
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA Breakthrough Therapy Designation: 32 And Counting
On February 3rd, GlaxoSmithKline (GSK) announces that Promacta (US)/Revolade (Europe) (Eltrombopag) receives the coveted FDA Breakthrough Therapy Designation (BTD) for cytopenias in patients with Severe Aplastic Anemia (SAA), who have had insufficient response to Immunosuppressive Therapy (IST). The drug is not approved or licensed anywhere in the world for this indication.
SAA is a rare disorder where the bone marrow fails to make enough new blood cells. There are currently no therapies approved for this indication. About forty percent (40%) of patients who do not respond to initial IST die within 5 years of diagnosis.
Regulatory Actions
• Receives FDA ODD in November 2013 for Aplastic Anemia
• Receives FDA BTD in February 2014 for Aplastic Anemia
• Receives FDA ODD in May 2008 & FDA approval in November 2008 for Idiopathic Thrombocytopenia Purpura.
This is the 32nd BTD that is announced by a sponsor company since…
View original post 150 more words
SUMATRIPTAN …Avanir files new drug application for migraine drug

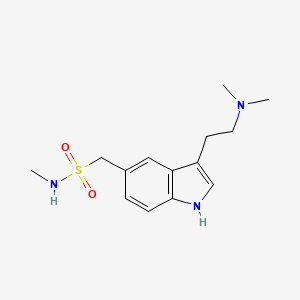
SUMATRIPTAN, GR-43175
1-[3-(2-dimethylaminoethyl)-1H-indol-5-yl]- N-methyl-methanesulfonamide
3-[2-(Dimethylamino)ethyl]-N-methyl-1H-indole-5-methanesulfonamide
| Formula | C14H21N3O2S |
|---|---|
| Mol. mass | 295.402 g/mol |
| CAS number | 103628-46-2 |
|---|
Avanir Pharmaceuticals has filed a new drug application (NDA) with the US Food and Drug Administration (FDA) for approval of its new breath-powered investigational drug-device combination product, ‘AVP-825’, for the acute treatment of migraines. click on title Avanir files new drug application for migraine drug
 SUMATRIPTAN
SUMATRIPTAN
SUMATRIPTAN SUCCINATE

AVP-825 is an investigational drug-device combination product consisting of low-dose sumatriptan powder delivered intranasally utilizing a novel Breath Powered delivery technology. If approved, AVP-825 would be the first and only fast-acting, dry-powder intranasal form of sumatriptan for the treatment of migraine.
The Breath Powered delivery technology is activated by user’s breath to propel medications deep into the nasal cavity where absorption is more efficient and consistent than through most other routes. A user exhales into the device, automatically closing the soft palate and sealing off the nasal cavity completely. Through a sealing nosepiece placed into the nostril, the exhaled breath carries medication from the device directly into one side of the nose. Narrow nasal passages are gently expanded and medication is dispersed deep into the nasal cavity reaching areas where it can be rapidly absorbed. As the medication is delivered, the air flows around to the opposite side of the nasal cavity and exits through the other nostril. Closure of the soft palate helps prevent swallowing or inhalation of sumatriptan powder into the lungs.
| Canada | 2469019 | APPROVED 2005-09-13 | EXP 2022-12-04 |
| United States | 6135979 | 1997-03-21 | 2017-03-21 |
| United States | 5705520 | 1994-12-10 | 2011-12-10 |
| Canada | 2098302 | 2001-10-16 | 2011-12-10 |
| Patent No | PatentExpiry | use code |
|---|---|---|
| 5307953 | Dec 2, 2012 | |
| 5307953*PED | Jun 2, 2013 | |
| 5554639 | Sep 10, 2013 | U-232…METHOD OF TREATING MIGRAINE |
| 5554639*PED | Mar 10, 2014 |
Sumatriptan is a synthetic drug belonging to the triptan class, used for the treatment of migraine headaches. Structurally, it is an analog of the naturally occurring neuro-active alkaloids dimethyltryptamine (DMT), bufotenine, and 5-methoxy-dimethyltryptamine, with an N-methyl sulfonamidomethyl- group at position C-5 on the indole ring.[1]
Sumatriptan is produced and marketed by various drug manufacturers with many different trade names such as Sumatriptan, Imitrex, Treximet, Imigran, Imigran recovery.
Large doses of sumatriptan can cause sulfhemoglobinemia, a rare condition in which the blood changes from red to greenish-black, due to the integration of sulfur into the hemoglobin molecule.[2] If sumatriptan is discontinued, the condition reverses within a few weeks.
Serious cardiac events, including some that have been fatal, have occurred following the use of sumatriptan injection or tablets. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation.
The most common side-effects[3] reported by at least 2% of patients in controlled trials of sumatriptan (25, 50, and 100 mg tablets) for migraine are atypical sensations (paresthesias and warm/cold sensations) reported by 4% in the placebo group and 5–6% in the sumatriptan groups, pain and other pressure sensations (including chest pain) reported by 4% in the placebo group and 6–8% in the sumatriptan groups, neurological events (vertigo) reported by less than 1% in the placebo group and less than 1% to 2% in the sumatriptan groups. Malaise/fatigue occurred in less than 1% of the placebo group and 2–3% of the sumatriptan groups. Sleep disturbance occurred in less than 1% in the placebo group to 2% in the sumatriptan group.
 SUMATRIPTAN
SUMATRIPTAN
Sumatriptan is structurally similar to serotonin (5HT), and is a 5-HT (types 5-HT1D and 5-HT1B[4]) agonist. The specific receptor subtypes it activates are present on the cranial arteries and veins. Acting as an agonist at these receptors, sumatriptan reduces the vascular inflammation associated with migraines.
The specific receptor subtype it activates is present in the cranial and basilar arteries. Activation of these receptors causes vasoconstriction of those dilated arteries. Sumatriptan is also shown to decrease the activity of the trigeminal nerve, which, it is presumed, accounts for sumatriptan’s efficacy in treating cluster headaches. The injectable form of the drug has been shown to abort a cluster headache within fifteen minutes in 96% of cases.[5]
Sumatriptan is administered in several forms; tablets, subcutaneous injection, and nasal spray. Oral administration (as succinate) suffers from poorbioavailability, partly due to presystemic metabolism—some of it gets broken down in the stomach and bloodstream before it reaches the target arteries. A new rapid-release tablet formulation has the same bioavailability, but the maximum concentration is achieved on average 10–15 minutes earlier. When injected, sumatriptan is faster-acting (usually within 10 minutes), but the effect lasts for a shorter time. Sumatriptan is metabolised primarily by monoamine oxidase A into an indole acetic acid analogue, part of which is further conjugated with glucuronic acid. These metabolites are excreted in the urine and bile. Only about 3% of the active drug may be recovered unchanged.
There is no simple, direct relationship between sumatriptan concentration (pharmacokinetics) per se in the blood and its anti-migraine effect (pharmacodynamics). This paradox has, to some extent, been resolved by comparing the rates of absorption of the various sumatriptan formulations, rather than the absolute amounts of drug that they deliver.[6][7]
Sumatriptan was the first clinically available triptan (in 1991). In the United States, it is available only by medical prescription. However, it can be bought over the counter in the UK and Sweden in 50 mg dosage. Several dosage forms for sumatriptan have been approved, including tablets, solution for injection, and nasal inhalers.
On April 15, 2008, the US FDA approved Treximet, a combination of sumatriptan and naproxen, an NSAID.[8] This combination has shown a benefit over either medicine used separately.[9]
In July 2009, the US FDA approved a single-use jet injector formulation of sumatriptan. The device delivers a subcutaneous injection of 6 mg sumatriptan, without the use of a needle.Autoinjectors with needles have been previously available in Europe and North America for several years.[10]
Phase III studies with a iontophoretic transdermal patch (Zelrix/Zecuity) started in July 2008.[11] This patch uses low voltage controlled by a pre-programmed microchip to deliver a single dose of sumatriptan through the skin within 30 minutes.[12][13]Zecuity was approved by the US FDA in January 2013.[14]

