
Home » Posts tagged 'NEW DRUGS' (Page 5)
Tag Archives: NEW DRUGS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GT Biologics obtains FDA orphan drug designation for paediatric Crohn’s drug
GT Biologics, a developer of live biotherapeutics for the treatment of autoimmune diseases, has received orphan drug designation from the US Food and Drug Administration (FDA) for its lead product candidate, Thetanix.
read all at
read all on
Bacteroides thetaiotaomicron
http://microbewiki.kenyon.edu/index.php/Bacteroides_thetaiotaomicron
Molidustat (BAY 85-3934), Bayer’s drug under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease.
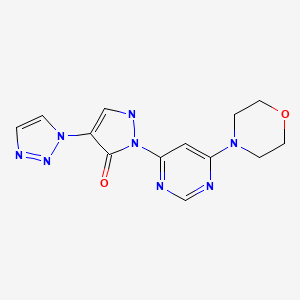
Molidustat
UNII-9JH486CZ13, cas no 1154028-82-6, MW: 314.3076
2-(6-morpholin-4-ylpyrimidin-4-yl)-4-(triazol-1-yl)-1H-pyrazol-3-one
Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
For the cardio-renal syndrome, a Phase IIb program with the investigational new drug Molidustat (BAY 85-3934) is under initiation in patients with anemia associated with chronic kidney disease and/or end-stage renal disease. Molidustat is a novel inhibitor of hypoxia-inducible factor (HIF) prolyl hydroxylase (PH) which stimulates erythropoietin (EPO) production and the formation of red blood cells. Phase I data have shown that inhibition of HIF-PH by Molidustat results in an increase in endogenous production of EPO.
About Bayer HealthCare
The Bayer Group is a global enterprise with core competencies in the fields of health care, agriculture and high-tech materials. Bayer HealthCare, a subgroup of Bayer AG with annual sales of EUR 18.6 billion (2012), is one of the world’s leading, innovative companies in the healthcare and medical products industry and is based in Leverkusen, Germany. The company combines the global activities of the Animal Health, Consumer Care, Medical Care and Pharmaceuticals divisions. Bayer HealthCare’s aim is to discover, develop, manufacture and market products that will improve human and animal health worldwide. Bayer HealthCare has a global workforce of 54,900 employees (Dec 31, 2012) and is represented in more than 100 countries. More information at www.healthcare.bayer.com.
molidustat
…………………………………………………………..
molidusat sodium
Sodium 1-[6-(morpholin-4-yl)pyrimidin-4-yl]-4-(1H-1,2,3-triazol-1-yl)-1H-pyrazol-5-olate
Molidustat sodium is an orally-available hypoxia-inducible factor prolyl hydroxylase inhibitor in phase I clinical trials at Bayer for the treatment of patients suffering from renal anemia due to chronic kidney disease.
WO 2008067871
WO 2012065967
WO 2013167552
…………………………
2-Heteroaryl-4-aryl-1,2-dihydropyrazolones having a bactericidal and/or fungicidal action are disclosed in EP 165 448 and EP 212 281. The use of 2-heteroaryl-4-aryl-1,2-dihydropyrazolones as lipoxygenase inhibitors for treatment of respiratory tract, cardiovascular and inflammatory diseases is claimed in EP 183 159. 2,4-Diphenyl-1,2-dihydropyrazolones having a herbicidal activity are described in DE 2 651 008.
The preparation and pharmacological properties of certain 2-pyridyl-1,2-dihydropyrazolones are reported in Helv. Chim. Acta 49 (1), 272-280 (1966). WO 96/12706, WO 00/51989 and WO 03/074550 claim compounds having a dihydropyrazolone partial structure for treatment of various diseases, and hydroxy- or alkoxy-substituted bipyrazoles for treatment of neuropsychiatric diseases are disclosed in WO 2006/101903.
Heteroaryl-substituted pyrazole derivatives for treatment of pain and various CNS diseases are furthermore described in WO 03/051833 and WO 2004/089303. WO 2006/114213 has meanwhile disclosed 2,4-dipyridyl-1,2-dihydropyrazolones as inhibitors of HIF prolyl 4-hydroxylases.
The x-ray crystal structure of the compound 3-methyl-1-(pyridin-2-yl)-4-(1-pyridin-2-yl-3-methyl-1H-pyrazol-5-yl)-2H-3-pyrazolin-5 (114)-one (other name: 5,5′-dimethyl-2,2′-di-pyridin-2-yl-1′,2′-dihydro-2H,3′H-3,4′-bipyrazol-3′-one) is reported inActa Crystallogr., Section E: Structure Reports Oμline E57 (11), o1126-o1127 (2001) [Chem. Abstr. 2001:796190].
The synthesis of certain 3′,5-dimethyl-2-phenyl-1′-(1,3-thiazol-2-yl)-1′H,2H-3,4′-bipyrazol-5′-ol derivatives is described inIndian J. Heterocyclic Chem. 3 (1), 5-8 (1993) [Chem. Abstr. 1994:323362].
The preparation and tautomerism of individual 4-(pyrazol-5-yl)-pyrazolin-5-one derivatives is reported in J. Heterocyclic Chem. 27 (4), 865-870 (1990) [Chem. Abstr. 1991:428557]. A therapeutic use has not hitherto been described for the compounds mentioned in these publications. The compound 2-tert-butyl-1′-[4-(4-chlorophenyl)-1,3-thiazol-2-yl]-3′,5-dimethyl-1′H,2H-3,4′-bipyrazol-5′-ol is listed as a test example in WO 2007/008541.
………………………….
https://www.google.co.in/patents/US20100305085
Example 3A 3-(Dimethylamino)-2-(1H-1,2,3-triazol-1-yl)acrylic acid ethyl ester

The preparation of the starting compound is carried out analogously to 2A starting from 1.00 g (6.45 mmol) 2-(1H-1,2,3-triazol-1-yl)acetic acid ethyl ester.
Yield: 1.4 g (100% of th.)
1H-NMR (400 MHz, DMSO-d6): δ=8.10 (d, 1H), 7.78 (d, 1H), 7.65 (s, 1H), 4.03 (q, 2H), 3.06 (br. s, 3H), 2.10 (br. s, 3H), 1.12 (t, 3H).
LC-MS (Method 5): Rt=1.40 min; MS (ESIpos): m/z=211 [M+H]+.
Example 16A 4-(6-Hydrazinopyrimidin-4-yl)morpholine

