
Home » 2013 (Page 57)
Yearly Archives: 2013
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cyclospora outbreak spreads to Texas
http://www.foxnews.com/health/2013/07/19/cyclospora-outbreak-spreads-to-texas/
Forty-eight people have been diagnosed with cyclospora infections in Texas within the past week, Medical Daily reports.
Cyclosporiasis is an intestinal illness caused by the parasite Cyclospora cayetanensis. It is believed to be contracted by consuming food or drink that has been contaminated with feces. Symptoms include diarrhea, loss of appetite and stomach cramps – and signs of illness usually appear two to 11 days after people contract the parasite.
Over 100 people have also been infected with cyclospora in Iowa and Nebraska, according to reports earlier this week. It is unknown whether the outbreaks are connected.
The Texas Department of State Health Services is currently investigating 50 new cases of cyclospora in the Dallas/Fort Worth area.
It is highly unusual for such a large quantity of newly reported cases to appear within the span of one week, and investigators are looking into a possible source of food…
View original post 41 more words
Breast Cancer Drugs in Late-Stage Development/Recently Approved
The article is 2012-2013 based and reader discretion is sought to ascertian the stage of approval

Afinitor® (everolimus)
https://newdrugapprovals.wordpress.com/2013/04/27/drug-spotlight-afinitor-everolimus-novartis/
Sponsor: Novartis
Method of Action: Mammalian target of rapamycin (mTOR) inhibitor
Indications/Phase of Trial: Hepatocellular carcinoma; human epidermal growth factor receptor 2-positive (HER2+) breast cancer first-line and second-line; lymphoma; nonfunctional carcinoid tumor (Phase III; all new indications)
Approved in July in U.S., EU for advanced hormone-receptor-positive (HR+) and human epidermal growth factor Receptor 2-negative (HER2-) metastatic breast cancer with exemestane in postmenopausal women who have already received certain other medicines for their cancer
Approved earlier for adults with pancreatic neuroendocrine tumors (PNET) that cannot be treated with surgery; adults with advanced renal cell carcinoma (RCC) when certain other medicines have not worked; adults with angiomyolipoma, seen with tuberous sclerosis complex (TSC), when surgery is not required immediately; and adults and children with TSC who have a brain tumor called subependymal giant cell astrocytoma (SEGA) that cannot be removed completely by surgery
Avastin (Bevacizumab; RG435)
Sponsor: Roche/Genentech
Method of Action: Monoclonal antibody; Vascular endothelial growth factor (VEGF) inhibitor
Indications/Phase of Trial: U.S.: Relapsed ovarian cancer, platinum-sensitive (Registration); first-line metastatic breast cancer and first-line metastatic ovarian cancer (both Phase III).
EU: Relapsed platinum-resistance ovarian cancer (Phase III)
Metastatic colorectal cancer, treatment beyond progression (Registration); adjuvant breast cancer, HER2- and HER2+; adjuvant NSCLC; first-line glioblastoma (GBM) multiforme; high-risk carcinoid (all Phase III)
Approved for metastatic colorectal cancer (mCRC) when started with the first or second intravenous 5-FU–based chemotherapy for metastatic cancer; advanced nonsquamous non-small-cell lung cancer (NSCLC) with carboplatin and paclitaxel in people who have not received chemotherapy for their advanced disease; metastatic RCC (mRCC) with interferon alfa; and GBM in adult patients whose cancer has progressed after prior treatment. Effectiveness based on tumor response, as no data have shown whether Avastin improves disease-related symptoms or survival in people previously treated for GBM
Approval conditionally granted in 2008 and withdrawn November 2011 for HER2- metastatic breast cancer (mBC) with Paclitaxel

Buparlisib (BKM120)
Sponsor: Novartis
Method of Action: Pan-PI3K inhibitor
Indications/Phase of Trial: mBC (Phase III and confirmatory Phase I/II); with Fulvestrant, in postmenopausal women with hormone receptor-positive HER2- locally advanced or mBC which progressed on or after aromatase inhibitor (AI) treatment (Phase III; BELLE-2 study recruiting as of November 2012); with Fulvestrant, in postmenopausal women with hormone receptor-positive HER2- AI-treated, locally-advanced or mBC who progressed on or after mTOR inhibitor-based treatment (Phase III; BELLE-3 study, recruiting as of October 2012); with Paclitaxel in patients with HER2- inoperable locally advanced or mBC, with or without PI3K pathway activation (Phase III; BELLE-4 study, recruiting as of November); metastatic castration-resistant prostate cancer (CRPC; Phase II; recruiting as of October); recurrent glioblastoma (Phase II; recruiting as of November); recurrent/metastatic head and neck squamous cell carcinoma (Phase II; recruiting as of October); endometrial cancer (Phase I/II); NSCLC (Phase I/II); prostate cancer (Phase I/II); GBM multiforme (Phase I/II); with Fulvestrant in postmenopausal women with estrogen receptor-positive metastatic breast cancer (Phase I); previously treated advanced colorectal cancer (Phase I)