Sumatriptan vials 100 5509
On November 6, 2008, Par Pharmaceutical announced that it would begin shipping generic versions of sumatriptan injection (sumatriptan succinate injection) 4 mg and 6 mg starter kits and 4 mg and 6 mg pre-filled syringe cartridges to the trade immediately. In addition, Par anticipates launching the 6 mg vials early in 2009.[15]
Mylan Laboratories Inc., Ranbaxy, Sandoz, Dr. Reddy’s Pharmaceuticals and other companies have received FDA approval for generic versions of Imitrex tablets in 25-, 50-, and 100-milligram doses since 2009. The drug is available in U.S. and European markets, since Glaxo’s patent protections have expired in those jurisdictions. However, sales of a generic delivered via nasal spray are still restricted in the United States.
See also Sumavel DosePro (above).[10]
Chemistry
hydrogenation of nitrile with pd/c in presence of dimethyl amine
…………………


The diazotation of 4-amino-N-methylbenzenemethanesulfonamide (I) with NaNO2-HCl followed by reduction with SnCl2 gives the 4-hydrazino compound (II), which is condensed with (phenylthio)acetaldehyde (III) in ethanol yielding the ethylideneamino compound (IV). The cyclization of (IV) with HCl in ethanol affords N-methyl-3-(phenylthio)-1H-indole-5-methansulfonamide (V), which is desulfurized with RaNi in refluxing ethanol-water to give N-methyl-1H-indole-5-methanesulfonamide (VI). The reaction of (VI) with oxalyl chloride and dimethylamine yields the oxalyl derivative (VII), which is finally reduced with LiAlH4 in refluxing THF.

The condensation of hydrazine (II) with 4,4-dimethoxy-N,N-dimethylbutylamine (VIII) by means of HCl in water gives the butylidenehydrazino compound (IX), which is cyclized with polyphosphate ester (PPE) in CHCl3.

