
Home » Posts tagged 'PHASE 1' (Page 10)
Tag Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK 2269557 In Phase 1….Asthma , COPD, is it COMPD A OR B?
 COMPD A
COMPD A
Compd A OR B IS GSK 2269557
DATA FOR COMPD A
6-(1H-indol-4-yl)-4-[5-[[4-(1-methylethyl)-1-piperazinyl]methyl]-2-oxazolyl]-1H-Indazole,
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole
EMAIL ME amcrasto@gmail.com
Phosphoinositide 3ΌΗ kinases (hereinafter PI3Ks) are a family of signal transducer enzymes which are involved in various cellular functions including cell growth, proliferation and differentiation. A wide variety of retroviruses and DNA-based viruses activate the PI3K pathway as a way of preventing host cell death during viral infection and ultimately exploiting the host cell synthesis machinery for its replication (Virology 344(1) p. 131-8 (2006) by Vogt et al.; and Nat. Rev. Microbiol. 6(4) p. 265-75 (2008) by Buchkovich et al). It has therefore been postulated that PI3K inhibitors may have potential therapeutic benefit in the treatment of viral infections such as influenza virus infection, in addition to the more established treatment of cancer and inflammatory diseases.
The Influenza NS1 protein activates Class la PI3Ks by binding to their regulatory subunit p85beta but not to other Class la regulatory subunits such as p85alpha. The recent crystal structure of the NS1-p85beta complex (Hale et al. Proc. Natl. Acad. Sci. U S A. 107(5) p.1954-1959 (2010)) is also suggestive of an interaction with the p110 kinase subunit providing a mechanism for catalytic activation of the kinase domain. This observation provides a rationale for isoform specificity not only with the p85 regulatory subunit but also potentially with the p110 catalytic subunit too. The function of PI3K during influenza virus infection has also been investigated by, for example, Ehrhardt et al. (Cell. Microbiol. 8(8) p. 1336-1348 (2006)), and the role of PI3K5 signalling in morbidity and lung pathology induced by influenza virus infection has been reported in WO 2010/083163.
There remains a need to provide compounds which are inhibitors of the activity or function of PI3K5 which may be useful in the treatment or prevention of influenza virus infection.
GSK 2269557 is an inhaled phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor in early clinical trials at GlaxoSmithKline for the treatment of patients with asthma and also for the treatment of chronic obstructive pulmonary disease (COPD) in patients who smoke cigarettes.
- 18 Nov 2014GlaxoSmithKline plans a phase II trial in Chronic obstructive pulmonary disease in Belgium, Denmark, the Netherlands and Russia (NCT02294734)
- 01 Jun 2014Phase-II clinical trials in Chronic obstructive pulmonary disease in Germany (Inhalation)
- 01 May 2014GlaxoSmithKline plans a phase II trial for Chronic obstructive pulmonary disease in Germany (NCT02130635)
EMAIL ME amcrasto@gmail.com
CLICK ON IMAGES TO VIEW SIMILAR ROUTES FOR COMPD A AND B
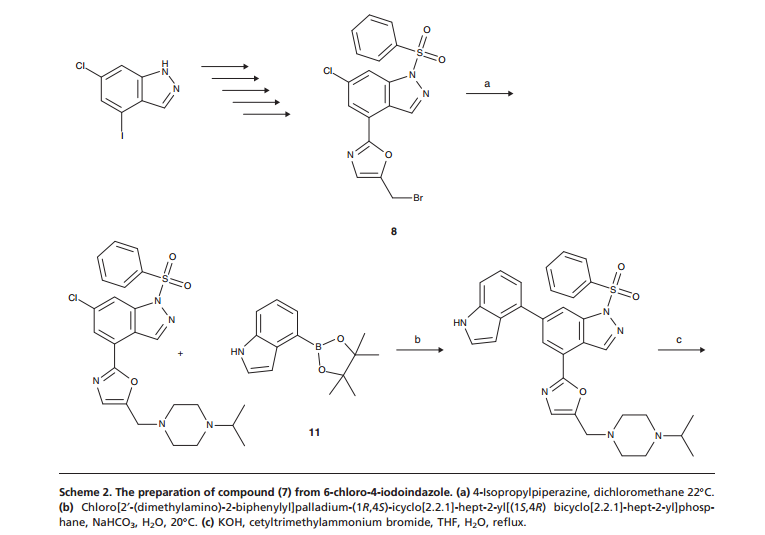
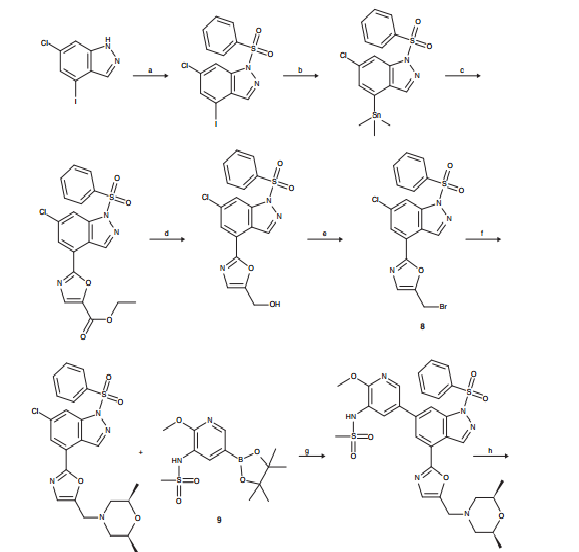
CLICK ON IMAGE TO VIEW
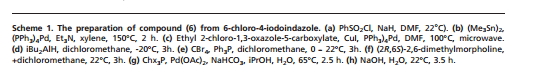
…………………………………………………………………….
COMPD A
WO 2012032065
http://www.google.com/patents/WO2012032065A1?cl=en
Example 68
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole

Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1/-/-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 100°C for 30 min. Additional 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 10°C for 30 min, then 140°C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 : 1 , v/v) and purified by MDAP (method H). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml_) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 120°C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 45°C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 : 1). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 : 1). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane: DCM (1 : 1) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 50°C overnight. This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 : 1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441.
1 H NMR (400MHz ,DMSO-d6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
Method C
Potassium hydroxide (145.6 g) was added to a suspension of 6-(1 H-indol-4-yl)-4-(5-{[4-(1- methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (300.7 g) and cetyltrimethylammonium bromide (9.3 g) in tetrahydrofuran (6.0 L) and water (30 ml) stirring under nitrogen at ambient temperature. The mixture was heated at reflux for 17 hours and was then cooled to 20-25°C. Ethyl acetate (3.0 L) and water (3.0 L) were added, stirred for 10 minutes and then separated. The organic layer was extracted with hydrochloric acid (1 M, 1 x 3.0 L, 2 x 1.5L) and the acidic extracts combined and basified to ~pH 8 by the addition of saturated sodium carbonate solution (2.1 L). After ageing for 30 minutes the resultant suspension was filtered, washed with water (300 ml) and the solid dried under vacuum at 65°C to give the title compound as a pale yellow solid (127.9 g).
LCMS (Method B): Rt 2.44 min, MH+ 441.
…………………………………………………………………………
WO 2010125082
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 6
6-(1 H-lndol-4-yl)-4-(5-{[4-(1 -methylethyl)-1 -piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1 H- indazole

Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1H-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 1000C for 30 min. Additional 4-(4, 4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 1O0C for 30 min, then 14O0C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and purified by MDAP (method A). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml.) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 1200C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 450C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 :1 ). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 :1 ). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane:DCM (1 :1 ) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 500C overnight.
This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 :1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441. 1H NMR (400MHz ,DMSOd6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
EMAIL ME amcrasto@gmail.com
COMPD B
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 1
Λ/-[5-[4-(5-{[(2/?,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-
6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide

Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1 ,3-oxazol-2- yl)-1-(phenylsulfonyl)-1 H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1 ,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium- 1 (1 /?,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 12O0C under microwave irradiation for 1 h. Additional chloroP’^dimethylamino^-biphenylyOpalladium-^I R^S^bicycloP^.ilhept^- yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 12O0C under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2x 2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3ml, 1 :1 , v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2x 25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 4O0C for 3 h to give the title compound as a white solid (26 mg). LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4- morpholinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1 ,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 800C. Chloro[2′-(dimethylamino)-2- biphenylyl]palladium-1 (1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)-bicyclo[2.2.1]hept-2- yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 800C.
The reaction mixture was cooled to 450C, sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45 0C for 4 hours. The mixture was cooled to RT and diluted with water (610 ml_). Dichloromethane (920 ml.) was added, and the mixture was filtered twice through Celite (washed with 200 ml. 1 ,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1 ,4-dioxane/DCM 2:1 (500 ml_). The aqueous phase was neutralised with hydrochloric acid to pH -7 and extracted with 1 ,4- dioxane/DCM 2:1 (1 L), then 1 ,4 dioxane/DCM 1 :1 (2×500 ml_). The organics were washed with brine (500 ml_), and filtered through Celite (washed with 200 ml. 1 ,4 dioxane/DCM 2:1 ), and evaporated to yield a dark black solid, which was purified in 4 batches:
Batch 1 : 28g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
Batch 2: 3Og was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
Batch 4: 29g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1 100 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 600C for 5hrs to give the title compound as an off-white solid (45.51 g). LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield -23 g of a solid residue that was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 ml_). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65°C overnight to give the title compound as an off-white solid (11.9O g). LCMS (Method A): Rt 0.62 mins, MH+ 513.
………………………………………..
http://www.google.co.in/patents/US8735390
Example 1N-[5-[4-(5-{[(2R,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide

Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 120° C. under microwave irradiation for 1 h. Additional chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 120° C. under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2×2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3 ml, 1:1, v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1:1, v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2×25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 40° C. for 3 h to give the title compound as a white solid (26 mg).
LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B
N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 80° C. Chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 80° C.
The reaction mixture was cooled to 45° C., sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45° C. for 4 hours. The mixture was cooled to RT and diluted with water (610 mL). Dichloromethane (920 mL) was added, and the mixture was filtered twice through Celite (washed with 200 mL 1,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1,4-dioxane/DCM 2:1 (500 mL). The aqueous phase was neutralised with hydrochloric acid to pH ˜7 and extracted with 1,4-dioxane/DCM 2:1 (1 L), then 1,4 dioxane/DCM 1:1 (2×500 mL). The organics were washed with brine (500 mL), and filtered through Celite (washed with 200 mL 1,4 dioxane/DCM 2:1), and evaporated to yield a dark black solid, which was purified in 4 batches:
- Batch 1: 28 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
- Batch 2: 30 g was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
- Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
- Batch 4: 29 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1100 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 60° C. for 5 hrs to give the title compound as an off-white solid (45.51 g).
LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield ˜23 g of a solid residue that was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 mL). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65° C. overnight to give the title compound as an off-white solid (11.90 g).
LCMS (Method A): Rt 0.62 mins, MH+ 513.
Method C
10M Sodium hydroxide solution (0.70 ml) was added to a stirred suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (1.17 g) in water (5.8 ml). The resulting mixture was stirred at room temperature for 3.75 hours and was then washed with ethyl acetate (2×6 ml). The layers were separated and the aqueous phase was acidified to pH 6 with 2M hydrochloric acid (0.8 ml). The acidified aqueous layer was extracted twice with ethyl acetate (11 ml then 5 ml). The combined ethyl acetate extracts were dried by azeotropic distillation and diluted with further ethyl acetate (11 ml). The misture was stirred at room temperature for 112 hours. The slurry was seeded and then stirred at room temperature for 48 hours. The resultant suspension was filtered, washed with ethyl acetate (2×2 ml) and the solid dried under vacuum at 40° C. to give the title compound as a pale yellow solid (0.58 g).
LCMS (Method B): Rt 1.86 min, MH+ 513.
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
Preparation of Polymorphs of Compound A
Form (II)
Ethyl acetate (15 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (2.1 g) and was stirred at ambient conditions overnight. The resultant slurry was filtered and dried under vacuum at 50° C. to give a new solid state form (91 ckw/w).
1H NMR (400 MHz, DMSO d6) d=13.49 (br s, 1H), 9.39 (s, 1H), 8.58 (s, 1H), 8.42 (d, J=2.2 Hz, 1H), 7.99 (d, J=2.2 Hz, 1H), 7.93 (d, J=1.2 Hz, 1H), 7.88 (s, 1H), 7.35 (s, 1H), 4.00 (s, 3H), 3.74 (s, 2H), 3.58 (m, 2H), 3.11 (s, 3H), 2.80 (d, J=10.3 Hz, 2H), 1.78 (t, J=10.3 Hz, 2H), 1.05 (d, J=6.4 Hz, 6H)
SODIUM SALT OF COMPD B
http://www.google.com/patents/US20140256721
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
http://www.google.com/patents/US20140256721
Preparation of Salts of Compound ASodium Salt
Methanol (2 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (0.3 g) followed by aqueous sodium hydroxide (0.129 ml) to give a solution. Tert-butylmethylether (4 ml) was added to the solution followed by seed crystals of the sodium salt and this suspension was stirred overnight at ambient conditions. The suspension was filtered, washed with tert-butylmethylether (2 ml) and air dried to give the sodium salt (0.2312 g) as a hydrate.
NMR: Consistent with salt formation
1H NMR (400 MHz, DMSO d6) d=13.35 (br s, 1H), 8.53 (s, 1H), 7.90 (d, J=1.2 Hz, 1H), 7.73 (s, 1H), 7.65 (d, J=2.5 Hz, 1H), 7.62 (d, J=2.2 Hz, 1H), 7.33 (s, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.59 (m, 2H). 2.83 (d, J=10.3, 2H), 2.61 (s, 3H), 1.78 (t, J=10.5 Hz, 2H), 1.05 (d, J=6.1 Hz, 6H)
EMAIL ME amcrasto@gmail.com
EMAIL ME amcrasto@gmail.com
| US20100280029 * | 28 Apr 2010 | 4 Nov 2010 | Julie Nicole Hamblin | Novel compounds |
| WO2010125082A1 | 28 Apr 2010 | 4 Nov 2010 | Glaxo Group Limited | Oxazole substituted indazoles as pi3-kinase inhibitors |
| US20140256721 * | 14 Apr 2014 | 11 Sep 2014 | Glaxosmithkline Intellectual Property Development Limited | Novel Polymorphs and Salts |
| WO2012032065A1 | 6 Sep 2011 | 15 Mar 2012 | Glaxo Group Limited | Indazole derivatives for use in the treatment of influenza virus infection |
| WO2012032067A1 | 6 Sep 2011 | 15 Mar 2012 | Glaxo Group Limited | Polymorphs and salts of n- [5- [4- (5- { [(2r,6s) -2, 6 – dimethyl – 4 -morpholinyl] methyl} – 1, 3 – oxazol – 2 – yl) – 1h- inda zol-6-yl] -2- (methyloxy) – 3 – pyridinyl] methanesulfonamide |
| WO2012055846A1 | 25 Oct 2011 | 3 May 2012 | Glaxo Group Limited | Polymorphs and salts of 6-(1h-indol-4-yl)-4-(5- { [4-(1-methylethyl)-1-pi perazinyl] methyl} -1,3-oxazol-2-yl)-1h-indazole as pi3k inhibitors for use in the treatment of e.g. respiratory disorders |
| WO2012064744A2 * | 8 Nov 2011 | 18 May 2012 | Lycera Corporation | Tetrahydroquinoline and related bicyclic compounds for inhibition of rorϒ activity and the treatment of disease |
| WO2013088404A1 | 14 Dec 2012 | 20 Jun 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2014068070A1 | 31 Oct 2013 | 8 May 2014 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for preventing antiphospholipid syndrome (aps) |
| US8524751 | 5 Mar 2010 | 3 Sep 2013 | GlaxoSmithKline Intellecutual Property Development | 4-oxadiazol-2-YL-indazoles as inhibitors of P13 kinases |
| US8536169 | 3 Jun 2009 | 17 Sep 2013 | Glaxo Group Limited | Compounds |
| US8575162 | 28 Apr 2010 | 5 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8580797 | 28 Apr 2010 | 12 Nov 2013 | Glaxo Smith Kline Intellectual Property Development Limited | Compounds |
| US8586583 | 2 Oct 2012 | 19 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8586590 | 2 Oct 2012 | 19 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8609657 | 2 Oct 2012 | 17 Dec 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8658635 | 3 Jun 2009 | 25 Feb 2014 | Glaxosmithkline Intellectual Property Development Limited | Benzpyrazol derivatives as inhibitors of PI3 kinases |
| US8735390 | 6 Sep 2011 | 27 May 2014 | Glaxosmithkline Intellectual Property Development Limited | Polymorphs and salts |
| US8765743 | 3 Jun 2009 | 1 Jul 2014 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
…..

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA

.




 .
.



