
Home » 2013 (Page 33)
Yearly Archives: 2013
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
US FDA grants breakthrough therapy designation to Boehringer Ingelheim’s volasertib to treat patients with AML

Volasertib
755038-65-4
CHEMICAL NAMES
1. Benzamide, N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-
ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-
methoxy-
2. N-{trans-4-[4-(cyclopropylmethyl)piperazin-1-yl]cyclohexyl}-4-{[(7R)-7-ethyl-5-methyl-8-
(1-methylethyl)-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxybenzamide
CODE DESIGNATION BI 6727
| Ingelheim, Germany Thursday, September 19, 2013, 16:00 Hrs [IST] |
|
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to Boehringer Ingelheim’s volasertib, a selective and potent polo-like kinase (Plk) inhibitor, for the treatment of patients with acute myeloid leukaemia (AML), a type of blood cancer. |
http://www.pharmabiz.com/NewsDetails.aspx?aid=77733&sid=2

Volasertib (also known as BI 6727) is a small molecule inhibitor of the PLK1 (polo-like kinase 1) protein being developed byBoehringer Ingelheim for use as an anti-cancer agent. Volasertib is the second in a novel class of drugs called dihydropteridinone derivatives.[1]
Mechanism of action
Volasertib is a novel small-molecule targeted therapy that blocks cell division by competitively binding to the ATP-binding pocket of the PLK1 protein. PLK1 proteins are found in the nuclei of all dividing cells and control multiple stages of the cell cycle and cell division.[2] [3] [4] The levels of the PLK1 protein are tightly controlled and are raised in normal cells that are dividing. Raised levels of the PLK1 protein are also found in many cancers including; breast, non-small cell lung, colorectal, prostate, pancreatic, papillary thyroid, ovarian, head and neck and Non-Hodgkin’s Lymphoma.[5] [3] [6] [4] [7] [8] Raised levels of PLK1 increase the probability of improper segregation of chromosomes which is a critical stage in the development of many cancers. Raised levels of PLK1 have been associated with a poorer prognosis and overall survival in some cancers[4][9] [10] In addition to its role in cell division, there is evidence that PLK1 also interacts with components of other pathways involved in cancer development including the K-Ras oncogene and the retinoblastoma and p53 tumour suppressors[11] These observations have led to PLK1 being recognised as an important target in the treatment of cancer.
Volasertib can be taken either orally or via intravenous infusion, once circulating in the blood stream it is distributed throughout the body, crosses the cell membrane and enters the nucleus of cells where it binds to its target; PLK1. Volasertib inhibits PLK1 preventing its roles in the cell-cycle and cell division which leads to cell arrest and programmed cell death.[2] Volasertib binds to and inhibits PLK1 at nanomolar doses however, it has also been shown to inhibit other PLK family members; PLK2 and PLK3 at higher; micromolar doses. The roles of PLK2 and PLK3 are less well understood; however they are known to be active during the cell cycle and cell division.[12]
Volasertib inhibits PLK1 in both cancer and normal cells; however it only causes irreversible inhibition and cell death in cancer cells, because inhibition of PLK1 in cancer cells arrests the cell cycle at a different point to normal, non-cancer cells. In cancer cells PLK1 inhibition results in G2/M cell cycle arrest followed by programmed cell death, however, in normal cells inhibition of PLK1 only causes temporary, reversible G1 and G2 arrest without programmed cell death.[13] This specificity for cancer cells improves the efficacy of the drug and minimizes the drug related toxicity.
Clinical uses
Volasertib is currently undergoing investigation in phase 1 and 2 trials and has yet to be licensed by the FDA. Volasertib may be effective in several malignancies evidenced by the fact that its target PLK1 is overexpressed in up to 80% of malignancies, where it has been associated with a poorer treatment outcome and reduced overall survival.[1][4][9]Further phase 1 and 2 trials are active, investigating the effects of Volasertib both as a single agent and in combination with other agents in solid tumours and haematological malignancies including; ovarian cancer, urothelial cancer and acute myeloid leukaemia.[14]
Studies
Preclinical studies on volasertib have demonstrated that it is highly effective at binding to and blocking PLK1 function and causing programmed cell death in colon and non-small cell lung cancer cells both in vitro and in vivo. Volasertib can also cause cell death in cancer cells that have are no longer sensitive to existing anti-mitotic drugs such as vinca alkaloids and taxanes.[13] This suggests that volasertib may be effective when used as a second line treatment in patients who have developed resistance to vinca alkaloid and taxane chemotherapeutics.
A first in man trial of volasertib in 65 patients with solid cancers reported that the drug is safe to administer to patients and is stable in the bloodstream. This study also reported favourable anti-cancer activity of the drug; three patients achieved a partial response, 48% of patients achieved stable disease and 6 patients achieved progression free survival of greater than 6 months.[15] A further phase 1 trial of volasertib in combination with cytarabine in patients with relapsed / refractory acute myeloid leukaemiareported that 5 of 28 patients underwent a complete response, 2 achieved a partial response and a further 6 patients no worsening of their disease.[16]
- Schoffski, P. (2009). “Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology”. Oncologist 14 (6): 559–70. ISSN (Electronic) 1083-7159 (Linking) 1549-490X (Electronic) 1083-7159 (Linking).
