
Home » Posts tagged 'organic chemistry' (Page 47)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New Route to Anticancer Agent Quinocarcin

Quinocarcin,
Antibiotic DC 52, DC 52, CHEBI:554143, CID158486, LS-80981, 3,6-Imino-1H-2-oxa-11c-azanaphth(1,2,3-cd)azulene-5-carboxylic acid, 2a,3,4,5,6,6a,7,11b-octahydro-11-methoxy-12-methyl-, (2a-alpha,3-alpha,5-alpha,6-alpha,6a-alpha,11b-alpha)-, (-)-, 84573-33-1
Synthesis of quinocarcin through a convergent strategy based on Sonogashira coupling and gold(I)-catalyzed hydroamination
Takeda’s Ixazomib, Multiple Myeloma Drug

CAS#: 1201902-80-8
Synonym: Ixazomib; MLN-9708.
IUPAC/Chemical name:
4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid
UPDATES AT THE BOTTOM OF PAGE
CAMBRIDGE, Mass., May 23, 2013 – Takeda Pharmaceutical Company Limited (TSE:4502) today announced the initiation of an international phase 3 clinical trial evaluating once a week MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma who are not candidates for transplant. The multi-center study with MLN9708, an investigational, oral proteasome inhibitor, will be conducted in Europe and North America.———————-READ MORE AT
http://www.pharmalive.com/takeda-begins-phase-iii-trial-of-multiple-myeloma-drug
Description of Ixazomib: ixazomib is an orally bioavailable second generation proteasome inhibitor (PI) with potential antineoplastic activity. Ixazomib inhibits the activity of the proteasome, blocking the targeted proteolysis normally performed by the proteasome, which results in an accumulation of unwanted or misfolded proteins; disruption of various cell signaling pathways may follow, resulting in the induction of apoptosis. Compared to first generation PIs, second generation PIs may have an improved pharmacokinetic profile with increased potency and less toxicity. Proteasomes are large protease complexes that degrade unneeded or damaged proteins that have been ubiquinated
MLN9708 is an investigational proteasome inhibitor that, compared with bortezomib, has improved pharmacokinetics, pharmacodynamics, and antitumor activity in preclinical studies. MLN9708 rapidly hydrolyzes to MLN2238, the biologically active form. MLN9708 has a shorter proteasome dissociation half-life and improved pharmacokinetics, pharmacodynamics, and antitumor activity compared with bortezomib.MLN9708 has a larger blood volume distribution at steady state, and analysis of 20S proteasome inhibition and markers of the unfolded protein response confirmed that MLN9708 has greater pharmacodynamic effects in tissues than bortezomib. MLN9708 showed activity in both solid tumor and hematologic preclinical xenograft models, and we found a correlation between greater pharmacodynamic responses and improved antitumor activity. Moreover, antitumor activity was shown via multiple dosing routes, including oral gavage. Taken together, these data support the clinical development of MLN9708 for both hematologic and solid tumor indications. (source: Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16.).
| References |
1: Mullard A. Next-generation proteasome blockers promise safer cancer therapy. Nat Med. 2012 Jan 6;18(1):7. doi: 10.1038/nm0112-7a. PubMed PMID: 22227650.
2: Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445-52. Epub 2012 Jan 3. PubMed PMID: 22215754.
3: Appel A. Drugs: More shots on target. Nature. 2011 Dec 14;480(7377):S40-2. doi: 10.1038/480S40a. PubMed PMID: 22169800.
4: Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011 Dec 1;17(23):7313-23. Epub 2011 Sep 8. PubMed PMID: 21903769.
5: Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011 Aug 15;17(16):5311-21. doi: 10.1158/1078-0432.CCR-11-0476. Epub 2011 Jun 30. PubMed PMID: 21724551; PubMed Central PMCID: PMC3156932.
6: Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16. Erratum in: Cancer Res. 2010 May 1;70(9):3853. Hales, Paul [added]. PubMed PMID: 20160034.
7: Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 Mar;15(5-6):243-9. Epub 2010 Jan 29. Review. PubMed PMID: 20116451.8: Marblestone JG. Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs. 2009 Dec;12(12):750-3. PubMed PMID: 19943215.

http://www.cancernetwork.com/conference-reports/ash2012/content/article/10165/2119611
Nasopharyngeal cancer is a sub-type of head and neck cancer that arises from the epithelial cells that cover the surface and line the nasopharynx. The incidence of nasopharyngeal cancer has been reported at approximately 0.5 to 2 new cases per year per 100,000 in Europe and the USA. Rottey et ah, Curr. Opin. Oncol., 23(3): 254-258 (201 1). There are three subtypes of nasopharyngeal cancer recognized in the World Health Organization (WHO) classification: (i) Type 1 – squamous cell carcinoma, typically found in the older adult population; (ii) Type 2 non-keratinizing carcinoma; and (iii) Type 3 – undifferentiated carcinoma. Treatment for nasopharyngeal cancer often involves radiotherapy and/or chemotherapy. There remains a continuing need for new and improved treatments for patients with nasopharyngeal cancer. There remains a further need to identify nasopharyngeal patients most likely to benefit from treatment with a proteasome inhibitor.
Proteasome inhibition represents an important new strategy in cancer treatment. King et al. , Science 274: 1652-1659 ( 1996), describes an essential role for the ubiquitin-proteasome pathway in regulating cell cycle, neoplastic growth and metastasis. The authors teach that a number of key regulatory proteins, including cyclins, and the cyclin-dependent kinases p21 and p27K,P ! , are temporally degraded during the cell cycle by the ubiquitin-proteasome pathway. The ordered degradation of these proteins is required for the cell to progress through the cell cycle and to undergo mitosis.
The proteasome inhibitor VELCADE© (bortezomib; N-2-pyrazinecarbonyl-L -phenylalanine -L- leucineboronic acid) is the first proteasome inhibitor to achieve regulatory approval. Mitsiades et ai, Current Drug Targets, 7: 1341 (2006), reviews the clinical studies leading to the approval of bortezomib for the treatment of multiple myeloma patients who have received at least one prior therapy. Fisher et ai , J. Clin. Oncol, 30:4867, describes an international multi-center Phase II study confirming the activity of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Ishii et al, Anti-Cancer Agents in Medicinal Chemistry, 7:359 (2007), and Roccaro et al., Curr. Pharm. Biotech., 7: 1341 (2006), discuss a number of molecular mechanisms that may contribute to the antitumor activities of bortezomib. The proteasome inhibitor MLN9708 [2,2′-{2-[(lR)- l -( {[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3- methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid] is currently undergoing clinical evaluation for hematological and solid cancers. MLN9708 is a citrate ester which rapidly hydrolyzes to the active form [(lR)-l -({[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3-methylbutyl]boronic acid (MLN2238) on exposure to aqueous solution or plasma. MLN9708 has demonstrated anti-tumor activity in a range of hematological and solid tumor xenograft models (Kupperman et al. (2010) Cancer Res. 70: 1970- 1980),
Summary
The invention relates to the discovery that patients with nasopharyngeal cancer respond to treatment with MLN9708. In one aspect, the invention relates to the discovery of the increased expression of Nuclear Factor Kappa-B RelA 65,000 dalton subunit (NFKB p65) in biological samples comprising cells obtained from patients with nasopharyngeal cancer and responsive to MLN9708.
Accordingly, the invention features treating nasopharyngeal cancer patients withMLN9708 if a sample from the patient demonstrates an elevated expression of NFKB p65.
PATENT

or a pharmaceutically acceptable salt or a pharmaceutical composition or a boronic acid anhydride thereof.
[048| The compound of formula (II), [( l R)-l -( } [(2,5-dichlorobenzoyl)amino]acetyl} amino)-3- methylbutyljboronic acid (MLN2238) is disclosed in Olhava and Danca, U .S. Patent No. 7,442,830, herein incorporated by reference in its entirety. [049] In some other embodiments, Z and Z together form a moiety derived from a compound having at least two hydroxyl groups separated by at least two connecting atoms in a chain or ring, said chain or ring comprising carbon atoms and, optionally, a heteroatom or heteroatoms which can be N, S, or O, wherein the atom attached to boron in each case is an oxygen atom.
In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A) or (III-B):