Stage a): 4-(6-Chloropyrimidin-4-yl)morpholine

45.0 g (302.1 mmol) 4,6-dichloropyrimidine are initially introduced into 450 ml water. 26.3 g (302.1 mmol) morpholine are added and the mixture is stirred at 90° C. for 16 h. Thereafter, it is cooled to 0° C. and the precipitate formed is filtered off. The precipitate is washed once with 50 ml water and dried in air.
Yield: 51.0 g (85% of th.)
LC-MS (Method 4): Rt=1.09 min; MS (ESIpos): m/z=200 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.35 (s, 1H), 6.95 (s, 1H), 3.62 (s, 8H).
Stage b) 4-(6-Hydrazinopyrimidin-4-yl)morpholine
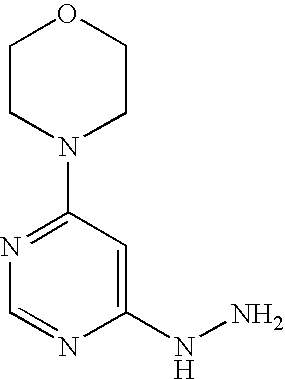
53.0 g (2.7 mmol) 4-(6-chloropyrimidin-4-yl)morpholine are initially introduced into 260 ml ethanol. 132.9 g (2.7 mol) hydrazine hydrate are added and the mixture is stirred under reflux for 16 h. Thereafter, it is cooled to RT and approx. half of the solvent is removed by distillation. The mixture is cooled to 0° C. and the solid formed is filtered off. It is rinsed with cold ethanol and the solid is dried first in air and then in vacuo.
Yield: 35.0 g (68% of th.)
LC-MS (Method 1): Rt=0.17 min; MS (ESIpos): m/z=196 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=7.94 (s, 1H), 7.70 (s, 1H), 5.91 (s, 1H), 4.15 (s, 2H), 3.66-3.60 (m, 4H), 3.45-3.37 (m, 4H).
Example 71 2-(6-Morpholin-4-ylpyrimidin-4-yl)-4-(1H-1,2,3-triazol-1-yl)-1,2-dihydro-3H-pyrazol-3-one

1.9 g (8.8 mmol) of the compound from Example 3A and 1.9 g (9.7 mmol) of the compound from Example 16A are initially introduced into 25 ml ethyl acetate and 504 mg (4.4 mmol) TFA are added at RT. The mixture is stirred under reflux for 16 h, then cooled to 5° C. and subsequently stirred for a further 2 h. The solid formed is filtered off, washed with ethyl acetate and dried first in air and thereafter under a high vacuum. 1.7 g of product are obtained.
The mother liquor is combined with the wash solution and the solvent is removed. According to LC-MS, the residue (2.4 g) still contains the intermediate 3-[2-(6-morpholin-4-ylpyrimidin-4-yl)hydrazino]-2-(1H-1,2,3-triazol-1-yl)prop-2-enoic acid ethyl ester (intermediate stage of the cyclization), which is used directly for the preparation of Example 72 (see there).
Yield: 1.7 g (61% of th.)
LC-MS (Method 9): Rt=0.90 min; MS (ESIpos): m/z=315 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.42 (s, 1H), 8.38 (s, 1H), 8.01 (s, 1H), 7.73 (s, 1H), 7.70 (s, 1H), 3.71-3.65 (m, 4H), 3.57-3.51 (m, 4H).
hydrochloride
Example 72 2-(6-Morpholin-4-ylpyrimidin-4-yl)-4-(1H-1,2,3-triazol-1-yl)-1,2-dihydro-3H-pyrazol-3-one hydrochloride

Batch 1: 7.5 ml of a 4 N solution of hydrogen chloride in dioxane are added to 1.7 g (5.4 mmol) of the compound from Example 71. The mixture is stirred at RT, 5 ml dioxane are added and the mixture is stirred at RT for 16 h. The solid is filtered off and washed with 5 ml dioxane. The mixture is dried under a high vacuum for 16 h, 10 ml methanol are then added and the mixture is stirred at RT for 1 h. The solid is filtered off, washed with 4 ml methanol and dried under a high vacuum. 1.6 g of the title compound are obtained.
Batch 2: A further amount of the title compound is obtained as follows: The residue (2.4 g) obtained from the mother liquor during the synthesis of Example Compound 71, which contains the open-ring intermediate state of the cyclization, 3-[2-(6-morpholin-4-ylpyrimidin-4-yl)hydrazino]-2-(1H-1,2,3-triazol-1-yl)prop-2-enoic acid ethyl ester, is dissolved in 12 ml ethanol and 1.5 ml 30% strength sodium methylate solution in methanol are added at RT, while stirring. The mixture is subsequently stirred at RT for 45 min, then adjusted to pH 5 with 2 N hydrochloric acid and subsequently stirred at RT for a further 16 h. The mixture is cooled to 10° C. and the solid is filtered off and washed with 3.5 ml dioxane. The mixture is dried under a high vacuum for 16 h, 5 ml methanol are then added and the mixture is subsequently stirred at RT for 1 h. The solid is filtered off, washed with 2 ml methanol and dried under a high vacuum to give a further 997 mg of the title compound in this way.
Yield: together 2.6 g (83% of th.)
LC-MS (Method 6): Rt=0.89 min; MS (ESIpos): m/z=315 [M+H]+;
1H-NMR (400 MHz, DMSO-d6): δ=8.54 (s, 1H), 8.39 (s, 1H), 8.28 (s, 1H), 7.88 (s, 1H), 7.42 (s, 1H), 3.71 (s, 8H).


amcrasto@gmail.com
email me if u like my posts
Finerenone (BAY 94-8862), BAYER’S next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone
CAS Number: 1050477-31-0, UNII-DE2O63YV8R
MW: 378.4298, C21-H22-N4-O3
Finerenone (BAY 94-8862) is a next generation oral, non-steroidal Mineralocorticoid Receptor antagonist which blocks the deleterious effects of aldosterone.
Currently available steroidal MR antagonists have proven to be effective in reducing cardiovascular mortality in patients with heart failure but have significant side effects that limit their utilization.
Finerenone is currently in clinical Phase IIb development for the treatment of worsening chronic heart failure, as well as diabetic nephropathy.
Glaxo Plans to File for Malaria Vaccine Approval Next Year

Malaria vaccine candidate reduces disease over 18 months of follow-up in late-stage study of more than 15,000 infants and young children
Malaria is a significant public health burden, claiming 660,000 lives a year – mostly children in sub-Saharan Africa
-Data support plan to submit regulatory application in 2014
Multilateral Initiative on Malaria Pan African Conference, Durban, South Africa — Results from a large-scale Phase III trial, presented today in Durban, show that the most clinically advanced malaria vaccine candidate, RTS,S, continued to protect young children and infants from clinical malaria up to 18 months after vaccination. Based on these data, GSK now intends to submit, in 2014, a regulatory application to the European Medicines Agency (EMA). The World Health Organization (WHO) has indicated that a policy recommendation for the RTS,S malaria vaccine candidate is possible as early as 2015 if it is granted a positive scientific opinion by EMA.
READ ALL AT
http://www.pharmalive.com/glaxo-plans-to-file-for-malaria-vaccine-approval-next-year
![]()
Current Oncology Pipeline Trends

Oncology drug development far outpaces drug development for other therapeutic areas and the magnitude of that difference is significant. Here’s a current review of what is in the pipeline and an analysis of where oncology research is headed.

