")
Anna Popova, Head of the Federal Service for Supervision of Consumer Protection and Welfare (Rospotrebnadzor)
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » 2014 (Page 27)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Anna Popova, Head of the Federal Service for Supervision of Consumer Protection and Welfare (Rospotrebnadzor)
MOSCOW, August 5 (RIA Novosti) – A Russian vaccine against Ebola hemorrhagic fever is now undergoing preclinical tests, Russian consumer rights watchdog, Rospotrebnadzor, head Anna Popova told journalists.
Russian Ebola Vaccine in Preclinical Trials
RIA Novosti
As of today, it is in a stage of preclinical drug trials. The works are intensified now,” Popova said. She added that there are currently no licensed drugs …
see link
http://en.ria.ru/russia/20140805/191739156/Russian-Ebola-Vaccine-in-Preclinical-Trials.html




Identifying target product profile (TPP). TPP has been defined as a “prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and thus the safety and efficacy, of a drug product is realized”. This includes dosage form and route of administration, dosage form strength(s), therapeutic moiety release or delivery and pharmacokinetic characteristics (e.g., dissolution and aerodynamic performance) appropriate to the drug product dosage form being developed and drug product-quality criteria (e.g., sterility and purity) appropriate for the intended marketed product. The concept of TPP in this form and its application is novel in the QbD paradigm.
Identifying CQAs. Once TPP has been identified, the next step is to identify the relevant CQAs. A CQA has been defined as “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality”10. Identification of CQAs is done through risk assessment as per the ICH guidance Q9 . Prior product knowledge, such as the accumulated laboratory, nonclinical and clinical experience with a specific product-quality attribute, is key in making these risk assessments. Such knowledge may also include relevant data from similar molecules and data from literature references. Taken together, this information provides a rationale for relating the CQA to product safety and efficacy. The outcome of the risk assessment would be a list of CQAs ranked in order of importance. Use of robust risk assessment methods for identification of CQAs is novel to the QbD paradigm.
Defining product design space. After CQAs for a product have been identified, the next step is to define the product design space (that is, specifications for in-process, drug substance and drug product attributes). These specifications are established based on several sources of information that link the attributes to the safety and efficacy of the product, including, but not limited to, the following:
The difference between the actual experience in the clinic and the specifications set for the product would depend on our level of understanding of the impact that the CQA under consideration can have on the safety and efficacy of the product. For example, taking host cell proteins as a CQA, it is common to propose a specification that is considerably broader than the clinical experience. This is possible because of a greater ability to use data from other platform molecules to justify the broader specifications. On the other hand, in the case of an impurity that is unique to the product, the specifications would rely solely on clinical and nonclinical studies.
In QbD, an improved understanding of the linkages between the CQA and safety and efficacy of the product is required. QbD has brought a realization of the importance of the analytical, nonclinical and animal studies in establishing these linkages and has led to the creation of novel approaches.
Defining process design space. The overall approach toward process characterization involves three key steps. First, risk analysis is performed to identify parameters for process characterization. Second, studies are designed using design of experiments (DOE), such that the data are amenable for use in understanding and defining the design space. And third, the studies are executed and the results analyzed to determine the importance of the parameters as well as their role in establishing design space.
Failure mode and effects analysis (FMEA) is commonly used to assess the potential degree of risk for every operating parameter in a systematic manner and to prioritize the activities, such as experiments, necessary to understand the impact of these parameters on overall process performance. A team consisting of representatives from process development, manufacturing and other relevant disciplines performs an assessment to determine severity, occurrence and detection. The severity score measures the seriousness of a particular failure and is based on an estimate of the severity of the potential failure effect at a local or process level and the potential failure effect at end product use or patient level. Occurrence and detection scores are based on an excursion (manufacturing deviation) outside the operating range that results in the identified failure. Although the occurrence score measures how frequently the failure might occur, the detection score indicates the probability of timely detection and correction of the excursion or the probability of detection before end product use. All three scores are multiplied to provide a risk priority number (RPN) and the RPN scores are then ranked to identify the parameters with a high enough risk to merit process characterization. FMEA outcome for a process chromatography step in a biotech process. RPN scores are calculated and operating parameters with an RPN score >50 are characterized using a qualified scaled-down model. For the case study presented here, these include gradient slope, temperature, flow rate, product loading, end of pool collection, buffer A pH, start of pool collection, volume of wash 1, buffer B pH, buffer C pH and bed height. Process characterization focused on parameters such as temperature, that have a high impact on the process (severity = 6), occur frequently in the manufacturing plant (occurrence = 6) and are difficult to quickly correct if detected (detection = 7). In contrast, parameters such as equilibration volume, with a low impact on the process (severity = 3), low occurrence (occurrence = 2) and a limited ability to detect and correct (detection = 5), were not examined in process characterization.
I liked this pic
![]()
SEE AT
http://www.tevapharm.com/Media/EventsUpdates/Pages/Quality-by-Design.aspx


Shanghai Natural Bio-engineering Co., Ltd, export branch of Hunan Keyuan Bio-products co., Ltd, established in 2003, is a professional large-scale high-tech manufacturer of raw materials for nutraceuticals, nutritional supplements, and pharmaceuticals. Plant extracts, Active Pharmaceutical Ingredient (API) & intermediates are our focused areas.Key products include resveratrol, curcumin,artemisinin,artemether,artesunate,dihydroartemisinin,Lumefantrine,etc
link is
posted by
Sales Manager at Shanghai Natural Bio-engineering Co., Ltd
| synonyms | Japanese knotweed extract, Polygonum cuspidatum, red wine extract, trans-3,5,4′-trihydroxystilbene, trans-Resveratrol, cis-resveratrol |
|---|---|
| CAS number | 501-36-0 |
| Latin Name | Polygonum cuspidatum |
| Botanical source | 1.Japanese knotweed plant Polygonum cuspidatum 2. red wine 3. red grape extracts |
| Molecular Formula | C14H12O3 |
| Molecular weight | 228.24 |
| Appearance | white powder with slight yellow |
| Solubility in water | 0.03 g/L |
| Dosage | 500mg |
| Key benefits | Anti-aging, Anti-Cancer, cardiovascular support, regulate estrogen level, weight loss |
| Applied industry | Sports nutrition, nutraceuticals, cosmetics |
When talk about resveratrol, we have to mention red wine since resveratrol is first popularly known in red wine. In fact, resveratrol was actually first isolated in 1940 from white hellebore roots by the Japanese scientist Michio Takaoka. Red wine, in moderation, has long been thought of as heart healthy. However, the most popular source of resveratrol is from Japanese knotweed extract (Latin name:Polygonum cuspidatum)
Resveratrol (3,5,4′-trihydroxystilbene) is a polyphenolic phytoalexin. It is a stilbenoid, a derivate of stilbene, and is produced in plants with the help of the enzyme stilbene synthase.
Resveratrol exists as two geometric isomers: “cis-” (“Z”) and “trans-” (“E”). The ”trans-” form can undergo isomerisation to the “cis-” form when exposed to ultraviolet irradiation. Trans-resveratrol in the powder form was found to be stable under “accelerated stability” conditions of 75% humidity and 40 degrees C in the presence of air. Resveratrol content also stayed stable in the skins of grapes and pomace taken after fermentation and stored for a long period.
The resveratrol in red wine comes from the skin of grapes used to make wine. Because red wine is fermented with grape skins longer than is white wine, red wine contains more resveratrol. Simply eating grapes, or drinking grape juice, has been suggested as one way to get resveratrol without drinking alcohol. Red and purple grape juices may have some of the same heart-healthy benefits of red wine.
Other foods that contain some resveratrol include peanuts, blueberries and cranberries. It’s not yet known how beneficial eating grapes or other foods might be compared with drinking red wine when it comes to promoting heart health. The amount of resveratrol in food and red wine can vary widely.
Numerous studies have been conducted regarding various purported resveratrol benefits. Studies have primarily been conducted on laboratory animals, and while human search is very promising, is still in its earliest stages. Current research into resveratrol benefits points to resveratrol having amazing anti-aging properties, hence dubbed “The Fountain of Youth.” Many other key benefits such as cardiovascular effects, anti-cancer, estrogen regulating effects are mentioned here.
1.Resveratrol and its anti-aging benefits
The study by Harvard Medical School researchers shows that resveratrol stimulates production of SIRT1, a serum that blocks diseases by speeding up the cell’s energy production centers known as mitochondria.
Resveratrol affects the activity of enzymes called sirtuins. Sirtuins control several biological pathways and are known to be involved in the aging process. Resveratrol is only one of many natural and synthetic sirtuin-activating compounds (STACs) now known. Certain metabolic diseases, including type 2 diabetes and heart disease, tend to strike as we age. In animal studies, severely restricting calories can help prevent some of these diseases. Over a decade ago, researchers found that resveratrol can mimic calorie restriction in some ways and extend the lifespans of yeast, worms, flies and fish.
2.Resveratrol and cardiovascular benefits
Resveratrol is famous for its Cardioprotective effects.According to Wikipedia, moderate drinking of red wine has long been known to reduce the risk of heart disease. This is best known as “the French paradox”.
Studies suggest resveratrol in red wine may play an important role in this phenomenon. It achieves the effects by the following functions: (1) inhibition of vascular cell adhesion molecule expression;(2) inhibition of vascular smooth muscle cell proliferation;(3) stimulation of endolethelial nitric oxide synthase (eNOS) activity;(4) inhibition of platelet aggregation;and (5) inhibition of LDL peroxidation.
The cardioprotective effects of resveratrol also are theorized to be a form of preconditioning—the best method of cardioprotection, rather than direct therapy.Study into the cardioprotective effects of resveratrol is based on the research of Dipak K. Das, however, who has been found guilty of scientific fraud and many of his publications related to resveratrol have been retracted. A 2011 study concludes, “Our data demonstrate that both melatonin and resveratrol, as found in red wine, protect the heart in an experimental model of myocardial infarction via theSAFE pathway.”
Resveratrol, a polyphenol in red wine, induces nitric oxide (NO) synthase, the enzyme responsible for the biosynthesis of NO, in cultured pulmonary artery endothelial cells, suggesting that Resveratrol could afford cardioprotection by affecting the expression of nitric oxide synthase.
3.Reveratrol and anti-cancer benefits
Experts already claim it can help you beat cancer – from brain tumours to breast, colon, prostate cancers and many more. Resveratrol is being studied to see how it affects the initiation, promotion, and progression of cancer. With regard to tumor initiation, it has been shown to act as an antioxidant by inhibiting free radical formation and as an anti-mutagen in rat models. Studies related to progression have found that resveratrol induced human promyelocytic leukemia cell differentiation, inhibited enzymes that promote tumor growth, and exerted antitumor effects in neuroblastomas. Noting that in animal studies, resveratrol was effective against tumors of the skin, breast, gastrointestinal tract, lung, and prostate gland. Memorial Sloan-Kettering, the American pillar of cancer treatment, conducted research on theinflammatory effects on cells leading to cancer. It is widely known that an enzyme, COX-2, lies behind the stimulation of localised hormones (eicosanoids) causing inflammation, the precursor to cancer. In the research Resveratrol completely turned off the COX-2 driver. MD Anderson´s studies have shown this same anti-inflammatory benefit too. Plus, after conversion in the liver to a sulphated form the compound can attack several of the steps in the cancer process even killing cancer cells.
4. The Benefits of Resveratrol Weight Loss
Resveratrol is actually a very popular nutrient that has been shown on Dr. Oz, Oprah, Barbara Walters, and a number of other national television shows. It is quickly becoming one of the country’s best natural supplements.
How does Resveratrol help you lose weight? Resveratrol on its own will not be effective at helping you to lose weight, but you have to use it in conjunction with exercise and a proper diet if you really want to obtain the maximum benefits from the supplement.
However, the vitamin, when in concentrated form, has been proven to help speed up the metabolism. This speeding up of the metabolism causes the body to metabolize and process to food consumed faster, which causes the calories in the food to be used more effectively. When the body metabolizes food faster, there is less risk of excess calories being stored in the body in the form of fat.
However, in order to ensure that Resveratrol actually works, you need to take sufficient amounts of the vitamin. The supplement is effective because it is a concentrated form of the helpful vitamin, and taking the supplement is the best way to ensure that Resveratrol works effectively in helping you shed those excess pounds.
Another way Resveratrol helps you to lose weight is through reducing the amounts of estrogen that your body produces. Estrogen increases body fat and decreases muscle mass, so reducing the amounts of estrogen produced by your body will help you lose weight and build muscle. Taking Resveratrol can be a good way to ensure that your body doesn’t produce the amounts of estrogen that will keep it from building muscle.
Because there have been very few studies conducted on resveratrol in humans, doctors still can’t confirm what adverse effects these supplements might have on people over the long term. So far, studies have not discovered any severe side effects, even when resveratrol is taken in large doses. However, resveratrol supplements might interact with blood thinners such as warfarin (Coumadin), and nonsteroidal anti-inflammatory medications such as aspirin and ibuprofen, increasing the risk for bleeding.
Like other supplements, resveratrol isn’t regulated by the FDA, so it’s difficult for consumers to know exactly what they’re getting when they buy a bottle, or whether the product is actually effective.
There also isn’t any specific dosage recommendation, and dosages can vary from supplement to supplement. The dosages in most resveratrol supplements are typically far lower than the amounts that have been shown beneficial in research studies. Most supplements contain 250 to 500 milligrams of resveratrol. To get the equivalent dose used in some animal studies, people would have to consume 2 grams of resveratrol (2,000 milligrams) or more a day.
Fallopia japonica, commonly known as Japanese knotweed, is a large, herbaceous perennial plant of the family Polygonaceae, native toEastern Asia in Japan, China and Korea. In North America and Europe the species is very successful and has been classified as aninvasive species in several countries. Japanese knotweed has hollow stems with distinct raised nodes that give it the appearance ofbamboo, though it is not closely related. While stems may reach a maximum height of 3–4 m each growing season, it is typical to see much smaller plants in places where they sprout through cracks in the pavement or are repeatedly cut down. The leaves are broad oval with a truncated base, 7–14 cm long and 5–12 cm broad,[1] with an entire margin. The flowers are small, cream or white, produced in erectracemes 6–15 cm long in late summer and early autumn.
Closely related species include giant knotweed (Fallopia sachalinensis, syn. Polygonum sachalinense) and Russian vine (Fallopia baldschuanica, syn. Polygonum aubertii, Polygonum baldschuanicum).
Other English names for Japanese knotweed include fleeceflower, Himalayan fleece vine, monkeyweed, monkey fungus, Hancock’s curse, elephant ears, pea shooters, donkey rhubarb (although it is not a rhubarb), sally rhubarb, Japanese bamboo, American bamboo, and Mexican bamboo (though it is not a bamboo). In Chinese medicine, it is known as Huzhang (Chinese: 虎杖; pinyin: Hǔzhàng), which translates to “tiger stick.” There are also regional names, and it is sometimes confused with sorrel. In Japanese, the name is itadori (虎杖, イタドリ?).[2]