……………………
Beilstein J. Org. Chem. 2011, 7, 442–495.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S9
ref are below article
Indoles
The neuroamine transmitter serotonin contains an indole ring, so it is not surprising that indoles are a recurring theme in many drugs affecting central nervous system (CNS) function including antidepressants, antipsychotics, anxiolytics and antimigraine drugs, as well as psychedelic agents. Indole is also one of the best represented heterocyclic motifs present in the top selling pharmaceuticals, being found in eight of the top 200 drugs, with five of these belonging to the triptan family of antimigraine treatments. The classical Fischer indole synthesis is usually reported as one of the first choice routes to prepare these scaffolds. Drugs such as GSK’s serotonin receptor modulators sumatriptan (49, Imitrex) and zolmitriptan (50, Zomig) use the Fischer indole synthesis at a late stage in order to form the desired compound albeit in only low to moderate yields (Scheme 9).
![[1860-5397-7-57-i9]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i9.png)
However, in sumatriptan the indole product resulting from the Fischer synthesis can still react further which leads to the formation of by-products and significantly reduced yields. One way to minimise this was to protect the nitrogen of the sulfonamide group prior to indole formation [11]. This leads not only to an increased yield in the indole forming step (to 50%) but also facilitates chromatographic purification. The dimethylamino group can be present from the beginning of the synthesis or can be introduced via displacement of chloride or reduction of a cyano moiety. Alternatively, the dimethyl ethylene amine side chain can be introduced in position 3 via a Friedel–Crafts-type acylation. The resulting acid chloride is transformed in situ to the corresponding amide which on reduction with lithium aluminium hydride affords sumatriptan (Scheme 10) [12].
![[1860-5397-7-57-i10]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i10.png)
In the standard Fischer indole synthesis a hydrazine, which is most commonly derived from the corresponding diazonium salt, is reacted with a suitable carbonyl compound. Alternatively, the Japp–Klingemann reaction can be used to directly couple the diazonium salt with a β-ketoester to obtain a hydrazone which can then undergo indole ring formation (Scheme 11) [13].
![[1860-5397-7-57-i11]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i11.png)
As can be seen from Scheme 11 the indole 59 prepared via the Japp–Klingemann reaction is substituted at position 2 by an ester group which prevents reaction with electrophiles, thereby reducing the amount of undesired by-products. A simple sequence of hydrolysis and decarboxylation then affords sumatriptan [14].
All the reported methods for the synthesis of sumatriptan begin with the sulfonamide group already present on the aromatic ring and several routes are possible to introduce this functional group. The scalable route to the sulfonamides inevitably involves the preparation of the sulfonyl chloride intermediate which is then trapped with the desired amine. The sulfonyl chloride can also be prepared from the corresponding hemithioacetal 61 by treatment with NCS in wet acetic acid (Scheme 12). This efficient oxidation produces only methanol and formaldehyde as by-products [15].
![[1860-5397-7-57-i12]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i12.png)
- 11. Pete, B.; Bitter, I.; Szántay, C., Jr.; Schön, I.; Töke, L. Heterocycles 1998, 48, 1139–1149. doi:10.3987/COM-97-8087
- 12…Oxford, A. W. Indole Derivative. U.S. Patent 5,037,845, Aug 6, 1991.
- 13…Japp, F. R.; Klingemann, F. Chem. Ber. 1887, 20, 2942–2944. doi:10.1002/cber.188702002165
- Pete, B.; Bitter, I.; Harsányi, K.; Töke, L. Heterocycles 2000, 53, 665–673. doi:10.3987/COM-99-8815
- Kim, D.-W.; Ko, Y. K.; Kim, S. H. Synthesis 1992, 12, 1203–1204. doi:10.1055/s-1992-26333
[15
References for full article
- The presence of the sulfonamide group in the molecule does not make sumatriptan a “sulfa drug”, since it does not have any anti-microbial properties.
- “Patient bleeds dark green blood”. BBC News. 8 June 2007. Retrieved 6 March 2010.
- Tablets
- Razzaque Z, Heald MA, Pickard JD, et al. (1999). “Vasoconstriction in human isolated middle meningeal arteries: determining the contribution of 5-HT1B- and 5-HT1F-receptor activation”.Br J Clin Pharmacol 47 (1): 75–82. doi:10.1046/j.1365-2125.1999.00851.x. PMC 2014192.PMID 10073743.
- Treatment of acute cluster headache with sumatriptan. The Sumatriptan Cluster Headache Study Group. N Engl J Med 1991;325:322-6.
- Fox, A. W. (2004). “Onset of effect of 5-HT1B/1D agonists: a model with pharmacokinetic validation”. Headache 44 (2): 142–147. doi:10.1111/j.1526-4610.2004.04030.x.PMID 14756852. edit
- Freidank-Mueschenborn, E.; Fox, A. (2005). “Resolution of concentration-response differences in onset of effect between subcutaneous and oral sumatriptan”. Headache 45 (6): 632–637. doi:10.1111/j.1526-4610.2005.05129a.x. PMID 15953294. edit
- GSK press release – Treximet (sumatriptan and naproxen sodium) tablets approved by FDA for acute treatment of migraine
- Brandes JL, Kudrow D, Stark SR, et al. (April 2007). “Sumatriptan-naproxen for acute treatment of migraine: a randomized trial”. JAMA 297 (13): 1443–54.doi:10.1001/jama.297.13.1443. PMID 17405970.
- Brandes, J.; Cady, R.; Freitag, F.; Smith, T.; Chandler, P.; Fox, A.; Linn, L.; Farr, S. (2009). “Needle-free subcutaneous sumatriptan (Sumavel DosePro): bioequivalence and ease of use.”. Headache 49 (10): 1435–1444. doi:10.1111/j.1526-4610.2009.01530.x.PMID 19849720. edit
- ClinicalTrials.gov NCT00724815 The Efficacy and Tolerability of NP101 Patch in the Treatment of Acute Migraine (NP101-007)
- SmartRelief -electronically assisted drug delivery (iontophoresis)
- Pierce, M; Marbury, T; O’Neill, C; Siegel, S; Du, W; Sebree, T (2009). “Zelrix: a novel transdermal formulation of sumatriptan”. Headache 49 (6): 817–25. doi:10.1111/j.1526-4610.2009.01437.x. PMID 19438727.
- Zecuity Approved by the FDA for the Acute Treatment of Migraine
- “PAR PHARMACEUTICAL BEGINS SHIPMENT OF SUMATRIPTAN INJECTION”. Par Pharmaceutical. 2008-11-06. Retrieved 2008-11-25.
- Serotonin 5HT1-receptor agonist. Prepn: M. D. Dowle, I. H. Coates, DE 3320521; eidem, US 4816470; A. W. Oxford, GB 2162522 (1983, 1989, 1986 all to Glaxo).
- Receptor binding studies: P. P. A. Humphrey et al., Br. J. Pharmacol.94, 1123 (1988); P. Schoeffter, D. Hoyer, Arch. Pharmacol. 340, 135 (1989).
- LC-MS determn in plasma: J. Oxford, M. S. Lant, J. Chromatogr. 496, 137 (1989).
- Clinical evaluations in migraine: A. Doenicke et al., Lancet 1, 1309 (1988);
- Subcutaneous Sumatriptan International Study Group, N. Engl. J. Med. 325, 316 (1991); in acute cluster headache: Sumatriptan Cluster Headache Study Group, ibid. 322.
- Review of pharmacology and clinical experience: S. J. Peroutka, Headache 30 (Suppl. 2), 554-560 (1990).
- Drugs Fut 1989,14(1),35
|
1-4-2012
|
Noncardiotoxic pharmaceutical compounds
|
|
|
7-9-2010
|
NON-MUCOADHESIVE FILM DOSAGE FORMS
|
|
|
1-22-2010
|
Fixed Combination Dosage Forms for the Treatment of Migraine
|
|
|
12-11-2009
|
ACTIVE AGENT DELIVERY SYSTEMS AND METHODS FOR PROTECTING AND ADMINISTERING ACTIVE AGENTS
|
|
|
10-9-2009
|
PHARMACEUTICAL COMPOSITIONS COMPRISING A TRIPTAN AND A NONSTEROIDAL ANTI-INFLAMMATORY DRUG
|
|
|
10-9-2009
|
ACTIVE AGENT DELIVERY SYSTEMS AND METHODS FOR PROTECTING AND ADMINISTERING ACTIVE AGENTS
|
|
|
5-7-2009
|
Patient controlled drug delivery device
|
|
|
3-20-2009
|
DEUTERIUM-ENRICHED SUMATRIPTAN
|
|
|
3-13-2009
|
Rapid dissolution of combination products
|
|
|
2-19-2009
|
A METHOD OF IDENTIFYING MODULATORS OF CELL SURFACE MEMBRANE RECEPTORS USEFUL IN THE TREATMENT OF DISEASE
|
|
4-8-1992
|
PREPARATION OF INDOLE DERIVATIVES
|
|
|
1-10-1992
|
PHARMACEUTICAL PREPARATIONS
|
|
|
10-32-1991
|
SYSTEM AND METHOD FOR DETERMINING THREE-DIMENSIONAL STRUCTURES OF PROTEINS
|
|
|
8-7-1991
|
Indole derivative
|
|
|
7-4-1990
|
Pharmaceutical formulations
|
|
|
8-8-1984
|
Fuel and water homogenizer
|
Avanir Pharmaceuticals, Inc. is a biopharmaceutical company focused on bringing innovative medicines to patients with central nervous system disorders of high unmet medical need. As part of our commitment, we have extensively invested in our pipeline and are dedicated to advancing medicines that can substantially improve the lives of patients and their loved ones. For more information about Avanir, please visit http://www.avanir.com.
AVANIR® is a trademark or registered trademark of Avanir Pharmaceuticals, Inc. in the United States and other countries. All other trademarks are the property of their respective owners.
Avanir Pharmaceuticals, Inc. licensed exclusive rights for the development and commercialization of AVP-825, a novel Breath Powered intranasal system containing a low-dose sumatriptan powder from OptiNose Inc. of Yardley, PA.
IMITREX Tablets contain sumatriptan succinate, a selective 5-HT1B/1D receptor agonist. Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole- 5-methanesulfonamide succinate (1:1), and it has the following structure:
IMITREX Tablets contain sumatriptan succinate, a selective 5-HT1B/1Dreceptor agonist. Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole- 5-methanesulfonamide succinate (1:1), and it has the following structure:
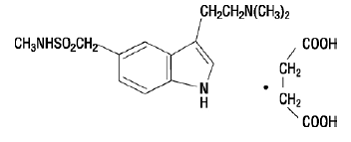 |
The empirical formula is C14H21N3O2S•C4H6O4, representing a molecular weight of 413.5. Sumatriptan succinate is a white to off-white powder that is readily soluble in water and in saline.
Each IMITREX Tablet for oral administration contains 35, 70, or 140 mg of sumatriptan succinate equivalent to 25, 50, or 100 mg of sumatriptan, respectively. Each tablet also contains the inactive ingredients croscarmellose sodium, dibasic calcium phosphate, magnesium stearate, microcrystalline cellulose, and sodium bicarbonate. Each 100-mg tablet also contains hypromellose, iron oxide, titanium dioxide, and triacetin.
Topiroxostat 托匹司他 for gout and hyperuricemia


