
Home » Posts tagged 'EMCURE'
Tag Archives: EMCURE
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
(S)-Atenolol

(S)-atenolol
- Description
Selective β1 adrenoceptor antagonist
- Biological descriptionSelective β1 adrenoceptor antagonist. Orally active. Limited ability to cross the blood-brain barrier. Antihypertensive activity in vivo.
Properties
- Chemical name(S)-(-)-4-[2-Hydroxy-3-[(1-methylethyl)amino]propoxy]benzeneacetamide
- Molecular Weight 266.34
- Molecular formula C14H22N2O3
- CAS Number 93379-54-5
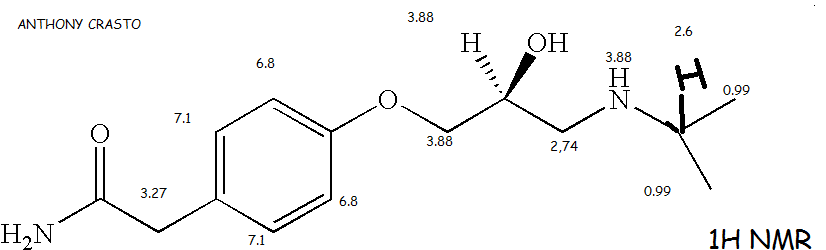
1H NMR (DMSO-d6): δ 0.99 (d, J=7 Hz, 6H, 2×CH3), 2.60 (m, 1H, CH), 2.74 (m, 2H, CH2), 3.27 (s, 2H, CH2), 3.88 (m, 4H, CH2, CH, NH), 6.83 (d, J=8 Hz, 2H, Ar—H), 7.14 (d, J=8 Hz, 2H, Ar—H), 7.40 (bs, 1H).
22.01, 22.09,
41.26, 48.39, 49.38, 67.73, 70.58, 114.16, 128.41, 129.93, 157.17, 172.59 ppm.

The compound (R,S)-atenolol (4-[2-hydroxy-3-[(1-methylethyl)amino]propoxy]-benzeneacetamide) is useful as a β-adrenegic blocker for the treatment of angina pectoris, arrhythmia and hypertension. It is known that atenolol is a 1-aryloxy-3-aminopropane-2-ol derivative wherein the hydroxy bearing carbon is an asymmetric carbon and hence exists as R- and S-isomers. It is also known that the S-isomer is particularly useful as a β-adrenegic blocker in view of its superior pharmacological activities. It is reported that S-atenolol has hypotensive activity and activity on brachycardia (A. A. Pearson, T. E. Gaffney, T. Walle, P. J. Privitera; J. Pharmacol. Exp. Ther., 250(3), 759, 1989).
In prior art, the optical resolution of racemic atenolol has been studied to obtain the desired optically active atenolol, however, any practical method has not been reported so far. It is also reported that the diastereomers of atenolol having high purity is obtained from racemic mixture by using (R,R)-O,O-di-toluoyltartaric acid anhydride (M. J. Wilson et al., J. Chromatogr. (NLD) 431 (1), 222–227, 1988). However, this method is not suitable for large scale production of optically active atenolol as it requires a large volume of solvent and further it is technically very troublesome to recycle (R,R)-O,O-di-toluoyltartaric acid anhydride.
Another method of preparing optically active atenolol has been proposed in JP-A-50-77331 and DE-A-2453324:

Wherein Z is halogen atom or sulphonyloxy group, and * means asymmetric carbon.
However, this process has some disadvantages as this process requires several steps for obtaining optically active S-atenolol stating from D-manitol; moreover the yield of S-atenolol by this process is less than 50% and the optical purity is just about 80% ee.
Another method for the preparation of S-atenolol has been reported in U.S. Pat. No. 5,223,646 which consists of reacting sodium salt of 4-carbamoylmethylphenol with R-epichlorohydrin at 0° to 35° C. to obtain an intermediate—an optically active glycidyl ether and then reacting the optically active intermediate glycidyl ether with isopropylamine to obtain S-atenolol (see also EP-435068 A2; EP-605384; JP 03077856 A2).
It has also been reported that the above procedure gives optically active glycidyl ether and atenolol of 90–96% ee optical purity. According to this report, the optical purity of atenolol may be enhanced to 98% or higher, if the intermediate optically active glycidyl ether is repeatedly recrystallised from a suitable solvent.
It has also been reported that the optically active atenolol in an optical purity of 98% or higher can be produced from atenolol of lower optical purity by converting it to its salt with Bronsted’s acid (K. Kazuhiro; T. Yosikazu; F. Yoshiro; Y. Hiroshi; O. Junzo, Chem. Pharm. Bull., 46(3), 505–507, 1998).
The separation of the atenolol salt having higher optical purity (>98% ee) is carried out by dissolving the atenolol salt having lower optical purity in a solvent, precipitating solid materials having a high content of racemic atenolol salt, and then isolating the desired atenolol salt having higher optical purity (>98% ee) by solid-liquid separation method. The optically active salt having high optical purity is then subjected to removal of acid moiety to isolate the desired optically active atenolol in free form. Though this process yields atenolol of higher optical purity, it involves salt formation and tedious separation of racemic salt from an optically active salt, which leads to the lower yields of desired optically active atenolol. Further, the salt has to be converted to free atenolol either by neutralisation or using ion exchange resins. Thus, this process gives lower overall yield of the desired optically active atenolol is low.
There is therefore a need to provide a process whereby S-atenolol may be obtained in high yield and high optical purity.
Emcure Pharmaceuticals Limited
http://www.google.com/patents/US6982349
Satish Ramanlal Mehta, Baburao Manikroa Bhawal, Vishnu Hari Deshpande, Mukund Keshav Gurjar
Accordingly, the present invention provides a process for the preparation of (S)-atenolol (1), which comprises the steps of:
-
- a) reacting a phenol of formula 2:

with an (R)-epichlorohydrin of formula (3):

in presence of an alkali metal hydroxide and a quaternary ammonium salt as phase transfer catalyst (PTC) in an aqueous solution at a temperature in a range of −10° C. to 0° C. to obtain optically active intermediate glycidyl ether of formula 4:

- b) reacting the optically active intermediate glycidyl ether (4) with isopropylamine at 10° to 40° C. to obtain (S)-atenolol of the formula 1:

in good chemical yield and high optical purity (>99 ee).
- a) reacting a phenol of formula 2:




One major advantage of this process is that S-atenolol may be obtained directly without going through the cumbersome step of recrystallization or additional salt formation step, as in the prior art.
The aqueous alkali metal hydroxide used in the process is selected from sodium hydroxide or potassium hydroxide and is used as aqueous solution in 1 to 1.5 moles to 1 mole of the phenol 2. The (R)-epichlorohydrin (3) used in the process is preferably of high optical purity and used in an amount of 1 to 3 moles, more preferably 1 to 1.6 moles, to 1 mole of phenol (2).
The quaternary ammonium salt has the formula:
R1R2R3R4N+X−
Wherein R1, R2, R3 and R4 are same or different, each an alkyl group having 1 to 16 carbon atoms (e.g. methyl, ethyl, propyl butyl etc), phenyl or benzyl, X is chlorine, bromine, iodine, hydrogen sulphate or hydroxyl group. The amount of quaternary ammonium salt used is 0.001 to 2% by weight of phenol (2).
The Applicant studied the reaction temperature extensively and found that it plays an important role in deciding optical purity of (S)-atenolol (1) formed via optically active glycidyl ether. When the reaction of phenol (2) and (R)-epichlorohydrin is carried out at 5° C. or at any other higher temperature, (S)-atenolol (1) of a lower optical purity was obtained via optically active glycidyl ether, as for example in EP 435068.
The Applicant, after studying the prior art processes found that during the course of these reactions, the phenoxide (or phenol) attacks the C-1 carbon atom of (R)-epichlorohydrin with the expulsion of chloride to yield (R)-glycidyl ether, which on reaction with isopropyl amine gives (R)-atenolol. The original epoxide ring remains unchanged in the reaction.