Old stems remain in place as new growth appears

Erect inflorescence
It is listed by the World Conservation Union as one of the world’s worst invasive species.[3]
The invasive root system and strong growth can damage concrete foundations, buildings, flood defences, roads, paving, retaining walls and architectural sites. It can also reduce the capacity of channels in flood defences to carry water.[4]
It is a frequent colonizer of temperate riparian ecosystems, roadsides and waste places. It forms thick, dense colonies that completely crowd out any other herbaceous species and is now considered one of the worst invasive exotics in parts of the eastern United States. The success of the species has been partially attributed to its tolerance of a very wide range of soil types, pH and salinity. Its rhizomes can survive temperatures of −35 °C (−31 °F) and can extend 7 metres (23 ft) horizontally and 3 metres (9.8 ft) deep, making removal by excavation extremely difficult.
The plant is also resilient to cutting, vigorously resprouting from the roots. The most effective method of control is by herbicideapplication close to the flowering stage in late summer or autumn. In some cases it is possible to eradicate Japanese knotweed in one growing season using only herbicides. Trials in the Queen Charlotte Islands (Haida Gwaii) of British Columbia using sea water sprayed on the foliage have demonstrated promising results, which may prove to be a viable option for eradication where concerns over herbicide application are too great.[citation needed]
Two biological pest control agents that show promise in the control of the plant are the psyllid Aphalara itadori[5] and a leaf spotfungus from genus Mycosphaerella.[6]
It is classed as an unwanted organism in New Zealand and is established in some parts of the country.[7]
In the UK, Japanese Knotweed is established in the wild in many parts of the country and creates problems due to the impact on biodiversity, flooding management and damage to property. It is an offence under section 14(2) of the Wildlife and Countryside Act 1981 to “plant or otherwise cause to grow in the wild” any plant listed in Schedule nine, Part II to the Act, which includes Japanese knotweed. It is also classed as “controlled waste” in Britain under part 2 of the Environmental Protection Act 1990. This requires disposal at licensed landfill sites. The species is expensive to remove; Defra‘s Review of Non-native Species Policy states that a national eradication programme would be prohibitively expensive at £1.56 billion.[8]
The decision was taken on 9 March 2010 in the UK to release into the wild a Japanese psyllid insect, Aphalara itadori.[9] Its diet is highly specific to Japanese knotweed and shows good potential for its control.[10][11]
In Scotland, the Wildlife and Natural Environment (Scotland) Act 2011 came into force in July 2012 that superseded the Wildlife and Countryside Act 1981. This act states that is an offence to spread intentionally or unintentionally Japanese knotweed (or other non-native invasive species).
The weed can be found in 39 of the 50 United States[12] and in six provinces in Canada. It is listed as an invasive weed in Maine,Ohio, Vermont, Virginia, West Virginia, New York, Alaska, Pennsylvania, Michigan, Oregon and Washington state.[13]

A variegated variety of Japanese Knotweed, used as a landscape plant
Japanese knotweed flowers are valued by some beekeepers as an important source of nectar for honeybees, at a time of year when little else is flowering. Japanese knotweed yields a monofloral honey, usually called bamboo honey by northeastern U.S. beekeepers, like a mild-flavored version of buckwheat honey (a related plant also in the Polygonaceae).
The young stems are edible as a spring vegetable, with a flavor similar to extremely sour rhubarb. In some locations, semi-cultivating Japanese knotweed for food has been used as a means of controlling knotweed populations that invade sensitive wetland areas and drive out the native vegetation.[14] It is eaten in Japan as sansai or wild foraged vegetable.
Similarly to rhubarb, knotweed contains oxalic acid, which when eaten may aggravate conditions such as rheumatism, arthritis, gout, kidney stones or hyperacidity.[15]
Both Japanese knotweed and giant knotweed are important concentrated sources of resveratrol and its glucoside piceid,[16] replacing grape byproducts. Many large supplement sources of resveratrol now use Japanese knotweed and use its scientific name in the supplement labels. The plant is useful because of its year-round growth and robustness in different climates.[17]

This antique locomotive at Beekbergen,Netherlands is overgrown by knotweed. A few years before, it was free of knotweed
Japanese knotweed has a large underground network of roots (rhizomes). To eradicate the plant the roots need to be killed. All above-ground portions of the plant need to be controlled repeatedly for several years in order to weaken and kill the entire patch. Picking the right herbicide is essential, as it must travel through the plant and into the root system below. Glyphosate is the best active ingredient in herbicide for use on Japanese knotweed as it is ’systemic’; it penetrates through the whole plant and travels to the roots.
Digging up the rhizomes is a common solution where the land is to be developed, as this is quicker than the use of herbicides, but safe disposal of the plant material without spreading it is difficult; knotweed is classed as controlled waste in the UK, and disposal is regulated by law.Digging up the roots is also very labor-intensive and not always efficient. The roots can go to up to 10 feet (3 meters) deep, and leaving only a few inches of root behind will result in the plant quickly growing back.
Covering the affected patch of ground with a non-translucent material can be an effective follow-up strategy. However, the trimmed stems of the plant can be razor sharp and are able to pierce through most materials. Covering with non-flexible materials such as concrete slabs has to be done meticulously and without leaving even the smallest splits. The slightest opening can be enough for the plant to grow back.
More ecologically-friendly means are being tested as an alternative to chemical treatments. Soil steam sterilization [18] involves injecting steam into contaminated soil in order to kill subterranean plant parts. Research has also been carried out on Mycosphaerella leafspot fungus, which devastates knotweed in its native Japan. This research has been relatively slow due to the complex life cycle of the fungus.[19]
Research has been carried out by not-for-profit inter-governmental organisation CABI in the UK. Following earlier studies imported Japanese knotweed psyllid insects (Aphalara itadori), whose only food source is Japanese knotweed, were released at a number of sites in Britain in a study running from 1 April 2010 to 31 March 2014. In 2012, results suggested that establishment and population growth were likely, after the insects overwintered successfully.[20][21]