Topiroxostat
托匹司他
FUJI YAKUHIN ……..INNOVATOR
Approved in japan PMDA JUNE 28 2013
Xanthine oxidase inhibitor
FOR GOUT AND HYPERURICEMIA
Launched – 2013, Fuji YakuhinSanwa, Topiloric Uriadec
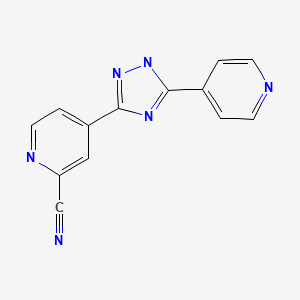
IUPAC Name: 4-(5-pyridin-4-yl-1H-1,2,4-triazol-3-yl)pyridine-2-carbonitrile
CAS Registry Number: 577778-58-6
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1)
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole
Synonyms: 4-(5-PYRIDIN-4-YL-1H-1,2,4-TRIAZOL-3-YL)PYRIDINE-2-CARBONITRILE,
AC1NRB9T, Topiroxostat (JAN/INN), DB01685, D09786, FYX-051
SK-0910
4-[5-PYRIDIN-4-YL-1H-[1,2,4]TRIAZOL-3-YL]-PYRIDINE-2-CARBONITRILE,
C13H8N6 MF,248.2482 MW
 TOPIROXOSTAT
TOPIROXOSTAT
托匹司他
A xanthine oxidase inhibitor used to treat gout and hyperuricemia.

PATENT EXP 3/12/22, US /EU/CN

FYX-051, TOPIROXOSTAT is a xanthine oxidase inhibitor. This agent was approved in Japan by Fuji Yakuhin and Sanwa for the treatment of gout and hyperuricemia in 2013 and launched at the same year. In 2009, the compound was licensed to Sanwa by Fuji Yakuhin in Japan for the codevelopment and commercialization of gout.
The number of patients with hyperuricemia in Japan is reported to be 1.25 million and the number suffering from asymptomatic hyperuricemia is estimated to reach several millions. Hyperuricemia is becoming a popular disease.
Presently, hyperuricemia and gout due to hyperuricemia are treated by improving the living environment and administering various drug therapies for each period when an attack of gout is predicted to occur (presymptomatic period), when an attack of gout occurs, or when an attack of gout subsides. That is, preventive therapy is conducted in the presymptomatic period by administering colchicines as well as controlling the daily living environment. When an attack occurs, drug therapy using non-steroidal or steroidal anti-inflammatory agents is mainly conducted. After the attack subsides, patients are given guidance to improve their lifestyle. When improvement is judged insufficient, an assessment is made as to whether hyperuricemia is caused by reduced excretion of uric acid or by increased production of uric acid followed by treatment with drugs, which exhibit a uricosuric effect, such as probenecid and benzbromarone, those which inhibit resorption of uric acid, such as sulfinpyrazone, those which improve acidurea conditions, such as citrates, and xanthine oxidase inhibitors which inhibit production of uric acid, such as allopurinol. Colchicine is said to be able to prevent about 90% of attacks through inhibiting chemotaxis and phagocytosis of leukocytes, such as neutrophils, if administration thereof has been completed within a few hours before the attack. Since colchicine has various adverse effects, however, the use thereof is limited to the minimum and it is therefore difficult to timely administer it.
Accordingly, drug therapies are mainly adopted, but only allopurinol is available for the treatment of a disease caused by increased production of uric acid. However, a metabolite of allopurinol, oxypurinol, tends to accumulate and may cause calculi formation. Furthermore, this drug has been reported to induce adverse events such as rash, a decreased renal function and hepatitis, and it is not easy to administer.
Examples of compounds having xanthine oxidase inhibiting activity that can be used for treating gout caused by increased production of uric acid and that are effective for hyperuricemia and gout due to hyperuricemia have been described in J. Medicinal Chemistry, 1975, Vol. 18, No. 9, pp. 895–900, Japanese Patent Publication No. 49-46622 and Japanese Patent Publication No. 50-24315, which disclose some 1,3,5-substituted or 3,5-substituted 1,2,4-triazole compounds.
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1) has a xanthine oxidase inhibitory activity and serum uric acid level known as the agent that reduces (Patent Document 1).

