


如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » Posts tagged 'organic chemistry' (Page 18)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide,
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl] -N’-propylsulfamide
CAS NO 441798-33-0
ACT-064992, Opsumit,UNII-Z9K9Y9WMVL
Mechanism of Action: Endothelin receptor antagonist (ERA)
Date of Approval: October 18, 2013(US)
Indication: Pulmonary Hypertension (PAH)
Company: Actelion Pharmaceuticals Ltd
PCT patent application: WO2002053557
FDA N204410, MACITENTANTABLET; ORAL10MG, OPSUMIT, ACTELION PHARMS LTD
Macitentan is achiral
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan (ACT-064992) is a tissue-targeting dual ET(A)/ET(B) endothelin (ET) receptor antagonist designed for tissue targeting. Macitentan inhibited ET-1-induced contractions in isolated endothelium-denuded rat aorta (ET(A) receptors) and sarafotoxin S6c-induced contractions in isolated rat trachea (ET(B) receptors). In diabetic rats, chronic administration of macitentan decreased blood pressure and proteinuria and prevented end-organ damage. Treatment with macitentan enhanced the cytotoxicity mediated by paclitaxel as measured by the degree of apoptosis in tumor cells and tumor-associated endothelial cells. A Phase III clinical trial of macitentan was successfully completed in 2012.


Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.

Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

SYNTHESIS
The synthesis begins with the reaction of chlorosulfonyl isocyanate (1) (dissolved in dichloromethane at 0 ° C) with one equivalent of tert-butanol. This produces a by BOC protected Aminosulfonylchlorid (2). With one equivalent of n-propylamine (dissolved in 3 eq. Of triethylamine, dichloromethane, at 0 ° C, RT 16 h) is produced by a hydrochloric acid elimination BOC-protected sulfamide (3). This is dissolved in 5 M HCl and dioxane (4-8 h), the BOC protecting group is cleaved. The sulfamide formed (4) is potassium tert-butoxide-(dissolved in MeOH, 3h) is converted to the potassium salt (5). Tert-butoxide potassium acts as a very strong base for deprotonation. This sulfamide potassium salt reacts with the nucleophilic substituents on the heteroaromatic Dichlorpyrimidinderivat (6) (dissolved in dimethyl sulfoxide, at room temperature, RT 42-72 h) under KCl-cleavage to a Monochlorpyrimidin intermediate (7). By treatment with ethylene glycol (dissolved in dimethyl ether, potassium-tert-butoxide,), the ethylene glycol side chain is generated (8). With 2-chloro-5-bromo-pyrimidine (dissolved in tetrahydrofuran, close, at 60-75 ° C) is formed under elimination of HCl in an S N 1 reaction Macitentan (9)…………Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .

Synthesis of Macitentan
…………………………………………….
SYNTHESIS


YOU CAN READ AT YAOPHA.COM, lovely site to see for drugs


如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
………………………….
SYNTHESIS
(WO2006/051502A2, JMC2012, 7849). Chlorosulfonyl isocyanate ( 1 ) reaction with tert-butyl alcohol 2 , which is then reacted with n-propylamine 3 . 3 de-boc protected through the acid after reaction with potassium t-butoxide 4 . Another compound 5 with NaH after acidic protons off with dimethyl carbonate ( 6 ) to obtain 7 . 7 and formamidine hydrochloride ( 8 ) to ring chlorinated later POCl3 9 . 9 and 4 SNAr reaction occurs 10 . 10under basic conditions with ethylene glycol SNAr reaction occurs again in alkaline conditions with11 SNAr reaction occurs MACITENTAN.

………………………
http://www.google.com/patents/WO2014155304A1?cl=en
LC-MS (Agilent MS detector G1956B with Agilent 1200 Binary Pump and DAD).
Parameters of the LC-MS method:
Injection volume: 2 |jL
Column: Kinetex C18, 2.6 μιη, 2.1 x 50 mm
Column flow rate: 1 mL/min
Eluents: Eluent A: water + 0.08% TFA
Eluent B: MeCN + 0.012% TFA
Gradient: 2.0 min 95% B
2.8 min 95% B
3.0 min 5% B
Temperature: 40°C Detector wavelength 210 nm

Preparation B: N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]- 4-pyrimidinyl] -N’-propylsulfamide (macitentan):
N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)propane- 1-sulfamide (200 g; 0.46 mol; see Example 2 or 3) and 5-bromo-2-chloropyrimidine (117 g; 0.60 mol; 1.3 eq) were dissolved in toluene (3 L) and DMF (400 mL). The reaction mixture was warmed up to 50°C and toluene (approx. 400 mL) was distilled our under reduced pressure. The mixture was cooled to 0 °C and tBuOK (156 g, 3 eq, 1.38 mol) was added portionwise. It was stirred at 20 °C for 1 h. Water (1 L) was added and the pH of the solution was adjusted to 3-5 using 33% aq. HC1. The mixture was heated to 50°C and the layers were separated. The org. phase was treated with charcoal at 50°C and filtered over Celite. The filter cake was rinsed with toluene. At 50°C, water (1 L) was added to the org. layer. The layers were separated. The org. layer was concentrated under reduced pressure to a total volume of 1 L and cooled to 0°C. The solid obtained was filtered off. It was rinsed with toluene and MeOH. The crude material was suspended in EA (1 L) and heated to 50°C. 300 mL of EA were distilled out and MeOH (400 mL) was added. The suspension was cooled down to 0°C. The solid was filtered off, rinsed with MeOH and dried under reduced pressure to afford the title compound as a white solid (225 g; 83% yield).

