
Home » Posts tagged 'DIABETES' (Page 4)
Tag Archives: DIABETES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
澳格列汀, SP2086, Retagliptin

 澳格列汀, SP2086, Retagliptin 1174122-54-3(Retagliptin), 1174038-86-8 (Retagliptin Hydrochloride), 1256756-88-3(Retagliptin Phosphate) (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester Methyl (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo [1,5-a]pyrazine-1-carboxylate, DPP-4 inhibitor Type II diabetes
澳格列汀, SP2086, Retagliptin 1174122-54-3(Retagliptin), 1174038-86-8 (Retagliptin Hydrochloride), 1256756-88-3(Retagliptin Phosphate) (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester Methyl (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo [1,5-a]pyrazine-1-carboxylate, DPP-4 inhibitor Type II diabetes
| Jiangsu Hengrui Medicine Co., Ltd |
 Nanjing Changao Pharmaceutical 澳格列汀 is a novel DPP-4 inhibitor (gliptin) for the treatment of type II diabetes. Because Shanghai Sun Sail Pharmaceutical, a wholly owned subsidiary of Nanjing Changao Pharmaceutical, has filed two patents to protect DPP-4 inhibitors (WO2011147207 and CN101786978), it is unknown which one covers this drug. Relevant data’s from WHO showed morbidity rate, disability rate, death rate of diabetes mellitus and overall health level of diabetes mellitus patients have already ranked the third place in non-infectious diseases, diabetes, together with tumors and cardiovascular diseases were the three main diseases which threats human health. Diabetes mellitus is usually classified into type 1 and type 2, there are more than 240 million diabetes patients, and 90% of them are suffering from type 2 diabetes, which also has a 1% growth rate every year, so, type 2 diabetes will be the main new growth point of diabetes drug market. The incidence of diabetes in China is about 5%, the number of patients of which ranks second place in the world just behind India. There are many antidiabetic drugs on the market, insulin injection, metformin, rosiglitazone, pioglitazone are representations of them. However, there is no drug alone can keep the HbA1c level of type 2 diabetes patients within the aimed range in a long term. Even though used in combination, the effect of the drugs will go down year by year after 3-4 years. Adverse reaction is one of the problems of many hypoglycemic drugs, wherein the fatal hypoglycemia is most worried by clinicians; secondly, many oral hypoglycemic drugs, such as sulfonylureas, α-glycosidase inhibitors and thiazolidinediones may all induce weight gain to patients, some of the drugs may also induce cardiovascular diseases. Therefore, developing new type hypoglycemic drugs with brand new mechanism of action, higher safety and effectiveness is an important task that should be completed quickly for the scientists. In the process of constantly finding new methods endocrine hormones were found to play an important role in the pathology and physiology of type 2 diabetes. Dipeptidyl peptidase-IV (DPP-IV) is an important enzyme related to diabetes, inhibiting the action of which to treat type 2 diabetes is a new method with good prospect. DPP-IV inhibitors can indirectly stimulate the secretion of insulin, the action of which is generated by inhibit DPP-IV to stabilize endocrine hormones such as incretin hormones, glucagons-like-peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a production expressed by glucagon protogene after eating, and mainly secreted by intestinal mucosa L-cell, and it can stimulate the secretion of insulin by pancreatic β-cells, which plays a significant role in the stability of blood sugar. Experiments prove that GLP-1 has physiological functions as following: acting on pancreatic β-cells in a glucose-dependent manner, facilitating the transcription of insulin genes, increasing the biosynthesis and secretion of insulin, stimulating the proliferation and differentiation of β-cells, inhibiting the apoptosis of β-cells to increasing the number of pancreatic β-cells; inhibiting the secretion of glucagon; inhibiting the appetite and food intake; retarding the emptying of gastric contents, etc., all of these functions are helpful to reduce blood sugar after food intake and to keep blood sugar within constant level. In addition, it won’t cause the danger of severe hypoglycemia. GLP-1 well controlled the blood sugar of type 2 diabetes animal models and patients by multiple mechanisms. However, GLP-1 may lose biological activity through quick degradation by DPP-IV, and the half life of it is shorter than 2 minutes, which utterly limits the clinical use of GLP-1. It was found in researches that DPP-IV inhibitors can totally protect endogenous and even extraneous GLP-1 from inactivation by DPP-IV, improve activated GLP-llevel, and reduce the antagonistic effect of GLP-1 metabolites. Moreover, DPP-IV inhibitors can also delay the incidence of diabetes through stimulating the regeneration of pancreatic β-cells and the improving the glucose tolerance and insulin sensitivity. Dipeptidyl peptidase-IV (DPP-IV) inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with Type 2 diabetes. For reviews on the application of DPP-IV inhibitors for the treatment of Type 2 diabetes, reference is made to the following publications: (1) H.-U.Demuth.et al. “Type 2 diabetes-Therapy with dipeptidyl peptidase IV inhibitors“, Biochim.Biophvs. Acta. 1751:33-44 (2005) and (2) K.Augustyns. et al. “Inhibitors of proline-specific dipeptidyl peptidases: DPP4 inhibitors as a novel approach for the treatment of Type 2 diabetes“, Expert Opin. Ther. Patents, 15:1387-1407 (2005). At present, some DPP-IV inhibitors have been disclosed ( US5462928 , US5543396 , WO9515309 ,WO2003004498 , WO2003082817 , WO2004032836 , WO2004085661 ), including MK-0431 as an DPP-IV inhibitor made by Merck which showed good inhibition activity and selectivity, and which has been on the market by 2006.
Nanjing Changao Pharmaceutical 澳格列汀 is a novel DPP-4 inhibitor (gliptin) for the treatment of type II diabetes. Because Shanghai Sun Sail Pharmaceutical, a wholly owned subsidiary of Nanjing Changao Pharmaceutical, has filed two patents to protect DPP-4 inhibitors (WO2011147207 and CN101786978), it is unknown which one covers this drug. Relevant data’s from WHO showed morbidity rate, disability rate, death rate of diabetes mellitus and overall health level of diabetes mellitus patients have already ranked the third place in non-infectious diseases, diabetes, together with tumors and cardiovascular diseases were the three main diseases which threats human health. Diabetes mellitus is usually classified into type 1 and type 2, there are more than 240 million diabetes patients, and 90% of them are suffering from type 2 diabetes, which also has a 1% growth rate every year, so, type 2 diabetes will be the main new growth point of diabetes drug market. The incidence of diabetes in China is about 5%, the number of patients of which ranks second place in the world just behind India. There are many antidiabetic drugs on the market, insulin injection, metformin, rosiglitazone, pioglitazone are representations of them. However, there is no drug alone can keep the HbA1c level of type 2 diabetes patients within the aimed range in a long term. Even though used in combination, the effect of the drugs will go down year by year after 3-4 years. Adverse reaction is one of the problems of many hypoglycemic drugs, wherein the fatal hypoglycemia is most worried by clinicians; secondly, many oral hypoglycemic drugs, such as sulfonylureas, α-glycosidase inhibitors and thiazolidinediones may all induce weight gain to patients, some of the drugs may also induce cardiovascular diseases. Therefore, developing new type hypoglycemic drugs with brand new mechanism of action, higher safety and effectiveness is an important task that should be completed quickly for the scientists. In the process of constantly finding new methods endocrine hormones were found to play an important role in the pathology and physiology of type 2 diabetes. Dipeptidyl peptidase-IV (DPP-IV) is an important enzyme related to diabetes, inhibiting the action of which to treat type 2 diabetes is a new method with good prospect. DPP-IV inhibitors can indirectly stimulate the secretion of insulin, the action of which is generated by inhibit DPP-IV to stabilize endocrine hormones such as incretin hormones, glucagons-like-peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a production expressed by glucagon protogene after eating, and mainly secreted by intestinal mucosa L-cell, and it can stimulate the secretion of insulin by pancreatic β-cells, which plays a significant role in the stability of blood sugar. Experiments prove that GLP-1 has physiological functions as following: acting on pancreatic β-cells in a glucose-dependent manner, facilitating the transcription of insulin genes, increasing the biosynthesis and secretion of insulin, stimulating the proliferation and differentiation of β-cells, inhibiting the apoptosis of β-cells to increasing the number of pancreatic β-cells; inhibiting the secretion of glucagon; inhibiting the appetite and food intake; retarding the emptying of gastric contents, etc., all of these functions are helpful to reduce blood sugar after food intake and to keep blood sugar within constant level. In addition, it won’t cause the danger of severe hypoglycemia. GLP-1 well controlled the blood sugar of type 2 diabetes animal models and patients by multiple mechanisms. However, GLP-1 may lose biological activity through quick degradation by DPP-IV, and the half life of it is shorter than 2 minutes, which utterly limits the clinical use of GLP-1. It was found in researches that DPP-IV inhibitors can totally protect endogenous and even extraneous GLP-1 from inactivation by DPP-IV, improve activated GLP-llevel, and reduce the antagonistic effect of GLP-1 metabolites. Moreover, DPP-IV inhibitors can also delay the incidence of diabetes through stimulating the regeneration of pancreatic β-cells and the improving the glucose tolerance and insulin sensitivity. Dipeptidyl peptidase-IV (DPP-IV) inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with Type 2 diabetes. For reviews on the application of DPP-IV inhibitors for the treatment of Type 2 diabetes, reference is made to the following publications: (1) H.-U.Demuth.et al. “Type 2 diabetes-Therapy with dipeptidyl peptidase IV inhibitors“, Biochim.Biophvs. Acta. 1751:33-44 (2005) and (2) K.Augustyns. et al. “Inhibitors of proline-specific dipeptidyl peptidases: DPP4 inhibitors as a novel approach for the treatment of Type 2 diabetes“, Expert Opin. Ther. Patents, 15:1387-1407 (2005). At present, some DPP-IV inhibitors have been disclosed ( US5462928 , US5543396 , WO9515309 ,WO2003004498 , WO2003082817 , WO2004032836 , WO2004085661 ), including MK-0431 as an DPP-IV inhibitor made by Merck which showed good inhibition activity and selectivity, and which has been on the market by 2006.
-
 sitagliptin
sitagliptin(R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester of the following formula is compound A, the code of which is SP2086.


 courtesy yaopha see enlarged image at http://www.yaopha.com/2014/02/10/chemical-structure-and-synthesis-of-hengrui-medicines-diabetes-drug-retagliptin/ …………………………………………………………..
courtesy yaopha see enlarged image at http://www.yaopha.com/2014/02/10/chemical-structure-and-synthesis-of-hengrui-medicines-diabetes-drug-retagliptin/ …………………………………………………………..
- EP2436684A1
- Example 1. Preparation of hydrochloride of compound A (SP2086-HCL)
- (R)-7-[3-t-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester (SM2086-15) (1.35kg, 2.40mol), HCL-ethyl acetate (greater than 2M) (12.3kg) were added into a 100L reaction kettle and stirred to dissolved. The mixture was reacted for more than 2 hours at normal temperature. Detected with TLC to reaction completely before evaporated and pumped to dryness with oil pump to give 1.15∼1.20kg of white to light yellow solid product with [α]
D20
- -28.0∼-33.0° (C=1, methanol), yield 96.0∼100%. The product was hydrochloride of (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester (SP2086-HCL). (TLC detection: silica gel GF254plate; developing reagent: chloroform: methanol: ammonia= 40: 1: 0.1; raw material 15: Rf=0.80, product 1: Rf=0.50; ultraviolet visualization).
Example 2. Preparation of phosphate of compound A (SP2086-HPO4)
-
SP2086-HCL(1.20kg, 2.40mol) was added into 100L reaction kettle, and dissolved in dichloromethane (15.2kg), then washed with saturated sodium bicarbonate solution (5.8kg). The aqueous layer was extracted once with dichloromethane ( 6.0 kg). The organic layers were combined and washed once with water (5kg), dried with anhydrous sodium sulphate. The mixture was filtrated and concentrated to dryness under reduced pressure at 40°C to give 1.12 kg of oil. The oil was stirred and dissolved with 30 times amount of isopropanol (26.0kg). A solution of 85% phosphoric acid (305.2g, 2.65mol) in isopropanol (1.22kg) was added immidiately after the oil completely dissolved. The solid was separated out, filtered after stirring for 2 hours and washed with cold isopropanol. The wet product was dried under reduced pressure at 40°C to give 1.16∼1.24kg of white to light yellow solid with a yield of 86.0∼92.0% (the wet product could be directly suspended in isopropanol without drying).
……………………………………… http://www.google.com/patents/EP2230241A1?cl=en Example 1(R)-7-[3-Amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester hydrochloride