The method for producing the compound (1), for example, 2 by Reissert Henze reaction isonicotinic acid methyl N-oxide – is a cyano isonicotinate, and the hydrazide which is then, 4 – this condensed cyanopyridine After obtaining a hydrazide of isonicotinic acid N-oxide (Patent Document 1, Example 12) and method, a cyano group after introduction, 4 by Reissert Henze reaction – method of condensing a cyano pyridine is known (Patent Document 1, Example 39).Further, 4 – as a starting material cyano-N-oxide, a triazole ring after construction (Patent Document 3), Reissert Henze unprotected or (Patent Document 2) to protect the ring condensed with isonicotinic acid hydrazide method of obtaining the compound (1) by introducing a cyano group by the reaction have also been reported.
The crystalline polymorph, yet the same molecule with the same chemical composition, the molecular arrangement in the crystal are different, and are different crystalline states. The pharmaceutical compounds having crystal polymorphism such the differences in physicochemical properties, affect pharmacological activity, solubility, bioavailability, stability and the like are known.Therefore, when the crystal polymorphism is present in a pharmaceutically useful compound, producing compounds of the crystalline form highly useful from polymorphs thereof is desirable.
WO 2003/064410 discloses WO 2005/009991 discloses Japanese Patent Publication No. 2005-41802
However, 4 of the above Patent Document – no description about the presence of crystalline polymorph on carbonitrile – pyridine-2-[yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol] It has not been, to these manufacturing methods, it is disclosed a method for the purpose of improving the chemical purity and yield, there is no description of the crystallographic plane.
Method of producing topiroxostat, useful for preventing or treating gout; and its intermediates. Picks up from WO2012060308, claiming the use of this topiroxostat for treating renal dysfunction. Along with the concurrently published WO2014017515, claiming crystalline Forms I and II of this compound, which, Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017516
Crystalline Forms I and II of topiroxostat, useful for preventing or treating gout. Along with the concurrently published WO2014017516, claiming a method of producing this compound. Picks up from WO2012060308, claiming a method of treating renal dysfunction using topiroxostat, which Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017515
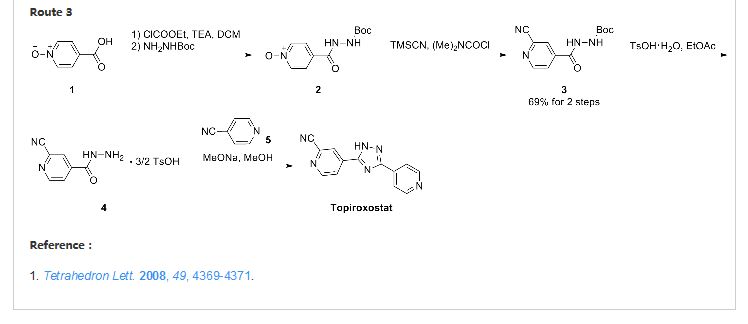
novel 1,2,4-triazole compounds having an optionally substituted 2-cyanopyridin-4-yl group at 3-position and an optionally substituted aromatic group at 5-position inhibit a xanthine oxidase and are useful for treatment of gout and hyperuricemia, and have previously filed a patent application (Patent Document 1). The compounds can be prepared according to a method shown by the following reaction scheme:
-
 wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
Patent Document 1: JP-A-2002-017825 -
-
A compound represented by formula (1) which is a starting material may be prepared by a method described in, for example, JP-A-47-7120, JP-A-61-152661A, JP-A-62-149673, JP-A-2002-528447, or European Patent Application No. 559363 specification. However, it is preferable to prepare compound (1) according to the following reaction scheme:
-