……………………
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm3009103

Starting from the structure of bosentan (1), we embarked on a medicinal chemistry program aiming at the identification of novel potent dual endothelin receptor antagonists with high oral efficacy. This led to the discovery of a novel series of alkyl sulfamide substituted pyrimidines. Among these, compound 17 (macitentan, ACT-064992) emerged as particularly interesting as it is a potent inhibitor of ETA with significant affinity for the ETB receptor and shows excellent pharmacokinetic properties and high in vivo efficacy in hypertensive Dahl salt-sensitive rats. Compound 17 successfully completed a long-term phase III clinical trial for pulmonary arterial hypertension
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (17)
……………
WO 2015004265 click
Example 3 : N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)pr opane- 1- sulfamide (reaction in and work-up with MIBK):
EG (124 mL, 3.7 mol, 6.0 eq.) was added to a warm (40-50°C) suspension of the compound of Preparation A (150 g, 0.37 mol) in MIBK (600 mL). Solid KOtBu (114 g, 1.11 mol, 3.0 eq.) was added portionwise so that IT < 60°C. The mixture was stirred for
2- 3 h at 100-105°C. After completion of the reaction (LC-MS control), it was cooled to 50 °C. A 40%) aq. solution of citric acid monohydrate (300 mL) was added until pH 4 was reached. The layers were separated. The org. phase was washed with water (450 mL) and the layers were separated. Water (450 mL) was added and the mixture was warmed to 50°C. It was stirred at 50°C for 5 min. The layers were separated. The org. phase was concentrated under vacuum at 50°C until 200 mL of MIBK were removed. Hept (800 mL) was added dropwise at 70-75°C until turbidity was observed. The mixture was seeded with an analytically pure sample of N-(5-(4-bromophenyl)-6-(2 hydroxy ethoxy)pyrimidin-4-yl)propane-l-sulfamide and stirred at 60-65°C for 30 min. It was allowed to cool to 5°C within 5 h. It was filtered off, rinsed with a cold MIBK/Hept mixture (300 mL, 1 : 1) and dried under vacuum at 50°C to yield the title compound as a white solid (121 g; 76% yield).
The product had NMR data equivalent to those reported in Bolli et al, J. Med. Chem. (2012), 55, 7849-7861. [M+H]+ = 430 and 432. LC-MS: tR = 1.46 min; purity: 98.4% a/a. Residual ethylene glycol (GC-FID): 530 ppm.
…….
CN 104447572 click
(l) Martin H. Bolli et al. Reported the synthesis of Marcy cefotetan follows:

[0008] The method W 5- (4- desert phenyl) -4,6-dichloro-chewing clever as a starting material, N- propyl amine Lai ugly bell in DMS0 as a reaction solvent, an alcohol bell as t a base under substitution reaction conditions, the reaction temperature needs of 24-7 to give


The intermediate compound 15, compound 15 in hexylene glycol dimethyl off as the reaction solvent, a tertiary alcohol under conditions with a strong base clock as hexanediol substitution reaction, l〇 (TC Reaction of 18-2 to give compound 17, Compound 17 was then reacted with 5-chloro-chewing desert -2 clever substitution reaction at tetraammine Qiao Nan as a reaction solvent, ammoniated axis as the alkali conditions, the reaction to give the final product of Marcy cefotetan The route every step the higher the yield, the experimental use of N- propyl amine Lai ugly bell hygroscopic, unstable and a long time before the two-step reaction, the reaction at the second step requires l〇 (TC high temperature 18-2 technology is not suitable for industrial production.
[0009] International Patent W02002 / 053557 discloses some preparation methods and other Massey cefotetan column derivative method at each step of the preparation of the reaction times are longer, some reactions up to 4 days, and the resulting intermediate are purified by column chromatography method is not suitable for industrial production.



[00 pairs (3) N- [5- (4- desert) -6-mouth – [(5-desert -2- chew clever-yl) oxy] hexyl oxy] -4-chewing clever yl] -N ‘- Lai ugly propyl amine (Formula I) Synthesis
[0036] Weigh 20gN-5- (4- desert) -6- (2-2- light hexyl group -) 4- chew clever group -N ‘- Lai ugly propyl amine, 200ml dried DMS0 added to 1L H jar, add 20g of alcohol t-clock was added in portions, then add 17. 7g5- desert – dichloro chew clever, 30-4 (TC reduction reaction, the reaction and the reaction solution. a 10% sample skillfully acid to adjust PH value 3 to 4, the reaction mixture was added to 1000ml water, olive mix, suction. suction Massey cefotetan get wet crude product 42g, 450ml of methanol was added at room temperature and then beating 20min, filtration and dried 45C to give white solid was dried under vacuum to give 23.2 Marcy cefotetan yield;.. 85%
[0037] The compound (Formula I) relating to the physical and chemical properties, spectroscopic data are as follows:
[0038] branded point; 135-136 ° C; we NMR (300MHz, DMS0) 5 (egg m):… 9 8 (s, lH), 8 7 (s, 2H), 8 5 (s, l H,) 7. 5 (s, 2H), 7. 2 (s, IH), 7. 1 (s, 2H,) 4. 7 (s, 2H), 4. 6 (s, 2H,) 2. 8 (s, 2H,), 1. 5 (m, 2H,), 0. 81 (m, 3H), MS Qiaoqiao m / z 589 ([M + Tin +).
…………
see
WO 2002053557
http://www.google.com/patents/WO2002053557A1?cl=en
………..