Thus, the reaction of phenol (2) at carbon centre C-1 of (R)-epichlorohydrin by nucleophilic displacement of chlorine leads to the formation of undesired (R)-atenolol via optically active (R)-glycidyl ether as a side product, which accounts for the low yield of optically active S-atenolol in the prior art.
The Applicant then conducted this reaction at a lower temperature and found to their surprise that S-atenolol could be obtained in high yield. The reason is that during the course of reaction, the phenoxide (or phenol) ion attacks the C-3 carbon atom of (R)-epichlorohydrin and opens the epoxide ring. The new epoxide ring formation takes place by the attack of O− on C-3 carbon with expulsion of chloride to give (S)-glycidyl ether, which on reaction with isopropyl amine gives (S)-atenolol. Thus, the reaction of phenol (2) at carbon centre C-3 of (R)-epichlorohydrin leads to the formation desired (S)-atenolol (1) as a major product via optically active glycidyl ether (4).

The lower optical purity in (S)-atenolol formation in the prior art may therefore be on account of the slow reaction rate at carbon atom 1 and the high yield of S-atenolol obtained by the process of the present invention may be due to the reaction at carbon atom 3 of (R)-epichlorohydrin (3). Both these reactions occurring on different atoms are shown as path ‘a’ and path ‘b’ in the following scheme herebelow.
Path ‘a’ is the process of the present invention whereas path ‘b’ is the process of the prior art.

EXAMPLE 1A mixture of (R)-epichlorohydrin ([α]D 25: −35.1 (neat), 138.75 g, 1.5 mole) and water (82 ml) was cooled to −7° C. and to this cold reaction mixture is added a solution of 4-hydroxyphenyl acetamide of formula 1 (151.00 g, 1 mole) and benzyltrimethylammonium chloride (1.3 g) in sodium hydroxide [40 g, 1 mole; dissolved in water (670 ml)] with stirring over a period of 3 hrs. maintaining the temperature at −7° C. to −5° C. The reaction mixture is then stirred further at −7° C. to −5° C. for 50 hrs. The precipitated solid is filtered, washed with water and dried at 60° C. to give 176 g of a mixture of S-glycidyl ether of formula 4 and S-chlorohydrin of formula 5 in about 3:2 ratio. m.p. 159–161° C.
EXAMPLE 2A mixture of isopropylamine (1.1 kg) and water (200 ml) is cooled to 10° C. and a mixture of S-glycidyl ether of formula 4 and S-chlorohydrin of formula 5 obtained in Example 1 (176 g) is added to it in lots maintaining temperature between 10 to 15° C. over a period of 3 hrs. The reaction is then stirred further for another 10 hr. The excess of isopropylamine is removed by distillation and the residue was treated with the water. The slurry so obtained is acidified with 5N HCl to pH 2.0. The resulting solution is then filtered, washed with water. The filtrate is basified with 2N NaOH to pH 11.7 and precipitated solid is filtered washed with water and dried to get (S)-atenolol (206 g, 91%) in 99.1% ee when analysed by using Chiracel OD column.
m.p. 152–153° C.
[α]D 25: −17.2 (c=1.0, 1N HCl).
IR: νmax 3352, 3168, 1635, 1242 cm−1.
1H NMR (DMSO-d6): δ 0.99 (d, J=7 Hz, 6H, 2×CH3), 2.60 (m, 1H, CH), 2.74 (m, 2H, CH2), 3.27 (s, 2H, CH2), 3.88 (m, 4H, CH2, CH, NH), 6.83 (d, J=8 Hz, 2H, Ar—H), 7.14 (d, J=8 Hz, 2H, Ar—H), 7.40 (bs, 1H).
13C NMR (DMSO-d6): 22.01, 22.09, 41.26, 48.39, 49.38, 67.73, 70.58, 114.16, 128.41, 129.93, 157.17, 172.59 ppm.
OTHER INFO
Despite both optical isomers being bioactive, as briefly mentioned in the Physical section, recent studies have shown that the S-ATENOLOL isomer was found to avoid the occasional side effect of an excessively lowered heart rate sometimes encountered with the racemate. The following steps are involved with the isolation of each enantiomer from 1-[p-[ (butoxy-carbonyl)methyl]phenoxy]-3-chloropropan-2-ol (7) using Lipase Catalysis.
The subsequent step highlights the production of S-ATENOLOL via (S-)1-[p-[ (butoxy-
carbonyl)methyl]phenoxy]-3-chloropropan-2-ol (8). This is fundamentally achieved using lipase from Pseudomonas Cepacia in a mixture of acetic anhydride and DIPE.
STEP 4 (b)