- Barr, F. A.; H. H. Sillje, E. A. Nigg (2004). “Polo-like kinases and the orchestration of cell division”. Nat Rev Mol Cell Biol 5 (6): 429–40. ISSN (Print) 1471-0072 (Linking) 1471-0072 (Print) 1471-0072 (Linking).
- Garland, L. L.; C. Taylor, D. L. Pilkington, J. L. Cohen, D. D. Von Hoff (2006). “A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors”. Clin Cancer Res 12 (17): 5182–9. ISSN (Print) 1078-0432 (Linking) 1078-0432 (Print) 1078-0432 (Linking).
- Santamaria, A.; R. Neef, U. Eberspacher, K. Eis, M. Husemann, D. Mumberg, S. Prechtl, V. Schulze, G. Siemeister, L. Wortmann, F. A. Barr, E. A. Nigg (2007). “Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis”. Mol Biol Cell 18 (10): 4024–36. ISSN (Print) 1059-1524 (Linking) 1059-1524 (Print) 1059-1524 (Linking).
- Fisher, R.A.H.; D.K. Ferris (2002). “The functions of Polo-like kinases and their relevance to human disease.”. Curr Med Chem 2: 125–134.
- Holtrich, U.; G. Wolf, A. Brauninger, T. Karn, B. Bohme, H. Rubsamen-Waigmann, K. Strebhardt (1994). “Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors”. Proc Natl Acad Sci U S A 91 (5): 1736–40. doi:10.1073/pnas.91.5.1736. ISSN (Print) 0027-8424 (Linking) 0027-8424 (Print) 0027-8424 (Linking). PMC 43238. PMID 8127874.
- Steegmaier, M.; M. Hoffmann, A. Baum, P. Lenart, M. Petronczki, M. Krssak, U. Gurtler, P. Garin-Chesa, S. Lieb, J. Quant, M. Grauert, G. R. Adolf, N. Kraut, J. M. Peters, W. J. Rettig (2007). “BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo”. Curr Biol 17 (4): 316–22. doi:10.1016/j.cub.2006.12.037. ISSN (Print) 0960-9822 (Linking) 0960-9822 (Print) 0960-9822 (Linking). PMID 17291758.
- Winkles, J. A.; G. F. Alberts (2005). “Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues”. Oncogene 24 (2): 260–6.doi:10.1038/sj.onc.1208219. ISSN (Print) 0950-9232 (Linking) 0950-9232 (Print) 0950-9232 (Linking). PMID 15640841.
- Eckerdt, F.; J. Yuan, K. Strebhardt (2005). “Polo-like kinases and oncogenesis”. Oncogene 24 (2): 267–76. doi:10.1038/sj.onc.1208273. ISSN (Print) 0950-9232 (Linking) 0950-9232 (Print) 0950-9232 (Linking). PMID 15640842.
- Weichert, W.; A. Ullrich, M. Schmidt, V. Gekeler, A. Noske, S. Niesporek, A. C. Buckendahl, M. Dietel, C. Denkert (2006). “Expression patterns of polo-like kinase 1 in human gastric cancer”. Cancer Sci 97 (4): 271–6. ISSN (Print) 1347-9032 (Linking) 1347-9032 (Print) 1347-9032 (Linking).
- Liu, X.; R. L. Erikson (2003). “Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells”. Proc Natl Acad Sci U S A 100 (10): 5789–94. doi:10.1073/pnas.1031523100.ISSN (Print) 0027-8424 (Linking) 0027-8424 (Print) 0027-8424 (Linking). PMC 156279. PMID 12732729.
- Schmit, T. L.; N. Ahmad (2007). “Regulation of mitosis via mitotic kinases: new opportunities for cancer management”. Mol Cancer Ther 6 (7): 1920–31. ISSN (Print) 1535-7163 (Linking) 1535-7163 (Print) 1535-7163 (Linking).
- Rudolph, D.; M. Steegmaier, M. Hoffmann, M. Grauert, A. Baum, J. Quant, C. Haslinger, P. Garin-Chesa, G. R. Adolf (2009). “BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity”. Clin Cancer Res 15 (9): 3094–102. ISSN (Print) 1078-0432 (Linking) 1078-0432 (Print) 1078-0432 (Linking).
- ClinicalTrials.gov (2011). “Clinical Trials.gov Search of: Volasertib”. Missing or empty
|url=(help) - Gil, T.; P. Schöffski, A. Awada, H. Dumez, S. Bartholomeus, J. Selleslach, M. Taton, H. Fritsch, P. Glomb, Munzert G.M. (2010). “Final analysis of a phase I single dose-escalation study of the novel polo-like kinase 1 inhibitor BI 6727 in patients with advanced solid tumors”. J Clin Oncol 28.
- Bug, G.; R. F. Schlenk, C. Müller-Tidow, M. Lübbert, A. Krämer, F. Fleischer, T. Taube, O. G. Ottmann, H. Doehner (2010). “Phase I/II Study of BI 6727 (volasertib), An Intravenous Polo-Like Kinase-1 (Plk1) Inhibitor, In Patients with Acute Myeloid Leukemia (AML): Results of the Dose Finding for BI 6727 In Combination with Low-Dose Cytarabine”. 52nd ASH Annual Meeting and Exposition. Orange County Convention Centre, Florida: American Society of Haematology.
VOLASERTIB TRIHYDROCHLORIDE
CHEMICAL NAMES
1. Benzamide, N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-
ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-
methoxy-, hydrochloride (1:3)
2. N-{trans-4-[4-(cyclopropylmethyl)piperazin-1-yl]cyclohexyl}-4-{[(7R)-7-ethyl-5-methyl-8-
(1-methylethyl)-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxybenzamide
trihydrochloride
MOLECULAR FORMULA C34H50N8O3 . 3 HCl
MOLECULAR WEIGHT 728.2
SPONSOR Boehringer Ingelheim Pharmaceuticals, Inc.
CODE DESIGNATION BI 6727 CL3
CAS REGISTRY NUMBER 946161-17-7
Volasertib is a highly potent and selective inhibitor of the serine-threonine Polo like kinase 1 (Plk1), a key regulator of cell-cycle progression. Volasertib is a dihydropteridinone derivative with distinct pharmacokinetic (PK) properties. The problem underlying this invention was to develop improved dosage schedules for combination therapy of advanced and/or metastatic solid tumours.
Volasertib (I) is known as the compound N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-methoxy-benzamide,

This compound is disclosed in WO 04/076454. Furthermore, trihydrochloride salt forms and hydrates thereof are known from WO 07/090844. They possess properties which make those forms especially suitable for pharmaceutical use. The above mentioned patent applications further disclose the use of this compound or its monoethanesulfonate salt for the preparation of pharmaceutical compositions intended especially for the treatment of diseases characterized by excessive or abnormal cell proliferation.
U.S. 8,188,086
Several dihydropteridione derivatives effectively prevent cell proliferation. G. Linz and co-inventors report a comprehensive method for preparing pharmacologically active crystalline and anhydrous forms of compound 1 (Figure 1) that are suitable for drug formulations.

