
Home » Posts tagged 'organic chemistry' (Page 13)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BAYER- sPRM (BAY 1002670) Vilaprisan is a novel oral progesterone receptor modulator that holds the promises of long-term treatment of patients with symptomatic uterine fibroids
http://www.who.int/medicines/publications/druginformation/issues/Proposed-List_109.pdf str is available in this link
20,20,21,21,21-pentafluoro-17-hydroxy-11β-[4-
(methanesulfonyl)phenyl]-19-nor-17α-pregna-4,9-dien-3-one
progesterone receptor antagonist
BAY 1002670, vilaprisan
1262108-14-4
C27H29F5O4S, 544.574
http://www.who.int/medicines/publications/druginformation/issues/Proposed-List_109.pdf str is available in this link
Bayer has also made good progress in the development of new treatment options for patients with gynecological diseases: sPRM (BAY 1002670) is a novel oral progesterone receptor modulator that holds the promises of long-term treatment of patients with symptomatic uterine fibroids. Based on promising early clinical data the initiation of a Phase III study is planned for mid-2014.
A selective progesterone receptor modulator (SPRM) is an agent that acts on the progesterone receptor. A characteristic that distinguishes such substances from receptor full agonists (such as progesterone) and full antagonists (such as mifepristone) is that their action differs in different tissues (agonist in some while antagonist in others). This mixed agonist/antagonist profile of action leads to selective stimulation or inhibition progesterone-like action in different tissues and furthermore raises the possibility of dissociation of desirable therapeutic effects from undesirable side effects in synthetic progesterone receptor drug candidates


amcrasto@gmail.com
email me if u like my posts
Molidustat (BAY 85-3934), Bayer’s drug under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease.
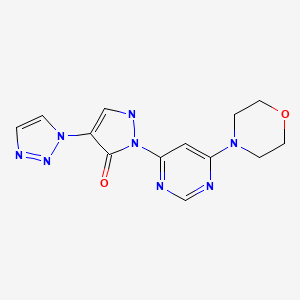
Molidustat
UNII-9JH486CZ13, cas no 1154028-82-6, MW: 314.3076
2-(6-morpholin-4-ylpyrimidin-4-yl)-4-(triazol-1-yl)-1H-pyrazol-3-one
Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
For the cardio-renal syndrome, a Phase IIb program with the investigational new drug Molidustat (BAY 85-3934) is under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease. Molidustat is a novel inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase (PH) which stimulates erythropoietin (EPO) production and the formation of red blood cells. Phase I data have shown that inhibition of HIF-PH by Molidustat results in an increase in endogenous production of EPO.
About Bayer HealthCare
The Bayer Group is a global enterprise with core competencies in the fields of health care, agriculture and high-tech materials. Bayer HealthCare, a subgroup of Bayer AG with annual sales of EUR 18.6 billion (2012), is one of the world’s leading, innovative companies in the healthcare and medical products industry and is based in Leverkusen, Germany. The company combines the global activities of the Animal Health, Consumer Care, Medical Care and Pharmaceuticals divisions. Bayer HealthCare’s aim is to discover, develop, manufacture and market products that will improve human and animal health worldwide. Bayer HealthCare has a global workforce of 54,900 employees (Dec 31, 2012) and is represented in more than 100 countries. More information at www.healthcare.bayer.com.
molidustat
…………………………………………………………..
molidusat sodium
Sodium 1-[6-(morpholin-4-yl)pyrimidin-4-yl]-4-(1H-1,2,3-triazol-1-yl)-1H-pyrazol-5-olate
Molidustat sodium is an orally-available hypoxia-inducible factor prolyl hydroxylase inhibitor in phase I clinical trials at Bayer for the treatment of patients suffering from renal anemia due to chronic kidney disease.
WO 2008067871
WO 2012065967
WO 2013167552
…………………………
2-Heteroaryl-4-aryl-1,2-dihydropyrazolones having a bactericidal and/or fungicidal action are disclosed in EP 165 448 and EP 212 281. The use of 2-heteroaryl-4-aryl-1,2-dihydropyrazolones as lipoxygenase inhibitors for treatment of respiratory tract, cardiovascular and inflammatory diseases is claimed in EP 183 159. 2,4-Diphenyl-1,2-dihydropyrazolones having a herbicidal activity are described in DE 2 651 008.
The preparation and pharmacological properties of certain 2-pyridyl-1,2-dihydropyrazolones are reported in Helv. Chim. Acta 49 (1), 272-280 (1966). WO 96/12706, WO 00/51989 and WO 03/074550 claim compounds having a dihydropyrazolone partial structure for treatment of various diseases, and hydroxy- or alkoxy-substituted bipyrazoles for treatment of neuropsychiatric diseases are disclosed in WO 2006/101903.
Heteroaryl-substituted pyrazole derivatives for treatment of pain and various CNS diseases are furthermore described in WO 03/051833 and WO 2004/089303. WO 2006/114213 has meanwhile disclosed 2,4-dipyridyl-1,2-dihydropyrazolones as inhibitors of HIF prolyl 4-hydroxylases.
The x-ray crystal structure of the compound 3-methyl-1-(pyridin-2-yl)-4-(1-pyridin-2-yl-3-methyl-1H-pyrazol-5-yl)-2H-3-pyrazolin-5 (114)-one (other name: 5,5′-dimethyl-2,2′-di-pyridin-2-yl-1′,2′-dihydro-2H,3′H-3,4′-bipyrazol-3′-one) is reported inActa Crystallogr., Section E: Structure Reports Oμline E57 (11), o1126-o1127 (2001) [Chem. Abstr. 2001:796190].
The synthesis of certain 3′,5-dimethyl-2-phenyl-1′-(1,3-thiazol-2-yl)-1′H,2H-3,4′-bipyrazol-5′-ol derivatives is described inIndian J. Heterocyclic Chem. 3 (1), 5-8 (1993) [Chem. Abstr. 1994:323362].
The preparation and tautomerism of individual 4-(pyrazol-5-yl)-pyrazolin-5-one derivatives is reported in J. Heterocyclic Chem. 27 (4), 865-870 (1990) [Chem. Abstr. 1991:428557]. A therapeutic use has not hitherto been described for the compounds mentioned in these publications. The compound 2-tert-butyl-1′-[4-(4-chlorophenyl)-1,3-thiazol-2-yl]-3′,5-dimethyl-1′H,2H-3,4′-bipyrazol-5′-ol is listed as a test example in WO 2007/008541.
………………………….
https://www.google.co.in/patents/US20100305085
Example 3A 3-(Dimethylamino)-2-(1H-1,2,3-triazol-1-yl)acrylic acid ethyl ester

