors may be acquired at significant capital cost for reactions up to 100 atm. Especially for an API in the early stages of chemical development, such an investment clearly bears a large risk.
An additional concern was that the hydrogenation product has an unfavorable eutectic point, so it was impossible to isolate the crude intermediate in more than 94 percent ee by batch process. Because of this limitation, the process chemistry route toward LY500307 necessarily involved a kinetically controlled crystallization step after the hydrogenation to upgrade the enantiopurity of this penultimate intermediate to >99 percent ee.
The process chemistry team at Eli Lilly successfully developed a fully continuous process to this penultimate intermediate, including reaction, workup and kinetically controlled crystallization modules (the engineering considerations implicit in these efforts are beyond the scope of this article). An advantage of flow reactors is that high-pressure tubing can be utilized for hydrogenation and other hyperbaric reactions. Because the head space of a batch reactor is eliminated, however, many of the safety concerns associated with running high-pressure reactions are obviated by the use of a continuous process reactor. Additionally, a two-stage mixed suspension-mixed product removal (MSMPR) module was designed for the scalable, continuous, kinetically controlled crystallization of the product, so it was possible to isolate in >99 percent ee, eliminating the need for an additional batch crystallization step.
This continuous process afforded 144 kg of the key intermediate in 86 percent yield, comparable with a 90 percent isolated yield using the batch process. This 73-liter pilot-scale flow reactor (occupying less than 0.5 m3 space) achieved the same weekly throughput as theoretical batch processing in a 400-liter reactor. Therefore, the continuous flow process demonstrates advantages in safety, efficiency (eliminates the need for batch crystallization) and throughput, compared with a theoretical batch process.
US scientists have found a way to stop solid byproducts clogging channels in continuous flow reactors, a problem that has hampered their progress for use in manufacturing pharmaceuticals.
Klavs Jensen, Stephen Buchwald and their team at the Massachusetts Institute of Technology believe that flow methods will become increasingly important in the future of pharmaceuticals and chemical manufacturing. ‘One of the biggest hurdles is handling solids,’ says group member Timothy Noël. ‘Precipitates can form during the reactions, which usually lead to irreversible clogging of microchannels in the reactors.’ Previous methods suggested to overcome this problem include introducing another solvent to dissolve the solids, but this can reduce the overall efficiency of the reactions. Now, the team have used an ultrasound bath to break up the byproducts to prevent clogging.
Traditionally, pharmaceutical manufacture is done in a batch-based system, but the process suffers from interruptions and the need to transport material between batch reactors. Performing these reactions in a continuous flow system would speed up the process and reduce chemical waste.
Reagents were introduced into a tube, which was then placed in an ultrasonic bath heated to 60 degrees Celsius. When the reagents exited the reactor, the reaction was mixed with a quench of water and ethyl acetate in a larger tube, allowing plenty of time for salt byproducts to dissolve
The team tested the method on palladium-catalysed C-N cross-coupling reactions, making amines that are common in biologically active molecules. The reactions couple aryl halides to nitrogen nucleophiles and form byproducts – inorganic salts – that are insoluble in the solvents used.
As a result, says Noël, they were able to obtain diarylamine products with reaction times ranging from 20 seconds to 10 minutes. At very short residence times (time in the reactor under reaction conditions) they observed a significantly higher rate for the reaction in flow compared to the equivalent batch experiments. With high conversions in short reaction times, they were able to reduce the catalyst loading in flow to just 0.1 mol per cent. ‘Extremely low catalyst loadings such as these are of particular interest to the pharmaceutical industry,’ says Noël.
Noël believes that in the future microfluidics will be used to construct increasingly complex molecules. Different devices will automate and integrate many synthetic steps that are currently performed using the more traditional and time-consuming batch-based practices.
Oliver Kappe, from the Christian Doppler Laboratory for Microwave Chemistry, Institute of Chemistry, Karl-Franzens-University Graz says: ‘Jensen and Buchwald clearly demonstrate that immersing a flow device into an ultrasound bath can prevent clogging problems that unfortunately are all too familiar to the flow/microreactor community.’
Direct Fluorination and Microreactor Technology
Elemental fluorine has long been considered to be too reactive and uncontrollable for use as a reagent in organic synthesis and this perception still predominates. Prof. Poliakoff’s comments on the popular Periodic Table video series (www.PeriodicVideos.com), ‘It was much more exciting than I thought …you see the flames,’ and general comments in standard advanced organic chemistry textbooks (J. March, Advanced Organic Chemistry, ‘Direct fluorination of aromatic rings with F2 is not feasible at room temperature because of the extreme reactivity of F2….not yet of preparative significance) are typical.