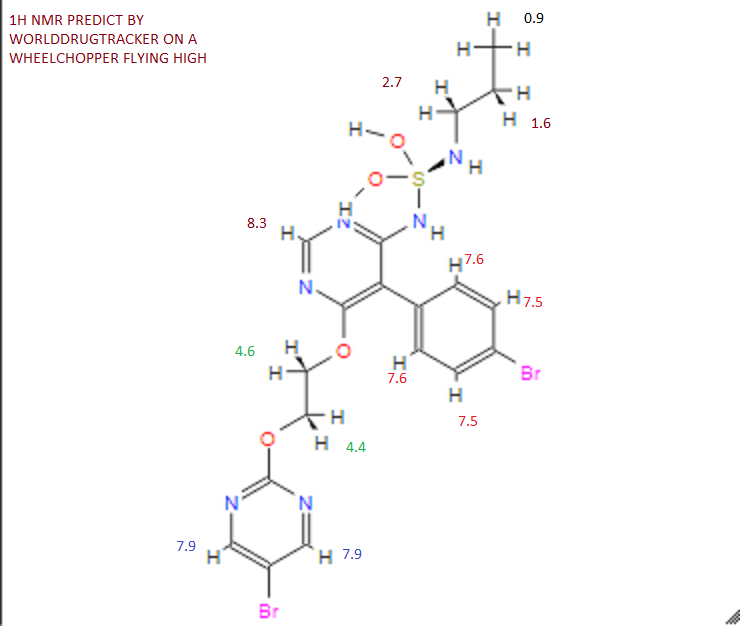
Assignment of the signals mentioned in the text of the H-NMR spectrum of the drug Macitentan
Solvent: CDCl 3
δ 8.51 (s, 2H, CH) 11 , 8.49 (s, 1 H, CH) 10 , 7.58 to 7.63 (m, 2H, CH) 9 , 7.16 to 7.21 ( m, 2H, CH) 8 , 6.88 (s, 1H, NH) 7 , 5.61 (t, J = 6.2 Hz, 1H, NH) 6 , 4.72 to 4.76 (m, 2H , CH 2 ) 5 , 4.62 to 4.66 (m, 2H, CH 2 ) 4 , 2.99 (q, J = 6.8 Hz, 2H, CH 2 ) 3 , 1.61 (h, J = 7.3 Hz, 2H, CH2 ) 2 , 0.97 (t, J = 7.4 Hz, 3H, CH 3 ) 1 . [Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .]

Solvent: CDCl 3
δ 11.6, 22.7, 46.1, 65.3, 65.9, 104.8, 112.4, 123.7, 128.0, 131.7, 133.0, 155.7, 156 , 4, 159.7, 163.5, 166.3. [ Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 . ]
NMR PREDICT BY ME
1H NMR PREDICT

13C NMR PREDICT BY ME


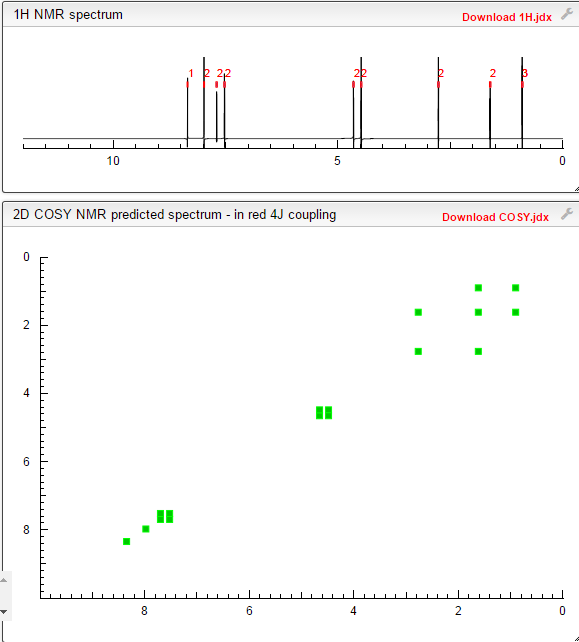
COSY PREDICT BY ME, WORLDDRUGTRACKER ON A WHEELCHOPPER SCALING NEW HEIGHTS
REFERENCES
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide
|
|
| Clinical data | |
| Trade names | Opsumit |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hydrolysis, oxidation (CYP3A4) |
| Excretion | 2/3 urine, 1/3 faeces |
| Identifiers | |
| CAS Registry Number | 441798-33-0 |
| ATC code | C02KX04 |
| PubChem | CID: 16004692 |
| ChemSpider | 13134960 |
| ChEBI | CHEBI:76607 |
| Synonyms | ACT-064992 |
| Chemical data | |
| Formula | C19H20Br2N6O4S |
| Molecular mass | 588.273 g/mol |
| Patent | Submitted | Granted |
|---|---|---|
| Sulfamides and their use as endothelin receptor antagonists [US7094781] | 2004-04-22 | 2006-08-22 |
| Sulfamides and their use as endothelin receptor antagonists [US7285549] | 2006-08-10 | 2007-10-23 |
| Stable Pharmaceutical Compositions Comprising a Pyrimidine – Sulfamide [US2008233188] | 2008-09-25 | |
| Combination Comprising Paclitaxel for Treating Ovarian Cancer [US2010311774] | 2010-12-09 | |
| Stable pharmaceutical compositions comprising a pyrimidine-sulfamide [US2010004274] | 2010-01-07 | |
| SULFONYLUREA MODULATORS OF ENDOTHELIN RECEPTOR [US2011082151] | 2011-04-07 | |
| ENDOTHELIN RECEPTOR ANTAGONISTS FOR EARLY STAGE IDIOPATHIC PULMONARY FIBROSIS [US2010022568] | 2007-04-12 | 2010-01-28 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2011136818] | 2011-06-09 | |
| Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor [US2009318459] | 2009-12-24 |
Patent and Exclusivity
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
|
|---|---|---|---|---|---|---|---|
| N204410 | 001 | US7094781 | Oct 12, 2022 | Y | Y | ||
| N204410 | 001 | US8268847 | Apr 18, 2029 | U – 1446 | |||
| N204410 | 001 | US8367685 | Oct 4, 2028 | Y | U – 1445 |
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N204410 | 001 | ODE | Oct 18, 2020 |
| N204410 | 001 | NCE | Oct 18, 2018 |
U1446 METHOD OF TREATING PULMONARY HYPERTENSION COMPRISING ADMINISTERING MACITENTAN IN COMBINATION WITH A COMPOUND HAVING PHOSPHODIESTERASE-5 INHIBITORY PROPERTIES
U1445 METHOD OF TREATING PULMONARY ARTERIAL HYPERTENSION BY ADMINISTERING A PHARMACEUTICAL COMPOSITION COMPRISING MACITENTAN AND A POLYSORBATE, WHERIN THE POLYSORBATE REPRESENTS 0.1 TO 1% OF THE WEIGHT OF SAID PHARMACEUTICAL COMPOSITION
OPSUMIT (macitentan) is an endothelin receptor antagonist. The chemical name of macitentan is N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide. It has a molecular formula of C19H20Br2N6O4S and a molecular weight of 588.27. Macitentan is achiral and has the following structural formula:
 |
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
OPSUMIT is available as a 10 mg film-coated tablet for once daily oral administration. The tablets include the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80, povidone, and sodium starch glycolate Type A. The tablets are film-coated with a coating material containing polyvinyl alcohol, soya lecithin, talc, titanium dioxide, and xanthan gum.
//////