.
 MANUDEVI
MANUDEVI

GSK 2793660, Trying to crack the structure
 COMPD A
COMPD A
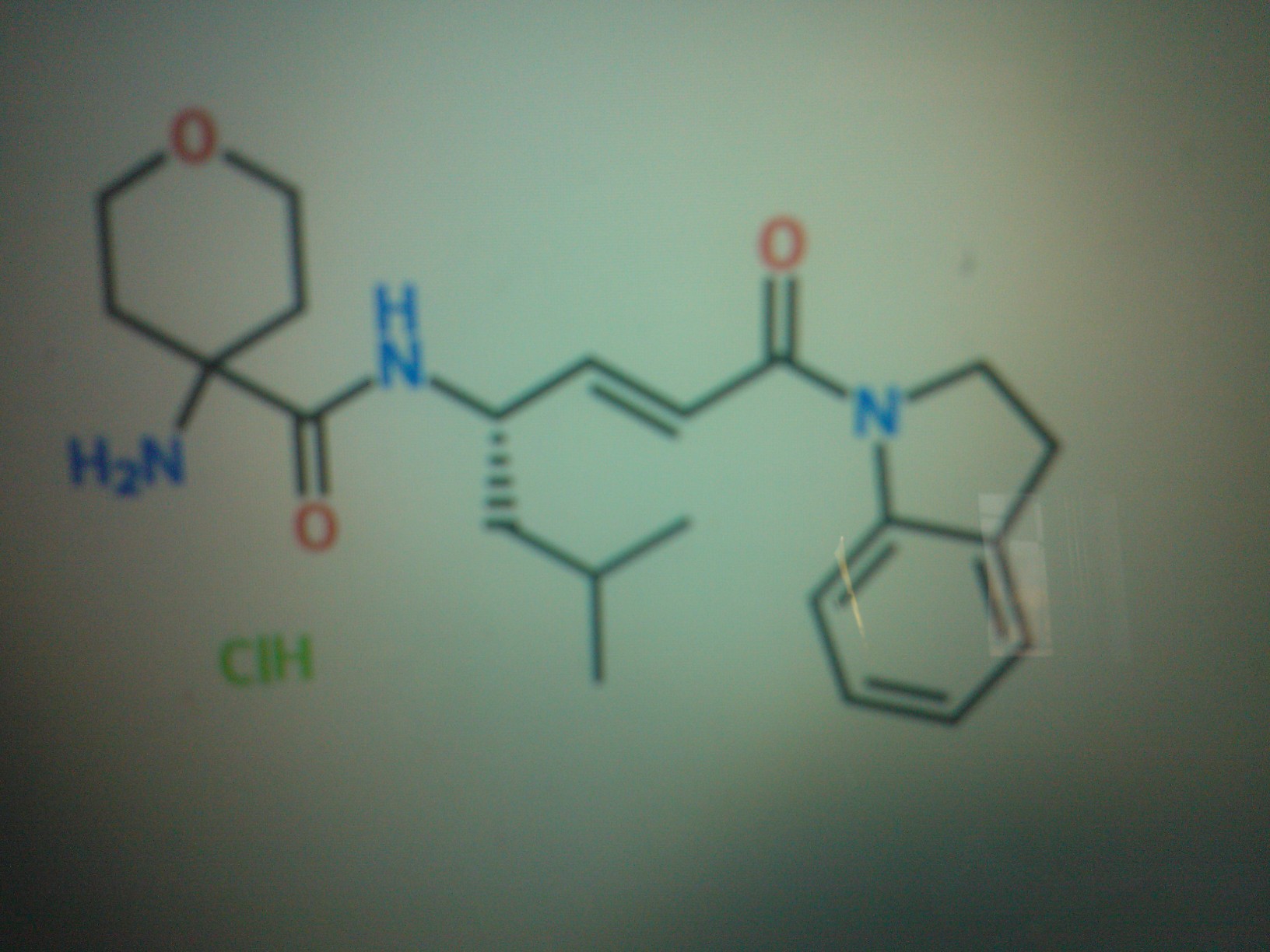 COMPD B
COMPD B
 COMPD C
COMPD C
 COMPD D
COMPD D
 A
A B
B C
C D
DGSK 2793660
DATA FOR A
HCL SALT CAS 1613458-78-8
BASE CAS 1613458-70-0
C20 H27 N3 O3 . Cl H
MW OF BASE…..357.45
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
2H-Pyran-4-carboxamide, 4-amino-N-[(1S,2E)-4-(2,3-dihydro-1H-indol-1-yl)-1-ethyl-4-oxo-2-buten-1-yl]tetrahydro-, hydrochloride (1:1)
DATA FOR B
1613458-79-9 HCL SALT
1613458-71-1 BASE
C22 H31 N3 O3 . Cl H
MW 385.50 OF BASE
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
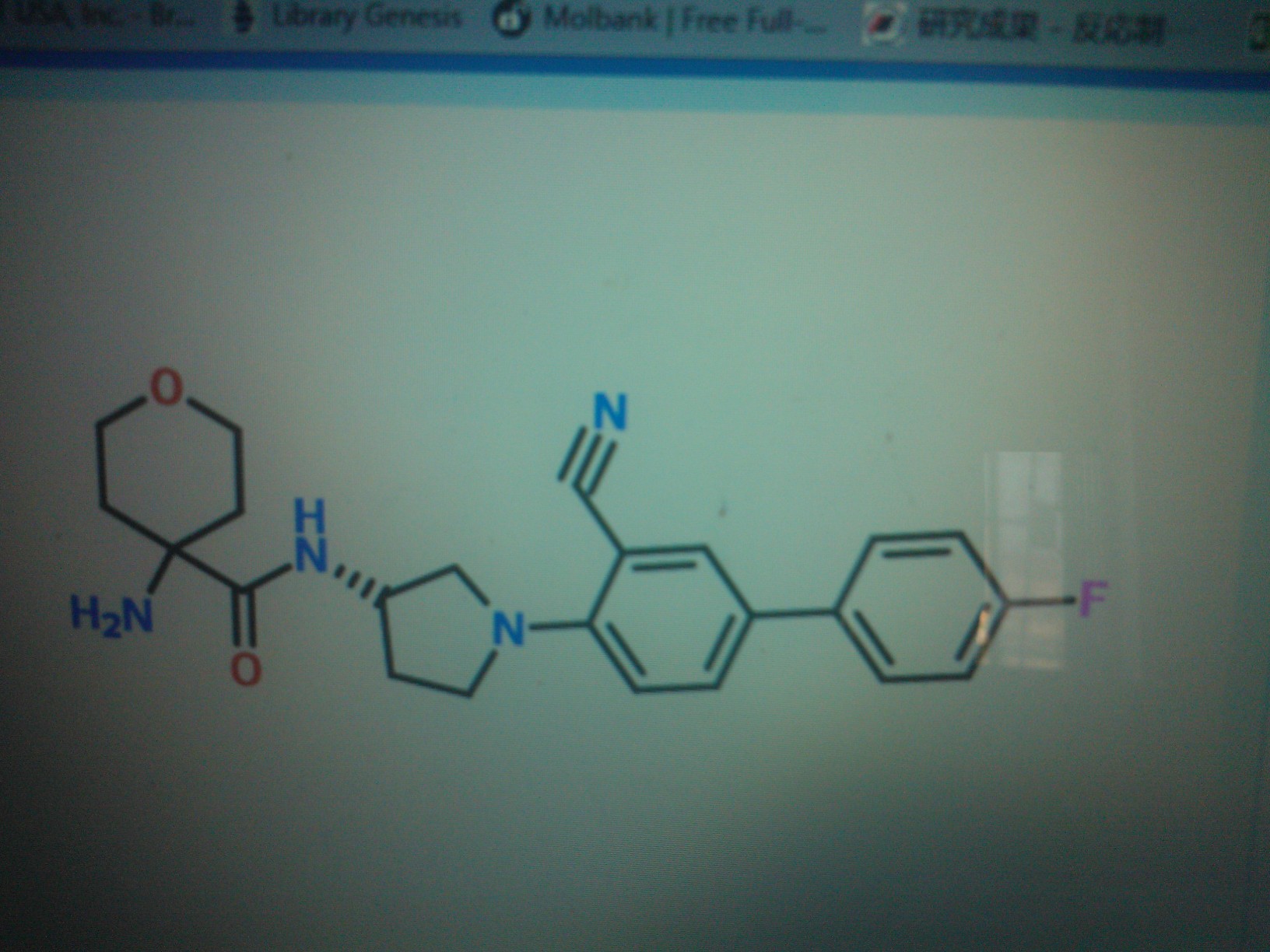
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
C24 H27 F N4 O . Cl H, MW 442.957
CAS OF BASE 1394001-73-0
CAS OF HCL 1394001-71-8
DATA FOR D
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride
CAS OF BASE 1394001-74-1
CAS OF HCL 1394001-72-9
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Bronchiectasis
Dipeptidyl peptidase I inhibitor
http://www.gsk.com/media/280387/product-pipeline-2014.pdf
This study is the first administration of GSK2793660 to humans and will evaluate the safety, tolerability, PK and PD of single oral ascending doses of GSK2793660, and of repeat oral doses of GSK2793660 in healthy subjects. The study will comprise two parts (Part A and Part B). Part A will consist of two cohorts of subjects, each taking part in a three-way cross over study, with ascending doses of GSK2793660 and placebo. Available safety, PK and PD data will be reviewed before each dose escalation. This will be followed by a food-effect arm in the cohort that received what is deemed to be the target clinical dose. Part B is planned to consist of up to two cohorts of subjects, each taking part in one 14 day repeat dose study period. Subjects will be dosed on Day 1 and then on Days 3-15. It is planned that two doses will be evaluated. The dose(s) to be tested will be selected based on safety, PK, and PD from Part A. The study is intended to provide sufficient confidence in the safety profile of the molecule and information on target engagement to allow progression to further studies………..https://clinicaltrials.gov/ct2/show/NCT02058407
Cathepsin C inhibitors for treating cystic fibrosis, non-cystic fibrosis bronchiectasis, and ANCA-associated vasculitis
Cathepsins are a family of enzymes included in the papain superfamily of cysteine proteases. Cathepsins B, C, F, H, K, L, S, V, and X have been described in the scientific literature. Cathepsin C is also known in the literature as Dipeptidyl Peptidase I or “DPPI.”
A number of recently published studies have begun to describe the role cathepsin C plays in certain inflammatory processes. See e.g. Adkison et al., The Journal of Clinical Investigation 109:363-371 (2002); Tran et al., Archives of Biochemistry and Biophysics 403 : 160-170 (2002); Thiele et al., The Journal of Immunology 158: 5200-5210 (1997);
Bidere et al., The Journal of Biological Chemistry 277: 32339-32347 (2002); Mabee et al., The Journal of Immunology 160: 5880-5885 (1998); McGuire et al., The Journal of
Biological Chemistry, 268: 2458-2467 (1993); and Paris et al., FEBS Letters 369: 326-330 (1995). From these studies, it appears that cathepsin C is co-expressed in granules of neutrophils and other leukocytes with certain serine proteases and cathepsin C functions to process the pro-forms of the serine proteases to active forms. Serine proteases are released from the granules of leukocytes recruited to sites of inflammation. Once activated, these proteases have a number of functions including degradation of various extracellular matrix components, which together can propagate tissue damage and chronic inflammation.
Studies in both cathepsin C deficient mice, and the human cathepsin C deficiency
Papillon-Lefevre syndrome clearly demonstrate that cathepsin C is required for the
activation of the neutrophil serine proteases in azurophilic granules such as neutrophil elastase (NE), cathepsin G, and proteinase 3. See Pham, C. T. et al., J. Immunol. 173 :
7277-7281 (2004).
A number of respiratory diseases are associated with an overabundant
acculumation of neutrophils and the presence of increased levels of at least some
neutrophil serine proteases. These enzymes are believed to play a role in the pathology of several respiratory diseases, such as Chronic Obstructive Pulmonary Disease (“COPD”), cystic fibrosis (CF), and non-cystic fibrosis (non-CF) bronchiectasis. Each of these diseases is associated with increased levels of E in particular, and E at least is considered to play a role in the progression of disease. See Ranes, J. and Stoller, J. K., Semin. Respir. Crit. Care Med 26: 154-166 (2005); Saget, S. D. et al., Am. J. Resp. Crit. Care Med. 186: 857-865 (2012); Tsang, K. W. et al., Chest 117: 420-426 (2000).
Additional roles of the other proteases is emerging. See Hartl, D. et al., Nature Med. 13 : 1423-1430 (2007); Korkmaz, B. et al., Pharm. Rev. 62: 726-759 (2010).
Cigarette smoking is a significant risk factor for developing COPD. Exposure to cigarette smoke and other noxious particles and gases may result in chronic inflammation of the lung. In response to such exposure, inflammatory cells such as CD8+ T cells, macrophages, and neutrophils are recruited to the area. These recruited inflammatory cells release proteases, which are believed to play a major role in the disease etiology by a number of mechanisms. Proteases released from recruited cells include the serine proteases NE as above; granzymes A and B, released from cytotoxic T cells or natural killer cells; and chymases, released from mast cells. Cathepsin C appears to be involved in activating all of these enzymes to some extent.
A number of studies with cathepsin C deficient mice have suggested roles for cathepsin C in disease models. Cathepsin C knockout mice are resistant to lung airspace enlargement and inflammatory cell infiltration in both cigarette smoke and ozone exposure models of COPD. See Guay et al., Current Topics in Medicinal Chemistry, 2010, 10, 708- 716; See also Podolin et al. (2008), Inflammation Research, 57(Suppl 2) S104.
In a model of rheumatoid arthritis (“RA”), another chronic inflammatory disease where cathepsin C may play a role, neutrophils are recruited to the site of joint
inflammation and release cathepsin G, NE, and proteinase 3, which are believed to be responsible in part for cartilage destruction associated with RA (Hu, Y. and Pham, C. T. Arthritis Rheum. 52: 2553-2558 (2005); Zen, K. et al, Blood 117:4885-4894 (2011)). Other models where cathepsin C may play a role include osteoarthritis, asthma, Multiple Sclerosis, and Anti-Neutrophil Cytoplasmic Autoantibody (ANCA)-related diseases (e.g. ANCA-associated vasculitis). See e.g. Matsui, K., Yuyama, N., Akaiwa, M., Yoshida, N. L., Maeda, M., Sugita, Y., Izuhara, K., Gene 293(1-2): 1-7 (2002); Wolters, P. J., Laig- Webster, M., Caughey, G. H., American Journal of Respiratory Cell & Molecular Biology 22(2): 183-90 (2000); Schreiber et al., J. Am. Soc. Nephrol. 23 :470-482 (2012). Cathepsin C has been demonstrated to have a role in neutrophil migration in the development of aortic aneurysms by a mechanism which has not been clearly elucidated (Pagano, M. B. et al., PNAS 104: 2855-2860 (2007)).
One approach to treating these conditions is to inhibit the activity of the serine proteases involved in the inflammatory process, especially NE activity. See e.g.,
Ohbayashi, Expert Opin. Investig. Drugs 11(7): 965-980 (2002); Shapiro, Am. J. Respir. Cell Mol. Biol. 26: 266-268 (2002). Indeed, a potent and selective inhibitor of NE was found to improve lung function in patients with bronchiectasis (Stockley, R. et al. Respir. Med. 107, 524-533 (2013)). In light of the role cathepsin C plays in activating certain serine proteases, especially NE, it is desirable to prepare compounds that inhibit its activity, which thereby inhibit serine protease activity. Thus, there is a need to identify compounds that inhibit cathepsin C, which can be used in the treatment of a variety of conditions mediated by cathepsin C.
There are additional activities of cathepsin C that may also be related to disease etiology. Cathepsin C is highly expressed in the lung epithelium where it may play a role in the processing of other enzymes not yet identified. Cathepsin C has also been reported to cleave kallikrein-4, which is believed to play a role in dental enamel maturation (Tye, C. E. et al. J. Dental Res. 88: 323-327 (2009)). Finally, cathepsin C is itself released from cells and may play a direct role in the degradation of matrix proteins.
DATA FOR A
WO 2014091443
http://www.google.com/patents/WO2014091443A1?cl=en

synthesis







Intermediate 1
1,1-dimethylethyl ((l -l-{[methyl(methyloxy)amino]carbonyl}propyl)carbamate
To a solution of (2,S)-2-({[(l,l-dimethylethyl)oxy]carbonyl}amino)butanoic acid (2.50 g, 12.3 mmol) in THF (15.0 mL) was added Ι,Γ-carbonyldiimidazole (2.39 g, 14.8 mmol) portionwise over about 10 min. After stirring 30 min at RT, a solution of Ν,Ο- dimethylhydroxylamine hydrochloride (1.32 g, 13.5 mmol) and DIPEA (2.36 mL, 13.5 mmol) in DMF (4.0 mL) was added. The reaction mixture was stirred for 2 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HC1 (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.60 g, 88%) as a clear, colorless oil. LC-MS m/z 247 (M+H)+, 0.94 min (ret time).
Intermediate 2
1,1-dimethylethyl [(lS -l-formylpropyl] carbamate
To a solution of L1AIH4 (0.453 g, 11.9 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution of 1, 1-dimethylethyl ((l,S)-l-{[methyl(methyloxy)amino]carbonyl}- propyl)carbamate (2.67 g, 10.8 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6.5 mL) followed by 5% aq. potassium bisulfate (6.5 mL). The reaction mixture was washed with 1 M aq. HC1 (3 x 10 mL), saturated aq. NaHC03 (3 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil.
Intermediate 3
methyl (2E V)-4-({ [(1 , l-dimethylethyl)oxy] car bonyl} amino)-2-hexenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (4.35 g, 13.0 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 2 in Et20 (15 mL). The reaction mixture was stirred at RT overnight. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.44 g, 55% over two steps) as a clear, colorless oil. LC-MS m/z 244 (M+H)+, 0.98 min (ret time). Intermediate 4
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-2-hexenoic acid
Li OH (2.95 g, 123 mmol) was added to a solution of methyl (2£, S 4-({[(1, 1- dimethylethyl)oxy]carbonyl}amino)-2-hexenoate (6 g, 24.66 mmol) in THF (50 mL), MeOH (10.00 mL), and water (50.0 mL). The reaction was stirred overnight at RT. After 18.5 h, the reaction mixture was concentrated under reduced pressure to remove the THF and MeOH. Water (40 mL) was added, and aqueous mixture was adjusted to pH = 3 with 6 M aq. HC1, as measured by pH paper. EtOAc (80 mL) was added, the layers were separated, and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were dried over Na2S04, concentrated under reduced pressure, and dried under high vacuum, giving 6.09 g of the title compound. LC-MS m/z 230 (M+H)+, 0.77 min (ret time).
Intermediate 5
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl] carbamate
A solution of 50 wt% *T3P in EtOAc (22.00 mL, 37.0 mmol) was added dropwise via addition funnel to a solution of (2£,,4,S)-4-({[(l, l-dimethylethyl)oxy]carbonyl}- amino)-2-hexenoic acid (5.65 g, 24.64 mmol), 2,3-dihydro-lH-indole (2.76 mL, 24.64 mmol), and Et3N (11 mL, 79 mmol) in CH2C12 (90 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 30 min, the reaction was quenched by dropwise addition of saturated aq. NaHC03 (50 mL). The layers were separated, and the reaction was washed with 10% citric acid (1 x 50 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 7.21 g (89%) of the title compound. LC-MS m/z 331 (M+H)+, 1.05 (ret time). Intermediate 6
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine
trifluoroacetate
TFA (25 mL, 324 mmol) was added to a solution of 1, 1-dimethylethyl [(1^,2£)-4- (2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]carbamate (7.21 g, 21.82 mmol) in CH2C12 (25 mL). The reaction was stirred at RT. After 3.5 h, CH2C12 (200 mL) was added, and the reaction was concentrated under reduced pressure and dried under high vacuum. LC-MS m/z 231 (M+H)+, 0.69 (ret time).
Intermediate 7
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino carbonyl)tetrahydro-2H-pyran-4-yl]carbamate
A solution of 50 wt% UT3P in EtOAc (1.3 mL, 2.184 mmol) was added dropwise to a solution of [(l,S’,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l-yl]amine trifluoroacetate (500 mg, 1.452 mmol), 4-((tert-butoxycarbonyl)amino)tetrahydro-2H- pyran-4-carboxylic acid (356 mg, 1.452 mmol), and Et3N (1 mL, 7.21 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction mixture was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 251 mg (38%) of the title compound. LC-MS m/z 458 (M+H)+, 0.96 (ret time).
Example 1
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten-l- yl]tetrahydr -2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.23 mL, 2.76 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-ethyl-4-oxo-2-buten- l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.549 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp) for 1 h 45 min. The solvent was evaporated under reduced pressure. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 193.3 mg (89%) of the title compound. LC-MS m/z 358 (M+H)+, 0.68 (ret time).
1H MR (400 MHz, METHANOL-^) δ ppm 8.14 (br. s., 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.53 Hz, 1 H); 7.02 – 7.09 (m, 1 H); 6.83 (dd, J=15.18, 6.65 Hz, 1 H); 6.49 (d, 7=14.8 Hz, 1 H); 4.56 (d, 7=7.28 Hz, 1 H); 4.22 (br. s., 2 H); 3.95 (d, 7=7.53 Hz, 1 H); 3.88 – 3.94 (m, 1 H); 3.71 – 3.78 (m, 2 H); 3.23 (br. s., 2 H); 2.39 – 2.46 (m, 2 H); 1.79 – 1.86 (m, 2 H); 1.75 (s, 1 H); 1.72 (d, 7=8.28 Hz, 1 H); 1.00 (t, 7=7.40 Hz, 3 H)
DATA FOR B
4-Amino-N-[(2E,4S)-1-(2,3-dihydro-1H-indol-1-yl)-6-methyl-1-oxohept-2-en-4-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride







http://www.google.com/patents/WO2014091443A1?cl=en
Intermediate 8
N -{[(l,l-dimethylet leucinamide
To a solution ofN-(tert-butoxycarbonyl)-L-leucine (3.00 g, 13.0 mmol) in THF (25.0 mL) was added Ι,Γ-carbonyldiimidazole (2.52 g, 15.6 mmol) portionwise over about 10 min. After stirring 1 h at RT, a solution of N,O-dimethylhydroxylamine hydrochloride (1.39 g, 14.3 mmol) and DIPEA (2.49 mL, 14.3 mmol) in DMF (6.0 mL) was added. The reaction mixture was stirred for 2.5 h at RT, followed by concentration in vacuo. The residue was diluted with EtOAc (50 mL) and washed with 1 M aq. HCl (2 x 20 mL), saturated aq. NaHC03 (2 x 20 mL), and brine (20 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound (2.34 g, 66%) as a clear, colorless oil. LC-MS m/z 275 (M+H)+, 1.17 min (ret time).
Intermediate 9
1,1-dimethylethyl [(lS -l-formyl-3-methylbutyl]carbamate
To a solution of L1AIH4 (0.356 g, 9.38 mmol) in Et20 (20 mL) at 0 °C was added dropwise a solution ofN2-{[(l, l-dimethylethyl)oxy]carbonyl}-N1-methyl-N1-(methyloxy)-L- leucinamide (2.34 g, 8.53 mmol) in Et20 (15 mL). The reaction mixture was stirred for 30 min at 0 °C and quenched with EtOAc (6 mL) followed by 5% aq. potassium bisulfate (6 mL). The reaction mixture was washed with 1 M aq. HCl (2 x 10 mL), saturated aq. NaHC03 (2 x 10 mL), and brine (10 mL). The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to afford the title compound as a clear, colorless oil. Intermediate 10
methyl (2E 4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoate
To a stirred solution of methyl (triphenylphosphoranylidene) acetate (3.42 g, 10.2 mmol) in Et20 (25 mL) at RT was added a solution of Intermediate 9 in Et20 (15 mL). The reaction mixture was stirred for 15 h at RT. The solid was removed by filtration and the solution was concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) afforded the title compound (1.74 g, 75% over two steps) as a clear, colorless oil. LC-MS m/z 272 (M+H)+, 1.22 min (ret time).
Intermediate 11
(2E,4S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2-heptenoic acid
To a solution of methyl (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6- methyl-2-heptenoate (5.00 g, 18.43 mmol) in THF (15 mL), MeOH (15.0 mL), and water (15 mL) was added Li OH (2.206 g, 92.00 mmol). After stirring for 2 h at RT, the reaction mixture was concentrated in vacuo. The reaction mixture was acidified with 6 M aq. HC1 to pH = 5 and then extracted with EtOAc. The organic layer was washed with water, dried over Na2SC”4, filtered, and concentrated in vacuo to afford the title compound (4.7 g, 99%) as a white semi-solid. LC-MS m/z 158 (M+H-Boc)+, 0.94 min (ret time).
Intermediate 12
1,1-dimethylethyl [(lS,2E)-4-(2,3-dihydro-li -indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]carbamate
To a solution of (2£,,4,S)-4-({[(l,l-dimethylethyl)oxy]carbonyl}amino)-6-methyl-2- heptenoic acid (4.70 g, 18.26 mmol) in DMF (30.0 mL) were added BOP reagent (8.08 g, 18.26 mmol) and DIPEA (6.38 mL, 36.5 mmol). After stirring at RT for 5 min, 2,3-dihydro- lH-indole (2.053 mL, 18.26 mmol) was added and stirring continued overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04, filtered, concentrated in vacuo and purified by flash column chromatography (0-20% EtOAc/hexanes) to afford the title compound (4.83 g, 74%) as a white solid. LC-MS m/z 359 (M+H)+, 1.18 min (ret time).
Intermediate 13
[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten-l-yl]amine trifluoroacetate
To a solution of 1, 1-dimethylethyl [(l^,2£)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2- methylpropyl)-4-oxo-2-buten-l-yl]carbamate (3.21 g, 8.95 mmol) in CH2C12 (10.0 mL) was added TFA (10 mL, 130 mmol). The reaction mixture was stirred for 17.5 h at RT and then concentrated under reduced pressure and dried under high vacuum to afford the title compound. LC-MS m/z 259 (M+H)+, 0.76 min (ret time).
Intermediate 14
1,1-dimethylethyl [4-({[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l- l]amino}carbonyl)tetrahydro-2H- ran-4-yl]carbamate
A solution of 50 wt% ¾P in EtOAc (1.2 mL, 2.016 mmol) was added dropwise to a solution of [(15′,2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2- buten-l-yl]amine trifluoroacetate (500 mg, 1.343 mmol), 4-((tert- butoxycarbonyl)amino)tetrahydro-2H-pyran-4-carboxylic acid (329 mg, 1.343 mmol), and Et3N (0.93 mL, 6.71 mmol) in CH2C12 (5 mL) at 0 °C (bath temp). The ice bath was removed, and the reaction was stirred at RT. After 1 h 20 min, the reaction was washed with saturated aq. NaHC03 (1 x 5 mL) and 10% citric acid (1 x 5 mL). The organic layer was concentrated under a stream of nitrogen, and the residue was purified by flash column chromatography, giving 204 mg (31%) of the title compound. LC-MS m/z 486 (M+H)+, 1.07 min (ret time).
Example 2
4-amino-N-[(lS,2E)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4-oxo-2-buten- l-yl]tetrahydro-2H-pyran-4-carboxamide hydrochloride
A solution of concentrated aq. HCI (0.22 mL, 2.64 mmol) was added to a solution of 1,1-dimethylethyl [4-({[(1^2JE)-4-(2,3-dihydro-lH-indol-l-yl)-l-(2-methylpropyl)-4- oxo-2-buten-l-yl]amino}carbonyl)tetrahydro-2H-pyran-4-yl]carbamate (251 mg, 0.517 mmol) in isopropanol (2.5 mL). The reaction flask was fitted with an air condenser, and the reaction mixture was heated to 65 °C (bath temp). After 1 h 45 min, the solvent was evaporated under reduced pressure at 60 °C. Water (5 mL) was added to the residue, and the mixture was concentrated under reduced pressure at 65 °C. Water (2 mL) was added to the residue, and the mixture was lyophilized, giving 130.6 mg (60%) of the title compound. LC-MS m/z 386 (M+H)+, 0.79 (ret time). 1H MR (400 MHz, METHANOL- d4) δ ppm 8.15 (d, J=7.03 Hz, 1 H); 7.25 (d, J=7.03 Hz, 1 H); 7.18 (t, J=7.65 Hz, 1 H); 7.06 (t, J=7.91 Hz, 1 H); 6.81 (dd, J=15.18, 6.40 Hz, 1 H); 6.49 (br. s., 1 H); 4.73 – 4.85 (m, 2 H); 4.21 (t, J=8.28 Hz, 2 H); 3.91 – 3.97 (m, 2 H); 3.70 – 3.77 (m, 2 H); 3.25 – 3.21 (m, 2 H); 2.35 – 2.48 (m, 2 H); 1.82 (d, J=14.31 Hz, 2 H); 1.63 – 1.71 (m, 2 H); 1.50 – 1.57 (m, 1 H); 0.98 (dd, J=11.92, 6.40 Hz, 6 H).
DATA FOR C
1-Amino-N-[(3S)-1-(3-cyano-4′-fluorobiphenyl-4-yl)pyrrolidin-3-yl]cyclohexanecarboxamide hydrochloride
http://www.google.im/patents/WO2012112733A1?cl=en
Example 1
l-amino-N-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidin l] cyclohexanecarboxamide hydrochloride

HCI salt
A solution of 1,1-dimethylethyl [l-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl]amino}carbonyl)cyclohexyl]carbamate (44 mg, 0.087 mmol) in HCI (4 M solution in 1,4-dioxane, 1.0 mL, 4.00 mmol) was stirred at RT for 1 h. The reaction mixture was diluted with Et20 (5 mL), and the mixture was filtered and washed with Et20 (2 x 2 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (33.5 mg, 87%). LC-MS m/z 407 (M+H)+, 0.94 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.65 – 7.72 (m, 2 H), 7.52 – 7.59 (m, 2 H), 7.10 – 7.17 (m, 2 H), 6.89 (d, J=8.53 Hz, 1 H), 4.50 – 4.58 (m, 1 H), 3.94 (dd, J=10.29, 6.53 Hz, 1 H), 3.80 (dt, J=9.41, 7.09 Hz, 1 H), 3.67-3.71 (m, 1 H), 3.64 (dd, J=10.29, 4.52 Hz, 1 H), 2.29 – 2.37 (m, 1 H), 2.04 – 2.16 (m, 3 H), 1.78 – 1.88 (m, 5 H), 1.45 – 1.62 (m, 3 H).
DATA FOR D
http://www.google.im/patents/WO2012112733A1?cl=en
Example 2
4-amino- V-[(3S)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3-pyrrolidinyl]tetrahydro-2H- pyr -4-carboxamide hydrochloride
HCI salt
A solution of 1,1-dimethylethyl [4-({[(35)-l-(3-cyano-4′-fluoro-4-biphenylyl)-3- pyrrolidinyl] amino }carbonyl)tetrahydro-2H-pyran-4-yl] carbamate (183 mg, 0.360 mmol) in HC1 (4 M solution in 1,4-dioxane, 2.0 mL, 8.00 mmol) was stirred at RT for 0.5 h. The reaction mixture was diluted with Et20 (10 mL), and the mixture was filtered and washed with Et20 (2 x 5 mL). Residual solid was dissolved in MeOH and concentrated under a stream of nitrogen at 50 °C and dried under high vacuum. Water (2 mL) was added to the residue, and the mixture was lyophilized with a Genevac® HT-4X to afford the title compound (122.8 mg, 77%). LC-MS m/z 409 (M+H)+, 0.87 min (ret time). 1H NMR (400 MHz, METHANOL-^) δ ppm 7.66 – 7.72 (m, 2 H), 7.53 – 7.60 (m, 2 H), 7.11 – 7.18 (m, 2 H), 6.89 (d, J=8.78 Hz, 1 H), 4.53 – 4.60 (m, 1 H), 3.87 – 3.97 (m, 3 H), 3.78 – 3.84 (m, 1 H), 3.64 – 3.76 (m, 4 H), 2.30 – 2.44 (m, 3 H), 2.11 – 2.19 (m, 1 H), 1.77 – 1.84 (m, 2 H).
| WO2004002491A1 * | 25 Jun 2003 | 8 Jan 2004 | David J Aldous | Morpholine and tetrahydropyran drivatives and their use as cathepsin inhibitors |
| WO2008121065A1 * | 28 Mar 2008 | 9 Oct 2008 | Astrazeneca Ab | Novel pyrrolidine derivatives as antagonists of the chemokine receptor |
| US20070032484 * | 25 Jul 2006 | 8 Feb 2007 | Roche Palo Alto Llc | Cathepsin K inhibitors |
| US20020107266 * | Dec 11, 2001 | Aug 8, 2002 | Marguerita Lim-Wilby | Amides used particularly in the treatment, prevention or amelioration of one or more symptoms of malaria or Chagas’ disease; inhibiting the activity of falcipain or cruzain |
| US20100286118 * | May 6, 2010 | Nov 11, 2010 | Rhonan Ford | Substituted 1-cyanoethylheterocyclylcarboxamide compounds 750 |
| WO2012109415A1 | Feb 9, 2012 | Aug 16, 2012 | Glaxosmithkline Llc | Cathepsin c inhibitors |
Dacinostat (LAQ-824, NVP-LAQ824,)
C22H25N3O3
Exact Mass: 379.18959
Molecular Weight: 379.45