Despite this background, research into the use of elemental fluorine for organic synthesis at Durham has overcome the many problems of using fluorine gas for the safe synthesis of fine chemicals, in particular, by use of dilute fluorine gas in nitrogen, appropriate solvent choice (high dielectric constant media such as formic acid, sulfuric acid or acetonitrile), reactor vessel design, gas flow regulator systems and stainless steel/monel fluorine gas handling lines have developed over the years to allow selective direct fluorination of a range of aliphatic, dicarbonyl, aromatic, heteroaromatic, heterocyclic, steroid and carbohydrate derivatives to be established and the mechanism (regiochemistry, stereochemistry, selectivity, etc.) of these processes to be assessed. Indeed, direct fluorination of aromatic rings is feasible at room temperature ! Research expanding the use of fluorine gas continues to develop new selective fluorination methodology for the synthesis of a range of aromatic, heterocylic and aliphatic systems.2,3
In particular, a process for the synthesis of a fluoroketoester first carried out in Durham was developed by our industrial collaborators, F2 Chemicals Ltd (UK), for the Pfizer company and forms a key starting material in the multi step synthesis of the widely used anti-fungal agent V-Fend (Voriconazole) throughout the clinical trial, launch and commercialization periods. In the period from January 2008 to March 2011 approximately 17 tonnes of the fluoroketoester were manufactured for Pfizer by F2 Chemicals Ltd. Global sales of V-Fend in the 2008-2010 period total $2.4 billion (Pfizer annual financial reports) and in 2010 was 17th position in Pfizer’s best selling products and it is one of the global top 100 best selling pharmaceutical products.
Further reaction control in selective fluorination reactions was achieved by the design, fabrication and commissioning of single and multi-channel continuous flow reactor systems, establishing the use of convenient, inexpensive flow reactors for gas – liquid processes using flow regimes in the laboratory. Techniques for the supply of individual gas and liquid reagents from single sources to a parallel array of many flow channels at the same flow rate and pressure whilst maintaining laminar flow within the reactor channels and telescoped gas – liquid / liquid – liquid processes involving fluorination and ring formation in one continuous flow process have been developed.
References
Roughley, S. D.; Jordan, A. M. (2011). “The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates”. J. Med. Chem.54: 3451.
Dach, R.; Song, J. J.; Roschangar, F.; Samstag, W.; Senanayake, C. H. (2012). “The eight criteria defining a good chemical manufacturing process”. Org. Process Res. Dev.16: 1697.
Trost, B. M. (1991). “The atom economy – a search for synthetic efficiency”. Science254: 1471.
Van Aken, K.; Strekowski, L.; Patiny, L. (2006). “EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters”. Beilstein J. Org. Chem.2 (No. 3).
Faucher, A-M.; Bailey, M. D.; Beaulieu, P. L.; Brochu, C.; Duceppe, J-S.; Ferland, J-M.; Ghiro, E.; Gorys, V.; Halmos, T.; Kawai, S. H.; Poirier, M.; Simoneau, B.; Tsantrizos, Y. S.; Llinas-Brunet, M. (2004). “Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans”. Org. Lett.6: 2901.
Yee, N. K.; Farina, V.; Houpis, I. N.; Haddad, N.; Frutos, R. P.; Gallou, F.; Wang, X-J.; Wei, X.; Simpson, R. D.; Feng, X.; Fuchs, V.; Xu, Y.; Tan, J.; Zhang, L.; Xu, J.; Smith-Keenan, L. L.; Vitous, J.; Ridges, M. D.; Spinelli, E. M.; Johnson, M. (2006). “Efficient large-scale synthesis of BILN 2061, a potent HCV protease inhibitor, by a convergent approach based on ring-closing metathesis”. J. Org. Chem.71: 7133.
Zeng, X.; Wei, X.; Farina, V.; Napolitano, E.; Xu, Y.; Zhang, L.; Haddad, N.; Yee, N. K.; Grinberg, N.; Shen, S.; Senanayake, C. H. (2006). “Epimerization reaction of a substituted vinylcyclopropane catalyzed by ruthenium carbenes: mechanistic analysis”. J. Org. Chem.71: 8864.
Grela, K.; Harutyunyan, S.; Michrowska, A. (2002). “A highly efficient ruthenium catalyst for metathesis reactions”. Angew. Chem. Int. Ed.41: 4038.
Wei, X.; Shu, C.; Haddad, N.; Zeng, X.; Patel, N. D.; Tan, Z.; Liu, J.; Lee, H.; Shen, S.; Campbell, S.; Varsolona, R. J.; Busacca, C. A.; Hossain, A.; Yee, N. K.; Senanayake, C. H. (2013). “A highly convergent and efficient synthesis of a macrocyclic hepatitis C virus protease inhibitor BI 201302”. Org. Lett.15: 1016.
Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K. (2012). “Engineering the third wave of biocatalysis”. Nature485: 185.
Savile, C. K.; Janey, J. M.; Mundorff, E. C.; Moore, J. C.; Tam, S.; Jarvis, W. R.; Colback, J. C.; Krebber, A.; Fleitz, F. J.; Brands, J.; Devine, P. N.; Huisman, G. W.; Hughes, G. J. (2010). “Biocatalytic asymmetric synthesis of chiral amines applied to sitagliptin manufacture”. Science329: 305.