BENAZEPRIL HYDROCHLORIDE, CAS NO 86541-74-4
Benazepril, brand name Lotensin (Novartis), is a medication used to treat high blood pressure (hypertension), congestive heart failure, and chronic renal failure. Upon cleavage of its ester group by the liver, benazepril is converted into its active form benazeprilat, a non-sulfhydryl angiotensin-converting enzyme (ACE) inhibitor.
Benazepril is available as oral tablets, in 5-, 10-, 20-, and 40-mg doses.
Benazepril is also available in combination with hydrochlorothiazide, under the trade name Lotensin HCT, and with amlodipine(trade name Lotrel).

Most commonly, headaches and cough can occur with its use. Anaphylaxis, angioedema and hyperkalemia, the elevation of potassium levels, can also occur.
Benazepril may cause harm to the fetus during pregnancy.
| “ | ACE inhibitors can pose a potential threat to kidneys as well. The key question was whether damaged kidneys would worsen if patients took ACE inhibitors. In a nutshell, concerns centered on blood levels of potassium andcreatinine, waste products that are excreted by the kidneys. Testing creatinine levels in the blood is used as a way to monitor kidney function (…) kidney problems worsened more slowly in those taking Lotensin. Overall, there were no major differences in side effects between patients taking Lotensin or the placebo.[2] | ” |
This study marks the first indication that benazepril, and perhaps other ACE inhibitors, may actually be beneficial in the treatment of hypertension in patients with kidney disease.
The Benazepril hydrochloride, with the CAS registry number 86541-74-4, is also known as (3S)-3-(((1S)-1-Carboxy-3-phenylpropyl)amino)-2,3,4,5-tetrahydro-2-oxo-1H-1-benzazepine-1-acetic acid, 3-ethyl ester, monohydrochloride; Benazepril HCl; Cibacen; Cibacen CHF; Labopol. It belongs to the product categories of Intermediates & Fine Chemicals; Pharmaceuticals; Amines; Aromatics; Heterocycles. This chemical’s molecular formula is C24H29ClN2O5 and molecular weight is 460.96. What’s more, its IUPAC name 2-[(3S)-3-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]-2-oxo-4,5-dihydro-3H-1-benzazepin-1-yl]acetic acid hydrochloride. In addition, Benazepril hydrochloride (CAS 86541-74-4) is crystalline solid which is soluble in DMSO. It is used in high blood pressure and congestive heart failure. When you are using this chemical, you should not breathe dust and avoid contact with skin and eyes.
Under the brand names Fortekor (Novartis) and VetACE (Jurox Animal Health), benazepril hydrochloride is used to treat congestive heart failure in dogs and chronic renal failure in dogs and cats.

| Benazepril hydrochloride, TWT-8154, CGS-14824A, Cibacene, Briem, Cibacen, Lotensin | |
| 1-Carboxymethyl-3(S)-[1(S)-ethoxycarbonyl-3-phenylpropylamino]-2,3,4,5-tetrahydro-1H-1-benzazepin-2-one monohydrochloride; 3(S)-[1(S)-Ethoxycarbonyl-3-phenylpropylamino]-2-oxo-2,3,4,5-tetrahydro-1-benzazepine-1-acetic acid monohydrochloride | |
| 【CAS】 | 86541-74-4, 86541-75-5 (free base) |
| MF | C24-H28-N2-O5.Cl-H |
| MW | 460.9551rot–[Alpha] 20 D -141.0 °. (C = 0.9, ethanol) |
| Cardiovascular Drugs, Hypertension, Treatment of, Angiotensin-I Converting Enzyme (ACE) Inhibitors | |
| Launched-1990 | |
| Novartis (Originator), Pierre Fabre (Licensee), Andrx (Generic), Eon Labs (Generic), KV Pharmaceutical (Generic), Mylan (Generic) |

Above Preparation of Benazepril hydrochloride (CAS 86541-74-4): The reaction of 2(R)-hydroxy-4-phenyl butyric acid ethyl ester (I) with trifluoromethanesulfonic anhydride in dichloromethane gives the corresponding triflate (II), which is then condensed with the amino benzazepinone (III) by means of NMM in the same solvent to provide the target benazepril.