The preparation of the starting compound is carried out analogously to 2A starting from 1.00 g (6.45 mmol) 2-(1H-1,2,3-triazol-1-yl)acetic acid ethyl ester.
Yield: 1.4 g (100% of th.)
1H-NMR (400 MHz, DMSO-d6): δ=8.10 (d, 1H), 7.78 (d, 1H), 7.65 (s, 1H), 4.03 (q, 2H), 3.06 (br. s, 3H), 2.10 (br. s, 3H), 1.12 (t, 3H).
LC-MS (Method 5): Rt=1.40 min; MS (ESIpos): m/z=211 [M+H]+.
Example 16A 4-(6-Hydrazinopyrimidin-4-yl)morpholine

Stage a): 4-(6-Chloropyrimidin-4-yl)morpholine

45.0 g (302.1 mmol) 4,6-dichloropyrimidine are initially introduced into 450 ml water. 26.3 g (302.1 mmol) morpholine are added and the mixture is stirred at 90° C. for 16 h. Thereafter, it is cooled to 0° C. and the precipitate formed is filtered off. The precipitate is washed once with 50 ml water and dried in air.
Yield: 51.0 g (85% of th.)
LC-MS (Method 4): Rt=1.09 min; MS (ESIpos): m/z=200 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.35 (s, 1H), 6.95 (s, 1H), 3.62 (s, 8H).
Stage b) 4-(6-Hydrazinopyrimidin-4-yl)morpholine
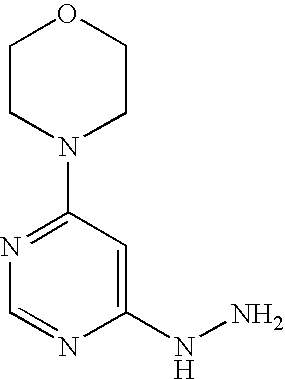
53.0 g (2.7 mmol) 4-(6-chloropyrimidin-4-yl)morpholine are initially introduced into 260 ml ethanol. 132.9 g (2.7 mol) hydrazine hydrate are added and the mixture is stirred under reflux for 16 h. Thereafter, it is cooled to RT and approx. half of the solvent is removed by distillation. The mixture is cooled to 0° C. and the solid formed is filtered off. It is rinsed with cold ethanol and the solid is dried first in air and then in vacuo.
Yield: 35.0 g (68% of th.)
LC-MS (Method 1): Rt=0.17 min; MS (ESIpos): m/z=196 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=7.94 (s, 1H), 7.70 (s, 1H), 5.91 (s, 1H), 4.15 (s, 2H), 3.66-3.60 (m, 4H), 3.45-3.37 (m, 4H).
Example 71 2-(6-Morpholin-4-ylpyrimidin-4-yl)-4-(1H-1,2,3-triazol-1-yl)-1,2-dihydro-3H-pyrazol-3-one