Desai, A. A. (2011). “Sitagliptin manufacture: a compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis”. Angew. Chem. Int. Ed.50: 1974.
Busacca, C. A.; Fandrick, D. R.; Song, J. J.; Sananayake, C. H. (2011). “The growing impact of catalysis in the pharmaceutical industry”. Adv. Synth. Catal.353: 1825.
Johnson, M. D.; May, S. A.; Calvin, J. R.; Remacle, J.; Stout, J. R.; Dieroad, W. D.; Zaborenko, N.; Haeberle, B. D.; Sun, W-M.; Miller, M. T.; Brannan, J. (2012). “Development and scale-up of a continuous, high-pressure, asymmetric hydrogenation reaction, workup, and isolation”. Org. Process Res. Rev.16: 1017.
The European Medicines Agency (EMA) and Australia’s Therapeutic Goods Administration (TGA) have announced that they are to share the full assessment reports related to marketing authorisations (MA) for orphan drugs.
‘It could still be a life-changer for them,’ experts say, but they also add that while this breakthrough is promising, ‘there is no miracle cure on the way’ for paralysis.
Electricity may provide hope to men and women who suffer paralysis.
Three years ago, doctors reported that zapping a paralyzed man’s spinal cord with electricity allowed him to stand and move his legs. Now they’ve done the same with three other patients, suggesting their original success was no fluke.
“This is wonderful news. Spinal cord injury need no longer be a lifelong sentence of paralysis,” said Dr. Roderic Pettigrew, director of the National Institute of Biomedical Imaging and Bioengineering, one of the National Institutes of Health, according to NBC News. “It is just downright marvelous.”
“The big message here is that people with spinal cord injury of the type these men had no longer need to think they have…
Dacomitinib has advanced to several Phase III clinical trials. The results of the first trials were disappointing, with a failure to meet the study goals,[2][3][4] Additional Phase III trials are ongoing.[2]
Dacomitinib is a HER (erbB) inhibitor in clinical trial development at Pfizer for the treatment of advanced non-small cell lung cancer (NSCLC) and for the treatment of relapsed/recurrent glioblastoma.
No recent development has been reported for research into the treatment of recurrent and/or metastatic head and neck squamous cell cancer. In 2012, Pfizer and SFJ Pharmaceuticals signed a codevelopment agreement for dacomitinib for the treatment of patients with locally advanced or metastatic NSCLC with activating mutations of epidermal growth factor receptor.
Substituted 4-phenylamino-quinazolin-6-yl-amides useful in the treatment of cancer have been described in the art, including those of U.S. Pat. No. 5,457,105 (Barker), U.S. Pat. No. 5,760,041 (Wissner et al.), U.S. Pat. No. 5,770,599 (Gibson), U.S. Pat. No. 5,929,080 (Frost), U.S. Pat. No. 5,955,464 (Barker), U.S. Pat. No. 6,251,912 (Wissner et al.), U.S. Pat. No. 6,344,455 (Bridges et al.), U.S. Pat. No. 6,344,459 (Bridges et al.), U.S. Pat. No. 6,414,148 (Thomas et al.), U.S. Pat. No. 5,770,599 (Gibson et al.), U.S. patent application 2002/0173509 (Himmelsbach et al.), and U.S. Pat. No. 6,323,209 (Frost).
Dacomitinib is a pan-human epidermal growth factor receptor (pan-HER) inhibitor developed by Pfizer, as ー small molecules targeting ffiR-1, HER-2 and HER-4 tyrosine kinase inhibitor by irreversibly binding to HER-l, HER-2, HER-4 and anti-tumor effect.Ni-line treatment of non-small cell lung cancer (NSCLC) display, Dacomitinib in non-small cell lung cancer Dinner erlotinib compared to some extend on progression-free survival and quality of life have mentioned the smell.
_4] Structural formula for Dacomitinib
[0005] U.S. patent US7772243 Dacomitinib first proposed a synthesis method, first, a fluorine-2_ _4_ amino acid and formamidine ring closure reaction to give 7 – fluoro-4 – quinazolinone, nitration and then successively chlorination reaction, to give 4 – chloro-7 – fluoro-6 – nitro-quinazoline; another aspect ー 3 – chloro-4 – amino-substituted on a fluoroaniline to give 3 – chloro – # – (3,4 – ni section yl methoxy)-4_ fluoro-aniline, obtained after the coupling of both an amino-protected N-(3 – chloro-4 – fluorophenyl)-7 – fluoro-6 – nitro-quinazoline -4 – amine, protected amino N-(3 – chloro-4 – fluorophenyl
Yl)-7_ fluoro-6 – nitro-quinazolin-4 – amine is of formula
Followed by a methoxy group, an amidation reaction and hydrogenation, the final deprotection ko under the action of trifluoroacetic acid to give the final product Dacomitinib.Throughout the reaction as follows:
Synthesis ー kind EGFR inhibitors Dacomitinib, synthetic route for
A synthetic method EGFR inhibitors Dacomitinib, concrete steps are as follows:
Step I, 7 – fluoro-4 – Synthesis of quinazolinone:
30 g (0.1934mol) 2 – fluoro-amino acid was dissolved in 250 ml _4_ formamide among the reaction was heated to 150 ° C for 6 inch, TLC plates to determine the point of completion of the reaction.The reaction was poured hot into 2000 ml of ice water, filtered, the filter cake was washed with water, vacuum dried at 50 ° C for 14 hours to give a pale brown solid powder 7 – fluoro-4 – quinazolinone, 28 g, yield 88%.