The inventors list several criteria for the properties of 1 and its manufacturing procedure:
- favorable bulk characteristics such as drying times, filterability, solubility in biologically acceptable solvents, and thermal stability;
- purity of the pharmaceutical composition;
- low hygroscopicity;
- no or low tendency toward polymorphism; and
- scalability to a convenient commercial process.
They describe their finding that the tri-HCl salt of 1 satisfies these criteria as “surprising”.
Free base 1 is prepared by condensing cyclopropylmethylpiperazine derivative 2 with pteridinone 3 in the presence of p-toluenesulfonic acid (TsOH), as shown in Figure 1. After the reaction is complete, the crude free base 1 is recovered as a viscous oil. It is then treated with HCl in an organic solvent to form 1·3HCl, isolated in 91% yield. Alternatively, the free base is not isolated; instead, concd HCl is added to the reaction mixture, followed by acetone. The crude salt is recovered in 92% yield.
The salt is purified by crystallization from refluxing EtOH, adding water, and cooling to precipitate the crystals. The inventors do not report the purity of this or any other reaction product.
The inventors obtained a hydrated form of the tri-HCl salt by dissolving the free base in EtOH at room temperature, followed by adding concd HCl and cooling to 2 °C. An anhydrous form can be recovered by drying the hydrate at 130 °C. The solubility of the hydrated salt in aqueous and organic media is reported, as are X-ray diffraction data for the hydrated form. The hydrated salt has good solid-state stability.
The patent also contains the syntheses of reactants 2 and 3 (Figures 2 and 3). The preparation of 2 begins with the formation of amide 7. Acid 4 is treated with SOCl2–DMF to form acid chloride 5; the crude product is added to a suspension of chiral difunctionalized cyclohexane 6 in THF and aq K2CO3 to produce 7. The crude product is recovered in 98% yield and oxidized to 8 with RuCl3 and N-methylmorpholine N-oxide (NMMO) in 91% yield.

Amide 8 reacts with cyclopropylmethylpiperazine 9 in the presence of methanesulfonic acid (MsOH). The solvent is evaporated, and the reaction mixture is treated with NaBH4. After further workup, product 10 is isolated in 46% yield. The nitro group is then hydrogenated over Raney Ni to give 2 in 90% yield. An alternative method for preparing10 is also described.
To prepare 3, readily available amino acid 11 is esterified and alkylated to form 12. In a multistep, one-pot procedure, 11 is first treated with HC(OMe)3 and SOCl2. Further reaction with NaBH(OAc)3, acetone, and NH4OH produces 12 as its HCl salt in 90% yield. The salt is treated with aq NaOH to form the free base, which reacts with pyrimidine 13 in the presence of NaHCO3 to form 14 in 79% isolated yield.

The pteridinone system is formed by hydrogenating 14 over a Pt/C catalyst in the presence of V(acac)3. Precursor 15 is recovered in 90% yield and methylated with (MeO)2CO and K2CO3 to give 3 in 82% isolated yield.
The inventors succeeded in developing a route for making a crystalline salt that is suitable for preparing pharmaceutical formulations. The many synthetic steps, however, use a large number of solvents that are frequently evaporated to dryness. [This observation implies that the processes have a significant environmental burden. —Ed.] (Boehringer Ingelheim International [Ingelheim am Rhein, Germany]. US Patent U.S. 8,188,086,
Sernova’s Cell Pouch (TM) and Sertolin (TM) Hold Promise for Treating Diabetes
 A major advance for the treatment of Type 1 diabetes has been the development of a procedure for transplanting islet cells, which are responsible for producing insulin, called the Edmonton Protocol. However, while this procedure has had success in treating diabetics, it is limited by several factors.
A major advance for the treatment of Type 1 diabetes has been the development of a procedure for transplanting islet cells, which are responsible for producing insulin, called the Edmonton Protocol. However, while this procedure has had success in treating diabetics, it is limited by several factors.
During this procedure many of the islet cells die due to their placement into a harsh environment, which is not ideal as the only current source of these cells are deceased donors, and their loss potentially results in the need for additional operations. The Edmonton Protocol is also very expensive (approximately $100,000), and patients must take immunosuppressant drugs indefinitely following the procedure.
To overcome these limitations, Sernova has developed a device that provides a natural environment for the islet cells, called a Cell PouchTM. Approximately the size of a matchbook, this device promotes the survival of the islet cells and is…
View original post 102 more words
FDA OKs Teva’s Injectable Treanda

FDA OKs Teva’s Injectable Treanda
FDA Approves Teva’s Injectable Treanda

bendamustine
Sept. 17, 2013 (GLOBES)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA; TASE: TEVA) has announced that the US Food and Drug Administration (FDA) has approved a new injectable version Treanda for treatment of indolent B-cell non-Hodgkin lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen, and chronic lymphocytic leukemia. read all at
http://www.pharmalive.com/fda-oks-tevas-injectable-treanda
Bendamustine (INN, trade names Treakisym, Ribomustin, Levact and Treanda; also known as SDX-105) is a nitrogen mustard used in the treatment of chronic lymphocytic leukemia[1] and lymphomas. It belongs to the family of drugs called alkylating agents. It is also being studied for the treatment of sarcoma.[2]
History
Bendamustine was first synthesized in 1963 by Ozegowski and Krebs in East Germany(the former German Democratic Republic). Until 1990 it was available only in East Germany. East German investigators found that it was useful for treating chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myelomaand lung cancer.
Bendamustine received its first marketing approval in Germany, where it is marketed under the tradename Ribomustin, by Astellas Pharma GmbH’s licensee, Mundipharma International Corporation Limited. It is indicated as a single-agent or in combination with other anti-cancer agents for indolent non-Hodgkin’s lymphoma, multiple myeloma, and chronic lymphocytic leukemia. SymBio Pharmaceuticals Ltd holds exclusive rights to develop and market bendamustine HCl in Japan and selected Asia Pacific Rim countries.
In March 2008, Cephalon received approval from the United States Food and Drug Administration to market bendamustine in the US, where it is sold under the tradename Treanda, for treatment of chronic lymphocytic leukemia.[3]
In October 2008, the FDA granted further approval to market Treanda for the treatment of indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. [4]
Bendamustine, 4-{5-[Bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric acid:

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there under the tradename Cytostasan®. See, e.g., W. Ozegowski and D. Krebs, IMET 3393 γ-[1-methyl-5-bis-(β-chloroethyl)-aminobenzimidazolo-(2)]-butyryl chloride, a new cytostatic agent of the group of benzimidazole nitrogen mustards. Zbl. Pharm. 110, (1971) Heft 10, 1013-1019, describing the synthesis of bendamustine hydrochloride monohydrate. Since that time, it has been marketed in Germany under the tradename Ribomustin®. Bendamustine is an alkylating agent that has been shown to have therapeutic utility in treating diseases such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to technical difficulties in its preparation and administration. Researchers, therefore, have investigated methods of improving the preparation and stability of bendamustine and its formulations. For example, German (GDR) Patent No. 159877 discloses a method for preparing bendamustine free base by reaction of the bis-hydroxyl precursor with thionyl chloride followed by recrystallization from water.
German (GDR) Patent No. 34727 discloses a method of preparing derivatives of bendamustine. The described derivatives differ from bendamustine in the substitution at the 1-position.
German (GDR) Patent No. 80967 discloses an injectable preparation of bendamustine hydrochloride monohydrate, ascorbic acid, and water. GDR 80967 describes that lyophilization of compounds such as bendamustine is only possible if the compound is of sufficient stability that it can withstand the processing conditions. The preparation described in GDR 80967 is not lyophilized.
German (GDR) Patent No. 159289 discloses a ready-to use, injectable solution of bendamustine hydrochloride that avoids lyophilization. GDR 159289 describes an anhydrous solution of bendamustine hydrochloride in 1,2-propylene glycol or ethanol.
U.S. application Ser. No. 11/330,868, filed Jan. 12, 2006, assigned to Cephalon, Inc., Frazer, P A, discloses methods of preparing lyophilized pharmaceutical compositions comprising bendamustine hydrochloride.
Chemotherapeutic uses
Bendamustine has been used both as sole therapy and in combination with other agents including etoposide, fludarabine, mitoxantrone,methotrexate, prednisone, rituximab, vincristine and 90Y-ibritumomab tiuxetan.
One combination for stage III/IV relapsed or refractory indolent lymphomas and mantle cell lymphoma (MCL), with or without prior rituximab-containing chemoimmunotherapy treatment, is bendamustine with mitoxantrone and rituximab.[5] In Germany in 2012 it has become the first line treatment of choice for indolent lymphoma.[6] after Trial results released in June 2012 showed that it more than doubled disease progression-free survival when given along with rituximab. The combination also left patients with fewer side effects than the older R-CHOP treatment.[7]
Common adverse reactions are typical for the class of nitrogen mustards, and include nausea, fatigue, vomiting, diarrhea, fever, constipation, loss of appetite, cough, headache, unintentional weight loss, difficulty breathing, rashes, and stomatitis, as well as immunosuppression, anemia, and low platelet counts. Notably, this drug has a low incidence of hair loss (alopecia) unlike most other chemotherapy drugs.[8]
References
- Kath R, Blumenstengel K, Fricke HJ, Höffken K (January 2001). “Bendamustine monotherapy in advanced and refractory chronic lymphocytic leukemia”. J. Cancer Res. Clin. Oncol. 127 (1): 48–54. doi:10.1007/s004320000180. PMID 11206271.
- Bagchi S (August 2007). “Bendamustine for advanced sarcoma”. Lancet Oncol. 8 (8): 674. doi:10.1016/S1470-2045(07)70225-5.PMID 17726779.
- “Cephalon press release – Cephalon Receives FDA Approval for TREANDA, a Novel Chemotherapy for Chronic Lymphocytic Leukemia”. Retrieved 2008-03-23.
- “Cephalon press release -Cephalon Receives FDA Approval for TREANDA to Treat Patients with Relapsed Indolent Non-Hodgkin’s Lymphoma”. Retrieved 2008-11-03.
- Weide R, Hess G, Köppler H, et al. (2007). “High anti–lymphoma activity of bendamustine/mitoxantrone/rituximab in rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A muticenter phase II study of the German Low Grade Lymphoma Study Group (GLSG)”. Leuk. Lymphoma. 48 (7): 1299–1306. doi:10.1080/10428190701361828. PMID 17613757.
- New Combo Replaces CHOP for Lymphoma. Dec 2012
- “‘Rediscovered’ Lymphoma Drug Helps Double Survival: Study”. June 3, 2012.
- Tageja, Nishant; Nagi, Jasdeepa; “Bendamustine: something old, something new”; Cancer Chemotherapy and Pharmacology, 2010 Aug;66(3):413-23. doi: 10.1007/s00280-010-1317-x.
External links
- Manufacturer’s official website intended for US patients
more info
Bendamustine hydrochloride, 4-{5-[Bis(2-chloroethyl) amino]- l-methyl-2- benzimidazolyl} butyric acid hydrochloride, of the formula (VI) :

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there, as the hydrochloride salt, under the trade name Cytostasan®. Since that time, it has been marketed in Germany under the trade name Ribomustin®. Bendamustine Hydrochloride as injection is available in the United States under the tradename Treanda®. Bendamustine hydrochloride is an alkylating agent that is approved for the treatment of non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia.
Bendamustine hydrochloride is a benzimidazole analog. While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to formation of non-bendamustine products (i.e. “degradation impurities”) which leads to technical difficulties in its preparation and administration. In light of its instability in aqueous solution, bendamustine is supplied as a lyophilized cake of bendamustine hydrochloride salt. US2006/159713, US 2006/128777 and WO2010/036702 disclose various impurities of Bendamustine hydrochloride which are as follows:

PC-1 PC-2
Jena et al. were the first to disclose the synthesis of Bendamustine hydrochloride in German (GDR) Patent No. 34727. Krueger et al. in German (GDR) Patent No. 159877 recite a method as summarized in scheme-1, for the synthesis of bendamustine hydrochloride comprising the reaction of the 4-[l-methyl-5-bis-(2- hydroxyethyl)-benzimidazolyl-2]butyric acid ethyl ester (4) (or the corresponding methyl, propyl or butyl ester) with thionyl chloride in chloroform at 0-5°C to form 4-[l- methyl-5-bis-(2-chloroethyl)-benzimidazolyl-2]butyric acid ethyl ester (5). Excess of thionyl chloride is destroyed by stirring the reaction mixture in aqueous HCl. Finally chloroform is distilled off and stirred at 95°C for 3 hours. The reaction mixture is partially concentrated and the residue is diluted with water and stirred upto crystallization. Further purification is done by recrystallization from water.
Scheme-1: Method disclosed by Krueger et al. in DD159877 for the synthesis of Bendamustine hydrochloride

Bendamustine hydrochloride (6)
Ozegowski et al in Zentralblatt fuer Pharmazie, Pharmakotherapie und Laboratoriumsdiagnostik 1 10 (10), 1013-1019 (1971) discloses a process for the preparation of bendamustine hydrochloride monohydrate. The Chinese journal “Chinese journal of New Drugs “, 2007, No. 23, Vol. 16, 1960-61 and J. Prakt. Chem. 20, 178-186 (1963) disclose another method for the synthesis of Bendamustine hydrochloride monohydrate starting from 2,4-dinitrochlorobenzene as summarized in scheme-2.

The crucial conversions are reaction of l-methyl-2-(4′-ethyl butyrate)-5- amino]-lH-benzimidazole 6 with ethylene oxide in the presence of water, sodium acetate and acetic acid, by maintaining at 5°C for 5 hours and overnight at 20°C to give 4-{5-[bis-(2-hydroxy-ethyl)-amino]-l-methyl-lH-benzimidazol-2-yl}-butyric acid ethyl ester (dihydroxy ester) 7 as a jelly mass, which on chlorination using thionyl chloride in chloroform and subsequent in situ hydrolysis with concentrated HCI gave bendamustine hydrochloride. It also discloses a process for the recrystallization of bendamustine hydrochloride from water and the product obtained is a monohydrate with a melting point of 148-151°C.
IP.com Journal 2009, 9(7B), 21 discloses another process as shown below for the preparation of ethyl-4-[5-[bis(2-hydroxyethyl) amino]- l-methylbenzimidazol-2- yl]butanoate (III) wherein ethyl-4-(5 -amino- 1 -methyl- lH-benzo[d]imidazol-2-yl) butanoate (II) is reacted with 2-halo ethanol in the presence of an inorganic base selected from the group consisting potassium carbonate, potassium bicarbonate, sodium

The PCT application WO 2010/042568 assigned to Cephalon discloses the synthesis of Bendamustine hydrochloride as summarized in schem-3 starting from 2,4- dintroaniline in six steps. The crucial step is reductive alkylation of Il-a, using borane- tetrahydrofuran and chloroacetic acid at ambient temperature, producing compound of formula I-a. Acid mediated hydrolysis of I-a using concentrated hydrochloric acid at reflux produced bendamustine hydrochloride which has a purity of 99.1%. The above PCT Patent application also discloses a method of purification of Bendamustine hydrochloride by agitating the Bendamustine hydrochloride in a mixture of DMF and THF at 75°C for about 30 minutes followed by cooling to ambient temperature and isolating the solid by filtration.
Scheme-3:


iil-a


Bemdamuatine hydrochloride
The PCT application WO 2011/079193 assigned to Dr. Reddy’s Laboratories discloses the synthesis of Bendamustine hydrochloride as summarized in schem-4 starting from compound of formula (II). The crucial step is alkylation of compound of formula II with 2-haloethanol in the presence of an organic base to give a compound of formula (III) which on chlorination with a chlorinating agent affords a compound of formula (IV). Compound of formula (IV) on hydrolysis in acidic medium gives bendamustine hydrochloride. It further discloses purification of bendamustine hydrochloride using aqueous hydrochloric acid and acetonitrile.
Scheme-4:

Bendamustine hydrochloride (Pure)
The most of the prior art processes described above involve
• The use of ethylene oxide for the preparation of bendamustine hydrochloride, which is often not suitable for industrial scale processes due to difficulty in handling ethylene oxide, since it is shipped as a refrigerated liquid.
• Further, the known processes involve the use of strongly acidic conditions and high temperatures for the hydrolysis of ethyl ester of bendamustine and subsequent in-situ formation of bendamustine hydrochloride, thereby resulting in increased levels of various process-related impurities IMP. -A (RRT-0.46), IMP. -B (RRT-1.27) and IMP. -C (RRT-1.31) whose removal is quite difficult and make the process less economically viable.

