

Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
Home » Posts tagged 'phase 2' (Page 14)
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
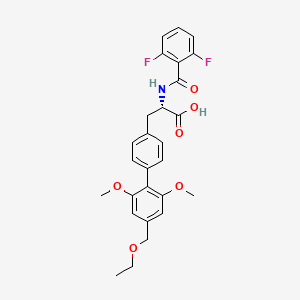
Zydus-Cadila is developing ZYH-7, a PPAR alpha modulator for the potential treatment of dyslipidemia

ZYH-7
Prediction of ZYH 7 below……..If it does not match then ZYH 7 will be a very close structure

1014989-63-9
Acetic acid, 2-[2-methyl-4-[1-[[[4-methyl-2-[4-(trifluoromethyl)phenyl]- 5-thiazolyl]methoxy]imino]ethyl]phenoxy]-
Zydus-Cadila is developing ZYH-7, a PPAR alpha modulator for the potential treatment of dyslipidemia .
By January 2012, phase II trials had begun ; in January 2014, the drug was still listed as being in phase II development
By January 2012 phase II trials had begun for Diabetes type 2 Lipoprotein disorders
Obesity
In August 2007, an IND was filed , and by March 2008, a phase I trial was underway ; by April 2011, the trial had been completed
| Zydus Cadila has filed an Investigational New Drug (NID) application for seeking DCGI’s permission for conducting clinical trials for its New Molecular Entity (NME) ZYH7. |
 |
| According to a company release, it claims that ZYH7 is a novel drug candidate for treating dyslipidemia and metabolic disorders. The company inform that ZYH7 had been conceptualised and developed by its scientists from Zydus Research Centre. |
| The company has its in-house research centre and it had recently concluded pre-clinical studies on ZYH7, which have reported interesting and encouraging finding which indicate a novel molecule to treat dyslipidemia and associated metabolic disorders. |
| Commenting on the new development, Pankaj Patel, chairman and managing director, Zydus Cadila said, “We have been building a promising pipeline of new molecular entities at the Zydus Research Centre and ZYH7 is an important step in this direction”. |
| Starting with its first IND filing in 2005, Zydus today has four INDs in various stages of clinical trials. NME – ZYH1 for treating dyslipidemia and ZYI1 for treating pain and inflammation are undergoing Phase II trials. ZYH2 for treating diabetes and the novel CB-1 antagonist, ZYO1 for treating obesity, are undergoing Phase I trials. |
| Diabetes, a worldwide health problem, affects more than 150 million people, a number expected to double to 300 million by 2025. People with diabetes are at especially high risk for dyslipidemia, particularly high triglyceride levels and low HDL levels. |
| Dyslipidemia is also a key independent risk factor for cardiovascular disease (CVD), which is the largest therapeutic segment in the world pharmaceutical market. |
| With an increasing correlation between several endocrine and metabolic disorders, there has been considerable emphasis in recent times on metabolic syndrome. The metabolic components of cardiovascular disease, diabetes and obesity, are linked in numerous ways with each having an impact on the other. |
| For instance, it is also well known that patients with Type 2 diabetes have a two to four-fold excess risk of coronary heart disease and that these patients very often have increased cardiovascular risk factors even before the onset of their diabetes. |
Dyslipidemia is an abnormal amount of lipids (e.g. cholesterol and/or fat) in the blood. In developed countries, most dyslipidemias are hyperlipidemias; that is, an elevation of lipids in the blood. This is often due to diet and lifestyle. Prolonged elevation of insulin levels can also lead to dyslipidemia. Likewise, increased levels of O-GlcNAc transferase (OGT) may cause dyslipidemia.
| Dyslipidemia | |
|---|---|
| Classification and external resources | |
| ICD–10 | E78 |
| ICD–9 | 272 |
| DiseasesDB | 33452 |
| MeSH | D050171 |
Classification
Physicians and basic researchers classify dyslipidemias in two distinct ways:
- Phenotype, or the presentation in the body (including the specific type of lipid that is increased)
- Etiology, or the reason for the condition (genetic, or secondary to another condition). This classification can be problematic, because most conditions involve the intersection of genetics and lifestyle issues. However, there are a few well-defined genetic conditions that are usually easy to identify.
Fredrickson Classification:[1]
For more a detailed version, see Hyperlipidemia#Classification.
| Phenotype | I | IIa | IIb | III | IV | V |
|---|---|---|---|---|---|---|
| Elevated Lipoprotein | Chylomicron | LDL | LDL and VLDL | IDL | VLDL | VLDL and chylomicrons |
WO 2008035359
https://www.google.com/patents/WO2008035359A2?cl=en
Scheme 1 below which comprises:

Scheme 2 below which comprises

| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009021740A2 | Aug 14, 2008 | Feb 19, 2009 | Sanofis Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010049946A2 * | Oct 22, 2009 | May 6, 2010 | Cadila Healthcare Limited | Thyroid receptor ligands |
| WO2010084512A1 * | Dec 22, 2009 | Jul 29, 2010 | Cadila Healthcare Limited | Novel oxime derivatives |
| WO2010110479A1 * | Mar 24, 2010 | Sep 30, 2010 | Nippon Chemiphar Co., Ltd. | Activator for peroxisome proliferator-activated receptor |
| WO2011157827A1 | Jun 17, 2011 | Dec 22, 2011 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2013037390A1 | Sep 12, 2011 | Mar 21, 2013 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014192023A1 * | May 20, 2014 | Dec 4, 2014 | Cadila Healthcare Limited | Novel compounds suitable for the treatment of dyslipidemia |
| EP2567959A1 | Sep 12, 2011 | Mar 13, 2013 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8742117 | Dec 22, 2009 | Jun 3, 2014 | Cadila Healthcare Limited | Oxime derivatives |
| US8822414 * | Dec 26, 2011 | Sep 2, 2014 | Cadila Healthcare Limited | Heterocyclic compounds suitable for the treatment of dyslipidemia |
………….
PARIS

Firategrast, T-0047

 Japan
Japan
Firategrast, 402567-16-2;
Firategrast, MS, Alpha4beta1 integrin
PHASE 2 GSK
Mitsubishi Tanabe Pharma INNOVATOR
Glaxo Group Limited, Mitsubishi Tanabe Pharma Corporation
SB 683699, SB-683699, UNII-OJY3SK9H5F
Firategrast; UNII-OJY3SK9H5F; SB-683699; Firategrast (USAN); 402567-16-2; SB683699; T-0047
Molecular Formula: C27H27F2NO6
Molecular Weight: 499.503186 g/mol
SYSTEMATIC NAME:
1,1′-Biphenyl)-4-propanoic acid, alpha-((2,6-difluorobenzoyl)amino)-4′-(ethoxymethyl)-2′,6′-dimethoxy-, (alphaS)-
N-(2,6-Difluorobenzoyl)-4-[4-(ethoxymethyl)-2,6-dimethoxyphenyl]-L-phenylalanine
N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
2S)-2-((2,6-Difluorobenzoyl)amino)-3-(4′-(ethoxymethyl)-2′,6′-dimethoxybiphenyl-4- yl)propanoic acid
(2S)-2-{[(2,6- difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4- biphenylyl]propanoic acid
(2S)-2-[[2,6-bis(fluoranyl)phenyl]carbonylamino]-3-[4-[4-(ethoxymethyl)-2,6-dimethoxy-phenyl]phenyl]propanoic acid
Pharmacological half-life is 2.5 – 4.5 hours, compared to 11 days for natalizumab, a drug in the same class
Orally bioavailable small molecule α4-integrin antagonist
see
http://www.msdiscovery.org/node/1377#node-biblio-1338
http://multiple-sclerosis-research.blogspot.com/2012/01/research-oral-tysabri-analogue.html
SB683699 is an alpha4 integrin antagonist that had been studied in phase II trials at GlaxoSmithKline under a license from Mitsubishi Tanabe Pharma for the oral treatment of multiple sclerosis (MS) in Europe. GlaxoSmithKline and Tanabe Seiyaku (now Mitsubishi Tanabe Pharma) had been studying the drug candidate for the treatment of asthma, rheumatoid arthritis (RA) and Crohn’s disease
MECHANISMS/EFFECTS
HUMAN:
Similar mechanism of action to natalizumab (α4-integrin blocker), but its faster elimination could improve safety profile

Firategrast
SYNTHESIS
………………….
PATENT
Scheme 1

Scheme 2

In a further aspect the present invention provides for a process for the preparation of compound of formula (II) which comprises coupling the compound of formula (V)

Suitable coupling conditions for the compound of formula (V) and the compound of formula (VI) include those shown in Scheme 2. In a further aspect of the invention there is provided the compound of formula (V):

1H NMR characterisation data for the compound of formula (V) were generated on an isolated and purified batch. 1H-NMR spectra were recorded on a Bruker Avance 400 at 400MHz, using TMS as an internal reference.1H NMR (400 MHz, DMSO-D6) δ ppm 1.17 (t, J=7.09 Hz, 3 H) 2.96 (dd, J=13.82, 9.90 Hz, 1 H) 3.1 1 (dd, J=13.82, 5.26 Hz, 1 H) 4.12 (q, J=7.09 Hz, 2 H) 4.63 (ddd, J=9.78, 7.82, 5.38 Hz, 1 H) 7.15 (t, J=7.95 Hz, 2 H) 7.25 (d, J=8.31 Hz, 2 H) 7.47 – 7.55 (m, 3 H) 9.23 (d, J=7.83 Hz, 1 H).
The present invention provides a process for the preparation of the compound of formula

which process comprises the steps: a) hydrolysis of an ester of formula (I la):

Recrvstallisation of (2S)-2-{r(2,6-difluorophenyl)carbonyllamino)-3-r4′-r(ethyloxy)methyll- 2′,6′-bis(methyloxy)-4-biphenylyllpropanoic acid
(2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)- 4-biphenylyl]propanoic acid (9.38Kg) was charged into a clean reactor, followed by ethyl acetate (46.9L). The solution was heated to 50°C and filtered into the pre-warmed (35°C) crystallizing vessel. A line-wash with ethyl acetate (9.4L) was carried out. The combined ethyl acetate solutions were heated to 50°C, stirred to ensure complete dissolution. Filtered heptane (9.4L) was added maintaining the temperature at 50°C then the solution cooled to 30°C and seeded with (2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4 – [(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4-biphenylyl]propanoic acid (47g) slurried in 1 :9 ethyl acetate:heptane (0.47L). The slurry was aged for 2 hours at 30°C. Filtered heptane (75L) was added over 3 hours. The slurry was then cooled to 0°C over 1 hour. The mixture was aged at 0°C for 1 hour then the solid was filtered off, washed with isopropyl ether (29.6L and dried under vacuum at 50±3°C to give the product (8.55Kg, 91 %). Characterised by having an infrared absorption spectrum with significant absorption bands at about 754, 768, 800, 820, 849, 866, 1006, 1 100, 1 122, 1 157, 1 188, 1225, 1242, 1268, 1292, 1317, 1352, 1417, 1466, 1530, 1580, 1624, 1650, 1662, 171 1 , 1728, 2938, 3302cm“
…………………………………..
PATENT
Example 10: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine ethyl ester.
(1) The product obtained in Example l-(4) (2.1 g) was acylated with 2 , 6-difluorobenzoyl chloride in a similar manner as described in Example 1 -(5) to give N- (2, 6-difluorobenzoyl) – 4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L-phenylalanine ethyl ester (2.75 g) . mp . 70-72 °C; IR (Nujol) 3400, 3263, 1735, 1654, 1624 cm“1; MS (APCI) m/z 500 (M+H) . (2) To a solution of the product obtained above (1.72 g) in DMSO (20 ml) were added Et3N (4.8 ml) and S03«pyridine (5.6 g) successively at room temperature. The whole mixture was stirred at room temperature for 25 minutes. The reaction mixture was poured into ice-water, and then the mixture was extracted with EtOAc. The organic layer was sequentially washed with 5% aqueous HCl, H20 and brine, dried (Na2S04) and then evaporated. The residue was purified by column chromatography (silica gel; eluent: n-hexane/EtOAc 5:1 to 1:1) to yield N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-formylphenyl) -L- phenylalanine ethyl ester (1.54 g) . mp. 114-116°C; IR (Nujol)
3332, 1735, 1695, 1657, 1644, 1623 cm“1; MS (APCI) m/z 498 (M+H) .
(3) The product obtained above (716 mg) was converted into the title compound (428 mg) in a similar manner as described in Example 1- (7) . mp . 87-89°C; IR (Neat+CHC13) 3300, 1739, 1668 cm“ 1; MS (APCI) m/z 528 (M+H) .
Example 11: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl ) -L-phenylalanine methyl ester.
(1) The product obtained in Example 2- (4) (1.00 g) was acylated with 2 , 6-difluorobenzoyl chloride to give N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L- phenylalanine methyl ester (873 mg) in a similar manner as described in Example l-(5). IR (Nujol) 3257, 1743, 1655, 1624 cm“ 1; MS (APCI +Q1MS) m/z 503 (M+NH4) , 486 (M+H) . (2) The product obtained above (860 mg) was converted into the title compound (220 mg) in a similar manner as described in Example 2- (6) and (7).
Example 12: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
The product obtained in Example 10 (200 mg) was hydrolyzed in a similar manner as described in Example 3 to give the title compound (160 mg) . The product obtained in Example 11 (220 mg) was also hydrolyzed in a similar manner as described in Example 3 to give the title compound (167 mg) . mp. 156-158°C; IR (Nujol) 1735, 1655 cm“1; MS (ESI) m/z 498 (M-H) .
…………………….
PATENT
https://www.google.com/patents/WO2003072536A1?cl=en
OUT LINE
phenylalanine derivative of the formula (I) :

wherein X1 is a halogen atom, X2 is a halogen atom, Q is a group of the formula -CH2– or -(CH2)2– and Y is a lower alkyl group, or a pharmaceutically acceptable salt thereof, which has excellent inhibitory activity against α4 integrin-mediated cell adhesion.
Thus, the present invention relates to a process for preparing a compound of the formula (I) :

wherein the symbols are the same as defined above, or a pharmaceutically acceptable salt thereof, comprising : (1) coupling a compound of the formula (VI) :

wherein Z is a leaving group, R1NH is a protected amino group and C02R is a protected carboxyl group with a compound of the formula (V) :

wherein the symbols are the same as defined above, removing the protecting group from the protected amino group, and if necessary, converting the resulting compound into a salt, to yield a compound of the formula (IV) :

wherein the symbols are the same as defined above, or a salt thereof,
(2) condensing the compound (IV) or a salt thereof with a compound of the formula (III) :

wherein the symbols are the same as defined above, a salt or a reactive derivative thereof to yield a compound of the formula (II) :

Ethyl (ocS) – – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4- hydroxybenzene propionate and ethyl (otS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzene propionate are described in J. Med. Chem. , 33: 1620 (1990) and JP-A-7- 157472, respectively. 4-Bromo-3, 5-dimethoxybenzyl alcohol is described in, for example, J. Med. Chem. , 20: 299 (1977), and can also be prepared according to the following process.

Firstly, 4-bromo-3, 5-dihydroxybenzoic acid is methylated to give methyl 4-bromo-3, 5-dimethoxybenzoate, which is then reduced to yield 4-bromo-3, 5-dimethoxy benzyl alcohol. The methylation can be carried out by reacting with dimethyl sulfate in the presence of a base in a suitable solvent (e.g., ethyl acetate). The reduction can be carried out by reacting with an reducing agent (e.g., lithium alminium hydride, sodium borohydride and calcium borohydride) in a suitable solvent (e.g., tetrahydrofuran) .
EXAMPLES
The following Examples are provided to further illustrate the process of preparation according to the present invention. In the following examples, some compounds may be referred to by different compound name depending on the nomenclature, as illustrated below.
Ethyl (αS) -α-amino-4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate
Another name: ethyl (2S) -2-amino-3- [4- (4-ethoxymethyl- 2, 6-dimethoxyphenyl) phenyl]propanoate
Ethyl (αS) – [ [1, 1-dimethylethoxy] carbonyl] amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate
Another name 1: ethyl (2S) -2- [ (t-butoxycarbonyl) – amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) – phenyl]propanoate
Another name 2: Ethyl N- (t-butoxycarbonyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
Ethyl (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionate Another name 1: Ethyl (2S) -2- [ (2, 6- difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6- di ethoxyphenyl) phenyl] propanoate
Another name 2: Ethyl N- [2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
(ocS) – – [ (2, 6-Difluorobenzoyl) amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionic acid
Another name 1: (2S) -2- [ (2, 6-difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) phenyl]propanoic acid
Another name 2: N- [ 2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
EXAMPLE 1 (1) Under nitrogen atmosphere, pyridine (130.3 g) and trifluoromethanesulfonic anhydride (170.4 g) were added dropwise to a solution of ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-hydroxybenzenepropionate
(170.0 g) in dichloromethane (1.7 L) at 10 ° C or below. After stirring for 1 hour at the same temperature, water
(850 ml) was added dropwise to the mixture and the mixture was stirred for 2 hours at the same temperature. The organic layer was washed with 10 % aqueous citric acid solution and aqueous saturated sodium hydrogen carbonate solution, and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy)benzenepropionate (242.5 g) as oil . MS (m/z) : 441 (M+) (2) Under nitrogen atmosphere, to a mixture of ethyl (αS)- – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzenepropionate (66.2g), 4- ethoxymethyl-2, 6-dimethoxyphenylboric acid (54.0 g) , triphenylphosphine (9.83 g) and N-methylpyrrolidone (330 ml) were added palladium acetate (1.68 g) and diisopropylamine (24.9 g ), and the mixture was heated at 90 °C. After stirring for 1 hour at the same temperature, the mixture was cooled and toluene and water were added. The organic layers were washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [[ (1, 1-dimethylethoxy) carbonyl] amino] – 4′ -ethoxymethyl-2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionate (90.1 g) as oil.
The product was dissolved in ethanol (330 ml) , and after addition of p-toluenesulfonic acid monohydrate (28.5 g) , the mixture was stirred for 2 hours at 75 °C. After cooling to room temperature, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS)-α- amino-4′ -ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4- propionate p-toluenesulfonate (63.4 g) .
MS (m/z) : 387 (M+-p-toluenesulfonic acid), M.p. 127-129°C
(3) To a mixture of ethyl (αS) -α-amino-4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate p- toluenesulfonate (29.0 g) , sodium hydrogen carbonate (15. 2 g) , water (290 ml) and ethyl acetate (290 ml) was added dropwise 2, 6-difluorobenzoyl chloride (9. 6 g) at 15 °C or below and the mixture was stirred for 30 minutes at the same temperature. The ethyl acetate layer was washed with saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo. The residue was recrystallized from isopropanol-water to yield ethyl (αS) -oi- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (26.4 g) . MS (m/z) : 527 (M+) , M.p. 87-89°C (4) To a solution of sodium hydroxide (2.9 g) in water- tetrahydrofuran (317 ml-159 ml) was added ethyl (oιS)-α- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate (31.7 g) at 15°C and the mixture was stirred for 4 hours at the same temperature. After neutralizing with IN HC1, the organic solvent was removed in vacuo. The aqueous layer was cooled, the crystalline precipitates were collected by filtration and recrystallized from ethanol-water to yield (αS) -a- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionic acid (28.8 g) . MS (m/z): 499 (M+) , M.p. 154-155°C
EXAMPLE 2 (1) Under nitrogen atmosphere, a mixture of ethyl (oιS)-o:- [[ (1, 1-dimethylethoxy) carbonyl] amino] -4-bromobenzene propanoate (11.17 g) , 4-ethoxymethyl-2, 6- dimethoxyphenylboronic acid (10.80 g ), palladium acetate (0.34 g), triphenylphosphine (1.57 g) , anhydrous potassium carbonate (12.44 g) , iV-methylpyrrolidone (56 ml) and water (11 ml) was stirred for 50 minutes at 80 °C. After completion of the reaction, the mixture was cooled to room temperature and extracted with ethyl acetate and water. The organic layer was washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution, dried over magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure to yield ethyl (αS)-α- [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate (20.4 g) as oil. The product was dissolved in ethanol (100 ml) , and after addition of p-toluenesulfonic acid monohydrate (5.7 g) , the mixture was stirred for 1.5 hours at 75 °C. After cooling, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was suspended in toluene with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS) – -amino-4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate p- toluenesulfonate (13.80 g) . (2) The compound obtained in the above step (1) was treated in the same manner as described in Example 1 (2) to (4) to yield (αS) -a- [ [2 , 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionic acid. The physicochemical data were the same as that obtained in Example 1.
EXAMPLE 3
To a solution of ethyl (αS) -α- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (500 g ) in water (12.6 ml) and dioxane (50 ml) was added hydrochloric acid (12.4 g) and the mixture was stirred for 60 hours at 60 “C. The organic solvent was removed in vacuo and the aqueous layer was cooled. The crystalline precipitates were collected by filtration and recrystallized from ethanol- water to yield (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionic acid (426 mg) . The physicochemical data were the same as that obtained in Example 1.
REFERENCE EXAMPLE 1
(1) To a mixture of 4-bromo-3, 5-dimethoxybenzylalcohol (44.5 g) , triethylammonium benzyl chloride (2.05 g) and 20% aqueous sodium hydroxide solution (288 g) was added diethyl sulfate (41.7 g) under ice-cooling, and the mixture was stirred overnight at 25-30 °C. After stirring for 1 hour at 70 °C, the mixture was cooled and extracted with toluene. The toluene layer was washed with water and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield 4-bromo-3, 5- dimethoxybenzyl ethyl ether (49.5 g) as colorless oil. MS (m/z): 276 (M++2) , 274 (M+)
(2) Under nitrogen atmosphere, to a solution of 4-bromo- 3, 5-dimethoxybenzyl ethyl ether (440.0 g) in tetrahydrofuran (4.0 L) was added dropwise n-butyl lithium (1.6 M n-hexane solution, 1.1 L) at -60°C. After stirring for 15 minutes at the same temperature, trimethyl borate (249.3 g) was added. The temperature of the mixture was gradually elevated, followed by stirring for 1 hour under ice-cooling. To the mixture was added dropwise 10% aqueous sulfuric acid solution (835 g ) . The mixture was extracted with ethyl acetate and the organic layer was washed with water and saturated aqueous NaCl solution. After drying over magnesium sulfate, the solvent was removed in vacuo. The residue was dissolved in isopropyl ether with heating and cooled. The crystalline precipitates were collected by filtration and dried to yield 4-ethyoxymethyl-2, 6- dimetoxyphenylboronic acid (312.9 g) . M.p. 59-61°C
REFERENCE EXAMPLE 2
(1) To a suspension of 4-bromo-3, 5-dihydroxybenzoic acid (95.0 kg) in ethyl acetate (950 L) were added anhydrous potassium carbonate (270.8 kg) and dimethyl sulfate (174.7 kg) . The mixture was heated at 50-80 ‘C for about 4 hours and partitioned by adding water. The organic layer was washed with water and saturated aqueous NaCl solution and concentrated under reduced pressure. The residue was suspended into methanol, stirred under heating and cooled. The crystalline precipitates were collected by filtration and dried to yield methyl 4-bromo-3, 5-dimethoxybenzoate (98.8 kg) as pale yellow crystals. MS (m/z): 277 (M++2) , 275 (M+) , M.p. 120-122°C
(2) To a solution of calcium chloride (46.5 kg) in ethanol (336 L) were added tetrahydrofuran (672 L) and methyl 4- bromo-3, 5-dimethoxybenzoate (96.0 kg) to obtain a suspension. To the suspension was added sodium borohydride
(31.7 kg) by portions at room temperature, and the mixture was stirred for about 9 hours at temperature of room temperature to 45 °C. The reaction mixture was added dropwise to aqueous HC1 solution and stirred for about 16 hours at room temperature. Organic solvent was removed in vacuo, and water (1440 L) was added to the residue and stirred for 1 hour at 50 °C. After cooling, the crystalline precipitates were collected by filtration and dried to yield 4-bromo-3, 5-dimethoxybenzyl alcohol (83.3 kg) as colorless crystals. MS (m/z): 249 (M++2), 247 (M+) , M.p. 100-102°C.
INDUSTRIAL APPLICABILITY The process for preparation of the present invention makes it possible to afford a compound of the formula (I) or a pharmaceutically acceptable salt thereof with high- purity, in a high yield and inexpensively, and, therefore, the process of the present invention is industrially very useful.
References
5. Firategrast
| WO2002018320A2 | 27 Ago 2001 | 7 Mar 2002 | Tanabe Seiyaku Co | INHIBITORS OF α4 MEDIATED CELL ADHESION |
| WO2003072536A1 | 27 Fev 2003 | 4 Set 2003 | Tanabe Seiyaku Co | A process for preparing a phenylalanine derivative and intermediates thereof |
| WO2003072537A2 | 6 Fev 2003 | 4 Set 2003 | Abbott Lab | Selective protein tyrosine phosphatatase inhibitors |
Mitsubishi Tanabe Pharma Corporation


Mitsubishi Tanabe Pharma Corporation
Pharmacological research building

 |
||
| ■Mitsubishi Tanabe Pharma Corporation Pharmacological research building |

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Flupirtine Revisited

Flupirtine, D 9998
2-amino-6-(4-fluoro-benzylamino)- pyridin-3-yl)-carbamic acid ethyl ester, is unique as a non-opioid, non-NSAID, non-steroidal analgesic with a favorable tolerability. It first became available in Europe in 1984, and was sold mainly under the names Katadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor
PHASE 2
MS
- Neuronal potassium channels (7)
- Membrane resting potential (6)
- NMDA receptor channels (indirectly)(14)
- Originally developed by Asta Medica (1) (4)
- Being developed and commercialized to treat fibromyalgia by Synthetic Biologics (1)
Flupirtine

75507-68-5 maleate
56995-20-1 (free base)
56995-20-1 (free base)
| LAUNCHED | 1986 NEUROPATHIC PAIN |
Flupirtine maleate is the INN for 2-amino-3-ethylcarbamato-6- (4-fluoro-benzylamino) maleate, CAS: 75507-68-5, molar mass 420.40 g / mol, molecular formula C1 5 H17FN4O2 • C4H4O4, and corresponds to the structure of formula I.