(III-B), or a mixture thereof or a pharmaceutical composition thereof.
[054] In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A):

or a pharmaceutical composition thereof.
[055] The compound of formula (III-A), 2,2′- {2-[( l i?)- l -( { [(2,5-dichlorobenzoyl)amino]acetyl } amino)- 3-methylbutyl]-5-oxo- l ,3,2-dioxaborolane-4,4-diyl} diacetic acid (MLN9708) is disclosed in Elliott et al. , WO 09/ 154737, herein incorporated by reference in its entirety
PATENT
http://www.google.com/patents/WO2009154737A1?cl=en
Example 1: Synthesis of 4-(/?,S)-(carboxymethyl)-2-( (R)-I -(2-(2,5- dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-l,3,2-dioxaborinane-4- carboxylic acid (1-1)

Step l: 2,5-r(dichlorobenzoyI)aminolacetic acid
[0310] To a mixture of NaOH (12 g, 300 mmol) and glycine (18 g, 239 mmol) in water (120 mL) was added dropwise over 45 min a solution of 2,5-dichlorobenzoyl chloride (10 g, 48 mmol) in THF (15 mL) keeping the internal temperature below about 25 0C. After 1 h, the mixture was acidified with 2.0 M HCl (125 mL) keeping the internal temperature below about 5 0C. The resulting precipitate was collected by vacuum filtration. The crude product was recrystallized from water to give 2,5-[(dichlorobenzoyl)amino]acetic acid as a white, crystalline solid (6.1 g, 52%). mp 173.3 0C. 1H NMR (300 MHz, DMSOd6, δ): 12.72 (bs, IH), 8.89 (t, J = 6.0 Hz, IH), 7.54 (m, 2H), 7.48 (m, IH), 3.93 (d, J = 6.0 Hz). 13C NMR (75 MHz, DMSO-Ci6, δ): 41.6, 129.3, 129.6, 131.4, 132.2, 138.2, 171.4, 165.9. MS (ni/z): [M+H] calculated for C9H8Cl2NO3, 248.0; found, 248.0; [M+Na] calculated for C9H7Cl2NNaO3, 270.0; found 270.2.
2,5-[(dichlorobenzoyl)amino]acetic acid was also be prepared via the following procedure: To a mixture of glycine (21.5 g, 286 mmol) in water (437 mL), was added 2.0 M NaOH (130 mL) and the resulting solution was cooled to 0 0C. A solution of 2,5-dichlorobenzoyl chloride (50.0 g, 239 mmol) in THF (75 mL) was added dropwise at such a rate that the internal temperature was maintained at 0 ± 1 0C. During the addition, the pH was controlled at 11.0 ± 0.2 using a pH controller titrated with 2.0 M NaOH. After complete addition, the mixture was stirred at 0 ± 1 0C for an additional 2 h. The mixture was then acidified with 2.0 M HCl (176 mL) to a final pH of 2.5. The resulting precipitate was collected by filtration, washed with cold water (125 mL), and dried at 45 0C in a vacuum oven to afford 2,5-[(dichlorobenzoyl)amino]acetic acid as a white solid (57.6 g, 97.3%). Step 2: 2,5-dichloro-N-f2-(( (lR’)-3-niethyl-l-r(3aS,4S.6S.7aR)-3a,5,5-trimethylhexahvdro-
4,6-methano-l,3,2-benzodioxaborol-2-yllbutyl }amino)-2-oxoethvπbenzamide
To a solution of 2,5-[(dichlorobenzoyl)amino]acetic acid (6.10 g, 24.6 mmol) and TBTU (8.34 g, 26.0 mmol) in DMF (40 mL) with an internal temperature below about 5 0C was added (IR)- 3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5-trimethylhexahydro-4,6-methano-l,3,2-benodioxaborol-2- yl]butan-l-amine»TFA (9.35 g, 24.7 mmol). DIPEA (13 mL, 75 mmol) was then added dropwise over 2 h keeping the internal temperature below about 5 0C. After 40 min, the mixture was diluted with EtOAc (90 mL), washed with 5% NaCl (150 mL), twice with 10% NaCl (2 x 40 mL), once with 2% K2CO3 (1 x 40 mL), once with 1% H3PO4 (1 x 40 mL), and once with 10% NaCl (1 x 40 mL). The resulting organic layer was concentrated to a thick oil, diluted with heptane (40 mL) and evaporated to yield 2,5-dichloro-N-[2-({ (lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l ,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide as a white solid which was used in the next step without purification.
Step 3: N,N\N’Wboroxin-2A6-triyltrisir(lR)-3-methylbutane-l J-diyllimino(2-oxoethane- 2,l-diyl)^ ^tris(2,5-dichlorobenzamide)
To a solution of 2,5-dichloro-N-[2-({(lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide (12.2 g, 24.6 mmol) in methanol/hexane (1 :1) (250 mL) were added IN HCl (30 mL, 30 mmol) and (2-methylpropyl)boronic acid (6.5 g, 64 mmol). The reaction mixture was allowed to stir overnight. The phases were separated and the methanol layer was washed twice with additional heptane (2 x 55 mL). The resulting organic layer was concentrated to about 10 mL and partitioned between 2.0M NaOH (30 mL) and DCM (25 mL). The DCM layer was washed once with additional 2.0M NaOH (5 mL). The basic aqueous layers were then combined, washed twice with DCM (2 x 25 mL) and acidified with IM HCl (60 mL). The resulting mixture was diluted with DCM (40 mL), the layers were separated, and the resulting aqueous layer was washed three times with DCM (3 x 10 mL). The combined DCM extracts were dried over MgSO4 (25 g) and evaporated to a thick oil. The product was precipitated with heptane (50 mL) and collected by filtration to yield N,N’,N”-{boroxin-2,4,6- -riyltris[[(lR)-3-methylbutane-l,l-diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) as a white solid (6.6 g, 74%). 1H NMR (300 MHz, DMSO-Cl6, δ): 8.93 (t, J – 6.0 Hz, IH), 8.68 (bs, IH), 7.63 (m, IH), 7.52 (m, 2H), 4.00 (d, J = 6.0 Hz, 2H), 2.62 (m, IH), 1.59 (m, IH), 1.33 (m, IH), 1.24 (m, IH), 0.81 (d, / = 5.9 Hz, 6H). 13C NMR (125 MHz, DMSO-Cl6, δ): 23.2, 25.8, 40.1, 40.7, 43.0, 129.0, 130.0, 131.0, 137.5, 165.0, 172.5. MS (m/z) in CH3CN: [M+H] calculated for C42H52B3Cl6N6O9, 1027.2; found, 1027.3; [M+Na] calculated for C42H51B3Cl6N6NaO9, 1049.2; found 1049.5.
Step 4: 4-(/?.S)-(carboxymethyl)-2-((/?)-l-(2-(2,5-dichlorobenzamido)acetamido)-3- methylbutyl)-6-oxo-l,3,2-dioxaborinane-4-carboxylic acid (1-1)
Form 1: To a solution of citric acid (2.75 g, 14.3 mmol) in EtOAc (85 mL) with an internal temperature of about 74 0C was added N,N’,N”-{boroxin-2,4,6-triyltris[[(lR)-3-methylbutane-l,l- diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) (5.00 g, 4.87 mmol) as a solid. The solution was cooled uncontrolled until the internal temperature was about 25 0C and the mixture was stirred overnight. The resulting precipitate was collected by filtration to yield 2,2′-{2-[(lR)-l-({ [(2,5- dichlorobenzoyl)amino]acetyl }amino)-3-methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid Form 1 as a crystalline solid (6.65 g, 88 %). 1H NMR (500 MHz, DMSOd6, δ 110 0C): 10.08 (s, IH), 8.69 (s, IH), 7.61 (s, IH), 7.52 (d, J = 1.3 Hz, 2H), 4.26 (d, J = 5.5 Hz, 2H), 2.70 (q, J = 14.5 Hz, 4H), 2.70 (bs, IH), 1.72 (sept, J – 6.5 Hz, IH), 1.42 (ddd, J = 5.2 Hz, J = 8.6 Hz, J = 13.9 Hz, IH), 1.28 (ddd, J = 5.3, J = 9.4 Hz, J = 14.3 Hz, IH), 0.91 (dd, J = 3.3 Hz, J = 6.6 Hz, 6H). MS (m/z) in CH3CN: [M+Na] calculated for C20H23BCl2N2NaO9, 539.1; found, 539.1.
Ixazomib citrate [USAN]
1,3,2-Dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- [ACD/Index Name]
1,3,2-Dioxaborolane-4,4-diacetic acid,2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-
1239908-20-3 [RN]
2,2′-{2-[(1R)-1-{[N-(2,5-Dichlorbenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4,4-diyl}diessigsäure [German] [ACD/IUPAC Name]
2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacetic acid [ACD/IUPAC Name]
2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diacetic acid
2-[4-(carboxymethyl)-2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methyl-butyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
Acide 2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-méthylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacétique [French] [ACD/IUPAC Name]
MLN9708
UPDATES………..
Ixazomib (trade name Ninlaro) is a drug for the treatment of multiple myeloma, developed by Takeda Pharma. It acts as aproteasome inhibitor and has orphan drug status in the US. In November 2015, the U.S. Food and Drug Administration approved ixazomib for use in combination with lenalidomide and dexamethasone for the treatment of multiple myeloma after at least one prior therapy.[2]
Mechanism
Ixazomib is a peptide analogue that reversibly inhibits the protein proteasome subunit beta type-5 (PSMB5), which is part of the 20Sproteasome complex.[3]
Chemistry