ABOVE SCHEME-EP 1891014 B1
BACKGROUND



SYNTHETIC SCHEMES
| Benzazepin-2-ones, process for their preparation, pharmaceutical preparations containing these compounds and the compounds for therapeutical use | |
| Watthey, J.W.H. (Novartis AG) | |
| EP 0072352; GB 2103614; JP 8338260 | |
 |
|
| The reaction of 2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (I) with PCl5 in hot xylene gives 3,3-dichloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (II), which is treated with sodium acetate and reduced with H2 over Pd/C in acetic acid yielding 3-chloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (III). The reaction of (III) with sodium azide in DMSO affords 3-azido-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (IV), which is condensed with benzyl bromoacetate (V) by means of NaH in DMF giving 3-azido-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VI). The treatment of (VI) with Raney-Ni in ethanol-water yields 3-amino-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VII), which is debenzylated by hydrogenation with H2 over Pd/C in ethanol affording 3-amino-1-(carboxymethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VIII). Finally, this compound is condensed with ethyl 3-benzylpyruvate (IX) by means of sodium cyanoborohydride in methanol acetic acid. | |
| Process for the preparation of benazepril | |
| Kumar, Y.; De, S.; Thaper, R.K.; Kumar, D.S.M. (Ranbaxy Laboratories Ltd.) | |
| WO 0276375 | |
 |
|
| The reaction of 2(R)-hydroxy-4-phenyl butyric acid ethyl ester (I) with trifluoromethanesulfonic anhydride in dichloromethane gives the corresponding triflate (II), which is then condensed with the amino benzazepinone (III) by means of NMM in the same solvent to provide the target benazepril. | |
| CGS-14824 A | |
| Casta馿r, J.; Serradell, M.N. | |
| Drugs Fut 1984,9(5),317 | |
|
|
| The reaction of 2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (I) with PCl5 in hot xylene gives 3,3-dichloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (II), which is treated with sodium acetate and reduced with H2 over Pd/C in acetic acid yielding 3-chloro-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (III). The reaction of (III) with sodium azide in DMSO affords 3-azido-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (IV), which is condensed with benzyl bromoacetate (V) by means of NaH in DMF giving 3-azido-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VI). The treatment of (VI) with Raney-Ni in ethanol-water yields 3-amino-1-(benzyloxycarbonylmethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VII), which is debenzylated by hydrogenation with H2 over Pd/C in ethanol affording 3-amino-1-(carboxymethyl)-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VIII). Finally, this compound is condensed with ethyl 3-benzylpyruvate (IX) by means of sodium cyanoborohydride in methanol acetic acid. | |
| Synthesis of 14C-labeled 3-([1-ethoxycarbonyl-3-phenyl-(1S)-propyl]amino)-2,3,4,5-tetrahydro-2-oxo-1H-1-(3S)-benzazepine-1-acetic acid hydrochloride ([14C]CGS 14824A) | |
| Chaudhuri, N.K.; Patera, R.; Markus, B.; Sung, M.-S. | |
| J Label Compd Radiopharm 1987,24(10),1177-84 | |
 |
|
| A new synthesis of CGS-14824A is given: The reaction of 3-bromo-1-phenylpropane (I) with KCN gives 4-phenylbutyronitrile (II), which is hydrolyzed to the corresponding butyric acid (III). The cyclization of (III) with polyphosphoric acid affords 1-tetralone (IV), which is brominated to 2-bromo-1-tetralone (V) and treated with hydroxylamine to give the oxime (VI). The Beckman rearrangement of (VI) yields 3-bromo-2,3,4,5-tetrahydro-1H-(1)benzazepin-2-one (VII), which is treated with sodium azide to afford the azide derivative (VIII). The N-alkylation of (VIII) with ethyl bromoacetate (IX) by means of KOH and tetrabutylammonium bromide in THF gives the N-alkylated azide (X), which is reduced by catalytic hydrogenation to the corresponding amine (XI). The hydrolysis of the ester group of (XI) with NaOH yields the free acetic acid derivative (XII), which is finally reductocondensed with ethyl 2-oxo-4-phenylbutyrate (XIII) by means of sodium cyanoborohydride. | |