Reversible acetylation of histones is a major regulator of gene expression that acts by altering accessibility of transcription factors to DNA. In normal cells, histone deacetylase (HDA) and histone acetyltrasferase together control the level of acetylation of histones to maintain a balance. Inhibition of HDA results in the accumulation of hyperacetylated histones, which results in a variety of cellular responses.
Inhibitors of HDA have been studied for their therapeutic effects on cancer cells. For example, butyric acid and its derivatives, including sodium phenylbutyrate, have been reported to induce apoptosis in vitro in human colon carcinoma, leukemia and retinoblastoma cell lines. However, butyric acid and its derivatives are not useful pharmacological agents because they tend to be metabolized rapidly and have a very short half-life in vivo. Other inhibitors of HDA that have been widely studied for their anti-cancer activities are trichostatin A and trapoxin. Trichostatin A is an antifungal and antibiotic and is a reversible inhibitor of mammalian HDA. Trapoxin is a cyclic tetrapeptide, which is an irreversible inhibitor of mammalian HDA.
Although trichostatin and trapoxin have been studied for their anti-cancer activities, the in vivo instability of the compounds makes them less suitable as anti-cancer drugs. There remains a need for an active compound that is suitable for treating tumors, including cancerous tumors, that is highly efficacious and stable

|
References |
1: Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A, Sotomayor EM. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011 Apr 1;186(7):3986-96. doi: 10.4049/jimmunol.1001101. Epub 2011 Mar 2. PubMed PMID: 21368229.
2: Schwarz K, Romanski A, Puccetti E, Wietbrauk S, Vogel A, Keller M, Scott JW, Serve H, Bug G. The deacetylase inhibitor LAQ824 induces notch signalling in haematopoietic progenitor cells. Leuk Res. 2011 Jan;35(1):119-25. doi: 10.1016/j.leukres.2010.06.024. Epub 2010 Jul 31. PubMed PMID: 20674020.
3: Cho YS, Whitehead L, Li J, Chen CH, Jiang L, Vögtle M, Francotte E, Richert P, Wagner T, Traebert M, Lu Q, Cao X, Dumotier B, Fejzo J, Rajan S, Wang P, Yan-Neale Y, Shao W, Atadja P, Shultz M. Conformational refinement of hydroxamate-based histone deacetylase inhibitors and exploration of 3-piperidin-3-ylindole analogues of dacinostat (LAQ824). J Med Chem. 2010 Apr 8;53(7):2952-63. doi: 10.1021/jm100007m. PubMed PMID: 20205394.
4: Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, Craft N, Economou JS, Marincola FM, Wang E, Ribas A. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009 Nov 15;69(22):8693-9. doi: 10.1158/0008-5472.CAN-09-1456. Epub 2009 Oct 27. PubMed PMID: 19861533; PubMed Central PMCID: PMC2779578.
5: Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, Scott CL, Strasser A, Atadja P, Lowe SW, Johnstone RW. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. 2009 Jul 9;114(2):380-93. doi: 10.1182/blood-2008-10-182758. Epub 2009 Apr 21. PubMed PMID: 19383971.
6: de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, Karavasilis V, Mita M, Shaw H, Workman P, Kaye S, Rowinsky EK, Aherne W, Atadja P, Scott JW, Patnaik A. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008 Oct 15;14(20):6663-73. doi: 10.1158/1078-0432.CCR-08-0376. PubMed PMID: 18927309.
7: Chung YL, Troy H, Kristeleit R, Aherne W, Jackson LE, Atadja P, Griffiths JR, Judson IR, Workman P, Leach MO, Beloueche-Babari M. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of a novel histone deacetylase inhibitor, LAQ824, in human colon carcinoma cells and xenografts. Neoplasia. 2008 Apr;10(4):303-13. PubMed PMID: 18392140; PubMed Central PMCID: PMC2288545.
8: Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs. 2007 Aug;18(7):793-800. PubMed PMID: 17581301.
9: Kato Y, Salumbides BC, Wang XF, Qian DZ, Williams S, Wei Y, Sanni TB, Atadja P, Pili R. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther. 2007 Jan;6(1):70-81. PubMed PMID: 17237267.
10: Leyton J, Alao JP, Da Costa M, Stavropoulou AV, Latigo JR, Perumal M, Pillai R, He Q, Atadja P, Lam EW, Workman P, Vigushin DM, Aboagye EO. In vivo biological activity of the histone deacetylase inhibitor LAQ824 is detectable with 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography. Cancer Res. 2006 Aug 1;66(15):7621-9. PubMed PMID: 16885362.
SEE MORE AT……….http://drugsynthesisint.blogspot.in/p/nostat-series.html
SEE MORE AT……….http://drugsynthesisint.blogspot.in/p/nostat-series.html

US Orphan status for Bexion’s brain tumour drug BXQ-350
SDVYCEVCEFLVKEVTKLIDNNKTEKEILDAFDKMCSKLPKSLSEECQEVVDTYGSSILSILLEEV SPELVCSMLHLCSG [SEQ ID NO: 2].
BXQ-350
Cincinnati Children’s Hospital ……..innovator
Bexion Pharmaceuticals……….under license
In February 2015, the US FDA granted saposin C Orphan designation for the treatment of glioblastoma multiforme

SAPOCIN C
Recombinant human Saposin C (SapC) bound to a liposomal formulation of the dioleoylphosphatidylserine
Bexion’s Saposin C – the active ingredient in the brain tumour therapy BXQ-350 – has been awarded Orphan Drug status by US regulators.
Read more at: http://www.pharmatimes.com/Article/15-02-17/US_Orphan_status_for_Bexion_s_brain_tumour_drug.aspx#ixzz3S3zXdHlO
Bexion Pharmaceuticals, under license from the Cincinnati Children’s Hospital, is investigating a human saposin C (SapC)/liposomal dioleoylphosphatidylserine (DOPS) conjugate, SapC-DOPS (BXQ-350), a nanovesicle-formulated pro-apoptotic sphingomyelinase activating molecular imaging agent and anticancer agent, for the potential diagnosis and treatment of cancer , . In October 2013, Bexion was planning a phase I first-in-human trial for the therapy of glioblastoma multiforme

Bexion Pharmaceuticals LLC announced today that the U.S. Food and Drug Administration (FDA) has granted the company Orphan Drug designation for Saposin C, active ingredient in its proprietary drug BXQ-350 for the potential treatment of glioblastoma multiforme.
The FDA’s Office of Orphan Drug Products Development reviews applications for Orphan Drug status to support development of medicines for underserved patient populations, or rare disorders that affect fewer than 200,000 people in the United States. The successful application submitted by Bexion and the FDA granting of Orphan Drug status entitles the company to a seven-year period of marketing exclusivity in the United States for BXQ-350, if it is approved by the FDA for the treatment of glioblastoma multiforme. Orphan Drug status also enables the company to apply for research grant funding for Phase I and II Clinical Trials, tax credits for certain research expenses, and a waiver from the FDA’s application user fee, as well as additional support from FDA and a potentially faster regulatory process.
Bexion was previously awarded a prestigious Phase II Bridge Award (Small Business Innovation Research Grant; SBIR) from the National Cancer Institute (NCI) to support the manufacture and clinical testing of BXQ-350.
“Orphan Drug status for BXQ-350 is an important milestone in the development of this new treatment modality,” stated Dr. Ray Takigiku, founder and CEO of Bexion. “Few treatment options are available for patients suffering from glioblastoma multiforme and this designation recognizes the unmet need that exists with this disease, as well as the unique attributes of BXQ-350. In addition, orphan designation allows Bexion to benefit from important financial, regulatory and commercial considerations and we have seen recently that products with orphan designation have become sought after assets.”

About Orphan Drug Designation
Orphan Drug designation is a status assigned to a medicine intended for use in rare diseases. In the U.S., the Orphan Drug Designation program confers Orphan Drug status to successful applicants for medicines intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the U.S. or that are not expected to recover the costs of developing and marketing a treatment.1
The approval of an orphan designation request does not alter the standard regulatory requirements and process for obtaining marketing approval for investigational use. Sponsors must establish safety and efficacy of a compound in the treatment of a disease through adequate and well-controlled studies. However, the FDA review process may be speedier for Orphan Drugs than those which do not receive Orphan Drug designation.
About BXQ-350
In pre-clinical studies, Bexion’s first-in-class biologic, BXQ-350 has shown promising results in selectively inducing cell death in the laboratory. BXQ-350 is a proprietary nanovesicle formulation of Saposin C (sphingolipid activator protein C, or SapC) and the phospholipid dioleoylphosphatidylserine (DOPS).
About Bexion Pharmaceuticals
Bexion Pharmaceuticals is a privately held biotech company focused on the development and commercialization of innovative cures for cancer. Initial products are based on a proprietary platform technology licensed from Cincinnati Children’s Hospital Medical Center. The technology has demonstrated potential for development as a therapeutic, diagnostic and surgical imaging reagent, and as a carrier for other pharmaceutical agents, such as oligonucleotides. For more information, visit www.bexionpharma.com or contact Margaret van Gilse atmvangilse@bexionpharma.com.
1 U.S. Food and Drug Administration web site. “Regulatory Information: Orphan Drug Act.”http://www.fda.gov/regulatoryinformation/legislation/federalfooddrugandcosmeticactfdcact/significantamendmentstothefdcact/orphandrugact/default.htm.
Margaret van Gilse859-757-1652mvangilse@bexionpharma.com
SOURCE Bexion Pharmaceuticals LLC
Glioblastoma is the most common primary CNS malignant neoplasm in adults, and accounts for nearly 75% of the cases. Although there has been steady progress in their treatment due to improvements in neuro-imaging, microsurgery, and radiation, glioblastomas remain incurable. The average life expectancy is less than one year from diagnosis, and the five-year survival rate following aggressive therapy, including gross tumor resection, is less than 10%. Glioblastomas cause death due to rapid, aggressive, and infiltrative growth in the brain. The infiltrative growth pattern is responsible for the un-resectable nature of these tumors. Glioblastomas are also relatively resistant to radiation and chemotherapy, and therefore post-treatment recurrence rates are high. In addition, the immune response to the neoplastic cells is mainly ineffective in completely eradicating residual neoplastic cells following resection and radiation therapy.
One problem in treating glioblastoma is the tumor’s protection behind the blood-brain tumor barrier (BBTB). A significant obstacle in the development of therapeutics for glioblastoma is the inability of systemic therapies to efficiently cross the BBTB. Saposin C (SapC) is a sphingolipid- activating protein that functions to catabolize glycosphingolipids. SapC-DOPS forms stable nanovesicles which can efficiently cross the blood-brain tumor barrier and fuse with GBM cells inducing cell death.
Rapamycin is a macrolide antibiotic produced by Streptomyces hygroscopicus, which was discovered first for its properties as an antifungal agent. Streptomyces hygroscopicus has also been implicated as a cancer agent.
There remains a need in the art for new therapeutics for the treatment of glioblastoma.
…………………………………………………………………..
https://www.google.com/patents/US20040229799?cl=en22
Example 1Purification of Recombinant Saposin C
[0106] Recombinant saposin C was overexpressed in E. coli cells by using the isopropyl-1-thio-β-D-galactopyranoside inducing pET system (Qi et al. (1994) J. Biol. Chem. 269:16746-16753, herein incorporated by reference in its entirety). Expressed polypeptides with a His-tag were eluted from nickel columns. After dialysis, the polypeptides were further purified by HPLC chromatography as follows. A C4 reverse phase column was equilibrated with 0.1% trifluoroacetic acid (TFA) for 10 minutes. The proteins were eluted in a linear (0-100%) gradient of 0.1% TFA in acetonitrile over 60 minutes. The major protein peak was collected and lyophilized. Protein concentration was determined as previously described (Qi et al. (1994) J. Biol. Chem. 269:16746-16753).
Example 2Bath Sonication of Sanosin C and Dioleoylphosphatidylserine
[0107] Dioleoylphosphatidylserine (DOPS) was obtained from Avanti Polar Lipids (Alabaster AL). Twenty to thirty imoles of DOPS in chloroform were dried under N2 and vacuum to lipid films. Five to ten μmoles saposin C polypeptide was added to the dried films and suspended in 50 μl McIlvanine buffer (pH 4.7). The suspension was then brought to a 1 ml volume with either cell culture medium or phosphate buffered saline (PBS) (Ausubel et al. (2002) Current Protocols in Molecular Biology. John Wiley & Sons, New York, New York, herein incorporated by reference). The mixture was sonicated in a bath sonicator for approximately 20 minutes. Ice was added as needed to prevent overheating the samples.

………………………………………………………………
http://www.google.com/patents/WO2014078522A1?cl=en
The SapC-DOPS composition comprises a phospholipid, an isolated saposin C-related polypeptide, wherein the polypeptide comprises an amino acid sequence at least 75% identical to the entire length of SEQ ID NO: 2, and a pharmaceutically acceptable carrier, wherein the phospholipid forms a nano vesicle incorporating the polypeptide. In certain embodiments, the polypeptide comprises an amino acid sequence at least 85% identical to the entire length of SEQ ID NO: 2. In certain embodiments, the polypeptide comprises an amino acid sequence at least 95% identical to the entire length of SEQ ID NO: 2. In certain embodiments, the polypeptide comprises an amino acid sequence at least 99% identical to the entire length of SEQ ID NO: 2.
The Sequence Listing, filed electronically and identified as SEQ_LIST_OSIF-2013- 102.txt, was created on November 12, 2013, is 5,548 in size, and is hereby incorporated by reference.
[0004] SEQ ID NO: 1
siy
J su c n 61y &n
*8 a 210 2iS
t n«
:?e
<H ¾■ yts ca« ¾»* **u v ΆΧ» s?s ass ¾«¾
:»o
L st S«x ri» r s
SEQ ID NO: 2

BEXION PHARMA

-
-
Russell Street, Covington, KY 41011, United States
 $2.9 Million Grant Awarded to Covington-Based Bexion for Next Step in Cancer Fight
$2.9 Million Grant Awarded to Covington-Based Bexion for Next Step in Cancer Fight 921 Spring Street Covington, Kentucky 41016 United States
921 Spring Street Covington, Kentucky 41016 United States 112 East 4th Street, Covington, KY 41011.
112 East 4th Street, Covington, KY 41011. 1182 Riverhouse Way Covington KY : 427657
1182 Riverhouse Way Covington KY : 427657
-
LUCITANIB a VEGFR/FGFR dual kinase inhibitor in Phase 2 trials
LUCITANIB
6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide
6-(7-((l-aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)- N-methyl- 1 -naphthamide
6-(7-((l- aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-N-methyl-l- naphthamide
1058137-23-7 (E-3810 free base); 1058137-84-0 (E-3810 HCl salt)
| Patent | Submitted | Granted |
|---|---|---|
| Spiro Substituted Compounds As Angiogenesis Inhibitors [US8163923] | 2008-09-18 | 2012-04-24 |
A 4-(3-methoxypropoxy)-3-methylpyridinyl derivative of timoprazole that is used in the therapy of STOMACH ULCERS and ZOLLINGER-ELLISON SYNDROME. The drug inhibits H(+)-K(+)-EXCHANGING ATPASE which is found in GASTRIC PARIETAL CELLS.
For in advanced solid tumors.
Lucitanib (E-3810): Lucitanib, also known as E-3810, is a novel dual inhibitor targeting human vascular endothelial growth factor receptors (VEGFRs) and fibroblast growth factor receptors (FGFRs) with antiangiogenic activity. VEGFR/FGFR dual kinase inhibitor E-3810 inhibits VEGFR-1, -2, -3 and FGFR-1, -2 kinases in the nM range, which may result in the inhibition of tumor angiogenesis and tumor cell proliferation, and the induction of tumor cell death. Both VEGFRs and FGFRs belong to the family of receptor tyrosine kinases that may be upregulated in various tumor cell type

Overview

http://www.clovisoncology.com/products-companion-diagnostics/lucitanib/
Lucitanib is an oral, potent inhibitor of the tyrosine kinase activity of fibroblast growth factor receptors 1 through 3 (FGFR1-3), vascular endothelial growth factor receptors 1 through 3 (VEGFR1-3) and platelet-derived growth factor receptors alpha and beta (PDGFR α-ß). We own exclusive development and commercial rights to lucitanib on a global basis, excluding China. Lucitanib rights to markets outside of the U.S. and Japan have been sublicensed to Les Laboratoires Servier (Servier). We are collaborating with Servier on the global clinical development of lucitanib.

A Phase I/IIa clinical trial of lucitanib was initiated in 2010 and has demonstrated multiple objective responses in FGFR1 gene-amplified breast cancer patients, and objective responses were also observed in patients with tumors often sensitive to VEGFR inhibitors, such as renal cell and thyroid cancer. FGFR amplification is common in a number of tumor types, including breast cancer and squamous non-small cell lung cancer, and we intend to study lucitanib in these cancers as well as other solid tumors exhibiting FGFR pathway activation. A broad Phase II development program has been initiated by us and Servier in multiple indications, including advanced breast cancer and squamous NSCLC. For more information or to participate in the trials, contact the Clovis Oncology Clinical Trial Navigation Service at 1-855-262-3040, or 303-625-5010, or clovistrials@emergingmed.com.

http://www.asianscientist.com/2013/09/pharma/servier-license-lucitanib-simm-china-2013/
WO 2008/112408 Al and US 2008/0227812 Al disclose angiogenesis inhibitors with quinoline structure, useful for the treatment of neoplasias. One of the disclosed products is 6-(7-((l-aminocyclopropyl)methoxy)-6- methoxyquinolin-4-yloxy)-N-methyl-l-naphthamide of formula (I), described in example 3 of the above mentioned patent applications.

According to said documents, compound (I) is prepared by removing the benzyloxycarbonyl protective group from the compound benzyl l-((6- methoxy-4-(5-(methylcarbamoyl)-naphthalen-2-yloxy)quinolin-7- yloxy)methyl)cyclopropyl carbamate (II):

in acid medium or by hydrogenolysis, to give compound (I).
Compound (II) is obtained in a number of steps with different processes in which the benzyloxycarbonyl protected 1 -amino- 1-cyclopropylmethyl moiety is introduced by subjecting the acyl azide obtained from l-((6- methoxy-4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7- yloxy)methyl)cyclopropanecarboxylic acid of formula (III):

to Curtius rearrangement, in the presence of benzyl alcohol, or by alkylation of 6-(7-hydroxy-6-methoxyquinolin-4-yloxy)-N- methyl-1-naphthamide of formula (IV):

with 1 -benzyloxy carbony lamino- 1 -methylsolfonyloxymethyl- cyclopropane of formula (V):

The above mentioned applications do not provide yields concerning both the preparation of compound (II) by the two above mentioned reactions, and the conversion of compound (II) to (I).
Compound (III) is prepared by a process in which the 1-carboxy-l- cyclopropylmethyl moiety is introduced in 4-hydroxy-3-methoxyacetophenone as in the form of the ethyl ester, followed by formation of the 4- hydroxyquinoline ring and, finally, by the introduction of the 1- naphthylcarboxyamido fragment.
It is well known that the reactions requiring the use of azides, such as the formation of acyl azides, or Curtius rearrangement of the latter, are potentially hazardous as they involve risk of explosions, therefore they are not suitable for use in preparations on large scale. The synthetic methods reported in WO 2008/1 12408 and US
2008/0227812 include, inter alia, a general synthetic scheme in which the cycloalkyl-alkyl portion of the products is introduced by reaction between a cycloalkyl-alkyl mesylate and an hydroxy or amino acetophenone, followed by nitration to give a nitroacetofenone, reduction of the nitro group to amino group, formation of the 4-hydroxyquinoline ring and further work up of the latter to the final products. The above mentioned applications do not provide examples of the use of this process for compound (I) or the other described products.