Step 1
-
2,2-Dimethyl-5-[2-(2,4,5-trifluoro-phenyl)-acetyl]-[1,3]dioxane-4,6-dione 2,2-Dimethyl-[1,3]dioxane-4,6-dione (5.69 g, 39.5 mmol) was dissolved in 400 mL of dichloromethane under stirring, followed by addition of (2,4,5-trifluoro-phenyl)-acetic acid 1a (7.15 g, 37.6 mmol) and 4-dimethylaminopyridine (7.35 g, 60.2 mmol) in an ice-water bath. Then a suspension of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (8.28 g, 43.2 mmol) in 250 mL of dichloromethane was added dropwise slowly. After stirring at room temperature for 36 hours, the reaction mixture was washed with the solution of 5% potassium bisulfate (250 mL×7) and saturated brine (250 mL×2), dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to obtain the title compound 2,2-dimethyl-5-[2-(2,4,5-trifluoro-phenyl)-acetyl]-[1,3]dioxane-4,6-dione 1b (11.4 g, yield 96%) as a white solid. MS m/z (ESI): 315.5 [M-1]
Step 23-Oxo-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester
-
2,2-Dimethyl-5-[2-(2,4,5-trifluoro-phenyl)-acetyl]-[1,3]dioxane-4,6-dione 1b (15.72 g, 49.6 mmol) was dissolved in 280 mL of ethanol under stirring, then the reaction mixture was heated to 70 °C in an oil bath overnight. After cooling, the mixture was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 3-oxo-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1c (12 g, yield 88%) as a yellow oil. MS m/z (ESI): 259 [M-1]
Step 33-Amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoic acid ethyl ester
-
3-Oxo-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1c (24.6 g, 94.5 mmol) was dissolved in 240 mL of methanol, and ammonium acetate (36.4 g, 473 mmol) was added to the solution. The reaction mixture was heated to reflux for 3 hours and monitored by thin layer chromatography until the disappearance of the starting materials. The reaction mixture was concentrated under reduced pressure, then 100 mL of water was added to the residue. The mixture was extracted with ethyl acetate (200 mL×3), and the combined organic phase was washed with 200 mL of saturated brine, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to obtain a light yellow solid. The resulting solid was dissolved in 50 mL of ethyl acetate at 80 °C, then 50 mL of n-hexane and seed-crystal were added to the solution. The mixture was cooled to room temperature, half an hour later, 100 mL of n-hexane was added. The mixture was stored in refrigerator overnight and then filtered under reduced pressure to obtain the title compound 3-amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoic acid ethyl ester 1d(19.5 g, yield 80%) as a white solid. MS m/z (ESI): 260.1 [M+1]Step 43-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester
-
3-Amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoic acid ethyl ester 1d (4.1 g, 15.8 mmol) was added into an autoclave, followed by addition of 70 mL of methanol, di-tert-butyl dicarbonate (3.8 g, 17.4 mmol), chloro(1, 5-cyclooctadiene)rhodium( I ) dimer (32 mg, 0.0632 mmol) and (R)-1-[(S)-2-(diphenyl phosphino)ferrocenyl]-ethyl-tert-butylphosphine (68 mg, 0.126 mmol). The reaction mixture was hydrogenated for 24 hours under 6.67 atmosphere at 30 °C. The mixture was filtered and the filtrate was concentrated under reduced pressure. Then 34 mL of methanol was added to the residue at 50 °C, followed by addition of 12 mL of water until all dissolved. After cooling to room temperature, the mixture was stored in the refrigeratory overnight and then filtered. The solid product was washed with the solvent mixture of methanol/water (v:v = 3:2), dried in vacuo to obtain the title compound 3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1e (4 g, yield 70%) as a light yellow solid. MS m/z (ESI): 362.4 [M+1]Step 5(R)-3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid
-
3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1e (10 g, 27.7 mmol) and sodium hydroxide (3.32 g, 83.1 mmol) were dissolved in the solvent mixture of 100 mL of methanol and 50 mL of water under stirring. The reaction mixture was reacted at 40-45 °C for 1-1.5 hours, then part of the solution was evaporated under reduced pressure. The residue was added with some water, then pH was adjusted to 2-3 with 1 N hydrochloric acid in an ice-water bath. The mixture was extracted with ethyl acetate (200 mLx3), and the combined organic phase was washed with 200 mL of saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure, and then recrystallized from ethyl acetate/n-hexane to obtain the title compound (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid 1f (9.2 g) as a white solid, which was directly used in the next step. MS m/z (ESI): 332.3 [M-1] Reference: Tetrahedron Asymmetry, 2006, 17(2), 205-209
Step 6C-Pyrazin-2-yl-methylamine
-
Pyrazine-2-carbonitrile 1g (10.5 g, 100 mmol) was dissolved in 150 mL of 1,4-dioxane under stirring, then Raney nickel (1.0 g) was added into a 250 mL autoclave. The reaction mixture was hydrogenated for 8 hours under 40 atmosphere at 60 °C, filtered and concentrated under reduced pressure to obtain the title compound C-pyrazin-2-yl-methylamine 1h (10.7 g, yield 98%) as a brown oil. MS m/z (ESI): 110 [M+1]
Step 72,2,2-Trifluoro-N-pyrazin-2-ylmethyl-acetamide
-
C-Pyrazin-2-yl-methylamine 1h (10.9 g, 100 mmol) was added into a reaction flask, then 20 mL of trifluoroacetic anhydride was added dropwise slowly within an hour at 0 °C in an ice-water bath. The reaction mixture was reacted at room temperature for 2 hours and monitored by thin layer chromatography until the disappearance of the starting materials. Then it was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 2,2,2-trifluoro-N-pyrazin-2-ylmethyl-acetamide 1i (21.0 g) as a brown oil. MS m/z (ESI): 206.1 [M+1]
Step 83-Trifluoromethyl-imidazo[1,5-a]pyrazine
-
2,2,2-Trifluoro-N-pyrazin-2-ylmethyl-acetamide 1i (21.0 g, 100 mmol) was added into a reaction flask at room temperature, followed by addition of 100 mL of phosphorus oxychloride. After stirring at room temperature for 30 minutes, phosphorous pentoxide (17.8 g, 125 mmol) was added to the solution. The reaction mixture was heated to reflux for 5 hours and monitored by thin layer chromatography until the disappearance of the starting materials. Phosphorus oxychloride was removed, and the reaction system was quenched with deionized water. The mixture was adjusted to pH 5-6 with 20% sodium hydroxide solution in an ice-water bath. The mixture was extracted with ethyl acetate (250 mL×4), and the combined organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 3-trifluoromethyl-imidazo[1,5-a]pyrazine 1j (12.0 g, yield 65%) as a yellow solid. MS m/z (ESI): 188.0 [M+1] 1H NMR (400 MHz, CDCl3): δ 9.15 (s, 1H), 8.06 (d, 1H), 7.92 (s, 1H), 7.81 (d, 1H)
Step 93-Trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine
-
3-Trifluoromethyl-imidazo[1,5-a]pyrazine 1j (12.0 g, 64.2 mmol) was dissolved in 150 mL of anhydrous ethanol under stirring, then 10% Pd/C (500 mg) was added to the solution. The reaction mixture was stirred at room temperature under a hydrogen atmosphere overnight. The reaction solution was filtered through a pad of coarse silica gel and concentrated under reduced pressure to obtain the title compound 3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine 1k (12.2 g, yield 99%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 6.84 (s, 1H), 4.10 (m, 4H), 3.26 (m, 2H), 1.81 (s, 1H)
Step 10(R)-[3-Oxo-1-(2,4,5-trifluoro-benzyl)-3-(3-trifluoromethyl-5,6-dihydro-8H-imidazo [1,5-a]pyrazin-7-yl)-propyl]-carbamic acidtert-butyl ester
-
Under a nitrogen atmosphere, 3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid 1k (8.6 g, 45 mmol) and 9.4 mL of triethylamine were dissolved in 300 mL of dichloromethane under stirring. After stirring at room temperature for 5 minutes, 3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine 1f (15.0 g, 45 mmol) and bis(2-oxo-3-oxazolidinyl)phosphonic chloride (17.1 g, 67.3 mmol) were added to the solution successively. The reaction mixture was reacted at room temperature for 2 hours and monitored by thin layer chromatography until the disappearance of the starting materials and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound (R)-[3-oxo-1-(2,4,5-trifluoro-benzyl)-3-(3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-propyl]-carbamic acid tert-butyl ester 1l (20.0 g, yield 88%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.25 (m, 1H), 7.11 (m, 1H), 7.032 (s, 1H), 4.93 (m, 2H), 4.35 (m, 3H), 4.05 (m, 2H), 2.99 (m, 2H), 2.73 (m, 2H), 1.34 (s, 9H)
Step 11(R)-[3-(1-Bromo-3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-3-oxo-1-(2,4,5-trifluoro-benzyl)-propyl]-carbamic acidtert-butyl ester
-
(R)-[3-Oxo-1-(2,4,5-trifluoro-benzyl)-3-(3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-propyl]-carbamic acid tert-butyl ester 11 (20.0 g, 39.6 mmol) was dissolved in 300 mL of anhydrous ethanol under stirring, and 1-bromo-2,5-pyrolidinedione (14.1 g, 79.2 mmol) was then added to the solution at room temperature. After stirring for an hour, potassium carbonate (10.9 g, 79.2 mmol) and di-tert-butyl dicarbonate (8.6 g, 39.6 mmol) were added to the mixture, and the mixture was stirred for an hour and monitored by thin layer chromatography until the disappearance of the starting materials. The reaction mixture was filtered through a pad of coarse silica gel to remove potassium carbonate, and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound (R)-[3-oxo-1-(2,4,5-trifluoro-benzyl)-3-(1-bromo-3-trifluoromethyl-5,6-dihydro-8H-i midazo [1,5-a]pyrazin-7-yl)-propyl]-carbamic acid tert-butyl ester 1m (20.0 g, yield 86%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.063 (m, 1H), 6.88 (m, 1H), 4.72 (s, 1H), 4.56 (s, 1H), 4.13 (m, 3H), 3.88 (m, 2H), 2.94 (m, 2H), 2.62 (m, 2H), 1.36 (s, 9H)
Step 12(R)-7-[3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester
-
Octacarbonyldicobalt (4.02 g, 11.76 mmol), ethyl chloroacetate (0.71 g, 5.88 mmol), potassium carbonate (1.62 g, 11.76 mmol) and 50 mL of methanol were added into the reaction flask. After stirring for 5 minutes, (R)-[3-oxo-1-(2,4,5-trifluoro-benzyl)-3-(1-bromo-3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-propyl]-carbamic acidtert-butyl ester 1m (2.3 g, 3.92 mmol) was added. The reaction mixture was reacted at 60 °C in an oil bath, and the colour of the reaction mixture turned from puce to purple. 2 hours later, Electro-Spray Ionization (ESI) mass spectrometry showed the starting material disappeared. The reaction mixture was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound (R)-7-[3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester 1n (1.1 g, yield 50%) as a white solid. MS m/z (ESI): 565.0 [M+1] Reference: Journal of Organometallic Chemistry, 1985, 285(1-3), 293-303
Step 13(R)-7-[3-Amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester hydrochloride
-
[0064](R)-7-[3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester 1n (0.12 g, 2.12 mmol) was added to a solution of 2.2 N hydrochloric acid in 5 mL of ethyl acetate. The reaction mixture was reacted at room temperature for 5 hours and monitored by thin layer chromatography until the disappearance of the starting materials. The reaction mixture was concentrated under reduced pressure to obtain the title compound (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester hydrochloride 1 (0.12 g, yield 94.3%) as a light yellow solid. MS m/z (ESI): 465.2 [M+1] 1H NMR (400 MHz, CD3OD): δ 7.101-7.08 (m, 1H), 6.906-6.864 (m, 1H), 5.343-4.995 (m, 2H), 4.221-4.093 (m, 5H), 3.954 (s, 3H), 2.978-2.937 (m, 2H), 2.71-2.643 (m, 2H), 2.061 (s, 2H)
| EP2230241A1 * | Nov 27, 2008 | Sep 22, 2010 | Jiangsu Hengrui Medicine Co., Ltd. | Tetrahydro-imidazoý1,5-a¨pyrazine derivatives, preparation methods and medical uses thereof |
| WO2003004498A1 * | Jul 5, 2002 | Jan 16, 2003 | Merck & Co Inc | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2005003135A1 * | Jun 18, 2004 | Jan 13, 2005 | Alex Minhua Chen | Phosphoric acid salt of a dipeptidyl peptidase-iv inhibitor |
CARMEGLIPTIN………….a DPP-4 inhibitor

(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
(2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one
813452-14-1 (di-HCl)
916069-91-5 (mono-HCl)
Roche…….innovator

CARMEGLIPTIN
813452-18-5
(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one
(S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| 分子式: | C20H28FN3O3 |
| 分子量: | 377 |
813452-18-5, Carmegliptin, R-1579;carmegliptin, Carmegliptin (USAN/INN), SureCN419289, UNII-9Z723VGH7J, CHEMBL591118, CHEBI:699093, Ro-4876904, D08631, R-1579, B1Q
Type 2 diabetes is a chronic, progressive metabolic disease, affecting about 4% of the world population. The main goal of the management of type 2 diabetes is to achieve glycemic control as close to the nondiabetic range as practicable, in order to reduce the risk of late-stage complications.However, the therapeutic effect provided by existing medications is often not sustainable, since the multi-organ defects responsible for the disease are only insufficiently addressed.
Dipeptidyl peptidase-IV (DPP-IV) inhibitors have emerged as a new therapeutic option to treat type 2 diabetes.
Their rapid rise in popularity is due to the favourable safety profile (no hypoglycemia, no weight gain, no gastrointestinal problems—typical side effects associated with established anti-diabetic agents). DPP-IV is a ubiquitous serine protease, the inhibition of which prevents the degradation of glucagon-like peptide 1 (GLP-1). The resulting higher levels of GLP-1 have a beneficial impact on major players involved in the pathogenesis of type 2 diabetes: β-cells, liver, α-cells, gut, and brain.
Long-term studies with DPP-IV inhibitors in patients are underway in order to confirm the safety and sustainability of these effects, and, in particular, their ability to prevent the progressive loss of β-cell function.
SYNTHESIS

aReagents and conditions: a) HCO2Me, Δ; b) POCl3, MeCN; c) HO2CCH2CO2Et, neat, 120 °C; d) ethyl acrylate, neat; e) t-BuOK, neat (5 steps); f) NH4OAc, MeOH; g) NaBH4, TFA, THF; h) Boc2O, CH2Cl2; i) KOH, aq THF; j) DPPA, Et3N, TMSCH2CH2OH, PhMe, 80 °C; k) Et4NF, MeCN; l) chiral HPLC; m) Et3N, CH2Cl2; n) NaH, DMF; o) HCl, dioxane; p) HCl, 2-PrOH.

Scheme 2.
Reagents and conditions: (a) NH4OAc, MeOH, rt, 95%; (b) NaBH4, TFA, THF, 0 °C; (c) Boc2O, CH2Cl2, 83% over 2 steps; (d) KOH, aq THF, rt; (e) DPPA, Et3N, 2-(trimethylsilyl)ethanol, toluene, 80 °C; (f) Et4NF, CH3CN, 50 °C, 56% over 3 steps; (g) Et3N, CH2Cl2, (h) NaH, cat. NaI, DMF; (i) HCl, 1,4-dioxane.
Carmegliptin (2.70) is an anti-diabetes drug which is currently in late stage clinical trials. It represents a further structural advancement from the other existing marketed drugs in this class, sitagliptin (2.71, Januvia) and vildagliptin (2.72, Zomelis, Figure 7). These compounds are all members of the dipeptidyl peptidase 4 class (DPP-4), a transmembrane protein that is responsible for the degradation of incretins; hormones which up-regulate the concentration of insulin excreted in a cell. As DPP-4 specifically cleaves at proline residues, it is unsurprising that the members of this drug class exhibit an embedded pyrrolidine ring (or mimic) and additional decoration (a nitrile or fluorinated alkyl substituent is present in order to reach into a local lipophilic pocket). One specific structural liability of the 2-cyano-N-acylpyrrolidinyl motif (2.73) is its inherent susceptibility towards diketopiperazine formation (2.74, Scheme 29) [80], however, one way to inhibit this transformation is to position a bulky substituent on the secondary amine nucleophile as is the case in vildagliptine (2.72).
![[1860-5397-9-265-7]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-7.png)
Figure 7: Structures of DPP-4 inhibitors of the gliptin-type.
![[1860-5397-9-265-i29]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i29.png)
Scheme 29: Formation of inactive diketopiperazines from cis-rotameric precursors.
A single crystal X-ray structure of carmegliptin bound in the human DPP-4 active site has been published indicating how the fluoromethylpyrrolidone moiety extends into an adjacent lipophilic pocket [81]. Additional binding is provided by π–π interaction between the aromatic substructure and an adjacent phenylalanine residue as well as through several H-bonds facilitated by the adjacent polar substituents (Figure 8).
![[1860-5397-9-265-8]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-8.png)
Figure 8: Co-crystal structure of carmegliptin bound in the human DPP-4 active site (PDB 3kwf).
The reported synthesis of carmegliptin enlists a Bischler-Napieralski reaction utilising the primary amine 2.76 and methyl formate to yield the initial dihydroquinoline 2.77 as its HCl salt (Scheme 30) [82]. This compound was next treated with 3-oxoglutaric acid mono ethyl ester (2.78) in the presence of sodium acetate. Decarboxylation then yields the resulting aminoester 2.79 which was progressed through an intramolecular Mannich-type transformation using aqueous formaldehyde to allow isolation of enaminoester 2.80 after treatment of the intermediate with ammonium acetate in methanol.
The next step involves a very efficient crystallisation-induced dynamic resolution of the racemic material using the non-natural (S,S)-dibenzoyl-D-tartaric acid ((+)-DBTA). It is described that the desired (S)-enantiomer of compound 2.81 can be isolated in greater than 99% ee and 93% overall yield. This approach is certainly superior to the original separation of the two enantiomers (at the stage of the final product) by preparative chiral HPLC that was used in the discovery route (albeit it should be noted that both enantiomers were required for physiological profiling at the discovery stage).
Next, a 1,2-syndiastereoselective reduction of enaminoester 2.81 occurs with high diastereocontrol imposed by the convexed presentation of the substrate for the formal conjugate addition and subsequent protonation steps. This is followed by Boc-protection and interconversion of the ethyl ester to its amide derivative 2.82 in 80% overall yield for this telescoped process. The primary amide in 2.82 was then oxidised via a modern variant of the classical Hoffmann rearrangement using phenyliodine diacetate (PIDA).
Following extensive investigation it was found that slowly adding this reagent in a mixture of acetonitrile/water to a suspension of amide 2.82 and KOH gave clean conversion to the amine product in high yield. This new procedure was also readily scalable offering a cleaner, safer and more reliable transformation when compared to other related rearrangement reactions. During a further telescoped procedure amine 2.83 was treated with lactone 2.84 to regenerate the corresponding lactam after mesylate formation. Finally, removal of the Boc-group with aqueous hydrochloric acid furnished carmegliptin as its HCl salt.
![[1860-5397-9-265-i30]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i30.png)
Scheme 30: Improved route to carmegliptin.
- Peters, J.-U. Curr. Top. Med. Chem. 2007, 7, 579–595……………..80
- Mattei, P.; Boehringer, M.; Di Gorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U.Bioorg. Med. Chem. Lett. 2010, 20, 1109–1113. doi:10.1016/j.bmcl.2009.12.024………..81
- Albrecht, S.; Adam, J.-M.; Bromberger, U.; Diodone, R.; Fettes, A.; Fischer, R.; Goeckel, V.; Hildbrand, S.; Moine, G.; Weber, M. Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207……….82
………………………………………………………………………………………………………………..
Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207
http://pubs.acs.org/doi/full/10.1021/op2000207

A short and high-yielding synthesis of carmegliptin (1) suitable for large-scale production is reported. The tricyclic core was assembled efficiently by a decarboxylative Mannich addition−Mannich cyclization sequence. Subsequent crystallization-induced dynamic resolution of enamine 7 using (S,S)-dibenzoyltartaric acid was followed by diastereoselective enamine reduction to give the fully functionalized tricyclic core with its three stereogenic centers. The C-3 nitrogen was introduced by Hofmann rearrangement of amide 28, and the resulting amine 10was coupled with (S)-fluoromethyl lactone 31. Following cyclization to lactam 13 and amine deprotection, 1 was obtained in 27−31% overall yield with six isolated intermediates.
Preparation of (2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride (1) CARMEGLIPTIN
A suspension of carbamate 13 (136 kg, 285 mol) in a mixture of H2O (112 kg) and acetone (122 kg) was treated at 50 °C within 60 min with 37% aq HCl (98.0 kg). After 90 min at 47−52 °C the solution was polish filtered through a 5 μm filter. The first reactor and the transfer lines were washed with a hot (47−52 °C) mixture of H2O (13.0 kg) and acetone (116 kg). The filtrate was cooled to 25 °C and treated at this temperature within 80 min with acetone (1600 kg) whereupon the product crystallized out. The resulting suspension was stirred for 1 h at 25 °C and subsequently centrifuged. The crystals were washed in two portions with acetone (391 kg) and dried at 50 °C and <30 mbar until constant weight to afford 122.4 kg (95%) of the title compound as colorless crystals with an assay (HPLC) of 98.8% (w/w).
1H NMR (400 MHz, D2O) δ 2.11−2.22 (m, 1H); 2.45 (dd, J = 17.6 Hz, 6.7 Hz; 1H); 2.76 (dd, J = 17.6 Hz, 9.55 Hz, 1H); 2.90−3.05 (m, 1H); 3.08−3.19 (m, 2H); 3.24−3.36 (m, 1H); 3.43 (dd, J = 9.8 Hz, 5.75 Hz, 1H); 3.49−3.58 (m, 1H); 3.70−3.84 (m, 4H); 3.87 (s, 3H); 3.88 (s, 3H); 4.12 (td, J = 11.6 Hz, 4.5 Hz, 1H); 4.45−4.55 (m, 1H); 4.56−4.68 (m, 3H); 6.91 (s, 1H), 6.95 (s, 1H).
IR (cm−1): 3237, 2925, 1682, 496, 478.
MS (ESI): m/z 378.3 ([M + H]+ (free amine)).
Anal. Calcd for C20H30Cl2FN3O3: C, 53.34; H, 6.71; N, 9.33; Cl, 15.74; F 4.22; O, 10.66. Found: C, 53.04; H, 6.43; N, 9.45; Cl, 15.66; F, 4.29; O, 11.09.
REF FOR ABOVE
Mattei, P.; Böhringer, M.; Di Giorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Kocer, B.; Kuhn, B.; Löffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U. Bioorg. Med. Chem. Lett. 2010, 20, 1109
Böhringer, M.; Kuhn, B.; Lübbers, T.; Mattei, P.; Narquizian, R.; Wessel,H. P. (F. Hoffmann-La Roche AG). U.S. Pat. Appl. 2004/0259902, 2004.
…………………………………………………..
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
Bioorg Med Chem Lett 2010, 20(3): 1109
Bioorg Med Chem Lett 2010, 20(3): 1109
http://www.sciencedirect.com/science/article/pii/S0960894X09017296
-
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
- Pages 1109-1113
- Patrizio Mattei, Markus Boehringer, Patrick Di Giorgio, Holger Fischer, Michael Hennig, Joerg Huwyler, Buelent Koçer, Bernd Kuhn, Bernd M. Loeffler, Alexander MacDonald, Robert Narquizian, Etienne Rauber, Elena Sebokova, Urs Sprecher
-

