
Home » Posts tagged 'NEW DRUGS' (Page 26)
Tag Archives: NEW DRUGS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
sNDA – FDA accepts AMAG Feraheme (Ferumoxytol) sNDA for review

Feraheme (ferumoxytol)
Iron(II,III) oxide
Fe3O4
CUT PASTE OF INFO….
7 MAR 2013
The US Food and Drug Administration (FDA) has accepted for review AMAG Pharmaceuticals’ supplemental new drug application (sNDA) for Feraheme (ferumoxytol) injection for Intravenous (IV) use.
The sNDA filed is to expand the indication for ferumoxytol for the treatment of iron deficiency anemia (IDA) in adult patients with chronic kidney disease (CKD), who have failed or could not take oral iron treatment.
Ferumoxytol is currently indicated for oral use for the treatment of IDA in adult patients with CKD, according to the company.
The sNDA included the data from a global phase III program, which included two phase III clinical trials such as as IDA-301 (placebo comparator) and IDA-302 (active comparator).
The trials, which enrolled 1,400 patients, evaluated the use of ferumoxytol in a broad range of adult IDA patients, all of whom had failed or could not take oral iron treatment.
Both studies achieved the primary efficacy endpoints with statistically significant improvements in hemoglobin from baseline to the 35-day.
The studies, which also included patient-reported outcomes data as pre-specified secondary and exploratory endpoints, found no new safety signals, outside of those described in the current Feraheme (ferumoxytol) label, were observed with ferumoxytol treatment in these studies, claims the company.
In response to the application, the FDA said it will complete the review of Feraheme sNDA by 21 October 2013.
Feraheme, an iron replacement product, is a non-stoichiometric magnetite (superparamagnetic iron oxide) coated with polyglucose sorbitol carboxymethylether. The overall colloidal particle size is 17-31 nm in diameter. The chemical formula of Feraheme is Fe5874O8752-C11719H18682O9933Na414 with an apparent molecular weight of 750 kDa.
Feraheme injection is an aqueous colloidal product that is formulated with mannitol. It is a black to reddish brown liquid, and is provided in single use vials containing 510 mg of elemental iron. Each mL of the sterile colloidal solution of Feraheme injection contains 30 mg of elemental iron and 44 mg of mannitol, and has low bleomycin-detectable iron. The formulation is isotonic with an osmolality of 270-330 mOsm/kg. The product contains no preservatives, and has a pH of 6 to 8.
Ferumoxytol
STRUCTURE SOURCE http://chem.sis.nlm.nih.gov/chemidplus/rn/1309-38-2
Molecular Formulas
-
Fe.O
-
Fe3-O4
Molecular Weight
- 231.531
Ferumoxytol [USAN]
RN: 1309-38-2
Polyglucose sorbitol carboxymethyl ether-coated non-stoichiometric magnetite. Ferumoxytol is a superparamagnetic iron oxide that is coated with a low molecular weight semi-synthetic carbohydrate, polyglucose sorbitol carboxymethyl ether. The iron oxide is a superparamagnetic form of non-stoichiometric magnetite with crystal size of 6.2 to 7.3 nm. In solution, the colloidal particle of ferumoxytol has a Stokes diameter of 18-20 nm. Molecular weight is approximately 308,000
Iron oxide (Fe3O4). It is a black ore of IRON that forms opaque crystals and exerts strong magnetism. The NANOPARTICLES; and MICROSPHERES of its mineral form, magnetite, have many biomedical applications.
Ferumoxytol is the generic ingredient in one branded drug marketed by Amag Pharms Inc and is included in one NDA. There are six patents protecting this compound and one Paragraph IV challenge. Additional information is available in the individual branded drug profile pages.
This ingredient has eleven patent family members in ten countries.
There is one drug master file entry for ferumoxytol. One supplier is listed for this compound.
Phase II
Cas 722492-56-0
Launched – 2009, Anemia, iron deficiency
7228
AMI-7228
Code-7228
A superparamagnetic iron oxide (non-stoichiometric magnetite) coated with a low molecular weight semi-synthetic carbohydrate polyglucose carboxymethyl ether; USAN (OO-74) (Advanced Magnetics, Cambridge, MA, USA)
Other Names
- C 7228
- Code 7228
- Cytogen
- Feraheme
- Rienso
Superparamagnetic iron oxide coated with a low molecular weight semi-synthetic carbohydrate polyglucose sorbitol carboxymethyl ether. The iron oxide is a superparamagnetic form of non-stoichiometric magnetite with crystal size of 6.2 to 7.3 nm. In solution, the colloidal particle has a Stokes diameter of 18-20 nm
CLICK ON IMAGE

CLICK O IMAGE

Feraheme, an iron replacement product, is a non-stoichiometric magnetite (superparamagnetic iron oxide) coated with polyglucose sorbitol carboxymethylether. The overall colloidal particle size is 17-31 nm in diameter. The chemical formula of Feraheme is Fe5874O8752C11719H18682O9933Na414 with an apparent molecular weight of 750 kDa.
Feraheme Injection is an aqueous colloidal product that is formulated with mannitol. It is a black to reddish brown liquid, and is provided in single use vials containing 510 mg of elemental iron. Each mL of the sterile colloidal solution of Feraheme Injection contains 30 mg of elemental iron and 44 mg of mannitol, and has low bleomycin-detectable iron. The formulation is isotonic with an osmolality of 270-330 mOsm/kg. The product contains no preservatives, and has a pH of 6 to 8.
Ferumoxytol is AMAG Pharmaceuticals’ lead investigational compound. In 2007, the company filed a regulatory application seeking approval in the U.S. for use as an intravenous iron replacement therapeutic in patients who may be on dialysis and are suffering from anemic chronic kidney disease (CKD). In 2009, FDA approval was assigned and the product became available on the market. A regulatory application was filed in the E.U. in 2010 for this indication and a positive opinion was received in 2012. Final E.U. approval was obtained in June 2012. In 2012, AMAG Pharmaceuticals submitted a supplemental NDA to the FDA for the treatment of patients with iron-deficiency anemia (IDA) who are not candidates for oral iron, for which they received a complete response letter in January 2014. In 2013, Takeda filed for approval for this indication in the E.U. This application was withdrawn in 2015 due to safety concerns.
In terms of clinical studies, phase II trials are underway at AMAG and at Oregon Health and Science University for use in magnetic resonance angiography (MRA). The National Cancer Institute is also conducting phase II trials for the imaging of primary high-grade brain tumors and/or cerebral metastases from lung or breast cancer. Phase I clinical trials are ongoing at Dana-Farber Cancer Institute for use in magnetic resonance imaging in pediatric and adult patients with malignant sarcoma.
The drug consists of intravenously administered bioavailable iron which allows for more efficient replenishment of the body’s iron stores than oral iron supplements, without their associated common side effects. Ferumoxytol is a blood pool agent, a true intravascular contrast agent that remains in the blood stream for an extended period of time. Based on this quality, the product may be useful as a contrast agent in a wide range of applications in MRI.
In 2008, fast track designation was received in the U.S. as a diagnostic agent for vascular-enhanced magnetic resonance imaging (VE-MRI) to improve the assessment of peripheral arterial disease in patients with known or suspected chronic kidney disease. In 2010, a license, development and commercialization agreement was established between Takeda and AMAG Pharmaceuticals in Asia Pacific countries (excluding Japan, China and Taiwan), Canada, Europe, the Commonwealth of Independent States and Turkey. However, in December 2014, both companies announced the termination of this license agreement. In 2011, orphan drug designation was assigned by the FDA for use in magnetic resonance imaging in brain metastases. This designation was assigned in 2012 for use in magnetic resonance imaging to assess, and monitor treatment of solid tumor malignancies previously diagnosed in pediatric patients (age 16 years and younger).
SFDA