US 6548665 B2– above

see translated vesrsion————-First, 2,3,4,5 – tetrahydro-1H-[1] azepin-2 phenyl – one (2) Preparation of
the dry reaction flask, add α- tetralone 20g (0.137mol), stacked acid 7.36g (0.171mol) and chloroform 140ml, was stirred at 40 ℃ in 1h concentrated sulfuric acid was slowly added dropwise 36ml, acid layer was separated and poured into 900ml water to give a creamy solid. Recrystallization with hot water to give white crystals (2) 15.5g (70%), mp141 ℃. (Acidic filtrate and after a small amount of product can be obtained.)
Second, 3,3 – dichloro-2, 3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one (3) of the prepared
in a dry reaction flask, (2) 48.3g (0.3mol) and xylene solution of 1300ml, phosphorus pentachloride 188g (0.9mol), stirred and gradually heated to at 0.5h 90 ℃, (Caution! When phosphorus pentachloride dissolved hydrogen chloride gas had severe.) 90 ℃ the reaction was continued for 0.5h, filtered to remove a small amount of suspended solids, solvent recovery under reduced pressure, to the residue was added saturated sodium bicarbonate solution, 100ml, stirred until a solid precipitate complete, filtered and the cake washed with ethanol (100ml × 2), diethyl ether (50ml) and dried to give (3) 69.0g (90%), mp185 ~ 187 ℃.
III.3 – chloro-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one (4) Preparation of
the reaction flask (3) 10g (0.087mol), Sodium acetate 77g (0.11mol), acetic acid 460ml and 5% Pd-C 0.86g, under atmospheric pressure at room temperature to a hydrogen-absorbing up total of 950ml (about 0.5h). Filtration, recycling the catalyst recovered solvent, the residue was dried under reduced pressure, and then added 900ml of 10% sodium bicarbonate solution and dichloromethane 300ml, stirring, standing, the organic layer was separated and the aqueous layer extracted with dichloromethane (300ml × 3) extracted organic layers were combined, dried over anhydrous sodium sulfate, the solvent recovered under reduced pressure. Diethyl ether was added to the cured 350ml, and mashed, filtered and dried to give (4) 8.19g (95%), mp163 ~ 167 ℃.
4 (3) – azido-2, 3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one (5) Preparation of
the dry reaction flask (4) 15.9g ( 0.08mol), sodium azide 6.4g (0.10mol) and 320ml solution of dimethyl sulfate, the reaction was stirred at 80 ℃ 3h, cooled to room temperature, poured into ice-water (1L) to precipitate a pale yellow solid , filtered and dried under reduced pressure at 75 ℃ to give (5) 14.7g (90%), mp142 ~ 145 ℃.
V.3 – azido-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (6) Preparation of
the dry reaction flask, (5) 3.0g (0.015mol), tetrabutylammonium bromide, 0.5g (0.0015 mol), powdered potassium hydroxide 1.1g (0.016mol) and 30ml of tetrahydrofuran solution of ethyl bromoacetate was added 1.9ml ( 0.016mol), stirred rapidly at room temperature for 1.5h (nitrogen). Water was added: dichloromethane (50:100 ml), stirred, allowed to stand, the organic layer separated. Washed with water, dried over anhydrous sodium sulfate, the solvent recovered under reduced pressure to give a pale yellow oil (6) 4.1g (96%) (can be used directly in the next step).
VI.3 – amino-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (7) Preparation of
the dry reaction flask, (6 ) 20.0g (0.070mol), ethanol 100ml, 10% Pd-C 1.0g stirring, at room temperature, 303.9kPa hydrogenated under a hydrogen pressure 1.5h, intermittent deflated to remove the generated nitrogen gas, after the reaction was collected by filtration Pd / C, recovery of solvents under reduced pressure to give a yellow oil, add ether l00ml, mashed, filtered and dried to give a white solid (7) 17.0g (93%) mp101 ~ 102 ℃.
Seven, (3S) -3 – amino-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (8) Preparation of
the reaction flask, adding (7) 25.1g (0.096mol), L – tartaric acid 14.4g (0.096mol) and hot ethanol 200ml, stirring to dissolve, cooled at room temperature overnight, filtered and dried under reduced pressure to give a white powder 30.7g, with ethanol Recrystallization twice (each 200ml), to give (8) tartaric acid salt of 13.6g (34%), mp168 ~ 169 ℃, with 10% ammonium hydroxide, to give a white solid (8) 8.0g (95%) mp104 ~ 106 ℃.
Eight, (3S) -3 – amino-2 ,3,4,5 – tetrahydro-1H-[1] benzene azepin-2 – one-1 – acetate (9) Preparation of
the reaction flask, (8) 4.0g (0.056mol) and 150ml of methanol solution of sodium hydroxide 2.1g (0.056moI) and a solution of 5ml of water, stirred at room temperature for 2h, the solvent recovered under reduced pressure, the residue was dried and diethyl ether was added 100ml, trace broken, filtered, and dried to give (9) 12.9g (89%) (used directly in the next step).
IX benazepril (1) Synthesis of
the reaction flask (9) 12.9g (0.050mol), 2 – oxo-4 – phenylbutyrate 31.0g (0.15mol), acetic acid and 100ml methanol 75ml, the reaction was stirred at room temperature for 1h (nitrogen). Of sodium borohydride cyanide was slowly added dropwise 3.8g (0.062mol) and 30ml of methanol solution of (4h was completed within), stirred overnight, heat. Concentrated hydrochloric acid 10ml, 1h stirring at room temperature, the solvent was recovered under reduced pressure, water was added to the residue and diethyl ether 400ml l00ml, dissolved with concentrated ammonium hydroxide and the pH adjusted to 9.3, the organic layer was separated and the aqueous layer acidified with concentrated hydrochloric to pH 4.3, extracted with ethyl acetate (100ml × 3) extracted organic layers were combined, dried over anhydrous magnesium sulfate, the solvent recovered under reduced pressure, to the residue was added methylene chloride (150ml) to dissolve. And pass into dry hydrogen chloride after 5min recovered solvent under reduced pressure, to the residue was added hot ethyl ketone 100ml, stirring to dissolve, cooled and precipitated solid was filtered to give crude product (1). A 3 – amyl ketone / methanol (volume ratio 10:1) (110ml) was recrystallized (1) 5.8 g, mp 188 ~ 190 ℃, [alpha] D 20 -141.0 (C = 0.9, C 2 H 5 OH )
[Spectral Data] (free base) [2]
MS: m / Z (%) 424 (M + , 2), 351 (100), 190 (22), 91 (65)
] [other synthetic routes
described in the reference literature.
[References]
[1] Briggs LH et al. J Chem Soc, 1937, 456
[2] Watthey WH et al. J Med Clmm, 1985, 28:1511
[3] EP 1986, 206933 (CA, 1987, 107: 77434e)
[4] EP 1983, 72352 (CA, 1983, 99:53621 d)
[5] package insert: Lotensin
[6] property protection case I: Lotensin
[7] property protection case II: Lotensin
[8] Drug Monograph information: BENAZEPRIL
more info

read all at
………

Aimee Tharaldson, PharmD, is a senior clinical consultant in the emerging therapeutics department at Express Scripts. She is responsible for monitoring and analyzing the specialty pharmaceutical pipeline. The emerging therapeutics department produces several proprietary reports on the pipeline for use by Express Scripts’ employees and clients. It is also responsible for the safety program that alerts patients, physicians, and clients to important information regarding serious drug safety alerts and market withdrawals. She contributes to Express Scripts’ Drug Trend Report and plays a key role in developing and maintaining Express Scripts’ specialty drug list. She received her doctor of pharmacy degree from the University of Minnesota, College of Pharmacy, and completed a pharmacy practice residency at the Minneapolis VA Medical Center. –
lixisenatide
Sanofi Provides Update on Lixisenatide New Drug Application in U.S.
Paris, France – September 12, 2013 – Sanofi (EURONEXT: SAN and NYSE: SNY) announced today its decision to withdraw the lixisenatide New Drug Application (NDA) in the U.S., which included early interim results from the ongoing ELIXA cardiovascular (CV) outcomes study. The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
The decision to withdraw the lixisenatide application follows discussions with the U.S. Food and Drug Administration (FDA) regarding its proposed process for the review of interim data. Sanofi believes that potential public disclosure of early interim data, even with safeguards, could potentially compromise the integrity of the ongoing ELIXA study. Sanofi’s decision is not related to safety issues or deficiencies in the NDA………………………read all at
http://www.pharmalive.com/sanofi-pulls-diabetes-drug-nda
EU