Concentrated sulfuric acid (50 ml) and fuming nitric acid (50 ml) mixture was cooled with an ice bath to (TC hereinafter under stirring slowly added 25 g (0.1523mol) 7 – fluoro-4 – quinazolinone , the addition was complete, the reaction mixture was stirred at room temperature for I hour and then the reaction was heated to 110 ° C for 2 inch, TLC plates to determine the point of completion of the reaction the reaction was cooled to room temperature, 300 ml of ice water, the precipitated solid was stirred for 30 minutes , filtered, the filter cake was washed with water, vacuum dried at 50 ° C in 14 hours to give a yellow solid powder 7 – fluoro-6 – nitro-4 – (hydrogen) – quinazolinone, 26 g, yield 82%.
24 g (0.1148mol) 7 – fluoro-6 – nitro _4_ (hydrogen) – quinazolinone was dissolved in 400 ml of methanol was added 2 g of palladium / carbon catalyst was added 8 ml of concentrated hydrochloric acid, and hydrogen was 2 small inch atmospheric reaction, TLC plates to determine the point of the reaction is complete.The catalyst was removed by suction filtration through celite, washed several fitness methanol, and the filtrate was concentrated by rotary evaporation to dryness to give 6 – amino-7_ fluoro-4 – (hydrogen) – quinazolinone, yellow powder, 20 g, yield 97%.
18 g (0.1006mol) 4 – bromo-methyl crotonate dissolved in 180 ml of methylene chloride ni added 27.9 g (0.2019mol) potassium carbonate, cooled to ice-bath (TC, was slowly added dropwise 10 ml (0.1012mol ) piperidine, (I reaction was stirred under a small inch TC, TLC plates to determine the point of completion of the reaction was concentrated by rotary evaporation to dryness, to give (E) -4 – (piperidin-1 – yl) – 2 – butenoic acid methyl Cool as a yellow solid, 17.1 g, yield 93%.
16 g (0.0873mol) of W) -4 – (piperidin-_1_ yl) -2 – butenyl acetate and 80 ml of concentrated hydrochloric acid was added to 250 ml of 1,4 – ni oxygen dioxane, heated under reflux 20 hours inch, TLC plate point the reaction was determined complete, the reaction solution was concentrated by rotary evaporation to dryness surplus was recrystallized from isopropanol to give a pale yellow solid, Buddhist) _4-(piperidin-1 – yl) -2 – butene acid hydrochloride, 14.5 g, yield 81%.
13 g (0.0632mol) of (K) ~ 4 ~ (piperidin-1 – yl) -2 – butene acid hydrochloride was dissolved in 750 ml of methylene chloride ni, 5 ml of DMF, was slowly added dropwise 8 ml ( 0.0933mol) of oxalyl chloride, the reaction was stirred at room temperature for I h, TLC plates to determine the point of completion of the reaction, the reaction solution was concentrated to dryness by rotary evaporation to give a pale yellow oil, Buddhist) _4-(piperidin-1 – yl) -2 – butyl allyl chloride, 11.8 g, yield 99%.
11 g (0.0586mol) of the) -4 – (piperidin-1 – yl) – 2 – butenyl chloride ni chloride (50 ml) was slowly added dropwise to 6 – amino-1 – fluoro-4 – ( hydrogen) – quinazolinone (7 g, 0.0391mmol), three ko amine (14 ml) and the mixture was ni chloride (200 ml), the reaction mixture was stirred at room temperature for 2 hours the reaction inch, TLC determined the completion of reaction points board , was added 800 liters of halo ni halo chloroformate and 500 liters of burning the separated organic phase was washed with 500 liters of halo, halo and then with 500 liters of brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness was subjected to silica gel surplus Column chromatography (30% acid ko ko acetate / hexane) to give (M)-N-(7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butenamide, as a pale yellow solid, 12.3 g, yield 95%.
I ^ xN MeONa N. Under nitrogen atmosphere, to 100 ml of anhydrous methanol was slowly added 1.52 g of sodium metal (0.0661mol), stirred for 10 minutes to dissolve all of the sodium metal to the completion of the reaction, to obtain a freshly prepared solution of sodium methoxide, and the The sodium methoxide solution was added 11 g (0.0333mol) of (receive) (7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) 2_ butene-amide, the reaction was heated to reflux for 3 inch, TLC plates to determine completion of the reaction point, cooled to room temperature, acidified with 2N hydrochloric acid solution to pH = 3 ~ 4, and concentrated by rotary evaporation to dryness, the residue was washed with water beating, filtration, The filter cake was washed with water, vacuum dried at 50 ° C in 14 hours to give (article) – # – (7 – methoxy – 4 – oxo – ni hydrogen quinazolin-6 – yl) – 4_ (piperidin-1 – yl)-2_ butenamide yellow solid, 10.6 g, yield 93%.