As announced in May 2008, we entered into a development and commercialization agreement with AMAG Pharmaceuticals, Inc. (“AMAG”) (NASDAQ:AMAG), a US biopharmaceutical company, for ferumoxytol, an intravenous iron replacement therapeutic agent being developed to treat iron deficiency anemia in CKD patients and in patients requiring hemodialysis.
Under the terms of the agreement, AMAG granted us exclusive rights to develop and commercialize ferumoxytol in the PRC, initially for CKD, and with an option to expand into additional indications. We will be responsible for the clinical development, registration, and commercialization of ferumoxytol in the PRC. We and AMAG will form a joint steering committee, with equal representation from both parties, to oversee and guide the development and commercialization of ferumoxytol in China. The agreement has an initial duration of 13 years and will be automatically renewed for a set term if minimum sales thresholds are achieved. AMAG will retain all manufacturing rights for ferumoxytol and will provide, under a separate agreement, commercial supply to us at a predetermined supply price.
Ferumoxytol was approved in June 2009 by the U.S. Food and Drug Administration to treat iron deficiency anemia in CKD patients and launched commercially in the U.S. by AMAG in July 2009. Ferumoxytol received marketing approval in Canada in December 2011 and a positive recommendation for approval from the Committee for Medicinal Products for Human Use of the European Medicines Agency in April 2012.
We have submitted the application for a registrational clinical trial for ferumoxytol to SFDA, as announced in January 2010. Once approved by the SFDA, we will commence a multi-center randomized efficacy and safety study in China with approximately 200 CKD patients, measuring the mean change in hemoglobin from baseline at Day 35 after first dose.
https://www.google.com/patents/US20100266644
Ferumoxytol is a newer parenteral iron formulation but limited information is available as to its efficacy and administration. See e.g., Landry et al. (2005) Am J Nephrol 25, 400-410, 408; and Spinowitz et al. (2005) Kidney Intl 68, 1801-1807; U.S. Pat. No. 6,599,498.
Another example of a preferred iron carbohydrate complex for use in the methods described herein is a carboxyalkylated reduced polysaccharide iron oxide complex (e.g., ferumoxytol, described in U.S. Pat. No. 6,599,498).
Another preferred iron carbohydrate complex for use in the methods described herein is a polyglucose sorbitol carboxymethyl ether-coated non-stoichiometric magnetite (e.g., “ferumoxytol”). Ferumoxytol is known in the art to be effective for treating anemia (at single unit doses lower than described herein). See e.g., Spinowitz et al. (2005) Kidney Intl 68, 1801-1807. Ferumoxytol is a superparamagnetic iron oxide that is coated with a low molecular weight semi-synthetic carbohydrate, polyglucose sorbitol carboxymethyl ether. Ferumoxytol and its synthesis are described in U.S. Pat. No. 6,599,498, incorporated herein by reference. Safety, efficacy, and pharmacokinetics of ferumoxytol are as described, for example, in Landry et al. (2005) Am J Nephrol 25, 400-410, 408; and Spinowitz et al. (2005) Kidney Intl 68, 1801-1807.
The iron oxide of ferumoxytol is a superparamagnetic form of non-stoichiometric magnetite with a crystal size of 6.2 to 7.3 nm. Average colloidal particle size can be about 30 nm, as determined by light scattering. Molecular weight is approximately 750 kD. The osmolarity of ferumoxytol is isotonic at 297 mOsm/kg and the pH is neutral. The blood half-life of ferumoxytol is approximately 10-14 hours. It has been previously reported that ferumoxytol can be given by direct intravenous push over 1-5 minutes in doses up to 1,800 mg elemental iron per minute, with maximal total dose up to 420 mg per injection. Landry et al. (2005) Am J Nephrol 25, 400-410, 408.
About Feraheme® (ferumoxytol)/Rienso
In the United States, Feraheme (ferumoxytol) Injection for Intravenous (IV) use is indicated for the treatment of iron deficiency anemia (IDA) in adult patients who have failed oral iron therapy. Feraheme received marketing approval from the FDA on June 30, 2009 for the treatment of IDA in adult chronic kidney disease (CKD) patients and was commercially launched by AMAG in the U.S. shortly thereafter.
Ferumoxytol is protected in the U.S. by five issued patents covering the composition and dosage form of the product. Each issued patent is listed in the FDA’s Orange Book. These patents are set to expire in March 2020; a request for patent term extension has been filed, which, if granted, may extend the patent term to June 2023 for one of the patents.
Ferumoxytol received marketing approval in Canada in December 2011, where it is marketed by Takeda as Feraheme, and in the European Union in June 2012 and Switzerland in August 2012, where it is marketed by Takeda as Rienso.
For additional U.S. product information, including full prescribing information, please visit www.feraheme.com.
AMAG now has five Orange Book-listed patents for ferumoxytol, with patent protection through March 2020, without patent term extension. AMAG has applied for a patent term extension for an Orange Book-listed ferumoxytol patent, which would lengthen that patent term through June 2023.
//////////Ferumoxytol, AMAG Pharmaceuticals, Phase II, 722492-56-0, Launched, 2009, Anemia, iron deficiency, 7228 , AMI-7228 , Code-7228
[Fe](O[Fe]=O)O[Fe]=O
NDA FDA-Nuvo reports FDA response to PENNSAID 2% , diclofenac sodium topical solution, 2% w/w

DICLOFENAC

PENNSAID 2%
7 MAR 2013
The US Food and Drug Administration (FDA) has issued a Complete Response Letter (CRL) to Nuvo Research’s US licensing partner, Mallinckrodt, following the review of Mallinckrodt’s New Drug Application (NDA) for diclofenac sodium topical solution, 2% w/w (PENNSAID 2%).
FDA in the letter mentioned that it requires Mallinckrodt’s complete pharmacokinetic study comparing PENNSAID 2% to original PENNSAID 1.5%.
FDA denied to review the similar pharmacokinetic studies submitted by Mallinckrodt with the NDA, as the reserve samples were not retained at the clinical site.
Pharmacokinetic studies are standard studies conducted during a drug development program to identify the total exposure or the amount of drug that reaches the blood stream after a patient receives both single and multiple doses of the product.
Mallinckrodt has suggested Nuvo that it expects to complete the study and submit the results to the FDA in the third quarter of 2013, and that it anticipates the FDA will provide a formal response to the filing within 6 months thereafter.
Nuvo’s Pain Group president Dr. Bradley Galer said with the new FDA’s letter the firm was disappointed that PENNSAID 2% will not be approved in this review cycle.
“We are pleased that the FDA has outlined a clear pathway to approval that we believe can be completed in a relatively short time frame,” Galer added.
“Upon approval, PENNSAID 2% will be the first and only topical NSAID in the U.S. featuring twice per day dosing and a metered dose pump bottle.”
Takeda Submits Marketing Authorisation Application for Vedolizumab in Moderately to Severely Active Ulcerative Colitis and Crohn’s Disease in the European Union