Faslodex (Fulvestrant Injection)
Sponsor: AstraZeneca
Method of Action: Estrogen receptor antagonist
Indications/Phase of Trial: First line HR+ mBC (Phase III; FALCON study commenced Oct. 29)
Approved for HR+ mBC in women who have experienced menopause and whose breast cancer has worsened after they were treated with antiestrogen medications

Herceptin (Trastuzumab; RG597)
Sponsor: Roche, in partnership with Halozyme
Method of Action: Humanized monoclonal antibody designed to target and block the function of HER2+
Indications/Phase of Trial: EU: Early HER2+ breast cancer, subcutaneous formulation (Registration)
Approved for early-stage HER2+ breast cancer that has spread into the lymph nodes, and HER2+ breast cancer that has not spread into the lymph nodes and is estrogen receptor/progesterone receptor-negative (ER-/PR-) or have one high-risk feature. High-risk is defined as estrogen receptor/progesterone receptor-positive (ER+/PR+) with one of the following features: tumor size >2 cm, age <35 years, or tumor grade 2 or 3. Can be used with Adriamycin® (doxorubicin), Cytoxan® (cyclophosphamide), and either Taxol® (paclitaxel) or Taxotere® (docetaxel); or with Taxotere and Paraplatin® (carboplatin); or alone after treatment with multiple other therapies, including an anthracycline (Adriamycin)-based chemotherapy
Also approved alone for the treatment of HER2+ breast cancer in patients who have received one or more chemotherapy courses for metastatic disease; and with paclitaxel for first-line treatment of HER2+ mBC

Iniparib (Tivolza; BSI-201; SAR240550)
Sponsor: Sanofi, through acquisition of original developer BiPar Sciences
Method of Action: Poly (ADP-ribose) polymerase 1 (PARP1) inhibitor
Indications/Phase of Trial: Stage IV squamous NSCLC (Phase III; NME); solid tumors such as sarcoma and breast, uterine, lung, and ovarian cancers (Phase I/II)
Phase III trial in breast cancer failed January 2011 by failing to improve survival and progression-free survival (PFS) in breast cancer patients

Nexavar® (Sorafenib)
https://newdrugapprovals.wordpress.com/2013/07/16/nexavar-sorafenib/
Sponsor: Onyx Pharmaceuticals
Method of Action: Dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases
Indications/Phase of Trial: Liver cancer adjuvant (Phase III; STORM study); kidney cancer adjuvant (Phase III; SORCE/ASSURE study); thyroid cancer monotherapy (Phase III; DECISION study); breast cancer with capecitabine (Phase III; RESILIENCE study)
Approved for hepatocellular carcinoma (HCC) and RCC

Perjeta (Pertuzumab; RG1273)
Sponsor: Roche/Genentech
Method of Action: HER2/neu receptor antagonist
Indications/Phase of Trial: EU: With Herceptin and docetaxel chemotherapy for previously-untreated HER2+ mBC or locally recurrent, inoperable breast cancer in patients who have not received previous treatment or whose disease has returned after treatment in the early-stage setting (Registration)
U.S.: Approved June 2012 for HER2+ mBC with Herceptin (trastuzumab) and docetaxel, in patients who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease
Switzerland: Approved August 2012 for HER2+ breast cancer with Herceptin (trastuzumab) and docetaxel in patients with advanced or locally recurring breast cancer that has not previously been treated with chemotherapy