Ixazomib citrate—a prodrug for ixazomib
U.S. FDA Approves Takeda’s NINLARO® (ixazomib), the First and Only Oral Proteasome Inhibitor to Treat Multiple Myeloma
NINLARO Provides a New Option for Patients Living with Multiple Myeloma Who Have Received at Least One Prior Therapy
Cambridge, Mass. and Osaka, Japan, November 20, 2015 – Takeda Pharmaceutical Company Limited (TSE: 4502) today announced that the U.S. Food and Drug Administration (FDA) has approved NINLARO®(ixazomib) capsules, the first and only oral proteasome inhibitor, indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is a once-weekly pill. More information is available at www.NINLARO.com.
Takeda submitted a New Drug Application for NINLARO to the FDA in July 2015, and in September NINLARO was granted Priority Review status with a PDUFA date of March 10, 2016, reflecting the profound and continuing unmet need for new treatments for multiple myeloma, a devastating, relapsing and incurable rare cancer.
“With the approval of NINLARO, we can now offer patients a once-weekly oral proteasome inhibitor as part of a highly active triplet therapy,” said Paul Richardson, M.D., Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center Institute Physician at Dana-Farber Cancer Institute, and investigator for TOURMALINE-MM1, the pivotal Phase 3 trial on which today’s approval is based. “We, as investigators of the TOURMALINE-MM1 trial, felt it was vital to conduct a comprehensive ‘real world’ evaluation of this combination that included some of the most common patient types in the relapsed/refractory multiple myeloma setting, such as older patients, patients with moderate renal impairment, light chain disease, and high risk cytogenetics. Further, we treated patients until disease progression to determine the sustainability of NINLARO in treating their relapsed/refractory disease. The TOURMALINE-MM1 data demonstrate convincingly that oral NINLARO-based triplet treatment is effective at extending progression-free survival, over and above the clinical benefit seen with lenalidomide and dexamethasone, with a tolerable safety profile.”
“We introduced the first proteasome inhibitor for multiple myeloma, VELCADE, into clinical research approximately 20 years ago. Since that time, we’ve significantly advanced scientific understanding of this rare cancer, culminating in the introduction of NINLARO,” said Andy Plump, M.D., Ph.D, Takeda Chief Medical and Scientific Officer. “NINLARO is an entirely new molecule that offers the efficacy of this proteasome inhibitor in a convenient once-weekly pill with a tolerable safety profile. Takeda is delighted to bring this significant innovation to multiple myeloma patients today, and we continue to examine the potential of NINLARO through a robust clinical development program.”
Dr. Brian Durie, Chairman of the International Myeloma Foundation, said, “The IMF is pleased by the approval of ixazomib. This opens the door for a fully oral proteasome inhibitor-based triplet combination therapy. Having worked in multiple myeloma for decades, I’ve seen notable progress, yet significant unmet needs remain. With today’s approval, we now have another attractive option for many patients living with multiple myeloma.”
The FDA approval of NINLARO is based on results from the TOURMALINE-MM1 Phase 3 clinical trial, the first double-blind, placebo-controlled trial with a proteasome inhibitor. TOURMALINE-MM1 is the first of five ongoing Phase 3 clinical trials with study results available. The TOURMALINE program has enrolled approximately 3,000 patients to date in 40 countries. Data from the NINLARO Phase 3 TOURMALINE-MM1 pivotal trial will be presented at the upcoming 57th Annual Meeting of the American Society of Hematology on December 7, 2015.
“The approval of ixazomib offers a much-needed additional option in the multiple myeloma treatment landscape. It is developments such as these that help us to better understand the disease and provide continued hope for patients,” said Kathy Giusti, Founder and Executive Chairman of the Multiple Myeloma Research Foundation (MMRF). “A cancer diagnosis today is different from what it was just a few years ago and it’s exciting to see continued progress. As a patient, I understand the urgent need for advancing research through partnerships that bring new treatment options, as we’ve done with Takeda.”
“NINLARO is a first-of-its-kind innovation that is supported by a global development program, unprecedented for us at Takeda Oncology, and we would like to express our immense appreciation for all patients involved for their incredible strength and invaluable participation. The introduction of NINLARO marks an important step forward, as its efficacy and safety profile – coupled with its completely oral administration – potentially can reduce some logistical burdens, and help enable patients to reap the full benefits of this sustainable therapy,” explained Christophe Bianchi, M.D., President, Takeda Oncology. “As part of our unwavering 20-year commitment, Takeda will continue to pursue advances for these patients, and we look forward to introducing and expanding access to NINLARO in other markets around the world.”
About the TOURMALINE-MM1 Trial
TOURMALINE-MM1 is an international, randomized, double-blind, placebo-controlled clinical trial of 722 patients, designed to evaluate NINLARO plus lenalidomide and dexamethasone compared to placebo plus lenalidomide and dexamethasone in adult patients with relapsed and/or refractory multiple myeloma. Results showed NINLARO is effective in extending Progression Free Survival (PFS) and has a manageable safety profile. The trial achieved its primary endpoint and demonstrated a clinically meaningful and statistically significant prolongation in PFS at this analysis, which showed that patients treated in the NINLARO arm lived without their disease worsening for a significantly longer time compared to patients in the control arm. Patients continue to be treated to progression in this trial and will be evaluated for long term outcomes.
In the TOURMALINE-MM1 trial, the most common adverse reactions (≥20%) in patients receiving NINLARO included diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting and back pain. Serious adverse reactions reported in ≥2% patients included thrombocytopenia (2%) and diarrhea (2%).
Efficacy and safety data were reviewed by an Independent Data Monitoring Committee (IDMC), who recommended the study be continued in blinded fashion to allow further maturation of long term outcomes, including overall survival (OS) and long-term safety.
About NINLARO (ixazomib) capsules
NINLARO (ixazomib) is the first and only oral proteasome inhibitor indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is administered orally, once-weekly on days 1, 8, and 15 of a 28-day treatment cycle. NINLARO is currently under review by the European Medicines Agency (EMA) and was granted an accelerated assessment by the Committee for Medicinal Products for Human Use (CHMP). NINLARO also received Breakthrough Therapy status by the U.S. FDA for relapsed or refractory systemic light-chain (AL) amyloidosis, a related ultra orphan disease, in 2014.
The TOURMALINE clinical development program further reinforces Takeda’s ongoing commitment to developing innovative therapies for people living with multiple myeloma worldwide and the healthcare professionals who treat them. Five global Phase 3 trials are ongoing:
- TOURMALINE-MM1, investigating ixazomib vs. placebo, in combination with lenalidomide and dexamethasone in relapsed and/or refractory multiple myeloma
- TOURMALINE-MM2, investigating ixazomib vs. placebo, in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma
- TOURMALINE-MM3, investigating ixazomib vs. placebo as maintenance therapy in patients with newly diagnosed multiple myeloma following induction therapy and autologous stem cell transplant (ASCT)
- TOURMALINE-MM4, investigating ixazomib vs. placebo as maintenance therapy in patients with newly diagnosed multiple myeloma who have not undergone ASCT
- TOURMALINE-AL1, investigating ixazomib plus dexamethasone vs. physician choice of selected regimens in patients with relapsed or refractory AL amyloidosis
In addition to the TOURMALINE program, a large number of investigator initiated studies are evaluating ixazomib for patients globally.
For additional information on the ongoing Phase 3 studies please visit www.clinicaltrials.gov. To learn more about NINLARO, please visit www.NINLARO.com or call 1-844-N1POINT (1-844-617-6468).
![]()
References
- “Ninlaro (ixazomib) Capsules, for Oral Use. Full Prescribing Information” (PDF). NINLARO (ixazomib) For Healthcare Professionals. Takeda Pharmaceutical Company Limited Cambridge, MA 02139. Retrieved 21 November 2015.
- “FDA Okays Ixazomib, Another Multiple Myeloma Drug”. November 20, 2015.
- KEGG: Ixazomib
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N2-(2,5-Dichlorobenzoyl)-N-[(1R)-1-(dihydroxyboryl)-3-methylbutyl]glycinamide
|
|
| Clinical data | |
| Trade names | Ninlaro |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 58%[1] |
| Protein binding | 99% |
| Metabolism | hepatic, CYP3A4 (42%),CYP1A2 (26%) and others |
| Biological half-life | 9.5 days |
| Excretion | urine (62%), feces (22%) |
| Identifiers | |
| CAS Number | 1072833-77-2 |
| ATC code | L01XX50 |
| PubChem | CID 25183872 |
| ChemSpider | 25027391 |
| UNII | 71050168A2 |
| KEGG | D10130 |
| ChEBI | CHEBI:90942 |
| Synonyms | MLN2238 |
| Chemical data | |
| Formula | C14H19BCl2N2O4 |
| Molar mass | 361.03 g·mol−1 |