IMP.-B
International Application Publication No. WO 2009/120386 describes various solid forms of bendamustine hydrochloride designated as bendamustine hydrochloride Form 1, bendamustine hydrochloride Form 2, bendamustine hydrochloride Form 3, bendamustine hydrochloride Form 4, amorphous bendamustine hydrochloride or a mixture thereof, processes for their preparation and lyophilized composition comprising the solid forms. According to the disclosure, monohydrate of bendamustine hydrochloride has been prepared previously. The monohydrate has a reported melting point of 152-156°C which is similar to that of the observed melting point of bendamustine hydrochloride Form 2.
It is known that synthetic compounds can contain extraneous compounds or impurities resulting from their synthesis or degradation. The impurities can be unreacted starting materials, by-products of the reaction, products of side reactions, or degradation products. Generally, impurities in an active pharmaceutical ingredient (API) may arise from degradation of the API itself, or during the preparation of the API. Impurities in Bendamustine hydrochloride or any active pharmaceutical ingredient (API) are undesirable and might be harmful.
Regulatory authorities worldwide require that drug manufacturers isolate, identify and characterize the impurities in their products. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurity is present in the lowest possible levels, even if structural determination is not possible. The product mixture of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. Side products and byproducts of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product mixture. At certain stages during processing of the active pharmaceutical ingredient, the product is analyzed for purity, typically, by HPLC, TLC. or GC analysis, to determine if it is suitable for continued processing and, ultimately, for use in a pharmaceutical product. Purity standards are set with the intention of ensuring that an API is as free of impurities as possible, and, thus, are as safe as possible for clinical use. The United States Food and Drug Administration guidelines recommend that the amounts of some impurities are limited to less than 0.1 percent.
Generally, impurities are identified spectroscopically and by other physical methods, and then the impurities are associated with a peak position in a chromatogram (or a spot on a TLC plate). Thereafter, the impurity can be identified by its position in the chromatogram, which is conventionally measured in minutes between injection of the sample on the column and elution of the particular component through the detector, known as the “retention time” (“RT”). This time period varies daily based upon the condition of the instrumentation and many other factors. To mitigate the effect that such variations have upon accurate identification of an impurity, practitioners use “relative retention time” (“RRT”) to identify impurities. The RRT of an impurity is its retention time divided by the retention time of a reference marker.
It is known by those skilled in the art, the management of process impurities is greatly enhanced by understanding their chemical structures and synthetic pathways, and by identifying the parameters that influence the amount of impurities in the final product.
Therefore, there remains a need for improved process for the preparation of bendamustine hydrochloride, producing high yield and purity, and well-suited for use on an industrial scale. Despite the existence of various polymorphic forms of bendamustine hydrochloride, there exists a need for a simple process for the preparation of the stable form of bendamustine hydrochloride which is amenable to scale up and results in high yield and purity.
Bendamustine, (4-{5-[bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric

Bendamustine
is an atypical structure with a benzimidazole ring, which structure includes an active nitrogen mustard. Bendamustine was initially synthesized in 1963 in the German Democratic Republic and was available from 1971 to 1992 in that location under the name Cytostasan®. Since that time, it has been marketed in Germany under the tradename Ribomustin®. It is currently available for use in the United States under the tradename Treanda® (Cephalon, Inc., Frazer, Pa.). It has been widely used to treat chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
Like other nitrogen mustards, bendamustine hydrolyzes in aqueous solution, with the major degradant being the primary alcohol HP1 (See U.S. application Ser. No. 11/330,868, the entirety of which is incorporated herein):
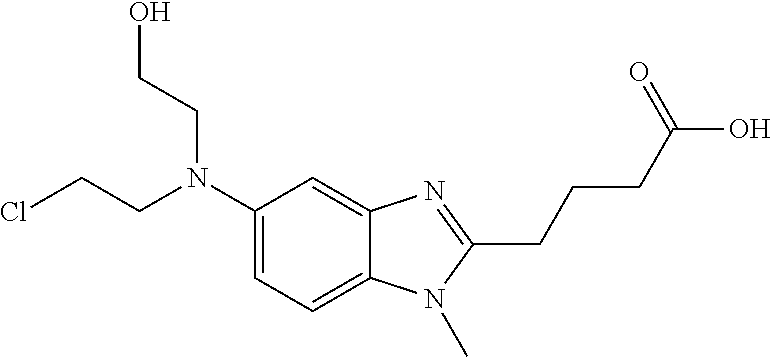
In light of its instability in aqueous solution, bendamustine is currently supplied as a lyophilized powder for injection. Just prior to its infusion, the medical practitioner reconstitutes the powder with Sterile Water for Injection. Reconstitution should yield a clear, colorless to pale yellow solution and the powder should completely dissolve in about 5 minutes. If particulate matter is observed, the reconstituted product should not be used and should be discarded. The reconstituted product is then transferred to a 0.9% Sodium Chloride Injection infusion bag within 30 minutes of reconstitution. This admixture should be a clear and colorless to slightly yellow solution. If the admixture comprises particulate matter or is discolored, it should be discarded and a fresh sample prepared.
The salt bendamustine hydrochloride is an alkylating agent, originally synthesized in 1963 at the Institute for Microbiology & Experimental Therapy in Jena, German Democratic Republic, with the intent to produce an agent with both alkylating and antimetabolite properties. Jenapharm (now Schering AG) formerly marketed it in Germany under the trade name Cytostasan from 1971 to 1992. Cytostasan was a lyophilised powder for solution for injection (vials) conatining 25 mg of Bendamustine HCI. It was widely used but never studied systematically in patients until the 1990s, then German investigators demonstrated its clinical activity in a number of malignancies. Since 1993, Ribosepharm was marketing bendamustine in Germany under the brand name Ribomustin RBO. Ribomustin is available as a lyophilized powder for injection, containing 100 mg of drug in each 50 ml_ vial, or 25 mg of drug in each 20 ml_ vial, also comprising mannitol, and indicated for the treatment of chronic lymphocytic leukemia. The lyophilized powder is reconstituted as close to the time of patient administration as possible with 40 ml_ (for a 100 mg product) or 10 mL (for a 25 mg product) of sterile water for injection. The reconstituted product then is further diluted to 500 mL with 0.9% sodium chloride for injection. The route of administration is by intravenous infusion over 30 to 60 minutes.
Another bendamustine product is sold in the United States by Cephalon, Inc. as TREANDA® for Injection, a lyophilized powder in a single-use vial indicated for the treatment of patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin’s lymphoma. A 25 mg dose vial contains 25 mg of bendamustine hydrochloride and 42.5 mg of mannitol, and a 100 mg dose vial contains 100 mg of bendamustine hydrochloride and 170 mg of mannitol.
TREANDA is intended for intravenous infusion only after reconstitution with Sterile Water for Injection USP, and then further dilution with either 0.9% Sodium
Chloride Inj.ection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Inj.ection, USP. The pH of the reconstituted solution is 2.5-3.5. TREANDA is supplied as a sterile non-pyrogenic white to off-white lyophilized powder, in a single-use vial.
Bendamustine hydrochloride is very unstable in an aqueous solution. The bis-2-chlorethylamino bond is hydrolyzed in weak acid, neutral, or alkaline solution. Monohydroxybendamustine [HP-1 ] is formed rapidly in the presence of water. Bendamustine ethyl ester [BM1 EE] is formed when bendamustine reacts with ethyl alcohol. BM1 EE can be formed during drug substance manufacturing, e.g., during recrystalization and/or purification processes. BM1 EE is a more potent cytotoxic drug than bendamustine.
FDA accepts new drug application for investigational compound Epanova for the treatment of severe hypertriglyceridaemia
LONDON, Sept. 18, 2013 – AstraZeneca today announced that the US Food and Drug Administration (FDA) has accepted for review a New Drug Application (NDA) for EpanovaTM, an investigational compound for the treatment for patients with severe hypertriglyceridaemia (triglyceride levels greater than or equal to 500mg/dL). The NDA submission for Epanova was filed by Omthera Pharmaceuticals, now a wholly-owned subsidiary of AstraZeneca, as a 505(b)(1) application in July 2013. The Prescription Drug User Fee Act (PDUFA) goal date for the FDA is 5 May 2014.http://www.pharmalive.com/fda-accepts-astrazeneca-nda-for-epanova
Forigerimod, (Rigerimod) also known as Lupuzor, CEP-3345 for treatment of systemic lupus erythematosus (SLE)