Ridaforolimus (MK-8669; AP23573; formerly Deforolimus)
Sponsor: Merck, under exclusive worldwide license agreement with Ariad Pharmaceuticals
Method of Action: Oral inhibitor of mammalian target of rapamycin inhibitor (mTOR)
Indications/Phase of Trial: Maintenance therapy for metastatic soft-tissue sarcoma and bone sarcomas after at least four chemotherapy cycles (under review after receiving Complete Response letter from FDA in June; NME); breast cancer with exemestane, compared to breast cancer with dalotuzumab and exemestane (Phase II; recruiting as of November); advanced head and neck cancer, NSCLC and colon cancer, with cetuximab (Phase II); pediatric patients with advanced solid tumors (Phase I; recruiting as of September); with dalotuzumab in pediatric patients with advanced solid tumors (Phase I; recruiting as of August); advanced RCC, with vorinostat (Phase I; recruiting as of October 2012); breast cancer, with dalotuzumab (Phase I: recruiting as of September); endometrial and ovarian cancers, with paclitaxel and carboplatin (Phase I; recruiting as of September 2012); advanced cancer, with MK-2206 and MK-0752 (Phase I: recruiting as of September 2012); advanced cancer, with dalotuzumab, MK-2206 and MK-0752 (Phase I: recruiting as of August 2012)
Tivozanib (ASP4130; AV-951)
Sponsor: Aveo Oncology and Astellas
Method of Action: Tyrosine kinase inhibitor; inhibits VEGF receptor 1, 2, and 3
Indications/Phase of Trial: U.S.: Advanced RCC (Registration; NDA filed September 2012); tivozanib biomarkers in solid tumors (Phase II; BATON study); stage IV metastatic colorectal cancer (mCRC), with mFOLFOX6, and compared with bevacizumab and mFOLFOX6 (Phase II; recruiting as of November); additional data as first-line therapy for advanced RCC, followed by sunitinib (Phase II; TAURUS study, enrollment initiated in October 2012); advanced solid tumors, with capecitabine (Xeloda®; Phase I; recruiting as of October)
EU: Advanced RCC (Phase III)
Trastuzumab-DM1 (T-DM1; Trastuzumab emtansine; RG3502)
Sponsor: Roche, with linker technology developed by ImmunoGen
Method of Action: Antibody-drug conjugate, consisting of the antibody trastuzumab and the chemotherapy DM1 attached via a stable linker
Indications/Phase of Trial: U.S.: HER2+, unresectable locally-advanced or mBC who have received prior treatment with Herceptin (trastuzumab) and a taxane chemotherapy (Registration; Priority review approved Nov. 7; action date Feb. 26, 2013)
EU: Marketing Authorization Application for HER2+ mBC accepted for review by European Medicines Agency

Tyverb/Tykerb (lapatinib)
Sponsor: GlaxoSmithKline
Method of Action: Human epidermal growth factor receptor-2 (Her2) and epidermal growth factor receptor (EGFR) dual kinase inhibitor
Indications/Phase of Trial: mBC with trastuzumab (Registration); breast cancer, adjuvant therapy (Phase III); Gastric cancer (Phase III); head & neck squamous cell carcinoma, resectable disease (Phase III)
Xgeva (denosumab)
Sponsor: Amgen, with commercialization by GlaxoSmithKline in countries where Amgen has no presence
Method of Action: Fully human monoclonal antibody that specifically targets a ligand known as RANKL that binds to a receptor known as RANK
Indications/Phase of Trial: Delay or prevention of bone metastases in breast cancer (Phase III); delay or prevention of bone metastases in prostate cancer (Phase III)
Approved for prevention of fractures in men with advanced prostate cancer
Rejected in April for supplemental Biologics License Application to treat men with CRPC at high risk of developing bone metastases

Yondelis® (trabectedin)
Sponsor: Johnson & Johnson; developed in collaboration with PharmaMar
Method of Action: Binds to minor groove of DNA, interfering with the cell division and gene transcription processes, as well as DNA’s repair machinery
Indications/Phase of Trial: U.S.: Locally advanced or metastatic soft tissue sarcoma excluding leiomyosarcoma and liposarcoma who have relapsed or are refractory to standard-of-care treatment (Phase III; recruiting as of November); soft tissue sarcoma, excluding liposarcoma and leiomyosarcoma (L-type sarcoma), in previously-treated patients who cannot be expected to benefit from currently available therapeutic options (Phase III; recruiting as of November); locally advanced or metastatic L-sarcoma (liposarcoma or leiomyosarcoma) who were previously treated with at least an anthracycline and ifosfamide-containing regimen, or an anthracycline-containing regimen and one additional cytotoxic chemotherapy regimen, compared with dacarbazine group (Phase III; recruiting as of November); breast cancer and pediatric tumors (Phase II); Advanced malignancies and liver dysfunction (Phase I; recruiting as of November)
EU: Approved for advanced or metastatic soft tissue sarcoma, and for relapsed platinum-sensitive ovarian cancer, with DOXIL®/Caelyx®