//////////
see….http://apisynthesisint.blogspot.in/2016/02/takedas-ixazomib-multiple-myeloma-drug.html
The FDA has opened the inside track to Novartis’ experimental lung cancer drug, LDK378, which gained “Breakthrough Therapy” designation

The FDA has opened the inside track to Novartis’ experimental lung cancer drug, which gained “Breakthrough Therapy” designation that speeds the development and review schedules for new treatments. The Swiss drug giant plans to file for approval the drug, now in mid-stage clinical trials, in early 2014. Since clinical development began in 2011, the program has advanced with lightning speed compared with those that take 10 years or so to trial before submitted for approval.
While there are no guarantees of an FDA approval for Novartis’ compound, code-named LDK378, the “breakthrough” tag provides an early nod to the potential of the candidate to improve treatment for patients with metastatic non-small cell lung cancer with anaplastic lymphoma kinase (ALK) mutations.
The “breakthrough” designation is also important because Novartis’ compound and others with the coveted status have a shot to be approved by the FDA without completing all three phases of clinical trials typically required before an approval decision.
Novartis’ LDK378 joined the “breakthrough” club after showing an 80% response rate in patients studied in Phase I trial of 88 subjects with advanced cases of ALK-positive NSCLC. The company has already begun a pair of Phase II studies of the compound for patients with the same kind of ALK-positive cancers, which account for about 3% to 8% of cases of NSCLC. And plans call for kicking off Phase III development of the new drug later this year.
“LDK378 is a strong example of our research approach, which focuses on identifying the underlying cause of disease pathways,” said Alessandro Riva, Novartis’ global head of oncology development, in a statement. “This Breakthrough Therapy designation will allow us to collaborate more closely with the FDA and potentially to expedite the availability of an important new treatment option for patients with ALK+ NSCLC.”
Biogen submits haemophilia A drug to FDA