9 g (0.0263mol) of (receive) – # – (7 – methoxy _4_ oxo – ni hydrogen quinazolin-6 – yl)-4_ (piperidin-1 – yl) – 2_ butenamide were added to 40 ml of phosphorus oxychloride was heated under reflux for 2 inch, TLC plates to determine the point of completion of the reaction, the reaction solution was concentrated to dryness by rotary evaporation, ice water was added surplus, beating, filtered, the cake washed with washed with water, vacuum dried at 50 ° C in 14 hours to give {W,-N-(4 – chloro-7 – methoxy-quinazolin-6 – yl) -4 – (piperidin-1 – yl) – 2 – butene amide as a yellow solid, 7 g, yield 74%
0.0166mol), 3 – chloro-4-fluoro-aniline (2.6 g, 0.0179mol) and three ko amine (2.6 ml, 0.0186mol) was added to 140 ml of isopropanol and the reaction was heated to reflux for 3 inch, TLC plates to determine the point completion of the reaction, cooled to room temperature, filtered, the filter cake washed with methanol, vacuum dried at 50 ° C in 14 hours to give the final product Dacomitinib, a yellow solid, 6.6 g, yield 84%.
Scheme 1, wherein the 4-position aniline group is represented a 4-fluoro-3-chloro aniline group.
4-Chloro-7-fluoro-6-nitroquinazoline (7) can be prepared by methods similar to those described in J.Med. Chem. 1996, 39, 918-928. Generally, 2-amino-4-fluoro-benzoic acid (1) can be reacted with formamidine (2) and acetic acid (3) in the presence of 2-methoxyethanol to provide 7-Fluoro-3H-quinazolin-4-one (4). The 7-fluoro-3H-quinazolin-4-one (4) can be nitrated to 7-fluoro-6-nitro-3H-quinazolin-4-one (5), which can be treated with thionyl chloride to yield 4-chloro-6-nitro-7-fluoro-3H-quinazoline (6). The 4-chloro-quinazoline compound (6) can be combined with a desirably substituted aniline, represented above by 4-fluoro-3-chloro-aniline, in the presence of a tertiary amine and isopropanol to provide the 4-anilino-6-nitro-7-fluoro-quinazoline (7).
The 4-anilino-6-nitro-7-fluoro-quinazoline (7) may be reacted with an alcohol of the formula R3OH, wherein R3 is as defined above, to yield the 7-alkoxylated compound (8). Reduction of the 6-nitro compound (8) provides the 6-amino analog (9).
The 6-position amino compound (9) may be reacted with a haloalkenoyl chloride (12), such as a 4-bromo-but-2-enoyl chloride, 5-bromo-pent-2-enoyl chloride, 4-chloro-but-2-enoyl chloride, or 5-chloro-pent-2-enoyl chloride, to provide an alkenoic acid[4-anilino]-7-alkoxylated-quinazolin-6-yl-amide (13). Haloalkenoyl chloride agents useful in this scheme may be prepared by methods known in the art, such as the treatment of a relevant haloalkenoic acid, represented by bromoalkenoic acid ester (10), with a primary alcohol, yielding the corresponding haloalkenoic acid (11), which may in turn be treated with oxalyl chloride to provide the desired haloalkenoyl chloride (12).
Finally, the quinazoline-6-alkanoic acid compound (13) may be treated with a cyclic amine, such as piperidine, piperazine, etc., to provide the desired final compound (14).
The title compound and other 7-methoxy analogs of this invention can be prepared as described in Example 1 by replacing the 2-fluoroethanol used in Example 1 with stoichiometric amount of methanol.
EXAMPLE 34-Piperidin-1 -yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxv -quinazolin-6-yl]-amide (Synthetic Route No. 2)
An alternative synthetic route for compounds of this invention involves preparing the 6-position substituent chain as a Het-alkenoyl chloride as depicted in Scheme 2, below.
It will be understood that other compounds within this invention may be prepared using Het-butenoyl halide, Het-pentenoyl halide and Het-hexenoyl halide groups of the formula:
wherein R4 is as described herein and halo represents F, Cl, Br or I, preferably Cl or Br. One specific group of these Het-alkenoyl halides includes those compounds in which halo is Cl or Br, R4 is —(CH2)m-Het, m is an integer from 1 to 3, and Het is piperidine or the substituted piperidine moieties disclosed above.