SYNTHESIS


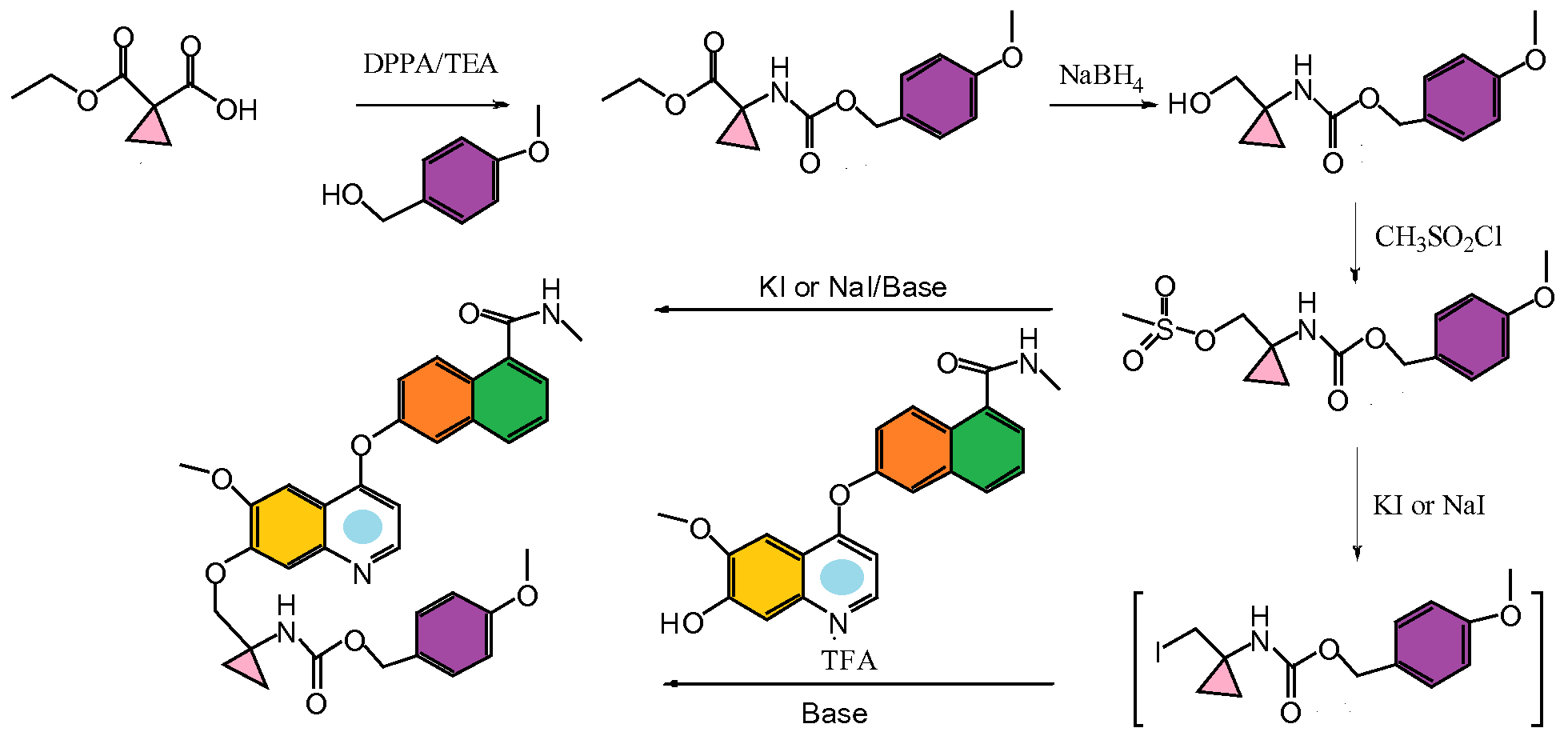
Patent
http://www.google.com/patents/WO2008112408A1?cl=en
Example 1
Benzyl l-((6-methoxy-4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7-yloxy)methyl)cyclo- propylcarbamate Method A:
6-Hydroxy- 1 -naphthoic acid (1 g) was mixed with acetic anhydride (5 ml) and sulfuric acid (5 drops). The mixture was refluxed for 3 hours and cooled at RT for 10 hours then mixed with water (15 ml). The solid was filtered and washed with water and cold MeOH to give the product as 6-acetoxy-l -naphthoic acid (900 mg) that was mixed with EDC (1.5 eq), HOBt (1 eq), MeNH2-HCl (2.5 eq, methylamine hydrochloride) and DIPEA (2.5 eq) in DCM (25 ml). The reaction was stirred at RT overnight and washed with NaHCO3 solution, dried. The solution was evaporated and mixed with 15% KOH (2 ml) in MeOH (10 ml) further stirred at RT for 30 minutes. The solvent was evaporated and the residue was adjusted to weak acidic with 2N HCl, the solid was filtered and washed with water twice and cold MeOH to give 6-Hydroxy-N-methyl- 1 -naphthamide (720 mg).
7-Benzyloxy-6-methoxy-quinolin-4-ol (WO2006108059) (1 g) was refluxed with POCl3 (8 ml) for 3 hours. The reaction was evaporated and dissolved into DCM (80 ml) that was washed with ice water followed by brine. The organic layer was dried with Na2Sθ4 and evaporated to dryness to give a dark yellow solid as 4-chloro-7-benzyloxy-6-methoxy-quinoline that was mixed with 6-Hydroxy-N-methyl-l -naphthamide (600 mg), DMAP (1.5 eq) in dioxane (40 ml). The reaction was refluxed for three days and diluted with EtOAc, water and extracted with EtOAc three times. The combined organic layer was washed with water, brine and dried. The solution was evaporated and purified with silica gel column to give 6-(7-(benzyloxy)-6-methoxyquinolin-4- yloxy)-N-methyl-l -naphthamide (210 mg). This product was mixed with Pd/C (120 mg, 10%), HCONH4 (210 mg) in EtOH (20 ml). The mixture was refluxed for 1 hour and evaporated then mixed with water (2 ml). The solid was filtered and washed with water twice and cold MeOH as 6- (7-hydroxy-6-methoxyquinolin-4-yloxy)-N-methyl-l -naphthamide for next step without further purification.
N-CBZ-amino-l-(hydroxymethyl)cyclopropane (similarly prepared according to JMC 31, 2004, 1998) (250 mg) was dissolved into DCM (25 ml) with DIPEA (250 1) and stirred at O0C for 15 minutes. To the reaction was added MsCl (1.1 eq) and stirred for 30 minutes. The reaction was washed with NaHCO3 solution, water, brine and dried with Na2SO4. The solution was evaporated to give N-CBZ-ammo-l^methylsulfonyloxymethyFjcyclopropane as an off white solid. This solid was mixed with above 6-(7-hydroxy-6-methoxyquinolin-4-yloxy)-N-methyl- 1 – naphthamide and Cs2CO3 (250 mg) in DMA (4 ml). The reaction was heated at 1000C for 10 hours and mixed with EtOAc and water, then filtered, further extracted with EtOAc. The combined organic layer was evaporated and purified with silica gel column to give the titled product. Mass: (M + 1), 578 Method B:
4-Chloro-7-benzyloxy-6-methoxy-quinoline (3 g) was mixed with 6-Hydroxy- 1 -naphthoic acid (2 g) and KOH (2.5 g) in DMSO (11 ml). The mixture was heated at 130oC for 5 hours and cooled to RT. The reaction was then poured into a stirred water (60 ml) solution slowly to give a precipitate that was filtered to give 6-(7-(benzyloxy)-6-methoxyquinolin-4-yloxy)- 1 -naphthoic acid (2.8 g). This product was mixed with MeNH2-HCl (2 g), EDC (3.3 g), HOBt (2 g) and DIPEA (4 ml) in DCM (80 ml). The reaction was stirred at RT overnight and washed with NaHCO3 solution, dried. The solution was evaporated and purified with silica gel column to give 6-(7-(benzyloxy)-6- methoxyquinolin-4-yloxy)-N-methyl- 1 -naphthamide. The title compound then was prepared according to the same procedures described in Method A. Method C:
Dimethyl l^-cyclopropanedicarboxylate (5 ml) was mixed with NaOH (1.4 g) in MeOH (40 ml)/water (4 ml). The reaction mixture was stirred at RT overnight and the solvent was evaporated. To the residue was added ether (50 ml), water (50 ml) and extracted once. The aqueous layer was acidified with 6N HCl and extracted three times with ether, the combined organic layer was washed with brine, dried and evaporated to give l-(methoxycarbonyl)cyclopropanecarboxylic acid (4 g).
The above product was mixed with DIPEA (1.2 eq) in THF and stirred at 00C for 10 minutes, to the reaction was added ethyl chloro formate (1 eq) slowly and further stirred for 1.5 hours from 00C to RT. To the reaction cooled at 00C was added NaBH4 (1.5 eq) slowly followed by MeOH (2 eq) and stirred for 2 hours from 00C to RT. The reaction was diluted with EtOAc, water and extracted with EtOAc three times. The combined organic layer was washed with water, brine and dried. The solution was evaporated and purified with silica gel column to give methyl 1 -(hydroxymethyl)cyclo- propanecarboxylate (2.5 g).
The above product was dissolved into DCM (40 ml) with DIPEA (4 ml) and stirred at O0C for 15 minutes. To the reaction was added MsCl (1.1 eq) and stirred for 30 minutes. The reaction was washed with NaHCθ3 solution, water, brine and dried with Na2SO4. The solution was evaporated and mixed with 4-hydroxy-3-methoxy-acetophenone (0.9 eq) and K2CO3 (1.5 eq) in DMF (20 ml). The reaction was heated at 1000C for 6 hours and diluted with EtOAc, water and extracted with EtOAc three times. The combined organic layer was washed with water, brine and dried further evaporated to give methyl l-((4-acetyl-2-methoxyphenoxy)methyl)cyclopropane-carboxylate (1.8 g). This product was dissolved into acetic acid (5 ml) and stirred at RT, to the reaction was very slowly added nitric acid (8 ml, 60%) and stirred at RT for 1 hour. The reaction was poured into ice-water and extracted with EtOAc three times. The combined organic layer was washed with water, brine and dried.
The solution was evaporated and mixed with iron powder (1.5 g) and NH4Cl (150 mg) in EtOH/H2O (80 ml, 9/1). The reaction was refluxed for 3 hours and filtered through Celite followed by evaporation. The residue was mixed with EtOAc/H2O and extracted with EtOAc three times. The combined organic layer was washed with water, brine and dried. The solution was evaporated and purified with silica gel column to give methyl l-((5-amino-4-acetyl-2-methoxyphenoxy)methyl)- cyclopropanecarboxylate (1 g).
The above product was mixed with fresh prepared NaOMe (2 eq) in ethylene glycol dimethyl ether (30 ml) and stirred at RT for 1 hour. To the mixture was added HCOOEt (3 eq), the reaction was stirred at RT overnight and neutralized with 6N HCl. The reaction was evaporated with silica gel to dryness and purified on silica gel column with DCM/MeOH as eluent to give methyl l-((4- hydroxy-6-methoxyquinolin-7-yloxy)methyl)cyclopropanecarboxylate (600 mg). This product was refluxed with POCI3 (4 ml) for 3 hours and evaporated, then dissolved into DCM. The solution was washed with ice water followed by brine. The organic layer was dried with Na2SC^ and evaporated to give methyl l-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropanecarboxylate (500 mg).
The above product was mixed with DMAP (1.5 eq), 6-Hydroxy-N-methyl- 1 -naphthamide (300 mg) in dioxane (20 ml). The reaction was refluxed for three days and diluted with EtOAc, water and extracted with EtOAc three times. The combined organic layer was washed with water, brine and dried. The solution was evaporated and purified with silica gel column to give methyl 1 -((6-methoxy- 4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7-yloxy)methyl)-cyclopropanecarboxylate (200 mg). This product of was mixed with 15% NaOH (3 eq) in MeOH (15 ml) and refluxed for 30 minutes. The reaction was evaporated and adjusted to PH=6, then diluted with EtOAc, water and extracted with EtOAc three times. The combined organic layer was washed with water, brine, dried and evaporated to give l-((6-methoxy-4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7- yloxy)methyl)cyclopropanecarboxylic acid (120 mg).
The above product was mixed with DIPEA (0.3 ml) in acetone (5 ml) at 00C. To the reaction was slowly added C1COOCH2CH(CH3)2 (100 1) and stirred for 2 hours from 00C to RT. NaN3 (0.2 g)/H2O (0.5 ml) was added to the reaction and stirred for 30 minutes. The reaction was diluted with EtOAc, water and extracted with EtOAc three times. The combined organic layer was washed with water, brine, dried and evaporated without further purification. The residue was mixed with benzyl alcohol (150 1) in toluene (10 ml) and refluxed for 1.5 hour. The reaction was evaporated and purified with silica gel column to give the titled product. Mass: (M + 1), 578
…………………………….
http://www.google.com/patents/WO2010105761A1?cl=en
sequence of intermediates……………






 LUCITANIB
LUCITANIB
Example 1: Preparation of l-[(4-acetyl-2-methoxyphenoxy)methyl]- N-benzyloxycarbonyl-1-aminocyclopropane
A 10 L reactor equipped with mechanical stirrer was loaded with triphenylphosphine (340.0 g, 1.296 mol) and THF (2 L) and the suspension was cooled with an ice bath. The stirred suspension was then slowly added with DIAD (264 g, 1.296 mol) over 30 minutes. After stirring for 30 min at 00C, the stirred suspension was added dropwise with a solution of 4-hydroxy- 3-methoxyacetofenone (180 g, 1.08 mol) and DIPEA (210 g, 1.62 mol) in THF (1500 mL). The suspension was left under stirring for 45 min at 00C, then added dropwise with a solution of 1-benzyloxycarbonylamino-l- hydroxymethylcyclopropane (China Gateway) (240 g, 1.08 mol) in THF (1500 mL). After Ih, LC-MS analysis of a sample from the reaction mixture showed the complete disappearance of 1-benzyloxycarbonylamino-l- hydroxymethylcyclopropane. The reaction mixture was evaporated and the crude product was recrystallized with EtOH 95% (4000 mL) to give l-[(4- acetyl-2-methoxyphenoxy)methyl]-N-benzyloxycarbonyl- 1 – aminocyclopropane (214 g, yield: 53.5%) as a white solid.
1H-NMR (300 MHz, CDCl3): δ: 7.41-7.45 (m, 2 H), 7.26 (s, 5 H), 6.77 (d, 1 H), 5.43 (s, 1 H), 5.00 (s, 2 H), 4.04 (s, 2 H), 3.82 (s, 3 H), 2.49 (s, 3H), 0.92 (m, 4 H).
LC-MS: M+H+: 370.4
Example 2: Preparation of l-[(4-acetyl-2-methoxy-5- nitrophenoxy)methyl]-N-benzyloxycarbonyl-l-aminocycIopropane

A solution of HNO3 (65%, 3 mL) in Ac2O (2 mL) at 0°C was slowly added with a suspension of the compound of Example 1 (1.1 g, 2.9 mmol) in
Ac2O (3 mL). After stirring at 00C for 2 h, the reaction mixture was poured into 50 mL of ice/water and the precipitate was recovered by filtration. The resulting yellow solid was recrystallized with 95% EtOH (5 mL) to give l-[(4- acetyl-2-methoxy-5-nitrophenoxy)methyl]-N-benzyloxycarbonyl-l- aminocyclopropane (0.69 g, yield: 56%) as a yellow solid.
1H-NMR (300 MHz, CDCl3): δ: 7.52 (s, 1 H), 7.26 (s, 5 H), 6.67 (s, 1 H), 5.36 (s, IH), 5.02 (s, 2 H), 4.05 (s, 2 H), 3.86 (s, 3 H), 2.42 (s, 3 H), 0.94 (m, 4 H).
LC-MS: M+H+: 414.41
Example 3: Preparation of l-[(4-(3-dimethylaminopropenoyl)-2- methoxy-5-nitrophenoxy)methyl]-N-benzyloxycarbonyl-l- aminocyclopropane
A mixture of the compound of Example 2 (1.7 g, 4.1 mmol) and N5N- dimethylformamide dimethylacetal (0.9 g, 8.2 mmol) in DMF (6 mL) was stirred at 1000C for 2 h. After cooling at room temperature, the reaction mixture was diluted with water (30 mL) and extracted with AcOEt (3 x 50 mL). The combined organic phases were washed with brine (2 x 50 mL), dried and evaporated to give l-[(4-(3-dimethylaminopropenoyl)-2-methoxy-5- nitrophenoxy)methyl]-N-benzyloxycarbonyl-l -aminocyclopropane (1.9 g, yield: 95%) as a yellow solid. 1H-NMR (300 MHz, CDCl3): δ: 7.50 (s, 1 H), 7.27 (s, 5 H), 6.75 (s, 1
H), 5.44 (s, 1 H), 5.23 (s, 1 H), 5.1 1 (br, 1 H), 5.01 (s, 2 H), 4.04 (s, 2 H), 3.83 (s, 3 H), 2.78-3.00 (m, 6 H), 0.94 (m, 4 H) LC-MS: M+H+: 470.49
Example 4: Preparation of l-[(4-hydroxy-6-methoxyquinolin-7- yloxy)methyl]-N-benzyloxycarbonyl-l-aminocyclopropane
A mixture of the compound of Example 3 (1.5 g, 3.2 mmol) and powder iron (1.8 g, 32 mmol) in AcOH (15 mL) was stirred a 800C for 2 h. The reaction mixture was cooled at room temperature, diluted with AcOEt (150 mL), filtered and washed with 50 ml of AcOEt. The filtration liquors were combined, washed with water (2 x 100 mL) and an NaHCO3 saturated solution (2 x 100 mL), dried and evaporated to give l-[(4-hydroxy-6-methoxyquinolin-7-yloxy)methyl]-N- benzyloxycarbonyl-1 -aminocyclopropane (1.2 g, yield: 95%) as a yellow solid.
1H-NMR (300 MHz, MeOD): δ: 7.75 (d, 1 H), 7.51 (s, 1 H), 7.15 (m, 5 H), 6.80 (br, 1 H), 6.20 (d, 1 H), 4.97 (s,2 H), 4.05 (s, 2 H), 3.84 (s, 3 H), 0.87 (m, 4 H).
LC-MS: M+H+: 395.2
Example 5: Preparation of l-[(4-chloro-6-methoxyquinolin-7- yloxy)methyl]-N-benzyIoxycarbonyl-l-aminocyclopropane
a) By chlorination of the compound of Example 4
A 50 ml round-bottom flask fitted with magnetic stirrer, thermometer, condenser and kept under nitrogen atmosphere, was loaded at 20°/25°C with 3.90 g (9.89 mmol) of the compound of Example 4 and 25 ml of POCl3. The resulting suspension became a solution after stirring for a few minutes. The solution was heated at 85°C inner T and after 30 minutes the reaction was monitored by TLC, showing the disappearance of the starting product. The solution was cooled and dropwise added, over about 30 minutes and keeping the temperature below 100C, to a mixture of 250 ml of DCM and 250 ml of water, cooled at 00C. After completion of the addition, stirring was maintained for 30 minutes at 0°-10°C. The phases were separated and the aqueous phase was washed with 150 ml of DCM; the phases were separated and the organic phases combined. The combined organic phase was added with 150 ml of water, stirred at 20°/25°C for 15 minutes and pH was adjusted to 7-8 with a sodium bicarbonate saturated solution. The phases were separated and the organic phase was washed with 150 ml of water; the phases were separated, the organic phase was dried with sodium sulfate, filtered and the solvent evaporated off by distillation under vacuum. Stripping with ethyl ether afforded 3.8 g of a brownish solid. The solid residue was dissolved in 20 ml of tert-butyl methyl ether, stirring at 20°/25°C for an hour; filtered and washed with ter /-butyl methyl ether, then dried to obtain l-[(4-chloro-6- methoxyquinolin-7-yloxy)methyl]-N-benzyloxycarbonyl- l- aminocyclopropane (3.4 g; yield: 87%) having (1H-NMR) titre of 95%.
1H-NMR (500 MHz, DMSO-d6) δ ppm: 8.61 (d, 1 H), 7.91 (s, 1 H), 7.56 (s, 1 H), 7.44 (s, 1 H), 7.38 (s, 1 H), 7.29 (m, 5 H), 4.99 (s, 2 H), 4.23 (s, 2 H), 3.97 (s, 3 H), 0.87 (m, 4 H). b) by Mitsunobu reaction between 4-chloro-7-hydroxy-6- methoxyquinoline and 1 -benzyloxycarbonylamino- 1 – hydroxymethylcyclopropane 20 ml of DCM were added with 4-chloro-7-hydroxy-6- methoxyquinoline (300 mg, 1.43 mmol; from China Gateway),
1 -benzyloxycarbonylamino- 1 -hydroxymethylcyclopropane (412 mg,
1.87 mmol, 1.3 eq; from China Gateway) and triphenylphosphine (490 mg,
1.87 mmol, 1.3 eq). The resulting solution was dropwise added with a solution of DEAD (378 mg, 1.87 mmol, 1.3 eq) in 3 ml of DCM, keeping the temperature at 00C for 2 hours. The mixture was then left at 100C for 20 hours, then filtered to recover the unreacted 4-chloro-7-hydroxy-6- methoxyquinoline. The filtrate was evaporated under vacuum and the resulting residue was added with 20 ml of 95% EtOH and left under stirring for 30 min. The solid was collected by filtration, washed with 5 ml of 95% EtOH and dried under vacuum to give l-[(4-chloro-6-methoxyquinolin-7-yloxy)methyl]-
N-benzyloxycarbonyl-1-aminocyclopropane (273 mg; yield: 46%).
LC-MS: M+H+: 413.1
Example 6: Preparation of benzyl l-[(6-methoxy-4-(5- (methylcarbamoyl)naphthalen-2-yloxy)quinolin-7-yloxy)methyl)]cyclopropyl carbamate (II)
A solution of 0.51 g (2.53 mmol) of 6-hydroxy-N-methyl- 1 – naphthamide prepared according to WO2008/112408, 2, 7 ml of 2,6-lutidine and 0.3 g (2.42 mmol) of DMAP, kept at 20°/25°C and under nitrogen atmosphere, was added with the compound of Example 5 (1.0 g, NMR titre 95%, 2.30 mmol). The suspension was heated to 1400C inner temperature for
6 hours; then cooled to 20°/25°C and added with 80 ml of water and kept under stirring a 20°/25°C for 1 hour; the suspension was filtered and washed with water, to afford 0.88 g (yield: 66%) of benzyl l-[(6-methoxy-4-(5-
(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7-yloxy)methyl)]cyclopropyl carbamate (II).
1H-NMR (500 MHz, DMSO-d6) δ ppm: δ: 8.56 (d, 1 H), 8.50 (d, 1 H), 8.39 (d, 1 H), 8.04 (d, 1 H), 7.94 (s, 1 H), 7.87 (s, 1 H), 7.59 (m, 4 H), 7.41 (s, 1 H), 7.44 (s, 1 H), 7.30 (m, 5 H), 6.56 (d, 1 H), 5.01 (s, 2 H), 4.48 (s, 2 H), 4.23 (s, 2 H), 3.95 (s, 3 H), 0.87 (m, 4 H). LC-MS: M+H+: 578.3
Example 7: Preparation of 6-(7-((l-aminocyclopropyl)methoxy)-6- methoxyquinolin-4-yloxy)-N-methyl-l-naphthamide (I)
A mixture of the compound of Example 6 (0.24 g, 0.42 mmol) in 2 ml of a solution of 40% HBr in acetic acid was stirred at 300C for 3h, then added with 10 ml of water and the reaction mixture was extracted with AcOEt (2 x 10 mL). The organic phases were removed. The aqueous solution was dropwise added with a solution of 50% NaOH to reach pH 10. The mixture was extracted with DCM (3 x 20 mL) and the combined organic phases were dried and evaporated to give a crude containing 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxyquinolin-4- yloxy)-N-methyl-l-naphthamide (I) with purity higher than >94% by LC-MS analysis. This crude was further purified by chromatography on a silica gel column eluting with DCM/MeOH 10: 1), to afford 6-(7-((l- aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-N-methyl-l- naphthamide (I) having purity higher than 98% by LC-MS analysis (140 mg, yield: 76%).
1H-NMR (500 MHz, DMSO-d6) δ ppm: 8.47 (d, 2 H), 7.87 (d, 1 H), 7.53 (m, 3 H), 7.51 (m, 1 H), 7.44 (d, 1 H), 7.38 (s, 1 H), 6.50 (d, 1 H), 6.16 (d, 1 H), 5.01 (s, 2 H), 4.05 (s, 2 H), 4.03 (s, 3 H), 3.12 (d, 3 H), 2.09 (m, 2 H), 0.80 (m, 4 H).
LC-MS: M+H+: 444.0
…………………………………………
http://www.google.com/patents/WO2014113616A1?cl=en
6-(7-((l -Aminocyclopropyl)-methoxy)-6-methoxyquinolin-4-yloxy)-N-methyl- 1 -naphthamide (AL3810), or a pharmaceutically acceptable salt (such as hydrochloride salt) thereof, has been developed as an anti-tumor agent also named as E3810 and lucitanib, see “Journal of Cellular and Molecular Medicine vol. 16 issue 10 October 2012. p. 2321-2330 “, “Cancer Res February 15, 2011 vol. 71 no A 1396-1405 “.
This compound has been structurally disclosed in WO20081 12408 as an agiogenesis inhibitor with few preparation methods. A new process has been disclosed in WO2010105761 with the removal of use of sodium azide. Both above disclosed processes have involved a deprotection of benzyl carbmate protected precursor by HBr/Acetic acid solution that is a strong, fuming and high corrosive acidic condition. No crystalline form has been disclosed.
A

B

Process C

Formula III Scheme IV


Example 1
Representation of Process A and Process B
Process for preparation of 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxy-quinolin-4- yloxy)-N-methyl- 1 -naphthamide (AL3810)
To a stirred mixture of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)- naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II (150 g) in DCM (1.5 L) was added TFA (150 ml) through an additional funnel for about 30 min at RT. The reaction was stirred at 30°C for 4 hours and added into water (3 L). The aqueous layer was extracted with DCM twice (1.5L X 2) and basified with 3N NaOH (620 ml) to adjust pH 1 1-12 with a fine white solid precipitation. The solid was filtered and washed with water, further suction dry. The solid was dissolved into a mixture of chloroform/methanol (5 L, 3.5L/1.5L) and further washed with brine (2 L). It was dried with MgS04 and filtered. The solution was evaporated with EtOAc (2 L) three times to a slurry solution and cooled to RT. It was filtered and the filter cake was washed with ether, further air dried to give the crude titled compound 105g, yield: 95.9%. MS: (M+l) 444.
Example 2
Representation of Process A and Process B
Process for preparation of 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxy-quinolin-4- yloxy)-N-methyl- 1 -naphthamide (AL3810)
To a stirred mixture of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)naph- thalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II (1 g) in ACN (15 ml) was added TSA.H20 (3 eq). The reaction was stirred at RT for 24 hours and it was basified with 3N NaOH. The solution was extracted with DCM three times, washed with brine and dried with MgS04. The solution then was filtered and evaporated, further recrystalized from IPA to give pure titled compound 550 mg, yield: 75%. MS: (M+l) 444. Example 3
Representation of Process C
Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)- naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II

To a stirred mixture of 6-hydroxy- 1 -naphthoic acid (19 g, formula 10) in DMF (150 ml) was added CDI (22 g). The reaction was heated at 80°C for 30 min and CH3NH2.HC1 (40 g) was added into the reaction. The reaction was heated for 3 hours at 80°C and cooled to RT and further diluted with water (300 ml). It was acidified with IN HC1 to pH 2-3 and extracted three times with EtOAc (150 ml). The combined organic layer was washed with saturated NaHC03 solution followed by water and brine. The solution was dried with Na2S04 and evaporated to give the 4- rmula 1 1 compound 12 g.

(i) To a mixture of formula 1 1 (6.5 g), formula 12 (6.5 g) and DMAP (5.5 g) was added 1,6- lutidine (20 ml). The reaction was stirred and heated at 135°C for 5 hours from heterogeneous to homogeneous. The reaction was cooled and IPA (35 ml) was added into the reaction under slow stirring for 2 hours at RT. The solid was filtered and further washed with IPA, dried to give the formula 13 compound 5.8 g as a gray solid, yield 57%, or
(ii) To a mixture of formula 1 1 (500 mg), formula 12 (500 mg), Cul (80 mg), Cs2C03 (1 g) and 1-picolinic acid (150 mg) was added DMF (0.5 ml). The reaction was stirred and heated at 120 °C for 24 hours. It was directed loaded on silica gel column to purify to give the formula 13 compound 370 mg, yield 48%, or (iii) To a mixture of formula 1 1 (500 mg), formula 12 (500 mg), Cul (80 mg), CS2CO3 (1 g) and 2,4-pentanedione (10 mg) was added DMF (0.5 ml). The reaction was stirred and heated at 120 °C for 24 hours. It was directed loaded on silica gel column to purify to give the formula 13

A mixture of formula 13 (5.8 g) and TFA (12 ml) was heated at 90°C for one hour. The reaction was evaporated under reduced pressure and triturated with EtOAc. The solid was filtered e formula 14 as a TFA salt 5.5 g, yield 95%.