Flupirtine maleate is used, for example, under the trade name Katadolon® as an analgesic.
56995-20-1
CAS Name: [2-Amino-6-[[(4-fluorophenyl)methyl]amino]-3-pyridinyl]carbamic acid ethyl ester
Additional Names: 2-amino-6-[(p-fluorobenzyl)amino]-3-pyridinecarbamic acid ethyl ester
Trademarks: D-9998
Molecular Formula: C15H17FN4O2
Molecular Weight: 304.32
Percent Composition: C 59.20%, H 5.63%, F 6.24%, N 18.41%, O 10.51%
Properties: Crystals from isopropanol, mp 115-116°. 5% ethanol soln is colorless, turns green on exposure to air for 20 hours. LD50 orally in mice, rats: 617, 1660 mg/kg (Jakovlev).
Melting point: mp 115-116°
Toxicity data: LD50 orally in mice, rats: 617, 1660 mg/kg (Jakovlev)
Derivative Type: Hydrochloride
Molecular Formula: C15H17FN4O2.HCl
Molecular Weight: 340.78
Percent Composition: C 52.87%, H 5.32%, F 5.57%, N 16.44%, O 9.39%, Cl 10.40%
Properties: Crystals from water, mp 214-215°. When prepd industrially contains intensely blue by-product.
Melting point: mp 214-215°
Derivative Type: Maleate
CAS Registry Number: 75507-68-5
Trademarks: Katadolon (AWD)
Molecular Formula: C15H17FN4O2.C4H4O4
Molecular Weight: 420.39
Percent Composition: C 54.28%, H 5.04%, F 4.52%, N 13.33%, O 22.84%
Properties: Colorless crystals from isopropanol, mp 175.5-176°. Formed as mixture of two crystalline forms A and B; mixtures containing 60-90% A are preferred.
Melting point: mp 175.5-176°
Therap-Cat: Analgesic.
TARGET:
Neuronal potassium channels
Membrane resting potential
NMDA receptor channels (indirectly)
STATUS FOR MS:
Phase II
COMMERCIAL:
Originally developed by Asta Medica
Being developed and commercialized to treat fibromyalgia by Synthetic Biologics
Marketed for pain indications in various European countries by Meda
TRADE NAME:
Effirma (US)
Katadolon (Brazil, Germany, Latvia, Estonia, Slovakia, Lithiania, Russian Federation)
SYNONYMS:
EINECS 260-503-8,UNII-MOH3ET196H, Effirma (US), Katadolon (Brazil, Germany, Latvia, Estonia, Slovakia, Lithiania, Russian Federation)
SYSTEMATIC NAME:
Carbamic acid, (2-amino-6-(((4-fluorophenyl)methyl)amino)-3-pyridinyl)-, ethyl ester
PROPERTIES:
Molecular weight: 304
MECHANISMS/EFFECTS
HUMAN:
Stabilizes membrane resting potential by activating neuronal Kv7 potassium channels
Indirectly antagonizes NMDA receptors
Reduces muscle spasticity in humans
Prevents apoptosis and reduced formation of reactive oxygen species by in cultured human retinal pigment epithelial cells

Structures of flupirtine, D13223, and retigabine.
Regulatory and Commercial Status
STATUS FOR MS:
Phase II
HIGHEST STATUS ACHIEVED (FOR ANY CONDITION):
Approved in Europe
ADMINISTRATION:
Oral
COMMERCIAL:
Originally developed by Asta Medica
Being developed and commercialized to treat fibromyalgia by Synthetic Biologics
Marketed for pain indications in various European countries by Meda
Flupirtine is an aminopyridine that functions as a centrally acting non-opioid analgesic. It first became available in Europe in 1984, and is sold mainly under the names Katadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor.[5] Flupirtine is sold by Intas Pharma under the brand name Pruf in India. Like nefopam, it is unique among analgesics in that it is a non-opioid, non-NSAID, non-steroidal centrally acting analgesic. In 2010 the chemically related drug (the difference being that the pyridine group in flupirtine is replaced with a phenyl group) retigabine (INN; ezogabine [USAN]) was approved by the FDA as an anticonvulsant for the treatment of refractory partial-onset seizures in treatment-experienced patients.[6] Retigabine also works by opening the neuronal KCNQ/Kv7 potassium channel, just like flupirtine.
History
Flupirtine was originally developed by Asta Medica, with the synthesis of the compound and the development of the drug described in patents from the 1970s to the 2000s.[7][8][9][10][11][12]
It was approved for the treatment of pain in 1984 in Europe. However, it has never been introduced to the United States market for any indication. In 2008, Adeona Pharmaceuticals, Inc. (now called Synthetic Biologics, Inc.) obtained an option to license issued and patent pending applications relating to flupirtine’s use in the treatment of ophthalmic indications, particularly retinitis pigmentosa.[13]
Mechanism of Action
Flupirtine is a selective neuronal potassium channel opener that also has NMDA receptor antagonist and GABAA receptor modulatory properties.[14]
Uses
Flupirtine is used as an analgesic for acute and chronic pain, in moderate-to-severe cases.[15] Its muscle relaxant properties make it popular for back pain and other orthopedic uses, but it is also used for migraines, in oncology, postoperative care, and gynecology.
Flupirtine has been noted for its neuroprotective properties, and it is being investigated for possible use in Creutzfeldt–Jakob disease, Alzheimer’s disease, and multiple sclerosis.[16][17] It has also been proposed as a possible treatment for Batten disease.[18]
Flupirtine underwent a clinical trial as a treatment for multiple sclerosis[19] and fibromyalgia.[20] Flupirtine showed promise for fibromyalgia due to its different action than the three approved by U.S. FDA drugs: Lyrica (pregabalin), Savella (milnacipran), and Cymbalta (duloxetine).[21] Additionally, there are case reports regarding flupirtine as a treatment for fibromyalgia.[22] Adeona Pharmaceuticals (now called Synthetic Biologics) sub-licensed its patents for using flupirtine for fibromyalgia to Meda AB in May 2010.[21]
Side Effects
The most serious side effect is frequent hepatotoxicity which prompted regulatory agencies to issue several warnings and restrictions.[23][24]
Flupirtine is devoid of negative psychological or motor function effects, or effects on reproductive function.[25][26]
Abuse and Dependence
Although some studies have reported flupirtine has no addictive properties,[27][28] there was suggestion that it may possess some abuse potential and liability.[29] There were at least two registered cases of flupirtine abuse.[30] Drug tolerance does not develop in most cases; however, tolerance may develop in single cases.[30]
Flupirtine is 2-amino-3-carbethoxyamino-6-(p-fluorobenzylamino) pyridine; CAS No: 56995-20-1 , an aminopyridine that functions as a centrally acting non-opioid analgesic. It first became available in Europe in 1984, and is sold mainly under the names atadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor. It is unique as a non- opioid, non-NSAID, non-steroidal analgesic. Flupirtine is used as an analgesic for acute and chronic pain, in moderate to severe cases. Its muscle relaxant properties make it popular for back pain and other orthopaedic uses, but it is also used for migraines, in oncology, postoperative care, and gynaecology. Flupirtine has been noted for its neuro-protective properties, as well as its possible uses for Creutzfeld- Jakob disease, Alzheimer’s disease, and multiple sclerosis are being investigated. It has also been proposed as a possible treatment for Batten disease. Flupirtine also acts as an antioxidant and prevent free radical- mediated structural damage.
US3481943 (hereinafter referred as ‘943) discloses the process for the preparation of flupirtine hydrochloride of formula (T) wherein p- fluorobenzylamine (formula R) is reacted with 2-amino-3-nitro-6- chloropyridine (Q) in n-propanol using potassium carbonate to prepare 2-amino-3-nitro-6-p-fluorobenzylamino-pyridine of formula (S) which is hydrogenated in dioxane using raney nickel at 50 C under a gauge pressure of 30 atmospheres. Solution is filtered off to remove the catalyst and then reacted with chloroformic acid ethyl ester (ethyl chloroformate) while stirring. The product is filtered off and recrystallized from water to give flupirtine hydrochloride salt of formula (T). The process disclosed therein in ‘943 is depicted as given below

Drawbacks associated with the process disclosed in ‘943 are:
1) The yield of 2-amino-3-nitro-6-p-fluorobenzylamino-pyridine of formula S obtained is around 40% only. ‘943 does not disclose the preparation of maleate salt of flupirtine.
2) During the preparation hydrochloride salt of flupirtine on an industrial scale, intensely blue colored by products are formed which are either difficult to remove or can not be removed completely.
3) Use of n-propanol as reaction solvent is expensive. reaction mass thereby hindering the progress of the reaction. Another most probable reason attributed for getting poor yield of 40% in the said process could be masking of hydrochlorides of both the reactants of formulae (Q’) and (R’) (as both reactants are amino compounds and form hydrochlorides) over potassium carbonate making it unavailable for further reaction posing problem towards the completion of reaction thereby adversely affecting the yield.

DE3133519 (US4481205) discloses the preparation of flupirtine maleate of formula (IA), wherein 2-amino-3-nitro-6-chloro-pyridine of formula (S) is prepared by taking 2,6-dichloro-3-nitropyridine of formula (P) (90%, water wet) in isopropanol at 20°-30°C and purging ammonia gas (or dropping liquid ammonia) into the said reaction mixture and then resulting 2-amino-3-nitro-6-chloro-pyridine of formula (Q) is reacted with p-fluorobenzylamine (R) in isopropanol using triethyl amine as a base ; the reaction mixture is refluxed for 6 hours. Thereupon after addition of a large volume of water the compound 2-amino-3-nitro-6-(p- fluorobenzylamino)-pyridine of formula (S) precipitates.
2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine of formula (S) is then hydrogenated in the presence of raney nickel at 5 bar at 60°C to give 2,3- diamino-6-(4-fluorobenzylamino) pyridine using 2-methoxy ethanol as hydrogenating solvent. This is followed by acylation with ethyl chloroformate using triethylamine as a base under inert gas atmosphere to obtain flupirtine base of formula (I). The catalyst is filtered off and filtrate containing dissolved triethyi amine hydrochloride is directly added to solution of maleic acid in isopropanol resulting into formation of crude flupirtine maleate (IA). It also discloses the importance of the exclusion of atmospheric oxygen by an intensive supply of inert gas and closed reactor system to avoid development of troublesome coloured complexes.
The purification of crude flupirtine maleate is carried out by converting crude flupirtine maleate into crude flupirtine base by contacting with ammonia or sodium hydroxide solution. Then the crude flupirtine base is recrystallized from isopropanol and, after contacting with activated carbon/kieselguh’r, it is reacted with a solution of maleic acid in isopropanol to give flupirtine maleate of formula (IA). The reaction scheme of DE3133519 is depicted herein below.

Drawbacks associated with the process disclosed in DE3133519 (US4481205) are:
1) Use of gaseous ammonia or liquid ammonia for the preparation of 2-amino-3-nitro-6-chloro-pyridine of formula (Q) starting from 2, 6- dichloro-3-nitropyridine of formula (P) contributes towards increased level of impurities of formulae X and Y as the gaseous ammonia and liquid ammonia as sources of ammonia are in concentrated forms and it is not easy to control the purging or addition in appropriate quantities and as a consequence it results in the formation of higher amounts of impurities and poor yield of the desired compound.

Another disadvantage of using ammonia gas is that it is classified as a hazardous material and is subject to strict regulations and risk management procedures for transport, storage, and handling. These requirements result in additional costs and may generate local community concerns over transporting and storing hazardous materials. While aqueous ammonia used by the inventors requires minimal special handling, social and regulatory requirements.
2) Preparation of 2-amino-3-nitro-6-(p-fluorobenzylamino)-pyridine of formula (S comprises reaction between 2-amino-3-nitro-6-chloro- pyridine of formula (Q) and p-fluorobenzylamine of formula (R) using isopropanol as solvent and triethyl amine as base. To induce separation of 2-amino-3-nitro-6-(p-fluorobenzylamino)-pyridine of formula (S from the reaction mixture in IP A a large volume of water is required which makes reaction mass highly voluminous therefore, not preferred at industrial scale. 3) Basification of crude flupirtine maleate comprising the process of liberating free flupirtine base using ammonia or sodium hydroxide produces an ammonium or sodium salt which pollutes the water.
4) Use of activated charcoal and kieselgulir during the purification of flupirtine base (that contains three amino groups known for their light and colour sensitive nature) takes prolonged time for filtration through filtering bed thereby exposing to environment producing high coloration.
5) The crude flupirtine maleate remains trapped with triethyl amine hydrochloride.
US59591 15A (hereinafter referred as Ί 15) discloses a process for the preparation of flupirtine maleate (IA) as discussed under DE3133519 (US4481205). It also discloses crystalline form “A” of flupirtine maleate by the use of water soluble alcohols (such as ethanol or isopropanol) during synthesis and/or purification. There are three proposed variants in Ί 15 as shown below: process variant:

A: ANFP (S)→hydrogenation→acylation→crude flupirtine base.
B: crude flupirtine base→maleic acid→crude flupirtine maleate
C-E (as shown in scheme-II): not applicable F: crude maleate→pure maleate.
1 s process variant comprises synthesis of oxygen sensitive crude base in situ in process step A and it was converted by a “very rapid” suction filtration process into an aqueous maleic acid solution from which coloured crude flupirtine maleate (IA) is obtained, which is to be purified by recrystallization from isopropanol-water.
2″ process variant:

A: ANFP (S)→hydrogenation→acylation→crude flupirtine base.
B: flupirtine base→maleic acid→crude flupirtine maleate.
C-F (as shown in scheme-II): Not applicable.
G: without isolation of the crude maleate→pure maleate.
As compared to the process step F in 1st variant, process step G in 2nd variant represents substantially shorter alternative process in which the precipitation of crude flupirtine maleate from the flupirtine base formed in situ in isopropanol is effected by Alteration with suction into an aqueous maleic acid solution at 50-60°C and, after that without isolation of the crude maleate, colourless pure material is obtained.
3rd process variant:

A: ANFP (S)→hydrogenation→acylation→cmde flupirtine base (isolated)
B: pure flupirtine base→maleic acid→pure flupirtine maleate.
Herein, after acylation, the flupirtine base (I) is precipitated preferably in ethanol or water and is purified by recrystallization and than treated with maleic acid to prepare pure flupirtine maleate (IA).
Ί 15 disclose hydrogenation of ANFP (S), acylation and precipitation in water-soluble alcohols, such as ethanol or isopropanol.
1) In 1st process variant “very rapid” suction filtration process is a great limitation at plant scale.
2) 2nd process variant also does not produce colorless pure maleate.
3) In 3 process variant, after acylation, the flupirtine base is precipitated preferably in ethanol or water and is purified by recrystallization and than treated with maleic acid to prepare pure flupirtine maleate salt (IA).

It also discloses that although the treatment of final product with activated carbon and recrystallization is known as a reasonably successful procedure to remove impurities. This approach is reluctantly accepted because of the losses in overall yield as it is applied in the last production step of a drug and particularly in the case of flupirtine, it is not a preferred/desirable procedure as it may result into the formation of colored impurities.
US47851 10A discloses a process for the preparation of 2-amino-3-nitro- 6-fluorobenzylamino pyridine of formula (S) comprising reaction of 2- amino-3-nitro-6-methoxypyridine of formula (T) (1 mole) with 4-fluoro- benzylamine of formula R (2-4 mole) optionally as a mineral acid salt in water at a temperature between 70°C and 150°C; preferably between 90° and 120°C. The said condensation is also performed in autoclave as the temperature is above 100°C.It also discloses the necessity of using basic material suitably as an aqueous solution in case when acid addition salts of 4-fluoro-benzylamine of formula (R) is used to liberate the free base for the reaction. It also discloses subsequent reduction of nitro group of 2-amino-3-nitro-6-methoxypyridine by various modes with preference to catalytic hydrogenation optionally in the presence of carriers selected from barium sulphate, calcium sulphate, magnesium sulphate, sodium sulphate etc.

The drawbacks associated with the process described in US47851 10A are: 1) As per the experimental section of the said process of condensation for the preparation of 2-amino-3-nitro-6-fluorobenzylamino pyridine of formula (S) discloses heating at boiling for ten hours. The temperature would be around 100°C as water is used as solvent. However, inventors of the subject invention disclose herein the same process comprising using 6-chlorpyridine instead of 6-methoxy pyridine and water as solvent’, wherein the reaction is carried out at temperature much below boiling point of water and reaction gets completed in 3 hrs compare to 10 hrs at temperature of boiling water as in’ 1 10. Furthermore, the said reaction disclosed herein in the present invention does not require autoclave. There is no teaching or anticipation on this aspect from Ί 10.
2) Excessive use of 2-4 moles of 4-fluoro-benzylamine of formula ( ) for the preparation of 2-amino-3-nitro-6-fluorobenzylamino pyridine of the formula (S) comprising the reaction of 2-amino-3-nitro-6- methoxypyridine of formula (T)with 4-fluoro-benzylamine of formula (R).Unreacted 4-fluoro-benzylamine is then removed by steam distillation which is not only time and energy consuming but also increase in an extra unit operation.
3) In case when acid addition salts of 4-fluoro-benzylamine are used that requires another additional operation of basification to liberate free base to enable 4-fluoro-benzylamine to be available to react further with 2- amino-3-nitro-6-methoxypyridine forming 2-amino-3-nitro-6- fluorobenzylamino pyridine of the formula (S)
DE 31 33 519 describes a process for the preparation of flupirtine maleate as a mixture of polymorphic forms A and B, wherein A is present in a proportion> 60%. The key reaction steps are the hydrogenation of 2-amino-6- (4-fluorobenzylamino) -3-nitropyridine (Formula II) shown in Figure 1, hereinafter also referred to as ANFP, by means of Ra-Nickel for 2,3-diamino- 6- (4-fluoro-benzylamino) -pyridine (Formula III) and subsequent regioselective acylation with chloroformate for free flupirtine base. By precipitation as maleate to blue contaminants that are incurred in the production of HCl salt, are eliminated. Purification of flupirtine maleate is obtained as maleate by releasing the base from the maleate, treatment with activated carbon and reprecipitation. Despite this lengthy and economically expensive purification strategy traces of colored impurities can be difficult to remove.
In WO 98/47872 a process for the preparation of flupirtine maleate is described, in which, in water-soluble alcohols (IPA) is carried out. There are three proposed variants. Option 1 includes an implementation of ANFP to Ra-nickel in the IPA is directly attached to the acylation and the precipitation of a product by Rohmaleat called “very fast” extraction process in an aqueous solution of maleic acid. It falls on a colored Rohmaleat which is to be purified by recrystallization from isopropanol / water. However, the enactment of this variant in the laboratory showed a colored product. In variant 2 should already be colorless an image obtained by aspiration in 50 to 60 0 C warm maleic Rohmaleat. This also could not be confirmed. According to the third variant, the Flupirtinbase formed after acylation is not converted in situ but precipitated in ethanol or water and recrystallized before further reaction with maleic acid. Even with the procedure referred to in this document is a pure white flupirtine maleate is not readily available.
………………….
PATENT
http://www.google.com.tr/patents/WO2010136113A1?cl=en&hl=tr
Example 3 Preparation of flupirtine maleate
50 g of 2-amino-6- (4-fluorobenzylamino) -3-nitropyridine, 2.5 g of palladium on activated carbon and 267 g of isopropanol were hydrogenated with hydrogen at 4.5 bar and 70 0 C. After completion of the reaction was additionally hydrogenated for 8 hours at 70 0 C. Then 20.2 g of ethyl chloroformate, 24.8 g of triethylamine and 4.96 g of ethyl chloroformate at 20 0 C was added. Thereafter, the reaction mixture was stirred for 1.5 h at 55 0 C. It was then filtered at room temperature. The filtrate was then added to a solution of 35.6 g of maleic acid in 1000 g of water at room temperature slowly. The resulting suspension was stirred for 1 h at room temperature. The precipitate was filtered off and washed with water and isopropanol. Dried filter cake (HPLC purity 91.5%) was dissolved in 828 g of isopropanol / water mixture (mass ratio 5.3: 1), and heated to 70 0 C. The resulting clear solution was cooled to room temperature and stirred at room temperature. The precipitate was filtered off and washed with isopropanol / water mixture. The filter cake was dried at 50 0 C. 43 g flupirtine maleate (HPLC purity 97.8%) was obtained as a white-gray solid. The yield was 55%.
………………
PATENT
http://www.google.com/patents/WO2013080215A1?cl=en
The invention relates to an improved process for the preparation of flupirtine of formula (I) and its pharmaceutically acceptable salts, particularly flupirtine maleate of formula (IA) preferably pure crystal modification A of flupirtine maleate.
A process for the preparation of the compound of formula (I)

and pharmaceutically acceptable acid addition salts thereof comprising the steps of:
(a) contacting 2, 6-dichloro-3-nitro pyridine of formula (P) with aqueous ammonia solution in a compatible solvent to produce 2-amino-3-nitro-6- chloro-pyridine of formula (Q);

(b) contacting said compound of formula (Q) with p-fluorobenzylamine taking water as a solvent in presence of a base to produce 2-amino-3- nitro-6-p-fluorobenzylamino-pyridine of formula (S);

(c) reducing nitro group of 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine of formula (S) in a solvent base combination as solvent system in the presence of a catalyst;

(d) contacting 2,3-diamino-6-p-fluorobenzyl amino pyridine produced in step c with an ethyl chloroformate in presence of a base optionally insitu without isolation to produce flupritine base of formula (I);

(e) contacting the said flupritine base of formula (I) with acid solution to produce corresponding acid addition salt.



Scheme (I):


EXAMPLE 1 : Preparation of 2-amino-3-nitro-6-chloro-pyridine.
A solution of 100 gm. 2, 6-dichloro-3-nitro-pyridine in 800 ml isopropyl alcohol is taken in round bottom flask. 300 ml of aqueous ammonia solution (20-25%) is added at 20-25°C. The reaction mass is stirred for 20-24 hours at 20-25°C. After completion of the reaction
The solid is filtered and washed with 100 ml isopropyl alcohol then dried to obtain 70-75 gm 2-amino-3-nitro-6-chloro-pyridine.
EXAMPLE 2: Preparation of 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine.
100 gm of 2-amino-3-nitro-6-chloro-pyridine is taken in 800 ml of water. 90 gm of p-fluorobenzylamine is added dropwise into the reaction mixture at 20-25°C. Then 87 gm triethylamine is also added dropwise into the reaction mixture at 20-25°C. After complete addition, the reaction mass is stirred at 40-45°C for half an hour again the reaction mass is heated to 80-85°C and stirred at this temperature for 3-4 hours. After completion of the reaction, the reaction mass is cooled to 20-25°C and stirred at this temperature for 2-3 hours and then stirred at 15-20°C for 3-4 hours. The solid mass is filtered and then washed with 200 ml of water and 100 ml isopropyl alcohol and then dried in air oven till constant weight to get 140-150 gm. of 2-amino-3-nitro-6-p- fluorobenzylamino-py ridine .
EXAMPLE 3: Preparation of flupirtine maleate.
In an autoclave, 100 gm. 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine is taken in 500 ml. 1, 4-dioxane and 20 ml aqueous ammonia solution. 10 gm of raney nickel is added under nitrogen atmosphere and hydrogenated at 75-80°C for 2-3 hours under 4-5 kg pressure. After completion of the reaction, the reaction mass is cooled and filtered at 40- 45°Cthen in filtrate 45 ml of ethyl chloroformate is added slowly at 5- 10°C. The temperature is raised to 25-30°C and 80 ml triethyl amine is added under nitrogen atmosphere. The reaction mass, is heated at 55- 60°C under stirring for 3-4 hours. After completion of the reaction, the reaction mass is distilled up to 70-80% under vacuum. This concentrated reaction mass is added into aqueous solution of maleic acid (72 gm in 2000 ml DM water at 65-70°C and maintained at 65-70°C for 2 hours under nitrogen to get crude Flupirtine Maleate as a solid. The reaction mass is cooled to 25-30°C in 5-6 hours and maintained at this temperature for next 2-3 hours then filtered. The wet cake is washed with 200 ml water and dried to get 145 gm of flupirtine maleate.
EXAMPLE 4: Preparation of pure flupirtine maleate crystalline modification A.
1 15 gm crude Flupirtine maleate obtained in example 3 is taken in 1 150 ml methanol and 58 ml water. This mixture is heated to reflux and 58 ml water is added slowly to get a clear solution and refluxed for about half an hour. The reaction mixture is cooled slowly to 60°C and seeded with crystals of modification A. Then it is cooled slowly to 20-25°C and maintained at this temperature for 2 hours. The crystalline mass is filtered and washed with 100 ml chilled methanol and dried to give 92 gm. flupirtine maleate crystalline modification A.
………………….
PATENT
http://www.google.com.tr/patents/WO1998047872A1?cl=en
1. Example
Preparation of flupirtine maleate
75 g (0.286 mol) ANFP be in a suspension of 12.5 g of Raney nickel in 400 ml of isopropanol was hydrogenated at 65 ° C and 5 bar hydrogen pressure. After hydrogenation, the solution is then mixed with 26.4 ml of ethyl chloroformate and 50.6 ml of triethylamine. After adding a further 6.3 ml of ethyl chloroformate the reaction solution is stirred at 60 ° C. for 1 hour. Then sucks the hot solution with stirring in a 50 – 60 ° C heated solution of 53.3 g of maleic acid in 1, 5 IH 2 O and washed the catalyst with little isopropanol.
The flupirtine maleate is precipitated in colorless crystal suspension is cooled with further stirring at 20 ° C and maintained at this temperature for 20 minutes. It is suctioned off, washed with 500 ml of water and dried flupirtine maleate in vacuo at 35 ° C.
Yield: 107.55 g (89.6% of theory, based on ANFP.) Example 2
Preparation of flupirtine maleate
18.5 g (0.07 mol) ANFP be analogous to Example 1 in a suspension of 2.0 g of Raney nickel in 140 ml of ethanol 60 – 70 ° C and 5 bar hydrogen pressure After hydrogenation, the further reaction takes place at 40 – 50 ° C with 9.3 g of ethyl chloroformate (0.86 mol) of triethylamine and 9.2 g (0.91 mol) The separated from the catalyst reaction solution is added with stirring to 540 ml of water After 2 hours of stirring at room temperature suctioned the failed base off and washed with water and isopropanol and crystallized in the 3.7-fold amount of isopropanol to yield 18.4 g (86.0% of theory)
The precipitation and modification of pure flupirtine maleate is carried out according to the Examples 7 and 8
………………….
PATENT
CN104086481 (A) – Synthesis method of flupirtine maleate
http://worldwide.espacenet.com/publicationDetails/biblio?CC=CN&NR=104086481A&KC=A&FT=D
The invention provides a synthesis method of flupirtine maleate. Recrystallization by use of methanol is carried out in the refining step of the crude product of the flupirtine maleate so that the product is white in appearance and high in purity, and the crystal form of the product is pure A crystal and same as the crystal form of the commercial products. The optimal reaction solvent, reaction time and reaction temperature are explored and found out by use of a simplified process flow, and a method for preparing the flupirtine maleate in the pure A crystal form, which is high in yield, low in cost and simple to operate, uses easily available raw materials and is applicable to the industrial production is found.
………………
PATENT
http://www.google.com/patents/CN103333103A?cl=en
The preparation of a comprehensive literature about the ratio of maleic acid fluoride Jie Ting to 2_-amino-3-nitro-6-chloro-Jie ratio 唳 as a starting material, by condensation, reduction, acylation, salt and other processes for The most common route, however, due to the reduction, acylation, salt formation method of a three-step operation is different, not only the yield of the synthesis varies widely, and also on the flupirtine maleate product quality. This is mainly because of the intermediate 2,3-diamino-6-fluoro-benzyl amino pyridine and flupirtine multi-aminopyridine derivative, is easy to oxidative deterioration. So the use of continuous operation, not only simple steps, and can improve product quality and yield.
Chinese patent CN102241626 reported to 2,6_ dichloro _3_ nitropyridine as raw material by selective ammonia solution to give 2-amino-3-nitro-6-chloro-approved Li, then with amine fluoride Festival to afford a yellow solid 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine. After vacuum drying, the use of hydrogenation, and then under nitrogen and ethyl chloroformate acylation catalyst is filtered off and then a salt with maleic acid to give a pale green crude product yield was about 37% (2-amino-3-nitro-6-chloro-pyridin-meter).
Patent No. CN102838534 reported 2-amino-3-nitro-6-chloro-pyridine as starting material, the use of sub-step processing method, in a first reactor, and a condensation-fluorobenzyl amine, and dried in vacuo to give the intermediate 2-amino-3-nitro-6-p-fluorobenzyl-aminopyridine, in a second reactor to Raney nickel as the catalyst, the catalytic hydrogenation of hydrazine hydrate, after filtration the solvent was evaporated to give the intermediate 2,3-solid – diamino-6-p-fluorobenzyl-aminopyridine, in a third reactor with ethyl chloroformate acylated intermediate distillation under reduced pressure to give solid form of flupirtine with an aqueous solution of a salt of maleic acid, after purification, the total Yield 25% ~ 30%.
Patent W02012004391 discloses a method for preparing a high yield of flupirtine maleate method. In 2_-amino-3-nitro-6-chloro-fluoro-section batch Li and amines as raw material for condensation to give 2-amino-3-nitro-6-fluoro-section based on the amino pyridine granted, then using high-pressure hydrogenation the reduction, acylation step in a high pressure hydrogenation reactor concentrated completed, after the catalyst was filtered off and then the salt, the crude yield of greater than 70%. The preparation method using high-pressure hydrogenation apparatus, there are security risks, and takes too long, is not suitable for industrial production.
Patent No. CN102260209 discloses a 2_ amino _3_ _6_ fluorobenzyl nitro-pyridine as starting material, the reduction, acylation and salt-forming step of the continuous operation, the synthetic yield was improved to 58% so, no mention of product purity. Since the acylation step taken ethyl chloroformate, while an organic base is added, so that an increase in a side reaction, the product yield decreases; the same time, 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine as the raw material, the production cost high.
In the present invention, we consider the key intermediate 2,3-diamino-6-p-fluorobenzyl-aminopyridine and chemical properties of flupirtine, condensation, reduction, acylation, salt formation reaction is concentrated to the same conventional the reactor is completed, each step without intermediate separation, simplifying the process route and operations, improve efficiency, reduce costs, improve the overall yield of the crude by 40 percent following the step by step operation for more than 70% crude purity of more than 99% suitable for industrial scale production.