Detail of the stalk
In the United Kingdom, Japanese Knotweed has received a lot of attention in the press as a result of very restrictive lending policies by banks and other mortgage companies. Several lenders have refused mortgage applications on the basis of the plant being discovered in the garden or neighbouring garden.[22] The Royal Institution of Chartered Surveyors published a report in 2012 in response to lenders refusing to lend “despite [knotweed] being treatable and rarely causing severe damage to the property.” [23]
There is a real lack of information and understanding of what Japanese Knotweed is and the actual damage it can cause. Without actual advice and guidance, surveyors have been unsure of how to assess the risk of Japanese Knotweed, which can result in inconsistent reporting of the plant in mortgage valuations. RICS hopes that this advice will provide the industry with the tools it needs to measure the risk effectively, and provide banks with the information they require to identify who and how much to lend to at a time when it is essential to keep the housing market moving.
—Philip Santo, RICS Residential Professional Group[23]
In response to this guidance, several lenders have relaxed their criteria in relation to discovery of the plant. As recently as 2012, the policy at the Woolwich (part of Barclays plc) was “if Japanese Knotweed is found on or near the property then a case will be declined due to the invasive nature of the plant.”[24][25] Their criteria have since been relaxed to a category-based system depending on whether the plant is discovered on a neighbouring property (categories 1 and 2) or the property itself (categories 3 and 4) incorporating proximity to the property curtilage and the main buildings. Even in a worst-case scenario (category 4), where the plant is “within 7 metres of the main building, habitable spaces, conservatory and/or garage and any permanent outbuilding, either within the curtilage of the property or on neighbouring land; and/or is causing serious damage to permanent outbuildings, associated structures, drains, paths, boundary walls and fences” Woolwich lending criteria now specify that this property may be acceptable if “remedial treatment by a Property Care Association (PCA) registered firm has been satisfactorily completed. Treatment must be covered by a minimum 10-year insurance-backed guarantee, which is property specific and transferable to subsequent owners and any mortgagee in possession.” [26] Santander have relaxed their attitude in a similar fashion (citation needed).
Property Care Association chief executive Steve Hodgson, whose trade body has set up a task force to deal with the issue, said: “japanese knotweed is not “house cancer” and could be dealt with in the same way qualified contractors dealt with faulty wiring or damp.”[27]
The plant is known as itadori (イタドリ, 虎杖?). The kanji expression is from the Chinese meaning “tiger staff”, but as to the Japanese appellation, one straightforward interpretation is that it comes from “remove pain” (alluding to its painkilling use),[28][29] though there are other etymological explanations offered.
It grows widely throughout Japan and is foraged as a wild edible vegetable (sansai), though not in sufficient quantities to be included in statistics.[30] They are called by such regional names as: tonkiba (Yamagata),[30] itazuiko (Nagano, Mie),[30] itazura (Gifu, Toyama, Nara, Wakayama, Kagawa),[30] gonpachi (Shizuoka, Nara, Mie, Wakayama),[30]sashi (Akita, Yamagata),[30] jajappo (Shimane, Tottori, Okayama),[30] sukanpo (many areas).
Young leaves and shoots, which look like asparagus, are used. They are extremely sour; the fibrous outer skin must be peeled, soaked in water for half a day raw or after parboiling, before being cooked.
Places in Shikoku such as central parts of Kagawa Prefecture [31] pickle the peeled young shoots by weighting them down in salt mixed with 10% nigari (magnesium chloride).Kochi also rub these cleaned shoots with coarse salt-nigari blend. It is said (though no authority is cited) that the magnesium of the nigari binds with the oxalic acid thus mitigating its hazard.[32]
A novel use for a related species known as oh-itadori (Polygonum sachalinense) in Hokkaido is feeding it to larvae of sea urchins in aquaculture.[33]
| Japanese Knotweed | |
|---|---|
 |
|
| Scientific classification | |
| Kingdom: | Plantae |
| (unranked): | Angiosperms |
| (unranked): | Eudicots |
| (unranked): | Core eudicots |
| Order: | Caryophyllales |
| Family: | Polygonaceae |
| Genus: | Fallopia |
| Species: | F. japonica |
| Binomial name | |
| Fallopia japonica (Houtt.) Ronse Decr. |
|
| Synonyms | |
| Polygonum cuspidatum Siebold & Zucc. Reynoutria japonica Houtt. |
|
 Carfilzomib
Carfilzomib
Amgen’s Multiple Myeloma Drug Shows Promise in Phase 3 Trial
https://finance.yahoo.com/video/amgens-multiple-myeloma-drug-shows-195603222.html
The drug maker is seeing great signs in the development of treatment for multiple myeloma, a bone marrow cancer. The results from its Phase 3 of Kyprolis’ clinical trial shows that patients can live almost nine months longer without worsening symptoms. According to Amgen, about 70,000 people in the U.S. are living with the disease and 24,000 new cases are diagnosed every year. With the good clinical trial result, Amgen plans to begin regulatory submissions around the world next year. Dr. Pablo Cagnoni, president of Amgen’s subsidiary Onyx Pharmaceuticals said, “The results demonstrate that Kyprolis can significantly extend the time patients live without their disease progressing. The ability of novel therapies to produce deep and durable responses may, one day, transform this uniformly fatal disease to one that is chronic and manageable.” Male patients over the age of 65 have the highest risk of developing it.
Carfilzomib (marketed under the trade name Kyprolis, Onyx Pharmaceuticals, Inc.) is an anti-cancer drug acting as a selectiveproteasome inhibitor. Chemically, it is a tetrapeptide epoxyketone and an analog of epoxomicin.[1]
The U.S. Food and Drug Administration (FDA) approved it on 20 July 2012 for use in patients with multiple myeloma who have received at least two prior therapies, including treatment with bortezomib and an immunomodulatory therapy and have demonstrated disease progression on or within 60 days of completion of the last therapy. Approval is based on response rate. Clinical benefit, such as improvement in survival or symptoms, has not been verified.[2]
The abbreviation CFZ is common for referring to carfilzomib, but abbreviating drug names is not best practice in medicine.
Carfilzomib is derived from epoxomicin, a natural product that was shown by the laboratory of Craig Crews at Yale University to inhibit the proteasome.[3] The Crews laboratory subsequently invented a more specific derivative of epoxomicin named YU101,[4] which was licensed to Proteolix, Inc. Craig Crews, Raymond Deshaies from Caltech, Phil Whitcome, the former CEO of Neurogen and Larry Lasky, a venture capitalist, founded Proteolix, and along with other researchers and scientists, advanced YU101. The scientists at Proteolix invented a new, distinct compound that had potential use as a drug in humans, known as carfilzomib. Proteolix advanced carfilzomib to multiple Phase 1 and 2 clinical trials, including a pivotal Phase 2 clinical trial designed to seek accelerated approval.[5]Clinical trials for carfilzomib continue under Onyx Pharmaceuticals, which acquired Proteolix in 2009.[5]
In January 2011, the FDA granted carfilzomib fast-track status, allowing Onyx to initiate a rolling submission of its new drug application for carfilzomib.[6] In December 2011, the FDA granted Onyx standard review designation,[7][8] for its new drug application submission based on the 003-A1 study, an open-label, single-arm Phase 2b trial. The trial evaluated 266 heavily-pretreated patients with relapsed and refractory multiple myeloma who had received at least two prior therapies, including bortezomib and either thalidomide or lenalidomide.[9] It costs approximately $10,000 per 28-day cycle, making it the most expensive FDA-approved drug for multiple myeloma.[10]
Carfilzomib irreversibly binds to and inhibits the chymotrypsin-like activity of the 20S proteasome, an enzyme that degrades unwanted cellular proteins. Inhibition of proteasome-mediated proteolysis results in a build-up of polyubiquinated proteins, which may cause cell cycle arrest, apoptosis, and inhibition of tumor growth.[1]
A single-arm, Phase II trial (003-A1) of carfilzomib in patients with relapsed and refractory multiple myeloma showed that single-agent carfilzomib demonstrated a clinical benefit rate of 36 percent in the 266 patients evaluated and had an overall response rate of 22.9 percent and median duration of response of 7.8 months. The FDA approval of carfilzomib was based on results of the 003-A1 trial.[11]
In a Phase II trial (004), carfilzomib had a 53 percent overall response rate among patients with relapsed and/or refractory multiple myeloma who had not previously received bortezomib. This study also included a bortezomib-treated cohort. Results were reported separately.[12] This study also found prolonged carfilzomib treatment was tolerable, with approximately 22 percent of patients continuing treatment beyond one year. The 004 trial was a smaller study originally designed to investigate the impact of carfilzomib treatment in relationship to bortezomib treatment in less heavily pretreated (1-3 prior regimens) patients.[13]
A Phase II trial (005), which assessed the safety, pharmacokinetics, pharmacodynamics and efficacy of carfilzomib, in patients with multiple myeloma and varyi ng degrees of renal impairment, where nearly 50 percent of patients were refractory to both bortezomib and lenalidomide, demonstrated that pharmacokinetics and safety were not influenced by the degree of baseline renal impairment. Carfilzomib was tolerable and demonstrated efficacy.[14]
In another Phase II trial (006) of patients with relapsed and/or refractory multiple myeloma, carfilzomib in combination with lenalidomide and dexamethasone demonstrated an overall response rate of 69 percent.[15]
A Phase II trial (007) for multiple myeloma and solid tumors showed promising results.[16][17]
In Phase II trials of carfilzomib, the most common grade 3 or higher treatment-emergent adverse events were thrombocytopenia, anemia, lymphoenia, neutropenia, pneumonia, fatigue and hyponatremia.[18]
In a frontline Phase I/II study, the combination of carfilzomib, lenalidomide, and low-dose dexamethasone was highly active and well tolerated, permitting the use of full doses for an extended time in newly-diagnosed multiple myeloma patients, with limited need for dose modification. Responses were rapid and improved over time, reaching 100 percent very good partial response.[19]
A phase III confirmatory clinical trial, known as the ASPIRE trial, comparing carfilzomib, lenalidomide and dexamethasone versus lenalidomide and dexamethasone in patients with relapsed multiple myeloma is ongoing.[20] It is no longer recruiting and should report in 2014.


| Systematic (IUPAC) name | |
|---|---|
| (S)-4-Methyl-N-((S)-1-(((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)pentanamide | |
| Clinical data | |
| Trade names | Kyprolis |
| Licence data | US FDA:link |
| Pregnancy cat. | D (US) |
| Legal status | ℞-only (US) |
| Routes | Intravenous |
| Identifiers | |
| CAS number | 868540-17-4 |
| ATC code | L01XX45 |
| PubChem | CID 11556711 |
| ChemSpider | 9731489 |
| KEGG | D08880 |
| ChEMBL | CHEMBL451887 |
| Synonyms | PX-171-007 |
| Chemical data | |
| Formula | C40H57N5O7 |
| Mol. mass | 719.91 g mol |


http://pubs.rsc.org/en/content/articlelanding/2013/np/c3np20126k/unauth#!divAbstract
The initial enthusiasm following the discovery of a pharmacologically active natural product is often fleeting due to the poor prospects for its ultimate clinical application. Despite this, the ever-changing landscape of modern biology has a constant need for molecular probes that can aid in our understanding of biological processes. After its initial discovery by Bristol-Myers Squibb as a microbial anti-tumor natural product, epoxomicin was deemed unfit for development due to its peptide structure and potentially labile epoxyketone pharmacophore. Despite its drawbacks, epoxomicin’s pharmacophore was found to provide unprecedented selectivity for the proteasome. Epoxomicin also served as a scaffold for the generation of a synthetic tetrapeptide epoxyketone with improved activity, YU-101, which became the parent lead compound of carfilzomib (Kyprolis™), the recently approved therapeutic agent for multiple myeloma. In this era of rational drug design and high-throughput screening, the prospects for turning an active natural product into an approved therapy are often slim. However, by understanding the journey that began with the discovery of epoxomicin and ended with the successful use of carfilzomib in the clinic, we may find new insights into the keys for success in natural product-based drug discovery.




| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Pregnancy cat. | Not to be used |
| Routes | Intravenous infusion |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 27% (manganese) Negligible (DPDP) |
| Half-life | 20 minutes (manganese) 50 minutes (DPDP) |
| Excretion | Renal and fecal (manganese) Renal (DPDP) |
| Identifiers | |
| ATC code | V08CA05 |
| PubChem | CID 3086672 |
| ChemSpider | 2343239 |
| UNII | N02W67RKJS |
| Chemical data | |
| Formula | C22H28MnN4O14P2 |
| Mol. mass | 689.362 g/mol |
Mangafodipir (sold under the brand name Teslascan as mangafodipir trisodium) is a contrast agent delivered intravenously to enhance contrast in magnetic resonance imaging (MRI) of the liver. It has two parts, paramagnetic manganese (II) ions and thechelating agent fodipir (dipyridoxyl diphosphate, DPDP). Normal liver tissue absorbs the manganese more than abnormal or cancerous tissue. The manganese shortens the longitudinal relaxation time (T1), making the normal tissue appear brighter in MRIs. This enhanced contrast allows lesions to be more easily identified.

The condensation of pyridoxal 5-phosphate (I) with ethylenediamine (II) in methanol by means of NaOH gives the corresponding diimine (III), which is reduced with hydrogen over Pt/C in methanol/water yielding the expected diamine (IV). The reaction of (IV) with bromoacetic acid (V) by means of NaOH in methanol/water affords the N,N’-diacetic acid derivative (VI), which is finally treated with MnCl2 in water containing NaOH.

C22H27MnN4Na3O14P2 ![]()
![]()
![]()
![]()
![]() 757.33
757.33
Trisodium trihydrogen (OC-6-13)-[[N,N¢-1,2-ethanediylbis[N-[[3-hydroxy-2-methyl-5-[(phosphonooxy)methyl]-4-pyridinyl]methyl]glycinato]](8-)] manganate(6-).
Trisodium trihydrogen (OC-6-13)-[[N,N¢-ethylenebis[N-[[3-hydroxy-5-(hydroxymethyl)-2-methyl-4-pyridyl]methyl]glycine] 5,5¢-bis(phosphato)](8-)]manganate(6-) ![]()
![]()
![]() [140678-14-4].
[140678-14-4].

For the treatment of patients with progressive radioiodine-refractory, differentiated thyroid cancer (RR-DTC).
European regulators have agreed to undertake an accelerated assessment of Eisai’s lenvatinib as a treatment for progressive radioiodine-refractory, differentiated thyroid cancer.
The drug, which carries Orphan Status in the EU, is to be filed “imminently” and could become the first in a new class of tyrosine kinase inhibitors, the drugmaker said.

Read more at: http://www.pharmatimes.com/Article/14-07-31/Eisai_s_lenvatinib_to_get_speedy_review_in_Europe.aspx#ixzz39OGhRHas
Lenvatinib was granted Orphan Drug Designation for thyroid cancer by the health authorities in Japan in 2012, and in Europe and the U.S in 2013. The first application for marketing authorization of lenvatinib in the world was submitted in Japan on June 2014. Eisai is planning to submit applications for marketing authorization in Europe and the U.S. in the second quarter of fiscal 2014.
Lenvatinib is an oral multiple receptor tyrosine kinase (RTK) inhibitor with a novel binding mode that selectively inhibits the kinase activities of vascular endothelial growth factor receptors (VEGFR), in addition to other proangiogenic and oncogenic pathway-related RTKs including fibroblast growth factor receptors (FGFR), the platelet-derived growth factor (PDGF) receptor PDGFRalpha, KIT and RET that are involved in tumor proliferation. This potentially makes lenvatinib a first-in-class treatment, especially given that it simultaneously inhibits the kinase activities of FGFR as well as VEGFR.
LENVATINIB BASE
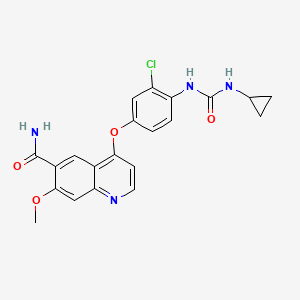
COSY PREDICT
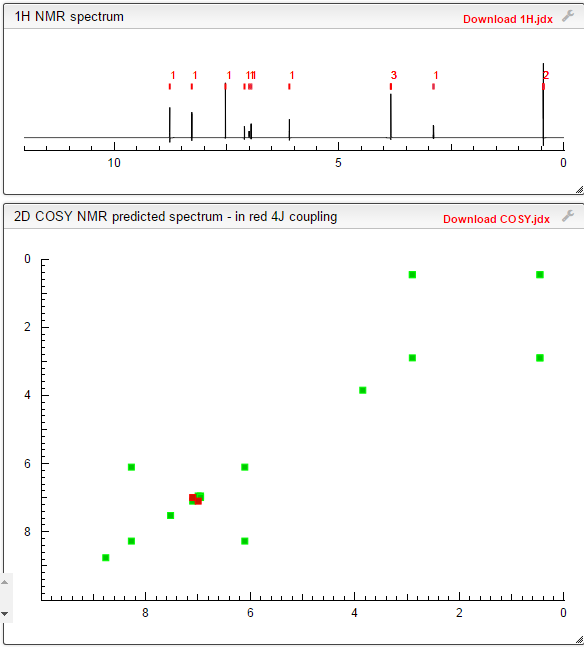
| Systematic (IUPAC) name | |
|---|---|
| 4-[3-chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxy-quinoline-6-carboxamide | |
| Clinical data | |
| Legal status | ℞ Prescription only |
| Identifiers | |
| CAS number | |
| ATC code | None |
| PubChem | CID 9823820 |
| ChemSpider | 7999567 |
| UNII | EE083865G2 |
| Chemical data | |
| Formula | C21H19ClN4O4 |
| Mol. mass | 426.853 g/mol |
Lenvatinib (E7080) is a multi-kinase inhibitor that is being investigated for the treatment of various types of cancer by Eisai Co. It inhibits both VEGFR2 and VEGFR3 kinases.[1]
The substence was granted orphan drug status for the treatment of various types of thyroid cancer that do not respond toradioiodine; in the US and Japan in 2012 and in Europe in 2013[2] and is now approved for this use.

Lenvatinib has had promising results from a phase I clinical trial in 2006[3] and is being tested in several phase II trials as of October 2011, for example against hepatocellular carcinoma.[4] After a phase II trial testing the treatment of thyroid cancer has been completed with modestly encouraging results,[5] the manufacturer launched a phase III trial in March 2011.[6]

Lenvatinib Mesilate
Molecular formula: C21H19ClN4O4,CH4O3S =523.0.
CAS: 857890-39-2.
UNII code: 3J78384F61.
About the Lenvatinib (E7080) Phase II Study
The open-label, global, single-arm Phase II study of multi-targeted kinase inhibitor lenvatinib (E7080) in advanced radioiodine (RAI)-refractory differentiated thyroid cancer involved 58 patients with advanced RAI refractory DTC (papillary, follicular or Hurthle Cell) whose disease had progressed during the prior 12 months. (Disease progression was measured using Response Evaluation Criteria in Solid Tumors (RECIST).) The starting dose of lenvatinib was 24 mg once daily in repeated 28 day cycles until disease progression or development of unmanageable toxicities.
2. About Thyroid Cancer
Thyroid cancer refers to cancer that forms in the tissues of the thyroid gland, located at the base of the throat or near the trachea. It affects more women than men and usually occurs between the ages of 25 and 65.
The most common types of thyroid cancer, papillary and follicular (including Hurthle Cell), are classified as differentiated thyroid cancer and account for 95 percent of all cases. While most of these are curable with surgery and radioactive iodine treatment, a small percentage of patients do not respond to therapy.
3. About Lenvatinib (E7080)
Lenvatinib is multi-targeted kinase inhibitor with a unique receptor tyrosine kinase inhibitory profile that was discovered and developed by the Discovery Research team of Eisai’s Oncology Unit using medicinal chemistry technology. As an anti-angiogenic agent, it inhibits tyrosine kinase of the VEGF (Vascular Endothelial Growth Factor) receptor, VEGFR2, and a number of other types of kinase involved in angiogenesis and tumor proliferation in balanced manner. It is a small molecular targeting drug that is currently being studied in a wide array of cancer types.
4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (additional name: 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]-7-methoxyquinoline-6-carboxamide) is known to exhibit an excellent angiogenesis inhibition as a free-form product, as described in Example 368 of Patent Document 1. 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide is also known to exhibit a strong inhibitory action for c-Kit kinase (Non-Patent Document 1, Patent Document 2).
However, there has been a long-felt need for the provision of a c-Kit kinase inhibitor or angiogenesis inhibitor that has high usability as a medicament and superior characteristics in terms of physical properties and pharmacokinetics in comparison with the free-form product of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide.
[Patent Document 1] WO 02/32872
[Patent Document 2] WO 2004/080462
[Non-Patent Document 1] 95th Annual Meeting Proceedings, AACR (American Association for Cancer Research), Volume 45, Page 1070-1071, 2004
………………………..
PATENT
http://www.google.co.in/patents/US8058474
EXAMPLES
Examples will now be described to facilitate understanding of the invention, but the invention is not limited to these examples.
Example 1Phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

After suspending 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) and adding pyridine (23.4 mL) while cooling on ice, phenyl chloroformate (23.2 ml) was added dropwise below 20° C. Stirring was performed at room temperature for 30 minutes, and then water (400 mL), ethyl acetate (300 mL) and 6N HCl (48 mL) were added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. The solvent was removed to give 46 g of the title compound as a solid.
1H-NMR (CDCl3): 5.12 (1h, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s)
Example 21-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

After dissolving phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL), cyclopropylamine (22.7 mL) was added while cooling on ice and the mixture was stirred overnight at room temperature. Water (400 mL), ethyl acetate (300 mL) and 6N HCl (55 mL) were then added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. Prism crystals obtained by concentrating the solvent were filtered and washed with heptane to give 22.8 g of the title compound (77% yield from 4-amino-3-chlorophenol).
1H-NMR (CDCl3): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8H)
Example 34-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide

To dimethylsulfoxide (20 mL) were added 7-methoxy-4-chloro-quinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), followed by heating and stirring at 70° C. for 23 hours. After the reaction mixture was allowed to cool down to room temperature, water (50 mL) was added, and the produced crystals were collected by filtration to give 1.56 g of the title compound (88% yield).
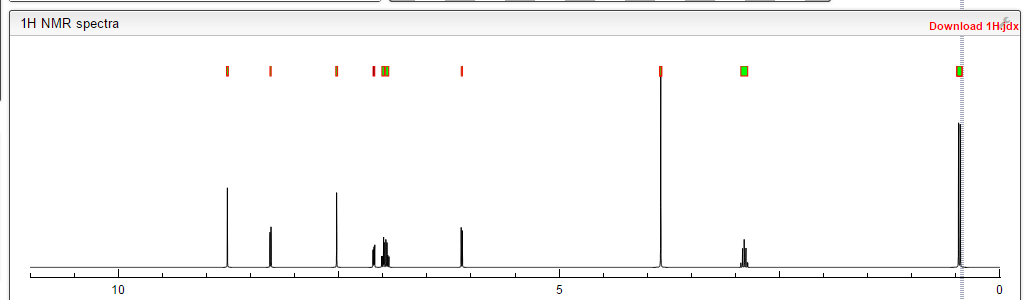
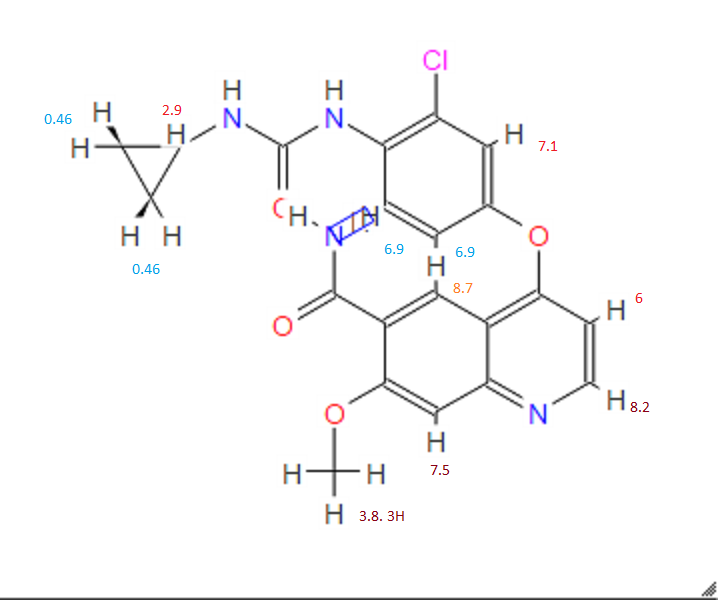
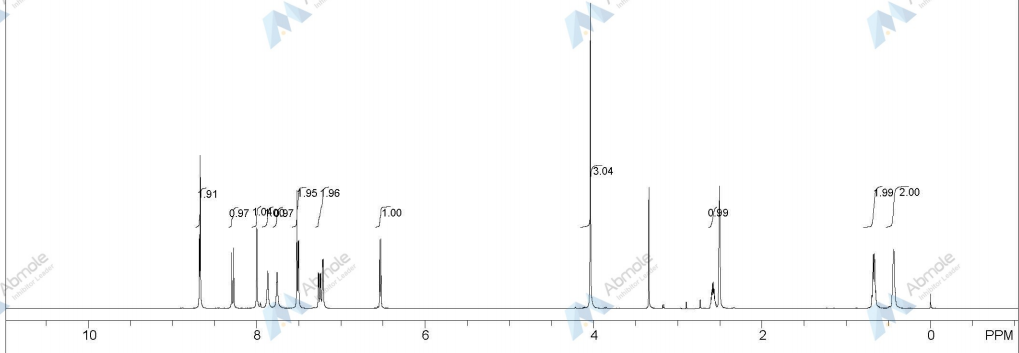
1H-NMR (d6-DMSO): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz)
Example 44-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
In a reaction vessel were placed 7-methoxy-4-chloro-quinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethylsulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) in that order, under a nitrogen atmosphere. After stirring at 20° C. for 30 minutes, the temperature was raised to 65° C. over a period of 2.5 hours. After stirring at the same temperature for 19 hours, 33% (v/v) acetone water (5.0 L) and water (10.0 L) were added dropwise over a period of 3.5 hours. Upon completion of the dropwise addition, the mixture was stirred at 60° C. for 2 hours, and 33% (v/v) acetone water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or higher over a period of 1 hour. After then stirring at 40° C. for 16 hours, the precipitated crystals were collected by filtration using a nitrogen pressure filter, and the crystals were washed with 33% (v/v) acetone water (33.3 L), water (66.7 L) and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum drier to give 7.78 kg of the title compound (96.3% yield).
…………………………
SYNTHESIS
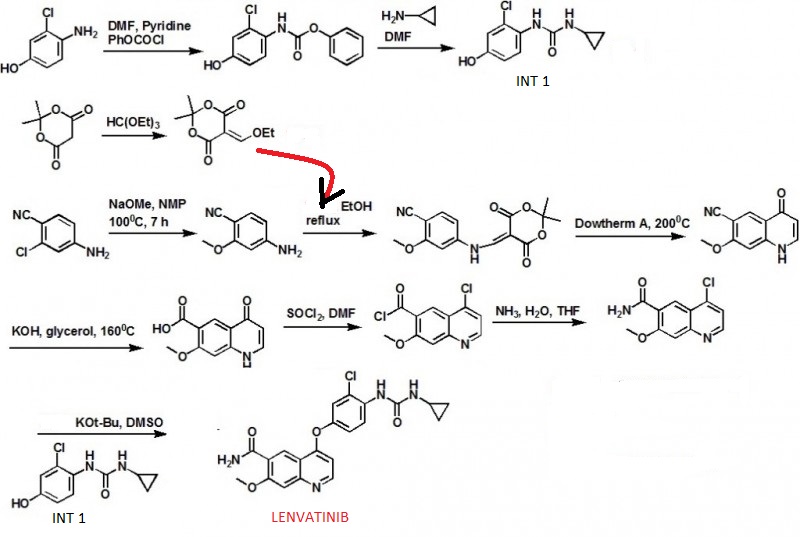
1H NMR PREDICT



PATENT
http://www.google.co.in/patents/US7253286
EX 368

Example 368
4-(3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
The title compound (22.4 mg, 0.052 mmol, 34.8%) was obtained as white crystals from phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (70 mg, 0.15 mmol) and cyclopropylamine, by the same procedure as in Example 11.
1H-NMR Spectrum (DMSO-d6) δ (ppm): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz).
The starting material was synthesized in the following manner.
Production Example 368-1Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate
The title compound (708 mg, 1.526 mmol, 87.4%) was obtained as light brown crystals from 4-(4-amino-3-chlorophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.745 mmol), by the same procedure as in Production Example 17.
1H-NMR Spectrum (CDCl3) δ (ppm): 4.14 (3H, s), 5.89 (1H, br), 6.50 (1H, d, J=5.6 Hz), 7.16 (2H, dd, J=2.4, 8.8 Hz), 7.22–7.30 (4H, m), 7.44 (2H, m), 7.55 (1H, s), 7.81 (1H, br), 8.31 (1H, d, J=8.8 Hz), 8.68 (1H, d, J=5.6 Hz), 9.27 (1H, s).
……………………
CRYSTALLINE FORM
http://www.google.co.in/patents/US7612208
Preparation Example 1
Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (1)
Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (17.5 g, 37.7 mmol) disclosed in WO 02/32872 was dissolved in N,N-dimethylformamide (350 mL), and then cyclopropylamine (6.53 mL, 94.25 mmol) was added to the reaction mixture under a nitrogen atmosphere, followed by stirring overnight at room temperature. To the mixture was added water (1.75 L), and the mixture was stirred. Precipitated crude crystals were filtered off, washed with water, and dried at 70° C. for 50 min. To the obtained crude crystals was added ethanol (300 mL), and then the mixture was heated under reflux for 30 min to dissolve, followed by stirring overnight to cool slowly down to room temperature. Precipitated crystals was filtered off and dried under vacuum, and then further dried at 70° C. for 8 hours to give the titled crystals (12.91 g; 80.2%).
Preparation Example 2Preparation of 4-(3-cloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (2)
(1) Preparation of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

To a suspension of 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) was added pyridine (23.4 mL) while cooling in an ice bath, and phenyl chloroformate (23.2 mL) was added dropwise below 20° C. After stirring at room temperature for 30 min, water (400 mL), ethyl acetate (300 mL), and 6N-HCl (48 mL) were added and stirred. The organic layer was separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give 46 g of the titled compound as a solid.