1.9 g (8.8 mmol) of the compound from Example 3A and 1.9 g (9.7 mmol) of the compound from Example 16A are initially introduced into 25 ml ethyl acetate and 504 mg (4.4 mmol) TFA are added at RT. The mixture is stirred under reflux for 16 h, then cooled to 5° C. and subsequently stirred for a further 2 h. The solid formed is filtered off, washed with ethyl acetate and dried first in air and thereafter under a high vacuum. 1.7 g of product are obtained.
The mother liquor is combined with the wash solution and the solvent is removed. According to LC-MS, the residue (2.4 g) still contains the intermediate 3-[2-(6-morpholin-4-ylpyrimidin-4-yl)hydrazino]-2-(1H-1,2,3-triazol-1-yl)prop-2-enoic acid ethyl ester (intermediate stage of the cyclization), which is used directly for the preparation of Example 72 (see there).
Yield: 1.7 g (61% of th.)
LC-MS (Method 9): Rt=0.90 min; MS (ESIpos): m/z=315 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.42 (s, 1H), 8.38 (s, 1H), 8.01 (s, 1H), 7.73 (s, 1H), 7.70 (s, 1H), 3.71-3.65 (m, 4H), 3.57-3.51 (m, 4H).
hydrochloride
Example 72 2-(6-Morpholin-4-ylpyrimidin-4-yl)-4-(1H-1,2,3-triazol-1-yl)-1,2-dihydro-3H-pyrazol-3-one hydrochloride

Batch 1: 7.5 ml of a 4 N solution of hydrogen chloride in dioxane are added to 1.7 g (5.4 mmol) of the compound from Example 71. The mixture is stirred at RT, 5 ml dioxane are added and the mixture is stirred at RT for 16 h. The solid is filtered off and washed with 5 ml dioxane. The mixture is dried under a high vacuum for 16 h, 10 ml methanol are then added and the mixture is stirred at RT for 1 h. The solid is filtered off, washed with 4 ml methanol and dried under a high vacuum. 1.6 g of the title compound are obtained.
Batch 2: A further amount of the title compound is obtained as follows: The residue (2.4 g) obtained from the mother liquor during the synthesis of Example Compound 71, which contains the open-ring intermediate state of the cyclization, 3-[2-(6-morpholin-4-ylpyrimidin-4-yl)hydrazino]-2-(1H-1,2,3-triazol-1-yl)prop-2-enoic acid ethyl ester, is dissolved in 12 ml ethanol and 1.5 ml 30% strength sodium methylate solution in methanol are added at RT, while stirring. The mixture is subsequently stirred at RT for 45 min, then adjusted to pH 5 with 2 N hydrochloric acid and subsequently stirred at RT for a further 16 h. The mixture is cooled to 10° C. and the solid is filtered off and washed with 3.5 ml dioxane. The mixture is dried under a high vacuum for 16 h, 5 ml methanol are then added and the mixture is subsequently stirred at RT for 1 h. The solid is filtered off, washed with 2 ml methanol and dried under a high vacuum to give a further 997 mg of the title compound in this way.
Yield: together 2.6 g (83% of th.)
LC-MS (Method 6): Rt=0.89 min; MS (ESIpos): m/z=315 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.54 (s, 1H), 8.39 (s, 1H), 8.28 (s, 1H), 7.88 (s, 1H), 7.42 (s, 1H), 3.71 (s, 8H).
amcrasto@gmail.com
email me if u like my posts
Copanlisib (BAY 80-6946), Bayer’s novel, oral phosphatidylinositol-3 kinases (PI3K) inhibitor