Example 4:
The 4Kg2_ amino-3-nitro-6-chloropyridine, 4.5Kg triethylamine, 40L of isopropanol into the reactor, stirred and heated to reflux for turn; the 4.4Kg of benzylamine was added to the fluorine reactor, the reaction under reflux conditions for 3 hours. After heating was stopped, the reaction solution was added to 40L of purified water, a lot of yellow solid was precipitated was filtered and the resulting wet product remains in the reaction vessel. To the reaction kettle was added 1.8Kg Raney nickel, 40L of isopropanol, stirred and heated to reflux for open, 7Kg80% hydrazine hydrate was added dropwise, the reaction was refluxed for 3 hours, after completion of the reaction down to room temperature in a nitrogen atmosphere, was added rapidly 3.6Kg chloro carboxylic acid ethyl ester, the reaction at room temperature for 3 hours. 3Kg of triethylamine was added, free 2 hours, filtered and the filtrate was added to 5Kg / 100L of maleic acid in isopropanol, cooling crystallization to give an off-white solid, 50 ° C blast drying, weight 7.8Kg, the yield was 80.5 %, purity 99.6%.
A sub-step treatment process research and data [0034] Comparative Example
The method according to Chinese patent CN102838534 disclosed flupirtine maleate was prepared, and a number of specific steps
………….
PATENT
FIG. 1 is flupirtine maleate 1H NMR.
[0021] FIG. 2 is flupirtine maleate A crystal X-ray diffraction pattern



Example 3
2-Amino-3-nitro-6-chloropyridine 246g, and 254g of triethylamine were added to 800ml of ethanol-necked flask and stirred under heating to reflux, fluorine was slowly added dropwise benzylamine 80g, reaction of 6 hours, the reaction was completed After the dropwise addition of purified water 500ml, cooled slowly with stirring to room temperature, filtered, dried to give 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine.
[0033] The ferric chloride hexahydrate was dissolved in purified water 41g 200ml, adding activated charcoal 20g, heated to 50 ° C, a saturated solution of sodium hydroxide was added 45g (24g of sodium hydroxide dissolved in 21ml water), 60 ° C with stirring I hours, cooled to room temperature, filtered, and dried to give ferric hydroxide / activated carbon catalyst.
[0034] A mixture of 2-amino-3-nitro-6-p-fluorobenzyl-aminopyridine 104.Sg, ferric hydroxide / activated carbon catalyst was added to 20g 2L reaction flask was added 95% ethanol 1200ml, heated with stirring to 90 ° C. Insulation 60% hydrazine hydrate was added dropwise 250g. Drops Bi insulation response to 3h. Completion of the reaction, the reaction solution is filtered hot with concentrated hydrochloric acid to 240ml and 95% ethanol IOOOml reaction flask. (TlO ° C crystallization I h, filtered, dried to give 2,3-amino-6-fluoro-benzyl-aminopyridine on
Hydrochloride.
[0035] A mixture of 2,3-diamino-6-p-fluoro-benzylamino-pyridine hydrochloride 132g, 800ml of isopropanol was added to a 2L reaction flask, the temperature control to 28 至 30 ° C, was slowly added dropwise acetic acid ester 39g. Stirred for 0.5 hours, was slowly added dropwise triethylamine 70g, after stirring for 0.5 hours, complement ethyl chloroformate 5g, stirred for 15 minutes, additional triethylamine remaining 10g. Continue stirring for I hour. The reaction solution was concentrated under reduced pressure to about 800ml of distillate was distilled out. The remaining reaction solution was poured into an aqueous solution of maleic acid with a good (39g of maleic acid was dissolved in purified water IlOOml), stirred for 30 minutes at room temperature, (T5 ° C was stirred for 5 ~ 8 hours, filtered, dried to give the maleic acid flupirtine crude.
[0036] The crude flupirtine maleate product 100g, 2000ml of ethanol into the reaction flask and heated to 70~80 ° C, was added 5g of activated carbon and dissolved, and incubated I hour, filtered hot, O~5 ° C CRYSTALLIZATION 3 hours, filtered and dried to give crude I. The crude product I 90g, 450ml of ethanol into the reaction flask and heated 20h, and then slowly cooled to room temperature, O~5 ° C for 2 hours, filtered, and dried to give crystal form A of flupirtine maleate product.
…………………..
PATENT
http://www.google.com/patents/CN102838534A?cl=en
flupirtine maleate is a non-opioid analgesic effects on the central nervous system drugs, which is a selective neuronal potassium channel opener (Selective Neuronal Potassium Channel Opener, SNEPCO), has analgesic, muscle relaxant and neuroprotective triple effect. Acute pain treatment is mainly used for various types of moderate, such as surgery, trauma-induced pain and headache / migraine and abdominal spasms.
flupirtine maleate English name: Flupirtine Maleate, chemical name: 2_ amino-6 – [((4-fluorophenyl) methyl) amino] pyridine-3-carboxylic acid ethyl ester maleic salt; Chemical Abstracts (CAS) number = 75507-68-5; formula = C15H17FN4O2 · C4H4O4; molecular weight: 420.39; its structural formula is:

From a structural perspective, flupirtine maleate molecular compounds, the derivatives of benzene and pyridine derivatives synthetically produced flupirtine, flupirtine and then forming an organic salt with maleic acid. Comprehensive literature, synthetic routes flupirtine maleate there are two major, now its main synthetic steps described below.
Route 1 (W0 98 / 47872Α1): The route to 2,6_ dichloro _3_ nitropyridine as raw material substitution, ammoniated, high-pressure hydrogenation, acylation, a process salt, refined and so on. The reaction formula is as follows:

Route 2 (US5959115A) to 2_ amino _3_ nitro _6_ methoxypyrido as the starting material, and on fluorobenzylamine substitution reaction to produce 2-amino-3-nitro–6 – fluorobenzyl amine of pyridine, the yield was 95.2%, and the high-pressure hydrogenation, the catalyst was filtered off, and then the occurrence of an acylation reaction with ethyl chloroformate to give the hydrochloride salt of flupirtine, three-step total yield of 53.3%. The reaction formula is as follows:

Route 1 starting material is different, but relatively speaking, the route I easily controlled reaction conditions, and 2-amino-3-nitro-6-chloro-pyridine is a common chemical raw materials, easy to buy on the market, This can shorten the reaction route. Route 2 two-step reaction process route is short, but the starting 2-amino-3-nitro-6-methoxy-approved Li expensive, hydrogenation, acidification two steps yield only 56.0%.
Chinese Patent Application Publication No. CN102241626A are disclosed and CN102260209A flupirtine maleate preparation method, but the application of these two methods for the preparation of a laboratory scale, for the industrial mass production were not optimized.
The method for purifying of flupirtine maleate in the final product are as follows:
650C ± 5 ° C under the flupirtine maleate crude and ethanol mass ratio of 1: 30-40 mixed, crude completely dissolved, then add 680g of activated carbon and stirred for 15–30 minutes, and hot filtration, the filtrate, stirring down to room temperature, and then cooled to 0 ° C crystallization 5–10 hours, filtered and the filter cake to take the filter cake can be dried.
BELOW AS FREE BASE
| Ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate | |
| CAS No.: | 56995-20-1 |
|---|---|
| Synonyms: |
|
| Formula: | C15H17FN4O2 |
| Exact Mass: | 304.13400 |
1H NMR INTERPRETATIONS/PREDICTIONS
![ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate NMR spectra analysis, Chemical CAS NO. 56995-20-1 NMR spectral analysis, ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-18/000/006/679/56995-20-1-1h.png)
13C NMR INTERPRETATIONS/PREDICTIONS
![ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate NMR spectra analysis, Chemical CAS NO. 56995-20-1 NMR spectral analysis, ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-18/000/006/679/56995-20-1-13c.png)
…….
PAPER
Helvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
 AND
AND  GIVES PRODUCT
GIVES PRODUCT
 ALSO AN INTHelvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
ALSO AN INTHelvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
…………..
HPLC
Instrumentation An HPLC system (Agilent HPLC Model-1200) equipped with a C18 (Agilent BDS, 250 mm x 4.6 mm, 5µ) column, binary pump, rheodyne loop injector with 20 μL, and a photodiode array detector was used. The software used for HPLC data acquisition was EZChrome Elite. A flash chromatograph equipped with silica gel as the column material, and VWD-UV detection (using the software Analogix IF 280 V 5.10) was used for the isolation and purification of degradation products. 1 H-NMR was recorded on the Varian Unity Inova at 400 MHz (using TMS as internal standard and DMSO-d6 as solvent), 13C-NMR (Mercury Plus at (abundance 100 MHz), using DMSO-d6 as solvent), and mass spectral studies were performed on the API 3000 ABPCIES instrument.
Method Development and Optimization of the Chromatographic Conditions In preliminary experiments, the drug was subjected to the reversed-phase mode using a C18 column (Agilent, 250 x 4.6 mm, 5µ) and mobile phases consisting of water (pH 3.0 adjusted with orthophosphoric acid) and methanol by varying the % aqueous phase from 10% to 30%. The drug was retained on the column, but the peak shape was not good. It was noted that increasing the % aqueous phase in the mobile phase composition increases the retention time of flupiritine maleate. Based on the suitable retention time for SIAM, the 20% aqueous phase was optimized. To reduce the tailing effect, 0.2% triethylamine (TEA) was added and the pH was adjusted to 3.0 with orthophosphoric acid and the corresponding retention of FLU was 10.3 ± 0.3 min. Finally, the mobile phase of 0.2% v/v TEA (pH-adjusted to 3.0 with OPA) and methanol in the ratio of 20:80% v/v was optimized. The flow rate was 1.0 mLmin−1. The injection volume was 20 µL and the PDA detection wavelength was at 254 nm. The chromatogram obtained in the optimized condition is shown in Fig. 2. It was observed that eight degradation products were formed with retention times 3.9 ± 0.2 min (D1), 4.8 ± 0.2 min (D2), 6.4 ± 0.1 min (D3), 6.8 ± 0.2 min (D4), 8.2 ± 0.2 min(D5), 12.0 ± 0.2 min (D6), 14.1 ± 0.1 min (D7), and 15.0 ± 0.1 min (D8), respectively. The chromatographic resolution among all of the peaks was more than 2. The % degradation was about 5–30% depending on stress conditions.
………………..
paper
J Pharm Biomed Anal. 2014 Mar;90:27-34. doi: 10.1016/j.jpba.2013.11.015. Epub 2013 Nov 27.
Flupirtine maleate is a centrally acting, non-opioid, nonsteroidal antiinflammatory analgesic. During the manufacturing of flupirtine maleate, two unknown impurities present in the laboratory batches in the range of 0.05-1.0% along with the known impurities in HPLC analysis. These unknown impurities were obtained from the enriched mother liquor by column chromatography. Based on the complete spectral analysis (MS, (1)H, (13)C, 2D NMR and IR) and knowledge of the synthetic scheme of flupirtine maleate, these two new impurities were designated as diethyl 5-((4-fluorobenzyl)amino)-2-oxo-1H-imidazo[4,5-b]pyridine-1,3(2H)-dicarboxylate (impurity-I) and diethyl(6-((4-fluorobenzyl)amino)pyridine-2,3-diyl)dicarbamate (impurity-II). Impurity isolation, identification, structure elucidation and the formation of impurities were also discussed. Preparation and structure elucidation of impurity-III were also first reported in this paper.
…………………
journal of pharmaceutical and biomedical analysis, 90, 2014, 27-34
References: Substituted pyridine with central analgesic properties. Prepn: K. Thiele, W. von Bebenburg, ZA 6902364(1970 to Degussa); W. von Bebenburg et al., Chem. Ztg. 103, 387 (1979); eidem, ibid. 105, 217 (1981).
Prepn of maleate: W. von Bebenburg, S. Pauluhn, BE 890331; eidem, US 4481205 (1980, 1984 both to Degussa).
Comparison of pharmacology with other analgesics: V. Jakovlev et al., Arzneim.-Forsch. 35, 30 (1985).
Pharmacokinetic studies: K. Obermeier et al., ibid. 60.
Effect on driving ability: B. Biehl, ibid. 77.
Clinical trials in treatment of cancer pain: W. Scheef, D. Wolf-Gruber, ibid. 75.
Efficacy in treatment of pain after hysterectomy: R. A. Moore et al., Br. J. Anaesth. 55, 429 (1983).
Symposium on pharmacology and clinical efficacy: Postgrad. Med. J. 63, Suppl. 3, 1-113 (1987).
References
1
- Abrams, SML; L.R.I. Baker, P. Crome, A.S.T. White, A. Johnston, S.I. Ankier, S.J. Warrington, P. Turner, G. Niebch (1988). “Pharmacokinetics of flupirtine in elderly volunteers and in patients with moderate renal impairment”. The Fellowship of Postgraduate Medicine 64 (751): 361–363. doi:10.1136/pgmj.64.751.361. PMC 2428663. PMID 3200777.
- 2
- Narang, PK; Tourville JF; Chatterji DC; Gallelli JF (1984). “Quantitation of flupirtine and its active acetylated metabolite by reversed-phase high-performance liquid chromatography using fluorometric detection”. Journal of Chromatography 305 (1): 135–143. doi:10.1016/S0378-4347(00)83321-6. PMID 6707137.
- 3
- Methling, K; Reszka P; Lalk M; Vrana O; Scheuch E; Siegmund W; Terhaag B; Bednarski PJ (2008). “Investigation of the in Vitro Metabolism of the Analgesic Flupirtine”. Drug Metabolism and Disposition 37: 479–493. doi:10.1124/dmd.108.024364.
- 4
- Blackburn-Munro, G; W. Dalby-Brown; N. R. Mirza; J. D. Mikkelsen; R. E. Blackburn-Munro (2005). “Retigabine: Chemical Synthesis to Clinical Application”. CNS Drug Reviews 11 (1): 1–20. doi:10.1111/j.1527-3458.2005.tb00033.x. PMID 15867950.
- 5
- Flupirtine Drugs.com. Accessed 20 September 2011.
- 6
- “POTIGA® (ezogabine) Tablets, CV. Full Prescribing Information”. GlaxoSmithKline and Valeant Pharmaceuticals. Revised: September, 2013. Initial U.S. Approval: 2011. Retrieved 2 June 2014. Check date values in:
|date=(help) - 7
- http://www.freepatentsonline.com/5721258.html Primary and secondary neuroprotective effect of flupirtine in neurodegenerative diseases The synthesis of flupirtine and its pharmaceutically acceptable salts is described in EP 160 865 and 199 951. EP0199951 December, 1986 Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino) pyridine.
- 8
- http://patent.ipexl.com/EP/EP0199951.html#reference Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of EPO Patent EP0199951 1986 German.
- 9
- http://www.patentfish.com/2-amino-3-carbethoxyamino-6-4-fluoro-benzylamino Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino) pyridine EP 0199951 B1 1986. English.
- 10
- http://patent.ipexl.com/US/4481205.html 2-Amino-3-carbethoxyamino-6-(p-fluoro-benzylamino)-pyridine-maleate United States Patent 4481205. 1981
- 11
- http://www.freepatentsonline.com/3998834.html Novel N-(4-piperidinyl)-N-phenylamides and -carbamates having very potent analgesic activity, methods of preparing same and useful intermediates therefor. Patent 3998834. 1976.
- 12
- http://www.faqs.org/patents/app/20090306150 CARBOXYLIC ACID SALTS OF 2-AMINO-3-CARBETHOXYAMINO-6-(4-FLUORO-BENZYLAMINO)-PYRIDINE patent 20090306150. 2009. Flupirtine is commonly used in the form of pharmaceutically acceptable acid addition salts. Commercially, flupirtine is available as its maleate addition salt under the trademark Katadolon®. There are two known polymorphs of flupirtine maleate, designated in the art as flupirtine maleate A and B. European patentEP 0 977 736 discloses pure flupirtine maleate crystalline form A and a process for its preparation. Flupirtine and mixtures of flupirtine maleate polymorphs A and B can be synthesised according to EP 0 199 951.
- 13
- “Adeona Pharmaceuticals and National Neurovision Research Institute (NNRI) Collaborate to Test Flupirtine for Retinitis Pigmentosa”. Ann Arbor, MI and Owings Mills, MD: Synthetic Biologics, Inc. December 2, 2008. Retrieved 2 June 2014.
- 14
- Kornhuber, J.; Bleich, S.; Wiltfang, J.; Maler, M.; Parsons, C. G. (1999). “Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication”. Journal of neural transmission (Vienna, Austria : 1996) 106 (9–10): 857–867. doi:10.1007/s007020050206. PMID 10599868.
- 15
- McMahon, FG; Arndt WF Jr, Newton JJ, Montgomery PA, Perhach JL. (1987). “Clinical experience with flupirtine in the U.S”. Postgraduate Medical Journal 63 (3): 81–85. PMID 3328854.
- 16
- Klawe, C; Maschke, M (2009). “Flupirtine: pharmacology and clinical applications of a nonopioid analgesic and potentially neuroprotective compound”. Expert opinion on pharmacotherapy 10 (9): 1495–500. doi:10.1517/14656560902988528. PMID 19505216.
- 17
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR (September 1988). “Pharmacological mechanisms of action of flupirtine: a novel, centrally acting, nonopioid analgesic evaluated by its discriminative effects in the rat”. J. Pharmacol. Exp. Ther. 246 (3): 1067–74. PMID 2901483.
- 18
- Dhar S, Bitting RL, Rylova SN et al. (April 2002). “Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons”. Ann. Neurol. 51 (4): 448–66. doi:10.1002/ana.10143. PMID 11921051.
- 19
- Flupirtine as Oral Treatment in Multiple Sclerosis (FLORIMS) Clinical Trials.gov Accessed 20 September 2011.
- 20
- Pipex Pharmaceuticals (PPXP)’ Oral Flupirtine Receives IND With FDA for Phase II Clinical Trial for Fibromyalgia 4/21/2008
- 21
- “Partnered Program. Effirma™ for Fibromyalgia”. Synthetic Biologics, Inc. Retrieved 2 June 2014.
- 22
- Stoll AL, Belmont MA. (2000) “Fibromyalgia Symptoms Relieved by Flupirtine: An Open-Label Case Series” Psychosomatics 41:371-372. Accessed 20 September 2011.
- 23
- EMA information about flupirtine
- 24
- article in Deutsches Ärzteblatt
- 25
- Singal, Rikki; Parveen Gupta; Nidhi Jain; Samita Gupta (2012). “Role of Flupirtine in the Treatment of Pain – Chemistry and its Effects”. Mædica — a Journal of Clinical Medicine 7 (2): 163–166. PMID 23401726.
- 26
- “DRUGDEX® Evaluations – Flupirtine”. Retrieved 24 March 2013.
- 27
- Preston, KL; Funderburk, FR; Liebson, IA; Bigelow, GE (Mar 1991). “Evaluation of the Abuse Potential of the Novel Analgesic Flupirtine Maleate”. Drug and Alcohol Dependence 27 (2): 101–113. doi:10.1016/0376-8716(91)90027-v. PMID 2055157.
- 28
- Sofia, RD; Diamantis, W; Gordon, R (1987). “Abuse Potential and Physical Dependence Liability Studies with Flupirtine Maleate in Laboratory Animals”. Postgraduate Medical Journal. 63 Suppl 3: 35–40. PMID 3447127.
- 29
- Gahr, M; Freudenmann, RW; Connemann, BJ; Hiemke, C; Schönfeldt–Lecuona, C (Dec 2013). “Abuse Liability of Flupirtine Revisited: Implications of Spontaneous Reports of Adverse Drug Reactions”. Journal of Clinical Pharmacology 53 (12): 1328–1333. doi:10.1002/jcph.164. PMID 24037995.
- 30
Stoessel, C; Heberlein, A; Hillemacher, T; Bleich, S; Kornhuber, J (Aug 16, 2010). “Positive Reinforcing Effects of Flupirtine—Two Case Reports”. Progress in Neuro-psychopharmacology & Biological Psychiatry 34 (6): 1120–1121. doi:10.1016/j.pnpbp.2010.03.031. PMID 20362025. Retrieved 2 June 2014.
References
2. Flupirtine
Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| ethyl {2-amino-6-[(4-fluorobenzyl)amino]pyridin-3-yl}carbamate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Pharmacokinetic data | |
| Bioavailability | 90% (oral), 70% (rectal)[1] |
| Metabolism | Hepatic to 2-amino-3-acetylamino-6-(para-fluorobenzylamino) pyridine (which has 20-30% the analgesic potential of its parent compound), para-fluorohippuric acid[2] and a mercapturic acid metabolite, presumably formed from a glutathione adduct[3] |
| Half-life | 6.5 hrs (average), 11.2-16.8 hrs (average 14 hrs) (elderly), 8.7-10.9 hrs (average 9.8 hrs) (in those with moderate-level renal impairment)[1] |
| Excretion | 72% of flupirtine and its metabolites appear in urine and 18% appear in feces[4] |
| Identifiers | |
| 56995-20-1 |
|
| N02BG07 | |
| PubChem | CID 53276 |
| IUPHAR ligand | 2598 |
| ChemSpider | 48119 |
| UNII | MOH3ET196H |
| KEGG | D07978 |
| ChEMBL | CHEMBL255044 |
| Chemical data | |
| Formula | C15H17FN4O2 |
| 304.32 g/mol | |
Uprosertib (GSK-2141795)


Uprosertib (GSK-2141795)
GSK 2141795C
N-[(1S)-1-(aminomethyl)-2-(3,4-difluorophenyl)ethyl]-5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)furan-2-carboxamide
N-[(2S)-1-amino-3-(3,4-difluorophenyl)propan-2-yl]-5-chloro-4-(4-chloro-2-methylpyrazol-3-yl)furan-2-carboxamide
2-Furancarboxamide, N-[(1S)-2-amino-1-[(3,4-difluorophenyl)methyl]ethyl]-5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-
Λ/-{(1 S)-2-amino-1-r(3,4-difluorophenyl)methyllethyl}-5-chloro-4-(4- chloro-1-methyl-1H-pyrazol-5-yl)-2-furancarboxamide
N-{(1S)-2-amino-1-[(3,4-difluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1-methyl-1Hpyrazol-5-yl)-2-furancarboxamide.
Cas 1047634-65-0 (GSK-2141795); BASE
CAS 1047635-80-2 (GSK-2141795 HCl salt)
Synonym: GSK-2141795; GSK2141795; GSK 2141795; GSK795; GSK-795; GSK 795. Uprosertib. UNII ZXM835LQ5E
IUPAC/Chemical name:
N-((S)-1-amino-3-(3,4-difluorophenyl)propan-2-yl)-5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)furan-2-carboxamide
C18H16Cl2F2N4O2
Exact Mass: 428.06184
Molecular Weight: 429.25
Elemental Analysis: C, 50.37; H, 3.76; Cl, 16.52; F, 8.85; N, 13.05; O, 7.45
Mechanims of Action:Akt inhibitor
Indication:Cancer Treatment
Drug Company:GlaxoSmithKline
PHASE 2… CANCER
Uprosertib, also known as GSK2141795 and GSK795, is an orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) with potential antineoplastic activity.
The National Cancer Institute (NCI) is evaluating the compound in phase II clinical studies for the treatment of endometrial carcinoma and multiple myeloma in combination with trametinib.
GSK-2141795, an oral AKT inhibitor, is in early clinical trials at GlaxoSmithKline for the treatment of solid tumors and lymphoma. The company is conducting phase II clinical trials for the treatment of patients with BRAF wild-type mutation melanoma and for the treatment of recurrent or persistent cervical cancer in combination with trametinib.
Akt inhibitor GSK2141795 binds to and inhibits the activity of Akt, which may result in inhibition of the PI3K/Akt signaling pathway and tumor cell proliferation and the induction of tumor cell apoptosis. Activation of the PI3K/Akt signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K/Akt signaling may contribute to tumor resistance to a variety of antineoplastic agents.
|
QC data: |
|

PATENT
| Patent | Submitted | Granted |
|---|---|---|
| Inhibitors of AKT Activity [US2011071182] | 2011-03-24 | |
| INHIBITORS OF Akt ACTIVITY [US2010267759] | 2010-10-21 | |
| INHIBITORS OF AKT ACTIVITY [US2009209607] | 2009-08-20 | |
| INHIBITORS OF Akt ACTIVITY [US2010041726] | 2010-02-18 |
More information about this drug
The chemical structures of Afuresertib (GSK-2110183) and GSK-2141795 are very similar as shown below:
 |
Fig 1. chemical structures of Afuresertib (GSK-2110183) and GSK-2141795
PATENT
WO 2008098104 OR EP2117523
http://www.google.com/patents/EP2117523A1?cl=en
Scheme 2

11-1 I-2

II-3 II-4
Reagents: (a) PyBrop, (i-Pr)2NEt, 1 ,1-dimethylethyl (2-amino-3- phenylpropyl)carbamate, DCM, RT; (b) 5-(5,5-dimethyl-1 ,3,2-dioxaborinan-2-yl)-1- methyl-1 H-pyrazole, K2CO3, Pd(PPh3)4, dioxane/H2O; (c) TFA / DCM, RT.
Preparation 7

Preparation of 5-(5,5-dimethyl-1 ,3,2-dioxaborinan-2-yl)-1 -methyl-1 H-pyrazole
To a solution of 1 -methyl pyrazole (4.1 g, 50 mmole) in THF (100 ml.) at 00C was added n-BuLi (2.2M in THF, 55 mmole). The reaction solution was stirred for 1 hour at RT and then cooled to -78°C [J. Heterocyclic Chem. 41 , 931 (2004)]. To the reaction solution was added 2-isopropoxy-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane (12.3 ml_, 60 mmole). After 15 min at -78°C, the reaction was allowed to warm to 00C over 1 hour. The reaction was diluted with saturated NH4CI solution and extracted with DCM. The organic fractions were washed with H2O (2 x 100 ml_), dried over Na2SO4 and concentrated under vacuum to afford a tan solid (8.0 g, 77%) which was used without further purification. LCMS (ES) m/z 127 (M+H)+ for [RB(OH)2]; 1H NMR (CDCI3, 400 MHz) δ 7.57 (s, 1 H), 6.75 (s, 1 H), 4.16 (s, 3H), and 1.41 (s, 12H).
Example . .24
UPROSERTIBPreparation Λ/-{(1 S)-2-amino-1-r(3,4-difluorophenyl)methyllethyl}-5-chloro-4-(4- chloro-1-methyl-1H-pyrazol-5-yl)-2-furancarboxamide
a) methyl 4-(1-methyl-1H-pyrazol-5-yl)-2-furancarboxylate

A solution of methyl 4-bromo-2-furancarboxylate (470 mg, 2.29 mmol), potassium carbonate (1584 mg, 11.46 mmol), 1-methyl-5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazole (525 mg, 2.52 mmol)[prepared according to Preparation 7] and bis-(tri-t-butylphosphine)Palladium (0) (58.6 mg, 0.12 mmol) in 1 ,4-dioxane (9.55 ml) and water (1.9 ml) was stirred at 80 0C. After 1 hr, the solution was partitioned between H2O-DCM and the aqueous phase was washed several times with DCM. The combined organic fractions were dried over I^^SOφ concentrated and purified via column chromatography (30% EtOAc in hexanes) affording the title compound (124 mg, 0.60 mmol, 26 % yield) as a white powder: LCMS (ES) m/e 206 (M+H)+.
b) methyl 5-chloro-4-(4-chloro-1-methyl-1 H-pyrazol-5-yl)-2-furancarboxylate

A solution of methyl 4-(1-methyl-1 H-pyrazol-5-yl)-2-furancarboxylate (412 mg, 2.0 mmol) and N-chlorosuccinimide (267 mg, 2.0 mmol) in DMF (10 ml.) was heated at 75 0C for 30 minutes. Another batch of N-chlorosuccinimide (267 mg, 2.0 mmol) was added. After 1 hr, the mixture was concentrated and purified using silica gel and eluting with 0-55% ethyl acetate / hexane to afford the title compound as a white solid (225 mg, 0.82 mmol, 71 % yield) : LCMS (ES) m/e 276 (M+H)+.
c) 5-chloro-4-(4-chloro-1-methyl-1 H-pyrazol-5-yl)-2-furancarboxylic acid

A solution of methyl 5-chloro-4-(4-chloro-1-methyl-1 H-pyrazol-5-yl)-2- furancarboxylate (224 mg, 0.82 mmol) in 6N sodium hydroxide (1.36 ml, 8.2 mmol) and tetrahydrofuran (5 ml) was stirred at 70 0C in a sealed tube for 1 h. The resulting solution was cooled and then partitioned between H2O-DCM. The aqueous phase was adjusted to pH ~4 and then washed several times with DCM. The combined organic fractions were dried over Na2SO4 and concentrated affording the title compound (201 mg, 0.77 mmol, 94 % yield) as a yellow oil: LCMS (ES) m/e 262 (M+H)+.
d) 5-chloro-4-(4-chloro-1-methyl-1 H-pyrazol-5-yl)-N-{(1S)-2-(3,4-difluorophenyl)-1- [(1 ,3-dioxo-1 ,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}-2-furancarboxamide