Stacey Ness, PharmD, RPh, MSCS, AAHIVP, has worked in both national specialty pharmacy and payer organizations and has experience in clinical management, adherence, and persistency programs, as well as chronic disease cost optimization strategies. Dr. Ness is active in the Consortium of Multiple Sclerosis Centers, Academy of Managed Care Pharmacy, National Home Infusion Association, National Association of Specialty Pharmacy, Specialty Pharmacy Certification Board, and Hematology and Oncology Pharmacy Association, and has served on the Minnesota Medicaid Drug Formulary Committee since 2008. She is a multiple sclerosis certified specialist, a credentialed HIV pharmacist, and currently serves as the director of specialty clinical services at Managed Health Care Associates, Inc, a health care services organization based in Florham Park, New Jersey.
OCRELIZUMAB

Ocrelizumab is a humanized anti-CD20 monoclonal antibody. It targets mature B lymphocytes[1] and hence is an immunosuppressive drug candidate. It is under development by Hoffmann–La Roche‘s subsidiary Genentech, and Biogen Idec.
It had reached Phase III clinical trials for rheumatoid arthritis[2] and lupus erythematosus,[3]and Phase II for multiple sclerosis[4] and hematological cancer.[5]
In March 2010, Roche announced the suspension of clinical trials in rheumatoid arthritis and lupus erythematosus. This step followed excess deaths due to opportunistic infections. Development for multiple sclerosis continues.[6]
In October 2010 Roche announced 24 week results from the PhII study in relapse remittingMS. The drug demonstrated a statistically significant reduction in disease activity as measured by brain lesions (measured by MRI scans) and relapse rate compared to placebo. Both doses (200 mg & 600 mg) were well tolerated.Anti-B cell therapy with rituximab has been shown to be safe and beneficial for RA treatment. Rituximab is approved and marketed for the treatment of RA in patients who have failed other therapies. Ocrelizumab, a fully human monoclonal antibody against CD20, may have less immunogenicity and less complement activation than rituximab which, theoretically, may reduce the development of drug neutralizing antibodies and infusion reactions. Here, Genovese at al report the results of a Phase I/II dose finding study of Ocrelizumab in RA patients who have failed other DMARDs (including prior TNF inhibitors).
- K. John Morrow Jr (2008-06-15). “Methods for Maximizing Antibody Yields”. Genetic Engineering & Biotechnology News (Mary Ann Liebert, Inc.). p. 36. Retrieved 2008-07-06. (Note: information included in this article only found in table present in print version of article.)
- Kausar, F; Mustafa, K; Sweis, G; Sawaqed, R; Alawneh, K; Salloum, R; Badaracco, M; Niewold, TB et al. (2009). “Ocrelizumab: a step forward in the evolution of B-cell therapy”. Expert opinion on biological therapy 9 (7): 889–95. doi:10.1517/14712590903018837.PMID 19463076.
- http://www.clinicaltrials.gov/ct2/show/NCT00539838 A Study to Evaluate Two Doses of Ocrelizumab in Patients With Active Systemic Lupus Erythematosus (BEGIN)
- http://www.clinicaltrials.gov/ct2/show/NCT00676715 A Study of the Efficacy and Safety of Ocrelizumab in Patients With Relapsing-Remitting Multiple Sclerosis.
- Hutas, G (2008). “Ocrelizumab, a humanized monoclonal antibody against CD20 for inflammatory disorders and B-cell malignancies”.Current opinion in investigational drugs (London, England : 2000) 9 (11): 1206–15. PMID 18951300.
- Katie Reid (2010-03-08). Update 2. Roche suspends arthritis treatment after deaths. Reuters. Retrieved 2010-03-08
Orexigen files obesity drug Contrave for approval in Europe

Orexigen files obesity drug Contrave for approval in Europe
Orexigen Therapeutics has submitted the Marketing Authorization Application (MAA) for Contrave, an investigational weight-loss drug to the European Medicines Agency (EMA).
 The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
read all at
http://www.pharmatopics.com/2013/10/orexigen-files-obesity-drug-contrave-approval-europe/
Aerial Biopharma announce positive Phase II results for narcolepsy drug
ADX-N05, ARL-N05, SKL-N05
Aerial Biopharma announce positive Phase II results for narcolepsy drug
A new drug to treat excessive daytime sleepiness associated with narcolepsy has shown positive results from a phase 2b clinical trial, US-based Aerial Biopharma has announced this week.
read all at
Vortioxetine ボルチオキセチン 臭化水素酸塩 – FDA Approves Brintellix to Treat Major Depressive Disorder

Vortioxetine
ボルチオキセチン 臭化水素酸塩
1-[2-(2,4-dimethylphenyl)sulfanylphenyl]piperazine
Lu AA21004
| VORTIOXETINE; CAS 508233-74-7;
1-(2-((2,4-Dimethylphenyl)thio)phenyl)piperazine; Lu AA21004; UNII-3O2K1S3WQV; C18H22N2S; |
|
| Molecular Formula: | C18H22N2S |
|---|---|
| Molecular Weight: | 298.44568 g/mol |
Vortioxetine Hydrobromide

C18H22N2S.HBr : 379.36
[960203-27-4] HYDROBROMIDE
Vortioxetine is an atypical antipsychotic and antidepressant indicated for the treatment of major depressive disorder (MDD). It is classified as a serotonin modulator and simulator (SMS) as it has a multimodal mechanism of action towards the serotonin neurotransmitter system whereby it simultaneously modulates one or more serotonin receptors and inhibits the reuptake of serotonin. More specifically, vortioxetine acts via the following biological mechanisms: as a serotonin reuptake inhibitor (SRI) through inhibition of the serotonintransporter, as a partial agonist of the 5-HT1B receptor, an agonist of 5-HT1A, and an antagonist of the 5-HT3, 5-HT1D, and 5-HT7 receptors. SMSs were developed because there are many different subtypes of serotonin receptors, however, not all of these receptors appear to be involved in the antidepressant effects of SRIs. Some serotonin receptors seem to play a relatively neutral or insignificant role in the regulation of mood, but others, such as 5-HT1A autoreceptors and 5-HT7 receptors, appear to play an oppositional role in the efficacy of SRIs in treating depression.
Sept. 30, 2013 — The U.S. Food and Drug Administration today approved Brintellix (vortioxetine) to treat adults with major depressive disorder.
Major depressive disorder (MDD),
 Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
READ ALL AT
http://www.drugs.com/newdrugs/fda-approves-brintellix-major-depressive-disorder-3918.html
The disease: Major depression
The developers: Lundbeck, Takeda
Vortioxetine (vor-tye-OX-e-teen, code name Lu AA21004) is an experimental drug currently under development by Lundbeck and Takeda for the treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD).Commercial names chosen are Brintellix and Rexulti.
Regulatory approval for the treatment of MDD for the European market has been filed in September 2012, for the United States in October 2012, and filing for Canada should follow. Filing for the Japanese market is expected in 2013.