Xtandi® Capsules (Enzalutamide; formerly MDV3100)
Sponsor: Medivation in collaboration with Astellas
Method of Action: Androgen receptor inhibitor
Indications/Phase of Trial: Prechemotherapy CRPC in patients who have failed luteinizing hormone-releasing hormone (LHRH) analog treatment only, as well as patients who have failed both LHRH analog and anti-androgen treatment. (Phase III; PREVAIL study); prostate cancer neoadjuvant therapy (Phase II); prechemo metastatic prostate cancer in Europe (Phase II; TERRAIN); prechemo metastatic and nonmetastatic prostate cancer patients in U.S. (Phase II; STRIVE); prostate cancer Hormone-naïve (Phase II; ASPIRE); prostate cancer with docetaxel (Phase I); breast cancer (Phase I)
EU: Marketing Authorization Application submitted June 2012 to European Medicines Agency, for patients with metastatic CRPC who have received docetaxel-based chemotherapy
Japan: Metastatic CRPC who have received docetaxel-based chemotherapy (Phase II)
Approved Aug. 31 for patients with metastatic CRPC who have previously received docetaxel. As a post-marketing requirement, Medivation and Astellas agreed to conduct an open-label safety study of Xtandi (160 mg/day) in patients at high risk for seizure, with data to be submitted to FDA in 2019
FDA gives tentative approval to Perrigo s ANDA for generic version of Prandin Tablets

repaglinide

Perrigo Company (Nasdaq: PRGO; TASE) today announced that it has received tentative approval from the U.S. Food & Drug Administration (FDA) for its abbreviated new drug application (ANDA) for repaglinide tablets, the generic equivalent to Prandin® Tablets (repaglinide tablets).
Prandin® tablets (repaglinide tablets), are indicated as an adjunct to diet and exercise to improve glycemic control in adults with type-2 diabetes mellitus and have annual sales of approximately $250 million, as measured by Symphony Health Solutions.
Repaglinide is an antidiabetic drug in the class of medications known as meglitinides, and was invented in 1983. It is sold byNovo Nordisk under the name of Prandin in the U.S., GlucoNorm in Canada, Surepost in Japan, Repaglinide in Egypt by EIPICO, and NovoNorm elsewhere. In Japan it is produced by Dainippon Sumitomo Pharma.
Repaglinide lowers blood glucose by stimulating the release of insulin from the pancreas. It achieves this by closing ATP-dependent potassium channels in the membrane of the beta cells. This depolarizes the beta cells, opening the cells’ calcium channels, and the resulting calcium influx induces insulin secretion.

SIMPONI® ARIA™ (golimumab),For Infusion Receives FDA Approval For Treatment Of Moderately To Severely Active Rheumatoid Arthritis

July 18, 2013 /PRNewswire/ — Janssen Biotech, Inc. announced today the U.S. Food and Drug Administration (FDA) approval of SIMPONI® ARIATM (golimumab) for infusion for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) in combination with methotrexate. SIMPONI ARIA, the only fully-human anti-tumor necrosis factor (TNF)-alpha infusible therapy, has been shown to significantly improve signs and symptoms and physical function, and inhibit the progression of structural damage. The SIMPONI ARIA dose regimen is 2 mg/kg given as an intravenous infusion at weeks 0 and 4, then every 8 weeks thereafter. The infusion is given over a 30-minute period, providing a short infusion time for patients. Approximately 1.3 million people in the United States are living with RA,[i] a chronic, systemic inflammatory condition that is often characterized by symptoms that include pain, stiffness and inflammation, and in some cases, joint destruction and disability.[ii]
read all at

Golimumab (CNTO 148)is a human monoclonal antibody which is used as an immunosuppressive drug and marketed under the brand name Simponi. Golimumab targets tumor necrosis factor alpha (TNF-alpha), a pro-inflammatory moleculeand hence is a TNF inhibitor.
Golimumab was developed by Centocor and is approved in Canadaand the United Statesas a once monthly subcutaneous treatment for adults with moderately to severely active rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis.Golimumab is pending FDA approval for Ulcerative Colitis.
In the same year, Johnson & Johnson, the parent company of Centocor, also received an approval from European Medicines Agency (EMEA) for the use of golimumab as a treatment for rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis.Golimumab is marketed by Merck & Co, Inc. in Europe, pending final arbitration between J&J and Merck