To a solution of 5-chloro-4-(4-chloro-1-methyl-1 H-pyrazol-5-yl)-2- furancarboxylic acid (200 mg, 0.77 mmol)[prepared according to the procedure of Preparation 6], 2-[(2S)-2-amino-3-(2,4-difluorophenyl)propyl]-1 H-isoindole-1 ,3(2H)- dione (254 mg, 0.80 mmol) and N,N-diisopropylethylamine (0.40 ml, 2.30 mmol) in DCM (10 ml) was added bromo-tris-pyrrolidino-phosphonium hexafluorophosphate (536 mg, 1.15 mmol). After stirring at ambient temperature for 20 hrs, the mixture was concentrated and purified with silica gel column eluting with gradient (0-50% ethyl acetate/hexanes) to afford the title compounds as an off-white foamy solid (304 mg, 0.54 mmol, 71 % yield): LCMS (ES) m/e 560(M+H)+.
e) Λ/-{(1 S)-2-amino-1-[(3,4-difluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1- methyl-1 /-/-pyrazol-5-yl)-2-furancarboxamide
To a solution of 5-chloro-4-(4-chloro-1-methyl-1 H-pyrazol-5-yl)-N-{(1S)-2- (3,4-difluorophenyl)-1 -[(1 ,3-dioxo-1 ,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}-2- furancarboxamide (304 mg, 0.54 mmol) in methanol (5 ml) at 25 0C was added hydrazine (0.08 ml, 2.7 mmol) dropwise. After 12h, the solution was concentrated, dry loaded onto silica and purified by column chromatography (5% MeOH in DCM (1 % NH4OH)). The free base was converted to the HCI salt by addition of excess 4M HCI in dioxane (1 ml) to the residue in MeOH (2 ml) affording the HCI salt of the title compound as a yellow solid:
LC-MS (ES) m/z 430(M+H)+,
1H NMR (400 MHz, MeOD) δ ppm 2.91 – 3.05 (m, 2 H) 3.17 – 3.28 (m, 2 H) 3.81 (s, 3 H) 4.57 (d, J=9.60 Hz, 1 H) 7.12 (br. s., 1 H) 7.18-7.28 (m., 2 H) 7.36-7.39 (m, 1 H) 7.58 (s, 1 H).
SYNTHESIS ELABORATED

STEP A

4,5-dibromo-2-furancarboxylic acid in methanol , sulfuric acid methyl 4,5-dibromo-2-furancarboxylate LCMS (ES) m/e 283 (M+H)+
STEP B
methyl 4,5-dibromo-2-furancarboxylate and isopropylmagnesium chloride ,to give methyl 4-bromo-2-furancarboxylate
LCMS (ES) m/e 204,206 (M, M+2)+
STEP C

methyl 4-bromo-2-furancarboxylate and NCS in N,N-dimethylformamide methyl 4-bromo-5-chloro-2-furancarboxylate LCMS (ES) m/e 238,240,242 (M, M+2, M+4)+
STEP D

methyl 4-bromo-5-chloro-2-furancarboxylate , 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole prepared according toPreparation 7], potassium carbonate and bis(tri-t-butylphosphine)paliadium(0) in 1,4-dioxane (19.14 ml) and water ……methyl 5-chloro-4-(1-methyl-1H-pyrazol-5-yl)-2-furancarboxylate obtained. LCMS m/e ES 240, 242 (M, M+2)+
STEP E
a) 5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-2-furancarboxylic acid.
A solution of methyl 5-chloro-4-(1-methyl-1H-pyrazol-5-yl)-2-furancarboxylate [prepared according to Example
127] and n-chlorosuccinimide (166 mg, 1.25 mmol) yielding 5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-2-furancarboxylic acid. LCMS (ES) m/e 261,263 (M, M+2)+
STEP F
Reacting 5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-2-furancarboxylic acid [prepared according to the procedure of Preparation 6], 2-[(2S)-2-amino-3-(2,4-difluorophenyl)propyl]-1H-isoindole-1,3(2H)-dione and N,N-diisopropylethylamine in DCM was added bromo-tris-pyrrolidino-phosphonium hexafluorophosphate …….obtd
5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-N-{(1S)-2-(3,4-difluorophenyl)-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}-2-furancarboxamide. the uproserib precursor
LCMS (ES) m/e 560(M+H)+
NOTE STRUCTURE OF 2-[(2S)-2-amino-3-(2,4-difluoro phenyl)propyl]-1H-isoindole-1,3(2H)-dione

SEE http://www.google.com/patents/WO2010093885A1?cl=en
Preparation 1

Preparation of 2-[(2S)-2-amino-3-(3,4-difluorophenyl)propyl1-1 /-/-isoindole-1 ,3(2H)-dione a) 1 ,1-dimethylethyl [(1 S)-2-(3,4-difluorophenyl)-1-(hydroxymethyl)ethyl]carbamate

To a solution of Λ/-{[(1 ,1-dimethylethyl)oxy]carbonyl}-3,4-difluoro-L-phenylalanine (2.0 g, 6.7 mmol) in THF (35 ml.) at 0 0C stirred was added BH3-THF (30 ml_, 30 mmol- 1 M in THF). After 12h, the reaction was quenched with AcOH:MeOH (1 :4, 20 ml.) and partitioned between saturated aqueous NaHCO3 and CHCI3. The aqueous phase was then extracted several times with CHCI3. The combined organic fractions were concentrated and the resulting white solid (7.0 g, 74%) used without further purification: LCMS (ES) m/e 288 (M+H)+.
b) 1 ,1-dimethylethyl {(1 S)-2-(3,4-difluorophenyl)-1-[(1 ,3-dioxo-1 ,3-dihydro-2/-/-isoindol-2- yl)methyl]ethyl}carbamate

To a solution of 1 ,1-dimethylethyl [(1 S)-2-(3,4-difluorophenyl)-1-
(hydroxymethyl)ethyl]carbamate (2.65 g, 9.22 mmol), polymer bound triphenylphosphine (5.33 g, 1 1.5 mmol, 2.15 mmol/g) and phthalimide (1.63 g, 10.9 mmol) in THF (50 ml.) at 25 0C was added diisopropyl azodicarboxylate (1.85 ml_, 11.3 mmol). After stirring at RT for 1 h, the reaction solution was filtered and concentrated. The residue was adsorbed onto silica and purified via column chromatography to yield product (0.33 g) as a white solid: LCMS (ES) m/z 417 (M+H)+.
c) 2-[(2S)-2-amino-3-(3,4-difluorophenyl)propyl]-1 H-isoindole-1 ,3(2H)-dione
To a solution of 1 ,1-dimethylethyl {(1S)-2-(3,4-difluorophenyl)-1-[(1 ,3-dioxo-1 ,3- dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (0.33 g, 0.79 mmol) in CHCI3:MeOH (10:3, 13 mL) at RT was added 4M HCI in dioxane (5 mL, 20 mmol). After 12h, the solvents were removed and affording the title compound (0.29 g, quant.) as a white HCI salt which was used without further purification: LCMS (ES) m/z 317 (M+H)+.
FINAL STEP
conversion of precursor to uprosertb
UPROSERTIB PRECURSOR GIVES UPROSERTIBN-{(1S)-2-amino-1-[(3,4-difluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1-methyl-1Hpyrazol-5-yl)-2-furancarboxamide.
5-chloro-4-(4-chloro-1-methyl-1Hpyrazol-5-yl)-N-{(1S)-2-(3,4-difluorophenyl)-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-
yl)methyl]ethyl}-2-furancarboxamide in methanol (5 ml) AND hydrazine …..N-{(1S)-2-amino-1-[(3,4-difluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1-methyl-1Hpyrazol-5-yl)-2-furancarboxamide.
yl)methyl]ethyl}-2-furancarboxamide in methanol (5 ml) AND hydrazine …..N-{(1S)-2-amino-1-[(3,4-difluorophenyl)methyl]ethyl}-5-chloro-4-(4-chloro-1-methyl-1Hpyrazol-5-yl)-2-furancarboxamide.
SYNTHESIS OF INTERMEDIATES
Example 127
a) methyl 4,5-dibromo-2-furancarboxylate
To a solution of 4,5-dibromo-2-furancarboxylic acid (25 g, 93 mmol) in methanol (185 ml) was added sulfuric acid (24.7 ml, 463 mmol). The resulting solution stirred at 50 0C over 12h. The solution was partitioned between H2O-DCM and the aqueous phase was washed several times with DCM. The combined organic fractions were dried over I^^SOφ concentrated and used directly without further purification providing methyl 4,5-dibromo-2-furancarboxylate (23.67 g, 83 mmol, 90 % yield), LCMS (ES) m/e 283, 285, 287 (M, M+2, M+4)+.b) methyl 4-bromo-2-furancarboxylate Br
To a solution of methyl 4,5-dibromo-2-furancarboxylate (3.3 g, 1 1.62 mmol) in tetrahydrofuran (46 ml) at -40 0C was added isopropylmagnesium chloride (6.97 ml, 13.95 mmol). After 1 h, Water (11 ml) was added and the solution warmed to 25 0C. The reaction mixture was then partitioned between H2O-DCM and the aqueous phase was washed several times with DCM. The combined organic fractions were dried over Na2SOφ concentrated and purified by column chromatography (3% EtOAc in hexanes) affording methyl 4-bromo-2-furancarboxylate (1.4 g, 6.49 mmol, 56 % yield) as a yellow solid: LCMS (ES) m/e 205, 207 (M, M+2)+.
c) methyl 4-bromo-5-chloro-2-furancarboxylate
A solution of methyl 4-bromo-2-furancarboxylate (1.4 g, 6.83 mmol) and NCS (0.912 g, 6.83 mmol) in N,N-dimethylformamide (13.7 ml) was stirred in a sealed tube for 1 h at 100 0C. After 1 h, the solution was partitioned between DCM- H2O and the aqueous phase was washed several times with DCM. The combined organic fractions were dried over I^^SOφ concentrated and purified via column chromatography (2-10% EtOAc in hexanes) affording methyl 4-bromo-5-chloro-2- furancarboxylate (1.348 g, 5.12 mmol, 75 % yield) as a white solid: LCMS (ES) m/e 238, 240, 242 (M, M+2, M+4)+.
d) methyl 5-chloro-4-(1-methyl-1 H-pyrazol-5-yl)-2-furancarboxylate
A solution of methyl 4-bromo-5-chloro-2-furancarboxylate (1.1 g, 4.59 mmol), 1-methyl-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1.05 g, 5.05 mmol)[prepared according to Preparation 7], potassium carbonate (3.17 g, 22.97 mmol) and bis(tri-t-butylphosphine)palladium(0) (0.117 g, 0.23 mmol) in 1 ,4- dioxane (19.14 ml) and water (3.83 ml) was stirred at 80 0C in a sealed tube for 1 h. The reaction mixture was partitioned between H2O-DCM and the aqueous phase was washed several times with DCM. The combined organic fractions were dried over Na2SOφ concentrated and purified via column chromatography (silica, 4-25% EtOAc in hexanes) yielding methyl 5-chloro-4-(1-methyl-1 H-pyrazol-5-yl)-2- furancarboxylate (800 mg, 2.53 mmol, 55 % yield) as a yellow oil: LCMS m/e ES 240, 242 (M, M+2)+.
e) 5-chloro-4-(1-methyl-1 H-pyrazol-5-yl)-2-furancarboxylic acid

A solution of methyl 5-chloro-4-(1-methyl-1 H-pyrazol-5-yl)-2- furancarboxylate (300 mg, 1.25 mmol) in 6N sodium hydroxide (4.16 ml, 24.93 mmol) and tetrahydrofuran (5.4 ml) was stirred at 70 0C in a sealed tube for 1 h. The resulting solution was cooled and then partitioned between H2O-DCM. The aqueous phase was adjusted to pH ~4 and then washed several times with DCM. The combined organic fractions were dried over Na2SO4 and concentrated affording 5-chloro-4-(1-methyl-1 H-pyrazol-5-yl)-2-furancarboxylic acid (267 mg, 0.59 mmol, 47 % yield) as a white foam: LCMS (ES) m/e 265 (M+H)+.
|
References |
1: Dumble M, Crouthamel MC, Zhang SY, Schaber M, Levy D, Robell K, Liu Q, Figueroa DJ, Minthorn EA, Seefeld MA, Rouse MB, Rabindran SK, Heerding DA, Kumar R. Discovery of Novel AKT Inhibitors with Enhanced Anti-Tumor Effects in Combination with the MEK Inhibitor. PLoS One. 2014 Jun 30;9(6):e100880. doi: 10.1371/journal.pone.0100880. eCollection 2014. PubMed PMID: 24978597; PubMed Central PMCID: PMC4076210.
2: Pachl F, Plattner P, Ruprecht B, Médard G, Sewald N, Kuster B. Characterization of a chemical affinity probe targeting Akt kinases. J Proteome Res. 2013 Aug 2;12(8):3792-800. doi: 10.1021/pr400455j. Epub 2013 Jul 3. PubMed PMID: 23795919.
3: Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010 Nov;19(11):1355-66. doi: 10.1517/13543784.2010.520701. Epub 2010 Sep 16. Review. PubMed PMID: 20846000; PubMed Central PMCID: PMC3244346.
Probable GSK 2245035
 GSK 2245035 PROBABLE
GSK 2245035 PROBABLE8H-Purin-8-one, 6-amino-2-butoxy-7,9-dihydro-9-[[1-(2-hydroxyethyl)-4-piperidinyl]methyl]-
CAS NO 1264370-20-8
GSK 2245035
PHASE 2, Allergic asthma; Allergic rhinitis
Toll-like receptor 7 agonist
Immunomodulators; Interferon alfa 2a stimulants; Toll-like receptor 7 agonists
- 01 Aug 2014 GlaxoSmithKline completes a phase II trial in Allergic asthma and allergic rhinitis in Canada (NCT01788813)
- 31 Jul 2013 GlaxoSmithKline completes a phase II trial in Allergic asthma and allergic rhinitis in Canada (NCT01607372)
- 29 Mar 2013 GlaxoSmithKline initiates enrolment in a phase II trial for Allergic asthma and allergic rhinitis in Canada (NCT01788813)
Patent
https://www.google.com/patents/WO2011098451A1?cl=en
Example 2: 6-Amino-2-(butyloxy)-9-([1 -(2-hvdroxyethyl)-3-piperidinyllmethyl|-7,9-dihydro-8/-/-purin-8- one
2-(Butyloxy)-8-(methyloxy)-9-(-piperidinylmethyl)-9/-/-purin-6-amine (for example, as prepared for Intermediate 14) (33.4 mg, 0.1 mmol) was suspended in DMF (0.3 mL) was added to 2- bromoethanol (commercially available, for example, from Aldrich) (0.0071 mL, 0.100 mmol). DIPEA (0.040 mL, 0.23 mmol) was added. The reaction was shaken in a stoppered vial at ambient temperature overnight. The reaction mixture was diluted with DMSO (0.4 mL) and the resultant solution purified by MDAP (Method A). Appropriate fractions were combined and evaporated in vacuo. The residues was dissolved in 4M HCI in dioxane (0.4 mL) and allowed to stand at room temperature overnight. The solvent was dried under a stream of nitrogen in the Radleys blowdown apparatus. The residue was redissolved in methanol (0.5 mL) and applied to the top of a 0.5 g aminopropyl SPE (preconditioned with methanol, 2 CV). The cartridge was washed with methanol (2 mL). The solvent was dried under a stream of nitrogen in the Radleys blowdown apparatus to give the title compound (0.022 g).
LCMS (System A): tRET = 0.57min; MH+ 365
REF
pdf (892 KB), English, Pages 211
hrcak.srce.hr/file/138695
by K BENDELJA – 2012 – Related articles
titis B vaccine both manufactured by GlaxoSmithKline. MPL is a nontoxic derivate … GSK2245035 compound that is a highly selective TLR7 agonist. Intranasal …
KHK 7580 structure cracked……Evocalcet
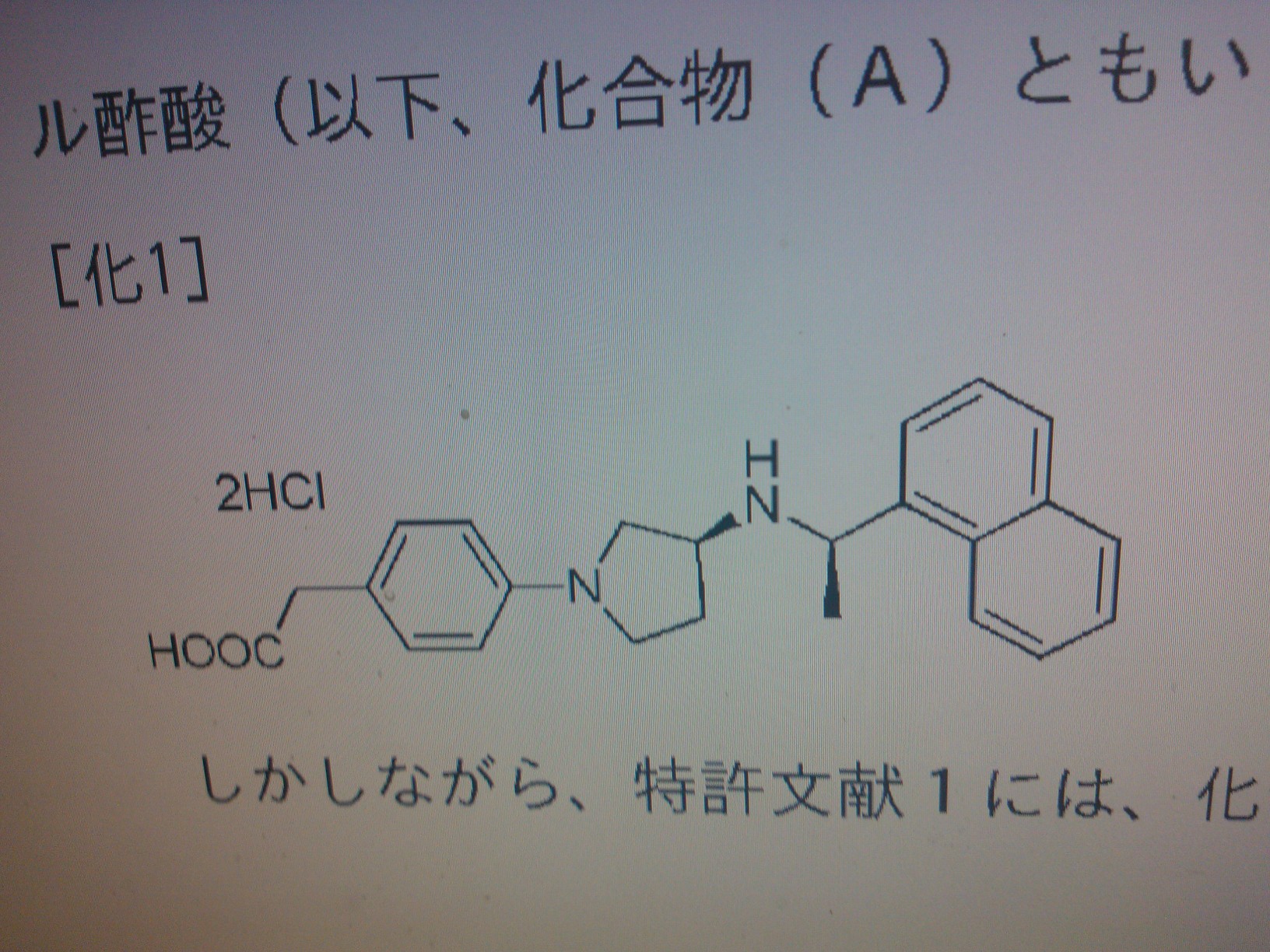
KHK 7580 …..example
| 3.008 |
 |
|
 |
2HCl | MS · APCI: 375[M + H]+ |

in EP1757582
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid
4-[(3S)-3-[[(1R)-1-(1-naphthalenyl)ethyl]amino]-1-pyrrolidinyl]-Benzeneacetic acid,
cas will be updated
BASE ….870964-67-3
DI HCL SALT …….870856-31-8
MF C24 H26 N2 O2 BASE
MW 374.48 BASE
KHK-7580
KHK-7580; MT-4580
Mitsubishi Tanabe Pharma Corp… innovator
Kyowa Hakko Kirin Co Ltd.. licencee
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1-yl)phenylacetic acid,
useful as calcium-sensitive receptor (CaSR) agonists for treating hyperparathyroidism. a CaSR agonist, being developed by Kyowa Hakko Kirin, under license from Mitsubishi Tanabe, for treating secondary hyperparathyroidism (phase 2 clinical, as of March 2015).
WILL BE UPDATED
WO2005115975,/EP1757582
http://www.google.co.in/patents/EP1757582A1?cl=en
Example no
| 3.008 |
|
|
|
2HCl | MS · APCI: 375[M + H]+ |
Mitsubishi Tanabe Pharma Corporation
The present invention provides a novel crystal form of an arylalkylamine
compound. Specifically, a novel crystal form of
4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid has
excellent stability, and is therefore useful as an active ingredient for
a medicine. The present invention also provides an industrially
advantageous method for producing an arylalkylamine compound.

http://worldwide.espacenet.com/publicationDetails/biblio?DB=worldwide.espacenet.com&II=0&ND=3&adjacent=true&locale=en_EP&FT=D&date=20150312&CC=WO&NR=2015034031A1&KC=A1
Mitsubishi Tanabe Pharma Corporation
The present invention provides a novel crystal form of an arylalkylamine compound. Specifically, a novel crystal form of 4-(3S-(1R-(1-naphthyl)ethylamino)pyrrolidin-1- yl)phenylacetic acid has excellent stability, and is therefore useful as an active ingredient for a medicine. The present invention also provides an industrially advantageous method for producing an arylalkylamine compound.
………………….
Reference Example 3.001

(1) To a mixed solution containing 33.5 g of 3-hydroxypiperidine and 62.7 ml of triethylamine dissolved in 250 ml of methylene chloride was added dropwise a solution of 55.7 ml of benzyloxycarbonyl chloride in 150 ml of methylene chloride, and the mixture was stirred at room temperature for 16 hours. To the reaction mixture were added a saturated aqueous citric acid and chloroform, the mixture was stirred and the liquids were separated. The organic layer was dried, the solvent was evaporated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 75.5 g of benzyl 3-hydroxypiperidine-1-carboxylate.
MS•APCI (m/z): 236 [M+H]+
(2) 800 ml of a solution of 52.4 ml of oxalyl chloride in methylene chloride was cooled to −78° C., 53.2 ml of DMSO was added dropwise to the solution, and the mixture was stirred at −78° C. for 0.5 hour. A solution of 75.5 g of benzyl 3-hydroxypiperidine-1-carboxylate dissolved in 200 ml of methylene chloride was added dropwise to the mixture, and further 293 ml of triethylamine was added dropwise to the same, and the mixture was stirred for 16 hours while a temperature thereof was gradually raised to room temperature. To the reaction mixture were added a saturated aqueous sodium bicarbonate solution and chloroform, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated to obtain 83.7 g of 1-benzyloxycarbonyl-3-piperidone. MS•APCI (m/z): 234 [M+H]+
(3) To a solution of 83.7 g of 1-benzyloxycarbonyl-3-piperidone dissolved in 1.2 liters of methylene chloride was added 55.0 g of (R)-(+)-1-(1-naphthyl)ethylamine, and after the mixture was stirred at room temperature for 2 hours, 69 ml of acetic acid and 160 g of sodium triacetoxy borohydride were added to the mixture, and the mixture was stirred at room temperature for 15 hours. To the reaction mixture was added an aqueous sodium hydroxide to make the mixture basic, and then, chloroform was added to the mixture, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 98.7 g of benzyl 3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
(4) To a solution of 40.95 g of triphosgene dissolved in 800 ml of methylene chloride was added dropwise a mixed solution containing 80.6 g of benzyl 3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate and 86.6 ml of triethylamine dissolved in 200 ml of methylene chloride at 0° C., and the mixture was stirred at room temperature for 16 hours. To the reaction mixture was added water, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was washed with 200 ml of diethyl ether, and the crystal collected by filtration was recrystallized from chloroform and diethyl ether to obtain 48.9 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate.
Further, the filtrate was purified by silica gel column chromatography (hexane:ethyl acetate=8:1→0:1) to obtain 5.82 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate and 14.5 g of benzyl (S)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate.
(5) To a solution containing 54.6 g of benzyl (R)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate dissolved in 700 ml of tetrahydrofuran was added 350 ml of water, and the mixture was stirred under reflux for 15 hours. After tetrahydrofuran was evaporated, a saturated aqueous sodium bicarbonate solution and chloroform were added thereto, the mixture was stirred and the liquids were separated. The organic layer was dried and concentrated, and the residue was purified by silica gel column chromatography (hexane:ethyl acetate=4:1→0:1) to obtain 24.3 g of benzyl (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
(6) To a solution containing 24.2 g of benzyl (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate dissolved in 250 ml of methanol was added 2.5 g of palladium carbon (10% wet), and the mixture was shaked under hydrogen atmosphere at 3 atm at room temperature for 40 hours. Palladium carbon was removed, and the solvent was evaporated, the residue was washed with ethyl acetate-chloroform (10:1), and collected by filtration to obtain 15.3 g of (R)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine (the following Reference example Table, Reference example 3.001(a)). MS•APCI (m/z): 255 [M+H]+
(7) By using 14.5 g of benzyl (S)-3-[chlorocarbonyl-(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate, the same treatment was carried out as in the above-mentioned (5) to obtain 4.74 g of benzyl (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate. MS•APCI (m/z): 389 [M+H]+
Moreover, by using 4.7 g of benzyl (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine-1-carboxylate, the same treatment was carried out as in the above-mentioned (6) to obtain 2.89 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine. MS•APCI (m/z): 255 [M+H]+
(8) To a solution of 3.46 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine dissolved in 15 ml of methanol was added dropwise 20 ml of a solution of 4M hydrochloric acid in ethyl acetate, and the mixture was stirred. The reaction mixture was concentrated under reduced pressure, diethyl ether was added to the residue, washed and dried to obtain 3.33 g of (S)-3-[(R)-1-(naphthalen-1-yl)ethylamino]piperidine dihydrochloride
| 3.008 |
|
|
|
2HCl | MS · APCI: 375[M + H]+ |
| TABLE A3 | |||||
 |
|||||
| Example No. | R1—X— |
|
—Ar | Salt | Physical properties, etc. |
…………………..
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
see all at http://drugpatentsint.blogspot.in/2015/03/wo-2015034031.html
do not miss out on above click
http://www.kyowa-kirin.com/research_and_development/pipeline/
KHK7580 -Secondary Hyperparathyroidism
![]()
![]()
![]()
![]()
JP
| Company | Mitsubishi Tanabe Pharma Corp. |
| Description | Calcium receptor agonist |
| Molecular Target | |
| Mechanism of Action | Calcium-sensing receptor (CaSR) agonist |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Thyroid disease |
| Indication Details | Treat hyperparathyroidism in patients receiving hemodialysis; Treat secondary hyperparathyroidism (SHPT) |
| Regulatory Designation | |
| Partner |
August 29, 2014
Kyowa Hakko Kirin Announces Commencement of Phase 2b Clinical Study of KHK7580 in Patients with Secondary Hyperparathyroidism in Japan
Tokyo, Japan, August 29, 2014 — Kyowa Hakko Kirin Co., Ltd. (Tokyo: 4151, President and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) today announced the initiation of a phase 2b clinical study evaluating KHK7580 for secondary hyperparathyroidism patients receiving hemodialysis in Japan.
This randomized, placebo-controlled, double-blind, parallel-group, multi-center study is designed to evaluate efficacy and safety in cohorts comprising KHK7580, its placebo and cinacalcet and initial dose of KHK7580 for secondary hyperparathyroidism patients receiving hemodialysis.
KHK7580 is a small molecular compound produced by Mitsubishi Tanabe Pharma Corporation (President & Representative Director, CEO: Masayuki Mitsuka, “Mitsubishi Tanabe Pharma”). Kyowa Hakko Kirin signed a license agreement of KHK7580 with Mitsubishi Tanabe Pharma for the rights to cooperative research, develop, market and manufacture the product in Japan and some part of Asia on March 2008.
The Kyowa Hakko Kirin Group is contributing to the health and prosperity of the world’s people by pursuing advances in life sciences and technology and creating new value.
Outline of this study
| ClinicalTrials.gov Identifier | |
|---|---|
| Target Population | Secondary hyperparathyroidism patients receiving hemodialysis |
| Trial Design | Randomized, placebo-controlled, double-blind (included open arm of cinacalcet), parallel-group, multi-center study |
| Administration Group | KHK7580, Placebo, cinacalcet |
| Target Number of Subjects | 150 |
| Primary Objective | Efficacy |
| Trial Location | Japan |
| Trial Duration | Jul. 2014 to Jun. 2015 |
Contact:
Kyowa Hakko Kirin
Media Contact:
+81-3-3282-1903
or
Investors:
+81-3-3282-0009
Update on march 2016
New comment waiting approval on New Drug Approvals |
|