 Scheme 3.
Scheme 3.Reagents and conditions: (a) preparative HPLC (Chiralpak® AD column), heptane/2-propanol 85:15, 37% (b) BH3.Me2S, THF, 0 °C; (c) (MeOCH2CH2)2NSF3, CH2Cl2, 67% (2 steps); (d), SOCl2, ZnCl2, 80 °C, 72 h, 61%; (e) Et3N, CH2Cl2; (f) NaH, DMF, 56% (2 steps); (g) HCl, 1,4-dioxane, 91%; (h) HCl, 2-propanol, 86%.
The synthesis of 8p is outlined ABOVE and required the enantiopure building blocks (S,S,S)-5 and 12. (S,S,S)-5 was obtained from the racemate by preparative chiral HPLC. Acid chloride 12 was prepared starting from (S)-paraconic acid (9). Reduction of 9 with borane–dimethyl sulfide complex afforded hydroxymethyl lactone 10. Since 10 is known to racemise rather readily, it was immediately treated with bis(2-methoxyethyl)aminosulfur trifluoride, thereby affording fluoromethyl lactone 11. This was converted to 12 by reaction with thionyl chloride in the presence of zinc chloride. The (S)-4-fluoromethyl-pyrrolidinone 8p was isolated as the dihydrochloride salt, a highly water soluble white crystalline solid, mp >275 °C.
…………………………………………………….
US 2013109859
The most preferred product is (2S,3S,11bS)-2-tert.-Butoxycarbonylamino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H pyrido[2,1-a]isoquinoline-3-carboxylic acid amide having the following structure:

It has been found that during the amidation of the ester epimerization takes place at position 3 and thus the 3R-epimer of the formula IVb is transformed to a larger extent in the 3S-epimer of formula V.

e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
A 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
…………………………………………………….
US 2008071087







(2S,3S,11bS)-(3-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester (8)
Example 8
Transformation of (2S,3S,11bS)-(3-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl) ]-carbamic acid tert-butyl ester into (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl) -4-fluoromethyl-pyrrolidin-2-one.a)
Preparation of 4-fluoromethyl-5H-furan-2-oneA 6 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 500 g (4.38 mmol) 4-hydroxymethyl-5H-furan-2-one and 2.0 L dichloromethane. The solution was cooled to −10° C. and 1.12 kg (4.82 mol) bis-(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) was added during 50 min, maintaining the temperature at −5 to −10° C. with a cooling bath. During the addition a yellowish emulsion formed, which dissolved to an orange-red solution after completed addition. This solution was stirred for 1.5 h at 15-20° C., then cooled to −10° C. A solution of 250 ml water in 1.00 L ethanol was added during 30 min, maintaining the temperature between −5 and −10° C., before the mixture was allowed to reach 15-20° C. It was then concentrated in a rotatory evaporator to a volume of ca. 1.6 L at 40° C./600-120 mbar. The residue was dissolved in 2.0 L dichloromethane and washed three times with 4.0 L 1N hydrochloric acid. The combined aqueous layers were extracted three times with 1.4 L dichloromethane. The combined organic layers were evaporated in a rotatory evaporator to give 681 g crude product as a dark brown liquid. This material was distilled over a Vigreux column at 0.1 mbar, the product fractions being collected between 71 and 75° C. (312 g). This material was re-distilled under the same conditions, the fractions being collected between 65 and 73° C., to give 299 g 4-fluoromethyl-5H-furan-2-one (58% yield; assay: 99%).MS: m/e 118 M+, 74,59,41.b) Preparation of (S)-4-fluoromethyl-dihydro-furan-2-oneA 2 L autoclave equipped with a mechanical stirrer was charged with a solution of 96.0 g 4-fluoromethyl-5H-furan-2-one (8.27×10−1 mol) in 284 mL methanol. The autoclave was sealed and pressurized several times with argon (7 bar) in order to remove any traces of oxygen. At ˜1 bar argon, a solution of 82.74 mg Ru(OAc)2((R)-3,5-tBu-MeOBlPHEP) (6.62×10−5 mol) (S/C 12500) in 100 mL methanol was added under stirring from a catalyst addition device previously charged in a glove box (O2 content <2 ppm) and pressurized with argon (7 bar). The argon atmosphere in the autoclave was replaced by hydrogen (5 bar). At this pressure, the reaction mixture was stirred (˜800 rpm) for 20 h at 30° C. and then removed from the autoclave and concentrated in vacuo. The residue was distilled to afford 91.8 g (94%) (S)-4-fluoromethyl-dihydro-furan-2-one. The chemical purity of the product was 99.7% by GC-area.c) Preparation of (2S,3S,11bS)-3-(3-Fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 50 g (128 mmol) (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester, 500 mL toluene and 2.51 g (25.6 mmol) 2-hydroxypyridine. To this slightly brownish suspension, 22.7 g (192 mmol) of (S)-4-fluoromethyl-dihydro-furan-2-one was added dropwise at RT. No exothermy was observed during the addition. The dropping funnel was rinsed portionwise with totally 100 mL toluene. The suspension was heated to reflux, whereas it turned into a dear solution starting from 60° C., after 40 min under reflux a suspension formed again. After totally 23 h under reflux, the thick suspension was cooled to RT, diluted with 100 mL dichloromethane and stirred for 30 min at RT. After filtration, the filter cake was washed portionwise with totally 200 mL toluene, then portionwise with totally 100 mL dichloromethane. The filter cake was dried at 50° C./10 mbar for 20 h, to give 60.0 g product (94% yield; assay: 100%).


 UNDESIRED
UNDESIRED

 DESIRED
DESIRED
 UNDESIRED
UNDESIRED


MS: m/e 496 (M+H)+, 437.
d) Preparation of (2S,3S,11bS)-3-((4S)-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel, a cooling bath and a nitrogen inlet was charged with 28 g (56.5 mmol) of (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino) -9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and 750 mL THF. The mixture was cooled to 0° C. and a solution of 6.17 mL (79 mmol) methanesulfonic acid in 42 mL THF was added during 10 min, maintaining the temperature at 0-5° C. At 0° C. a solution of 12.6 mL (90.2 mmol) triethylamine in 42 mL THF was added during 15 min. The resulting suspension was stirred for 80 min at 0-5° C., whereas it became gradually thicker. Then 141 mL (141 mmol) 1 M lithium-bis(trimethylsilyl)amide were added to the mixture during 15 min, whereas the suspension dissolved. The solution was allowed to reach RT during 60 min under stirring. 500 mL water was added without cooling, the mixture was extracted and the aqueous phase was subsequently extracted with 500 mL and 250 mL dichloromethane. The organic layers were each washed with 300 mL half saturated brine, combined and evaporated on a rotatory evaporator. The resulting foam was dissolved in 155 mL dichloromethane, filtered and again evaporated to give 30.5 g crude product as a slightly brownish foam. This material was dissolved in 122 mL methanol, resulting in a thick suspension, which dissolved on heating to reflux. After 20 min of reflux the solution was allowed to gradually cool to RT during 2 h, whereas crystallization started after 10 min. After 2 h the suspension was cooled to 0° C. for 1 h, followed by −25° C. for 1 h. The crystals were filtered off via a pre-cooled glass sinter funnel, washed portionwise with 78 mL TBME and dried for 18 h at 45° C./20 mbar, to give 21.0 g of the title product as white crystals (77% yield; assay: 99.5%).
MS: m/e 478 (M+H)+, 437, 422.
e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one dihydrochlorideA 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
These compounds are useful intermediates for the preparation of DPP-IV inhibitors as disclosed in PCT International Patent Appl. WO 2005/000848. More preferably, the invention relates to a process for the preparation of (2S,3S,11bS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester.
XXXXXXX
According to still another embodiment (Scheme 2, below) the (S)-4-fluoromethyl-dihydro-furan-2-one (VII) is directly coupled with the amino-pyrido[2,1-a]isoquinoline derivative (VI) to form the hydroxymethyl derivative of the pyrido[2,1-a]isoquinoline (VIII), which is then subsequently cyclized to the fluoromethyl-pyrrolidin-2-one derivative (IX). The latter can be deprotected to yield the desired pyrido[2,1-a]isoquinoline derivative (I).

In a further preferable embodiment, the process for the preparation of (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one or of a pharmaceutically acceptable salt thereof comprises the subsequent steps:
- e) coupling of the (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (amine of formula VI, wherein R2 and R3 are methoxy, R4 is hydrogen and Prot is Boc) with the (S)-4-fluoromethyl-dihydro-furan-2-one of formula

- f) cyclization of the obtained (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester in the presence of a base, and
- g) deprotecting the obtained (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester.

………………………………………………………….
PATENT
http://www.google.com.ar/patents/US7122555?cl=pt-PT
Example 23
RACEMIC
1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

a) 4-Fluoromethyl-dihydro-furan-2-one
A solution of 4-hydroxymethyl-dihydro-furan-2-one (Tetrahedron 1994, 50, 6839; 1.02 g, 8.78 mmol) and bis(2-methoxyethyl)aminosulfur trifluoride (3.88 g, 17.6 mmol) in chloroform (4.4 mL) was stirred at 40° C. for 1 h, then poured onto ice and partitioned between sat. aq. sodium hydrogencarbonate solution and dichloromethane. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, heptane-ethyl acetate gradient) afforded the title compound (576 mg, 56%). Colourless liquid, MS (EI) 118.9 (M+H)+.
b) 3-Chloromethyl-4-fluoro-butyryl chloride
A mixture of 4-fluoromethyl-dihydro-furan-2-one (871 mg, 7.37 mmol), thionyl chloride (4.39 g, 36.9 mmol), and zinc chloride (60 mg, 0.44 mmol) was stirred 72 h at 80° C., then excess thionyl chloride was removed by distillation. Kugelrohr distillation of the residue (85° C., 0.2 mbar) afforded the title compound (450 mg, 35%). Colourless liquid, 1H-NMR (300 MHz, CDCl3): 4.65–4.55 (m, 1H), 4.50–4.40 (m, 1H), 3.70–3.60 (m, 2H), 3.25–3.05 (m, 2H), 2.80–2.60 (m, 1H).
c) (RS,RS,RS)-[3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (RS,RS,RS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 5b) and 3-chloromethyl-4-fluoro-butyryl chloride. White solid, MS (ISP) 514.5 (M+H)+.
d) (RS,RS,RS)-[3-(4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5d from (RS,RS,RS)-[3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Off-white foam, MS (ISP) 478.5 (M+H)+.
e) 1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
The title compound was produced in accordance with the general method of Example 1e from (RS,RS,RS)-[3-(4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Light yellow oil, MS (ISP) 378.5 (M+H)+.
Examples 28 and 29
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
 UNDESIRED
UNDESIREDand
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

The title compounds were produced from 1-((RS,RS,RS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one (Example 23) by chromatographic separation (SiO2, CH2Cl2/MeOH/NH4OH 80:1:0.2, then 95:5:0.25).
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Yellow oil, Rf=0.45 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Light yellow solid, Rf=0.40 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
Example 30
(S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one Dihydrochloride
 DESIRED
DESIREDa) [(S,S,S)-3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (S,S,S)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 16b) and 3-chloromethyl-4-fluoro-butyryl chloride (Example 23b). Off-white solid.
b) [(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
Sodium hydride (55–65% dispersion in oil, 1.14 g, 28.5 mmol) was added to a suspension of [(S,S,S)-3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (6.72 g, 13.1 mmol) in N,N-dimethylformamide (95 mL) at r.t., then after 1 h the reaction mixture was poured onto ice and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, cyclohexane/2-propanol 4:1) afforded [(S,S,S)-3-((S)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 38%) and the epimer, [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.73 g, 44%).
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.6 (SiO2, cyclohexane/2-propanol 1:1).
[(S,S,S)-3-((R)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.4 (SiO2, cyclohexane/2-propanol 1:1).
-
- c) (S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 5.02 mmol) was converted to (S)-1-((S,S,S)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one in accordance with the general method of Example 1e. The product was dissolved in 2-propanol (10 mL) and treated with hydrogen chloride (5–6 M in 2-propanol, 37 mL). The suspension formed was stirred for 64 h at r.t., then the precipitate was collected by filtration and dried, to afford the title compound (2.04 g, 91%). White solid, m.p. >300° C.
Example 31(R)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
 UNDESIRED
UNDESIREDThe title compound was produced in accordance with the general method of Example 30c from [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (Example 30b). White solid, m.p. >300° C.

DR ANTHONY MELVIN CRASTO
MY BLOGS ON MED CHEM
New Drug Approvals ALL ABOUT DRUGS WORLD DRUG TRACKER MEDICINAL CHEMISTRY INTERNATIONALDRUG SYN INTERNATIONALSCALEUP OF DRUGS ALL FOR DRUGS ON WEBEUREKAMOMENTS
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS, BRAVESITES, GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO, YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN, FRIENDFEED, NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING
FDA ALLOWS MARKETING OF FIRST ZNT8AB AUTOANTIBODY TEST TO HELP DIAGNOSE TYPE 1 DIABETES
FDA allows marketing of first ZnT8Ab autoantibody test to help diagnose type 1 diabetes
Today, August 20, 2014, the U.S. Food and Drug Administration allowed marketing of the first zinc transporter 8 autoantibody (ZnT8Ab) test that can help determine if a person has type 1 diabetes and not another type of diabetes. When used with other tests and patient clinical information, the test may help some people with type 1 diabetes receive timely diagnosis and treatment for their disease.
Type 1 diabetes is the most common type of diabetes diagnosed in children and adolescents, but in some instances it may also develop in adults. People with the disease produce little or no insulin because their immune system attacks and destroys the cells in the pancreas that produce insulin, a hormone that converts sugars (glucose) in food to the energy the bodyneeds. People with type 1 diabetes mustinject insulinto regulate their blood glucose because proper regulation is critical to lower their risk of long-term complications such as blindness, kidney failure and cardiovascular disease.
The immune system of many people with type 1 diabetes produces ZnT8Ab, but patients with other types of diabetes (type 2 and gestational) do not. The KRONUS Zinc Transporter 8 Autoantibody (ZnT8Ab) ELISA Assay detects the presence of the ZnT8 autoantibody in a patient’s blood.
“Early treatment of type 1 diabetes is important in helping to prevent further deterioration of insulin producing cells,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the Center for Devices and Radiological Health at the FDA. “This test can help patients get a timely diagnosis and help start the right treatment sooner.”
The KRONUS ZnT8Ab ELISA Assay was reviewed through the de novo premarket review pathway, a regulatory pathway for some low- to moderate-risk medical devices that are not substantially equivalent to an already legally marketed device.

The agency reviewed data from a clinical study of 569 blood samples — 323 from patients with diagnosed type 1 diabetes and 246 samples from patients diagnosed with other kinds of diabetes, other autoimmune diseases, and other clinical conditions. The test was able to detect the ZnT8 autoantibody in 65 percent of the samples from patients with diagnosed type 1 diabetes and incorrectly gave a positive result in less than two percent of the samples from patients diagnosed with other disease.
A negative result from the test does not rule out a diagnosis of type 1 diabetes. The test should not be used to monitor the stage of disease or the response to treatment.
KRONUS Zinc Transporter 8 Autoantibody (ZnT8Ab) ELISA Assay is manufactured by KRONUS Market Development Associates, Inc. in Star, Idaho.

– See more at: http://worlddrugtracker.blogspot.in/#sthash.RfMvYLLf.dpuf
Lupin launches insulin glargine in India

Lupin launches insulin glargine in India:
Indian pharma company, Lupin Limited announced a strategic distribution agreement with LG Life Sciences of South Korea to launch Insulin Glargine, a novel insulin analogue under the brand name Basugine™.
According to the agreement, Lupin would be responsible for marketing and sales of Basugine™ in India.