To a mixture of acid-ester (8.2 g, formula 15) and 4-methoxybenzyl alcohol (9.5 g) in toluene (50 ml) was added DPPA (15 g), the reaction was stirred and TEA was added into the reaction through an additional funnel at RT. The reaction then was refluxed for 20 hours and cooled to RT. To the reaction was added 2N NaOH (30 ml) and followed by extraction with EtOAc three times. The combined organic layer was washed with water to neutral and dried with Na2SOzt. The solution was filtered and evaporated followed by addition of EtOAc/PE (petroleum ether) and stored in a refrigerator overnight. The crystals were filtered and washed with cold EtOAc/PE to give an off white powder. The product formula 15b was vacuum oven dried at 30°C to give 8.0 g as ethyl l-((4-methoxybenzyloxy)carbonylamino)cyclopropanecarboxylate (formula
294.

To a mixture of formula 15b (8.0 g) and THF (50 ml) was added NaBH4 (8 g). The reaction was refluxed for 12 hours. Methanol (15 ml) was slowly added to the reaction and refluxed for 4 hour. The solvent was evaporated and cooled. NH4C1 (6.3 g) and water (60 ml) were added and stirred. The mixture was extracted with DCM three times and dried with Na2S04. The solution was filtered and evaporated followed by addition of ethanol to recrystalize overnight. The crystal was filtered to give an off white powder and further dried in oven to give the product 4.0 g as 4-methoxybenzyl l-(hydroxymethyl)cyclopropylcarbamate (formula 15c),

To a stirred mixture of formula 15c (100 g) and DCM (400 ml) was added DIPEA (78g). The result solution was cooled to 0-5°C with ice/water and further stirred under this temperature for 15 min. MsCl (60g) was added via an addition funnel dropwise keeping temperature below 5°C for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5°C for 30 min and quenched with saturated NaHC03 (300 ml). The solution was extracted with 200 ml DCM twice. The combined DCM layer was washed with 0.1 N HC1 (400 ml) followed by brine. It was dried over Na2S04 and concentrated to obtain an off- white solid 123 g
330.

To a stirred mixture of formula 15d (3.3 g) and KI (3.3 g) was added acetone (30 ml), the reaction was refluxed for 2 hours and cooled. The reaction was evaporated and extracted with EtOAc (30 ml) twice and washed with brine, further evaporated under reduced pressure to give the crude product 2.3 g of formula 15e, MS: (M+l) 362.

Formula II Method A:
To a stirred mixture of formula 14 (500 mg), formula 15d (450 mg), K2C03 (400 mg) and Nal (180 mg) was added acetone (10 ml), the reaction suspension was heated to reflux for 20 hours as one pot reaction. The reaction was evaporated and purified on silica gel column to give the product 510 mg of Formula II. MS: (M+1) 608. lH NMR (DMSO-d6): δ: 8.53-8.54 (m, 2H), 8.37-8.39 (d, 1H), 8.00-8.02 (d, 1H), 7.83-7.88 (m, 2H), 7.53-7.61 (m, 4H), 7.42 (s, 1H), 7.22- 7.24 (d, 2H), 6.83-6.85 (d, 2H), 6.61-6.62 (d, 1H), 4.91 (s, 2H), 4.23 (s, 2H), 3.95 (s, 3H), 3.70 (s, 3H), 2.86-2.87 (d, 3H), 0.83-0.93 (d, 4H).
Method B:
To a stirred mixture of formula 14 (500 mg), formula 15e (500 mg) and K2C03 (400 mg) was added acetone (10 ml), the reaction suspension was heated to reflux for 20 hours. The reaction was evaporated and purified on silica gel column to give the product 560 mg of Formula II. MS: (M+1) 608. ¾ NMR conforms to Formula II from above Method A.
Method C:
To a stirred mixture of formula 14 (33 g), formula 15d (43 g), K2C03 (41 g) and KI (16.6 g) was added acetone (400 ml). The reaction suspension was heated to reflux for about 30 hr. The reaction was concentrated and to the residue was added water (700 ml). The result suspension was stirred for 1 hour slowly to get a brown solid. The solid was filtered and rinsed with water twice further rinsed with ethanol. The crude product was dried in oven at 40°C for 2-3 hours. The product was purified with IPA by recrystalization to give 29 g of Formula II. MS: (M+1) 608. lH NMR conforms to Formula II from above Method A.
Example 4
Representation of Process D
Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarba- moyl)naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropyl-carbamate Formula II
A mixture of 2-(l-((6-methoxy-4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quino-lin-7- yloxy)methyl)cyclopropyl)acetyl azide formula 17 (WO2008112408, 150 mg) and 4-methoxybenzyl alcohol (0.15 ml) in toluene (10 ml) was refluxed for 1.5 hour. The reaction was evaporated and purified with silica gel column to give the titled product. Mass: (M + 1), 608
Example 5
Representation of Process E Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarba- moyl)naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II

A mixture of 6-Hydroxy- 1 -naphthoic acid (1 g) and H2SO4 (0.2ml) in EtOH (25 ml) was refluxed overnight and evaporated, followed by dissolving into EtOAc. The solution was washed with water, IN NaHC03 solution and brine, further dried by Na2S04. The solution was evaporated to give crude ethyl 6-hydroxy- 1 -naphthoate 0.9 g which was reacted with formula 12 at similar preparation conditions to formula 13 of Example 3 to give the above product of formula 18. Formula 19 was similarly prepared to formula 14 of Example 3.
A reaction between formula 19 and formula 15d similarly to the preparation of Formula II of Method A gave ethyl 6-(6-methoxy-7-((l-((4-methoxybenzyloxy)carbonylamino)cyclopro- pyl)methoxy)quinolin-4-yloxy)- 1 -naphthoate which was hydro lyzed with 10% NaOH in EtOH at RT to give 6-(6-methoxy-7-((l-((4-methoxybenzyloxy)carbonylamino)cyclopropyl)methoxy)- quinolin-4-yloxy)-l -naphthoic acid. The resulting acid was acylated similarly to the preparation of formula 1 1 of Example 3 with CH3NH2.HC1 under the heat pre- activation at the presence of CDI to give the titled product.
Example 6
Representation of Process F
Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)- naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II

To a mixture of 4-chloro-6-methoxyquilolin-7-ol (formula 21, 5.2g), l-((4-methoxyben- zyloxy)carbonylamino)cyclopropanecarboxylate (formula 15b, 8.3g) and triphenylphosphine (9.8 g) in THF (250 ml) was added DEAD (6.5 g) dropwise at RT in 1.5 hours, the reaction was further stirred for 20 hours at RT and evaporated. The residue was purified with silica gel column to give the 4-methoxybenzyl 1 -((4-chloro-6-methoxy-quinolin-7-yloxy)methyl)cyclopropylcarba- mate formula 21b product 6.5 g.
The titled compound of Formula II was then similarly prepared by using formula 21b to react with 4-hydroxy-N-methyl-naphamide formula 1 1 according to formula 13 of Example 3.
Example 7
Preparation of the crystalline form of 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxy- quinolin-4-yloxy)-N-methyl- 1 -naphthamide (AL3810)
The crude product from Example 1 (105 g) was mixed with isopropanol (2.5 L) and active carbon (5 g), the mixture was heated to reflux for 0.5 hour to dissolve all crude product followed by filtration while it was hot, then the filtrate was refluxed again for 10 minutes and it was cooled to room temperature overnight under a slow stirring condition. The precipitate was filtered and washed with ethyl ether (500 ml x 2), further dried under high vacuum at 80°C to give the pure product (85 g) with melting point at 192°C- 196°C.
HI NMR shown in Fig 1.
DSC shown in Fig 2 having observable endotherm from about 193°C-202°C
TGA shown in Fig 3 demonstrating as an unsolvated material with weight loss at about 230°C
………………………………………….
synthesis…….will be updated
|
References |
1: Colzani M, Noberini R, Romanenghi M, Colella G, Pasi M, Fancelli D, Varasi M, Minucci S, Bonaldi T. Quantitative chemical proteomics identifies novel targets of the anti-cancer multi-kinase inhibitor E-3810. Mol Cell Proteomics. 2014 Jun;13(6):1495-509. doi: 10.1074/mcp.M113.034173. Epub 2014 Apr 2. PubMed PMID: 24696502; PubMed Central PMCID: PMC4047469.
2: Zangarini M, Ceriani L, Bello E, Damia G, Cereda R, Camboni MG, Zucchetti M. HPLC-MS/MS method for quantitative determination of the novel dual inhibitor of FGF and VEGF receptors E-3810 in tumor tissues from xenograft mice and human biopsies. J Mass Spectrom. 2014 Jan;49(1):19-26. doi: 10.1002/jms.3305. PubMed PMID: 24446259.
3: Bello E, Taraboletti G, Colella G, Zucchetti M, Forestieri D, Licandro SA, Berndt A, Richter P, D’Incalci M, Cavalletti E, Giavazzi R, Camboni G, Damia G. The tyrosine kinase inhibitor E-3810 combined with paclitaxel inhibits the growth of advanced-stage triple-negative breast cancer xenografts. Mol Cancer Ther. 2013 Feb;12(2):131-40. doi: 10.1158/1535-7163.MCT-12-0275-T. Epub 2012 Dec 27. PubMed PMID: 23270924.
4: Damia G, Colella G, Camboni G, D’Incalci M. Is PDGFR an important target for E-3810? J Cell Mol Med. 2012 Nov;16(11):2838-9. doi: 10.1111/j.1582-4934.2012.01601.x. PubMed PMID: 22805298.
5: Sala F, Bagnati R, Livi V, Cereda R, D’Incalci M, Zucchetti M. Development and validation of a high-performance liquid chromatography-tandem mass spectrometry method for the determination of the novel inhibitor of angiogenesis E-3810 in human plasma and its application in a clinical pharmacokinetic study. J Mass Spectrom. 2011 Oct;46(10):1039-45. doi: 10.1002/jms.1985. PubMed PMID: 22012670.
6: Bello E, Colella G, Scarlato V, Oliva P, Berndt A, Valbusa G, Serra SC, D’Incalci M, Cavalletti E, Giavazzi R, Damia G, Camboni G. E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 2011 Feb 15;71(4):1396-405. doi: 10.1158/0008-5472.CAN-10-2700. Epub 2011 Jan 6. PubMed PMID: 21212416.
7: Kawai T, Ikeda H, Harada Y, Saitou T. [Changes in the rat stomach after long-term administration of proton pump inhibitors (AG-1749 and E-3810)]. Nihon Rinsho. 1992 Jan;50(1):188-93. Japanese. PubMed PMID: 1311785.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008112408A1 * | Feb 24, 2008 | Sep 18, 2008 | Advenchen Lab Llc | Spiro substituted compounds as angiogenesis inhibitors |
| WO2010105761A1 * | Mar 11, 2010 | Sep 23, 2010 | Eos Ethical Oncology Science S.P.A. In Abbreviated Form Eos S.P.A. | A process for the preparation of 6-(7-((1-aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-n-methyl-1-naphthamide and synthetic intermediates thereof |
| Reference | ||
|---|---|---|
| 1 | * | BELLO, E. ET AL.: ‘E-3810 Is a Potent Dual Inhibitor of VEGFR and FGFR that Exerts Antitumor Activity in Multiple Preclinical Models‘ CANCER RES. vol. 71, no. 4, 2011, pages 1396 – 1405 |
| 2 | * | SALA, F. ET AL.: ‘Development and validation of a high-performance liquid chromatography-tandem mass spectrometry method for the determination of the novel inhibitor of angiogenesis E-3810 in human plasma and its application in clinical pharmacokinetic study‘ J. MASS. SPECTROM. vol. 46, 2011, pages 1039 – 1045 |
| 3 | * | ZHOU, Y. ET AL.: ‘AL3810, a multi-tyrosine kinase inhibitor, exhibits potent anti-angiogenic and anti-tumour activity via targeting VEGFR, FGFR and PDGFR‘ J. CELL . MOL. MED. vol. 16, no. 10, 2012, pages 2321 – 2330 |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008112408A1 | Feb 24, 2008 | Sep 18, 2008 | Advenchen Lab Llc | Spiro substituted compounds as angiogenesis inhibitors |
| US20080227812 | Feb 23, 2008 | Sep 18, 2008 | Advenchen Laboratories, Llc | Spiro Substituted Compounds As Angiogenesis Inhibitors |
| Reference | ||
|---|---|---|
| 1 | J. MED. CHEM. vol. 51, 2008, pages 5766 – 5779 | |
| 2 | ORG. REACT. vol. 42, 1992, pages 335 – 656 | |
| 3 | ORGANIC SYNTHESES vol. 63, 1985, page 314 | |
| 4 | SYNTHESIS 1981, pages 1 – 28 | |
| 5 | TETRAHEDRON LETT. vol. 38, 1997, page 191 | |
| 6 | TETRAHEDRON LETTERS vol. 46, 2005, pages 735 – 737 | |
| 7 | * | TOIS J ET AL: “Novel and convenient synthesis of 4(1H)quinolones” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, vol. 46, no. 5, 31 January 2005 (2005-01-31), pages 735-737, XP004705840 ISSN: 0040-4039 |
| 8 | * | WEILIN SUN ET AL: “Biososteric Replacement in the Design and Synthesis of Ligands for Nicotinic Acetylcholine Receptors” MEDICINAL CHEMISTRY RESEARCH, BIRKHÄUSER-VERLAG, BO, vol. 14, no. 5, 1 July 2005 (2005-07-01), pages 241-259, XP019428169 ISSN: 1554-8120 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014113616A1 * | Jan 17, 2014 | Jul 24, 2014 | Advenchen Pharmaceuticals, LLC | Process for preparing the anti-tumor agent 6-(7-((1-aminocyclopropyl) methoxy)-6-methoxyquinolin-4-yloxy)-n-methyl-1-naphthamide and its crystalline |
Patent Reference:
EOS ETHICAL ONCOLOGY SCIENCE S.p.A. in abbreviated form EOS S.p.A.; SPINELLI, Silvano; LIVI, Valeria Patent: WO2010/105761 A1, 2010 ; Location in patent: Page/Page column 21 ;
![6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide NMR spectra analysis, Chemical CAS NO. 1058137-23-7 NMR spectral analysis, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/567/717/1058137-23-7-1h.png) CAS NO. 1058137-23-7, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide H-NMR spectral analysis |
![6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide NMR spectra analysis, Chemical CAS NO. 1058137-23-7 NMR spectral analysis, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-09-06/001/567/717/1058137-23-7-13c.png) CAS NO. 1058137-23-7, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide C-NMR spectral analysis |


Advenchen Laboratories is a small pharmaceutical company focusing on pharmaceutical research and development involving small molecule cancer drug discovery …



Epelsiban being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.

Epelsiban
557296
GSK-557296
GSK-557296-B
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione
(3R, 6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[(1R)- 1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1-methylpropyl]-2,5- piperazinedione
Glaxo Group Limited INNOVATOR
Epelsiban (GSK-557,296-B)[1][2] is an oral drug which acts as a selective, sub-nanomolar (Ki=0.13 nM) oxytocin receptor antagonist with >31000-fold selectivity over the related vasopressin receptors and is being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.[3][4]


benzenesulfonic acid;(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione,CAS 1159097-48-9
UNII-H629P9T4UN, GSK557296B, Epelsiban besylate (USAN), Epelsiban besylate [USAN], 1159097-48-9, H629P9T4UN
GSK-557296 is being developed in early clinical studies at GlaxoSmithKline for enhancement of embryo and or blastocyst implantation in women undergoing IVF treatment. The product has been in phase II clinical development for the treatment of premature ejaculation.
Preterm labor is a major clinical problem leading to death and disability in newborns and accounts for 10% of all births and causes 70% of all infant mortality and morbidity.
Oxytocin (OT) is a potent stimulant of uterine contractions and is responsible for the initiation of labor via the interaction with the OT receptors in the mammalian uterus. OT antagonists have been shown to inhibit uterine contractions and delay preterm delivery. So there is increasing interest in OT antagonists because of their potential application in the prevention of preterm labor. Although several tocolytics have already been approved in clinical practice, they have harmful maternal or fetal side effects.
The first clinically tested OT antagonist atosiban has a much more tolerable side effect profile and has recently been approved for use in Europe. However, atosiban is a peptide and a mixed OT/vasopressin V1a receptor antagonist that has to be given by iv infusion and is not suitable for long-term maintenance treatment, as it is not orally bioavailable.
Hence there has been considerable interest in overcoming the shortcomings of the peptide OT antagonists by identifying orally active nonpeptide OT antagonists with a higher degree of selectivity toward the vasopressin receptors (V1a, V1b, V2) with good oral bioavailability. Although several templates have been investigated as potential selective OT antagonists, few have achieved the required selectivity for the OT receptor vs the vasopressin receptors combined with the bioavailability and physical chemical properties required for an efficacious oral drug.
Therefore our objective was to design a potent, orally active OT antagonist with high levels of selectivity over the vasopressin receptor with good oral bioavailability in humans that would delay labor safely by greater than seven days and with improved infant outcome, as shown by a reduced combined morbidity score.
| Patent | Submitted | Granted |
|---|---|---|
| Compounds [US7919492] | 2010-12-02 | 2011-04-05 |
| Piperazinediones as Oxytocin Receptor Antagonists [US7550462] | 2007-11-01 | 2009-06-23 |
| Compounds [US8202864] | 2011-06-23 | 2012-06-19 |
| Novel compounds [US2009247541] | 2009-10-01 |
………………………………………
PATENT
https://www.google.com/patents/US7919492
Example 3
Method A

(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
as a white lyophilisate (88 mg, 23%) after freeze-drying from 1,4-dioxane
HPLC Rt=2.70 minutes (gradient 2); m/z [M+H]+=519
1H NMR (CDCl3) δ 7.49 (d, 1H), 7.27-7.15 (m, 4H), 7.10 (d, 1H), 6.68 (s, 1H), 6.40 (d, 1H), 4.10 (dd, 1H), 4.01 (d, 1H), 3.74-3.52 (m, 5H), 3.28-3.07 (m, 5H), 2.97-2.84 (m, 2H), 2.79-2.71 (m, 1H), 2.62 (s, 3H), 2.59 (s, 3H), 1.65-1.53 (m, 1H), 0.98-0.80 (m, 2H), 0.70 (t, 3H), 0.45 (d, 3H).
Example 3
Method B

(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
A suspension of {(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-6-[(1S)-1-methylpropyl]-2,5-dioxo-1-piperazinyl}(2,6-dimethyl-3-pyridinyl)acetic acid hydrochloride (5.0 g, 10.3 mmol) (intermediate 5) in dry dichloromethane (50 ml) was treated with 1,1-carbonyldiimidazole (2.6 g, 16 mmol) and the reaction mixture was stirred under nitrogen for 18 hours. Morpholine (4.8 ml, 55 mmol) was added and the resultant solution was left to stand under nitrogen for 18 hours. The solvent was removed in vacuo and the residue was separated between ethyl acetate and water. The organic phase was washed with brine and dried over anhydrous magnesium sulphate. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. This was applied to a basic alumina cartridge (240 g) and eluted using a gradient of 0-7.5% methanol in diethyl ether (9CV), 7.5-10% methanol in diethyl ether (1CV) and 10% methanol in diethyl ether (1CV). The required fractions were combined and evaporated in vacuo to give (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione as a white solid (2.4 g, 45%).
HPLC Rt=2.72 minutes (gradient 2); m/z [M+H]+=519
………………………………………
WO 2011051814
http://www.google.com/patents/WO2011051814A1?cl=en
This invention relates to novel crystalline forms of (3R, 6R)-3-(2,3-dihydro-1 H- inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1 – methylpropyl]-2,5-piperazinedione benzenesulfonate salt, processes for their preparation, pharmaceutical compositions containing them and to their use in medicine. The benzenesulfonate salt of Compound A is represented by the following structure:

In one aspect, the present invention provides a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form provides an X-ray powder diffraction pattern substantially in accordance with Figure 1 .
In another aspect, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

In an additional aspect, the invention includes a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2.
In certain aspects, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2 In one aspect, the invention also provides a crystalline form of {3R, 6R)-3-(2,3- dihydro-1 H-inden-2-yl)-1-[(1 R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]- 6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

Experimental
Process Scheme

Stage 4
Acetone / Water Recrystallisation
Compound A-form I Ste8e 5 Besylate salt
MW 676.83 Acetone / Water
Recrystallisation MW 676.83 Process description for isolation of Compound A-Form 1
Stage 0
methyl d-alloisoleucinate hydrochloride (Compound 2) was charged to ethyl acetate. A solution of potassium carbonate in water was then added. The mixture was then stirred vigorously at room temperature for 1 hour. The two layers were separated and the aqueous layer further extracted with ethyl acetate. The organic layers were combined and washed with brine. The organic layers were then concentrated in vacuo and filtered to yield methyl D-alloisoleucinate (Compound 3) as a pale yellow oil.
Stage 1
2,6-dimethyl-3-pyridinecarbaldehyde (Compound 4) in methanol at ambient temperature was treated with D-alloisoleucinate (Compound 3) in methanol followed by 2,2,2- trifluoroethanol and the reaction mixture was warmed to 40°C. When formation of the intermediate imine (methyl A/-[(2,6-dimethyl-3-pyridinyl)methylidene]-D-alloisoleucine) was complete Compound 5 was added followed by 1-isocyano-2- [(phenylmethyl)oxy]benzene (Compound 6) and the reaction mixture was stirred at 40°C until formation of Compound 7 was deemed complete.
Stage 2
Palladium on carbon catalyst was treated with a solution of Compound 7 in methanol and 2,2,2-trifluoroethanol and diluted with acetic acid. The vessel was purged with nitrogen and the reaction mixture warmed to 50°C and hydrogenated at 4.0-4.5 barg. When the reaction was deemed complete it was cooled to ambient temperature and the catalyst removed by filtration and washed through with methanol. The organic solution of 2- {(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1-piperazinyl}- 2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) was concentrated at reduced pressure and then diluted with /‘so-propyl acetate and concentrated at reduced pressure.
The residue was diluted with /‘so-propyl acetate and washed with aqueous ammonia. The aqueous phase was separated and extracted into another portion of /‘so-propyl acetate. The combined organic phases were washed with water, concentrated by distillation at reduced pressure, diluted with /‘so-propyl acetate and concentrated by distillation at reduced pressure, to leave a concentrated solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8). The product was finally dissolved in 1 ,4-dioxane for the next stage and stored into drums.
Stage 3 Solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) in 1 ,4-dioxane was treated with 1 ,1 ‘-carbonyl diimidazole at ambient temperature to form a solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1 -[1-(2,6-dimethyl-3-pyridinyl)- 2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1 -methylpropyl]-2,5- piperazinedione (Compound 9).
In a separate vessel morpholine in 1 ,4-dioxane was heated to 80-85°C. The solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[1 – (2,6-dimethyl-3-pyridinyl)-2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1- methylpropyl]-2,5-piperazinedione (Compound 9) was slowly added to the morpholine in 1 ,4-dioxane. The reaction mixture was stirred for one hour at 80-85°C and cooled before concentration by distillation at reduced pressure.
The concentrated solution of Compound A was diluted with /‘so-propyl acetate and washed with aqueous sodium hydroxide followed by water. The /so-propyl acetate solution of COMPOUND A was then concentrated by distillation at reduced pressure and cooled to ambient temperature. The concentrated solution of Compound A was then diluted with acetone and treated with benzenesulfonic acid and seed crystals were added and the reaction mixture stirred until crystallisation occurred. The slurry of Compound A besylate was heated to 50°C, a temperature cycle was performed, and finally the slurry was cooled to -10°C and isolated by filtration. The filter cake was washed with cold acetone (-10°C) to give Compound A besylate (intermediate grade) as a wet cake.
Yield: 44% from Compound 5
39% from Compound 5
Stage 4
Compound A besylate (intermediate grade wet cake, Compound A besylate ) was suspended in acetone (17.4 vol including acetone content of wet cake) and heated to 55- 60°C. Water (0.66 vol) was added until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (3.2 vol). The temperature of the reaction mixture was adjusted to 45-50°C before the addition of seed crystals (0.00025wt). When crystallisation was complete the reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins.
The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to -3-2°C over 4.5 h and stirred for at least 1 h before the product was isolated by filtration. The wet cake was washed with acetone at 0°C (3 x 3.1 vol) and blown dry before being unloaded. COMPOUND A besylate was dried at 50°C under vacuum for 3 days. Compound A besylate was then milled. Yield: 66% Stage 5
Compound A besylate (OBU-D-02) was suspended in acetone (8 vol) and water (1 .1 vol) and heated to 48-52°C until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (2 vol). The reaction mixture was cooled to 20-25°C before the addition of Form 1 seed crystals (0.0025wt). When crystallisation was complete the reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins. The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins.
The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to -12— 8°C over 3.5 h and stirred for 15 h before the product was isolated by filtration. The wet cake was washed with acetone at -10°C (2 x 3 vol) and blown dry before being unloaded. Compound A besylate was dried at ambient temperature under vacuum for 6 days with a wet nitrogen bleed to afford Form 1 . Compound A besylate was then milled. Yield: 67%
Recrystallisation of Compound A besylate anhydrate (Form 2)