March 7, 2013
Pharmaceutical Company Limited (“Takeda”) today announced that a Marketing Authorisation Application (MAA) has been submitted to The European Medicines Agency (EMA) for vedolizumab, an investigational, gut-selective humanized monoclonal antibody for the treatment of adults with moderately to severely active ulcerative colitis (UC) and Crohn’s disease (CD), the two most common types of inflammatory bowel disease (IBD). If approved, vedolizumab would be the first and only gut-selective biologic agent for UC and CD on the market.
“Ulcerative colitis and Crohn’s disease are chronic debilitating diseases with important unmet medical needs, often affecting young people in the prime of their lives,” said Asit Parikh, M.D., Ph.D., vice president, general medicine, Takeda. “We are encouraged by the findings of GEMINI, the vedolizumab Phase 3 clinical development program, which studied approximately 3,000 patients in nearly 40 countries, making it the largest IBD clinical trial program conducted to date.”
Nearly four million people worldwide are affected by IBD, with UC affecting more than 500,000 people and CD affecting approximately 230,000 people in the EU. Crohn’s disease and ulcerative colitis are chronic diseases that cause inflammation of the lining of the digestive tract. Inflammation caused by CD can involve varying areas of the digestive tract, while UC impacts the colon only. CD and UC can be both painful and debilitating, which may sometimes lead to serious complications and can significantly impact the quality of life for patients.
The MAA submission was supported by Phase 3 clinical studies, GEMINI I, GEMINI II, GEMINI III and GEMINI LTS (Long-term Safety), which are part of the GEMINI Studies™, a four-study clinical research program to investigate the efficacy and safety of vedolizumab on clinical response and remission in moderately to severely active CD and UC patients, who had failed at least one conventional or anti-TNFα therapy.
“With a targeted mechanism of action, vedolizumab has clinical promise as a potential treatment option for people with moderate to severely active CD and UC,” said Paul Rutgeerts, M.D., Ph.D., F.R.C.P., professor of medicine, Catholic University of Leuven, Belgium. “While there is no known cure, there is a need for new CD and UC treatment options, in an effort to provide patients with additional choices for managing their disease, reducing symptoms and achieving remission.”
About Crohn’s disease and ulcerative colitis
Crohn’s disease (CD) and ulcerative colitis (UC) are the two most common forms of inflammatory bowel disease (IBD), which is marked by inflammation in the lining of the GI tract. CD can impact any part of the digestive tract, and common symptoms may include abdominal pain, diarrhea, rectal bleeding, weight loss, and/or fever. UC impacts the large intestine only, which includes the colon and the rectum. The most common symptoms of UC include abdominal discomfort and blood or pus in diarrhea. There is no known cause for CD or UC, although many researchers believe that the interaction of an outside agent, such as a virus or bacteria, with the body’s immune system may trigger them. No cure exists for CD or UC; the aim of IBD treatments is to induce and maintain remission, or achieve extended periods of time when patients do not experience symptoms.
About vedolizumab
Vedolizumab was developed for the treatment of CD and UC, as a gut-selective, humanized monoclonal antibody that specifically antagonizes the alpha4beta7 (α4β7) integrin, which is expressed on a subset of circulating white blood cells. These cells have been shown to play a role in mediating the inflammatory process in CD and UC. α4β7 binds with a specific adhesion molecule primarily expressed in the intestinal tract. Therefore, vedolizumab, by preventing this interaction, has a gut selective effect.
About Takeda Pharmaceutical Company Limited
Located in Osaka, Japan, Takeda is a research-based global company with its main focus on pharmaceuticals. As the largest pharmaceutical company in Japan and one of the global leaders of the industry, Takeda is committed to strive towards better health for patients worldwide through leading innovation in medicine. Additional information about Takeda is available through its corporate website, http://www.takeda.com.
Vedolizumab is a monoclonal antibody being developed by Millennium Pharmaceuticals, Inc. for the treatment of ulcerative colitis and Crohn’s disease.It binds to integrin α4β7(LPAM-1, lymphocyte Peyer’s patch adhesion molecule 1).[1][2]
The molecule was first identified by Dr. Andrew Lazarovits [1][2] as the murine MLN0002 homologue. His discovery of the mouse equivalent of this antibody—originally applied to anti-rejection strategies in kidney transplantation—was published in the journal Nature in 1996. The drug was then licensed to Millennium Pharmaceuticals of Boston for further development.
As of October 2009, vedolizumab is undergoing Phase III trials.[3] Clinical trials indicate that Vedolizumab was found safe and highly effective for inducing and maintaining clinical remission in patients with moderate to severe ulcerative colitis [3]. Dr. Brian Faegan, head researcher, reported an absence of any instances of progressive multifocal leukoencephalopathy (PML), which is a particularly important finding [4]. It looks like it will be an effective abiologic agent without some of the toxicity issues previously seen with anti-TNF drugs .
It is widely believed now that “vedolizumab can be used either as a first-line treatment or in case of anti-TNF failure”
- Statement On A Nonproprietary Name Adopted By The USAN Council – Vedolizumab, American Medical Association.
- Soler, D; Chapman, T; Yang, LL; Wyant, T; Egan, R; Fedyk, ER (2009). “The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases”. The Journal of Pharmacology and Experimental Therapeutics 330 (3): 864–75. doi:10.1124/jpet.109.153973. PMID 19509315.
- ClinicalTrials.gov NCT00790933 Study of Vedolizumab (MLN0002) in Patients With Moderate to Severe Crohn’s Disease (GEMINI II)

Phase 3-Trius Therapeutics will soon be reporting data from its second phase III trial of Tedizolid
Tedizolid
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
856866-72-3 cas no
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701”, while the active ingredient is called “TR-700”.[4][5]

March 5 2013
Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
Phase 1-Sangamo Presents New Clinical Data at CROI 2013 Demonstrating Persistent Immune System Improvements After Treatment With ZFN Therapeutic(R) SB-728-T