Copanlisib (BAY 80-6946)
1032568-63-0, cas no
MW: 480.5262
In oncology, Copanlisib (BAY 80-6946), a novel, oral phosphatidylinositol-3 kinases (PI3K) inhibitor, was selected for accelerated development. Copanlisib demonstrated a broad anti-tumor spectrum in preclinical tumor models and promising early clinical signals in a Phase I study in patients with follicular lymphoma. A Phase II study in patients with Non-Hodgkin’s lymphoma is currently ongoing.
PI3K inhibitor BAY 80-6946
A phosphoinositide 3-kinase (PI3K) inhibitor with potential antineoplastic activity. PI3K inhibitor BAY 80-6946 inhibits the activation of the PI3K signaling pathway, which may result in inhibition of tumor cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K signaling may contribute to tumor resistance to a variety of antineoplastic agents.
Finerenone (BAY 94-8862), BAYER’S next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone
CAS Number: 1050477-31-0, UNII-DE2O63YV8R
MW: 378.4298, C21-H22-N4-O3
Finerenone (BAY 94-8862) is a next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone.
Currently available steroidal MR antagonists have proven to be effective in reducing cardiovascular mortality in patients with heart failure but have significant side effects that limit their utilization.
Finerenone is currently in clinical Phase IIb development for the treatment of worsening chronic heart failure, as well as diabetic nephropathy.
Glaxo Plans to File for Malaria Vaccine Approval Next Year

Malaria vaccine candidate reduces disease over 18 months of follow-up in late-stage study of more than 15,000 infants and young children
Malaria is a significant public health burden, claiming 660,000 lives a year – mostly children in sub-Saharan Africa
-Data support plan to submit regulatory application in 2014
Multilateral Initiative on Malaria Pan African Conference, Durban, South Africa — Results from a large-scale Phase III trial, presented today in Durban, show that the most clinically advanced malaria vaccine candidate, RTS,S, continued to protect young children and infants from clinical malaria up to 18 months after vaccination. Based on these data, GSK now intends to submit, in 2014, a regulatory application to the European Medicines Agency (EMA). The World Health Organization (WHO) has indicated that a policy recommendation for the RTS,S malaria vaccine candidate is possible as early as 2015 if it is granted a positive scientific opinion by EMA.
READ ALL AT
http://www.pharmalive.com/glaxo-plans-to-file-for-malaria-vaccine-approval-next-year
![]()
The U.S. Food and Drug Administration approved Adempas (riociguat) to treat adults with two forms of pulmonary hypertension.

October 8, 2013 — The U.S. Food and Drug Administration today approved Adempas (riociguat) to treat adults with two forms of pulmonary hypertension.
Pulmonary hypertension is caused by abnormally high blood pressure in the arteries of the lungs. It makes the right side of the heart work harder than normal. In its various forms, pulmonary hypertension is a chronic, progressive, debilitating disease, often leading to death or need for lung transplantation
read all at
http://www.drugs.com/newdrugs/fda-approves-adempas-pulmonary-hypertension-3927.html
In the area of pulmonary hypertension Adempas (Riociguat) is the first member of a novel class of compounds – so-called ‘soluble guanylate cyclase (sGC) stimulators’ – being investigated as a new and specific approach to treating different types of pulmonary hypertension (PH). Adempas has the potential to overcome a number of limitations of currently approved treatments for pulmonary arterial hypertension (PAH) and addresses the unmet medical need in patients with chronic thromboembolic pulmonary hypertension (CTEPH). It was approved for the treatment of CTEPH in Canada in September 2013, making it the world’s first drug approved in this deadly disease.
Riociguat has already shown promise as a potential treatment option beyond these two PH indications. An early clinical study was conducted in PH-ILD (interstitial lung disease), a disease characterized by lung tissue scarring (fibrosis) or lung inflammation which can lead to pulmonary hypertension, and, based on positive data, the decision was taken to initiate Phase IIb studies in PH-IIP (idiopathic pulmonary fibrosis), a subgroup of PH-ILD. Moreover, scientific evidence was demonstrated in preclinical models that the activity may even go beyond vascular relaxation. To prove the hypothesis Bayer is initiating clinical studies in the indication of systemic sclerosis (SSc), an orphan chronic autoimmune disease of the connective tissue affecting several organs and associated with high morbidity and mortality. If successful, Riociguat has the potential to become the first approved treatment for this devastating disease.
synthesis
Generic Name: Riociguat
Trade Name: Adempas
Synonym: BAY 63-2521
CAS number: 625115-55-1
Chemical Name: Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
Mechanism of Action: soluble guanylyl cyclase (sGC) stimulator
Date of Approval: October 8, 2013(US)
Indication: Pulmonary Hypertension
Company: Bayer AG