http://www.medkoo.com/products/6729

Name: Evocalcet
CAS#: 870964-67-3
Chemical Formula: C24H26N2O2
Exact Mass: 374.19943
Evocalcet is a calcium-sensing receptor agonist. The calcium-sensing receptor (CaSR) is a Class C G-protein coupled receptor which senses extracellular levels of calcium ion. The calcium-sensing receptor controls calcium homeostasis by regulating the release of parathyroid hormone (PTH). CaSR is expressed in all of the organs of the digestive system. CaSR plays a key role in gastrointestinal physiological function and in the occurrence of digestive disease. High dietary Ca2+ may stimulate CaSR activation and could both inhibit tumor development and increase the chemotherapeutic sensitivity of cancer cells in colon cancer tissues. (Last update: 12/15/2015).
Synonym: MT-4580; MT 4580; MT4580; KHK-7580; KHK7580; KHK 7580; Evocalcet
IUPAC/Chemical Name: 2-(4-((S)-3-(((R)-1-(naphthalen-1-yl)ethyl)amino)pyrrolidin-1-yl)phenyl)acetic acid
2
https://tripod.nih.gov/ginas/app/substance/f580b9fd

http://www.drugspider.com/drug/evocalcet
| INN name |
Evocalcet
|
||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lab Code(s) |
MT-4580
KHK-7580
|
||||||||||||||||||||||||||||||||||||||||
| Chemical name |
{4-[(3S)-3-{[(1R)-1-(Naphthalen-1-yl)ethyl]amino}pyrrolidin-1-yl]phenyl}acetic acid
|
||||||||||||||||||||||||||||||||||||||||
| Chemical structure |
 |
||||||||||||||||||||||||||||||||||||||||
| Molecular formula |
C24H26N2O2
|
||||||||||||||||||||||||||||||||||||||||
| SMILES |
O=C(O)CC1=CC=C(N2C[C@@H](N[C@@H](C3=C4C=CC=CC4=CC=C3)C)CC2)C=C1
|
||||||||||||||||||||||||||||||||||||||||
| CAS registry number |
870964-67-3
|
||||||||||||||||||||||||||||||||||||||||
| Orphan Drug Status |
No
|
||||||||||||||||||||||||||||||||||||||||
| On Fast track |
No
|
||||||||||||||||||||||||||||||||||||||||
| New Molecular Entity |
Yes
|
||||||||||||||||||||||||||||||||||||||||
| Originator | |||||||||||||||||||||||||||||||||||||||||
| Developer(s) | |||||||||||||||||||||||||||||||||||||||||
| Class | |||||||||||||||||||||||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||||||||||||||||||||||
| Clinical trial(s) |
|
||||||||||||||||||||||||||||||||||||||||
| Updated on |
11 Oct 2015
|
///////////////
SMILES Code: O=C(O)CC1=CC=C(N2C[C@@H](N[C@@H](C3=C4C=CC=CC4=CC=C3)C)CC2)C=C1
C[C@H](c1cccc2c1cccc2)N[C@H]3CCN(C3)c4ccc(cc4)CC(=O)O
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease
CS-3150, (XL550)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
CAS 1632006-28-0 for S conf
MF C22 H21 F3 N2 O4 S
MW 466.47
(S)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
CAS 1632006-28-0 for S configuration
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide, CAS 880780-76-7
(+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-82-4
(-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-83-5
WO2008 / 126831 (US Publication US2010-0093826)http://www.google.co.in/patents/EP2133330A1?cl=en
WO 2006012642..compound A;..http://www.google.com/patents/WO2006012642A2?cl=en
WO2006 / 012642 (US Publication US2008-0234270)
WO 2015030010…http://www.google.com/patents/WO2015030010A1?cl=en
- Originator Exelixis
- Developer Daiichi Sankyo Company..
Daiichi Sankyo Company,Limited, 第一三共株式会社 - Class Antihypertensives; Small molecules
- Mechanism of Action Mineralocorticoid receptor antagonists

JAPAN PHASE 2……….Phase 2 Study to Evaluate Efficacy and Safety of CS-3150 in Patients with Essential Hypertension
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-121921
Phase II Diabetic nephropathies; Hypertension
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
- 01 May 2013 Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
-
Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.


Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
| Description | Small molecule antagonist of the mineralocorticoid receptor (MR) |
| Molecular Target | Mineralocorticoid receptor |
| Mechanism of Action | Mineralocorticoid receptor antagonist |
| Therapeutic Modality | Small molecule |
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
Several patents WO-2014168103,
WO-2015012205 and WO-2015030010
XL-550, claimed in WO-2006012642,
………………………………………………………………….
http://www.google.co.in/patents/EP2133330A1?cl=en
(Example 3)(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
-
After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
-
Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid.
1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s). -
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained.
1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
Optical Resolution of Compound of Example 3

 OLMESARTAN
OLMESARTAN

-
Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
-
Isomer C: (+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: +7.1° (c = 1.0, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.91 (s, 1H), 7.87-7.79 (m, 5H), 7.67-7.58 (m, 2H), 7.51 (s, 1H), 7.35 (d, 1H, J = 7.0 Hz), 3.78-3.65 (m, 4H), 3.05 (s, 3H), 2.07 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1260.
Retention time: 4.0 min. -
Isomer D: (-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: -7.2° (c = 1.1, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.88-7.79 (m, 6H), 7.67-7.58 (m, 2H), 7.50 (s, 1H), 7.36 (d, 1H, J = 7.5 Hz), 3.79-3.65 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1257.
Retention time: 4.5 min.
……………………………………………….
WO 2014168103
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014168103
Step B: pyrrole derivative compounds (A ‘)
[Of 16]

(Example 1) 2-bromo-1- [2- (trifluoromethyl) phenyl] propan-1-one
[Of 19]

1- [2- (trifluoromethyl) phenyl] propan-1-one 75 g (370 mmol) in t- butyl methyl ether (750 mL), and I was added bromine 1.18 g (7.4 mmol). After confirming that the stirred bromine color about 30 minutes at 15 ~ 30 ℃ disappears, cooled to 0 ~ 5 ℃, was stirred with bromine 59.13 g (370 mmol) while keeping the 0 ~ 10 ℃. After stirring for about 2.5 hours, was added while maintaining 10 w / v% aqueous potassium carbonate solution (300 mL) to 0 ~ 25 ℃, was further added sodium sulfite (7.5 g), was heated to 20 ~ 30 ℃. The solution was separated, washed in the resulting organic layer was added water (225 mL), to give t- butyl methyl ether solution of the title compound and the organic layer was concentrated under reduced pressure (225 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.91 (3H, D, J = 4.0 Hz), 4.97 (1H, Q, J = 6.7 Hz), 7.60 ~ 7.74 (4H, M).
(Example 2) 2-cyano-3-methyl-4-oxo-4- [2- (trifluoromethyl) phenyl] butanoate
[Of 20]

2-bromo-1- [2- (trifluoromethyl) phenyl] propan-1 / t- butyl methyl ether solution (220 mL) in dimethylacetamide (367 mL), ethyl cyanoacetate obtained in Example 1 53.39 g (472 mmol), potassium carbonate 60.26 g (436 mmol) were sequentially added, and the mixture was stirred and heated to 45 ~ 55 ℃. After stirring for about 2 hours, 20 is cooled to ~ 30 ℃, water (734 mL) and then extracted by addition of toluene (367 mL), washed by adding water (513 mL) was carried out in the organic layer (2 times implementation). The resulting organic layer was concentrated under reduced pressure to obtain a toluene solution of the title compound (220 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.33 ~ 1.38 (6H, M), 3.80 ~ 3.93 (2H, M), 4.28 ~ 4.33 (2H, M), 7.58 ~ 7.79 (4H, M).
(Example 3) 2-chloro-4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl
[Of 21]

The 20 ~ 30 ℃ 2-cyano-3-methyl-4-oxo-4 was obtained [2- (trifluoromethyl) phenyl] butanoate in toluene (217 mL) by the method of Example 2 ethyl acetate (362 mL) Te, after the addition of thionyl chloride 42.59 g (358 mmol), cooled to -10 ~ 5 ℃, was blown hydrochloric acid gas 52.21 g (1432 mmol), further concentrated sulfuric acid 17.83 g (179 mmol) was added, and the mixture was stirred with hot 15 ~ 30 ℃. After stirring for about 20 hours, added ethyl acetate (1086 mL), warmed to 30 ~ 40 ℃, after the addition of water (362 mL), and the layers were separated. after it separated organic layer water (362 mL) was added for liquid separation, and further 5w / v% was added for liquid separation aqueous sodium hydrogen carbonate solution (362 mL).
Subsequently the organic layer was concentrated under reduced pressure, the mixture was concentrated under reduced pressure further added toluene (579 mL), was added toluene (72 mL), and cooled to 0 ~ 5 ℃. After stirring for about 2 hours, the precipitated crystals were filtered, and washed the crystals with toluene which was cooled to 0 ~ 5 ℃ (217 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (97.55 g, 82.1% yield).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.38 (3H, t, J = 7.1 Hz), 2.11 (3H, s), 4.32 (2H, Q, J = 7.1 Hz), 7.39 (1H, D, J = 7.3 Hz), 7.50 ~ 7.62 (2H, m), 7.77 (1H, d, J = 8.0 Hz), 8.31 (1H, br).
(Example 4) 4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl
[Of 22]

Example obtained by the production method of the three 2-chloro-4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 97.32 g (293 mmol) in ethanol (662 mL), tetrahydrofuran (117 mL), water (49 mL), sodium formate 25.91 g (381 mmol) and 5% palladium – carbon catalyst (water content 52.1%, 10.16 g) was added at room temperature, heated to 55 ~ 65 ℃ the mixture was stirred. After stirring for about 1 hour, cooled to 40 ℃ less, tetrahydrofuran (97 mL) and filter aid (KC- flock, Nippon Paper Industries) 4.87 g was added, the catalyst was filtered and the residue using ethanol (389 mL) was washed. The combined ethanol solution was used for washing the filtrate after concentration under reduced pressure, and with the addition of water (778 mL) was stirred for 0.5 hours at 20 ~ 30 ℃. The precipitated crystals were filtered, and washed the crystals with ethanol / water = 7/8 solution was mixed with (292 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (86.23 g, 98.9% yield).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.35 (3H, t, J = 7.1 Hz), 2.18 (3H, s), 4.29 (2H, M), 7.40 ~ 7.61 (4H, M), 7.77 (1H, d, J = 7.9 Hz), 8.39 (1H, br).
(Example 5) (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl
[Of 23]

N to the fourth embodiment of the manufacturing method by the resulting 4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 65.15 g (219 mmol), N- dimethylacetamide ( 261 mL), ethylene carbonate 28.95 g (328.7 mmol), 4- dimethylaminopyridine 2.68 g (21.9 mmol) were sequentially added at room temperature, and heated to 105 ~ 120 ℃, and the mixture was stirred. After stirring for about 10 hours, toluene was cooled to 20 ~ 30 ℃ (1303 mL), and the organic layer was extracted by adding water (326 mL). Subsequently, was washed by adding water (326 mL) to the organic layer (three times). The resulting organic layer was concentrated under reduced pressure, ethanol (652 mL) was added, and was further concentrated under reduced pressure, ethanol (130 mL) was added to obtain an ethanol solution of the title compound (326 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.35 (3H, t, J = 7.1 Hz), 1.84 (1H, Broad singlet), 2.00 (3H, s), 3.63 ~ 3.77 (4H, M), 4.27 (2H , m), 7.35 ~ 7.79 (5H, m).
(Example 6) (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid
[Of 24]

Obtained by the method of Example 5 (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl / ethanol (321 mL) solution in water (128.6 mL), was added at room temperature sodium hydroxide 21.4 g (519 mmol), and stirred with heating to 65 ~ 78 ℃. After stirring for about 6 hours, cooled to 20 ~ 30 ℃, after the addition of water (193 mL), and was adjusted to pH 5.5 ~ 6.5, while maintaining the 20 ~ 30 ℃ using 6 N hydrochloric acid. was added as seed crystals to the pH adjustment by a liquid (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 6.4 mg , even I was added to water (193mL). Then cooled to 0 ~ 5 ℃, again, adjusted to pH 3 ~ 4 with concentrated hydrochloric acid and stirred for about 1 hour. Then, filtered crystals are precipitated, and washed the crystals with 20% ethanol water is cooled to 0 ~ 5 ℃ (93 mL). The resulting wet product crystals were dried under reduced pressure at 40 ℃, to give the title compound (64.32 g, 95.0% yield). 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.87 (3H, s), 3.38 ~ 3.68 (4H, M), 7.43 ~ 7.89 (5H, M).
(Example 7)
(S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
Quinine 31.05 g (96 mmol) in N, N- dimethylacetamide (25 mL), ethyl acetate (350 mL), was heated in water (15 mL) 65 ~ 70 ℃ was added, was added dropwise a solution 1. After about 1 hour stirring the mixture at 65 ~ 70 ℃, and slowly cooled to 0 ~ 5 ℃ (cooling rate standard: about 0.3 ℃ / min), and stirred at that temperature for about 0.5 hours. The crystals were filtered, 5 ℃ using ethyl acetate (100 mL) which was cooled to below are washed crystals, the resulting wet product crystals was obtained and dried under reduced pressure to give the title compound 43.66 g at 40 ℃ (Yield 42.9%). Furthermore, the diastereomeric excess of the obtained salt was 98.3% de. 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.30 ~ 2.20 (10H, M), 2.41 ~ 2.49 (2H, M), 2.85 ~ 3.49 (6H, M), 3.65 ~ 3.66 (1H, M), 3.88 (3H, s), 4.82 (1H, broad singlet), 4.92 ~ 5.00 (2H, m), 5.23 ~ 5.25 (1H, m), 5.60 (1H, br), 5.80 ~ 6.00 (1H, m), 7.36 ~ 7.92 (9H, M), 8.67 (1H, D, J = 4.6 Hz) (7-2) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3 diastereomeric excess of the carboxylic acid quinine salt HPLC measurements (% de) that the title compound of about 10 mg was collected, and the 10 mL was diluted with 50v / v% aqueous acetonitrile me was used as a sample solution.
Column: DAICEL CHIRALPAK IC-3 (4.6 mmI.D. × 250 mm, 3 μm)
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
[Table 1]

Detection: UV 237 nm
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(Example 8)
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxamide (Compound (A))
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
(8-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
After completion concentration under reduced pressure, acetonitrile (200 mL) was added and cooled to 10 ~ 15 ℃ (reaction 1).
Acetonitrile (240mL), pyridine 12.39 g (157 mmol), 4- were successively added (methylsulfonyl) aniline 26.85 g (157 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
The resulting reaction solution in acetonitrile (40 mL), 2 N hydrochloric acid water (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. Again, 2N aqueous hydrochloric acid to the organic layer (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. After filtering the resulting organic layer was concentrated under reduced pressure (400 mL). Water (360 mL) was added to the concentrated liquid, after about 1 hour stirring, the crystals were filtered, washed with 50v / v% aqueous acetonitrile (120 mL), wet product of the title compound (undried product, 62.02 g) and obtained. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(8-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 8-2), t- butyl methyl ether (200 mL), acetonitrile (40 mL), 48w / w potassium hydroxide aqueous solution (16 g) and water (200 mL) was added, I was stirred for about 2 hours at 25 ~ 35 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (120 mL), ethanol (240 mL) was added and further concentrated under reduced pressure (120 mL). After completion concentration under reduced pressure, ethanol (36 mL), and heated in water (12 mL) was added 35 ~ 45 ℃, while maintaining the 35 ~ 45 ℃ was added dropwise water (280 mL), and was crystallized crystals. After cooling the crystal exudates to room temperature, I was filtered crystal. Then washed with crystals 30v / v% aqueous ethanol solution (80 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystalline (26.26 g, 89.7% yield). Moreover, the enantiomers of the resulting crystals was 0.3%.
1 H NMR (400 MHz, CDCl 3 ) delta: 1.74 (1H, Broad singlet), 2.08 (3H, s), 3.04 (3H, s), 3.63 ~ 3.80 (4H, M), 7.36 (1H, D, J = 7.2 Hz), 7.48 (1H, s), 7.58 ~ 7.67 (2H, M), 7.77 ~ 7.90 (6H, M).
(8-4) (S)-1-(2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3- HPLC method for measuring the amount enantiomer carboxamide (%) and collected the title compound of about 10 mg is, what was the 10 mL was diluted with 50v / v% aqueous acetonitrile to obtain a sample solution.
see
(Example 12) (S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxamide (Compound (A)) Preparation of 2
(12-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H – pyrrole-3-carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
(12-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
After concentration under reduced pressure end, is added acetonitrile (10 mL) and oxalyl chloride 0.10 g (1 mmol), and cooled to 0 ~ 5 ℃ (reaction 1).
Acetonitrile (30mL), pyridine 3.15 g (40 mmol), 4- were successively added (methylsulfonyl) aniline 6.71 g (39 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
Insolubles from the resulting reaction solution was filtered, washed with acetonitrile (10 mL), and stirred for about 2 hours the addition of water (15 mL), followed by dropwise addition of water (75 mL) over about 1 hour . After about 1 hour stirring the suspension was filtered crystals were washed with 50v / v% aqueous acetonitrile (20 mL), wet product of the title compound (undried product, 15.78 g) to give a. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(12-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 12-2), t- butyl methyl ether (50 mL), acetonitrile (10 mL), 48w / w potassium hydroxide aqueous solution (4 g) and water (50 mL) was added, 15 I was about 2 hours of stirring at ~ 25 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (30 mL), was added ethanol (60 mL), was further concentrated under reduced pressure (30 mL). After completion concentration under reduced pressure, ethanol (14 mL), after addition of water (20 mL), was added a seed crystal, and was crystallized crystals. After dropwise over about 1 hour water (50 mL), and about 1 hour stirring, and crystals were filtered off. Then washed with crystals 30v / v% aqueous ethanol solution (10 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystal (6.36 g, 87.0% yield). Moreover, the enantiomers of the resulting crystals was 0.05%. Enantiomers amount, I was measured by the method of (Example 8-4). 1 H NMR (400 MHz, CDCl 3 ) delta: 1.74 (1H, Broad singlet), 2.08 (3H, s), 3.04 (3H, s), 3.63 ~ 3.80 (4H, M), 7.36 (1H, D, J = 7.2 Hz), 7.48 (1H, s), 7.58 ~ 7.67 (2H, m), 7.77 ~ 7.90 (6H, m).
………………………………………………
Patent literature
Patent Document 1: International Publication WO2006 / 012642 (US Publication US2008-0234270)
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Angiotensin II receptor 桔抗 agent
Angiotensin II receptor 桔抗 agent used as the component (A), olmesartan medoxomil, olmesartan cilexetil, losartan, candesartan cilexetil, valsartan, biphenyl tetrazole compounds such as irbesartan, biphenyl carboxylic acid compounds such as telmisartan, eprosartan, agile Sultan, and the like, preferably, a biphenyl tetrazole compound, more preferably, olmesartan medoxomil, is losartan, candesartan cilexetil, valsartan or irbesartan, particularly preferred are olmesartan medoxomil, losartan or candesartan cilexetil, Most preferably, it is olmesartan medoxomil.
Olmesartan medoxomil, JP-A-5-78328, US Patent No. 5,616,599
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
OLMESARTAN Losartan (DUP-753) is, JP 63-23868, is described in US Patent No. 5,138,069 JP like, and its chemical name is 2-butyl-4-chloro-1- [2 ‘ – The (1H- tetrazol-5-yl) biphenyl-4-ylmethyl] -1H- is imidazol-5-methanol, application of losartan includes its pharmacologically acceptable salt (losartan potassium salt, etc.).
LOSARTAN
Candesartan cilexetil, JP-A-4-364171, EP-459136 JP, is described in US Patent No. 5,354,766 JP like, and its chemical name is 1- (cyclohexyloxycarbonyloxy) ethyl-2 ethoxy-1- [2 ‘one (1H- tetrazol-5-yl) -4-Bife~eniru ylmethyl] -1H- benzimidazole-7-carboxylate is a salt application of candesartan cilexetil, which is a pharmacologically acceptable encompasses.
Valsartan (CGP-48933), the JP-A-4-159718, are described in EP-433983 JP-like, and its chemical name, (S) -N- valeryl -N- [2 ‘- (1H- tetrazol – It is a 5-yl) biphenyl-4-ylmethyl) valine, valsartan of the present application includes its pharmacologically acceptable ester or a pharmacologically acceptable salt thereof.
Irbesartan (SR-47436), the Japanese Patent Publication No. Hei 4-506222, is described in JP WO91-14679 publication, etc., its chemical name, 2-N–butyl-4-spiro cyclopentane-1- [2′ The (tetrazol-5-yl) biphenyl-4-ylmethyl] -2-imidazoline-5-one, irbesartan of the present application includes its pharmacologically acceptable salts.
Eprosartan (SKB-108566) is described in US Patent No. 5,185,351 JP etc., the chemical name, 3- [1- (4-carboxyphenyl-methyl) -2-n- butyl – imidazol-5-yl] The 2-thienyl – methyl-2-propenoic acid, present in eprosartan, the carboxylic acid derivatives, pharmacologically acceptable ester or a pharmacologically acceptable salt of a carboxylic acid derivative (eprosartan mesylate, encompasses etc.).
Telmisartan (BIBR-277) is described in US Patent No. 5,591,762 JP like, and its chemical name is 4 ‘- [[4 Mechiru 6- (1-methyl-2-benzimidazolyl) -2 – is a propyl-1-benzimidazolyl] methyl] -2-biphenylcarboxylic acid, telmisartan of the present application includes its carboxylic acid derivative, a pharmacologically acceptable ester or a pharmacologically acceptable salt thereof of carboxylic acid derivatives .
Agile Sultan, is described in Patent Publication No. 05-271228 flat JP, US Patent No. 5,243,054 JP like, and its chemical name is 2-ethoxy-1 {[2 ‘- (5-oxo-4,5-dihydro 1,2,4-oxadiazole-3-yl) biphenyl-4-yl] methyl} -1H- benzo [d] imidazole-7-carboxylic acid (2-Ethoxy-1 {[2 ‘- (5- oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) biphenyl-4-yl] is a methyl} -1H-benzo [d] imidazole-7-carboxylic acid).
Burixafor 布利沙福

Burixafor is a potent and selective chemokine CXCR4 antagonist developed by TaiGen Biotechnology (www.taigenbiotech.com.tw).
The SDF1/CXCR4 pathway plays key roles in homing and mobilization of hematopoietic stem cells and endothelial progenitor cells. In a mouse model, burixafor efficiently mobilizes stem cells (CD34+) and endothelial progenitor cells (CD133+) from bone marrow into peripheral circulation. It can be used in hematopoietic stem cell transplantation, chemotherapy sensitization and other ischemic diseases.
Because TaiGen has filed an IND (CXHL1200371) for burixafor as a chemotherapy sensitizer in October 2012, the new application (CXHL1400844) may supplement a new indication. Phase II clinical trials (NCT02104427) are currently underway in the US, with Phase IIa (NCT01018979, NCT01458288) already completed.
TaiGen plans to initiate clinical trials of burixafor as a chemotherapy sensitizer in China shortly. Burixafor’s annual sales are estimated at $1.1 billion by consultancy company JSB. This compound is protected by patent WO2009131598.

英文名称Burixafor
TG-0054
(2-{4-[6-amino-2-({[(1r,4r)-4-({[3-(cyclohexylamino)propyl]amino}methyl)cyclohexyl]methyl}amino)pyrimidin-4-yl]piperazin-1-yl}ethyl)phosphonic acid
[2-[4-[6-Amino-2-[[[trans-4-[[[3-(cyclohexylamino)propyl]amino]methyl]cyclohexyl]methyl]amino]pyrimidin-4-yl]piperazin-1-yl]ethyl]phosphonic acid
1191448-17-5
C27H51N8O3P, 566.7194
chemokine CXCR 4 receptor antagonist;
| Taigen Biotechnology Co., Ltd. |

ScinoPharm to Provide Active Pharmaceutical Ingredient to F*TaiGen for Novel Stem Cell Drug
MarketWatch
The drug has received a Clinical Trial Application from China’s FDA for the initiation of … In addition, six products have entered Phase III clinical trials.
read at

TAINAN, June 8, 2014 — ScinoPharm Taiwan, Ltd. (twse:1789) specializing in the development and manufacture of active pharmaceutical ingredients, and TaiGen Biotechnology (4157.TW; F*TaiGen) jointly announced today the signing of a manufacturing contract for the clinical supply of the API of Burixafor, a new chemical entity discovered and developed by TaiGen. The API will be manufactured in ScinoPharm’s plant in Changshu, China. This cooperation not only demonstrates Taiwan’s international competitive strength in new drug development, but also sees the beginning of a domestic pharmaceutical specialization and cooperation mechanisms, thus establishing a groundbreaking milestone for Taiwan’s pharmaceutical industry.
Dr. Jo Shen, President and CEO of ScinoPharm said, “This cooperation with TaiGen is of representative significance in the domestic pharmaceutical companies’ upstream and downstream cooperation and self-development of new drugs, and indicates the Taiwanese pharmaceutical industry’s cumulative research and development momentum is paving the way forward.” Dr. Jo Shen emphasized, “ScinoPharm’s Changshu Plant provides high-quality API R&D and manufacturing services through its fast, flexible, reliable competitive advantages, effectively assisting clients of new drugs in gaining entry into China, Europe, the United States, and other international markets.”
![]()