Luseogliflozin, TS 071…………. strongly inhibited SGLT2 activity,

LUSEOGLIFLOZIN, CAS 898537-18-3
An antidiabetic agent that inhibits sodium-dependent glucose cotransporter 2 (SGLT2).
(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol
(1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
Taisho Pharmaceutical Co., Ltd
Taisho (Originator), PHASE 3
Click to access 2013041801-e.pdf
TS-071
| Taisho Pharmaceutical Holdings Co. Ltd. | |
| Description | Oral sodium-glucose cotransporter 2 (SGLT2) inhibitor |

TS-071, an SGLT-2 inhibitor, is in phase III clinical development at Taisho for the oral treatment of type 1 and type 2 diabetes
In 2012, the product was licensed to Novartis and Taisho Toyama Pharmaceutical by Taisho in Japan for comarketing for the treatment of type 2 diabetes.
Diabetes is a metabolic disorder which is rapidly emerging as a global health care problem that threatens to reach pandemic levels. The number of people with diabetes worldwide is expected to rise from 285 million in 2010 to 438 million by 2030. Diabetes results from deficiency in insulin because of impaired pancreatic β-cell function or from resistance to insulin in body, thus leading to abnormally high levels of blood glucose.
Diabetes which results from complete deficiency in insulin secretion is Type 1 diabetes and the diabetes due to resistance to insulin activity together with an inadequate insulin secretion is Type 2 diabetes. Type 2 diabetes (Non insulin dependent diabetes) accounts for 90-95 % of all diabetes. An early defect in Type 2 diabetes mellitus is insulin resistance which is a state of reduced responsiveness to circulating concentrations of insulin and is often present years before clinical diagnosis of diabetes. A key component of the pathophysiology of Type 2 diabetes mellitus involves an impaired pancreatic β-cell function which eventually contributes to decreased insulin secretion in response to elevated plasma glucose. The β-cell compensates for insulin resistance by increasing the insulin secretion, eventually resulting in reduced β-cell mass. Consequently, blood glucose levels stay at abnormally high levels (hyperglycemia).
Hyperglycemia is central to both the vascular consequences of diabetes and the progressive nature of the disease itself. Chronic hyperglycemia leads to decrease in insulin secretion and further to decrease in insulin sensitivity. As a result, the blood glucose concentration is increased, leading to diabetes, which is self-exacerbated. Chronic hyperglycemia has been shown to result in higher protein glycation, cell apoptosis and increased oxidative stress; leading to complications such as cardiovascular disease, stroke, nephropathy, retinopathy (leading to visual impairment or blindness), neuropathy, hypertension, dyslipidemia, premature atherosclerosis, diabetic foot ulcer and obesity. So, when a person suffers from diabetes, it becomes important to control the blood glucose level. Normalization of plasma glucose in Type 2 diabetes patients improves insulin action and may offset the development of beta cell failure and diabetic complications in the advanced stages of the disease.
Diabetes is basically treated by diet and exercise therapies. However, when sufficient relief is not obtained by these therapies, medicament is prescribed alongwith. Various antidiabetic agents being currently used include biguanides (decrease glucose production in the liver and increase sensitivity to insulin), sulfonylureas and meglitinides (stimulate insulin production), a-glucosidase inhibitors (slow down starch absorption and glucose production) and thiazolidinediones (increase insulin sensitivity). These therapies have various side effects: biguanides cause lactic acidosis, sulfonylurea compounds cause significant hypoglycemia, a-glucosidase inhibitors cause abdominal bloating and diarrhea, and thiazolidinediones cause edema and weight gain. Recently introduced line of therapy includes inhibitors of dipeptidyl peptidase-IV (DPP-IV) enzyme, which may be useful in the treatment of diabetes, particularly in Type 2 diabetes. DPP-IV inhibitors lead to decrease in inactivation of incretins glucagon like peptide- 1 (GLP-1) and gastric inhibitory peptide (GIP), thus leading to increased production of insulin by the pancreas in a glucose dependent manner. All of these therapies discussed, have an insulin dependent mechanism.
Another mechanism which offers insulin independent means of reducing glycemic levels, is the inhibition of sodium glucose co-transporters (SGLTs). In healthy individuals, almost 99% of the plasma glucose filtered in the kidneys is reabsorbed, thus leading to only less than 1% of the total filtered glucose being excreted in urine. Two types of SGLTs, SGLT-1 and SGLT-2, enable the kidneys to recover filtered glucose. SGLT-1 is a low capacity, high-affinity transporter expressed in the gut (small intestine epithelium), heart, and kidney (S3 segment of the renal proximal tubule), whereas SGLT-2 (a 672 amino acid protein containing 14 membrane-spanning segments), is a low affinity, high capacity glucose ” transporter, located mainly in the S 1 segment of the proximal tubule of the kidney. SGLT-2 facilitates approximately 90% of glucose reabsorption and the rate of glucose filtration increases proportionally as the glycemic level increases. The inhibition of SGLT-2 should be highly selective, because non-selective inhibition leads to complications such as severe, sometimes fatal diarrhea, dehydration, peripheral insulin resistance, hypoglycemia in CNS and an impaired glucose uptake in the intestine.
Humans lacking a functional SGLT-2 gene appear to live normal lives, other than exhibiting copious glucose excretion with no adverse effects on carbohydrate metabolism. However, humans with SGLT-1 gene mutations are unable to transport glucose or galactose normally across the intestinal wall, resulting in condition known as glucose-galactose malabsorption syndrome.
Hence, competitive inhibition of SGLT-2, leading to renal excretion of glucose represents an attractive approach to normalize the high blood glucose associated with diabetes. Lower blood glucose levels would, in turn, lead to reduced rates of protein glycation, improved insulin sensitivity in liver and peripheral tissues, and improved cell function. As a consequence of progressive reduction in hepatic insulin resistance, the elevated hepatic glucose output which is characteristic of Type 2 diabetes would be expected to gradually diminish to normal values. In addition, excretion of glucose may reduce overall caloric load and lead to weight loss. Risk of hypoglycemia associated with SGLT-2 inhibition mechanism is low, because there is no interference with the normal counter regulatory mechanisms for glucose.
The first known non-selective SGLT-2 inhibitor was the natural product phlorizin
(glucose, 1 -[2-P-D-glucopyranosyloxy)-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)- 1 – propanone). Subsequently, several other synthetic analogues were derived based on the structure of phlorizin. Optimisation of the scaffolds to achieve selective SGLT-2 inhibitors led to the discovery of several considerably different scaffolds.
C-glycoside derivatives have been disclosed, for example, in PCT publications
W.O20040131 18, WO2005085265, WO2006008038, WO2006034489, WO2006037537, WO2006010557, WO2006089872, WO2006002912, WO2006054629, WO2006064033, WO2007136116, WO2007000445, WO2007093610, WO2008069327, WO2008020011, WO2008013321, WO2008013277, WO2008042688, WO2008122014, WO2008116195, WO2008042688, WO2009026537, WO2010147430, WO2010095768, WO2010023594, WO2010022313, WO2011051864, WO201 1048148 and WO2012019496 US patents US65151 17B2, US6936590B2 and US7202350B2 and Japanese patent application JP2004359630. The compounds shown below are the SGLT-2 inhibitors which have reached advanced stages of human clinical trials: Bristol-Myers Squibb’s “Dapagliflozin” with Formula A, Mitsubishi Tanabe and Johnson & Johnson’s “Canagliflozin” with Formula B, Lexicon’s “Lx-421 1″ with Formula C, Boehringer Ingelheim and Eli Lilly’s “Empagliflozin” with Formula D, Roche and Chugai’s “Tofogliflozin” with Formula E, Taisho’s “Luseogliflozin” with Formula F, Pfizer’ s “Ertugliflozin” with Formula G and Astellas and Kotobuki’s “Ipragliflozin” with Formula H.

Formula G Formula H
In spite of all these molecules in advanced stages of human clinical trials, there is still no drug available in the market as SGLT-2 inhibitor. Out of the potential candidates entering the clinical stages, many have been discontinued, emphasizing the unmet need. Thus there is an ongoing requirement to screen more scaffolds useful as SGLT-2 inhibitors that can have advantageous potency, stability, selectivity, better half-life, and/ or better pharmacodynamic properties. In this regard, a novel class of SGLT-2 inhibitors is provided herein
………………………
SYNTHESIS
- Example 5

Synthesis of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose
-
Five drops of 1,2-dibromoethane were added to a mixture of magnesium (41 mg, 1.67 mmol), 1-bromo-3-(4-ethoxybenzyl)-6-methoxy-4-methylbenzene (0.51 g, 1.51 mmol) and tetrahydrofuran (2 mL). After heated to reflux for one hour, this mixture was allowed to stand still to room temperature to prepare a Grignard reagent. A tetrahydrofuran solution (1.40 mL) of 1.0 M i-propyl magnesium chloride and the prepared Grignard reagent were added dropwise sequentially to a tetrahydrofuran (5 mL) solution of 2,3,4,6-tetra-O-benzyl-5-thio-D-glucono-1,5-lactone (0.76 g, 1.38 mmol) while cooled on ice and the mixture was stirred for 30 minutes. After the reaction mixture was added with a saturated ammonium chloride aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate =4:1) to obtain (0.76 g, 68%) a yellow oily title compound.
1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.37 (t, J=6.92 Hz, 3 H) 2.21 (s, 3 H) 3.51 – 4.20 (m, 12 H) 3.85 – 3.89 (m, 3 H) 4.51 (s, 2 H) 4.65 (d, J=10.72 Hz, 1 H) 4.71 (d, J=5.75 Hz, 1 H) 4.78 – 4.99 (m, 3 H) 6.59 – 7.43 (m, 26 H)
Example 6
-
[0315]


Synthesis of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol
-
An acetonitrile (18 mL) solution of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose (840 mg, 1.04 mmol) was added sequentially with Et3SiH (0.415 mL, 2.60 mmol) and BF3·Et2O (0.198 mL, 1.56 mmol) at -18°C and stirred for an hour. After the reaction mixture was added with a saturated sodium bicarbonate aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and then dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate=4:1) to obtain the title compound (640 mg, 77%).
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.35 (t, J=6.88 Hz, 3 H) 2.21 (s, 3 H) 3.02 – 3.21 (m, 1 H) 3.55 (t,J=9.40 Hz, 1 H) 3.71 (s, 1 H) 3.74 – 3.97 (m, 10 H) 4.01 (s, 1 H) 4.45 – 4.56 (m, 3 H) 4.60 (d, J=10.55 Hz, 2 H) 4.86 (s, 2 H) 4.90 (d, J=10.55 Hz, 1H) 6.58 – 6.76 (m, 5 H) 6.90 (d, J=7.34 Hz, 1 H) 7.09 – 7.19 (m, 5 H) 7.23 – 7.35 (m, 15 H).
ESI m/z = 812 (M+NH4).
Example 7

Synthesis of (1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
-
A mixture of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol (630 mg, 0.792 mmol), 20% palladium hydroxide on activated carbon (650 mg) and ethyl acetate (10 mL) – ethanol (10 mL) was stirred under hydrogen atmosphere at room temperature for 66 hours. The insolubles in the reaction mixture were filtered off with celite and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography (chloroform:methanol =10:1) to obtain a colorless powdery title compound (280 mg, 81%) as 0.5 hydrate. 1H NMR (600 MHz, METHANOL- d4) δ ppm 1.35 (t, J=6.9 Hz, 3 H) 2.17 (s, 3 H) 2.92 – 3.01 (m, 1 H) 3.24 (t, J=8.71 Hz, 1 H) 3.54 – 3.60 (m, 1 H) 3.72 (dd, J=11.5, 6.4 Hz, 1 H) 3.81 (s, 3 H) 3.83 (s, 2 H) 3.94 (dd, J=11.5, 3.7 Hz, 1 H) 3.97 (q, J=6.9 Hz, 2 H) 4.33 (s, 1 H) 6.77 (d, J=8.3 Hz, 2 H) 6.76 (s, 1 H) 6.99 (d, J=8.3 Hz, 2 H) 7.10 (s, 1 H). ESI m/z = 452 (M+NH4+), 493 (M+CH3CO2-). mp 155.0-157.0°C. Anal. Calcd for C23H30O6S·0.5H2O: C, 62.28; H, 7.06. Found: C, 62.39; H, 7.10.
………………………………..
PAPER
(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (TS-071) is a Potent, Selective Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for Type 2 Diabetes Treatment
(Journal of Medicinal Chemistry) Saturday March 20th 2010
Author(s): ,
DOI:10.1021/jm901893x
GO TO: [Article]
http://pubs.acs.org/doi/abs/10.1021/jm901893x

(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (3p)
Compound 3p (0.281 g, 81%) was prepared as a colorless powder from 21p (0.630 g, 0.792 mmol) according to the method described for the synthesis of 3a. (Method A)
mp 155.0−157.0 °C.
1H NMR (600 MHz, MeOH-d4) δ 1.35 (t, J = 6.9 Hz, 3 H), 2.17 (s, 3 H), 2.92−3.01 (m, 1 H), 3.24 (t, J = 8.7 Hz, 1 H), 3.54−3.60 (m, 1 H), 3.72 (dd, J = 6.4, 11.5, Hz, 1 H), 3.81 (s, 3 H), 3.83 (s, 2 H), 3.94 (dd, J = 3.7, 11.5 Hz, 1 H), 3.97 (q, J = 6.9 Hz, 2 H), 4.33 (brs, 1 H), 6.77 (d, J = 8.3 Hz, 2 H), 6.76 (s, 1 H), 6.99 (d, J = 8.3 Hz, 2 H), 7.10 (s, 1 H).
MS (ESI) m/z 452 (M+NH4).
Anal. Calcd for (C23H30O6S·0.5H2O) C, 62.28; H, 7.06. Found C, 62.39; H, 7.10.
 3p is compd
3p is compd
| cmpds | R1 | R2 | R3 | SGLT2 (nM) mean (95% CI) | SGLT1 (nM) mean (95% CI) | T1/T2 selectivity |
|---|---|---|---|---|---|---|
| 1 | 27.8 (21.8−35.3) | 246 (162−374) | 8.8 | |||
| 3a | H | H | OEt | 73.6 (51.4−105) | 26100 (20300−33700) | 355 |
| 3b | H | OH | OEt | 283 (268−298) | 14600 (11500−18500) | 51.6 |
| 3c | H | OMe | OEt | 13.4 (11.3−15.8) | 565 (510−627) | 42.2 |
| 3d | H | F | OEt | 9.40 (5.87−15.0) | 7960 (7180−8820) | 847 |
| 3e | H | Me | OEt | 2.29 (1.76−2.99) | 671 (230−1960) | 293 |
| 3f | H | Cl | OEt | 1.77 (0.95−3.30) | 1210 (798−1840) | 684 |
| 3g | OH | H | OEt | 17.4 (15.9−19.0) | 4040 (1200−13600) | 232 |
| 3h | OMe | H | OEt | 37.9 (26.4−54.4) | 100000 (66500−151000) | 2640 |
| 3i | OMe | OMe | OEt | 10.8 (6.84−17.1) | 4270 (1560−11600) | 395 |
| 3j | H | Cl | OMe | 1.68 (1.08−2.60) | 260 (72.5−931) | 155 |
| 3k | H | Cl | Me | 1.37 (0.97−1.95) | 209 (80.2−545) | 153 |
| 3l | H | Cl | Et | 1.78 (0.88−3.63) | 602 (473−767) | 338 |
| 3m | H | Cl | iPr | 4.01 (1.75−9.17) | 8160 (4860−13700) | 2040 |
| 3n | H | Cl | tBu | 18.8 (11.0−32.1) | 35600 (31900−39800) | 1890 |
| 3o | H | Cl | SMe | 1.16 (0.73−1.85) | 391 (239−641) | 337 |
| 3p | OMe | Me | OEt | 2.26 (1.48−3.43) | 3990 (2690−5920) | 1770 |
| 3q | OMe | Me | Et | 1.71 (1.19−2.46) | 2830 (1540−5200) | 1650 |
| 3r | OMe | Me | iPr | 2.68 (2.15−3.34) | 17300 (14100−21100) | 6400 |
| 3s | OMe | Cl | Et | 1.51 (0.75−3.04) | 3340 (2710−4110) | 2210 |
PATENT
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2004014930A1 * | Aug 8, 2003 | Feb 19, 2004 | Asanuma Hajime | PROCESS FOR SELECTIVE PRODUCTION OF ARYL 5-THIO-β-D- ALDOHEXOPYRANOSIDES |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | AL-MASOUDI, NAJIM A. ET AL: “Synthesis of some novel 1-(5-thio-.beta.-D-glucopyranosyl)-6-azaur acil derivatives. Thio sugar nucleosides” NUCLEOSIDES & NUCLEOTIDES , 12(7), 687-99 CODEN: NUNUD5; ISSN: 0732-8311, 1993, XP008091463 |
| 2 | * | See also references of WO2006073197A1 |
| EP2419097A1 * | Apr 16, 2010 | Feb 22, 2012 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| EP2455374A1 * | Oct 15, 2009 | May 23, 2012 | Janssen Pharmaceutica N.V. | Process for the Preparation of Compounds useful as inhibitors of SGLT |
| EP2601949A2 * | Apr 16, 2010 | Jun 12, 2013 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| EP2668953A1 * | May 15, 2009 | Dec 4, 2013 | Bristol-Myers Squibb Company | Pharmaceutical compositions comprising an SGLT2 inhibitor with a supply of carbohydrate and/or an inhibitor of uric acid synthesis |
| WO2009143020A1 | May 15, 2009 | Nov 26, 2009 | Bristol-Myers Squibb Company | Method for treating hyperuricemia employing an sglt2 inhibitor and composition containing same |
| WO2010043682A2 * | Oct 15, 2009 | Apr 22, 2010 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of sglt |
| WO2010119990A1 | Apr 16, 2010 | Oct 21, 2010 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| WO2013152654A1 * | Mar 14, 2013 | Oct 17, 2013 | Theracos, Inc. | Process for preparation of benzylbenzene sodium-dependent glucose cotransporter 2 (sglt2) inhibitors |
-
Luseogliflozin: Phase III data 10/21/2013
Week in Review, Clinical ResultsTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Molecular target: Sodium-glucose cotransporter 2 (SGLT2) Description: Oral sodium-glucose… -
Luseogliflozin: Phase III data 10/21/2013
Week in Review, Clinical ResultsTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Molecular target: Sodium-glucose cotransporter 2 (SGLT2) Description: Oral sodium-glucose… -
Luseogliflozin regulatory update 05/13/2013
Week in Review, RegulatoryTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Last month, Taisho’s Taisho Pharmaceutical Co. Ltd. subsidiary submitted a regulatory … -
Strategy: Doubling down in diabetes 05/06/2013
Merck shoring up slowing diabetes franchise with Pfizer, Abide dealsBioCentury on BioBusiness, StrategyAs sales flatten for Merck’s sitagliptin franchise and a new class of oral diabetes drugs comes to market, the pharma has tapped Pfizer and Abide to shore up its position.
see
SEE
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-132352