FORIGERIMOD
CHEMICAL NAMES
1. L-Tyrosine, L-arginyl-L-isoleucyl-L-histidyl-L-methionyl-L-valyl-L-tyrosyl-L-seryl-L-lysyl-L-arginyl-O-phosphono-L-serylglycyl-L-lysyl-L-prolyl-L-arginylglycyl-L-tyrosyl-L-alanyl-L-phenylalanyl-L-isoleucyl-L-α-glutamyl-
2. O3,140-phosphono(human U1 small nuclear ribonucleoprotein 70 kDa (snRNP70))-(131-151)-peptide
MOLECULAR FORMULA C117H181N34O32PS
MOLECULAR WEIGHT 2639
TRADEMARK Lupuzor
SPONSOR Cephalon, Inc.
CODE DESIGNATION IPP 201101
CAS REGISTRY NUMBER 497156-60-2
STRUCTURAL FORMULA

stucture, http://www.ama-assn.org/ama1/pub/upload/mm/365/forigerimod.pdf
-
Forigerimod nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/forigerimod.pdfSTATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN. FORIGERIMOD.
…………………………………………………………………………………………………………..
FORIGERIMOD ACETATE
CAS REGISTRY NUMBER 1160237-55-7 of acetate
http://www.ama-assn.org/resources/doc/usan/forigerimod-acetate.pdf
-
Forigerimod acetate nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/forigerimod-acetate.pdfSTATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN. FORIGERIMOD ACETATE
str is FORIGERIMOD ACETATE
FORIGERIMOD ACETATE
Forigerimod, also known as Lupuzor or CEP-33457, (SyB L-1001) is a CD4 T-cell modulator being investigated for the treatment of systemic lupus erythematosus (SLE). In the Phase II trials, Lupuzor was administered subcutaneously at a dose of 200 mcg once a month for 3 months. The Phase III study is anticipated to be complete in September 2012 and will measure the proportion of patients achieving a combined clinical response using the SLE responder index.

Positive final Lupuzor trial results. Marketwire. www.marketwire.com/press-release/Positive-Final-Lupuzor-Trial-Results-AIM-IMM-1176375.htm. Published November 19, 2009. Accessed June 18, 2011.
Rigerimod (IPP-201101, Lupuzor) is a polypeptide corresponding to the sequence 131-151 of the 70k snRNP protein with a serine phosphorylated in position 140.[1]
It gave encouraging results in a phase IIb trial for severe lupus.[1] Another phase IIb trial has started recruiting in the US.[2]
References
Lupuzor™ is a potential treatment for lupus, currently given the approval by the US FDA to start Phase III with a Special Protocol Assessment (SPA) and Fast Track designation. ImmuPharma holds all worldwide rights in this lead compound.
Background
Lupus (or Systemic Lupus Erythematosus) is a chronic, potentially life-threatening autoimmune disease. An estimated 1.4 million people are diagnosed in the 7 major world markets (the USA, Japan, Germany, France, Spain, the UK and Italy). Lupus is an inflammatory disease, which attacks multiple organs such as the skin, joints, kidneys, blood cells, heart and lungs. There is currently no cure.
The development of ImmuPharma’s Lupuzor™
ImmuPharma’s compound Lupuzor™ (previously known as IPP-201101 and also referred to as rigerimod or P140) has a novel mechanism of action aimed at modulating the body’s immune system so it does not attack healthy cells, without causing adverse side effects. It has the potential to halt the progression of the disease in a substantial proportion of patients.
Lupuzor™ has successfully completed Phase I, Phase IIa and Phase IIb studies and has now been given the approval by the US FDA to enter Phase III, the final testing phase.
The latest highlights of Lupuzor’s™ development as a treatment for lupus include:
- An ‘End of Phase 2’ meeting package with ImmuPharma’s Phase IIb data was submitted to the FDA and the FDA responded to all the questions
- The Investigational Medicinal Product Dossier (IMPD) submitted via the Voluntary Harmonized Procedure (VHP) in the EU was approved
- The Scientific Advice meeting with the European Medicines Evaluation Agency (EMEA) was held; the recommendations were very similar to those in the FDA’s ‘End Of Phase 2’ responses. Recommendations were incorporated into the Phase III pivotal programme
- The Japanese equivalent authorities (PMDA) have agreed to the initiation of clinical trials in Japan
- The FDA has granted Lupuzor™ the approval to start Phase III with a Special Protocol Assessment (SPA)
- The FDA has granted Lupuzor™ Fast Track designation
How Lupuzor™ works in the treatment of lupus
Lupuzor™ is a drug that specifically modulates the immune system of lupus patients by modifying the behaviour of some of the key cells involved in the pathogenesis of the disease. The clinical profile of lupus patients is generally assessed by standardised scales such as SLEDAI (SLE Disease Activity Index): the lower the score, the better the condition of the patient. During this Phase II study, the SLEDAI scores were assessed on multiple occasions even though the study was not designed or powered to demonstrate clinical benefit as the primary endpoint due to the short treatment period.
| forigerimod | IPP-201101 | oligopeptide | therapeutic | nucleolin |
| forigerimod acetate | CEP-33457, P-140, IPP-201101 | oligopeptide (salt) | therapeutic | nucleolin |
GLENMARK- ELOVERA , for dry skin disorders