The gene therapy diminished the levels of virus and eradicated in patients having naturally occurring mutation of gene, found a preliminary trail of HIV treatment. The first phase of very small trail tested the SB-728-T gene treatment that is intended to interrupt theCCR5 gene used by HIV to contaminate immune system cells.
The first clinical trial using zinc-finger nucleases to provide long-term resistance to HIV-1 infection has been given the go-ahead by the US Food and Drug Administration. Sangamo BioSciences of Richmond, California, and its clinical partner, the University of Pennsylvania, have begun enrolling the first 12 people in a phase 1 clinical trial to evaluate SB-728-T, a novel zinc-finger DNA-binding nuclease that permanently disrupts the CCR5 gene on CD4+ T cells (Nat. Biotechnol. 26, 808–816, 2008
Data Demonstrate that SB-728-T Possesses Necessary Immunologic Properties to Support a ‘Functional Cure’ for HIV/AIDS
RICHMOND, Calif., March 6, 2013
Sangamo BioSciences, Inc. announced new data from its program to develop a ‘functional cure’ for HIV/AIDS in two presentations at the 20th Conference on Retroviruses and Opportunistic Infections (CROI), held in Atlanta from March 3 to 6, 2013.
The first presentation described data from the SB-728-T Phase 1 study (SB-728-902, Cohorts 1-3) demonstrating that SB-728-T treatment of HIV-infected subjects leads to durable reconstitution of the immune system driven by increases in total CD4+ central memory T-cells (TCM) and CCR5-protected TCM. TCM are long-lived, self-renewing cells that have the ability to remember and react against foreign antigens including HIV. The data also showed that certain cell surface marker and gene expression profiles may predict which patients will likely respond best to SB-728-T treatment.
About Sangamo
Sangamo BioSciences, Inc. is focused on research and development of novel DNA-binding proteins for therapeutic gene regulation and genome editing. The Company has ongoing Phase 2 clinical trials to evaluate the safety and efficacy of a novel ZFP Therapeutic® for the treatment of HIV/AIDS. Sangamo’s other therapeutic programs are focused on monogenic diseases, including hemophilia, Huntington’s disease and hemoglobinopathies such as beta-thalassemia and sickle cell anemia. Sangamo’s core competencies enable the engineering of a class of DNA-binding proteins known as zinc finger DNA-binding proteins (ZFPs). Engineering of ZFPs that recognize a specific DNA sequence enables the creation of sequence-specific ZFP Nucleases (ZFNs) for gene modification and ZFP transcription factors (ZFP TFs) that can control gene expression and, consequently, cell function. Sangamo has entered into a strategic collaboration with Shire AG to develop therapeutics for hemophilia, Huntington’s disease and other monogenic diseases and has established strategic partnerships with companies in non-therapeutic applications of its technology including Dow AgroSciences and Sigma-Aldrich Corporation. For more information about Sangamo, visit the company’s website atwww.sangamo.com.
Phase III Study of Oral Laquinimod for Relapsing-Remitting Multiple Sclerosis

Laquinimod
5-chloro-N-ethyl-4-hydroxy-1-methyl-2-oxo-
N-phenyl-1,2-dihydroquinoline-3-carboxamide
Laquinimod is an experimental immunomodulator developed by Active Biotech and Teva. It is currently being investigated as an oral treatment for multiple sclerosis (MS).
Laquinimod is the successor of Active Biotech’s failed experimental immunomodulator linomide.[1]
The compound has been investigated in two Phase II trials using successive magnetic resonance scans (MRI). Laquinimod seems to be able to reduce the MS disease activity on MRI.[2][3] However, the response to a given dose was discrepant between both studies.[4]
Phase III studies for MS started in December 2007.[5] In 2011, Teva announced its clinical trials involving laquinimod had failed, being unable to significantly reduce relapses into MS among patients beyond a placebo.[6] However, the final results of above mentioned phase III trial proved oral laquinimod administered once daily slowed the progression of disability and reduced the rate of relapse in patients with relapsing–remitting multiple sclerosis [7]
Mar 6, 2013 –
CONCERTO Study Enrolling Patients Globally to Evaluate Impact of Laquinimod on Disability Progression
Teva Pharmaceutical Industries Ltd. and Active Biotech announced today enrollment of the first patient in the CONCERTO study – the third Phase III placebo-controlled study designed to evaluate the efficacy, safety and tolerability of once-daily oral laquinimod in patients with relapsing-remitting multiple sclerosis (RRMS). The primary outcome measure of CONCERTO will be confirmed disability progression as measured by the Expanded Disability Status Scale (EDSS).
“Previous Phase III studies in more than 2,400 people with RRMS suggest a unique profile of laquinimod, directly affecting the neurodegenerative processes that lead to disability progression, the main concern in the treatment of RRMS,” said CONCERTO principal investigator, Dr. Timothy Vollmer, Professor of Neurology, University of Colorado Denver, Medical Director of the Rocky Mountain Multiple Sclerosis Center, and Co-Director of the RMMSC at Anschutz. “We are currently enrolling patients in this third pivotal study to further examine the clinical benefits of laquinimod on disability progression, the primary endpoint of the CONCERTO trial, and brain atrophy, at both the previously studied 0.6 mg dose, and now a higher 1.2 mg dose.”
The multinational, randomized, double blind placebo-controlled study will aim to enroll approximately 1,800 patients at more than 300 sites globally (http://clinicaltrials.gov/show/NCT01707992). Along with the primary endpoint of time to confirmed disability progression, the study will also examine the impact of laquinimod on endpoints such as percent change in brain volume and other clinical and MRI markers of disease activity.
“For nearly 30 years, Teva has been focused on improving the lives of people with multiple sclerosis by delivering innovative treatment options that address this complex disease,” said Dr. Michael Hayden, President of Global R&D and Chief Scientific Officer at Teva Pharmaceutical Industries Ltd. “The CONCERTO study demonstrates our commitment to collaborating with MS communities worldwide to further develop laquinimod and address unmet patient needs.”
ABOUT LAQUINIMOD
Laquinimod is an oral, once-daily CNS-active immunomodulator with a novel mechanism of action being developed for the treatment of MS. In animal models laquinimod crosses the blood brain barrier to potentially have a direct effect on resident CNS inflammation and neurodegeneration. The global Phase III clinical development program evaluating oral laquinimod in MS includes two pivotal studies, ALLEGRO and BRAVO.
In addition to the MS clinical studies, laquinimod is currently in clinical development for Crohn’s disease and Lupus.
ABOUT CONCERTO
CONCERTO is a multinational, multicenter, randomized, double-blind, parallel-group, placebo-controlled study followed by an active treatment phase, to evaluate the efficacy, safety and tolerability of two doses of oral administration of laquinimod 0.6 mg/day or 1.2 mg/day in subjects with RRMS. This third Phase III laquinimod study will evaluate laquinimod in approximately 1,800 patients for up to 24 months, after which patients will continue to an active treatment period with laquinimod for an additional 24 months. The primary outcome measure will be time to confirmed disability progression as measured by the Expanded Disability Status Scale (EDSS). The study will also examine the impact of laquinimod on endpoints such as percent change in brain volume, as well as other clinical and MRI markers of disease activity.
ABOUT MULTIPLE SCLEROSIS
MS is the leading cause of neurological disability in young adults. It is estimated that more than 400,000 people in the United States are affected by the disease and that two million people may be affected worldwide. Multiple sclerosis is a degenerative disease of the central nervous system in which inflammation and axonal damage and loss result in the development of progressive disability.
ABOUT TEVA
Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) is a leading global pharmaceutical company, committed to increasing access to high-quality healthcare by developing, producing and marketing affordable generic drugs as well as innovative and specialty pharmaceuticals and active pharmaceutical ingredients. Headquartered in Israel, Teva is the world’s leading generic drug maker, with a global product portfolio of more than 1,000 molecules and a direct presence in about 60 countries. Teva’s branded businesses focus on CNS, oncology, pain, respiratory and women’s health therapeutic areas as well as biologics. Teva currently employs approximately 46,000 people around the world and reached $20.3 billion in net revenues in 2012.
ABOUT ACTIVE BIOTECH
Active Biotech AB is a biotechnology company with focus on autoimmune/inflammatory diseases and cancer. Projects in or entering pivotal phase are laquinimod, an orally administered small molecule with unique immunomodulatory properties for the treatment of multiple sclerosis, TASQ for prostate cancer as well as ANYARA for use in cancer targeted therapy, primarily of renal cell cancer. In addition, laquinimod is in Phase II development for Crohn’s and Lupus. Further projects in clinical development comprise the two orally administered compounds, 57-57 for SLE & Systemic Sclerosis and RhuDex(TM) for RA. Please visit http://www.activebiotech.com for more information.
- Tan IL, Lycklama à Nijeholt GJ, Polman CH et al. (April 2000). “Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trials”. Mult Scler 6 (2): 99–104. PMID 10773855.
- Comi G, Pulizzi A, Rovaris M et al. (June 2008). “Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study”. Lancet 371 (9630): 2085–2092. doi:10.1016/S0140-6736(08)60918-6. PMID 18572078.
- Polman C, Barkhof F, Sandberg-Wollheim M et al. (March 2005). “Treatment with laquinimod reduces development of active MRI lesions in relapsing MS”. Neurology 64 (6): 987–91. doi:10.1212/01.WNL.0000154520.48391.69. PMID 15781813.
- Keegan BM, Weinshenker BG (June 2008). “Laquinimod, a new oral drug for multiple sclerosis”. Lancet 371 (9630): 2059–2060. doi:10.1016/S0140-6736(08)60894-6. PMID 18572062.
- ClinicalTrials.gov NCT00509145 Safety and Efficacy of Orally Administered Laquinimod Versus Placebo for Treatment of Relapsing Remitting Multiple Sclerosis (RRMS) (ALLEGRO)
- Kresege, Naomi (1 August 2011). “Teva’s Copaxone Successor Fails in Latest Clinical Trial”. Bloomberg. http://www.bloomberg.com/news/2011-08-01/teva-s-oral-multiple-sclerosis-drug-fails-to-meet-goal-of-clinical-trial.html. Retrieved 2 August 2011. “Teva Pharmaceutical Industries Ltd. (TEVA)’s experimental multiple sclerosis pill failed to reduce relapses more than placebo in a clinical trial, dealing a blow to the company’s effort to find a successor to an older drug.”
- (Comi et al. N Engl J Med 2012;366:1000).