EXAMPLE 44-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide (Synthetic Route No. 3)
3-Chloro-4-fluoro-phenylamine 15 (50.31, 345.6 mmole) and 3,4-Dimethoxy-benzaldehyde 16 (57.43 g, 345.6 mmole) were mixed in 500 ml of IPA and cooled in an ice-water. The glacial acetic acid was added (20.76 g, 345.6 mole) and then sodium cyanoborohydride in one portion. The reaction was stirred at room temperature (RT) for 24 hrs. 250 mL of 10% NaOH was added dropwise at RT after the reaction was completed. The mixture was stirred for ½ hr. The slurry was then filtered and washed with IPA and dried in vacuo. The mass weight 88.75 g (17, 87%).
Compounds 6 (3 g, 13.18 mmole) and 17 (3.9 g, 13.18 mmole) were combined in CH3CN (25 mL) and heated for one hr. Mass spectroscopy indicated no starting material. Saturated K2CO3 was added and the reaction was extracted 3× with EtOAc. The organic layers were combined, washed with brine and concentrated in vacuo to give 6.48 g of 7 (78.4%).
Compound 7 (72.76 g, 149.4 mmole) was added to a cool solution of NaOMe in 1.5 L of dry MeOH under N2. The cooling bath was removed and the mixture was heated to reflux and stirred for 1 hr. The reaction was cooled to room temperature and quenched with water until the product precipitated out. The solid was filtered and washed with water and hexanes. The product was slurred in refluxing EtOAc and filtered hot to provide 68.75 g of yellow soled 8 (73%).
Compound 8 (63.62 g, 127.5 mole) was hydrogenated using Raney/Ni as catalyst to obtain 43.82 g of 9 (100%). Oxalyl chloride (6.5 g, 51.18 mmole) was added slowly to a suspension of 13 (10.5 g, 51.2 mmole) in 200 ml of dichloromethane containing 8 drops of DMF, after the reaction become homogeneous, the solvent was removed and the residual light yellow solid was slurred in 200 ml of DMAC and 9 (20 g, 42.65 mmole) was added gradually as a solid. The reaction was stirred for 15 min. and poured slowly into 1N NaOH. The mixture was extrated 3× EtOAc. The combined organic layers were washed with brine, filtered and concentrated in vacuo to obtain 28.4 g (100%) 10.
Compound 10(13.07 g, 21.08 mmole) was dissolved in trifluoroacetic acid (TFA) (74 g, 649 mmole) and heated to 30° C. for 24 hrs. The reaction was cooled to RT and poured gradually into a cooled 1 N NaO H-brine solution. Precipitate formed and was filtered and washed with 3X water then dried. The precipitate was recrystallized from toluene to obtain pure 4-Piperidin-1-vl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino) -7-methoxv-puinazolin-6-yl]-amide (9.90 g, 89%).
Example 1 is similar but not same…caution
EXAMPLE 1 4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-(2-fluoro-ethoxy)-quinazolin-6-yl]-amide
7-fluoro-6-nitro-4-chloroquinazoline (14.73,g, 65 mmol) was combined with 3-choro-4-fluoroaniline (9.49 g, 65 mmol) and triethylamine (10 mL, 72 mmol) in 150 mL of isopropanol. The reaction was stirred at room temperature for 1.5 hours, resulting in a yellow slurry. The solid was collected by filtration, rinsing with isopropanol and then water. The solid was dried in a 40° C. vacuum oven overnight to give 19.83 g (91%) of the product as an orange solid.
MS (APCI, m/z, M+1): 337.0
NaH (60% in mineral oil, 3.55 g, 88 mmol) was added, in portions, to a solution of 2-fluoroethanol (5.19 g, 80 mmol) in 200 mL THF. The reaction was stirred for 60 minutes at room temperature. To the reaction was added 7-fluoro-6-nitro-4-(3-chloro-4-fluoroaniline)quinazoline (18.11 g, 54 mmol) as a solid, rinsing with THF. The reaction was heated to 65° C. for 26 hours. The reaction was cooled to room temperature and quenched with water. THF was removed in vacuo. The resulting residue was sonicated briefly in water then the solid collected by filtration. The solid was triturated with MeOH, filtered and dried in a 40° C. vacuum oven overnight to 12.63 g of the product. Additional product was obtained by concentrating the MeOH filtrate to dryness and chromatography eluting with 50% EtOAc/hex. The isolated material was triturated with MeOH (2×), filtered and dried. 3.90 g
Total yield: 16.53 g, 81%
MS (APCI, m/z, M+1): 381.0
7-(2-fluoroethoxy)-6-nitro-4-(3-chloro-4-fluoroaniline)quinazoline (0.845 g, 2.2 mmol) in 50 mL THF was hydrogenated with Raney nickel (0.5 g) as the catalyst over 15 hours. The catalyst was filtered off and the filtrate was evaporated to give 0.77 g of product. (99%)
MS (APCI, m/z, M+1): 351.2
Methyl 4-bromocrotonate (85%, 20 mL, 144 mmol) was hydrolyzed with Ba(OH)2 in EtOH/H2O as described in J.Med.Chem. 2001, 44(17), 2729-2734.