Whereas lipase from Pseudomonas Cepacia was used to isolate the S-ATENOLOL ISOMER, lipase from Candida Cylindracea is utilised in the production of the R + ATENOLOL ISOMER. The same principle applies in each isomeric scenario as demonstated below.
STEP 5 (a)

Acetic anhydride may also be used as a substitute for 1-butanol however, its inherent toxicity led to one opting for 1-butanol. Even though 1-butanol is harmful in its own right, on a relative scale is was the most suitable and effective alternative evolving an approximate 94 %conversion.
STEP 5 (b)

Yet again, greater than 95 % conversion is achieved after purifying the precipitate by treating(R) 1-[p-[(butoxy-carbonyl)methyl]phenoxy]-3-chloropropan-2-ol (10) with 2-methyl-ethanamine. This is then followed by addition of ammonium hydroxide in methanol and finally single recrystallisation in ethyl acetate.
References for (S)-(-)-Atenolol (ab120856)
This product has been referenced in:
- Agon P et al. Permeability of the blood-brain barrier for atenolol studied by positron emission tomography. J Pharm Pharmacol 43:597-600 (1991). Read more (PubMed: 1681079) »
- Tsuchihashi H et al. Characteristics of 125I-iodocyanopindolol binding to beta-adrenergic and serotonin-1B receptors of rat brain: selectivity of beta-adrenergic agents. Jpn J Pharmacol 52:195-200 (1990). Read more (PubMed: 1968985) »
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4085136 | Jun 9, 1976 | Apr 18, 1978 | Imperial Chemical Industries Limited | Adrenergic blocking agents |
| US5223646 | Apr 21, 1992 | Jun 29, 1993 | Daiso Company, Ltd. | Process for producing optically active atenolol and intermediate thereof |
| JPH0377856A | Title not available | |||
| JPH01102072A * | Title not available | |||
| JPH04198175A * | Title not available |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4085136 | Jun 9, 1976 | Apr 18, 1978 | Imperial Chemical Industries Limited | Adrenergic blocking agents |
| US5223646 | Apr 21, 1992 | Jun 29, 1993 | Daiso Company, Ltd. | Process for producing optically active atenolol and intermediate thereof |
| JPH0377856A | Title not available | |||
| JPH01102072A * | Title not available | |||
| JPH04198175A * | Title not available |
Tie up with Emcure…..Roche to launch cheaper cancer drugs in India
Reuters | Updated On: June 06, 2012 12:36 (IST)
Mumbai:
Swiss drugmaker Roche Holding AG plans to offer cut-price versions of two blockbuster cancer drugs for the Indian market soon, a company spokesman said on Friday, days after New Delhi moved to slash the price of a rival cancer treatment.
India stripped German’s Bayer AG of its exclusive rights to Nexavar earlier this month and licensed a local drugs company to produce a cheap, generic version, on the grounds that poor Indians could not otherwise afford the life-saving drug.
Roche, the world’s biggest maker of cancer drugs, said it would offer “significantly” cheaper, locally branded versions of its two cancer drugs, Herceptin and MabThera, by early next year, under an alliance with India’s Emcure Pharmaceuticals Ltd.
http://profit.ndtv.com/news/corporates/article-roche-to-launch-cheaper-cancer-drugs-in-india-300344
EMCURE-A SUCESS STORY