EP 1073639; JP 2002513006; US 6077851; WO 9955678
5-Chloroisatoic anhydride (I) is alkylated with iodomethane and NaH to afford (II). Subsequent condensation of anhydride (II) with the malonic monoamide (III) in the presence of NaH in hot DMA furnishes the target quinoline carboxamide.
…

Reaction of 2-amino-6-chlorobenzoic acid (I) with phosgene and NaHCO3 in dioxane gives 5-chloroisatoic anhydride (II), which is methylated by means of iodomethane and NaH in DMF to yield 5-chloro-1-methylisatoic anhydride (III). Finally, anhydride (III) is condensed with the malonic monoamide (IV) by means of NaH in hot dimethylacetamide. Alternatively, condensation of anhydride (III) with ethoxy malonyl chloride (V) by means of NaOMe and triethylamine in dichloromethane affords 5-chloro-4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3- carboxylic acid ethyl ester (VI), which is finally condensed with N-ethylaniline (VII) in refluxing toluene. Alternatively, ester (VI) is hydrolyzed by means of concentrated HCl in hot Ac2O to give the carboxylic acid (VIII), which is finally condensed with N-ethylaniline (VII) by means of SOCl2 and TEA in dichloromethane
PHASE1,Progenics Pharmaceuticals’ Novel Small Molecule Drugs Targeting PSMA Successfully Visualize Prostate Cancer, 123-I-MIP-1095

Name: 123-I-MIP-1095
Synonym: 123-I-MIP-1095; [123I]-MIP-1095; iodine I 123 IMP-1095; 2-(3-{l-carboxy-5-[3-(4-iodo-phenyl)-ureido]-pentyl}-ureido)-pentanedioic acid.; [123I]-(S)-2-(3-((S)-1-carboxy-5-(3-(4-iodophenyl)ureido)pentyl)ureido)pentanedioic acid
IUPAC/Chemical name:
2-(3-(1-carboxy-5-(3-(4-iodophenyl)ureido)pentyl)ureido)pentanedioic acid
Chemical Formula: C19H25123IN4O8
Exact Mass: 560.07284
Molecular Weight: 560.33
123-I-MIP-1095
An iodine 123-radiolabled small molecule that exhibits high affinity for prostate-specific membrane antigen (PSMA) with potential use in molecular imaging. 123-I-MIP-1095, a radiolabeled glutamate-urea-lysine analogue, selectively binds PSMA, which allows imaging of PSMA-expressing prostate cancer cells with gamma scintigraph. PSMA is a transmembrane glycoprotein highly expressed by malignant prostate epithelial cells and vascular endothelial cells of various solid tumors.
![]()
| Synonym: | iodine I 123 IMP-1095 | ||
| Chemical structure: | 2-(3-{l-carboxy-5-[3-(4-iodo-phenyl)-ureido]-pentyl}-ureido)-pentanedioic acid | ||
March 5, 2013
Progenics Pharmaceuticals, Inc. (Nasdaq:PGNX) reported positive clinical data from a study of two novel radiolabeled small molecules targeting prostate-specific membrane antigen (PSMA). The imaging agents — 123I-MIP-1072 and 123I-MIP-1095 — had a high sensitivity of lesion detection in bone, tissue and the prostate gland with minimal retention in non-target tissue. The research was published as the cover article in the March issue of The Journal of Nuclear Medicine.
“Existing imaging techniques are limited in their ability to diagnose and stage prostate cancer,” said John J. Babich, Ph.D., senior author of the article “First-in-Man Evaluation of Two High-Affinity PSMA-Avid Small Molecules for Imaging Prostate Cancer.” “The approach described in this paper has the potential to assess disease status more accurately. It could help clinicians select optimal treatments and lead to better patient outcomes.”
Separate phase 1 studies were conducted under an exploratory investigational new drug (IND) application to measure the potential effectiveness of the small molecules in diagnosing and staging prostate cancer. In the first study, seven patients with documented prostate cancer were administered doses of 123I-MIP-1072 and 123I-MIP-1095, two weeks apart. In the second study, six healthy volunteers received 123I-MIP-1072 only. Whole body planar imaging and single photon emission computed tomography (SPECT)/computed tomography (CT) were performed for each group, and pharmacokinetics, tissue distribution, excretion, safety and organ radiation dose were analyzed.
Based on the data reported, Progenics is conductinga global, multi-center phase 2 trial investigating a next generation radiolabeled small molecule targeting PSMA, MIP-1404.
Mark R. Baker, chief executive officer of Progenics, said, “We recently acquired all of the rights to the compounds described in this Journal of Nuclear Medicine paper, as well as to the phase 2 stage imaging agent MIP-1404, through Progenics’ acquisition of Molecular Insight Pharmaceuticals. It is gratifying to see this expansion of our oncology pipeline demonstrating progress so soon.”
Robert J. Israel, M.D., Progenics’ senior vice president of medical affairs and clinical research, said, “We believe that MIP-1404 has excellent potential as a diagnostic radiopharmaceutical. Results to date from the study compounds and MIP-1404 show PSMA as a robust target for prostate cancer molecular imaging, and that a radiolabeled small molecule, which binds PSMA with high affinity, has the potential to detect prostate cancer throughout the body. Cancer treatment guidelines call for imaging prostate cancer with conventional bone scans or MRI. A more accurate method of imaging prostate cancer could be of great value.”
Mr. Baker further added, “Thought leaders in prostate cancer care are focused on avoiding unnecessary surgery and other invasive procedures due to the complications associated with them. Clinicians generally prefer “watchful waiting” when the cancer appears to be indolent. At the same time, some therapeutics to treat aggressive prostate cancer have recently been approved or are under development, such as Progenics’ own PSMA ADC, which currently is in phase 2 testing. Patients and their physicians would benefit from feedback on how therapeutic agents are impacting the course of cancer, and guidance on how and when to use therapeutic agents. It is clear that an improved way to visualize prostate cancer, with a high degree of specificity and sensitivity, would better inform both “watchful waiting” and the treatment of aggressive disease. We believe that data from the ongoing phase 2 trial of MIP-1404 will demonstrate its capabilities to assist prostate cancer patients and their physicians in making these critical decisions.”
About Prostate Cancer
Prostate cancer is the most common form of cancer affecting men in the United States and is the second leading cause of cancer deaths among men each year. The American Cancer Society estimates that in 2013, 238,590 new cases of prostate cancer will be diagnosed and approximately 29,720 American men will die from the disease. Accurate diagnosis and staging of prostate cancer is critical to determining appropriate patient management.
About Progenics
Progenics Pharmaceuticals, Inc. is discovering and developing innovative medicines for oncology, with a pipeline that includes product candidates in preclinical through late-stage development. Progenics’ first commercial product, Relistor® (methylnaltrexone bromide) for opioid-induced constipation, is marketed and in further development by Salix Pharmaceuticals, Ltd. for markets worldwide other than Japan, where Ono Pharmaceutical Co., Ltd. holds an exclusive license for the subcutaneous formulation. For additional information, please visit http://www.progenics.com.
Ziopharm Oncology will be releasing its Phase III results for its drug Palifofsamide towards the end of March 2013