To a solution of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL) was added cyclopropylamine (22.7 mL) while cooling in an ice bath, and the stirring was continued at room temperature overnight. Water (400 mL), ethyl acetate (300 mL), and 6N-HCl (55 mL) were added thereto, and the mixture was stirred. The organic layer was then separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give prism crystals, which were filtered off and washed with heptane to give 22.8 g of the titled compound (yield from 4-amino-3-chlorophenol: 77%).
To dimethyl sulfoxide (20 mL) were added 7-methoxy-4-chloroquinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), and the mixture was heated and stirred at 70° C. for 23 hours. The reaction mixture was cooled to room temperature, and water (50 mL) was added, and the resultant crystals were then filtered off to give 1.56 g of the titled compound (yield: 88%).
Preparation Example 3Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (3)
7-Methoxy-4-chloroquinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethyl sulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea 5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) were introduced in this order into a reaction vessel under a nitrogen atmosphere. The mixture was stirred for 30 min at 20° C., and the temperature was raised to 65° C. over 2.5 hours. The mixture was stirred at the same temperature for 19 hours. 33% (v/v) acetone-water (5.0 L) and water (10.0 L) were added dropwise over 3.5 hours. After the addition was completed, the mixture was stirred at 60° C. for 2 hours. 33% (v/v) acetone-water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or more over 1 hour. After stirring at 40° C. for 16 hours, precipitated crystals were filtered off using a nitrogen pressure filter, and was washed with 33% (v/v) acetone-water (33.3 L), water (66.7 L), and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum dryer to give 7.78 kg of the titled compound (yield: 96.3%).
1H-NMR chemical, shift values for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamides obtained in Preparation Examples 1 to 3 corresponded to those for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide disclosed in WO 02/32872.
Example 5
A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A)
(Preparation Method 1)
In a mixed solution of methanol (14 mL) and methanesulfonic acid (143 μL, 1.97 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (700 mg, 1.64 mmol) at 70° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, the reaction mixture was cooled to room temperature over 5.5 hours, further stirred at room temperature for 18.5 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (647 mg).
(Preparation Method 2)
In a mixed solution of acetic acid (6 mL) and methanesulfonic acid (200 μL, 3.08 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.41 mmol) at 50° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, ethanol (7.2 mL) and seed crystals of a crystalline form of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A) (12 mg) were added in this order to the reaction mixture, and ethanol (4.8 mL) was further added dropwise over 2 hours. After the addition was completed, the reaction mixture was stirred at 40° C. for 1 hour then at room temperature for 9 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (545 mg).
Example 6A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form B)
A crystalline form of the acetic acid solvate of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form I) (250 mg) obtained in Example 10 was dried under aeration at 30° C. for 3 hours and at 40° C. for 16 hours to give the titled crystals (240 mg)…………MORE IN PATENT
……………………………..
PATENT
https://www.google.com/patents/WO2014098176A1?cl=en
According to the present invention 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide amorphous is excellent in solubility in water.
Example 1 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide manufacture of amorphous amide
4- (3-chloro-4- (cyclopropylamino-carbonyl) amino phenoxy) -7-methoxy-6-quinolinecarboxamide B-type crystals (Patent Document 2) were weighed to 300mg, is placed in a beaker of 200mL volume, it was added tert- butyl alcohol (tBA) 40mL. This was heated to boiling on a hot plate, an appropriate amount of tBA to Compound A is dissolved, water was added 10mL. Then, the weakened heated to the extent that the solution does not boil, to obtain a sample solution. It should be noted, finally the solvent amount I was 60mL. 200mL capacity eggplant type flask (egg-plant shaped flask), and rotated in a state of being immersed in ethanol which had been cooled with dry ice. It was added dropwise a sample solution into the interior of the flask and frozen. After freezing the sample solution total volume, to cover the opening of the flask in wiping cloth, and freeze-dried. We got an amorphous A of 290mg.
Patent Document 2: US Patent Application Publication No. 2007/0117842 Patent specification 
………………………..
Paper
ACS Medicinal Chemistry Letters (2015), 6(1), 89-94
http://pubs.acs.org/doi/full/10.1021/ml500394m
……………..
Paper
Journal of Pharmaceutical and Biomedical Analysis (2015), 114, 82-87
http://www.sciencedirect.com/science/article/pii/S0731708515002940
KEEP WATCHING WILL BE UPDATED………….most of my posts are updated regularly
UPDATES
EXTRAS……………
Martin Schlumberger et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib(E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT). 2014 ASCO Annual Meeting. Abstract Number:LBA6008. Presented June 2, 2014. Citation: J Clin Oncol 32:5s, 2014 (suppl; abstr LBA6008). Clinical trial information: NCT01321554.
Bando, Masashi. Quinoline derivative-containing pharmaceutical composition. PCT Int. Appl. (2011), WO 2011021597 A1
Tomohiro Matsushima, four Nakamura, Kazuhiro Murakami, Atsushi Hoteido, Yusuke Ayat, Naoko Suzuki, Itaru Arimoto, Pinche Hirose, Masaharu Gotoda.Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor. US patent number US7612208 Also published as: CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1 , US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date: Aug 7, 2007 Original Assignee: Eisai Co., Ltd
Funahashi, Yasuhiro et al.Preparation of urea derivatives containing nitrogenous aromatic ring compounds as inhibitors of angiogenesis. US patent number US7253286, Also published as:CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1, US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date:Aug 7, 2007. Original Assignee:Eisai Co., Ltd
Sakaguchi, Takahisa; Tsuruoka, Akihiko. Preparation of amorphous salts of 4-[3-chloro-4-[(cyclopropylaminocarbonyl)amino]phenoxy]-7-methoxy-6-quinolinecarboxamide as antitumor agents. PCT Int. Appl. (2006), WO2006137474 A1 20061228.
Naito, Toshihiko and Yoshizawa, Kazuhiro. Preparation of urea moiety-containing quinolinecarboxamide derivatives. PCT Int. Appl., WO2005044788, 19 May 2005
Itaru Arimoto et al. Crystal of salt of 4-[3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy]-7-methoxy-6-quinolinecarboxamide or solvate thereof and processes for producing these. PCT Int. Appl. (2005), WO2005063713 A1 20050714.
|
10-23-2009
|
ANTITUMOR AGENT FOR UNDIFFERENTIATED GASTRIC CANCER
|
|
|
10-2-2009
|
ANTI-TUMOR AGENT FOR MULTIPLE MYELOMA
|
|
|
8-21-2009
|
ANTITUMOR AGENT FOR THYROID CANCER
|
|
|
8-14-2009
|
THERAPEUTIC AGENT FOR LIVER FIBROSIS
|
|
|
2-27-2009
|
USE OF COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND c-kit KINASE INHIBITOR
|
|
|
9-5-2008
|
Medicinal Composition
|
|
|
8-8-2007
|
Nitrogen-containing aromatic derivatives
|
|
|
5-25-2007
|
Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same
|
|
|
7-21-2006
|
Nitrogen-containing aromatic derivatives
|
|
|
6-23-2006
|
Use of sulfonamide-including compounds in combination with angiogenesis inhibitors
|
|
11-16-2011
|
UREA DERIVATIVE AND PROCESS FOR PREPARING THE SAME
|
|
|
8-10-2011
|
c-Kit kinase inhibitor
|
|
|
7-6-2011
|
Nitrogen-Containing Aromatic Derivatives
|
|
|
12-24-2010
|
COMBINED USE OF ANGIOGENESIS INHIBITOR AND TAXANE
|
|
|
9-24-2010
|
COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND ANTI-TUMOR PLATINUM COMPLEX
|
|
|
4-30-2010
|
METHOD FOR PREDICTION OF THE EFFICACY OF VASCULARIZATION INHIBITOR
|
|
|
4-16-2010
|
METHOD FOR ASSAY ON THE EFFECT OF VASCULARIZATION INHIBITOR
|
|
|
3-24-2010
|
Urea derivative and process for preparing the same
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF PANCREATIC CANCER
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF UNDIFFERENTIATED GASTRIC CANCER
|
| US7253286 * | 18 Apr 2003 | 7 Aug 2007 | Eisai Co., Ltd | Nitrogen-containing aromatic derivatives |
| US20040053908 | 18 Apr 2003 | 18 Mar 2004 | Yasuhiro Funahashi | Nitrogen-containing aromatic derivatives |
| US20040242506 | 9 Aug 2002 | 2 Dec 2004 | Barges Causeret Nathalie Claude Marianne | Formed from paroxetine hydrochloride and ammonium glycyrrhyzinate by precipitation, spray, vacuum or freeze drying, or evaporation to glass; solid or oil; masks the bitter taste of paroxetine and has a distinctive licorice flavor; antidepressants; Parkinson’s disease |
| US20040253205 | 10 Mar 2004 | 16 Dec 2004 | Yuji Yamamoto | c-Kit kinase inhibitor |
| US20070004773 * | 22 Jun 2006 | 4 Jan 2007 | Eisai R&D Management Co., Ltd. | Amorphous salt of 4-(3-chiloro-4-(cycloproplylaminocarbonyl)aminophenoxy)-7-method-6-quinolinecarboxamide and process for preparing the same |
| US20070078159 | 22 Dec 2004 | 5 Apr 2007 | Tomohiro Matsushima | Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor |
| US20070117842 * | 22 Apr 2004 | 24 May 2007 | Itaru Arimoto | Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same |
| EP0297580A1 | 30 Jun 1988 | 4 Jan 1989 | E.R. SQUIBB & SONS, INC. | Amorphous form of aztreonam |
| JP2001131071A | Title not available | |||
| JP2005501074A | Title not available | |||
| JPS6422874U | Title not available | |||
| WO2002032872A1 | 19 Oct 2001 | 25 Apr 2002 | Itaru Arimoto | Nitrogenous aromatic ring compounds |
| WO2003013529A1 | 9 Aug 2002 | 20 Feb 2003 | Barges Causeret Nathalie Claud | Paroxetine glycyrrhizinate |
| WO2004039782A1 | 29 Oct 2003 | 13 May 2004 | Hirai Naoko | QUINOLINE DERIVATIVES AND QUINAZOLINE DERIVATIVES INHIBITING AUTOPHOSPHORYLATION OF Flt3 AND MEDICINAL COMPOSITIONS CONTAINING THE SAME |
| WO2004080462A1 | 10 Mar 2004 | 23 Sep 2004 | Eisai Co Ltd | c-Kit KINASE INHIBITOR |
| WO2004101526A1 | 22 Apr 2004 | 25 Nov 2004 | Itaru Arimoto | Polymorphous crystal of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-qunolinecarboxamide and method for preparation thereof |
| WO2005044788A1 | 8 Nov 2004 | 19 May 2005 | Eisai Co Ltd | Urea derivative and process for producing the same |
| WO2005063713A1 | 22 Dec 2004 | 14 Jul 2005 | Itaru Arimoto | Crystal of salt of 4-(3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy)-7-methoxy-6-quinolinecarboxamide or of solvate thereof and processes for producing these |
| WO2006030826A1 | 14 Sep 2005 | 23 Mar 2006 | Eisai Co Ltd | Medicinal composition |
UPDATE………….
1H NMR PREDICT OF LENVATINIB BASE