Recent Progress in the Synthesis of Tamiflu

| ABOVE PICTURE-The synthetic route to tamiflu reported by M. Shibasaki starting from 1,4-cyclohexadiene. See JACS 2006, 128, 6312-6313 |
|
|
||||||
 |
|||||||
| Abstract Tamiflu, one of the most common orally drugs for the treatment and prevention of influenza, has attracted extensive interests of synthetic chemists all over the world.Concise, efficient, and scalable synthetic approaches toward this molecule have been a very active field in recent years, and many diverse synthetic routes have been developed to date.In this review, representative synthetic routes employing chiral starting material or catalytic asymmetric reactions are briefly summarized. |
|
||||
| Fund:Project supported by the National Natural Science Foundation of China (Nos.20972059, 21290180), the Program for Changjiang Scholars and Innovative Research Team in University (No.IRT1138) and the Fundamental Research Funds for the Central Universities (No.lzujbky-2013-ct02). | |||||
| Cite this article: |
| Zhang Tiancai,Lu Hui,Zhang Fu-Min et al. Recent Progress in the Synthesis of Tamiflu[J]. Chin. J. Org. Chem., 2013, 33(06): 1235-1243. |
| http://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract342132.shtml# |
| URL: |
| http://sioc-journal.cn/Jwk_yjhx/EN/10.6023/cjoc201303044 OR http://sioc-journal.cn/Jwk_yjhx/EN/Y2013/V33/I06/1235 |

Oseltamivir total synthesis concerns the total synthesis of the antiinfluenza drug oseltamivirmarketed by Hoffmann-La Roche under the trade name Tamiflu. Its commercial production starts from the biomolecule shikimic acid harvested from Chinese star anise with a limited worldwide supply. Due to its limited supply, searches for alternative synthetic routes preferably not requiring shikimic acid are underway and to date several such routes have been published. Control of stereochemistry is important: the molecule has three stereocenters and the sought-after isomer is only 1 of 8 stereoisomers.
Commercial production
The current production method is based on the first scalable synthesis developed by Gilead Sciences [1] starting from naturally occurring quinic acid or shikimic acid. Due to lower yields and the extra steps required (because of the additional dehydration), the quinic acid route was dropped in favour of the one based on shikimic acid, which received further improvements by Hoffmann-La Roche.[2][3] The current industrial synthesis is summarised below:

Karpf / Trussardi synthesis
The current production method includes two reaction steps with potentially hazardous azides. A reported azide-free Roche synthesis of tamiflu is summarised graphically below:[4]

The synthesis commences from naturally available (−)-shikimic acid. The 3,4-pentylidene acetal mesylate is prepared in three steps: esterification with ethanol and thionyl chloride; ketalization with p-toluenesulfonic acid and 3-pentanone; and mesylation with triethylamine and methanesulfonyl chloride. Reductive opening of the ketal under modified Hunter conditions[5] in dichloromethane yields an inseparable mixture of isomeric mesylates. The corresponding epoxide is formed under basic conditions withpotassium bicarbonate. Using the inexpensive Lewis acid magnesium bromide diethyl etherate (commonly prepared fresh by the addition of magnesium turnings to 1,2-dibromoethane in benzene:diethyl ether), the epoxide is opened with allyl amine to yield the corresponding 1,2-amino alcohol. The water-immiscible solvents methyl tert-butyl ether and acetonitrile are used to simplify the workup procedure, which involved stirring with 1 M aqueous ammonium sulfate. Reduction on palladium, promoted byethanolamine, followed by acidic workup yielded the deprotected 1,2-aminoalcohol. The aminoalcohol was converted directly to the corresponding allyl-diamine in an interesting cascade sequence that commences with the unselective imination of benzaldehyde with azeotropic water removal in methyl tert-butyl ether. Mesylation, followed by removal of the solid byproduct triethylamine hydrochloride, results in an intermediate that was poised to undergo aziridination upon transimination with another equivalent of allylamine. With the librated methanesulfonic acid, the aziridine opens cleanly to yield a diamine that immediately undergoes a second transimination. Acidic hydrolysis then removed the imine. Selective acylation with acetic anhydride (under buffered conditions, the 5-amino group is protonated owing to a considerable difference in pKa, 4.2 vs 7.9, preventing acetylation) yields the desired N-acetylated product in crystalline form upon extractive workup. Finally, deallylation as above, yielded the freebase of oseltamivir, which was converted to the desired oseltamivir phosphate by treatment with phosphoric acid. The final product is obtained in high purity (99.7%) and an overall yield of 17-22% from (−)-shikimic acid. It is noted that the synthesis avoids the use of potentially explosive azide reagents and intermediates; however, the synthesis actually used by Roche uses azides. Roche has other routes to oseltamivir that do not involve the use of (−)-shikimic acid as a chiral pool starting material, such as a Diels-Alder route involving furan and ethyl acrylate or an isophthalic acid route, which involves catalytic hydrogenation and enzymatic desymmetrization.
Corey synthesis
In 2006 the group of E.J. Corey published a novel route bypassing shikimic acid starting from butadiene and acrylic acid.[6] The inventors chose not to patent this procedure which is described below.