MS (APCI, m/z, M−1): 163.0
To a solution of 4-bromocrotonic acid (4.17 g, 25 mmol) in CH2Cl2 (20 mL) was added oxalyl chloride (33 mL, 38 mmoL) and several drops of DMF. The reaction was stirred at room temperature for 1.5 hours. The solvent and excess reagent was removed in vacuo. The resulting residue was dissolved in 10 mL THF and added to a 0° C. mixture of 6-amino-7-(2-fluoroethoxy)-4-(3-chloro-4-fluoroaniline)quinazoline (5.28 g, 15 mmol) and triethylamine (5.2 mL, 37 mmol). The reaction was stirred at 0° C. for 1 hour. Water was added to the reaction and the THF removed in vacuo. The product was extracted into CH2Cl2 (400 mL). The organic layer was dried over MgSO4, filtered and concentrated. The crude material was chromatographed on silica gel eluting with 0-4% MeOH/CH2Cl2. An isolated gold foam was isolated. Yield: 4.58 g, 61%
MS (APCI, m/z, M−1): 497.1
Piperidine (0.75 mL, 6.7 mmol) was added to a solution of the above compound (3.35 g, 6.7 mmol) and TEA (2.80 mL, 20 mmol) in 10 mL DMA at 0° C. The reaction was stirred at 0° C. for 17 hours. Water was added to the reaction until a precipitate was evident. The reaction was sonicated for 40 minutes