TA 1887 a highly potent and selective hSGLT2 inhibitor
 6a-4 is TA 1887
6a-4 is TA 1887![]()

TA 1887![]()
CAS 1003005-29-5![]()
Deleted CAS Registry Numbers: 1274890-87-7
C24 H26 F N O5
1H-Indole, 3-[(4-cyclopropylphenyl)methyl]-4-fluoro-1-β-D-glucopyranosyl-![]()
3-(4-cyclopropylbenzyl)-4-fluoroindole-N-glucoside![]()
(2R,3R,4S,5S,6R)-2-(3-(4-cvclopropylbenzyl)-4-fluoro-1 H-indol- 1 -yl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol,![]()
(TA-1887), a highly potent and selective hSGLT2 inhibitor, with pronounced antihyperglycemic effects in high-fat diet-fed KK (HF-KK) mice. Our results suggest the potential of indole-N-glucosides as novel antihyperglycemic agents through inhibition of renal SGLT2
Mitsubishi Tanabe Pharma Corp,![]()
Glucagon-like peptide-1 (GLP-I) is an incretin hormone that is released from L-cells in lower small intestine after food intake. GLP-I has been shown to stimulate glucose-dependent insulin secretion from pancreatic β-cells and increase pancreatic β-cell mass. GLP-I has also been shown to reduce the rate of gastric emptying and promote satiety. However, GLP-I is rapidly cleaved by dipeptidyl peptidase 4 (DPP4) leading to inactivation of its biological activity. Therefore, DPP4 inhibitors are considered to be useful as anti-diabetics or anti-obesity agents.
Sodium-glucose co-transporters (SGLTs) , primarily found in the intestine and the kidney, are a family of proteins involved in glucose absorption. Plasma glucose is filtered in the glomerulus and is reabsorbed by SGLTs in the proximal tubules. Therefore, inhibition of SGLTs cause excretion of blood glucose into urine and leads to reduction of plasma glucose level. In fact, it is confirmed that by continuous subcutaneous administration of an SGLT inhibitor, phlorizin, to diabetic animal models, the blood glucose level thereof can be normalized, and that by keeping the blood glucose level normal for a long time, the insulin secretion and insulin resistance can be improved [cf., Journal of Clinical Investigation, vol. 79, p. 1510 (1987); ibid., vol. 80, p. 1037 (1987); ibid., vol. 87, p. 561 (1991) ] .
In addition, by treating diabetic animal models with an SGLT inhibitor for a long time, insulin secretion response and insulin sensitivity of the animal models are improved without incurring any adverse affects on the kidney or imbalance in blood levels of electrolytes, and as a result, the onset and progress of diabetic nephropathy and diabetic neuropathy are prevented [cf., Journal of Medicinal Chemistry, vol. 42, p. 5311 (1999); British Journal of Pharmacology, vol. 132, p. 578 (2001)].
In view of the above, SGLT inhibitors are expected to improve insulin secretion and insulin resistance by decreasing the blood glucose level in diabetic patients and to prevent the onset and progress of diabetes mellitus and diabetic complications
DPP4 inhibitors are well known to those skilled in the art, and examples of DPP4 inhibitors can be found in the following publications: (1) TANABE SEIYAKU Co., Ltd.: WO 02/30891 or the corresponding U.S. patent (No. 6,849,622); and WO 02/30890 or the corresponding U.S. patent (No. 7,138,397); .
(2) Ferring BV: WO 95/15309, WO 01/40180, WO 01/81304, WO
01/81337, WO 03/000250, and WO 03/035057; (3) Probiodrug: WO 97/40832, EP1082314, WO 99/61431, WO
03/015775; (4) Novartis AG: WO 98/19998, WO 00/34241, WO 01/96295, US 6,107,317, US 6,110,949, and US 6,172,081;
(5) GlaxoSmithKline: WO 03/002531, WO 03/002530, and WO 03/002553; (6) Bristol Myers Squibb: WO 01/68603, WO 02/83128, and WO 2005/012249;
(7) Merck & Co.: WO 02/76450, and WO 03/004498;
(8) Srryx Inc.: WO 2005/026148, WO 2005/030751, WO 2005/095381, WO 2004/087053, and WO 2004/103993; (9) Mitsubishi Pharma Corp.: WO 02/14271, US 7,060,722, US
7,074,794, WO 2003/24942, Japan Patent Publication No.
2002-265439, Japan Patent Publication No. 2005-170792, and
WO 2006/088129;
(10) Taisho Pharma Co., Ltd.: WO 2004/020407; (12) Yamanouchi Pharmaceutical Co., Ltd.: WO 2004/009544,-
(13) Kyowa Hakko Kogyo : WO 02/051836;
(14) Kyorin Seiyaku: WO 2005/075421, WO 2005/077900, and WO 2005/082847;
(15) Alantos Pharmaceuticals: WO 2006/116157; (16) Glenmark Pharmaceuticals: WO 2006/090244, and WO 2005/075426;
(17) Sanwa Kagaku Kenkyusho : WO 2004/067509; and
(18) LG lifescience: WO 2005/037828, and WO 2006/104356.
In a preferable embodiment of the present invention, DPP4 inhibitors are the aliphatic nitrogen-containing 5- membered ring compounds disclosed in US 6,849,622, which are represented by Formula (29) :

…………………………………………..
WO 2012162115![]()
http://www.google.com/patents/EP2712359A2?cl=en![]()
The present invention is further directed to a process for the preparation of a compound of formula (l-S)
(l-S)
(also known as 3-(4-cyclopropylbenzyl)-4-fluoro-1 -p-D-glucopyranosyl- 1 /-/-indole); or a pharmaceutically acceptable salt or prodrug thereof;
comprising

reacting a compound of formula (V-S), wherein PG1 is an oxygen protecting group with an acylating reagent; wherein the acylating reagent is present in an amount in the range of from about 1 .5 to about 3.0 molar equivalents; in the presence of a carbonyl source; in a first organic solvent; at a temperature in the range of from about room temperature to about 40°C; to yield the corresponding compound of formula (Vl-S);

reacting the compound of formula (Vl-S) with a compound of formula (Vll-S), wherein A1 is MgBr or MgCI; in an anhydrous organic solvent; to yield the corresponding compound of formula (Vlll-S);

reacting the compound of formula (Vlll-S) with a reducing agent; in the presence of a Lewis acid; in a second organic solvent; to yield the
corresponding compound of formula (IX-S);

Scheme 2.

Example 1 : f2R.3R.4S.5R.6R)-2-facetoxymethyl)-6-f4-fluoro-3-formyl-1 H- indol-1 -yl)tetrahvdro-2H-pyran-3,4,5-triyl triacetate

A 5-L 4-neck round bottom flask equipped with a thermocouple controller, mechanical stirrer, addition funnel, condenser, heating mantle, and a nitrogen inlet adapter was (2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(4-fluoro-1 H- indol-1 -yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (225.0 g, 0.459 mol), DCE (1 .5 L) and DMF (50.2 ml_, 0.643 mol). The resulting mixture was warmed to 25°C, then phosphoryl chloride (107.8 ml_, 1 .15 mol) was added slowly via an addition funnel over 75 min. The resulting mixture was stirred for 30 min after the addition was completed, then slowly warmed to 40°C over 30 min, and then agitated at 40°C for an additional 12 h. The resulting solution was slowly poured into a rapidly stirred warm (40°C) 3M aqueous NaOAc (3.0 L) solution over 45 min. After the addition was completed, CH2CI2 (4.0 L) was added and the phases were separated. The aqueous phase was back extracted with CH2CI2 (1 .0 L) and the organic phases were combined, washed with 0.05 M HCI (2.0 L) and deionized water (2.0 L), then dried over MgS04. After filtration, the solvents were concentrated to dryness in vacuo to yield a solid, which was flushed with ethanol (1 .0 L) and re-evaporated. The resulting solid was transferred into a vacuum oven and dried at 40°C for 20 h to yield the title compound as a slightly yellow-brown solid.
1 H NMR (DMSO-d6, 300 MHz) δ 10.1 (s, 1 H), 8.53 (s, 1 H), 7.66 (d, J = 7.3 Hz, 1 H), 7.38 (m, 1 H), 7.10(dd, J = 6.7, 6.9 Hz, 1 H), 6.38 (d, J = 7.5 Hz, 1 H), 5.68 (dd, J = 6.5, 6.6 Hz, 1 H), 5.56 (t, J = 7.1 Hz, 1 H), 5.32 (t, J = 7.2 Hz, 1 H) 4.41 – 4.28 (m, 1 H), 4.24 – 4.06 (m, 2 H), 2.05 (s, 3H), 2.0 (s, 3H), 1 .98 (s, 3H), 1 .64 (s, 3H) 1JC NMR (DMSO-c(6, 75.47 MHz) £183.8, 169.9, 169.5, 169.3, 168.4, 155.8, 139.2, 135.7, 124.8, 1 17.7, 1 13.1 , 108.3, 107,9, 81 .9, 73.5, 72.1 , 70.3, 67.6, 61 .9, 20.4, 20.3, 20.1 , 19.6
LC-MS mlz MH+ = 494 (MH+), 516 [M+Na]+, 1009 [2M+Na]+
[a]D 25 = -0.099 (c = 0.316, CHCI3).
Example 2: f2R.3R.4S.5R.6R)-2-facetoxymethyl)-6-f3-ff4-cvclopropyl- phenyl)(hvdroxy)methyl)-4-fluoro-1 H-indol-1 -yl)tetrahydro-2H-pyran-3,4,5- triyl triacetate

A 12-L 4-neck round bottom flask equipped with a mechanical stirrer, a thermocouple, a septum and nitrogen inlet adapter was charged with the compound prepared as in Example 1 (230 g, 0.457 mol) and anhydrous THF (4.2 L), and the resulting solution was cooled to 0°C with stirring under N2. A solution of freshly prepared (4-cyclopropylphenyl)magnesium bromide in THF (530 mL) was added dropwise via a double-tipped needle under gentle positive nitrogen pressure over 20 min, while the internal temperature was maintained between 0-8°C by adjusting the rate of addition. The resulting mixture was stirred at 0°C for 30 min. The reaction was quenched with saturated aqueous NH4CI solution (5.4 L) and then extracted with EtOAc (4 L, 3 L). The combined organic phase was washed with brine (2.7 L) and dried over MgS04. After filtration, the filtrate was concentrated at 66°C under house vacuum (-120 mmHg) followed by hi-vacuum (-20 mmHg) to yield a residue which contained a large amount of EtOAc, which residue was chased with ΟΗ2ΟΙ2 (800 mL) to yield the title compound as a yellowish solid, which was used in next step without further purification.
1 H NMR (DMSO-cfe, 300 MHz) δ 7.53 (dd, J = 7.9, 1 .1 Hz, 1 H), 7.41 (dd, J = 8.0, 1 .0 Hz, 1 H), 7.10-6.92 (m, 3 H), 6.78 (m, 1 H), 6.15 (m, 1 H), 5.92 (dd, J = 5.0, 4.1 Hz, 1 H), 5.65 (dd, J = 5.1 , 4.2 Hz, 1 H), 5.50 (m, 1 H), 5.24 (dd, J = 7.9, 8.3 Hz, 1 H), 4.38 – 4.22 (m, 1 H), 4.20-4.0 (m, 2 H), 2.05 (s, 3 H), 2.01 (s, 3 H), 1 .98 (s, 3 H), 1 .84 (m, 1 H), 0.92 (m, 2 H), 0.61 (m, 2 H)
13C NMR (DMSO-c/6, 75.47 MHz): £ 170.1 , 170.0, 169.9, 169.3, 156.1 , 140.9 139.0, 137.9, 128.0 (2 C), 125.2 (2 C), 124.2, 122.6, 1 16.3, 1 14.6, 107.4, 105.2, 81 .5, 76.8, 73.0, 72.6, 70.1 , 68.2, 62.0, 20.6, 20.4, 20.2, 19.8, 14.8, 8.96 (2 C)
LC-MS mlz MH+ = 612 (MH+), 634 [M+Na]+.
Example 3: (2R.3R.4S.5R.6R)-2-(acetoxymethyl)-6-(3-(4- cvclopropylbenzyl)-4-fluoro-1H-indol-1 -yl)tetrahvdro-2H-pyran-3,4.5-triyl triacetate

OAc
A 3-L 4-neck round bottom flask equipped with a mechanical stirrer, a thermocouple, a septum and nitrogen inlet adapter, was charged with the product prepared as in Example 2 above (82%, 334.6 g, 0.449 mol), DCE (1 .14 L), CH3CN (2.28 L), and Et3SiH (108.6 mL, 0.671 mol) and the resulting mixture was stirred and cooled to 0°C under N2. Boron trifluoride etherate (68.8 mL; 0.539 mol) was added dropwise over 10 min and the resulting mixture was stirred at 0°C for 30 minutes. After completion, saturated aqueous NaHCC>3 solution (4.2 L) was added to the mixture, which was extracted with EtOAc (5 L, 4 L) and the combined organic phase was dried over MgS04. After filtration, the filtrate was concentrated under house vacuum at 60°C to yield the title compound as a slightly yellowish solid.
The slightly yellowish solid (315.0 g) was triturated with EtOH (2.1 L, 200 proof) in a 4-L heavy duty Erlenmeyer flask at 76°C (with sonication x 3), and then gradually cooled to 20°C and stirred under N2 for 1 h. The solid was then collected by filtration and washed with cold (0°C) EtOH (200 ml_), dried by air- suction for 30 min, and then placed in a vacuum oven under house vacuum with gentle of N2 stream at 60°C for 18 h, to yield the title compound as an off- white crystalline solid.
1 H NMR (DMSO-de, 300 MHz) δ 7.47 (d, J = 8.3 Hz, 1 H), 7.22 (s, 1 H),
7.20-7.10 (m, 1 H), 7.06 (d, J = 8.1 , 2 H), 6.95 (d, J = 8.1 Hz, 1 H), 6.78 (dd, J = 7.1 , 7.0 Hz, 1 H), 6.16 (d, J = 7.1 Hz, 1 H), 5.61 -5.44 (m, 2 H), 5.21 (t, J = 7.3, 7.1 Hz, 1 H), 4.34 – 4.21 (m, 1 H), 4.18-4.04 (m, 2 H), 4.0 (s, 2 H), 2.04 (s, 3 H), 1 .97 (s, 3 H), 1 .95 (s, 3 H), 1 .84 (m, 1 H), 1 .63 (s, 3 H), 0.89 (m, 2 H), 0.61 (m, 2 H)
13C NMR (DMSO-d6, 75.47 MHz): £ 169.9, 169.5, 169.3, 168.3, 156.2, 140.9, 139.0, 137.9, 128.0 (2 C), 125.2 (2 C), 124.2, 122.7, 1 16.1 , 1 14.1 , 107.2, 105.0, 81 .7, 73.0, 72.5, 69.8, 68.0, 62.0, 31 .2, 20.4, 20.3, 20.2, 19.7, 14.6, 8.93 (2 C)
LC-MS mlz MH+ = 596 (MH+), 618 [M+Na]+, 1213 [2M+Na]+
[a]D 25 = -0.008 (c = 0.306, CHCI3).
Example 4: (2R.3R.4S.5S.6R)-2-(3-(4-cvclopropylbenzyl)-4-fluoro-1 H-indol- 1 -yl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, ethanolate

OH
A 12-L 4-neck round bottom flask equipped with a mechanical stirrer, a thermocouple, a septum and nitrogen inlet adapter, was charged with the compound prepared as in Example 3 above (250 g, 0.413 mol), MeOH (1 .2 L) and THF (2.4 L), and the resulting mixture was stirred at 20°C under N2.
Sodium methoxide (2.5 ml_, 0.012 mol) solution was added dropwise and the resulting mixture was stirred at 20°C for 3 h. The solvent was concentrated at 60°C under house vacuum to yield a residue, which was dissolved in EtOAc (8.0 L), washed with brine (800 mL x 2) (Note 2), and dried over MgS04. The insoluble materials were removed by filtration, and the filtrate was concentrated at 60-66°C under hi-vacuum (20 mmHg) to yield the title compound as a slightly yellowish foamy solid.
The above obtained slightly yellowish foamy solid (195.1 g) was dissolved in EtOH (900 mL) at 76°C, and deionized H20 (1800 mL) was added slowly in a small stream that resulted in a slightly yellowish clear solution, which was then gradually cooled to 40°C with stirring while seeded (wherein the seeds were prepared, for example, as described in Example 5, below). The resulting slightly white-yellowish suspension was stirred at 20°C for 20 h, the solids were collected by filtration, washed with cold (0°C) EtOH/H20 (1 :4), and dried by air-suction for 6 h with gentle stream of N2 to yield the title compound as an off-white crystalline solid, as its corresponding EtOH/H20 solvate.
The structure of the EtOH/H20 solvate was confirmed by its 1H-NMR and LC-MS analyses. 1H-NMR indicated strong H20 and EtOH solvent residues, and the EtOH residue could not be removed by drying process. In addition, p-XRD of this crystalline solid showed a different pattern than that measured for a hemi-hydrate standard.
Example 5: (2R,3R,4S,5S,6R)-2-(3-(4-cvclopropylbenzyl)-4-fluoro-1 H-indol- 1 -yl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, ethanolate