ScinoPharm President, CEO and Co-Founder Dr. Jo Shen
According to Dr. Ming-Chu Hsu, Chairman and CEO of TaiGen, “R&D is the foundation of the pharmaceutical industry. Once a drug is successfully developed, players at all levels of the value chain could reap the benefit. Burixafor is a 100% in-house developed product that can be used in the treatment of various intractable diseases. The cooperation between TaiGen and ScinoPharm will not only be a win-win for both sides, but will also provide high-quality novel dug for patients from around the world.”
Burixafor is a novel stem cell mobilizer that can efficiently mobilize bone marrow stem cells and tissue precursor cells to the peripheral blood. It can be used in hematopoietic stem cell transplantation, chemotherapy sensitization and other ischemic diseases. The results of the ongoing Phase II clinical trial in the United States are very impressive. The drug has received a Clinical Trial Application from China’s FDA for the initiation of a Phase II clinical trial in chemotherapy sensitization under the 1.1 category. According to the pharmaceutical consultancy company JSB, with only stem cell transplant and chemotherapy sensitizer as the indicator, Burixafor’s annual sales are estimated at USD1.1 billion.
ScinoPharm currently has accepted over 80 new drug API process research and development plans, of which five new drugs have been launched in the market. In addition, six products have entered Phase III clinical trials. Through the Changshu Plant’s operation in line with the latest international cGMP plant equipment and quality management standards, the company provides customers with one stop shopping services in professional R&D, manufacturing, and outsourcing, thereby shortening the customer development cycle of customers’ products and accelerating the launch of new products to the market.
TaiGen’s focus is on the research and development of novel drugs. Besides Burixafor, the products also include anti-infective, Taigexyn®, and an anti-hepatitis C drug, TG-2349. Taigexyn® is the first in-house developed novel drug that received new drug application approval from Taiwan’s FDA. TG-2349 is intended for the 160 million global patients with hepatitis C with huge market potential. TaiGen hopes to file one IND with the US FDA every 3-4 years to expand TaiGen’s product line.
About ScinoPharm
ScinoPharm Taiwan, Ltd. is a leading process R&D and API manufacturing service provider to the global pharmaceutical industry. With research and manufacturing facilities in both Taiwan and China, ScinoPharm offers a wide portfolio of services ranging from custom synthesis for early phase pharmaceutical activities to contract services for brand companies as well as APIs for the generic industry. For more information, please visit the Company’s website at http://www.scinopharm.com
About TaiGen Biotechnology
TaiGen Biotechnology is a leading research-based and product-driven biotechnology company in Taiwan with a wholly-owned subsidiary in Beijing, China. The company’s first product, Taigexyn®, have already received NDA approval from Taiwan’s FDA. In addition to Taigexyn®, TaiGen has two other in-house discovered NCEs in clinical development under IND with US FDA: TG-0054, a chemokine receptor antagonist for stem cell transplantation and chemosensitization, in Phase 2 and TG-2349, a HCV protease inhibitor for treatment of chronic hepatitis infection, in Phase 2. Both TG-0054 and TG-2349 are currently in clinical trials in patients in the US.
SOURCE ScinoPharm Taiwan Ltd.
TG-0054 is a potent and selective chemokine CXCR4 (SDF-1) antagonist in phase II clinical studies at TaiGen Biotechnology for use in stem cell transplantation in cancer patients. Specifically, the compound is being developed for the treatment of stem cell transplantation in multiple myeloma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma and myocardial ischemia.
Preclinical studies had also been undertaken for the treatment of diabetic retinopathy, critical limb ischemia (CLI) and age-related macular degeneration. In a mouse model, TG-0054 efficiently mobilizes stem cells (CD34+) and endothelial progenitor cells (CD133+) from bone marrow into peripheral circulation.
BACKGROUND
Chemokines are a family of cytokines that regulate the adhesion and transendothelial migration of leukocytes during an immune or inflammatory reaction (Mackay C.R., Nat. Immunol, 2001, 2:95; Olson et al, Am. J. Physiol. Regul. Integr. Comp. Physiol, 2002, 283 :R7). Chemokines also regulate T cells and B cells trafficking and homing, and contribute to the development of lymphopoietic and hematopoietic systems (Ajuebor et al, Biochem. Pharmacol, 2002, 63:1191). Approximately 50 chemokines have been identified in humans. They can be classified into 4 subfamilies, i.e., CXC, CX3C, CC, and C chemokines, based on the positions of the conserved cysteine residues at the N-terminal (Onuffer et al, Trends Pharmacol ScI, 2002, 23:459). The biological functions of chemokines are mediated by their binding and activation of G protein-coupled receptors (GPCRs) on the cell surface.
Stromal-derived factor- 1 (SDF-I) is a member of CXC chemokines. It is originally cloned from bone marrow stromal cell lines and found to act as a growth factor for progenitor B cells (Nishikawa et al, Eur. J. Immunol, 1988, 18:1767). SDF-I plays key roles in homing and mobilization of hematopoietic stem cells and endothelial progenitor cells (Bleul et al, J. Exp. Med., 1996, 184:1101; and Gazzit et al, Stem Cells, 2004, 22:65-73). The physiological function of SDF-I is mediated by CXCR4 receptor. Mice lacking SDF-I or CXCR4 receptor show lethal abnormality in bone marrow myelopoiesis, B cell lymphopoiesis, and cerebellar development (Nagasawa et al, Nature, 1996, 382:635; Ma et al, Proc. Natl. Acad. ScI, 1998, 95:9448; Zou et al, Nature, 1998, 393:595; Lu et al, Proc. Natl. Acad. ScI, 2002, 99:7090). CXCR4 receptor is expressed broadly in a variety of tissues, particularly in immune and central nervous systems, and has been described as the major co-receptor for HIV- 1/2 on T lymphocytes. Although initial interest in CXCR4 antagonism focused on its potential application to AIDS treatment (Bleul et al, Nature, 1996, 382:829), it is now becoming clear that CXCR4 receptor and SDF-I are also involved in other pathological conditions such as rheumatoid arthritis, asthma, and tumor metastases (Buckley et al., J. Immunol., 2000, 165:3423). Recently, it has been reported that a CXCR4 antagonist and an anticancer drug act synergistically in inhibiting cancer such as acute promuelocutic leukemia (Liesveld et al., Leukemia
Research 2007, 31 : 1553). Further, the CXCR4/SDF-1 pathway has been shown to be critically involved in the regeneration of several tissue injury models. Specifically, it has been found that the SDF-I level is elevated at an injured site and CXCR4-positive cells actively participate in the tissue regenerating process.
………………………………………………………………………..
http://www.google.com/patents/WO2009131598A1?cl=en


Compound 52
Example 1 : Preparation of Compounds 1

1-1 1-Ii 1-m
^ ^–\\ Λ xCUNN H ‘ ‘22.. P rdu/’C^ ^. , Λ>\V>v
Et3N, TFAA , H_, r [ Y I RRaanneeyy–NNiicckkeell u H f [ Y | NH2
CH2CI2, -10 0C Boc^ ‘NNA/ 11,,44–ddιιooxxaannee B Boocer”1^”–^^ LiOH, H2O, 50 0C
1-IV 1-V

Water (10.0 L) and (BoC)2O (3.33 kgg, 15.3 mol) were added to a solution of trans-4-aminomethyl-cyclohexanecarboxylic acid (compound 1-1, 2.0 kg, 12.7 mol) and sodium bicarbonate (2.67 kg, 31.8 mol). The reaction mixture was stirred at ambient temperature for 18 hours. The aqueous layer was acidified with concentrated hydrochloric acid (2.95 L, pH = 2) and then filtered. The resultant solid was collected, washed three times with water (15 L), and dried in a hot box (60 0C) to give trα/?5-4-(tert-butoxycarbonylamino-methyl)-cyclo-hexanecarboxylic acid (Compound l-II, 3.17 kg, 97%) as a white solid. Rf = 0.58 (EtOAc). LC-MS m/e 280 (M+Na+). 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, IH), 2.98 (t, J= 6.3 Hz, 2H), 2.25 (td, J = 12, 3.3 Hz, IH), 2.04 (d, J= 11.1 Hz, 2H), 1.83 (d, J= 11.1 Hz, 2H), 1.44 (s, 9H), 1.35-1.50 (m, 3H), 0.89-1.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 181.31, 156.08, 79.12, 46.41, 42.99, 37.57, 29.47, 28.29, 27.96. M.p. 134.8-135.0 0C. A suspension of compound l-II (1.0 kg, 3.89 mol) in THF (5 L) was cooled at
-10 0C and triethyl amine (1.076 L, 7.78 mol) and ethyl chloroformate (0.441 L, 4.47 mol) were added below -10 0C. The reaction mixture was stirred at ambient temperature for 3 hours. The reaction mixture was then cooled at -100C again and NH4OH (3.6 L, 23.34 mol) was added below -10 0C. The reaction mixture was stirred at ambient temperature for 18 hours and filtered. The solid was collected and washed three times with water (10 L) and dried in a hot box (6O0C) to give trans-4- (tert-butoxycarbonyl-amino-methyl)-cyclohexanecarboxylic acid amide (Compound l-III, 0.8 kg, 80%) as a white solid. Rf= 0.23 (EtOAc). LC-MS m/e 279, M+Na+. 1H NMR (300 MHz, CD3OD) δ 6.63 (brs, IH), 2.89 (t, J= 6.3 Hz, 2H), 2.16 (td, J = 12.2, 3.3 Hz, IH), 1.80-1.89 (m, 4H), 1.43 (s, 9H), 1.37-1.51 (m, 3H), 0.90-1.05 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 182.26, 158.85, 79.97, 47.65, 46.02, 39.28, 31.11, 30.41, 28.93. M.p. 221.6-222.0 0C.
A suspension of compound l-III (1.2 kg, 4.68 mol) in CH2Cl2 (8 L) was cooled at -1O0C and triethyl amine (1.3 L, 9.36 mol) and trifluoroacetic anhydride (0.717 L, 5.16 mol) were added below -10 0C. The reaction mixture was stirred for 3 hours. After water (2.0 L) was added, the organic layer was separated and washed with water (3.0 L) twice. The organic layer was then passed through silica gel and concentrated. The resultant oil was crystallized by methylene chloride. The crystals were washed with hexane to give £rαns-(4-cyano-cyclohexylmethyl)-carbamic acid tert-butyl ester (Compound 1-IV, 0.95 kg, 85%) as a white crystal. Rf = 0.78 (EtOAc). LC-MS m/e 261, M+Na+. 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, IH), 2.96 (t, J = 6.3 Hz, 2H), 2.36 (td, J= 12, 3.3 Hz, IH), 2.12 (dd, J= 13.3, 3.3 Hz, 2H), 1.83 (dd, J = 13.8, 2.7 Hz, 2H), 1.42 (s, 9H), 1.47-1.63 (m, 3H), 0.88-1.02 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.96, 122.41, 79.09, 45.89, 36.92, 29.06, 28.80, 28.25, 28.00. M.p. 100.4~100.6°C.
Compound 1-IV (1.0 kg, 4.196 mol) was dissolved in a mixture of 1 ,4-dioxane (8.0 L) and water (2.0 L). To the reaction mixture were added lithium hydroxide monohydrate (0.314 kg, 4.191), Raney-nickel (0.4 kg, 2.334 mol), and 10% palladium on carbon (0.46 kg, 0.216 mol) as a 50% suspension in water. The reaction mixture was stirred under hydrogen atmosphere at 5O0C for 20 hours. After the catalysts were removed by filtration and the solvents were removed in vacuum, a mixture of water (1.0 L) and CH2Cl2 (0.3 L) was added. After phase separation, the organic phase was washed with water (1.0 L) and concentrated to give £rα/?s-(4-aminomethyl- cyclohexylmethyl)-carbamic acid tert- butyl ester (compound 1-V, 0.97 kg, 95%) as pale yellow thick oil. Rf = 0.20 (MeOH/EtOAc = 9/1). LC-MS m/e 243, M+H+. 1H NMR (300 MHz, CDCl3) δ 4.67 (brs, IH), 2.93 (t, J= 6.3 Hz, 2H), 2.48 (d, J= 6.3 Hz, 2H), 1.73-1.78 (m, 4H), 1.40 (s, 9H), 1.35 (brs, 3H), 1.19-1.21 (m, IH), 0.77-0.97 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 155.85, 78.33, 48.27, 46.38, 40.80, 38.19, 29.87, 29.76, 28.07. A solution of compound 1-V (806 g) and Et3N (1010 g, 3 eq) in 1-pentanol
(2.7 L) was treated with compound 1-VI, 540 g, 1 eq) at 900C for 15 hours. TLC showed that the reaction was completed. Ethyl acetate (1.5 L) was added to the reaction mixture at 25°C. The solution was stirred for 1 hour. The Et3NHCl salt was filtered. The filtrate was then concentrated to 1.5 L (1/6 of original volume) by vacuum at 500C. Then, diethyl ether (2.5 L) was added to the concentrated solution to afford the desired product 1-VII (841 g, 68% yield) after filtration at 250C .
A solution of intermediate 1-VII (841 g) was treated with 4 N HCl/dioxane (2.7 L) in MeOH (8.1 L) and stirred at 25°C for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated to 1.5 L (1/7 of original volume) by vacuum at 500C. Then, diethyl ether (5 L) was added to the solution slowly, and HCl salt of 1-VIII (774 g) was formed, filtered, and dried under vacuum (<10 torr). For neutralization, K2CO3 (2.5 kg, 8 eq) was added to the solution of HCl salt of 1-VIII in MeOH (17 L) at 25°C. The mixture was stirred at the same temperature for 3 hours (pH > 12) and filtered (estimated amount of 1-VIII in the filtrate is 504 g). Aldehyde 1-IX (581 g, 1.0 eq based on mole of 1-VII) was added to the filtrate of 1-VIII at 0-100C. The reaction was stirred at 0-100C for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (81 g, 1.0 eq based on mole of 1-VII) was added at less than 100C and the solution was stirred at 10-150C for Ih. The solution was concentrated to get a residue, which then treated with CH2Cl2 (15 L). The mixture was washed with saturated aq. NH4Cl solution (300 mL) diluted with H2O (1.2 L). The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (short column, EtOAc as mobile phase for removing other components; MeOH/28% NH4OH = 97/3 as mobile phase for collecting 1-X) afforded crude 1-X (841 g). Then Et3N (167 g, leq) and BoC2O (360 g, leq) were added to the solution of
1-X (841 g) in CH2Cl2 (8.4 L) at 25°C. The mixture was stirred at 25°C for 15 hours. After the reaction was completed as evidenced by TLC, the solution was concentrated and EtOAc (5 L) was added to the resultant residue. The solution was concentrated to 3L (1/2 of the original volume) under low pressure at 500C. Then, n-hexane (3 L) was added to the concentrated solution. The solid product formed at 500C by seeding to afford the desired crude product 1-XI (600 g, 60% yield) after filtration and evaporation. To compound 1-XI (120.0 g) and piperazine (1-XII, 50.0 g, 3 eq) in 1- pentanol (360 niL) was added Et3N (60.0 g, 3.0 eq) at 25°C. The mixture was stirred at 1200C for 8 hours. Ethyl acetate (480 mL) was added to the reaction mixture at 25°C. The solution was stirred for Ih. The Et3NHCl salt was filtered and the solution was concentrated and purified by silica gel (EtOAc/MeOH = 2:8) to afforded 1-XIII (96 g) in a 74% yield.
A solution of intermediate 1-XIII (100 mg) was treated with 4 N HCl/dioxane (2 mL) in CH2Cl2 (1 mL) and stirred at 25°C for 15 hours. The mixture was concentrated to give hydrochloride salt of compound 1 (51 mg). CI-MS (M+ + 1): 459.4
Example 2: Preparation of Compound 2

Compound 2 Intermediate 1-XIII was prepared as described in Example 1.
To a solution of 1-XIII (120 g) in MeOH (2.4 L) were added diethyl vinyl phosphonate (2-1, 45 g, 1.5 eq) at 25°C. The mixture was stirred under 65°C for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2 = 8/92) to get 87 g of 2-11 (53% yield, purity > 98%, each single impurity <1%) after analyzing the purity of the product by HPLC.
A solution of 20% TFA/CH2C12 (36 mL) was added to a solution of intermediate 2-11 (1.8 g) in CH2Cl2 (5 mL). The reaction mixture was stirred for 15 hours at room temperature and concentrated by removing the solvent to afford trifluoracetic acid salt of compound 2 (1.3 g). CI-MS (M+ + 1): 623.1
Example 3 : Preparation of Compound 3
TMSBr H H


s U
Intermediate 2-11 was prepared as described in Example 2. To a solution of 2-11 (300 g) in CH2Cl2 (1800 mL) was added TMSBr (450 g, 8 eq) at 10-150C for 1 hour. The mixture was stirred at 25°C for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 400C.
CH2Cl2 was added to the mixture to dissolve the residue. TMSBr and solvent were removed under vacuum again to obtain 36O g crude solid after drying under vacuum (<1 torr) for 3 hours. Then, the crude solid was washed with 7.5 L IPA/MeOH (9/1) to afford compound 3 (280 g) after filtration and drying at 25°C under vacuum (<1 torr) for 3 hours. Crystallization by EtOH gave hydrobromide salt of compound 3 (19Og). CI-MS (M+ + 1): 567.0.
The hydrobromide salt of compound 3 (5.27 g) was dissolved in 20 mL water and treated with concentrated aqueous ammonia (pH=9-10), and the mixture was evaporated in vacuo. The residue in water (30 mL) was applied onto a column (100 mL, 4.5×8 cm) of Dowex 50WX8 (H+ form, 100-200 mesh) and eluted (elution rate, 6 mL/min). Elution was performed with water (2000 mL) and then with 0.2 M aqueous ammonia. The UV-absorbing ammonia eluate was evaporated to dryness to afford ammonia salt of compound 3 (2.41 g). CI-MS (M+ + 1): 567.3.
The ammonia salt of compound 3 (1.5 g) was dissolved in water (8 mL) and alkalified with concentrated aqueous ammonia (pH=l 1), and the mixture solution was applied onto a column (75 mL, 3×14 cm) of Dowex 1X2 (acetate form, 100-200 mesh) and eluted (elution rate, 3 mL/min). Elution was performed with water (900 mL) and then with 0.1 M acetic acid. The UV-absorbing acetic acid eluate was evaporated, and the residue was codistilled with water (5×50 mL) to afford compound 3 (1.44 g). CI-MS (M+ + 1): 567.4. Example 4: Preparation of Compound 4

Compound 4
Intermediate 1-XIII was obtained during the preparation of compound 1. To a solution of diethyl vinyl phosphonate (4-1, 4 g) in CH2Cl2 (120 mL) was added oxalyl chloride (15.5 g, 5 eq) and the mixture was stirred at 300C for 36 hours. The mixture were concentrated under vacuum on a rotatory evaporated to give quantitatively the corresponding phosphochloridate, which was added to a mixture of cyclohexyl amine (4-II, 5.3 g, 2.2 eq), CH2Cl2 (40 mL), and Et3N (6.2 g, 2.5 eq). The mixture was stirred at 35°C for 36 hours, and then was washed with water. The organic layer was dried (MgSO4), filtered, and evaporated to afford 4-III (4.7 g, 85% yield) as brown oil.
Compound 4-III (505 mg) was added to a solution of intermediate 1-XIII (500 mg) in MeOH (4 mL). The solution was stirred at 45°C for 24 hours. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/ MeOH = 4: 1) to afford intermediate 4-IV (420 mg) in a 63% yield.
A solution of HCl in ether (5 mL) was added to a solution of intermediate 4- IV (420 mg) in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 12 hours at room temperature and concentrated by removing the solvent. The resultant residue was washed with ether to afford hydrochloride salt of compound 4 (214 mg). CI-MS (M+ + 1): 595.1
Preparation of compound 51

TMSBr

Intermediate l-II was prepared as described in Example 1. To a suspension of the intermediate l-II (31.9 g) in toluene (150 mL) were added phosphorazidic acid diphenyl ester (51-1, 32.4 g) and Et3N (11.9 g) at 25°C for 1 hour. The reaction mixture was stirred at 800C for 3 hours and then cooled to 25°C. After benzyl alcohol (51-11, 20 g) was added, the reaction mixture was stirred at 800C for additional 3 hours and then warmed to 1200C overnight. It was then concentrated and dissolved again in EtOAc and H2O. The organic layer was collected. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with 2.5 N HCl, saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (EtOAc/Hexane = 1 :2) to give Intermediate 51-111 (35 g) in a 79% yield. A solution of intermediate 51-111 (35 g) treated with 4 N HCl/dioxane (210 rnL) in MeOH (350 mL) was stirred at room temperature overnight. After ether (700 mL) was added, the solution was filtered. The solid was dried under vacuum. K2CO3 was added to a suspension of this solid in CH3CN and ώo-propanol at room temperature for 10 minutes. After water was added, the reaction mixture was stirred at room temperature for 2 hours, filtered, dried over anhydrous MgSO4, and concentrated. The resultant residue was purified by column chromatography on silica gel (using CH2Cl2 and MeOH as an eluant) to give intermediate 51-IV (19 g) in a 76% yield. Intermediate 1-IX (21 g) was added to a solution of intermediate 51-IV (19 g) in CH2Cl2 (570 mL). The mixture was stirred at 25°C for 2 hours. NaBH(OAc)3 (23 g) was then added at 25°C overnight. After the solution was concentrated, a saturated aqueous NaHCO3solution was added to the resultant residue. The mixture was then extracted with CH2Cl2. The solution was concentrated and the residue was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 51-V (23.9 g) in a 66% yield.
A solution of intermediate 51-V (23.9 g) and BoC2O (11.4 g) in CH2Cl2 (200 mL) was added to Et3N (5.8 mL) at 25°C for overnight. The solution was then concentrated and the resultant residue was purified by column chromatography on silica gel (using EtOAc and Hexane as an eluant) to give intermediate 51-VI (22 g) in a 77% yield.
10% Pd/C (2.2 g) was added to a suspension of intermediate 51-VI (22 g) in MeOH (44 mL). The mixture was stirred at ambient temperature under hydrogen atmosphere overnight, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 51-VII (16.5 g) in a 97% yield.
Intermediate 51-VII (16.5 g) and Et3N (4.4 mL) in 1-pentanol (75 mL) was allowed to react with 2,4-dichloro-6-aminopyrimidine (1-VI, 21 g) at 1200C overnight. The solvent was then removed and the residue was purified by column chromatography on silica gel (using EtOAc and hexane as an eluant) to afford intermediate 51-VIII (16.2 g) in a 77% yield.
A solution of intermediate 51-VIII (16.2 g) and piperazine (1-XII, 11.7 g) in 1-pentanol (32 mL) was added to Et3N (3.3 mL) at 1200C overnight. After the solution was concentrated, the residue was treated with water and extracted with CH2Cl2. The organic layer was collected and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc/ MeOH to 28% NH40H/Me0H as an eluant) to afford Intermediate 51-IX (13.2 g) in a 75% yield. Diethyl vinyl phosphonate (2-1) was treated with 51-IX as described in
Example 3 to afford hydrobromide salt of compound 51. CI-MS (M+ + 1): 553.3


………………………………….
Preparation of Compound 1


Water (10.0 L) and (Boc)2O (3.33 kgg, 15.3 mol) were added to a solution of trans-4-aminomethyl-cyclohexanecarboxylic acid (compound 1-I, 2.0 kg, 12.7 mol) and sodium bicarbonate (2.67 kg, 31.8 mol). The reaction mixture was stirred at ambient temperature for 18 hours. The aqueous layer was acidified with concentrated hydrochloric acid (2.95 L, pH=2) and then filtered. The resultant solid was collected, washed three times with water (15 L), and dried in a hot box (60° C.) to give trans-4-(tert-butoxycarbonylamino-methyl)-cyclo-hexanecarboxylic acid (Compound 1-II, 3.17 kg, 97%) as a white solid. Rf=0.58 (EtOAc). LC-MS m/e 280 (M+Na+). 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, 1H), 2.98 (t, J=6.3 Hz, 2H), 2.25 (td, J=12, 3.3 Hz, 1H), 2.04 (d, J=11.1 Hz, 2H), 1.83 (d, J=11.1 Hz, 2H), 1.44 (s, 9H), 1.35˜1.50 (m, 3H), 0.89˜1.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 181.31, 156.08, 79.12, 46.41, 42.99, 37.57, 29.47, 28.29, 27.96. M.p. 134.8˜135.0° C.
A suspension of compound 1-II (1.0 kg, 3.89 mol) in THF (5 L) was cooled at 10° C. and triethyl amine (1.076 L, 7.78 mol) and ethyl chloroformate (0.441 L, 4.47 mol) were added below 10° C. The reaction mixture was stirred at ambient temperature for 3 hours. The reaction mixture was then cooled at 10° C. again and NH4OH (3.6 L, 23.34 mol) was added below 10° C. The reaction mixture was stirred at ambient temperature for 18 hours and filtered. The solid was collected and washed three times with water (10 L) and dried in a hot box (60° C.) to give trans-4-(tert-butoxycarbonyl-amino-methyl)-cyclohexanecarboxylic acid amide (Compound 1-III, 0.8 kg, 80%) as a white solid. Rf=0.23 (EtOAc). LC-MS m/e 279, M+Na+. 1H NMR (300 MHz, CD3OD) δ 6.63 (brs, 1H), 2.89 (t, J=6.3 Hz, 2H), 2.16 (td, J=12.2, 3.3 Hz, 1H), 1.80˜1.89 (m, 4H), 1.43 (s, 9H), 1.37˜1.51 (m, 3H), 0.90˜1.05 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 182.26, 158.85, 79.97, 47.65, 46.02, 39.28, 31.11, 30.41, 28.93. M.p. 221.6˜222.0° C.
A suspension of compound 1-III (1.2 kg, 4.68 mol) in CH2Cl2 (8 L) was cooled at 10° C. and triethyl amine (1.3 L, 9.36 mol) and trifluoroacetic anhydride (0.717 L, 5.16 mol) were added below 10° C. The reaction mixture was stirred for 3 hours. After water (2.0 L) was added, the organic layer was separated and washed with water (3.0 L) twice. The organic layer was then passed through silica gel and concentrated. The resultant oil was crystallized by methylene chloride. The crystals were washed with hexane to give trans-(4-cyano-cyclohexylmethyl)-carbamic acid tent-butyl ester (Compound 1-IV, 0.95 kg, 85%) as a white crystal. Rf=0.78 (EtOAc). LC-MS m/e 261, M+Na+. 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, 1H), 2.96 (t, J=6.3 Hz, 2H), 2.36 (td, J=12, 3.3 Hz, 1H), 2.12 (dd, J=13.3, 3.3 Hz, 2H), 1.83 (dd, J=13.8, 2.7 Hz, 2H), 1.42 (s, 9H), 1.47˜1.63 (m, 3H), 0.88˜1.02 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.96, 122.41, 79.09, 45.89, 36.92, 29.06, 28.80, 28.25, 28.00. M.p. 100.4˜100.6° C.
Compound 1-IV (1.0 kg, 4.196 mol) was dissolved in a mixture of 1,4-dioxane (8.0 L) and water (2.0 L). To the reaction mixture were added lithium hydroxide monohydrate (0.314 kg, 4.191), Raney-nickel (0.4 kg, 2.334 mol), and 10% palladium on carbon (0.46 kg, 0.216 mol) as a 50% suspension in water. The reaction mixture was stirred under hydrogen atmosphere at 50° C. for 20 hours. After the catalysts were removed by filtration and the solvents were removed in vacuum, a mixture of water (1.0 L) and CH2Cl2 (0.3 L) was added. After phase separation, the organic phase was washed with water (1.0 L) and concentrated to give trans-(4-aminomethyl-cyclohexylmethyl)-carbamic acid tert-butyl ester (compound 1-V, 0.97 kg, 95%) as pale yellow thick oil. Rf=0.20 (MeOH/EtOAc=9/1). LC-MS m/e 243, M+H+. 1H NMR (300 MHz, CDCl3) δ 4.67 (brs, 1H), 2.93 (t, J=6.3 Hz, 2H), 2.48 (d, J=6.3 Hz, 2H), 1.73˜1.78 (m, 4H), 1.40 (s, 9H), 1.35 (brs, 3H), 1.19˜1.21 (m, 1H), 0.77˜0.97 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 155.85, 78.33, 48.27, 46.38, 40.80, 38.19, 29.87, 29.76, 28.07.
A solution of compound 1-V (806 g) and Et3N (1010 g, 3 eq) in 1-pentanol (2.7 L) was treated with compound 1-VI, 540 g, 1 eq) at 90° C. for 15 hours. TLC showed that the reaction was completed.
Ethyl acetate (1.5 L) was added to the reaction mixture at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was filtered. The filtrate was then concentrated to 1.5 L (1/6 of original volume) by vacuum at 50° C. Then, diethyl ether (2.5 L) was added to the concentrated solution to afford the desired product 1-VII (841 g, 68% yield) after filtration at 25° C.
A solution of intermediate 1-VII (841 g) was treated with 4 N HCl/dioxane (2.7 L) in MeOH (8.1 L) and stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated to 1.5 L (1/7 of original volume) by vacuum at 50° C. Then, diethyl ether (5 L) was added to the solution slowly, and HCl salt of 1-VIII (774 g) was formed, filtered, and dried under vacuum (<10 ton). For neutralization, K2CO3 (2.5 kg, 8 eq) was added to the solution of HCl salt of 1-VIII in MeOH (17 L) at 25° C. The mixture was stirred at the same temperature for 3 hours (pH>12) and filtered (estimated amount of 1-VIII in the filtrate is 504 g).
Aldehyde 1-IX (581 g, 1.0 eq based on mole of 1-VII) was added to the filtrate of 1-VIII at 0-10° C. The reaction was stirred at 0-10° C. for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (81 g, 1.0 eq based on mole of 1-VII) was added at less than 10° C. and the solution was stirred at 10-15° C. for 1 h. The solution was concentrated to get a residue, which then treated with CH2Cl2 (15 L). The mixture was washed with saturated aq. NH4Cl solution (300 mL) diluted with H2O (1.2 L). The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (short column, EtOAc as mobile phase for removing other components; MeOH/28% NH4OH=97/3 as mobile phase for collecting 1-X) afforded crude 1-X (841 g).
Then Et3N (167 g, 1 eq) and Boc2O (360 g, 1 eq) were added to the solution of 1-X (841 g) in CH2Cl2 (8.4 L) at 25° C. The mixture was stirred at 25° C. for 15 hours. After the reaction was completed as evidenced by TLC, the solution was concentrated and EtOAc (5 L) was added to the resultant residue. The solution was concentrated to 3 L (1/2 of the original volume) under low pressure at 50° C. Then, n-hexane (3 L) was added to the concentrated solution. The solid product formed at 50° C. by seeding to afford the desired crude product 1-XI (600 g, 60% yield) after filtration and evaporation.
To compound 1-XI (120.0 g) and piperazine (1-XII, 50.0 g, 3 eq) in 1-pentanol (360 mL) was added Et3N (60.0 g, 3.0 eq) at 25° C. The mixture was stirred at 120° C. for 8 hours. Ethyl acetate (480 mL) was added to the reaction mixture at 25° C. The solution was stirred for 1 h. The Et3NHCl salt was filtered and the solution was concentrated and purified by silica gel (EtOAc/MeOH=2:8) to afforded 1-XIII (96 g) in a 74% yield.
To a solution of 1-XIII (120 g) in MeOH (2.4 L) were added diethyl vinyl phosphonate (1-XIV, 45 g, 1.5 eq) at 25° C. The mixture was stirred under 65° C. for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2=8/92) to get 87 g of 1-XV (53% yield, purity>98%, each single impurity<1%) after analyzing the purity of the product by HPLC.
A solution of 20% TFA/CH2Cl2 (36 mL) was added to a solution of intermediate 1-XV (1.8 g) in CH2Cl2 (5 mL). The reaction mixture was stirred for 15 hours at room temperature and concentrated by removing the solvent to afford trifluoracetic acid salt of compound 1 (1.3 g).
CI-MS (M++1): 623.1.
(2) Preparation of Compound 2