Mar 14 2013
Biogen Idec has filed the first long-lasting Factor VIII treatment for haemophilia A with the US Food and Drug Administration.
The US biotech major has submitted recombinant factor VIII Fc fusion protein (rFVIIIFc), the first haemophilia A product candidate “in a new class of long-lasting clotting factor therapies being developed with the goal of reducing the burden of treatment for this condition”. If approved, rFVIIIFc will be the first major advance in haemophilia A treatment in more than two decades, Biogen claims.
The regulatory submission is based on results from A-LONG, the largest Phase III study in haemophilia A to date. Glenn Pierce, Biogen’s head of global medical affairs, noted that in that trial, patients were able to inject rFVIIIFc once-weekly to twice-weekly, “which creates the potential for those currently on prophylactic treatment to reduce injections by 50 to 100 per year”. Moreover, patients currently treating bleeding episodes could potentially dose once per week “and maintain significant protection from bleeding with about the same total number of injections each year they use to treat bleeding episodes today”, he added.
Earlier this month, the FDA accepted for review the company’s BLA for its factor IX candidate, rFIXFc, for use in patients with haemophilia B.
Links
Phase III trial of Merck’s Vytorin passes vital safety test
mar 13 2013
Merck & Co’s stock enjoyed a boost yesterday after it revealed it has been given permission to continue a late-stage trial of its cholesterol buster Vytorin.
The Whitehouse Station, New Jersey-based firm must have a breathed a sigh of relief when the Data Safety Monitoring Board issued a green light for the Phase III IMPROVE for a second time, having found no significant safety concerns raised by the data.
After an earlier planned review of data last year, the Board, rather unusually, said it would undertake a second interim analysis at a later date, which had led to some concerns that there may be issues that could lead to the trial being halted, according to media reports.
However, it seems these fears are unfounded at this point, as the18,000-plus patient study – which is designed to determine whether Vytorin is more effective at reducing the risk of heart attack, stroke and death in patients with heart disease than simvastatin alone – has been cleared to conclude.
The drugmaker said the trial should finish in September next year, and it will no doubt be hoping for a positive outcome to prove the benefits of Vytorin – a combination of the generic simvastatin and the still-patented Zetia (ezetimibe) – and breathe a little new life into its heart franchise.
Citi Investment Research analyst Andrew Baum, however, expressed doubt in a research note the final analysis will show Merck’s drug is more effective than generic competition, according to the Associated Press.
 |
|
|---|---|
 |
|
| Combination of | |
| Ezetimibe | via Niemann-Pick C1-Like 1 protein |
| Simvastatin | Statin HMG-CoA reductase inhibitor |
Cangrelor, AR-C69931MX Shows Improvement Over Plavix in Phase III Trial

Cangrelor, AR-C69931MX
[dichloro-[[[(2R,3S,4R,5R)-3,4-dihydroxy-5-[6-(2-methylsulfanylethylamino)-2-(3,3,3-trifluoropropylsulfanyl)purin-9-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]methyl]phosphonic acid
N-[2-(Methylthio)ethyl]-2-[(3,3,3-trifluoropropyl)thio]-5¢-adenylic acid monoanhydride with (dichloromethylene)bis[phosphonic acid]
163706-06-7 cas no
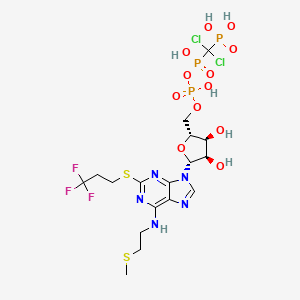 Cangrelor
CangrelorUPDATE
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Kengreal (cangrelor)
MAR 09, 2013
The Medicines Company said yesterday it will pursue marketing approvals for its anti-clotting drug candidate Cangrelor after it met its primary efficacy endpoint in a Phase III clinical trial of improvement compared with Plavix (clopidogrel).
The intravenous small molecule antiplatelet agent reduced by 22% the likelihood of patients experiencing death, myocardial infarction, ischemia-driven revascularization, or stent thrombosis within 48 hours of taking it—to 4.7% from 5.9% of subjects randomized during CHAMPION PHOENIX. The Phase III trial compared Cangrelor to oral Plavix in 11,145 patients undergoing percutaneous coronary intervention.
Cangrelor also showed a 38% reduction (0.8% compared with 1.4%) over Plavix in the likelihood of patients experiencing the key secondary endpoint, incidence of stent thrombosis at 48 hours.
Cangrelor is designed to prevent platelet activation and aggregation that leads to thrombosis in acute care settings, including in patients undergoing percutaneous coronary intervention. During CHAMPION PHOENIX, Cangrelor made its best showing in patients with Q-wave myocardial infarction (QMI), lowering by 39% (to 0.2% compared with 0.3%) the incidence of QMI. Cangelor’s most disappoint showing was its inability to lower the odds of death compared with Clopidogrel; both drugs showed a likelihood of 0.3%.
“Our next step is to submit for market approvals in the U.S. and Europe. We anticipate submitting these data for a new drug application to the U.S. Food and Drug Administration in the second quarter with findings of prior trials, including the BRIDGE trial in patients awaiting open heart surgery,” Simona Skerjanec, PharmD, senior vp and innovation leader for antiplatelet therapies at The Medicines Company, said in a statement.
Cangrelor is a P2Y12 inhibitor under investigation as an antiplatelet drug[1] for intravenous application. Some P2Y12 inhibitors are used clinically as effective inhibitors of adenosine diphosphate-mediated platelet activation and aggregation.[1] Unlike clopidogrel (Plavix), which is a prodrug, cangrelor is an active drug not requiring metabolic conversion.
Poor interim results led to the abandonment of the two CHAMPION clinical trials in mid 2009.[2] The BRIDGE study, for short term use prior to surgery, continues.[3] The CHAMPION PHOENIX trial was a randomized study of over 11,000 patients published in 2013. It found usefulness of cangrelor in patients getting cardiac stents. Compared with clopidogrel given around the time of stenting, intravenous ADP-receptor blockade with cangrelor significantly reduced the rate of stent thrombosis and myocardial infarction.[4] Reviewers have questioned the methodology of the trial.[5]

One particularly preferred example of a reversible, short-acting P2Y12 inhibitor is cangrelor. Cangrelor is a potent, direct, and reversible antagonist of the platelet P2Y12 receptor. Cangrelor has a half-life of approximately less than 10 minutes, allowing for a return to normal platelet function in a very short period of time upon discontinuation of the drug. By reducing the need for a compound to be metabolized for activity, and by having a relatively short half-life, reversible, short-acting P2Y12 inhibitors are considered “reversible,” meaning that full platelet functionality may return rather quickly as compared to thienopyridines.
The binding of cangrelor to the P2Y12 receptor inhibits platelet activation as well as aggregation when mediated in whole or in part via this receptor. Cangrelor can be derived completely from synthetic materials, and is an analogue of adenosine triphosphate (ATP). ATP is a natural antagonist of the P2Y12 receptor sites and is found in humans.
The chemical structure for cangrelor is depicted below as Formula I.

Cangrelor is clinically well tolerated and safe and has no drug-drug interaction with aspirin, heparin or nitroglycerin. Unlike orally dosed thienopyridines, cangrelor can be administered intravenously and binds directly to P2Y12 receptor sites of platelets. In each of the embodiments of the present invention, the term “cangrelor” encompasses the compound of Formula I as well as tautomeric, enantiomeric and diastereomeric forms thereof, and racemic mixtures thereof, other chemically active forms thereof, and pharmaceutically acceptable salts of these compounds, including a tetrasodium salt. These alternative forms and salts, processes for their production, and pharmaceutical compositions comprising them, are well known in the art and set forth, for example, in U.S. Pat. No. 5,721,219. Additional disclosure relevant to the production and use of cangrelor may be found in U.S. Pat. Nos. 5,955,447, 6,130,208 and 6,114,313, as well as in U.S. Appln. Publication Nos. 2006/0270607 and 2011/0112030.
Invasive procedures means any technique where entry to a body cavity is required or where the normal function of the body is in some way interrupted by a medical procedure and/or treatment that invades (enters) the body, usually by cutting or puncturing the skin and/or by inserting instruments into the body. Invasive procedures can include coronary artery bypass grafting (CABG), orthopedic surgeries, urological surgeries, percutaneous coronary intervention (PCI), other general invasive procedures, such as endarterectomy, renal dialysis, cardio-pulmonary bypass, endoscopic procedures or any medical, surgical, or dental procedure that could result in excessive bleeding or hemorrhage to the patient.
Perioperative means the period of a patient’s invasive procedure which can occur in hospitals, surgical centers or health care providers’ offices. Perioperative includes admission, anesthesia, surgery, to recovery.
Thrombosis is the formation of a blood clot (thrombus) inside a blood vessel obstructing the flow of blood through the circulatory system. When a blood vessel is injured, the body uses platelets and fibrin to form a blood clot to prevent blood loss. Some examples of the types of thrombosis include venous thrombosis which includes deep vein thrombosis, portal vein thrombosis, renal vein thrombosis, jugular vein thrombosis, Budd-Chiari syndrome, Paget-Schroetter disease, cerebral venous sinus thrombosis, cerebral venous sinus thrombosis and arterial thrombosis which includes stroke and myocardial infarction.
The compound cangrelor from the Medicines Company is represented by the structure