-


SYNTHESIS



PATENT
- Example 2
-
To the toluene solution obtained in Example 1 (2) was added 2-propanol (700 mL), and the mixture was stirred. To the resulting solution was added p-toluenesulfonic acid monohydrate (151.16 g) and the resulting mixture was stirred for 8 hours at an internal temperature of 80°C. The mixture was brought to room temperature, and the precipitated crystals were taken out and washed with 2-propanol (210 mL×2). The white crystals were dried under reduced pressure at 60°C for 15 hours to give 106.0 g of the captioned compound as white crystals. Subsequently, 90.0 g of the crystals was suspended in a mixture of 2-butanol (49 mL) and water (491 mL) and heated to an internal temperature of 80°C for 1 hour. The internal temperature was brought to room temperature, and the crystals were filtered and washed with a mixture of 2-butanol and water (1:10) (270 mL×3). The resulting crystals were dried under reduced pressure at 60°C for 15 hours to give 75.7 g of the captioned compound in a high purity.
-
1H―NMR(DMSO-d6)δppm:2.29(s,3H), 7.11 (m,2H), 7.48 (dd, 2H, J=6.48, 1.62Hz) , 8.32-8.35(m, 3H) , 8.57(dd, 1H, J=1.62, 0.81Hz) , 8.94-8.98(m, 3H)
- Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonate
Example 3
Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
-
To the white crystals (50.5g) obtained in Example 2 was added 2-propanol (937.5 mL) and water (312.5 mL), and the resulting mixture was heated and dissolved at an internal temperature of 80°C. Immediately thereafter, the solution was filtered and the filtrate was cooled to an internal temperature of 20°C. To the resulting suspension was added dropwise 0.52 mol/l of an aqueous sodium hydrogen carbonate solution (250 mL), and the mixture was stirred at room temperature for 2 hours. Then the crystals were filtered and washed with water (150 mL×3) and 2-butanol (150 mL×2). The crystals were dried under reduced pressure at 80°C for 15 hours to give 29.4 g of the captioned compound as pale yellow crystals.
-
1H―NMR(DMSO-d6)δppm:8.02(dd, 2H, J=4.59, 1.62Hz),8.32(dd, 1H, J=5.13, 1.62Hz), 8.55(dd, 1H, J=1.62, 1.08Hz), 8.80(dd, 2H, J=4.59, 1.62Hz), 8.93 (dd, 1H, J=5.13, 1.08Hz)
SYNTHESIS
Example 12
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of methyl isonicotinate N-oxide
13.9 g of isonicotinic acid N-oxide was added to 209 ml of methylene chloride, 29.7 g of 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline was further added thereto, and the mixture was stirred under argon atmosphere at room temperature for one hour. 32.1 g of methanol was added to this mixture, which was stirred at room temperature for 17 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (3:1) was used as an eluent to yield 11.1 g of a white powder.
1H-NMR (CDCl3) δppm: 3.95 (3H, s), 7.88 (2H, d, J=7.25 Hz), 8.22 (2H, J=7.25 Hz)
2) Production of Methyl 2-cyanoisonicotinate
11.1 g of the crystal obtained in 1) was dissolved in 170 ml of acetonitrile, 14.6 g of triethylamine and 21.5 g of trimethylsilylnitrile were added thereto, and the mixture was refluxed under argon atmosphere for 16 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (95:5) was used as an eluent to yield 8.44 g of a pale yellow powder.
1H-NMR (CDCl3) δppm: 4.01 (3H, s), 8.08 (1H, d, J=5.45 Hz), 8.24 (1H, s), 8.90 (1H, d, J=5.45 Hz)
3) Production of 2-cyanoisonicotinic acid hydrazide
8.44 g of the crystal obtained in 2) was added to 85 ml of methanol, 1.84 g of hydrazine was further added thereto, and the mixture was stirred under argon temperature for 2 hours. After the solvent was evaporated under reduced pressure, chloroform was added to the residue, which was stirred at room temperature for one hour. The precipitated crystal was filtered, washed with chloroform and dried with a vacuum pump to yield 4.15 g of a pale yellow powder.
1H-NMR (DMSO-d6) δppm: 4.72 (2H, s), 8.05 (1H, d, J=5.12 Hz), 8.31 (1H, s),8.90 (1H, d, J=5.12 Hz), 10.23 (1H, s)
4) Production of the Object Compound
2.67 g of 4-cyanopyridine was dissolved in 40 ml of methanol, 0.83 g of sodium methoxide was added thereto, and the mixture was stirred at room temperature for one hour. Then 4.15 g of the crystal obtained in 3) was added and the mixture was refluxed for 37 hours. After the reaction completed, the precipitated solid was filtered, washed with methanol and dried with a vacuum pump to yield 3.66 g of the object compound as a yellow powder.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54, 1.57 Hz), 8.31 (1H, dd, J=5.11, 1.65 Hz), 8.53 (1H, dd, J=1.65, 0.50 Hz), 8.80 (2H, dd, J=4.54, 1.57 Hz), 8.93 (1H, dd, J=5.11, 0.50 Hz)
Example 39
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of isonicotinic acid (N-2-tert-butoxycarbonyl)hydrazide-1-oxide
585 ml of methylene chloride was added to 39.0 g of isonicotinic acid N-oxide, and after 34.0 g of triethylamine was further added thereto, the mixture was cooled under argon atmosphere to −15° C. 33.5 g of ethyl chlorocarbonate in 117 ml of methylene chloride was added dropwise to this mixture, which was stirred at a temperature from −5 to −10° C. for one hour. Then 44.4 g of tert-butyl ester of carbamic acid in 117 ml of methylene chloride was added dropwise to this mixture and it was allowed to slowly rise to room temperature while it was stirred. The precipitated solid was filtered after 15 hours, washed with methylene chloride, and dried with a vacuum pump to yield 49.7 g of white crystal.
1H-NMR (DMSO-d6) δppm: 1.42 (9H, s), 7.82 (2H, d, J=7.09 Hz), 8.33 (2H, d, J=7.09 Hz), 9.02 (1H, s), 10.44 (1H, s)
Production of 2-cyanoisonicotinic acid hydrazine 1½ P-Toluenesulfonic acid salt
228 ml of dioxane was added to 30.4 g of the crystal obtained in 1), and after 13.1 g of trimethylsilyl cyanide and 38.8 g of N,N-dimethylcarbamoyl chloride were further added thereto, the mixture was stirred under argon atmosphere at 60° C. for 5 hours. After the solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and subsequently washed with 1.5 M sodium carbonate aqueous solution and a saturated saline solution and dried over magnesium sulfate. After the magnesium sulfate was filtered off, the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue, 68.5 g of p-toluenesulfonic acid monohydrate was added thereto, and the mixture was stirred at room temperature for 22 hours. The precipitated crystal was filtered, washed with ethyl acetate, and dried with a vacuum pump to yield 40.3 g of white crystal 2).
1H-NMR (DMSO-d6) δppm: 2.28 (4.5H, s), 7.12 (3H, dd, J=7.92 & 0.66 Hz), 7.48 (3H, dd, J=7.92 & 0.66 Hz), 8.10 (1H, dd, J=5.11 & 1.81 Hz), 8.39 (1H, dd, J=1.81 & 0.33 Hz), 8.99 (1H, dd, J=5.11 & 0.33 Hz)
3) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
9.98 g of 4-cyanopyridine was dissolved in 250 ml of methanol, and after 7.77 g of sodium methoxide was added thereto, the mixture was stirred at room temperature for one hour. Then 40.3 g of the crystal obtained in 2) was added and the mixture was refluxed for 24 hours. After the reaction completed, the precipitated crystal was filtered, washed with methanol, and dried with a vacuum pump to yield 16.3 g of yellow crystal.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54 & 1.57 Hz), 8.31 (1H, dd, J=5.11 & 1.65 Hz), 8.53 (1H, dd, J=1.65 & 0.50 Hz), 8.80 (2H, dd, J=4.54 & 1.57 Hz), 8.93 (1H, dd, J=5.11 & 0.50 Hz)
4) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
45 ml of ethanol and 15 ml of 1-methyl-2-pyrrolidone were added to 3.0 g of the crystal obtained in 3), and the mixture was heated and stirred at 80° C. for 19 hours. The crystal was filtered, subsequently washed with a mixture of ethanol and 1-methyl-2-pyrrolidone (3:1) and ethanol, and dried with a vacuum pump to yield 2.71 g of yellow crystal.
5) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonic acid salt
5 ml of ethanol and 30 ml of water were added to 2.48 g of the crystal obtained in 4), and after 3.8 g of p-toluenesulfonic acid monohydrate was further added thereto, the mixture was stirred at room temperature for 5 hours. The precipitated crystal was filtered, subsequently washed with a mixture of ethanol and water (1:6), water and then ethanol, and dried with a vacuum pump to yield 3.5 g of white crystal.
1H-NMR (DMSO-d6) δppm: 2.28 (3H, s), 7.12 (2H, dd, J=7.75 & 0.50 Hz), 7.48 (2H, dd, J=7.75 & 0.50 Hz), 8.33 (1H, dd, J=5.12 & 1.65 Hz), 8.45 (2H, d, J=6.11 Hz), 8.57 (1H, dd, J=1.65 & 0.66 Hz), 8.96˜9.02 (3H, m)
6) Production of the object compound
17 ml of ethanol and 17 ml of water were added to 3.36 g of the crystal obtained in 5), and the mixture was stirred at room temperature for 30 minutes. A solution of sodium carbonate (0.74 g of sodium carbonate in 17 ml of water) was further added, and the mixture was stirred at room temperature for 2 hours. The precipitated crystal was filtered, subsequently washed with water and ethanol, and dried with a vacuum pump to yield 1.89 g of the object compound as a pale yellow crystal.
TOPIROXOSTAT
SYNTHESIS