Besylate salt ………………………………………………………………Besylate salt
C30H38 4O4■ C6H603S C30H38 4O4■
MW 676.83 MW 676.83
COMPOUND A besylate is charged to the vessel and treated with methyl ethyl ketone (MEK) (8vol) and water (0.35vol) and the solution heated until dissolution is observed (ca. 55-60°C). The solution is then filtered and recharged to the vessel. Pressure is then reduced to 650mbar and the reaction mixture heated further to distil out solvent. MEK is added at the same rate as solvent is removed by distillation keeping the reaction mixture volume constant. After 4 volumes of MEK have been added the reaction mixture is treated with Form 2 seed crystals (2%wt) and the distillation continued in the same manner until another 7 volumes of MEK has been added. The vacuum is then released to an atmospheric pressure of nitrogen and the temperature of the reaction mixture adjusted to 65°C. The reaction mixture is then filtered and washed with pre heated MEK (2vol at 65°C). The purified COMPOUND A besylate anhydrate is then sucked dry and dried further in a vacuum oven at 65°C at l OOmbar with a nitrogen bleed. Yield 89%
NMR data is the same for Forms 1 and 2.
1 H NMR (500MHz, DMSO-d6) 5ppm 0.71-0.80(m, 6H) 0.87-0.98(m, 1 H) 1 .31 (br. S, 1 H) 1.69(br. S, 1 H) 2.68(s, 3H) 2.69(s, 3H) 2.72-2.79(m, 1 H) 2.80-2.87(m, 1 H) 2.88-3.01 (m, 3H) 3.18-3.25(m, 1 H) 3.27-3.33(m, 1 H) 3.38-3.46(m, 1 H) 3.47-3.52(m, 1 H)3.53-3.57(m, 1 H) 3.60-3.71 (m, 3H) 3.83(dd, J=9.46,3.15 Hz, 1 H) 3.89 (br. S, 1 H)6.10(br. S, 1 H) 7.1 1 – 7.14(m, 2H) 7.19-7.23(m, 2H) 7.30-7.35(m, 3H)7.59-7.63(m, 2H) 7.67(d, J=7.25Hz, 1 H) 8.12(br. S, 1 H) 8.50(d, J=3.78Hz, 1 H)
………………………………………..
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm201287w

A six-stage stereoselective synthesis of indanyl-7-(3′-pyridyl)-(3R,6R,7R)-2,5-diketopiperazines oxytocin antagonists from indene is described. SAR studies involving mono- and disubstitution in the 3′-pyridyl ring and variation of the 3-isobutyl group gave potent compounds (pKi > 9.0) with good aqueous solubility. Evaluation of the pharmacokinetic profile in the rat, dog, and cynomolgus monkey of those derivatives with low cynomolgus monkey and human intrinsic clearance gave 2′,6′-dimethyl-3′-pyridyl R–sec-butyl morpholine amide Epelsiban (69), a highly potent oxytocin antagonist (pKi = 9.9) with >31000-fold selectivity over all three human vasopressin receptors hV1aR, hV2R, and hV1bR, with no significant P450 inhibition. Epelsiban has low levels of intrinsic clearance against the microsomes of four species, good bioavailability (55%) and comparable potency to atosiban in the rat, but is 100-fold more potent than the latter in vitro and was negative in the genotoxicity screens with a satisfactory oral safety profile in female rats.
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione (69 EPELSIBAN)
References
- Borthwick AD, Liddle J, Davies DE, Exall AM, Hamlett C, Hickey DM, Mason AM, Smith IE, Nerozzi F, Peace S, Pollard D, Sollis SL, Allen MJ, Woollard PM, Pullen MA, Westfall TD, Stanislaus DJ (January 2012). “Pyridyl-2,5-diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists: synthesis, pharmacokinetics, and in vivo potency”. Journal of Medicinal Chemistry 55 (2): 783–96. doi:10.1021/jm201287w. PMID 205501.
- 2 Borthwick, A. D.; Liddle, J. (January 2013). “Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists”. In Domling, A. Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. ISBN 978-3-527-33107-9.
- 3 World Health Organization (2011). “International Nonproprietary Names for Pharmaceutical Substances (INN): Proposed INN: List 105”. WHO Drug Information 25 (2): 179.
- 4 USAN Council (2011). “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). Retrieved 2011-10-28.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-(morpholin-4-yl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]piperazine-2,5-dione | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 872599-83-2 1159097-48-9 (besylate) |
| ATC code | None |
| PubChem | CID 11634973 |
| ChemSpider | 9809717 |
| KEGG | D10117 |
| Chemical data | |
| Formula | C30H38N4O4 |
| Molecular mass | 518.6 g/mol |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| WO2003053443A1 | Dec 20, 2002 | Jul 3, 2003 | Glaxo Group Ltd | Substituted diketopiperazines as oxytocin antagonists | |
| WO2006000399A1 | Jun 21, 2005 | Jan 5, 2006 | Glaxo Group Ltd | Novel compounds | |
| EP2005006760W | Title not available | ||||
| US6914160 | Jul 31, 2003 | Jul 5, 2005 | Pfizer Inc | Oxytocin inhibitors | |
| US20070254888 | Jun 21, 2005 | Nov 1, 2007 | Glaxo Group Limited | Piperazinediones as Oxytocin Receptor Antagonists |
| US8202864 * | Feb 25, 2011 | Jun 19, 2012 | Glaxo Group Limited | Compounds |
| US8716286 | Oct 28, 2010 | May 6, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US8742099 | May 20, 2013 | Jun 3, 2014 | Glaxo Group Limited | Compounds |
| US8815856 | Mar 18, 2014 | Aug 26, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US20120202811 * | Apr 19, 2012 | Aug 9, 2012 | Glaxo Group Limited | Novel compounds |
APD 334 to treat to autoimmune diseases

APD 334
Arena Pharmaceuticals, Inc. innovator
2-[7-[4-Cyclopentyl-3-(trifluoromethyl)benzyloxy]-1,2,3,4-tetrahydrocyclopenta[b]indol-3(R)-yl]acetic acid
| Company | Arena Pharmaceuticals Inc. |
| Description | Sphingosine 1-phosphate receptor 1 (S1PR1; S1P1; EDG1) agonist |
| Molecular Target | Sphingosine 1-phosphate receptor 1 (S1PR1) (S1P1) (EDG1) |
| Mechanism of Action | Sphingosine 1-phosphate (S1P) receptor agonist |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase I |
| Standard Indication | Autoimmune (unspecified) |
| Indication Details | Treat autoimmune diseases |
APD334, an orally available agonist of the S1P1 receptor, is an internally discovered investigational drug candidate intended for the potential treatment of a number of conditions related to autoimmune diseases, including multiple sclerosis, psoriasis and rheumatoid arthritis. S1P1 receptors have been demonstrated to be involved in the modulation of several biological responses, including lymphocyte trafficking from lymph nodes to the peripheral blood. By isolating lymphocytes in lymph nodes, fewer immune cells are available in the circulating blood to effect tissue damage. We have optimized APD334 as a potent and selective small molecule S1P1 receptor agonist that reduces the severity of disease in preclinical autoimmune disease models.
Autoimmune diseases are characterized by an inappropriate immune response against substances and tissues that are normally present in the body. In an autoimmune reaction, a person’s antibodies and immune cells target healthy tissues, triggering an inflammatory response. Reducing the immune and/or inflammatory response is an important goal in the treatment of autoimmune disease.



The present invention relates to processes and intermediates useful in the preparation of of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol- 3-yl)acetic acid of Formula (la) or salts thereof, an SlPl receptor modulator that is useful in the treatment of SlPl receptor-associated disorders, for example, diseases and disorders mediated by lymphocytes, transplant rejection, autoimmune diseases and disorders, inflammatory diseases and disorders (e.g. , acute and chronic inflammatory conditions), cancer, and conditions characterized by an underlying defect in vascular integrity or that are associated with angiogenesis such as may be pathologic (e.g. , as may occur in inflammation, tumor development and atherosclerosis).
BACKGROUND OF THE INVENTION
SlPl receptor agonists have been shown to possess at least immunosuppressive, antiinflammatory, and/or hemostatic activities, e.g. by virtue of modulating leukocyte trafficking, sequestering lymphocytes in secondary lymphoid tissues, and/or enhancing vascular integrity. Accordingly, SlPl receptor agonists can be useful as immunosuppressive agents for at least autoimmune diseases and disorders, inflammatory diseases and disorders (e.g. , acute and chronic inflammatory conditions), transplant rejection, cancer, and/or conditions that have an underlying defect in vascular integrity or that are associated with angiogenesis such as may be pathologic (e.g., as may occur in inflammation, tumor development, and atherosclerosis) with fewer side effects such as the impairment of immune responses to systemic infection.
The sphingosine-1 -phosphate (SIP) receptors 1-5 constitute a family of G protein- coupled receptors containing a seven-transmembrane domain. These receptors, referred to as SlPl to S1P5 (formerly termed endothelial differentiation gene (EDG) receptor-1, -5, -3, -6, and -8, respectively; Chun et al., Pharmacological Reviews, 54:265-269, 2002), are activated via binding by sphingosine-1 -phosphate, which is produced by the sphingosine kmase-catalyzed phosphorylation of sphingosine. SlPl, S1P4, and S1P5 receptors activate Gi but not Gq, whereas S1P2 and S1P3 receptors activate both Gi and Gq. The S1P3 receptor, but not the SlPl receptor, responds to an agonist with an increase in intracellular calcium.
In view of the growing demand for S 1P1 agonists useful in the treatment of S 1P1 receptor-associated disorders, the compound (R)-2-(7-(4-cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid of Formula
(la):

has emerged as an important new compound, see PCT patent application, Serial No.
PCTVUS2009/004265 hereby incorporated by reference in its entirety. Accordingly, new and efficient routes leading to (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l, 2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid of Formula (la), salts, and intermediates related thereto are needed. The processes and compounds described herein help meet these and other needs.
Example 7: Preparation of (i?)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) and L-Arginine Salt of (JR)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)).
Method 1
Preparation of (/?)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) and L-Arginine Salt Thereof.
Step A: Preparation of (i?)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta [b] indol-3-yl)acetic acid.
To a solution of rac-ethyl 2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetate (20.00 g, 41.19 mmol) in acetonitrile (185 ml) in a 500 mL three-neck RBF equipped with magnetic stir bar, N2 inlet, thermocouple, and condenser was added potassium phosphate buffer (15 ml, 1.0 M, pH = 7.80) and followed by addition of lipase B, Candida antarctica, immobilized recombinant from yeast (1.0 g, 5865 U/g, 5865 U). The resultant yellow suspension was stirred at about 40 °C under N2 for 16 hours. To the mixture, 1 M citric acid was added to adjust the pH to 3.96 which was then filtered on a Whatman filter cup. The solids were washed with ACN (3 x 15 mL). The combined filtrate and washings were concentrated at about 30 °C under vacuum to give an orange residue, which was partitioned between EtOAc (60 mL) and brine (60 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were washed with H20 (2 x 80 mL), brine (2 x 80 mL), dried over Na2S04, decanted, and concentrated at 30 °C under vacuum to give an orange oil, which was dried under vacuum at room temperature overnight to give a light orange oil (22.203 g) containing (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta|¾]indol-3- yl)acetic acid. The crude was assayed to be 41.41 wt % (9.194 g) with 99.42% ee.
Step B: Preparation of L-Arginine Salt of (i?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)).
To the crude (21.837 g) (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (41.41 %w/w; 9.043 g, 19.77 mmol) containing the (5)-isomer as the ester impurity in a 200 mL round bottom flask was added IPA (150.72 mL). The mixture was heated at 60 °C under N2 till the oily residue dissolved completely. The resultant orange solution was heated at about 60 °C for 5 min. Seeds of L-arginine salt of (R)-2- (7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3- yl)acetate (362 mg) were added. The seeds were suspended in the orange solution. A 2.27 M aqueous solution of L-arginine (8.709 mL, 3.44 g, 19.77 mmol) pre-warmed to about 60 °C was added into the mixture dropwise over 30 min. A light yellow precipitate formed gradually during the addition. The suspension was stirred for about an additional 30 min. The temperature of the suspension was allowed to drop at about 0.4 °C per minute to room temperature. The mixture was agitated occasionally at room temperature overnight. The suspension was filtered and the cake was washed with IP A (3 6 mL) and EtOAc (3 x 15 mL). The filter cake was dried at room temperature under vacuum overnight to give L-arginine salt of (R)-2-(7-(4- cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate as a white solid (11.631 g, 44.7%): HPLC 99.38 Area %, 99.6 % ee. TGA, PXRD, PLM, SEM and DSC indicated the solid as a non-solvated, crystalline compound with an average aggregates size of 18.05 microns and a melting point of 202.69 °C.
Ή NMR (400 MHz, DMSO-d6) δ ppm 1.53-1.80 (m, 8H), 1.81-1.92 (m, 2H), 1.93-2.13 (m, 3H), 2.19 (dd, J= 15.12, 8.18 Hz, 1H), 2.46 (dd, J= 15.12, 6.61 Hz, 1H), 2.57-2.77 (m, 3H), 3.03-3.19 (m, 2H), 3.21-3.35 (m, 2H), 3.39-3.51 (m, 1H), 5.13 (s, 2H), 6.70 (dd, J= 8.75, 2.40 Hz, 1H), 6.93 (d, J= 2.40 Hz, 1H), 7.23 (d, 7= 8.75 Hz, 1H), 7.64 (d, J= 8.08 Hz, 1H), 7.72 (d, 7= 8.08 Hz, 1H), 7.74 (s, 1 H), 7.10-8.70 (br. s, 6H), 10.49 (s, 1H). LCMS m/z calcd for C32H40F3N5O5: 631.69, found: 632.1 (Msalt+H)+, 458.3 (100, (Macid+H)+).
Method 2
Preparation of (l?)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)).
Additional procedures to prepare (R)-2-(7-(4-Cyclopentyl-3- (1xiiluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) using other lipases were utilized, for example, the following were shown to hydrolyze rac-ethyl 2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetate to (R)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)). General hydrolysis conditions and % enantiomeric excess (% ee) are shown below for the following enzymes, lipase B Candida Antarctica, lipase Mucor miehei (MML), and P. fluorescens.

5% DMF inP. fluorescens 7.5 30 C 19-20 phosphate Buffer
Free enzyme (i.e., non-immoblized)
Each of the above enzymes provided the desired (R)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) with varying degrees of % ee.
Example 8: Preparation of L-Arginine Salt of (l?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid.
Method 1
(R)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (174.7 mg, 0.381 mmol) was dissolved in EPA (1.57 mL) and L-arginine (66.4 mg, 0.381 mmol) was added as a solution in water (263 μΕ,). The homogeneous solution was warmed to 40 °C. After 15 min at this temperature, a precipitate had formed. The reaction mixture was warmed to 70 °C causing the precipitate to dissolve. The heat bath was turned off. A precipitate began to form at 40 °C and the reaction mixture was allowed to cool to about 28 °C before collecting the solids by filtration. The solids were washed with 14% water in EPA to give the L-arginine salt of (R)-2-(7-(4-cyclopentyl-3- (1riiluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (130 mg).
Method 2
Example 8: Preparation of L-Arginine Salt of (i?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid.
Step A: Preparation of l-Cyclopentyl-2-(trifluoromethyl)benzene (Compound of Formula (lib)).

To a 50 L three-neck round-bottom flask equipped with a mechanical stirrer, thermocouple, and nitrogen inlet, was added dry THF (35 L) and cooled to 0-5 °C. To the flask was added Iron (III) chloride (2.7 kg, 0.15 eq) portion wise over 30-60 min. and stirred for 15- 30 min. resulting in a clear greenish solution. Under a nitrogen atmosphere in a dry 100 gallon glass lined reactor was added THF (87.5 L) and magnesium turnings (4.05 kg, 1.5 eq), and cooled to 0-5 °C. To the THF and magnesium mixture was added the solution of FeCl3 in THF at a rate to maintain the internal temperature below 10 °C. To the resulting yellow/green mixture was added TMEDA (15.5 kg, 1.2 eq) at a rate to maintain the internal temperature below 20 °C. The resulting reaction mixture was heated to 40-45 °C for 1 hour and a mixture of 1 bromo-2-
(trifluoromethyl) benzene (25 kg, 1.0 eq) and bromocyclopentane (19.9 kg, 1.2 eq) was added to the reaction mixture at a rate to maintain an internal temperature below 25 °C. The resulting reaction mixture was allowed to stir at room temperature overnight and subsequently cooled to an internal temperature of 0-5 °C. To the resulting mixture was added 6 N HC1 (100 L, 1.5 h) at such a rate as to maintain the internal temperature below 15 °C (caution, very exothermic). After the quench, MTBE (200 L) was added and the reactor contents was stirred for 30 min. The phases were separated and the aqueous layer back extracted with MTBE (75 L). The combined organic layers were washed with H20 (50 L), brine (50 L) and dried (MgS04). The mixture was filtered through an in-line (1 micron) filter cartridge followed by an additional in-line (0.45 micron) filter cartridge into a clean dry reactor. The solvent was evaporated under vacuum (jacket < 30 °C) and co-evaporated with heptanes (2 x 25 L) to provide a viscous liquid. The viscous liquid was dissolved in heptanes (100 L) and passed through a silica plug (25 kg). The silica plug was eluted with heptanes (TLC, Rf ~ 0.8, silica gel, heptanes) and the fractions containing the product were evaporated to provide the title compound as a yellow liquid, 11.7 kg (49.2%), purity as determined by HPLC was 94.1%. Ή NMR conforms to reference standard.
Step B: Preparation of 4-(Chloromethyl)-l-cyclopentyl-2-(trifluoromethyl)benzene (Compound of Formula (He)).

To a 100 gallon glass lined reactor equipped with a stirrer was added concentrated sulphuric acid (48.6 L) and cooled to an internal temperature between about -5 to -10 °C under an atmosphere of N2. To the sulfuric acid was added thionyl chloride (26.99 kg, 2 eq) at a rate to maintain the internal temperature below -5 °C. To the resulting mixture 1,3,5-trioxane (15.3 kg, 1.5 eq) was added portion wise at a rate to maintain the internal temperature below -5 °C. After the addition of 1,3,5-trioxane, l-cyclopentyl-2-(trifluoromethyl) benzene (24.0 kg) was added drop wise over a period of approximately 2-3 hours. The reaction mixture was stirred at 0 °C for approximately 3-4 hours, allowed to warm to room temperature overnight and subsequently cooled to an internal temperature of 0-5 °C. To the resulting mixture was added water (316 L) drop wise over a period of approximately 5-6 hours (Note: Very exothermic). After the quench with water, the resulting aqueous mixture was extracted with MTBE (243 L and 123 L). The combined organics were washed with saturated NaHC03 (100 L), brine (100 L), water (100 L), brine (100 L), and dried (MgS04). The mixture was filtered through an in-line (1 micron) filter cartridge followed by an additional in-line (0.45 micron) filter cartridge into a clean dry reactor. The solvent was evaporated under vacuum (jacket < 30 °C) and further evaporated under vacuum at 35-40 °C. The resulting oil was distilled under high vacuum to provide the title compound as a yellow liquid, 24.8 kg (83%), purity as determined by HPLC was 99.47%. Ή
NMR conforms to reference standard.
Step C: Preparation of Ethyl 2-(2-Morpholinocyclopent-2-enylidene)acetate (Compound of Formula (Kg), Whe

Cyclopentanone (22.00 kg), morpholine (22.88 kg) and cyclohexane (43.78 kg) were charged to a 400 L glass-lined reactor equipped with overhead agitation, jacket temperature control, a nitrogen inlet, and a Dean-Stark trap. The reactor contents were heated to about 85 °C to 95 °C for approximately 26 h while removing water using the Dean-Stark trap. The reaction to form the enamine (i.e., 4-cyclopentenylmorpholine, Compound of Formula (lie) wherein R1 and R2 together with the nitrogen atom form a morpholine ring) is deemed complete when the morpholine amount is verified to be < 3% by GC peak area.
The reactor contents were cooled to about 60 °C and ethyl glyoxalate (Compound of Formula (ΠΤ) wherein R3 is ethyl; 58.74 kg, 50% solution in toluene) was added to the mixture slowly so as to maintain an internal temperature of < 80 °C. The reactor contents were heated to about 85 °C to 95 °C for at least 25 hours while removing water using the Dean-Stark trap. The reaction was deemed complete when the eneamine (i.e., 4-cyclopentenylmorpholine) amount by GC was verified to be less than 0.5% by GC peak area. The cyclohexane/toluene mixture was distilled under vacuum, ethanol (261.80kg) was charged to the reactor, and the resulting solution was again distilled under vacuum. Ethanol (34.76 kg) and water 44.00 kg) were charged to the reactor and the reactor contents stirred at 25 °C. The mixture was stirred further for 6 h at about 0-5 °C.
The resulting product slurry was collected by filtration, washed with aqueous ethanol (34.76 kg ethanol dissolved in 176.00 kg water). The filter-cake was further washed with water (110.00 kg), dried initially at approximately 36 °C for 1 hour under vacuum and subsequently at approximately 50 °C under vacuum for 17 h. The title compound was obtained as a tan solid (23.48 kg, 37.8% yield).
Step D: Preparation of Ζί/ZEthyl 2-(7-(Benzyloxy)-l,2-dihydrocyclopenta[b]indol- 3(4H)-ylidene)acetate

To a 400 L glass-lined reactor equipped with overhead agitation, jacket temperature control, and a nitrogen inlet was added (4-(benzyloxy)phenyl)hydrazine hydrochloride (21.08 kg, 1.000 mole equiv.), ethyl 2-(2-mo holinocyclopent-2-en lidene)acetate (22.02 kg, 1.104 mole equiv.), ethanol (51.2 kg, 2.429 mass equiv.), and acetic acid (36.8 kg, 1.746 mass eq.). After the reactor contents are allowed to stand for 10 minutes, agitation and then heating to 60°C to 65°C (60°C target) was started. While stirring at that temperature, samples of the reaction mixture were taken over intervals of approximately 30 minutes and analyzed by HPLC for (4-
(benzyloxy)phenyl)hydrazine, ethyl 2-(2-morpholinocyclopent-2-enylidene)acetate, and hydrazone content. When (4-(benzyloxy)phenyl)hydrazine HPLC % area was < 1, TFA (11.6 kg, 101.7 mol, 1.200 mole equiv., 0.550 mass equiv.) was charged over approximately 1 hour while the stirred reaction mixture was maintained at 60°C ± 5°C with reactor jacket cooling. As stirring at 60°C to 65°C was continued, the hydrazone and imine content of the reaction mixture was monitored by HPLC. After stirring at 60°C to 65°C for at least 12 hours the imine content of the reaction mixture was < 5% area by HPLC, and the stirred reaction mixture was cooled to 20°C to 25°C over approximately 3 hours. Stirring was maintained at that temperature to allow isomerization of the Z isomer to the desired E isomer. The E isomer crystallizes from the reaction mixture. The Z isomer and E isomer % area content of the reaction mixture was monitored by HPLC during this period of stirring at 20°C to 25°C, which was continued until the Z-isomer content of the reaction mixture was < 15% area by HPLC.
The stirred reaction mixture was cooled (0°C to 5°C) over at least 2 hours and then filtered. The reactor was charged with ethanol (27.4 kg, 1.300 mass equiv.), which was stirred and chilled to 0°C to 5°C and then used in two approximately equal portions to slurry-wash the product filter cake twice. The reactor was charged with ethanol (13.8 kg, 0.655 mass equiv.), which was stirred and chilled to 0°C to 5°C and then used to wash the product filter cake by displacement. The reactor was charged with USP purified water (100 kg, 4.744 mass equiv.), and the temperature was adjusted to 20°C to 25°C. The USP purified water was then used in three approximately equal portions to wash the product filter cake three times, the first two by reslurrying and the third by displacement. The reactor was charged with ethanol (16.4 kg, 0.778 mass equiv.), stirred and chilled to 0°C to 5°C, and then used to wash the product filter cake by displacement. The washed product filter cake was dried under full vacuum first with a jacket temperature of 35°C for 1 hour and then with a jacket temperature of 50°C. While drying continues with a jacket temperature of 50°C, the product solids are turned over every 1 hour to 3 hours, and product samples are analyzed for loss on drying (LOD) every >4 hours. When LOD was < 1%, the product was cooled to < 30°C. The yield of the title compound was 13.06 kg (37.59 mol, 44.7%). Step E: Preparation of Ethyl 2-(7-Hydroxy-l,2,3,4-tetrahydrocyclopenta[b]indol-3- yl)acetate.