 OR
ORRN: 163706-36-3



…………
J. Med. Chem., 1999, 42 (2), pp 213–220
http://pubs.acs.org/doi/full/10.1021/jm981072s
10l (AR-C69931MX)
N6–(2-Methylthioethyl)-2-(3,3,3-trifluoropropylthio)-5‘-adenylic Acid, Monoanhydride withDichloromethylenebis(phosphonic acid) (10l). Prepared as the triammonium salt in 4% yield from 3l: 1H NMR δ(D2O) 8.30 (1H, s, H8), 5.97 (1H, d, J = 5.5 Hz, H1‘), 4.65 (1H, m, H2‘), 4.47 (1H, m, H3‘), 4.28 (1H, m, H4‘), 4.17 (2H, m, H5‘a and H5‘b), 3.67 (br s, NHCH2), 3.21 (2H, t, J = 7.6 Hz, SCH2), 2.72 (2H, t, J = 6.6 Hz, SCH2CH2CF3), 2.58 (2H, m, NCH2CH2), 2.04 (3H, s, SCH3);31P NMR δ(D2O) 8.80 (d, 1P, J = 18.6 Hz, Pγ), 0.42 (dd, 1P, J1 = 18.9 Hz, J2 = 28.9 Hz, Pβ), −9.41 (d, 1P, J = 29.0 Hz, Pα). Anal. (C17H34Cl2F3N8O12P3S2·3H2O) H, N, S; C: calcd, 23.16; found, 23.66.
References
- Cangrelor Attenuates Coated-Platelet Formation
- CHAMPION Trials With Cangrelor Stopped for Lack of Efficacy
- What Cangrelor Failure Means to Medicines
- Effect of Platelet Inhibition with Cangrelor during PCI on Ischemic Events (2013) Bhatt, DL etal. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMoa1300815 (published initially online).
- The Duel between Dual Antiplatelet Therapies (2013) Lange, RA and Hillis, LD. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMe1302504
- 15th European Federation for Medicinal Chemistry International Symposium on Medicinal Chemistry (Sept 6 1998, Edinburgh)1998,:Abst P.281
- Specific P2Y12 purinoceptor antagonist; inhibits ADP-induced platelet aggregation. Prepn: A. H. Ingall et al., WO 9418216 (1994 to Fisons); eidem, US 5721219 (1998 to Astra); and in vivo antithrombotic activity: idem et al., J. Med. Chem. 42, 213 (1999).
- In vivo antithrombotic effects in canine arterial thrombosis: J. Huang et al., J. Pharmacol. Exp. Ther. 295, 492 (2000).
- Mechanism of action study: A. Ishii-Watabe et al., Biochem. Pharmacol. 59, 1345 (2000).
- Clinical safety assessment and evaluation in acute coronary syndromes: R. F. Storey et al., Thromb. Haemostasis 85, 401 (2001); in angina pectoris and non-Q-wave myocardial infarction: F. Jacobsson et al., Clin. Ther. 24, 752 (2002).
- Clinical pharmacodynamics compared with clopidogrel: R. F. Storey et al., Platelets 13, 407 (2002).
- Review of clinical development: S. C. Chattaraj, Curr. Opin. Invest. Drugs2, 250-255 (2001).
- WO2013/103567 A2,
- Journal of Medicinal Chemistry, 1999 , vol. 42, 2 p. 213 – 220
Health Canada approval-aHUS Canada applauds approval of Soliris® (eculizumab) first treatment for fatal, ultra-rare disease affecting children and adults