(Zymafos; ZIO-201) is a cytotoxic, active metabolite of the alkylating agent ifosfamide, which causes irreparable DNA interstrand cross-linking in cancer cells. This prevents DNA replication and cell division, leading to cell death.
In contrast to ifosfamide, palifosfamide is not metabolised to the toxins acrolein and chloracetaldehyde, which are associated with haemorrhagic cystitis, and neuro- and nephro-toxicities respectively. Also, palifosfamide is not a substrate for aldehyde dehydrogenase (ALDH), an important mediator of drug resistance

Cyclophosphamide and ifosfamide are nitrogen mustard alkylating agents that act by crosslinking DNA strands at the guanine N-7 position, resulting in cell death. Both of these are prodrugs that are metabolised in the liver to phosphoramide mustard active metabolites, but their use is limited by toxic side-effects. They are also prone to tumour resistance, which results from numerous mechanisms, including DNA repair.
In an attempt to overcome some of these problems, Ziopharm Oncology has developed palifosfamide tromethamine, which is a salt formulation of isophosphoramide mustard, the active metabolite of isofosfamide.1

isofosamide
In a Phase I trial, it was given in combination with doxorubicin to 13 patients with advanced refractory tumours – eight with soft tissue sarcoma and the remainder with small cell lung cancer – for whom there was no available standard therapy.2 It was given on the first three days of a three-week cycle, with a starting dose of 150mg/m2, and doxorubicin given on the first day at a starting dose of 60mg/m2. The doses were escalated to a maximum tolerated dose of 150mg/m2 for palifosfamide and 75mg/m2 for doxorubicin. It was well tolerated, and three of the 12 assessable patients had a partial response, two of whom were from the sarcoma group, and the median progression free survival was 20 weeks.
references
1. S. Jung and B. Kasper, IDrugs 2010, 13, 38
2. L.J. Camacho et al. J. Clin. Oncol. 2009, 27 (Suppl.), Abst. 10577
3. C.F. Verschraegen et al. J. Clin. Oncol. 2010, 28 (Suppl.), A
Palifosfamide, A Novel Molecule for the Treatment of Soft Tissue Sarcoma
Palifosfamide (Zymafos™ or ZIO-201) references a novel composition (tris formulation) that is the functional active metabolite of ifosfamide (IFOS), a bi-functional DNA alkylator being investigated as a potential therapy for the treatment of soft tissue sarcoma (STS). Palifosfamide is formulated by combining the tris (hydroxymethyl) amino methane (tris) salt of palifosfamide and a number of excipients to create the final drug product. Preclinical development of palifosfamide has included in vitro and in vivo studies demonstrating activity against various sarcomas, breast cancers, other solid tumors and leukemias, including several that are resistant to IFOS. Several clinical studies have been initiated in a variety of cancer types. A Phase I study in advanced cancers, using the original lysine formulation, has been completed. A two-stage Phase I/II Study in advanced sarcomas, introducing the tris salt formulation, has completed enrollment and data retrieval is ongoing. A Phase I study in combination with doxorubicin evaluating patients with advanced, refractory solid tumors for whom treatment with doxorubicin is considered medically acceptable, has completed enrollment and data retrieval is ongoing. Based on the result of the Phase I combination study, an international randomized Phase II study comparing palifosfamide in combination with doxorubicin versus doxorubicin alone in 1st and 2nd line patients with advanced STS has been completed. phase 3 on now also
What is Soft Tissue Sarcoma, and what are the currently available treatments?
Soft-tissue sarcomas (STS) represent a rare and diverse group of tumors that are not very well understood. Although soft-tissue sarcomas account for <1% of all cancers, they represent a high percentage of cancer-related deaths worldwide (Ref. 3, Ref. 4, Ref. 5). STS tumors can occur anywhere within the body, originating in various soft tissues including fat, smooth or striated muscle, nerve/nerve sheath, vascular tissue, and other connective tissues; the extremities are the most common site of origin, accounting for approximately 50% of cases
Pfizer receives FDA approval for the use of Prevnar 13® , Pneumococcal 13-valent Conjugate Vaccine