To a 200 liter Hastelloy reactor was added ethyl 2-(7-(benzyloxy)-l ,2- dihydrocyclopenta[b]indol-3(4H)-ylidene)acetate (E/Z mixture, 12 kg), 10% Pd/C (50% wet with H20; 1.80 kg) and ethyl acetate (108 kg). The suspension was degassed 3x with N2 and triethylamine (1.76 kg) was added. To the resulting mixture was added formic acid (3.34 kg) while maintaining the internal temperature at below 35 °C. The reaction progression was followed by HPLC to monitor the complete consumption of starting material (i.e., E/Z mixture of ethyl 2-(7-(benzyloxy)-l ,2-dihydrocyclopenta[b]indol-3(4H)-ylidene)acetate) and the debenzylated intermediate. After approximately 30 minutes an additional amount of formic acid (0.50 kg) was added and the combined peak area of ethyl 2-(7-(benzyloxy)-l ,2- dihydrocyclopenta[b]indol-3(4H)-ylidene)acetate and the related debenzylated intermediate was determined to be < 1 % area by HPLC. The reactor contents were filtered through a 1.2 μιη cartridge filter followed by an in-line 0.2 μπι inline polishing filter. To the filtrate was added water (60 kg) and the biphasic mixture was partitioned. The organics were separated and concentrated under vacuum at approximately 60°C ± 5°C to a minimum stir volume, ethyl acetate (21.6 kg) was added and the mixture was further concentrated under vacuum to a minimum stir volume. Once again ethyl acetate (16.8 kg) was charged to the crude mixture and the resulting solution was heated to approximately 60 °C. Heptanes (37.2 kg) were charged maintaining the internal temperature at 60 °C. The solution was slowly cooled to approximately 0 to 5 °C and approximately 2-3 hr to facilitate crystallization. The slurry was filtered, the filter cake was reslurried in heptanes (27.12 kg) and ethyl acetate (7.08 kg). The resulting suspension was filtered and the solids dried under vacuum at approximately 40 ± 5 °C (until the loss on drying (LOD) is < 1%) to afford the title compound (6.23 kg, 70.3 % yield) as a solid.
Step F: Preparation of ( ft^-Ethyl 2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate (Compound of Formula (Ilk), Wher

To a 50 liter glass reactor containing ethyl 2-(7 -hydroxy- 1 ,2,3, 4- tetrahydrocyclopenta[b]indol-3-yl)acetate (2.000 kg, 1.000 equiv.) was added cesium carbonate
(3.266 kg, 1.300 equiv.) and acetonitrile (15.720 kg) under nitrogen. To the resulting mixture was added 4-(chloromethyl)-l-cyclopentyl-2-(trifluoromethyl)benzene (2.228 kg, 1.100 equiv.) over approximately one hour while maintaining the stirred reactor contents at 40°C ± 5°C. After the addition of 4-(chloromethyl)-l-cyclopentyl-2-(trifluoromethyl)benzene the reactor contents were heated to 65°C ± 5°C with stirring until the concentration of ethyl 2-(7-hydroxy-l , 2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetate in the reaction mixture was less than 2.0 % area by
HPLC. The reaction mixture was cooled to 50°C ± 5°C and filtered under nitrogen through a fine filter cloth with suction to remove cesium salts (Note: ethyl 2-(7-(4-cyclopentyl-3-
(trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate may precipitate below 30 °C). The filter cake was washed with fresh hot (50°C ± 5 °C) acetonitrile (5.658 kg divided in approximately three equal portions). The filtrates were returned to the reactor. The combined filtrates were concentrated by vacuum distillation with a jacket temperature of 60°C ± 10°C. To the reactor was added ethyl alcohol (3.156 kg) and once again concentrated with stirring by vacuum distillation with a jacket temperature of 60°C ± 10 °C. Once again, ethyl alcohol (3.156 kg) was added to the reactor and the contents were concentrated by vacuum distillation with a jacket temperature of 60 °C ± 10 °C to a reactor volume of approximately 14 L. The stirred reactor contents were cooled to 0 °C ± 5°C and the temperature maintained for 4 hours to facilitate the crystallization of the product. The resulting slurry was filtered. The filter cake was washed with cold 0 °C ± 5 °C ethyl alcohol (2 x 3.156 kg). The filter cake was dried under vacuum at 35 °C ± 5 °C until the weight loss over >1 hour was <2% to provide 3.0943 kg (81.0% yield) of the title compound as a solid.
Step G: Preparation of (!?)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)- l,2,3,4-tetrahydrocyclo

A 1.0 M buffer solution was prepared containing potassium phosphate monobasic (29.1 g, 0.0335 equiv.) in USP purified water (213 g) and potassium phosphate dibasic (368.2 g, 0.331 equiv.) in USP purified water (2.107 g). To a 50 liter glass reactor was added ethyl 2-(7-(4- cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate
(3.094 kg, 1.000 equiv.), Lipase B, Candida antarctica, immobilized (88.18 g, 293250 units/kg of ethyl ester starting material) and acetonitrile (22.32 kg). To the stirred contents of the reactor was added the previously prepared 1.0 M potassium phosphate buffer. The resulting mixture was stirred under nitrogen at a temperature of 40°C ± 5°C until the (R)-2-(7-(4-cyclopentyl-3-
(rrifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid concentration was >35% area as determined by HPLC (Note: although the reaction usually is complete after about 10 hours, the reaction mixture may be held at 40°C ± 5°C overnight). The stirred reactor contents were cooled to 25 °C ± 5°C and the pH was adjusted to between 4 and 5 by addition of a solution of citric acid (278.5 g, 0.228 equiv.) dissolved in USP purified water (1.454 kg). The reactor contents were filtered to remove immobilized lipase and phosphate and citrate salts. The reactor and solids were washed with acetonitrile (4.827 kg) and the combined filtrates were added backed into the reactor. The stirred reactor contents were concentrated to a volume of 1.0 L to 2.0 L by vacuum distillation at a jacket temperature of 55 °C ± 5°C. To the reactor was added ethyl acetate (5.582 kg) and USP purified water (6.188 kg). The contents were stirred at 20°C ± 5°C for at least 10 minutes and a solution of sodium chloride (1 kg) in USP purified water (1 kg) was added to facilitate phase separation. After phase separation was complete, the lower aqueous layer was drained. A solution of sodium chloride (5.569 kg) in USP purified water (12.38 kg) was divided in two approximately equal portions and the ethyl acetate phase was washed (2x). The ethyl acetate phase was transferred into a carboy and the reactor was rinsed with ethyl acetate (838.5 g) and added to the carboy containing the ethyl acetate phase. The reactor was washed sequentially with USP purified water (12.38 kg), acetone (4.907 kg), and ethyl acetate (838.5 g) and the ethyl acetate mixture from the carboy was transferred back to the reactor and concentrated with stirring to a volume of 1 L to 2 L by vacuum distillation at a jacket temperature of 55°C ± 5°C. To the reactor was added 2-propanol (14.67 kg) and after stirring the resulting mixture was concentrated to a volume of 1 L to 2 L by vacuum distillation at a jacket temperature of 55°C ± 5°C. To the reactor was added 2-propanol (7.333 kg) and heated with stirring at 60°C ± 5°C until the contents dissolved. The stirred reactor contents were cooled to 20°C ± 5°C and filtered through a medium-porosity fritted-glass filter to remove any inorganic solids to provide a 2-propanol solution containing 1.3188 kg of the title compound.
Step H: Preparation of L-Arginine Salt of (i?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3?4-tetrahydrocyclopenta [b] indol-3-yl)acetic acid
(Compound of For

To a 50 liter glass reactor containing the 2-propanol solution prepared in Step G of (R)- 2-(7-(4-cyclopen1yl-3-(trifluoromethyl)ben2yloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3- yl)acetic acid (1.3188 kg, 1.000 equiv.) was added an additional amount of 2-propanol (6.3389 kg) to adjust the total volume to approximately 16.7 L/kg of (R)-2-(7-(4-cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid. The reactor contents were stirred and heated to 60 °C ± 5 °C. To the reactor was added seed material (L- arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l , 2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid, 26.4 g, 0.0145 equiv.). The reactor contents were stirred for approximately 5 minutes at 60 °C ± 5 °C and a solution of L-arginine (502.5 g, 1.000 equiv.) in USP purified water (1.27 kg) preheated to 60°C ± 5°C was added over approximately
1 hour while maintaining the stirred reactor contents at 60°C ± 5°C. The stirring of the reactor contents at 60°C ± 5°C was maintained for approximately 1 hour and then allowed to cool at an approximate rate of 0.2°C/min to 1.0°C/min. to a temperature of 25°C ± 5°C. Once at approximately 25°C the contents of the reactor were stirred for approximately 1 hour maintaining the temperature of 25°C ± 5°C. The resulting slurry was filtered and the filter cake was washed with 2- propanol (6.2511 kg divided in three approximately equal portions) and with ethyl acetate (13.560 kg divided in six approximately equal portions. The filter cake was dried under vacuum at 40°C ± 5°C (until the weight loss over >1 hour is <2%) to provide 1.657 kg of the title compound (32.9% yield) as a crystalline solid.
HPLC purity: 99.64 Area %; Enantiomeric purity: 99.3%; DSC melting onset temperature 203.46 °C; TGA Weight Loss out to ~1 10 °C was 0.05%. NMR confirms the structure of the L-salt.
Five additional lots of the L-arg salt have been prepared using substantially this same synthetic method as described above, the DSC melting onset temperatures for a sample from each of the lots is as follows: 203.96 °C, 203.00 °C, 203.11 °C, 203.79 °C and 203.97 °C; the TGA Weight Loss out to ~1 10 °C for a sample from each of the lots is as follows: 0.04%, 0.04%, 0.03%, 0.10%, and 0.12%.
| WO2009078983A1 * | Dec 15, 2008 | Jun 25, 2009 | Arena Pharm Inc | Tetrahydrocyclopenta[b]indol-3-yl carboxylic acid derivatives useful in the treatment of autoimmune and inflammatory disorders |
| WO2010011316A1 * | Jul 22, 2009 | Jan 28, 2010 | Arena Pharmaceuticals, Inc. | SUBSTITUTED 1,2,3,4- TETRAHYDROCYCLOPENTA[b]INDOL-3-YL) ACETIC ACID DERIVATIVES USEFUL IN THE TREATMENT OF AUTOIMMUNE AND INFLAMMATORY DISORDERS |
| US20090004265 | Jan 19, 2006 | Jan 1, 2009 | Bayer Healthcare Ag | Prevention and Treatment of Thromboembolic Disorders |
TAK-733……. clinical studies for cancer treatment.

TAK-733
CAS: 1035555-63-5
Synonym: TAK-733; TAK 733; TAK733.
IUPAC/Chemical name:
(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Chemical Formula: C17H15F2IN4O4
Exact Mass: 504.01060
Molecular Weight: 504.23
Elemental Analysis: C, 40.49; H, 3.00; F, 7.54; I, 25.17; N, 11.11; O, 12.69
Phase I clinical studies for cancer treatment.Takeda Pharmaceutical,
Solid Tumors Therapy
Description of TAK-733: TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
Current developer: Millennium Pharmaceuticals, Inc./Takeda Pharmaceutical Company Limited.
TAK-733 is being developed at Millennium Pharmaceuticals for the treatment of adult patients with advanced non-hematological malignancies. Phase I clinical trials are ongoing for the treatment of advanced metastatic melanoma. In preclinical studies, the compound has been shown to bind to and potently inhibit MEK.

………………………………….
Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer
- Takeda San Diego;10410 Science Center Drive, San Diego, CA 92121, United States
http://www.sciencedirect.com/science/article/pii/S0960894X11000941

Synthesis of compounds 26 and 27 (Route 4). Reagents and conditions: (a) 1-chloro-2,4-dinitrobenzene, K2CO3, DMF; (b) (R)-O-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)hydroxylamine or 2,2-dimethyl-1,3-dioxan-5-amine, K2CO3 or Cs2CO3, DMF; (c) HCl, THF; (d) Selectfluor, CH3CN,DMF.
TAK-733 exhibited potent enzymatic and cell activity with an IC50 of 3.2 nM against constitutively active MEK enzyme and an EC50 of 1.9 nM against ERK phosphorylation in cells. TAK-733 did not inhibit any other kinases, receptors or ion channels that were tested with inhibitor concentrations up to 10 μM. TAK-733 was found to bind plasma protein moderately (ca. 97% for human and 96% for mouse), and exhibit high permeability and high microsomal stability across species. It did not inhibit P450s up to 30 μM.
The co-crystal structure of TAK-733 in the MEK1 allosteric site has been solved (Fig. 3). As predicted, the pyridone oxygen makes a hydrogen bond with the backbone NH of Ser212. The 2-fluoro-4-iodoaniline moeity sits in the deep lipophilic pocket. The pyrimidinone oxygen makes a hydrogen bond with Lys97, and the propanediol terminal hydroxyl interacts with both Lys97 and the ADP phosphate.

-
Figure 3.
The X-ray co-crystal structure of TAK-733 in the MEK1 allosteric site.

(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
| Molecular Weight: | 504.23 |
| TAK-733 Formula: | C17H15F2IN4O4 |
| CAS Number: | 1035555-63-5 |
TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
BRAF L597 mutations in melanoma are associated with sensitivity to MEK inhibitors.
Dahlman et al. Cancer Discov. 2012 Jul 13. PMID: 22798288.Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer.
Dong et al. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. PMID: 21310613.

16: 1652-1659


MEK kinases regulate the pathway that mediates proliferative and anti-apoptotic signaling factors that promote tumor growth and metastasis. TAK-733 is an MEK kinase inhibitor that entered phase I clinical trials for the treatment of cancer. A noteworthy feature of this short synthesis (25% yield overall) is the one-pot, three-step synthesis of the fluoropyridone D, in which the fluorine atom is present at the outset.
The reaction of F with the nosylate G gave a mixture of N- and O-alkylation products (8:1) from which the desired N-alkylation product was isolated by crystallization. The mixture of N-methyl pyrrolidine (NMP) and methanol used in the final deprotection step, helped to ensure formation of the desired polymorph. The nine-step discovery synthesis (3% overall yield) is also presented.
…………………………….
http://www.google.com/patents/WO2008079814A2?cl=en
Example 18: (i?)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione

[0364] To a solution of (i?)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 6) (1 g, 2.06 mmol, 1 eq) in DMF (19 mL) was added dropwise a mixture of Selectfluor (801 mg, 2.26 mmol, 1.1 eq) in acetonitrile (9 niL) and DMF (5 niL) at ambient temperature while stirring under nitrogen. The reaction mixture was stirred for 10 minutes at ambient temperature and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (i?)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 18) as an off-white solid. The recovered starting material was subjected to the same reaction conditions to produce a second crop of product. The total yield of product was 347 mg (33% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.40 (m, 1 H) 3.43 – 3.50 (m, 1 H) 3.58 (s, 3 H) 3.61 – 3.72 (m, 1 H) 3.72 – 3.82 (m, 1 H) 4.27 – 4.37 (m, 1 H) 4.78 – 4.87 (m, 1 H) 5.14 (d, J=5.81 Hz, 1 H) 6.93 – 7.03 (m, 1 H) 7.53 (d, J=8.84 Hz, 1 H) 7.65 – 7.74 (m, 1 H) 8.52 (s, 1 H) 10.25 (d, J=LOl Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
OTHER ISOMER
Example 19: (5)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione
 CAREFUL PLEASE
CAREFUL PLEASE[0365] To a solution of (5)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 5) (1.25 g, 2.58 mmol, 1 eq) in DMF (26 mL) was added a mixture of Selectfluor (1.187 g, 3.35 mmol, 1.3 eq) in acetonitrile (26 mL). The reaction mixture was irradiated with microwave at 82 0C for 10 minutes and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (5)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 19) as an off-white solid (331 mg, 25% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.42 (m, 1 H) 3.42 – 3.51 (m, 1 H) 3.59 (s, 3 H) 3.63 – 3.72 (m, 1 H) 3.73 – 3.81 (m, 1 H) 4.32 (dd, J=13.14, 3.03 Hz, 1 H) 4.79 (br. s., 1 H) 5.10 (br. s., 1 H) 6.98 (td, J=8.46, 5.31 Hz, 1 H) 7.52 (dd, J=8.46, 1.14 Hz, 1 H) 7.68 (dd, J=10.36, 1.77 Hz, 1 H) 8.51 (s, 1 H) 10.24 (d, J=2.27 Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
……………………………………….
http://www.google.com/patents/US8436200?cl=en

The synthetic process of the present invention allows for the preparation of (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione while avoiding the use of a fluorinating reagent in the last step. That is, the present invention provides a valuable process for making (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione characterized by the reaction of a compound of formula (8) with 2-fluoro-4-iodoaniline to give a compound of formula (9). Such a process avoids the use of costly and possible hazardous fluorinating regents in later steps which has significant advantages in large-scale manufacture.
Example 8.1(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Combine (R)-3-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (24.75 g, 45.58 mmol) and ethanol (250 mL). Add aqueous 9N sulfuric acid (50 mL) over 5 minutes. Heat to 75° C. After 2 hour, cool to ambient temperature and then cool in an ice bath to give a solid. Collect the solid by filtration, rinse with ethanol (3×30 mL), and dry to give the title compound. 1H NMR (400 MHz, DMSO-d6) δ10.24 (s, 1H), 8.52 (s, 1H), 7.69 (dd, 1H, J=10.4, 1.8 Hz), 7.52 (d, 1H, J=8.6 Hz), 6.98 (m, 1H), 5.14 (brs, 1H), 4.83 (brs, 1H), 4.32 (dd, 1H, J=12.9, 2.5 Hz), 3.76 (m, 1H), 3.67 (dd, 1H, J=13.1, 12.9 Hz), 3.58 (s, 3H), 3.46 (ddd, 1H, J=10.9, 5.3, 5.1 Hz), 3.38 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ161.3 (d, J=4.0 Hz), 155.6 (d, J=22.8 Hz), 154.6 (d, J=250 Hz), 152.0, 150.6, 134.3 (d, J=231 Hz), 133.8 (d, J=7.1 Hz), 133.1 (d, J=3.0 Hz), 127.8 (d, J=10.3 Hz), 125.3 (d, J=7.0 Hz), 123.9 (d, J=21.5 Hz), 95.0 (d, J=4.0 Hz), 87.1 (d, J=7.8 Hz), 68.0, 63.8, 50.1, 28.8; 19F NMR (376 MHz, DMSO-d6) δ-124.4, −149.8; MS (M+H)+m/z calcd 505.0. found 505.0.
Google+
|
Information about this agent |
TAK-733 is currently in Phase I clinical trials and is being developed by Millennium Pharmaceuticals, Inc. (a part of Takeda Pharmaceutical Company Limited).
|
References |
1: Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther. 2014 Feb;13(2):353-63. doi: 10.1158/1535-7163.MCT-13-0481. Epub 2014 Jan 7. PubMed PMID: 24398428.
2: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
3: Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013 Dec 1;73(23):7043-55. doi: 10.1158/0008-5472.CAN-13-1825. Epub 2013 Oct 11. PubMed PMID: 24121489.
4: Garraway LA, Baselga J. Whole-genome sequencing and cancer therapy: is too much ever enough? Cancer Discov. 2012 Sep;2(9):766-8. doi: 10.1158/2159-8290.CD-12-0359. PubMed PMID: 22969114.
5: Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle PL, Hicks DJ, Bozon V, Glaspy JA, Rosen N, Solit DB, Netterville JL, Vnencak-Jones CL, Sosman JA, Ribas A, Zhao Z, Pao W. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012 Sep;2(9):791-7. Epub 2012 Jul 13. PubMed PMID: 22798288; PubMed Central PMCID: PMC3449158.
6: von Euw E, Atefi M, Attar N, Chu C, Zachariah S, Burgess BL, Mok S, Ng C, Wong DJ, Chmielowski B, Lichter DI, Koya RC, McCannel TA, Izmailova E, Ribas A. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012 Apr 19;11:22. PubMed PMID: 22515704; PubMed Central PMCID: PMC3444881.
7: Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T, O’Connell SM, Scorah N, Shi L, Wallace MB, Zhou F. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. doi: 10.1016/j.bmcl.2011.01.071. Epub 2011 Jan 22. PubMed PMID: 21310613.
| US8030317 | Dec 18, 2007 | Oct 4, 2011 | Takeda Pharmaceutical Company Limited | MAPK/ERK kinase inhibitors |
| US20080255160 | Dec 18, 2007 | Oct 16, 2008 | Qing Dong | Mapk/erk kinase inhibitors |
| WO2008000020A1 | Jun 27, 2007 | Jan 3, 2008 | Gary L Corino | Improved process |
| EP1894932A1 | Jun 10, 2005 | Mar 5, 2008 | Japan Tobacco, Inc. | 5-amino-2,4,7-trioxo-3,4,7,8-tetrahydro-2H-pyrido[2,3-d]pyrimidine derivatives and related compounds for the treatment of cancer |
| US20050222177 * | Jul 29, 2004 | Oct 6, 2005 | Irm Llc | Diseases with abnormal activation of the Abl, BCR-Abl, Bmx, CSK, TrkB, FGFR3, Fes, Lck, B-RAF, C-RAF, MKK6, alpha and beta SAPK2 kinases; antiproliferative; pyrrolo[2,3-d]pyrimidine-7-carboxylic acid [3-phenylcarbamoyl-phenyl]-amides and pyrrolo[3,2-c]pyridine analogs |
Fosravuconazole in phase 1 for the treatment of fungal infections.

Fosravuconazole
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane
BEF-1224
BMS-379224
E-1224
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester bis(L-lysine) salt is used as drug
The azole antifungal agent E-1224 is a prodrug of ravuconazole. In 2009, originator Eisai licensed E-1224 to Drugs for Neglected Diseases Initiative for the treatment of American trypanosomiasis (Chagas disease) in Latin America and the Caribbean. DNDi was conducting phase II clinical trials with the prodrug for this indication, however, development of the compound has been discontinued due to lack of sustained efficacy. Ravuconazole was originally licensed by Eisai to Bristol-Myers Squibb (BMS). BMS developed the drug’s prodrug, referred to by BMS as BMS-379224. For strategic reasons, BMS did not pursue development of the compound. In 2010, E-1224 was licensed exclusively to Brain Factory for development, commercialization and sublicense in Japan for the treatment of fungal infections.
About Ravuconazole and Ravuconazole Prodrug
The compound on the left is ravuconazole; the compound on the right is the dihydrogen phosphonoxy methoxy derived ravuconazole prodrug which has improved solubility and bioavailability.

……………………………………………………………
WO 2001052852
http://www.google.com/patents/WO2001052852A1?cl=en
Triazole antifungal compounds are well known in the prior art. Of the several classes known, one particularly potent class contains a tertiary hydroxyl group. For example, U. S. Patent 5,648,372 discloses that (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)- butan-2-ol has anti-fungal activity.