Butadiene 1 reacts in an asymmetric Diels-Alder reaction with the esterfication product of acrylic acid and 2,2,2-Trifluoroethanol 2 catalysed by the CBS catalyst. The ester 3 is converted into an amide in 4 by reaction with ammonia and the next step to lactam 5 is an iodolactamization with iodine initiated by trimethylsilyltriflate. The amide group is fitted with a BOC protective group by reaction with Boc anhydride in 6 and the iodine substituent is removed in an elimination reaction with DBU to the alkene 7. Bromine is introduced in 8 by an allylic bromination with NBS and the amide group is cleaved with ethanol and caesium carbonate accompanied by elimination of bromide to the diene ethyl ester 9. The newly formed double bond is functionalized with N-bromoacetamide 10 catalyzed with Tin(IV) bromide with complete control of stereochemistry. In the next step the bromine atom in 11 is displaced by the nitrogen atom in the amide group with the strong base KHMDS to the aziridine 12 which in turn is opened by reaction with 3-pentanol 13 to the ether 14. In the final step the BOC group is removed with phosphoric acid and the oseltamivir phosphate 15 is formed.
Shibasaki synthesis
Also in 2006 the group of Masakatsu Shibasaki of the University of Tokyo published a synthesis again bypassing shikimic acid.[7][8]
 |
 |
|
| Shibasaki Tamiflu synthesis Part I | Part II |
An improved method published in 2007 starts with the enantioselective desymmetrization of aziridine 1 with trimethylsilyl azide (TMSN3) and a chiral catalyst to the azide 2. Theamide group is protected as a BOC group with Boc anhydride and DMAP in 3 and iodolactamization with iodine and potassium carbonate first gives the unstable intermediate 4and then stable cyclic carbamate 5 after elimination of hydrogen iodide with DBU.
The amide group is reprotected as BOC 6 and the azide group converted to the amide 7 by reductive acylation with thioacetic acid and 2,6-lutidine. Caesium carbonateaccomplishes the hydrolysis of the carbamate group to the alcohol 8 which is subsequently oxidized to ketone 9 with Dess-Martin periodinane. Cyanophosphorylation withdiethyl phosphorocyanidate (DEPC) modifies the ketone group to the cyanophosphate 10 paving the way for an intramolecular allylic rearrangement to unstable β-allylphosphate 11 (toluene, sealed tube) which is hydrolyzed to alcohol 12 with ammonium chloride. This hydroxyl group has the wrong stereochemistry and is therefore inverted in a Mitsunobu reaction with p-nitrobenzoic acid followed by hydrolysis of the p-nitrobenzoate to 13.
A second Mitsunobu reaction then forms the aziridine 14 available for ring-opening reaction with 3-pentanol catalyzed by boron trifluoride to ether 15. In the final step the BOC group is removed (HCl) and phosphoric acid added to objective 16.
Fukuyama synthesis
An approach published in 2007 [9] like Corey’s starts by an asymmetric Diels-Alder reaction this time with starting materials pyridine and acrolein.
 |
 |
|
| Fukuyama Tamiflu synthesis Part I | Part II |
Pyridine (1) is reduced with sodium borohydride in presence of benzyl chloroformate to the Cbz protected dihydropyridine 2. The asymmetric Diels-Alder reaction with acrolein3 is carried out with the McMillan catalyst to the aldehyde 4 as the endo isomer which is oxidized to the carboxylic acid 5 with sodium chlorite, Monopotassium phosphate and 2-methyl-2-butene. Addition of bromine gives halolactonization product 6 and after replacement of the Cbz protective group by a BOC protective group in 7 (hydrogenolysis in the presence of Di-tert-butyl dicarbonate) a carbonyl group is introduced in intermediate 8 by catalytic ruthenium(IV) oxide and sacrificial catalyst sodium periodate. Addition ofammonia cleaves the ester group to form amide 9 the alcohol group of which is mesylated to compound 10. In the next step iodobenzene diacetate is added, converting the amide in a Hofmann rearrangement to the allyl carbamate 12 after capturing the intermediate isocyanate with allyl alcohol 11. On addition of sodium ethoxide in ethanol three reactions take place simultaneously: cleavage of the amide to form new an ethyl ester group, displacement of the mesyl group by newly formed BOC protected amine to anaziridine group and an elimination reaction forming the alkene group in 13 with liberation of HBr. In the final two steps the aziridine ring is opened by 3-pentanol 14 and boron trifluoride to aminoether 15 with the BOC group replaced by an acyl group and on removal of the other amine protecting group (Pd/C, Ph3P, and 1,3-dimethylbarbituric acid in ethanol) and addition of phosphoric acid oseltamivir 16 is obtained.
Trost synthesis
In 2008 the group of Barry M. Trost of Stanford University published the shortest synthetic route to date.[10]