A 500-mL 3-neck round bottom flask equipped with a mechanical stirrer was charged with the compound prepared as in Example 3 above (4.67 g, 0.00784 mol), MeOH (47 mL) and THF (93 mL), and the resulting mixture was stirred at room temperature under argon atmosphere. Sodium methoxide (catalytic amount) solution was added dropwise and the resulting mixture was stirred at room temperature for 1 h. The solvent was concentrated at 30°C under reduced pressure. The residue was purified by silica gel column chromatography (chloroform : methanol = 99 : 1 – 90 : 10) to yield a colorless foamy solid (3.17 g).
First Crystallization
A portion of the colorless foamy solid prepared as described above (0.056 g) was crystallized from EtOH/H20 (1 :9, 5mL), at room temperature, to yield the title compound, as its corresponding EtOH solvate, as colorless crystals (0.047 g).
Second Crystallization
A second portion of the colorless foamy solid prepared as described above (1 .21 g) was dissolved in EtOH (6 mL) at room temperature. H20 (6 mL) was added, followed by addition of seeds (the colorless crystals, prepared as described in the first crystallization step above). The resulting suspension was stirred at room temperature for 18 h, the solids were collected by filtration, washed with EtOH/H20 (1 :4), and dried under reduced pressure to yield the title compound t, as its corresponding EtOH solvate, as an colorless crystalline solid (0.856 g).
The structure for the isolated compound was confirmed by 1H NMR, with peaks corresponding to the compound of formula (l-S) plus ethanol. Example 6: f2R.3R.4S.5S.6R)-2-f3-f4-cvclopropylbenzyl)-4-fluoro-1H-indol- 1 -yl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol hemihydrate

OH
A 5-L 4-neck round bottom flask equipped with a mechanical stirrer, a thermocouple, a septum and nitrogen inlet adapter was charged with the ethanolate (solvate) compound prepared as in Example 4 above (198.5 g, 0.399 mol) and deionized H20 (3.2 L). After the off-white suspension was warmed to 76°C in a hot water bath, along with sonication (x 4), it was gradually cooled to 20°C. The white suspension was stirred for 20 h at 20°C and then at 10°C for 1 h. The solid was collected by filtration, washed with deionized H20 (100 mL x 2), dried by air-suction for 2 h, and then placed in an oven under house vacuum with gentle stream of N2 at 50°C for 20 h, then at 60°C for 3 h to yield the title compound as an off-white crystalline solid.1 H NMR showed no EtOH residue and the p-XRD confirmed that the isolated material was a crystalline solid. TGA and DSC indicated that the isolated material contained about 2.3% of water (H20). M.P. = 108-1 1 1 °C.
1 H NMR (DMSO-c(6, 300 MHz) δ 7.36 (d, J = 8.2 Hz, 1 H), 7.22 (s, 1 H), 7.14 (d, J = 8.1 , 2 H), 7.10-7.0 (m, 1 H), 6.96 (d, J = 8.1 Hz, 2 H), 6.73 (dd, J = 7.5, 7.7 Hz, 1 H), 5.38 (d, J = 7.7 Hz, 1 H), 5.21 (d, J = 6.9 Hz, 1 H), 5.18 (d, J = 6.8 Hz, 1 H), 5.10 (d, J = 6.9 Hz, 1 H), 4.54 (t, J = 6.9, 1 .8 Hz, 1 H), 4.04 (s, 2 H), 3.75-3.60 (m, 2 H), 3.52-3.30 (m, 3 H), 3.20-3.17 (m, 1 H), 1 .84 (m, 1 H), 0.89 (m, 2 H), 0.61 (m, 2 H)
13C NMR (DMSO-de, 75.47 MHz): £ 156.2, 140.8, 139.4, 138.2, 128.2 (2 C), 125.2 (2 C), 124.4, 121 .8, 1 15.9, 1 12.8, 107.4, 104.2, 84.8, 79.3, 77.4, 71 .7, 69.8, 60.8, 31 .3, 14.6, 8.92 (2 C) LC-MS mlz MH+ = 428 (MH+), 450 [M+Na]+, 877 [2M+Na]+
[a]D 25 = -0.026 (c = 0.302, CH3OH)
Elemental Analysis: C2 H26NF05 + 0.54 H20 (MW = 437.20):
Theory: %C, 65.93; %H, 6.24; %N, 3.20; %F, 4.35, %H20, %2.23. Found: %C, 65.66; %H, 6.16; %N, 3.05; %F, 4.18, %H20, %2.26.
…………………………..
SEE
JP 2009196984![]()
……………………………………………..
WO 2008013322![]()
http://www.google.com/patents/WO2008013322A1?cl=en
Scheme 1 :

( III ) (ID
Scheme 2 :

( In the above scheme , R4 is bromine , or iodine , and the other symbols are the same as defined above.
The starting compounds of formula (V) can be prepared in accordance with the following scheme:

(V) (In the above scheme, the symbols are the same as defined above. )
The compounds of formula (XII ) can be prepared in accordance with the following scheme :

(In the above scheme, R5 is alkyl, and the other symbols are the same as defined above.)
Example 1 :
3- (4-Cyclopropylphenylmethyl) -4-fluoro-1- (β-D-gluco- pyranosyl) indole

OH
(1) A mixture of 4-fluoroindoline (185 mg) and D-glucose (267 mg) in H2O (0.74 ml) – ethyl alcohol (9 ml) was refluxed under argon atmosphere for 24 hours. The solvent was evaporated under reduced pressure to give crude 4-fluoro-1- (β-D-glucopyranosyl) indoline, whichwas used in the subsequent step without furtherpurification.
(2) The above compound was suspended in chloroform (8 ml) , and thereto were added successively pyridine (0.873 ml), acetic anhydride (1.02 ml) and 4- (dimethylamino) pyridine (a catalytic amount) . After being stirred at room temperature for 21 hours, the reaction solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate , and the solution was washed witha 10 % aqueous copper (II) sulfate solutiontwice anda saturated aqueous sodium hydrogen carbonate solution, and dried over magnesium sulfate. The insoluble materials were filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane : ethyl acetate = 90 : 10 – 60 : 40) to give 4-fluoro-1- (2, 3, 4, 6- tetra-O-acetyl-β-D-glucopyranosyl) indoline (365 mg) as colorless amorphous. APCI-Mass m/Z 468 (M+H) . 1H-NMR (DMSO-d6) δ 1.93 (s, 3H) , 1.96 (S1 3H) , 1.97 (s, 3H) , 2.00 (s, 3H) , 2.83 (ddd, J = 15.5, 10.5 and 10.3 Hz, IH) , 2.99 – 3.05 (m, IH) , 3.49 – 3.57 (m, 2H), 3.95 – 3.99 (m, IH), 4.07 – 4.11 (m, 2H), 4.95 (t, J = 9.5 Hz, IH) , 5.15 (t, J = 9.4 Hz, IH) , 5.42 (t, J= 9.6Hz, IH) , 5.49 (d, J= 9.3 Hz, IH) , 6.48 (t, J = 8.6 Hz, IH) , 6.60 (d, J = 8.0 Hz, IH) , 7.05 – 7.10 (m, IH) .
(3) The above compound (348 mg) was dissolved in 1,4-dioxane (14 ml), and thereto was added 2, 3-dichloro-5, 6-dicyano-l, 4- benzoquinone (306 mg) . After being stirred at room temperature for 33 hours , thereto was added a saturated aqueous sodium hydrogen carbonate solution (20 ml) , and the organic solvent was evaporated under reduced pressure. The residue was extracted with ethyl acetate twice, and the combinedorganic layerwas washedwithbrine, dried over magnesium sulfate and treated with activated carbon. The insoluble materials were filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane : ethyl acetate = 90 : 10 – 60 : 40) and recrystallization from ethyl alcohol to give 4-fluoro-1- (2,3,4, 6-tetra-O-acetyl-β-D-glucopyranosyl) indole (313 mg) as colorless crystals, mp 132-135°C. APCI-Mass m/Z 483 (M+NH4) . 1H-NMR (DMSO-d6) δ 1.64 (s, 3H), 1.97 (s, 3H), 1.99 (s, 3H), 2.04 (S, 3H), 4.10 (ABX, J = 12.4, 2.7 Hz, IH), 4.14 (ABX, J = 12.4, 5.2 Hz, IH) , 4.31 (ddd, J = 10.0, 5.2 and 2.7 Hz, IH) , 5.25 (t, J = 9.7 Hz, IH) , 5.53 (t, J = 9.5 Hz, IH) , 5.61 (t, J = 9.3 Hz, IH) , 6.22 (d, J = 9.0 Hz, IH) , 6.58 (d, J = 3.4 Hz, IH) , 6.88 (dd, J = 10.8, 7.9 Hz, IH) , 7.19 (td, J = 8.1, 5.3 Hz, IH) , 7.51 (d, J“ = 8.5 Hz, IH) , 7.53 (d, J = 3.4 Hz, IH) . (4) The above compound (3.50 g) and N, N-dimethylformamide (3.49 ml) were dissolved in 1, 2-dichloroethane (70 ml) , and thereto was added dropwise phosphorus (III) oxychloride (2.10 ml) . The mixture was stirred at 7O0C for 1 hour, and thereto was added water (100 ml) at 00C. The resultant mixture was extracted with ethyl acetate (200 ml) twice, and the combined organic layer was washed with brine (40 ml) and dried over magnesium sulfate. The insoluble materials were filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane : ethyl acetate = 90 : 10 – 50 : 50) and recrystallization from ethyl alcohol (20 ml) to give
4-fluoro-1- (2,3,4, 6-tetra-O-acetyl-β-D-glucopyranosyl) – indole-3 -carboxaldehyde (2.93 g) as colorless crystals, tnp 190 – 192°C. APCI-Mass m/Z 511 (M+NH4) . 1H-NMR (DMSO-de) δ 1.64 (s,
3H), 1.98 (s, 3H), 2.00 (s, 3H), 2.05 (s, 3H), 4.12 (A part of
ABX, J = 12.4, 2.5 Hz, IH) , 4.17 (B part of ABX, «7 = 12.4, 5.5
Hz, IH) , 4.33 (ddd, J= 10.0, 5.5 and 2.5 Hz, IH) , 5.32 (t, J= 9.8 Hz, IH) , 5.56 (t, J = 9.6 Hz, IH) , 5.66 (t, J = 9.3 Hz, IH) ,
6.36 (d, J = 9.0 Hz, IH) , 7.11 (dd, J = 10.6, 8.0 Hz, IH) , 7.38
(td, J = 8.1, 5.1 Hz, IH) , 7.65 (d, J = 8.3 Hz, IH) , 8.53 (s, IH) ,
10.0 (d, J = 2.9 Hz, IH) .
(5) To a mixture of magnesium turnings (664 mg) and 1, 2-dibromoethane (one drop) in tetrahydrofuran (40 ml) was added dropwise a solution of l-bromo-4-cyclopropylbenzene (see WO 96/07657) (5.2Ig) in tetrahydrofuran (12 ml) over 25 minutes under being stirred vigorously, and the mixture was vigorously stirred for 30 minutes at room temperature. The resultant mixture was then dropwise added to a solution of the above 4-fluoro-1- (2 , 3 , 4, 6- tetra-O-acetyl-β-D-glucopyranosyl) indole-3 -carboxaldehyde (4.35 g) in tetrahydrofuran (130 ml) over 15 minutes at -780C under argon atmosphere . The mixture was stirred at same temperature for 30 minutes, and thereto was added a saturated aqueous ammonium chloride solution (200 ml) . The resultant mixture was extracted with ethyl acetate (150 ml) twice, and the combined organic layer was dried over magnesium sulfate. The insoluble materials were filtered off, and the filtrate was evaporated under reduced pressure to give crude 4-cyclopropylphenyl 4-fluoro-l- (2,3,4, 6-tetra-O-acetyl-β-D-glucopyranosyl) indol-3-yl methanol, which was used in the subsequent step without further purification.
(6) To a stirred solution of the above compound and triethylsilane (2.11 ml) in dichloromethane (44 ml) – acetonitrile (87 ml) was added boron trifluoride -diethyl ether complex (1.34 ml) at O0C under argon atmosphere . The mixture was stirred at same temperature for 20 minutes, and thereto was added a saturated aqueous sodium





m/Z 479/481 (M+NH4) . 1H-NMR (DMSO-d6) δ 0.59 – 0.62 (m, 2H) , 0.88
– 0.91 (m, 2H) , 1.83 – 1.87 (m, IH) , 3.21 – 3.50 (m, 4H) , 3.57
– 3.63 (m, IH) , 3.65 – 3.71 (m, IH) , 4.18 (s, 2H) , 4.54 (t, J = 5.5 Hz, IH) , 5.10 (d, J = 5.3 Hz, IH) , 5.16 (d, J = 5.0 Hz, IH) , 5.23 (d, J“ = 5.8 Hz, IH) , 5.38 (d, J“ = 9.0 Hz, IH) , 6.97 (d, J“ = 8.2 Hz, 2H) , 7.01 (dd, J“ = 9.4, 2.0 Hz, IH) , 7.08 (d, J“ = 8.0 Hz, 2H) , 7.22 (s, IH) , 7.47 (dd, J = 10.1, 2.1 Hz, IH) .
……………………………………………………………..
US 20110065200![]()
http://www.google.com/patents/US20110065200![]()
Glucose analogs have long been used for the study of glucose transport and for the characterization of glucose transporters (for review, see Gatley (2003) J Nucl Med. 44(7):1082-6). Alpha-methylglucoside (AMG) is often the analog of choice for cell-based assays designed to study the activity of SGLT1 and/or SGLT2.


……………………………………..
WO 2009091082![]()
http://www.google.com/patents/WO2009091082A1?cl=en![]()
 R1 = FLUORO, R2= H
R1 = FLUORO, R2= H![]()
……………….
Novel Indole-N-glucoside, TA-1887 As a Sodium Glucose Cotransporter 2 Inhibitor for Treatment of Type 2 Diabetes ![]()
(ACS Medicinal Chemistry Letters) Thursday November 21st 2013![]()
Author(s): Sumihiro Nomura, Yasuo Yamamoto, Yosuke Matsumura, Kiyomi Ohba, Shigeki Sakamaki, Hirotaka Kimata, Keiko Nakayama, Chiaki Kuriyama, Yasuaki Matsushita, Kiichiro Ueta, Minoru Tsuda-Tsukimoto,
DOI:10.1021/ml400339b
GO TO: [Article]
http://pubs.acs.org/doi/full/10.1021/ml400339b![]()
………………
Organic Process Research & Development (2012), 16(11), 1727-1732.![]()