Human glucosidase, prepro-α-[199-arginine,223-histidine] [1]
Alglucosidase alfa
C4435H6739N1175O1279S32
105270.8020
August 1, 2014
The U.S. Food and Drug Administration today announced the approval of Lumizyme (alglucosidase alfa) for treatment of patients with infantile-onset Pompe disease, including patients who are less than 8 years of age. In addition, the Risk Evaluation and Mitigation Strategy (REMS) known as the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program is being eliminated.
Pompe disease is a rare genetic disorder and occurs in an estimated 1 in every 40,000 to 300,000 births. Its primary symptom is heart and skeletal muscle weakness, progressing to respiratory weakness and death from respiratory failure.
The disease causes gene mutations to prevent the body from making enough of the functional form of an enzyme called acid alpha-glucosidase (GAA). This enzyme is necessary for proper muscle functioning. GAA is used by the heart and muscle cells to convert a form of sugar called glycogen into energy. Without the enzyme action, glycogen builds up in the cells and, ultimately, weakens the heart and muscles. Lumizyme is believed to work by replacing the deficient GAA, thereby reducing the accumulated glycogen in heart and skeletal muscle cells.
Lumizyme, a lysosomal glycogen-specific enzyme, was approved by the FDA in 2010 with a REMS to restrict its use to treatment of patients with late (non-infantile) onset Pompe disease who are 8 years of age and older. The REMS was required to mitigate the potential risk of rapid disease progression in the infantile-onset Pompe disease patients and patients with late onset disease less than 8 years of age, and to communicate the risks of anaphylaxis, severe allergic reactions and severe skin and systemic immune mediated reactions to prescribers and patients.
At the time of Lumizyme’s approval, there were insufficient data to support the safety and efficacy of Lumizyme in the infantile-onset Pompe population, so Lumizyme was approved for use only in late onset Pompe disease patients who are at least 8 years of age. Pompe patients with infantile-onset disease and patients younger than 8 years of age continued treatment with Myozyme, which was already approved. Myozyme and Lumizyme, both manufactured by Genzyme Corporation, are produced from the same cell line at different production scales.
This approval provides access to Lumizyme for all Pompe disease patients, regardless of their age.
The FDA reviewed newly available information and determined that Lumizyme and Myozyme are chemically and biochemically comparable. Consequently, the safety and effectiveness of Lumizyme and Myozyme are expected to be comparable. In addition, a single-center clinical study of 18 infantile-onset Pompe disease patients, aged 0.2 to 5.8 months at the time of first infusion, provides further support that infantile-onset patients treated with Lumizyme will have a similar improvement in ventilator-free survival as those treated with Myozyme.
Because data were submitted supporting approval of Lumizyme for all Pompe patients, a REMS restricting its use to a specific age group is no longer necessary. While the risk of anaphylaxis, severe allergic reactions, and severe cutaneous and immune mediated reactions for Lumizyme still exist, these risks are comparable to Myozyme and are communicated in labeling through the Warnings and Precautions, and a Boxed Warning.
“REMS continue to be vital tools for the agency to employ as we work with companies to address the serious risks associated with drugs and monitor their appropriate and safe use in various health care settings,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “The agency remains committed to exercising a flexible and responsible regulatory approach that ensures REMS programs are being effectively and efficiently used and not resulting in an unnecessary burden on health care professionals and patients.”
Health care professionals and patients should also be aware:
The most commonly reported side effects for Lumizyme were infusion-related reactions and included severe allergic reactions, hives, diarrhea, vomiting, shortness of breath, itchy skin, skin rash, neck pain, partial hearing loss, flushing, pain in extremities, and chest discomfort.
Myozyme and Lumizyme are marketed by Cambridge, Massachusetts-based Genzyme.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2416492 | 2008-04-29 | 2021-07-10 |

>Alglucosidase alfa AHPGRPRAVPTQCDVPPNSRFDCAPDKAITQEQCEARGCCYIPAKQGLQGAQMGQPWCFF PPSYPSYKLENLSSSEMGYTATLTRTTPTFFPKDILTLRLDVMMETENRLHFTIKDPANR RYEVPLETPHVHSRAPSPLYSVEFSEEPFGVIVRRQLDGRVLLNTTVAPLFFADQFLQLS TSLPSQYITGLAEHLSPLMLSTSWTRITLWNRDLAPTPGANLYGSHPFYLALEDGGSAHG VFLLNSNAMDVVLQPSPALSWRSTGGILDVYIFLGPEPKSVVQQYLDVVGYPFMPPYWGL GFHLCRWGYSSTAITRQVVENMTRAHFPLDVQWNDLDYMDSRRDFTFNKDGFRDFPAMVQ ELHQGGRRYMMIVDPAISSSGPAGSYRPYDEGLRRGVFITNETGQPLIGKVWPGSTAFPD FTNPTALAWWEDMVAEFHDQVPFDGMWIDMNEPSNFIRGSEDGCPNNELENPPYVPGVVG GTLQAATICASSHQFLSTHYNLHNLYGLTEAIASHRALVKARGTRPFVISRSTFAGHGRY AGHWTGDVWSSWEQLASSVPEILQFNLLGVPLVGADVCGFLGNTSEELCVRWTQLGAFYP FMRNHNSLLSLPQEPYSFSEPAQQAMRKALTLRYALLPHLYTLFHQAHVAGETVARPLFL EFPKDSSTWTVDHQLLWGEALLITPVLQAGKAEVTGYFPLGTWYDLQTVPVEALGSLPPP PAAPREPAIHSEGQWVTLPAPLDTINVHLRAGYIIPLQGPGLTTTESRQQPMALAVALTK GGEARGELFWDDGESLEVLERGAYTQVIFLARNNTIVNELVRVTSEGAGLQLQKVTVLGV ATAPQQVLSNGVPVSNFTYSPDTKVLDICVSLLMGEQFLVSWC
| Systematic (IUPAC) name | |
|---|---|
| Human glucosidase, prepro-α-[199-arginine,223-histidine] [1] | |
| Clinical data | |
| AHFS/Drugs.com | monograph |
| Legal status | FDA approved for children[2] |
| Routes | Intravenous[2] |
| Identifiers | |
| CAS number | 420794-05-0 |
| ATC code | A16AB07 |
| DrugBank | DB01272 |
| UNII | DTI67O9503 |
| KEGG | D03207 |
| Chemical data | |
| Formula | C4758H7262N1274O1369S35[1] |
| Mol. mass | 105338 [1] |
Alglucosidase alfa (Lumizyme, Myozyme, Genzyme) is an enzyme replacement therapy (ERT) orphan drug for treatment of Pompe disease (Glycogen storage disease type II), a rare lysosomal storage disorder (LSD).[3] Chemically speaking, the drug is ananalog of the enzyme that is deficient in patients affected by Pompe disease, alpha-glucosidase. It is the first drug available to treat this disease.[2]
Orphan drug pharmaceutical company, Genzyme, markets alglucosidase alfa as “Myozyme”. In 2006, the U.S. Food and Drug Administration (FDA) approved Myozyme as a suitable ERT treatment for children.[2] Some health plans have refused to subsidize Myozyme for adult patients because it lacks approval for treatment in adults, as well as its high cost (US$300,000/yr for life).[4]
On August 1, 2014 the U.S. Food and Drug Administration announced the approval of Lumizyme (alglucosidase alfa) for treatment of patients with infantile-onset Pompe disease, including patients who are less than 8 years of age. In addition, the Risk Evaluation and Mitigation Strategy (REMS) known as the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program is being eliminated. [5]
Common observed adverse reactions to alglucosidase alfa treatment are pneumonia, respiratory complications, infections and fever. More serious reactions reported includeheart and lung failure and allergic shock. Myozyme boxes carry warnings regarding the possibility of life-threatening allergic response.[2]
MYOZYME (alglucosidase alfa), a lysosomal glycogen-specific enzyme, consists of the human enzyme acid α-glucosidase (GAA), encoded by the most predominant of nine observed haplotypes of this gene. MYOZYME is produced by recombinant DNA technology in a Chinese hamster ovary cell line. The MYOZYME manufacturing process differs from that for LUMIZYME®, resulting in differences in some product attributes. Alglucosidase alfa degrades glycogen by catalyzing the hydrolysis of α-1,4- and α-1,6- glycosidic linkages of lysosomal glycogen.
Alglucosidase alfa is a glycoprotein with a calculated mass of 99,377 daltons for the polypeptide chain, and a total mass of approximately 110 kilo Daltons, including carbohydrates. Alglucosidase alfa has a specific activity of 3 to 5 U/mg (one unit is defined as that amount of activity that results in the hydrolysis of 1 μmole of synthetic substrate per minute under the specified assay conditions). MYOZYME is intended for intravenous infusion. It is supplied as a sterile, nonpyrogenic, white to off-white, lyophilized cake or powder for reconstitution with 10.3 mL
Sterile Water for Injection, USP. Each 50 mg vial contains 52.5 mg alglucosidase alfa, 210 mg mannitol, 0.5 mg polysorbate 80, 9.9 mg sodium phosphate dibasic heptahydrate, 31.2 mg sodium phosphate monobasic monohydrate. Following reconstitution as directed, each vial contains 10.5 mL reconstituted solution and a total extractable volume of 10 mL at 5.0 mg/mL alglucosidase alfa. MYOZYME does not contain preservatives; each vial is for single use only.

Empagliflozin
For synthesis see https://newdrugapprovals.org/2013/12/19/empagliflozin/
August 1, 2014
The U.S. Food and Drug Administration today approved Jardiance (empagliflozin) tablets as an addition to diet and exercise to improve glycemic control in adults with type 2 diabetes.
Type 2 diabetes affects approximately 26 million people and accounts for more than 90 percent of diabetes cases diagnosed in the United States. Over time, high blood sugar levels can increase the risk for serious complications, including heart disease, blindness, and nerve and kidney damage.
“Jardiance provides an additional treatment option for the care of patients with type 2 diabetes,” said Curtis J. Rosebraugh, M.D., M.P.H., director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research. “It can be used alone or added to existing treatment regimens to control blood sugar levels in the overall management of diabetes.”
Jardiance is a sodium glucose co-transporter 2 (SGLT2) inhibitor. It works by blocking the reabsorption of glucose (blood sugar) by the kidney, increasing glucose excretion, and lowering blood glucose levels in diabetics who have elevated blood glucose levels. The drug’s safety and effectiveness were evaluated in seven clinical trials with 4,480 patients with type 2 diabetes receiving Jardiance. The pivotal trials showed that Jardiance improved hemoglobin A1c levels (a measure of blood sugar control) compared to placebo.
Jardiance has been studied as a stand-alone therapy and in combination with other type 2 diabetes therapies including metformin, sulfonylureas, pioglitazone, and insulin. Jardiance should not be used: to treat people with type 1 diabetes; in those who have increased ketones in their blood or urine (diabetic ketoacidosis); and in those with severe renal impairment, end stage renal disease, or in patients on dialysis.
The FDA is requiring four postmarketing studies for Jardiance:
Jardiance can cause dehydration, leading to a drop in blood pressure (hypotension) that can result in dizziness and/or fainting and a decline in renal function. The elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk.
The most common side effects of Jardiance are urinary tract infections and female genital infections.
Jardiance is distributed by Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, Connecticut.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
synthesis see https://newdrugapprovals.org/2013/12/19/empagliflozin/