Intermediate 1-XV was prepared as described in Example 1.
To a solution of 1-XV (300 g) in CH2Cl2 (1800 mL) was added TMSBr (450 g, 8 eq) at 10-15° C. for 1 hour. The mixture was stirred at 25° C. for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 40° C. CH2Cl2 was added to the mixture to dissolve the residue. TMSBr and solvent were removed under vacuum again to obtain 360 g crude solid after drying under vacuum (<1 torr) for 3 hours. Then, the crude solid was washed with 7.5 L IPA/MeOH (9/1) to afford compound 2 (280 g) after filtration and drying at 25° C. under vacuum (<1 ton) for 3 hours. Crystallization by EtOH gave hydrobromide salt of compound 2 (190 g). CI-MS (M++1): 567.0.
The hydrobromide salt of compound 2 (5.27 g) was dissolved in 20 mL water and treated with concentrated aqueous ammonia (pH=9-10), and the mixture was evaporated in vacuo. The residue in water (30 mL) was applied onto a column (100 mL, 4.5×8 cm) of Dowex 50WX8 (H+ form, 100-200 mesh) and eluted (elution rate, 6 mL/min). Elution was performed with water (2000 mL) and then with 0.2 M aqueous ammonia. The UV-absorbing ammonia eluate was evaporated to dryness to afford ammonia salt of compound 2 (2.41 g). CI-MS (M++1): 567.3.
The ammonia salt of compound 2 (1.5 g) was dissolved in water (8 mL) and alkalified with concentrated aqueous ammonia (pH=11), and the mixture solution was applied onto a column (75 mL, 3×14 cm) of Dowex 1×2 (acetate form, 100-200 mesh) and eluted (elution rate, 3 mL/min). Elution was performed with water (900 mL) and then with 0.1 M acetic acid. The UV-absorbing acetic acid eluate was evaporated, and the residue was codistilled with water (5×50 mL) to afford compound 2 (1.44 g). CI-MS (M++1): 567.4.
(3) Preparation of Compound 3

Intermediate 1-XIII was obtained during the preparation of compound 1.
To a solution of diethyl vinyl phosphonate (3-I, 4 g) in CH2Cl2 (120 mL) was added oxalyl chloride (15.5 g, 5 eq) and the mixture was stirred at 30° C. for 36 hours. The mixture were concentrated under vacuum on a rotatory evaporated to give quantitatively the corresponding phosphochloridate, which was added to a mixture of cyclohexyl amine (3-II, 5.3 g, 2.2 eq), CH2Cl2 (40 mL), and Et3N (6.2 g, 2.5 eq). The mixture was stirred at 35° C. for 36 hours, and then was washed with water. The organic layer was dried (MgSO4), filtered, and evaporated to afford 3-III (4.7 g, 85% yield) as brown oil.
Compound 3-III (505 mg) was added to a solution of intermediate 1-XIII (500 mg) in MeOH (4 mL). The solution was stirred at 45° C. for 24 hours. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/MeOH=4:1) to afford intermediate 3-IV (420 mg) in a 63% yield.
A solution of HCl in ether (5 mL) was added to a solution of intermediate 3-IV (420 mg) in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 12 hours at room temperature and concentrated by removing the solvent. The resultant residue was washed with ether to afford hydrochloride salt of compound 3 (214 mg).
CI-MS (M++1): 595.1.
(4) Preparation of Compound 4

Compound 4 was prepared in the same manner as that described in Example 2 except that sodium 2-bromoethanesulfonate in the presence of Et3N in DMF at 45° C. was used instead of diethyl vinyl phosphonate. Deportations of amino-protecting group by hydrochloride to afford hydrochloride salt of compound 4.
CI-MS (M++1): 567.3
(5) Preparation of Compound 5

Compound 5 was prepared in the same manner as that described in Example 2 except that diethyl-1-bromopropylphosphonate in the presence of K2CO3 in CH3CN was used instead of diethyl vinyl phosphonate.
CI-MS (M++1): 581.4
(6) Preparation of Compound 6

Compound 6 was prepared in the same manner as that described in Example 5 except that 1,4-diaza-spiro[5.5]undecane dihydrochloride was used instead of piperazine.
CI-MS (M++1): 649.5
(7) Preparation of Compound 7


Intermediate 1-II was prepared as described in Example 1.
To a suspension of the intermediate 1-II (31.9 g) in toluene (150 mL) were added phosphorazidic acid diphenyl ester (7-I, 32.4 g) and Et3N (11.9 g) at 25° C. for 1 hour. The reaction mixture was stirred at 80° C. for 3 hours and then cooled to 25° C. After benzyl alcohol (7-II, 20 g) was added, the reaction mixture was stirred at 80° C. for additional 3 hours and then warmed to 120° C. overnight. It was then concentrated and dissolved again in EtOAc and H2O. The organic layer was collected. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with 2.5 N HCl, saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (EtOAc/Hexane=1:2) to give Intermediate 7-III (35 g) in a 79% yield.
A solution of intermediate 7-III (35 g) treated with 4 N HCl/dioxane (210 mL) in MeOH (350 mL) was stirred at room temperature overnight. After ether (700 mL) was added, the solution was filtered. The solid was dried under vacuum. K2CO3 was added to a suspension of this solid in CH3CN and iso-propanol at room temperature for 10 minutes. After water was added, the reaction mixture was stirred at room temperature for 2 hours, filtered, dried over anhydrous MgSO4, and concentrated. The resultant residue was purified by column chromatography on silica gel (using CH2Cl2 and MeOH as an eluant) to give intermediate 7-IV (19 g) in a 76% yield.
Intermediate 1-IX (21 g) was added to a solution of intermediate 7-IV (19 g) in CH2Cl2 (570 mL). The mixture was stirred at 25° C. for 2 hours. NaBH(OAc)3(23 g) was then added at 25° C. overnight. After the solution was concentrated, a saturated aqueous NaHCO3 solution was added to the resultant residue. The mixture was then extracted with CH2Cl2. The solution was concentrated and the residue was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 7-V (23.9 g) in a 66% yield.
A solution of intermediate 7-V (23.9 g) and Boc2O (11.4 g) in CH2Cl2 (200 mL) was added to Et3N (5.8 mL) at 25° C. for overnight. The solution was then concentrated and the resultant residue was purified by column chromatography on silica gel (using EtOAc and Hexane as an eluant) to give intermediate 7-VI (22 g) in a 77% yield. 10% Pd/C (2.2 g) was added to a suspension of intermediate 7-VI (22 g) in MeOH (44 mL). The mixture was stirred at ambient temperature under hydrogen atmosphere overnight, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 7-VII (16.5 g) in a 97% yield.
Intermediate 7-VII (16.5 g) and Et3N (4.4 mL) in 1-pentanol (75 mL) was allowed to react with 2,4-dichloro-6-aminopyrimidine (1-VI, 21 g) at 120° C. overnight. The solvent was then removed and the residue was purified by column chromatography on silica gel (using EtOAc and hexane as an eluant) to afford intermediate 7-VIII (16.2 g) in a 77% yield.
A solution of intermediate 7-VIII (16.2 g) and piperazine (1-XII, 11.7 g) in 1-pentanol (32 mL) was added to Et3N (3.3 mL) at 120° C. overnight. After the solution was concentrated, the residue was treated with water and extracted with CH2Cl2. The organic layer was collected and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc/MeOH to 28% NH4OH/MeOH as an eluant) to afford Intermediate 7-IX (13.2 g) in a 75% yield.
Diethyl vinyl phosphonate (2-I) was treated with 7-IX as described in Example 3 to afford hydrobromide salt of compound 7.
CI-MS (M++1): 553.3
(8) Preparation of Compound 8


Cis-1,4-cyclohexanedicarboxylic acid (8-I, 10 g) in THF (100 ml) was added oxalyl chloride (8-II, 15.5 g) at 0° C. and then DMF (few drops). The mixture was stirred at room temperature for 15 hours. The solution was concentrated and the residue was dissolved in THF (100 ml). The mixture solution was added to ammonium hydroxide (80 ml) and stirred for 1 hour. The solution was concentrated and filtration to afford crude product 8-III (7.7 g).
Compound 8-III (7.7 g) in THF (200 ml) was slowly added to LiAlH4 (8.6 g) in THF (200 ml) solution at 0° C. The mixture solution was stirred at 65° C. for 15 hours. NaSO4.10H2O was added at room temperature and stirred for 1 hours. The resultant mixture was filtered to get filtrate and concentrated. The residue was dissolved in CH2Cl2 (100 ml). Et3N (27 g) and (Boc)2O (10 g) were added at room temperature. The solution was stirred for 15 h, and then concentrated to get resultant residue. Ether was added to the resultant residue. Filtration and drying under vacuum afforded solid crude product 8-IV (8.8 g).
A solution of compound 8-IV (1.1 g) and Et3N (1.7 g) in 1-pentanol (10 ml) was reacted with 2,4-dichloro-6-aminopyrimidine (1-VI, 910 mg) at 90° C. for 15 hours. TLC showed that the reaction was completed. Ethyl acetate (10 mL) was added to the reaction mixture at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was removed. The filtrate was concentrated and purified by silica gel (EtOAc/Hex=1:2) to afford the desired product 8-V (1.1 g, 65% yield).
A solution of intermediate 8-V (1.1 g) was treated with 4 N HCl/dioxane (10 ml) in MeOH (10 ml) and stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated, filtered, and dried under vacuum (<10 ton). For neutralization, K2CO3 (3.2 g) was added to the solution of HCl salt in MeOH (20 ml) at 25° C. The mixture was stirred at the same temperature for 3 hours (pH>12) and filtered. Aldehyde 1-IX (759 mg) was added to the filtrate at 0-10° C. The reaction was stirred at 0-10° C. for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (112 mg) was added at less than 10° C. and the solution was stirred at 10-15° C. for 1 hour. The solution was concentrated to get a residue, which was then treated with CH2Cl2 (10 mL). The mixture was washed with saturated NH4Cl (aq) solution. The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (MeOH/28% NH4OH=97/3) to afford intermediate 8-VI (1.0 g, 66% yield).
Et3N (600 mg) and Boc2O (428 mg) were added to the solution of 8-VI (1.0 g) in CH2Cl2 (10 ml) at 25° C. The mixture was stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The solution was concentrated and purified by chromatography on silica gel (EtOAc/Hex=1:1) to afford intermediate 8-VII (720 mg, 60% yield).
To a solution compound 8-VII (720 mg) and piperazine (1-XII, 1.22 g) in 1-pentanol (10 mL) was added Et3N (1.43 g) at 25° C. The mixture was stirred at 120° C. for 24 hours. TLC showed that the reaction was completed. Ethyl acetate (20 mL) was added at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was removed and the solution was concentrated and purified by silica gel (EtOAc/MeOH=2:8) to afford 8-VIII (537 mg) in 69% yield.
To a solution of 8-VIII (537 mg) in MeOH (11 ml) was added diethyl vinyl phosphonate (2-I, 201 mg) at 25° C. The mixture was stirred under 65° C. for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2=1:9) to get 8-IX (380 mg) in a 57% yield.
To a solution of 8-IX (210 mg) in CH2Cl2 (5 ml) was added TMSBr (312 mg) at 10-15° C. for 1 hour. The mixture was stirred at 25° C. for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 40° C., then, CH2Cl2 was added to dissolve the residue. Then TMSBr and solvent were further removed under vacuum and CH2Cl2 was added for four times repeatedly. The solution was concentrated to get hydrobromide salt of compound 8 (190 mg).
CI-MS (M++1): 566.9
To do a job well is one thing, but to consistently deliver a product that is nearly flawless is quite a different challenge. For its new molecule burixafor, the Taiwanese drug discovery firm TaiGen Biotechnology instructed its contract manufacturing partners to achieve 99.8% purity in the production of the active pharmaceutical ingredient (API).

Discovered in TaiGen’s labs in 2006, burixafor is in Phase II clinical trials in both the U.S. and China for use in stem cell transplants and cancer chemotherapy. Avecia, a unit of Japan’s Nitto Denko, manufactures the drug substance in the U.S., where burixafor was tested for the first time on human patients. When TaiGen later initiated clinical trials in China, it chose the Taiwanese firm ScinoPharm to produce the drug at its plant in Changshu, near Shanghai. Under Chinese law, only drugs made domestically can be tested in China.


NITTO DENKO Avecia Inc.
It is rare for a drug discovery firm to select two companies to scale up the production of a new molecule. TaiGen went one step further by paying both contract manufacturers to reach an extremely high level of purity.
“We are trying to avoid any unwanted side effects during the trials,” says C. Richard King, TaiGen’s senior vice president of research. Drug regulators in the U.S. and China “need very tight specifications these days for new drugs,” he adds.
TaiGen registered burixafor with the U.S. Food & Drug Administration in 2007. When it contracted Girindus America (bought by Avecia in 2013) to manufacture it that year, TaiGen specified purification by column chromatography, a cumbersome and relatively expensive procedure when carried out on a large scale. “Our process development efforts were racing against the clinical trials launch schedule,” King recalls. Column chromatography, he points out, is a “tedious approach, but it works.”
By the time ScinoPharm was hired last year, TaiGen’s process development team had come up with a simpler and more elegant process. But its purity demands hadn’t changed.
“Usually, clients are satisfied with a purity level of 98% to 99%,” says Koksuan Tang, head of operations at ScinoPharm’s Changshu plant. “To go from 99% to 99.8% is very different.” The manufacturing of burixafor, he adds, involves five chemical steps and two purification steps. Upstream of the API, ScinoPharm also produces burixafor’s starting material.
Purity level aside, burixafor is not a particularly difficult compound to make, Tang says. Nonetheless, the process supplied by TaiGen had to be adjusted for larger-scale production. “If you heat up 10 g in the lab, it takes two minutes, but in a plant, it could take as long as two hours,” he says.
Although, while hydrogen chloride gas can be controlled effectively when making minute quantities of a compound in the lab, it’s another challenge to handle large volumes of the toxic substance at the plant level. To safely execute one reaction step, ScinoPharm dissolved HCl in a special solvent that does not affect the purity profile of burixafor.
TaiGen selected ScinoPharm as its China contractor after a careful process that involved two visits to Changshu by TaiGen’s senior managers, Tang recalls. ScinoPharm’s track record of meeting regulatory requirements in different countries, including China, was a plus, Tang believes. Its ability to produce both for clinical trials and in larger quantities after commercial launch was also decisive.
Operational since 2012, ScinoPharm’s Changshu site can deliver products under Good Manufacturing Practices in quantities ranging from grams to kilograms. It employs 220 people.

ScinoPharm (Changshu) Pharmaceuticals, Ltd.
“Moving from the single-kilogram quantities we make now to hundreds of kilograms will require some adjustment to the process, but we believe we can deliver,” says Tang’s colleague Sing Ping Lee, senior director of product technical support in Changshu. One thing to keep in mind, he notes, is that Chinese regulatory standards for drug production are actually more restrictive than those in the U.S. or Europe, going so far as specifying what equipment manufacturers need to use.
Other than complying with Chinese regulators, one reason TaiGen needed to carefully select its China contractor is that the two companies could well be long-term partners, since TaiGen believes it has the ability to market the drug on its own in China, Taiwan, and Southeast Asia. In the event of approvals elsewhere, TaiGen plans to license the compound to a large drug company, which may or may not stick with ScinoPharm or Avecia.
Relatively unknown outside Taiwan, TaiGen was formed in 2001 by Ming-Chu Hsu, the founder of the Division of Biotechnology & Pharmaceutical Research at Taiwan’s National Health Research Institutes. The holder of a Ph.D. in biochemistry from the University of Illinois, Urbana-Champaign, she headed oncology and virology research at Roche for more than 10 years before returning to Taiwan in 1998.

Ming-Chu Hsu, Chairman & CEO, TaiGen Biotechnology, Taiwan
TaiGen employs about 80 people, three-quarters of whom are in R&D. The company develops its own drugs in-house and also in-licenses molecules that are in early stages of development. The company licenses out the molecules for the European Union and U.S. markets but seeks to retain Asian marketing rights. Burixafor was discovered in TaiGen’s own labs in Taipei. To come up with it, researchers used a high-throughput screening approach that involved 130,000 compounds, including the design and synthesis of 1,500 new compounds. “It went back and forth between chemistry and biology many times,” recalls King, TaiGen’s research head.
A so-called CXCR4 chemokine receptor antagonist, burixafor mobilizes hematopoietic stem cells and endothelial progenitor cells in human bone marrow and channels them into the peripheral blood within three hours of ingestion, according to results of Phase I and Phase II trials.
In the U.S., burixafor is undergoing clinical trials for use during stem cell transplantation in patients with multiple myeloma, non-Hodgkin’s lymphoma, or Hodgkin’s disease. In China, TaiGen is testing it as a chemotherapy sensitizer in relapsed or refractory adult acute myeloid leukemia.
Owing to its activity on CXCR4 chemokine receptors, the drug could also fight age-related macular degeneration and diabetic retinopathy diseases, as well as find use in tissue repair, King says. For clinical trials in the U.S., TaiGen has partnered with Michael W. Schuster, a medical doctor who conducts research at Stony Brook University Hospital in New York.

Dr. Michael Schuster is Gift of Life’s Medical Director, as well as the Director of the Hematopoietic Stem Cell Transplantation Program and Hematologic Malignancy Program of Stony Brook University Hospital in New York

Typical structure of a chemokine receptor
TaiGen sees particular potential for burixafor in stem cell applications. For example, patients undergoing hematopoietic stem cell transplantation often must take a granulocyte colony-stimulating factor plus a Sanofi drug called Mozobil to stimulate stem cell production. TaiGen says burixafor could accomplish this goal on its own in multiple myeloma patients. It cites one consulting firm forecast that puts eventual sales at more than $1 billion per year.

Sanofi drug called Mozobil to stimulate stem cell production
With that kind of potential, the company is counting on significant interest among licensors, any one of which might want to engage its own contract producer of burixafor. If that happens, a third manufacturer will have to learn to reach 99.8% purity.


TaiGen Biotechnology Co., Ltd.
7F,138 Shin Ming Rd. Neihu Dist., Taipei, Taiwan 114 R.O.C
Tel: 886-2-81777072 | 886-2-27901861
Fax: 886-2-27963606

Taipei Railway Station front

Taipei Songshan Airport

Scinopharm
ScinoPharm (Changshu) Pharmaceuticals, Ltd.
ScinoPharm (Changshu) Pharmaceuticals, Ltd.
ScinoPharm is currently expanding its manufacturing and process development capabilities by adding significant production and technical capacity in Mainland China at its new Changshu site.
ScinoPharm Changshu is located in the Changshu Economic Development Zone (CEDZ), near Suzhou City, Jingsu Province, China on a 6.6-hectare site.
The facilities will include a R&D centre and production plants fully compliant with U.S. and international GMP standards. The Changshu plant, slated to be fully completed by 2012, will be used for the production of GMP grade pharmaceutical intermediates initially, and later be equipped to handle API production. China’s market for better quality APIs has grown considerably, and local formulation companies are encouraged to utilize APIs from companies having DMFs filed in advanced countries. ScinoPharm had closed its site in Kunshan and relocated the production and R&D groups to Changshu in the 4th quarter of 2011. These groups will continue to be expanded to meet growing demand for ScinoPharm products by both multinational and local formulation companies.
The small and medium-sized production units had been operational in the 4th quarter of 2011. The large production Bays plus a peptide purification unit, a high potency unit and a physical property processing facility will be operational by the end of 2012. Using advanced engineering designs, this site will also have the capability to process high potency, injectable grade products.
ScinoPharm Changshu will adopt the same quality systems as ScinoPharm Taiwan, and will therefore comply with ICH guidelines and FDA 21 CFR Parts 210 & 211.
TAIPEI


Clockwise from top: Taipei skyline, Grand Hotel, Far Eastern Plaza, National Palace Museum,Chiang Kai-shek Memorial Hall, Jiantan Station

Old street in Taipei. 2013
|
|||
| Nickname(s): The City of Azaleas | |||
.svg) |
|||
 Satellite image of Taipei City |
|||
Coordinates:  25°02′N 121°38′E 25°02′N 121°38′E |
|||


GIVINOSTAT

GIVINOSTAT, ITF2357, UNII-5P60F84FBH, ITF-2357, Gavinostat,
[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate,
diethyl-[6-(4-hydroxycarbamoyl-phenylcarbamoyloxymethyl)-naphthalen-2-yl-methyl]-amine
4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzohydroxamic acid
CAS 497833-27-9 FREE BASE
199657-29-9 HCL SALT
Molecular Formula: C24H27N3O4
Molecular Weight: 421.48888 g/mol
PHASE 2 Italfarmaco (INNOVATOR)
DESCRIBED IN U.S. Pat. No. 6,034,096 or in U.S. Pat. No. 7,329,689.

Givinostat (INN[1]) or gavinostat (originally ITF2357) is a histone deacetylase inhibitor with potential anti-inflammatory, anti-angiogenic, and antineoplastic activities.[2] It is a hydroxamate used in the form of its hydrochloride.
Givinostat is in numerous phase II clinical trials (including for relapsed leukemias and myelomas),[3] and has been granted orphan drug designation in the European Union for the treatment of systemic juvenile idiopathic arthritis[4] and polycythaemia vera.[5]
In 2010, orphan drug designation was assigned in the E.U. for the treatment of systemic-onset juvenile idiopathic arthritis and for the treatment of polycythemia vera. In 2013, this designation was assigned by the FDA for the treatment of Duchenne’s muscular dystrophy and for the treatment of Becker’s muscular dystrophy.
ITF2357 was discovered at Italfarmaco of Milan, Italy. It was patented in 1997 and first described in the scientific literature in 2005.[6][7]
Givinostat hydrochloride, an orally active, synthetic inhibitor of histone deacetylase, is being evaluated in several early clinical studies at Italfarmaco, including studies for the treatment of myeloproliferative diseases, polycythemia vera, Duchenne’s muscular dystrophy and periodic fever syndrome. The company was also conducting clinical trials for the treatment of Crohn’s disease and chronic lymphocytic leukemia; however, the trials were terminated.
No recent development has been reported for research into the treatment of juvenile rheumatoid arthritis, for the treatment of multiple myeloma and for the treatment of Hodgkin’s lymphoma.
Muscular dystrophies (MDs) include a heterogeneous group of genetic diseases invariably leading to muscle degeneration and impaired function. Mutation of nearly 30 genes gives rise to various forms of muscular dystrophy, which differ in age of onset, severity, and muscle groups affected (Dalkilic I, Kunkel LM. (2003) Muscular dystrophies: genes to pathogenesis. Curr. Opin. Genet. Dev. 13:231-238). The most common MD is the Duchenne muscular dystrophy (DMD), a severe recessive X-linked disease which affects one in 3,500 males, characterized by rapid progression of muscle degeneration, eventually leading to loss of ambulation and death within the second decade of life.
Attempts to replace or correct the mutated gene, by means of gene or cell therapy, might result in a definitive solution for muscular dystrophy, but this is not easy to achieve. Alternative strategies that prevent or delay muscle degeneration, reduce inflammation or promote muscle metabolism or regeneration might all benefit patients and, in the. future, synergize with gene or cell therapy. Steroids that reduce inflammation are currently the only therapeutic tool used in the majority of DMD patients (Cossu G, Sampaolesi M . (2007) New therapies for Duchenne muscular dystrophy: challenges, prospects and clinical trials. TRENDS Mol . Med. 13:520-526).
Diethyl- [ 6- ( 4-hydroxycarbamoyl-phenyl-carbamoyloxy- methyl ) -naphthalen-2-yl-methyl ] -ammonium chloride , which is described in WO 97/43251 (anhydrous form) and in WO 2004/065355 (monohydrate crystal form), herein both incorporated by reference, is an anti-inflammatory agent which is able to inhibit the synthesis of the majority of pro-inflammatory cytokines whilst sparing anti-inflammatory ones. Diethyl- [ 6- ( 4-hydroxycarbamoyl-phenyl-carbamoyloxy- methyl ) -naphthalen-2-yl-methyl ] -ammonium chloride is also known as ITF2357.
The monohydrate crystal form of diethyl- [ 6- ( 4- hydroxycarbamoyl-phenyl-carbamoyloxy-methy1 ) – naphthalen-2-yl-methyl ] -ammonium chloride is known as Givinostat .
Givinostat is being evaluated in several clinical studies, including studies for the treatment of myeloproliferative diseases, polycythemia vera, periodic fever syndrome, Crohn’s disease and systemic- onset juvenile idiopathic arthritis. Orphan drug designation was assigned in the E.U. for the treatment of systemic-onset juvenile idiopathic arthritis and for the treatment of polycythemia vera.
Givinostat has been recently found to act also as a Histone Deacetylase inhibitor (WO 2011/048514).
Histone deacetylases ( HDAC ) are a family of enzymes capable of removing the acetyl group bound to the lysine residues in the N-terminal portion of histones or in other proteins.
HDACs can be subdivided into four classes, on the basis of structural homologies. Class I HDACs (HDAC 1, 2, 3 and 8) are similar to the RPD3 yeast protein and are located in the cell nucleus. Class II HDACs (HDAC 4, 5, 6, 7, 9 and 10) are similar to the HDA1 yeast protein and are located both in the nucleus and in the cytoplasm. Class III HDACs are a structurally distinct form of NAD-dependent enzymes correlated with the SIR2 yeast protein. Class IV (HDAC 11) consists at the moment of a single enzyme having particular structural characteristics. The HDACs of classes I, II and IV are zinc enzymes and can be inhibited by various classes of molecule: hydroxamic acid derivatives, cyclic tetrapeptides , short-chain fatty acids, aminobenzamides , derivatives of electrophilic ketones, and the like. Class III HDACs are not inhibited by hydroxamic acids, and their inhibitors have structural characteristics different from those of the other classes .
The expression “histone deacetylase inhibitor” in relation to the present invention is to be understood as meaning any molecule of natural, recombinant or synthetic origin capable of inhibiting the activity of at least one of the enzymes classified as histone deacetylases of class I, class II or class IV.
Although HDAC inhibitors, as a class, are considered to be potentially useful as anti-tumor agents, it is worth to note that, till now, only two of them (Vorinostat and Romidepsin) have been approved as drugs for the cure of a single tumor form (Cutaneous T-cell lymphoma ) .
It is evident that the pharmaceutical properties of each HDAC inhibitor may be different and depend on the specific profile of inhibitory potency, relative to the diverse iso-enzymes as well as on the particular pharmacokinetic behaviour and tissue distribution.
Some HDAC inhibitors have been claimed to be potentially useful, in combination with other agents, for the treatment of DMD (WO 2003/033678, WO 2004/050076, Consalvi S. et al. Histone Deacetylase Inhibitors in the Treatment of Muscular Dystrophies: Epigenetic Drugs for Genetic Diseases. (2011) Mol. Med. 17 : 457-465 ) .
The potential therapeutic use of HDAC inhibitors in DMD may however be hampered by the possible harmful effects of these relatively toxic agents, especially when used for long-term therapies in paediatric patients .
Givinostat, as anti-inflammatory agent, has been already used in a phase II study in children with Systemic Onset Juvenile Idiopathic Arthritis; Givinostat administered at 1.5 mg/kg/day for twelve weeks achieved ACR Pedi 30, 50 and 70 improvement of approximately 70% (Vojinovic J, Nemanja D. (2011) HDAC Inhibition in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis. Mol. Med 17:397-403) showing only a limited number of mild or moderate but short lasting, adverse effects.
To date more than 500 patients (including 29 children) have been treated with Givinostat. Repeated dose toxicity studies were carried out in dogs, rats and monkeys. Oral daily doses of the drug were administered up to nine consecutive months. The drug was well tolerated with no overt toxicity at high doses. The “no adverse effect levels” (NOAEL) ranged from 10 to 25 mg/kg/day depending on the animal species and the duration of treatment.
In juvenile animals Givinostat at 60 mg/kg/day did not affect the behavioural and physical development and reproductive performance of pups.
No genotoxic effect was detected for Givinostat in the mouse lymphoma assay and the chromosomal aberration assay in vitro and in the micronucleus test and UDS test in vivo.
| Patent | Submitted | Granted |
|---|---|---|
| Monohydrate hydrochloride of the 4-hydroxycarbamoyl-phenyl)-carbamic acid (6-diethylaminomethyl-naphtalen-2-yl) ester [US7329689] | 2005-11-03 | 2008-02-12 |
Adverse effects
In clinical trials of givinostat as a salvage therapy for advanced Hodgkin’s lymphoma, the most common adverse reactions were fatigue (seen in 50% of participants), mild diarrhea or abdominal pain (40% of participants), moderate thrombocytopenia (decreased platelet counts, seen in one third of patients), and mild leukopenia (a decrease in white blood cell levels, seen in 30% of patients). One-fifth of patients experienced prolongation of the QT interval, a measure of electrical conduction in the heart, severe enough to warrant temporary suspension of treatment.[8]
Mechanism of action
Givinostat inhibits class I and class II histone deacetylases (HDACs) and several pro-inflammatory cytokines. This reduces expression of tumour necrosis factor (TNF), interleukin 1α and β, and interleukin 6.[7]
It also has activity against cells expressing JAK2(V617F), a mutated form of the janus kinase 2 (JAK2) enzyme that is implicated in the pathophysiology of many myeloproliferative diseases, including polycythaemia vera.[9][10] In patients with polycythaemia, the reduction of mutant JAK2 concentrations by givinostat is believed to slow down the abnormal growth of erythrocytes and ameliorate the symptoms of the disease.[5]
………………….