and the liquid decanted. The residue was dissolved in CH2Cl2, dried over MgSO4, filtered and concentrated. The material was chromatographed on silica gel eluting with 4-10% MeOH/CH2Cl2. The isolated residue was triturated with acetonitrile (2×) and collected by filtration. Impurity found: Michael addition of piperidine (2.2% in first trituration of acetonitrile). Additional material can be obtained from the acetonitrile filtrates.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






































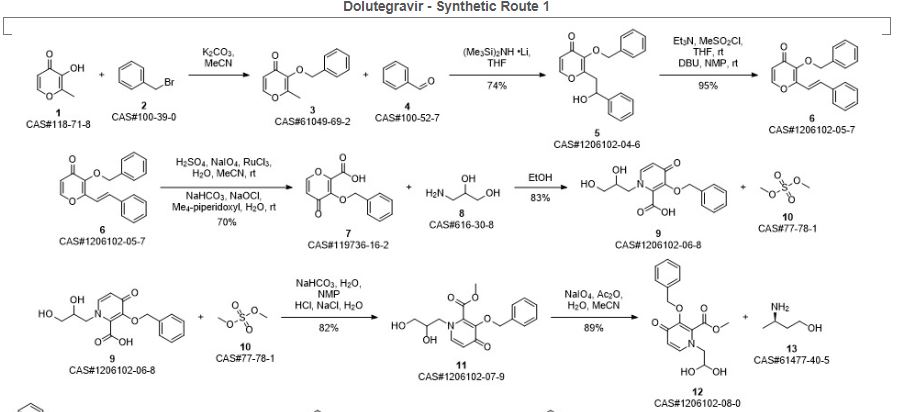
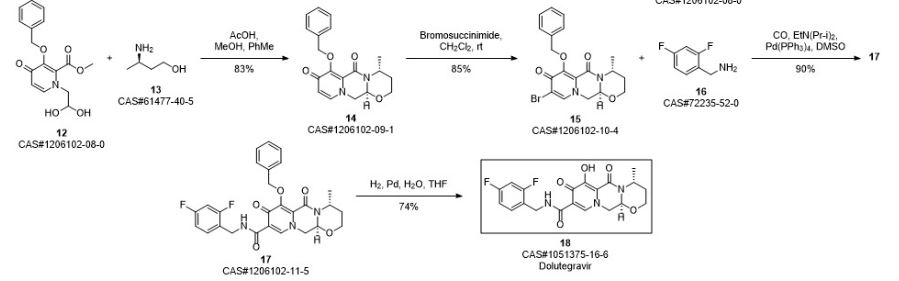







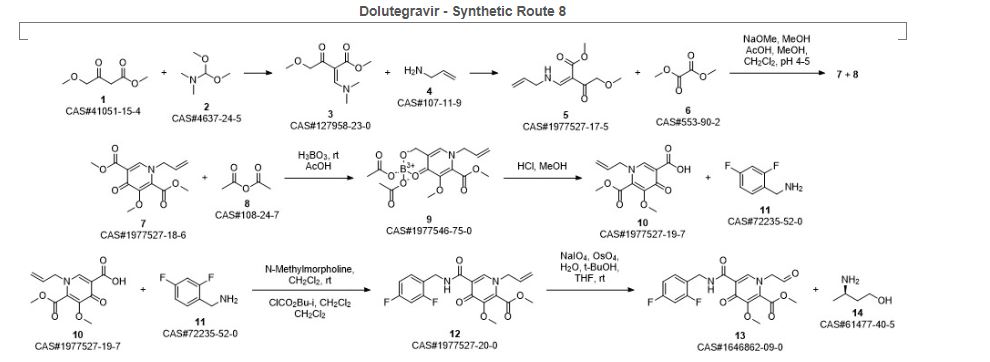

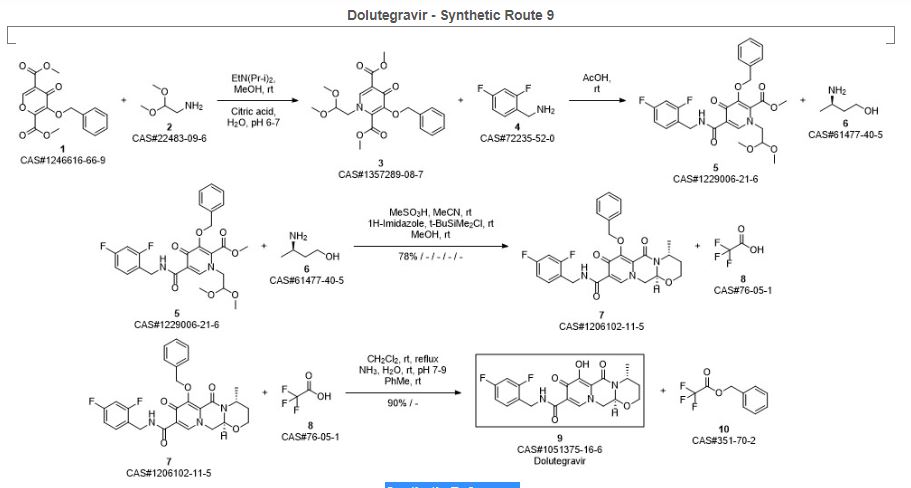

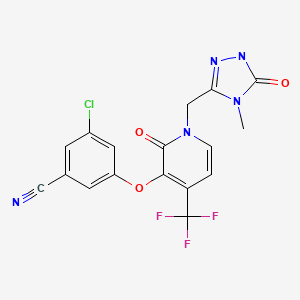





























 apigenin
apigenin






![VTO=\frac{\text{nominal volume of all reactors} [m^3]*\text{time per batch} [h]}{\text{output per step} [kg]}](https://i0.wp.com/upload.wikimedia.org/math/7/9/a/79aca4fd95405c01c04df9181fb3ee63.png)















 Photo by Rob Hill/MU Publications
Photo by Rob Hill/MU Publications


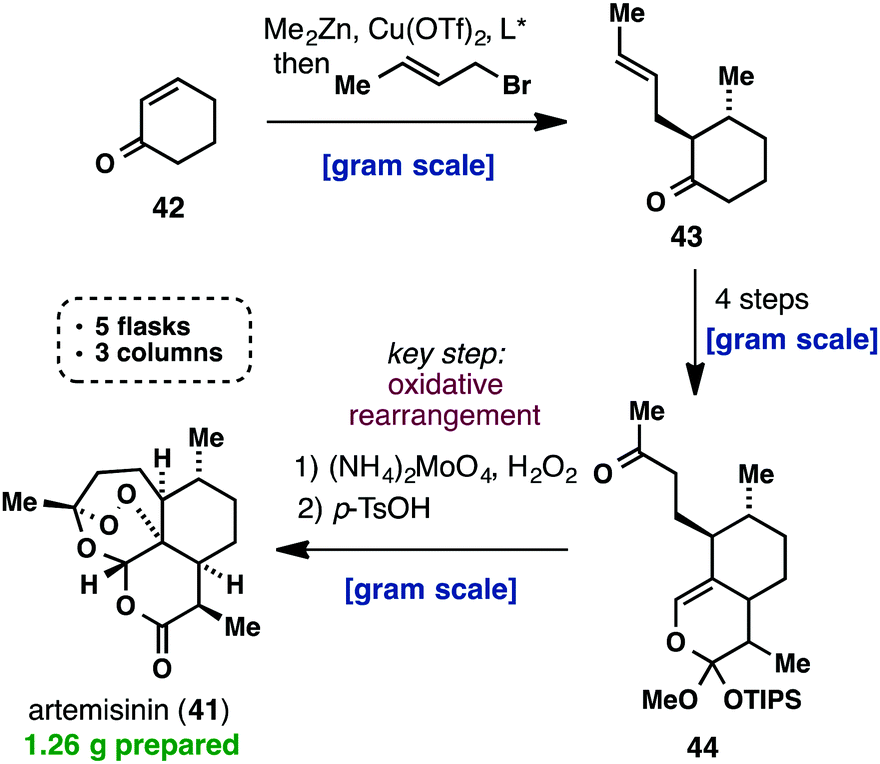





























.svg){kind=link}
{kind=link}