1)J. Mittendorf.; S. Weigand.; C. Alonso-Alija.; E. Bischoff.; A. Feurer.; M. Gerisch.; A. Kern.; A. Knorr.; D. Lang.; K. Muenter.; M. Radtke.; H. Schirok.; K.-H. Schlemmer.; E. Stahl.; A. Straub.; F. Wunder.; J.-P. Stasch. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension, ChemMedChem. 2009, 4, 853-865.
2)Cristina Alonso-Alija, Bayer Ag, Erwin Bischoff, Achim Feurer, Klaus Muenter, Elke Stahl, Johannes-Peter Stasch, Stefan Weigand, Carbamate-substituted pyrazolopyridines, WO2003095451 A1
3)Franz-Josef Mais, Joachim Rehse, Winfried Joentgen, Konrad SIEGEL, Process for preparing methyl methylcarbamate and its purification for use as pharmaceutically active compound,US20110130410
4)Claudia Hirth-Dietrich, Peter Sandner, Johannes-Peter Stasch, Andreas Knorr, Degenfeld Georges Von, Michael Hahn, Markus Follmann, The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc), WO 2011147810A1
5)Li Liang, Li Xing-zhou, Liu Ya-dan, Zheng Zhi-bing, Li Song, Synthesis of riociguat in treatment of pulmonary hypertension, Chinese Journal of Medicinal Chemistry(Zhongguo Yaowu Huaxue Zazhi), 21(2),120-125; 2011

Jens Ackerstaff, Lars BÄRFACKER, Markus Follmann, Nils Griebenow, Andreas Knorr, Volkhart Min-Jian Li, Gorden Redlich, Johannes-Peter Stasch, Stefan Weigand, Frank Wunder, Bicyclic aza heterocycles, and use thereof, WO2012028647 A1
2)Claudia Hirth-Dietrich, Peter Sandner, Johannes-Peter Stasch, Andreas Knorr, Degenfeld Georges Von, Michael Hahn, Markus Follmann, The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc), WO 2011147810A1

Jin Li, Xiaoyu Yang, Jingwei ZHU, Minmin Yang, Xihan Wu, Method for synthesizing 1-(2-fluorobenzyl)-1H -pyrazolo[3,4-b]pyridin -3-formamidine hydrochloride, WO2013086935 A1

veerareddy Arava, Surendrareddy Gogireddy, An expeditious synthesis of riociguat, A pulmonary hypertension drug, Der Pharma Chemica, 2013, 5(4):232-239
cut paste from my earlier post

RIOCIQUAT
CAS NO 625115-55-1
Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
9 APRIL2013
Bayer has been boosted by the news that regulators in the USA are fast-tracking the German group’s investigational pulmonary arterial hypertension riociguat.
The US Food and Drug Administration has granted priority review to the New Drug Application for riociguat, which Bayer filed in February on both sides of the Atlantic for PAH and a related condition, inoperable chronic thromboembolic pulmonary hypertension (CTEPH). The FDA bestows a priority review on medicines that offer major advances in care or that provide a treatment where no adequate therapy exists. The agency aims to complete its assessment within eight months from the submission of the NDA, rather than the standard 12 months.
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators
The submissions are based on two Phase III studies and riociguat, the first member of a novel class of compounds called stimulators of soluble guanylate cyclase (sGC), met its primary endpoint in both trials, a change in exercise capacity after 12- or 16 weeks respectively. The drug was generally well tolerated, with a good safety profile.

If approved, riociguat would be going up against Actelion’s Tracleer (bosentan) and Gilead Sciences/GlaxoSmithKline’s Letairis/Volibris (ambrisentan). Actelion, which has dominated the PAH market, has already filed its follow-up to Tracleer, Opsumit (macitentan).

Current Oncology Pipeline Trends

Oncology drug development far outpaces drug development for other therapeutic areas and the magnitude of that difference is significant. Here’s a current review of what is in the pipeline and an analysis of where oncology research is headed.