- Rohloff John C., Kent Kenneth M., Postich Michael J., Becker Mark W., Chapman Harlan H., Kelly Daphne E., Lew Willard, Louie Michael S., McGee Lawrence R. et al. (1998). “Practical Total Synthesis of the Anti-Influenza Drug GS-4104”. J. Org. Chem. 63 (13): 4545–4550. doi:10.1021/jo980330q.
- Federspiel M., Fischer R., Hennig M., Mair H.-J., Oberhauser T., Rimmler G., Albiez T., Bruhin J., Estermann H. et al. (1999). “Industrial Synthesis of the Key Precursor in the Synthesis of the Anti-Influenza Drug Oseltamivir Phosphate (Ro 64-0796/002, GS-4104-02) Ethyl (3R,4S,5S)-4,5-epoxy-3-(1-ethyl-propoxy)-cyclohex-1-ene-1-carboxylate”. Org. Process Res. Dev. 3: 266–274. doi:10.1021/op9900176.
- Abrecht S., Federspiel M. C., Estermann H., Fischer R., Karpf M., Mair H.-J., Oberhauser T., Rimmler G., Trussardi R. et al.. “The Synthetic-Technical Development of Oseltamivir Phosphate Tamiflu™: A Race against Time Chimia”. 2007; 61: 93–99. doi:10.2533/chimia.2007.93.
- New, Azide-Free Transformation of Epoxides into 1,2-Diamino Compounds: Synthesis of the Anti-Influenza Neuraminidase Inhibitor Oseltamivir Phosphate (Tamiflu) Martin Karpf and René Trussardi J. Org. Chem.; 2001; 66(6) pp 2044 – 2051; (Article) doi:10.1021/jo005702l PMID 11300898.
- Birgit Bartels and Roger Hunter (1993). “A selectivity study of activated ketal reduction with borane dimethyl sulfide”. J. Org. Chem. 58 (24): 6756. doi:10.1021/jo00076a041.
- A Short Enantioselective Pathway for the Synthesis of the Anti-Influenza Neuramidase Inhibitor Oseltamivir from 1,3-Butadiene and Acrylic Acid Ying-Yeung Yeung, Sungwoo Hong, and E. J. Corey J. Am. Chem. Soc.; 2006; 128(19) pp 6310 – 6311; (Communication) doi:10.1021/ja0616433
- De Novo Synthesis of Tamiflu via a Catalytic Asymmetric Ring-Opening of meso-Aziridines with TMSN3 Yuhei Fukuta, Tsuyoshi Mita, Nobuhisa Fukuda, Motomu Kanai, and Masakatsu Shibasaki J. Am. Chem. Soc.; 2006; 128(19) pp 6312 – 6313; doi:10.1021/ja061696k
- Second Generation Catalytic Asymmetric Synthesis of Tamiflu: Allylic Substitution Route Tsuyoshi Mita, Nobuhisa Fukuda, Francesc X. Roca, Motomu Kanai, and Masakatsu Shibasaki Org. Lett.; 2007; 9(2) pp 259 – 262; (Letter) doi:10.1021/ol062663c
- A Practical Synthesis of (-)-Oseltamivir Nobuhiro Satoh, Takahiro Akiba, Satoshi Yokoshima, Tohru Fukuyama Angew. Chem. Int. Ed. 2007, 46, 5734 –5736doi:10.1002/anie.200701754
- A Concise Synthesis of (−)-Oseltamivir Barry M.Trost, Ting Zhang Angew. Chem. Int. Ed. 2008, 47, 1-4 doi:10.1002/anie.200800282
UK launch for Astellas’ prostate cancer drug
http://www.pharmatimes.com/Article/13-07-19/UK_launch_for_Astellas_prostate_cancer_drug.aspx
UK patients with advanced prostate cancer have been given access to a new treatment that could prolong survival following the launch of Astella’s Xtandi in the country.
Xtandi (enzalutamide) was licensed in Europe this month for the treatment of men with advanced prostate cancer whose disease has become resistant to first-line hormonal treatments and has progressed following docetaxel chemotherapy.
Enzalutamide is an androgen receptor inhibitor. The chemical name is 4-{3-[4-cyano-3-(trifluoromethyl)phenyl]-5,5dimethyl-4-oxo-2-sulfanylideneimidazolidin-1-yl}-2-fluoro-N-methylbenzamide.
The molecular weight is 464.44 and molecular formula is C21H16F4N4O2S. The structural formula is:
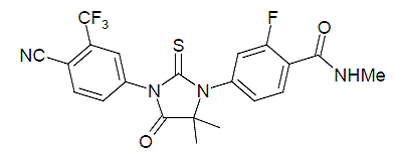 |
Enzalutamide is a white crystalline non-hygroscopic solid. It is practically insoluble in water.
XTANDI is provided as liquid-filled soft gelatin capsules for oral administration. Each capsule contains 40 mg of enzalutamide as a solution in caprylocaproyl polyoxylglycerides. The inactive ingredients are caprylocaproyl polyoxylglycerides, butylated hydroxyanisole, butylated hydroxytoluene, gelatin, sorbitol sorbitan solution, glycerin, purified water, titanium dioxide, and black iron oxide.
Menarini launches premature ejaculation drug in Singapore