(First step)
The first step, 4 – is a step of obtaining a compound (3) is reacted in the presence of an alkali metal alkoxide, cyano-N-oxide and (2), and isonicotinic acid hydrazide.
4 used in this reaction – isonicotinic acid hydrazide and (2) a cyano-N-oxide is a known compound both, I can be prepared by known means.
The alkali metal alkoxide is used, 6 alkoxide alkali metal C 1-C are preferred, sodium methylate, sodium ethylate and the like can be given as specific examples. The reaction is preferably carried out in a solvent, as the solvent, alcohol solvents such as methanol, ethanol and the like are preferable.
The reaction is preferably first in a solvent, is treated with an alkali metal alkoxide compound (2) and then to react the isonicotinic acid hydrazide. First, heated to reflux under cooling, at 80 ℃ from 15 ℃ preferably, 30 minutes and 12 hours in general, the reaction temperature in the reaction with an alkali metal alkoxide (2) with the compound is reacted 1-4 hours, preferably about. Under the temperature conditions, using an excess amount or one equivalent of 30 minutes to 12 hours usually, reaction with isonicotinic acid hydrazide Subsequent to reaction for 1 to 5 hours, preferably.
Example 1:
Synthesis 4 oxide (3) – – – (4 – pyridin-carbonyl) -4 – N “pyridine hydrazide imide -1 was suspended in 40mL of methanol cyanopyridine-N-oxide and (2) 5.00g, sodium was added to methylate 22.4mg, and the mixture was stirred for 2 hours under 40 ℃ nitrogen atmosphere. was cooled to room temperature. reaction solution was stirred for 4 hours at 40 ℃ was added isonicotinic acid hydrazide 5.71g at the same temperature, precipitated The filtrated crystals were, washed with methanol 15mL, and dried 15 hours at 80 ℃, N “- to give (3) 9.60g oxide – (4 – pyridin) -4 – pyridine-hydrazide imide -1.
1 H-NMR (DMSO-d 6) δ (ppm): 6.98 (br, 2H), 7.81 (d, 2H, J = 5.77Hz), 7.85 (d, 2H, J = 7 .09 Hz), 8.29 (d, 2H, J = 7.09Hz), 8.73 (d, 2H, J = 5.77Hz), 10.37 (br, 1H)
MS m / z: 256 [M-H] –
(Second step)
The second step is a step of obtaining compound (4) by cyanation agent cyano compound (3).
As the cyanation agent used, trialkyl cyanide alkali metal cyanide, sodium cyanide, potassium cyanide and the like, zinc cyanide, trimethylsilyl cyanide and the like.
The cyanation reaction is preferably, for example, be carried out (Heterocycles, Vol.22, No.5, 1994) by Reissert Henze reaction. This reaction, for example, to give compound (4) by an organic solvent in the compound (3), and after activation with carbamoyl halide, and reacting the cyano agent. The alkylcarbamoyl halide used in the carbamoylation is a first step in Reissert Henze reaction, 6 alkylcarbamoyl halide di C 1-C dimethylcarbamoyl chloride, and di-propyl carbamoyl chloride can be used, preferably, dimethylcarbamoyl is chloride. The solvent used in this reaction, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran and acetonitrile can be used, however, N, N-dimethylformamide is preferred. Further, 15 ~ 60 ℃, more preferably 30 ~ 50 ℃ reaction temperature. The reaction time is preferably 1 to 24 hours, more preferably 1 to 3 hours. As the cyanation agent used in the cyanation reaction followed, cyano agents above can be used, sodium cyanide, potassium cyanide, zinc cyanide, and trimethylsilyl cyanide, and more preferably, it is sodium cyanide . -20 ~ 60 ℃ is preferred, more preferably -10 ~ 40 ℃, reaction temperature is 1-4 hours.
Is a novel compound (4) The compound obtained in this second step, it is useful as an intermediate for the production of compound (1). If through Compound (4) can be synthesized in good yield and easily without the need for purification in the second step is also possible, and can be produced (1) Compound industrially efficiently compound (4).
Synthetic N “hydrazide (4) – (4 – pyridine carbonyl) -4 – pyridine carboxylic acid N’-(carboxylic imidoyloxy – 2 – – cyano-4)
Example 2
4 pyridine hydrazide imide -1 – oxide ( was suspended in N, N-dimethylformamide 48mL and 3) 10.0g, under nitrogen atmosphere, followed by stirring for 1 hour was added dimethylcarbamoyl chloride 9.20g at 40 ℃. was added sodium cyanide 2.48g at the same temperature, After cooling to 5 ℃ below. reaction mixture was stirred for 1 hour, the crystals were collected by filtration. precipitate was successively added dropwise a 5% aqueous sodium bicarbonate solution 100mL, and 100mL water, and washed with water 100mL, at 80 ℃ for 15 h and dried under reduced pressure to give 4 – hydrazide (4) 9.28g of pyridine-carboxylic acid N’-(carboxylic imide yl – 2 – cyano-4).
1 H-NMR (DMSO-d 6) δ (ppm): 7.15 (br, 2H), 7.82 (d, 2H, J = 5.61Hz), 8.14 (d, 1H, J = 5 .11 Hz), 8.37 (s, 1H), 8.75 (d, 2H, J = 5.61Hz), 8.86 (d, 1H, J = 5.11Hz), 10.47 (br, 1H )
MS m / z: 265 [M-H] –