A practical synthesis of two N-glycoside indoles 1 and 2, identified as highly potent sodium-dependent glucose transporter (SGLT) inhibitors is described. Highlights of the synthetic process include a selective and quantitative Vilsmeier acylation and a high-yielding Grignard coupling reaction. The chemistry developed has been applied to prepare two separate SGLT inhibitors 1 and 2 for clinical evaluation without recourse to chromatography.
http://pubs.acs.org/doi/abs/10.1021/op3001355![]()
Preparation of (2R,3R,4S,5S,6R)-2-(3-(4-Cyclopropylbenzyl)-4-fluoro-1H-indol-1-yl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1)
To a solution of compound 6 (250 g, 0.413 mol) in MeOH (1.2 L) and THF (2.4 L) was added sodium methoxide (2.5 mL, 0.012 mol), ………….DELETED………………….. There was obtained 198.5 g (97.5% isolated yield based on free base form; 98.8 LCAP) of 1 EtOH/H2O solvate as an off-white crystalline solid. A slurry of the EtOH/H2O solvate 1 (198.5 g, 0.399 mol) in de-ionized H2O (3.2 L,) was warmed to 76 °C, and then the slurry was gradually cooled to 20 °C over 30 min. The white suspension was stirred at 20 °C for 20 min and then at 10 °C for 1 h. The solid was collected by filtration, washed with de-ionized H2O (100 mL × 2), dried in an oven at 50 °C for 20 h and further at 60 °C for 3 h to afford 177.4 g, (99.8% isolated yield, 98.6 LCAP) of 1 hemihydrate as an off-white crystalline solid, of which the 1H NMR showed no EtOH residue and the powder X-ray diffraction (pXRD) confirmed that it was a crystalline solid. TGA indicated it contained 2.3% of water.
Mp = 108–111 °C.
1H NMR (DMSO-d6, 300 MHz) δ 7.36 (d, J = 8.2 Hz, 1 H), 7.22 (s, 1 H), 7.14 (d, J = 8.1, 2 H), 7.10–7.0 (m, 1 H), 6.96 (d, J = 8.1 Hz, 2 H), 6.73 (dd, J = 7.5, 7.7 Hz, 1 H), 5.38 (d, J = 7.7 Hz, 1 H), 5.21 (d, J = 6.9 Hz, 1 H), 5.18 (d, J = 6.8 Hz, 1 H), 5.10 (d, J = 6.9 Hz, 1 H), 4.54 (t, J = 6.9, 1.8 Hz, 1 H), 4.04 (s, 2 H), 3.75–3.60 (m, 2 H), 3.52–3.30 (m, 3 H), 3.20–3.17 (m, 1 H), 1.84 (m, 1 H), 0.89 (m, 2 H), 0.61 (m, 2 H).
13C NMR (DMSO-d6, 75.47 MHz): δ 156.2, 140.8, 139.4, 138.2, 128.2 (2 C), 125.2 (2 C), 124.4, 121.8, 115.9, 112.8, 107.4, 104.2, 84.8, 79.3, 77.4, 71.7, 69.8, 60.8, 31.3, 14.6, 8.92 (2 C). LC–MS m/z MH+ = 428 (MH+), 450 [M + Na]+, 877 [2M + Na]+.
[α]25D = −0.026 (c = 0.302, CH3OH).
Anal. Calc’d for C24H26NFO5·0.54 H2O: C, 65.93; H, 6.24; N, 3.20; F, 4.35, H2O, 2.23. Found: C, 65.66; H, 6.16; N, 3.05; F, 4.18, H2O, 2.26.
…………………
Journal of Medicinal Chemistry (2010), 53(24), 8770-8774
http://pubs.acs.org/doi/abs/10.1021/jm101080v![]()
………………….
TETRAACETYL COMPD![]()
Organic Process Research & Development (2012), 16(11), 1727-1732.
http://pubs.acs.org/doi/full/10.1021/op3001355![]()
1003005-35-3
- C32 H34 F N O9
- 1H-Indole, 3-[(4-cyclopropylphenyl)methyl]-4-fluoro-1-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-
-
Preparation of (2R,3R,4S,5R,6R)-2-(acetoxymethyl)-6-(3-(4-cyclopropylbenzyl)-4-fluoro-1H-indol-1-yl)tetrahydro-2H-pyran-3,4,5-triyl Triacetate (6)To a stirred solution of 5 (82%, 334.6 g, 0.449 mol) in DCE (1.14 L) and MeCN (2.28 L) at 0 °C was added Et3SiH (108.6 mL, 0.671 mol) followed by the addition of boron trifluoride etherate (68.8 mL, 0.539 mol) ———DELETE………………….. There was obtained 228.6 g (85% isolated yield, 98.4 LCAP) of pure 6 as an off-white crystalline solid. Mp 168–169 °C. 1H NMR (DMSO-d6, 300 MHz) δ 7.47 (d, J = 7.2 Hz, 1H), 7.22 (s, 1H), 7.20–7.10 (m, 1H), 7.06 (d, J = 8.1, 2H), 6.95 (d, J = 8.1 Hz, 2H), 6.78 (dd, J = 7.3, 7.0 Hz, 1H), 6.16 (d, J = 7.1 Hz, 1H), 5.61–5.48 (m, 2H), 5.21 (t, J = 7.3, 7.1 Hz, 1H), 4.34 – 4.25 (m, 1H), 4.18–4.04 (m, 2H), 4.0 (s, 2H), 2.04 (s, 3H), 1.97 (s, 3H), 1.95 (s, 3H), 1.84 (m, 1H), 1.61 (s, 3H), 0.89 (m, 2H), 0.61 (m, 2H). 13C NMR (DMSO-d6, 75.47 MHz): δ 169.9, 169.5, 169.3, 168.3, 156.2, 140.9, 139.0, 137.9, 128.0 (2 C), 125.2 (2 C), 124.2, 122.7, 116.1, 114.1, 107.2, 105.0, 81.7, 73.0, 72.5, 69.8, 68.0, 62.0, 31.2, 20.4, 20.3, 20.2, 19.7, 14.6, 8.93 (2 C). HRMS: m/z = 596.2261 [M – 1]+. [α]25D = −0.008 (c = 0.306, CHCl3).

WO2005012326A1 * Jul 30, 2004 Feb 10, 2005 Tanabe Seiyaku Co Novel compounds having inhibitory activity against sodium-dependant transporter WO2006035796A1 * Sep 28, 2005 Apr 6, 2006 Norihiko Kikuchi 1-(β-D-GLYCOPYRANOSYL)-3-SUBSTITUTED NITROGENOUS HETEROCYCLIC COMPOUND, MEDICINAL COMPOSITION CONTAINING THE SAME, AND MEDICINAL USE THEREOF WO2010092124A1 * Feb 11, 2010 Aug 19, 2010 Boehringer Ingelheim International Gmbh Pharmaceutical composition comprising linagliptin and optionally a sglt2 inhibitor, and uses thereof WO2010092125A1 * Feb 11, 2010 Aug 19, 2010 Boehringer Ingelheim International Gmbh Pharmaceutical composition comprising a sglt2 inhibitor, a dpp-iv inhibitor and optionally a further antidiabetic agent and uses thereof WO2011143296A1 * May 11, 2011 Nov 17, 2011 Janssen Pharmaceutica Nv Pharmaceutical formulations comprising 1 – (beta-d-glucopyranosyl) – 2 -thienylmethylbenzene derivatives as inhibitors of sglt US8163704 Oct 18, 2010 Apr 24, 2012 Novartis Ag Glycoside derivatives and uses thereof US8466114 Mar 21, 2012 Jun 18, 2013 Novartis Ag Glycoside derivatives and uses thereof WO2009091082A1 * Jan 16, 2009 Jul 23, 2009 Mitsubishi Tanabe Pharma Corp Combination therapy comprising sglt inhibitors and dpp4 inhibitors WO2009117421A2 * Mar 17, 2009 Sep 24, 2009 Kalypsys, Inc. Heterocyclic modulators of gpr119 for treatment of disease WO2011048148A2 Oct 20, 2010 Apr 28, 2011 Novartis Ag Glycoside derivative and uses thereof WO2012089633A1 * Dec 22, 2011 Jul 5, 2012 Sanofi Novel pyrimidine derivatives, preparation thereof, and pharmaceutical use thereof as akt(pkb) phosphorylation inhibitors WO2012162113A1 * May 18, 2012 Nov 29, 2012 Janssen Pharmaceutica Nv Process for the preparation of compounds useful as inhibittors of sglt-2 WO2012162115A2 * May 18, 2012 Nov 29, 2012 Janssen Pharmaceutica Nv Process for the preparation of compounds useful as inhibitors of sglt-2 WO2013090550A1 * Dec 13, 2012 Jun 20, 2013 National Health Research Institutes Novel glycoside compounds US7666845 Dec 3, 2007 Feb 23, 2010 Janssen Pharmaceutica N.V. Compounds having inhibitory activity against sodium-dependent glucose transporter US8394772 Oct 20, 2010 Mar 12, 2013 Novartis Ag Glycoside derivative and uses thereof US8697658 Dec 13, 2012 Apr 15, 2014 National Health Research Institutes Glycoside compounds
A New Way to Treat Diabetes?

Type-2 diabetes is a severe metabolic disease caused by the loss of the cells producing the hormone insulin. Since this molecule controls the up-take of glucose from the circulation, diabetic patients accumulate pathological levels of sugar in their blood.
A New Way to Treat Diabetes?
A novel synthetic macrocycle inhibiting insulin-degrading enzyme shows potent anti-diabetic effects
Read more
http://www.chemistryviews.org/details/news/6210821/A_New_Way_to_Treat_Diabetes.html
Discovery of Imigliptin, a Novel Selective DPP-4 Inhibitor for the Treatment of Type 2 Diabetes


compd 27 (AS ABOVE) IN http://pubs.acs.org/doi/abs/10.1021/ml5001905
Imigliptin
- CAS OF FREE BASE 1314944-07-4
- C21 H24 N6 O
- Benzonitrile, 2-[[7-[(3R)-3-amino-1-piperidinyl]-2,3-dihydro-3,5-dimethyl-2-oxo-1H-imidazo[4,5-b]pyridin-1-yl]methyl]-
Sihuan Pharmaceutical
Imigliptin dihydrochloride is an orally-available dipeptidyl peptidase IV (CD26; DPP-IV; DP-IV) inhibitor in phase I clinical trials at Sihuan Pharmaceutical for the treatment of type 2 diabetes.
………………………………………………………………
http://www.google.com/patents/EP2524917A1?cl=en

(R)-2-[[7-(3-aminopiperidin-1-yl)-3,5-dimethyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl]methyl]benzonitrile AS TFA SALT
- 1314944-08-5 CAS
- C21 H24 N6 O . C2 H F3 O2
- Benzonitrile, 2-[[7-[(3R)-3-amino-1-piperidinyl]-2,3-dihydro-3,5-dimethyl-2-oxo-1H-imidazo[4,5-b]pyridin-1-yl]methyl]-, 2,2,2-trifluoroacetate (1:1)
………………………………………………………………………….
LEAD compd 1 as above ……….cas ………1314943-88-8
- C19 H19 N5 O2
- Benzonitrile, 2-[[7-[(3R)-3-amino-1-piperidinyl]-2-oxooxazolo[5,4-b]pyridin-1(2H)-yl]methyl]-
………………………………………
SEE POLYMORPHS
CN 102863440
http://www.google.com/patents/CN102863440A?cl=en
Dipeptidyl peptidase-IV (DPP-IV) inhibitors are a new generation of oral treatment of type 2 diabetes by enhancing the role of incretin activity, a non-insulin therapy. With conventional medicine for treating diabetes compared, DPP-IV inhibitors have not weight gain and edema and other adverse reactions. [0003] The compound shown in formula ⑴ (R) -2 – [[7 – (3 – amino-piperidine-I-yl) -3,5 – dimethyl-2 – oxo-2 ,3 – dihydro- -IH-imidazo [4,5-b] pyridin-I-yl] methyl] benzonitrile (referred to as the specification of compound A, in the patent application CN201010291056. 9 already described) is a DPP-IV inhibitor compounds , the DPP-IV has a strong inhibitory effect and high selectivity.
V
[0004] formula ⑴

[0005] In the crystalline drug development research is very important, compound crystal form, will result in its stability, solubility and other properties are different. Therefore, the inventors of the compound or its salt polymorph A lot of research carried out, whereby it was confirmed, and the invention of the compound A crystalline salt.
3, Invention
[0006] The object of the present invention is to solve the above problems and to provide better stability, better maneuverability, good bioavailability and solubility of the compound A or a salt thereof and method for preparing the crystalline form.
[0007] The present invention provides formula (I), the compound A dihydrochloride salt polymorph I: using Cu-K α radiation, to angle 2 Θ (°) represents an X-ray powder diffraction at 8. 7 ± 0. 2 °, 19.4 ± 0.2 °, 23. 5 ± 0. 2 °, 27. 2 ± 0. 2 ° at a characteristic peaks.
Butterfly NC N
[0008] formula ⑴

[0009] A compound of the dihydrochloride salt polymorph I, with Cu-Ka radiation, to angle 2 Θ (°) represents an X-ray powder diffraction peaks in addition to the features described above, it also at 12. 5 ± 0. 2 °, 22. 5 ± 0. 2 °, 25. 5 ± 0.2 ° at a characteristic peaks.
[0010] A compound of the dihydrochloride salt polymorph I, with Cu-κα exposed to radiation angle 2 Θ (°) represents an X-ray powder diffraction peaks in addition to the features described above, it also at 11.7 ± 0.2 °, 14.6 ± 0.2 °,
26. O ± 0.2 ° at a characteristic peak.
[0011] The present invention also provides the compound A dihydrochloride Preparation of polymorph I.
[0012] Compound A was dissolved in an organic solvent, and temperature, was added dropwise a stoichiometric ratio of hydrochloric acid, after the addition was complete stirring, filtered and dried to give the dihydrochloride salt of Compound A crystalline form I.
……………………………………………….
http://www.google.com/patents/EP2524917A1?cl=en
0r
WO 2011085643
-
Diabetes mellitus is a systemic chronic metabolic disease caused by a blood glucose level higher than normal level due to loss of blood glucose control. It is basically classified into four categories, including: type I (insulin-dependent) and type II (non-insulin-dependent), the other type and gestational diabetes. Type I and type II diabetes are primary diabetes, which are the two most common forms caused by the interaction of genetic and environmental factors. The cause of diabetes is very complicated, but in the final analysis, is due to absolute or relative insulin deficiency, or insulin resistance. It is characterized by the metabolic disorder of carbohydrate, protein, fat, electrolytes and water caused by absolute or relative insulin deficiency and the reduced sensitivity of target cells to insulin.
-
In recent years, because of the improvement of living level, changes in the diet structure, the increasingly intense pace of life and lifestyle of less exercise and many other factors, the global incidence of diabetes is rapidly increasing, so that diabetes has become the third chronic disease which has a serious threat to human health next to tumor and cardiovascular diseases. Presently, the number of the patients suffering from diabetes has exceeded 120 million in the world, and the number in our country is the second largest in the world. According to statistics, up to 40 million people have been diagnosed as diabetes in China, and the number of the patients is increasing at a rate of 1 million per year. Among them, patients having type I and type II diabetes accounted for 10% and 90% respectively. Diabetes has become the increasingly concerned public health issue.
-
The main drugs currently used for the treatment of type I diabetes are insulin preparations and their substitutes; for the treatment of type II diabetes, the main drugs are oral hypoglycemic agents, generally divided into sulfonylureas, biguanides, traditional Chinese medicine preparations, other hypoglycemic agents, and auxiliary medication. Although these drugs have good effects, they can not maintain long-term efficacy in reducing the high blood glucose, and can not effectively alleviate the condition against the cause of diabetes. Many of the anti-diabetic drugs can well control the blood glucose at the beginning, but their efficacy can not be maintained when the treatment using such drugs are continuously used. It is one of the main reasons why combination therapies or drugs in different classes are used. However, the existing anti-diabetic drugs is lack of long-term efficacy mainly because their mechanism of action is to increase the sensitivity of target tissues to insulin action or improve insulin-producing activity of pancreas, but these drugs have no targeted effect to the reduced function of the pancreatic β cell, which is the fundamental cause of diabetes.
-
Dipeptidyl peptidase-IV (DPP-IV) is widely present in the body, and is a cell surface protein involved in a variety of biological functions. It can degrade many active enzymes in vivo, such as glucagon like peptide-1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), neuropeptide, substance P, and chemokines and the like. The deficiency of GLP-1 and GIP is the main cause resulting in type II diabetes (i.e., non-insulin-dependent diabetes). DPP-IV inhibitor is a new generation of anti-diabetic drug. It protects the activity of GLP-1, GIP and the like, stimulates the secretion of insulin, lowers blood glucose level by inhibiting the activity of DPP-IV, and does not cause hypoglycemia, weight gain, edema and other side effects. Its effect for lowering blood glucose level stops when a normal blood glucose level has been reached, and hypoglycemia will not occur. It can be used for a long term, and can repair the function of β-cells.
-
Sitagliptin is the first marketed DPP-IV inhibitor. It rapidly became a “blockbuster” drug after marketed in 2006 by Merck. The FDA approved the saxagliptin developed by AstraZeneca and Bristol-Myers Squibb on July 31, 2009. SYR-322 developed by Takeda has an activity and selectivity better than that of sitagliptin and saxagliptin, and is currently in the phase of pre-registration. In addition, there are three drugs in clinical phase III: BI-1356 (linagliptin) developed by Boehringer Ingelheim, PF-734200 (gosogliptin) developed by Pfizer Inc, and PHX1149 (dutogliptin) developed by Phenomix Inc. Nine drugs are in the clinical phase II, and seven drugs are in clinical phase I.



-
However, the limited varieties of drugs can not satisfy the clinical requirements. Accordingly, there is an urgent need for development of many DPP-IV inhibitor drugs to satisfy the clinical use.



- Example 17 The preparation of (R)-2-[[7-(3-aminopiperidin-1-yl)-3,5-dimethyl-2-oxo-2,3-dihydro-1
- -imidazo[4,5-b]pyridin-1-yl]methyl]benzonitrile (Compound 17) trifluoroacetate

(1)2,4-dichloro-6-methyl-3-nitropyridine
-

-
6-methyl-3-nitropyridin-2,4-diol (1.7 g, 10 mmol) was dissolved in 10 mL POCl3, heated to 95°C, and stirred for 1.5 h. The excess POCl3 was removed through centrifugation. 100 mL ice water was carefully added. The reaction solution was extracted with ethyl acetate (80 mL×3). The organic phase was combined, washed with saturated brine, dried with anhydrous Na2SO4 and spinned to dryness to afford 1.773 g yellow powder with a yield of 85.7 %.

(2) (R)-1-(2-chloro-3-nitro-6-methylpyridin-4-yl)piperidin-3-yl tert-butyl carbamate
-

-
[0216]The specific operation referred to the step (1) described in Example 1 for details. 0.96 g 2,4-dichloro-6-methyl-3-nitropyridin (4.64 mmol), and 0.933 g R-tert-butylpiperidin-3-yl-carbamate (4.66 mmol) were charged to afford 1.1 g titled product with a yield of 63.9 %.

(3) (R)-1-(2-methylamino-3-nitro-6-methylpyridin-4-yl)piperidin-3-yl tert-butyl carbamate
-

-
The specific operation referred to the step (2) described in Example 1 for details, 1.1 g (R)-1-(2-chloro-3-nitro-6-methylpyridin-4-yl)piperidin-3-yl tert-butyl carbamate (2.97 mmol), and 5 mL 27 % solution of methylamine in alcohol were charged to afford 1.0 g titled product with a yield of 92.1 %.

(4) (R)-1-(2-methylamino-3-amino-6-methylpyridin-4-yl)piperidin-3-yl tert-butyl carbamate
-

-
The specific operation referred to the step (3) described in Example 1 for details. 1.0 g (R)-1-(2-methylamino-3-nitro-6-methylpyridin-4-yl)piperidin-3-yl tert-butyl carbamate (2.74 mmol), and 0.1 g 10% Pd-C were charged to afford 0.873 g titled product with a yield of 95 %.