WEDNESDAY, September 11, 2013 — The U.S. Food and Drug Administration today approved a new use for Botox Cosmetic (onabotulinumtoxinA) for the temporary improvement in the appearance of moderate to severe lateral canthal lines, known as crow’s feet, in adults. Botox Cosmetic is the only FDA approved drug treatment option for lateral canthal lines.
The FDA approved Botox Cosmetic in 2002 for the temporary improvement of glabellar lines (wrinkles between the eyebrows, known as frown lines), in adults. Botox Cosmetic works by keeping muscles from tightening so wrinkles are less prominent
READ ALL AT

BOTOX Cosmetic (onabotulinum toxin A) For Injection, is a sterile, vacuum-dried purifiedbotulinum toxin type A, produced from fermentation of Hall strain Clostridium botulinumtype A grown in a medium containing casein hydrolysate, glucose, and yeast extract, intended for intramuscular use. It is purified from the culture solution by dialysis and a series of acid precipitations to a complex consisting of the neurotoxin, and several accessory proteins. The complex is dissolved in sterile sodium chloride solution containing Albumin Human and is sterile filtered (0.2 microns) prior to filling and vacuum-drying.
The primary release procedure for BOTOX Cosmetic uses a cell-based potency assay to determine the potency relative to a reference standard. The assay is specific to Allergan’s products BOTOX and BOTOX Cosmetic. One Unit of BOTOX Cosmetic corresponds to the calculated median intraperitoneal lethal dose (LD50) in mice. Due to specific details of this assay such as the vehicle, dilution scheme and laboratory protocols, Units of biological activity of BOTOX Cosmetic cannot be compared to nor converted into Units of any other botulinum toxin or any toxin assessed with any other specific assay method. The specific activity of BOTOX Cosmetic is approximately 20 Units/nanogram of neurotoxin protein complex.
Each vial of BOTOX Cosmetic contains either 50 Units of Clostridium botulinum type A neurotoxin complex, 0.25 mg of Albumin Human, and 0.45 mg of sodium chloride; or 100 Units of Clostridium botulinum type A neurotoxin complex, 0.5 mg of Albumin Human, and 0.9 mg of sodium chloride in a sterile, vacuum-dried form without a preservative.
Since the approval of BOTOX® Cosmetic by the U.S. Food and Drug Administration in 2002, Allergan has virtually changed the face of medical aesthetics. Men and women between the ages of 18 to 65 now have the ability to choose science-based, non-invasive medical aesthetic solutions, including BOTOX® Cosmetic and the JUVÉDERM® family of dermal fillers, to achieve their own results. Over the last seven years, there have been nearly 11.8 million BOTOX® Cosmetic treatments recorded in the United States.1 More importantly, its 97 percent satisfaction rating (survey of 117 patients)2,3 is just one indication of the trust consumers have placed in Allergan.
BOTOX® Cosmetic is a simple, non-surgical procedure for temporarily reducing the appearance of moderate to severe glabellar lines – the vertical frown lines between the eyebrows that look like an “11” – in adult women and men aged 18 to 65. BOTOX® Cosmetic reduces the activity of the muscles that cause the “11s” to form by blocking nerve impulses that trigger wrinkle-causing muscle contractions, creating an improved appearance between the brows. Results can last up to four months and may vary with each patient. Ask your doctor if BOTOX® Cosmetic is right for you.
![]()

Healthy, Wealthy & Wise
For over 25 years the Sami Group has been unlocking the mystery of herbs, extracting the goodness, and gifting the world with good health.
Now the Sami Group provides YOU an opportunity to unlock the mystery of Success, Wealth & Better living.
FREE OF COST CONSULTATION on Diabetes, Cancer, Arthritis, Osteoporosis, Heart- Liver -Lung & Kidney problems, Low Immunity, Alzheimer, Weight Management, Weak Memory, Neutritional Deficiency, UTI problems etc.
(REVOLUTIONARY AYURVEDIC SOLUTIONS with ISO 22000 Certified Indian MNC after 25 years of R & D by 120 Scientists.
Numberless Testimonials.)
http://www.samidirect.com/products/
Do visit the website www.samidirect.com & have the study in detail. Have a look at the Sami Direct Corporate Video on you tube. If you get the wonderful potential of the brightest future…do call me for ‘How to get started?’




…..









DISTRIBUTOR ENQUIRY WELCOME
CONTACT MR JAY DESAI
REGARDS
+91 9699952526
Mumbai, INDIA
email-jaydesai1502@gmail.com
 |
Bussines Sami Direct Seminar ppt.ppt1.pdf 9204K View Download |
samidirect corporate video