NS 398
N-[2-(Cyclohexyloxy)-4-nitrophenyl]methanesulfonamide
Taisho (Originator)
N-(2-cyclohexyloxy-4-nitrophenyl)methanesulfonamide.
123653-11-2, 123653-43-0 (Ca salt), 123653-44-1 (Na salt)
Cerebrovascular Diseases, Treatment of, NEUROLOGIC DRUGS, Stroke, Treatment of, Cyclooxygenase-2 Inhibitors
NS-398 is a COX-2 inhibitor used in the study of the function of cyclooxygenases.[2]
Selective cyclooxygenase-2 inhibitor (IC50 values are 3.8 and > 100 μM for COX-2 and COX-1 respectively). Orally active. Anti-inflammatory, anti-pyretic, analgesic and non-ulcerogenic in vivo. Induces apoptosis and cell cycle arrest
Cyclooxygenase (COX-2) has been recently suggested to play a role in hepatocarcinogenesis. However, the exact pathway by which COX-2 affects the growth of hepatocellular carcinoma (HCC) is not clear. This study investigated the effects of a specific COX-2 inhibitor, NS-398, on the cell proliferation and apoptosis of COX-2-expressing and non-expressing HCC cell lines.
In addition, the modulatory effect of NS-398 on apoptosis-regulating gene expression was examined. Semi-quantitative/quantitative reverse transcription-polymerase chain reaction and Western blot showed that Hep3B and HKCI-4 cells expressed COX-2 mRNA and protein, but HepG2 cells did not. NS-398 suppressed cell proliferation and induced apoptosis in the two COX-2-expressing cell lines in a dose-dependent manner, but not in HepG2 cells.
Fas ligand mRNA and protein expression were increased by the treatment with NS-398 (10 micro M) in COX-2-expressing cell lines. The expressions of Fas and Bcl-2 family genes (Bax, Bcl-2, Bcl-xL, Bcl-xS) were not affected by NS-398 treatment in all three cell lines. In conclusion, specific COX-2 inhibitor suppresses cell proliferation and induces apoptosis in HCC cell lines that express COX-2. Our finding suggests that COX-2 inhibition may offer a new approach for HCC chemoprevention.
| Identifiers | |
|---|---|
| CAS number | 123653-11-2 |
| PubChem | 4553 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C13H18N2O5S |
| Molar mass | 314.36 g mol−1 |
| Appearance | Off-white solid |
| Solubility in water | Insoluble |
| Solubility in DMSO | 5 mg/mL |
| Hazards | |
| S-phrases | S22 S24/25 |

The condensation of 2-fluoronitrobenzene (I) with cyclohexanol (II) by means of NaH gives 2-(cyclohexyloxy)nitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 2-(cyclohexyloxy)aniline (IV). The acylation of (IV) with methanesulfonyl chloride (V) in pyridine affords N-(2-cyclohexyloxy phenyl)methanesulfonamide (VI), which is finally nitrated with concentrated HNO3 in hot acetic acid.
EP 0317332
http://www.google.com/patents/EP0317332A2?cl=en
| EP0093591A1 * | Apr 29, 1983 | Nov 9, 1983 | Eli Lilly And Company | Selective sulfonation process |
| FR2244473A1 * | Title not available | |||
| US3725451 * | Apr 13, 1970 | Apr 3, 1973 | Riker Laboratories Inc | Substituted benzoylhaloalkanesulfonanilides |
| US3840597 * | Jul 3, 1972 | Oct 8, 1974 | Riker Laboratories Inc | Substituted 2-phenoxy alkane-sulfonanilides |
| US3856859 * | Jun 8, 1973 | Dec 24, 1974 | Riker Laboratories Inc | Selective nitration process |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP1535614A2 * | Aug 22, 1997 | Jun 1, 2005 | University OofFlorida | Materials and methods for detection and treatment of immune system dysfunctions |
……………………………………………………..
The cortical collecting duct (CCD) is a major site of intrarenal prostaglandin E2 (PGE2) synthesis. This study examines the expression and regulation of the prostaglandin synthesizing enzymes cyclooxygenase-1 (COX-1) and -2 in the CCD. By indirect immunofluorescence using isoform-specific antibodies, COX-1 and -2 immunoreactivity was localized to all cell types of the murine M-1 CCD cell line. By immunohistochemistry, both COX-1 and COX-2 were localized to intercalated cells of the CCD on paraffin-embedded mouse kidney sections. When COX enzyme activity was measured in the M-1 cells, both indomethacin (COX-1 and -2 inhibitor) and the specific COX-2 inhibitor NS-398 effectively blocked PGE2 synthesis. These results demonstrate that COX-2 is the major contributor to the pool of PGE2synthesized by the CCD. By Western blot analysis, COX-2 expression was significantly upregulated by incubation with either indomethacin or NS-398. These drugs did not affect COX-1 protein expression. Evaluation of COX-2 mRNA expression by Northern blot analysis after NS-398 treatment demonstrated that the COX-2 protein upregulation occurred independently of any change in COX-2 mRNA expression. These studies have for the first time localized COX-2 to the CCD and provided evidence that the intercalated cells of the CCD express both COX-1 and COX-2. The results also demonstrate that constitutively expressed COX-2 is the major COX isoform contributing to PGE2synthesis by the M-1 CCD cell line. Inhibition of COX-2 activity in the M-1 cell line results in an upregulation of COX-2 protein expression.
http://jasn.asnjournals.org/content/10/11/2261.abstract
…………………………………………….
NS398 inhibits the growth of OSCC cells by mechanisms that are dependent and independent of suppression of PGE2 synthesis. Molecular targeting of COX-2, PGE2 synthase, or PGE2 receptors may be useful as a chemopreventive or therapeutic strategy for oral cancer.
http://clincancerres.aacrjournals.org/content/9/5/1885.full
…………………………………
Futaki et al (1993) NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen.Pharmacol. 24 105. PMID: 8482483.
Futaki et al (1994) NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins 47 55. PMID: 8140262.
Elder et al (2002) The MEK/ERK pathway mediates COX-2-selective NSAID-induced apoptosis and induced COX-2 protein expression in colorectal carcinoma cells. Int.J.Cancer 99 323. PMID: 11992399.

Gemeprost, SC-37681, Ono-802, Cergem, Preglandin, Cervagem,
(E) -7 – [(1R, 2R, 3R-3-Hydroxy-2 – [(E) – (3R) -3-hydroxy-4,4-dimethyl-1-octenyl] -5-oxocyclopentyl] -2 -heptenoic acid methyl ester;
16,16-Dimethyl-DELTA2-trans-PGE1 methyl ester;
9-Oxo-11alpha, 15alpha-dihydroxy-16,16-dimethyl-2-trans, 13-trans-prostadiene-1-oic acid
Gemeprost (16, 16-dimethyl-trans-delta2 PGE1 methyl ester) is an analogue of prostaglandin E1.
It is used as a treatment for obstetric bleeding.
It is used with mifepristone to terminate pregnancy up to 24 weeks gestation. [1]
Vaginal bleeding, cramps, nausea, vomiting, loose stools or diarrhea, headache, muscle weakness; dizziness; flushing; chills; backache; dyspnoea; chest pain; palpitations and mild pyrexia. Rare: Uterine rupture, severe hypotension, coronary spasms with subsequent myocardial infarctions
 |
|
| Systematic (IUPAC) name | |
|---|---|
| methyl (2E,11α,13E,15R)-11,15-dihydroxy-16,16-dimethyl-9-oxoprosta-2,13-dien-1-oate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status | ? |
| Routes | Pessary |
| Identifiers | |
| CAS number | 64318-79-2 |
| ATC code | G02AD03 |
| PubChem | CID 5282237 |
| ChemSpider | 4445416 |
| UNII | 45KZB1FOLS |
| KEGG | D02073 |
| Synonyms | methyl (E)-7-[(1R,2S,3R)-3-hydroxy-2-[(E,3R)-3-hydroxy-4,4-dimethyl-oct-1-enyl]-5-oxo-cyclopentyl]hept-2-enoate |
| Chemical data | |
| Formula | C23H38O5 |
| Mol. mass | 394.545 g/mol |
………………………………
http://www.chemdrug.com/databases/8_0_oqxuqtwlqgeukaaa.html

The reaction of 3-bromopropionic acid (I) with triphenylphosphine (II) in refluxing acetonitrile gives (2-carboxyethyl) -triphenylphosphonium bromide (III), which by a Wittig reaction with 2-oxa-3-hydroxy-6-syn- ( 3alpha-tetrahydropyranyloxy-4,4-dimethyl-1-trans-octen-1-yl) -7-anti-tetrahydropyranyloxybicyclo- [3.3.0] cis-octane (IV) (prepared according to reference 2) by means of sodium dimethylsulfinate in DMSO yields 9alpha-hydroxy-11alpha, 15alpha-bis (tetrahydropyranyloxy) -16,16-dimethyl-alpha-dinorprosta-5-cis-13-trans-dienoic acid (V). The reduction of (V) with H2 over Pd / C in methanol affords the 13-trans-prostenoic acid (VI), which is methylated with CH2N2 in ether yielding the methyl ester (VII). The reduction of (VII) with diisobutyl aluminum hydride in toluene affords the corresponding aldehyde (VIII) , which by a Wittig reaction with triethyl phosphonoacetate (IX) by means of NaH in THF is converted into 9alpha-hydroxy-11alpha, 15alpha-bis (tetrahydropyranyloxy) -16,16-dimethylprosta-2-trans-dienoic acid ethyl ester (X .) The hydrolysis of the ester (X) with KOH in ethanol-water gives the corresponding acid (XI), which is oxidized with CrO3, MnSO4 and H2SO4 in ether – water yielding the protected ketoacid (XII) The hydrolysis of (XII. ) with acetic acid-water at 80 C gives 9-oxo-11alpha, 15alpha-dihydroxy-16,16-dimethyl-prosta-2-trans-13-trans-dienoic acid (16,16-dimethyl-DELTA2-trans-PGE1 ) (XIII), which is finally methylated with CH2N2 in ether
 |
|
: Gemeprost
CAS 64318-79-2
CAS Name: (2E,11a,13E,15R)-11,15-Dihydroxy-16,16-dimethyl-9-oxoprosta-2,13-dien-1-oic acid methyl ester
Additional Names: 16,16-dimethyl-trans-D2-PGE1 methyl ester
Manufacturers’ Codes: ONO-802
Trademarks: Cergem (Searle); Cervagem(e) (M & B); Preglandin (Ono)
Molecular Formula: C23H38O5
Molecular Weight: 394.54
Percent Composition: C 70.02%, H 9.71%, O 20.28%
Literature References:
Analog of prostaglandin E1, q.v. Prepn: M. Hayashi et al., DE 2700021; eidem, US 4052512 (both 1977 to Ono);
H. Suga et al., Prostaglandins 15, 907 (1978).
Effects on uterine contractility and steroid hormone plasma levels: K. Oshimaet al., J. Reprod. Fertil. 55, 353 (1979).
Effects on reproductive function: K. Matsumoto et al., Nippon Yakurigaku Zasshi 79, 15 (1982), C.A. 96, 98392 (1982).
Use in termination of first trimester pregnancy: O. Reiertsen et al., Prostaglandins Leukotrienes Med. 8, 31 (1982).
Therap-Cat: Abortifacient; oxytocic.
Keywords: Abortifacient/Interceptive; Oxytocic; Prostaglandin/Prostaglandin Analog
|