The utility of this class of compounds is limited by their low water solubility. For example, the solubility of the above triazole compound in water at pH 6.8 is 0.0006 mg/mL. This greatly impedes developing suitable parenteral dosage forms.
One method of addressing this problem was disclosed in European Patent Application 829478, where the water solubility of an azole antifungal agent was increased by attaching a linked amino-acid to the azole portion of the molecule

Alternatively, WO 97/28169 discloses that a phosphate moiety can be attached directly to the tertiary hydroxyl portion of the anti-fungal compound, e.g. the compound having the formula

U.S. Patent 5,707,977 and WO 95/19983 disclose water soluble prodrugs having the general formula

wherein X is OP(O)(OH)2 or an easily hydrolyzable ester OC(O)RNR l’rR>2.
WO 95/17407 discloses water-soluble azole prodrugs of the general formula

wherein X is P(O)(OH)2, C(O)-(CHR’)n-OP(O)(OH)2 or C(O)-(CHR’)π
-(OCHR,CHR1)mOR2.
WO 96/38443 discloses water-soluble azole prodrugs of the general formula

U.S. Patent 5,883,097 discloses water-soluble amino acid azole prodrugs such as the glycine ester

The introduction of the phosphonooxymethyl moiety into hydroxyl containing drugs has been disclosed as a method to prepare water-soluble prodrugs of hydroxyl containing drugs.
European Patent Application 604910 discloses phosphonooxymethyl taxane derivatives of the general formula

wherein at least one of R1 ‘, R2″, R3′, R6′ or R7′ is OCH2OP(O)(OH)2.
European Patent Application 639577 discloses phosphonooxymethyl taxane derivatives of the formula T-[OCH2(OCH2)mOP(O)(OH)2]n wherein T is a taxane moiety bearing on the C13 carbon atom a substituted 3-amino-2- hydroxypropanoyloxy group; n is 1, 2 or 3; m is 0 or an integer from 1 to 6 inclusive, and pharmaceutically acceptable salts thereof. WO 99/38873 discloses O-phosphonooxymethyl ether prodrugs of a diaryl 1,3,4-oxadiazolone potassium channel opener.
Golik, J. et al, Bioorganic & Medicinal Chemistry Letters, 1996, 6:1837- 1842 discloses novel water soluble prodrugs of paclitaxel such as


EXAMPLE 1
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane, sodium salt

(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol- 1 -yl)-2-[(di-tert-butyl phosphonoxy)methoxy1butane

To a solution of (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)-l-(lH-l,2,4-triazol-l-yl)butan-2-ol, II, (8.74 g, 20 mmol) in THF (40 mL) under a nitrogen atmosphere was added sodium hydride (0.80 g, 60% in oil, 20 mmol) at rt. The resulting mixture was stirred at rt for 0.25 h and then di- tert-butyl chloromethyl phosphate, III (10.3 g, 40 mmol) was added. The reaction mixture was heated at 50 °C for 16 h. The reaction mixture was then allowed to cool to rt and was concentrated under reduced pressure. The residue was dissolved in Et2O and was washed with H2O and brine. The organic layer was dried over MgSO4 and was concentrated under reduced pressure to obtain 17.0 g of crude subtitled compound. IV, as a gum. A small portion of this crude compound was purified by reverse phase chromatography on C- 18. The column was eluted with 30% CH3CN/H2O, 38% CH3CN/H2O, 45% CH3CN/H2O and then 50% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure in order to remove CH3CN. The resulting aqueous layer was then extracted with Et2O. The Et O layers were washed with brine, dried and concentrated under reduced pressure to afford purified subtitled compound, IV, as a white solid. 1H NMR (300 MHz, CDC13): δ 8.35 (s, 1H), 7.98 (d, 2H, J=9), 7.76 (s, 1H), 7.71 (d, 2H, J=9), 7.63 (s, 1H), 7.36-7.27 (m, 1H), 6.86-6.78 (m, 2H), 5.53 (dd, 1H, J=28,6), 5.53 (dd, 1H, J=9,6), 5.17 (d, 1H, J=15), 5.03 (d, 1H, J=15), 4.01 (q, 1H, J=7), 1.47 (s, 9H), 1.45 (s, 9H), 1.37 (d, 3H, J=7). MS [ESI+ (M+H)+] 660.2 obs. B. (2R,3R)-3-r4-(4-cyanoρhenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxy]butane, sodium saltdeprotection


The crude (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluoropheny 1)- 1 -( 1 H- 1 ,2 ,4-triazol- 1 -y l)-2- [(di-tert-buty 1 phosphonoxy)methoxy]butane, IV, (17 g) was dissolved in CH C1 (100 mL). To this solution was added TFA (50 mL) and the reaction mixture was stirred at rt for 0.25 h. The reaction mixture was then concentrated under reduced pressure. To the residue was added H2O (200 mL), Et2O (100 mL) and EtOAc (100 mL). The pH of the aqueous layer was adjusted to 7.6 by addition of solid Na2CO3 and then the organic and aqueous layers were separated. The aqueous layer was then subjected to reverse phase chromatography on 400 g of C-18 eluted with H2O to 5% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure, frozen and lyophilized to afford 1.5 g of the subtitled compound, I, as a white solid. (1.5 g, 12% over two steps). Η NMR (500 MHz, D2O) δ 8.91 (s, IH), 7.92 (s, IH), 7.81 (d, 2H, J=8), 7.80 (s, IH), 7.77 (d, 2H, J=8), 7.21 (dd, IH, J=15,9), 6.99 (ddd, IH, J=9,9,2), 6.91 (ddd, IH, J=9,9,2), 5.35 (dd, IH, J=6,6), 5.29 (d, IH, J=15), 5.21 (dd, IH, J=6,6), 5.19 (d, IH, J=15), 3.86 (q, IH, J=7), and 1.35 (d, 3H, J=7); MS [(ESI“ (M-HV 546.1]; Anal. Calcd for C23Hi8F2N5θ5SιPι Na2/3.5 H2O: C, 42.21 : H, 3.85: N, 10.70: Na, 7.03. Found: C, 42.32: H, 3.83: N, 10.60: Na, 7.04.
Di-tert-butyl chloromethyl phosphate, III:
Di-tert-butyl chloromethyl phosphate, III, may be made by any of the following methods.
Method 1
Silver di-t-butyl phosphate (6.34 g, 20 mmol), which was prepared by mixing di- t-butyl phosphate (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971 , 27, 3163) with one equivalent of silver carbonate in 50% aqueous acetonitrile and by lyophilizing to dryness, was placed together with chloroiodomethane (35 g, 200 mmol) in benzene and stirred at room temperature for 18 hrs. The reaction mixture was filtered and the filtrate concentrated under reduced pressure. The residue was chromatographed on silica and eluted with 2:1 hexanes-ethyl acetate. Appropriate fractions were concentrated to dryness to obtain the subtitled compound III (3.7 g, 71% yield): H NMR (CDCI3) δ 5.63 (d, 2H, J=17), 1.51 (s, 18H); MS (MH+ = 259).
Method 2
Tetrabutylammonium di-t-butyl phosphate was prepared by dissolving di-t-butyl phosphate [ 20g, 94 mmol (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971, 27, 3163)] in methanolic tetrabutylammonium hydroxide (47 mL of 1M solution, 47 mmol). The reaction mixture had a temperature of 23 °C and pH of 4.33. The pH of the reaction mixture was adjusted to 6.5-7.0 by addition of methanolic tetrabutylammonium hydroxide (48 mL of 1M solution, 48 mmol) over 0.2 h. The reaction mixture was stirred for 0.5 h at approximately 26 °C and then was concentrated under reduced pressure at a bath temperature below 40 °C. The crude residue was azeotroped three times by adding toluene (3×100 mL) and then the mixture was concentrated under reduced pressure. The crude residue was then triturated in cold hexanes (0°C) for 1 h and then the solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a first crop of tetrabutylammonium di-t-butyl phosphate as a white solid. (24. Og). The mother liquor was concentrated under reduced pressure and then triturated in cold hexanes (20 mL) for 1 h. The solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a second crop of tetrabutylammonium di-t-butyl phosphate as a white solid. [(8.5g), 32.5g total (77%)]. A solution of tetrabutylammonium di-t-butyl phosphate (218 g, 480 mmol) in benzene (200 mL) was added dropwise to stirred chloroiodomethane (800g, 4535 mmol) over 1.5 h at rt. The reaction mixture was stirred an additional 1.5 h at rt and then was concentrated under reduced pressure. The oily residue was dissolved in Et2O and filtered to remove white solids which had precipitated. The organic layer was washed with saturated NaHCO3 and H O/brine (1/1). The organic layer was then dried over magnesium sulfate, filtered and concentrated under reduced pressure to yield a red brown oil (320 g). The red brown oil was subjected to chromatography on silica gel (800g) eluted with 20% EtOAc/Hexanes, 25% EtOAc/Hexanes then 30% EtOAc/Hexanes. The product containing fractions were concentrated under reduced pressure to yield a golden oil. The oil was diluted with CH2C12 (30 mL) , concentrated under reduced pressure and then dried under vacuum to yield the subtitled compound III (61.3g, 49% yield). 1H NMR (Benzene-d6) δ 5.20 (2H, d, J=15), 1.22 (18H, s).
Method 3
Iodochloromethane (974 g, 402 mL, 5.53 mol) at 25°C was treated with tetrabutylammonium di-t-butylphosphate (250 g, 0.553 mol). The phosphate was added portion wise over 10 minutes. The heterogeneous mixture became a clear pink solution after approximately 15 minutes. The mixture was stirred for three hours, and the iodochloromethane was then removed by rotary evaporation with a bath temperature of <30°C. The residue was taken up in 1 L t-butyl methyl ether and stirred for 15 minutes to precipitate tetrabutylammonium iodide by-product. Tetrabutylammonium iodide was removed by vacuum filtration through a sintered glass funnel. The filtrate was concentrated by rotary evaporation to an oil which contained a 5:1 mixture of III and undesired dimer impurity

III”
The mixture can be purified by a silica gel chromatography to obtain III as pure compound in ~60% yield as an oil.
EXAMPLE 2
(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)-l-(lH-l,2,4- triazol- 1 -yl)-2- (dihydrogen phosphonoxy)methoxy]butane
A. An oven dried, 1L round-bottom flask equipped with a mechanical stirrer, nitrogen inlet adapter, pressure-equalizing addition funnel fitted with a rubber septum and temperature probe was charged with sodium hydride (2.89 g, 0.069 mol, 60%) and THF (50 mL). To this stirred suspension, (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)butan- 2-ol, II, (10 g, 0.023 mol) in 30 mL of THF was added dropwise over 20 minutes at room temperature. After stirring for 45 minutes, a solution of iodine (2.99 g, 0.0115 mol) in THF (30 mL)) was added dropwise over 10 minutes followed by dropwise addition of compound di tert butylchloromethyl phosphate, III (13.29 g, 0.035 mol, -68% purity) over 15 minutes. The reaction mixture was stirred for 4 hours at about 41 °C to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into ice cold water (100 mL). The aqueous phase was separated and extracted with ethyl acetate (3 x 50 mL) and the combined organic extract was washed with 10% sodium thiosulfite (50 mL), water (50 mL), brine (50 mL), dried over magnesium sulfate and concentrated under reduced pressure to give pale yellow oil (22.8 g, In-process HPLC: ~ 97% pure). The crude product was used “as is” in step B.
B. To a round-bottom flask equipped with magnetic stirrer, cooling bath, pH probe and N2 inlet-outlet was charged the product of Step A above (7.5 g) in CH2C12 (23 mL) and cooled to 0 °C. To this stirred solution, trifluoroacetic acid (8.8 mL) was added slowly and stirred for 3 h to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into a cold solution of 2N NaOH (64 mL). The reaction mixture was extracted with t-butyl acetate (2 x 65 mL) to remove all the organic impurities. The aqueous layer containing the title product as bis sodium salt was treated with activated charcoal (10 g) and filtered through a bed of Celite. The clear filtrate was acidified with IN HC1 to pH 2.5. The free acid, the title product, was extracted into ethyl acetate (2 x 50 mL). The combined organic layer was washed with water, dried over MgSO4) filtered, and the filtrate concentrated under reduced pressure to afford 3.39 g of crude title product.
EXAMPLE 3
Bis lysine salt of (2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)-2-[(dihydrogen phosphonoxy)methoxy]butane
The above obtained title product from Example 2 was dissolved in methanol (75 mL) and to this L-lysine (1.8 g) was added and heated at 60 °C for 4.5 h. The hot reaction mixture was filtered through a bed of Celite. The filtrate was concentrated to about 5 mL, mixed with ethanol (100 mL) and heated to 65 °C to crystallize the bis lysine salt. The salt was collected on a Buchner funnel and dried under vacuum to afford 3.71 g of the title compound as an off white crystalline solid.
About Eisai Co., Ltd.
Eisai Co., Ltd. is a research-based human health care (hhc) company that discovers, develops, and markets products throughout the world. Eisai focuses its efforts in three therapeutic areas: integrative neuroscience, including neurology and psychiatric medicines; integrative oncology, which encompasses oncotherapy and supportive-care treatments; and vascular and immunological reactions. Eisai contributes to the well-being of people around the world through a global network of research facilities, manufacturing sites and marketing subsidiaries. For more information about Eisai Co., Ltd., please visit http://www.eisai.co.jp/index-e.html.
ref
BMS-379224, a water-soluble prodrug of ravuconazole
42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-817
| WO2000030655A1 * | Nov 17, 1999 | Jun 2, 2000 | Squibb Bristol Myers Co | Water soluble prodrugs of azole compounds |
| WO2006118351A1 | May 1, 2006 | Nov 9, 2006 | Eisai Co Ltd | Mono-lysine salts of azole compounds |
| WO2012060448A1 | Nov 4, 2011 | May 10, 2012 | Eisai R&D Management Co., Ltd. | Combined pharmaceutical composition as antifungal agent |
| CN101341160B | Dec 20, 2006 | Jan 25, 2012 | 卫材R&D管理有限公司 | Process for production of water-soluble azole prodrug |
| EP1345915A1 * | Oct 18, 2001 | Sep 24, 2003 | Bristol-Myers Squibb Company | Improved process for water soluble azole compounds |
| EP2291084A1 * | May 20, 2009 | Mar 9, 2011 | Neurogesx, Inc. | Carbonate prodrugs and methods of using the same |
| US7230023 | Aug 20, 2003 | Jun 12, 2007 | Sankyo Company, Limited | Triazole compound containing a phosphonate group |
| US8735376 | May 20, 2009 | May 27, 2014 | Acorda Therapeutics, Inc. | Carbonate prodrugs and methods of using the same |
some animations
![]()



DARA BioSciences receives FDA orphan drug designation for KRN5500 (SPK 241) …..Antitumor agent

KRN5500
Antitumor agent
151276-95-8 cas
IUPAC/Chemical name:
(2E,4E)-N-(2-(((2R,3R,4R,5R,6S)-6-((7H-purin-6-yl)amino)-2-((S)-1,2-dihydroxyethyl)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)amino)-2-oxoethyl)tetradeca-2,4-dienamide
C28H43N7O7
Exact Mass: 589.32240
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-((((1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-, (E,E)-
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-(((((2E,4E)-1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-
-
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:

- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).
- EP 0525479; JP 1993186494; US 5461036; US 5631238

DARA BioSciences receives FDA orphan drug designation for KRN5500
DARA BioSciences has received orphan drug designation from the US Food and Drug Administration’s (FDA) Office of Orphan Products Development for KRN5500, for treating multiple myeloma
Multiple myeloma is a hematologic cancer or cancer of the blood.
KRN5500 is a non-opioid, non-narcotic compound that is currently being tested in Phase I clinical trial.
Earlier this year, KRN5500 received orphan status to be developed for the parenteral treatment of painful, chronic, chemotherapy-induced peripheral neuropathy (CCIPN) that is refractory to conventional analgesics in patients with cancer.
“We believe this myeloma-specific orphan designation enhances both the viability and the future market opportunity for this valuable pipeline product.”
DARA BioSciences MD, CEO and chief medical officer David J Drutz said: “It is noteworthy in this regard that up to 20% of myeloma patients have intrinsic peripheral neuropathy, an incidence that increases to the range of 75% in patients treated with neurotoxic drugs such as thalidomide or bortezomib.
KRN5500 is a semisynthetic derivative of the nucleoside-like antineoplastic antibiotic spicamycin, originally isolated from the bacterium Streptomyces alanosinicus. KRN 5500 inhibits protein synthesis by interfering with endoplasmic reticulum and Golgi apparatus functions. This agent also induces cell differentiation and caspase-dependent apoptosis.
KRN5500 is available as a solution for intravenous (IV) administration. KRN5500 was discovered in an effort to identify new agents that induced differentiation of myeloid leukemia cells.
Safety and efficacy data from Phase I trials have been leveraged to support DARA Therapeutics’ active IND and ongoing Phase 2a clinical trial. The objective of this Phase 2a feasibility study is to determine the potential of KRN5500 (a spicamycin analogue) to be a breakthrough medicine for the treatment of neuropathic pain in cancer patients.
Four clinical trials have been conducted in cancer patients, including one in Japan and 3 in the United States. Three of these studies are complete; the fourth was closed to patient accrual and treatment in December 2004.
A total of 91 patients with solid tumors have been treated with single IV KRN5500 doses of up to 21 mg/m2 and weekly doses of up to 42 mg/m2. While KRN5500 has not shown anti-cancer efficacy in any trial, its use in pain elimination is encouraging. (source: http://www.darabiosciences.com/krn5500.htm).

Chemical structures of KRN5500 and its known metabolites.
………………..
http://www.google.com/patents/EP0525479A1?cl=en
spk 241
- 6-[4′-N-(N’-trans,trans-2,4-tridecadienylglycyl)spicamynyl-amino]purine,
- (20) SPK241:


Example 52: Preparation of SPK241
-
[0214]To trans-2-dodecenal (4.5 g) dissolved in methylene chloride (80 ml) was added (carbomethoxymethylene)triphenylphosphorane (8.3 g), and the mixture was stirred for 2 hours. The reaction mixture was subjected to chromatography on a silica gel column with eluent systems of n-hexane- ethyl acetate (from 100:1 to 20:1) to give the methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g). Potassium hydroxide (6.5 g) was dissolved in a mixed solvent of ethanol-water (1:1) (100 ml). The methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g) was added to the mixture, and the resulting mixture was stirred at 60 °C for 40 minutes. After the reaction mixture was cooled, it was adjusted to the weak acidic range of pH with citric acid and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous sodium sulfate and concentrated to give trans,trans-2,4-tetradecadienoic acid (4.4 g). Hereafter, the title compound can be synthesized by the two methods described below.
-
[0215]In the first method, trans,trans-2,4-tetradecadienoic acid (4.3 g) is first dissolved in N,N-dimethylformamide (DMF, 50 ml). Para-nitrophenol (2.67 g) and N,N’-dicyclohexylcarbodiimide (3.9 g) were added to trans,trans-2,4-tetradecadienoic acid solution, and the mixture was stirred for 12 hours. After precipitates produced were removed by filtration and the solvent (DMF) was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of n-hexane-ethyl acetate (from 200:1 to 50:1) to give the active ester of trans,trans-2,4-tetradecadienoic acid (5.1 g). To the active ester (500 mg) dissolved in DMF (30 ml) were added 6-(4′-N-glycyl-spicamynyl-amino)purine hydrochloride (556 mg) and triethylamine (1.2 ml). The mixture was stirred for 12 hours. After the solvent was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 398 mg.
-
[0216]In the second method, trans,trans-2,4-tetradecadienoic acid (99.6 g) was dissolved in thionyl chloride (87 ml), and the mixture was stirred at room temperature. The excessive thionyl chloride was removed by distillation to give trans,trans-2,4-tetradecadienoic acid chloride (102.0 g). To glycine (66.8 g) dissolved in an aqueous 2N sodium hydroxide solution (540 ml) were added at the same time trans,trans-2,4-tetradecadienoic acid chloride (102.0 g) and 2N sodium hydroxide (270 ml) with 1/10 portions at a 3 minute interval. After the addition was completed, the mixture was warmed to room temperature, stirred for 15 minutes and acidified with concentrated hydrochloric acid (140 ml) under ice-cooling. Precipitates thus produced were collected by filtration and desiccated to give trans,trans-2,4-tetradecadienoyl glycine (75.0 g). To the solution of trans,trans-2,4-tetradecadienoyl glycine (4.7 g) and 6-(4′-N-glycyl-spicamynyl-amino)-purine (5.1 g) in N,N-dimethylformamide (DMF, 60 ml) was added N-hydroxysuccinimide (2.1 g), and the mixture was ice-cooled. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (3.4 g) dissolved in DMF (100 ml) was added dropwise to the mixture. After the addition was completed, the mixture was heated to room temperature and stirred for 12 hours. Water (500 ml) was added to the reaction mixture, and precipitates produced were collected by filtration and desiccated. Sodium methoxide (3.1 g) was added to a suspension of the precipitates in methanol (100 ml), and the mixture was stirred at room temperature, then ice-cooled and acidified by adding dropwise thereto a 10% methanolic hydrochloric acid solution. Precipitates produced were filtered, dried and subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 5.00 g.
Physicochemical properties of SPK241
-
[0217]
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:
- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).

| DE3407979A1 * | Mar 3, 1984 | Sep 6, 1984 | Kirin Brewery | Spicamycin sowie verfahren zu seiner herstellung |
| JPS59161396A | Title not available | |||
| US3155647 | Jul 24, 1963 | Nov 3, 1964 | Olin Mathieson | Septaciding and derivatives thereof |
| WO1990015811A1 | Jun 14, 1990 | Dec 27, 1990 | Kirin Brewery | Spicamycin x and its use |
| EP1328236A2 * | Sep 20, 2001 | Jul 23, 2003 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| EP2305264A1 * | Sep 20, 2001 | Apr 6, 2011 | The General Hospital Corporation | Spicamycin derivatives for use in decreasing or preventing pain |
| EP2349285A2 * | Oct 9, 2009 | Aug 3, 2011 | Dara Biosciences, Inc. | Methods for treating or preventing pain using spicamycin derivatives |
| EP2597082A1 | Nov 24, 2011 | May 29, 2013 | Symrise AG | Compounds for masking an unpleasant taste |
| US5905069 * | Jan 26, 1998 | May 18, 1999 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin or derivatives thereof |
| US7196071 | Sep 20, 2001 | Mar 27, 2007 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| US7375094 | Mar 15, 2007 | May 20, 2008 | The General Hospital Corporation | Produced via Streptomyces; antitumor agents; time-release agents; for opiod-resistant pain; drug screening |
| US7632825 | Apr 30, 2008 | Dec 15, 2009 | Bayer Pharmaceuticals Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
|
References 1: Mizumura Y. [Spicamycin derivative]. Nippon Rinsho. 2006 Feb;64(2):322-8. Review. Japanese. PubMed PMID: 16454188. 2: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2004 Apr;26(3):211-44. PubMed PMID: 15148527. 3: Borsook D, Edwards AD. Antineuropathic effects of the antibiotic derivative spicamycin KRN5500. Pain Med. 2004 Mar;5(1):104-8. PubMed PMID: 14996243. 4: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Dec;25(10):831-55. PubMed PMID: 14735233. 5: Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Nov;25(9):747-71. PubMed PMID: 14685303. 6: Supko JG, Eder JP Jr, Ryan DP, Seiden MV, Lynch TJ, Amrein PC, Kufe DW, Clark JW. Phase I clinical trial and pharmacokinetic study of the spicamycin analog KRN5500 administered as a 1-hour intravenous infusion for five consecutive days to patients with refractory solid tumors. Clin Cancer Res. 2003 Nov 1;9(14):5178-86. PubMed PMID: 14613997. 7: Yamamoto N, Tamura T, Kamiya Y, Ono H, Kondoh H, Shirao K, Matsumura Y, Tanigawara Y, Shimada Y. Phase I and pharmacokinetic study of KRN5500, a spicamycin derivative, for patients with advanced solid tumors. Jpn J Clin Oncol. 2003 Jun;33(6):302-8. PubMed PMID: 12913085. 8: Kobierski LA, Abdi S, DiLorenzo L, Feroz N, Borsook D. A single intravenous injection of KRN5500 (antibiotic spicamycin) produces long-term decreases in multiple sensory hypersensitivities in neuropathic pain. Anesth Analg. 2003 Jul;97(1):174-82, table of contents. PubMed PMID: 12818962. 9: Gadgeel SM, Boinpally RR, Heilbrun LK, Wozniak A, Jain V, Redman B, Zalupski M, Wiegand R, Parchment R, LoRusso PM. A phase I clinical trial of spicamycin derivative KRN5500 (NSC 650426) using a phase I accelerated titration “2B” design. Invest New Drugs. 2003 Feb;21(1):63-74. PubMed PMID: 12795531. 10: Byrd JC, Lucas DM, Mone AP, Kitner JB, Drabick JJ, Grever MR. KRN5500: a novel therapeutic agent with in vitro activity against human B-cell chronic lymphocytic leukemia cells mediates cytotoxicity via the intrinsic pathway of apoptosis. Blood. 2003 Jun 1;101(11):4547-50. Epub 2003 Feb 20. PubMed PMID: 12595316. |

{kind=link}