Depression
In May 22 2011, Lundbeck presented the results of four phase III trials on vortioxetine at the 2011 Annual Meeting of the American Psychiatric Association. A statistically significant effect was shown in two of the studies (one for active treatment using the Hamilton Depression Rating Scale (HAM-D), the second as a maintenance treatment), vortioxetine failed to prove superiority over placebo in a third (again using the HAM-D) and the fourth was nullified by an exceptionally high placebo response (according to the Montgomery-Åsberg Depression Rating Scale (MADRS)).
In July 2011, Lundbeck published the results of a double-blind, randomized, placebo-controlled clinical trial with venlafaxine as an active reference. It was found to be superior to placebo in treating MDD while having fewer side effects than venlafaxine. Similarly, in May 2012, Lundbeck published the results of a double-blind, randomized, placebo-controlled clinical trial with duloxetine evaluating vortioxetine in elderly depressed patients, and it was found superior to placebo, with fewer side effects than duloxetine.
In May 2012, Lundbeck disclosed the results of three phase III clinical trials, showing vortioxetine’s superiority over placebo according to the MADRS.
In August 2012, a randomized, double-blind trial confirms the superiority of vortioxetine over placebo according to all measures, excepted the Sheehan Disability scale.
In September 2012, a randomised, double-blind trial reveals that a dose of 5mg shows superiority over placebo only in patients that suffer from comorbid anxiety.This is consistent with results from another trial published in December 2012, demonstrating that 2.5 mg and 5 mg doses are ineffective.
Anxiety
August 2012, contradictory results of two randomized, double-blind trial were published. While the first demonstrated vortioxetine’s superiority over the placebo, the second showed that the drug had no efficacy, leading the authors to question the designs of the different trials.