Stacey Ness, PharmD, RPh, MSCS, AAHIVP, has worked in both national specialty pharmacy and payer organizations and has experience in clinical management, adherence, and persistency programs, as well as chronic disease cost optimization strategies. Dr. Ness is active in the Consortium of Multiple Sclerosis Centers, Academy of Managed Care Pharmacy, National Home Infusion Association, National Association of Specialty Pharmacy, Specialty Pharmacy Certification Board, and Hematology and Oncology Pharmacy Association, and has served on the Minnesota Medicaid Drug Formulary Committee since 2008. She is a multiple sclerosis certified specialist, a credentialed HIV pharmacist, and currently serves as the director of specialty clinical services at Managed Health Care Associates, Inc, a health care services organization based in Florham Park, New Jersey.
OCRELIZUMAB

Ocrelizumab is a humanized anti-CD20 monoclonal antibody. It targets mature B lymphocytes[1] and hence is an immunosuppressive drug candidate. It is under development by Hoffmann–La Roche‘s subsidiary Genentech, and Biogen Idec.
It had reached Phase III clinical trials for rheumatoid arthritis[2] and lupus erythematosus,[3]and Phase II for multiple sclerosis[4] and hematological cancer.[5]
In March 2010, Roche announced the suspension of clinical trials in rheumatoid arthritis and lupus erythematosus. This step followed excess deaths due to opportunistic infections. Development for multiple sclerosis continues.[6]
In October 2010 Roche announced 24 week results from the PhII study in relapse remittingMS. The drug demonstrated a statistically significant reduction in disease activity as measured by brain lesions (measured by MRI scans) and relapse rate compared to placebo. Both doses (200 mg & 600 mg) were well tolerated.Anti-B cell therapy with rituximab has been shown to be safe and beneficial for RA treatment. Rituximab is approved and marketed for the treatment of RA in patients who have failed other therapies. Ocrelizumab, a fully human monoclonal antibody against CD20, may have less immunogenicity and less complement activation than rituximab which, theoretically, may reduce the development of drug neutralizing antibodies and infusion reactions. Here, Genovese at al report the results of a Phase I/II dose finding study of Ocrelizumab in RA patients who have failed other DMARDs (including prior TNF inhibitors).
- K. John Morrow Jr (2008-06-15). “Methods for Maximizing Antibody Yields”. Genetic Engineering & Biotechnology News (Mary Ann Liebert, Inc.). p. 36. Retrieved 2008-07-06. (Note: information included in this article only found in table present in print version of article.)
- Kausar, F; Mustafa, K; Sweis, G; Sawaqed, R; Alawneh, K; Salloum, R; Badaracco, M; Niewold, TB et al. (2009). “Ocrelizumab: a step forward in the evolution of B-cell therapy”. Expert opinion on biological therapy 9 (7): 889–95. doi:10.1517/14712590903018837.PMID 19463076.
- http://www.clinicaltrials.gov/ct2/show/NCT00539838 A Study to Evaluate Two Doses of Ocrelizumab in Patients With Active Systemic Lupus Erythematosus (BEGIN)
- http://www.clinicaltrials.gov/ct2/show/NCT00676715 A Study of the Efficacy and Safety of Ocrelizumab in Patients With Relapsing-Remitting Multiple Sclerosis.
- Hutas, G (2008). “Ocrelizumab, a humanized monoclonal antibody against CD20 for inflammatory disorders and B-cell malignancies”.Current opinion in investigational drugs (London, England : 2000) 9 (11): 1206–15. PMID 18951300.
- Katie Reid (2010-03-08). Update 2. Roche suspends arthritis treatment after deaths. Reuters. Retrieved 2010-03-08
GSK and Genmab seek alternative approval for leukaemia drug Arzerra

Arzerra
GlaxoSmithKline and Genmab A/S have announced the submission of leukaemia drug Arzerra to the European Medicines Agency (EMA) for a variation in marketing authorisation.
The companies are seeking authorisation for the drug to be used in combination with an alkylator-based therapy for treatment of Chronic Lymphocytic Leukemia (CLL) patients who have not received prior treatment and are inappropriate for fludarabine-based therapy.
READ ALL AT
Ofatumumab(trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.
Chronic lymphocytic leukemia (CLL) is a slowly progressing cancer of the blood and bone marrow. Arzerra (ofatumumab) has been approved by the FDA for treating CLL.
Patients with CLL whose cancer is no longer being controlled by other forms of chemotherapy can be prescribed Arzerra.
People older than fifty are mainly affected by CLL. A group of white blood cells known as B-cells that are part of the body’s immune system is the source of CLL. Every year, about ¼ of people diagnosed with CLL die from the disease.