CAS number 219685-50-4
Soliris is a formulation of eculizumab which is a recombinant humanized monoclonal IgG2/4;κ antibody produced by murine myeloma cell culture and purified by standard bioprocess technology. Eculizumab contains human constant regions from human IgG2 sequences and human IgG4 sequences and murine complementarity-determining regions grafted onto the human framework light- and heavy-chain variable regions. Eculizumab is composed of two 448 amino acid heavy chains and two 214 amino acid light chains and has a molecular weight of approximately 148 kDa.
TORONTO, March 7, 2013
Atypical Hemolytic Uremic Syndrome (aHUS) Canada is thrilled by Health Canada’s recent approval of Soliris® (eculizumab) for the treatment of patients with atypical Hemolytic Uremic Syndrome (aHUS), 1 a very rare, chronic and life-threatening genetic condition affecting fewer than 60 patients in Canada.
aHUS leaves a part of the immune system (known as the complement system) uncontrolled and always active. As a result, the immune system attacks the body’s unhealthy and healthy cells which can cause blood vessel damage, abnormal blood clotting 2,3 and progressive damage to the body’s major organs, leading to heart attack, stroke, kidney failure and death.4
The management of aHUS has relied on plasma infusion and plasma exchange therapies with variable results.5 These lifelong therapies are costly, painful and time-consuming, and have not been studied or approved for the treatment of aHUS.6 If kidney failure has already occurred as a result of aHUS, dialysis is required, though it is not a curative treatment.7 Within a year of diagnosis, over half of patients will need dialysis, will have irreversible kidney damage, or will not survive.6 The majority of patients progress to end-stage kidney failure within three years of diagnosis.8,9
With the approval of Soliris, aHUS patients and their families finally have a reason for hope.
Sonia DeBortoli knows all too well the destructive force of the disease. Sonia’s 11-year-old son Joshua was diagnosed with aHUS in March 2012 and experienced kidney failure, internal bleeding and a blood clot in his groin as a result. He endured several painful hours of daily dialysis and plasma therapy, and was on prednisone and oxygen. Then, a chance to join a clinical trial for Soliris restored Joshua’s health so that he no longer needed the other therapies.
“Our whole world changed when Joshua was given Soliris – we now believe he has a long and healthy future. He is back at school, taking karate lessons and playing soccer,” Sonia says. “We got our little boy back, he got his life back, and we want the same for anyone who has to deal with this rare and devastating disease.”
A groundbreaking treatment advance for aHUS patients
Soliris (eculizumab) is the first and only pharmaceutical treatment for aHUS, and is being hailed by experts worldwide as a critical breakthrough in treating the disease. It has been shown to significantly improve patients’ health and quality of life.10 In clinical trials, Soliris has been proven effective in preventing blood vessel damage and abnormal blood clotting,11,12 leading to remission and significant improvement in kidney function.5,4 Soliris has also allowed patients to discontinue dialysis and plasma exchange therapies.10
Soliris is also indicated, and proven safe and effective, for the treatment of another rare and life-threatening disorder called paroxysmal nocturnal hemoglobinuria (PNH).13 Canadians living with PNH already have access to Soliris through private health insurance and provincial drug plans.
Immediate and sustained access to treatment urgently needed
Now that this new treatment option has been approved for the small number of Canadians living with the devastating symptoms of aHUS, Soliris must be made immediately accessible to all aHUS patients whose lives depend on this treatment.
“We are so hopeful that the Common Drug Review will recognize the urgent need for access to Soliris, and that provincial governments will act swiftly to provide reimbursement to patients who are in urgent need of this life-saving treatment,” says Tracy MacIntyre, a founder of aHUS Canada whose daughter is living with aHUS. “Immediate access to the drug would have a profoundly positive impact on the few Canadians living with aHUS, while any delay in funding treatment could lead to devastating consequences.”
About aHUS Canada
aHUS Canada was formed in November 2012 to support Canadian patients and families living with aHUS. In addition to establishing a Canadian aHUS community, the group is committed to building public awareness and understanding of aHUS and advocating for the best possible care and treatment for patients. For more information, please visit http://www.ahuscanada.org.
Eculizumab (INN and USAN; trade name Soliris®) is a humanized monoclonal antibody that is a first-in-class terminal complement inhibitor and the first therapy approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH), a rare, progressive, and sometimes life-threatening disease characterized by excessive destruction of red blood cells (hemolysis).[1] It costs £400,000 ($US 600,000) per year per patient.[1]
Eculizumab also is the first agent approved for the treatment of atypical hemolytic uremic syndrome (aHUS), an ultra-rare genetic disease that causes abnormal blood clots to form in small blood vessels throughout the body, leading to kidney failure, damage to other vital organs and premature death.[2][3]
In clinical trials in patients with PNH, eculizumab was associated with reductions in chronic hemolysis, thromboembolic events, and transfusion requirements, as well as improvements in PNH symptoms, quality of life, and survival.[1][4][5][6] Clinical trials in patients with aHUS demonstrated inhibition of thrombotic microangiopathy (TMA),[7] the formation of blood clots in small blood vessels throughout the body,[1][3][4] including normalization of platelets and lactate dehydrogenase (LDH), as well as maintenance or improvement in renal function.[7]
Eculizumab was discovered and developed by Alexion Pharmaceuticals and is manufactured by Alexion. It was approved by the United States Food and Drug Administration (FDA) on March 16, 2007 for the treatment of PNH, and on September 23, 2011 for the treatment of aHUS. It was approved by the European Medicines Agency for the treatment of PNH on June 20, 2007, and on November 24, 2011 for the treatment of aHUS. Eculizumab is currently being investigated as a potential treatment for other severe, ultra-rare disorders
- Hillmen, Young, Schubert, P, N, J, et al (2006). “The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria”. N Engl J Med 355 (12): 1233–1243. doi:10.1056/NEJMMoa061648. PMID 16990386.
- Noris, Caprioli, Bresin, M, J, E, et al. (2010). “Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype”. Clin J Am Soc Nephrol 5: 1844–1859.
- Caprioli, Noris, Brioschi, J, M, S, et al (2006). “Genetics of HUS: the impact of MPC, CFH, and IF mutations on clinical presentation, response to treatment, and outcome”. Blood 108: 1267–1279.
- Hillman, Hall, Marsh, P, C, JC, et al (2004). “Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria”. N Eng J Med 350: 552–559.
- Ray, Burrows, Ginsberg, Burrows, JG, RF, JS, EA (2000). “Paroxysmal nocturnal hemoglobinuria and the risk of venous thrombosis: review and recommendations for management of the pregnant and nonpregnant patient”. Haemostasis 30: 103–107.
- Kelly, Hill, Arnold, RJ, A, LM, et al (2011). “Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival”. Blood 117: 6786–6792.
- .Soliris® (eculizumab) prescribing information (2011). Cheshire, CT: Alexion Pharmaceuticals. http://www.soliris.net/sites/default/files/assets/soliris)pi.pdf.
Phase 2, Sarepta Therapeutics, Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
s excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Clinical studies
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
References
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods.. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303.. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
Phase 2 Drug: Ustekinumab A monoclonal antibody against the p40 subunit of IL-12/23 Other Name: Stelara
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-12 and IL-23
|