Prevnar 13
Publication date: 25 January 2013
Author: Pfizer
Pfizer Inc. announced today that the U.S. Food and Drug Administration (FDA) has granted approval for the expansion of the company’s pneumococcal conjugate vaccine, Prevnar 13®* (Pneumococcal 13-valent Conjugate Vaccine [Diphtheria CRM197 Protein]), for use in older children and adolescents aged 6 years through 17 years for active immunization for the prevention of invasive disease caused by the 13 Streptococcus pneumoniae serotypes contained in the vaccine. For this age group, Prevnar 13 is administered as a one-time dose to patients who have never received Prevnar 13.1
“As a global leader in pneumococcal disease prevention, extending the impact of Prevnar 13 to older children and adolescents aged 6 through 17 years is a reflection of our dedication to improving public health worldwide,”said Susan Silbermann, president, vaccines, Pfizer. “We continue to work tirelessly to make this vaccine available to people at risk for invasive pneumococcal disease.”
The FDA approval followed submission and review of a Phase 3, open-label trial of Prevnar 13 in 592 older children and adolescents, including those with asthma.2 The study met all endpoints, demonstrating immunogenicity and establishing a safety profile in children aged 6 years through 17 years consistent with the safety profile established in previous trials in infants and young children.2
About Prevnar 13
Prevnar 13 was first introduced for use in infants and young children in December 2009 in Europe and in February 2010 in the U.S., and it is now approved for such use in nearly 120 countries worldwide. It is the most widely used pneumococcal conjugate vaccine in the world, and more than 500 million doses of Prevnar/Prevnar 13 have been distributed worldwide. Currently, Prevnar 13 is included as part of a national or regional immunization program in more than 60 countries, offering coverage against invasive pneumococcal disease to nearly 30 million children per year.3
Prevnar 13 is also approved for use in adults 50 years of age and older in more than 80 countries and it is the first and only pneumococcal vaccine to be granted World Health Organization prequalification in the adult population.3
About Pneumococcal Disease
Pneumococcal disease (PD) is a group of illnesses caused by the bacterium Streptococcus pneumoniae (S. pneumoniae), also known as pneumococcus.4 PD is associated with significant morbidity and mortality.4 Invasive manifestations of the disease include bacteremia (bacteria in the blood) and meningitis (infection of the tissues surrounding the brain and spinal cord).4 Invasive pneumococcal disease can affect people of all ages, although older adults and young children are at heightened risk.4,5,6
us fda data
STN#: 125324
Proper Name: Pneumococcal 13-valent Conjugate Vaccine (Diphtheria CRM197 Protein)
Tradename: Prevnar 13
Manufacturer: Wyeth Pharmaceuticals, Inc, License #0003
Indications:
- Active immunization for the prevention of pneumonia and invasive disease caused by S. pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F in persons 50 years of age or older.
- Active immunization for the prevention of invasive disease caused by Streptococcus pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F, for use in children 6 weeks through 17 years of age.
- Active immunization for the prevention of otitis media caused by Streptococcus pneumoniae serotypes 4, 6B, 9V, 14, 18C, 19F, and 23F for use in children 6 weeks through 5 years of age.
Pfizer presents Phase 3 safety and immunogenicity data on Prevnar 13® in adults with HIV
Publication date: 4 March 2013
Author: Pfizer
Pfizer Inc. (NYSE:PFE) presented today the results from a Phase 3 study demonstrating the immunogenicity, tolerability and safety of Prevnar 13®(Pneumococcal 13-valent Conjugate Vaccine [Diphtheria CRM197 Protein])in adults infected with human immunodeficiency virus (HIV). The results were presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in Atlanta, Ga.
These data support planned regulatory submissions seeking to include data on HIV-infected immunocompromised adults in the Prevnar 13 label in the United States, the European Union, and other countries around the world.
Phase 3 , breast cancer, Ridaforolimus (MK-8669; AP23573; formerly Deforolimus) Merck, license,Ariad Pharmaceuticals

Ridaforolimus
572924-54-0
(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl dimethylphosphinate
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
42-(dimethylphosphinate) Rapamycin
- AP 23573
- AP23573
- Deforolimus
- MK 8669
- MK-8669
- MK8669
- Ridaforolimus
- Taltorvic
- UNII-48Z35KB15K
An mTOR inhibitor for the treatment of cancer.
Ridaforolimus (MK-8669; AP23573; formerly Deforolimus)
Merck, under exclusive worldwide license agreement with Ariad Pharmaceuticals
Method of Action: Oral inhibitor of mammalian target of rapamycin inhibitor (mTOR)
Indications/Phase of Trial: Maintenance therapy for metastatic soft-tissue sarcoma and bone sarcomas after at least four chemotherapy cycles (under review after receiving Complete Response letter from FDA in June; NME)
Ridaforolimus is an investigational small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN, known to be important to malignancy.
TARGET- mTOR
Ridaforolimus (also known as AP23573 and MK-8669; formerly known as Deforolimus[1]) is an investigational targeted and small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN known to be important to malignancy. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism, and angiogenesis.
It has had promising results in a clinical trial for advanced soft tissue and bone sarcoma.
RIDAFOROLIMUS
NMR….http://file.selleckchem.com/downloads/nmr/S102201-Deforolimus-HNMR-Selleck.pdf
HPLC . http://file.selleckchem.com/downloads/hplc/S102201-Deforolimus-HPLC-Selleck.pdf
MSDS..http://www.selleckchem.com/msds/Deforolimus-MSDS.html
| Commercial arrangements |
Ridaforolimus is being co-developed by Merck and ARIAD Pharmaceuticals. On May 5, 2010, Ariad Pharmaceuticals and Merck & Company announced a clinical development and marketing agreement. With this agreement, Ariad received $125 million in upfront payments from Merck and $53 million in milestone payments. Future payments are triggered upon acceptance of the NDA by the FDA with another payment when the drug receives marketing approval. There are similar milestones for acceptance and approval in both Europe and Japan. Other milestone payments are tied to revenue goals for the drug.[2] ARIAD has opted to co-promote ridaforolimus in the U.S. Merck plans to submit a New Drug Application (NDA) for ridaforolimus to the U.S. Food and Drug Administration (FDA) and a marketing application in the European Union in 2011.[3]
Clinical trials
Phase III SUCCEED
On June 6, 2011, Ariad and Merck announced detailed results from the largest randomized study ever in the soft tissue and bone sarcoma population, the Phase III SUCCEED clinical trial. SUCCEED evaluated oral ridaforolimus, in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. In this patient population, ridaforolimus improved progression-free survival (PFS) compared to placebo, the primary endpoint of the study. The complete study results were presented by Sant P. Chawla, M.D., director, Sarcoma Oncology Center, Santa Monica, CA, during the 2011 American Society of Clinical Oncology (ASCO) annual meeting.
The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind study of oral ridaforolimus administered at 40 mg/day (five of seven days per week) in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. Oral ridaforolimus was granted a Special Protocol Assessment (SPA) by the FDA for the SUCCEED trial.
Based on 552 progression-free survival (PFS) events in 711 patients, (ridaforolimus (N=347), placebo (N=364) determined by an independent radiological review committee, the study achieved its primary endpoint of improvement in PFS, with a statistically significant (p=0.0001) 28 percent reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo (hazard ratio=0.72).
Median PFS was 17.7 weeks for those treated with ridaforolimus compared to 14.6 weeks in the placebo group. Furthermore, based on the full analysis of PFS determined by investigator assessment, there was a statistically significant (p<0.0001) 31 percent reduction by ridaforolimus in the risk of progression or death compared to placebo (hazard ratio=0.69). In the investigator assessment analysis, median PFS was 22.4 weeks for those treated with ridaforolimus compared to 14.7 weeks in the placebo group [4
EU WITHDRAWAL IN NOV 2012
Merck, known as MSD outside the U.S. and Canada, announced today that it has formally notified the European Medicines Agency (EMA) of Merck’s decision to withdraw the Marketing Authorisation Application (MAA) for ridaforolimus.
The application for Marketing Authorisation for ridaforolimus was accepted by the EMA in August 2011. At the time of the withdrawal it was under review by the Agency’s Committee for Medicinal Products for Human Use (CHMP). In its letter to the EMA, Merck said that the withdrawal of ridaforolimus was based on the provisional view of the CHMP that the data available to date and provided in the Marketing Authorisation Application were not sufficient to permit licensure of ridaforolimus in the European Union for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor.
Although the application for these uses was withdrawn, Merck is studying ridaforolimus in combination with other drugs in other tumor types. The withdrawal of the European application of ridaforolimus for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor does not change Merck’s commitment to the ongoing clinical trials with ridaforolimus.
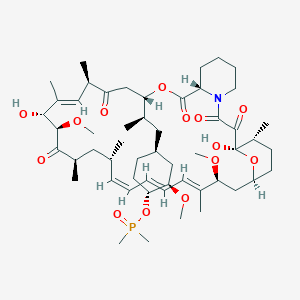
Description
42-(dimethylphosphinate) Rapamycin (Ridaforolimus) represented by the following formula I:

2. Description of RelatedArt
The mammalian target of Rapamycin (mTOR) is known as a mechanistic target of Rapamycin (H), which is found in the studies of Rapamycin. On the other hand, 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) is a derivative of Rapamycin (II), which is also a kind of mTOR inhibitor. Ridaforolimus (I) can inhibit cell division and possibly lead to tumor cell death. Hence, there are many studies related to solid tumor treatments and blood cancer treatments using Ridaforolimus (I). In addition, in 2011, Merck also applied a certification of this compound against soft tissue and bone cancer.
U.S. Pat. No. 7,091,213 discloses a process for preparing 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I), and the process thereof is shown in the following Scheme I.