Arzerra is an anti-CD20 monoclonal antibody that targets a membrane-proximal (which means close to the cell surface), small loop epitope, which is a portion of a molecule to which an antibody binds, on the CD20 molecule on B-cells. This epitope isn’t similar to binding sites that are targeted by other CD20 antibodies that are currently available. The CD20 molecule is highly expressed in most B-cell malignancies, making it a key target in CLL therapy.

MECHANISM OF ACTION:
Binding specifically to both the small and large extracellular loops of the CD20 molecule, Arzerra is an anti-CD20 monoclonal antibody. The CD20 molecule is expressed on normal B lymphocytes (pre-B- to mature B-lymphocyte) and on B-cell CLL. The CD20 molecule isn’t internalized following antibody binding and it isn’t shed from the cell surface. The Fc domain of ofatumumab mediates immune effector functions to result in B-cell lysis in vitro, while the Fab domain binds to the CD20 molecule. Complement-dependent cytotoxicity and antibody-dependent, cell-mediated cytotoxicity has been suggested as the possible mechanisms of cell lysis.
Products receive accelerated approval based on a surrogate endpoint, such as a reduction in the size of the tumor or decrease in the number of cancerous white cells or in an enlarged spleen or lymph nodes. These indirect measures for clinical outcomes are considered reasonably likely to predict that the drug will allow patients to live with fewer side effects of a disease or to live longer. Arzerra was approved under the FDA’s accelerated approval process, which allows earlier approval of drugs that meet unmet medical needs.