United States Patent Number: 7,144,884 , 8,476,279
related to Chinese patent: CN1319958 C , CN1561336 A; CN1319958C, CN1561336A
patent validity: January 9, 2023 (U.S. Patent Number: 7,144,884), October 2, 2022 (U.S. Patent No.: 8,476,279)
peak annual sales (estimated): $ 2 billion
drug companies: Lundbeck (Lundbeck), Takeda (Takeda)
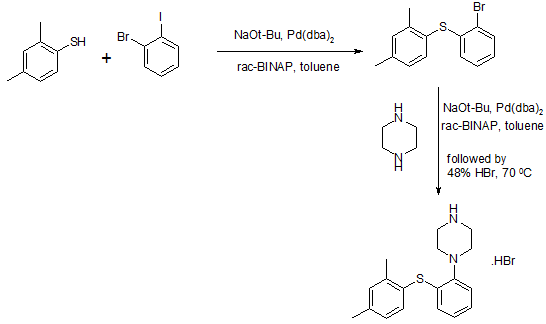
Wal antidepressant drug Paxil (Brintellix, Vortioxetine) for – 1 – Preparation – [2 – (2,4 methyl) phenyl] piperazine process
Method II:
815g of the NaOBut (8,48 mo1), 844 g of piperazine Qin (9,8 mol), 6,6 g of Pd (dba) 2 (11,48 mmol) and 13,6 g of rac-BINAP (21, 84 mmol) was stirred for 50 minutes with 4L ofbenzene. Then, 840 g 2 – bromo – iodobenzene (2,97 mol) and 1.5L of Yue added to the mixture with benzene, and the stirring was continued for 30 minutes. Finally, 390.8g of 2,4 -thiophenol (2,83 mol) was added together with 1.5L toluene. The resulting suspension was heated to reflux and reflux was continued for 5 hours. The reaction mixture was cooled overnight. 2L of water was added and stirred for l hour and then filtered through a filter aid, the resulting mixture. Then, the filtrate was washed with brine 3xlL. Subsequently, the combined aqueous phase extracted with 600ml of benzene. Then, Yue The combined benzene phase was heated to 70 ° C, then adding 329.2ml 48-wt. / HBr (aq.) and 164.6ml water o’s. The mixture was cooled to room temperature overnight. Final product was collected by filtration (l-[2 – (2,4 – di曱group – phenylsulfanyl) – phenyl] – piperazine hydrobromide Qin), and dried under vacuum (60 0 C), to give 895g of product (84% yield).
Method III:
The benzene is placed 500ml three-necked 1L round bottom flask equipped with a mechanical stirrer and add 809mg Pd2dba3 (0.88mmol; 0.5 mol%) and 952 mg DPEPhos (1.77 mmol; 0.5mol-%). The deep red solution was purged with nitrogen for 5 minutes, then add 100g2-bromo-iodobenzene (353 mmol) and 48.9 g 2,4 – bis thiophenol (353 mmol). Add 43.6g KOBut (389 mmol) caused an exothermic reaction, so that the temperature rise of 20 ° C 42 ° C, while forming a non-uniform mixture, and the color changed from deep red to orange / brown. The force of the suspension under nitrogen was heated to port 100 ° C. After only 20 minutes, HPLC showed complete conversion to have l-(2 – bromo – phenylsulfanyl) -2,4 – Yue group – benzene. The mixture was cooled to 40 ° C, was added to 600ml 15-wt% NaCl, and stirred for 5 minutes. The organic phase was separated, and the aqueous phase was washed 2xl00mwith benzene. The combined organic phase was washed with HCl (aq) NaCl and washed with 100ml 2M 100ml 15-wt%, and then Na 2 S04 dried by activated charcoal (10 g) at reflux for 15 minutes, filtered twice and evaporated to 107.3 g of orange-red oil (103%), the oil was found by HPLC purity of 98%.
To 90 g of the orange-red oil (307 mmol) in 500ml of anhydrous toluene was added 57 g boc-piperazine Qin (307 mmol), degassed with nitrogen for 5 minutes, was added 1.4g Pd2dba3 (1.53 mmol- %; 0.5 mol%) and 2.9g mc-BINAP (4.6 mmol; 1.5 mol-%), degassed and then another 2 minutes, then add 35.4 g of NaOtBu (368 mmol), and heated to 80 ° C for 18 hours. HPLC showed complete conversion to have the reaction mixture was cooled to RT, filtered, and the filter cake was washed with 2 x 100ml of曱benzene. % NaCl, washed twice in Na2S04 dried, added charcoal, refluxed for 30 minutes, filtered twice and evaporated to 140.7 g of a brown oil (4 – – The combined filtrates with 2 x 150ml 15 [2 – (2, 4 – di曱group – phenylsulfanyl) -. phenyl]-BOC-piperazine Qin). The resulting crude oil was dissolved in 300ml MeOH and 200ml 6MHCl (aq.) and refluxed for l hour, after which HPLC showed complete deprotection. After cooling to RT, the vacuum on a rotary evaporator to remove曱alcohol was added 20ml of concentrated NaOH (pH was measured to 13-14), after which the mixture with 1000ml EtOAc – 15 minutes from stirring. The organic phase was collected and dried 300ml 15wtQ /. Saline extraction in Na2S04 dried, and added 46.3 g of fumaric acid in 300mlMeOH (399 mmol) was added. The mixture was heated to reflux, cooled to room temperature and then placed in the tank (-18. C) overnight. The precipitate was collected, washed with 100ml and 100ml of acetone with EtOAc, and dried in vacuo (50 ° C), to give 103.2g of l-[2 – (2,4 – di group – phenylsulfanyl) – phenyl] – piperazine. Qin fumarate (249mmo1), as a white powder, overall yield 81%, determined by LC-MS and the purity was 99% fumarate. Use EtOAc/H20 / concentrated NaOH to the fumarate salt into the free base (l-[2 – (2,4 – dimethyl – phenylsulfanyl) – phenyl] – piperazine Qin), The organic phase was washed with brine, dried over Na 2 S04 sulfate, filtered and to the filtrate was added 34ml48-wto / o of HBr (aq.), to form a white solid precipitated. The solid was collected, and the solid was washed with 1000ml H20 boiling process, the resultant was cooled to room temperature and purified by forming a slurry. The final product was collected by filtration (l-[2 – (2,4 – digroup – phenylsulfanyl) – phenyl] – piperazine hydrobromide Qin Kr), and dried in vacuo (50 ° C), to produce 83g of white powder (total yield 71%).
Source:
1) Bang-Andersen B, Ruhland T, Jørgensen M, Smith G, Frederiksen K, Jensen KG, Zhong H, Nielsen SM, Hogg S, Mørk A, Stensbøl TB “Discovery of 1 -. [2 – (2,4 – dimethylphenylsulfanyl) phenyl] PIPERAZINE (Lu AA21004): a novel multimodal Major Compound for the treatment of depressive disorder. ” Journal of Medicinal Chemistry 54 (9): 3206-21.
2) Thomas Ruhland, Garrick Paul Smith, Benny Bang-Andersen, Ask Puschl, Ejner Knud Moltzen, Kim Andersen,; Phenyl-piperazine derivatives as serotonin reuptake inhibitors; US patent number 7144884 ; also published as CA2462110A1, CA2462110C , CN1319958C, CN1561336A, DE60225162D1, DE60225162T2, DE60233608D1, EP1436271A1, EP1436271B1, EP1749818A2, EP1749818A3, EP1749818B1, US7138407, US7148238, US7683053, US8110567, US8476279, US20050014740, US20060084662, US20060089368, US20070060574, US20110009423, US20120302553, WO2003029232A1; H. Lundbeck A / S;
T · Rouland, G · P · Smith, B · Bang – Anderson, A · Pi Shier, E · K · Moore Cen, K · Anderson; as serotonin reuptake inhibitors phenyl piperazine derivatives matter; CN 1319958 C
T · Rouland, G · P · Smith, B · Bang – Anderson, A · Pi Shier, E · K · Moore Cen, K · Anderson; as serotonin reuptake inhibitors phenyl piperazine derivatives; CN 1561336 A
3) Benny Bang-Andersen; Phenyl-piperazine derivatives as serotonin reuptake inhibitors; US patent number 8476279 B2 ; Also published as CA2462110A1, CA2462110C, CN1319958C, CN1561336A, DE60225162D1, DE60225162T2, DE60233608D1, EP1436271A1, EP1436271B1, EP1749818A2, EP1749818A3, EP1749818B1, US7138407, US7144884, US7148238, US7683053, US8110567, US20050014740, US20060084662, US20060089368, US20070060574, US20110009423, US20120302553, WO2003029232A1; H. Lundbeck A / S;
4) Kim Lasse Christensen; Process for the manufacture of 1 – [2 – (2,4-dimethyl-phenylsulfanyl)-phenyl]-piperazine; PCT application, WO2013102573 A1
5) Benny Bang-Andersen, Joergen Brodersen, Andre Faldt, Rene Holm, Morten Joergensen, De Diego Heidi Lopez, Michael J Mealy, Arne Moerk, Nicholas Moore, Lone Munch Ringgaard, Michael Harold Rock, Tine Bryan Stensboel; 1 – [2 – (2, 4-dimethylphenylsulfanyl)-phenyl] piperazine as a compound with combined serotonin reuptake, 5-ht3 and 5-ht1a activity for the treatment of cognitive impairment; WO2007144005 A1
Updated oct 2015…………….