In this process, a solution of Rapamycin (II) in dichloromethane (DCM) was respectively added with 2,6-di-tert-butyl-4-methylpyridine or 3,5-lutidine as a base, and followed by the addition of a solution of dimethylphosphinic chloride (DMP-Cl) to perform a phosphorylation reaction at 0° C., under a stream of N2(g). The crude product was purified by flash chromatography (eluted with MeOH/DCM/EtOAc/hexane=1:10:3:3) to provide 42-(dimethyl- phosphinate) Rapamycin (Ridaforolimus) (I), which is a phosphorylated compound at 42-hydroxyl position of Rapamycin (II). In addition, this patent also disclosed a side product of 31,42-bis(dimethyl phosphinate) Rapamycin (III), which is a phosphorylated compound at both 31- hydroxyl position and 42- hydroxyl position of Rapamycin (II).
…………………..
SYNTHESIS
Some additional transformations of potential interest to the practitioner are shown below, including the preparation of reagents for generating the described C-43 phosphorus-containing rapalogs:
Preparation of Diakyl/diaryl Chlorophoshates

Preparation of Alkyl Halide Phosphonates

Illustrative routes for using the foregoing sorts of reagents to prepare certain rapalogs of this invention are shown below.

The synthesis of compounds of this invention often involves preparation of an activated form of the desired moiety “J”, such as a phosphoryl chloride as shown above (e.g. (R)(RO)P—Cl or RR′P(═O)—Cl, etc), and reaction of that reagent with rapamycin (or the appropriate rapalog) under conditions yielding the desired product, which may then be recovered from residual reactants and any undesired side products. Protecting groups may be chosen, added and removed as appropriate using conventional methods and materials.
Purification of Compounds of the Invention
A variety of materials and methods for purifying rapamycin and various rapalogs have been reported in the scientific and patent literatures and may be adapted to purification of the rapalogs disclosed herein. Flash chromatography using a BIOTAGE prepacked cartridge system has been particularly effective. A typical protocol is disclosed in the Examples which follow.
Physicochemical Characterization of Compounds of the Invention
The identity, purity and chemical/physical properties of the rapalogs may be determined or confirmed using known methods and materials, including HPLC, mass spectral analysis, X ray crystallography and NMR spectroscopy. High resolution 1D 1H and 31P NMR spectra acquired using a typical relaxation delay of 3 seconds have proved useful, as has reverse phase HPLC analysis (analytical column, 3 micron particle size, 120 ansgstrom pore size, thermostatted to 50° C. with a mobile phase of 50% acetonitrile, 5% methanol and 45% water (all % s by volume), for example, in an isocratic elution system, with elution of product and impurity peaks followed by UV detection at 280 nanometers). Normal phase HPLC may also be used, especially to evaluate the level of residual rapamycin or rapalog by-products. The presence of residual solvent, heavy metals, moisture and bioburden may be assessed using conventional methods.
Example 9
Dimethyl-phosphinic Acid C-43 Rapamycin Ester

Dimethyl-phosphinic Acid C-43 Rapamycin Ester
To a cooled (0° C.) solution of rapamycin (0.1 g, 0.109 mmol) in 1.8 mL of dichloromethane was added 0.168 g (0.82 mmol) of 2,6-di-t-butyl-4-methyl pyridine, under a stream of N2, followed immediately by a solution of dimethylphosphinic chloride (0.062 g, 0.547 mmol) in 0.2 mL of dichloromethane. The slightly yellow reaction solution was stirred at 0° C., under an atmosphere of N2, for 3.5 h (reaction monitored by TLC). The cold (0° C.) reaction solution was diluted with ˜20 mL EtOAc then transferred to a separatory funnel containing EtOAc (150 mL) and saturated NaHCO3 (100 mL). Upon removing the aqueous layer, the organic layer was washed successively with ice cold 1N HCl (1×100 mL), saturated NaHCO3 (1×100 mL), and brine (1×100 mL), then dried over MgSO4 and concentrated. The crude product was purified by silica gel flash chromatography (eluted with 1:10:3:3 MeOH/DCM/EtOAc/hexane) to provide 0.092 g of a white solid:
1H NMR (300 MHz, CDCl3) d 4.18 (m, 1H), 4.10 (m, 1H), 3.05 (m, 1H), 1.51 (m, 6H);
31P NMR (121 MHz, CDCl3) d 53.6; 1013 m/z (M+Na).
Example 9
Alternative Synthesis
Rapamycin and dichloromethane are charged into a nitrogen-purged reaction flask. The stirred solution is cooled to approximately 0° C. (an external temperature of −5±5° C. is maintained throughout the reaction). A solution of dimethylphosphinic chloride (2.0 molar equivalents) in dichloromethane is then added over a period of approximately 8–13 minutes.
This is followed immediately by the addition of a solution of 3,5-lutidine (2.2 molar equivalents) in dichloromethane over a period of approximately 15–20 minutes. Throughout both additions, the internal temperature of the reaction sssstays below 0° C. The cooled reaction solution is stirred for 1 hour and then transferred, while still cold, to an extractor containing saturated aqueous NaHCO3 and methyl-t-butyl ether (MTBE), ethyl acetate or diethyl ether. In-process samples are removed at 30 and 60 minute time points.
Samples are prepared in a similar fashion to that described for the reaction workup. Reaction progress is monitored by TLC (1:10:3:3 MeOH/DCM/EtOAc/hexanes) and reverse-phase HPLC analyses. The isolated organic layer is successively washed with ice cold 1N HCl, saturated aqueous NaHCO3 (2×), saturated aqueous NaCl, and dried over sodium sulfate. Upon filtration and solvent removal, the residue undergoes solvent exchange with acetone followed by concentration in vacuo to provide crude product, which may be analyzed for purity by normal- and reversed-phase HPLC.
…………………….
SYNTHESIS
The process of the present invention is shown in the following Scheme II.


- “ARIAD Reports First Quarter 2009 Development Progress and Financial Results- Ridaforolimus New USAN Name to Replace Deforolimus”. ARIAD Pharmaceuticals. 2009. Retrieved 2009-05-07.
- “ARIAD – News release”. Phx.corporate-ir.net. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-03-17. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-06-06. Retrieved 2012-10-07.
|
7-11-2012
|
FULLY HUMAN ANTI-VEGF ANTIBODIES AND METHODS OF USING
|
|
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
4-16-2004
|
Phosphorus-containing compounds and uses thereof
|