(5)(R)-1-(3,5-dimethyl-2-oxo-2,3-dihydro-1
H
-

-
The specific operation referred to the step (4) described in Example 1 for details. 873 mg (R)-1-(2-methytamino-3-amino-6-methylpyridin-4-yl)piperidin-3-yl tert-butyl carbamate (2.60 mmol), 849 mg triphosgene (2.86 mmol), and 1.39 mL triethylamine (10.4 mmol) were charged to afford 0.813 g titled product with a yield of 86.5 %.
- -imidazo[4,5-b]pyridin-7-yl)piperidin-3-yl tert-butyl carbamate

(6)(R)-1-[1-(2-cyanobenzyl)-3,5-dimethyl-2-oxo-2,3-dihydro-1
H
-

-
The specific operation referred to the step (5) described in Example 1 for details.813 mg (R)-1-(3,5-dimethyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-7-yl)piperidin-3-yl tert-butyl carbamate (2.25 mmol), 441 mg 2-(bromomethyl)benzonitrile (2.25 mmol), and 621 mg potassium carbonate (4.50 mmol) were charged to afford 0.757 g titled product with a yield of 70.5%.
- -imidazo[4,5-b] pyridin-7-yl]piperidin-3-yl tert-butyl carbamate

(7)(R)-2-[[7-(3-aminopiperidin-1-yl)-3,5-dimethyl-2-oxo-2,3-dibydro-1-imidazo [4,5-b]pyridin-1-yl]methyl]benzonitrile trifluoroacetate
-

-
The specific operation referred to the step (6) described in Example 1 for details. 750 mg (R)-1-[1-(2-cyanobenzyl)-3,5-dimethyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin -7-yl]piperidin-3-yl tert-butyl carbamate (1.57 mmol), and 8.5 mL trifluoroacetic acid were charged to afford 0.680 g titled product with a yield of 88.3%.
Molecular formula: C21H24N6O Molecular weight: 376.45 Mass spectrum (M+H): 377.2
1H-NMR(D2O, 400 MHz): δ 7.64 (d, 1H), 7.42 (t, 1H), 7.29 (d, 1H), 6.93(d, 1H), 6.76(s, 1H), 5.39(d, 1H), 5.25(d, 1H), 3.27(s, 3H), 3.04(m, 1H), 2.90(m, 2H), 2.80-2.60 (m, 2H), 2.48 (m, 1H), 2.32 (s, 3H), 1.90 (m, 1H), 1.54 (m, 1H), 1.32 (m, 1H).
…………………….
PAPER
We report our discovery of a novel series of potent and selective dipeptidyl peptidase IV (DPP-4) inhibitors. Starting from a lead identified by scaffold-hopping approach, our discovery and development efforts were focused on exploring structure–activity relationships, optimizing pharmacokinetic profile, improving in vitro and in vivo efficacy, and evaluating safety profile. The selected candidate, Imigliptin, is now undergoing clinical trial.
Discovery of Imigliptin, a Novel Selective DPP-4 Inhibitor for the Treatment of Type 2 Diabetes
† Department of Project Management, Medicinal Chemistry, Process, Pharmacology, Drug Metabolism and Pharmacokenetics, Toxicology, XuanZhu Pharma, 2518 Tianchen Street, Jinan, Shandong, The People’s Republic of China
‡ School of Pharmaceutical Sciences & Institute of Human Virology, Sun Yat-Sen University, 132 East Circle Road at University City, Guangzhou, The People’s Republic of China
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/ml5001905
http://pubs.acs.org/doi/abs/10.1021/ml5001905
synthesis………http://pubs.acs.org/doi/suppl/10.1021/ml5001905/suppl_file/ml5001905_si_001.pdf
data for LEAD compd 1
mono-TFA solvate (160mg, 71%).
1H NMR (d-DMSO + D2O, 600 MHz):
δ
8.01 (d, 1 H), 7.89 (d, 1 H), 7.69 (t, 1 H),
7.53 (t, 1 H), 7.40 (d, 1 H), 7.13 (d, 1 H),
5.41 (d, 1 H), 5.30 (d, 1 H), 3.25 (d, 1 H), 3.05
(m, 1 H), 2.93 (d, 1 H), 2.77 (m, 1 H),
2.65 (m, 1H), 1.95 (m, 1 H), 1.66 (m, 1 H),
1.46-1.26 (m, 2 H).
Molecular Formula C19H19N5O2:(M+H) 350.2
compd 27
mono-TFA solvate (680 mg, 88%).1H NMR (D2O, 400 MHz):δ7.64 (d, 1 H), 7.42 (t, 1 H), 7.29 (d, 1 H), 6.93(d, 1 H),










6.76 (s, 1 H), 5.39 (d, 1 H), 5.25 (d, 1 H), 3.27(s, 3 H), 3.04 (m, 1 H), 2.90 (m, 2 H),
2.80-2.60 (m, 2 H), 2.48 (m, 1 H), 2.32 (s, 3 H), 1.90 (m, 1 H), 1.54 (m, 1 H), 1.32 (m,1 H).
Molecular Formula C21H24N6O: (M+H) 377.2.
……………………………………………………………………………………….
Start of the first 4 volunteers in Imigliptin Dihydrochloride Phase I clinical trial
2013-10-18 16:31:08 Copyfrom: Sihuan Pharmaceutical Holdings Group Ltd.
Sihuan R&D clinical research centre (based in Beijing) announced that four healthy volunteers (human subjects) were administrated Imigliptin Dihydrochloride at first dosage of 5mg this morning around 8:00 am on 18 Oct 2013, and they all are in good conditions without any observed adverse effects so far.This is the first category 1.1 innovative drug independently developed by Sihuan Group which has now officially entered into clinical trials; that is from laboratory research into human studies. The preclinical studies of Imigliptin Dihydrochloride, a novel DPP-4 inhibitor treating type II diabetes, demonstrate excellent in vitro and in vivo activities and selectivities. In animal studies, it can protect pancreatic β–cells in long-term treatment. Pharmacokinetic studies of Imigliptin Dihydrochloride show attractive profile of good oral bioavailability, fast absorption and onset, and longer half-life compatible with the once daily dosing. We anticipate the above mentioned preclinical profiles be confirmed in our ongoing clinical trials.
………………………..
CN102127070A, CN102127071A,CN102741251A, EP2524917A1, US8680089,US20120289497, WO2011085643A1,WO2011085643A8,
Sitagliptin (sitagliptin) is the first one listed on the DPP-IV inhibitor, in 2006 after the listing quickly became a blockbuster for Merck. July 31, 2009, FDA has approved AstraZeneca and Bristol-Myers Squibb developed saxagliptin (saxagliptin) listed. Takeda (Taketa)’s SYR-322 activity and selectivity are superior to sitagliptin and saxagliptin, is currently in pre-registration. In addition, there are three stages of drug is in phase III: Bo Mingge Yan Gehan’s BI-1356 (Iinagliptin), Pfizer’s PF-734200 (gosogliptin), phenomix company PHX 1149 (dutogliptin) [0007]
In phase II drug has nine, in phase I of seven.
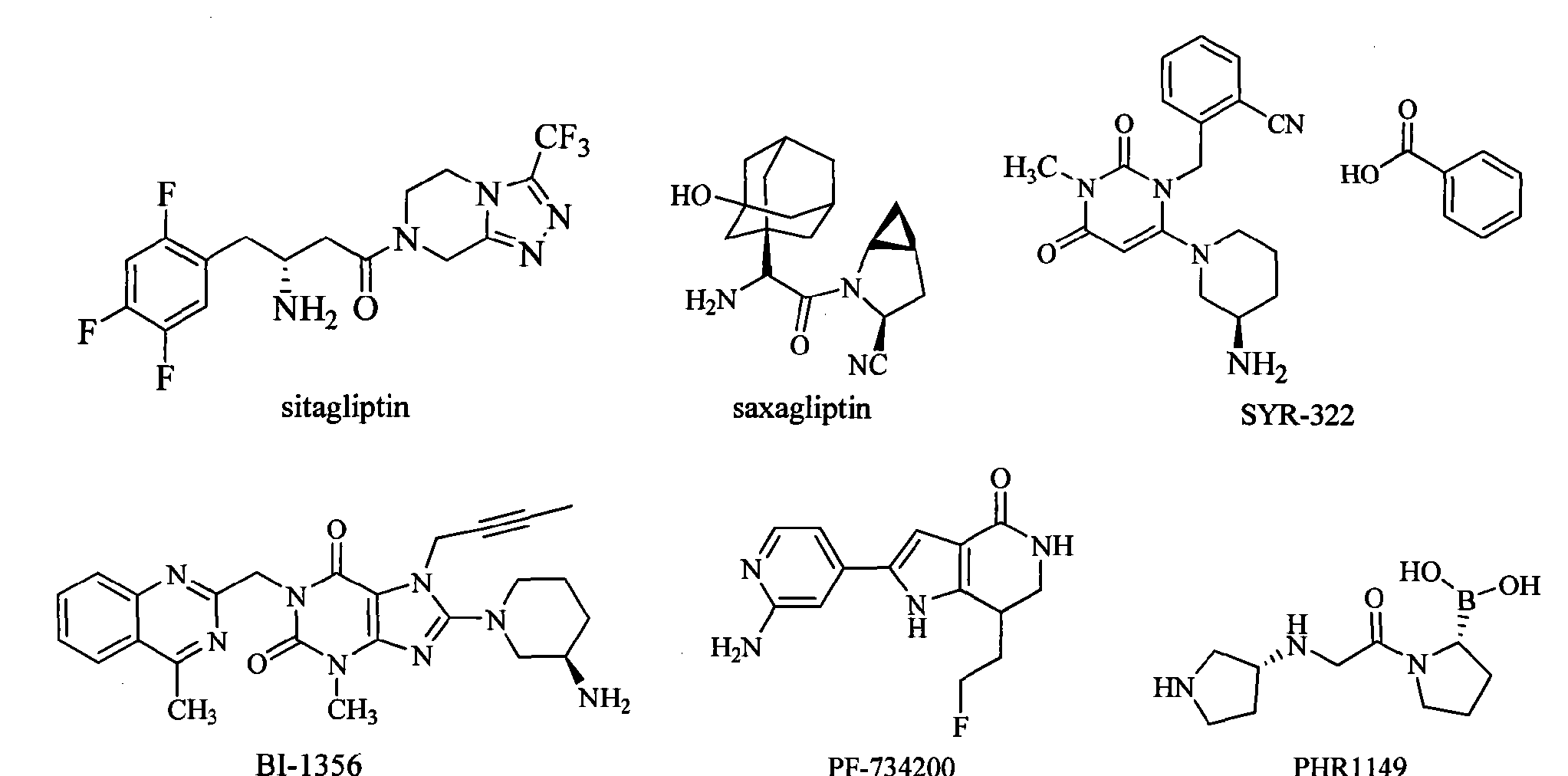
[0008] However, the limited varieties of drugs, can not meet the clinical needs, the urgent need to develop more of the DPP-IV inhibitor drugs to meet the clinical medication.
Example 17 (R)-2-ΓΓ7-(3 ~ amino-piperidin-yl) -3, 5_ dimethyl _2_ oxo, 3_ dihydro-IH-blind half and P “4,5 Pyridine-b1-i-a] benzonitrile Jiamou 1 (Compound 17) The system of the
[0451]

[0452] (1) 2,4 – dichloro-6 – methyl-nitropyridine _3_
[0453]

[0454] A mixture of 6 – methyl-3 – nitropyridine 2,4 – diol (1. Lg, IOmmol) dissolved in IOmL POCl3, heated to 95 ° C, stirred for 1.5 hours, rotating to excess POCl3 , ice water was added carefully IOOmL, extracted with ethyl acetate (80mLX3), the combined organic phases washed with saturated brine, dried over anhydrous Na2SO4, rotary done 1. 773g yellow powder, yield 85.7%.
[0455] (2) (R)-I-(2 – chloro-nitro _6_ _3_ _4_ picoline) piperidin-_3_ t-butyl carbamate
[0456]

[0457] Specific operation in Reference Example 1 (1), cast _ 2,4 dichloro-6 – methyl-_3_ nitropyridine 0. 96g (4. 64mmol), R-tert-butyl piperidin-_3_ yl – carbamate 0. 933g (4. 66mmol), to give the product 1. Ig, yield 63.9%.
[0458] (3) (R)-I-(2 – methylamino-nitro _6_ _3_ _4_ picoline) piperidin-_3_ t-butyl carbamate
[0459]

[0460] Specific operation in Reference Example 1 (2), cast (R) -1 – (2 – chloro-nitro _6_ picoline _3_ _4_ yl)-piperidin-3 – tert-butyl imino ester 1. Ig (2. 97mmol), 27% methylamine alcohol solution 5mL, to give the product 1. Og, yield 92.1%.
[0461] (4) (R)-I-(2 – methyl amino -3 – diamino-6 – methylpyridine _4_ yl) piperidin-_3_ t-butyl carbamate
[0462]

[0463] Specific operation in Reference Example 1 (3), cast (R)-l_ (2 – methylamino-methyl-4 _3_ nitro _6_ – yl) piperidin-3 – tert- butyl carbamate 1.0g (2. 74mmol), 10% Pd-C 0. lg, to give the product 0. 873g, 95% yield.
[0464] (5) (R)-I-(3,5 – dimethyl-2 – oxo-2 ,3 – dihydro-IH-imidazo [4,5 _b] pyridin _7_ yl)
Piperidin-3 – t-butyl carbamate
[0465]

[0466] Specific operation in Reference Example 1 (4), cast ((R)-l_ (2 – methylamino-4 _3_ methyl amino _6_ – yl) piperidin-3 – yl t-butyl carbamate 873mg (2. 60mmol), triphosgene 849mg (2. 86mmol), triethylamine 1. 39mL (10. 4mmol), to give the product 0. 813g, yield 86.5% 0
[0467] (6) (R)-l-[l_ (2 – cyano-benzyl) -3,5 _ dimethyl-2 – oxo-2 ,3 – dihydro-IH-imidazo [4, 5 -b] pyridin-7 – yl] piperidin-3 – t-butyl carbamate
[0468]

[0469] Specific operation in Reference Example 1 (5), cast (R)-I-(3,5 – dimethyl-2 – oxo-2 ,3 – dihydro-IH-imidazo [4, 5-b] pyridin-7 – yl) piperidin-3 – t-butyl carbamate 813mg (2. 25mmol), 2_ (bromomethyl) benzonitrile 441mg (2. 25mmol), potassium carbonate 621mg (4. 50mmol), to give the product 0. 757g, yield 70.5%.
[0470] (7) (R) -2 – [[7 – (3 – amino-piperidin-1 – yl) -3,5 – dimethyl-2 – oxo-2 ,3 – dihydro-IH- imidazo [4,5-b] pyridin-1 – yl] methyl] benzonitrile
[0471]

[0472] Specific operation in Reference Example 1 (6), cast (R)-l-[l_ (2 – cyano-benzyl) -3,5-dimethyl-2-_ – oxo – two H-IH-imidazo [4,5-b] pyridin-7 – yl] piperidin-3 – t-butyl carbamate 750mg (l. 57mmol), trifluoroacetic acid 8. 5mL, 0 to give the product . 680g, yield 88.3%.
[0473] MF = C21H24N6O MW: 376 * 45 MS (M + H): 377. 2
[0474] 1H-NMR (D2OdOOMHz): δ 1. 32 (1Η, m), 1. 54 (1H, m), 1. 90 (1H, m), 2. 32 (3H, s), 2. 48 (1H, m), 2. 80-2. 60 (m, 2H), 2. 90 (2H, m), 3. 04 (1H, m), 3. 27 (3H, s), 5. 25 ( 1H, d), 5. 39 (1H, d), 6. 76 (1H, s), 6. 93 (1H, d), 7. 29 (1H, d), 7. 42 (1H, t), 7. 64 (1H, d) ·
| WO2004050658A1 * | Dec 3, 2003 | Jun 17, 2004 | Boehringer Ingelheim Pharma | Novel substituted imidazo-pyridinones and imidazo-pyridazeiones, the production and use thereof as medicaments |
| WO2009099594A1 * | Feb 2, 2009 | Aug 13, 2009 | Luke W Ashcraft | Certain chemical entities, compositions and methods |
| WO2011085643A1 * | Jan 17, 2011 | Jul 21, 2011 | Kbp Biomedical Co., Ltd. | Fused pyridine derivatives |
| CN101228164A * | May 11, 2006 | Jul 23, 2008 | 布里斯托尔-迈尔斯·斯奎布公司 | Pyrrolopyridine-based inhibitors of dipeptidyl peptidase IV and methods |
How leptin, the ‘satiety hormone,’ reverses diabetes
Treatment with leptin, the hormone associated with fullness or satiety, reverses hyperglycemia in animals models of poorly controlled type 1 (T1D) and type 2 (T2D) diabetes by suppressing the neuroendocrine pathways that cause blood glucose levels to soar, a Yale-led team of researchers has found. The study appears in the Advance Online Publication of Nature Medicine.
The leptin hormone regulates metabolism, appetite, and body weight. The researchers discovered that, in a fasting state, rats with poorly controlled T1D and T2D diabetes had lower plasma insulin and leptin concentrations and large increases in concentrations of plasma corticosterone—a stress hormone made in the adrenal glands that raises levels of blood glucose.
The researchers then found that normalizing plasma leptin concentrations in the T1D rats with a leptin infusion resulted in marked reductions in plasma glucose concentrations, which could mostly be attributed to reduction in rates of liver conversion of…
View original post 180 more words
Lilly’s diabetes drug peglispro outshines world’s most popular insulin
| |
|---|
|
|||||||||||||||||||||||||

Lilly’s diabetes drug outshines world’s most popular insulin
World News | May 13, 2014
Late-stage clinical data has shown Lilly’s experimental diabetes drug Peglispro to be better at reducing blood sugar in patients with type II diabetes than Sanofi’s Lantus – the world’s most prescribed insulin.
The US drugmaker says it expects to file for approval of its basal insulin (BIL) by the first quarter of next year, after three Phase III studies showed it induced “a statistically superior reduction in HbA1c” compared with Lantus.
………….