Compositions:
Elovera extract 10% cream, Vitamin E 0.5%
Category–Locally Acting Skin Preparations
Description
* Aqueeze adequate amount of elovera moisturizing body wash onto wet hands or wet loran and work into a creamy lather. apply it all ovr the body, keep for some time and then rinse with water.
| Products Name : | Elovera Moisturizing Body Wash 150ml – (Glenmark) |
Elovera Cream, manufacture by Glenmark pharmaceuticals limited , is cream enriched with vitamin E and Aloe Vera. It’s a very special cream specially for treating scars and other minor pimple spots on the face.
reviews from net
My skin is very much oily hence I get these ugly Pimples very profoundly. On top of it i have the habit of bursting out the puss from these pimples. I always play it with my hands and as a result forms some very ugly scars on my face which are visible from distant away.Though I am bit dark with my completion ,even then It’s clearly visible and my mother scolds me like hell for bursting the pimples out.Honestly I just can’t stop my hands reaching out for them no matter how busy I am so Finally has to resort to some ointments to reduce the visibility of the scars.
I did try few popular products but were of no use basically. The spots didn’t get reduced but instead effected the completion of my face.Finally my mother came to my rescue. She had hear about this Elovera Cream from some one and bought home one for me.Initially i was a bit skeptic but finally I thought of trying it. For the first few days it had no effect what-so-ever , but slowly it started clearing the skin blemishes. My skin started showing it’s effects and the scars became less visible. Not only does it clear the scars but it helped me to fight the ugly pimples as well.
My face became much more glowing and healthy and i use the cream regularly even now.It’s really a magical product and should try it for clearing the blemishes and other skin problem.
EMA approves biosimilar Somatropin from Biopartners Gmbh
![OMNITROPE® (somatropin [rDNA origin] injection) Structural Formula Illustration](https://i0.wp.com/images.rxlist.com/images/rxlist/omnitrope1.gif)
SOMATROPIN
The European Medicine agency has approved a biosimilar somatropin from Biopartners GMBH. Somatropin biopartner would be the third biosimilar version of somatropin the European market. Other players selling somatropin inlcude Sandoz and Roche. Sandoz sells under the brand Omnitrope, while Roche which is the innovator of somatropin sells it under the brand name NutropinAq.
READ AT
DETAILS OF OMNITROPE
Omnitrope® (somatropin-[rDNA] origin) is a polypeptide hormone of recombinant DNA origin. It has 191 amino acid residues and a molecular weight of 22,125 daltons. The amino acid sequence of the product is identical to that of human growth hormone of pituitary origin (somatropin). Omnitrope® is synthesized in a strain of. Escherichia coli that has been modified by the addition of the gene for human growth hormone. Omnitrope® Cartridge is a clear, colorless, sterile solution for subcutaneous injection. Omnitrope® for Injection is a lyophilized powder that is reconstituted for subcutaneous injection.
Figure 1: Schematic amino acid sequence of human growth hormone including the disulfide bonds
|
Each Omnitrope® Cartridge or vial contains the following (see Table 4):
Table 4. Contents of Omnitrope® Cartridges and Vial
| Product | Cartridge 5 mg/1.5 mL | Cartridge 10 mg/1.5 mL | For Injection 5.8 mg/vial |
| Component | |||
| Somatropin | 5 mg | 10 mg | 5.8 mg |
| Disodium hydrogen phosphate heptahydrate | 1.3 mg | 1.70 mg | 2.09 mg |
| Sodium dihydrogen phosphate dihydrate | 1.6 mg | 1.35 mg | 0.56 mg |
| Poloxamer 188 | 3.0 mg | 3.0 mg | – |
| Mannitol | 52.5 mg | – | – |
| Glycine | – | 27.75 mg | 27.6 mg |
| Benzyl alcohol | 13.5 mg | – | – |
| Phenol | – | 4.50 mg | – |
| Water for Injection | to make 1.5 mL | to make 1.5 mL | – |
| Diluent (vials only) | Bacteriostatic Water for Injection | ||
| Water for injection | to make 1.14 mL | ||
| Benzyl alcohol | 17 mg | ||
Genzyme’s multiple sclerosis treatment approved by European Commission

Alemtuzumab
Sanofi and its subsidiary Genzyme have been given marketing approval by the European Commission for Lemtrada (alemtuzumab), a treatment for multiple sclerosis. read all at
click on title below
Genzyme’s multiple sclerosis treatment approved by European Commission
Sanofi wins EU approval for second MS treatment Lemtrada
September 17,2013 | By Márcio Barra

Sanofi won today a marketing approval from the European commission for their second multiple sclerosis treatment, the injectable drug Lemtrada, following the approval of the pill Aubagio (teriflunomide) on August 30. This was the drug’s first regulatory approval worldwide.
View original post 283 more words