Mukund K Gurjar
| Emcure Pharmaceuticals Limited | ||
| ITBT Park Phase II | ||
| Hinjwadi, PUNE, INDIA |
Among the vast ocean of literature on organic chemistry , you will find a pearl in the form of Emcure
we are treated to excellent reading material and important communications in our field
hats off to this team


Dr. Mukund K. Gurjar serves as the Chief Scientific Officer of Emcure Pharmaceuticals Limited and serves as its Director of Research & Development.
Dr. Gurjar has been closely associated with Drugs and Pharmaceutical Sciences since 1975 and is a distinguished Researcher in the country. He has carried out extensive work in the field of new chemical entities (NCEs). Dr. Gurjar has been an Executive Director of Emcure Pharmaceuticals Ltd. since 2001.
He serves as Deputy Director at National Chemical Laboratory, Pune. Dr. Gurjar served as Non-Executive Director of Cipla Limited since January 19, 2002 until August 27, 2007.
Dr. Gurjar has the distinction of being one of the 43 scientists from India who have been mentioned in the Institute of Scientific Information of Chemists and has more than 500 citations. Dr. Gurjar has obtained Master of Science degree in Organic Chemistry and Ph.D. in chemistry from the Nagpur University. He also obtained the second Ph.D. degree from the London University, UK.
He is a leading Fellow at various National and International Academies
Board Members Memberships
Education
Other Affiliations

LINKS
http://www.emcure.co.in/bod.asp
http://www.ias.ac.in/php/fell_detail.php3?name=Gurjar&intials=Mukund&year=28-08-1952
http://www.researchgate.net/profile/Mukund_Gurjar/
| About EMCURE : Company Profile as quoted by COMPANY WEBSITE |
|
The Company was incorporated as Emcure Pharmaceuticals Private Limited on April 16, 1981 as a private limited company under the Companies Act, 1956.Emcure Pharmaceuticals is a fast growing Indian pharmaceutical company engaged in developing, manufacturing and marketing a broad range of pharmaceutical products globally. Our core strength lies in developing and manufacturing differentiated pharmaceutical products in-house, which we commercialize through our marketing infrastructure across geographies and relationships with multi-national pharmaceutical companies.Emcure Pharmaceuticals is ranked as the 14th largest pharmaceutical company (Source: IMS Health India, Secondary Stockist Audit (“SSA”), March 2013) in India in terms of market share based on the domestic sales of pharmaceutical products. We believe that our competitive advantage in the domestic market lies in our established presence in all major therapeutic areas including blood related, cardiology, pain and analgesics, HIV, gynecology, nephrology, anti-infective, and vitamins, minerals and nutrients products. We have also recently entered the oncology and diabetes therapeutic areas.Emcure Pharmaceuticals have a well-diversified income base thanks to our business in the international markets. We have our own sales and marketing infrastructure in the United States through our subsidiary, Heritage. We sell our portfolio of branded generic products to the Rest of World. Our products are currently shipped to over 65 countries, where we have established our presence by focusing on important alliances with local and multi-national companies that enjoy a leadership position in the therapeutic areas on which we focus. We have subsidiaries in Dubai, Brazil, South Africa, Singapore and Nigeria and branch offices in Russia and Morocco.Emcure Pharmaceuticals…….quote………. are focus our research and development efforts on developing a portfolio of differentiated products across several platforms, including chiral molecules and biosimilars, and novel drug delivery systems. We have a portfolio of 11 chiral molecules, eight of which we launched for the first time in India. We also have capabilities to develop complex products, including difficult iron preparations, oncology drugs and controlled release products. Our portfolio of in-house manufactured five commercialized biosimilars including TNK-tPA, which we launched for the first time in India, and our brand Vintor is ranked no. 1 in erythropoietin market (Epoetin Alfa Recombinant) (Source: IMS Health India, SSA, March 2013).
|
..