Ustekinumab, CAS number 815610-63-0, is also known by it’s brand name Stelara, which is marketed by Janssen Biotech, Inc. Developed as a treatment for adults with moderate to severe plaque psoriasis
Rockefeller University, MAR 2013
http://clinicaltrials.gov/ct2/show/NCT01806662
Atopic dermatitis (AD) is a chronic disease associated with intense itching, which affects most aspects of everyday life in the majority of patients. Acute inflammation and extensor/facial involvement is common in infants, whereas chronic inflammation increases in prevalence with age, as do localization to flexures. AD has a complex background characterized by immune activation, increased epidermal thickness in chronic diseased skin, and defective barrier function. In normal, healthy skin, the outer layer of the epidermis, the stratum corneum is made up flattened dead cells called corneocytes held together by a mixture of lipids and proteins. The stratum corneum and, in particular, the lipid layer are vital in providing a natural barrier function that locks water inside the skin and keeps allergens and irritants out. In people with AD, the barrier function is defective, which leads to dry skin. As the skin dries out, it cracks allowing allergens and irritants to penetrate.
Mild AD can be controlled with emollients and topical medications. However, moderate to severe AD is extremely difficult to control and requires systemic treatment that is often unsatisfactory due to impracticality and lack of effectiveness. Only three therapeutic options exist for moderate to severe AD, including: 1) oral steroids 2) cyclosporine A (CsA), that is not widely used in the US as it is not FDA approved for AD and 3) ultraviolet phototherapy. Oral steroids and CsA treatments have major side effects and UV radiation therapy is highly inconvenient for patients. Several biologic medications, such as TNF-alpha inhibitors, are effective, convenient, and relatively safe therapies for psoriasis, but have thus far not shown efficacy in AD. Ustekinumab is a unique biologic medication that may specifically target AD.
The investigators study will determine whether there is a reversal of the skin thickness and the immune pathways involved in the disease during treatment with Ustekinimab and what specific immune cells are involved. The investigators are also interested to understand how the clinical reversal of the disease will correlate with tissue reversal of the disease.
Detailed Description:
In psoriasis, epidermal hyperplasia is driven by underlying immune activation, whether as a direct response to IL-20 family cytokines that induces hyperplasia and inhibits keratinocyte terminal differentiation or as an indirect response to immune-mediated injury to keratinocytes. The epidermal reaction in psoriasis is largely restored to normal with selective immune suppression. Hence, one might hypothesize that similar epidermal responses should occur in the presence of “generalized” cellular immune activation, in diseases with similar inflammatory infiltrate and epidermal hyperplasia, such as AD. In fact, psoriasis and AD share features of dense T-cells and dentritic cell infiltrates, as well as over-expression of IL-22 in skin lesions. These diseases also share similar epidermal hyperplasia in their chronic phases.
Work from the investigators group showed that IL-22 is a key cytokine in the pathogenesis of both AD and psoriasis. The investigators have demonstrated that in psoriasis, ustekinumab suppresses the production of IL-12, IL-23, and IL-22. Additionally, by RT-PCR the investigators demonstrated that the mRNA expression of p40 cytokine and the IL23R is up-regulated in AD as compared to both normal skin and psoriasis. The investigators therefore hypothesize that ustekinumab will suppress IL-22 and possibly also p40 production in AD lesions and reverse both the epidermal growth/differentiation defects and the underlying immune activation, and hence will suppress disease activity. Interestingly, p40 was also found to be significantly up-regulated in non-lesional AD skin as compared with normal skin.
Although AD is thought to be predominately a disease of Th2-type cells, in the chronic stage, there is large Th1 component. To date, the precise mechanism by which sequential activation of Th2 and Th1 cells in AD is achieved remains unknown. IL-12 induces the differentiation and maturation of human Th cells into Th1-type cells. Recent circumstantial evidence suggests that in AD patients IL-12 may facilitate a change from the Th2-type to a Th1 cytokine profile. IL-12 was recently shown to be highly elevated in pediatric AD and its levels were strongly associated with disease severity.
Expression of IL-12 p40 mRNA is significantly enhanced in lesional skin from AD, suggesting that the enhanced local production of IL-12 in dendritic cells and macrophages may be responsible for the increased production of IFN-γ in chronic lesions potentially suggesting that IL-12 may have a pivotal role in promoting inflammation in atopic dermatitis. Topical steroids which constitute a mainstay of therapy in AD are known to strongly down-regulate IL-12 expression, possibly also indicating that targeted anti IL-12 therapy might important role in treating AD.
Recently, the Th1/Th2 paradigm in autoimmunity and allergy has been revisited to include a role for a new population of IL-17-producing Th cells known as Th17. Th17 cells are characterized by the production of inflammatory cytokines such as IL-17A, IL-17F, IL-22, and IL-26. One of the key factors involved in naive Th-cell commitment to a Th17 phenotype is IL-23.
Patients with acute AD were found to have increased Th17 T-cells in peripheral blood by flow cytometry and intracellular cytokine staining 26 as well as by immunohistochemistry (IHC) in lesions. Since IL-23 is the major inducer of Th17 T-cells, as well as “T22” T-cells, neutralization of IL-23 could potentially result in both decreased Th17 signal in acute AD as well as decreased “T22/IL22″ signal. Therefore the investigators postulate that ustekinumab in AD will act both inhibiting the IL-12-dependent Th1 shift in chronic AD stage as well as the pathogenic IL-22/”T22” axis in this disease.
Ustekinumab [1] (INN, experimental name CNTO 1275, proprietary commercial name Stelara,[2] Centocor) is a human monoclonal antibody. It is directed against interleukin 12 and interleukin 23, naturally occurring proteins that regulate the immune system and immune-mediated inflammatory disorders.[3]
In two Phase III trials for moderate to severe psoriasis, the longest >76 weeks, ustekinumab was safe and effective.[4][5]
A third Phase III trial, ACCEPT, compared the efficacy and safety of ustekinumab with etanercept in the treatment of moderate to severe plaque psoriasis.[6] This trial found a significantly higher clinical response with ustekinumab over the 12-week study period compared to high-dose etanercept.[6] It also demonstrated the clinical benefit of ustekinumab among patients who failed to respond to etanercept.[6]
Ustekinumab is approved in Canada, Europe and the United States to treat moderate to severe plaque psoriasis.[7]
As of November 2009, the drug is being investigated for the treatment of psoriatic arthritis.[8][9] It has also been tested in Phase II studies for multiple sclerosis[10] and sarcoidosis, the latter versus golimumab (Simponi).[11]
- Cingoz, Oya (2009). “Ustekinumab”. MAbs 1 (3): 216–221. doi:10.4161/mabs.1.3.8593. PMC 2726595. PMID 20069753.
- ^ European Medicines Agency, 20 November 2008, http://www.emea.europa.eu/pdfs/human/opinion/Stelara_58227008en.pdf
- ^ Reddy M, Davis C, Wong J, Marsters P, Pendley C, Prabhakar U (May 2007). “Modulation of CLA, IL-12R, CD40L, and IL-2Ralpha expression and inhibition of IL-12- and IL-23-induced cytokine secretion by CNTO 1275”. Cell. Immunol. 247 (1): 1–11. doi:10.1016/j.cellimm.2007.06.006. PMID 17761156.
- ^ Leonardi CL, Kimball AB, Papp KA, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1)”. Lancet 371 (9625): 1665–74. doi:10.1016/S0140-6736(08)60725-4. PMID 18486739.
- ^ Papp KA, Langley RG, Lebwohl M, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2)”. Lancet 371 (9625): 1675–84. doi:10.1016/S0140-6736(08)60726-6. PMID 18486740.
- ^ a b c Griffiths C, Strober B, van de Kerkhof P et al. (2010). “Comparison of Ustekinumab and Etanercept for Moderate-to-Severe Psoriasis”. N Engl J Med 362 (2): 118–28. doi:10.1056/NEJMoa0810652. PMID 20071701.
- ^ Medarex to Receive Milestone Payment for Approval of STELARA(TM) (Ustekinumab) for the Treatment of Moderate to Severe Plaque Psoriasis
- ^ ClinicalTrials.gov NCT00267956 A Study of the Safety and Efficacy of CNTO 1275 in Patients With Active Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT01009086 A Study of the Safety and Efficacy of Ustekinumab in Patients With Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT00207727 A Safety and Efficacy Study of CNTO1275 in Patients With Multiple Sclerosis
- ^ ClinicalTrials.gov NCT00955279 A Study to Evaluate the Safety and Effectiveness of Ustekinumab or Golimumab Administered Subcutaneously (SC) in Patients With Sarcoidosis
- ^ http://www.empr.com/stelara-approved-for-moderate-to-severe-psoriasis/article/149760/
- ^ a b Centocor 12/19/08 Press Release, http://www.centocor.com/centocor/i/press_releases/FDA_ISSUES_COMPLETE_RESPONSE_LETTER_TO_CENTOCOR_FOR_USTEKINUMAB_BIOLOGIC_LICENSE_APPLICATION_
- ^ Johnson LL. “Study: Drug for serious psoriasis tops competition” The Associated Press. 18 Sept 2008.[dead link]
- ^ Wild, David (November 2011), “Novel IL-12/23 Antagonist Shows Potential in Severe Crohn’s”, Gastroenterology & Endoscopy News 62 (11), retrieved 2011-12-04
- ^ a b c Weber J, Keam SJ (2009). “Ustekinumab”. BioDrugs 23 (1): 53–61. doi:10.2165/00063030-200923010-00006. PMID 19344192.
- ^ Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH (September 2008). “Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study”. Lancet Neurol 7 (9): 796–804. doi:10.1016/S1474-4422(08)70173-X. PMID 18703004.
- ^ “Important Safety Information”. STELARA® (ustekinumab). Janssen Biotech.
External links
- Centocor Ortho Biotech official site
- CNTO 1275 research studies registered with U.S. National Institutes of Health:
- ClinicalTrials.gov NCT00207727 Phase II Study on Multiple Sclerosis
- ClinicalTrials.gov NCT00320216 Phase II Study on Psoriasis
- ClinicalTrials.gov NCT00267969 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00307437 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00267956 Phase II Study on Psoriatic Arthritis
- Sylvester, Bruce (2006-03-06). “CNTO 1275 Shows Efficacy for Psoriasis: Presented at AAD”. Doctor’s Guide Publishing. Retrieved 2007-01-25.

{kind=link}