PATENT
https://www.google.com/patents/WO2004065355A1?cl=en
Hydrochloride of (6-diethylaminomethyl-naphthalen-2-yl)- methyl ester of (4-hydroxycarbamoylphenyl)-carbamic acid (II)

has been described in US patent 6,034,096 as a derivative of hydroxamic acid having anti-inflammatory and immunosuppressive activity, probably owing to the ability thereof to inhibit the production of pro-inflammatory cyto ines. This compound is obtained according to
Example 12 of the above-mentioned patent as an anhydrous, amorphous, hygroscopic, deliquescent solid which is difficult to handle.
crystalline form of monohydrous hydrochloride of
(6-diethylaminomethyl-naphthalen-2-yl)-methyl ester of
(4~hydroxycarbamoylphenyl)-carbamic acid (I).

This form is particularly advantageous from the industrial perspective because it is stable and simpler to handle than the anhydrous and amorphous form described above.
………………
PATENT
http://www.google.co.in/patents/US7329689
Hydrochloride of (6-diethylaminomethyl-naphthalen-2-yl)-methyl ester of (4-hydroxycarbamoylphenyl)-carbamic acid (II)

has been described in U.S. Pat. No. 6,034,096 as a derivative of hydroxamic acid having anti-inflammatory and immunosuppressive activity, probably owing to the ability thereof to inhibit the production of pro-inflammatory cytokines. This compound is obtained according to Example 12 of the above-mentioned patent as an anhydrous, amorphous, hygroscopic, deliquescent solid which is difficult to handle.
The 4-(6-diethylaminomethyl-naphthalen-2-ylmethoxycarbonylamino)-benzoic acid can be prepared as described in Example 12, point C, of U.S. Pat. No. 6,034,096.
The acid (1.22 kg, 3 moles) was suspended in THF (19 l) and the mixture was agitated under nitrogen over night at ambient temperature. The mixture was then cooled to 0° C. and thionyl chloride (0.657 l, 9 moles) was added slowly, still under nitrogen, with the temperature being maintained below 10° C. The reaction mixture was heated under reflux for 60 minutes, DMF (26 ml) was added and the mixture was further heated under reflux for 60 minutes.
The solvent was evaporated under vacuum, toluene was added to the residue and was then evaporated. This operation was repeated twice, then the residue was suspended in THF (11.5 l) and the mixture was cooled to 0° C.
The mixture was then poured into a cold solution of hydroxylamine (50% aq., 1.6 l, 264 moles) in 5.7 l of water. The mixture was then cooled to ambient temperature and agitated for 30 minutes. 6M HCl was added until pH 2 was reached and the mixture was partially evaporated under vacuum in order to eliminate most of the THF. The solid was filtered, washed repeatedly with water and dissolved in a solution of sodium bicarbonate (2.5%, 12.2 l). The solution was extracted with 18.6 l of a mixture of THF and ethyl acetate (2:1 v/v). 37% HCl (130 ml) were added to the organic layer in order to precipitate the monohydrate of the (6-diethylaminomethyl-naphthalen-2-yl)-methyl ester hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid. If necessary, this operation can be repeated several times to remove any residues of the original acid.
Finally, the solid was dried under vacuum (approximately 30 mbar, 50° C.), producing 0.85 kg (60%) of compound (I).
HPLC purity: 99.5%; water content (Karl Fischer method): 3.8%; (argentometric) assay: 99.8%.
| Elemental analysis | |||||
| C % | H % | Cl % | N % | ||
| Calculated for | 60.56 | 6.35 | 7.45 | 8.83 | |
| C24H30ClN3O5 | |||||
| Found | 61.06 | 6.48 | 7.48 | 8.90 | |
…..
PATENT
http://www.google.co.in/patents/US20120302633
The hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphtalenyl) ester, also known as ITF 2357 and having the International Non Proprietary Name (INN) of Givinostat® is an organic compound with immunosuppressive and anti-inflammatory activity,

…………………..
http://www.google.com/patents/US6034096
EXAMPLE 12
4-[6-(Diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]-benzohydroxamic acid hydrochloride
A. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) (22.2 g, 115 mmol) was added to a solution of 2,6-naphthalenedicarboxylic acid (25 g, 115 mmol) and hydroxybenzotriazole (15.6 g, 115 mmol) in dimethylformamide (1800 ml) and the mixture was stirred at room temperature for 2 hours. Diethyl amine (34.3 ml, 345 mmol) was added and the solution was stirred overnight at room temperature. The solvent was then evaporated under reduced pressure and the crude was treated with 1N HCl (500 ml) and ethyl acetate (500 ml), insoluble compounds were filtered off and the phases were separated. The organic phase was extracted with 5% sodium carbonate (3×200 ml) and the combined aqueous solutions were acidified with concentrated HCl and extracted with ethyl acetate (3×200 ml). The organic solution was then washed with 1N HCl (6×100 ml), dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure yielding 18.5 g (Yield 60%) of pure 6-(diethylaminocarbonyl)-2-naphthalenecarboxylic acid; m.p.=122-124° C.
1 H-NMR d 8.67 (s, 1H), 8.25-8.00 (m, 4H), 7.56 (d, 1H), 3.60-3.20 (m, 4H), 1.30-1.00 (m, 6H).
B. A solution of 6-(diethylaminocarbonyl)-2- naphthalenecarboxylic acid (18 g, 66 mmol) in THF (200 ml) was slowly added to a refluxing suspension of lithium aluminium hydride (7.5 g, 199 mmol) in THF (500 ml). The mixture was refluxed for an hour, then cooled at room temperature and treated with a mixture of THF (25 ml) and water (3.5 ml), with 20% sodium hydroxide (8.5 ml) and finally with water (33 ml). The white solid was filtered off and the solvent was removed under reduced pressure. Crude was dissolved in diethyl ether (200 ml) and extracted with 1N HCl (3×100 ml). The aqueous solution was treated with 32% sodium hydroxide and extracted with diethyl ether (3×100 ml). The organic solution was dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure yielding 12.7 g (79% yield) of pure 6-(diethylaminomethyl)-2-naphthalenemethanol as thick oil.
1 H-NMR d 7.90-7.74 (m, 4H), 7.49 (m, 2H), 5.32 (t, 1H, exchange with D2 O), 4.68 (d, 2H), 3.69 (s, 2H), 2.52 (q, 4H), 1.01 (t, 6H).
C. A solution of 6-(diethylaminomethyl)-2-naphthalene-methanol (12.5 g, 51 mmol) and N,N’-disuccinimidyl carbonate (13.2 g, 51 mmol) in acetonitrile (250 ml) was stirred at room temperature for 3 hours, then the solvent was removed and the crude was dissolved in THF (110 ml). This solution was added to a solution of 4-amino benzoic acid (7.1 g, 51 mmol) and sodium carbonate (5.5 g, 51 mmol) in water (200 ml) and THF (100 ml). The mixture was stirred overnight at room temperature, then THF was removed under reduced pressure and the solution was treated with 1N HCl (102 ml, 102 mmol). The precipitate was filtered, dried under reduced pressure, tritured in diethyl ether and filtered yielding 13.2 g (yield 64%) of pure 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]-benzoic acid; m.p.=201-205° C. (dec.)
1 H-NMR d 10.26 (s, 1H), 8.13 (s, 1H), 8.05-7.75 (m, 6H), 7.63 (m, 3H), 5.40 (s, 2H), 4.32 (s, 2H), 2.98 (q, 4H), 1.24 (t, 6H).
D. A solution of 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzoic acid (13.1 g, 32 mmol) and thionyl chloride (7 ml, 96 mmol) in chloroform (300 ml) was refluxed for 4 hours, then the solvent and thionyl chloride were evaporated. Crude was dissolved in chloroform (100 ml) and evaporated to dryness three times. Crude was added as solid to a solution of hydroxylamine hydrochloride (2.7 g, 39 mmol) and sodium bicarbonate (5.4 g, 64 mmol) and 1N sodium hydroxide (39 ml, 39 mmol) in water (150 ml) and THF (50 ml). The mixture was stirred overnight at room temperature, then THF was removed under reduced pressure and the aqueous phase was extracted with ethyl acetate (3×100 ml). The combined organic phases were dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. Crude was dissolved in THF and treated with a 1.5 N etheric solution of HCl. The solid product was filtered and dried yielding 6 g (yield 41%) of pure 4-[6-(diethylaminomethyl)naphth-2-ylmethyloxycarbamoyl]benzohydroxamic acid hydrochloride as white solid; m.p.=162-165° C., (dec.)
1 H-NMR d 11.24 (s, 1H, exchange with D2 O), 10.88 (s, 1H, exchange with D2 O), 10.16 (s, 1H), 8.98 (bs, 1H, exchange with D2 O), 8.21 (s, 1H), 8.10-7.97 (m, 3H), 7.89 (d, 1H), 7.80-7.55 (m, 5H), 5.39 (s, 2H), 4.48 (d, 2H), 3.09 (m, 4H), 1.30 (t, 6H).

Some nmr predictions
CAS NO. 497833-27-9, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectral analysis
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_1h.png)
13 C NMR PREDICTIONS
![[6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate NMR spectra analysis, Chemical CAS NO. 497833-27-9 NMR spectral analysis, [6-(diethylaminomethyl)naphthalen-2-yl]methyl N-[4-(hydroxycarbamoyl)phenyl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-20/001/566/1566912_13c.png)

COSY NMR…..http://www.nmrdb.org/
HMBC /HSQC
NMR spectra of reference
References
1
- World Health Organization (2010). “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended INN: List 63”. WHO Drug Information 24 (1): 58–9.
- 2
- National Cancer Institute (2010). “Gavinostat”. NCI Cancer Dictionary. U.S. National Institutes of Health. Retrieved 2010-09-15.
- 3
- “Search results for ITF2357”. ClinicalTrials.gov.
- 4
- Committee for Orphan Medicinal Products (23 February 2010). “Public summary of opinion on orphan designation: Givinostat for the treatment of systemic-onset juvenile idiopathic arthritis”. European Medicines Agency. Retrieved 2010-09-15.
- 5
- Committee for Orphan Medicinal Products (3 March 2010). “Public summary of opinion on orphan designation: Givinostat for the treatment of polycythaemia vera”. European Medicines Agency.
- 6
- WO patent application 1997/043251, “Compounds with anti-inflammatory and immunosuppressive activities”, published 1997-11-20, assigned to Italfarmaco S.p.A.
- 7
- Leoni F, Fossati G. (2005). “The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo”. Molecular Medicine 11: 1. doi:10.2119/2006-00005.Dinarello. PMID 16557334.
- 8
- Tan J, Cang S, Ma Y, Petrillo RL, Liu D (2010). “Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents”. Journal of Hematology & Oncology 3: 5. doi:10.1186/1756-8722-3-5. PMID 20132536. Review.
- 9
- Vannucchi AM, Guglielmelli P, Pieri L, Antonioli E, Bosi A (2009). “Treatment options for essential thrombocythemia and polycythemia vera.” Expert Review of Hematology 2 (1): 41–55. doi:10.1586/17474086.2.1.41. Review.
- Guerini V, Barbui V, Spinelli O, et al. (April 2008). “The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F)”. Leukemia 22 (4): 740–7. doi:10.1038/sj.leu.2405049. PMID 18079739.
Further reading
- Job-Deslandre, C (January 2007). “Idiopathic juvenile-onset systemic arthritis”. Orphanet. Orphan number: ORPHA85414.
| US6034096 | 12 May 1997 | 7 Mar 2000 | Italfarmaco S.P.A. | Compounds with anti-inflammatory and immunosuppressive activities |
| WO1997043251A1 | May 12, 1997 | Nov 20, 1997 | Italfarmaco Spa | Compounds with anti-inflammatory and immunosuppressive activities |
| WO2004063146A1 | Jan 7, 2004 | Jul 29, 2004 | Italfarmaco Spa | Hydroxamic acid derivatives having anti-inflammatory action |
| WO2004065355A1 | Jan 8, 2004 | Aug 5, 2004 | Italfarmaco Spa | Monohydrate hydrochloride of the 4-hydroxycarbamoyl-phenyl)-carbamic acid (6-diethylaminomethyl-naphtalen-2-yl) ester |
| WO2006003068A2 | Jun 7, 2005 | Jan 12, 2006 | Italfarmaco Spa | Alpha-amino acid derivatives with antiinflammatory activity |
| WO2008097654A1 | Feb 8, 2008 | Aug 14, 2008 | Nancie M Archin | Methods of using saha for treating hiv infection |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8518988 * | 3 Dec 2010 | 27 Aug 2013 | Chemi Spa | Polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphthalenyl) ester |
| US20120302633 * | 3 Dec 2010 | 29 Nov 2012 | Chemi Spa | Novel polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphthalenyl) ester |
| WO2011092556A1 | 3 Dec 2010 | 4 Aug 2011 | Chemi Spa | Novel polymorph of the hydrochloride of the (4-hydroxycarbamoyl-phenyl)-carbamic acid (6-dimethylamino methyl-2-naphtalenyl) ester |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| {6-[(diethylamino)methyl]naphthalen-2-yl}methyl [4-(hydroxycarbamoyl)phenyl]carbamate | |
| Clinical data | |
|
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 497833-27-9 |
| ATC code | None |
| PubChem | CID 9804992 |
| ChemSpider | 7980752 |
| UNII | 5P60F84FBH |
| Chemical data | |
| Formula | C24H27N3O4 |
| Molecular mass | 421.489 g/mol |


| Italfarmaco S.p.A. | |
|---|---|
|
|
|
| Stato | |
| Tipo | Società per azioni |
| Fondazione | 1938 a Milano |
| Fondata da | Gastone De Santis |
| Sede principale | Milano |
| Filiali | |
| Persone chiave | Francesco De Santis, [Presidente Holding] |
| Settore | sanità |
| Prodotti | Farmaci |
| Fatturato | >500 milioni di Euro (gruppo) (2011) |
| Dipendenti | >1900 (gruppo) (2011) |
| Sito web | www.italfarmaco.com |
MILAN ITALY


LY-156735 (TIK-301, PD-6735)….for the treatment of sleep latency in patients with primary insomnia

N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide
LY-156735 (TIK-301, PD-6735) is a melatonin MT1 and MT2 agonist which is under development for the treatment of insomnia and other sleep disorders.[1]
Beta-methyl-6-chloromelatonin (PD-6735) is a melatonin MT1 and MT2 agonist which had been in phase II trials at Phase 2 Discovery for the treatment of sleep latency in patients with primary insomnia, however, no recent development has been reported.
The melatonin agonist exhibits high selectivity and provides a novel mode of action different from that of benzodiazepine receptor ligands currently on the market.
Furthermore, the drug candidate is believed to be non-addicting, therefore, offering an advantage over marketed sleep medications. Originally discovered by Lilly, PD-6735 was licensed to Phase 2 Discovery in 2002 for further development.
Orphan drug designation has been assigned in the U.S. for the treatment of circadian rhythm sleep disorders in blind people with no light perception and for the treatment of neuroleptic-induced tardive dyskinesia in schizophrenia patients.
In 2007, the product candidate was licensed to Tikvah Therapeutics by Phase 2 Discovery for worldwide development and commercialization for the treatment of sleep disorder, depression and circadian rhythm disorder.
beta -alkylmelatonins as ovulation inhibitors [US4997845]1991-03-05
BETA-ALKYLMELATONINS [EP0281242]1988-09-07 GRANT1992-08-12

The condensation of 6-chloro-5-methoxy-1H-indole (I) with Meldrum’s acid (II) and acetaldehyde (III) catalyzed by L-proline in acetonitrile gives the adduct (IV), which is treated with Cu and ethanol in refluxing pyridine to yield 3-(6-chloro-5-methoxy-1H-indol-3-yl)butyric acid ethyl ester (V). The reaction of (V) with hydrazine at 140 C affords the hydrazide (VI), which is treated with NaNO2 and Ac-OH to provide the corresponding azide that, without isolation, is thermolyzed and rearranged in toluene at 80?C to give 7-chloro-6-methoxy-4-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (VII). The cleavage of the lactam ring of (VII) with KOH in refluxing ethanol/water yields 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (VIII). The decarboxylation of (VIII) by means of refluxing aq. 3M HCl affords 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole (IX), which is finally acylated with acetic anhydride and pyridine in toluene to provide the target 6-chloromelatonin as a racemic compound.
EP 0281242;……….http://www.google.com/patents/EP0281242B1?cl=en
Example 3
- Preparation of β-Methyl-6-chloromelatonin
-
Following the procedure of Example 1, a solution of 10.0 g (0.055 mole) of 5-methoxy-6-chloroindole, 3.1 ml (2.44 g, 0.055 mole) of acetaldehyde, and 7.94 g (0.055 mole) of Meldrum’s acid in 90 ml of acetonitrile was stirred for 48 hours. The solvent was removed under vacuum, and the adduct thus prepared was recrystallized by dissolving in warm toluene and immediately cooling. The adduct was obtained as slightly pink crystals; m.p. = 145°C; yield = 16.5 g (85%). The elemental analysis of the product showed a slightly elevated percentage of carbon. However, the NMR spectrum indicated that the product was pure and had the indicated structure.
Analysis calc. for C₁₇H₁₈NO₅Cl- Theory:
- C, 58.04; H, 5.16; N, 3.98; Cl, 10.08
- Found :
- C, 59.34; H, 5.15; N, 3.84; Cl, 9.69
-
The solvolysis and decarboxylation of the adduct (11.0 g; 31.3 mmoles) using ethanol, pyridine, and copper dust was carried out by the procedure of Example 1. The yield of 3-(5-methoxy-6-chloro-1H-indol-3-yl)pentanoic acid ethyl ester, a pale yellow oil, after chromatography over silica gel using 10% EtOAc/90% toluene was 8.68 g (94%).
Analysis calc. for C₁₅H₁₈NO₃Cl- Theory:
- C, 60.91; H, 6.13; N, 4.74; Cl, 11.99
- Found :
- C, 60.67; H, 5.86; N, 4.93; Cl, 11.73
-
A mixture of 8.68 g (29.3 mmoles) of the above ethyl ester and 6 ml of hydrazine hydrate was heated at 140°C under nitrogen in a flask fitted with an air cooled condensor. After 6½ hours, the excess hydrazine hydrate was removed under vacuum. The 2-methyl-2-(5-methoxy-6-chloro-3-indolyl)-propionhydrazide thus prepared was recrystallized from ethyl acetate; Yield = 7.13 g (86%); m.p. = 154-155°C.
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.14; H, 5.51; N, 14.49; Cl, 12.78
-
The above hydrazide (7.13 g, 25 mmoles) was converted to the corresponding acyl azide, the azide thermolyzed and rearranged at 80° in toluene, and the rearranged product cyclized with HCl according to the procedure of Example 1. The yield of crude, light tan, lactam, 1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole, product, (m.p. = 249-252°C) was 4.77 g (72%).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58
- Found :
- C, 59.45; H, 4.77; N, 10.72
-
The crude lactam (4.77 g, 18 mmoles) was hydrolyzed with aqueous ethanolic KOH as described in Example 1. The yield of crude amino acid, 2-carboxy-3-(1-amino-2-propyl)-5-methoxy-6-chloroindole, was 3.98 g (78%). The crude product (3.0 g; 10.6 mmoles) was decarboxylated, using the procedure of Example 1, by refluxing in 100 ml of 3M HCl overnight. The acidic solution was decolorized with activated carbon and was then basified with 5M NaOH. The amine was extracted into diethyl ether. After drying the ether extract over Na₂SO₄, the diethyl ether was removed in vacuo leaving as a residue the crystallized tryptamine, 3-(1-amino-2-propyl)-5-methoxy-6-chloroindole; m.p. 133-4°C. The yield, after recrystallization from toluene/hexane, was 1.62 g (64%).
Analysis calc. for C₁₂H₁₅N₂OCl- Theory:
- C, 60.38; H, 6.33; N, 11.74; Cl, 14.85
- Found :
- C, 60.11; H, 6.05; N, 11.93; Cl, 15.06
-
A solution of 1.51 g (6.3 mmoles) of the above tryptamine in 10 ml of toluene and 2.5 ml of pyridine was treated with 1.5 ml of acetic anhydride. After allowing the reaction mixture to stand for three hours at room temperature, the volatile materials were removed under vacuum. The residue was dissolved in ethyl acetate, and washed with aqueous NaHCO₃, and brine. The ethyl acetate solution was dried over Na₂SO₄, and the solvent removed by evaporation. The residual oil was crystallized from toluene/hexane yielding 6-chloro-β-methylmelatonin, (m.p. = 133-5°C; 1.09 g, 61%).
Analysis calc. for C₁₄H₁₇N₂O₂Cl- Theory:
- C, 59.89; H, 6.10; N, 9.98; Cl, 12.63
- Found :
- C, 60.03; H, 6.22; N, 9.75; Cl, 12.92
…………………………………………….
PATENT
http://www.google.com/patents/EP0281242B1?cl=en

The intermediate diazonium salt (XIII) has been obtained as follows: the hydrogenation of 3-chloro-4-methoxynitrobenzene (XI) with H2 over Pt/Al2O3 in toluene gives the corresponding aniline (XII), which is diazotized with NaNO2/HCl and treated with sodium tetrafluoroborate to yield the target diazonium salt intermediate (XIII). The reduction of pulegone (I) with H2 over Pd/C gives the menthol (II), which is oxidized with CrO3/H2SO4 to yield 3(R),7-dimethyl-6-oxooctanoic acid (IV), which can also be obtained by direct oxidation of (l)-menthol (III) under the same conditions.
The oxidation of (IV) with trifluoroperacetic acid (trifluoroacetic anhydride/H2O2) in dichloromethane yields the 3(R)-methylhexanedioic acid isopropyl monoester (V), which is treated with NaOEt in ethanol to obtain the corresponding ethyl monoester (VI). The reaction of (VI) with diethyl carbonate, EtONa, and “Adogen 464” (a phase transfer catalyst) in ethanol affords 5,5-bis(ethoxycarbonyl)-3(S)-methylpentanoic acid (VII), which is treated with oxalyl chloride to provide the expected acyl chloride (VIII). The reaction of (VIII) with sodium azide and benzyl alcohol gives the intermediate azide that rearranges to the benzyl carbamate (IX).
The reductive cyclization of (IX) with H2 over Pd/C in ethanol yields 5(R)-methyl-2-oxopiperidine-3-carboxylic acid ethyl ester (X), which is condensed with the intermediate diazonium salt (XIII) to afford the hydrazono derivative (XIV). The cyclization of (XIV) in hot formic acid provides 7-chloro-6-methoxy-4(R)-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (XV), which is treated with KOH In refluxing ethanol/water to cleave the lactam ring, yielding 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (XVI). The decarboxylation of (XVI) by means of refluxing 3M HCl affords 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole (XVII), which is finally acylated with Ac2O and pyridine in toluene to provide the target 6-chloromelatonin as a pure enantiomer.
Example 7
- Preparation of S-(-)-β-methyl-6-chloromelatonin and R-(+)-β-methyl-6-chloromelatonin
-
A solution of 4.0 g (21 mmoles) of 3-chloro-4-methoxynitrobenzene in 200 ml of toluene was hydrogenated over 0.4 g of 5% platinum on alumina. The catalyst was removed by filtration and the solvent evaporated from the filtrate. The crude 3-chloroanisidine prepared was placed in solution in diethyl ether and treated with ethereal HCl to produce the hydrochloride salt, which was collected and dried; weight = 2.48 g (61% yield).
-
A mixture of 2.40 g (12.4 mmoles) of 3-chloroanisidine hydrochloride in 7 ml of 4M HCl was treated, at 0°C, with 0.86 g (12.5 mmoles) of sodium nitrite in 5 ml of water. After stirring at 0°C for an hour the solution was filtered and the filtrate added slowly to an ice cold solution of 2.6 g (24 mmoles) of sodium fluoroborate in 8 ml of water. After stirring at 0°C for an hour the salt was collected and washed successively with, cold 5% sodium fluoroborate solution, cold methanol, and ether. The dried 3-chloro-4-methoxybenzene diazonium fluoroborate thus prepared weighed 2.2 g (69% yield).
-
A mixture of 2.03 g (11.0 mmole) of (R)-(-)-3-ethoxycarbonyl-5-methyl-2-piperidone and 30 ml of 0.75M NaOH was stirred at room temperature (24°C) overnight. The solution was cooled to 0°C and the pH lowered to 3.5 with 3M hydrochloric acid. The diazonium salt (2.8 g, 10.9 mmoles) was added in small portions and the reaction mixture cooled to about 0°C overnight. The product, R-(-)-3-(3-chloro-4-methoxy)phenylhydrazono-5-methyl-2-piperidone, was collected, washed with water, and dried; weight = 2.30 g (75% yield); m.p. = 205°C. A small sample was further purified by chromatography over a short silica gel column using ethyl acetate as the eluant. [α]²⁵ = -58° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.79; H, 5.78: N, 14.72; Cl, 12.69
-
A mixture of 2.20 g (7.8 moles) of the R-(-) hydrazone and 20 ml of 90% formic acid was heated at 85° for three hours then slowly diluted with an equal volume of water. The mixture was allowed to cool and then chilled overnight. The dark precipitate was collected, washed with water, then recrystallized from acetone/water, yielding 1.20 g (60% yield) of S-(-)-1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole; m.p. = 248°C. [α]²⁵ = -12.2° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58; Cl, 13.39
- Found :
- C, 59.16; H, 4.88; N, 10.80; Cl, 13.15
-
The conversion of (S)-(-)-lactam to (S)-(-)-6-chloro-β-methylmelatonin was carried out as described previously in Example 3. The product, S-(-)-β-methyl-6-chloromelatonin, was spectroscopically identical to the racemate, but gave an optical rotation of [α]²⁵ = -13.2° (c = 10, MeOH).
-
(R)-(+)-6-chloro-β-methylmelatonin was synthesized from (S)-(+)-3-ethoxycarbonyl-5-methyl-2-piperidone in the same manner as described above. The stereoisomer was identical to the (S)-(-) material except for the sign of rotation.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-[(2R)-(6-Chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 118702-11-7 |
| ATC code | ? |
| PubChem | CID 219018 |
| ChemSpider | 189853 |
| Chemical data | |
| Formula | C14H17ClN2O2 |
| Molecular mass | 280.757 |

{kind=link}