(Third step)
The third step is a step of obtaining a compound (1) by the presence of an acid catalyst, the cyclization reaction of the compound (4).
As the acid, organic phosphoric acid, p-toluenesulfonic acid, such as hydrochloric acid, inorganic acids can be used, inorganic acids, phosphoric acid is particularly preferable. As the reaction solvent, water, 2 – butanol, 2 – mixed solvent of alcohol and water or alcohol, propanol, ethanol and the like can be used, but water and 2 – I was mixed 5:1 to 10:1 butanol solvent. The reaction temperature and time, 60 ~ 100 ℃, preferably 2 to 12 hours at 70 ~ 90 ℃, I want to 8-10 hours, preferably.
Intermediates and compounds of the present invention the method (1) can be isolated and purified from the washed reaction mixture, recrystallization, by means of various conventional chromatography.
Example 3:
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile 4 Synthesis of (1) – pyridine-carboxylic acid N’- (2 – cyano-4 – carboxylic imide yl) water 82mL, 2 hydrazide (4) 9.25g – butanol was added 8.2mL, phosphate 4.00g, was stirred for 8 h at 80 ℃. After cooling to room temperature, the reaction mixture was precipitated crystals were collected by filtration, water: 2 – were washed with a mixed solution of 92.5mL butanol = 10:1. The 13 h and dried under reduced pressure at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl) – 1 H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1 I got a) 7.89g.
Topiroxostat
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
MS m / z: 247 [M-H] –
PATENT
Synthetic water-carbonitrile p-toluenesulfonate – pyridine Example 1: 4 – [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol]: 2 – butanol = was added monohydrate 6.62g p-toluenesulfonic acid in a mixed solution of 55mL of 10:1, 4 at 80 ℃ – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl] pyridine-2 – – triazol-3 was added carbonitrile 7.85g, and the mixture was stirred at the same temperature for 1 hour. After cooling to room temperature, the reaction mixture, and the precipitated crystals were collected by filtration, and water: 2 – were washed with a mixed solution of 40mL of butanol = 10:1. The dried under reduced pressure for 10 hours at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile p-toluene I got a sulfonate 12.6g.
1 H-NMR (DMSO-d 6) δ (ppm): 2.29 (s, 3H), 7.11 (m, 2H), 7.48 (dd, 2H, J = 6.48,1.62 Hz ) ,8.32-8 .35 (m, 3H), 8.57 (dd, 1H, J = 1.62,0.81 Hz) ,8.94-8 .98 (m, 3H)
– [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazole and potassium carbonate 8.22g, 4 in a mixed solution of 80mL of ethanol = 9:1: preparation water of crystal form I: Example 2 I was dissolved carbonitrile p-toluenesulfonate 10.0g – -3 – yl] pyridine-2. After stirring for 5 hours plus 15mL 6M hydrochloric acid at 20 ℃, was the precipitated crystals were collected by filtration, and washed with water 100mL. The 23 h and dried under reduced pressure at 80 ℃, 4 – to obtain carbonitrile 5.78g – pyridin-2 [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazole. Having a DSC as shown in FIG 4 and the powder X-ray diffraction pattern shown in FIG 1, the resulting crystals were type-I crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
N, N carbonitrile 40.0g – preparation of 4 Form II – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl – triazol-3]-2: Example 3 – dimethylformamide was added 300mL, and stirred for 25 min at 150 ℃. After cooling to room temperature the solution, and the precipitated crystals were collected by filtration, and washed twice with water 200mL, 4 and dried under reduced pressure overnight at 80 ℃ the crystal – [5 – (pyridin-4 – yl)-1H-1 , 2,4 – I got carbonitrile 30.4g – yl] pyridine-2 – triazole-3. Having a DSC as shown in FIG 5 and powder X-ray diffraction pattern shown in FIG 2, the resulting crystals were type II crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
The 25 ℃, about 2g carbonitrile, – preparation of the hydrate 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2: Example 4 I was stored for 14 days under conditions of relative humidity 97%. Having a DSC as shown in FIG 7 and the powder X-ray diffraction pattern shown in FIG 3, the obtained crystal was a hydrate.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
Test Example: solubility test Type I crystal by crystal form, II-type crystal, and water solubility of the hydrate was calculated by absorbance measurement method, a saturated solution concentration of each sample. I Figure 8 shows the results.Whereas the 6.2μg/mL water solubility of crystalline Form I, II type crystal 4.2μg/mL, hydrate was 1.9μg/mL.
From Figure 8, the water solubility of Form II and Form I crystals is good, water-soluble type I crystal is particularly good.
NMR
BMCL Volume 19, Issue 21, 1 November 2009, Pages 6225–6229
http://www.sciencedirect.com/science/article/pii/S0960894X09012372?np=y
view compd 39 and ignore rest
 TOPIROXOSTAT, FYX O51
TOPIROXOSTAT, FYX O51
view compd 39 and ignore rest
| 1 | * | Baldwin, J.J., J. Med. Chem.; 1975; 18(9); 895-900, especially p. 898, lines 3-5. |
| 2 | * | Geldard, J.F. et al., J. Org. Chem.; 1965; 30(1); 318-319, especially p. 319, starting line 33. |
| 3 | * | Lever, A.B.P., Inorg. Chem; 1990; 29; 1271-1285, especially p. 1275, line 18 and 19. |
Nucleosides, Nucleotides and Nucleic Acids, 2008 , vol. 27, 6-7 pg. 888 – 893
Inoue, Tsutomu; Sato, Takahiro; Ashizawa, Naoki; Iwanaga, Takashi; Matsumoto, Koji; Nagata, Osamu; Nakamura, Hiroshi
Bioorganic and Medicinal Chemistry Letters, 2009 , vol. 19, 21 pg. 6225 – 6229
WO 2012060308
WO 2007148835
WO 2005009991
| WO2003064410A1 * | Dec 3, 2002 | Aug 7, 2003 | Naoki Ashizawa | Novel 1,2,4-triazole compound |
| US3882134 * | May 21, 1973 | May 6, 1975 | Merck & Co Inc | 1-Substituted-3,5-dipyridyl-1,2,4-triazoles |
| US3947577 * | Jan 8, 1975 | Mar 30, 1976 | Merck & Co., Inc. | Anti-hyperuricemia composition |
| US3984558 * | Nov 29, 1974 | Oct 5, 1976 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds used as bronchodilators |
| US4011218 * | Dec 3, 1974 | Mar 8, 1977 | Merck & Co., Inc. | 1,2,4-triazoles |
| US4104393 * | Sep 2, 1977 | Aug 1, 1978 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds |
| US5571897 * | Dec 5, 1991 | Nov 5, 1996 | Wallac Oy | Luminescent lanthanide chelates |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-9199970-B2 |
2015-12-01 |
|
|||
|
2.
US-20150322006-A1 |
2015-11-12 |
|
|||
|
3.
US-20150309021-A1 |
2015-10-29 |
|
|||
|
4.
US-20150291543-A1 |
2015-10-15 |
|
|||
|
5.
EP-2927219-A1 |
2015-10-07 |
EN
|
|
||
|
6.
US-20150274680-A1 |
2015-10-01 |
|
|||
|
7.
EP-2913053-A1 |
2015-09-02 |
EN
|
|
||
|
8.
EP-2511844-B1 |
2015-08-12 |
EN
|
|
||
|
9.
EP-2712861-B1 |
2015-07-29 |
EN
|
|
||
|
10.
US-20150203490-A1 |
2015-07-23 |
|
|||
|
11.
US-20150191463-A1 |
2015-07-09 |
|
|||
|
12.
US-20150166510-A1 |
2015-06-18 |
|
|||
|
13.
EP-2878594-A1 |
2015-06-03 |
EN
|
|
||
|
14.
EP-2878598-A1 |
2015-06-03 |
E
|
|
||
|
15.
EP-2878595-A1 |
2015-06-03 |
EN
|
|
||
|
16.
US-20150126558-A1 |
2015-05-07 |
|
|||
|
17.
US-8987473-B2 |
2015-03-24 |
|
|||
|
18.
EP-2842948-A1 |
2015-03-04 |
EN
|
|
||
|
19.
EP-2776028-A1 |
2014-09-17 |
EN
|
|
||
|
20.
US-20140256748-A1 |
2014-09-11 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2-pyridinecarbonitrile
|
|
| Clinical data | |
| Trade names | Topiloric, Uriadec |
| Legal status |
|
| Identifiers | |
| CAS Number | 577778-58-6 |
| ATC code | None |
| PubChem | CID: 5288320 |
| ChemSpider | 4450517 |
| Chemical data | |
| Formula | C13H8N6 |
| Molecular mass | 248.24 g/mol |
/////////////
C1=CN=CC=C1C2=NC(=NN2)C3=CC(=NC=C3)C#N