To confirm that the addition of Arzerra to standard chemotherapy delays the progression of the disease, the manufacturer of this medication is currently conducting a clinical trial in CLL patients. This is because the accelerated approval process requires further study of the drug.
Epratuzumab
Epratuzumab
Epratuzumab is a humanised anti-CD22 monoclonal antibody under investigation (clinical development phase III) for its efficacy in SLE. CD22 is a B cell specific surface protein that is considered to be involved in B cell function.
| Expected indication | Systemic lupus erythematosus |
| R&D stage | Phase 3 ongoing (started in December 2010) |
| Next milestone | Phase 3 results (H1 2014) |
| Quick facts |
|
Epratuzumab is a humanized monoclonal antibody. Potential uses may be found inoncology and in treatment of inflammatory autoimmune disorders, such as lupus (SLE).[1][2] The manufacturers in August 2009 announced success in early trials against SLE.[3]
Epratuzumab binds to the glycoprotein CD22 of mature and malignant B-cells.
- Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties Clinical Cancer Research Vol. 9, September 1, 2003 free full text
- Dose-Fractionated Radioimmunotherapy in Non-Hodgkin’s Lymphoma Using DOTA-Conjugated, 90Y-Radiolabeled, Humanized Anti-CD22 Monoclonal Antibody, Epratuzumab Clinical Cancer Research Vol. 11, July 15, 2005 free full text
- Reuters: UCB and Immunomedics Announce Positive Results for Epratuzumab Phase IIb Study in Systemic Lupus Erythematosus (SLE)
Epratuzumab is a humanized IgG1 antibody that acts as an antagonist of the CD22 receptor present on B cells. UCB is currently enrolling patients for the 2 Phase III trials, EMBODY-1 and EMBODY-2. The primary objective of both studies is to measure the percent of subjects meeting treatment response criteria at week 48 among those patients with moderate to severe SLE. Epratuzumab is dosed at either 600 mg per week or 1200 mg every other week administered over four 12-week treatment cycles.
The cumulative dose for both treatment arms is 2400 mg for each of the 4-week dosing periods. The estimated primary completion date is January 2014 for both EMBODY-1 and EMBODY-2. –
UCB pipeline. UCB Web site. www.ucb.com/rd/pipeline/new-development/epratuzumab. Published July 10, 2010. Accessed June 18, 2011
Brussels (Belgium), June 13th 2013, 0700 CEST – UCB today announced new data from an open-label extension (SL0008) of the EMBLEM™ phase 2b study evaluating the long-term effects of epratuzumab treatment in adult patients with moderate-to-severe systemic lupus erythematosus (SLE). The primary outcome of the open-label extension was to assess the safety of epratuzumab in patients with SLE.4
Relative to the 12 week, double-blind, placebo-controlled EMBLEM™ study, data from the open-label, long-term extension identified no new safety or tolerability signals.1 In addition, relative to EMBLEM™ baseline values, secondary outcome data indicated that the efficacy of epratuzumab as measured by reduction in disease activity was maintained over two years.2 Secondary outcome data also indicated that relative to EMBLEM™ baseline values, treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day.1 These data were presented this week at the European League Against Rheumatism 2013 Congress in Madrid, Spain.
Epratuzumab, licensed from Immunomedics Inc. (NASDAQ: IMMU), is an investigational medicine and the first CD-22/B-Cell receptor (BCR) targeted monoclonal antibody to be evaluated in clinical studies for the treatment of SLE. Also known as lupus, SLE is a complex, systemic autoimmune disease that affects many different organ systems, including the skin, joints, lungs, kidneys and blood.3,5
“In EMBLEM™, a dose-ranging, phase 2b study, reduction in disease activity was observed in patients treated with epratuzumab,” said Professor Daniel J Wallace MD, Clinical Professor of Medicine, Cedars-Sinai Medical Center, California, US. “This double-blind study had a relatively short 12-week, placebo-controlled, treatment period and it was important to accumulate long-term data on epratuzumab in the treatment of SLE. The phase 2b extension study adds new two year open-label data on epratuzumab to that already available from the 12-week, randomized, controlled study.”
EMBLEM™ was designed to identify a suitable dosing regimen for epratuzumab.6 A total of 227 patients with moderate-to-severe SLE received either: placebo, epratuzumab cumulative dose of 200 mg (100 mg every other week), 800 mg (400 mg every other week), 2400 mg (600 mg weekly), 2400 mg (1200 mg every other week) or 3600 mg (1800 mg every other week).3,6 In the open-label extension 203 patients from any arm of the EMBELM™ study received 1200 mg epratuzumab at weeks 0 and 2 of 12-week cycles.1,2,7
Data on epratuzumab presented at EULAR 2013
Evaluation of the safety profile of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Safety variables were primary outcome measures in SL0008 and included duration of exposure, adverse events, infusion reactions and infections.
Exposure to epratuzumab was a median 845 days over a median 10 treatment cycles. Adverse events (AEs) caused discontinuation in 29 (14.3%) patients. The most common serious AEs were SLE flare (3.4%), lupus nephritis (2%) and symptomatic cholelithiasis (1.5%). The most common infections/infestations were urinary tract infection (24.6%) and upper respiratory tract infection (23.2%). There were no opportunistic infections and no patterns of specific serious or severe infections.
Evaluation of long-term efficacy of epratuzumab as measured by reduction in disease activity in patients with moderate-to-severe SLE2
Secondary outcome measures in SL0008 included efficacy as measured by reduction in disease activity, and assessed by: British Isles Lupus Assessment Group (BILAG) improvement, SLE disease activity index (SLEDAI) score, Physician Global Assessment (PGA) score and combined treatment response defined as BILAG improvement without worsening, no SLEDAI worsening and no PGA worsening, relative to EMBLEM™ baseline.
The median BILAG total score was 25.0 at EMBLEM™ baseline and 9.0 at week 108. The score was 14.0 at SL0008 screening. Median SLEDAI score was 12.0 at EMBLEM™ baseline and 4.0 at week 108. The score was 10.0 at SL0008 screening. The median PGA score was 50.0 at EMBLEM™ baseline and 17.5 at week 108 with a score of 31.0 at SL0008 screening.
The proportion of patients achieving the combined treatment response was 32.5% at SL0008 screening (n=203) and 60.3% at week 108 (n=116).
Effect of corticosteroid use of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Corticosteroid doses were monitored throughout SL0008 and was a secondary outcome measure.
Median corticosteroid dose at EMBLEM™ baseline and SL0008 screening was 10.0 mg/day. At week 116, this was 5 mg/day (n=112). Data indicated that treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day with a corresponding increase in the proportion of patients receiving lower doses or no longer receiving corticosteroids.
The proportion of patients requiring 7.5-20 mg/day and >20 mg/day decreased (49.8% and 10.8% at baseline and 33.9% and 8.0% respectively, at week 116) and the proportion of patients receiving >0–7.5mg/day or no longer receiving corticosteroids increased (33.5% and 5.9% at baseline and 45.5% and 12.5% respectively, at week 116).