Vortioxetine (vor-tye-oks-e-teen, trade name Trintellix) is an atypical antidepressant (a serotonin modulator and stimulator) made by Lundbeck and Takeda.[1]
Vortioxetine [1-[2-(2,4-Dimethylphenyl-sulfanyl)-phenyl]-piperazine] is an orally administered small molecule developed as once-daily treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD). As a drug, Vortioxetine is a bis-aryl-sulphanyl amine compound that combines serotonin (5-HT) reuptake inhibition with other characteristics, including receptor activity modulation.
Animal and in vitro studies indicate that several neurotransmitter systems may be impacted by vortioxetine, with the drug enhancing levels of 5-HT, noradrenaline, dopamine, acetylcholine and histamine in certain areas of the brain, as well as modulating γ-aminobutyric acid and glutamate neurotransmission. Results from additional animal models suggest vortioxetine may also improve measures of cognitive function, such as memory. In healthy volunteers, single or repeated administration of vortioxetine (10 mg) did not impair cognitive function, psychomotor performance or driving ability in a placebo-controlled study.
Medical use
Vortioxetine is used as first-line treatment for major depressive disorder.[1][2][3][4][5]
Pharmacokinetics
Vortioxetine reaches peak plasma concentration (Cmax) within 7 to 11 hours post-administration (Tmax), and its mean terminal half-life (t½) is ≈ 66 hours. Steady-state plasma concentrations are typically reached within two weeks.[1] It has no active metabolites (i.e. it is not a prodrug).[1]
Research
Vortioxetine has been studied in several clinical trials as a potential treatment for general anxiety disorder but results were inconsistent.[9][10]
History
Vortioxetine was discovered by scientists at Lundbeck who reported the rationale and synthesis for the drug (then called Lu AA21004) in a 2011 paper.[7][11]
In 2007, the compound was in Phase II clinical trials, and Lundbeck and Takeda entered into a partnership in which Takeda paid Lundbeck $40 million upfront, with promises of up to $345 million in milestone payments, and Takeda agreed to pay most of the remaining cost of developing the drug. The companies agreed to co-promote the drug in the US and Japan, and that Lundbeck would receive a royalty on all such sales. The deal included another drug candidate, tedatioxetine (Lu AA24530), and could be expanded to include two other Lundbeck compounds.[12]
Vortioxetine was approved by the U.S. FDA for the treatment of major depressive disorder (MDD) in adults in September, 2013,[13] and it was approved in Europe later that year.[14]
Vortioxetine was previously trademarked as Brintellix in the United States, but on May 2, 2016, the US FDA approved a name change to Trintellix in order to avoid confusion with the blood-thinning medication ticagrelor (Brilinta).[15]
WO2015155153, SYNTHESIS OF VORTIOXETINE VIA (2,4-DIMETHYLPHENYL)(2-IODOPHENYL)SULFANE INTERMEDIATE
| LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI) | |
| Inventors:ZUPANCIC, Borut; (SI) |
Vortioxetine is disclosed as Example 1 e in WO 2003/029232 A1 and is described as being prepared analogously to Example 1 . The process used to prepare Example 1 involves the preparation of 1 -(2-((2-(trifluoromethyl)phenyl)thio)phenyl)piperazine on a solid polystyrene support, followed by decomplexation using visible light irradiation, and purification by preparative LC-MS and ion-exchange chromatography. The overall yield for the preparation of vortioxetine is described as 17%.
Several alternative palladium catalyzed processes for the preparation of vortioxetine are described in Examples 17 to 25 of WO 2007/144005 A1 . These processes describe the preparation of vortioxetine from 2,4-dimethylthiophenol and 2-bromoiodobenzene (or 1 ,2-dibromobenzene) starting materials via a 1 -(2-bromo-phenylsulfanyl)-2,4-dimethyl-benzene intermediate. Each of these processes involves the use of a palladium catalyst and a phosphine ligand.
The preparation of vortioxetine is also described by Bang-Andersen et al. in J. Med. Chem. (201 1 ), Vol. 54, 3206-3221 . Here, in a first step, te/t-butyl 4-(2-bromophenyl)piperazine-1 -carboxylate intermediate is prepared from Boc-piperazine and 2-bromoiodobenzene in a palladium catalyzed coupling reaction. te/t-Butyl 4-(2-bromophenyl)piperazine-1 -carboxylate is then reacted with 2,4-dimethylthiophenol, again in the presence of palladium catalyst and a phosphine ligand, to provide Boc-protected vortioxetine. In the final step, vortioxetine is deprotected using hydrochloric acid to give vortioxetine hydrochloride.
WO 2013/102573 A1 describes a reaction between 1 -halogen-2,4-dimethyl-phenyl, 2-halogen-thiophenol and an optionally protected piperazine in the presence of a base and a palladium catalyst consisting of a palladium source and a phosphine ligand.
Each of the above processes has disadvantages. The process described in WO 2003/029232 is low yielding and unsuitable for the large scale production of vortioxetine, whereas the processes described in WO 2007/144005 A1 , WO 2013/102573 A1 and by Bang-Andersen et al. require the use of expensive starting materials, palladium catalyst and phosphine ligand. In addition, the toxicity of palladium is well known, Liu et al. Toxicity of Palladium, Toxicology Letters, 4 (1979) 469-473, and the European Medicines Agency’ s Guideline on the Specification for Residues of Metal Catalysts sets clear limits on the permitted daily exposure to palladium arising from palladium residue within drug substances, http://www.ema.europa.eu. Thus it would be desirable to avoid the use of a palladium catalyst in the synthesis of vortioxetine and the subsequent purification steps required to remove palladium residue from the final pharmaceutical product.
The invention is described below in further detail by embodiments, without being limited thereto.
A general concept of the process of the present invention may be represented in Scheme 1 .

Scheme 1 : General representation of the basic synthetic concept of the present invention.
Scheme 2.

X = NH2: lb
Scheme 2: Representation of a particular synthetic embodiment of the present invention.
Compound III can also be prepared from 2,4-dimethylbenzenethiol (II) and 1 -fluoro-2-nitrobenzene (l”‘a) or 1 -chloro-2-nitrobenzene (l'”b). In the first step (2,4-dimethylphenyl)(2- nitrophenyl)sulfane (III’) is formed and in the second reaction step nitro group is reduced to ami

Z = F: l”‘a
Z = CI: l”‘b
Scheme 3: Representation of a particular synthetic embodiment of the present invention.
Example 7: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine vortioxetine, VII)

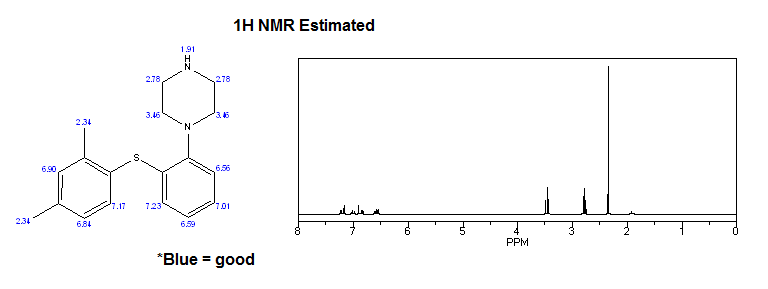
Mixture of (2,4-dimethylphenyl)(2-iodophenyl)sulfane V (0.34 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and 2-phenylphenol (68 mg, 0.4 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120°C for 20 h. Water (10 mL) is then added and product is extracted to EtOAc (3 x 10 mL). Combined organic layers were washed with water (3 x 10 mL) and brine (2 x 10 mL) and dried over Na2S04. After evaporation of the solvent crude product is purified by chromatography to afford title compound: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-
3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J= 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Example 8: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine (vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-iodophenyl)sulfane V (0.34 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and N,N-diethyl-2-hydroxybenzamide (39 mg, 0.2 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120 ^ for 20 h. Water (10 mL) is then added and product is extracted to EtOAc (3 x 10 mL). Combined organic layers were washed with water (3 x 10 mL) and brine (2 x 10 mL) and dried over Na2S04. After evaporation of the solvent crude product is purified by chromatography to afford title compound: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J= 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Example 9: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine hydrobromide
(vortioxetine HBr, VII.HBr)