DR MAJID, FOUNDER , SAMIDIRECT
Dear Friend, Congratulations on your decision!
A little over three decades ago I went from a small town in South India to the United States Of America seeking fulfillment of my dreams. Today with a business conglomerate spread across the globe, I can confidently say that the future belongs to those who believe in the beauty of their dreams.
The aspiration to dream and the conviction to follow their dreams is what sets apart the extraordinary from the ordinary. Congratulations for choosing to be among the extraordinary. Now we are in it together. You have chosen the right place and the right means. The awesome combination of extensively researched products and a revolutionary business plan is a definite formula for success. We are with you at every step to help you fulfill your dreams and reach greater heights.
Dr. Muhammed Majeed
Welcome home again!
– See more at: http://www.samidirect.com/about/founder-desk/#sthash.rrOCRiJ1.dpuf
Sami Direct, as a part of the Sami Group, is the culmination of relentless Research and Development for more than two decades. We at Sami Direct are committed to offer you an unrivalled range of nutraceuticals, soon to be followed by cosmeceutical products, which have been acknowledged by the world over for its highest quality and safety standards.
Sami Direct is supported by its very own R&D facility- SAMI LABS LTD., located in Bangalore. This state-of-the-art, multi-disciplinary division pursues diverse fields of research with over 120 scientists focusing all efforts towards creating effective and safe products. With six highly advanced cutting-edge manufacturing units adhering to the strictest quality and safety standards, Sami Direct ensures that the highest quality of products are being produced.
Today the Sami Group holds a strong intellectual property portfolio with over 70 US and International Patents to its credit including awards and recognitions worldwide.
With the perfect blend of world class products and a revolutionary business plan, it is a lifetime opportunity not just to enhance your health, but also a fruitful and lasting career heightening your income.
DISTRIBUTOR ENQUIRY WELCOME
CONTACT MR JAY DESAI
REGARDS
+91 9699952526
Mumbai, INDIA
email; jaydesai1502@gmail.com
……………………………..
Stem cells are undifferentiated biological cells, that can differentiate into specialized cells and can divide (through mitosis) to produce more stem cells. They are found in multicellular organisms. In mammals, there are two broad types of stem cells: embryonic stem cells, which are isolated from the inner cell mass of blastocysts, and adult stem cells, which are found in various tissues. In adult organisms, stem cells and progenitor cells act as a repair system for the body, replenishing adult tissues. In a developing embryo, stem cells can differentiate into all the specialized cells—ectoderm, endoderm and mesoderm (see induced pluripotent stem cells)—but also maintain the normal turnover of regenerative organs, such as blood, skin, or intestinal tissues.
There are three accessible sources of autologous adult stem cells in humans:
Stem cells can also be taken from umbilical cord blood just after birth. Of all stem cell types, autologous harvesting involves the least risk. By definition, autologous cells are obtained from one’s own body, just as one may bank his or her own blood for elective surgical procedures.
Highly plastic adult stem cells are routinely used in medical therapies, for example in bone marrow transplantation. Stem cells can now be artificially grown and transformed (differentiated) into specialized cell types with characteristics consistent with cells of various tissues such as muscles or nerves through cell culture. Embryonic cell lines and autologous embryonic stem cells generated through therapeutic cloning have also been proposed as promising candidates for future therapies. Research into stem cells grew out of findings by Ernest A. McCulloch and James E. Till at the University of Toronto in the 1960s
……………

Medical researchers believe that stem cell therapy has the potential to dramatically change the treatment of human disease. A number of adult stem cell therapies already exist, particularly bone marrow transplants that are used to treat leukemia. In the future, medical researchers anticipate being able to use technologies derived from stem cell research to treat a wider variety of diseases including cancer, Parkinson’s disease, spinal cord injuries, Amyotrophic lateral sclerosis, multiple sclerosis, and muscle damage, amongst a number of other impairments and conditions. However, there still exists a great deal of social and scientific uncertainty surrounding stem cell research, which could possibly be overcome through public debate and future research, and further education of the public.
One concern of treatment is the risk that transplanted stem cells could form tumors and become cancerous if cell division continues uncontrollably.
Stem cells are widely studied, for their potential therapeutic use and for their inherent interest.
Supporters of embryonic stem cell research argue that such research should be pursued because the resultant treatments could have significant medical potential. It has been proposed that surplus embryos created for in vitro fertilization could be donated with consent and used for the research.
The recent development of iPS cells has been called a bypass of the legal controversy. Laws limiting the destruction of human embryos have been credited for being the reason for development of iPS cells, but it is still not completely clear whether hiPS cells are equivalent to hES cells. Recent work demonstrates hotspots of aberrant epigenomic reprogramming in hiPS cells (Lister, R., et al., 2011).

Pluripotent, embryonic stem cells originate as inner cell mass (ICM) cells within a blastocyst. These stem cells can become any tissue in the body, excluding a placenta. Only cells from an earlier stage of the embryo, known as the morula, are totipotent, able to become all tissues in the body and the extraembryonic placenta

http://www.scripintelligence.com/awards/categories/
Deciding on a shortlist from so many deserving entries was never going to be an easy process for our independent judging panel, but they rose to the challenge and this list represents the best of the best.
Best Company in an Emerging Market – Sponsored by Clinigen Group
Best Technological Development in Clinical Trials
Best Partnership Alliance
Financing Deal of the Year
Best Advance in an Emerging Market
Clinical Advance of the Year – Sponsored by Quintiles
Licensing Deal of the Year
Executive of the Year
Biotech Company of the Year
Best Contract Research Organization
Management Team of the Year
Best New Drug – Sponsored by INC Research
Pharma Company of the Year – Sponsored by ICON
 By Jim Zhang, Ph.D., JZMed, Inc.
By Jim Zhang, Ph.D., JZMed, Inc.
The pharmaceutical markets of China and India have been experiencing such rapid growth in the past decade that they are widely recognized as two of the world’s most dynamic emerging markets. Consequently, they have attracted many drug companies around the world…………FULL ARTICLE
READ ALL AT
Jim Zhang, Ph.D., is president and managing director of JZMed, Inc., a market research company specializing in research on the Chinese pharmaceutical outsourcing industry. The company also provides consulting services for pharmaceutical outsourcing in China.

http://www.allfordrugs.com/2013/09/11/comparing-chinas-and-indias-pharmaceutical-manufacturing/

pertuzumab
TUESDAY Sept. 10, 2013 — A drug already used to treat advanced breast cancer also appears to shrink early stage breast tumors, potentially offering women a first-of-its-kind treatment option, U.S. health regulators say.
read all at
http://www.drugs.com/news/novel-shows-promise-early-stage-breast-cancer-47311.html