DAPOXETINE
Menarini has launched dapoxetine for premature ejaculation in Singapore, having recently published a survey highlighting the rising problem of sexual dissatisfaction in the Asia-Pacific region.
The Italian drugmaker acquired Priligy (dapoxetine)from Johnson & Johnson last year and the drug is now approved in over 50 countries. It estimates that PE affects 34% of men in Singapore at some point in their lives.
READ ALL AT
Dapoxetine, marketed as Priligy (among and other brands) is the first compound developed specially for the treatment of premature ejaculation (PE) in men 18–64 years old.Dapoxetine works by inhibiting the serotonin transporter, increasing serotonin’s action at the post synaptic cleft, and as a consequence promoting ejaculatory delay. As a member of selective serotonin reuptake inhibitor (SSRI) family, dapoxetine was initially created as an antidepressant. However, unlike other SSRIs, dapoxetine is absorbed and eliminated rapidly in the body. Its fast acting property makes it suitable for the treatment of PE but not as an antidepressant.[3]
Originally created by Eli Lilly pharmaceutical company, dapoxetine was sold to Johnson & Johnson in 2003 and submitted as a new drug application to the Food and Drug Administration (FDA) for the treatment of PE in 2004. Dapoxetine has been sold in several European and Asian countries, and lately in Mexico. In the US, dapoxetine is in phase III development and expected to be marketed soon. In 2012, Menarini acquired the rights to commercialise Priligy in Europe, most of Asia, Africa, Latin America and the Middle East.

Premature ejaculation
Randomized, double blind, placebo-controlled trials have confirmed the efficacy of dapoxetine for the treatment of PE. Different dosage has different impacts on different type of PE. Dapoxetine 60 mg significantly improves the mean intravaginal ejaculation latency time (IELT) compare to that of dapoxetine 30 mg in men with lifelong PE, but there is no different in men with acquired PE. Dapoxetine, given 1–3 hours before sexual episode, prolongs IELT, increases the sense of control and sexual satisfaction in men of 18 to 64 years of age with PE. Since PE is associated with personal distress, interrelationship difficulty, dapoxetine provides help for men with PE to overcome this condition.Because lack of specific approval treatment for PE in the US and some other countries, other SSRIs such as fluoxetine, paroxetine, sertraline, fluvoxamine, and citalopram have been used as off label drugs to treat PE. Waldinger’s meta analysis shows that the use of these conventional antidepressants increasing IELT from two to ninefold above base line in comparison of three to eightfold when dapoxetine is used. However, these SSRIs must be taken daily in order to achieve meaningful efficacy, and the long half-life increases the risk of the drug accumulation and as a consequence increased of adverse effects such as decreasing sexual libido and causing erectile dysfunction. Dapoxetine, on the other hand, is a fast-acting SSRI. It is rapidly absorbed and eliminated from the body within a few hours. This favorable pharmacokinetics minimizes the risk of the drug’s accumulation in the body, and therefore reducing side effects.
Sequella Acquires Exclusive Worldwide Rights To Pfizer’s Sutezolid, Currently In Clinical Development For Tuberculosis
sutezolid
Sequella, a clinical-stage pharmaceutical company commercializing novel antibiotics to treat life-threatening infectious diseases, today announced that it has licensed Pfizer Inc’s exclusive worldwide rights to develop and commercialize sutezolid, a Phase 2 oxazolidinone antibiotic currently in development for the treatment of tuberculosis (TB).
Sutezolid demonstrated potent antibacterial activity against Mycobacterium tuberculosis in the laboratory and in animal models of TB, an aerosol-transmitted infection with a prevalence of over 2 billion people worldwide. It also demonstrated encouraging activity in a Phase 2a Early Bactericidal Activity (EBA) study in TB patients in South Africa. Under the terms of the parties’ exclusive license agreement, Sequella will be solely responsible for completing clinical development and commercializing the product globally. Financial terms of the transaction were not disclosed.http://www.drugdiscoveryonline.com/Doc/sequella-acquires-exclusive-worldwide-rights-pfizer-s-sutezolid-0001