To a solution of vortioxetine VII (1 .80 g, 6.03 mmol) in iPrOAc (20 mL) at room temperature 48% HBr (0.68 mL, 6.03 mmol) was slowly added. Obtained mixture was stirred at room temperature for 1 h, white precipitate was then filtered off, washed with acetone (2 x 20 mL), and dried to afford title compound VII.HBr as a white powder (2.15 g, 94% yield): H NMR (DMSO-d6, 500 MHz) δ 2.23 (s, 3H), 2.32 (s, 3H), 3.15-3.27 (m, 8H), 6.40 (m, 1 H), 6.96 (m, 1 H), 7.08-7.17 (m, 3H), 7.24 (m, 1 H), 7.32 (d, J= 7.8 Hz, 1 H), 8.85 (br, 2H).
Reference Example 1 : Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of piperazine (1 .0 g, 1 1 .6 mmol), NaOtBu (1 .37 g, 13.8 mmol), Pddba2 (40 mg, 0.07 mmol), and 1 ,3-bis(2,6-di-i-propylphenyl)imidazolium chloride (24 mg, 0,07 mmol) in dry and degassed toluene (10 mL) is stirred at room temperature for 1 h. (2,4-Dimethylphenyl)(2-iodophenyl)sulfane V (1 .32 g, 3.86 mmol) is then added, reaction mixture is heated to l OO’C and stirred for 24 h. After cooling to room temperature to the reaction mixture water (5 mL) and Celite (0.4 g) is added. After stirring for 20 min salts are filtered off, organic layer is separated, washed with brine (2 x 10 mL), dried over Na2S04 and solvent is evaporated to afford crude product, which is then purified by chromatography to afford title compound as yellowish crystals: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H),
Reference Example 2: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of piperazine (1 .29 g, 15.0 mmol), NaOtBu (1 .77 g, 17.8 mmol), Pddba2 (52 mg, 0.09 mmol), and rac-BINAP (93 mg, 0,15 mmol) in dry and degassed toluene (10 mL) was stirred at room temperature for 1 h. (2,4-Dimethylphenyl)(2-iodophenyl)sulfane V (1 .70 g, 5.0 mmol) was then added, reaction mixture was heated to 100°C and stirred for 24 h. After cooled to room temperature to the reaction mixture water (5 mL) and Celite (0.4 g) were added. After stirring for 20 min salts were filtered off, organic layer was separated, washed with brine (2 x 10 mL), dried over Na2S04 and solvent was evaporated to afford product as an orange oil (1 .41 g, 95% yield): H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J = 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Comparative Example 1 : Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-bromohenyl)sulfane V” (0.29 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and 2-phenylphenol (68 mg, 0.4 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120°C for 20 h. Vortioxetine VII was not formed.
Comparative Example 2: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-bromophenyl)sulfane V (0.29 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and N,N-diethyl-2-hydroxybenzamide (39 mg, 0.2 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120 ^ for 20 h. Vortioxetine VII was not formed.
WO2007144005A1: Industrial process

Discovery of 1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A Novel Multimodal Compound for the Treatment of Major Depressive Disorder

The synthesis and structure−activity relationship of a novel series of compounds with combined effects on 5-HT3A and 5-HT1A receptors and on the serotonin (5-HT) transporter (SERT) are described. Compound 5m (Lu AA21004) was the lead compound, displaying high affinity for recombinant human 5-HT1A (Ki = 15 nM), 5-HT1B (Ki = 33 nM), 5-HT3A (Ki = 3.7 nM), 5-HT7 (Ki = 19 nM), and noradrenergic β1 (Ki = 46 nM) receptors, and SERT (Ki = 1.6 nM). Compound 5mdisplayed antagonistic properties at 5-HT3A and 5-HT7 receptors, partial agonist properties at 5-HT1B receptors, agonistic properties at 5-HT1A receptors, and potent inhibition of SERT. In conscious rats, 5m significantly increased extracellular 5-HT levels in the brain after acute and 3 days of treatment. Following the 3-day treatment (5 or 10 (mg/kg)/day) SERT occupancies were only 43% and 57%, respectively. These characteristics indicate that 5m is a novel multimodal serotonergic compound, and 5m is currently in clinical development for major depressive disorder.
1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine Hydrochloride (5m)
ALERT HYDROCHLORIDE DATA
References
- US Label Last updated July 2014 after review in September, 2014. Versions of label are available at FDA index page Page accessed January 19, 2016
- [No authors listed] Vortioxetine. Aust Prescr. 2015 Jun;38(3):101-2. PMID 26648632Free full text
- “Relative efficacy and tolerability of vortioxetine versus selected antidepressants by indirect comparisons of similar clinical studies.”. Curr Med Res Opin 30: 2589–606. Oct 10, 2014. doi:10.1185/03007995.2014.969566. PMID 25249164.
- Köhler S, Cierpinsky K, Kronenberg G, Adli M. The serotonergic system in the neurobiology of depression: Relevance for novel antidepressants. J Psychopharmacol. 2016 Jan;30(1):13-22. PMID 26464458
- Kelliny M, Croarkin PE, Moore KM, Bobo WV. Profile of vortioxetine in the treatment of major depressive disorder: an overview of the primary and secondary literature. Ther Clin Risk Manag. 2015 Aug 12;11:1193-212. PMID 26316764 Free full text
- “Lundbeck’s “Serotonin Modulator and Stimulator” Lu AA21004: How Novel? How Good? – GLG News”.
- ^ Jump up to:a b c Bang-Andersen B, Ruhland T, Jørgensen M, et al. (May 2011). “Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder”. Journal of Medicinal Chemistry 54 (9): 3206–21. doi:10.1021/jm101459g. PMID 21486038.
- N. Moore; B. Bang-Andersen; L. Brennum; K. Fredriksen; S. Hogg; A. Mork; T. Stensbol; H. Zhong; C. Sanchez; D. Smith (August 2008). “Lu AA21004: a novel potential treatment for mood disorders”. European Neuropsychopharmacology 18 (Supplement 4): S321.doi:10.1016/S0924-977X(08)70440-1.
- Pae CU et al. Vortioxetine, a multimodal antidepressant for generalized anxiety disorder: a systematic review and meta-analysis. J Psychiatr Res. 2015 May;64:88-98. PMID 25851751
- Reinhold JA, Rickels K. Pharmacological treatment for generalized anxiety disorder in adults: an update. Expert Opin Pharmacother. 2015;16(11):1669-81. PMID 26159446
- Sanchez C, Asin KE, Artigas F Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol Ther. 2015 Jan;145:43-57. PMID 25016186 Free full text
- Daniel Beaulieu for First Word Pharma. September 5th, 2007 Lundbeck, Takeda enter strategic alliance for mood disorder, anxiety drugs
- FDA approves new drug to treat major depressive disorder, U.S. Food and Drug Administration Press Announcement.
- EMA Brintellix page at EMA site Page accessed January 19, 2016
- Commissioner, Office of the. “Safety Alerts for Human Medical Products – Brintellix (vortioxetine): Drug Safety Communication – Brand Name Change to Trintellix, to Avoid Confusion With Antiplatelet Drug Brilinta (ticagrelor)”. http://www.fda.gov. Retrieved2016-05-02.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7683053 | 2010-03-23 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| US7148238 | 2006-12-12 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US7144884 | 2006-12-05 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US7138407 | 2006-11-21 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013184291 | 2013-07-18 | THERAPEUTIC USES OF 1-[2-(2,4-DIMETHYL-PHENYLSUFLANYL)PHENYL]PIPERAZINE |
| US8476279 | 2013-07-02 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US2013115292 | 2013-05-09 | ENTERIC TABLET |
| US2012189697 | 2012-07-26 | COMPOSITIONS OF 1-[2-(2,4-DIMETHYL-PHENYLSULFANYL)-PHENYL]PIPERAZINE |
| US2012035188 | 2012-02-09 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]-PIPERAZINE |
| US8110567 | 2012-02-07 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| US2012004409 | 2012-01-05 | Purification of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine |
| US2011201617 | 2011-08-18 | Therapeutic Uses Of Compounds Having Combined SERT, 5-HT3 And 5-HT1A Activity |
| US2011009422 | 2011-01-13 | 1- [2-(2,4-DIMETHYLPHENYLSULFANYL)-PHENYL] PIPERAZINE AS A COMPOUND WITH COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF PAIN OR RESIDUAL SYMPTOMS IN DEPRESSION RELATING TO SLEEP AND COGNITION |
| US2010297240 | 2010-11-25 | 1- [2- (2,4-DIMETHYLPHENYLSULFANYL)-PHENYL] PIPERAZINE AS A COMPOUND WITH COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF COGNITIVE IMPAIRMENT |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094316 | 2015-04-02 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]PIPERAZINE |
| US8969355 | 2015-03-03 | 1-[2-(2,4 dimethylphenylsulfanyl)-phenyl]piperazine as a compound with combined serotonin reuptake, 5-HT3 and 5-HT1a activity for the treatment of cognitive impairment |
| US2015005318 | 2015-01-01 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014371453 | 2014-12-18 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014343287 | 2014-11-20 | Process for the Manufacture of 1-[2-(2,4-Dimethyl-Phenylsulfanyl)-Phenyl]-Piperazine |
| US2014315921 | 2014-10-23 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014256943 | 2014-09-11 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014248355 | 2014-09-04 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014248356 | 2014-09-04 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014163043 | 2014-06-12 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016083359 | 2016-03-24 | 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)-PHENYL]PIPERAZINE AS A COMPOUND With COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF COGNITIVE IMPAIRMENT |
| US2016060215 | 2016-03-03 | New Process For The Synthesis Of 1-(2-((2,4-Dimethylphenyl)Thio)Phenyl)Piperazine |
| US2016015706 | 2016-01-21 | CRYSTALLINE FORMS OF AN ANTIDEPRESSANT COMPOUND |
| US2016009670 | 2016-01-14 | VORTIOXETINE MANUFACTURING PROCESS |
| US9211288 | 2015-12-15 | Compositions comprising vortioxetine and donepezil |
| US2015297585 | 2015-10-22 | COMPOSITIONS COMPRISING VORTIOXETINE AND DONEPEZIL |
| US2015266841 | 2015-09-24 | Novel Crystalline Form Of Vortioxetine Hydrobromide |
| US2015150867 | 2015-06-04 | COMPOSITIONS OF 1-[2-(2,4-DIMETHYL-PHENYLSULFANYL)-PHENYL]PIPERAZINE |
| US2015110873 | 2015-04-23 | ENTERIC TABLET |
| US2015094316 | 2015-04-02 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]PIPERAZINE |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
1-[2-(2,4-Dimethyl-phenylsulfanyl)-phenyl]piperazine
|
|
| Clinical data | |
| Trade names | Trintellix, Brintellix |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 75% (peak at 7–11 hours) |
| Protein binding | 98% |
| Metabolism | extensive hepatic, primarilyCYP2D6-mediated oxidation |
| Biological half-life | 66 hours |
| Excretion | 59% in urine, 26% in feces |
| Identifiers | |
| CAS Number | 508233-74-7 |
| ATC code | N06AX26 (WHO) |
| PubChem | CID 9966051 |
| IUPHAR/BPS | 7351 |
| ChemSpider | 8141643 |
| KEGG | D10184 |
| ChEBI | CHEBI:76016 |
| Synonyms | Lu AA21004 |
| Chemical data | |
| Formula | C18H22N2S |
| Molar mass | 298.45 g/mol (379.36 as hydrobromide) |
/////////////
-
CC(C=C(C)C=C1)=C1SC2=C(N3CCNCC3)C=CC=C2
Certolizumab pegol – FDA gave green light to UCB’s Cimzia to treat psoriatic arthritis

Certolizumab pegol
The US Food and Drug Administration has approved UCB’s Cimzia for the treatment of adults with psoriatic arthritis, the third indication approved by the agency.
The UCB’s biologic drug Cimzia is already on the market for rheumatoid arthritis and Crohn’s disease in both US and Europe. Cimzia, also known as Certolizumab pegol, is a monoclonal antibody directed against tumor necrosis factor alpha. It is a PEGylated Fab’ fragment of a humanized TNF inhibitor monoclonal antibody
read all at http://www.pharmatopics.com/2013/09/fda-gave-green-light-to-ucbs-cimzia-to-treat-psoriatic-arthritis/
Certolizumab pegol (CDP870, tradename Cimzia) is a therapeutic monoclonal antibody to tumor necrosis factor alpha (TNF-α), for the treatment of Crohn’s disease and rheumatoid arthritis, manufactured by UCB.

certolizumab pegol is a monoclonal antibody directed against tumor necrosis factor alpha. More precisely, it is a PEGylated Fab’fragment of a humanized TNF inhibitor monoclonal antibody.
Polyethylene glycol does not cross the placenta, so it should be safe in pregnancy.

Positive results have been demonstrated in two phase III trials (PRECiSE 1 and 2) of certolizumab pegol versus placebo in moderate to severe active Crohn’s disease. In addition, data from both trials suggest it is well tolerated. As yet its efficacy has not been directly compared to other anti-TNF-α agents.
Preliminary results of the RAPID 1 and 2 phase III studies were also reportedly positive.
In 2013, a phase 3 double blind randomized placebo-controlled study found significantly positive result in patient self-reported questionnaires, with rapid improvement of function and pain reduction.
On April 22, 2008, the U.S. Food and Drug Administration (FDA) approved Cimzia for use in the United States for the treatment of Crohn’s disease in people who did not respond sufficiently or adequately to standard therapy.
On June 26, 2009, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMEA) issued a positive opinion recommending that the European Commission grant a marketing authorisation for Cimzia for the treatment of rheumatoid arthritis only – the CHMP refused approval for the treatment of Crohn’s disease. The marketing authorisation was granted to UCB Pharma SA on October 1, 2009.



